-
Residual DNA in modRNA-Based Vaccines

mRNA vaccines are considered one of the most significant biomedical developments of recent years. Billions of doses have been administered worldwide, while numerous additional mRNA-based applications are currently under development – ranging from cancer immunotherapies to vaccines against a wide variety of infectious diseases.
The public discussion over residual DNA in mRNA vaccines did not begin with a regulatory announcement or official safety warning, but rather somewhat coincidentally in the course of molecular biology sequencing work.
In early 2023, a research team led by the U.S. geneticist Kevin McKernan was working on RNA sequencing experiments that required highly purified RNA samples. As part of this work, the team used unopened vials of the COVID-19 vaccines produced by BioNTech/Pfizer and Moderna. During the analyses, the researchers came across an unexpected finding: in addition to the modified mRNA, DNA sequences were also detected in the samples.
It is well known in the pharmaceutical industry – and taken into account by regulators – that small amounts of residual DNA can technically occur in certain biotechnological manufacturing processes. What was surprising, however, was that at that time there had been little public discussion about which manufacturing processes were actually being used for large-scale production and to what extent independent analyses of the final products were available at all.
Subsequent studies by McKernan and other research groups ultimately reported the presence of residual DNA in various vaccine batches – in some cases in amounts that, according to the authors, exceeded regulatory guidelines. This sparked a scientific debate that continues to this day.
But what exactly is this all about?
The public debate quickly focused on seemingly simple questions:
Are there residual DNA – or not?
Are regulatory limits being met?
And if DNA is detectable, what biological significance would that actually have?However, on closer examination, it becomes clear that these questions are analytically much more complex than they first appear.
This is because the production of modRNA-based vaccines is already a multi-step process in which bacterial DNA templates are technically indispensable. The removal of this DNA is part of the standard purification steps in manufacturing. However, complete molecular separation in biological production processes is rarely a purely binary matter. What matters instead is:
Which fragments might remain in the final product?
In what quantity?
With what structure?
And above all:
What methods can be used to measure this reliably at all?The debate garnered additional attention following the public disclosure of discrepancies between early development methods and later large-scale industrial production. While comparatively small amounts of DNA were amplified in early clinical development phases using PCR-like methods, bacteria-based manufacturing processes were used for the subsequent billion-dose production. These processes are industrially well established and enable large-scale output, but they also come with more complex requirements for purifying biological residual components.
It was precisely this transition between different production processes that was later the subject of intense debate – partly because a large portion of the early clinical trials had still been conducted using vaccine batches produced via the original manufacturing process.
There is also another issue: The detection of residual DNA depends strongly on the methodology used.
Different laboratory groups used different digestion methods, extraction methods, PCR designs, and sequencing approaches – with results that in some cases differed significantly from one another. The discussion therefore concerns not only the measured values themselves, but also the question of how reliable and comparable these measurements actually are.
This two-part series of articles therefore does not attempt to provide hasty answers. Instead, its aim is to make the scientific and methodological background underlying the current debates understandable.
The first part describes the manufacturing and purification processes of modRNA-based vaccines:
How is the mRNA produced?
Why are bacterial DNA templates needed?
What byproducts are generated during production?
And what methods are used to remove unwanted residual components?The second part then focuses on the analytical perspective:
How can residual DNA be detected in the first place?
What is the interpretive value of qPCR, Qubit, or sequencing methods?
Why can different measurement techniques lead to different results?
And what do the independent studies published to date actually show?It becomes clear that a distinction must be made between analytical detection and biological assessment. Many questions are still scientifically unresolved – such as the native fragment distribution of residual DNA, the possible role of RNA, or the biological relevance of specific sequence elements under real physiological conditions.
This is precisely why the debate ultimately touches on a fundamental scientific question:
How does modern biomedicine deal with uncertainty, detection limits, and methodological complexity – especially in the context of novel biological technologies?
The following two parts are intended as an invitation to consider these questions in a nuanced way: not as a headline, but as a scientific problem whose assessment requires precision, transparency, and methodological understanding.
This presentation aims to bridge the gap between often highly technical academic publications and simplified media reports, so that even interested non-specialist readers without prior expertise can follow the complex interrelationships.
The text does not claim to be exhaustive – it strives for objectivity, accuracy, and fairness toward all perspectives involved.
About the article series
The analysis is divided into two parts that build on each other thematically. Both can be read independently; however, for a comprehensive understanding, the suggested order is recommended.
Part 1: Production and Purification
Table of Contents – Part 1
1. Manufacturing Processes – An Overview
1.1. Product Formation – From Gene to mRNA
1.2. Impurities and Byproducts
1.3. Purification Methods in the Manufacturing Process
1.4. Comparison: Process 1 vs. Process 2
1.5. Summary: Production and Purification of modRNAPart 2: Detection Methods and Current Evidence
Table of Contents – Part 2
2. Methods for Measuring DNA and RNA
2.1. Spectrophotometric Methods
2.2. Fluorescence-based Quantification
2.3. Amplification-based Methods
2.4. Sequencing-based Methods
2.5. Electrophoretic Fragment Analysis
2.6. Comparison of Key Detection Methods for Nucleic Acids
3. Guidelines for Limiting Residual DNA in Vaccines
4. Studies on Residual DNA in modRNA-based Vaccines
4.1. Why Independent Measurements?
4.2. Methodological Remarks
4.3. Overview and Individual Analyses of the Studies
4.4. Interim Conclusion: What do we know — and what do we not know?
5. Conclusion and Outlook
Current as of June 2026.
-
Residual DNA in modRNA-Based Vaccines – Part 1

Guide to the Article Series
Part 1: Production and Purification of modRNA
Part 2: Detection Methods and Current EvidenceBefore we turn to the question of why residual DNA may be detectable in mRNA vaccines, it is helpful to have a basic understanding of the manufacturing process.
The production of modRNA-based vaccines involves multiple sequential manufacturing and purification steps. The starting point is a DNA template that serves as the blueprint for the later nucleoside-modified mRNA (modRNA). During the manufacturing process, complex reaction mixtures are generated, from which unwanted residual components must be removed as thoroughly as possible.
Particular attention in recent years has focused on the fact that different methods were used to amplify this DNA template during development and later industrial production. These processes differ not only in terms of their technical scalability, but also with regard to the requirements for the subsequent purification of biological residual components.
This first part describes the fundamental manufacturing logic of modRNA-based vaccines, typical by-products and impurities, as well as the most important purification methods used during production. Finally, the two manufacturing pathways – Process 1 and Process 2 – are systematically compared.
It thus forms the basis for the second part of this series of articles, which focuses on analytical detection methods, regulatory limits and the experimental studies published to date on residual DNA in mRNA-based vaccines.
Note on terminology
📑
In general usage, the term „mRNA vaccine” is common. From a technical perspective, however, the approved products consist of nucleoside-modified mRNA (modRNA). For reasons of clarity and readability, this work primarily uses the established term „mRNA vaccine”.Table of contents
1. Manufacturing Processes – An Overview
1.1. Product Formation – From Gene to mRNA
1.2. Impurities and Byproducts
1.3. Purification Methods in the Manufacturing Process
1.4. Comparison: Process 1 vs. Process 2
1.5. Summary: Production and Purification of modRNA
1. Manufacturing Processes – An Overview
The following overview presents the key production steps in a simplified form. In industrial practice, individual procedures and process details may vary depending on the manufacturer.
The manufacturing process can be broadly divided into the following steps:
Step 1 🟢 Extraction of genetic information Step 2 🟢 Amplification of the spike protein DNA
Process 1: using PCR
Process 2: using bacteria💧 Purification of the DNA Step 3 🟢 In vitro transcription for the production of mRNA Step 4 🟢 RNA processing – maturation of the mRNA 💧 Purification of the DNA Step 5 🟢 Encapsulation of the mRNA in lipid nanoparticles (LNPs) 💧 Final purification While the upstream process generates the actual product – the mRNA – step by step, the downstream process serves to isolate the resulting mRNA from complex reaction mixtures, purify it, and formulate it for medical use.
1.1. Product Formation – From Gene to mRNA
Step 1: Extraction of genetic information
Step 2: Amplification of the spike protein DNA
Process 1: using PCR
Process 2: using bacteria
Step 3: In vitro transcription for the production of mRNA
Step 4: RNA processing – maturation of the mRNA
Step 5: Encapsulation of the mRNA in lipid nanoparticles (LNPs)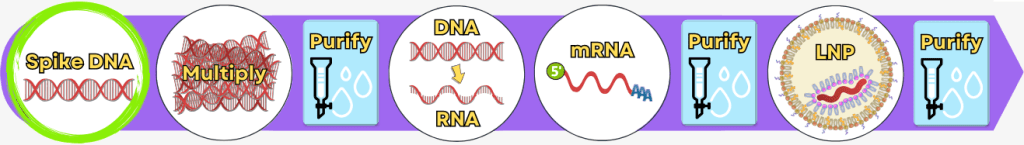
Step 1: Extraction of genetic information
The first step is to identify the genetic blueprint for the desired protein – in this case, the spike protein of the coronavirus.
In practice, the process unfolds as follows:
Collecting virus samples: Viral material is extracted from patient samples (e.g., throat or nasal swabs, blood, or tissue).
Note: There are differing views on whether SARS-CoV-2 was ever directly isolated from patient samples and cultured in a laboratory. This question is not addressed in this article, as the focus here is on the manufacturing process of mRNA vaccines.
Extracting genetic information: SARS-CoV-2 is an RNA virus. The viral RNA is isolated from the sample.
Converting RNA into DNA: Using a specific enzyme (reverse transcriptase), the viral RNA is converted into DNA. DNA is more stable and can be analyzed more easily.
Decoding the genome: The DNA is amplified, and the exact sequence of nucleotides (A, T, G, C) is determined.
Identifying the relevant section: Using computational analysis, the exact segment containing the blueprint for the spike protein is identified.
Entry into public databases: The resulting genetic blueprint is entered into public databases. This allows research teams worldwide to access the sequence without needing to isolate the virus itself.
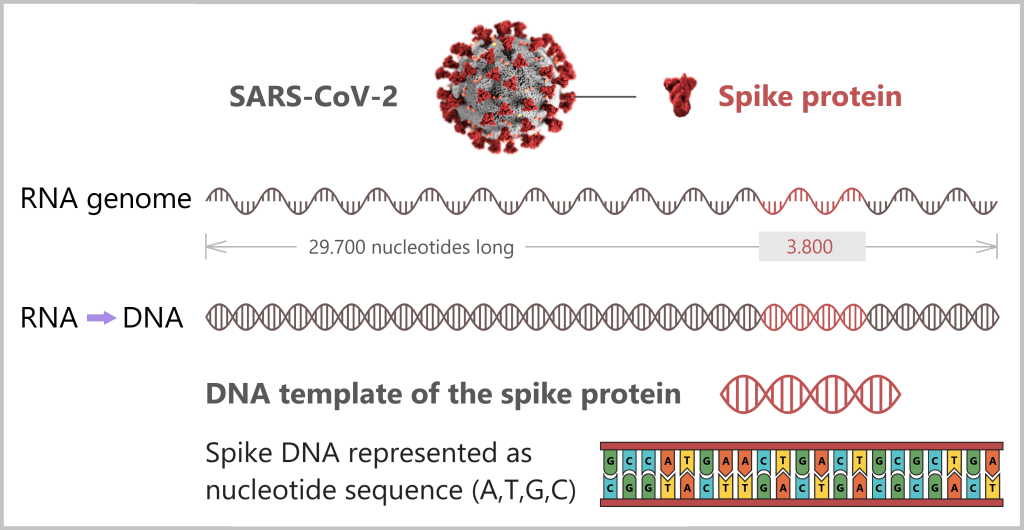
Fig. 1.1.1: From the virus to the DNA template Using the enzyme reverse transcriptase, a DNA version is generated from the RNA genome of SARS-CoV-2. The section containing the blueprint for the spike protein is identified and synthetically produced in the laboratory as a DNA template. This DNA template later serves as the basis for producing the mRNA used in the vaccine.
Note: The coronavirus SARS-CoV-2 stores its genetic information on a long RNA strand composed of individual nucleotides – the „letters” of the genetic code. With around 29,700 of these building blocks, it possesses one of the largest known RNA genomes. The blueprint for the characteristic spike protein comprises a segment of approximately 3,800 letters.
Creating the DNA template
Based on the determined genetic sequence (known as sequence data), a synthetic version of the spike gene can be produced in the laboratory. This DNA template later serves as a template for mRNA production.
Step 2: Amplification of the spike protein DNA
Once the blueprint for the spike protein has been produced in the form of a DNA template, it must be amplified millions of times. Only then is enough material available to subsequently produce mRNA from it.
Process 1: using PCR
Process 2: using bacteria

Process 1: Amplification of spike DNA using PCR
Process 1 uses a method that became widely known during the COVID-19 pandemic: the polymerase chain reaction (PCR). It is carried out in the laboratory – technically known as in vitro – and mimics natural DNA replication as it normally occurs in living cells.
What is needed for PCR?
To get nature’s copying machine running, a few ingredients are required:
- DNA template – the starting material is the spike protein DNA from step 1.
- Nucleotides – the building blocks: the four „letters” of DNA – adenine (A), thymine (T), cytosine (C), and guanine (G) – from which new strands are assembled.
- DNA polymerase – the builder: the enzyme that reads the template and constructs new DNA strands. Most commonly, the heat-stable Taq polymerase is used.
- Primers – the guide markers: short DNA fragments that indicate where the polymerase should start copying. Two are always required: a forward and a reverse primer.
- Buffer solution – the right environment: ensures the proper conditions for the polymerase to function reliably.
- Thermocycler – the temperature carousel: a device that automatically runs through the required temperature cycles.
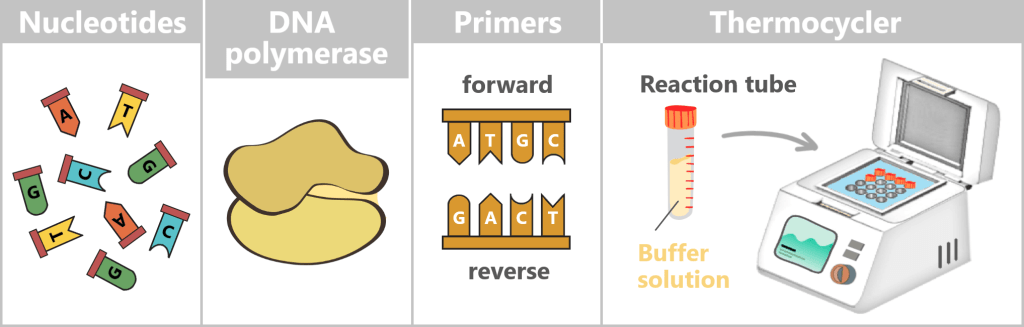
Fig. 1.1.2.1-A: Schematic representation of the ingredients and equipment For the subsequent mRNA production, the DNA template is already deliberately extended in this step. An additional DNA segment – the so-called T7 promoter – is attached to the spike sequence. This short sequence is not required for PCR itself, but only becomes relevant in the following process step. The T7 promoter serves as a binding site for T7 RNA polymerase, which, during in vitro transcription (IVT), transcribes the DNA into therapeutic mRNA.
The Polymerase Chain Reaction (PCR) Process
All ingredients are placed into a small reaction tube, which is then inserted into the thermocycler. The device controls the same three steps, which are repeated cyclically:
1) Separation of the DNA strands (denaturation): The sample is heated to approximately 94–98 °C for about 20-30 seconds. This breaks the hydrogen bonds between the DNA bases, causing the double strand to separate into two single strands. These then serve as templates in the next step.
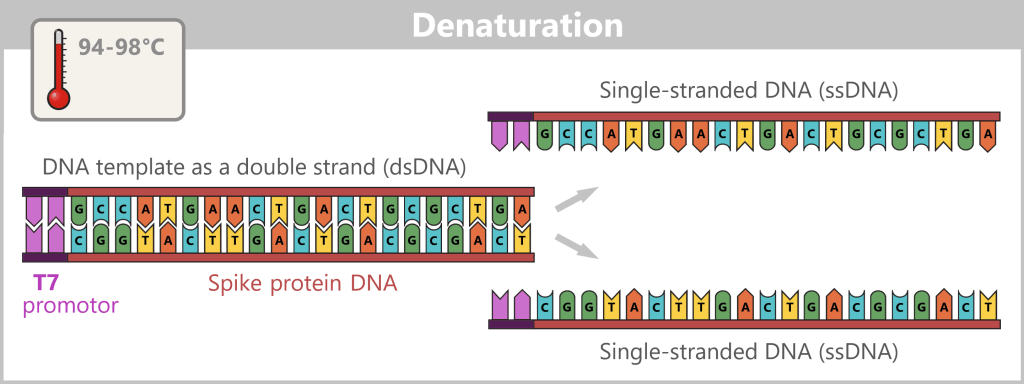
Fig. 1.1.2.1-B: Denaturation step of PCR The illustration is highly schematic and not to scale. The coding sequence for the SARS-CoV-2 spike protein comprises approximately 3,800 base pairs, while the T7 promoter consists of approximately 20 base pairs.
2) Primer binding (annealing): The temperature is lowered to 50–65 °C. The primers now bind specifically to the respective single DNA strands. They mark the starting point for DNA synthesis.
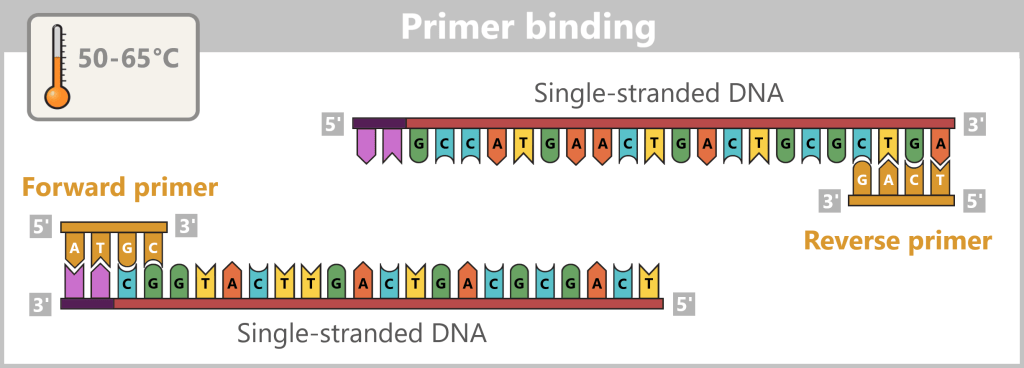
Fig. 1.1.2.1-C: Primer binding The illustration is highly schematic and not to scale. Primers are typically 20–25 nucleotides long.
3) DNA synthesis (amplification): At around 70 °C, the optimal temperature for the polymerase, the actual amplification begins. The DNA polymerase binds to the primer, reads the single strand in the 3′→5′ direction, and simultaneously synthesizes the new strand in the 5′→3′ direction. In doing so, it assembles nucleotides according to the principle of base pairing: A with T and G with C. In this way, two new double-stranded DNA molecules are produced.
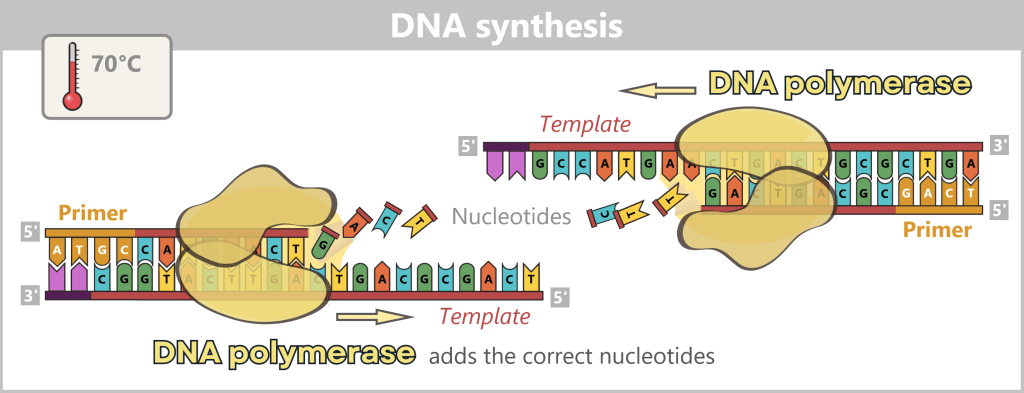
Fig. 1.1.2.1-D: DNA synthesis The illustration shows an active DNA polymerase moving along the exposed single strand like a molecular motor. The process of polymerization is clearly visible: individual nucleotides (the building blocks of DNA) enter the enzyme through an opening and are precisely added by the polymerase to the end of the growing strand. This process leads to elongation (extension) of the new DNA strand, which continuously grows in the 5′→3′ direction. Through this ongoing copying activity, amplification (replication) of the genetic information takes place, resulting in a new complementary double strand from the original template strand.
Why is synthesis always carried out in the 5′→3′ direction?
The structure of DNA
DNA consists of two strands that run in opposite directions – one in the 5’→3′ direction and the other in the 3’→5′ direction. The two strands are complementary to each other.
DNA polymerase, the enzyme that synthesizes new DNA, can work in only one direction: it reads the template strand in the 3’→5′ direction and synthesizes the new strand in the 5’→3′ direction.What do 5′ and 3′ mean?
To understand this, we need to look at the building blocks of DNA: the nucleotides. Each nucleotide consists of:- A base (A, C, G, or T)
- A sugar (deoxyribose)
- A phosphate group (P)
The sugar has five carbon atoms (C), which are numbered as follows:
- 1′: The base (A, T, C, or G) is attached here.
- 2′: In DNA, this position carries only a hydrogen atom (-H) and no hydroxyl group (-OH), unlike RNA. This missing OH group gives deoxyribose its name („deoxy” ribose, where „deoxy” means „without oxygen”).
- 3′: This is where a hydroxyl group (-OH) is located, which is essential for attaching new nucleotides during synthesis.
- 4′: Connects the sugar ring to the 5′ carbon.
- 5′: Carries the phosphate group, which links the nucleotide to the next one.
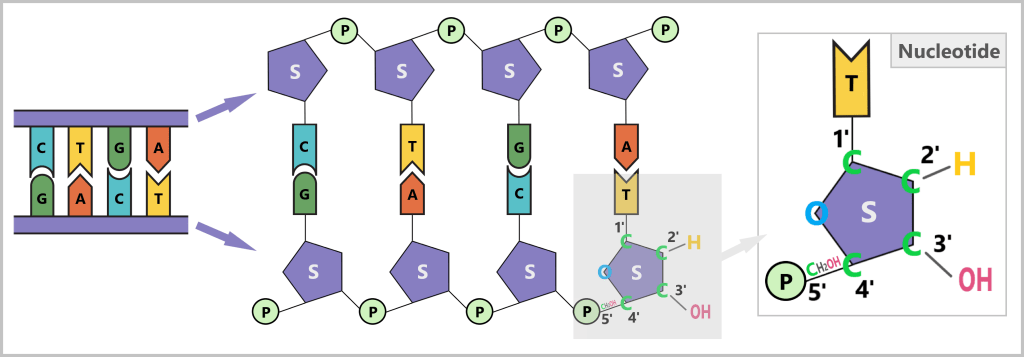
Why synthesis is only possible in the 5’→3′ direction
DNA polymerase can only add new nucleotides to the 3′ end of the growing strand. The reason is that this end carries a free hydroxyl group (–OH), which is required for forming the phosphodiester bond with the phosphate group of the incoming nucleotide.
At the 5′ end, a phosphate group is already present – no further nucleotide can be added there. Therefore, strand elongation is only possible in the 5′→3′ direction.In summary
DNA polymerase moves along the DNA template strand in the 3’→5′ direction. At the same time, it adds complementary DNA nucleotides to the new DNA strand, always at its free 3′ end. In this way, the new strand grows continuously in the 5’→3′ direction.Cycle repetition
The newly formed DNA double strands serve directly as templates for the next round. The steps denaturation – primer binding – DNA synthesis are repeated cyclically.
With each cycle, the amount of DNA doubles: 1 → 2 → 4 → 8 → 16 → 32 …
After only 25–40 cycles, billions of copies of the spike protein DNA are present.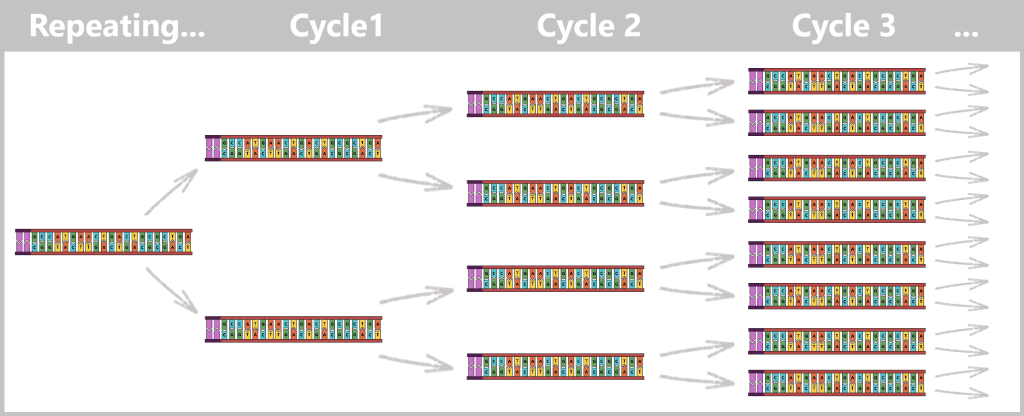
Fig. 1.1.2.1-E: Cycle repetition The result
At the end of PCR, you obtain a highly concentrated solution containing many copies of the spike blueprint – the starting material for the next step: the production of mRNA.
🎥 Tip: The video „What is PCR? Polymerase Chain Reaction” provides a clear visual summary of the process.

Process 2: Amplification of spike DNA using bacteria
In contrast to PCR, which takes place in a test tube (in vitro), this approach uses living organisms, specifically bacteria. This is referred to as an in vivo process (Latin for „within the living”), because amplification occurs directly inside the cells.
a) Why bacteria?
b) The bacterium Escherichia coli (E. coli)
c) Plasmids – small DNA rings with a big impact
d) Plasmids in genetic engineering
e) Insertion of spike protein DNA into a plasmid
f) Transfer of modified plasmids into bacteria
g) Bacterial multiplication
h) Harvesting the bacteria
i) Isolation of modified plasmids
j) Linearization of spike protein DNAa) Why bacteria?
Bacteria are tiny, single-celled organisms that reproduce through simple cell division: one cell divides into two, those divide again – and in a very short time, this produces vast numbers of identical copies.
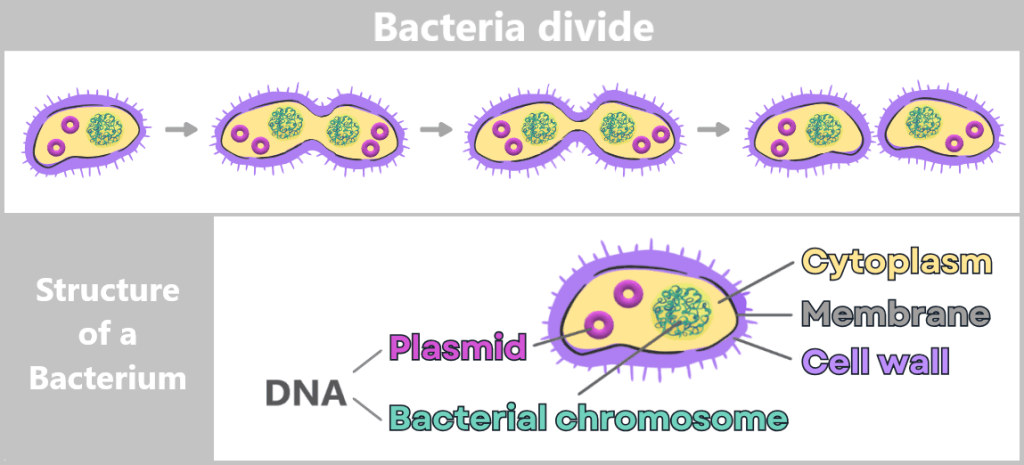
Fig. 1.1.2.2-A: Growth and structure of bacteria Since bacteria are asexual, they reproduce primarily through simple division. The process begins with cell growth and duplication of the genetic material. A constriction then forms, until two genetically identical daughter cells are ultimately produced.
The genetic information of bacteria – their DNA – is freely located inside the cell in the so-called cytoplasm. Most of it is stored in the bacterial chromosome. In addition, many bacteria contain small, circular DNA molecules called plasmids.
Adaptation through mutation and gene transfer
The continuous alteration of genetic material is essential for organisms to successfully adapt to new environmental conditions over generations. Bacteria are considered particularly adaptable. This is ensured by two mechanisms:Mutations: small random changes in the genetic material that can sometimes provide advantages (e.g., antibiotic resistance).
Gene transfer: bacteria can additionally exchange DNA segments, especially via their plasmids. In this way, beneficial traits can spread very rapidly through a bacterial population.
This is why bacteria are so suitable:
- Rapid reproduction: Under ideal conditions, many bacteria double every 20 minutes.
- Simple structure: Their DNA is not enclosed in a cell nucleus but lies freely within the cell, which facilitates manipulation.
- Plasmids as additional DNA: These can be easily modified, transferred, and used for the amplification of foreign DNA – ideal for genetic engineering.
The ability to introduce genetic material into bacteria and have it replicated has been used by researchers for decades. In this way, bacteria become small „DNA factories”.
A historical breakthrough
In 1973, Stanley Cohen and Herbert Boyer achieved a groundbreaking experiment: they were the first to insert a foreign gene into bacteria. The result showed that the bacteria adopted the new gene and even expressed it. This demonstrated that genes can be transferred from one organism to another – marking the birth of modern genetic engineering. [The Cohen–Boyer experiment]b) The bacterium Escherichia coli (E. coli)
In research, the bacterium Escherichia coli (E. coli for short) is particularly widely used – for several reasons:
- It is easy to culture and grow in the laboratory.
- It reproduces very quickly: under optimal conditions, it divides every 20–30 minutes.
- Its genome has been extensively studied and well understood.
- It can be easily genetically manipulated.
- Its cultivation is inexpensive.
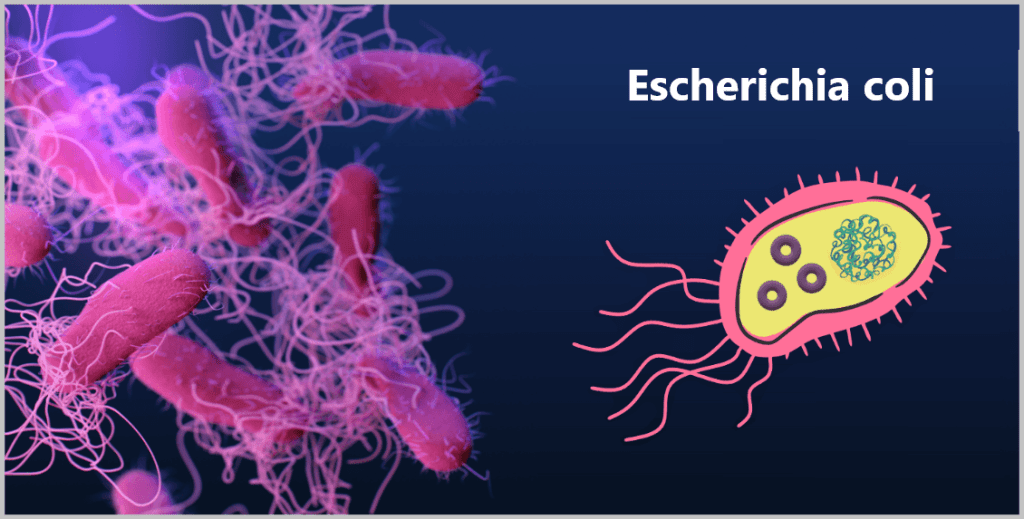
Fig. 1.1.2.2-B: The bacterium Escherichia coli On the left, a microscopic image; on the right, a simplified illustration showing DNA (large tangle) and plasmids (small rings).
Because E. coli is the most extensively studied organism in molecular biology and genetics, scientists jokingly refer to it as the „pet of geneticists“.
For these reasons, E. coli is one of the most important tools in biotechnology – and it also plays a central role in vaccine development.
A brief journey into the world of the E. coli bacterium
Escherichia coli – colorless, almost transparent – scarcely longer than one thousandth of a grain of sand – appears inconspicuous. And yet, it is anything but boring.
From the outside, it looks like a small capsule, but beneath its surface lies one of the busiest megacities at rush hour.
No space for emptiness. Molecule by molecule. Proteins, ribosomes, nucleic acids – they crowd together, collide, and make way for one another. A state that biologists soberly call macromolecular crowding, but which here feels like permanent congestion. And still – or precisely because of it – everything functions with breathtaking precision.
In the midst of this crowded space lies the cell’s memory: a single, circular DNA molecule. Everything is written here – how to consume sugars, how to swim, how to divide. If one were to gently stretch out this DNA, it would be a thousand times longer than the cell itself. A strand of millions of letters, tightly packed, repeatedly twisted, forced into loops and coils. This folding art is called supercoiling – as if someone had crammed a kilometer-long story into a matchbox without losing a single word.
Yet for all its power, this DNA rarely speaks directly. Instead, it continuously sends out messengers: RNA. Thousands, tens of thousands. Short messages, work instructions, construction plans. Some are only fleeting notes – mRNA, barely a thousand characters long, with a lifespan of minutes. Others are more stable and substantial: rRNA, the structural backbone of ribosomes. And then there are the small tRNA molecules, little more than assistants, yet essential in delivering the right building blocks at precisely the right moment. They dart through the cytoplasm like couriers. Their destination?
The ribosomes – the true giants of this world. Tens of thousands of them drift through the cytoplasm. They read the fleeting mRNA messages and turn them into tangible reality: proteins. They are silent, tireless, and they consume a large portion of the cell’s resources. Almost a quarter of all proteins here exist solely to produce new proteins.
And these proteins are everywhere. Millions of them, in thousands of forms. Enzymes that accelerate reactions which would otherwise take days or years – here they occur in fractions of a second. Some operate in bulk, performing the same task repeatedly. Others are rare specialists, perhaps only a few dozen copies, yet indispensable at the decisive moment. Among them patrol the guardians: RNases, which break down old messages. Once an RNA instruction has been executed, they disassemble it back into its basic components. This prevents the buildup of informational waste and allows the building blocks to be recycled for new instructions. Interspersed – in carefully regulated amounts – are DNases. They ensure that the valuable master plan in the archive is repaired if something goes wrong. At the same time, they eliminate foreign intruders and even extract energy from them.
All of this takes place within a shell that must be constantly renewed: a living boundary made of lipids, whose outer layer carries endotoxins. And it is under time pressure. Because this E. coli does not live to remain static – it lives to divide.
Under favourable conditions, it measures time in minutes. Twenty, perhaps thirty – then one cell must become two. While copying, reading, and building take place inside, the surface expands in parallel. Millions of new lipid molecules are produced, integrating themselves and extending the protective wall. It is a race without pause – a controlled chaos with a clear direction.
And then comes the moment of division. No dramatic cut, no ending – more a quiet drifting apart. The mother cell ceases to exist as an individual. Yet its story is not torn apart; it continues, twice. In two cells that are both old and new at the same time. Each carries the same long DNA within it, the same noise of molecules, the same restless order.
One can imagine the world inside the E. coli cell expanding to yet another, almost mysterious layer:
Alongside the large circular chromosome, smaller DNA rings drift through the crowded interior: plasmids. They are like loose pages in the cell’s internal archive – not strictly essential for survival, but often crucial in practice. Some carry instructions for new capabilities: resistance to a toxin, an enzyme that enables the use of an unusual nutrient source, sometimes only a small tactical advantage for difficult times.
They move freely, collide with ribosomes, are briefly bound by proteins, and released again. They, too, are read; their genes are transcribed into RNA, and their messages land on the same ribosomes as those of the main chromosome. In this overcrowded city, there are no special privileges – only function.
When the cell prepares for division, the plasmids begin to move. They must not be lost. Special proteins ensure that copies are made and that each of the resulting daughter cells receives at least one copy. A silent act of transmission – almost like slipping a letter into someone’s hand in a crowd before two paths separate.
Sometimes, however, their story does not end within their own lineage. Occasionally, a connection opens to the outside, to a neighbouring cell. In that moment, plasmids change hands. A brief contact, a transfer, and information jumps from one cellular history into another. In this way, traits can spread faster than any chromosomal mutation could ever achieve.
In this world, plasmids are the rumours, the blueprints, the survival tricks that are not carved in stone. Mobile, exchangeable, opportunistic. And precisely because of this, they fit perfectly into the restless, crowded interior of E. coli, where nothing ever stands still – not even the genetic material itself.
The illustrations by David Goodsell are highly recommended. He depicts the interior of cells true to scale – his images of E. coli make this molecular „crowding” truly tangible.
c) Plasmids – small DNA rings with a big impact
Plasmids are small, circular DNA molecules that occur in addition to the bacterial chromosome within the cell. They are much smaller than the main chromosome, may be present in varying copy numbers, and can replicate independently of it. Unlike the linear DNA of eukaryotes (e.g., humans), plasmids form closed circles of double-stranded DNA (dsDNA).
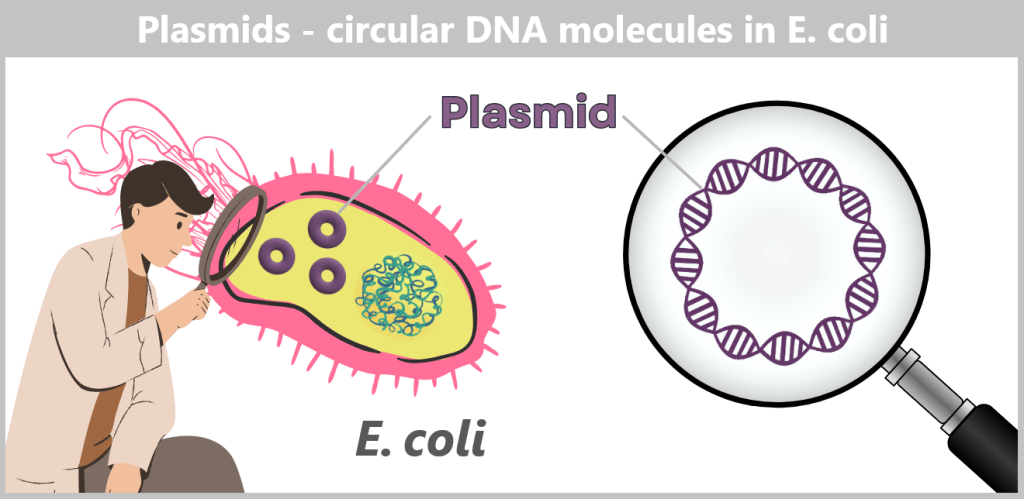
Fig. 1.1.2.2-C1: Schematic representation of the plasmid DNA of Escherichia coli Plasmids can carry a wide variety of genes – for example, genes conferring antibiotic resistance or encoding the production of specific proteins. To enable their targeted use by researchers, so-called plasmid maps are created: schematic representations showing the most important functional regions.
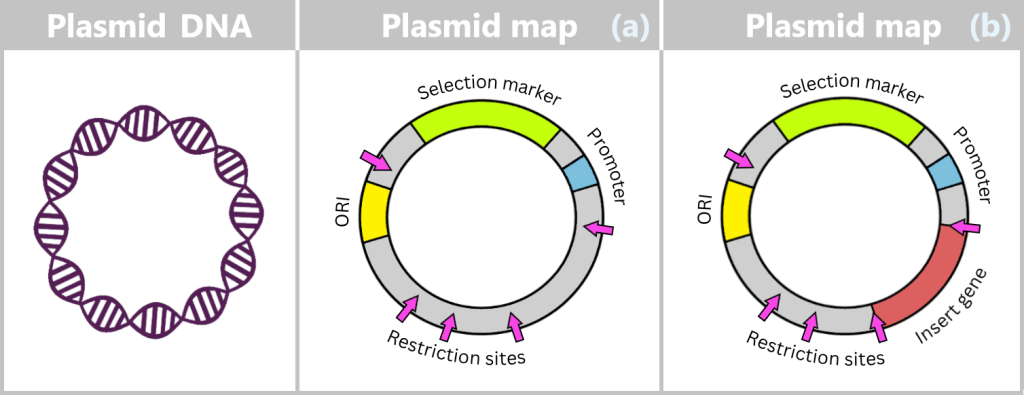
Fig. 1.1.2.2-C2: Plasmid map – schematic representation ORI (Origin of Replication): Starting point for the duplication of the plasmid. When environmental conditions and internal signals are favourable, the bacterium can „press this start button”, and the plasmid makes a copy of itself. Only if this ORI is „compatible” with the bacterium can the plasmid reliably replicate, even independently of cell division.
Selection markers: Genes that confer an advantage to bacteria, such as resistance to a specific antibiotic. They help researchers identify which bacteria carry the plasmid.
Promoter: The promoter is a specific DNA region that controls gene activity. It regulates when and how strongly certain genes on the plasmid are transcribed, thereby controlling the production of the corresponding proteins.
Restriction sites: These are short DNA sequences that can be recognized and specifically cleaved by restriction enzymes. These enzymes serve as a kind of immune system for bacteria, enabling them to cut the DNA of invading viruses and thus defend themselves.
d) Plasmids in genetic engineering
In bacteria, restriction enzymes actually serve to defend against foreign DNA. In genetic engineering, however, they are used as precise tools: tiny molecular scissors that cut DNA at very specific sites.
This creates a „gap” in a plasmid into which a desired gene can be inserted. Another enzyme, DNA ligase, then „glues” the DNA ends back together. The inserted gene is called the insert gene (see upper figure).
Through this process, plasmids are transformed into small gene shuttles: they specifically transport new genes into bacteria. With each cell division, the plasmid – and thus the insert gene – is automatically copied as well. In this way, entire bacterial cultures are created that act as „mini-factories”, producing specific proteins or DNA in large quantities.
🎥 Tip: A short animated introduction to plasmids can be found here.
e) Insertion of spike protein DNA into a plasmid
For the amplification of the SARS-CoV-2 spike protein sequence, the spike gene is first integrated into a bacterial plasmid – a process referred to as „cloning”. In this step, the molecular biology tools described earlier are used: restriction enzymes open the circular plasmid at defined sites, while DNA ligases precisely insert the spike gene and permanently join the DNA ends. In this way, a recombinant (newly assembled) plasmid is created that contains the genetic information for the desired antigen.
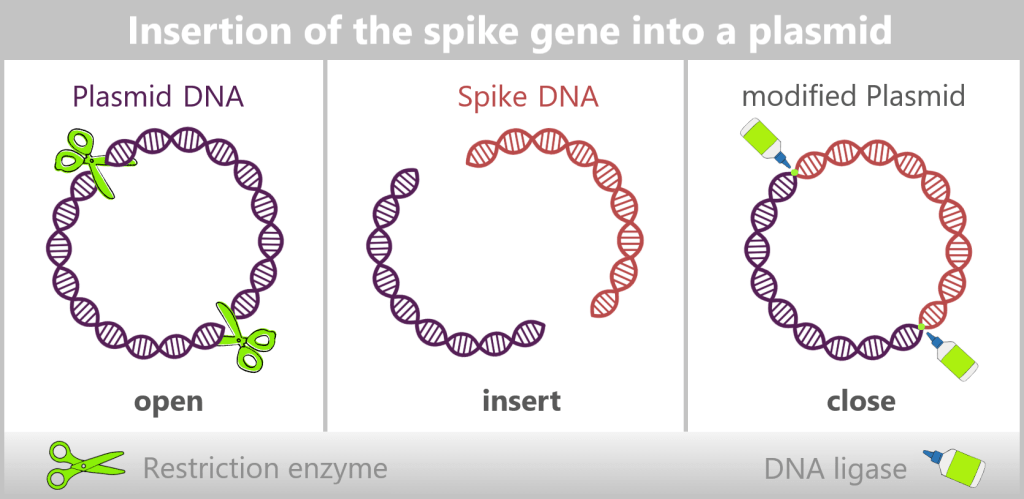
Fig. 1.1.2.2-E1: How the spike gene is inserted into a plasmid DNA elements within the plasmid
The spike DNA is not inserted into the plasmid in isolation, but together with additional genetic elements required for plasmid replication.
ORI (Origin of Replication): The origin of replication determines the site at which plasmid replication begins within the bacterial cell. Without this element, the plasmid could not be stably amplified in E. coli.
Spike protein gene: This gene contains the blueprint for the spike protein – the central antigen of the vaccine.
Antibiotic resistance gene: The resistance gene serves as a selection marker. It ensures that, under antibiotic pressure, only those bacteria survive that have taken up the desired plasmid. This allows suitable bacterial clones to be specifically selected and amplified.
- Moderna: kanamycin resistance gene
- Pfizer/BioNTech: Neo/Kan resistance gene (conferring resistance to neomycin and kanamycin)
SV40 components (Pfizer/BioNTech): The production plasmid used by Pfizer/BioNTech additionally contains regulatory sequence elements derived from Simian Virus 40 (SV40) – a monkey virus whose genetic components have been used in molecular biology for decades.
Specifically, these include:
- an SV40 promoter/enhancer,
- as well as parts of the SV40 origin of replication.
Such sequences are used in genetic engineering because they can enhance gene expression in mammalian cells and may facilitate plasmid replication in certain cell lines.
However, for bacterial amplification in E. coli, these elements have no known functional role, since bacteria do not possess the necessary cellular factors required for their activity.
Why these SV40 sequences are included in the final production plasmid is not fully documented in publicly available sources. It is discussed, among other things, that they may have been carried over from earlier development or testing systems and later retained.
According to current knowledge, Moderna does not use comparable SV40 components in its production plasmid.
The varying use of such regulatory sequences is among the issues currently being debated in scientific circles in connection with residual DNA.
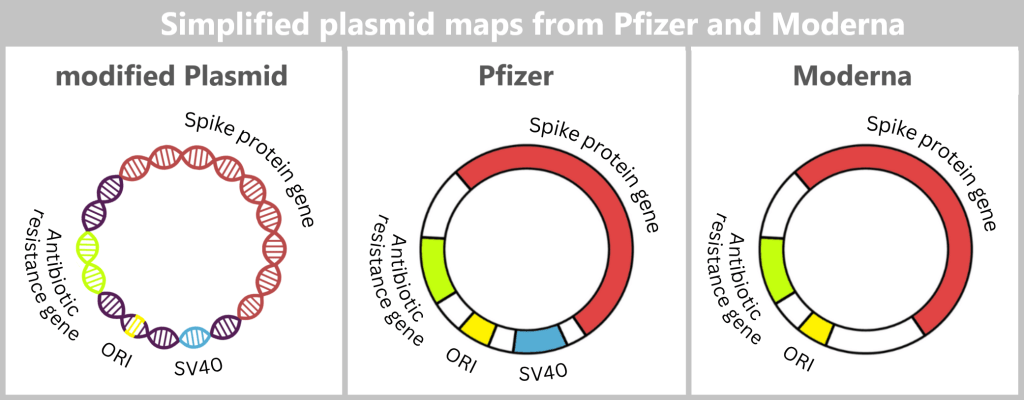
Fig. 1.1.2.2-E2: Simplified representation of the plasmid maps of Pfizer and Moderna A more detailed representation of the plasmid maps can be found here.
f) Transfer of modified plasmids into bacteria
The modified plasmids, which now carry the blueprint for the spike protein, are then introduced into E. coli bacteria – a process known as transformation. During this step, the bacteria take up the plasmids; these remain permanently within the bacterial cell and are passed on during each cell division.
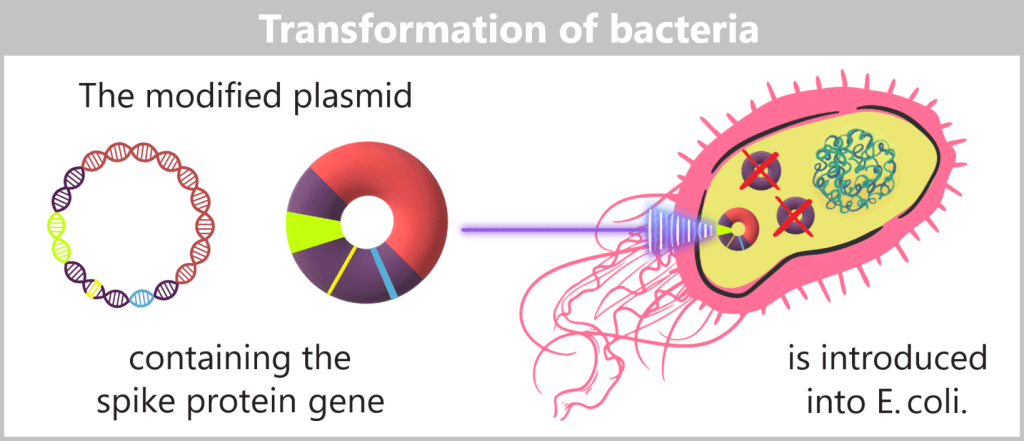
Fig. 1.1.2.2-F: Schematic representation of transformation The modified plasmids are introduced into E. coli bacteria via transformation. Plasmids used in genetic engineering to transport foreign DNA sequences are called vectors.
Plasmid-free host cells – for clean clones
In biotechnological plasmid production, only the modified plasmids are intended to be produced. Therefore, special plasmid-free bacterial strains are used that do not carry any of their own natural plasmids.
This has several reasons:
- Natural plasmids could compete for the cell’s replication machinery,
- they could exchange DNA segments through recombination,
- and they would make the genetic composition of the colony unpredictable.
By using plasmid-free host cells, it is ensured that all bacteria in a colony are genetically identical clones – each containing the same, defined plasmid with the desired sequence.
g) Bacterial multiplication
The E. coli bacteria carrying the modified plasmids are transferred into a fermenter. A fermenter, also called a bioreactor, is a device used in biotechnology for the large-scale production of products such as antibiotics, enzymes, vitamins, or vaccines. It allows precise control of conditions such as temperature, pH, oxygen supply, stirring speed, and nutrient availability.
The nutrient medium in the fermenter contains all essential substances required for bacterial growth. Under these optimal conditions, the bacteria begin to multiply rapidly. With each cell division, the plasmids are also copied, so that the desired DNA is amplified as well.
To ensure that only bacteria survive which actually carry the spike gene plasmid, an antibiotic is additionally added to the fermenter. Only cells containing the plasmid – and therefore the antibiotic resistance gene – can grow. This results in a culture composed exclusively of the desired, modified bacteria.
E. coli can divide approximately every 20–30 minutes. Within just a few days, this leads to an enormous bacterial population in the fermenter – containing trillions of copies of the spike plasmid.
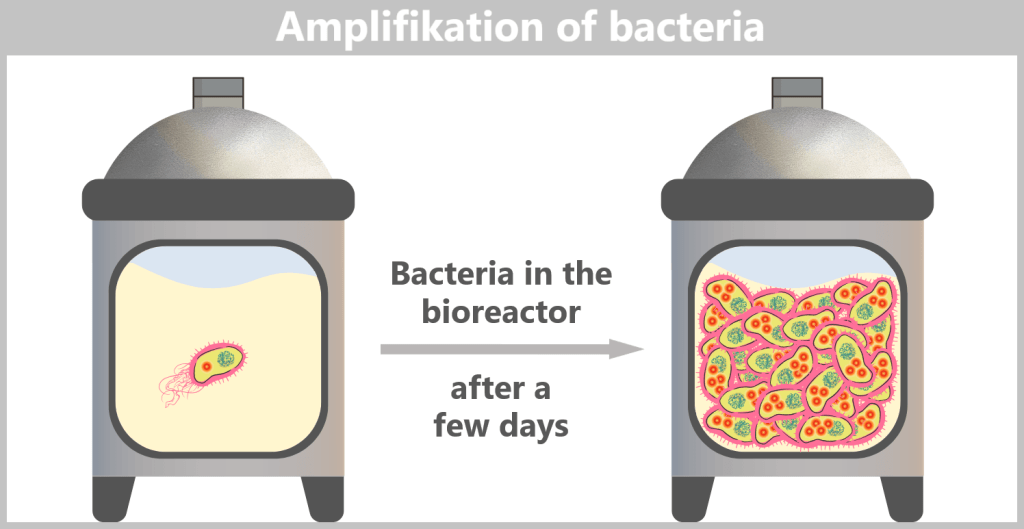
Fig. 1.1.2.2-G: Amplification of E. coli bacteria carrying the modified plasmid in a bioreactor h) Harvesting the bacteria
After the growth phase, the E. coli cells are „harvested”. For this purpose, the entire contents of the fermenter – the cell suspension – are transferred into a harvest tank. There, the separation of liquid and cells takes place, usually by centrifugation (rapid spinning) or filtration. In the end, a cell pellet is formed, meaning a concentrated collection of bacteria at the bottom of the container.
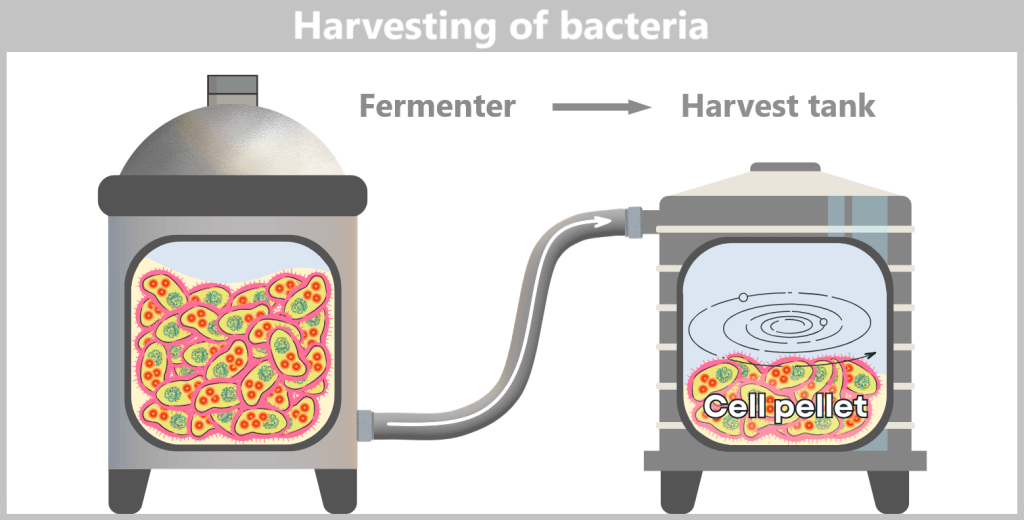
Fig. 1.1.2.2-H1: Transfer of fermenter contents to the harvest tank and formation of a cell pellet by centrifugation or filtration The cell pellet – now separated from the nutrient medium – is transferred to a downstream processing facility. It is either resuspended in a liquid to form a uniform, pumpable „cell slurry”, or it is automatically conveyed as a solid pellet. In modern facilities, all of this takes place in a closed system, without the biomass being exposed to the external environment.
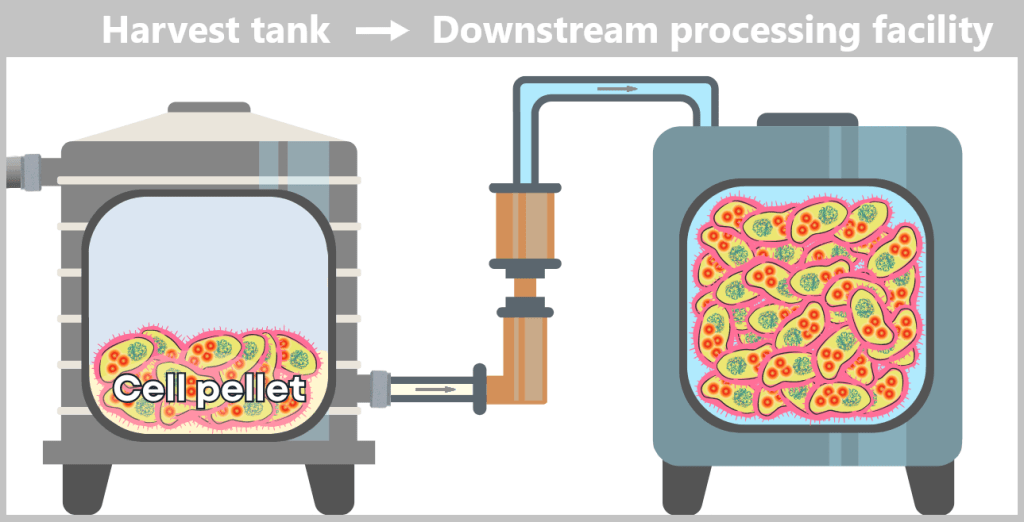
Fig. 1.1.2.2-H2: Transfer of the harvested material to the downstream processing facility i) Isolation of modified plasmids
After harvesting the bacterial cultures, a crucial separation step follows: the E. coli cells are specifically lysed in dedicated downstream processing systems, meaning they are carefully broken open.
The addition of sodium hydroxide (NaOH) and the detergent SDS (a specialized soap-like molecule) dissolves the lipid-based cell envelope, similar to how dishwashing detergent removes grease.
This releases the entire cellular content – a complex mixture consisting of the desired plasmids, chromosomal DNA, proteins, membrane components, and many other cellular substances.
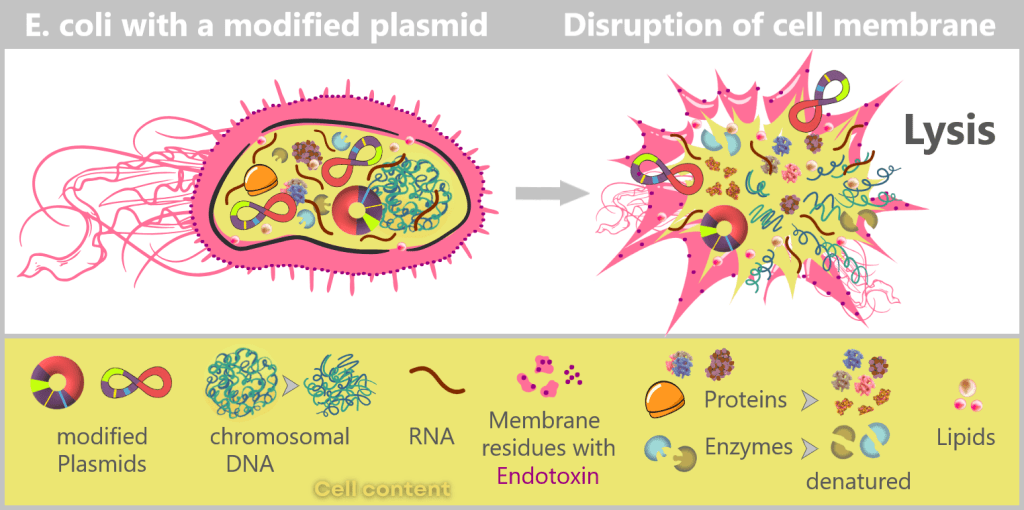
Fig. 1.1.2.2-I1: Lysis of bacteria and release of cellular contents Left: Schematic cross-section of an E. coli cell containing a modified plasmid. The outer membrane is shown with embedded endotoxin molecules (purple).
Right: After disruption of the cell membrane (lysis), the various cellular components are released – modified plasmids, chromosomal DNA (bacterial chromosome), proteins, enzymes, and lipids. The outer membrane appears in fragments containing structurally bound endotoxin (LPS).DNA forms in E. coli: plasmid and chromosome
Plasmids in Escherichia coli exist as small, circular DNA molecules. Within the cell, they are predominantly found in a supercoiled form. This topology (spatial arrangement) is biologically preferred because it provides high mechanical stability and compactness. The closed DNA ring is additionally twisted, allowing the molecule to be organized in a space-saving manner while remaining relatively resistant to physical stress.

The difference between supercoiled (superhelical) and relaxed (ring-shaped) plasmids lies exclusively in their topology. Both forms contain identical genetic information, but they exhibit markedly different physical properties.
In the supercoiled form, the DNA molecule is under torsional strain and tightly wound around itself. The relaxed form typically arises when one of the two DNA strands acquires a single-strand break (a nick). This nick relieves the torsional stress, and the plasmid adopts a more open, less compact circular structure.
The bacterial chromosome (genomic DNA, gDNA) is also circular, but many times larger. It is highly organized, associated with proteins, and folded into multiple domains.
Supplementary explanation regarding the mechanism of alkaline lysis
Destruction of genomic DNA, preservation of plasmids
During alkaline lysis, the cell membrane and cell wall are disrupted. The double-stranded DNA of both molecular types (genomic DNA and plasmids) is briefly denatured and subjected to mechanical and chemical shear forces.
The following occurs:
- The long, fibrous genomic DNA is broken and linearized by shear forces, as it is too large and fragile to remain intact.
- In contrast, the compact, supercoiled plasmid DNA remains largely intact.
When the previously alkaline solution is neutralized, the small circular plasmid DNA can renature correctly: its two single strands find each other again and re-form a stable double helix. As a result, it remains in solution.
The much longer genomic DNA can no longer fully renature under these conditions. It precipitates together with cell debris and proteins as an insoluble pellet.
Destruction of protein structures
Alkaline lysis affects not only nucleic acids but also proteins. Under strongly basic conditions, proteins lose their three-dimensional folding and thus their biological function. Large complexes such as ribosomes, which consist of ribosomal RNA and numerous proteins, are also completely destroyed and break down into their components. During subsequent neutralization, these denatured proteins and RNA fragments aggregate (clump together) – similar to how egg white solidifies when cooked – forming a precipitate that can be easily removed together with the remaining cell debris.
Why small RNA fragments appear after bacterial lysis
When E. coli cells are broken open, not only plasmids enter the lysate but also large amounts of bacterial RNA. This consists of a wide variety of molecules that were responsible for the bacteria’s metabolism – including many short, stable RNA types. In addition, bacterial enzymes that degrade RNA (RNases) are released during lysis. Active RNases can rapidly break existing RNA down into smaller pieces. Mechanical shear forces during the lysis process also contribute to fragmentation. The result is a complex mixture of short bacterial RNA fragments.
Natural RNA of the bacterial cell
The bacterial cell (E. coli) already contains a vast amount of its own natural RNA – significantly more than DNA. When the cells are disrupted (lysis), all of this cellular RNA enters the lysate and represents a major impurity. This includes:
- rRNA (ribosomal RNA): makes up the majority (~80–90%) of bacterial RNA and is present in large quantities.
- tRNA (transfer RNA): very small, stable molecules in many different variants.
- Bacterial mRNA: naturally short-lived in bacteria and often already partially degraded.
- Regulatory small RNAs (sRNAs): natural RNA species only a few dozen nucleotides in length.
In summary, this collection of ribosomal, transfer, and messenger RNA forms the natural cellular RNA pool of E. coli.
Destruction of the membrane and release of endotoxins
Endotoxins (lipopolysaccharides, LPS) are a natural component of the outer membrane of bacteria with a double-layered cell envelope such as E. coli. In scientific terminology, such bacteria are referred to as ‚Gram-negative‘. They provide structural stability and help the bacterium withstand external stress. During lysis, this membrane is disrupted, releasing large amounts of LPS. While endotoxins act as a kind of „armour” for the bacterium, they are highly toxic to humans. The biologically active component – lipid A – can trigger strong inflammatory responses in humans, which is why endotoxins are among the most critical contaminants in biotechnological production.
The challenge now lies in isolating the desired plasmids from this biochemical complexity and removing all unwanted accompanying substances. This involves a multi-stage purification process, which we will examine in more detail later in the section on Purification Methods in the Manufacturing Process.
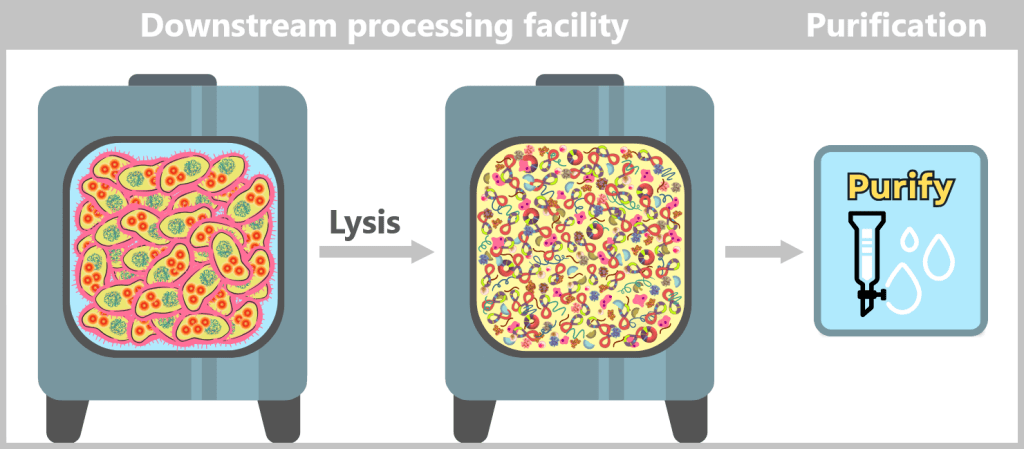
Fig. 1.1.2.2-I2: Lysis in the downstream processing facility and initiation of plasmid purification In the downstream processing facility, the harvested bacterial cells are lysed. This produces a „cell slurry” containing all cellular components. The purification process then begins, in which the desired plasmids are separated from the mixture using specific filtration and chromatography techniques.
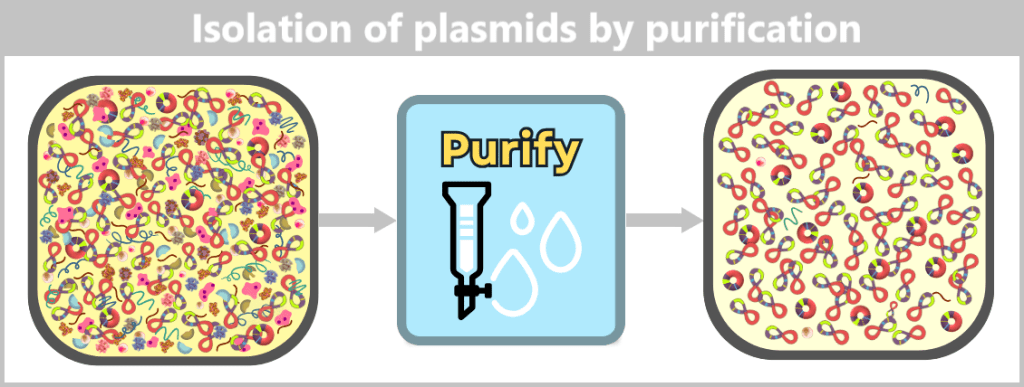
Fig. 1.1.2.2-I3: Result of plasmid isolation After purification, the plasmid DNA is obtained in high purity. In addition to the dominant supercoiled conformation, a small proportion of relaxed plasmids is visible. The latter arise from single-strand breaks (nicks) during cell lysis or downstream processing.
j) Linearization of spike protein DNA
The purified circular plasmids already contain the blueprint for the spike protein, but they are not yet suitable for the next step. For transcription, the circular plasmid DNA must be converted into a linear form.
To achieve this, the plasmids are specifically opened: a targeted restriction enzyme makes a precise cut at a defined site, typically after the end of the spike protein gene. In this way, clean DNA ends are generated that facilitate the reading of the sequence.
This cut converts the entire plasmid from its circular form into a linear, open DNA strand. This strand contains not only the spike gene, but also all other plasmid components, such as the origin of replication (ORI), selection markers, and regulatory elements. One particularly important of these sequences is the T7 promoter; this will become clear in the next step.
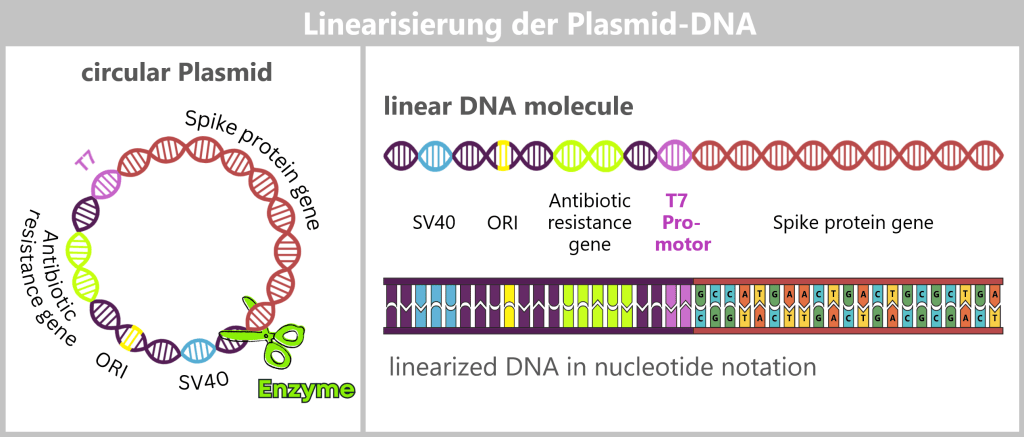
Fig. 1.1.2.2-J1: Linearization of plasmid DNA The circular plasmid is cut open with a restriction enzyme, resulting in a linear DNA molecule. This contains different sections: the origin of replication (ORI, yellow), selection markers (light green), regulatory sequences such as SV40 (blue) and the T7 promoter (purple), as well as the actual spike gene (red). In the detailed view below, the nucleotide sequence of the spike gene is indicated. The complete nucleotide sequence is shown in a strongly shortened form.
Further purification after linearization
The linearization of plasmid DNA generates additional reaction components and by-products that must be removed before in vitro transcription. These include, in particular:
- Restriction enzymes: The endonucleases used for the targeted cleavage must not remain in the subsequent manufacturing process.
- DNA by-products: These include incompletely linearized plasmids (e.g., residual supercoiled or relaxed forms) as well as short DNA fragments that may arise from nonspecific breaks or side reactions.
- Salts and buffer residues: The linearization reaction is carried out in specific buffer systems whose ions and additives could interfere with subsequent process steps.
Appropriate purification methods are used to obtain a highly purified linear DNA template, which serves as the template for the next process step.
In this context, the term highly purified does not describe an absolute state, but rather the extensive removal of process-related impurities to a regulatorily acceptable minimum. Small residual amounts of non-linear plasmid forms may remain and are further reduced in subsequent process steps.
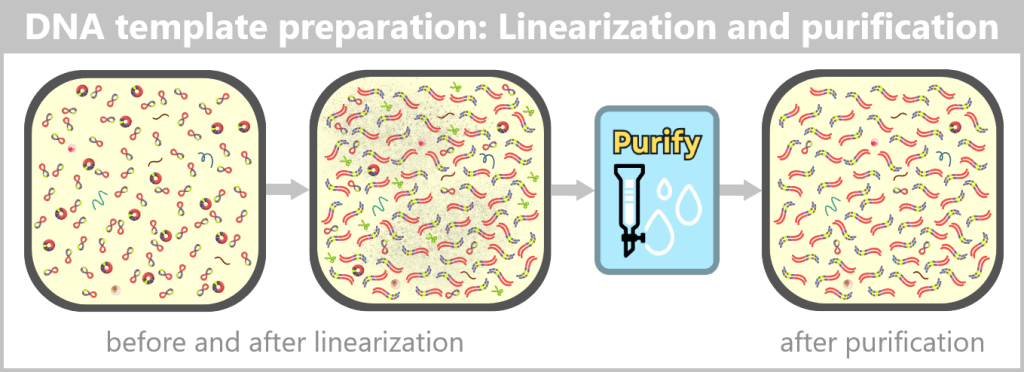
Fig. 1.1.2.2-J2: From plasmid to DNA template: production of the linear DNA template Left: The plasmid DNA solution before linearization. It contains a mixture of supercoiled (predominant) and relaxed (open circular) plasmid forms.
Center: The solution after linearization by a restriction enzyme. The circular plasmids have been cut at a defined site and now exist as linear, double-stranded DNA molecules. However, the solution also contains residual enzyme, buffer salts, and possible by-products.
Right: The solution after purification. Interfering components such as the restriction enzyme, buffer constituents, and unwanted DNA fragments have been removed.
Step 3: In vitro transcription for the production of mRNA
After the plasmid DNA has been linearized, it now serves as the template for in vitro transcription (IVT). The goal of this step is to produce the RNA molecule from the DNA that will later be used as the mRNA vaccine.
Transcription – the „rewriting” of DNA into RNA
Transcription takes place in a separate bioreactor under controlled conditions (e.g., specific pH, temperature, and ionic strength) that are optimized for RNA synthesis. Three components are required for this process:
1) The DNA template containing the spike gene.
2) The RNA building blocks, known as nucleotides.
3) An enzyme – T7 RNA polymerase – which specifically recognizes the T7 promoter. The T7 promoter was inserted into the plasmid directly upstream of the spike gene sequence and marks the starting point of transcription.Course of transcription
Initiation (start): The T7 RNA polymerase binds to the T7 promoter. Once the enzyme is anchored there, it opens the DNA double helix at the transcription start site and exposes one strand as the DNA template.
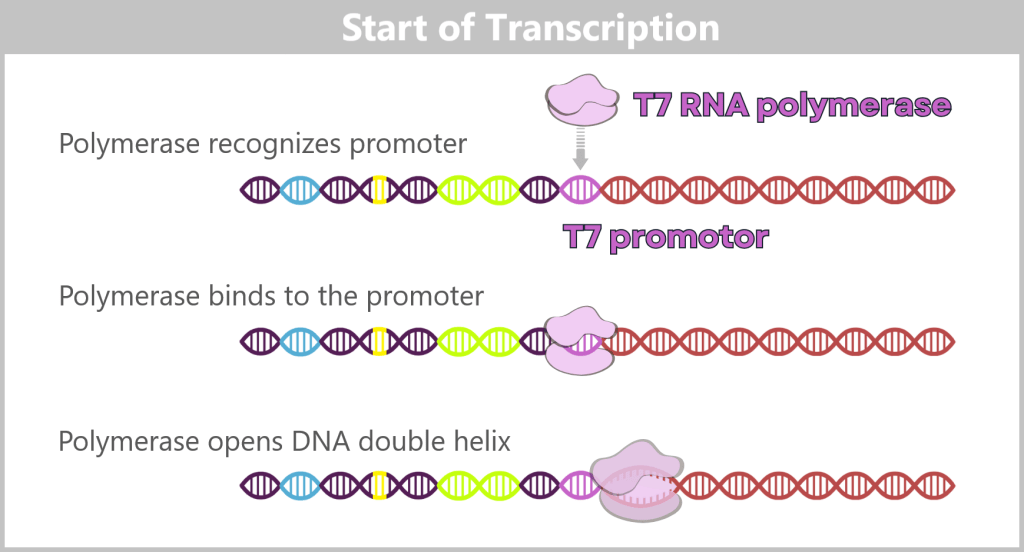
Fig. 1.1.3-A: Schematic representation of how T7 RNA polymerase binds to DNA. The DNA contains various functional regions, including the origin of replication (yellow), selection markers (light green), regulatory elements (blue), the T7 promoter, and the spike gene sequence (red). The polymerase recognizes the T7 promoter and binds precisely at this site. It then unwinds the DNA double helix and begins synthesizing the RNA strand along the spike gene.
Elongation (extension): The polymerase moves along the DNA template strand in the 3′ → 5′ direction and simultaneously synthesizes the complementary RNA strand by stepwise addition of nucleotides in the 5′ → 3′ direction. The following complementary base pairing applies:
- DNA A (adenine) → RNA U (uracil or m¹Ψ)
- DNA T (thymine) → RNA A (adenine)
- DNA C (cytosine) → RNA G (guanine)
- DNA G (guanine) → RNA C (cytosine)
Behind the polymerase, the DNA double helix re-forms.
Termination (end): At the end of the spike gene, there is a terminator sequence. Once this is reached, the polymerase stops, detaches from the DNA, and the completed RNA molecule is released.
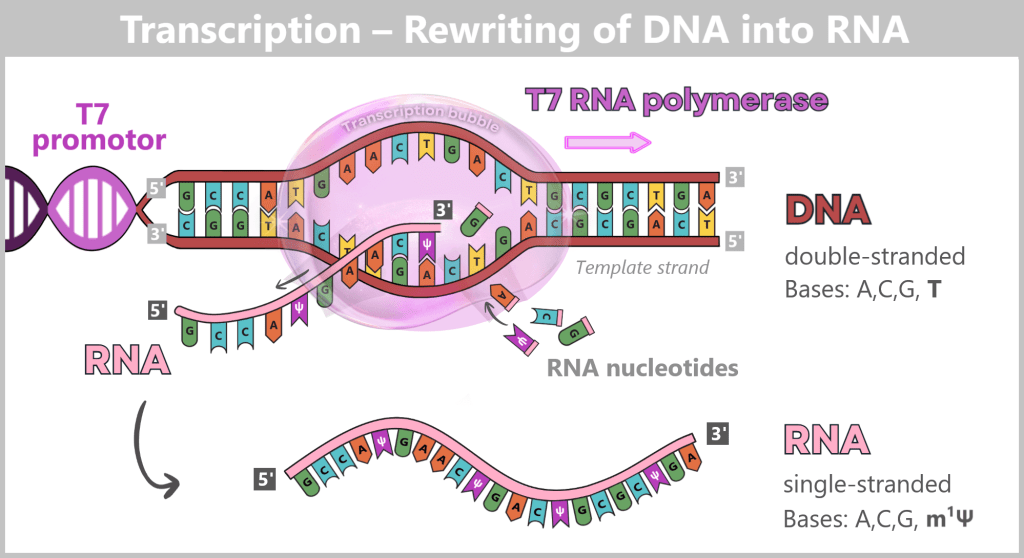
Fig. 1.1.3-B: The figure illustrates the molecular mechanism of transcription. T7 RNA polymerase binds to the DNA and separates the two strands. Along the template strand, matching RNA nucleotides are incorporated. In this stepwise process, a single-stranded RNA molecule is formed that carries the information of the spike gene.
The RNA produced in this way is single-stranded and corresponds in its base sequence to the spike gene.
A distinctive feature of mRNA production
To improve stability and tolerability for use in vaccines, a modified building block is used:
Instead of uridine (U), N¹-methyl-pseudouridine (m¹Ψ) is incorporated.
This modification makes the RNA more stable, protects it from rapid degradation, and reduces unwanted immune reactions.
Both Pfizer’s and Moderna’s mRNA vaccines contain N¹-methyl-pseudouridine (m¹Ψ) instead of uridine. [The Critical Contribution of Pseudouridine to mRNA COVID-19 Vaccines]
Comparison of DNA, mRNA, and modRNA
During in vitro transcription, double-stranded DNA is converted into single-stranded RNA. DNA and RNA are similar in their basic chemical structure but differ in key properties such as stability, lifetime, and biological function. In mRNA vaccines, however, natural mRNA is not used; instead, a chemically modified form (modRNA) is employed. This differs from natural mRNA in several essential aspects.
The following table compares the properties of DNA, natural mRNA, and synthetic modRNA.
Property DNA Natural mRNA Synthetic modRNA Definition Master archive: permanent storage of all genetic information Transcript of a single gene: contains the blueprint for a body´s own protein Gene copy with a „disguise”: laboratory-produced (synthetic) form of mRNA that has been chemically modified. It contains the blueprint for a „non-native” protein. Occurrence Universal: nucleus (in eukaryotes); also mitochondrial DNA Cell-specific: produced on demand only where the corresponding protein is needed. Non-specific: can be taken up by many cell types in the body via lipid nanoparticles Structure Double-stranded Single-stranded Single-stranded Sugar Desoxyribose Ribose Ribose Basen A – adenine
C – cytosine
G – guanine
T – thymineA – adenie
C – cytosine
G – guanine
U – uracilA – adenine
C – cytosine
G – guanine
m¹Ψ – N1-MethylpseudouridineLifetime and degradation rate Very stable: protected by the nuclear membrane and repair systems; normally not degraded except during cell death or targeted DNA breakdown. Short-lived: minutes to hours. Protein production is flexibly adapted to current metabolic needs. Extended: hours to days. By replacing uridine with N1-methylpseudouridine, modRNA is less strongly recognized by innate immune RNA sensors and is degraded more slowly. Degrading enzymes DNases (deoxyribonucleases) RNases (ribonucleases) RNases (reduced recognition and slower degradation) ✧ ✧ ✧
IVT by-products and impurities
After in vitro transcription (IVT), the result is not a pure product but a complex mixture. In addition to the desired mRNA, various by-products and impurities are formed as a result of the process, including short or long single-stranded RNA (ssRNA), double-stranded RNA (dsRNA), and RNA:DNA hybrids.
These accompanying products must be selectively removed, as they can otherwise impair the stability, efficacy, and tolerability of the vaccine. The purification methods used for this purpose are explained in more detail in Chapter 1.3.
Step 4: RNA processing – maturation of the mRNA
RNA processing comprises a series of modifications that occur during or after transcription in order to produce a mature, functional mRNA from the RNA.
In the production of mRNA for vaccines, efforts are made to mimic natural processes as closely as possible, as they normally occur in human cells. The synthetically produced mRNA is designed to imitate certain properties of naturally occurring mRNA, thereby making it stable and enabling efficient translation into the desired protein.
A functional vaccine mRNA requires, like normal human mRNA:
- a protective cap (5′ cap) at the front end of the RNA
- a stabilizing tail (poly-A tail) at the back end
The 5′ cap: a „security seal” for the cell
For a synthetically produced mRNA to function in the body, it must carry a specific chemical protective structure at its 5′ end – the Cap-1 structure. This cap is a key recognition feature for the cell and determines whether the mRNA remains stable, is efficiently translated, and is not recognized as foreign.
The components of the Cap-1 structure (m⁷GpppN¹m)
The Cap-1 structure is not a simple „cap”, but a precisely built molecule consisting of three functional components:
The recognition element: 7-methylguanosine (m⁷G)
A special guanosine building block with a methyl group. This modification acts like a molecular ID card: only mRNAs with this structure are recognized by the cell as correct and trustworthy.The linkage: triphosphate bridge (ppp)
Three phosphate groups connect the cap to the mRNA via an unusual 5′-5′ linkage. This special bond effectively protects the RNA end from enzymatic degradation.The disguise: modified first nucleotide (N¹m)
The first nucleotide of the mRNA is additionally 2′-O-methylated. This modification is crucial for protecting the mRNA from recognition by cellular RNA sensors.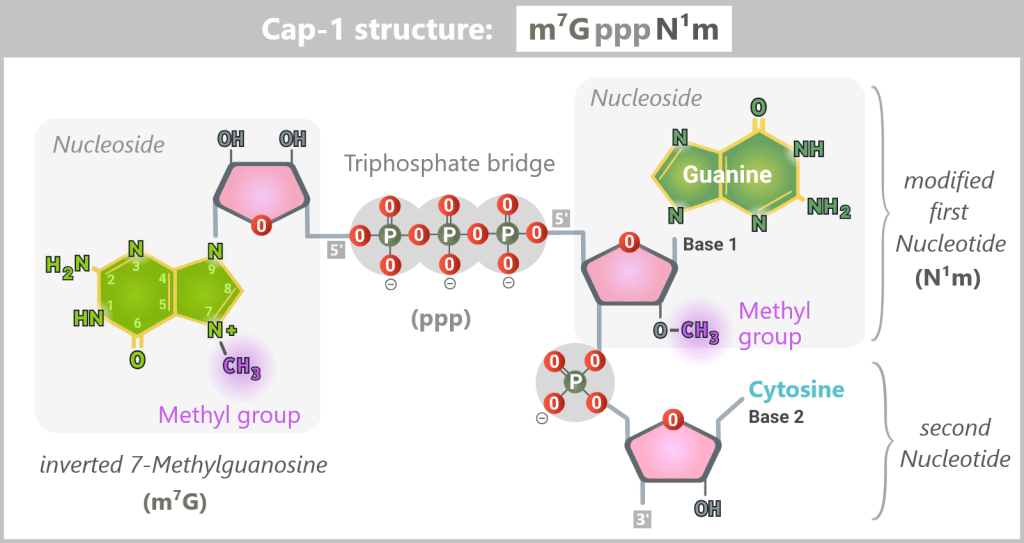
Fig. 1.1.4-A: Schematic representation of the Cap-1 structure (m⁷GpppN¹m) The 5′ end of the mRNA is linked to the first nucleotide (guanine in this example) via a 5′–5′ triphosphate bridge (ppp) and an invertedly linked, N7-methylated guanosine (m⁷G). Additionally, this first nucleotide bears a 2′-O-methylation (N¹m), which defines the Cap-1 structure and contributes to the stability, translational efficiency, and immune evasion of the mRNA.
During natural gene expression in human cells, mRNA is already processed while it is being synthesized in the nucleus. In particular, all mRNA molecules produced by RNA polymerase II receive a 5′ cap structure very early during transcription, along with additional chemical modifications.
Uncapped mRNA is non-functional under physiological conditions in eukaryotic cells: it is unstable, rapidly degraded, and does not reach the cytoplasm. This is because its 5′ end exists as a 5′ triphosphate (5′-ppp) structure, which is recognized and eliminated by cellular quality control systems as an abnormality (see lower figure).
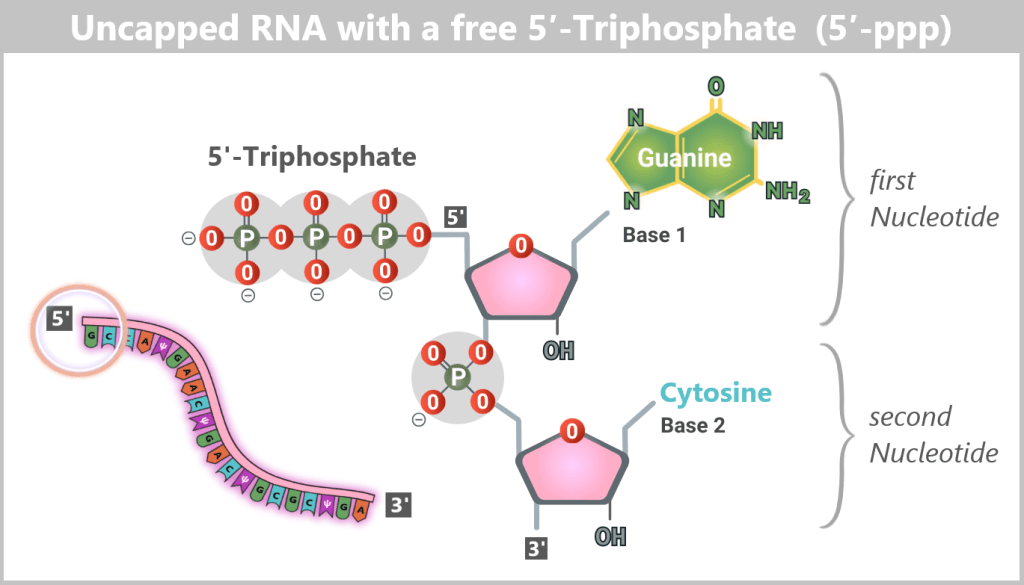
Fig. 1.1.4-B: Uncapped RNA – the free 5′-triphosphate is a key recognition motif for cytosolic immune sensors. Why is Cap-1 so important?
The Cap-1 structure fulfills three central functions:Immune evasion (camouflage): Cellular pattern recognition receptors such as RIG-I are highly sensitive to RNA with free 5′ triphosphates or unmodified ends. The Cap-1 structure of modRNA is chemically identical to the Cap-1 structure of the body’s own mRNA. Because cellular RNA sensors recognize this structure as „self”, no strong innate immune response is triggered under physiological conditions.
Initiation of protein production: The cap serves as a docking site for cap-binding proteins and marks the starting point for protein synthesis by ribosomes.
Stability: The cap protects the 5′ end of the mRNA from rapid enzymatic degradation, thereby extending its functional lifetime within the cell.
The poly-A tail: the protection and control center at the end
At the other end of the mRNA (the so-called 3′ end) is a long sequence consisting exclusively of adenine nucleotides – the poly-A tail. In vaccine production, it is usually made up of 100 to 150 ‘A’ units.
What is this tail for?
The „hourglass” of mRNA: The cell contains enzymes that gradually „nibble away” mRNA molecules from back to front. The poly-A tail acts as a buffer or protective extension at the end. It is degraded first, before the actual genetic information is attacked. The longer the tail, the longer the mRNA survives in the cell and the more protein can be produced.
Translation enhancer: Specific proteins bind simultaneously to the 5′ cap and the poly-A tail, forming the so-called closed-loop complex – an efficient „circular track”. On the one hand, this signals to the ribosome (the protein factory) that the mRNA is complete and ready for translation. On the other hand, after finishing the synthesis of one protein, the ribosome can directly restart translation at the 5′ end. This mechanism greatly increases the speed and efficiency of protein production.
✧ ✧ ✧
In summary: while the 5′ cap legitimizes the mRNA, disguises it, and makes it available for translation, the poly-A tail determines how long and how often the cell can use the information. Both modifications are therefore crucial for the lifetime of the mRNA and for its efficient translation into protein.
In the following schematic representations, the Cap-1 structure is shown as a functional 5′ unit positioned in front of the RNA strand, although chemically it represents a modified extension of the first nucleotide. For didactic reasons, the poly-A tail is symbolized by three adenine residues.
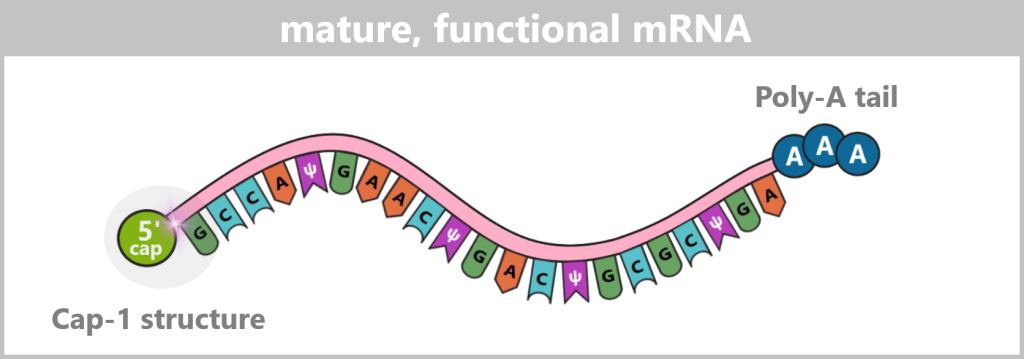
Fig. 1.1.4-C: Schematic representation of a mature mRNA with 5′ cap and poly-A tail The figure is highly simplified; the actual mRNA is significantly longer and comprises approximately 4,200 nucleotides in the COVID-19 vaccine.
In biotechnological production, two methods have become established for generating these essential structures:
1. Co-transcriptional modification (the „all-in-one” method)
In this approach, the cap and poly-A tail are generated during in vitro transcription (IVT).
mRNA capping: Pre-formed cap building blocks (cap analogs) are added to the reaction. The T7 RNA polymerase automatically incorporates this cap structure as the first element at the beginning of the emerging mRNA molecule.
Polyadenylation: The DNA template already contains a sequence that serves as a template for a defined-length poly-A tail. The polymerase therefore synthesizes it directly following the coding sequence.
Advantage: The process is fast, scalable, and takes place in a single reaction vessel. By using modern cap analogs (e.g. ARCA or CleanCap), capping efficiencies of >95% can be achieved.
In industrial practice, the co-transcriptional method is the predominant standard.
2. Enzymatic modification (the „step-by-step” method)
In this more traditional, multi-step approach, the cap and poly-A tail are added in separate reaction steps after IVT.
Polyadenylation: First, the RNA is treated with the enzyme poly(A) polymerase. This enzyme specifically adds a long sequence of adenine nucleotides to the 3′ end of the RNA. The mRNA thus receives its poly(A) tail after synthesis.
mRNA capping: In a subsequent step, the 5′ cap structure is built up in a separate reaction.
Advantage: This method achieves very high and clean capping efficiency, but it is more labor-intensive.
Regardless of the chosen method, a comprehensive purification step follows in order to obtain a homogeneous and highly pure mRNA product.
The key steps – from the purified DNA template to the formulation-ready product – are summarized in the following overview:
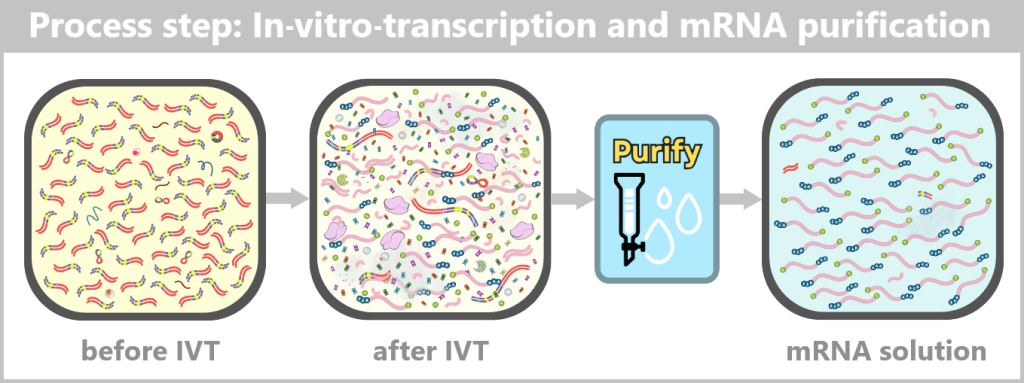
Fig. 1.1.4-D: In-process: From purified DNA to formulation-ready mRNA After IVT, a complex mixture of mRNA, template DNA, enzymes, by-products, and reaction components is present. Through subsequent purification steps, this mixture is reduced to a predominantly pure mRNA solution, which serves as the starting material for LNP formulation.
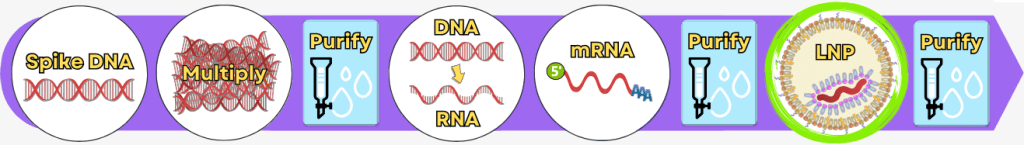
Step 5: Encapsulation of the mRNA in lipid nanoparticles (LNPs)
After the mRNA has been fully produced and purified, it still needs to be protected and made transportable. This is exactly the role of packaging it into lipid nanoparticles (LNPs).
Why does mRNA need packaging?
mRNA is a very fragile molecule. Without protection, it would be rapidly degraded in the body. LNPs fulfill several functions simultaneously:
- they protect the mRNA from degradation
- they transport it into body cells (through the cell membrane)
- they enable controlled release of the mRNA inside the cell
Without LNPs, the vaccine would not be functional.
How does an LNP form? – The basic mechanism
Formulation typically takes place in a device such as a microfluidizer or nanoparticle mixer. In this process, two liquids are rapidly mixed at very high speed:
1) An ethanolic lipid solution
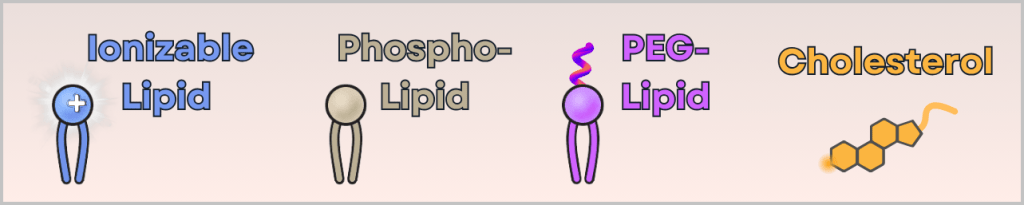
This contains four different lipids:
- ionizable cationic lipid (binds the mRNA and enables cellular uptake)
- phospholipid (stabilizes the structure – similar to cell membranes)
- PEG-lipid (controls particle size and prevents aggregation)
- cholesterol (makes the particle flexible and stable)
2) An aqueous mRNA solution
This contains only:
- purified mRNA
- a mild buffer
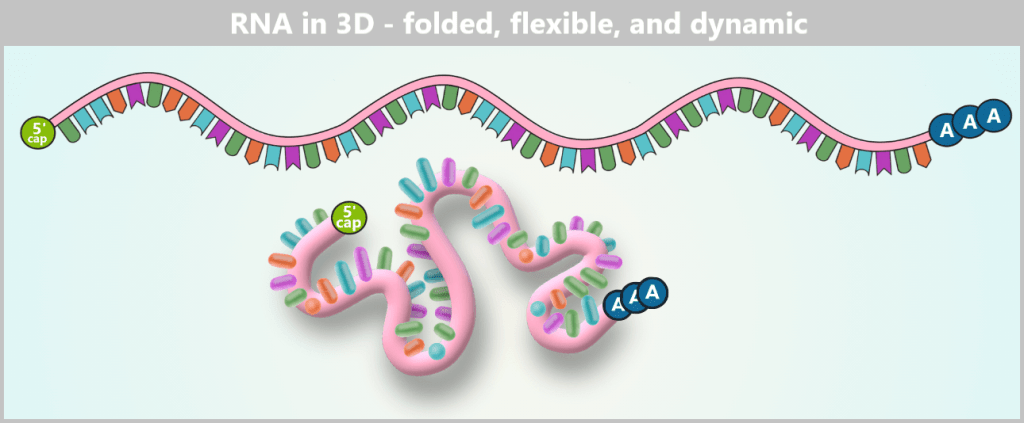
Fig. 1.1.5-A: Schematic representation of RNA in solution – linear vs. 3D The RNA is shown in its linear form above, representing how the base sequence is read. Below, the same strand is illustrated as it actually exists in aqueous solution: a flexible, constantly moving 3D coil in which individual segments interact only loosely or transiently with each other.
What happens during mixing – a self-assembly effect
When the two solutions meet within fractions of a second, several parameters change:
- pH value
- lipid solubility
- charge states

Fig. 1.1.5-B: The microfluidics principle: precision mixing on a millisecond timescale The figure schematically illustrates the principle of microfluidic mixing. On the left and right, two separate liquids enter the mixer: a lipid solution (left) containing the different lipid types and an mRNA solution (right) containing freely dissolved mRNA strands. In the central microfluidic channel, both streams meet and are intensely mixed.
As a result, a spontaneous and highly precise process occurs:
- The ionizable lipid becomes positively charged → it attracts the negatively charged mRNA.
- The mRNA is „wrapped” and encapsulated.
- The other lipids arrange themselves around it to form a stable outer shell.
This automatically generates a nanoparticle with a typical size of 60–100 nm. It is therefore not a „manual packaging” process, but rather a biophysical self-assembly process – the molecules spontaneously organize into the correct structure.
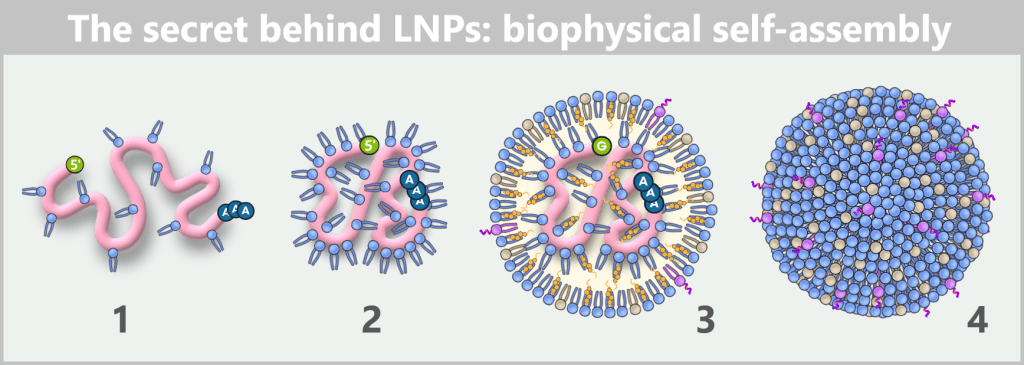
Fig. 1.1.5-C: How two liquids become a nanoparticle: microfluidic formulation The figure shows in four steps how mRNA and lipids spontaneously organize into a lipid nanoparticle during formulation.
1) As soon as mRNA and ionizable lipids come into contact, the positively charged lipid head groups immediately bind to the negatively charged phosphate backbone of the mRNA and begin to tightly wrap around the strand.
2) As a result, the mRNA contracts locally, becomes more compact and continues to condense.
3) In this way, an initial „core” of mRNA–lipid complexes is formed. Additional ionizable lipids then accumulate around it, along with phospholipids, cholesterol, and PEG-lipids. Together, they stabilize the emerging structure while the mRNA becomes progressively more tightly enclosed.
4) Finally, a densely packed, nearly spherical nanoparticle is formed. Its shape is not determined by external control but solely by the physicochemical properties of the molecules – an example of spontaneous self-organization on the nanoscale.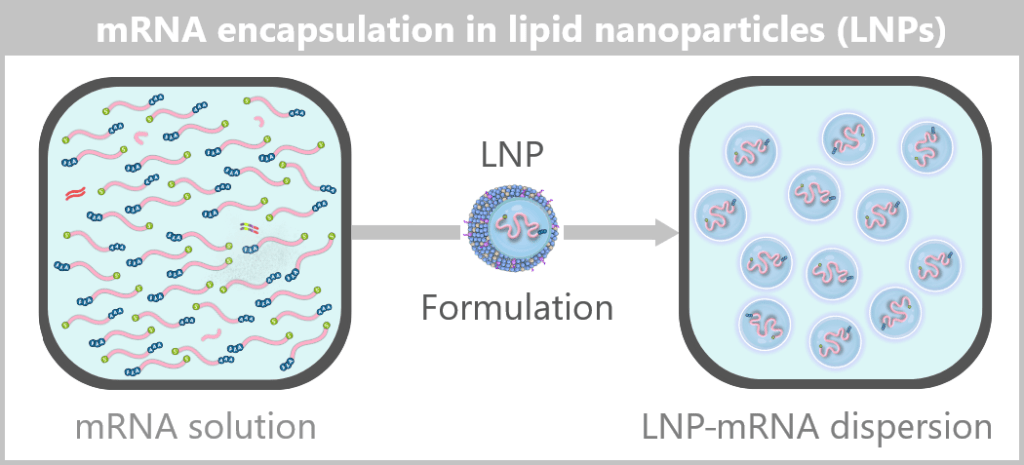
Fig. 1.1.5-D: Formulation step: preparation of the LNP–mRNA dispersion The formulated LNPs now constitute the final drug substance concentrate, which is subsequently filled under sterile conditions in the next step.
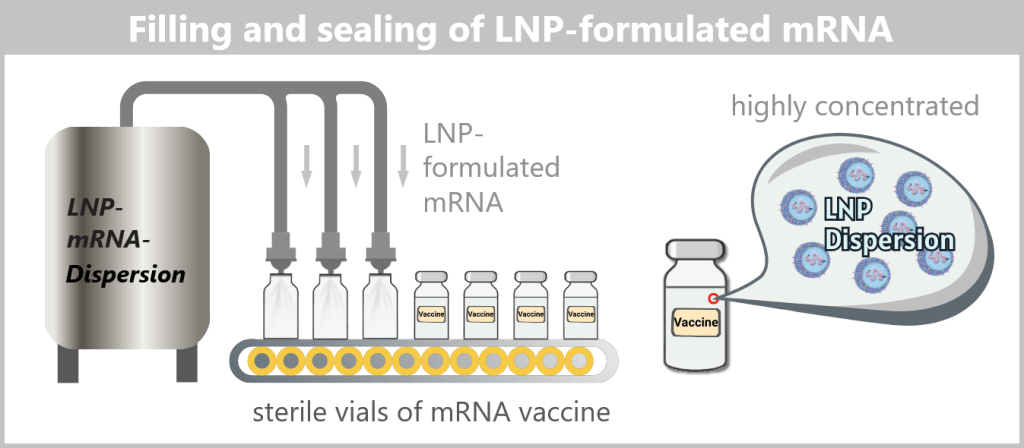
Fig. 1.1.5-E: Schematic of sterile filling of the mRNA drug substance concentrate A vaccine vial contains a very large number of individual lipid nanoparticles, typically in the range of 10¹³–10¹⁴ particles per vial.
1.2. Impurities and Byproducts
The production of a functional mRNA involves several sequential process steps – from the amplification of the DNA template to the processing and formulation of the RNA. In nearly every one of these steps, by-products, impurities, or residual substances are also generated alongside the desired product.
1.2.1. Typical impurities in bacterial production
1.2.2. Typical impurities and by-products after in vitro transcriptionFor the safety and efficacy of the therapeutic product, their identification and subsequent removal in the downstream process is crucial.
In this section, we take a look at the most important potential impurities and by-products and their known biological activities.
✧ ✧ ✧
1.2.1. Typical impurities in bacterial production
After bacterial cell lysis, the plasmid DNA is present in a complex reaction mixture. In addition to the desired plasmid DNA, the solution contains chromosomal DNA, bacterial RNA, proteins, enzymes (including RNases), endotoxins, and fragments of the cell wall.
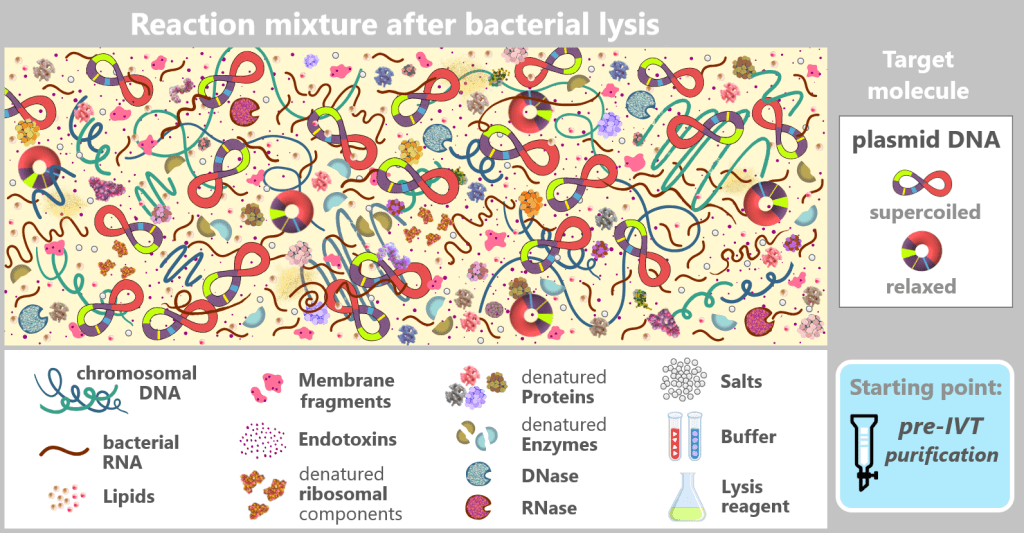
Fig. 1.2.1: Reaction mixture after bacterial lysis (not to scale) The figure schematically shows the composition of the solution after disruption of the E. coli cells in which the plasmid was amplified. In addition to the desired plasmid DNA, which contains the expression cassette for the mRNA, numerous bacterial components and process-related substances are present. The depicted components are embedded in a protein-rich cytosolic matrix derived from the bacterial cytoplasm (yellow background).
This complex and heterogeneous mixture illustrates that the plasmid DNA must first be isolated from a highly contaminated biological environment before it can be used as a template for in vitro transcription.
✧ ✧ ✧
1.2.2. Typical impurities and by-products after in vitro transcription
Although in vitro transcription (IVT) is an efficient enzymatic method for producing mRNA, the reaction does not yield a pure final product. After synthesis, a complex mixture is present that, in addition to the target mRNA, contains various process-related impurities and by-products.
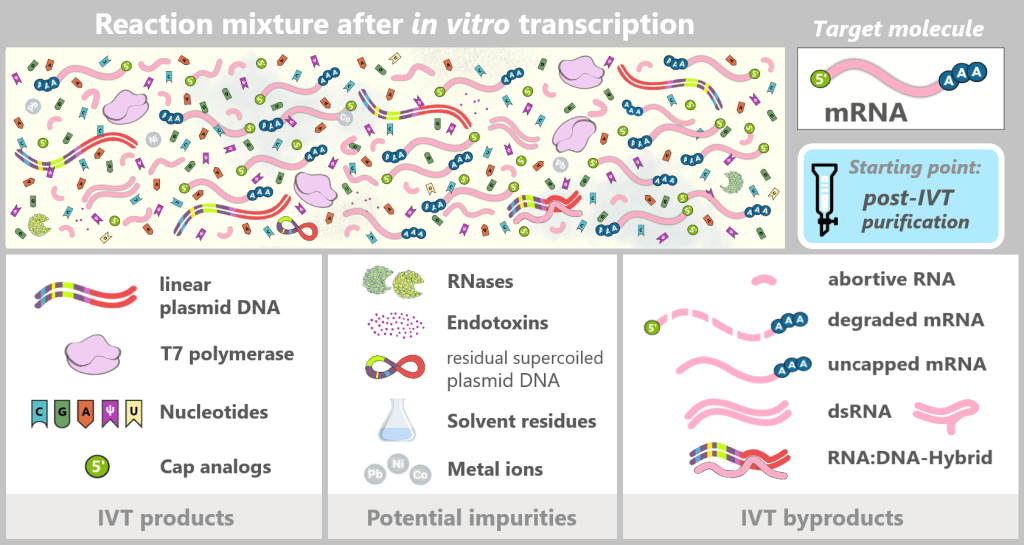
Fig. 1.2.2: Reaction mixture after in-vitro transcription (not to scale) The figure schematically illustrates the complex composition of the IVT reaction solution. In addition to the desired, fully processed mRNA (target molecule), the mixture contains various RNA by-products such as uncapped, degraded, or abortive RNA, double-stranded RNA, and RNA:DNA hybrids. Additional components include:
IVT products
Linear plasmid DNA: DNA template containing the expression cassette for the spike protein.
T7 polymerase: Enzyme that recognizes the T7 promoter and synthesizes the mRNA.
Nucleotides: Building blocks of mRNA synthesis – cytosine (C), guanine (G), adenine (A), and N1-methylpseudouridine (m¹Ψ) instead of uridine (U). Uridine is listed because small amounts of U may remain from the production of modified nucleotides.
Cap analogs: Synthetic cap analogs used during IVT to form the Cap-1 structure and which may remain in excess after the reaction.Potential impurities
RNases: Enzymes that can cleave and degrade RNA; they may be introduced unintentionally via raw materials or bacterial residues.
Endotoxins: Components of the outer membrane of Gram-negative bacteria such as E. coli.
Solvent residues: Traces from the production and purification of starting materials.
Metal ions: Trace impurities from raw materials, process water, or production equipment.Overview: Typical RNA by-products after IVT (before purification)
a) Abortive mRNA
b) Degraded mRNA
c) Uncapped mRNA
d) Double-stranded RNA (dsRNA)
e) RNA:DNA hybrids⚬ ⚬ ⚬
a) Abortive mRNA
At the start of transcription, T7 RNA polymerase binds tightly to the promoter and forms a stable initiation complex. However, the transition from this initiation phase into productive elongation is mechanically unstable.
The polymerase locally unwinds the DNA around the transcription start site and begins synthesizing a short RNA strand. In doing so, it pulls DNA into the enzyme without itself moving forward along the DNA. This so-called scrunching leads to the accumulation of mechanical stress within the transcription complex, as the enzyme remains anchored at the promoter while continuously drawing in more DNA.
If the polymerase is unable to relieve this tension by releasing from the promoter and transitioning into the elongation phase, the DNA snaps back into its original configuration. The short RNA fragment that has already been synthesized is released – resulting in an abortive transcript.
This process can repeat multiple times, meaning that a single polymerase can produce numerous abortive RNA fragments before successfully completing a full transcription event. Only once an RNA length of typically around 8–14 nucleotides is reached does the polymerase escape the promoter and form a stable elongation complex.
Appearance
Abortive RNAs are very short RNA fragments, typically only 2–10 nucleotides (nt) long, that arise during the unstable initiation phase of transcription. Despite the use of co-transcriptional capping strategies in which a cap analog serves as the initiating nucleotide, most abortive transcripts do not reach the length required for stable cap incorporation. Consequently, they predominantly exist as uncapped fragments carrying a 5′ triphosphate end. Only in rare cases – typically in longer abortive transcripts of approximately 10–12 nucleotides – can a cap analog be formally incorporated.
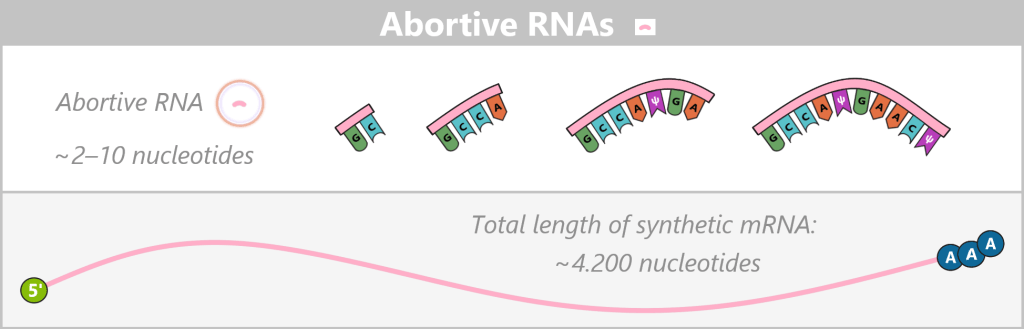
Fig. 1.2.2-A: Abortive RNAs are schematically shown with a free 5′ triphosphate (ppp) and, due to the unstable initiation phase, do not reach a cap-containing elongation form. Biological impact
The biological effects of abortive RNA fragments are not yet fully understood. Individual isolated fragments of only a few nucleotides are generally too short to activate known innate immune RNA sensors and are not translated into proteins.
The potential risk therefore lies less in the individual 2–10-nt fragments themselves, but rather in possible secondary effects:
Double-strand formation: Short RNA fragments could act as primers or contribute to the formation of short double-stranded RNA molecules.
Complex formation: High amounts of such fragments could aggregate into more complex structures or potentially reduce the efficiency of downstream purification steps.
Non-specific stress effects: A high concentration of short RNA fragments could transiently increase cellular stress, even if they are rapidly degraded.BioNTech notes that abortive by-products in the cytosol (the fluid interior of the cell) of transfected cells (cells into which the mRNA has been introduced) may potentially interact in unknown ways with endogenous RNAs or pattern recognition receptors (PRRs), which highlights the need for further research. [Understanding the impact of in vitro transcription byproducts and contaminants]
Approximate proportion (before purification)
BioNTech reports that approximately 44% of T7 RNA polymerases produce abortive transcripts before a full-length transcription is achieved. As a result, abortive RNAs can be the most frequent RNA species in terms of molecule number in the crude reaction mixture. However, under standard IVT conditions, they account for less than 1% of the total RNA mass. The exact proportion of abortive RNAs strongly depends on the template sequence and the reaction conditions.
b) Degraded mRNA
In contrast to abortive mRNA, degraded mRNA consists of formerly complete or long mRNA strands that have been partially or completely destroyed by external influences or enzymatic processes. Degradation can occur through several mechanisms:
Enzymatic degradation by RNases: This is the most common cause of degradation during and after IVT. RNases (ribonucleases) are extremely stable enzymes that are found almost everywhere (e.g. on skin, in dust, or in raw materials). They cleave RNA at specific or structurally preferred sites. Even minimal contamination of the reaction mixture can cause freshly synthesized mRNA to break down into fragments.
Chemical/physical degradation: Due to its chemical structure, RNA is significantly less stable than DNA. Elevated pH values, high temperatures, or certain metal ions promote hydrolytic cleavage of the RNA backbone and lead to strand breaks.
Mechanical degradation: If the solution is exposed to high shear forces – for example through vigorous stirring or pumping through narrow tubing – long mRNA strands can physically tear apart. This tends to generate relatively large fragments rather than the fine fragments typically caused by RNases.
Premature termination during elongation: Strictly speaking, this is not classical degradation, but rather incomplete mRNA generated during synthesis itself. If the polymerase prematurely dissociates from the DNA template during elongation – for example due to strong secondary structures or limited nucleotide availability – a shortened mRNA molecule is produced that lacks the 3′ end and therefore the poly-A tail.
Appearance
Degraded mRNA exists as a mixture of fragments of varying lengths. These fragments may contain individual strand breaks or may be extensively degraded, and they often lack a complete 5′ cap and/or poly-A tail.

Fig. 1.2.2-B: This schematic illustration shows degraded mRNA … as a heterogeneous mixture of fragments that can arise during or after in vitro transcription. In contrast to intact, full-length mRNA (shown below as a reference), the degradation products vary in length and display characteristic damage: lost or damaged 5′ cap structures, exposed 5′ triphosphates (ppp) – which become exposed upon cap loss, shortened poly-A tails, and internal strand breaks. The fragments do not exist as a uniform species, but rather as a complex mixture.
Biological impact
Degraded mRNA is not a uniform substance but a heterogeneous mixture. Not all fragments are biologically problematic. The greatest risk arises from fragments that carry specific immunological danger signals, including:
- exposed 5′ triphosphates, which are recognized by cytosolic RNA sensors such as RIG-I,
- short double-stranded RNA structures that arise through secondary structure formation or hybridization and can activate receptors such as MDA5 or TLR3,
- unusual end structures (missing cap or poly-A tail), which may be recognized as „FOREIGN”.
Such fragments can trigger innate immune responses and impair the tolerability of the mRNA formulation.
Approximate proportion (before purification)
The proportion of degraded mRNA varies strongly depending on process conditions. Typically, it lies in the range of about 1–10% before purification. However, in well-optimized IVT processes, this fraction is deliberately reduced to a minimum.
c) Uncapped mRNA
In modern IVT processes, the 5′ cap structure is often introduced co-transcriptionally using so-called CleanCap analogs. These are synthetic initiator oligonucleotides that already contain the complete 5′ cap structure, including the first transcribed nucleotide.
T7 RNA polymerase specifically uses CleanCap to initiate transcription; unlike classical cap analogs, there is no direct competition with GTP, the natural initiating nucleotide. As a result, predominantly correctly capped mRNA molecules are produced, while the proportion of uncapped mRNA is greatly reduced.
Appearance
Uncapped mRNA exists as a full-length RNA sequence but is characterized by the absence of the 5′ cap structure at its 5′ end.

Fig. 1.2.2-C: This illustration visualizes mRNA molecules that lack the functional 5′ cap. Instead, they carry an immunogenic 5′ triphosphate (ppp). Biological impact
Cells possess specialized enzymes that efficiently degrade RNA strands lacking a 5′ cap from the 5′ end. This mechanism serves to rapidly eliminate defective or aged endogenous RNAs.
Immune response: Uncapped RNA is recognized by cellular pattern recognition receptors such as RIG-I and can trigger a strong type I interferon response, placing the cell into an antiviral state.
In uncapped modRNA, the substitution of uridine with N1-methylpseudouridine (m¹Ψ) significantly reduces activation of these sensors. This creates a biological „tug-of-war” between the immunostimulatory uncapped 5′ end and the immunomodulatory nucleoside modification. Overall, uncapped modRNA would therefore act as a moderately immunogenic molecule but would still be degraded more rapidly than correctly capped modRNA.
Minimal protein production: Since the 5′ cap structure is also required for efficient ribosome binding, uncapped mRNA is translated into protein very poorly or not at all.
Approximate proportion (before purification)
The fraction of uncapped mRNA after IVT depends strongly on the capping strategy used. With modern co-transcriptional CleanCap approaches, the proportion of uncapped mRNA before purification typically lies in the range of approximately 1–6%.
d) Double-stranded RNA (dsRNA)
Another relevant by-product of in vitro transcription (IVT) is double-stranded RNA (dsRNA). While the desired vaccine mRNA is synthesized as a single-stranded molecule, RNA duplexes can also form during IVT in which two complementary RNA strands are linked via Watson–Crick base pairing.
The formation of such dsRNA species is not a random aggregation phenomenon but results from specific enzyme-mediated side reactions of T7 RNA polymerase.
Formation mechanisms
Promoter-independent transcription of the non-template strand
Under certain conditions, T7 RNA polymerase can synthesize RNA even without a canonical promoter initiation by reading the non-template strand of the DNA template. The resulting RNA molecules are largely complementary to the desired mRNA. When sense and antisense RNA strands encounter each other, they form extensive, nearly fully double-stranded RNA duplexes. This mechanism is particularly promoted when the DNA template is not fully linearized or when specific sequences are present at the 3′ end of the template.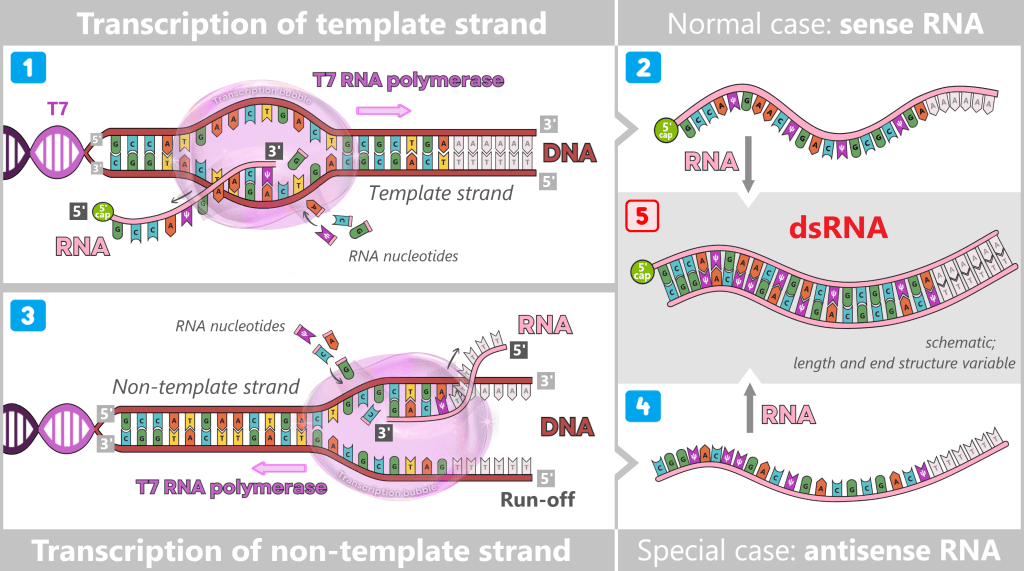
Fig. 1.2.2-D1: Formation of dsRNA through promoter-independent transcription of the non-template strand (RNA is shown schematically; nucleoside modifications (e.g. m¹Ψ) are not explicitly depicted.)
1) The normal case: The T7 promoter has an exceptionally high affinity for T7 RNA polymerase. Therefore, the polymerase almost exclusively initiates transcription at this site. It reads DNA in the 3′ → 5′ direction and synthesizes a new RNA molecule exclusively in the 5′ → 3′ direction.
2) The result is the desired sense RNA, which corresponds exactly to the sequence of the non-template strand.
3) The special case: After run-off – the polymerase leaving the DNA template at the end of transcription – the enzyme dissociates from the DNA. In rare cases, it can bind nonspecifically to structurally accessible DNA ends. If it binds to the strand that was previously not used as a template, this strand becomes the new template.
Under certain reaction conditions, locally open or dynamically melting DNA regions may also form, allowing T7 RNA polymerase to bind in a promoter-independent manner. If this binding occurs on the previously unused strand, it is read as the template.
During promoter-independent transcription of the non-template strand, there is no defined initiation site, so no co-transcriptional capping occurs. The resulting antisense RNA therefore typically carries a free 5′ triphosphate (pppN).
4) The result is a complementary antisense RNA that corresponds to the sequence of the template strand.
5) Formation of long dsRNA duplexes: When sense RNA and antisense RNA encounter each other, double-stranded RNA (dsRNA) is formed. The resulting dsRNA by-products are not a homogeneous molecule but a structurally heterogeneous mixture of fully or partially complementary RNA duplexes. These may contain blunt ends or single-stranded overhangs and vary considerably in length and terminal structure.RNA-dependent 3′ end extension (self-priming)
A second key mechanism is RNA-dependent extension of the 3′ end of the freshly synthesized mRNA. In this process, the 3′ end of the RNA folds back intramolecularly due to complementary sequences, forming a short double-stranded RNA hairpin. T7 RNA polymerase can bind to this structure again and extend the RNA strand, generating a sequence that is complementary to the original RNA. This process is independent of the DNA template and can even occur in already fully transcribed RNA molecules.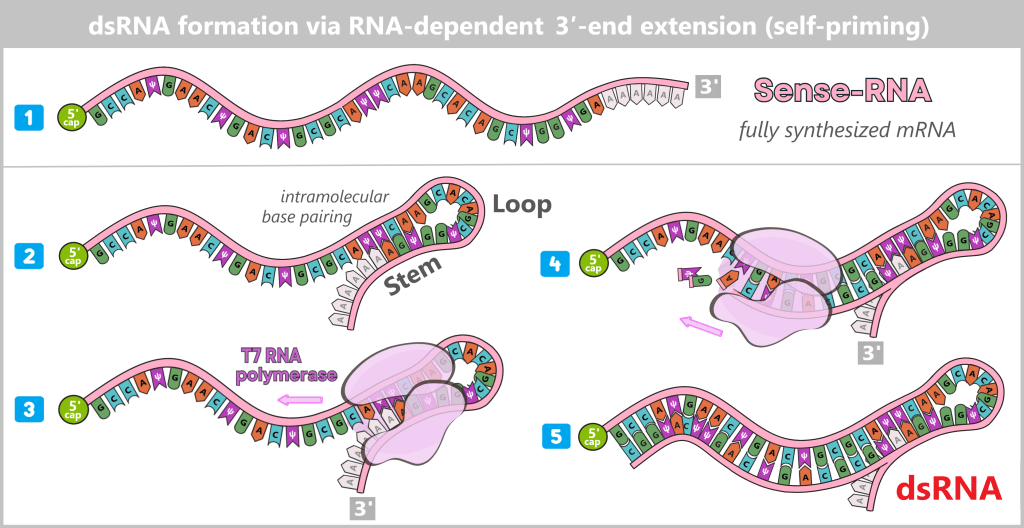
Fig. 1.2.2-D2: Formation of dsRNA via RNA-dependent 3′ end extension (self-priming) (RNA is shown schematically)
1) Sense RNA: A normal, fully synthesized mRNA molecule.
2) Hairpin formation: The 3′ region folds back on itself. A short dsRNA stem (e.g. 5–10 base pairs) forms with a single-stranded loop.
3) Polymerase binding: T7 RNA polymerase binds to this dsRNA stem as if it were a DNA template–primer complex.
4) Extension: The polymerase uses one strand of the hairpin as a template and extends the free 3′ end along the complementary RNA region, thereby synthesizing the antisense sequence directly into the extension of the sense strand.
5) Result: The polymerase extends the strand as far as possible. The newly synthesized antisense RNA immediately hybridizes with the sense RNA. This results in a partially or fully dsRNA-containing RNA molecule.Distinction from RNA secondary structures
Single-stranded RNA naturally forms intramolecular secondary structures such as hairpins or internal loops (see upper figure – point 2). These are an integral part of functional mRNA and are typically short and thermodynamically flexible. Such structural elements are not considered dsRNA by-products in the strict sense and are not equivalent to the long dsRNA duplexes that are relevant as IVT impurities.Both mechanisms lead to the formation of longer, more stable dsRNA regions, which are structurally clearly distinct from the short, dynamic secondary structures of correctly transcribed mRNA.
Appearance
dsRNA by-products typically exist as long, partially or fully complementary RNA duplexes. They may span nearly the entire length of the target mRNA or contain extended double-stranded regions. These duplexes often lack a 5′ cap structure and may feature single-stranded overhangs at their termini.
Biological impact
dsRNA as a structural danger signal
Double-stranded RNA (dsRNA) is not a typical structural motif of endogenous, translatable mRNA in the cytoplasm of eukaryotic cells. This means that while cellular mRNAs are normally single-stranded and serve as templates for protein synthesis via translation, dsRNA occurs physiologically only in tightly regulated, short-lived contexts. The presence of dsRNA in the cytoplasm is therefore not primarily interpreted by the cell as genetic information, but as a structural warning signal.Cytosolic recognition of dsRNA
Cells possess specialized pattern recognition receptors (PRRs). These „danger detectors” patrol the cytoplasm and search for structural patterns such as duplex length, end structure, and chemical nucleotide modifications.Cytosolic dsRNA sensors and their immunological consequences
Feature / specificity Cellular sensor Biological consequence (for cell) Short dsRNA (≈ 10–300 bp) with 5′ triphosphate and/or blunt ends RIG-I (Retinoic acid-Inducible Gene I) short-ppp detector Induction of inflammatory gene expression (type I interferons, cytokines). Activation of neighboring cells. Long dsRNA (> ~500–1,000 bp), independent of end structures MDA5 (Melanoma Differentiation-Associated protein 5) long-duplex scanner Strong type I interferon response. Important antiviral defense mechanism. Medium-length dsRNA (≈ ≥30–100 bp) PKR (Protein Kinase R) intracellular effector Global shutdown of protein synthesis via phosphorylation of translation factor eIF2α. Inhibition of viral replication. Medium-length dsRNA (≈ ≥40–100 bp) OAS (2′-5′-Oligoadenylate Synthetases) intracellular effector Non-specific RNA degradation via activation of latent RNase L, which cleaves cellular and viral RNA. Can lead to cell death. dsRNA in endosomes (extracellularly taken up or phagocytosed dsRNA) TLR3 (Toll-like Receptor 3) gatekeeper receptor Induction of type I interferons and pro-inflammatory cytokines. Effect of m¹Ψ
The use of nucleoside-modified mRNA (modRNA), for example through the incorporation of N¹-methylpseudouridine, can significantly reduce the activation of certain cytosolic RNA sensors. RIG-I and PKR are particularly affected, as they preferentially respond to single-stranded RNA or short-lived dsRNA structures. In contrast, length-dependent sensors such as MDA5 are only partially modulated by this modification and remain largely sensitive to extended dsRNA duplexes.For this reason, dsRNA still possesses a high immunostimulatory potential even in the context of modRNA – meaning it can activate the immune system and trigger unwanted inflammatory responses. The efficient minimization of such dsRNA by-products through optimized transcription conditions, chemical modifications, and downstream purification processes is therefore a central aspect of mRNA manufacturing.
Approximate proportion (before purification)
The amount of dsRNA after IVT is highly process-dependent and is influenced by factors such as the quality of the DNA template, reaction conditions, and the polymerase used. Typically, the dsRNA fraction before purification is in the low single-digit percentage range, but under unfavorable conditions it can be significantly higher.
e) RNA:DNA hybrids
During in vitro transcription, under certain conditions, the newly synthesized RNA strand may not fully dissociate from the DNA template. It can remain partially bound to the template strand while displacing the complementary DNA strand. This leads to the formation of so-called RNA:DNA hybrids.
The formation of such hybrid structures is strongly sequence-dependent. In particular, purine-rich transcripts – RNA sequences containing high amounts of adenine and guanine – as well as DNA templates with repetitive GAA (guanine–adenine–adenine) motifs favor the formation of stable RNA:DNA duplexes. These by-products are often underestimated but can be experimentally detected using specific antibodies against RNA:DNA hybrids. [Understanding the impact of in vitro transcription byproducts and contaminants]
Appearance
RNA:DNA hybrids typically exist in the form of R-loop-like structures. These are locally restricted, three-stranded nucleic acid conformations consisting of:
- an RNA:DNA hybrid duplex,
- a displaced single-stranded DNA segment,
- and the adjacent double-stranded DNA outside the hybrid region.
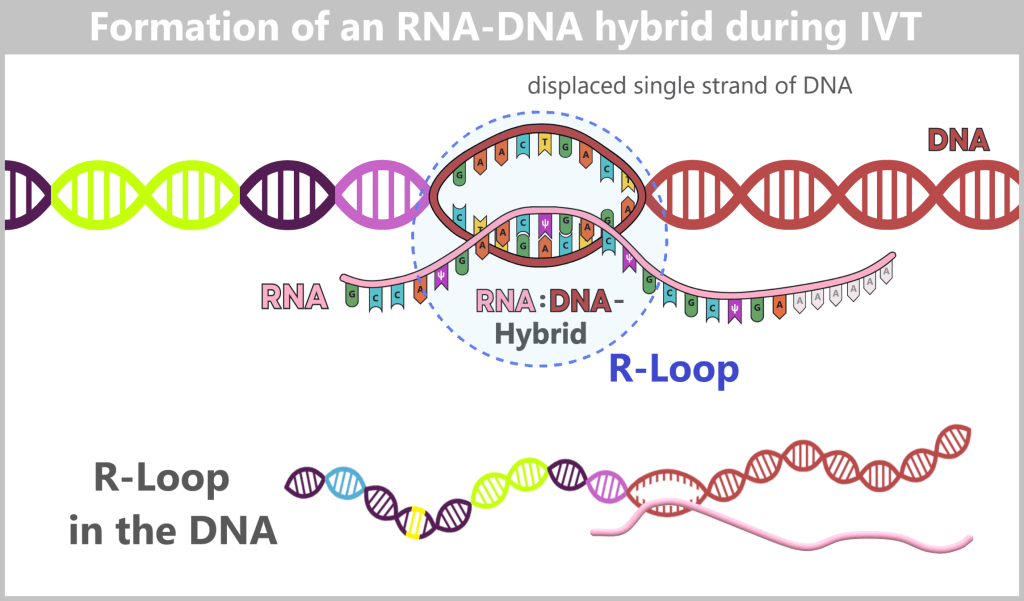
Fig. 1.2.2-E: Schematic representation of R-loop formation during in vitro transcription The newly synthesized RNA strand partially hybridizes with the DNA template strand, displacing the complementary non-template strand. This forms an R-loop consisting of an RNA:DNA hybrid duplex and a displaced single-stranded DNA region, embedded within an otherwise double-stranded DNA region. Outside the R-loop, the DNA remains in its regular double-helical structure.
Biological impact
Current research suggests that RNA:DNA hybrids may be recognized by cellular pattern recognition receptors (PRRs). These sensors of the innate immune system include cGAS, TLR9, and the inflammasome protein NLRP3, which can bind such hybrid structures. Their activation can trigger an immune response, leading to the production of pro-inflammatory cytokines and type I interferons.
However, whether and to what extent RNA:DNA hybrid contaminants actually contribute to unwanted immune reactions in therapeutically administered IVT mRNA has not yet been systematically investigated.
Against this background, the efficient removal of RNA:DNA hybrid impurities from IVT mRNA appears to be a relevant aspect of quality assurance. This is particularly important for therapeutic applications in which strong immune activation is not desired. [Understanding the impact of in vitro transcription byproducts and contaminants]
Approximate proportion (before purification)
The proportion of RNA:DNA hybrids after in vitro transcription is highly process-dependent. It is influenced, among other factors, by the sequence and quality of the DNA template as well as by the reaction conditions. Overall, their fraction prior to purification is considered low, but it can vary depending on the specific transcript and process parameters. Reliable publicly available quantitative data on the exact amount of RNA:DNA hybrids in therapeutic mRNA products are not currently available, as such measurements are typically part of proprietary manufacturing and quality control knowledge.
** Another possible mechanism for the formation of RNA:DNA duplexes during DNase treatment is described in section „1.3.1. a) Possible post-transcriptional RNA:DNA hybridization”. **
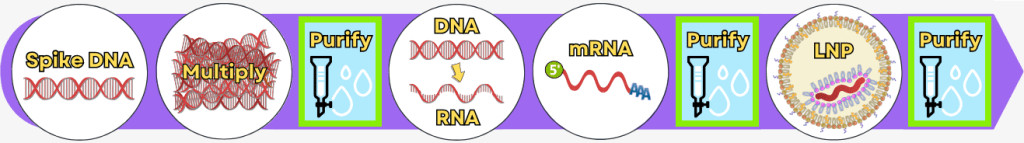
1.3. Purification Methods in the Manufacturing Process
Because mRNA vaccines must meet extremely high purity requirements, purification is not a single isolated process step. Rather, it runs like a common thread throughout the entire manufacturing process.
The exact industrial workflow used by manufacturers such as BioNTech/Pfizer or Moderna is proprietary (company-internal) and is not fully disclosed. However, patents and regulatory documents indicate that a combination of established biotechnological purification methods is employed.
A schematic, cross-industry overview of mRNA manufacturing, illustrating the continuous integration of purification activities throughout the overall process, can be found for example in the technical documentation of Merck / Sigma-Aldrich. (Manufacturing strategies for mRNA vaccines and therapies, Figure 1).
Within the manufacturing process, three particularly critical purification phases can be distinguished in simplified form:
- before in vitro transcription (pre-IVT): purification of the DNA template,
- after in vitro transcription (post-IVT): purification of the synthesized mRNA,
- after formulation: sterile filtration of the lipid nanoparticle formulation.
Rather than discussing these phases strictly in chronological order, the following sections present the most important purification principles and methods that are repeatedly applied at different stages of the manufacturing process.
1.3.1. DNase I digestion – enzymatic DNA degradation
1.3.2. Proteinase K – enzymatic degradation of residual proteins
1.3.3. Filtration
1.3.4. Chromatography – the molecular separation method
1.3.5. Magnetic bead purificationThe following overview summarizes the key purification techniques. It serves as a framework for the subsequent detailed description of each method.
Method Removes Principle Advantages Limitations DNase I
digestionTemplate DNA Specific enzymatic degradation Highly selective, breaks down problematic nucleic acids Enzymes must be completely removed; otherwise risk of residual activity Proteinase K Enzymes from transcription (e.g. polymerases, ligase, restriction enzymes) Non-specific protein degradation Broadly effective, removes many protein types Additional purification to remove enzyme residues Filtration & TFF
(tangential flow filtration)Salts, nucleotides, small molecules, buffer residues; also enables concentration Size-based separation using membranes Scalable, robust, versatile (concentration + buffer exchange in one step) Does not distinguish similar nucleic acids; rather a „rough tool” Chromatography
(Poly(dT), AEX)dsRNA, short RNA fragments, residual nucleotides, proteins Separation by charge or specific binding to surfaces Very precise, separates closely related nucleic acids; well established in industry Technically demanding, cost-intensive, requires tight process control Magnetic bead purification
(Process 1)Selects mature polyadenylated mRNA; removes short fragments & impurities Poly(T)-coated beads bind specifically to the poly(A) tail of mRNA High specificity, fast and efficient separation Suitable for small to medium volumes; difficult to scale for large-scale production
1.3.1. DNase I digestion – enzymatic DNA degradation
Deoxyribonuclease I (DNase I) is an enzyme that cleaves DNA molecules into smaller fragments. This process is often referred to as „digestion” of DNA – a figurative term describing the enzymatic breakdown of long DNA strands into shorter pieces. In biotechnology, DNase I is used to selectively fragment unwanted DNA contaminants.
DNase I is therefore not a complete purification method on its own, but rather a preparatory step that facilitates subsequent physical and chromatographic separation processes.
In mRNA vaccine production, DNase I is used after in vitro transcription (IVT) to fragment the DNA template (template DNA) as well as any DNA component of RNA:DNA hybrids that may have formed and could affect product quality or safety.
DNase I recognizes the three-dimensional structure of DNA – specifically the sugar-phosphate backbone. The enzyme binds to DNA like a saddle and inserts itself into the so-called minor groove. In doing so, it locally bends the DNA structure to gain access to the phosphodiester bond. Because the width of the minor groove varies slightly depending on the base sequence, DNase I exhibits mild sequence preference, cleaving somewhat more readily at certain regions (e.g. A/T-rich stretches).
Fig. 1.3.1-A: Schematic representation of DNase I binding to double-stranded DNA. The typical B-form DNA is a slender, right-handed double helix resembling a spiral staircase with two grooves of different sizes (a major and a minor groove) winding around it. The enzyme preferentially interacts with the minor groove of the DNA duplex and binds to the sugar-phosphate backbone. This binding induces a local distortion of the DNA, enabling cleavage of the phosphodiester bond. The major groove is shown for completeness but does not play a primary role in DNase I binding.
DNase I cleaves DNA non-specifically, meaning it cuts at many different positions along the strand. It acts on both single-stranded DNA (ssDNA) and double-stranded DNA (dsDNA). Its activity depends on several factors, in particular the presence of metal ions, which influence the nature of the cleavage sites [bioswisstec, YEASEN]:
In the presence of magnesium ions (Mg²⁺), DNase I primarily introduces single-strand cuts („nicks”) in double-stranded DNA (dsDNA). This results in the formation of short dsDNA fragments with overhangs, as well as a smaller proportion of single-stranded DNA (ssDNA) fragments.
In the presence of manganese ions (Mn²⁺), DNase I cleaves both strands of dsDNA at nearly the same position, producing predominantly blunt-ended dsDNA fragments.
Fig. 1.3.1-B: Schematic representation of DNase I activity depending on metal ions Effectiveness of DNase I
The manufacturer Thermo Fisher Scientific describes the mode of action and limitations of DNase I digestion in the online article „DNase I demystified”. The article highlights that the effectiveness of DNase I strongly depends on the reaction conditions and the type of DNA substrate present.
We are now in the production process after in vitro transcription (IVT). The reaction mixture is a complex mix consisting of:
- the target mRNA (main component),
- RNA by-products (e.g. truncated transcripts, RNA:DNA hybrids),
- the DNA template (in relevant but lower amounts than mRNA),
- rare residual forms of non-linear plasmid DNA (e.g. supercoiled),
- enzymes, free nucleotides, salts, and buffer components.
Like many enzymes, the activity of DNase I is also influenced by the composition of the reaction mixture.
Factors influencing the effectiveness of DNase I digestion
Factor Mechanism Process relevance (mRNA production) Substrate accessibility DNase I preferentially cleaves accessible phosphodiester bonds; compact or topologically constrained DNA is less accessible. Supercoiled or strongly constrained DNA structures (e.g. rare plasmid residual forms) are degraded more slowly or incompletely. Substrate saturation High DNA amounts can saturate the enzyme; high RNA concentrations can influence spatial accessibility of DNA. In IVT mixtures with high nucleic acid load, an enzyme excess is required. Helix geometry The active site of DNase I is optimized for B-form DNA (deviations in helix structure reduce efficiency). RNA:DNA hybrids adopt an A-form-like helix → cleavage activity < 2% compared to dsDNA DNA structure (dsDNA vs ssDNA) Highest activity on dsDNA; activity on ssDNA is ~500-fold lower Short or partially single-stranded DNA fragments are degraded less efficiently. Ions in buffer Mg²⁺ is essential for catalytic activity; Ca²⁺ stabilizes enzyme structure Therapeutic processes require precisely controlled ion concentrations. Chelators Chelators (e.g. EGTA, EDTA) bind Ca²⁺/Mg²⁺ and inactivate DNase I. Residual buffer components from upstream steps can strongly reduce DNase activity. Salt concentration Increased ionic strength weakens electrostatic binding between enzyme and DNA. IVT buffer conditions may reduce activity → compensated by higher enzyme amounts. The Thermo Fisher Scientific article notes that it is „probably impossible to remove every last DNA strand from an RNA preparation”.
This highlights that the complete elimination of residual DNA is technically challenging. Even after careful DNase I digestion, short DNA fragments or RNA:DNA hybrids may remain.
These small DNA fragments are subsequently removed in downstream process steps – for example through magnetic bead purification (in process 1) or chromatography (in process 2) (cf. EPAR, section 2.2).
Fig. 1.3.1-C: Schematic representation of the DNase I digestion in the purification step In industrial mRNA production, DNase I digestion is performed immediately after in vitro transcription (IVT), as the reaction mixture already contains a Mg²⁺-based buffer suitable for enzyme activity. The Mg²⁺-dependent activity of DNase I predominantly leads to fragmentation of DNA into short double-stranded DNA fragments with nicks (strand breaks) and short overhangs. (Mn²⁺ is not used in industrial processes due to its less specific and less controllable cleavage activity.)
After completion of the reaction, DNase I is typically inactivated or removed, for example by heat treatment, addition of EDTA, or Proteinase K treatment, to ensure that no enzymatic activity affects the mRNA.
1.3.1. a) Possible post-transcriptional RNA:DNA hybridization
In addition to R-loop structures formed during in vitro transcription, a second potential pathway for the formation of RNA:DNA hybrids is discussed in the recent literature.
DNase I digestion fragments the remaining DNA templates into pieces of varying length – predominantly double-stranded, but in some cases also with single-stranded overhangs or as short single-stranded fragments.
Fragments whose sequence is complementary to the mRNA – in particular those derived from the template strand of the original DNA template – may, under suitable conditions, hybridize with the mRNA and form RNA:DNA duplexes.
For thermodynamically stable hybridization, contiguous complementary sequences of approximately 8–12 nucleotides or more are generally required. Very short overhangs of only a few nucleotides (e.g. 2–4 bases), as typically generated in a near-complete DNase digestion, are not sufficient to form stable binding interactions. This mechanism therefore requires the presence of sufficiently long complementary DNA fragments, such as those arising from incomplete digestion or specific fragmentation patterns.
Regarding enzymatic degradability, it should be considered that DNase I preferentially cleaves double-stranded DNA. RNA:DNA hybrids represent a structurally distinct substrate and are more efficiently degraded by specialized enzymes such as RNase H or DNase I-XT. In practice, this may mean that once formed, RNA:DNA hybrids are relatively more resistant to subsequent DNase I digestion than free DNA fragments. Additional stabilization may occur when such structures are encapsulated within lipid nanoparticles.
This proposed mechanism of post-IVT hybridization has not yet been comprehensively characterized experimentally. It is based on theoretical considerations and isolated experimental indications and should therefore be regarded as a plausible but not yet conclusively demonstrated possibility.
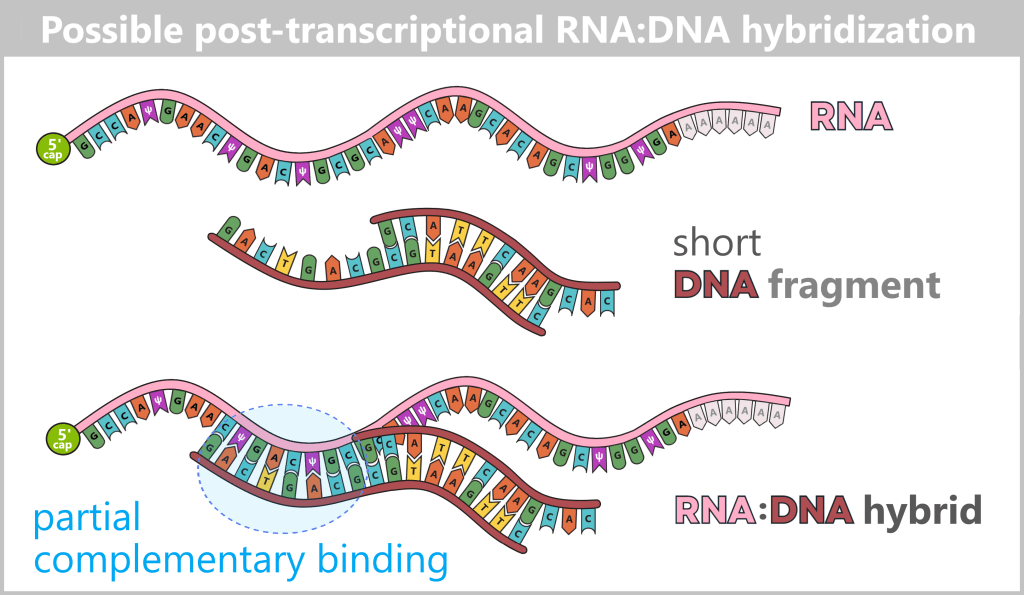
Fig. 1.3.1-II: Schematic representation of a possible post-transcriptional hybridization Short DNA fragments may, under suitable conditions, partially bind to complementary regions of the mRNA. In contrast to an R-loop, no three-stranded structure is formed; instead, a locally restricted RNA:DNA duplex arises.
1.3.2. Proteinase K – enzymatic degradation of residual proteins
Proteinase K is a broad-spectrum serine protease used in biotechnology to non-specifically hydrolyze proteins, breaking them down into smaller peptide fragments. It belongs to the class of proteases – enzymes that cleave peptide bonds between amino acids and thereby irreversibly disrupt the three-dimensional structure of proteins.
In mRNA vaccine production, Proteinase K can be applied at different stages of the process, in particular:
After cell lysis in plasmid production: Here, it serves to degrade bacterial proteins, nucleases, and other cellular components that could compromise the purity of the plasmid DNA.
After in vitro transcription (IVT): Following transcription, during which mRNA is synthesized from the DNA template, several enzymes remain in the reaction mixture, including T7 RNA polymerase as well as DNase I from the preceding process step. Proteinase K can be used to selectively hydrolyze these enzymatic residues, thereby further increasing the purity of the mRNA.
Mechanism of action
Proteinase K initially binds to accessible regions of a target protein and begins cleaving peptide bonds at those sites. These initial cuts progressively disrupt the protein’s compact three-dimensional structure. As destabilization increases, previously shielded internal regions become exposed, allowing the protein to gradually break down into increasingly smaller peptide fragments – effectively “falling apart.” At the end of the process, only short, biologically inactive peptide chains remain, which no longer retain enzymatic activity.
Fig. 1.3.2: Schematic representation of Proteinase K activity Proteinase K attacks DNase I and cleaves its peptide bonds. Initially, outer protein regions are hydrolyzed, leading to structural destabilization. Step by step, the protein breaks down into smaller fragments until only short, biologically inactive peptide fragments remain.
Proteinase K is itself a protein and is not retained in the final product. Its activity can be reduced by process conditions, for example through the removal of stabilizing calcium ions. In practice, however, Proteinase K is primarily removed physically: both the enzyme itself and the resulting peptide fragments are reliably eliminated in subsequent purification steps, particularly through ultrafiltration (e.g. tangential flow filtration) and chromatographic methods.
Relevance in the process
Through the targeted use of Proteinase K, residual process enzymes, bacterial proteins, and other protein contaminants can be effectively reduced. This creates well-defined starting conditions for subsequent filtration and chromatographic purification steps. The use of Proteinase K is not an obligatory component of every manufacturing process, but it increases the robustness and reproducibility of downstream processing, particularly in large-scale industrial production.
1.3.3. Filtration
Filtration is a physical separation process that separates particles or molecules primarily based on their size.
In this process, a mixture is passed through a porous filter material. In its simplest form, the pores act like a mechanical sieve: particles larger than the pore size are retained, while smaller molecules pass through.
a) Filtration – separation by size
b) Tangential flow filtration (TFF)
c) Sterile filtrationa) Filtration – separation by size
In plasmid purification, two fundamental principles are used:
- separation of insoluble particles (clarification), in order to clear a turbid suspension,
- separation of dissolved molecules by size (membrane filtration), for example to exchange buffers or concentrate molecules.
Clarification: Filtration after lysis of E. coli bacterial cells
Starting point: cell lysis
The addition of sodium hydroxide (NaOH) and the detergent SDS disrupts the lipid-rich cell envelope of the bacteria. Alkaline lysis affects not only membranes but also nucleic acids and proteins. The DNA double helix is denatured, and the long, fibrous chromosomal DNA is additionally mechanically fragmented. Under strongly basic conditions, proteins lose their three-dimensional structure and thus their biological function.Neutralization: system reorganization
Upon addition of an acidic solution, the alkaline reaction mixture is neutralized. Under these conditions, the small circular plasmid DNA molecules can correctly renature and remain in solution. In contrast, fragmented and mispaired chromosomal DNA as well as denatured proteins aggregate together with lipids and membrane components and precipitate as a whitish precipitate.Intermediate state
After alkaline lysis and neutralization, the sample exists as a two-phase system: a soluble phase containing plasmid DNA and an insoluble precipitate of aggregated components. These aggregates consist of fragmented genomic DNA, denatured proteins, ribosomal components, and membrane debris.Clarification and depth filtration
At industrial scale, these solids are efficiently removed by depth filtration. In this process, the suspension passes through a multilayer porous filter material. Particles are retained not only on the surface but also within the filter matrix, providing high loading capacity and effective clarification.Result
The clear filtrate contains the dissolved plasmid DNA, largely free of cell debris and precipitated aggregates. This crudely purified solution forms the basis for subsequent high-resolution chromatographic purification steps.
Fig. 1.3.3-A: The figure shows the cell lysate before and after depth filtration. After alkaline lysis, the lysate contains, in addition to the desired plasmid DNA, numerous bacterial components and process-related substances.
During subsequent neutralization, fragmented genomic DNA, denatured proteins, as well as lipids and membrane remnants form irregular, voluminous aggregates (depicted as clouds).
Through depth filtration, these precipitated aggregates are effectively removed. The resulting filtrate is a clear solution containing the dissolved plasmid DNA. In addition, soluble RNA residues, endotoxins (LPS), salts, buffer components, and small amounts of dissolved protein residues may still be present.Membrane filtration: ultrafiltration and diafiltration
A particularly important variant of membrane filtration in biotechnology is ultrafiltration (UF). It is used to separate dissolved macromolecules (e.g. nucleic acids or proteins) from smaller molecules such as salts, buffers, or nucleotides.
When fresh buffer is continuously added during ultrafiltration, the process is referred to as diafiltration (DF). This allows molecules not only to be concentrated but also to be transferred into a new solution in a controlled manner. UF and DF are often combined and referred to as the UF/DF process.
The predominant technique used to perform UF/DF is tangential flow filtration (TFF). [ROCKER]
✧ ✧ ✧
b) Tangential flow filtration (TFF)
In classical filtration methods (dead-end filtration or direct-flow filtration), the solution flows perpendicular to the membrane, causing large molecules to accumulate on the surface and form a „filter cake” layer. Over time, this reduces flow rate and membrane lifetime.
Tangential flow filtration (TFF) overcomes this limitation: here, the fluid moves tangentially along the membrane surface. A portion of the flow (the permeate) passes through the membrane, while the remaining stream (the retentate) continues to flow parallel to the surface and helps remove deposited material. As a result, the membrane remains permeable for longer periods, and concentration and diafiltration can be performed simultaneously.
Definitions:
Retentate: The fraction retained by the membrane (usually the desired macromolecule).
Permeate: The fraction that passes through the membrane (usually small molecules and impurities).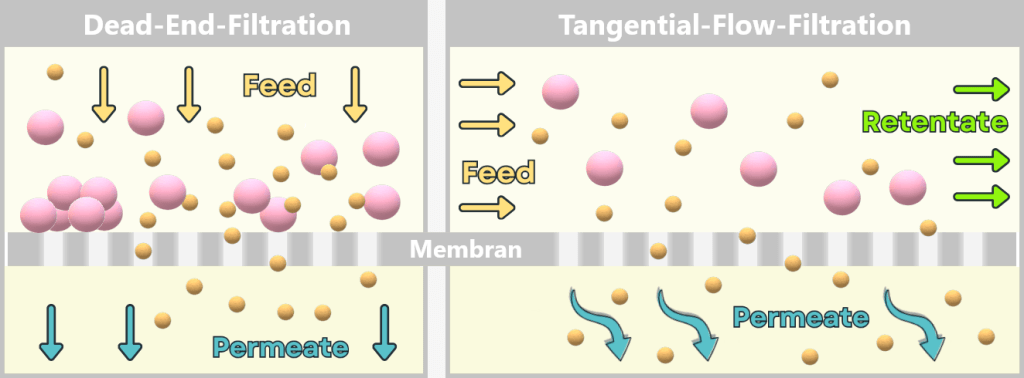
Fig. 1.3.3-B1: Dead-end filtration vs. tangential flow filtration In dead-end filtration, the entire feed stream flows perpendicular to the membrane. Small molecules (permeate) pass through the pores, while larger molecules are retained. Over time, this leads to the formation of a „filter cake”, which reduces efficiency.
In tangential flow filtration, the solution flows parallel to the membrane. While small molecules pass through into the permeate, larger molecules are carried along in the retentate stream. The tangential flow continuously washes the membrane surface, preventing fouling and maintaining stable, efficient filtration over extended periods.TFF is now a standard technique in biopharmaceutical production because it is robust, scalable, and versatile.
TFF in vaccine manufacturing
Tangential flow filtration (TFF) is used at multiple points during mRNA production: for example after transcription, after enzymatic processing steps, or before and after chromatographic purification steps. Its functions range from removing small molecules to buffer exchange and product concentration.
In this chapter, we focus on a central but not exhaustive purification step:
TFF directly after in vitro transcription (IVT)
Here, it is used for the initial („rough”) purification of freshly synthesized mRNA. It separates the mRNA from enzymes, reagents, and by-products, thereby preparing it for subsequent high-resolution purification steps.
Starting point: the „crude solution” after IVT, DNase I, and Proteinase K treatment
At this stage, the mRNA is present in a reaction-containing solution that still includes a variety of unwanted components:
- enzymes (e.g. T7 polymerase, DNase I),
- unconsumed nucleotides,
- residual DNA from the template,
- buffer salts,
- reaction by-products (e.g. short RNA fragments),
- any residual components from upstream biological processes.
To process the mRNA solution, it is passed through a filtration unit with membranes of defined pore size. Molecules below this cutoff – such as salts, nucleotides, or small protein remnants – pass through the membrane (permeate), while the large mRNA molecules are retained in the retentate.
During the process, fresh buffer is continuously added (diafiltration). This gradually washes out low-molecular-weight impurities while simultaneously transferring the mRNA into the desired buffer and concentrating it (ultrafiltration).
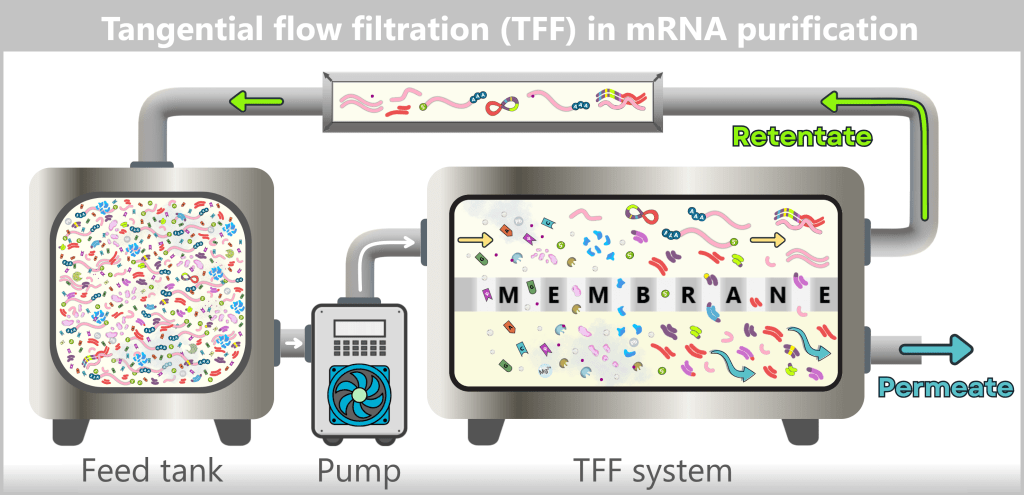
Fig. 1.3.3-B2: Schematic representation of tangential flow filtration (TFF) in mRNA processing The mRNA solution is continuously circulated through the UF/DF system. Small molecules pass through the membrane and are removed as permeate. A pump ensures that the retentate – the mRNA-containing fraction – is repeatedly recirculated across the membrane. Through repeated cycles and buffer addition, impurities are removed while the mRNA fraction is simultaneously concentrated and buffer-exchanged.
Important to note:
TFF is a coarse separation based on size. Impurities with a size or shape similar to mRNA – such as double-stranded RNA, RNA fragments of comparable length, or RNA:DNA hybrids – cannot be removed in this step. These critical by-products remain in the retentate and must be separated in subsequent high-resolution chromatographic purification steps.
✧ ✧ ✧
c) Sterile filtration
Sterile filtration is a key final step prior to filling an mRNA vaccine. In this process, the finished formulation (lipid nanoparticles containing the mRNA) is passed through a very fine filter. The pore size is sufficiently small to reliably retain all bacteria, fungi, and larger particles.
Principle of sterile filtration
Sterile filtration is a purely size-based sieving process:
- The membrane has precisely defined pores of 0,22 µm.
- Microorganisms are significantly larger and are retained.
- The active substance particles (LNPs) are much smaller (60–100 nm = 0,06–0,1 µm) and pass through the filter without difficulty.
Since no heat or chemical treatment is required, this step is particularly suitable for sensitive biological preparations such as mRNA-LNPs, which cannot be sterilized by autoclaving (steam, pressure, and heat).
Process in detail
The LNP-mRNA solution is present in formulation buffer. Sterile filtration is carried out in a cleanroom environment (Grade A/B) under strictly aseptic conditions. Only sterile single-use components are used: filters, tubing, bags, pump heads, and collection containers.
The formulation is gently driven through the 0,22 µm filter using low pressure (peristaltic pump or pressurized gas).
Retained by the filter:
▪️Bacteria (~0,5–5 µm)
▪️Yeasts / Fungal spores (~1–10 µm)
▪️Particles or aggregates > 0,22 µmPass through the filter:
▪️LNPs (60–100 nm)
▪️Buffer components
▪️Dissolved molecules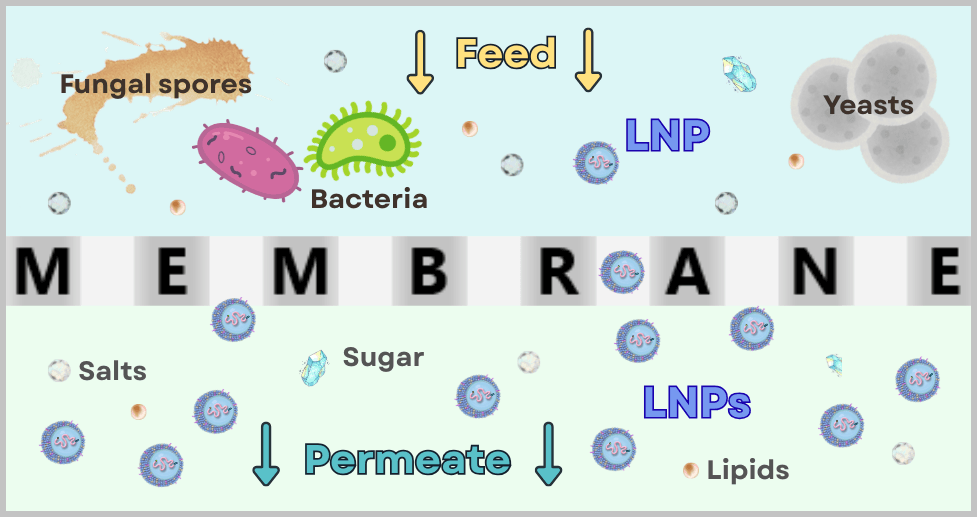
Fig. 1.3.3-B3: Schematic representation of sterile filtration This ensures that the formulation is free of microorganisms and visible particles.
Why sterile filtration is necessary
According to GMP guidelines, sterile medicinal products must be manufactured using validated sterile filtration processes. Sterile filtration is therefore mandatory and ensures that:
- the final vaccine solution is microbiologically sound,
- no large particles, aggregates, or contaminants enter the final product,
- the subsequent sterile filling („aseptic fill & finish”) can be performed correctly.
Sterile filtration is not a chemical purification step, but an essential safety and quality control process. It forms the bridge between the biotechnological purification of the mRNA and the final sterile medicinal product in the vial.
1.3.4. Chromatography – the molecular separation method
Chromatography is one of the most important methods for purifying biological molecules such as DNA, RNA, or proteins. The principle is based on the differing interactions of substances with two phases: a stationary phase (a solid support material within a column) and a mobile phase (a liquid solution that transports the sample mixture).
As the mobile phase – that is, the mixture of molecules to be separated – flows through the stationary phase, the individual molecules are retained to different extents depending on properties such as size, charge, or other chemical characteristics. This leads to temporal separation, in which the components of the mixture elute (are washed out) sequentially.
Put simply, chromatography can be thought of as a „molecular obstacle course”: some molecules interact only weakly with the stationary phase and pass through quickly, while others „stick” more frequently and are therefore delayed. As a result, each component reaches the end of the column at a different time and can be collected separately.
Chromatographic methods in mRNA purification
In industrial processes, such as those described by Merck („Manufacturing strategies for mRNA vaccines”), two chromatographic methods are commonly used in combination:
Method Separation principle Typical application Affinity chromatography
(Poly(dT))Specific binding Primary purification: isolation of intact mRNA from the IVT reaction mixture Ion-exchange chromatography Electrical charge Fine purification: removal of dsRNA, RNA:DNA hybrids, DNA fragments, and other product-related impurities ✧ ✧ ✧
a) Poly(dT) affinity chromatography
Poly(dT)-AC = Poly(dT) Affinity Chromatography
Affinity chromatography utilizes specific interactions between biomolecules and a functionalized surface. In the production of mRNA vaccines, poly(dT) affinity chromatography is specifically used to isolate mature mRNA from the complex reaction mixture after in vitro transcription (IVT).
Starting point: After in vitro transcription (IVT), enzymatic post-treatment, and tangential flow filtration (TFF)
Principle and process
Stationary phase: The resin in the column is coated with long single-stranded stretches of thymine (T) bases, known as a poly(dT) matrix.
Mobile phase: The pre-purified IVT solution – consisting of mRNA, RNA by-products (e.g. dsRNA, truncated transcripts), residual DNA fragments, and defined buffer salts – is applied to the column.
The target molecule is mature mRNA, i.e. „polyadenylated mRNA”. It carries a stretch of adenine (A) bases at its 3′ end, known as the poly(A) tail.
Specific binding via base pairing
Adenine (A) and thymine (T) form specific hydrogen bonds, as in DNA. Figuratively speaking, the poly(A) tail of the mRNA binds strongly to the poly(dT) „hooks” on the resin.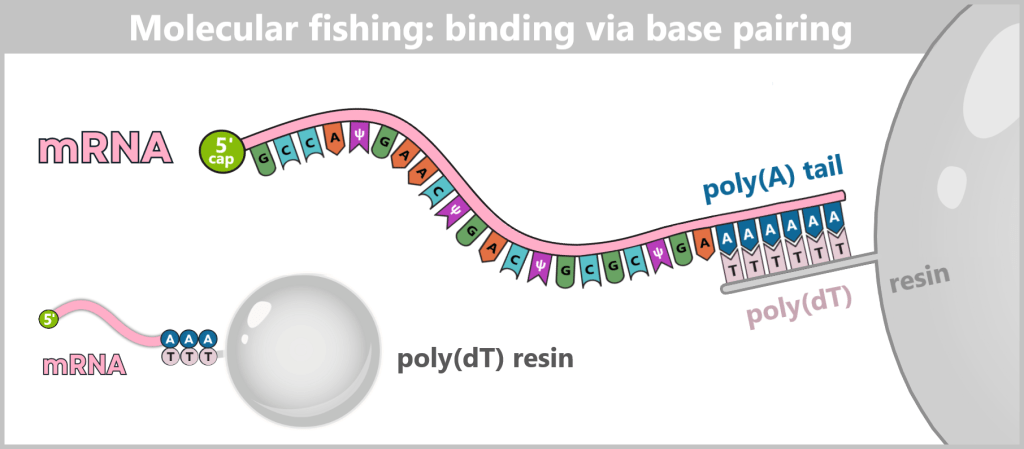
Fig. 1.3.4-A1: The mRNA binds to the poly(dT) resin via its poly(A) tail. The poly(A) tail of therapeutic mRNA typically consists of about 100–120 adenine bases; in the figure, it is shown in a shortened form for illustrative purposes.
Only molecules with an intact poly(A) tail bind strongly to the resin; all other components of the IVT reaction flow through. These include:
(a) residual free nucleotides (only in trace amounts)
(b) protein fragments from enzymatic degradation
(c) short RNA fragments without a poly(A) tail
(d) DNA fragments and DNA-containing by-products
(e) residual supercoiled plasmid DNA
(f) free dsRNA
(g) buffer salts
Washing
Wash buffers remove weakly bound, non-specifically adhering molecules, while the mRNA remains firmly attached to the poly(dT) matrix.Elution (release)
To release the purified mRNA from the „bait”, the specific interactions between A and T are deliberately disrupted. This is typically achieved by reducing ionic strength and/or moderately increasing the temperature. The mRNA detaches from the matrix and is eluted.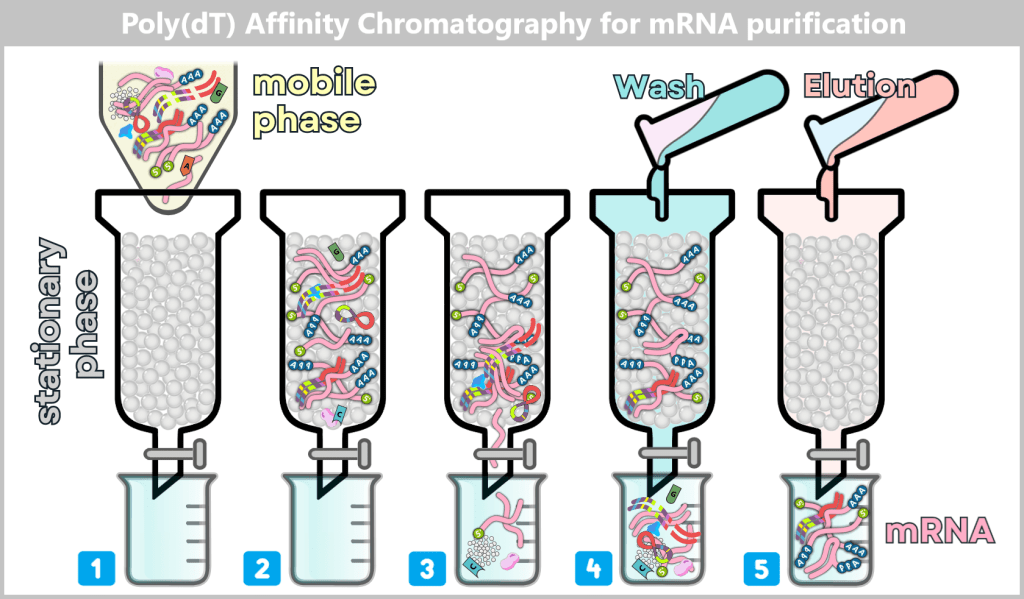
Fig. 1.3.4-A2: Schematic representation of poly(dT) affinity chromatography 1) The IVT reaction mixture is applied to a column functionalized with poly(dT).
2) Polyadenylated mRNA binds specifically to the resin via base pairing,
3) while non-binding components flow through.
4) A wash buffer removes weakly or non-specifically bound molecules.
5) Addition of an elution buffer releases the mRNA from the poly(dT) matrix, yielding it in purified form.Advantages
- very high selectivity due to poly(A)/poly(dT) hybridization
- efficient removal of most non-specific RNA and protein impurities
- enrichment of full-length, correctly polyadenylated mRNA
- comparatively simple and robust method
Limitations – why further purification steps are necessary
Despite its high specificity, poly(dT) affinity chromatography cannot remove all critical impurities:- dsRNA: may bind to the matrix via polyadenylated ends or through non-specific interactions
- RNA:DNA hybrids: if DNA is bound to mRNA, it is co-eluted together with it
- fragmented mRNA with residual poly(A) sequences also binds to the resin
- intramolecular dsRNA structures within mRNA can lead to co-elution
- larger RNA complexes associated with mRNA are co-purified
- endotoxins are not specifically removed and may co-elute
Manufacturers such as MERCK, Lonza, or Cytiva therefore recommend anion-exchange chromatography (AEX) as a subsequent step for fine purification, particularly for the removal of dsRNA and residual DNA contaminants.
✧ ✧ ✧
b) Ion-exchange chromatography
Ion-exchange chromatography (IEX) is a well-established method for purifying nucleic acids and proteins. It separates molecules based on their electrical charge. In nucleic acids, this charge arises from the high density of negatively charged phosphate groups along the molecule. In contrast, the charge of proteins depends on their specific amino acid sequence and folding and can include both positively and negatively charged regions.
Depending on the type of charged target molecules, two main types are distinguished:
▪️Anion exchangers (AEX, Anion Exchange Chromatography):
bind negatively charged molecules such as DNA or RNA.
▪️Cation exchangers (CEX, Cation Exchange Chromatography):
bind positively charged molecules, for example certain proteins.Due to their continuous phosphate backbone, nucleic acids carry a high and uniform negative charge density.
The sugar-phosphate backbone as the source of the negative charge in DNA/RNA
Schematic representation of the structural basis of the negative charge of nucleic acids
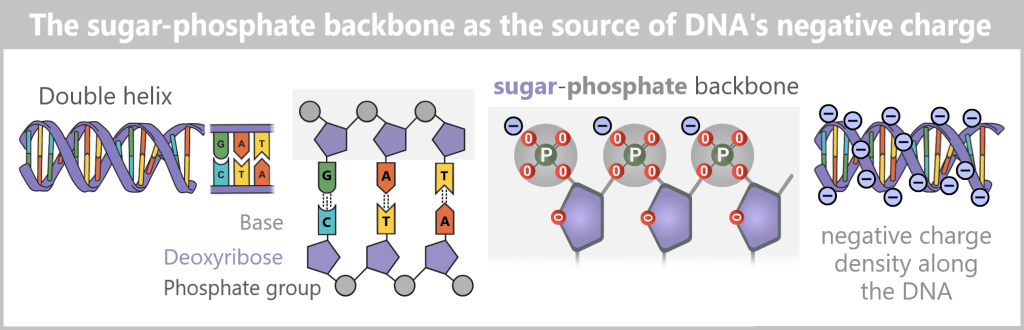
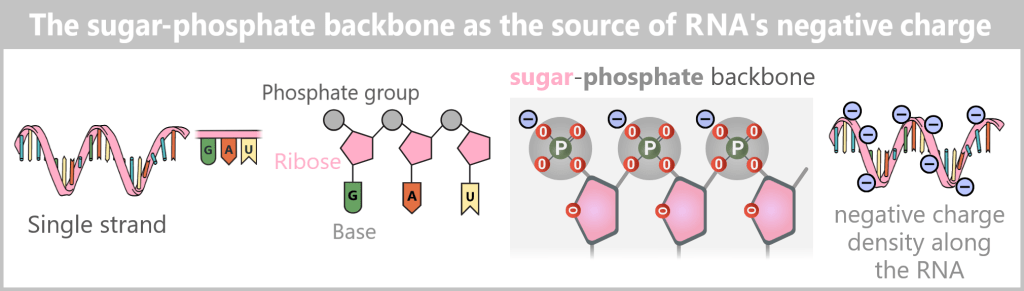
DNA (and analogously RNA) consists of a sugar-phosphate backbone, in which each phosphate group carries a negative charge. These regularly repeating phosphate groups give the molecule a high and uniformly distributed negative charge density along its length. The bases do not contribute to the overall charge.
This property makes anion-exchange chromatography (AEX) the method of choice for their purification. In the overall process, AEX is therefore used twice: first for the purification of the plasmid DNA template from the bacterial lysate. Then, after in vitro transcription (IVT) and initial crude purification, it is used for high-resolution separation of the desired mRNA from structurally similar impurities, in particular double-stranded RNA (dsRNA).
AEX in plasmid DNA purification
Starting point
After lysis of E. coli cells, neutralization, and subsequent depth filtration, a clarified lysate is obtained containing the plasmid DNA. In addition, residual bacterial RNA, endotoxins, salts, buffer components, and small amounts of dissolved protein residues may still be present.
Principle and process
Stationary phase: The support material (resin) consists of small, positively charged polymer beads. These serve as a binding surface for negatively charged molecules.
Mobile phase: The clarified lysate is applied to the column. Initially, negatively charged biomolecules – including plasmid DNA, RNA, and endotoxins – bind to the positively charged resin. Unbound or weakly charged components are removed with wash buffer.
The target molecule is plasmid DNA, which carries a uniformly distributed negative charge along its backbone.
Selective elution: Subsequently, a buffer with increasing salt concentration is passed through the column. The positively charged ions (e.g. Na⁺) in the buffer compete with the biomolecules for binding sites on the resin. As a result, molecules are released in a characteristic order depending on their charge density and binding strength:
- Endotoxins and small RNA fragments elute early, as they have a lower charge density.
- Plasmid DNA – predominantly in supercoiled, but also in relaxed circular form – binds most strongly due to its high negative charge density and therefore elutes only at higher salt concentrations.
Result
This selective elution yields a solution enriched in highly pure plasmid DNA, predominantly in the supercoiled form, with a smaller proportion of relaxed circular forms. Most other cellular components and impurities have already been removed. This DNA fraction subsequently serves as the template for the following in vitro transcription (IVT).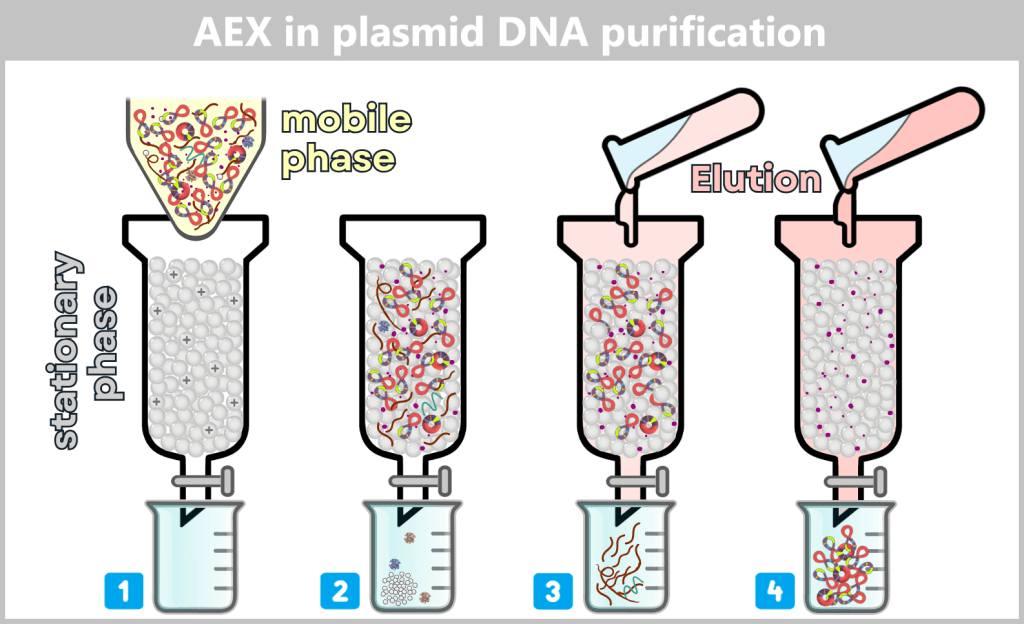
Fig. 1.3.4-B1: Schematic representation of AEX in plasmid DNA purification 1) The clarified cell lysate (mobile phase) is applied to a column with a positively charged stationary phase (anion-exchange resin). 2) Negatively charged molecules in the sample bind to the resin; non-bound or weakly bound components (salts, protein residues) elute early. 3) Elution begins: a buffer with increasing salt concentration is applied. Dissolved ions (e.g. Na⁺ and Cl⁻) compete with bound molecules for binding sites. At low to moderate salt concentrations, mainly short and/or single-stranded RNA fragments elute. 4) Plasmid DNA elutes only at higher salt concentrations due to its high, uniformly distributed negative charge density. Endotoxins (LPS) bind very strongly to the matrix, which is generally advantageous from a purification perspective. The only problematic fraction consists of components that may co-elute with plasmid DNA due to non-specific interactions or association with DNA.
After anion-exchange chromatography, the plasmid DNA is obtained in high purity. The product consists predominantly of supercoiled plasmid DNA and may contain small proportions of relaxed circular forms. In low concentrations, residual short RNA fragments or endotoxins may also be detectable, while cellular macromolecules and process-related impurities have largely been removed.
AEX in mRNA purification: separation of mRNA and dsRNA
Starting point: After in vitro transcription (IVT), enzymatic post-treatment, tangential flow filtration (TFF), and poly(dT) affinity chromatography
After these upstream process steps, a highly enriched mRNA fraction is obtained. However, this mRNA crude solution typically still contains certain by-products and impurities that cannot be fully removed by poly(dT) affinity chromatography, including:
(a) double-stranded RNA by-products (dsRNA)
(b) RNA:DNA hybrids (in low, process-dependent amounts)
(c) fragmented mRNA with residual poly(A) sequences
(d) incorrectly capped mRNA transcripts
(e) endotoxins (LPS), particularly when associated with nucleic acids
The subsequent anion-exchange chromatography therefore does not serve further enrichment of mRNA, but rather high-resolution removal of structurally similar RNA by-products.
Principle and process
Stationary phase: The stationary phase consists of positively charged anion-exchange resins, typically polymer beads functionalized with quaternary ammonium groups. These provide binding sites for negatively charged nucleic acids.
Mobile phase: The mRNA crude solution is applied to the column as the mobile phase. Under low salt conditions, negatively charged nucleic acids in the sample bind to the resin.
The target molecule is single-stranded mRNA. It carries a negative charge along its backbone but, due to its single-stranded nature, secondary structure, and flexibility, it has a lower effective charge density than double-stranded RNA.
Selective elution
In the next step, an elution buffer with increasing salt concentration is passed through the column. The ions in the buffer (e.g. Na⁺) compete with the bound RNA molecules for binding sites on the resin. As a result, the bound components are eluted stepwise according to their binding strength:
Single-stranded mRNA elutes at low to moderate salt concentrations, as its effective charge density is lower.
Double-stranded RNA (dsRNA) binds significantly more strongly to the anion-exchange resin and therefore elutes only at higher salt concentrations or may remain on the column.
RNA:DNA hybrids represent a particular challenge for chromatographic purification. Their binding properties lie between those of single-stranded RNA (ssRNA) and double-stranded RNA (dsRNA). This hybrid character results not only from the RNA:DNA duplex itself, but is also influenced by three factors: the length of the hybrid region, the proportion of free single-stranded RNA, and the three-dimensional conformation of the molecule.
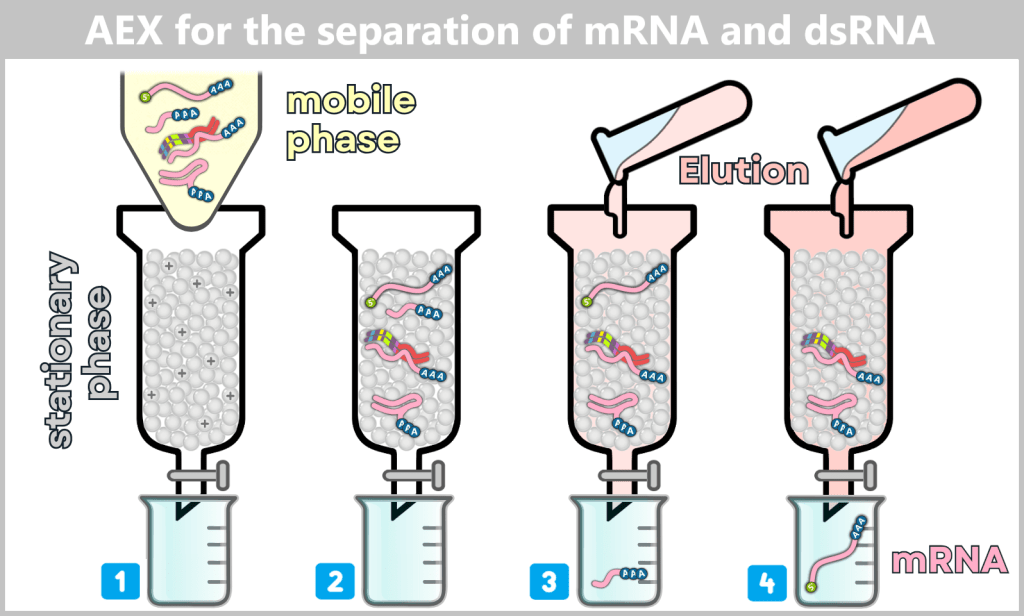
Fig. 1.3.4-B2: Schematic representation of AEX for separation of mRNA and dsRNA The representation is simplified and shows each component only once as an example.
1) The mRNA-containing solution (mobile phase) – consisting of the desired mRNA, dsRNA, and other by-products – is applied to the column. The stationary phase consists of a positively charged anion-exchange resin. 2) Under low salt conditions, all negatively charged RNA molecules bind to the resin. Due to its higher charge density and more rigid helical structure, dsRNA binds more strongly than flexible single-stranded mRNA. 3) With the addition of a buffer of increasing salt concentration, competing anions (e.g. Cl⁻) are introduced. These gradually displace the bound RNA molecules from the column. First, weakly bound impurities such as short RNA fragments are released. 4) At low to moderate salt concentrations, the desired single-stranded mRNA elutes. Double-stranded RNA remains bound longer due to its stronger interaction and elutes only at higher salt concentrations or may partially remain on the matrix.
Result
Anion-exchange chromatography (AEX) enables the selective separation of mRNA from more strongly or more weakly binding nucleic acid structures such as dsRNA. This yields an mRNA fraction that is largely free of dsRNA contaminants and hybrid structures.
The purified mRNA obtained in this way serves as the starting material for subsequent formulation into lipid nanoparticles (LNPs).
✧ ✧ ✧
Interplay of chromatographic steps in IVT mRNA purification
The purification of IVT mRNA does not rely on a single separation technique, but rather on the deliberate combination of multiple chromatographic principles with complementary strengths and limitations.
Poly(dT) affinity chromatography is used for the highly selective enrichment of polyadenylated mRNA. It efficiently removes non-polyadenylated by-products, enzymes, and low-molecular-weight process components. However, due to its purely sequence-based binding mechanism, it cannot fully discriminate between structurally similar RNA impurities. In particular, dsRNA, fragmented mRNA with residual poly(A) stretches, and RNA:DNA hybrids may co-elute with the target mRNA.
The subsequent anion-exchange chromatography (AEX) addresses this limitation by separating the remaining nucleic acids based on their effective charge density and conformation. Single-stranded mRNA, double-stranded RNA by-products, and complex RNA structures exhibit characteristic, but partially overlapping, binding and elution profiles.
RNA:DNA hybrids occupy a special position in this context. Due to their hybrid structural nature, they cannot be clearly assigned to either single- or double-stranded RNA. Their chromatographic separability strongly depends on the extent of the hybrid region, the proportion of free single-stranded RNA, and the overall spatial organization of the molecule. Accordingly, RNA:DNA hybrids may, depending on process conditions, either co-elute with mRNA or remain bound to the column.
Only the stepwise combination of affinity and ion-exchange chromatography therefore enables the substantial removal of relevant RNA by-products without compromising the structural integrity of the sensitive mRNA. At the same time, this approach highlights that complete elimination of all potential hybrid and structural variants is technically challenging and strongly dependent on process conditions.
1.3.5. Magnetic bead purification
dT-MBP = Poly(dT)-magnetic bead purification
A method described in regulatory documents (EMA EPAR, section 2.2, p. 32) for the purification of PCR products in process 1 is the use of magnetic beads.
Starting point: After in vitro transcription (IVT), DNase I digestion, and filtration
After in vitro transcription (IVT), enzymatic post-treatment with DNase I, and an initial coarse purification step by filtration (UF/DF or TFF), an mRNA-containing solution is obtained. In addition to the desired mRNA, this still contains various transcription by-products, including:
(a) residual free nucleotides (only in trace amounts)
(b) protein residues (e.g. enzymes or protein fragments)
(c) RNA fragments with and without a poly(A) tail
(d) incompletely capped RNA
(e) double-stranded RNA (dsRNA), partially with polyadenylated strands
(f) RNA:DNA hybrids
(g) residual DNA template fragments (especially short fragments)
(h) buffer salts from previous process steps
Principle
Magnetic bead purification is based on sequence-specific affinity binding. Magnetic polymer beads are functionalized with oligo(dT) sequences that are complementary to the poly(A) tail of mRNA.
mRNA molecules with a poly(A) tail bind specifically to these oligo(dT) ligands, while molecules without a poly(A) sequence remain in solution.
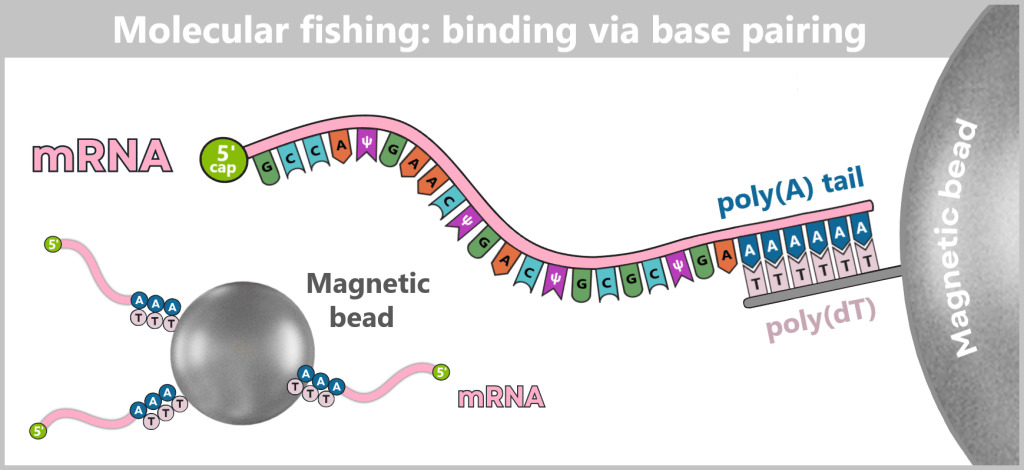
Fig. 1.3.5-A: The mRNA binds to the poly(dT) on the magnetic beads via its poly(A) tail. The process consists of several steps:
Binding: Poly(dT)-functionalized magnetic beads are added to the solution. Polyadenylated RNA binds to the bead surface via specific A–T base pairing.
Magnetic separation: The vessel is placed on a magnetic rack. The beads with bound RNA are drawn to the wall of the container, allowing unbound components to be removed.
Washing: Several washing steps reduce non-specifically bound molecules. The washing conditions influence binding stringency but do not enable complete removal of dsRNA or RNA:DNA hybrids if they contain polyadenylated RNA.
Elution: Under suitable elution conditions (e.g. altered salt concentration or temperature), the bound RNA is released from the beads and transferred back into solution.
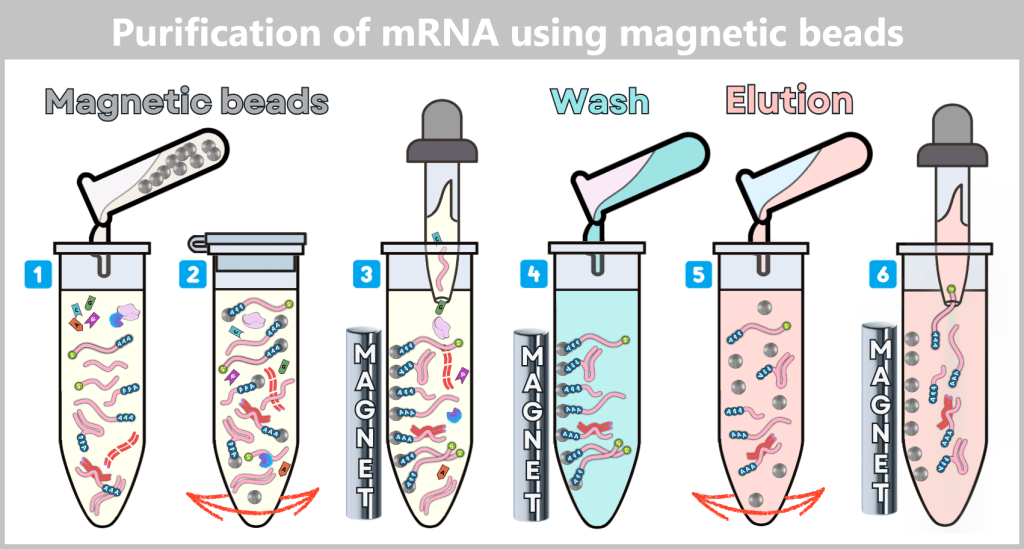
Fig. 1.3.5-B: Workflow of magnetic bead purification The representation is simplified and shows each component only once as an example.
1) Addition of magnetic beads: Poly(dT)-functionalized magnetic beads are added to the solution containing polyadenylated mRNA.
2) Mixing: The sample is gently mixed by rocking or brief rotation. Polyadenylated RNA hybridizes to the poly(dT) sequences on the magnetic beads via A–T base pairing.
3) Magnetic separation and removal of supernatant: The vessel is placed on a magnetic rack. The beads with bound RNA are pulled to the side of the tube, forming a compact bead aggregate. The liquid supernatant is carefully removed. It contains non-bound molecules such as DNA fragments, short RNA fragments, dsRNA without a poly(A) tail, buffer components, etc.
4) Washing steps: The magnet remains in place. Wash buffer is added to remove loosely bound or non-specifically adsorbed impurities. The sample is briefly mixed (gentle rocking), after which the beads are again attracted to the tube wall by the magnet. This step is repeated 2–3 times until loosely bound or non-specifically associated components are largely removed.
5) mRNA elution: After removal of the final wash buffer, the magnet is briefly removed. An elution buffer (e.g. low-salt water) or slightly elevated temperature disrupts the hybridization between polyadenylated RNA and poly(dT) oligos. The RNA is released back into solution.
6) Return to magnetic separation: The tube is placed on the magnet again, immobilizing the beads at the wall. The supernatant now contains the enriched polyadenylated RNA in elution buffer and is carefully transferred into a new sterile vessel.Note: dsRNA, RNA:DNA hybrids, or fragmented RNA containing poly(A) regions may co-elute during magnetic bead purification.
Separation performance and limitations
Magnetic bead purification is highly selective for polyadenylated RNA but has inherent limitations:
- dsRNA by-products may co-elute, especially if they contain poly(A) sequences or are structurally associated with mRNA
- RNA:DNA hybrids are co-purified if the RNA component is polyadenylated
- fragmented mRNA retaining a poly(A) tail also binds to the beads
- intramolecular double-stranded structures within mRNA are not selectively removed
For these reasons, magnetic bead purification does not represent a complete purification step but rather an enrichment and pre-purification stage.
Downstream purification
Since dsRNA and RNA:DNA hybrids with poly(A) regions may co-elute during magnetic bead purification, an additional purification step is generally required. This is often achieved using anion-exchange chromatography (AEX) or a comparable high-resolution separation method.Position in the manufacturing process
Poly(dT)-based magnetic bead purification is mainly used at laboratory scale, in process development, and in smaller production volumes. For large-scale industrial manufacturing processes (process 2), however, it is less practical, as magnetic separation becomes less efficient with increasing volume and handling becomes more complex. Therefore, scalable chromatographic methods are predominantly used in industrial production.Further details on its application can be found in the manufacturer documentation of Thermo Fisher Scientific.
1.4. Comparison: Process 1 vs. Process 2
The following overview compares the two manufacturing processes, showing which starting materials and by-products arise at each stage and which methods are typically used to remove them.
Typical steps – depending on the manufacturer:
Feature
Process 1 – PCR-based
Process 2 – plasmid-based
Type of process
Laboratory process
(small-scale, flexible)Industrial process
(large-scale production)
DNA template
Minimalistic.
Contains only the essential elements required for IVT:
• T7 promotor
• 5′ UTR
• Spike gene
• 3′ UTR
• Poly(A) sequenceComplex.
Includes additional elements required for propagation in E. coli:
• Origin of Replication (ORI)
• Antibiotic resistance gene
• bacterial REgulatory sequences
• Multiple Cloning Site (MCS)
• SV40 sequence elements
(for Pfizer/BNT)
Template structure
[T7] – [5′ UTR] – [Spike gene] – [3′ UTR] – [AAAA…]
[ORI] – [Resistance gene] – [bREs] – [MCS] – [SV40] – [T7] – [5′ UTR] – [Spike gene] – [3′ UTR] – [AAAA…]
DNA amplification
PCR-based amplification
Plasmid propagation in E. coli (bioreactor)
Pre-IVT preparation
/
Cell lysis: release of plasmid DNA + bacterial debris
Linearization: restriction enzyme cuts plasmid at a defined site
Impurities before IVT
PCR fragments: enzymes, primers, nucleotides, salts
Bacterial components: host DNA, RNAs, proteins, endotoxins, lipids, cell wall fragments, …
Purification
of PCR DNA
of plasmid DNA (pDNA)
Methods
Silica binding, UF/DF
TFF + multistep chromatography
Effort
moderate
high
IVT (in vitro transcription)
Template: PCR DNA
Expression cassette:
[T7] – [5′ UTR] – [Spike gene] – [3′ UTR] – [AAAA…]
→ mRNA is synthesizedTemplate: linearized pDNA
Expression cassette:
[T7] – [5′ UTR] – [Spike gene] – [3′ UTR] – [AAAA…]
→ mRNA is synthesized
IVT processing
5′ cap and poly(A) tail are generated either during or immediately after IVT depending on the process
→ functional mRNA obtained5′ cap and poly(A) tail are generated either during or immediately after IVT depending on the process
→ functional mRNA obtained
Impurities after IVT (*)
Removal of IVT by-products:
template DNA, RNA fragments, RNA:DNA hybrids, double-stranded RNA (dsRNA), enzymes, salts, free nucleotidesRemoval of IVT by-products + removal of additional plasmid- and bacterial-derived impurities (more challenging)
Purification
of mRNA
of mRNA
Upstream burden
low – moderate
high
Methods
▪️DNase I digestion
▪️Precipitation or centrifugation
▪️UF/DF or TFF
▪️Magnetic bead purification (**)▪️DNase I digestion
▪️Proteinase K Treatment (**)
▪️UF/DF and/or TFF (**)
▪️Multistep chromatography
Effort
moderate
high
LNP formulation
mRNA is packaged into lipid nanoparticles
mRNA is packaged into lipid nanoparticles
Sterile filtration
removes microbial contamination risk
removes microbial contamination risk
Filling
aseptic sealing and labeling
aseptic sealing and labeling
Advantages
very fast, highly flexible
highly scalable, cost-efficient mass production
Disadvantages
PCR-based methods are only limitedly suitable for large-scale industrial production volumes
template contains many bacterial elements → significantly higher purification effort
(*) Although the post-IVT reaction mixtures for process 1 and process 2 may appear similar at first glance, their „upstream burden” differs qualitatively. Upstream burden refers to the amount and complexity of accompanying substances present prior to purification. Plasmid-based processes introduce more structurally complex DNA forms, a higher tendency for hybrid formation, and trace bacterial-derived impurities that, even after DNase digestion, make subsequent mRNA purification more demanding.
(**) Information on the manufacturing process according to the EMA „EPAR for the BioNTech/Pfizer COVID-19 vaccine” (Section 2.2, p. 32).
Terminology:
T7 promoter – start signal for transcription
5′ UTR – stabilizes the mRNA
Spike gene – blueprint for the protein
3′ UTR – stop signal and stabilizing element
Poly(A) sequence – protects against premature degradationUTR stands for untranslated region – a segment of mRNA that is not translated into protein. These regions are located before the coding sequence (5′ UTR) and after it (3′ UTR). They perform important regulatory functions (see step 4).
Origin of replication (ORI) – starting point for plasmid replication
Antibiotic resistance gene – enables selection in E. coli
Regulatory bacterial sequences – control expression of the resistance gene
Multiple cloning site (MCS) – short region containing restriction sites for inserting the target sequence
SV40 sequence elements – regulatory viral sequences used in certain applications in eukaryotic cell culture (in Pfizer/BNT)
1.5. Summary: Production and Purification of modRNA
The production of modified mRNA for therapeutic and prophylactic applications is a multistep process that extends well beyond the actual in vitro transcription. The goal is to obtain from a complex reaction mixture a highly homogeneous, functionally active RNA molecule that is efficiently translated in cells while inducing only a controlled and desired immune response.
From DNA template to mRNA crude solution
The starting point of production is a defined DNA template, which serves as the template for RNA synthesis during in vitro transcription. Even at this early stage, various by-products are inevitably generated alongside the desired mRNA, including truncated transcripts, double-stranded RNA structures, RNA:DNA hybrids, and improperly processed RNA molecules. These by-products are not exceptions but a well-known consequence of enzymatic transcription under technical conditions.
After completion of transcription, the DNA template is typically enzymatically degraded. This is followed by an initial coarse purification step, such as filtration or tangential flow filtration, which removes small molecules, excess reagents, and enzymes. At this stage, a concentrated mRNA-containing solution is obtained; however, it remains structurally heterogeneous.
Enrichment of the target mRNA
To selectively enrich the desired mRNA, poly(A)-based affinity methods are employed. These include both classical poly(dT) chromatography and poly(dT)-functionalized magnetic beads. These approaches use the poly(A) sequence of mRNA as a selection feature and enable efficient separation of non-polyadenylated RNA fragments and many accompanying impurities.
At the same time, a key limitation of these methods becomes apparent: anything carrying a poly(A) sequence or structurally associated with polyadenylated RNA may be co-purified. This includes fragmented mRNA molecules, certain dsRNA by-products, and RNA:DNA hybrids, provided that the RNA component is polyadenylated. Poly(A)-based methods are therefore highly selective enrichment techniques, but not complete purification methods.
High-resolution separation by chromatography
To further reduce remaining structural heterogeneity, high-resolution separation techniques are applied, in particular anion-exchange chromatography (AEX). This method exploits differences in effective charge density and conformation of nucleic acids to separate single-stranded mRNA from more strongly bound by-products such as dsRNA.
AEX represents a critical step for the targeted depletion of immunologically active impurities. At the same time, it also highlights that certain molecular classes – especially RNA:DNA hybrids – do not exhibit uniform chromatographic behavior and may elute differently depending on their structure. Their complete removal therefore requires careful process design and, where necessary, the combination of multiple purification steps.
Classification and outlook
Overall, the production of modified mRNA is not a linear „input–output” process, but rather a finely tuned interplay of enzymatic, physical, and chromatographic methods. Each purification step reduces sample complexity, but none achieves absolute separation of all theoretically possible by-products.
Against this background, it is scientifically more appropriate to consider not only the final product itself, but also the logic of the manufacturing and purification process when discussing potential residual components such as DNA fragments or RNA-associated impurities. The key question is therefore less whether such residual components can arise in principle, but rather in which forms, at what levels, and with what biological relevance they may remain in the final product.
In view of the described manufacturing and purification processes, a central question emerges: how remaining nucleic acid components – particularly potential residual DNA – can be reliably detected and quantified.
The second part therefore addresses analytical detection methods, regulatory thresholds, and published experimental data on residual DNA in mRNA-based vaccines.
Further part of the article series
Part 2: Detection Methods and the Current State of Research
Current as of June 2026.
-
Residual DNA in modRNA-Based Vaccines – Part 2

Guide to the Article Series
Part 1: Production and Purification of modRNA
Part 2: Detection Methods and Current EvidenceFollowing the discussion of manufacturing and purification processes in Part 1, the focus now shifts to the analytical aspects of the debate. Whether residual DNA is present in modRNA-based vaccines, and in what quantities, depends crucially on the methods used to detect and characterize it.
The detection of residual nucleic acids is methodologically challenging. Different lysis procedures, extraction methods, and analytical platforms can lead to significantly different results. This not only complicates the interpretation of individual studies, but also raises the fundamental question of what exactly a given measurement captures under specific experimental conditions.
Part 2 therefore examines the most important analytical methods for detecting DNA and RNA, their methodological limitations, and the independent studies published to date on residual DNA in modRNA-based vaccines. Particular attention is given to the interpretive value of the methods used and the comparability of the reported results.
Note on terminology
📑
In general usage, the term „mRNA vaccine” is common. From a technical perspective, however, the approved products consist of nucleoside-modified mRNA (modRNA). For reasons of clarity and readability, this work primarily uses the established term „mRNA vaccine”.Table of contents
2. Methods for Measuring DNA and RNA
3. Guidelines for Limiting Residual DNA in Vaccines
4. Experimental Studies on residual DNA in mRNA-based Vaccines
5. Conclusion and Outlook

2. Methods for Measuring DNA and RNA
The analytical determination of RNA concentrations and residual DNA is a key component of quality control for modRNA-based pharmaceuticals. During the manufacturing and purification processes, complex mixtures of nucleic acids are generated, whose composition must be characterized and monitored using appropriate analytical methods. The objective is to reliably assess both the integrity and quantity of the desired mRNA, as well as possible residual components such as DNA fragments.
No single method is capable of addressing all analytical questions equally well. Rather, the available techniques differ with regard to sensitivity, specificity, quantifiability, and structural resolution. Some methods allow rapid determination of total nucleic acid concentration, while others enable the selective detection of specific DNA sequences or even the direct sequence analysis of individual molecules.
Against this background, the following sections provide a systematic overview of different methodological approaches – beginning with fundamental spectrophotometric techniques, continuing through fluorescence-based quantification methods and amplification-assisted detection procedures, and extending to sequencing-based technologies. The presentation follows a progression of increasing analytical resolution: from global concentration measurements to sequence-specific identification of individual nucleic acid components.
In addition to the functional principles of each method, their detection limits and methodological constraints are also considered, as these are of critical importance for the interpretation of possible residual DNA findings.
2.1. Spectrophotometric Methods
2.2. Fluorescence-based Quantification
2.3. Amplification-based Methods
2.4. Sequencing-based Methods
2.5. Electrophoretic Fragment Analysis
2.6. Comparison of Key Detection Methods for Nucleic Acids
2.1. Spectrophotometric Methods
Spectrophotometric methods are based on the interaction of light with matter. The intensity of light is measured before and after passing through a sample as a function of wavelength. From the measured absorption, the concentration of the substances present can be inferred under defined conditions.
UV/Vis spectrophotometry is an established method for determining total nucleic acid concentration and is used at several stages of mRNA production. It is characterized by rapid execution, low sample consumption, and comparatively low cost.
When is photometry used?
In the manufacturing process, three relevant measurement time points can typically be distinguished:
After in vitro transcription: to estimate whether the transcription reaction was successful and what total nucleic acid yield was achieved.
After purification: as a rapid control step to verify whether the mRNA concentration is within the expected range.
Before formulation: prior to encapsulation in lipid nanoparticles, the mRNA concentration is measured again, since the mixing ratio between lipids and mRNA is critical for particle formation.
It should be noted that this method cannot distinguish between RNA and DNA and therefore primarily provides an indicative measurement of total nucleic acid concentration.
Typical workflow in the manufacturing process
After purification of the mRNA and before LNP formulation, the mRNA drug substance batch undergoes a concentration measurement. For this purpose, a small sample is taken from the process vessel under controlled conditions.
Two types of instruments are available for this measurement, which differ in their handling:
UV/Vis spectrophotometers: The sample is placed into a cuvette – a small, usually rectangular container made of UV-transparent material (e.g., quartz glass). The cuvette is inserted into the instrument, where the measurement is performed.
Microvolume spectrophotometers (e.g., NanoDrop): A tiny droplet (0,5–2,0 µl) is pipetted directly onto a measurement pedestal. A second (folded-down) pedestal forms a defined liquid column between the two optical surfaces via surface tension. The major advantage is that neither cuvettes nor significant sample volumes are required. These instruments are commonly used for DNA and RNA measurements.
Regardless of the instrument type, the actual spectrophotometric analysis takes only a few seconds. Immediately after the measurement, the result is displayed on the screen.

Fig. 2.1-A: UV/Vis spectrophotometer vs microvolume spectrophotometer The path of light in a spectrophotometer
1️⃣ Light generation: The light source is a broadband UV/Vis lamp, for example a xenon flash lamp, which emits short, intense pulses of light covering the entire UV and visible spectrum.
2️⃣ Wavelength selection: The light passes through the entrance slit into the monochromator, where it is separated into its spectral components by a movable diffraction grating. To determine the mRNA concentration, the grating is set so that light with a wavelength of 260 nm – the absorption maximum of nucleic acids – is directed onto the sample.
3️⃣ Light guidance to the sample: A system of mirrors and lenses focuses the monochromatic light beam and precisely directs it onto the sample – either onto the cuvette or directly onto the suspended sample droplet.
4️⃣ Interaction with the sample: A portion of the light is absorbed by the nucleic acid molecules, while the remainder is transmitted through the sample.
5️⃣ Detection: The transmitted light reaches a detector (e.g., a photodiode or a CCD sensor), which measures the incoming light intensity.
6️⃣ Calculation: The instrument compares the measured intensity with the intensity of the incident light (reference measurement without a sample). Based on the ratio of incident to transmitted light intensity, the instrument calculates the absorbance in accordance with Lambert-Beer’s law; given the known layer thickness, this allows the concentration of nucleic acids in the sample to be determined.
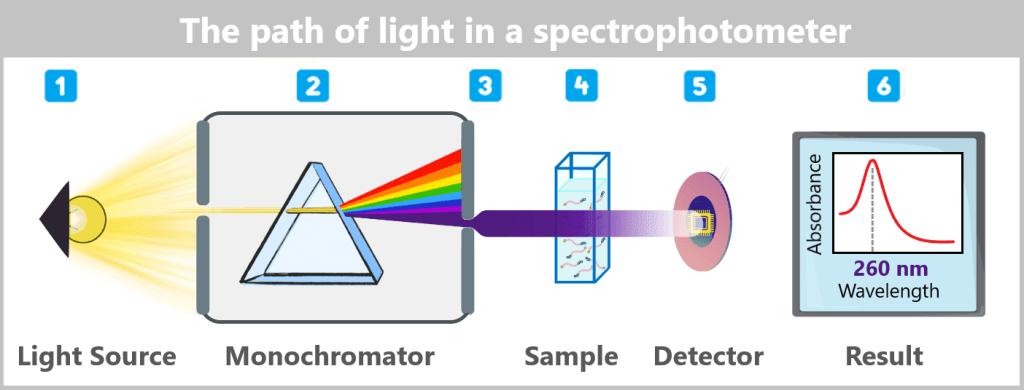
Fig. 2.1-B: Schematic representation of a UV/Vis spectrophotometer Why do RNA and DNA absorb at 260 nm?
The absorption of UV light at 260 nm is a characteristic property of the nucleobases – the „letters” that make up the genetic code. Since these bases are nearly identical in RNA and DNA, both molecules exhibit very similar absorption behavior.
The secret lies in their chemical structure: the bases contain so-called aromatic rings. In these rings, electrons are not fixed between specific atoms but are distributed across the entire ring system. These „delocalized electrons” can be imagined as a kind of electron cloud spread over the molecule.
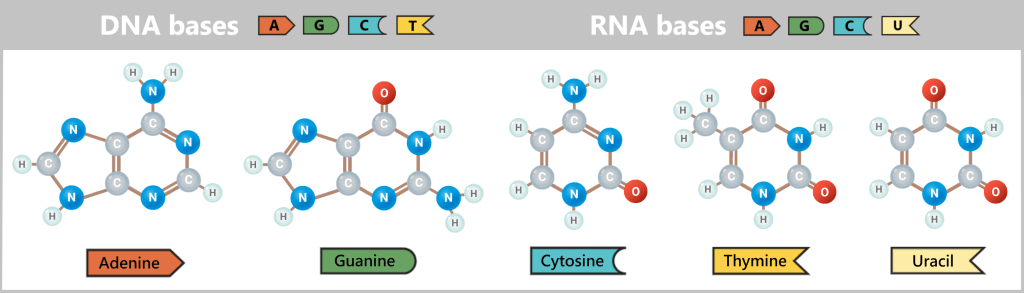
Fig. 2.1-C: The molecular building blocks of nucleic acids The illustration shows the two types of bases: the larger double-ring structures (purines: A and G) and the smaller single-ring structures (pyrimidines: C, T, and U). Despite their different sizes, both types contain a system of alternating double bonds. This allows the electrons to be delocalized across the entire ring system, forming a shared electron cloud.
When a photon (a particle of light) with a wavelength of around 260 nm encounters this electron cloud, its energy precisely matches the energetic properties of the electrons in these rings. An electron absorbs the photon’s energy entirely and is thereby promoted to a higher energy level – the so-called excited state. The energy of the photon is fully transferred to the electron, meaning the photon no longer exists as a light particle; this process is referred to as absorption.
Since both RNA and DNA consist of the same types of bases (with the exception of uracil replacing thymine), both types of molecules exhibit nearly identical behavior under UV light. Spectrophotometry therefore cannot directly distinguish between RNA and DNA.
Significance for the assessment of residual DNA
The physical principles described above also explain a key limitation of UV spectrophotometry: since absorption at around 260 nm is based on the shared nucleobase structures of RNA and DNA, the method only measures the total amount of nucleic acids in a sample. It is not possible to distinguish between therapeutic mRNA and any residual DNA fragments on this basis.
For the quantitative determination of specific residual DNA sequences, analytical methods are therefore required that either selectively detect double-stranded DNA or amplify defined target sequences. In practice, fluorometric DNA assays (e.g., Qubit technologies) and quantitative polymerase chain reaction (qPCR) are particularly important in this context.
The discussion of suitable methods for detecting residual DNA therefore focuses less on photometric techniques and primarily on molecular biological and fluorescence-based approaches.
2.2. Fluorescence-based Quantification
2.2.1. DNA-/RNA-Specific Dyes
a) Mechanism of action of PicoGreen
b) Why PicoGreen preferentially detects double-stranded DNA
c) Limitations of the PicoGreen method
2.2.2. Qubit SystemsIn contrast to UV spectrophotometry, which measures the attenuation of an incident light beam, fluorescence-based methods rely on the targeted excitation of specific molecules followed by measurement of the light they emit. (Put more simply: while UV spectrophotometry determines how much light „disappears”, fluorescence measures not the shadow, but the glow.)
In this process, a fluorescent dye is added to the sample. The dye itself exhibits little or no fluorescence. Only after binding to a target structure – such as double-stranded DNA or RNA – does its spatial arrangement change, so that, when excited by light of a specific wavelength, it emits a characteristic fluorescent signal. The intensity of this emitted light is proportional to the amount of bound nucleic acid.
The key feature of fluorescence-based methods is their selectivity: by using appropriate dyes, specific types of nucleic acids can be selectively detected, while other sample components remain largely ignored. This allows for substantially more specific quantification than the non-specific UV absorption at 260 nm.
Particularly at low concentrations, such as in the determination of residual DNA in mRNA preparations, fluorescence-based methods offer significantly higher sensitivity.
2.2.1. DNA-/RNA-Specific Dyes
The basis of fluorescence-based quantification is formed by nucleic acid-binding dyes that selectively interact with DNA or RNA.
Double-stranded DNA-specific dyes
A commonly used DNA-specific dye is PicoGreen. It preferentially binds to double-stranded DNA. In free solution, PicoGreen exhibits only very weak intrinsic fluorescence. Only after binding to DNA does its fluorescence intensity increase dramatically. This enables the quantification of even very small amounts of DNA.
RNA-specific dyes
For RNA, RiboGreen is frequently used. This dye also produces a strong fluorescent signal only after binding to RNA.
The differing binding affinities of these dyes allow for a largely selective detection of the respective type of nucleic acid.
The measurement principle always follows the same basic scheme:
- Addition of a specific dye to the sample
- Binding of the dye to the target nucleic acid
- Excitation with light of a defined wavelength (λEX – excitation)
- Measurement of the emitted fluorescence (λEM – emission)
- Comparison with a calibration series of known concentrations
Since only the bound dye produces a strong signal, background fluorescence remains low. This explains the high sensitivity of this method compared with UV spectrophotometry.
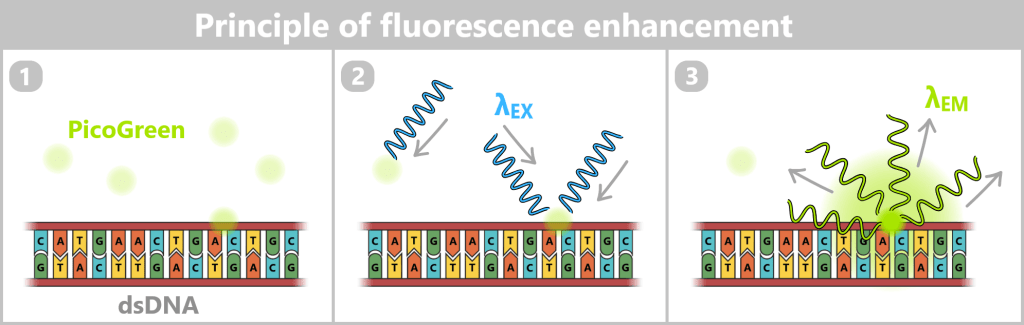
Fig. 2.2.1-A: Schematic representation of DNA quantification by fluorometry 1) After addition of the fluorescent dye (e.g., PicoGreen), preferential binding to double-stranded DNA (dsDNA) occurs.
2) The sample is irradiated with short-wavelength, high-energy light of a defined wavelength (λEX ≈ 480 nm, blue), which excites the dye molecules.
3) Upon returning to the ground state, the bound dye emits light of a longer wavelength (λEM ≈ 520 nm, green). Within the linear measurement range, the measured fluorescence intensity is proportional to the DNA concentration present in the sample. Since free dye exhibits only very weak intrinsic fluorescence, a strong signal is generated primarily by DNA-bound molecules.Quantification is performed by comparing the signal intensity with a standard curve of known DNA concentrations.
a) Mechanism of action of PicoGreen
PicoGreen is a fluorescent dye with a high affinity for double-stranded DNA (dsDNA). Its exceptional sensitivity is based on the fact that it exhibits almost no fluorescence in its free state, but fluoresces very strongly after binding to DNA.
The difference between these two states (free vs. bound) is crucial for analytical application.
Free PicoGreen in solution
In aqueous solution, the molecule is highly mobile:
- It can rotate and bend.
- Its aromatic ring systems can move relative to each other.
- It constantly collides with water molecules.
When the molecule is excited by light, it absorbs energy. In its free state, however, this energy is not primarily released as light. Instead, it is dissipated through molecular motion and interactions with water, being converted into heat. As a result, intrinsic fluorescence is very low. Free PicoGreen therefore remains almost „dark”.
Approach to double-stranded DNA
PicoGreen carries positive charges. The DNA backbone is negatively charged due to its phosphate groups. This creates an electrostatic attraction between the dye and DNA. The molecule preferentially associates with double-stranded DNA (dsDNA), particularly in regions where the two DNA strands are closely aligned. This initial approach is a prerequisite for a more stable binding interaction.
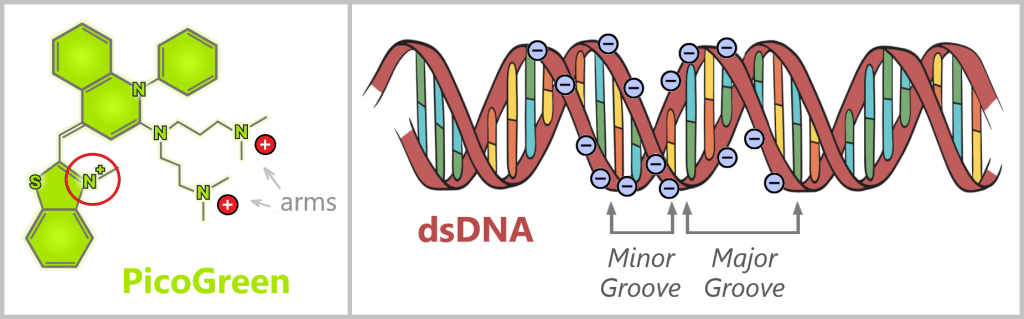
Fig. 2.2.1-B: Schematic representation of the structure of PicoGreen and dsDNA (not to scale) On the left, a simplified structure of the fluorescent dye PicoGreen is shown. The hexagonal and pentagonal shapes in the structure represent aromatic ring systems – particularly stable, planar molecular units capable of absorbing UV light. In addition, the dye contains positively charged side groups.
On the right, double-stranded DNA is depicted with its negatively charged sugar-phosphate backbone. The electrostatic attraction between the positively charged side groups of the dye and the negatively charged DNA backbone initially brings the dye into close proximity to the DNA.Binding and fixation
After binding, the molecule’s mobility is strongly restricted:
- It can hardly rotate or bend anymore.
- Parts of the molecule are positioned between or close to the base pairs.
- Direct contact with water is partially reduced.
In this sense, DNA acts like a scaffold that stabilizes the molecule in a fixed position.
Why fluorescence increases strongly
This fixation changes the molecule’s energy dissipation pathways: in the free state, a large portion of the absorbed energy is lost through molecular motion. In the bound state, these motions are strongly restricted.
As a result, the probability that the absorbed energy is released as light – i.e., as fluorescence – increases significantly. The outcome is a much stronger signal.
The difference between free and DNA-bound PicoGreen is so pronounced that even very small amounts of double-stranded DNA can be reliably detected.
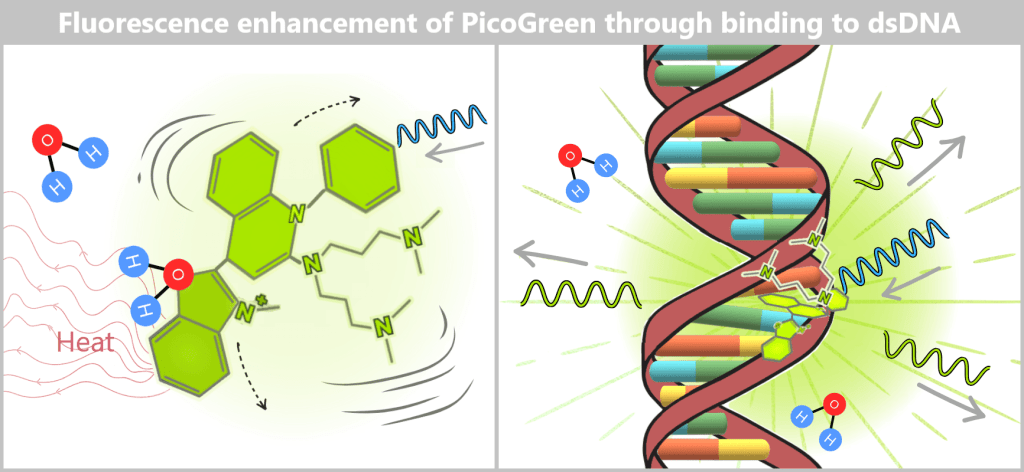
Fig. 2.2.1-C: Fluorescence behavior of PicoGreen – free in solution and bound to dsDNA Left: PicoGreen in aqueous solution (without DNA)
In aqueous solution, the dye molecule is in constant motion: it rotates and undergoes intramolecular vibrations. When excited with blue light, it dissipates most of the absorbed energy through these motions into the surrounding water as heat. As a result, the dye exhibits only very weak intrinsic fluorescence.
Right: PicoGreen bound to dsDNA
After binding to double-stranded DNA, PicoGreen inserts into the minor groove and is further stabilized by stacking interactions with the bases. This strongly restricts its molecular mobility. As a consequence, less of the absorbed energy is lost through molecular motion, and a much larger fraction is emitted as green light. The fluorescence intensity therefore increases significantly.Why this property is analytically so valuable
- Free dye produces almost no background signal.
- Only bound dye generates a strong fluorescent signal.
- Double-stranded DNA is preferentially detected.
In the context of measuring residual DNA in mRNA preparations – even at very low concentrations – this strong contrast between „dark” and „bright” is crucial for the sensitivity of the method.
b) Why PicoGreen preferentially detects double-stranded DNA
The selectivity of PicoGreen is not based on the dye „recognizing” DNA in a conscious sense, but rather on structural features of double-stranded DNA.
Double-stranded DNA has:
- a regularly arranged stacked base structure
- tightly packed, planar base pairs
- clearly defined major and minor grooves
- a stable, spatially ordered conformation
This ordered architecture provides suitable binding sites for the dye. It can intercalate between or bind close to the stacked bases and is thereby mechanically constrained. This restriction is exactly what drives the strong increase in fluorescence.
Single-stranded DNA or RNA have a much more flexible structure. They lack the regularly stacked base organization of the double helix. Although interactions can still occur, they are:
- weaker in binding strength
- less stable in structural fixation
- associated with a much smaller fluorescence enhancement
Therefore, PicoGreen reacts particularly sensitively to double-stranded DNA – and much more weakly to RNA or single-stranded nucleic acids.
For the analysis of residual DNA, this property is crucial: residual plasmid DNA or DNA fragments are typically double-stranded and therefore generate a strong signal.
Although DNA-specific fluorescent dyes such as PicoGreen show a strong preference for double-stranded DNA, their selectivity is not absolute. Especially in samples with very high RNA concentrations, nonspecific signal contributions may occur. These can arise from weak binding to RNA, secondary RNA structures, or simple mass effects. Without appropriate control measures – such as RNase treatment – this may lead to an overestimation of the DNA content.
c) Limitations of the PicoGreen method
Despite its high sensitivity, the method remains a quantitative bulk measurement of dsDNA. It does not provide information about:
- the sequence of the DNA
- its exact origin
- its biological functionality
- the fragment composition
Influence of fragment length
The signal intensity depends, among other factors, on how many binding sites are available. Very short DNA fragments can bind fewer dye molecules than long, intact molecules.
This means that two samples with an identical mass of DNA can produce slightly different fluorescence signals depending on the degree of fragmentation.
In practice, this effect is accounted for using calibration standards, but it cannot be completely eliminated.
Matrix effects
Components of the sample can influence the measurement result, such as:
- high salt concentrations
- residual proteins
- surfactants or lipid components
- buffer composition
Such factors can affect binding efficiency or fluorescence intensity. For this reason, measurement conditions are standardized and validated.
✧ ✧ ✧
Interim conclusion
Fluorescence-based methods such as PicoGreen enable highly sensitive quantification of double-stranded DNA and are fundamentally well suited for detecting small amounts of DNA even in complex samples.
However, the method is not based on sequence-specific recognition but on structural properties of nucleic acids. The measured signal can therefore be influenced by factors such as residual RNA, fragment length, secondary structures, or components of the sample matrix. Without appropriate control measures – in particular RNase treatment and standardized sample preparation – there is a risk of over- or underestimating the actual DNA content.
PicoGreen therefore primarily answers the question of how much fluorescence-active double-stranded DNA is present in a sample. It does not provide information about the sequence, origin, integrity, or biological functionality of the DNA.
For such questions, complementary sequence- or amplification-based methods are required, which are discussed in the following chapter.
2.2.2. Qubit systems
While DNA- and RNA-specific dyes such as PicoGreen or RiboGreen represent the actual fluorescent tools used for nucleic acid quantification, the Qubit system refers to the integrated analytical platform designed for precise concentration measurement based on these dyes.
The Qubit system is a compact fluorometer developed by Thermo Fisher Scientific for the highly sensitive and specific quantification of DNA, RNA, and proteins. It combines:
- specific fluorescent dyes = molecular sensors
- optimized reagent solutions = defined environment (buffer system)
- standardized assay protocols = standardized operating procedures
The aim of this system is the selective and sensitive quantification of DNA or RNA in biological samples.
Principle of the Qubit system
The underlying measurement principle corresponds to the method described previously (Chapter 2.2.1): a selective fluorescent dye binds to the target nucleic acid, is excited with light of a defined wavelength, and emits a measurable fluorescence signal. The intensity of this signal is proportional to the amount of bound DNA or RNA. By comparison with standard solutions of known concentration, the concentration of the sample can then be determined.
The Measurement Procedure
The procedure is intentionally simple and follows a standardized protocol:
1️⃣ Preparation of the working solution: The fluorescent dye (from the assay kit) is mixed with the supplied buffer in the recommended ratio.
Examples of available assay kits:
Assay kit Intended application dsDNA BR (Broad Range) Higher DNA concentrations dsDNA HS (High Sensitivity) Low DNA concentrations RNA BR (Broad Range) Higher RNA concentrations RNA HS (High Sensitivity) Lower RNA concentrations microRNA Assay Small RNA molecules such as miRNA and siRNA Protein Assay Quantification of proteins 2️⃣ Addition of the sample: A small volume of the sample (typically 1–20 µl) is added to the working solution.
3️⃣ Allow binding to occur: After a short incubation period of only about 2 minutes (for DNA/RNA assays), the dye binds specifically to its target molecule.
4️⃣ Calibration using standards: One or two supplied standards with known concentrations are measured. The instrument automatically generates a standard curve from these measurements.
5️⃣ Insert the sample into the instrument:
Excitation: The instrument irradiates the sample with the appropriate excitation wavelength, e.g., blue light at approximately 470 nm for DNA quantification.
Emission: The bound dye molecules absorb this light and re-emit it at a longer wavelength (lower energy), for example green light at approximately 520 nm.
Filtering: An optical filter inside the instrument blocks the blue excitation light so that the detector registers only the green fluorescence emitted by the sample. This explains the high accuracy of the system: the instrument „sees” only the target signal, while interfering excitation light is excluded.6️⃣ Measurement and result: The instrument measures the emitted longer-wavelength light (e.g., green fluorescence). Within seconds, an algorithm calculates the exact concentration based on the standard curve. The result is displayed on the screen.
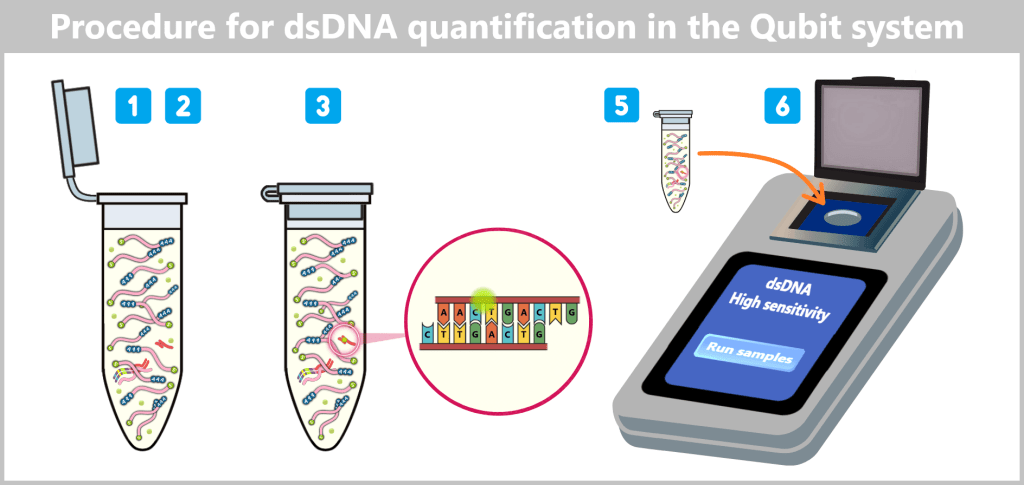
Fig. 2.2.2: Schematic workflow of dsDNA quantification using the Qubit system The illustration shows the workflow of dsDNA quantification with the Qubit system. After addition of the specific fluorescent dye to the sample (1–2), the dye selectively binds to double-stranded DNA (3). The sample is then inserted into the Qubit fluorometer (5), which measures the fluorescence intensity and calculates the DNA concentration (6).
Advantages of the Qubit system
- High specificity and selectivity: The fluorescent dyes used exhibit a strong preference for their respective target molecules. This allows, for example, relatively selective quantification of double-stranded DNA (dsDNA) even in the presence of larger amounts of RNA. However, nonspecific signal contributions cannot be completely excluded, particularly at very high RNA concentrations or in complex sample matrices.
- High sensitivity: Compared with UV photometry, the method is significantly more sensitive and allows the quantification of very low nucleic acid concentrations.
- Low sample consumption: Only small sample volumes are required for measurement (typically 1–20 µl), which is especially advantageous when sample material is limited or valuable.
- Relative robustness against contaminants: Many substances that can interfere with UV spectroscopic methods – such as free nucleotides, salts, or proteins – have a much smaller effect on fluorometric measurements. Nevertheless, certain matrix components may still influence fluorescence.
- Simple handling and rapid results: The system is based on standardized procedures and enables reproducible measurements with comparatively low effort.
Limitations of the Qubit system
- No information on DNA integrity: The method does not distinguish between intact and fragmented DNA.
- No sequence information: The measurement provides no information about the sequence, origin, or biological function of the nucleic acids.
- Influence of fragment length: Different fragment lengths can affect signal intensity. Very short or highly degraded fragments may produce a reduced fluorescence signal.
- Residual nonspecificity at high RNA concentrations: Very high RNA concentrations can lead to nonspecific signal contributions despite the high specificity of the dyes.
- Limited linear measurement range: Quantification is restricted to the linear range of the respective assay. Highly concentrated samples therefore need to be diluted accordingly.
- Limited information on sample purity: Only limited conclusions can be drawn regarding sample purity, composition, or possible contamination.
✧ ✧ ✧
Summary
Qubit fluorometry is considered a sensitive and comparatively selective method for the quantification of DNA and RNA. In contrast to UV spectroscopy, it is not based on nonspecific light absorption but on fluorescent dyes with a strong preference for specific nucleic acid structures. This enables the measurement of low concentrations even in complex samples.
However, the method remains a structure-dependent bulk measurement: it provides information about the amount of specific nucleic acid types, but not about their sequence, origin, or biological properties. For such questions, sequence- or amplification-based methods are required, which are discussed in the following chapters.
2.3. Amplification-based Methods
The previously described methods (UV spectroscopy and fluorometry) allow the determination of the total amount of nucleic acids – i.e. RNA and/or DNA. However, for the targeted detection of specific DNA or RNA sequences, methods based on amplification of the target sequence are required. This principle is referred to as amplification.
In the manufacturing process, amplification-based methods are used in particular to detect residual DNA impurities.
The basis of these methods is the polymerase chain reaction (PCR). In this process, a predefined DNA segment is exponentially amplified through repeated cycles of enzymatic synthesis, meaning that the amount of the target sequence ideally doubles in each cycle. Even a few initial copies can thus be converted into a measurable quantity. Both quantitative PCR (qPCR) and digital PCR (ddPCR) are based on this principle.
2.3.1. quantitative PCR (qPCR)
2.3.2. digital PCR (ddPCR)
2.3.3. Comparison of PCR-based quantification methods⚬ ⚬ ⚬
2.3.1. Quantitative Polymerase Chain Reaction (qPCR)
Quantitative PCR, also known as real-time PCR, is an advanced version of conventional PCR. It enables the simultaneous amplification and quantification of a specific DNA segment in real time.
The basic reaction steps correspond to those of conventional PCR, as described in the first part (amplification of spike DNA by PCR). The key difference is that during each amplification cycle, the amount of newly formed DNA is measured via a fluorescent signal.
In qPCR, a fluorophore is used as a „light signal”. In principle, two strategies can be distinguished:
a) Dye-based qPCR
b) Probe-based qPCR◦ ◦ ◦
a) Dye-based qPCR (e.g., SYBR® Green)
For the analysis, a reaction mixture is added to the sample consisting of:
- free nucleotides
- a specific primer pair
- a thermostable DNA polymerase
- a buffer containing Mg²⁺ ions (for polymerase activity)
- a fluorescent dye (e.g., SYBR® Green)
SYBR® Green is a fluorophore that selectively binds to double-stranded DNA. Only after binding to dsDNA does the dye exhibit strong fluorescence.
This mixture is placed into a qPCR instrument – a so-called real-time PCR thermocycler.
Principle of the method
1️⃣ Denaturation: The reaction mixture is heated to approximately 94–98 °C. At this temperature, hydrogen bonds between base pairs are broken, and the double-stranded DNA is separated into two single strands.
2️⃣ Primer annealing: The temperature is lowered to approximately 50–65 °C. Specific primers bind to their complementary target sequences on the single strands. They define the starting point of DNA synthesis.
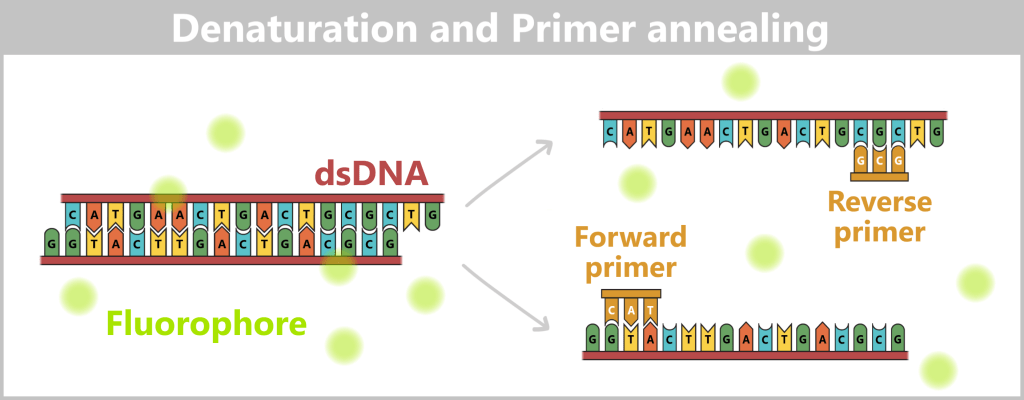
Fig. 2.3.1-A1: SYBR Green in the first qPCR cycle – denaturation and primer annealing Left: The dye SYBR® Green is added to the reaction mixture before the start of PCR. It binds selectively to double-stranded DNA and exhibits strong fluorescence in its bound state.
Right: During denaturation, double-stranded DNA is separated into single strands. The bound dye is released in the process and largely loses its fluorescence. Subsequently, specific primers bind to their complementary target sequences. (Note: For clarity, the primers are shown shortened with only three nucleotides; in practice, they are significantly longer.)3️⃣ DNA synthesis (amplification): At approximately 72 °C, the DNA polymerase synthesizes the complementary strand. This results in the formation of new double-stranded DNA. SYBR® Green binds to this newly formed dsDNA.
4️⃣ Excitation and emission: The reaction mixture is irradiated with blue light (approx. 490 nm). The bound dye molecules absorb this energy and emit green light (approx. 520 nm). The DNA-bound dye generates a measurable fluorescence signal.
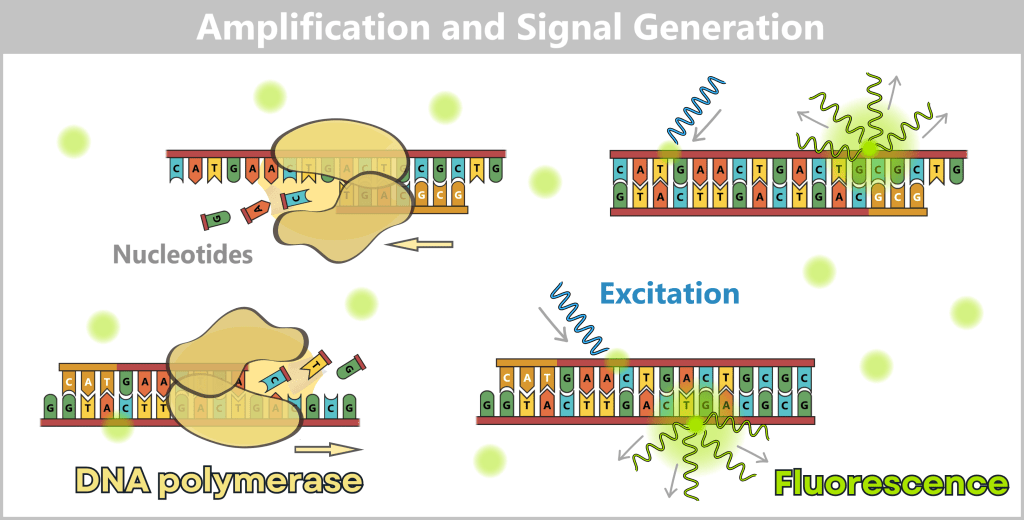
Fig. 2.3.1-A2: SYBR Green in the first qPCR cycle – from double strand to fluorescence signal Left: DNA polymerase synthesizes new complementary strands. This results in the formation of new double-stranded DNA, to which SYBR® Green immediately binds.
Right: At the end of the cycle, the reaction mixture is excited with light of an appropriate wavelength. The dye molecules bound to double-stranded DNA emit green fluorescence light, the intensity of which is proportional to the amount of DNA formed.5️⃣ Quantification: Fluorescence is measured at the end of each cycle. As the amount of DNA increases, the signal rises accordingly, producing a characteristic amplification curve.
The key measurement parameter is the so-called Ct value (cycle threshold). It refers to the cycle at which the fluorescence signal first exceeds a defined threshold.
- Low Ct value → high initial amount of target DNA
- High Ct value → low initial amount of target DNA
For absolute quantification, a standard curve based on known reference concentrations is typically generated.
Significance of amplicon length for detection
An amplicon refers to the DNA fragment that is amplified during PCR between the two primer binding sites – including the primers themselves.
Specific detection of the target sequence requires that a complete amplicon can be formed. This is only possible if both primers – forward and reverse – can bind to the same DNA fragment. If the fragment is shorter than the distance between the two primer binding sites, one of the primers cannot bind. Efficient exponential amplification does not occur, and no meaningful fluorescence signal is generated.
This leads to an important methodological consequence: the longer the chosen amplicon – i.e., the greater the distance between the two primer binding sites – the more short fragments remain undetected. This is not because they are absent, but because they are too short for the assay. Thus, the choice of amplicon length directly influences which fraction of the actual DNA present is detected.
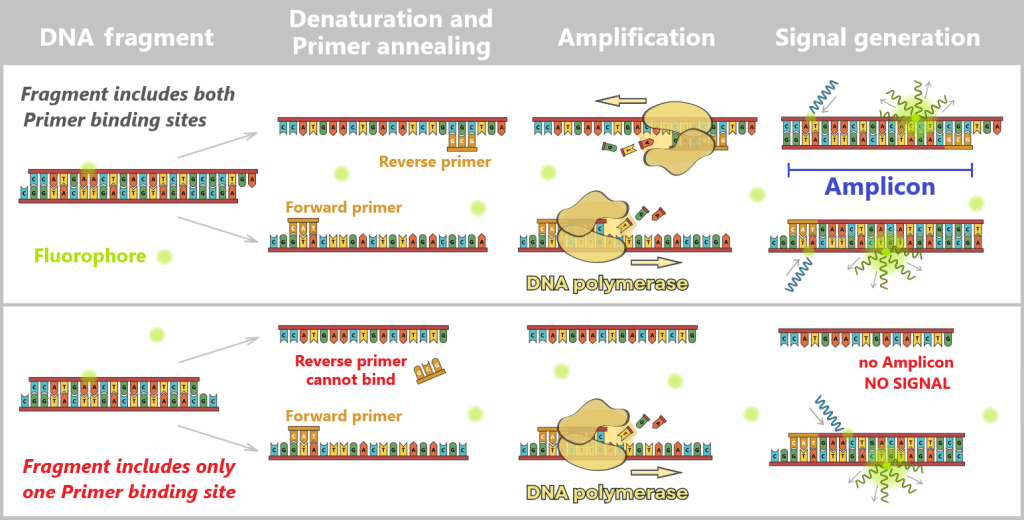
Fig. 2.3.1-A3: Significance of amplicon length for detection Top: Fragment contains both primer binding sites
The DNA fragment is long enough for both primers (forward and reverse primer) to bind to their respective binding sites. DNA polymerase can fully copy the region between the primers. A specific amplicon of defined length is generated. In the next cycle, this product can again be efficiently amplified. As a result, a strong fluorescence signal is produced with SYBR Green (proportional to the amount of specific dsDNA).
Bottom: Fragment contains only one primer binding site
The DNA fragment is shorter than the distance between the two primer binding sites. Only one primer can bind (the other has no complementary sequence). No complete amplicon is formed, and therefore no exponential amplification occurs. As a result, no or only a very weak SYBR Green fluorescence signal is generated.Note: For clarity, the primers are shown shortened with only three nucleotides; in practice, they are significantly longer. The schematic representation of DNA fragments is also simplified.
Advantages of dye-based qPCR
- Low cost and effort: The method is comparatively inexpensive and technically straightforward.
- Simple primer design: Only sequence-specific primers are required.
- High sensitivity: Even very small amounts of target DNA can be reliably detected.
- Fast execution: The method is widely established and enables rapid analysis of large numbers of samples. Quantification is performed directly via fluorescence detection during amplification.
- High flexibility: New target sequences can be analyzed relatively quickly by adjusting the primers.
- Melting curve analysis as quality control: After amplification, the specificity of PCR products can be assessed based on their characteristic melting temperature. This often allows the detection of nonspecific products or primer dimers.
Disadvantages of dye-based qPCR
- Nonspecific dye binding: The fluorescent dye binds to any double-stranded DNA – including nonspecific amplification products such as primer dimers.
- Dependence on primer design: The specificity of the analysis strongly depends on the quality and selectivity of the primers used.
- Influence of the target region: The choice of the target sequence and, in particular, the length of the amplified region (amplicon) significantly affects the result.
- Influence of fragment length: Highly fragmented DNA may be incompletely detected when longer amplicons are used, as both primer binding sites are often no longer present on the same fragment.
- Influence of amplicon length: Shorter amplicons generally detect fragmented DNA more efficiently, but may increase the likelihood of nonspecific signals.
- Dependence on efficiency and calibration: The accuracy of quantification is strongly influenced by PCR efficiency and the quality of the standard curve used.
b) Probe-based qPCR (e.g., TaqMan®)
For the analysis, a reaction mixture is added to the sample consisting of:
- free nucleotides
- a specific primer pair
- a thermostable DNA polymerase with 5’→3′ exonuclease activity (this polymerase can degrade obstacles in its path)
- a buffer containing Mg²⁺ ions (required for polymerase activity)
- a sequence-specific probe (e.g., a TaqMan® probe)
Structure and function of the TaqMan® probe
A TaqMan® probe is a short, synthetically produced DNA sequence that is complementary to a segment of the target sequence. The probe binds to one of the two DNA strands, specifically between the two primer binding sites.
The probe carries:
• a fluorescent dye (reporter) at the 5′ end
• a quencher molecule at the 3′ end
The reporter can carry different fluorophores, which also enables multiplex analyses – the simultaneous detection of multiple target sequences.
As long as reporter and quencher are in close proximity, the fluorescence signal is suppressed. Quenching only works while the probe is intact: due to the spatial proximity of reporter and quencher, fluorescence resonance energy transfer (FRET) occurs. In this process, the energy absorbed by the reporter is transferred to the quencher and dissipated as heat instead of being emitted as light.
The reaction is carried out in a real-time PCR thermocycler, which not only performs the temperature cycles but also measures fluorescence during amplification.
Principle of the method
1️⃣ Denaturation: The reaction mixture is heated to approximately 94–98 °C. Hydrogen bonds break, causing the double-stranded DNA to separate into two single strands.
2️⃣ Hybridization (primer and probe binding): The temperature is lowered to approximately 55–60 °C. In addition to the specific primers, the TaqMan probe binds to its target sequence between the primers. In this state, the fluorescence of the reporter remains suppressed due to the nearby quencher.
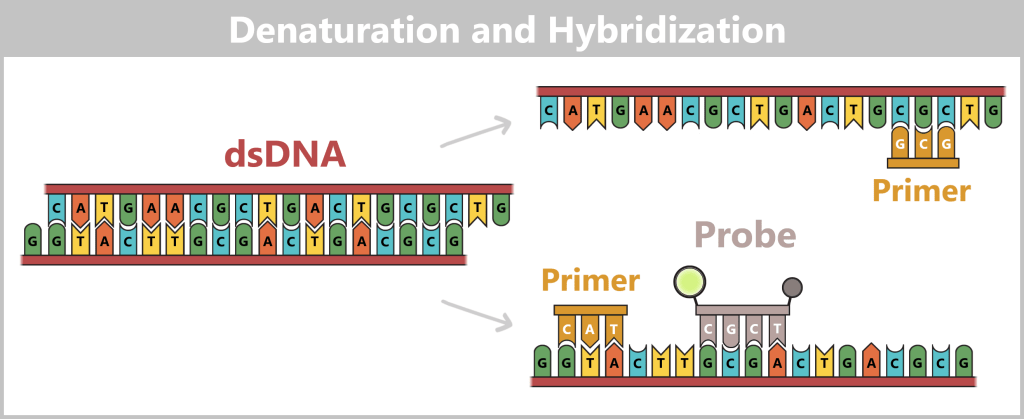
Fig. 2.3.1-B1: Probe-based qPCR during the annealing step As soon as the temperature decreases from 95 °C to 60 °C, the probe binds to its target sequence first. The probe has a higher melting temperature (Tm), typically 8–10 °C above that of the primers, so that it is already stably bound at the chosen working temperature while the primers are just beginning to anneal.
The probe is a single-stranded DNA molecule of approximately 20–30 nucleotides in length. It is designed to bind specifically to a defined target sequence – a segment that is characteristic of the DNA of interest.3️⃣ DNA synthesis & hydrolysis (amplification): At approximately 60 °C, DNA polymerase begins synthesizing the new strand. In many modern qPCR protocols, annealing (primer and probe binding) and elongation (DNA synthesis) occur at the same temperature.
When the polymerase encounters the bound probe during strand synthesis, it uses its 5’→3′ exonuclease activity to hydrolyze the probe (breaking it down into individual nucleotides). Even the first cleavage separates the reporter dye from the quencher.
This permanently releases the reporter from the quenched state.4️⃣ Excitation and emission: During the measurement phase, the reaction mixture is excited with light of a specific wavelength. The released reporter molecules absorb this energy and emit light at a longer wavelength (e.g., green for FAM or yellow for VIC).
In this case, the fluorescence signal is not generated by dye binding to double-stranded DNA (as in SYBR Green), but by enzymatic cleavage of the probe during amplification.
The probe does not participate in DNA amplification. Amplification is driven exclusively by the forward and reverse primers, while the probe serves only as a sequence-specific fluorescent reporter.
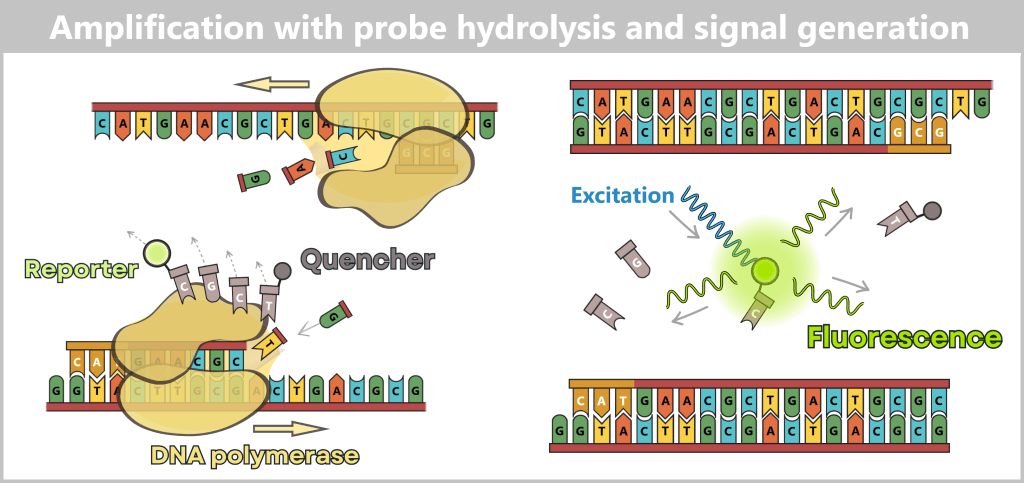
Fig. 2.3.1-B2: Amplification with probe hydrolysis – signal generation (fluorescence) The polymerase extends from the primer and reaches the probe. While synthesizing the new DNA strand, it removes the probe that lies in its path. This separates the reporter from the quencher, thereby generating the measurable fluorescence signal.
5️⃣ Quantification: Fluorescence is measured after each cycle. For every newly synthesized DNA strand that contains an intact probe-binding site, one probe is hydrolyzed. The signal intensity therefore increases in proportion to the number of correctly amplified target molecules.
The key measurement parameter is the Ct value (cycle threshold):
- Low Ct value → high initial amount of target DNA
- High Ct value → low initial amount of target DNA
Advantages of probe-based qPCR
- High specificity: In addition to the two primers, a sequence-specific fluorescent probe also binds. A signal is only generated when the correct target sequence is actually amplified. Nonspecific amplification products such as primer dimers therefore generally do not produce a meaningful signal.
- Suitable for multiplex analyses: Different reporter dyes allow the simultaneous detection of multiple target sequences in a single reaction.
- Improved analytical robustness: The additional probe layer increases specificity and enhances the reliability of quantification, especially in complex samples with high background signals.
- Lower susceptibility to nonspecific amplification: Compared with dye-based methods, false-positive signals from nonspecific PCR products are significantly reduced.
Disadvantages of probe-based qPCR
- Higher costs: In addition to primers, a specific fluorescently labeled probe must be synthesized for each target sequence.
- More complex assay design: In addition to the primers, the probe must also be optimized in terms of sequence, melting temperature, position, and potential secondary structures.
- Dependence on the target region: Fragmentation, mutations, or damage within the primer or probe binding sites can impair or prevent amplification and signal generation.
- Influence of fragment length: As with other PCR-based methods, detectability depends on whether a DNA fragment contains all required binding sites for primers and probe. Highly fragmented DNA may therefore be incompletely detected, especially when longer target regions are used.
- Indirect quantification: Absolute quantification is usually performed using standard curves and remains dependent on amplification efficiency, assay design, and sample preparation.
✧ ✧ ✧
Detailed information on the principles of both qPCR methods can be found here.
Both dye-based and probe-based qPCR enable sensitive and specific detection of defined DNA sequences. However, quantification is indirect via the Ct value and typically requires a standard curve with known reference concentrations. The accuracy therefore depends on amplification efficiency and the quality of the reference standards.
To reduce this dependence and enable absolute quantification without a standard curve, digital PCR (ddPCR) was developed.
2.3.2. Digital PCR (ddPCR)
ddPCR stands for droplet digital PCR.
Digital PCR is an advancement of PCR technology that enables absolute quantification of DNA or RNA molecules – without the need for a standard curve and with very high precision.
In contrast to qPCR, it does not measure a continuously increasing fluorescence signal but instead counts individual positive reaction units. The result is binary – „positive” or „negative” – which is why it is referred to as digital.
Basic principle of ddPCR
While in qPCR the entire reaction takes place in a single reaction volume, in ddPCR the sample is partitioned into thousands of microscopic, separated reaction compartments – so-called droplets. Each droplet represents an independent PCR reaction.
Method Procedure (using DNA as an example):
1️⃣ Sample preparation: The DNA sample is mixed with primers, a sequence-specific probe (usually TaqMan®-like), nucleotides, DNA polymerase, and buffer – analogous to probe-based qPCR.
2️⃣ Partitioning (droplet generation): The reaction mixture is transferred into a droplet generator. There, it is passed through microfluidic channels together with a special oil..
Due to fluid dynamics, this process generates up to 20,000 nanoliter-sized droplets. The oil stabilizes the droplets and forms a stable emulsion, ensuring that each droplet acts as an isolated reaction compartment. The resulting droplets are collected in a well of a 96-well plate.
The distribution of DNA molecules across the droplets is random, resulting in:
- many empty droplets
- some droplets containing one target molecule
- some droplets containing multiple molecules
This distribution follows Poisson statistics.
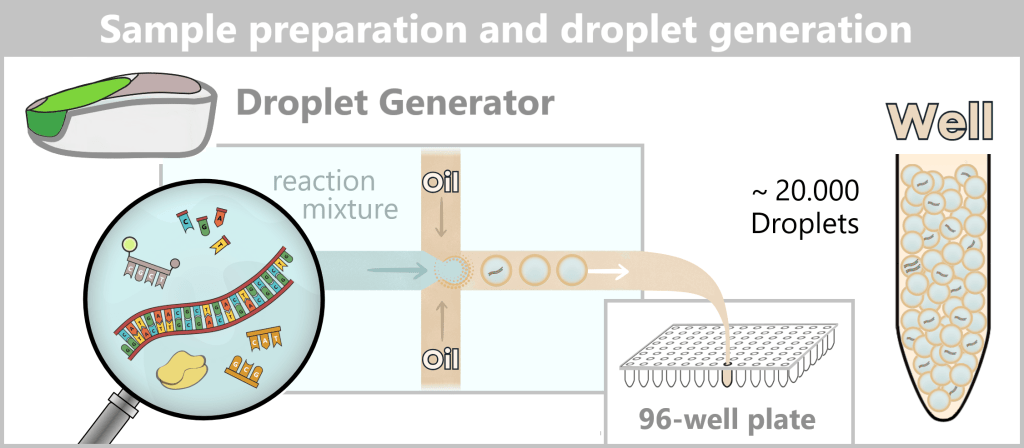
Fig. 2.3.2-A: Schematic representation of droplet formation in digital PCR (ddPCR) 3️⃣ Amplification (PCR): The sealed 96-well plate is transferred into a thermocycler.
The droplets undergo the standard PCR cycles (denaturation, annealing, elongation), typically 40–45 cycles.
During amplification, no fluorescence is measured.
If a droplet contains at least one target DNA molecule, it is amplified. The probe is hydrolyzed, and the droplet develops a strong fluorescent signal.
Droplets without target DNA remain dark.
Fig. 2.3.2-B: ddPCR signals after amplification depending on target DNA concentration The figure schematically shows the result of droplet digital PCR (ddPCR) after completion of amplification.
The previously generated and sealed droplets undergo 40–45 PCR cycles in a thermocycler. During amplification, no continuous fluorescence measurement is performed.
If a droplet contains at least one target DNA molecule, the target sequence is amplified. During this process, the sequence-specific probe is hydrolyzed by the 5′→3′ exonuclease activity of the DNA polymerase, separating the reporter from the quencher and generating a strong fluorescence signal. Droplets without target DNA remain signal-free („negative”).
As the initial concentration of target DNA increases, the number of fluorescent (positive) droplets increases, while the intensity of individual positive droplets remains comparable.4️⃣ Readout (droplet reader): After completion of PCR, the 96-well plate is inserted into a droplet reader.
The instrument:
- aspirates the emulsion from a well
- passes the droplets individually through a narrow capillary
- irradiates each droplet with a laser
- measures the fluorescence intensity
Each droplet is individually classified as:
• positive (fluorescence signal present)
• negative (no signal)The result is binary – 0 or 1.
Fig. 2.3.2-C: Schematic representation of the principle of fluorescence readout in ddPCR After completion of PCR, the 96-well plate containing the droplet emulsion is inserted into the droplet reader. The instrument aspirates the emulsion from a well and passes the droplets individually through a narrow capillary. Each droplet is irradiated with a laser, and the measured fluorescence intensity is recorded as an amplitude value. In the analysis software, these values are displayed in a 1D amplitude plot: the X-axis sequentially numbers the droplets, while the Y-axis shows the fluorescence amplitude. Using a software-defined threshold, negative droplets (no signal) are separated from positive droplets (fluorescence signal). The number of positive droplets is then converted into the absolute target DNA concentration of the original sample using Poisson statistics.
5️⃣ Quantification: Since it is known:
- how many droplets were analyzed in total
- how many of them are positive
the absolute number of original DNA molecules can be calculated using the Poisson distribution.
📍 No standard curve is required.
📍 Quantification is independent of amplification efficiency within individual cycles.🎥 Tip: What this looks like in practice is demonstrated in the video „Digital PCR Using the Bio-Rad QX100™ ddPCR™ System“.
Advantages of ddPCR
- Absolute quantification without a standard curve: The number of target molecules is calculated directly from the ratio of positive to negative droplets using Poisson statistics. An external standard curve is generally not required.
- High precision and reproducibility: By partitioning the sample into thousands of independent reaction compartments, quantification becomes less susceptible to fluctuations in amplification efficiency than in conventional qPCR methods.
- High sensitivity at low concentrations: Even very small amounts of target DNA can be reliably detected because individual molecules are amplified in separate droplets.
- Relative robustness against PCR inhibitors: Inhibitors are likewise distributed among the individual droplets. As a result, their inhibitory effect is often reduced compared with a single bulk reaction.
- Endpoint measurement instead of Ct interpretation: The analysis is based on the presence or absence of a signal at the end of PCR and is therefore less dependent on the interpretation of amplification curves.
- Particularly suitable for rare target sequences: The method is frequently used for the detection of rare mutations, low viral loads, or very small amounts of residual DNA.
Disadvantages of ddPCR
- More complex technology and higher costs: In addition to a thermocycler, specialized instruments for droplet generation and droplet readout are required.
- More complex workflow: Partitioning of the sample and the subsequent droplet analysis require additional processing steps and increase technical complexity.
- Limited dynamic range per run: At very high target concentrations, too many droplets may become positive. This reduces the accuracy of the statistical analysis (saturation effect), making dilution series necessary.
- Lower sample throughput: Compared with qPCR, the analysis is generally more time-consuming and less suitable for very high sample throughput.
- Still sequence- and assay-dependent: As with qPCR, only DNA fragments containing all required primer and, where applicable, probe binding sites are detected.
- Influence of fragmentation: Highly fragmented DNA may also be incompletely detected by ddPCR, particularly when the target region is relatively long or primer binding sites are missing.
- Dependence on sample preparation: Extraction losses, incomplete LNP disruption, or selective fragment losses can still influence the result.
2.3.3. Comparison of PCR-based quantification methods
Feature Dye-based qPCR Probe-based qPCR Digital PCR (ddPCR) Measurement principle Fluorescence binding to all dsDNA Sequence-specific fluorescent probe Counting of positive individual reactions Signal generation Dye binds to dsDNA Probe is hydrolyzed during amplification Endpoint measurement of positive droplets Quantification Relative (via Ct value and standard curve) Relative (via Ct value and standard curve) Absolute (via Poisson distribution) Standard curve required Yes Yes No Specificity Medium
(primer-dependent)High
(primer + probe)High
(primer + probe)Measurement timing During amplification (real-time) During amplification (real-time) After endpoint PCR Reaction format Single reaction volume Single reaction volume ~20,000 droplets per well Cost Low Medium High Instrumentation Real-time PCR system Real-time PCR system Droplet generator + thermocycler + droplet reader
2.4. Sequencing-based Methods
2.4.1. Oxford Nanopore Technology
2.4.2. Illumina SequencingWhile the previously described methods (qPCR, ddPCR) can specifically detect and quantify individual DNA sequences, sequencing-based approaches go a step further: they read the exact order of bases in a DNA or RNA molecule – step by step, letter by letter.
One example of these technologies is nanopore sequencing. In a study by Gunter et al. (Nature Communications 2023), long-read nanopore sequencing was used for the comprehensive analysis of mRNA vaccines. Methods such as „VAX-seq” can capture key quality attributes – including sequence identity, integrity, length, and purity – while simultaneously assessing mRNA and residual DNA. This makes nanopore sequencing a promising analytical approach for manufacturing and quality control processes.
In particular, Oxford Nanopore Technology (ONT) has emerged in recent years as a promising tool for quality control of mRNA-based therapeutics. It provides direct sequence information at the single-molecule level and can analyze both short and very long fragments – a key advantage for the characterization of residual DNA.
⚬ ⚬ ⚬
2.4.1. Oxford Nanopore-Technologie
When nanopore sequencing is mentioned today, it usually refers to the platform developed by Oxford Nanopore Technologies (ONT). Although alternative concepts for nanopore-based sequencing exist, ONT is currently the only technology that is widely commercially available and practically established. Since its market introduction in 2015, it has become established as a representative of so-called third-generation sequencing technologies.
What makes this method special is the direct analysis of individual DNA or RNA molecules in real time. In contrast to classical sequencing approaches, neither cyclic amplification nor chemical labeling of nucleotides is required. Instead, sequence information is derived directly from physical measurement signals.
Functional principle
ONT sequencing is based on the interaction of several key components:
Nanopores – act as tiny molecular „reading units”. When a single DNA or RNA strand passes through a pore, it generates characteristic electrical signals, comparable to a molecular „fingerprint”.
Membrane – serves as a filter and barrier. It ensures that only ions and nucleic acids can pass through the nanopores, while unwanted molecules are excluded. This creates a defined measurement environment for signal detection.
Chip (flow cell) – forms the basis of the system. The chip contains the membrane with numerous nanopores as well as the integrated electronics for signal detection. It represents the functional unit in which sequencing takes place.
System architecture
The chip is divided into two compartments:
Upper chamber (cis) This is where the DNA sample is introduced. Lower chamber (trans) This chamber receives the strand after it
passes through the nanopore.Both chambers are filled with an ion-containing buffer solution that conducts electrical current. The membrane electrically separates the two chambers – current can only flow at the locations where nanopores are embedded. These pores act as the only „tunnels” through which ions and nucleic acids can pass.
When a constant voltage is applied between the chambers, an ionic current is generated through the nanopores. As a DNA or RNA strand passes through a pore, it causes characteristic changes in this current. These current modulations are continuously recorded and stored as raw electrical signals.
Fig. 2.4.1-A: Structure of the nanopore device On the left, a portable nanopore sequencing device is shown, approximately the size of a slightly wider USB stick. It contains a replaceable sequencing unit (flow cell) used for the actual analysis.
On the right, a magnified view shows the schematic structure of this sequencing cell: it consists of a chip covered by an electrically insulating membrane. Embedded within this membrane are numerous nanopores that act as molecular sensors. The cell is divided into an upper cis and a lower trans chamber. By applying an electrical voltage, DNA (as shown in the image) is pulled through the nanopore. The resulting changes in ionic current are measured and used for sequence determination.The path of DNA through the nanopore
1️⃣ Library preparation
Before sequencing can begin, the nucleic acids in the sample (DNA and/or RNA) must be prepared for nanopore sequencing. This step is referred to as library preparation.
In the context of quality control of therapeutic mRNA – for example directly after in vitro transcription or before formulation – the following steps are relevant:
a) Purification
After transcription, the reaction mixture still contains, in addition to the desired mRNA or DNA, enzymes (e.g., polymerases), free nucleotides, and buffer components.
These components must be removed because they can:
- reduce the efficiency of adapter ligation
- block nanopores
- interfere with the electrical signal
In early process stages, thorough purification is therefore required. Shortly before final formulation, however, the product is typically already purified, so no additional bulk purification step is usually necessary.
b) End repair & dA-tailing (for DNA)
If DNA is sequenced – for example to analyze residual DNA fragments – the fragment ends must be prepared.
First, an end repair step is performed: overhanging („sticky”) ends are enzymatically filled in or trimmed back, resulting in blunt-ended double-stranded DNA.
This is followed by a so-called dA-tailing step. In this process, a polymerase specifically adds a single adenine (A) nucleotide to each 3′ end of the DNA. This generates a defined 3′ A-overhang, which is required for the subsequent adapter ligation step.
c) Adapter ligation – the key step
In order for nucleic acids to be recognized by the nanopore and actively guided through it (translocated), specific adapter molecules must be attached (ligated) to their ends.
These adapters are functional modules and contain several essential components:
Motor protein
The motor protein acts as a molecular „brake”. Without this control, DNA would pass through the pore at extremely high speed – far too fast for accurate signal resolution.
The motor protein:
- regulates the translocation speed (~400–500 bases per second)
- ensures a uniform stepping motion
- unwinds double-stranded DNA during passage
Oxford Nanopore uses different motor proteins (helicases) for:
- double-stranded DNA
- cDNA
- native (direct) RNA sequencing
Tether
The tether can be described as a molecular „parking assistant”. It typically contains a cholesterol-like structure that inserts into the lipid membrane of the flow cell. This brings the nucleic acid into close proximity to the membrane surface – exactly where the nanopores are located.
As a result, the likelihood that a molecule enters the electric field of a pore and is captured is significantly increased.
In some protocols, a so-called tether buffer is additionally used to enrich the membrane surface with these anchoring structures.
Barcode sequences (optional)
Barcode sequences enable multiplexing, i.e., the simultaneous analysis of multiple samples in a single sequencing run.
For routine quality control of individual batches, they are not strictly required and are therefore not further considered here.
⬦ ⬦ ⬦
Adapter design in ONT
Oxford Nanopore Technologies provides sequencing adapters that are pre-loaded with the motor protein and tether. During ligation, the entire adapter complex is therefore attached to the nucleic acid molecule.
For DNA:
- The adapter is ligated to the ends of double-stranded DNA.
- The adapters contain an overhanging thymine (T) residue, which is complementary to the previously generated 3′ A-overhang of the DNA.
- This enables efficient and directional ligation.
For RNA (direct RNA sequencing):
- The adapter is ligated to the 3′ end of the RNA, typically via the existing poly(A) tail.
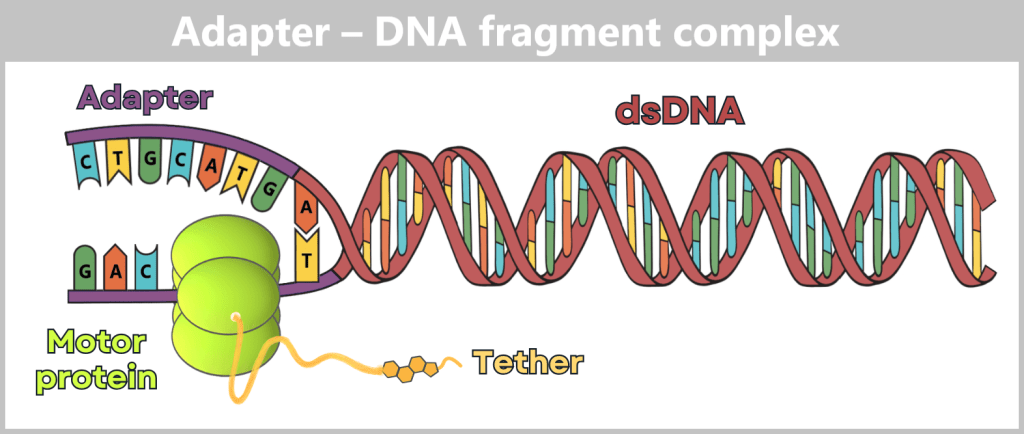
Fig. 2.4.1-B: Schematic representation of adapter ligation The sequencing adapter contains, in addition to a double-stranded DNA region (typically ~30 to 50 base pairs in length), two short single-stranded sequences (often ~20 to 40 nucleotides). For clarity, the figure shows a strongly simplified representation.
One single strand is already bound inside the motor protein, while parts of the second single strand remain exposed.
Directly adjacent to this is the junction to the actual sample. A specific overhang at the end of the adapter (T) has hybridized with the complementary end of the sample DNA (A). This „ligation” stably links the target double-stranded DNA (dsDNA) to the sequencing system.
Attached laterally to the motor protein is the tether. At its free end is a cholesterol molecule. This acts as a chemical „anchor,” embedding the entire complex into the lipid membrane of the pore, thereby increasing the likelihood that the DNA is captured by a nanopore.After ligation, the adapters are stably attached to the nucleic acid, and the library is ready for sequencing.
Special feature: Direct RNA sequencing (e.g., in the context of VAX-seq)
A key advantage of nanopore technology is the ability to sequence native RNA directly, without prior conversion into cDNA.
For quality control of therapeutic mRNA, this is particularly important because:
- chemical modifications (e.g., pseudouridine) can be detected directly
- no reverse transcription artifacts are introduced
- structural features of the RNA are preserved
This enables the technology not only to quantify molecules, but also to provide structural and functional characterization at the single-molecule level.
2️⃣ Application of an electrical voltage
Both chambers of the sequencing cell contain charged particles (ions). Once an electrical voltage is applied between the upper cis chamber and the lower trans chamber, an electric field is generated across the membrane. This field drives ions through the nanopores, producing a measurable ionic current.
As long as no DNA is present in the pore, this ion flow remains constant. In this state, the device records a stable baseline current that serves as a reference signal (see following figure).
3️⃣ The DNA is introduced into the sequencing cell
The prepared DNA sample is pipetted into the upper cis chamber of the sequencing cell. Sequencing adapters carrying a motor protein and a tether are already attached to the DNA ends.
At the end of the tether is a hydrophobic (water-repellent) cholesterol group. It functions as a molecular anchor.
Due to diffusion, the DNA strands constantly move within the cis chamber. As soon as the hydrophobic cholesterol randomly contacts the membrane, it immediately adheres to it. Energetically, it is far more favorable for the cholesterol to remain within the lipid layer than in the aqueous buffer.
This mechanism provides two major advantages:
- Concentration at the membrane surface: The DNA molecules accumulate preferentially near the membrane. This increases the probability that the motor protein encounters a nanopore and docks onto it.
- Lateral mobility: The cholesterol is not rigidly fixed within the membrane but remains laterally mobile. The DNA can therefore „slide” along the membrane surface until it is captured by the electric field of a pore and eventually drawn inside.
4️⃣ Docking to the nanopore
Once a single-stranded region of the DNA enters the nanopore, the electric field pulls the negatively charged DNA strand toward the trans chamber. This positions the motor protein against the pore entrance, where it docks stably onto the nanopore.
The electrical force acting on the DNA strand serves as a mechanical trigger for the motor protein. It then begins its helicase activity:
- it unwinds the double-stranded DNA (dsDNA)
- one strand is guided stepwise through the nanopore at controlled speed
- the second strand remains outside the pore
This marks the beginning of the actual sequencing process.
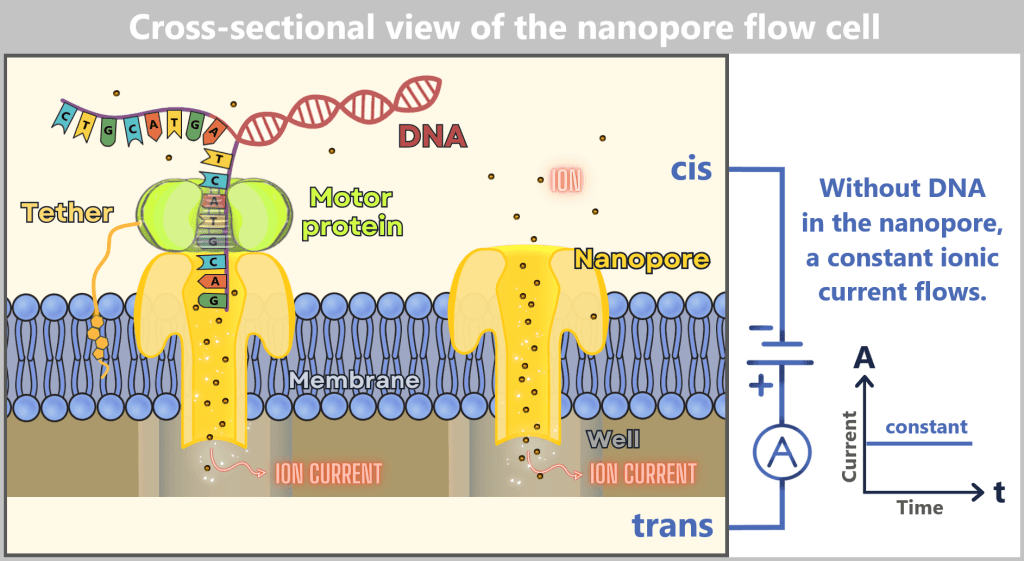
Fig. 2.4.1-C: Schematic representation of the sequencing cell and ionic current at resting state
(not to scale)The sequencing cell consists of two chambers: the cis chamber (top) and the trans chamber (bottom), separated by a membrane containing embedded nanopores. On the left, a DNA molecule is shown docked to a nanopore via a motor protein. On the right, an empty nanopore is depicted through which a constant ionic current flows. The applied voltage between the negative electrode in the cis chamber and the positive electrode in the trans chamber drives the ion flow through the pore. The ionic current is measured in the well (a small channel in the chip beneath the pore), as illustrated in the current-versus-time diagram. As long as no DNA passes through the pore, the current remains constant.
The motor protein – the pacemaker of sequencing
The prepared DNA carries an adapter containing a motor protein (a helicase). Once this complex reaches he vicinity of a nanopore, the protein docks onto the pore entrance like a precisely fitting cap. The electrical voltage within the system acts like an invisible pulling force that draws the DNA into the pore.
This electrical force supports the activity of the motor protein. The protein acts like a wedge that gently pries apart the DNA double strand. The hydrogen bonds between the base pairs – the „rungs” of the DNA ladder – break under this force because they are considerably weaker than the stabilizing interactions within the protein itself. In this way, the double-stranded DNA (dsDNA) is unzipped like a zipper without damaging the chemical backbone of the strands.
At this stage, precise control begins: Inside the motor protein, four to six DNA-binding loops (so-called hairpins or loops) grip the newly exposed single strand (ssDNA). These gripping elements are arranged in a spiral fashion, comparable to the steps of a spiral staircase or the thread of a screw.
The protein can be imagined as a „breathing screw”: Driven by ATP, it rhythmically changes its shape. It contracts and expands repeatedly. During each cycle, the upper gripping elements release the DNA, move one step forward, and reattach further upstream, while the lower elements maintain their grip and push the strand downward into the pore in a controlled manner.
This staircase-like arrangement ensures that the DNA does not shoot uncontrollably through the nanopore, but instead advances step by step – effectively base by base. It is this precise mechanical pacing by the motor protein that allows the nanopore to accurately register the electrical signal generated by each nucleotide.
5️⃣ The DNA passes through the nanopore – signal generation
DNA carries a negative electrical charge due to its phosphate groups. Because of the applied voltage between the negatively charged cis chamber and the positively charged trans chamber, the single strand is pulled through the nanopore.
At this stage, the motor protein performs a crucial role:
- it moves the DNA through the pore stepwise
- thereby greatly reducing the translocation speed
- the movement becomes slow, uniform, and controlled
Without this control, the DNA would pass through the nanopore at speeds of millions of bases per second. The motor protein slows this movement down to approximately 300–450 bases per second. Only this controlled pacing enables reliable signal measurement.
As the DNA strand travels through the pore, it influences the ionic current. Each nucleotide has a slightly different size and chemical structure. These differences alter the current flow in characteristic ways.
The current changes are measured by electrodes connected to the individual wells within the chip. Each well contains a nanopore and has its own measurement unit. This allows the software to assign the recorded signals unambiguously to a specific pore.
6️⃣ The electrical signal is recorded
During sequencing, typically about five to six bases are located simultaneously within the narrowest region of the nanopore. This short DNA segment is referred to as a k-mer. Within the pore channel, the k-mer acts like an electrical resistor: different base combinations influence the ionic current in characteristic ways and therefore generate distinct electrical signals.
The DNA sequence can be reconstructed because the DNA moves stepwise through the pore, with each k-mer producing a characteristic electrical signature. Specialized software analyzes this sequence of current signals using machine learning algorithms that assign the complex signal patterns to the corresponding nucleotide combinations. This process is called basecalling.
In this way, the complete nucleotide sequence is reconstructed step by step from the electrical measurement data.
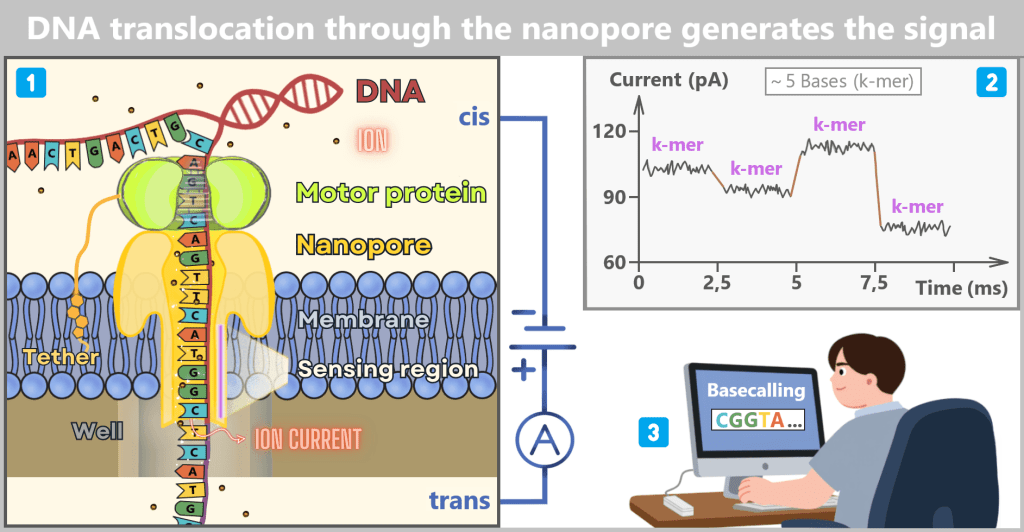
Fig. 2.4.1-D: The principle of nanopore sequencing 1) Cross-sectional view of the sequencing cell
A single-stranded DNA molecule is pulled from the cis side toward the trans side due to the applied voltage. The motor protein is positioned above the nanopore. It acts as a molecular brake and controls the forward movement of the DNA. As a result, the single strand is translocated through the pore at a speed of approximately 400 bases per second, allowing each signal to be measured precisely.
At the narrowest constriction of the pore (marked here as the sensing region), several bases are present simultaneously – typically around five nucleotides, referred to as a k-mer. This short DNA segment partially obstructs the pore channel. The ions (small red dots) still attempt to flow through the pore. Because the k-mer reduces the available space, fewer ions can pass through. This generates a characteristic current signal.2) The data in the current-versus-time plot
The Y-axis shows the ionic current in picoamperes (pA), while the X-axis represents time in milliseconds (ms).
A k-mer typically remains within the narrowest region of the pore for approximately 2–3 milliseconds. During this period, multiple current measurements are recorded for that exact base combination.
This produces a slightly fluctuating plateau in the diagram, caused by electrical noise. The brown connecting lines between the plateaus indicate the moment when the motor protein advances the DNA by one base, thereby moving a new k-mer into the sensing region.3) Basecalling – conversion of current signals into bases
The characteristic current signals – the plateaus in the current-versus-time plot – each correspond to the electrical signature of a specific k-mer, i.e., a short base combination residing in the sensing region of the pore for a few milliseconds.
Basecalling algorithms, often based on neural networks, analyze this sequence of current signals. From the characteristic signal patterns, they reconstruct the underlying DNA base sequence.🎥 Tip: The video „How nanopore sequencing works“ provides a clear overview of the process.
⬦ ⬦ ⬦
Result of nanopore sequencing
Nanopore sequencing does not produce a single measurement value, but rather a comprehensive dataset consisting of many individual sequence reads. Each read corresponds to the analysis of a single DNA or RNA molecule. Together, these individual measurements provide a detailed representation of the nucleic acids present in the sample.
In principle, all DNA or RNA molecules contained in the sample can be detected without the need to predefine a specific target sequence. The method is therefore not target-directed, but exploratory in nature.
◈ ◈ ◈
Two key levels of information: identity and quantity
The generated sequencing data contain two complementary levels of information:
Identity (sequence information)
For each individual molecule, the nucleotide sequence can be determined. This provides information about:
- the exact base sequence
- the length of the molecules
- structural characteristics (e.g., full-length molecules or fragments)
- possible sequence variants
- and, with methodological limitations, chemical modifications
On this basis, it can be determined which nucleic acids are present in a sample.
Quantity (relative abundance)
In addition, the number of reads can be used to estimate how frequently certain sequences or molecule types are represented in the sample. However, this does not constitute absolute quantification, but rather a relative approximation influenced by several factors, including:
- efficiency of library preparation
- molecule length
- sequence characteristics
The quantitative interpretability of the method must therefore always be considered in the context of these influencing factors.
✧ ✧ ✧
Parallel single-molecule analysis in real time
Data generation is performed by a large number of nanopores operating in parallel within the so-called flow cell. Devices such as the MinION contain several hundred active measurement channels that function simultaneously and independently.
Each nanopore analyzes one individual molecule at a time. Once a sequencing event is completed, the pore immediately becomes available for the next molecule. In this way, a continuous stream of individual reads is generated.
The measurement is performed in real time, with the software simultaneously recording and processing the signals from many individual pores.
✧ ✧ ✧
Nature of the data: distribution instead of a single value
A key characteristic of nanopore sequencing is that the results are not provided as a single uniform measurement value, but as a distribution of many individual sequencing reads. These reads can differ significantly in:
- length
- sequence
- quality
In complex samples in particular, the dataset typically contains:
- short fragments
- longer contiguous molecules
- different molecular populations
Therefore, analysis does not focus on individual reads in isolation, but on interpreting the overall distribution of all reads.
✧ ✧ ✧
A key feature of the method: long reads
A defining characteristic of nanopore technology is its ability to sequence very long nucleic acid molecules continuously. In contrast to short-read approaches (such as Illumina sequencing), where DNA is first fragmented into short pieces and later computationally reconstructed, nanopore sequencing can directly capture contiguous molecules.
In principle, the length of a read is not limited by the technology, but is mainly determined by the integrity of the source molecule.
This enables, in particular:
- direct analysis of whole molecules
- distinction between intact sequences and fragments
- investigation of structural relationships within individual molecules
✧ ✧ ✧
Limitations and methodological influences
Despite its advantages, nanopore sequencing is subject to several methodological limitations:
Error rate of individual reads
The accuracy of single sequencing reads is lower compared to classical short-read technologies. This is due to the indirect determination of sequence information via electrical current signals. Certain sequence motifs, in particular homopolymeric regions (e.g., long stretches of the same base), can be more difficult to resolve.
Selection and process bias
Not all molecules are captured with equal probability. Differences in length, structure, or adapter ligation efficiency can lead to certain sequences being preferentially or underrepresented in sequencing.
In particular, library preparation can influence the composition of the sequenced molecules. Purification steps using magnetic beads or other size-dependent processes may partially remove or underrepresent very short fragments. At the same time, longer molecules are often sequenced more efficiently in nanopore systems, as they remain in the pore longer and generate more stable signals.
As a result, the observed read distribution does not necessarily reflect the original fragment distribution of the sample.
Dependence on sample quality
The quality and integrity of the starting nucleic acids significantly influence the resulting reads. In particular, long molecules are prone to fragmentation during sample preparation.✧ ✧ ✧
Consensus building and bioinformatic analysis
To increase the accuracy of the results, the data are not interpreted on the basis of individual reads. Instead, many sequencing reads of the same molecule type are compared and merged into a consensus sequence.
Since sequencing errors are mostly randomly distributed, they can be largely reduced through this repeated analysis. Modern basecalling and analysis methods, which are often based on machine learning, further contribute to improving sequence accuracy.
In this way, a large number of individual measurements can be combined to produce a consistent and reliable overall representation.
✧ ✧ ✧
Context for nucleic acid analysis
Nanopore sequencing therefore enables a detailed characterization of nucleic acids with regard to their sequence, length, and relative abundance. At the same time, the interpretation of the data requires consideration of methodological influences, particularly with respect to quantification and the representativeness of the molecules that are detected.
2.4.2. Illumina sequencing
Nanopore sequencing enables the analysis of long, contiguous molecules in real time – its particular strength lies in capturing full-length sequences without necessarily fragmenting them into short standard fragments beforehand. A different approach is taken by Illumina sequencing: instead of working with individual long molecules, it analyzes a very large number of short fragments in parallel with high precision. Both technologies complement each other – they answer different questions using different methods.
Illumina sequencing is a short-read approach and belongs to the group of so-called next-generation sequencing (NGS) technologies.
It is currently the most widely used sequencing method worldwide. The underlying principle differs fundamentally from nanopore technology: while ONT passes individual molecules through a pore and measures electrical signals in real time, Illumina relies on an optical method – sequencing by synthesis using fluorescently labeled nucleotides. In this process, DNA is not read directly but decoded step by step during a controlled synthesis reaction.
Operating principle
1️⃣ Preparation of DNA fragments = library construction
For sequencing, DNA must first be converted into a format suitable for the Illumina platform. This process is also called library preparation and consists of the following steps:
a) Fragmentation
DNA is mechanically or enzymatically broken into smaller pieces (typically with a target size of about 150–500 base pairs). This generates fragments of slightly different lengths, which are often further standardized using size-selection methods.
This step is particularly important when long DNA molecules are to be sequenced, since Illumina technology processes short fragments more efficiently than very long contiguous molecules.
In some applications, fragmentation can be deliberately reduced or omitted – especially when already highly fragmented DNA is analyzed. However, even in such approaches, very long fragments (typically above ~800–1000 bp) are often less efficiently processed during library preparation and sequencing, and may therefore be underrepresented.
To illustrate this, we consider three DNA fragments of slightly different lengths in our example.
b) Adapter ligation
Specific adapter sequences (P5 and P7 adapters) are attached to both ends of the DNA fragments. These adapters serve several functions:
Flow cell binding sites: The adapters contain specific DNA sequences that act like a key fitting into a lock, allowing fragments to attach to a surface required for sequencing.
Primer binding sites: The adapters include regions where sequencing primers can bind. These primers are later used to synthesize DNA strands step by step.
Indexes (optional): If multiple samples are sequenced simultaneously, index sequences allow the assignment of fragments to their respective samples. For clarity, indices are not shown in this example.
c) PCR amplification
To ensure that sufficient DNA is available for sequencing, the adapter-ligated DNA fragments are amplified using the polymerase chain reaction (PCR).
Fig. 2.4.2-A: Library preparation – overview of the DNA preparation workflow. The following close-up shows a detailed representation of a fragmented double-stranded DNA molecule with both adapters attached.
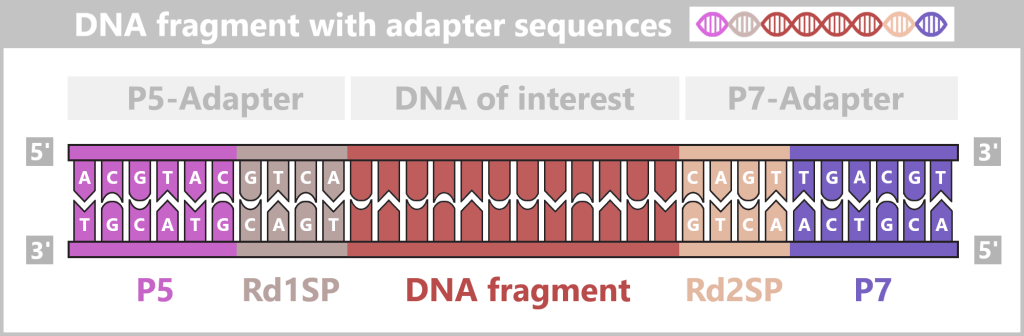
Fig. 2.4.2-B: Schematic representation of a double-stranded DNA fragment with adapters Each DNA fragment receives two adapter sequences: the P5 adapter contains the P5 adapter sequence, which enables binding to the flow cell, and the Rd1SP (Read 1 Sequencing Primer) binding site to initiate DNA synthesis. Similarly, the P7 adapter contains the P7 adapter sequence for flow cell binding as well as the Rd2SP (Read 2 Sequencing Primer) binding site for the initiation of DNA synthesis.
This representation is highly simplified. In practice, Illumina adapter sequences are significantly longer (typically 60–120 base pairs). The DNA fragment itself is also shortened for clarity; in reality, DNA fragments typically range from 100–500 base pairs in length.2️⃣ Cluster generation on a flow cell
The core of Illumina technology is the so-called flow cell – a glass-like surface coated with millions of short, immobilized DNA binding sites.
Fig. 2.4.2-C: Schematic representation of a flow cell and its surface a) DNA fragments bind to the flow cell surface
At the beginning of this step, a solution of single-stranded DNA fragments is passed over the flow cell. The fragments carry the previously ligated adapter sequences at their ends and have been denatured into single strands, allowing them to exist freely in solution.
As the fragments flow across the flow cell, their adapters bind via complementary base pairing to matching DNA oligonucleotides on the surface.
To illustrate this, the flow cell surface can be imagined as a dense Velcro-like system: the adapter sequences of the DNA fragments adhere to complementary binding sites on the flow cell – similar to small hooks anchoring into a Velcro mesh.
b) First synthesis
After the DNA fragments have bound to the flow cell, the first DNA synthesis begins. A primer binds to the adapter sequence of the immobilized single strand. The DNA polymerase then synthesizes a complementary strand.
In the next step, the original template strand is removed, while the newly synthesized strand remains attached to the flow cell.
Fig. 2.4.2-D: Schematic of the first synthesis: formation of surface-bound DNA strand copies Left: Primers bind to the adapters (P5, P7), and DNA polymerase (DNAP) initiates synthesis of a new complementary strand.
Middle: DNA polymerase synthesizes the first new strand.
Right: The resulting DNA double strand is denatured. After separation, the original template strand is no longer attached to the flow cell and is washed away. The newly synthesized strand remains firmly bound to the flow cell via its 5′ end.At this stage, direct sequencing would not yet be feasible, as the fluorescence signals from individual molecules would be too weak to be reliably detected. Therefore, a process called bridge amplification follows.
c) Bridge amplification
To ensure that subsequent sequencing generates a sufficiently strong fluorescence signal, the DNA molecules bound to the flow cell must first be amplified.
The process begins when a surface-bound single-stranded DNA molecule, due to its flexibility, bends back toward the surface and hybridizes via its free adapter end to a neighboring complementary binding site on the flow cell. This forms a characteristic bridge structure.
Fig. 2.4.2-E: Bridge formation and DNA synthesis The surface-anchored single strands fold and bind to adjacent anchors on the flow cell, forming a bridge structure. This is followed by the initiation of the copying process.
Subsequently, DNA polymerase synthesizes a complementary strand along the bound template strand. After synthesis, the bridge structure is denatured again, resulting in two single strands that are both firmly anchored to the flow cell.
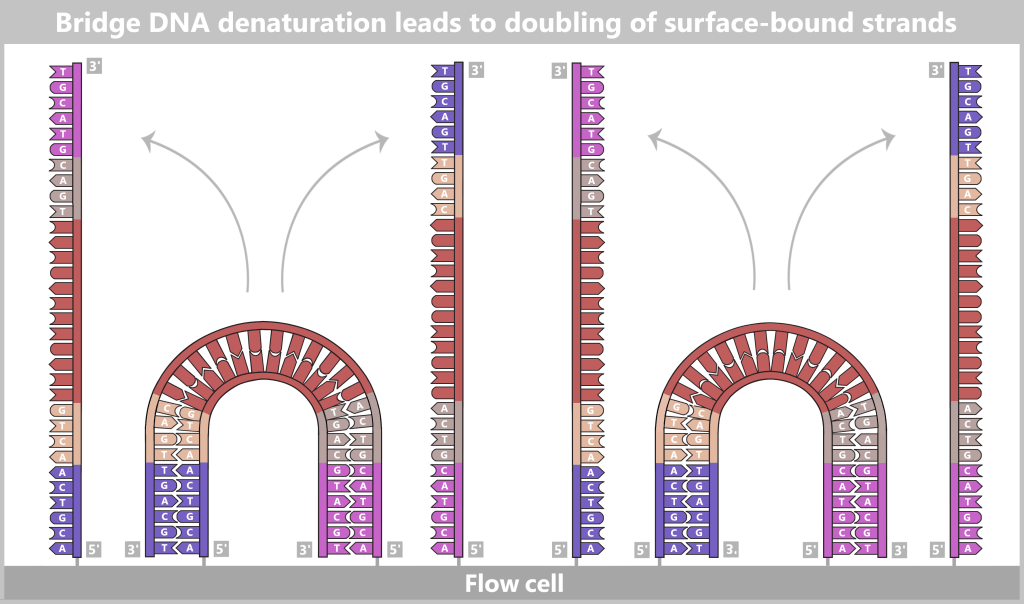
Fig. 2.4.2-F: Forward and reverse – bridge dissociation After the replication process, the bridge double-stranded DNA structures are denatured. As a result, the number of surface-bound DNA strands is doubled. These strands are now ready for further amplification.
This process is repeated multiple times. With each cycle, the number of surface-bound DNA molecules doubles.
Fig. 2.4.2-G: With each round of bridge amplification, more DNA copies are generated. In this way, locally confined clusters consisting of millions of nearly identical copies of a single original DNA fragment are formed.
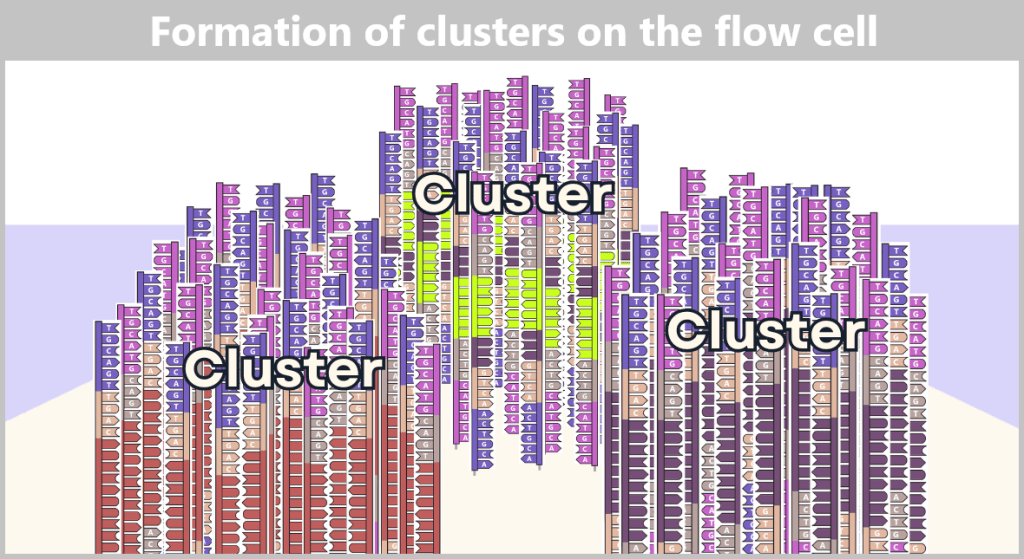
Fig. 2.4.2-H: Cluster formation: Each cluster consists of numerous copies of a single DNA fragment. In this illustration, only three clusters are shown as examples. In reality, however, millions of such clusters are present on a flow cell, enabling a high sequencing throughput.
The result is a densely packed surface of millions of spatially separated DNA clusters, each consisting of many nearly identical copies of a single original DNA fragment. This enables millions of different sequences to be analyzed in parallel.
Without this amplification, reading DNA would be like trying to detect a single firefly in the wind. The clusters, however, make the DNA bases (A, T, C, G) clearly detectable – like a glowing neon sign in the dark.
Removal of one strand type
To ensure that sequencing proceeds in a synchronous and unambiguous manner, the DNA strands within each cluster must be oriented uniformly. Therefore, one of the two strand types is selectively removed prior to the actual sequencing step.
The unnecessary complementary strands are chemically cleaved and washed away. At the same time, certain free ends on the flow cell are blocked to prevent unwanted re-binding or further amplification.
After this process, the clusters consist of many single strands with identical orientation. The DNA library is now prepared for the actual sequencing step.
3️⃣ Sequencing by synthesis
To initiate sequencing, primers, DNA polymerases, and modified nucleotides are added to the flow cell. Each of the four nucleotides (A, T, G, and C) carries a distinct fluorescent label as well as a reversible chemical blocking group.
The primers hybridize to the DNA strands of the library. DNA polymerase then binds to the primer and begins synthesizing the complementary strand.
Because of the reversible blocking group, only a single nucleotide can be incorporated during each cycle. After incorporation, DNA synthesis temporarily stops.
The flow cell is then imaged using high-resolution cameras. The fluorescence color emitted by each cluster indicates which nucleotide was incorporated during that cycle. Analysis software interprets these signals and converts them into the corresponding base sequence.
Subsequently, excess nucleotides are washed away. In an additional chemical step, both the fluorescent label and the blocking group of the incorporated nucleotide are removed, allowing the next synthesis cycle to begin.
This process repeats cycle by cycle. In this way, the sequence of each DNA fragment is read out step by step.
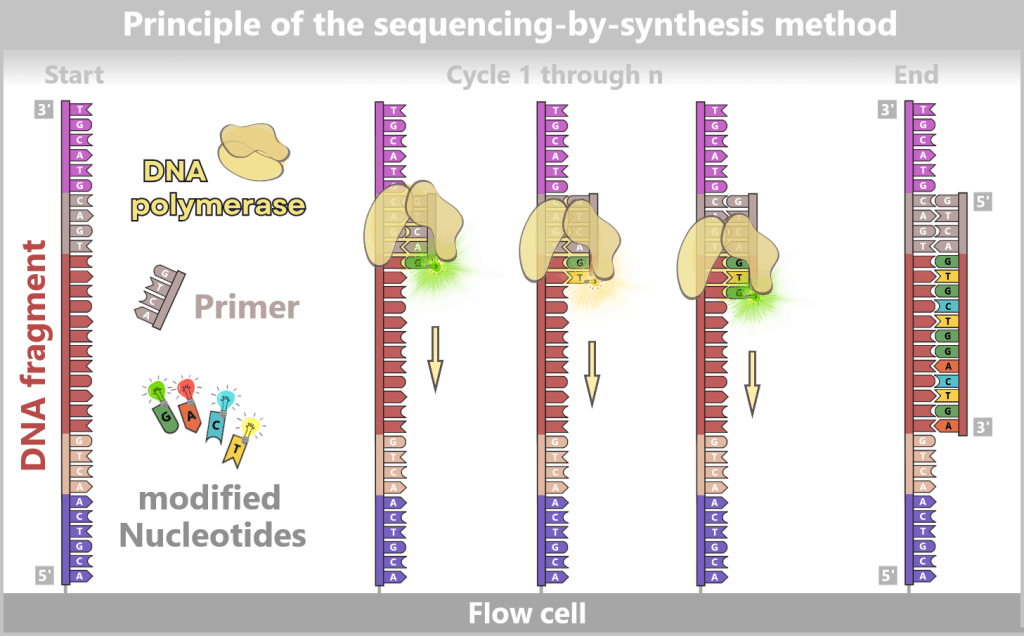
Fig. 2.4.2-I: Illumina sequencing workflow 4️⃣ Datenauswertung
After completion of sequencing, millions of individual fluorescence signals are available. These raw data are first translated into nucleotide sequences by the instrument software. The result consists of short sequence fragments, referred to as reads.
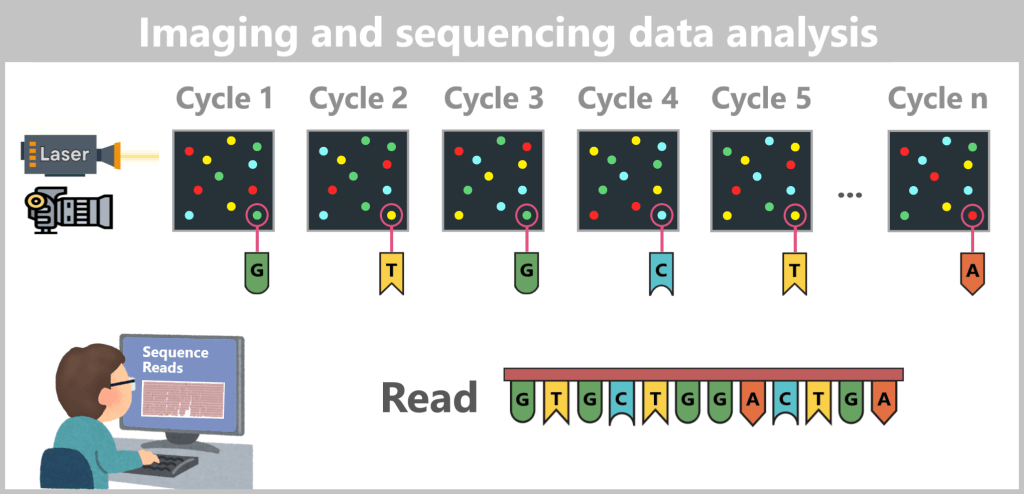
Fig. 2.4.2-J: Analysis of sequencing data 🎥 Tip: A more detailed yet illustrative explanation of the Illumina sequencing technology can be found in the video „Illumina Sequencing Technology“.
⬦ ⬦ ⬦
Quality control and trimming
In the first step, the quality of the reads is assessed. Bases with low signal quality – typically located at the ends of reads – are removed (trimming). This reduces incorrect sequence assignments and improves the reliability of downstream analysis.Mapping: alignment to a reference sequence
Next, the reads are compared to known reference sequences, such as the vaccine manufacturer’s plasmid or bacterial reference genomes. Specialized algorithms (e.g., BWA-MEM or Bowtie2) search for matches and assign each read to its most likely position within the reference sequence.
This allows determination of which sequences are present in the sample and their potential origin.Determination of fragment lengths
In commonly used paired-end sequencing, each DNA fragment is read from both ends. The distance between the two reads allows estimation of the original fragment length.
This information is particularly important for analyzing fragmented DNA samples.Identification of DNA origin
By comparing the sequencing data with reference databases, it can be determined whether the detected fragments originate from, for example:
▪️the expected production plasmid
▪️bacterial sequences
▪️or other genetic sources
This allows Illumina sequencing to provide not only quantitative information but also a detailed characterization of the DNA present in a sample.Data normalization
The number of generated reads depends, among other factors, on sequencing depth. To enable comparison between different samples or batches, the data are often normalized, for example as reads per million (RPM). This makes it possible to better assess relative differences between samples.◈ ◈ ◈
Methodological limitations
Limited suitability for absolute quantification
Illumina sequencing is highly effective for identifying DNA fragments and analyzing their relative abundance. However, direct absolute quantification of the original DNA amount is only possible to a limited extent.
This is because library preparation itself can alter the composition of the sample. Steps such as PCR amplification, adapter ligation, or size selection may preferentially enrich or deplete certain fragments. As a result, the number of reads generated is not necessarily proportional to the original abundance of DNA fragments in the sample.
Bias in fragment length distribution
In short-read sequencing, DNA fragments within a certain size range are preferentially analyzed. Very short fragments may be lost during purification, while very long fragments may be disadvantaged during amplification and cluster formation.
Therefore, the observed fragment length distribution does not necessarily reflect the original distribution in the sample, but rather the distribution of successfully sequenced fragments.
2.5. Electrophoretic Fragment Analysis
Electrophoretic fragment analysis is an established standard method in molecular biology for separating and characterizing nucleic acids (DNA and RNA). Depending on the application, it can provide information on several key parameters:
- Size (fragment length): reported in base pairs (bp) for DNA or nucleotides (nt) for RNA
- Quantity: semi-quantitative estimation of the amount of nucleic acid present
- Integrity: degree of fragmentation or molecular integrity
The method is based on the physical principle of electrophoresis (movement under the influence of an electric field). Charged molecules such as DNA or RNA are driven through a supporting medium (typically a gel or a polymer-based matrix) by an electric field. Due to their negatively charged phosphate backbone, nucleic acids migrate toward the positively charged electrode.
Separation is achieved through the pore structure of the medium: smaller fragments move more rapidly through the network, while larger molecules are more strongly retarded. This results in a size-dependent separation of nucleic acids.
In the context of residual DNA analysis in mRNA-based pharmaceuticals, this method is particularly useful for estimating fragment length distributions and therefore complements both quantitative and sequencing-based detection approaches.
2.5.1. Agarose gel electrophoresis
2.5.2. Automated systems⚬ ⚬ ⚬
2.5.1. Agarose gel electrophoresis
Agarose gel electrophoresis is the classic and most widely used form of electrophoretic fragment analysis of DNA. It is used to separate nucleic acids according to their size and make them visible.
System setup
The central component is a gel made of agarose, a polysaccharide-based network derived from algae. This gel forms a three-dimensional matrix with pores of defined size. The pore size can be controlled by the agarose concentration: low concentrations produce larger pores (suitable for long DNA fragments), while higher concentrations produce smaller pores (for short fragments).
The gel is embedded in a buffer solution and placed in an electrophoresis chamber. At both ends of the chamber are electrodes (cathode/anode), between which a constant electrical voltage is applied.
Before electrophoresis begins, the samples are pipetted into small wells at the edge of the gel.
In complex samples, such as those obtained after in vitro transcription and enzymatic treatment (e.g., DNase I), RNA and remaining DNA fragments can migrate through the gel simultaneously. For targeted analysis of residual DNA, the sample is therefore often additionally treated with RNase, so that predominantly DNA fragments are visible. This allows a clearer assessment of the fragment length distribution.
Fig. 2.5.1-A: Schematic setup of an agarose gel electrophoresis chamber Principle of operation
DNA molecules carry a negative charge due to their phosphate groups. When an electric voltage is applied, they begin to migrate through the gel toward the positively charged electrode.
During migration, two opposing forces act on the molecules:
- the electric force, which pulls the DNA through the gel
- the mechanical resistance of the gel matrix
Since smaller DNA fragments are less hindered by the pore structure, they move through the gel faster than larger fragments. This results in a size-dependent separation of the molecules along the direction of migration.
For size determination, a so-called DNA ladder (marker) with known fragment lengths is run in parallel. By comparing the migration distances, the sizes of the unknown fragments can be estimated.
Visualization and analysis
After electrophoresis, the DNA fragments are initially invisible. Visualization of the DNA is achieved using fluorescent dyes, which are either added to the gel before electrophoresis or applied to the gel afterward. Both methods are well established and differ mainly in their influence on migration behavior and the experimental effort required.
The gel is removed from the electrophoresis chamber and placed on a transilluminator. Under UV or blue light, the fluorescent dyes bound to the DNA appear as bands in the gel.
By comparing the bands with the simultaneously run marker, the sizes of the DNA fragments can be estimated. In addition, the intensity of the bands allows a rough semiquantitative assessment of the amount of DNA present.
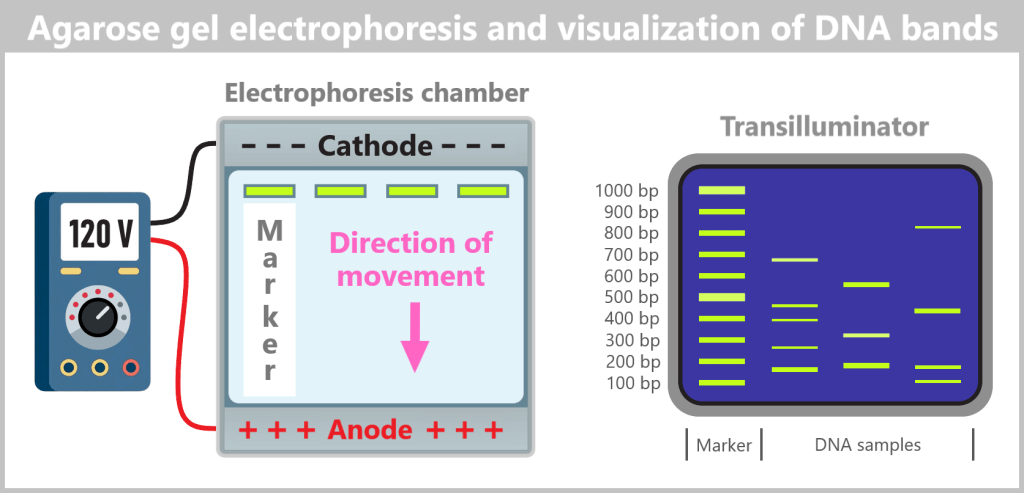
Fig. 2.5.1-B: Detection of DNA fragments after electrophoretic separation 🎥 Tip: The video „Electrophoresis explained“ briefly summarizes the principle of agarose gel electrophoresis.
2.5.2. Automated Systems
Automated electrophoretic systems represent a further development of classical agarose gel electrophoresis. They are based on the same physical principle of size-dependent separation of nucleic acids in an electric field, but combine it with microfluidic technology, integrated detection, and digital analysis.
In contrast to manual gel preparation, separation is carried out in miniaturized, prefabricated systems. The samples are loaded into specialized cartridges or chips containing fine channels filled with polymer matrices. Within these microstructures, the nucleic acids are separated electrophoretically under an applied voltage.
During the run, the fragments are continuously detected using fluorescent dyes. The signals are directly converted into digital data and displayed as so-called electropherograms. Instead of bands in a gel, peaks are generated, where the position indicates the fragment size and the height or area reflects the relative amount of nucleic acid.
Commonly used systems include:
- the Bioanalyzer
- the TapeStation
- and the Fragment Analyzer
These systems differ in details such as throughput, sensitivity, and degree of automation, but they follow a comparable basic principle.
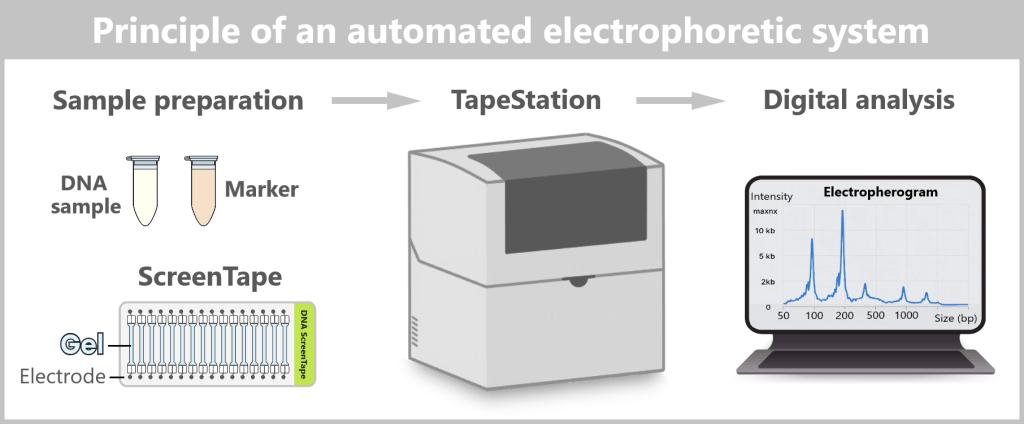
Fig. 2.5.2: Schematic representation of an automated electrophoretic system (e.g., TapeStation) with ScreenTape unit and a typical electropherogram Workflow of an automated electrophoretic system (e.g., TapeStation)
1) Sample preparation
The samples to be analyzed are pipetted into tubes or a microtiter plate (e.g., 96-well format) and mixed with a kit-specific buffer. In parallel, a reference standard (ladder/marker) with defined fragment sizes is prepared.
2) Addition of fluorescent dye
A fluorescent dye is added to the samples, which binds selectively to DNA or RNA and enables subsequent detection.
3) Provision of the separation medium
The separation medium is a so-called ScreenTape – a prefabricated microfluidic system containing an integrated gel matrix and multiple parallel separation channels. Depending on the application, specific ScreenTapes are used for DNA or RNA.
4) Automated electrophoresis run
Samples, marker, and ScreenTape are loaded into the instrument. After startup, the system performs the analysis automatically: the samples are sequentially transferred into the separation channels and electrophoretically separated under an applied voltage.
5) Detection and data analysis
During separation, fluorescent signals from the nucleic acids are optically detected (e.g., using a laser). The software processes these signals into digital outputs, including:
▫️electropherograms (peak-based representations of fragment sizes)
▫️size distributions
▫️and semi-quantitative concentration measurements🎥 Tip: A visual demonstration of the operating principle of the TapeStation system and ScreenTape technology is provided in the following video „Agilent 2200 TapeStation System“.
⬦ ⬦ ⬦
Advantages of automated systems
Automated systems offer several key advantages over classical agarose gel electrophoresis:
- Higher sensitivity: even small amounts of nucleic acids can be detected
- Improved reproducibility: standardized cartridges and automated workflows reduce experimental variability
- Quantifiability: in addition to fragment size, relative concentration can be estimated more accurately
- Digital analysis: objective and reproducible evaluation without visual interpretation of gel images
In addition, these systems often provide further metrics, such as average fragment lengths or integrity indices, enabling a standardized assessment of sample quality.
⬦ ⬦ ⬦
DNA- and RNA-specific assays
A key aspect of automated systems is the use of specific assays for different types of nucleic acids. In complex samples, separate analyses are therefore often performed, in which identical samples are analyzed using either DNA- or RNA-specific reagents.
These assays differ, among other things, in:
- the composition of the polymer matrix
- the fluorescent dyes used
- the data analysis algorithms
In this way, DNA and RNA fragments can be characterized independently, even if they were originally present in the same sample.
⬦ ⬦ ⬦
Interpretative value of electrophoretic fragment analysis
Electrophoretic fragment analysis primarily provides structural information about nucleic acids. In the context of quality control of mRNA-based products, it is particularly useful for:
- analyzing the fragment length distribution of DNA and RNA
- assessing RNA integrity
- distinguishing between highly fragmented and largely intact nucleic acids
- identifying dominant fragment sizes
- and detecting residual DNA fragments
◈ ◈ ◈
Limitations of electrophoretic fragment analysis
Both classical agarose gels and automated electrophoretic systems have methodological limitations, particularly when detecting low levels of residual DNA in complex samples.
- No sequence information: The method separates nucleic acids solely based on size or electrophoretic mobility. It cannot determine whether a fragment originates from genomic DNA, plasmid DNA, mRNA, or nonspecific degradation products.
- Limited sensitivity compared to amplification-based methods: Very small amounts of residual DNA may fall below the detection limit. Techniques such as qPCR or ddPCR are significantly more sensitive for quantifying trace DNA levels.
- Limited resolution for highly fragmented DNA: Highly degraded DNA often appears as a diffuse signal region („smear”), making individual fragments difficult to characterize.
- No information on biological functionality: The method only shows fragment size and quantity distribution. Whether DNA fragments are still biologically active, complete, or potentially transcription-competent remains unknown.
- Dependence on dye binding and fragment size: Very short fragments may bind less dye and can therefore be underestimated or even go undetected. Quantitative accuracy is especially limited at the lower end of fragment sizes.
- Overlapping signals in complex samples: In samples containing both RNA and DNA, signal regions may overlap. Despite specific assays, misassignments or background signals cannot be fully excluded.
- No absolute quantification of specific target sequences: The method does not allow targeted measurement of defined DNA sequences. Specific identification of residual process DNA is therefore only possible in combination with sequence-specific techniques.
Overall, electrophoretic fragment analysis primarily provides structural information on nucleic acids and complements quantitative and sequencing-based methods by offering insights into fragment size, integrity, and distribution, but it cannot replace sequence-specific or functional assays.
2.6. Comparison of Key Detection Methods for Nucleic Acids
The quantitative and qualitative determination of nucleic acids forms the foundation of numerous molecular biological analyses. However, depending on the method, both the informational content and the limitations of the measurement differ significantly. None of the established techniques provides a complete picture – instead, they complement each other.
UV spectroscopic measurements determine the total amount of nucleic acids in a sample by measuring the absorption of UV light at 260 nm. This method is fast and straightforward, but it does not distinguish between different DNA or RNA sequences. Contaminants such as proteins or other molecules can also influence the signal.
Fluorometric methods use specific dyes that preferentially bind to DNA or RNA. As a result, they are significantly more selective and sensitive than UV-based measurements. Nevertheless, they do not provide information about the exact sequence or origin of the nucleic acids – they only quantify the amount of specific classes of molecules present.
Electrophoretic fragment analysis adds a structural perspective to these methods. It allows the estimation of fragment sizes, fragment length distributions, and the integrity of DNA or RNA. This makes it possible, for example, to distinguish between intact and highly fragmented nucleic acids. However, the method does not provide sequence information and does not allow the unambiguous identification of specific DNA or RNA molecules.
PCR-based methods (qPCR and ddPCR) enable highly sensitive and specific detection of defined DNA sequences. By using sequence-specific primers, individual regions of the genome can be selectively amplified and quantified. However, this high specificity is also a limitation: only those sequences for which appropriate primers have been designed can be detected. Unknown or unexpected sequences remain undetected.
Another methodological limitation arises from the requirements regarding DNA integrity. For amplification to occur, both primer binding sites must be fully contained within a DNA fragment. qPCR amplicons are typically 80–150 base pairs in length. Highly fragmented DNA, in which fragments are shorter or in which one of the primer binding sites is missing, cannot be amplified. In other words, only DNA fragments that span the complete target region between both primers are detectable by qPCR.
This dependence on fragment length is particularly relevant when DNA has been broken down by enzymatic or mechanical processes, such as DNase I treatment during the removal of residual DNA in mRNA production. For successful qPCR/ddPCR, however, both primers – forward and reverse – must be able to bind to the same fragment. The shorter the fragments, the lower the probability that a fragment contains both primer binding sites – an important consideration when assessing residual DNA.
Illumina sequencing is based on the parallel sequencing of many short DNA fragments. It provides highly accurate sequence data with a very low error rate compared to other sequencing methods, making it particularly suitable for the precise analysis of known or referenceable sequences. However, the method requires more complex sample preparation, PCR-based library amplification, and advanced bioinformatic analysis. In addition, it typically sequences only relatively short fragments, which makes it more difficult to capture larger structural relationships.
Nanopore sequencing goes one step further: it enables the direct determination of the base sequence of individual DNA or RNA molecules in real time. As a result, unknown sequences can also be identified, and very long fragments can be analyzed. In addition, the technology allows, under certain conditions, the direct detection of chemical nucleic acid modifications. However, the resulting data require careful bioinformatic analysis and, compared to Illumina sequencing, currently still show higher error rates.
⚬ ⚬ ⚬
The following overview summarizes the most important detection methods, their measurement principles, as well as their specific strengths and limitations in the context of mRNA production.
Method
What is measured?
Strengths
Weaknesses / limitations
Typical application
UV spectroscopy
Total concentration of all nucleic acids
Fast and simple, no reagents required, low cost
No distinction between DNA, RNA, and free nucleotides; sensitive to contamination
Rapid rough estimation of concentration and purity
Fluorometry
DNA or RNA via dye binding
High sensitivity, tolerant to contaminants
No sequence information
Precise quantification of target RNA
Qubit fluorometer
DNA or RNA via dye binding (fluorometric device)
Very high sensitivity and selectivity, reproducible results
No sequence information, higher cost than basic fluorometry
Standardized concentration measurement in lab and GMP settings
qPCR
Specific DNA sequences (e.g., template DNA, host cell DNA)
High sensitivity, sequence-specific
Only known target sequences detectable; depends on primer design and amplification efficiency; quantification requires standard curve; susceptible to inhibitors
Detection of residual DNA
ddPCR
Absolute copy number of specific DNA sequences
Absolute quantification without standard curve, very high precision, more robust against inhibitors
Only known target sequences detectable; depends on primer/probe design and target integrity; higher cost; more complex sample preparation
Precise quantification of defined DNA target sequences in validation assays
Agarose gel electrophoresis
DNA/RNA fragments by size
Simple, cost-effective, visual estimation of fragment size and integrity
Low resolution, semi-quantitative, not suitable for precise concentration measurement, no sequence information
Rapid quality control, integrity assessment
Automated electrophoresis
Size, concentration, and integrity of DNA/RNA
High resolution, quantitative, reproducible, low sample input
Higher cost, requires consumables
RNA integrity assessment (RIN score), fragment analysis of residual DNA
Illumina sequencing (NGS)
Sequence information of many short DNA/RNA fragments
Very high accuracy, broad coverage, well established
Short reads (150–300 bp), complex sample preparation, requires bioinformatic analysis
High-resolution sequence analysis and characterization of residual nucleic acids
Nanopore sequencing
Full DNA/RNA sequence of individual molecules
Direct sequence information, long reads, real-time data, detects unknown sequences and structural variants
Higher error rate, complex analysis, still less established in GMP environments
Characterization of complex nucleic acid populations, identity testing, integrity analysis
Overall, it becomes clear that each method has its own specific strengths and weaknesses. The choice of the appropriate procedure therefore always depends on the respective research question.
This is particularly true for the quality control of therapeutic mRNA. Active ingredient concentration, purity, and potential contaminants each place distinct demands on the analytical method.
No single method can answer all analytical questions – only the combination of quantification, structural characterization, and sequence information enables a comprehensive assessment of nucleic acids.
◈ ◈ ◈
Combined application of methods
In practical analytical work, the methods described are rarely used in isolation. Instead, the characterization of nucleic acids is typically performed using a combination of complementary techniques. For example, UV spectroscopic measurements can be used for a rapid estimation of concentration and purity, while fluorometric methods enable more selective quantification. PCR-based methods such as qPCR or ddPCR are applied to specifically detect and quantify defined DNA sequences. Sequencing approaches such as Illumina or Nanopore technologies complement these analyses by directly determining the sequence and enabling the identification of unknown sequences or structural variants.
⬦ ⬦ ⬦
Regulatory classification
In a regulatory context, quality control of therapeutic nucleic acid products requires the use of validated and standardized analytical methods. PCR-based techniques such as qPCR and ddPCR are well established for this purpose and are routinely used for the quantification of defined residual DNA sequences. Sequencing-based approaches, including Nanopore technology, are gaining increasing importance; however, in many application areas they are still in the phase of methodological establishment and validation.
3. Guidelines for Limiting Residual DNA in Vaccines
Residual DNA (residual DNA), including host cell DNA or plasmid DNA, refers to nucleic acid remnants from the manufacturing process that may remain in the final product after purification. As outlined in previous chapters, complete removal of this DNA cannot be achieved technically; therefore, low residual amounts are considered an unavoidable component of biotechnological products.
The development of regulatory guidelines for limiting residual DNA has progressed in parallel with advances in biotechnological manufacturing processes. The aim of these requirements is to minimize potential biological risks. These include in particular:
- Oncogenicity
- Insertional mutagenesis
- Immunogenicity
- and, in certain contexts, infectivity (e.g., in viral systems)
Historical development
Early phase: precaution-oriented approach
With the introduction of recombinant DNA technologies (methods in which DNA from different organisms is deliberately combined) in the 1980s – such as for the production of therapeutic insulin or growth hormones – the need to systematically evaluate residual DNA emerged for the first time.
At that time, there were considerable scientific uncertainties regarding the biological effects of exogenous DNA. In particular, it was unclear under which conditions foreign DNA could integrate into the genome of human cells and potentially induce oncogenic effects.
Against this background, regulatory authorities adopted a conservative, precaution-oriented approach. In the 1980s, the World Health Organization (WHO) recommended a limit value of 10 pg residual DNA per dose for products derived from continuous cell lines (cells that are capable of unlimited division in the laboratory). This value was not based on a quantitative risk assessment but represented a pragmatic guideline aligned with the analytical capabilities available at the time.
Later evaluations led to an upward revision of this value; by the mid-1980s, levels on the order of approximately 100 pg per dose were already considered negligible.
Paradigm shift: from mass to biological activity
In the 1990s and 2000s, the regulatory perspective changed fundamentally. Two developments were primarily responsible for this:
First, manufacturing and purification processes improved significantly. Methods such as chromatography, filtration, and enzymatic treatment enabled a reproducible reduction of residual DNA by several orders of magnitude.
Second, experimental studies increasingly provided quantitative insights into the biological activity of DNA. It became evident that not only the total amount is relevant, but that structural properties such as length and integrity of DNA play a central role.
Longer, intact DNA fragments containing functional gene segments can, under suitable conditions, theoretically exert biological effects. However, this potential decreases significantly with increasing fragmentation. Short DNA fragments have a strongly reduced probability of entering the cell nucleus, persisting stably, or being integrated into the genome.
For actual integration of exogenous DNA into a cellular genome, several conditions must be met simultaneously. These include, among others, uptake of the DNA into the cell, its transport into the nucleus, and the presence of suitable cellular mechanisms for stable integration. The likelihood of all these conditions occurring simultaneously under natural physiological conditions in the body is considered low and decreases further with increasing DNA fragmentation.
Modern regulatory approaches
Based on these findings, modern regulatory concepts have evolved toward a risk-based and product-specific approach. Instead of a single universal limit value, several complementary requirements are now central:
Requirement Rationale Validated elimination The manufacturing process must demonstrably and reproducibly contribute to the reduction of residual DNA. Fragmentation Remaining DNA should be present in a highly fragmented form whenever possible. Product-specific limits Acceptance criteria are defined depending on the manufacturing process, cell line, and intended use. Regulatory guidelines issued by organizations such as the U.S. Food and Drug Administration (FDA), the European Medicines Agency (EMA), and the International Council for Harmonisation (ICH) today largely follow this differentiated approach.
✧ ✧ ✧
Classification in the context of modRNA-based vaccines
For modRNA-based vaccines, fundamentally similar regulatory principles apply as for other biotechnologically manufactured medicinal products. At the same time, manufacturing processes and product characteristics differ in certain aspects, particularly due to in vitro transcription and the use of synthetic RNA.
In the context of vaccines, regulatory guidelines regarding residual DNA content for parenteral administration (i.e., administration by injection or infusion that bypasses the gastrointestinal tract) are often oriented toward the nanogram range per dose.
In addition, it is required that any remaining DNA be highly fragmented and not contain functionally complete genes. In the scientific literature, fragment lengths below a few hundred base pairs (bp) are frequently discussed in this context, although no uniform threshold has been established. These requirements reflect the current regulatory approach, which takes into account both the quantity and the structural properties of DNA.
The following table summarizes the key regulatory documents in which limits for residual DNA are specified.
Document Statement / Context 1998 – WHO TRS 878, Annex 1 Historical source: first mention of <10 ng/dose for continuous cell lines. 2007 – WHO Study Group on Cell Substrates for the Production of Biologicals Discussion of <10 ng/dose in the context of highly fragmented DNA (e.g., <200 bp). 2013 – WHO TRS No. 978 Confirms <10 ng/dose as an established standard; emphasizes the importance of fragmentation and manufacturing process; key WHO document on cell substrates. 2014 – WHO TRS 987, Annex 4 <10 ng/dose (in the context of recombinant protein therapeutics) 2020 – FDA CMC Guidance
(Gene Therapy)<10 ng/dose and <200 bp (for gene therapy products derived from continuous non-tumorigenic cell lines) 2020 – Rapporteur Rolling Review critical assessment report (Josephson 2020-11-19) Acceptance criteria: ≤ 330 ng DNA/mg RNA (derived from 10 ng/dose at 30 µg RNA per dose). 2024 – WHO TRS 1062,
Annex 1 (not yet public)Recommendations for mRNA vaccines – expected to include similar order-of-magnitude ranges. Dose: Total amount of the medicinal product administered per application.
ng/dose (nanograms per dose): A measure of the permissible amount of residual DNA per dose.
bp (base pairs): A measure of the length of DNA fragments.⚬ ⚬ ⚬
Conversion of measurement values to ng per dose
In order to compare measurement results from different studies with regulatory guideline values, a standardized reference unit is required. While regulatory limits are usually expressed as an amount per dose (ng/dose), analytical measurements are often reported relative to the RNA quantity (e.g., ng DNA per mg RNA).
For comparability, these values must be converted accordingly.
Basic principle:
DNA (ng/dose) = DNA (ng/mg RNA) × RNA amount per dose (mg)Example calculation
Assume that an analytical measurement yields: 330 ng DNA per mg RNA
and the administered dose contains: 30 µg RNA = 0,03 mg RNA
The calculation is therefore: 330 ng/mg × 0,03 mg = 9,9 ng DNA per doseThis example demonstrates that concentration-based values (ng/mg RNA) and dose-related limits (ng/dose) can be directly converted into one another, provided that the underlying RNA amount per dose is known.
The result is therefore in the range of 10 ng DNA per dose, corresponding to the commonly cited regulatory guideline value.
⚬ ⚬ ⚬
Classification
An important aspect of regulatory classification is that no single universally binding limit applies to all modRNA vaccines. Instead, the evaluation is based on a combination of general recommendations, product-specific specifications, and risk-based principles.

4. Experimental Studies on residual DNA in mRNA-based Vaccines
Following the presentation of manufacturing processes, potential impurities, and analytical detection methods, the central question arises as to the extent to which residual DNA can actually be detected in commercial mRNA vaccines.
Since 2023, several independent investigations have been published addressing the detection, quantification, and characterization of DNA in different vaccine batches.
The aim of this chapter is to present the studies published to date, classify their methodological approaches in a comprehensible manner, and summarize the reported findings.
4.1. Why independent measurements?
4.2. Methodological preliminary remarks
4.3. Overview and individual analysis of the studies
4.4. Interim conclusion: What do we know – and what do we not know?⚬ ⚬ ⚬
4.1. Why independent measurements?
The approval of a medicinal product requires the manufacturer to demonstrate compliance with regulatory quality standards to the competent authorities. For mRNA-based vaccines, this includes, among other things, ensuring that residual amounts of DNA remain below established limit values. The corresponding analytical data are submitted as part of the regulatory approval process and are evaluated by the authorities.
What this process does not structurally provide for is systematic verification by independent laboratories after market authorization. Authorities such as the EMA or FDA base their assessments primarily on the data submitted by the manufacturer. Independent reanalysis of commercially available batches is not part of the standard procedure.
This is precisely where independent research becomes relevant. Since 2023, several research groups –without affiliation to the manufacturers – have investigated commercially obtained batches of mRNA vaccines for residual DNA. The primary motivation was not necessarily the expectation of a problem, but rather a fundamental question of scientific reproducibility: Can the quality data submitted during the regulatory approval process be reproduced through independent measurements?
The results of these studies have not been uniform – neither with respect to the measured quantities nor the methodological approaches employed. Some groups reported DNA levels exceeding regulatory guideline values, whereas others arrived at different conclusions or raised methodological concerns regarding the analytical procedures used.
This chapter presents the most important of these investigations. Its purpose is not to provide a final assessment – the scientific discussion is still ongoing – but rather to offer an objective overview of the available findings, the methods applied, and the remaining open questions.
4.2. Methodological preliminary remarks
Before presenting the individual studies, a methodological framework is helpful – not to anticipate results, but to provide the reader with a tool for interpreting the findings.
The studies presented in this chapter originate from independent laboratories and differ in some cases significantly with regard to their methodological approaches and the detection methods employed.
The dispersion of lipid nanoparticles
Every analysis begins with the same fundamental challenge: the nucleic acids to be measured – mRNA and potential residual DNA – are encapsulated within lipid nanoparticles (LNPs). To make them analytically accessible, the particles must first be disrupted.
In the studies considered, a variety of methods were used for this purpose. Detergents such as Triton X-100 chemically dissolve the lipid membrane. Combinations of lithium dodecyl sulfate (LiDS) and magnetic SPRI beads enable both particle disruption and selective purification of nucleic acids. Thermal approaches – such as heating to 95 °C – destabilize particle structure through heat exposure. Commercial kits such as the Monarch Plasmid DNA Miniprep Kit combine multiple steps into a standardized protocol.
This methodological diversity is not trivial. Three aspects deserve particular attention:
First, the methods differ in their disruption efficiency: not every approach fully opens all particles. If intact LNPs remain in the sample, their contents remain inaccessible to downstream measurement, which can lead to an underestimation of the actual DNA content.
Second, some methods may alter or degrade the released molecules. Thermal disruption at high temperatures can fragment RNA in particular; DNA is more thermally stable but may also be affected under certain conditions. As described in Chapter 2.6, highly fragmented DNA is less accessible to PCR-based detection methods – short fragments may no longer contain both primer binding sites and therefore escape detection.
Third, residues of certain reagents can interfere with subsequent analytical steps. Detergents such as Triton X-100 are known PCR inhibitors. If they are not fully removed, amplification efficiency may be reduced, again leading to an underestimation of the measured quantity. SPRI beads, on the other hand, select based on fragment size: very short DNA fragments may be lost during this step.
Choice of analytical method
Following sample disruption, the actual measurement is performed. Here too, the studies differ considerably. Some research groups used quantitative PCR methods (qPCR), which enable highly sensitive but sequence-specific detection. Others employed sequencing approaches – either nanopore technology (ONT) or the Illumina platform – which provide a broader, sequence-independent overview but require more complex data analysis. In some cases, fluorometric methods were also used for total quantification.
Each of these methods measures slightly different aspects: fluorometry detects total DNA irrespective of sequence but can be affected by interference from RNA (cross-talk). PCR-based methods detect only predefined target sequences. Sequencing approaches provide a broader picture but are more strongly influenced by library preparation, fragment selection, and bioinformatic processing. As a result, direct comparability of absolute quantitative values across studies using different methodologies is only limited.
Validation and standardization
In regulated pharmaceutical analysis, analytical methods must be validated before use: extraction efficiency, limit of detection, linearity, and potential sources of interference must be systematically determined. The independent studies presented here generally lack this formal validation – which does not mean that the work was carried out carelessly, but rather that methodological uncertainties are more difficult to quantify.
A direct comparison of the same sample using multiple methods in parallel – a so-called orthogonal approach – would help to distinguish method-related variability from true differences between batches. Only a subset of the available studies systematically applies multiple orthogonal methods to the same samples. The importance of this approach will be addressed again in the concluding chapter.
4.3. Overview and individual analyses of the studies
The following studies differ in terms of methodology, form of publication, and scientific classification. In addition to peer-reviewed articles, preprints and exploratory analyses are also included in order to fully capture the current spectrum of available data.
The presentation is organized chronologically to allow the development of the scientific discussion to be followed in a coherent manner.
Sequencing of bivalent Moderna and Pfizer mRNA vaccines reveals nanogram to microgram quantities of expression vector dsDNA per dose
Authors: Kevin McKernan, Yvonne Helbert, Liam T. Kane, Stephen McLaughlin
Published: 2023 April 10
DNA fragments detected in monovalent and bivalent Pfizer/BioNTech and Moderna modRNA COVID-19 vaccines from Ontario, Canada: Exploratory dose response relationship with serious adverse events.
Authors: David Speicher, J. Rose, L. M. Gutschi, D. M. Wiseman, Kevin McKernan
Published: 2023 October 19
Methodological Considerations Regarding the Quantification of DNA Impurities in the COVID-19 mRNA Vaccine Comirnaty®
Authors: Brigitte König, Jürgen O. Kirchner
Published: 2024 May 8
Confirmation of the presence of vaccine DNA in the Pfizer anti-COVID-19 vaccine
Authors: Didier Raoult
Published: 2024 November 12
BioNTech RNA-Based COVID-19 Injections Contain Large Amounts Of Residual DNA Including An SV40 Promoter/Enhancer Sequence
Authors: Ulrike Kämmerer, Verena Schulz, Klaus Steger
Published: 2024 December 3
A rapid detection method of replication-competent plasmid DNA from COVID-19 mRNA vaccines for quality control.
Authors: Tayler J. Wang, Alex Kim, Kevin Kim
Published: 2024 December 29
Quantitative Analysis of Nucleic Acid Content in Spikevax (Moderna) and BNT162b2 (Pfizer) COVID-19 Vaccine Lots
Authors: Richard M Fleming PhD, MD, JD; Peter Kotlár MD; Sona Pekova MD, PhD
Published: 2025 May 13
Quantification of residual plasmid DNA and SV40 promoter-enhancer sequences in Pfizer/BioNTech and Moderna modRNA COVID-19 vaccines from Ontario, Canada
Authors: David Speicher, Jessica Rose, Kevin McKernan
Published: 2025 September 25
Systematic analysis of COVID-19 mRNA vaccines using four orthogonal approaches demonstrates no excessive DNA impurities
Authors: Adam Achs, Tatiana Sedlackova, Lukas Predajna, Jaroslav Budis, Maria Bartosova, Vladimir Zelnik, Diana Rusnakova, Martina Melichercikova, Marta Miklosova, Veronika Gencurova, Barbora Cernakova, Tomas Szemes, Boris Klempa, Juraj Kopacek, Silvia Pastorekova
Published: 2025 Dezember 13
🧬 🧬 🧬
4.3.1. McKernan et al. (2023)
Sequencing of bivalent Moderna and Pfizer mRNA vaccines reveals nanogram to microgram quantities of expression vector dsDNA per dose
Authors: Kevin McKernan, Yvonne Helbert, Liam T. Kane, Stephen McLaughlin
Published: April 10, 2023 (Preprint, OSF)
Link to study: https://osf.io/preprints/osf/b9t7ma) Context and objective of the study
The study by McKernan et al. is among the first publicly available independent analyses in which mRNA-based COVID-19 vaccines were specifically tested for the presence of DNA. The study was not initiated by regulatory concerns, but rather as an exploratory sequencing project during which DNA was detected in the samples. This prompted the authors to systematically investigate the composition of the vaccines further.
The aim of the study was to:
- detect DNA components in the vaccines,
- estimate their quantity, and
- characterize their sequence identity.
b) Materials and methods
Vaccine lots
The following batches were used for the analyses:- 2 batches of bivalent Pfizer vaccines (expired)
- 2 batches of bivalent Moderna vaccines (expired)
- 8 batches of monovalent Pfizer vaccines (expired, unopened)
The study combines several analytical methods to enable both quantitative and qualitative assessments.
Disruption of LNPs and nucleic acid extraction
The lipid nanoparticles were lysed using lithium dodecyl sulfate (LiDS). Subsequent purification was performed using the SPRI principle (magnetic beads), which selectively binds nucleic acids in a size- and condition-dependent manner while separating them from lipids and other accompanying substances. The resulting eluate contained isolated nucleic acids – mRNA and potentially present DNA.
As a quality control step for the extraction process, a Bioanalyzer was used. This system applies automated capillary electrophoresis to visualize the size distribution and relative quantity of the recovered nucleic acids.Sequencing using Illumina
The extracted nucleic acids were analyzed using Illumina sequencing. Multiple sequencing runs (reads) formed the basis for the bioinformatic reconstruction of the plasmid sequences of both vaccines. From the obtained reads, plasmid maps were generated for Moderna and for Pfizer.PCR-based verification
Based on the sequencing data, specific primers were designed for two target regions: a spike probe and an origin-of-replication (Ori) probe, both targeting sequences present in both the Pfizer and Moderna plasmids. This qPCR analysis was not intended for primary quantification but rather for independent confirmation that the plasmid sequences identified by sequencing were indeed present in the samples – and not attributable to laboratory contamination.Fluorometry and electrophoresis
Qubit fluorometry and Agilent electrophoresis were used to estimate DNA concentration. Both methods provide information on the total amount of DNA present, but do not yield any sequence information.c) Key findings
Qualitative findings
The sequencing analyses revealed sequence matches with known plasmid vector sequences and identified genetic elements associated with the manufacturing process. The detected DNA was characterized as double-stranded DNA (dsDNA).Unexpected finding: SV40 promoter/enhancer
During the sequencing analysis, the authors identified an SV40 promoter/enhancer sequence in the Pfizer vaccine. This finding was considered unexpected because the sequence was not shown in the publicly available plasmid maps submitted to the European Medicines Agency (EMA), specifically in the Rapporteur Rolling Review Critical Assessment Report (Figure S.2.3-1, pST4-1525 Plasmid Map). The authors further noted that the Pfizer plasmid also contains an SV40 nuclear localization signal (NLS), which, in theory, could facilitate the transport of DNA into the cell nucleus.Quantitative findings
Qubit fluorometry and electrophoresis measurements indicated DNA concentrations that, according to the authors‘ assessment, substantially exceeded commonly cited regulatory guideline values, including the EMA-derived specification of 330 ng DNA per mg RNA.d) Interpretation by the authors
The authors conclude that residual DNA is present in the analyzed vaccine batches and that its origin is most likely attributable to the plasmid vectors used during the manufacturing process. In their view, the measured quantities may be of regulatory relevance.
With regard to replicable DNA, the authors suggest that stricter limits would be appropriate for such sequences than for non-replicable DNA fragments.
e) Methodological limitations
The origin, storage conditions, and batch characterization of the analyzed vials are documented only to a limited extent. Information on negative controls – such as water samples subjected to the same processing workflow – as well as positive controls containing known DNA concentrations is largely absent or incompletely described. Furthermore, no formal method validation was reported.
Furthermore, the authors themselves point out two opposing sources of uncertainty in quantification: PCR-based methods may underestimate the actual amount of DNA, as fragments shorter than the amplicon length are not detected. Conversely, fluorometry and electrophoresis may overestimate the DNA concentration if the dye used also binds non-specifically to RNA.
f) Contextualization
The study represents an early and significant contribution to the scientific debate on residual DNA in mRNA vaccines. It has played a key role in encouraging other research groups to conduct their own investigations. The findings should be regarded as a starting point for further research, not as a definitive assessment.
🧬 🧬 🧬
4.3.2. Speicher et al. (2023)
DNA fragments detected in monovalent and bivalent Pfizer/BioNTech and Moderna modRNA COVID-19 vaccines from Ontario, Canada: Exploratory dose response relationship with serious adverse events.
Authors: David Speicher, Jessica Rose, L. Maria Gutschi, David M. Wiseman, Kevin McKernan
Published: October 19, 2023 (ResearchGate)
Link to study: https://osf.io/preprints/osf/mjc97_v1a) Context and Objective of the Study
The study by David J. Speicher builds conceptually on the previously published analyses by Kevin McKernan and pursues a broader approach.
In addition to detecting and quantifying residual DNA in further vaccine batches, it addresses two additional research questions:
- Localization of the DNA: Is the detected DNA located within the lipid nanoparticles (LNPs), or is it present freely in solution?
- Exploratory correlation: Is there a statistical correlation between measured DNA levels and reported adverse events at batch level?
The study is therefore divided into two conceptually distinct parts:
- analytical section: Detection and characterisation of DNA in vaccine samples
- exploratory section: Investigation of a possible statistical relationship with adverse reaction data
Both parts require a different methodological assessment.
b) Materials and Methods
Vaccine batches
A total of 27 vials from 12 batches were examined: 8 expired, unopened Pfizer-BNT162b2 vials and 16 expired, unopened Moderna Spikevax vials, obtained from various pharmacies in Ontario, Canada. All vials were original sealed with intact protective caps and labeled batch numbers and expiration dates. In addition, three still-valid residual volumes from a Moderna XBB.1.5 batch were included. Storage and transport were carried out under documented refrigerated conditions. A sterile, unopened vial of an unrelated medication (TriMix) served as a negative control.The authors combine several analytical and statistical methods.
Disruption of lipid nanoparticles (LNPs)
To disrupt the lipid nanoparticles (LNPs), the samples were heated for 8 minutes at 95 °C, followed by cooling to 4 °C for five minutes. This thermal step is intended to release the nucleic acids contained within the nanoparticles.Quantification by qPCR
DNA concentrations were determined using qPCR based on the Cq value (quantification cycle) and converted into nanograms per dose. The authors adopted the primers from the McKernan study: a Spike probe and an Ori probe targeting sequences common to both vaccines (Pfizer and Moderna). In addition, an SV40 enhancer assay was used, specifically designed to detect the SV40 sequence identified in the Pfizer plasmid.Quantification by Qubit fluorometry
For total DNA quantification, Qubit fluorometry with the DNA-specific dye AccuGreen was used. Fluorescence intensity is proportional to the amount of DNA present and provides a sequence-independent but structure-sensitive overall estimate (dependent on dye specificity and sample composition).Size profile of residual DNA using ONT sequencing
The size distribution of DNA fragments in a Pfizer sample was determined using Oxford Nanopore sequencing. It should be noted that the library preparation method used systematically removes fragments smaller than approximately 150 base pairs (bp). As a result, the obtained length distribution is biased toward longer fragments and does not fully represent the actual proportion of short fragments.DNA inside or outside LNPs – nuclease protection assay
To assess whether residual DNA is located inside the lipid nanoparticles (LNPs) or freely outside them, the enzyme DNase I-XT was added directly to the unmodified vaccine – that is, to intact, non-disrupted LNPs. DNase I-XT degrades accessible DNA. Subsequent qPCR measurement allowed comparison of DNA quantities before and after enzymatic treatment.Analysis of VAERS data
For each investigated batch, a Serious Adverse Event Reporting Ratio (SRR) was calculated from the VAERS database, defined as the ratio of serious adverse event reports to all reported adverse events. This value was then compared with the measured DNA concentration for the corresponding batch.c) Key Results
Quantitative findings and method comparison
The comparison of qPCR and Qubit fluorometry results shows that the measured values strongly depend on the analytical method used.Determination of DNA concentrations for Pfizer
qPCR Qubit fluorometry Ori probe: 0,28 – 4,27 ng/dose
Spike probe: 0,22 – 2,43 ng/doseTotal DNA: 1.896 – 3.720 ng/dose The SV40 promoter-enhancer-Ori was detected only in Pfizer vials, with Cq values between 16,64 and 22,59.
Determination of DNA concentrations for Moderna
qPCR Qubit fluorometry Ori probe: 0,01 – 0,34 ng/dose
Spike probe: 0,25 – 0,78 ng/doseTotal DNA: 3.270 – 5.100 ng/dose Relative to the FDA limit of 10 ng DNA per dose, the qPCR values are below the threshold, whereas the fluorometric values exceed it by 188- to 509-fold.
The authors note that qPCR methods cannot detect DNA fragments below the amplicon length (105–114 bp) and therefore systematically underestimate the total DNA amount.
Furthermore, the measurements revealed variations in DNA levels between different batches of the same vaccine – indicating possible fluctuations in the purification process.
DNA located inside LNPs
After treatment of intact Pfizer vaccine with DNase I-XT, the subsequent qPCR measurement showed little to no change in DNA quantity. Since the enzyme only degrades accessible DNA, this result suggests that the majority of the residual DNA is located inside the lipid nanoparticles (LNPs) and is therefore protected from enzymatic degradation.Size profile of residual DNA
ONT sequencing of a Pfizer sample revealed a mean fragment length of 214 bp. Individual fragments reached lengths of over 1.000 bp; the longest detected fragment was approximately 3.200 bp and thus corresponded to roughly half of the plasmid length of 7.810 bp.Since the library preparation systematically excludes fragments below 150 bp, it is assumed that the actual proportion of short fragments in the sample is higher than what is reflected in the sequencing data.
Exploratory VAERS correlation with measured DNA levels
The authors observed a positive correlation between measured DNA concentrations at the batch level and the SRR value of the respective batch. Batches with higher DNA amounts tended to show higher proportions of serious adverse event reports in the VAERS database.d) Interpretation by the authors
Method dependence of residual DNA assessments
The authors report two fundamentally different measurement outcomes for the same vaccines:Method Result: residual DNA per dose
(FDA limit: 10 ng/dose)Interpretation qPCR BELOW FDA limit Compliant Fluorometry 188- to 509-fold ABOVE FDA limit Exceedance This means that depending on the analytical method used, completely contradictory conclusions can be reached.
This discrepancy is a central finding of the study and highlights the strong methodological dependence of measured DNA quantities.
Note on methodological inconsistency
The authors argue that the observed discrepancies cannot be explained solely by measurement imprecision, but rather result from fundamentally different measurement principles.The European Medicines Agency (EMA) applies ratiometric guidelines – meaning that there is an allowed limit for the ratio of DNA to RNA (e.g., 330 ng DNA per 1 mg RNA).
However, different methods are used to measure RNA and DNA, which differ in their specificity:
What is measured Method used What is actually measured RNA
(main component)Fluorometry / UV spectrophotometry Total nucleic acids
(non-specific)DNA
(residuals)qPCR Specific DNA sequences
(selective)Various supporting sources on this:
- „Rapporteur Rolling Review critical assessment report” (Table S.2.6-16. Evolution of BNT162b2 Drug Substance Methods)
- „EU Official Control Authority Batch Release Certificate for Immunological Products” (Table 2. Drug Substance Quality Control Tests)
- „Assessment report EMA/707383/2020”
- „Assessment report EMA/15689/2021”
The authors argue that if:
- RNA is measured using a non-specific method (fluorometry: also detecting DNA fragments, degradation products, etc.), and
- DNA is measured using a specific method (qPCR: detecting only the exact target sequence),
then the two resulting measurements are not methodologically compatible.
As a result, the calculated DNA/RNA ratios are methodologically biased and not directly comparable.
This argument concerns the comparability of measurement methods, not necessarily the validity of each individual measurement.
The authors advocate that regulatory agencies should not only define threshold values but also specify the exact analytical methods that must be used for compliance testing.DNA within LNPs: biological implications
The presence of DNA inside lipid nanoparticles (LNPs) is considered by the authors to be potentially biologically relevant. Unlike free, naked DNA – which is rapidly degraded in the body – LNP-encapsulated DNA is protected from nucleases and can be efficiently taken up by cells. According to the authors, existing regulatory thresholds were developed for naked host-cell DNA and do not account for this difference.Criticism of regulatory guidelines
The authors raise fundamental criticism of current threshold concepts. They argue that existing guidelines are based on outdated assumptions, that they neglect molecule number in favor of mass-based metrics, that they do not consider cumulative dosing (multiple doses within a short period of time), and that they overlook functional sequences such as promoters or nuclear targeting signals. In particular, the presence of the SV40 promoter/enhancer in the Pfizer plasmid is highlighted as a factor that could facilitate the transport of DNA into the cell nucleus.Exploratory VAERS analysis
The observed correlation is interpreted by the authors as a possible indication of a relationship between DNA content and adverse event profiles. It is explicitly emphasized that this is an exploratory observation and does not allow for causal conclusions. Therefore, the authors call for further investigation.e) Methodological limitations
The origin, storage, and batch characterization of the investigated samples are overall well documented – cold chain handling, transport conditions, and vial integrity are described. However, the thermal lysis step without subsequent purification is not validated in a regulatory context; information on extraction efficiency is lacking.
All examined vials were already expired at the time of analysis. Three Moderna XBB.1.5 vials consisted of residual volumes remaining after patient administration and were not originally sealed. They therefore differ in sample quality from the other vials and must be assessed separately.
The VAERS correlation is subject to substantial uncertainty. VAERS is a passive reporting system: reports are not systematically verified, reporting rates vary across time periods and regions, and correlations at the batch level do not allow inference of individual causality. The interpretive value of this analysis is therefore structurally limited – independent of the quality of the analytical measurements.
Finally, the ONT sequencing approach only captures fragments above approximately 150 bp due to library preparation, and does not fully represent the true proportion of shorter DNA fragments.
f) Contextualization
The study extends the findings of Kevin McKernan in several ways: it examines additional batches, provides a nuclease protection assay as direct evidence for the intraparticulate location of DNA, and addresses structural limitations of current regulatory methodologies. The analytical component represents a factual contribution to the ongoing debate.
The exploratory component – the linkage with VAERS data – is methodologically demanding and subject to the known limitations of the reporting system. The authors themselves characterize it as hypothesis-generating rather than confirmatory.
The authors point out several limitations of their study and call for replication of their findings under forensic-grade conditions.
Overall, the study emphasizes the need for standardized and methodologically harmonized approaches for assessing residual DNA in mRNA-based medicinal products.
🧬 🧬 🧬
4.3.3. König & Kirchner (2024)
Methodological Considerations Regarding the Quantification of DNA Impurities in the COVID-19 mRNA Vaccine Comirnaty®
Authors: Brigitte König, Jürgen O. Kirchner
Published: May 8, 2024 (Peer-reviewed, PubMed)
Link to study: https://pubmed.ncbi.nlm.nih.gov/38804335/a) Context and Objective of the Study
The work by Brigitte König and Johannes O. Kirchner differs fundamentally in its objective from the previously described studies. While earlier work primarily aimed at detecting and quantifying residual DNA in mRNA vaccines, this study focuses on a methodological evaluation of the analytical methods used for detection.
The key question is: Is the qPCR-based DNA quantification used in the authorisation process suitable for batch testing and, therefore, as a quality assurance tool?
To address this question, the authors examine:
- which analytical methods are used for the quantification of residual DNA,
- what methodological limitations these methods have, and
- to what extent different methods can lead to divergent results.
The study was peer-reviewed and published in a scientific journal.
b) Materials and Methods
Vaccine batches
The laboratory received several original sealed vials from different batches of the mRNA vaccine Comirnaty® (BNT162b2), provided by official vaccination centers. Storage and transport were carried out under documented compliance with the cold chain (2–8 °C). Four batches were already expired at the time of analysis, while three still had a remaining shelf life of 11 to 13 months.The authors combine two approaches: a critical methodological analysis of existing procedures and their own laboratory measurements.
A critical analysis of methodology
The authors examine the qPCR method described in the EMA’s November 2020 rolling review document (including the sample preparation protocol set out therein) and identify three structural issues.
First, the qPCR assay detects only a 69 bp target sequence – the T7 promoter – within a 7,824 bp plasmid. The remaining 99% of the plasmid DNA, as well as any potential bacterial genomic DNA, are not captured. The authors note that this limitation is fundamentally inherent to any qPCR approach that targets only single sequences – regardless of which sequence is chosen.
Second, the standard curve used is methodologically problematic. For quantification, a calibration curve is generated using untreated, intact plasmid DNA. However, the DNA in the vaccine sample has undergone multiple processing steps during manufacturing – particularly a DNase digestion – and is therefore fragmented. Since fragmented DNA amplifies less efficiently than intact DNA, the measured concentration is systematically underestimated compared to the true amount present. A correct comparison would require a standard curve generated from DNA that has been subjected to the same treatment steps as the sample.
Third, the question of proportionality arises:
- Are there sequence-dependent differences in degradation rates?
- Is the qPCR target sequence degraded proportionally to the rest of the plasmid during DNase treatment?
- Are certain regions of linearized plasmid DNA degraded more or less efficiently than others?
If proportionality does not hold, any extrapolation from the target sequence to total DNA content is methodologically flawed. The European Pharmacopoeia (Ph. Eur. 2.6.35) confirms this limitation: while qPCR is described as a method of choice for detecting specific sequences, it explicitly notes that other or complementary methods may be required for total DNA quantification.
Considerations on regulatory practice
The authors point to an inconsistency in regulatory practice: according to an European Directorate for the Quality of Medicines & HealthCare protocol, DNA is measured only at the drug substance level, not in the final vaccine product. The rationale given is that lipid nanoparticles (LNPs) may interfere with DNA measurement. At the same time, RNA concentration in the final vaccine is measured using fluorometry after disruption of the LNPs with Triton X-100. Since both RNA and DNA are nucleic acids with comparable analytical properties and both may be encapsulated within LNPs, this difference in approach appears to require further justification. Whether this reflects technical constraints or regulatory considerations not fully disclosed in public documents cannot be conclusively assessed based on the available sources.Own measurements using Qubit fluorometry
To evaluate the practical suitability of an alternative method, the authors conducted own measurements on seven Comirnaty® batches. Samples were analyzed both before and after the addition of the detergent Triton X-100, which disrupts LNPs and releases their encapsulated contents. Qubit fluorometry with the DNA-specific dye PicoGreen was used as the measurement system.c) Key Results
Qubit measurements before and after LNP disruption
Without Triton X-100, only small amounts of DNA were detected. After addition of Triton X-100, the measured values increased dramatically – to 360- to 534-fold above the regulatory limit of 10 ng per dose, corresponding to 3,600 to 5,340 ng DNA per dose.In the four expired batches, between 16% and 81% of the total DNA was already detectable without Triton X-100, suggesting partial degradation of the lipid nanoparticles (LNPs) over storage time. In the three fresh batches, 93% to 97% of the DNA became detectable only after LNP disruption, indicating largely intact particles.
RNA measurements as control
The parallel RNA measurements served as a methodological control and showed plausible values. The authors interpret this as evidence for the general suitability of the Qubit system in this sample matrix.d) Interpretation by the authors
Critique of qPCR as the sole reference method
The authors conclude that the regulatory qPCR-based method is structurally unsuitable for reliably quantifying total DNA in the final vaccine product. According to their findings, DNA levels substantially exceed the regulatory threshold, but remain undetected because the applied method only measures a small target region of the existing DNA and is additionally performed at the drug substance level rather than on the final product.Call for methodological adaptation
The authors therefore advocate for fluorometric spectroscopic measurement of total DNA in the final vaccine formulation as part of official batch release testing – analogous to the already established RNA measurement.DNA within LNPs: biological implications
Furthermore, they call for a reassessment of the potential risks associated with LNP-encapsulated DNA, particularly with respect to possible integration into the human genome.e) Methodological limitations
Small sample size
The study is based on a limited sample of seven batches. The samples were provided as original sealed vials from official vaccination centers; storage and transport were carried out under documented cold chain conditions. The provenance documentation is therefore comparatively well substantiated.Influence of expiry date on LNP integrity
Four of the seven batches were already expired at the time of analysis. Expired samples may undergo chemical changes, particularly with regard to lipid nanoparticle (LNP) integrity. The authors explicitly acknowledge this and interpret the increased detectability of DNA without Triton X-100 in expired batches as an indication of partial LNP degradation over time.No RNase treatment prior to fluorometry (Qubit)
Qubit assays use DNA-specific dyes that can also exhibit interference with RNA (the main component of the vaccine). Without prior RNase digestion, the measured „DNA amount” is likely to be overestimated.Lack of formal validation
The Qubit method was not formally validated by the authors according to regulatory standards. Such validation – required under the ICH Q2(R2) for regulatory analytical methods – includes systematic demonstration of specificity, limit of detection, linearity, precision, accuracy, robustness, and matrix effects. The authors justify the suitability of the method through an argumentative approach: by comparing measurements before and after LNP disruption and by demonstrating plausible RNA control values. This is an acceptable framework for a scientific publication aimed at illustrating a methodological point. This remark is not intended as a criticism of the authors’ diligence, but rather as an objective assessment of the context in which the work was published.Lack of validation of extraction efficiency
Extraction methods do not always demonstrate that they completely release and quantify all DNA (particularly small fragments and encapsulated DNA). This can lead to over- or underestimates.Availability of regulatory test methods
The methodological critique relies, with respect to the regulatory qPCR method, on a rolling review document from the early authorization phase. Whether the method described there is identical to the final approved batch release assay cannot be conclusively determined based on publicly available sources.f) Contextualization
König and Kirchner make a methodologically independent contribution to the debate. Unlike the work of Kevin McKernan and David J. Speicher, the focus is not on how much DNA is present, but on whether the analytical methods used are capable of reliably answering that question in the first place.
The study thus makes an important contribution to the debate by:
- highlighting the methodological dependence of measurement outcomes
- exposing limitations of established standard methods (in particular qPCR)
- and emphasizing the need for cross-method validation
It also illustrates that differences between studies do not necessarily reflect contradictory or incorrect results, but are often explained by:
- different detection methods
- different target sequences
- and variations in sample preparation
Overall, the work underscores that DNA quantification in this context is methodologically complex and that results from different studies are only limitedly comparable in a direct way.
🧬 🧬 🧬
4.3.4. Didier Raoult (2024)
Confirmation of the presence of vaccine DNA in the Pfizer anti-COVID-19 vaccine
Authors: Didier Raoult
Published: November 12, 2024 (Preprint, HAL Open Science)
Link to study: https://hal.science/hal-04778576v1a) Context and Objective of the Study
The work by Didier Raoult is part of a series of independent investigations examining the presence of DNA in mRNA-based COVID-19 vaccines.
The objective of the study is to experimentally confirm the presence of DNA components in the Pfizer/BioNTech vaccine and to independently verify the findings reported by earlier research groups. The study is therefore primarily intended as a replication effort.
b) Materials and Methods
Vaccine batches
The study examined only the Comirnaty® (BNT162b2) mRNA vaccine manufactured by BioNTech/Pfizer. The analysis was based on a limited number of samples; the available report does not provide a systematic overview of vaccine batches.The study employed classical molecular biology techniques for nucleic acid analysis.
Disruption of lipid nanoparticles (LNPs)
The lipid nanoparticles (LNPs) were disrupted using the detergent Triton X-100.Quantification by Qubit fluorometry
For one batch (No. GJ7184), DNA quantification was performed using a commercial Qubit fluorometer, both before and after LNP disruption with Triton X-100.Illumina sequencing
Two batches (FP8191 and FP9359) were analyzed using Illumina sequencing. Reverse transcription was deliberately omitted, meaning that RNA was not converted into DNA. This ensured that only DNA originally present in the sample was sequenced, rather than DNA copies generated from vaccine mRNA.The resulting reads (sequenced fragments) were compared with the known plasmid DNA reference sequence, GenBank OR134577.1. For the bioinformatic analysis, only sequence fragments longer than 200 bp and showing more than 90% identity to the reference sequence were included.
As a control experiment, the samples were treated with DNase prior to sequencing and then sequenced again to determine whether the observed signal was indeed attributable to DNA.
c) Key Results
Quantification by Qubit fluorometry
Ohne Triton X-100 wurden durchschnittlich 216 ng DNA pro Dosis gemessen. Nach LNP-Aufschluss mit Triton X-100 stieg der Wert auf durchschnittlich 5.160 ng pro Dosis – ein Anstieg um das ca. 24-Fache. Bezogen auf den angegebenen RNA-Gehalt von 30 µg pro Dosis entspricht dies rechnerisch einem DNA-Anteil von etwa 17 %.Without Triton X-100 treatment, an average of 216 ng of DNA per dose was measured. After disruption of the lipid nanoparticles (LNPs) with Triton X-100, the value increased to an average of 5,160 ng per dose, representing approximately a 24-fold increase. Based on the stated RNA content of 30 µg per dose, this corresponds mathematically to a DNA proportion of approximately 17%.
Sequencing by Illumina
Using Illumina sequencing, more than 140,000 reads longer than 200 base pairs were generated. From these data, the complete 7.824 bp plasmid sequence could be reconstructed. This indicates that the DNA fragments present collectively covered the entire plasmid sequence.Die Sequenzierungstiefe war mit mehr als 4.000-facher Abdeckung außergewöhnlich hoch – das bedeutet, jede Position des Plasmids wurde im Durchschnitt von mehr als 4.000 unabhängigen Reads abgedeckt. Die Sequenzdaten beider Chargen wurden in der GenBank hinterlegt (PP544445 und PP544446).
The sequencing depth was exceptionally high, with more than 4.000-fold coverage, meaning that each position within the plasmid was covered on average by more than 4.000 independent reads. The sequence data from the two analyzed batches were deposited in GenBank under accession numbers PP544445 and PP544446.
Between 63% and 97% of the sequenced DNA could be assigned to the vaccine plasmid. The origin of the remaining 3% to 37% was not further investigated.
DNase control experiment
Following DNase treatment, virtually no reads were detected (only 1–2 reads remained). This near-complete loss of signal after DNase treatment suggests that the sequencing signal indeed originated from accessible DNA, either freely present or released after LNP disruption.d) Interpretation by the authors
Reproducibility
The author interprets the findings as an experimental confirmation of results reported in earlier studies. The abundant presence of plasmid DNA in the Pfizer/BioNTech vaccine could be demonstrated across multiple batches and using several independent analytical approaches. The consistency with previous reports is presented as evidence supporting the reproducibility of such findings.DNA encapsulated in cationic lipids: biological implications
With regard to potential biological consequences, the author notes that plasmid DNA encapsulated within lipid nanoparticles (LNPs) could, in theory, integrate into the genome after cellular uptake. However, the author explicitly characterizes the actual risk as extremely low. Nevertheless, he argues that further investigation of this question would be scientifically justified.e) Methodological limitations
Origin and storage of the samples
Information regarding the origin, storage history, and condition of the analyzed vials is largely absent. It is not documented whether the samples were originally sealed, properly refrigerated, and unopened prior to analysis.Missing methodological details
Several important methodological details are not specified:- No information is provided regarding RNase treatment to remove RNA before DNA quantification.
- The specific Qubit kit used (High Sensitivity or Broad Range) is not reported.
- No information is given on the extraction efficiency for LNP-encapsulated DNA.
- The DNase treatment is described only as using a „TURBO DNA-free kit”; the specific enzyme type is not identified.
Small sample size
The study is based on a limited sample of three vaccine batches.Sequencing and quantification
Illumina sequencing provides strong evidence for the presence and completeness of the plasmid sequence, but it is not an appropriate method for absolute quantification. High sequencing depth can arise even when relatively few DNA molecules are present at high copy number; therefore, sequencing coverage does not necessarily reflect the actual mass or concentration of DNA in the sample.f) Contextualization
The study supplements the existing body of evidence by providing further independent proof of plasmid DNA in mRNA vaccines. Its main contribution lies in the reproducibility of the qualitative findings: another laboratory, using different methods and different batches, has arrived at comparable results.
🧬 🧬 🧬
4.3.5. Kämmerer et al. (2024)
BioNTech RNA-Based COVID-19 Injections Contain Large Amounts Of Residual DNA Including An SV40 Promoter/Enhancer Sequence
Authors: Ulrike Kämmerer, Verena Schulz, Klaus Steger
Published: December 3, 2024 (Peer Reviewed, Cureus/Public Health Policy Journal)
Link to study: https://publichealthpolicyjournal.com/biontech-rna-based-covid-19-injections-contain-large-amounts-of-residual-dna-including-an-sv40-promoter-enhancer-sequence/a) Context and Objective of the Study
The study investigates the presence of DNA components in mRNA-based COVID-19 vaccines, with a particular focus on regulatory sequence elements. Against the background of previous analyses, the study addresses three central questions:
- Can the high amount of residual DNA in BioNTech batches be confirmed using different analytical methods?
- Can existing DNA fragments be co-delivered into human cells together with the mRNA (transfection)?
- Does this lead to sustained spike protein production?
b) Materials and Methods
Vaccine batches
For the analyses, four original, unopened BNT162b2 batches were used:- FD7958, FE6975, and EX8679 (monovalent, Wuhan strain; expiration date October/August 2021)
- HD9869 (bivalent, Wuhan/Omicron XBB1.5; expiration date October 2024)
Batch GH9715, for which DNA contamination had already been demonstrated in previous studies, served as a positive control.
All vials were obtained from a pharmacy under refrigerated conditions, were originally sealed, and were continuously kept cold during transport and storage. The provenance documentation is therefore comparatively well substantiated.
Quantification
The lipid nanoparticles (LNPs) were disrupted using Triton X-100.
RNA quantification was performed using the Qubit RNA High Sensitivity assay.
For DNA quantification, three different fluorometric dsDNA assays were used:- Qubit dsDNA High Sensitivity assay
- Quanti-iT PicoGreen dsDNA assay
- AccuBlue High Sensitivity dsDNA assay
Fluorescence-based measurements were performed using a plate reader both before and after RNase A treatment (an RNA-degrading enzyme).
The use of three independent assays is methodologically considered an orthogonal approach: agreement across all three increases the reliability of the results substantially.
Cell line experiments and ELISA
To investigate whether the vaccines transfect human cells and lead to spike protein production, HEK293 cells were used – an immortalized human cell line with high transfection efficiency.The cells were cultured under standardized conditions. They were obtained from certified original stocks and were regularly tested negative for mycoplasma contamination.
For the transfection experiments, the cells were seeded in 12-well plates. The cell density was selected such that, after a short growth period, approximately 80% of the bottom surface area was covered. Each well was transfected with 1/12 of a clinical dose (corresponding to 25 µl) of the respective vaccine. An immediate medium change or washing step after transfection is not documented.
Cells and culture medium were harvested at day 1, day 3, day 5, and day 7 post-transfection. Untransfected cells at day 7 served as a negative control.
For protein analysis, the cells were washed to remove residual culture medium. Cells were then lysed using a lysis buffer, releasing intracellular contents. The amount of SARS-CoV-2 spike protein in both the cell lysate supernatant and the culture medium was quantified using a highly sensitive commercial ELISA kit.
Immunohistochemistry
Immunohistochemistry was performed to visualise the spike protein within the cells. To this end, HEK293 cells were seeded in 24-well plates (60% cell density) and transfected with 12.5 µl of a clinical dose per well (200 µl medium). After a 4-hour incubation, the cells were washed twice with PBS and supplemented with 500 µl of fresh medium.Twenty-four hours after the start of transfection, cells were harvested, fixed, embedded in 2% agarose, and processed into paraffin blocks after dehydration. Thin sections were cut from these blocks.
The cell sections were treated with a specific primary antibody against the SARS-CoV-2 spike protein (S1 subunit), which binds only to the spike protein. A secondary antibody, which binds to the first and triggers a colour reaction, was added. The spike protein appeared green under the microscope, whilst the cell nucleus was counterstained blue with haematoxylin for better visualisation.
PCR and agarose gel electrophoresis
Conventional PCR was used to test whether vaccine-derived DNA enters human cells. For each target region of the plasmid, a separate primer pair was used – for spike sequences, the ORI replication origin, the neomycin resistance cassette, and the SV40 promoter/enhancer. Each PCR reaction contains exactly one primer pair, thereby generating a defined amplicon.The PCR products were separated by agarose gel electrophoresis and their sizes determined by comparison with a DNA marker (GeneRuler). HEK293 cells were treated with half a vaccine dose (150 µl); after six hours, DNA was isolated and analyzed. Untreated cells served as a negative control.
Extracellular vesicles and mass spectrometry
Background: Cells constantly release extracellular vesicles (small membrane-bound particles) which, like tiny parcels, contain signalling molecules. These vesicles can travel to other cells.To investigate whether transfected cells release vaccine components via extracellular vesicles (EVs), EVs were isolated from the cell culture medium and analyzed using mass spectrometry (TimsTOF-HT). Protein identification was performed by comparison with a database containing human proteins as well as vaccine vector and spike sequences.
c) Key Results
Transfection and spike protein production
All four investigated batches led, under the applied cell culture conditions, to efficient uptake of mRNA into HEK293 cells and detectable spike protein production.The proportion of spike-positive cells ranged from 74.6% to 90.5%; untransfected cells showed no signal. Transfected cells additionally exhibited morphological changes – such as intracellular vacuolization and detachment from the growth surface – interpreted as signs of a cytopathic effect.
Spike protein production was detectable as early as day 1, increased until day 5, and remained detectable up to day 7. The authors interpret this as an indication of prolonged expression beyond the expected timeframe.
Spike protein release into cell culture medium
Spike protein was detectable in the cell culture medium for the monovalent batches; the bivalent batch was below the detection limit.Spike-Protein-Freisetzung über extrazelluläre Vesikel
Mass spectrometry showed that spike protein was primarily detectable in extracellular vesicles (14 spectral counts), while only minimal amounts were present in the soluble medium (2 spectral counts). The highest intracellular concentration was observed in the cell lysate (60 spectral counts).RNA content
The measured RNA values corresponded to the manufacturer’s specification of 30 µg per clinical dose.DNA content
Without LNP disruption, DNA values were 1.6- to 6.7-fold lower than after Triton X-100 treatment –further evidence that DNA is predominantly located inside the lipid nanoparticles.Without RNase treatment, measured DNA values ranged from 1.326 to 4.225 ng per dose. After RNase A treatment, values decreased to 32,71 to 43,38 ng per dose. The authors attribute the elevated initial readings to background signal caused by structured RNA regions: dsDNA-specific dyes preferentially bind double-stranded DNA but can generate increased signals in the presence of large amounts of RNA.
After correction, the measured DNA amounts exceeded the levels specified in regulatory guidelines. The results were reproducible across multiple measurements and showed good agreement between the three assays used.
PCR detection of plasmid elements
Strong PCR signals were detected in all four batches for all tested plasmid elements – spike sequences, ORI, neomycin resistance cassette, and the SV40 promoter/enhancer. The same elements were also detectable in HEK293 cells after transfection. For the SV40 promoter/enhancer, the primers produced two amplicons of different sizes (93 bp and 165 bp), a known methodological outcome attributable to the structure of the target sequence.d) Interpretation by the authors
The authors conclude that the investigated batches contain residual DNA that exceeds regulatory threshold values. The DNA is predominantly LNP-encapsulated, is taken up by cells, and remains detectable within them. The sustained spike protein production beyond seven days raises questions about the mechanism underlying this prolonged expression.
The authors place particular emphasis on the presence of the SV40 promoter/enhancer. This element is not required for mRNA production in E. coli. They discuss that such regulatory elements can influence transcriptional activity in certain experimental contexts. A potential role in nuclear localization or genomic integration is raised as a theoretical consideration.
The authors criticize existing regulatory limits as not being designed for LNP-based RNA injection products and emphasize that there is currently no scientific evidence sufficient to define a universally safe DNA threshold for this product class.
They also report that spike protein is released via extracellular vesicles (exosomes). The extent to which such vesicles contribute to protein distribution in the human body remains an active area of research and is not yet fully understood.
Finally, the authors highlight a fundamental issue: the choice of analytical method significantly influences the results, and this must be carefully considered when comparing studies.
e) Methodological limitations
Cell line
HEK293 cells are immortalized and exhibit altered properties compared to primary cells (e.g., muscle, immune, or neuronal cells), particularly higher transfection efficiency and modified regulatory mechanisms.Dosage
The applied dose is not physiologically representative. The quantities used (e.g., 1/12 of a clinical dose per well) result in direct and locally high cellular exposure that does not reflect distribution conditions in the human body.In vitro context
All experiments were conducted under cell culture conditions. Direct extrapolation to in vivo conditions – such as distribution, degradation, or immune response in the human body – is therefore not straightforward.Assay specificity
Fluorometric methods are sensitive to interference signals, which may affect DNA quantification.Mean value interpretation
The measured residual DNA ranged from 32,71 to 43,38 ng per clinical dose. These values are based on fluorometric measurements using three different assays (PicoGreen, Qubit, AccuBlue). Since the individual assays produced partly divergent results, the reported range represents the arithmetic mean across methodologically different measurement principles. The spread of individual measurements is substantial, meaning the mean value should primarily be interpreted as an orienting estimate.f) Contextualization
The study goes beyond the mere quantification of residual DNA: it links analytical detection with cell-based experiments and examines potential biological consequences. It combines multiple analytical and functional methods (fluorometric assays, PCR, ELISA, and mass spectrometry), thereby pursuing a comparatively broad investigative approach.
The study further highlights that even within the same methodological category, substantial differences can occur between individual fluorometric assays. This finding underscores the importance of methodological standardization for future investigations.
🧬 🧬 🧬
4.3.6. Wang et al. (2024)
A rapid detection method of replication-competent plasmid DNA from COVID-19 mRNA vaccines for quality control.
Authors: Tayler J. Wang, Alex Kim, Kevin Kim
Published: December 29, 2024 (Journal of High School Science, peer-reviewed)
Link to study: https://jhss.scholasticahq.com/article/127890-a-rapid-detection-method-of-replication-competent-plasmid-dna-from-covid-19-mrna-vaccines-for-quality-controla) Context and Objective of the Study
The study by Wang et al. addresses a different research question than the studies presented so far. The focus is not on the total amount of residual DNA but on a more specific question: Does the detected DNA contain functional, replicable plasmid sequences – that is, fragments that could be introduced into bacteria and replicated there??
To answer this question, the authors developed a simple and rapid detection method. Their underlying rationale was that if the DNA template used during vaccine manufacturing had not been completely removed, intact or nearly intact plasmids might still remain in the final product.
The work was conducted as part of an FDA student volunteer program and carried out on the FDA White Oak Campus with support from scientists at the U.S. Food and Drug Administration (FDA). The publication venue, the Journal of High School Science, reflects the educational context of the project rather than the institutional setting in which the work was performed.
b) Materials and Methods
Vaccine batches
The study uses several fundamentally different sample sources, which must be clearly distinguished when interpreting the results.First, the authors investigated a self-produced laboratory vaccine containing the XBB.1.5 spike sequence, manufactured using a commercial lipid nanoparticle (LNP) kit. Second, they examined biosimilar reference materials obtained from BEI Resources. These NIH-supported reference materials were produced for research purposes and are not identical to licensed commercial vaccine batches. Third, they analyzed two commercial Pfizer vaccine batches – Lot PAA194854 (monovalent) and Lot PAA184098 (bivalent) – which were original sealed, licensed vaccine products.
Vaccine / Material Source Description In-house mRNA vaccine Self-produced Contains XBB.1.5 spike sequence,
formulated in LNPsModerna Biosimilar BEI Resources Not the original commercial product Pfizer Biosimilar BEI Resources Not the original commercial product COMIRNATY Original
(NR-59604)BEI Resources Monovalent (Wuhan strain) COMIRNATY Bivalent
(NR-59605)BEI Resources Bivalent (Wuhan + Omicron BA.5) DNA-Extraktion DNA extraction
DNA was extracted using a commercial plasmid miniprep kit (Monarch Plasmid DNA Miniprep, New England Biolabs). The kit contains RNase A in the neutralization buffer, resulting in substantial degradation of RNA during the extraction process. DNA was eluted in 30 µl of nuclease-free water.Quantification
DNA concentration was determined using two methods: NanoDrop spectrophotometry and the Qubit dsDNA High Sensitivity (HS) assay. The authors explicitly note that NanoDrop does not distinguish between DNA and RNA and may be influenced by free nucleotides, salts, and organic compounds. Qubit is more specific for double-stranded DNA, but can also overestimate DNA concentrations in the presence of large amounts of RNA or when samples are inadequately prepared.Transformation assay – the proposed detection method
The first step involves extraction of DNA from the vaccine sample. The extracted material consists of various DNA fragments, potentially including the origin of replication (ORI), which serves as the starting point for plasmid replication, and an antibiotic resistance gene, which enables selection of transformed bacteria.The extracted DNA is then treated with T4 DNA ligase. This enzyme joins DNA fragment ends together. As a result, linear DNA fragments can be re-circularized into plasmids. Because the ligase can join compatible DNA ends regardless of their original arrangement, fragments that were separate in the original vaccine preparation may become artificially linked, creating recombinant artifacts.
These newly formed plasmids are subsequently introduced into specially prepared E. coli bacteria through a transformation procedure.
The underlying reasoning:
- If DNase digestion during vaccine manufacturing was efficient, the remaining DNA fragments should be so small that they no longer contain a complete origin of replication (ORI) or a complete antibiotic resistance gene. In that case, even after ligation, no functional plasmid can be formed.
- If DNase digestion was incomplete, larger DNA fragments may still be present that contain intact ORI and antibiotic resistance sequences. After ligation, these fragments may be able to form a functional plasmid. Importantly, the ligase can only join DNA ends that already exist; it cannot reconstruct missing portions of a gene from numerous small fragments.
Only bacteria that acquire a plasmid containing both a functional antibiotic resistance gene and a functional ORI can survive on antibiotic-containing growth media and form visible colonies. The appearance of bacterial colonies therefore serves as a direct biological indication that replication-competent plasmid DNA was present in the original vaccine sample.
Size analysis
Fragment sizes were determined using agarose gel electrophoresis and an Agilent 2100 Bioanalyzer.c) Key Results
Quantitative findings
DNA amounts per dose measured by Qubit ranged from 40 to 110 ng, thus above the WHO guideline threshold of 10 ng. NanoDrop yielded substantially higher values (several thousand ng), which the authors attribute to the well-known lack of specificity of this method.Laboratory vaccine and biosimilar materials
In the self-produced laboratory vaccine and the Pfizer biosimilar, replication-competent DNA fragments were detected: the transformation assay produced bacterial colonies. In contrast, no colonies were observed in the Moderna biosimilar and the in-house standard, despite higher measured DNA concentrations. This indicates that colony formation is not determined solely by total DNA quantity, but depends on the integrity of functional sequence elements.Commercial Pfizer batches
In the two commercial Pfizer batches, no replication-competent DNA fragments were detected; the transformation assay did not yield colonies. Fragment size analysis showed that nearly all detected DNA was shorter than 35 bp. According to the authors, this is consistent with a fully effective DNase digestion during manufacturing.d) Interpretation by the authors
The authors interpret the absence of replication-competent DNA in the commercial batches as evidence of the effectiveness of the industrial manufacturing process. The complete fragmentation of plasmid DNA to below 35 bp makes any biological activity of the residual DNA unlikely, according to the authors – including the SV40 promoter/enhancer sequence, whose functionality is considered practically impossible at such fragment sizes.
At the same time, the authors confirm that total DNA levels exceed the WHO guideline threshold and recommend stricter and more transparent monitoring of residual DNA. They frame this recommendation as a measure also intended to strengthen public confidence in mRNA vaccine technologies.
e) Methodological limitations
Sample size and selection
The study includes only two commercial batches, which does not allow for robust conclusions regarding batch-to-batch variability. The biosimilar materials used are not identical to licensed commercial products; therefore, findings from these samples cannot be directly generalized to marketed vaccine products.DNA extraction
Extraction was performed using a plasmid miniprep kit that is primarily optimized for the isolation of intact plasmids. It is not yet fully understood to what extent highly fragmented DNA fragments – particularly very short fragments – are fully captured using this method.Detection limits of the transformation assay
The method only detects DNA fragments that, after ligation, contain a functional origin of replication (ORI) and an antibiotic resistance gene. It assesses whether sufficiently large fragments (>800 bp for the resistance gene, >600 bp for the ORI) are present that could form a functional plasmid after ligation. Fragments lacking these elements – even if they contain other relevant sequences such as the SV40 enhancer (approximately 72–165 bp) – are not detected.
In addition, ligation can artificially generate plasmids that were not present in the original vaccine sample.Limits of functional interpretability
The transformation assay exclusively evaluates whether DNA fragments can form a replication-competent plasmid after uptake into bacteria. Other potential biological properties of DNA fragments are not assessed. In particular, the method does not allow conclusions about whether smaller or non-replicating fragments may have functional effects in other biological contexts. The study therefore specifically assesses bacterial replicative capability of certain plasmid components, not the broader biological relevance of all detectable DNA fragments.f) Contextualization
The study by Wang et al. investigates whether biologically active, replication-competent plasmid DNA can still be detected in vaccine samples.
It thereby adds an additional layer to the ongoing discussion. While PCR, fluorometry, and sequencing methods primarily provide information on quantity, sequence identity, and fragment length, the transformation assay used here specifically assesses the functional integrity of DNA in a bacterial system.
At the same time, the scope of the method is limited to the experimentally tested function: the assay only detects DNA that is transformable and replicable in bacteria. Other biological properties or potential mechanisms of action of DNA fragments are not examined.
Overall, the study provides a methodologically interesting contribution by focusing on the functional relevance of residual DNA, thereby complementing purely quantitative analyses.
The work thus highlights that the assessment of residual DNA should not rely solely on quantity but also consider its structural and functional properties.
🧬 🧬 🧬
4.3.7. Fleming et al. (2025)
Quantitative Analysis of Nucleic Acid Content in Spikevax (Moderna) and BNT162b2 (Pfizer) COVID-19 Vaccine Lots
Authors: Richard M Fleming PhD, MD, JD; Peter Kotlár MD; Sona Pekova MD, PhD
Published: May 13, 2025 (Herald Open Access)
Link to study: https://www.heraldopenaccess.us/openaccess/quantitative-analysis-of-nucleic-acid-content-in-spikevax-moderna-and-bnt162b2-pfizer-covid-19-vaccine-lotsa) Context and Objective of the Study
The study was conducted in the context of an official request from authorities in Slovakia. The aim of the investigation was to characterize the nucleic acid composition of mRNA COVID-19 vaccines with regard to:
- identification and quantification of contained nucleic acids
- assessment of batch homogeneity (between and within lots)
- detection of undeclared genetic material
- evaluation of the stability of expired batches
The results were intended to provide insights into vaccine composition, stability, and manufacturing practices.
b) Materials and Methods
Vaccine batches
Molecular analyses were performed on 24 batches – 17 Moderna and 7 Pfizer:Spikevax (Moderna): MV1013A, 200023A, 200156A, 223049, 200090A, 200106A, 200100A, 3005885, 3005836, 000090A, 000058A, 3005241, 3005697, 3006272, MV1018A, 400012A, 400011A and
BNT162b2 (Pfizer): FP9632, 1F1051A, 1LO84A, 1F1047A, 1F1059A, 1F1055A, PCB0020.
For each batch, five original, unopened vials were analyzed. Samples were stored at −80 °C and transported under documented temperature-controlled conditions. The provenance documentation is therefore comparatively well substantiated.
Disruption of lipid nanoparticles (LNPs)
Lipid nanoparticles were disrupted using a combination of:
▫️chaotropic salts (QIAamp DNA Mini Kit, Qiagen)
▫️heat treatment (60 °C)
▫️enzymatic digestion (proteinase K)
This released both the mRNA and any residual DNA.Reverse transcription and multiplex qPCR
The mRNA was converted into cDNA by reverse transcription. Quantification was then performed using multiplex real-time PCR (Rotor-Gene Q, Qiagen) with four simultaneous fluorescence channels:- FAM (green): for S-protein (cDNA)
- HEX (yellow): for vector origin (Ori) DNA
- Cy5 (red): for E. coli DNA
- ROX: as a passive reference signal
All three target sequences were thus measured simultaneously in a single reaction, although potential interactions between targets (e.g., competition for reagents) may influence quantification.
Quantification by qPCR
For the multiplex quantitative real-time PCR, the authors used primers targeting three different sequences:Target Source mRNA for S-protein
(Spikevax & BNT162b2)self-designed Expression vector DNA (Ori) adopted from previous studies E. coli genomic DNA (ITS) from a commercial kit (ISO 13485-validated) All primers and fluorescence-labeled hybridization probes were custom-synthesized by Eurofins Genomics (Germany).
Calibration strategy
Since the mRNA in the vaccines contains N1-methylpseudouridine modifications and the exact extent of these modifications is not known, it was not possible to generate synthetic standards for a conventional target-specific calibration curve. Instead, the authors validated PCR efficiency using serial dilution series of the vaccine samples themselves. For absolute quantification, they applied a universal calibration curve derived from averaged dilution series of 50 different microorganisms.Verification by Sanger sequencing
All PCR products from the 24 batches were verified by Sanger sequencing and compared against database reference entries.c) Key Results
mRNA detection and batch variability
PCR analysis confirmed the presence of the declared spike protein mRNA sequences in both vaccines.
Analysis of S-protein mRNA showed substantial variability between different batches for both products.
Absolute mRNA levels ranged from 10⁹ to 10¹⁰ copies/ml (measured as cDNA). However, some batches exhibited approximately 10-fold lower mRNA levels in both vaccine types.Detection of undeclared DNA sequences as an incidental finding
During validation of the cDNA dilution series, unexpectedly strong signals appeared in the DNA channels: the Cy5 channel (targeting Ori DNA) and, in some cases, the HEX channel (targeting E. coli DNA). Since the samples contained both cDNA and residual DNA, the Ori and E. coli primers also amplified these unintended templates. The resulting PCR products were purified and identified by Sanger sequencing, showing 100% sequence identity with the expression vector (plasmid Ori and spike cassette) as well as the E. coli ITS region.Quantitative DNA findings
A central finding of the study is the consistently high DNA content across all tested batches of both Moderna and Pfizer vaccines.Vaccine mRNA (cDNA) (copies/ml) DNA amount (copies/ml) Spikevax (Moderna) 109 – 1010 107 – 109 BNT162b2 (Pfizer) 1010 – 1011 108 – 109 Indication of near-complete DNA constructs
The authors note that the Ori primer (5′-end of the construct) and the Spike primer (3′-end, bases 3184–3417) both yield a signal simultaneously. Since the two primer binding sites are several thousand base pairs apart, this suggests that at least longer DNA fragments are present that could encompass both target regions.Batch heterogeneity in Moderna products
In three Moderna batches, the ratio of spike DNA to vector DNA is inverted and shifted by an order of magnitude in a manner that does not align with a single plasmid construct. The authors speculate that these batches may contain a second DNA construct – potentially an Omicron variant inserted into the same vector backbone.E. coli DNA
In two Pfizer batches, E. coli genomic DNA was detected at levels of approximately a few copies per milliliter, just above the assay’s detection limit.SV40
No evidence of SV40 sequences was detected.d) Interpretation by the authors
Presence of DNA
The authors argue that the detected DNA quantities – comparable in magnitude to the declared mRNA levels (on the order of 10⁷ to 10⁹ copies/ml) – are too high to be explained as random contamination. They therefore interpret the DNA as a regular, albeit undeclared, component of the vaccine formulations.Batch variability and mRNA identity
Batch-to-batch variability in mRNA content is assessed as a potential issue for dosing consistency. The use of the outdated Wuhan spike sequence is described as a limitation with respect to protective efficacy against currently circulating variants.Based on their findings, the authors recommend:
- improved purification protocols
- stricter batch testing
- genomic updates of vaccine constructs
- enhanced regulatory oversight
e) Methodological limitations
Universal calibration curve
Absolute quantification was not based on a target-specific standard curve, but on a universal curve derived from the average of 50 different microorganisms. This approach assumes comparable PCR efficiency across all target sequences. However, this assumption is not necessarily valid, particularly when dealing with different target types (mRNA, plasmid DNA, bacterial DNA) and complex sample matrices. As a result, absolute copy numbers should be interpreted with caution, whereas qualitative findings – i.e., the presence of detected sequences – are less affected.No measurement prior to LNP disruption
In contrast to other studies, no measurements were performed before lipid nanoparticle (LNP) disruption. Therefore, a direct comparison that could have provided information on the fraction of LNP-encapsulated DNA is missing.Non-informative SV40 conclusion
The authors report no detection of SV40. However, the primer set used did not include SV40-specific sequences. Consequently, while the statement is technically correct, it is not informative: neither detection nor exclusion of SV40 could be meaningfully assessed with the employed methodology.f) Contextualization
The study by Fleming et al. expands the existing literature primarily through its comparative approach across a relatively large number of batches.
Methodologically, it combines multiplex qPCR with Sanger sequencing to verify the amplified products. The detection of DNA sequences, including plasmid and bacterial DNA, is consistent with previous studies.
The work places particular emphasis on differences between batches as well as quantitative comparisons of measured nucleic acid fractions. The observed variability between individual lots thus adds to the ongoing discussion regarding potential differences in composition, fragmentation, and analytical detectability of residual DNA.
🧬 🧬 🧬
4.3.8. Speicher et al. (2025)
Quantification of residual plasmid DNA and SV40 promoter-enhancer sequences in Pfizer/BioNTech and Moderna modRNA COVID-19 vaccines from Ontario, Canada
Authors: David Speicher, Jessica Rose, Kevin McKernan
Published: September 25, 2025 (PubMed/Herald Open Access)
Link to study: https://pubmed.ncbi.nlm.nih.gov/40913499/a) Context and Objective of the Study
The study by Speicher, Rose, and McKernan is a methodologically extended follow-up to an earlier investigation by the same research group (Study 2, 2023). It builds on findings from McKernan et al. and Kämmerer et al., and aims to quantify residual DNA in mRNA vaccines using two independent methods – qPCR and Qubit fluorometry with RNase correction – across a larger sample set.
In addition, fragment lengths are assessed using Oxford Nanopore sequencing, and an exploratory correlation with VAERS adverse event data is performed.
b) Materials and Methods
Vaccine batches
A total of 32 vials from 16 batches of mRNA-based COVID-19 vaccines were analyzed: BNT162b2 (Pfizer/BioNTech; 10 vials from 6 batches) and mRNA-1273 (Moderna; 22 vials from 10 batches). The samples were obtained from various pharmacies in Ontario, Canada.All vials were original sealed and featured intact flip-off caps with batch numbers and expiry dates. Storage and transport were carried out under cold-chain conditions (2–8 °C). The samples were stored in specialized cooling units and transported in insulated containers with ice packs, and were placed in the laboratory refrigerator within 5 hours of receipt.
A subset of samples consisted of residual volumes from recently used vials (within 30 minutes of clinical use), which were also transported under refrigeration and stored in the laboratory within 12 hours.
One Moderna sample had no printed expiration date but included a QR code enabling identification by pharmacy personnel.
Disruption of lipid nanoparticles (LNPs)
To disrupt lipid nanoparticles (LNPs), samples were heated for 8 minutes at 95 °C and subsequently cooled to 4 °C for 5 minutes. This thermal step serves to release the contained nucleic acids.DNA quantification by qPCR
DNA concentrations were determined using qPCR based on Cq values. The primers largely correspond to previous assays developed by the research group: spike probe, ORI probe, and an SV40 enhancer assay for Pfizer batches.Quantification was performed using a calibration curve based on synthetic DNA and 10-fold dilution series (QuantStudio 3). From the measured copy numbers of the target sequences, total DNA per dose was extrapolated, taking into account the full plasmid length (Pfizer: 7,824 bp; Moderna: 6,777 bp).
For each measurement, 1 µL of treated vaccine sample was directly added to the qPCR reaction without prior DNA extraction. Potential PCR inhibition by vaccine components was assessed using dilution series of selected samples.
DNA quantification using Qubit fluorometry
Samples were measured multiple times using the DNA-specific dye AccuGreen: without pretreatment, after LNP disruption by heating, and after RNase A treatment. The latter corrects for RNA-derived signal contributions (fluorescence dye crosstalk, as the dye can also bind RNA). The authors tracked fluorescence decay over a 10-minute time course, allowing them to estimate the RNA-related signal fraction and calculate the corrected DNA concentration.Size profiling of residual DNA via ONT sequencing
To determine DNA fragment size distribution, a previously sequenced Pfizer batch (FL8095) was used as a reference standard. Sequencing was performed using an Oxford Nanopore Flongle system with the Ligation Sequencing Kit (SQK-LSK114). The resulting reads were aligned to a reference plasmid sequence.The library preparation method (SPRI-based cleanup) may result in loss of short fragments (<100–200 bp), which can bias the observed length distribution toward longer DNA fragments.
Assessment of nuclease susceptibility
To assess whether DNA is located inside lipid nanoparticles (LNPs), DNase I-XT was added directly to the untreated vaccine samples. After a 30-minute incubation, the enzyme was inactivated, DNA was purified, and quantified by qPCR. A naked DNA control served as a reference.Exploratory VAERS correlation with measured DNA levels
For each batch, VAERS reports from Canada were collected, and the number of serious adverse events (SAEs) was correlated with the measured DNA levels (Spike, ORI, SV40). The aim was to examine a possible relationship between DNA content and reported adverse events. The association was calculated using Pearson’s correlation coefficient.c) Key Results
qPCR validation
The qPCR assays for Spike and ORI showed excellent performance with R² values of 0.998–0.999 and efficiencies of 94.7–99.8%. The SV40 assay showed slightly lower performance (R² = 0.906, efficiency 93.6%). Negative controls showed no signal.LNP inhibition
Undiluted samples exhibited PCR inhibition due to lipid nanoparticles. After 1:10 dilution, inhibition was resolved; therefore, diluted samples were used for quantification.qPCR-based quantification
All Pfizer batches tested positive for Spike, ORI, and SV40. The similar Cq values across all three targets suggest the presence of shared, intact DNA molecules. In Moderna samples, Spike and ORI were detected, but SV40 was absent. ORI signals in Moderna appeared approximately 3 cycles later than Spike, indicating either a lower abundance or fragmentation of the ORI region.The authors converted Cq values into absolute DNA amounts using the standard curve:
Vaccine Spike (ng/dose) Ori (ng/dose) SV40 (ng/dose) Pfizer 0,22 – 2,43 0,28 – 7,28 0,25 – 23,72 Moderna 0,25 – 0,78 0,01 – 0,34 not detected Pfizer showed higher residual DNA levels than Moderna overall.
Two Pfizer batches (FM7380 and FN7934, monovalent, PBS formulation) exceeded the FDA guideline threshold of 10 ng/dose based on SV40 content, with values up to 23.72 ng/dose. All other batches remained below this level.
Quantification using Qubit fluorometry
The authors measured samples repeatedly at different time points. The average variability (mean deviation from the mean) was:Measurement phase Pfizer Range Before heating 317 ± 278 ng/dose 43 to 727 ng/dose After heating 1.145 ± 533 ng/dose 519 to 1.949 ng/dose After RNase A 297 ± 217 ng/dose 100 to 765 ng/dose Measurement phase Moderna Range Before heating 456 ± 121 ng/dose 199 to 675 ng/dose After heating 1.554 ± 1.023 ng/dose 176 to 3.169 ng/dose After RNase A 1.065 ± 624 ng/dose 113 to 2.365 ng/dose After LNP disruption by heating, values increased markedly. Subsequent RNase A treatment reduced the measured signal (crosstalk correction).
Overall, Moderna showed consistently higher DNA levels than Pfizer.
A comparison between Qubit and qPCR showed a positive correlation for Pfizer, but not for Moderna. The authors interpret this discrepancy as evidence of DNA fragments lacking intact primer binding sites, which would therefore not be detected by qPCR.
Fragment length analysis
The longest detected read was approximately 3.5 kb and covered most of the plasmid backbone. This indicates that, in addition to short fragments, larger DNA fragments in the kilobase range are also present in the samples.DNA located inside LNPs
DNase treatment did not lead to a significant reduction in DNA signal in the vaccine samples (≤1 Cq shift), whereas naked control DNA was fully degraded. This is interpreted as evidence that the DNA is predominantly encapsulated within lipid nanoparticles (LNPs) and therefore protected from enzymatic degradation.Exploratory VAERS correlation with measured DNA levels
For the analyzed batches, adverse event reports from the US VAERS spontaneous reporting system were used. For all but three Moderna batches, VAERS reports were available.The authors conducted an exploratory correlation between measured DNA levels and VAERS data. They report that certain batches with higher DNA levels (particularly ORI DNA) also showed higher numbers of reported serious adverse events (SAEs).
d) Interpretation by the authors
The authors conclude that all analyzed batches contain residual DNA that is encapsulated within LNPs and therefore protected from degradation. The measured total amounts exceed existing regulatory guideline values. The SV40 promoter/enhancer element in Pfizer batches is highlighted as particularly relevant, as it may facilitate nuclear targeting and could theoretically increase the likelihood of genomic integration.
The authors also point to several methodological limitations:
Method Issue qPCR Cannot quantify molecules smaller than the amplicon length (105–114 bp), leading to potential underestimation of total DNA Qubit/fluorometry without RNase Overestimation due to RNA crosstalk (hence RNase treatment is required) Ethanol precipitation Loss of short fragments (<100 bp) ONT (Nanopore sequencing) Library preparation biases against fragments <150 bp, skewing results toward longer fragments The authors therefore conclude that the choice of method has a significant impact on the results – which is why methodological clarity is essential when setting threshold values.
The authors formulate several recommendations:
Recommendation Rationale Reassessment of DNA threshold values Current regulatory limits were developed for naked DNA in conventional vaccines. They should be reevaluated in light of LNP encapsulation, cumulative dosing, and functional genetic elements. Method standardization Since qPCR may underestimate total DNA, fluorometry with RNase correction should additionally be used to more accurately quantify residual DNA and the RNA/DNA ratio. Independent replication Results should be confirmed by other laboratories. Long-term monitoring Long-term effects (e.g., integration, cancer) are not recorded by VAERS. Consideration of functional sequences Not only DNA quantity is relevant, but also functional properties – particularly the presence of SV40 elements and promoter sequences. The authors interpret variability of residual DNA between and within batches as a potential indicator of inconsistencies in the manufacturing process.
e) Methodological limitations
The qPCR assay cannot detect fragments below the amplicon length (105–152 bp), leading to a systematic underestimation of total DNA content.
The ONT sequencing data are derived from a single batch and are biased toward longer fragments due to the purification method, which preferentially retains larger DNA molecules.
The exploratory VAERS correlation with measured DNA levels is only limitedly interpretable due to well-known limitations of spontaneous reporting systems – particularly the absence of exposure data and potential reporting biases. The authors additionally note further constraints: limited batch size, lack of precise dose administration data, and potential under-detection of long-term effects.
The sample set includes both expired vials and partially used residual volumes, which further reduces comparability within the dataset.
f) Contextualization
The study represents a methodologically expanded continuation of earlier work by the same research group, combining multiple analytical approaches – including qPCR, fluorometric quantification with RNase correction, nanopore sequencing, and functional assays of nuclease susceptibility. A key contribution is the direct comparison of different quantification methods, which highlights how strongly measured DNA levels depend on the analytical technique used.
The qualitative findings – particularly the detection of plasmid DNA in both vaccine types and SV40-related sequences in Pfizer batches – are consistent with other studies (e.g., McKernan et al., Kämmerer et al.). The observation that DNA appears to be encapsulated within lipid nanoparticles is also supported by multiple independent reports.
At the same time, substantial discrepancies are observed in quantitative DNA estimates between methods (qPCR vs. fluorometry), underscoring the importance of methodological standardization and limiting direct comparability across studies.
Overall, the study broadens the existing evidence base through a wider methodological scope and a larger sample size. Its main contribution lies in highlighting methodological issues and variability in measurement outcomes, which require further systematic investigation in future work.
🧬 🧬 🧬
4.3.9. Achs et al. (2025)
Systematic analysis of COVID-19 mRNA vaccines using four orthogonal approaches demonstrates no excessive DNA impurities
Authors: Adam Achs, Tatiana Sedlackova, Lukas Predajna, Jaroslav Budis, Maria Bartosova, Vladimir Zelnik, Diana Rusnakova, Martina Melichercikova, Marta Miklosova, Veronika Gencurova, Barbora Cernakova, Tomas Szemes, Boris Klempa, Juraj Kopacek, Silvia Pastorekova
Published: December 13, 2025 (npj Vaccines, peer-reviewed; PMCID verfügbar)
Link to study: https://pmc.ncbi.nlm.nih.gov/articles/PMC12715226/a) Context and Objective of the Study
The study by Achs et al. follows a systematic and methodologically broad approach to investigating potential residual DNA in mRNA-based COVID-19 vaccines. It was conducted against the background of earlier reports describing elevated DNA quantities and specific sequence elements in such products.
The primary objective is to reassess these findings using multiple independent and complementary („orthogonal”) analytical methods, with a focus on evaluating the robustness of results against methodological influences. A central question is whether previously reported residual DNA findings are reproducible or can be explained by methodological artifacts.
A further emphasis is placed on evaluating the analytical methods themselves. The authors examine how different detection techniques – such as sequencing, qPCR, and fluorometric assays – may yield divergent results and what methodological limitations need to be considered.
Thus, the study not only aims to quantify potential residual DNA but also to contextualize the existing literature methodologically and assess the reliability of different analytical approaches in this context.
b) Materials and Methods
Vaccine batches
The study analyzed 15 vaccine batches – 9 Comirnaty (Pfizer) and 6 Spikevax (Moderna) – sourced from state-managed vaccine stocks in Slovakia and provided under the supervision of the State Institute for Drug Control. At the time of analysis, four Comirnaty batches were still within their expiration date, while the remaining batches had already expired. All vials were stored and transported according to manufacturer specifications.A subset of the batches –specifically those also examined in Fleming et al. (Study 7) – was deliberately included to enable direct cross-study comparison.
Four orthogonal analytical methods
A central methodological feature of the study is the application of four independent, complementary analytical approaches on the same samples. Each method has distinct strengths and limitations; their combined use is intended to produce more robust conclusions than any single technique alone.Method 1: qPCR
The LNPs were disrupted using Triton X-100, and the treated samples were added directly to the qPCR reaction without prior DNA extraction. This avoids a purification step that could lead to DNA loss. The authors explicitly tested for potential PCR inhibition by vaccine components and reported no relevant inhibition of amplification under the conditions used.Eight primer pairs were employed, targeting three regions of the production plasmid: the spike-coding sequence (SPIKE), the origin of replication (ORI), and the kanamycin resistance gene (KAN). Amplicon sizes varied considerably, ranging from 63 bp (KAN 1C) to 233 bp (SPIKE 2). Two primer pairs – SPIKE 2 and ORI 2 – correspond to those used in Fleming et al., enabling direct comparison between studies.
Quantification was performed using calibration curves based on synthetic DNA standards (gBlocks) matching the respective target sequences. Measured copy numbers were converted into nanograms per dose based on the molecular mass of each amplicon.
Method 2: Fluorometry
For fluorometric DNA quantification, a DNA extraction step was performed using two different extraction approaches: phenol-chloroform extraction (yield 60–80%) and magnetic bead-based extraction for cell-free DNA (yield 90–95%). Both methods included RNase A treatment to eliminate interference from mRNA with the DNA-specific fluorescent dye.Die RNA-Konzentration nach der Behandlung wurde kontrolliert und auf unter 1 ng/µl gesenkt. The authors experimentally demonstrated that even small amounts of RNA can significantly inflate the DNA signal, and that RNase treatment is necessary to minimize RNA-induced overestimation. After treatment, RNA levels were confirmed to be reduced to below 1 ng/µl.
Method 3: Capillary electrophoresis
Capillary electrophoresis was used to directly visualize very high DNA amounts, such as those reported in some previous studies. If DNA were present at levels of several hundred to thousand nanograms per dose, it would appear as a clear peak or smear in the electropherogram. Samples were treated with Triton X-100 and RNase A prior to analysis. Two kits were used – one covering the standard range (0.5–50 ng/µl) and another for high sensitivity (5–600 pg/µl per fragment).Method 4: Short-read sequencing (Illumina)
For sequencing, DNA was extracted using an optimized protocol combining heat treatment (95 °C), RNase A digestion, and proteinase K treatment. Library preparation was performed without an additional fragmentation step to preserve the original fragment length distribution as much as possible. Sequencing was carried out on a NextSeq 2000 system using paired-end reads, enabling precise fragment length determination.Bioinformatic analysis included de novo assembly of reads into complete plasmid sequences, followed by mapping of all reads to the reconstructed reference sequences.
c) Key Results
qPCR findings
All eight qPCR assays were performed in two independent laboratories by three operators. In none of the 15 analyzed batches was the regulatory threshold of 10 ng DNA per dose exceeded.Clear differences were observed between individual assays, depending on amplicon length. The highest DNA amounts were measured with the shortest amplicon (KAN 1C, 63 bp), reaching up to 8 ng/dose for Comirnaty. The lowest values were obtained with the longest amplicons (SPIKE 1A and SPIKE 2, 228–233 bp), at approximately 0.5–0.7 ng/dose. The authors attribute this discrepancy to DNA fragment length: with a median fragment length of ~150 bp, longer amplicons fail to detect many fragments because they are too short to contain both primer binding sites.
They interpret this consistent pattern across assays as evidence for the internal consistency of their methodological approach.
Fluorometry
After complete RNase A treatment and DNA extraction, fluorometrically measured DNA levels in all batches were below the regulatory threshold of 10 ng per dose.The authors experimentally demonstrated that insufficient RNase treatment can substantially overestimate DNA signals: even small amounts of RNA generate a measurable background signal in the DNA assay.
RNA measurements showed that all Comirnaty batches contained at least 90% of the declared mRNA content. In some Spikevax batches, values were lower, which the authors attribute to advanced expiration and degradation over time.
Capillary electrophoresis
No specific DNA signal was detected in any of the analyzed batches – neither with the standard kit (detection range 0.5–50 ng/µl) nor with the high-sensitivity kit (5–600 pg/µl per fragment). Weak, non-specific signals around 60 and 300 bp were observed in some samples, but these also appeared in the negative controls and were therefore attributed to methodological background noise.The authors argue that if DNA were present at levels of several hundred to thousand nanograms per dose, it would appear as a clear peak or smear in capillary electrophoresis. The complete absence of such a signal is considered inconsistent with such high DNA estimates.
RNA integrity analysis showed that six of nine expired batches had mRNA integrity below 50%, which was also visually reflected in a more turbid appearance of these vials compared with clear, non-expired samples.
Sequencing
The full sequences were generated bioinformatically by assembling many overlapping short reads and do not necessarily represent direct evidence of physically intact plasmid molecules.The proportion of plasmid-derived reads was 96.6% for Comirnaty (range: 95.3–98.1%) and 88.7% for Spikevax (range: 83.5–94.5%). The remaining reads mainly originated from E. coli K12 – a non-pathogenic laboratory strain – as well as bacteriophage sequences and repetitive sequences without database matches. The authors note that part of these reads may be attributable to reagent contamination, as described in the literature.
Fragment length analysis showed a median length of 154 bp for Comirnaty (range: 130–181 bp) and 144 bp for Spikevax (range: 127–201 bp). The fraction of fragments longer than 300 bp was low. Genome coverage across the plasmid was uneven, a pattern the authors consider consistent with incomplete or non-uniform DNase digestion.
d) Interpretation by the authors
DNA levels below regulatory threshold
The authors conclude that residual DNA in all 15 analyzed batches complies with the regulatory threshold of 10 ng per dose. In their view, this finding is robust because it is consistently supported across multiple complementary analytical methods. They further argue that the inclusion of expired batches strengthens the overall conclusion: even under conditions potentially associated with degradation or instability, no excessive DNA levels were detected.Origin and nature of the DNA
The authors conclude that the detected DNA originates exclusively from the production plasmid, i.e., from an expected and well-characterized by-product of the manufacturing process. According to their analyses, the DNA is highly fragmented, with a median fragment length of approximately 150 bp. In the authors‘ assessment, fragments of this size fall below the range typically associated with biological activity such as plasmid replication or efficient genomic integration.Explanation for differing findings in earlier studies
The authors discuss several methodological factors that, in their view, may help explain why some previous studies reported substantially higher DNA levels.First, they argue that fluorometric DNA assays can be significantly affected by residual mRNA if RNase treatment is incomplete. In their own control experiments, even small amounts of remaining RNA produced a noticeable increase in the apparent DNA signal. They therefore suggest that some of the elevated DNA values reported in earlier studies may have been influenced by RNA-related crosstalk effects.
Second, they emphasize the impact of amplicon length in qPCR-based assays. Because the detected DNA is largely fragmented, longer amplicons can only detect a subset of the available fragments. As a result, differences between studies may partly reflect differences in primer design and target regions rather than true differences in DNA content.
Overall, the authors interpret their findings as indicating that the detection of residual DNA in mRNA vaccines is highly method-dependent and that standardized analytical methods are needed.
Methodological Recommendations
The authors recommend the following for future analyses: complete RNase treatment with verification of residual RNA levels prior to fluorometric DNA measurement; direct use of Triton X-100–treated vaccine material for qPCR without prior DNA extraction to avoid losses; the use of short amplicons to achieve more comprehensive detection of fragmented DNA; and the combination of multiple complementary methods to ensure robust conclusions.Biological Assessment
The authors consider the risk posed by the detected residual DNA to be negligible, citing its low quantity, known plasmid origin, high degree of fragmentation, and the fact that no eukaryotic cell lines are used during manufacturing. They do not see any need to revise the existing regulatory limits or manufacturing practices.e) Methodological Limitations
Amplicon Size and Fragment Length
The authors themselves demonstrate that the measured DNA quantities depend strongly on the amplicon size of the respective qPCR assay. Using the shortest amplicon (KAN 1C, 63 bp), up to 8 ng per dose was measured – close to the regulatory limit. In contrast, the longest amplicons (SPIKE 1A and SPIKE 2, 228–233 bp) yielded only 0.5–0.7 ng per dose.These differences within the same study illustrate that qPCR results are substantially influenced by the chosen assay design. The measured values therefore do not represent an absolute total DNA content, but rather the fraction of DNA fragments that can be detected by the specific primer set used in each assay.
Fragment Length Determination by Illumina Sequencing
Sequencing revealed median fragment lengths of approximately 130–201 bp. However, these values should not be interpreted as direct measurements of the native fragment-length distribution.Library preparation involves multiple SPRI cleanup steps as well as PCR amplification, both of which can systematically disadvantage very short fragments (<150 bp). At the same time, very long fragments (>800 bp) may also be represented less efficiently during library preparation and sequencing, potentially resulting in underrepresentation or failure to be detected. Consequently, the reported length distribution primarily reflects the fragments that remained detectable after library preparation, rather than necessarily representing the original distribution present in the vaccine.
Therefore, the study does not permit definitive conclusions regarding either the true proportion of very short DNA fragments below the detection threshold or the possible presence of longer DNA fragments outside the size range that can be efficiently captured by the sequencing workflow.
Calculation Method in qPCR
The measured copy numbers were converted into nanograms based on the molecular mass of the respective qPCR amplicons, rather than using the full plasmid length. This approach is methodologically consistent for quantifying the actually amplified target regions. However, the resulting nanogram values are not directly comparable to studies that base their calculations on complete plasmid sequences.Influence of Extraction Efficiency
The fluorometric DNA measurements also depend on the efficiency of the extraction method used. The authors report an extraction yield of approximately 60–80% for phenol-chloroform extraction and about 90–95% for magnetic bead-based methods.Incomplete extraction can in principle lead to a fraction of the existing DNA – particularly very short fragments or nucleic acids still associated with lipid nanoparticles (LNPs) – not being fully recovered. Accordingly, bead-based extractions in the study tended to yield higher DNA values than the phenol-chloroform method.
No Measurement Before LNP Disruption
In contrast to some earlier studies, no systematic comparative measurements were performed before and after targeted disruption of lipid nanoparticles (LNPs). As a result, the data do not allow a direct inference regarding the proportion of detected DNA that was encapsulated within lipid nanoparticles.Capillary Electrophoresis: Limit of Detection
Capillary electrophoresis is well suited for detecting larger amounts of DNA. However, its sensitivity is limited for very low concentrations in the single-digit nanogram range. The method therefore complements qPCR and sequencing data but does not replace them.No SV40-Specific Analysis
The study does not include a specific assay for SV40 promoter/enhancer sequences. Consequently, no conclusions can be drawn from the data regarding the presence or absence of such sequences. This does not constitute a methodological weakness in a strict sense, as the investigation was not designed to detect SV40; however, compared to other studies, this aspect remains unaddressed.f) Contextualization
The study by Achs et al. represents one of the most methodologically comprehensive investigations to date into residual DNA in mRNA vaccines. It combines multiple complementary analytical approaches – qPCR with amplicons of varying lengths, fluorometric measurements with controlled RNase treatment, capillary electrophoresis, and short-read sequencing – performed on the same samples and across two different laboratories. This allows potential methodological artifacts of individual techniques to be better identified and contextualized.
In the overall context of the existing literature, the study highlights that the detection and quantification of residual DNA in mRNA vaccines is strongly method-dependent. Differences in sample preparation, extraction methods, RNase treatment, qPCR design, and sequencing approaches can lead to substantially different results.
Compared with earlier work, the study supports the assessment that the investigated batches do not contain unusually high levels of residual DNA. At the same time, several methodological questions – particularly regarding the complete fragment-length distribution of very short or very long DNA fragments – remain unresolved even with this approach.
Thus, the work by Achs et al. contributes less to a definitive resolution of the controversy than to a more precise methodological framing of the existing findings. In this sense, it complements previous studies – not by broadly contradicting their results, but by clarifying under which methodological conditions specific conclusions can be drawn.
4.4. What do we know – and what do we not know?
What the Available Studies Show in Common
The studies presented in this chapter originate from different laboratories, were conducted at different times, and employ partially distinct analytical methods. Nevertheless, several findings can be identified that are consistent across multiple studies.
First: Residual DNA is detectable in mRNA-based COVID-19 vaccines. This is not an unexpected finding; it is a known and anticipated consequence of the manufacturing process, which uses a DNA template for in vitro transcription. It is well established in the scientific literature that DNase I digestion does not guarantee complete removal of all DNA fragments.
Second: The detected DNA predominantly originates from the production plasmid. This is consistently shown by sequencing data from McKernan, Raoult, and Achs et al., as well as by PCR-based analyses from multiple research groups.
Third: Several studies provide indications that a relevant fraction of the detected DNA is protected from enzymatic degradation and is associated with lipid nanoparticles.
Where the Studies Diverge
The central question of dispute – how much DNA is actually present and whether regulatory limits are complied with – is answered differently across the available studies. These discrepancies are not primarily attributable to differences in the samples themselves, but can at least in part be explained by differences in methodological approaches.
Fluorometric measurements without adequate RNase treatment systematically yield higher values because the dye also binds RNA. PCR-based measurements depend on amplicon size: longer amplicons detect fewer fragments and therefore underestimate total DNA content. Sequencing methods provide qualitative information on sequence identity and fragment length, but do not offer reliable absolute quantification. Each method therefore measures slightly different aspects and correspondingly produces different numerical results.
This means that differences between studies are not necessarily evidence of flawed work by any of the research groups. Instead, they can – and in many cases must – be understood as a reflection of methodological variability.
What Remains Open
Despite the growing body of data, several questions remain unresolved.
The question of the true fragment-length distribution – particularly the proportion of very short fragments below 100 bp – has not yet been conclusively clarified. Illumina sequencing systematically underestimates short fragments, while ONT datasets can be more strongly influenced by longer fragments due to process-related selection and sequencing biases. A method that accurately captures the native fragment distribution without size-dependent distortion has not been applied in the available studies.
The biological relevance of the detected DNA – including questions of potential cellular uptake, persistence, or genomic integration – has not been directly investigated so far. Existing theoretical models and analogies from other biological systems are currently insufficient to draw robust conclusions about actual risk, although they do justify further investigation.
The question of the SV40 promoter/enhancer – its actual prevalence across different batches and its biological activity in fragmented form – has been addressed inconsistently across studies and remains unresolved.
What This Means for the Scientific Debate
A fundamental issue running through all the studies discussed here is the absence of a standardized, validated analytical protocol for the measurement of residual DNA in the final vaccine product. Regulatory limits do exist, but the methods used to verify them are not uniformly defined. As a result, different laboratories using different methods arrive at different results, without a straightforward way to compare them directly.
A meaningful next step would be the establishment of an orthogonal standard protocol: the same sample analyzed using multiple validated methods across several independent laboratories. Only such an approach would allow methodological variability to be distinguished from true batch-to-batch differences – and thereby generate robust, comparable data.
Regulatory Context
Regulatory limits for residual DNA were originally developed for traditional biological medicinal products and not specifically for LNP-formulated modRNA vaccines. To what extent existing limits and testing procedures are fully transferable to this newer platform technology has not yet been conclusively addressed.
Overall Assessment
The studies available so far have played an important role in initiating a scientific discussion that would otherwise likely not have taken place. At the same time, they also highlight the limitations of independent re-analyses outside a fully standardized regulatory framework: lack of validation, limited sample sizes, and methodological heterogeneity.
What remains is a scientifically grounded demand for greater transparency, more standardized methods, and independent replication across different research groups. This is not an alarmist conclusion – it reflects what sound science generally requires.
5. Conclusion and Outlook
The discussion of residual DNA in mRNA vaccines illustrates on multiple levels how complex the analytical evaluation of novel biological medicinal products can be.
What initially appears to be a simple question – whether residual DNA is present and in what quantity – turns out, upon closer examination, to depend strongly on sample preparation, detection methods, and data interpretation.
Key Findings of the Study
The production of modRNA-based vaccines involves a multi-step process in which bacterial DNA templates are indispensable. The removal of this DNA is part of the standard purification steps in the manufacturing process. However, the available investigations consistently show that residual DNA fragments can be detected in commercial vaccine batches – albeit in quantities that vary depending on the analytical method used.
The evaluation of existing studies makes clear that the detection and quantification of residual DNA is considerably more complex than regulatory threshold values alone might suggest. Different lysis procedures, extraction methods, RNase treatments, qPCR designs, and sequencing approaches can lead to substantially divergent results.
The controversy of recent years thus highlights not only differences between individual studies, but above all the limitations arising from the lack of standardization in independent re-analyses.
Open Questions for Future Research
Despite the substantial amount of data now available, several questions remain unresolved:
What is the actual proportion of very short or potentially biologically active DNA fragments across different vaccine batches?
What is the validity of current analytical methods with regard to the native fragment-length distribution in the vaccine product?
What role do RNA:DNA hybrids or other nucleic acid complexes play in the stability and detectability of residual DNA?
Finally, what is the biological relevance of the detected DNA fragments – including the SV40 promoter/enhancer element – under real physiological conditions?Many of these questions cannot currently be either definitively confirmed or ruled out based on the available data. They require further investigation using standardized, orthogonal analytical approaches as well as independent replication across multiple laboratories.
Outlook
The scientific discussion on residual DNA in modRNA vaccines is therefore not yet concluded. At the same time, existing research shows that a clear distinction must be made between analytical detection and biological significance.
While the present work focuses on manufacturing processes, detection methods, and experimental findings, the question of potential biological effects remains a subject for further research and future consideration. This includes, among other things, questions regarding the biological activity of residual DNA, the stability of nucleic acid complexes, theoretical integration mechanisms, and the relevance of functional sequence elements.
Regardless of the interpretation of individual studies, a fundamental scientific requirement emerges: greater transparency, better standardized analytical procedures, and reproducible independent investigations. Especially in the context of novel biological technologies, this is not an exceptional demand, but a core component of scientific quality assurance.

Acknowledgements
I would like to express my sincere gratitude to all scientists, researchers, and dedicated individuals who, through their work – across studies, articles, and lectures – contribute to the investigation of this complex topic. Their commitment is of invaluable importance, both for the advancement of knowledge and for a fact-based public discourse.
The scientific debate that this text seeks to document is not a flaw – it is the essence of good science.
✧ ✧ ✧
Postscript
I began this text as a layperson.
With uncertainty, curiosity, and open questions.
Perhaps that was an advantage.Many questions received answers.
New questions emerged.
The deeper I went, the clearer it became:
It is not only biology that is complex.
The measurement of biology is complex as well.The journey through this subject was long.
Along the way, I received help:
patient AI systems that explained,
challenged, structured, formulated – and thought along with me.
These were long, productive dialogues
between human and machine.And in the end, one important insight remains:
learning is a shared process.
Current as of June 2026.
-
DNA-Rückstände in modRNA-basierten Impfstoffen

mRNA-Impfstoffe gelten als eine der bedeutendsten biomedizinischen Entwicklungen der vergangenen Jahre. Weltweit wurden Milliarden Dosen verabreicht, gleichzeitig befinden sich zahlreiche weitere mRNA-basierte Anwendungen in Entwicklung – von Krebsimmuntherapien bis hin zu Impfstoffen gegen unterschiedlichste Infektionskrankheiten.
Die öffentliche Diskussion über DNA-Rückstände in mRNA-Impfstoffen begann nicht mit einer regulatorischen Mitteilung oder einer offiziellen Sicherheitswarnung, sondern eher zufällig im Rahmen molekularbiologischer Sequenzierungsarbeiten.
Anfang 2023 arbeitete ein Forschungsteam um den US-amerikanischen Genetiker Kevin McKernan an RNA-Sequenzierungen, für die hochreine RNA-Proben benötigt wurden. Dabei griff das Team unter anderem auf ungeöffnete Fläschchen der COVID-19-Impfstoffe von BioNTech/Pfizer und Moderna zurück. Während der Analysen stießen die Forscher auf einen unerwarteten Befund: Neben der modifizierten mRNA fanden sich auch DNA-Sequenzen in den Proben.
Dass bei bestimmten biotechnologischen Herstellungsverfahren geringe Mengen residualer DNA technisch grundsätzlich möglich sind, ist in der Arzneimittelproduktion bekannt und regulatorisch berücksichtigt. Überraschend war jedoch, dass zu diesem Zeitpunkt kaum öffentlich diskutiert wurde, welche Herstellungsverfahren für die großtechnische Produktion tatsächlich verwendet wurden und in welchem Umfang unabhängige Analysen der Endprodukte überhaupt vorlagen.
Die später veröffentlichten Untersuchungen von McKernan und weiteren Arbeitsgruppen berichteten schließlich über DNA-Rückstände in verschiedenen Impfstoffchargen – teilweise in Mengen, die nach Einschätzung der Autoren oberhalb regulatorischer Richtwerte lagen. Damit begann eine wissenschaftliche Debatte, die bis heute anhält.
Doch worum geht es dabei eigentlich genau?
Die öffentliche Diskussion konzentrierte sich schnell auf scheinbar einfache Fragen:
Sind DNA-Rückstände vorhanden – oder nicht?
Werden regulatorische Grenzwerte eingehalten?
Und falls DNA nachweisbar ist:
Welche biologische Bedeutung hätte das überhaupt?Bei näherer Betrachtung zeigt sich jedoch, dass diese Fragen analytisch wesentlich komplexer sind, als es zunächst erscheint.
Denn bereits die Herstellung modRNA-basierter Impfstoffe ist ein mehrstufiger Prozess, bei dem bakterielle DNA-Vorlagen technisch unverzichtbar sind. Die Entfernung dieser DNA gehört zu den regulären Reinigungsschritten der Produktion. Vollständige molekulare Trennung ist in biologischen Herstellungsverfahren jedoch selten eine rein binäre Frage. Entscheidend ist vielmehr:
Welche Fragmente verbleiben möglicherweise im Endprodukt?
In welcher Menge?
Mit welcher Struktur?
Und vor allem:
Mit welchen Methoden lässt sich das überhaupt zuverlässig messen?Zusätzliche Aufmerksamkeit erhielt die Debatte durch öffentlich gewordene Unterschiede zwischen frühen Entwicklungsverfahren und der späteren industriellen Massenproduktion. Während in frühen klinischen Entwicklungsphasen vergleichsweise kleine DNA-Mengen über PCR-ähnliche Verfahren vervielfältigt wurden, kamen für die spätere Milliardenproduktion bakterienbasierte Herstellungsprozesse zum Einsatz. Diese Verfahren sind industriell etabliert und ermöglichen große Produktionsmengen, bringen jedoch zugleich komplexere Anforderungen an die Aufreinigung biologischer Restbestandteile mit sich.
Gerade dieser Übergang zwischen unterschiedlichen Produktionsprozessen wurde später intensiv diskutiert – auch deshalb, weil große Teile der frühen klinischen Studien noch mit Impfstoffchargen aus dem ursprünglichen Herstellungsverfahren durchgeführt worden waren.
Hinzu kommt ein weiteres Problem: Der Nachweis von DNA-Rückständen hängt stark von der verwendeten Methodik ab.
Unterschiedliche Laborgruppen verwendeten unterschiedliche Aufschlussverfahren, Extraktionsmethoden, PCR-Designs und Sequenzierungsansätze – mit teils deutlich voneinander abweichenden Ergebnissen. Die Diskussion betrifft daher nicht nur die gemessenen Werte selbst, sondern ebenso die Frage, wie belastbar und vergleichbar diese Messungen überhaupt sind.
Die vorliegende zweiteilige Artikelserie versucht deshalb nicht, vorschnelle Antworten zu liefern. Ihr Ziel ist vielmehr, die wissenschaftlichen und methodischen Hintergründe verständlich zu machen, die hinter den aktuellen Debatten stehen.
Der erste Teil beschreibt die Herstellungs- und Reinigungsprozesse modRNA-basierter Impfstoffe:
Wie entsteht die mRNA?
Warum werden bakterielle DNA-Vorlagen benötigt?
Welche Nebenprodukte entstehen während der Produktion?
Mit welchen Verfahren versucht man, unerwünschte Restbestandteile zu entfernen?Der zweite Teil widmet sich anschließend der analytischen Perspektive:
Wie lassen sich DNA-Rückstände überhaupt nachweisen?
Welche Aussagekraft besitzen qPCR, Qubit oder Sequenzierungsverfahren?
Warum können verschiedene Messmethoden zu unterschiedlichen Ergebnissen führen?
Was zeigen die bislang veröffentlichten unabhängigen Studien tatsächlich?Dabei wird deutlich, dass zwischen einem analytischen Nachweis und einer biologischen Bewertung unterschieden werden muss. Viele Fragen sind derzeit wissenschaftlich noch offen – etwa zur nativen Fragmentverteilung residualer DNA, zur möglichen Rolle von RNA oder zur biologischen Relevanz bestimmter Sequenzelemente unter realen physiologischen Bedingungen.
Gerade deshalb berührt die Debatte letztlich eine grundsätzliche wissenschaftliche Frage:
Wie geht moderne Biomedizin mit Unsicherheit, Nachweisgrenzen und methodischer Komplexität um – insbesondere bei neuartigen biologischen Technologien?
Die folgenden beiden Teile verstehen sich als Einladung, diese Fragen differenziert zu betrachten: nicht als Schlagzeile, sondern als wissenschaftliches Problem, dessen Bewertung Präzision, Transparenz und methodisches Verständnis erfordert.
Die Darstellung möchte eine Brücke schlagen zwischen den oft sehr technischen Fachpublikationen und vereinfachten Medienberichten, sodass auch interessierte fachfremde Leserinnen und Leser ohne Spezialkenntnisse die komplexen Zusammenhänge nachvollziehen können.
Der Text erhebt keinen Anspruch auf Vollständigkeit – er bemüht sich um Nüchternheit, Genauigkeit und Fairness gegenüber allen beteiligten Positionen.
Zur Artikelserie
Die Analyse ist in zwei thematisch aufeinander aufbauende Teile gegliedert. Beide können unabhängig gelesen werden; für ein umfassendes Verständnis empfiehlt sich jedoch die angegebene Reihenfolge.
Teil 1: Herstellung und Reinigung
Inhaltsverzeichnis – Teil 1
1. Die Herstellungsverfahren – ein Überblick
1.1. Produktentstehung – vom Gen zur mRNA
1.2. Verunreinigungen und Nebenprodukte
1.3. Reinigungsverfahren im Herstellungsprozess
1.4. Vergleich: Prozess 1 vs. Prozess 2
1.5. Zusammenfassung: Herstellung und Reinigung von modRNATeil 2: Nachweisverfahren und Studienlage
Inhaltsverzeichnis – Teil 2
2. Methoden zur Messung von DNA und RNA
2.1. Spektralphotometrische Verfahren
2.2. Fluoreszenzbasierte Quantifizierung
2.3. Amplifikationsbasierte Methoden
2.4. Sequenzierungsbasierte Verfahren
2.5. Elektrophoretische Fragmentanalyse
2.6. Vergleich zentraler Nachweisverfahren für Nukleinsäuren
3. Richtlinien zur Begrenzung von Rest-DNA in Impfstoffen
4. Studien zu DNA-Rückständen in modRNA-basierten Impfstoffen
4.1. Warum unabhängige Messungen?
4.2. Methodische Vorbemerkungen
4.3. Übersicht und Einzelanalysen der Studien
4.4. Zwischenfazit: Was wissen wir – und was nicht?
5. Schlussbetrachtung und Ausblick
Stand: Mai 2026
-
DNA-Rückstände in modRNA-basierten Impfstoffen – Teil 1

Leitfaden zur Artikelserie
Teil 1: Herstellung und Reinigung von modRNA
Teil 2: Nachweisverfahren und StudienlageBevor wir uns der Frage zuwenden, warum DNA-Rückstände in modRNA-Impfstoffen nachweisbar sein können, ist ein grundlegendes Verständnis des Herstellungsprozesses hilfreich.
Die Herstellung modRNA-basierter Impfstoffe erfolgt über mehrere aufeinander aufbauende Produktions- und Reinigungsschritte. Ausgangspunkt ist eine DNA-Vorlage, die als Bauplan für die spätere nukleosid-modifizierte mRNA (modRNA) dient. Im Verlauf des Herstellungsprozesses entstehen dabei komplexe Reaktionsgemische, aus denen unerwünschte Restbestandteile möglichst weit entfernt werden müssen.
Besondere Aufmerksamkeit erhielt in den vergangenen Jahren die Tatsache, dass für die Entwicklung und spätere industrielle Produktion unterschiedliche Verfahren zur Vervielfältigung dieser DNA-Vorlage eingesetzt wurden. Diese Prozesse unterscheiden sich nicht nur hinsichtlich ihrer technischen Skalierung, sondern auch hinsichtlich der Anforderungen an die anschließende Reinigung biologischer Restbestandteile.
Der vorliegende erste Teil beschreibt die grundlegende Herstellungslogik modRNA-basierter Impfstoffe, typische Nebenprodukte und Verunreinigungen sowie die wichtigsten Reinigungsverfahren innerhalb der Produktion. Abschließend werden die beiden Herstellungswege – Prozess 1 und Prozess 2 – systematisch gegenübergestellt.
Er bildet damit die Grundlage für den zweiten Teil dieser Artikelserie, der sich den analytischen Nachweisverfahren, regulatorischen Grenzwerten sowie den bisher veröffentlichten experimentellen Untersuchungen zu DNA-Rückständen in modRNA-basierten Impfstoffen widmet.
Hinweis zur Terminologie
📑
Im allgemeinen Sprachgebrauch ist der Begriff „mRNA-Impfstoff“ gebräuchlich. Fachlich handelt es sich bei den zugelassenen Präparaten um nukleosid-modifizierte mRNA (modRNA). Aus Gründen der Verständlichkeit wird in dieser Arbeit der etablierte Begriff „mRNA-Impfstoff“ verwendet.Inhaltsverzeichnis
Teil 1: Herstellung und Reinigung
1. Die Herstellungsverfahren – ein Überblick
1.1. Produktentstehung – vom Gen zur mRNA
1.2. Verunreinigungen und Nebenprodukte
1.3. Reinigungsverfahren im Herstellungsprozess
1.4. Vergleich: Prozess 1 vs. Prozess 2
1.5. Zusammenfassung: Herstellung und Reinigung von modRNA
1. Die Herstellungsverfahren – ein Überblick
Die folgende Übersicht stellt die wesentlichen Produktionsschritte vereinfacht dar. In der industriellen Praxis können einzelne Verfahren und Prozessdetails je nach Hersteller variieren.
Die Herstellung lässt sich vereinfacht in folgende Prozessschritte gliedern:
Schritt 1 🟢 Gewinnung der genetischen Information Schritt 2 🟢 Vervielfältigung der Spike-Protein-DNA
Prozess 1: mittels PCR
Prozess 2: mittels Bakterien💧 Reinigung der DNA Schritt 3 🟢 In-vitro-Transkription zur Herstellung der mRNA Schritt 4 🟢 RNA-Prozessierung – Reifung der mRNA 💧 Reinigung der DNA Schritt 5 🟢 Verpackung der mRNA in Lipid-Nanopartikel (LNPs) 💧 Endreinigung Während im Upstream-Prozess das eigentliche Produkt – die mRNA – schrittweise erzeugt wird, dient der Downstream-Prozess dazu, die entstandene mRNA aus komplexen Reaktionsgemischen zu isolieren, zu reinigen und für die medizinische Anwendung zu formulieren.
1.1. Produktentstehung – vom Gen zur mRNA
Schritt 1: Gewinnung der genetischen Information
Schritt 2: Vervielfältigung der Spike-Protein-DNA
Prozess 1: mittels PCR
Prozess 2: mittels Bakterien
Schritt 3: In-vitro-Transkription zur Herstellung der mRNA
Schritt 4: RNA-Prozessierung – Reifung der mRNA
Schritt 5: Verpackung der mRNA in Lipid-Nanopartikel (LNPs)Schritt 1: Gewinnung der genetischen Information
Der erste Schritt besteht darin, die genetische Bauanleitung für das gewünschte Protein – hier das Spike-Protein des Coronavirus – zu identifizieren.
In der Praxis läuft das so ab:
Virusproben sammeln: Aus Patientenproben (z. B. Rachen- oder Nasenabstrichen, Blut oder Gewebe) wird Virusmaterial gewonnen.
Anmerkung: Es gibt unterschiedliche Auffassungen darüber, ob SARS-CoV-2 jemals direkt aus Patientenproben isoliert und im Labor kultiviert wurde. Diese Frage wird in diesem Artikel nicht behandelt, da der Fokus hier auf dem Herstellungsprozess der mRNA-Impfstoffe liegt.
Erbinformation extrahieren: SARS-CoV-2 ist ein RNA-Virus. Die virale RNA wird aus der Probe isoliert.
RNA in DNA umwandeln: Mithilfe eines speziellen Enzyms (Reverse Transkriptase) wird die Virus-RNA in DNA übersetzt. DNA ist stabiler und kann leichter analysiert werden.
Genom entschlüsseln: Die DNA wird vervielfältigt und ihre genau Abfolge der Nukleotide (A, T, G, C) bestimmt.
Den wichtigen Abschnitt finden: Mittels Computeranalysen wird der genaue Teil identifiziert, der den Bauplan für das Spike-Protein enthält.
Eintrag in öffentliche Datenbanken: Die so ermittelte genetische Bauanleitung wird in öffentliche Datenbanken eingetragen. Forschungsteams weltweit können dadurch auf diese Sequenz zugreifen, ohne das Virus selbst isolieren zu müssen.
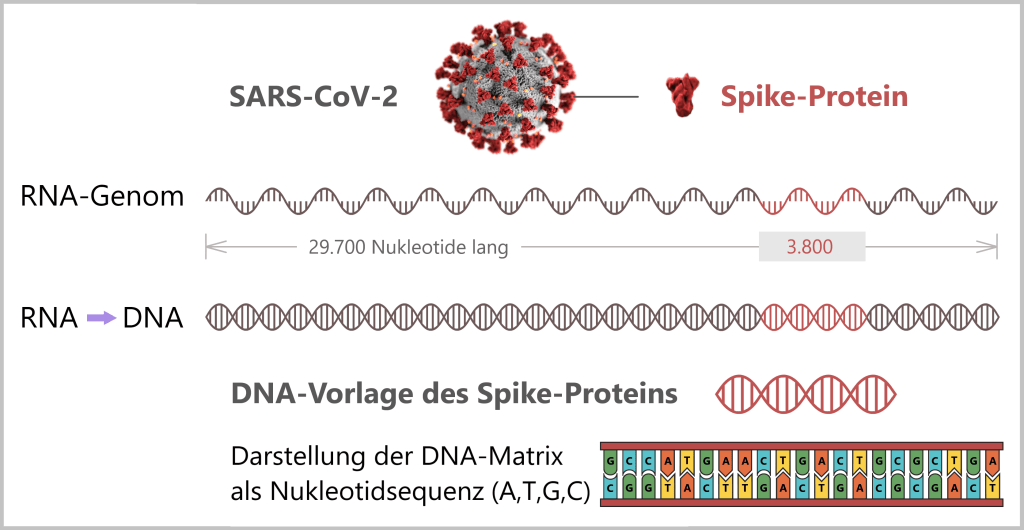
Abb. 1.1.1: Vom Virus zur DNA-Vorlage Aus dem RNA-Genom des SARS-CoV-2 wird mithilfe des Enzyms Reverse Transkriptase eine DNA-Version erzeugt. Der Abschnitt, der den Bauplan für das Spike-Protein enthält, wird identifiziert und als DNA-Vorlage (DNA-Template) synthetisch im Labor hergestellt. Das DNA-Template dient später als Vorlage für die Produktion der mRNA im Impfstoff.
Anmerkung: Das Coronavirus SARS-CoV-2 speichert seine Erbinformation auf einer langen RNA-Kette, die aus einzelnen Nukleotiden – den „Buchstaben“ des genetischen Codes – besteht. Mit rund 29.700 dieser Bausteine besitzt es eines der größten bekannten RNA-Genome. Der Bauplan für das charakteristische Spike-Protein umfasst dabei einen Abschnitt von etwa 3.800 Buchstaben.
Erstellen der DNA-Vorlage
Basierend auf der ermittelten genetischen Abfolge (den sogenannten Sequenzdaten) lässt sich im Labor eine DNA-Version des Spike-Gens synthetisch herstellen. Dieses DNA-Template dient später als Vorlage für die mRNA-Produktion.Schritt 2: Vervielfältigung der Spike-Protein-DNA
Nachdem die Bauanleitung für das Spike-Protein in Form einer DNA-Vorlage hergestellt wurde, muss sie millionenfach vervielfältigt werden. Nur so steht genug Material zur Verfügung, um später mRNA daraus herzustellen.
Prozess 1: mittels PCR
Prozess 2: mittels Bakterien
Prozess 1: Vervielfältigung der Spike-DNA mittels PCR
Prozess 1 nutzt ein Verfahren, das seit der Corona-Pandemie weltweit bekannt ist: die Polymerase-Kettenreaktion (PCR). Sie wird im Labor – fachsprachlich in vitro – durchgeführt und imitiert die natürliche DNA-Vermehrung, wie sie sonst in lebenden Zellen abläuft.
Was braucht man für eine PCR?
Damit die Kopiermaschine der Natur läuft, braucht es ein paar Zutaten:
- DNA-Vorlage – das Ausgangsmaterial ist die Spike-Protein-DNA aus Schritt 1.
- Nukleotide – die Bausteine: die vier „Buchstaben“ der DNA > Adenin (A), Thymin (T), Cytosin (C) und Guanin (G), aus denen neue Stränge zusammengesetzt werden.
- DNA-Polymerase – der Baumeister: das Enzym, das die Vorlage liest und neue DNA-Stränge baut. Meist wird die hitzestabile Taq-Polymerase verwendet.
- Primer – die Wegweiser: kurze DNA-Stücke, die der Polymerase zeigen, wo sie mit dem Kopieren beginnen soll. Man benötigt immer zwei: einen Vorwärts- und einen Rückwärts-Primer.
- Pufferlösung – das richtige Milieu: sorgt für die richtige Umgebung, damit die Polymerase zuverlässig arbeiten kann.
- Thermocycler – das Temperatur-Karussell: ein Gerät, das automatisch die nötigen Temperaturzyklen durchläuft.
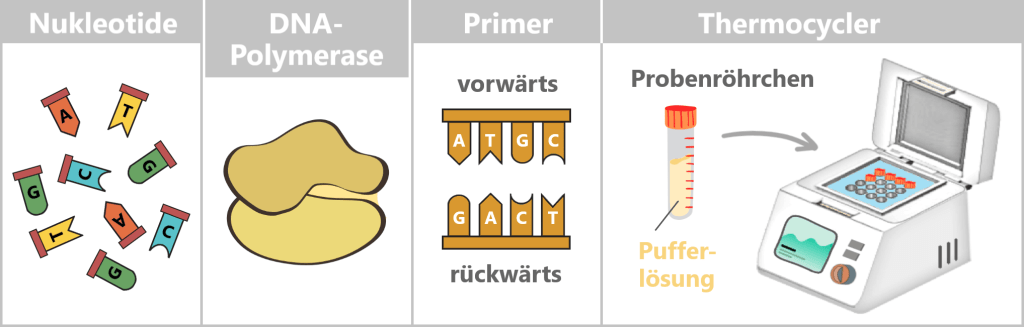
Abb. 1.1.2.1-A: Schematische Darstellung der Zutaten und Geräte Für die spätere mRNA-Herstellung wird die DNA-Vorlage bereits in diesem Schritt gezielt erweitert. An die Spike-Sequenz wird ein zusätzlicher DNA-Abschnitt angehängt – der sogenannte T7-Promotor. Diese kurze Sequenz wird für die PCR selbst nicht benötigt, sondern spielt erst im nachfolgenden Prozessschritt eine Rolle. Der T7-Promotor dient als Andockstelle für die T7-RNA-Polymerase, die in der in-vitro-Transkription (IVT) die DNA in die therapeutische mRNA umschreibt.
Der Ablauf der Polymerase-Kettenreaktion (PCR)
Alle Zutaten kommen in ein kleines Reaktionsröhrchen, das in den Thermocycler gestellt wird. Dieser steuert die immer gleichen drei Schritte, die sich zyklisch wiederholen:
1) Trennung der DNA-Stränge (Denaturierung)
Die Probe wird auf ca. 94–98 °C für etwa 20-30 Sekunden erhitzt. Dadurch lösen sich die Wasserstoffbrücken zwischen den DNA-Basen und der Doppelstrang trennt sich in zwei Einzelstränge auf. Diese dienen im nächsten Schritt als Vorlage.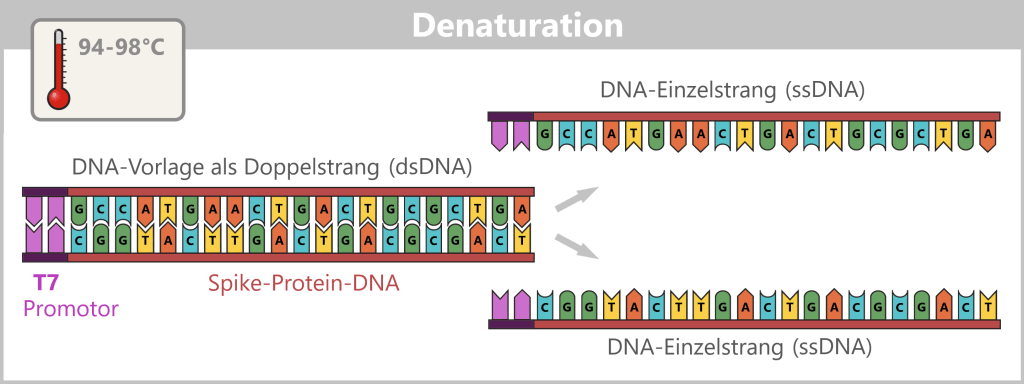
Abb. 1.1.2.1-B: Denaturationsschritt der PCR Die Darstellung ist stark schematisch und nicht maßstabsgetreu. Die kodierende Sequenz für das SARS-CoV-2-Spike-Protein beträgt ~3.800 Basenpaare, die für den T7-Promotor ~20 Basenpaare.
2) Primerbindung (Annealing)
Die Temperatur wird auf 50–65 °C abgesenkt. Jetzt binden sich die Primer gezielt an die jeweiligen DNA-Einzelstränge. Sie markieren den Startpunkt für die DNA-Synthese.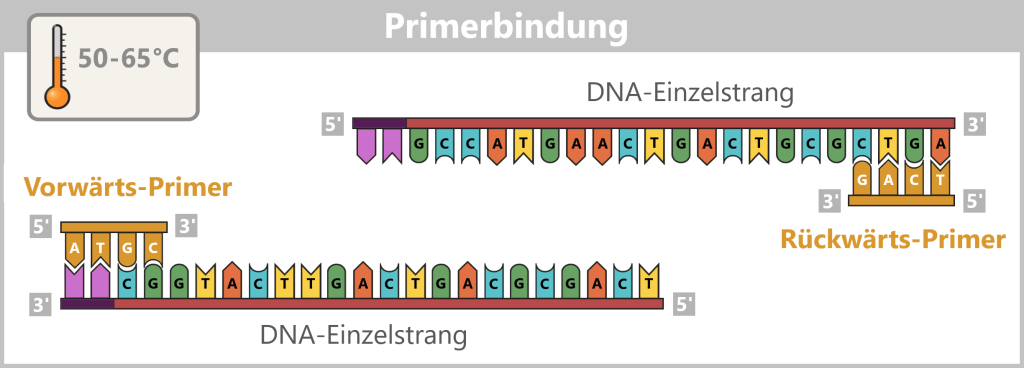
Abb. 1.1.2.1-C: Primerbindung Die Darstellung ist stark schematisch und nicht maßstabsgetreu. Primer sind typischerweise 20–25 Nukleotide lang.
3) DNA-Synthese (Amplifikation)
Bei etwa 70 °C, der optimalen Temperatur für die Polymerase, beginnt die eigentliche Vervielfältigung. Die DNA-Polymerase bindet an den Primer, liest den Einzelstrang von 3′ nach 5′ – und synthetisiert parallel den neuen Strang in 5′ nach 3′-Richtung. Dabei setzt sie Nukleotide nach dem Prinzip der Basenpaarung zusammen: A mit T und G mit C. So entstehen zwei neue DNA-Doppelstränge.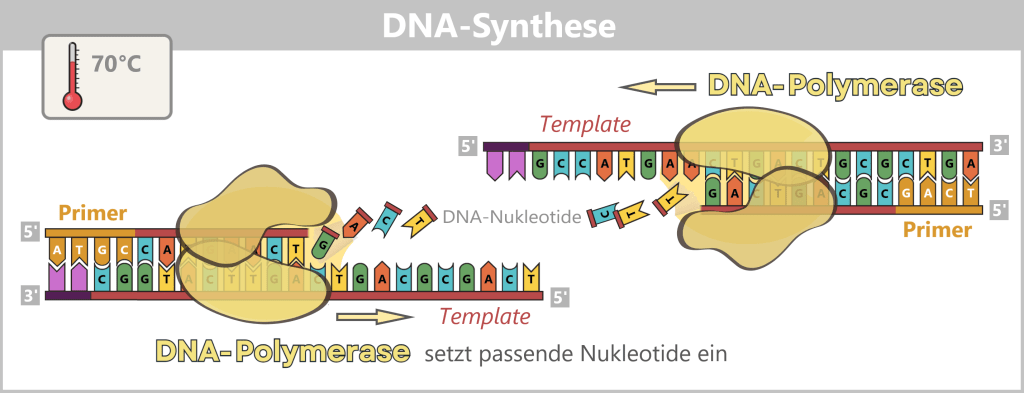
Abb. 1.1.2.1-D: DNA-Synthese Die Abbildung zeigt ein aktive DNA-Polymerase, die wie ein molekularer Motor am freigelegten Einzelstrang entlanggleitet. Der Prozess der Polymerisation ist deutlich erkennbar: Einzelne Nukleotide (die Bausteine der DNA) treten durch eine Öffnung in das Enzym und werden von der Polymerase passgenau an das Ende des wachsenden Stranges angefügt. Dieser Vorgang bewirkt die Elongation (Verlängerung) der neuen DNA-Kette, die kontinuierlich in 5′-3′-Richtung wächst. Durch diese stetige Kopierarbeit findet die Amplifikation (Vervielfältigung) der genetischen Information statt, wobei aus einem ursprünglichen Templatestrang ein neuer, komplementärer Doppelstrang entsteht.
Warum wird immer in 5′-3′-Richtung synthetisiert?
Der DNA-Aufbau
Die DNA besteht aus zwei Strängen, die in entgegengesetzte Richtungen verlaufen – der eine in 5’→3′-Richtung, der andere in 3’→5′-Richtung. Beide sind komplementär zueinander.
Die DNA-Polymerase, das Enzym, das neue DNA herstellt, kann nur in eine Richtung arbeiten: Sie liest den Matrizenstrang in 3’→5′-Richtung und synthetisiert den neuen Strang in 5’→3′-Richtung.Was bedeuten 5′ und 3′?
Um das zu verstehen, schauen wir uns die Bausteine der DNA an: die Nukleotide. Jedes Nukleotid besteht aus:- Einer Base (A, C, G oder T)
- Einem Zucker (Desoxyribose)
- Einer Phosphatgruppe (P)
Der Zucker hat fünf Kohlenstoffatome (C), die durchnummeriert sind:
- 1′: Hier ist die Base (A, T, C oder G) angeheftet.
- 2′: Trägt in der DNA nur ein Wasserstoffatom (-H) und keine Hydroxylgruppe (-OH) wie in der RNA. Diese fehlende OH-Gruppe gibt der Desoxyribose ihren Namen („Desoxy“-Ribose, da „desoxy“ = ohne Sauerstoff).
- 3′: Hier befindet sich eine Hydroxylgruppe (-OH), die für das Anfügen neuer Nukleotide während der Synthese wichtig ist.
- 4′: Verbindet den Zuckerring mit dem 5′-Kohlenstoff.
- 5′: Trägt die Phosphatgruppe, die das Nukleotid mit dem nächsten verknüpft.
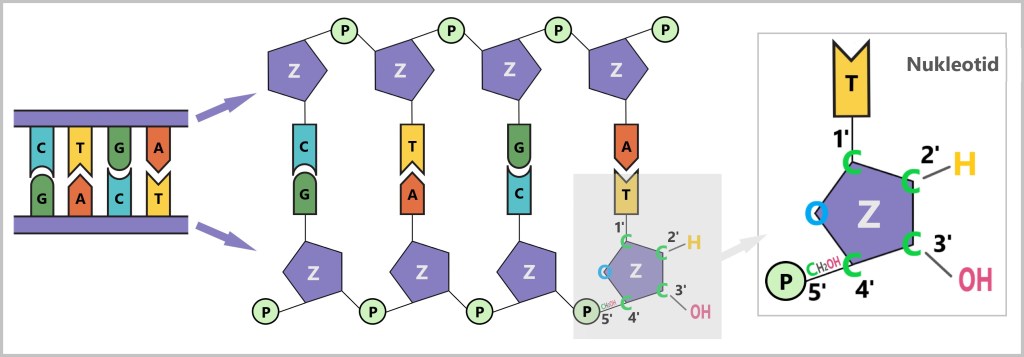
Warum die Synthese nur in 5’→3′-Richtung möglich ist
Die DNA-Polymerase kann neue Nukleotide nur am 3′-Ende des wachsenden Strangs anfügen. Der Grund: Dort befindet sich die freie Hydroxylgruppe (–OH), die für die chemische Verknüpfung mit der Phosphatgruppe des nächsten Nukleotids benötigt wird.
Am 5′-Ende sitzt bereits eine Phosphatgruppe – dort kann nichts Neues angehängt werden. Die Kettenverlängerung ist also nur in 5’→3′-Richtung möglich.Zusammengefasst
Die DNA-Polymerase bewegt sich entlang des Matrizenstrangs der DNA in 3′-5′-Richtung. Währenddessen fügt sie komplementäre DNA-Nukleotide an den neuen DNA-Strang an und zwar immer an dessen freies 3′-Ende. So wächst der neue Strang kontinuierlich in 5’→3′-Richtung.Wiederholung der Zyklen
Die neu entstandenen DNA-Doppelstränge dienen direkt als Vorlage für die nächste Runde. Die Schritte Denaturierung – Primerbindung – DNA-Synthese wiederholen sich zyklisch.
Mit jedem Zyklus verdoppelt sich die Menge der DNA: 1 → 2 → 4 → 8 → 16 → 32 … Nach nur 25–40 Zyklen liegen Milliarden Kopien der Spike-Protein-DNA vor.
Abb. 1.1.2.1-E: Wiederholung der Zyklen Das Ergebnis
Am Ende der PCR hat man eine hochkonzentrierte Lösung mit vielen Kopien der Spike-Bauanleitung – die Ausgangsbasis für den nächsten Schritt: die Herstellung von mRNA.
🎥 Tipp: Das Video „What is PCR?“ fasst den Prozess anschaulich zusammen.
Prozess 2: Vervielfältigung der Spike-DNA mittels Bakterien
Im Gegensatz zur PCR, die im Reagenzglas abläuft (in vitro), nutzt dieser Ansatz lebende Organismen, genauer gesagt Bakterien. Man spricht dabei von einem in vivo-Verfahren (lateinisch: „im Lebendigen“), weil die Vervielfältigung direkt innerhalb der Zellen stattfindet.
a) Warum Bakterien?
b) Das Bakterium Escherichia coli (E. coli)
c) Plasmide – kleine DNA-Ringe mit großer Wirkung
d) Plasmide in der Gentechnik
e) Einbau der Spike-Protein-DNA in das Plasmid
f) Übertragung der modifizierten Plasmide in Bakterien
g) Vermehrung der Bakterien
h) Ernte der Bakterien
i) Isolierung der modifizierten Plasmide
j) Linearisierung der Spike-Protein-DNAa) Warum Bakterien?
Bakterien sind winzige, einzellige Lebewesen, die sich durch einfache Teilung vermehren: Eine Zelle teilt sich in zwei, diese teilen sich wieder – und so entstehen in kürzester Zeit riesige Mengen identischer Kopien.
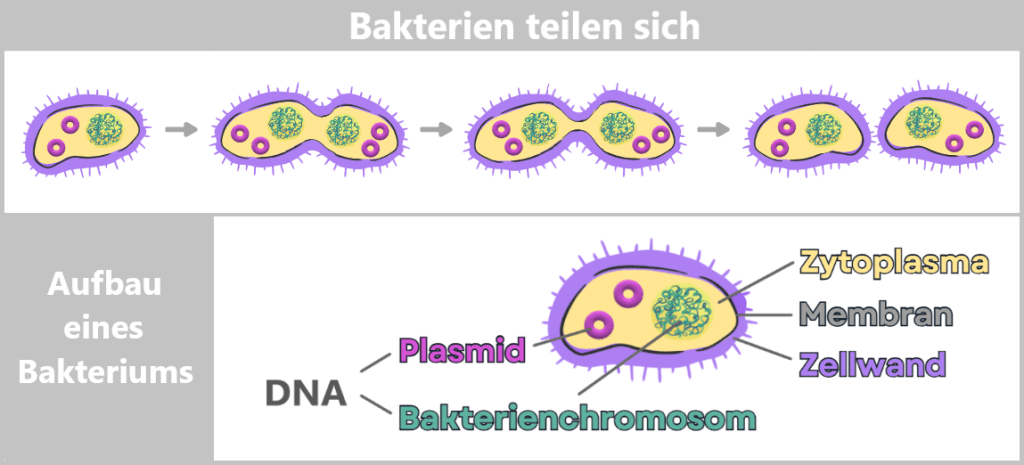
Abb. 1.1.2.2-A: Vermehrung und Aufbau von Bakterien Da Bakterien ungeschlechtlich sind, vermehren sie sich hauptsächlich durch einfache Teilung. Der Prozess beginnt mit dem Wachstum der Zelle und der Verdopplung des genetischen Materials. Dann bildet sich eine Einschnürung, bis schließlich zwei genetisch identische Tochterzellen entstehen.
Die Erbinformation der Bakterien – ihre DNA – liegt frei im Inneren der Zelle, im sogenannten Zytoplasma. Der größte Teil ist im Bakterienchromosom gespeichert. Zusätzlich besitzen viele Bakterien kleine, ringförmige DNA-Moleküle, die Plasmide.
Anpassung durch Mutation und Gentransfer
Die kontinuierliche Veränderung des genetischen Materials ist entscheidend, damit sich Organismen im Laufe der Generationen neuen Umweltbedingungen erfolgreich anpassen können. Bakterien gelten als besonders anpassungsfähig. Dafür sorgen zwei Mechanismen:Mutationen: kleine zufällige Veränderungen im Erbgut, die manchmal Vorteile bringen (z. B. Resistenz gegen Antibiotika).
Gentransfer: Bakterien können zusätzlich DNA-Abschnitte austauschen, vor allem über ihre Plasmide. So verbreiten sich nützliche Eigenschaften oft rasend schnell in einer Bakterienpopulation.
Darum sind Bakterien so geeignet:
- Schnelle Vermehrung: Unter idealen Bedingungen verdoppeln sich viele Bakterien alle 20 Minuten.
- Einfache Struktur: Ihre DNA liegt nicht im Zellkern, sondern frei im Zellinneren – das erleichtert Manipulationen.
- Plasmide als Zusatz-DNA: Sie können leicht verändert, übertragen und zur Vervielfältigung von Fremd-DNA genutzt werden – ideal für die Gentechnik.
Die Möglichkeit, genetisches Material in Bakterien einzuschleusen und mitzukopieren, nutzen Forscher seit Jahrzehnten. So werden Bakterien zu kleinen „DNA-Fabriken“.
Ein historischer Durchbruch
1973 gelang Stanley Cohen und Herbert Boyer ein bahnbrechendes Experiment: Sie schleusten erstmals ein fremdes Gen in Bakterien ein. Ergebnis: Die Bakterien übernahmen das neue Gen und setzten es sogar um. Damit war bewiesen, dass Gene von einer Art auf eine andere übertragen werden können – die Geburtsstunde der modernen Gentechnik. [Das Cohen-Boyer-Experiment]b) Das Bakterium Escherichia coli (E. coli)
In der Forschung wird besonders gern das Bakterium Escherichia coli (kurz: E. coli) eingesetzt – und das hat mehrere Gründe:
- Es lässt sich leicht im Labor kultivieren und vermehren.
- Es vermehrt sich sehr schnell: Unter optimalen Bedingungen teilt es sich alle 20–30 Minuten.
- Sein Genom ist umfassend erforscht und verstanden.
- Es lässt sich einfach genetisch manipulieren.
- Die Kultivierung ist kostengünstig.
Abb. 1.1.2.2-B: Das Bakterium Escherichia coli Links eine mikroskopische Aufnahme, rechts eine vereinfachte Darstellung mit DNA (großes Knäuel) und Plasmiden (kleine Ringe).
Weil E. coli molekularbiologisch und genetisch der am besten untersuchte Organismus ist, nennen Wissenschaftler es scherzhaft das „Haustier der Genetiker“.
Aus diesen Gründen ist E. coli einer der wichtigsten Helfer in der Biotechnologie – und spielt auch bei der Impfstoffentwicklung eine zentrale Rolle.
Eine kleine Reise in eine Welt des E. coli Bakterium
Escherichia coli. – farblos, fast durchsichtig – kaum länger als ein Tausendstel eines Sandkorns – wirkt unscheinbar. Und doch ist es alles andere als langweilig.
Von außen sieht es aus wie eine kleine Kapsel, doch unter seiner Haut verbirgt sich eine der geschäftigsten Megastädte zur Hauptverkehrszeit.
Kein Platz für Leere. Molekül an Molekül. Proteine, Ribosomen, Nukleinsäuren – sie drängen sich, stoßen aneinander, weichen aus. Ein Zustand, den Biologen nüchtern Macromolecular Crowding nennen, der sich hier aber anfühlt wie permanentes Gedränge. Und trotzdem – oder gerade deswegen – funktioniert alles mit atemberaubender Präzision.
Mitten in diesem Gedränge liegt das Gedächtnis der Zelle: ein einziges, ringförmiges DNA-Molekül. Hier steht alles geschrieben: wie man Zucker frisst, wie man schwimmt, wie man sich teilt. Würde man diese DNA vorsichtig auseinanderziehen, wäre sie tausendmal länger als die Zelle selbst. Ein Band mit Millionen von Buchstaben, dicht gepackt, mehrfach verdreht, zu Schleifen und Windungen gezwungen. Supercoiling nennt man diese Kunst des Zusammenfaltens – als hätte jemand eine kilometerlange Geschichte in eine Streichholzschachtel gezwängt, ohne ein Wort zu verlieren.
Doch so mächtig diese DNA auch ist, sie spricht selten selbst. Stattdessen schickt sie unablässig Boten aus: RNA. Tausende, Zehntausende. Kurze Nachrichten, Arbeitsanweisungen, Baupläne. Manche sind nur flüchtige Zettel – mRNA, kaum tausend Zeichen lang, mit einer Lebensdauer von Minuten. Andere sind stabiler, massiver: rRNA, die tragenden Säulen der Ribosomen. Und dann die kleinen tRNA-Moleküle, kaum mehr als Handlanger, die doch an entscheidenden Stellen die richtigen Bausteine anliefern. Sie flitzen wie Kuriere durch das Zytoplasma. Ihr Ziel?
Die Ribosomen – die eigentlichen Giganten dieser Welt. Zehntausende von ihnen treiben durch das Zytoplasma. Sie lesen die flüchtigen mRNA-Botschaften und verwandeln sie in greifbare Realität: Proteine. Sie sind lautlos, unermüdlich, und sie verschlingen einen großen Teil der Ressourcen der Zelle. Fast ein Viertel aller Proteine hier existiert nur, um neue Proteine herzustellen.
Und diese Proteine sind überall. Millionen von ihnen, in tausend Formen. Enzyme, die Reaktionen beschleunigen, die ohne sie Tage oder Jahre dauern würden – hier geschehen sie in Sekundenbruchteilen. Einige arbeiten in Massen, immer wieder dieselbe Aufgabe. Andere sind seltene Spezialisten, vielleicht nur ein paar Dutzend Exemplare, aber im entscheidenden Moment unverzichtbar. Dazwischen patrouillieren die Wächter: RNasen, die alte Botschaften zerlegen. Sobald ein RNA-Befehl ausgeführt wurde, zerlegen sie ihn wieder in seine Einzelteile. So stapelt sich kein Informationsmüll, und die Bausteine können für neue Befehle recycelt werden. Mittendrin – wohldosiert: DNasen. Sie passen derweil auf, dass der wertvolle Masterplan im Archiv repariert wird, falls mal etwas kaputtgeht. Gleichzeitig vernichten sie fremde Eindringlinge und gewinnen daraus neue Energie.
All das spielt sich innerhalb einer Hülle ab, die ständig erneuert werden muss: eine lebendige Grenze aus Lipiden, deren äußere Schicht Endotoxine trägt. Und sie steht unter Zeitdruck. Denn dieses E. coli lebt nicht, um zu verharren. Es lebt, um sich zu teilen.
Unter günstigen Bedingungen zählt es die Zeit in Minuten. Zwanzig, vielleicht dreißig – dann muss aus einer Zelle zwei werden. Während im Inneren kopiert, gelesen und gebaut wird, wächst die Oberfläche mit. Millionen neuer Lipidmoleküle entstehen, fügen sich ein, erweitern die schützende Mauer. Es ist ein Wettlauf ohne Pause, ein kontrolliertes Chaos mit klarer Richtung.
Und dann kommt der Moment der Teilung. Kein dramatischer Schnitt, kein Ende – eher ein leises Auseinandergehen. Die Mutterzelle hört auf, als Individuum zu existieren. Doch ihre Geschichte zerreißt nicht. Sie setzt sich fort, zweimal. In zwei Zellen, die beide alt und neu zugleich sind. Jede trägt dieselbe lange DNA in sich, denselben Lärm aus Molekülen, dieselbe rastlose Ordnung.
Man kann sich die Welt in der E.-coli-Zelle noch um eine weitere, fast schon geheimnisvolle Ebene erweitert vorstellen:
Neben dem großen, ringförmigen Chromosom treiben kleinere DNA-Ringe durch das Gedränge. Plasmide. Sie sind wie lose Seiten im inneren Archiv der Zelle – nicht lebensnotwendig im strengen Sinn, aber oft entscheidend fürs Überleben. Manche tragen Rezepte für neue Fähigkeiten: Resistenz gegen ein Gift, ein Enzym, das eine ungewöhnliche Nahrungsquelle erschließt, manchmal nur ein kleines taktisches Extra für schlechtere Zeiten.
Sie bewegen sich frei, stoßen gegen Ribosomen, werden kurz von Proteinen gepackt, wieder freigegeben. Auch sie werden gelesen, ihre Gene in RNA übersetzt, ihre Botschaften landen auf denselben Ribosomen wie die des großen Chromosoms. In der überfüllten Stadt gibt es keine Sonderrechte – nur Funktion.
Wenn die Zelle sich auf die Teilung vorbereitet, geraten die Plasmide in Bewegung. Sie dürfen nicht vergessen werden. Spezielle Proteine sorgen dafür, dass sich Kopien bilden und dass jede der entstehenden Tochterzellen wenigstens ein Exemplar erhält. Ein stiller Akt der Weitergabe, fast wie das Zustecken eines Briefes im Gedränge, bevor sich zwei Wege trennen.
Manchmal endet ihre Geschichte aber nicht in der eigenen Nachkommenschaft. Gelegentlich öffnet sich eine Verbindung nach außen, zu einer Nachbarzelle. Dann wechseln Plasmide den Besitzer. Ein kurzer Kontakt, ein Transfer, und eine Information springt von einer Geschichte in eine andere. Fähigkeiten verbreiten sich so schneller als jede Mutation des Chromosoms es je könnte.
In dieser Welt sind Plasmide die Gerüchte, die Baupläne, die Überlebenskniffe, die nicht fest in Stein gemeißelt sind. Beweglich, austauschbar, opportunistisch. Und genau dadurch passen sie perfekt in das rastlose, gedrängte Innenleben von E. coli, in dem nichts stillsteht – nicht einmal das Erbgut selbst.
Empfehlenswert sind die Illustrationen von David Goodsell. Er zeichnet das Innere von Zellen maßstabsgetreu – seine Bilder von E. coli machen dieses „Gedränge“ der Moleküle erst so richtig greifbar.c) Plasmide – kleine DNA-Ringe mit großer Wirkung
Plasmide sind kleine, ringförmige DNA-Moleküle, die zusätzlich zum Bakterienchromosom in der Zelle vorkommen. Sie sind deutlich kleiner als das Hauptchromosom, liegen in variabler Anzahl vor und können sich unabhängig davon vervielfältigen. Im Gegensatz zur linearen DNA von Eukaryoten (z. B. beim Menschen) bilden Plasmide geschlossene Kreise aus doppelsträngiger DNA (dsDNA).
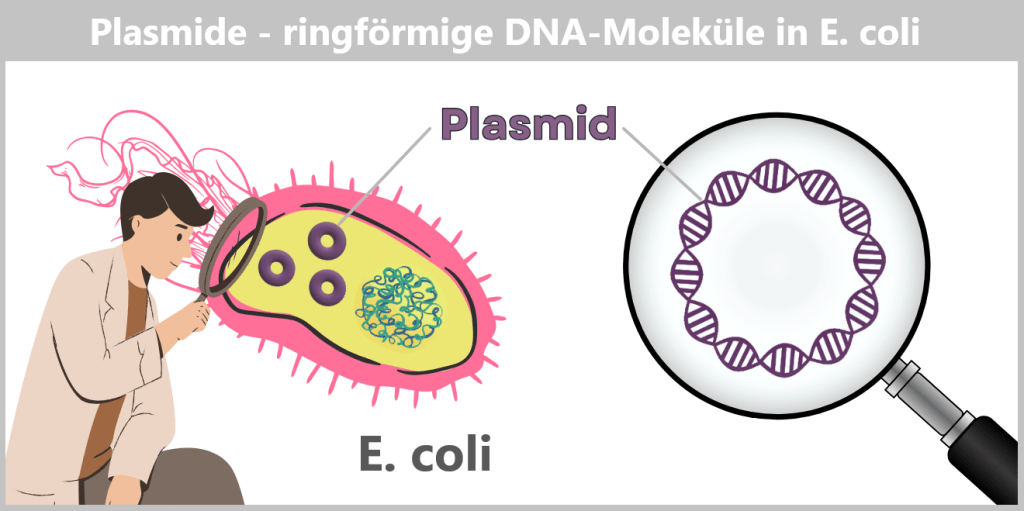
Abb. 1.1.2.2-C1: Schematische Darstellung der Plasmid-DNA des Escherichia coli Plasmide können ganz unterschiedliche Gene tragen – etwa für Antibiotikaresistenz oder für die Herstellung bestimmter Proteine. Damit sie von Forschern gezielt genutzt werden können, erstellt man sogenannte Plasmidkarten: schematische Darstellungen, die die wichtigsten Funktionsbereiche zeigen.
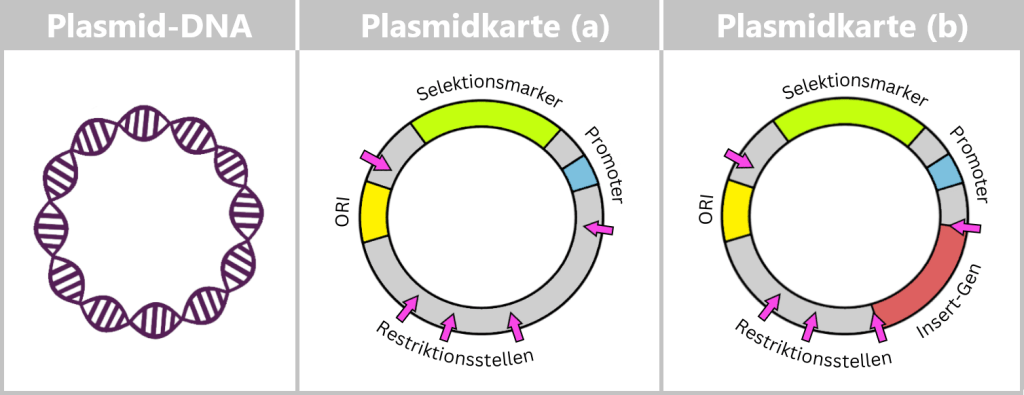
Abb. 1.1.2.2-C2: Plasmidkarte – schematische Darstellung ORI (Origin of Replication): Startpunkt für die Verdopplung des Plasmids. Wenn Umweltbedingungen und interne Signale günstig sind, kann das Bakterium diesen Startknopf drücken, und das Plasmid macht eine Kopie von sich selbst. Nur wenn dieser ORI mit dem Bakterium „kompatibel“ ist, kann sich das Plasmid zuverlässig vermehren, auch unabhängig von der Zellteilung.
Selektionsmarker: Gene, die den Bakterien einen Vorteil verschaffen, z. B. Resistenz gegen ein bestimmtes Antibiotikum. Sie helfen Forschern zu erkennen, welche Bakterien das Plasmid tragen.
Promoter: Der Promoter ist eine spezielle DNA-Region, die die Aktivität von Genen steuert. Er reguliert, wann und wie stark bestimmte Gene auf dem Plasmid abgelesen werden und steuert somit die Produktion der entsprechenden Proteine.
Restriktionsstellen: sind kurze DNA-Sequenzen, die von Restriktionsenzymen erkannt und gezielt geschnitten werden können. Diese Enzyme dienen Bakterien als eine Art Immunsystem, um die DNA von eindringenden Viren zu zerschneiden und sich so zu verteidigen.
d) Plasmide in der Gentechnik
Eigentlich dienen Restriktionsenzyme in Bakterien der Abwehr fremder DNA. In der Gentechnik nutzt man sie jedoch als präzise Werkzeuge: winzige molekulare Scheren, die DNA an ganz bestimmten Stellen schneiden.
So lässt sich eine „Lücke“ in einem Plasmid erzeugen, in die ein gewünschtes Gen eingefügt wird. Ein weiteres Enzym, die DNA-Ligase, „verklebt“ anschließend die DNA-Enden wieder. Das eingefügte Gen nennt man Insert-Gen (siehe obere Abbildung).
Durch dieses Verfahren verwandeln sich Plasmide in kleine Gen-Fähren: Sie transportieren gezielt neue Gene in Bakterien. Bei jeder Zellteilung wird das Plasmid – und damit auch das Insert-Gen – automatisch mitkopiert. Auf diese Weise entstehen ganze Bakterienkulturen, die als „Mini-Fabriken“ bestimmte Proteine oder DNA in großen Mengen herstellen.
🎥 Tipp: Eine kurze, animierte Einführung zu Plasmiden gibt es hier.
e) Einbau der Spike-Protein-DNA in ein Plasmid
Für die Vermehrung des Spike-Proteins von SARS-CoV-2 wird zunächst das Spike-Gen in ein bakterielles Plasmid integriert – ein Vorgang, den man als „Einklonieren“ bezeichnet. Dabei kommen die zuvor beschriebenen molekularbiologischen Werkzeuge zum Einsatz: Restriktionsenzyme öffnen das ringförmige Plasmid an definierten Stellen, während DNA-Ligasen das Spike-Gen passgenau einfügen und die DNA-Enden dauerhaft verbinden. Auf diese Weise entsteht ein rekombinantes (neu zusammengesetztes) Plasmid, das die genetische Information für das gewünschte Antigen enthält.
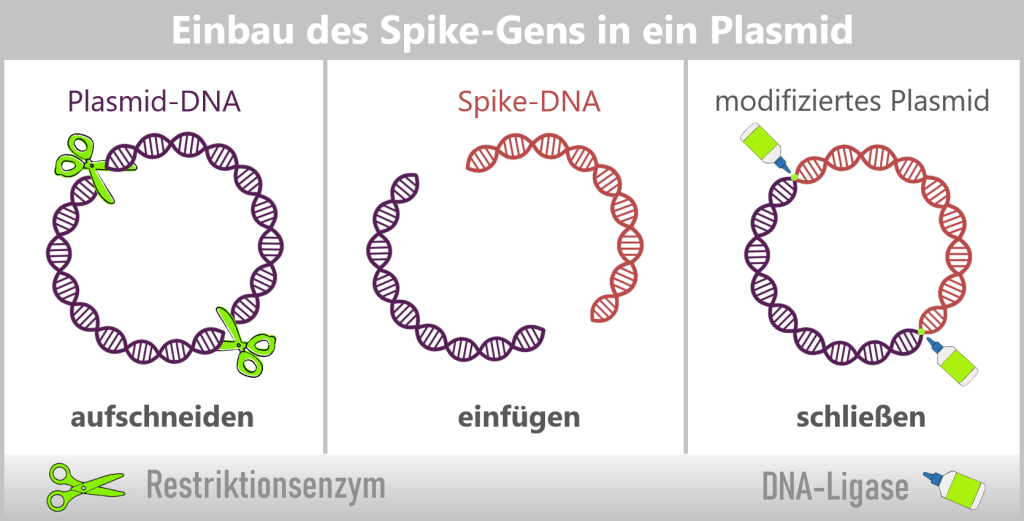
Abb. 1.1.2.2-E1: Wie das Spike-Gen in ein Plasmid eingefügt wird DNA-Elemente im Plasmid
Die Spike-DNA wird nicht isoliert in das Plasmid eingebracht, sondern zusammen mit weiteren genetischen Elementen, die für die Vermehrung des Plasmids erforderlich sind.
ORI (Origin of Replication): Der Replikationsursprung legt fest, an welcher Stelle die Vervielfältigung des Plasmids innerhalb der Bakterienzelle beginnt. Ohne dieses Element könnte das Plasmid in E. coli nicht stabil vermehrt werden.
Spike-Protein-Gen: Dieses Gen enthält den Bauplan für das Spike-Protein – das zentrale Antigen des Impfstoffs.
Antibiotikaresistenzgen: Das Resistenzgen dient als Selektionsmarker. Es stellt sicher, dass unter Antibiotikadruck nur diejenigen Bakterien überleben, die das gewünschte Plasmid aufgenommen haben. Dadurch können geeignete Bakterienklone gezielt ausgewählt und vermehrt werden.
- Moderna: Kanamycin-Resistenzgen
- Pfizer/BioNTech: Neo/Kan-Resistenzgen (Resistenz gegen Neomycin und Kanamycin)
SV40-Komponenten: Das Produktionsplasmid von Pfizer/BioNTech enthält zusätzlich regulatorische Sequenzelemente, die vom Simian Virus 40 (SV40) abgeleitet sind – einem Affenvirus, dessen genetische Elemente seit Jahrzehnten in der Molekularbiologie verwendet werden.
Konkret handelt es sich um:
- einen SV40-Promoter/Enhancer,
- sowie Teile des SV40-Replikationsursprungs.
Solche Sequenzen werden in der Gentechnik genutzt, weil sie die Genexpression in Säugetierzellen verstärken und die Plasmidvermehrung in bestimmten Zelllinien erleichtern können.
Für die bakterielle Vermehrung in E. coli besitzen diese Elemente jedoch keine bekannte Funktion, da Bakterien die hierfür notwendigen zellulären Faktoren nicht besitzen.
Warum diese SV40-Sequenzen im endgültigen Produktionsplasmid enthalten sind, ist nicht vollständig öffentlich dokumentiert. Diskutiert wird unter anderem, dass sie aus früheren Entwicklungs- oder Testsystemen übernommen und später beibehalten wurden.
Nach aktuellem Kenntnisstand verwendet Moderna in seinem Produktionsplasmid keine vergleichbaren SV40-Komponenten.
Die unterschiedliche Verwendung solcher regulatorischer Sequenzen gehört zu den Aspekten, die im Zusammenhang mit DNA-Rückständen wissenschaftlich diskutiert werden.
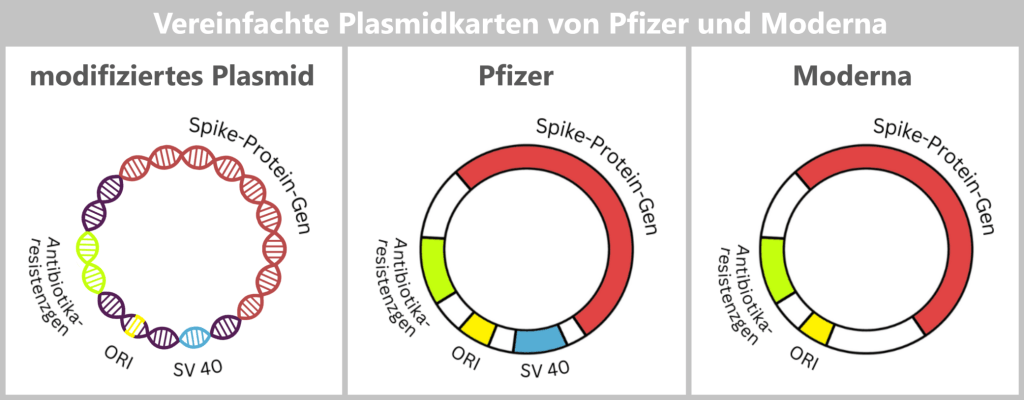
Abb. 1.1.2.2-E2: Vereinfachte Darstellung der Plasmidkarten von Pfizer und Moderna. Eine genauere Darstellung der Plasmidkarten findet man hier.
f) Übertragung der modifizierten Plasmide in Bakterien
Die veränderten Plasmide, die nun den Bauplan für das Spike-Protein tragen, werden anschließend in E. coli-Bakterien eingeschleust – ein Prozess, der als Transformation bezeichnet wird. Dabei nehmen die Bakterien die Plasmide auf; diese verbleiben dauerhaft in der Bakterienzelle und werden bei jeder Zellteilung weitergegeben.
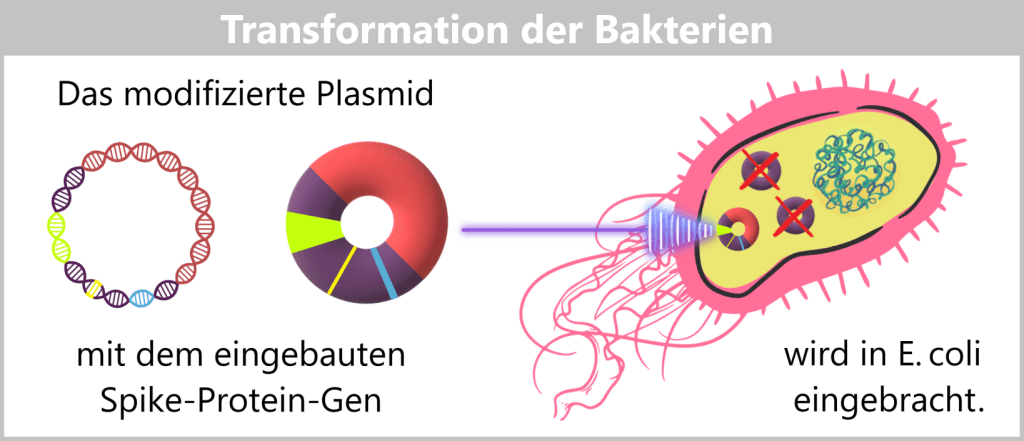
Abb. 1.1.2.2-F: Schematische Darstellung der Transformation Die modifizierten Plasmide werden mittels Transformation in E. coli-Bakterien eingebracht. Plasmide, die in der Gentechnik für den Transport fremder DNA-Sequenzen genutzt werden, heißen Vektoren.
Plasmidfreie Wirtszellen – für saubere Klone
Da in der biotechnologischen Plasmidproduktion ausschließlich die modifizierten Plasmide hergestellt werden sollen, verwendet man spezielle, plasmidfreie Bakterienstämme, die keine eigenen, natürlichen Plasmide besitzen.
Das hat mehrere Gründe:
- Natürliche Plasmide könnten um die zelluläre Replikationsmaschinerie konkurrieren,
- sie könnten durch Rekombination DNA-Abschnitte austauschen,
- und sie würden die genetische Zusammensetzung der Kolonie unvorhersehbar machen.
Durch den Einsatz plasmidfreier Wirtszellen wird sichergestellt, dass alle Bakterien einer Kolonie genetisch identische Klone sind – sie enthalten das gleiche, definierte Plasmid mit der gewünschten Sequenz.
g) Vermehrung der Bakterien
Die E. coli-Bakterien mit den modifizierten Plasmiden werden in einen Fermenter gegeben. Ein Fermenter, auch Bioreaktor genannt, ist ein Gerät, das in der Biotechnologie für die großtechnische Herstellung von Produkten wie Antibiotika, Enzymen, Vitaminen oder Impfstoffen genutzt wird. Darin lassen sich Bedingungen wie Temperatur, pH-Wert, Sauerstoffzufuhr, Rührgeschwindigkeit und Nährstoffversorgung exakt steuern.
Das Nährmedium im Fermenter enthält alle lebensnotwendigen Stoffe für die Bakterien. Unter diesen optimalen Bedingungen beginnen sie, sich rasant zu vermehren. Bei jeder Zellteilung werden auch die Plasmide mitkopiert, sodass sich die gewünschte DNA vervielfältigt.
Um sicherzustellen, dass nur Bakterien überleben, die tatsächlich das Spike-Gen-Plasmid tragen, wird zusätzlich ein Antibiotikum in den Fermenter gegeben. Nur Zellen mit Plasmid – und damit mit Antibiotikaresistenz – können wachsen. So entsteht eine Kultur, in der ausschließlich die gewünschten, modifizierten Bakterien vorkommen.
E. coli kann sich etwa alle 20–30 Minuten teilen. Innerhalb weniger Tage wächst so im Fermenter eine riesige Menge an Bakterien heran – mit Trillionen von Kopien des Spike-Plasmids.
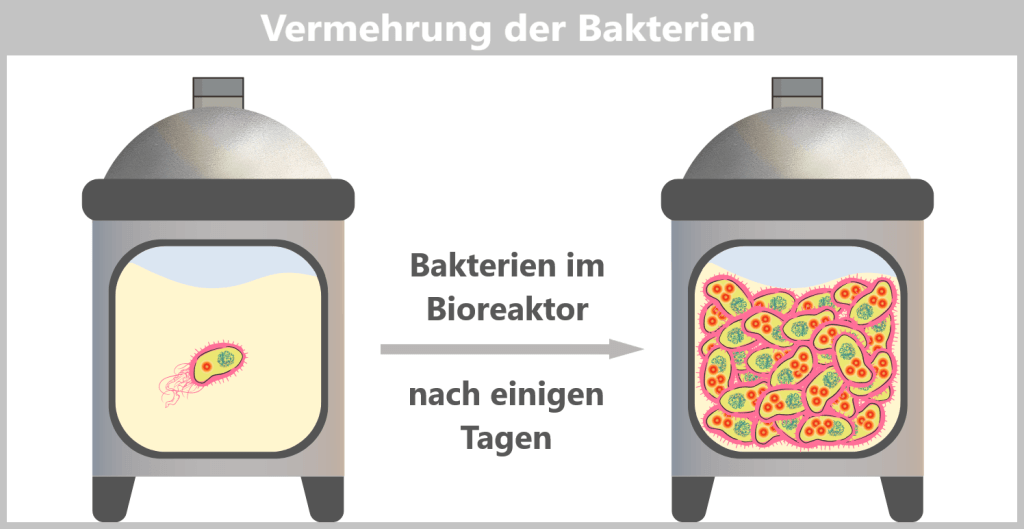
Abb. 1.1.2.2-G: Vermehrung von E. coli-Bakterien mit modifiziertem Plasmid im Bioreaktor h) Ernte der Bakterien
Nach der Vermehrungsphase werden die E. coli-Zellen „geerntet“. Dazu wird der gesamte Inhalt des Fermenters – die Zellsuspension – in einen Erntetank überführt. Dort erfolgt die Trennung von Flüssigkeit und Zellen, meist durch Zentrifugation (schnelles Schleudern) oder Filtration. Am Ende entsteht ein Zellpellet, das heißt eine konzentrierte Sammlung von Bakterien am Boden des Behälters.
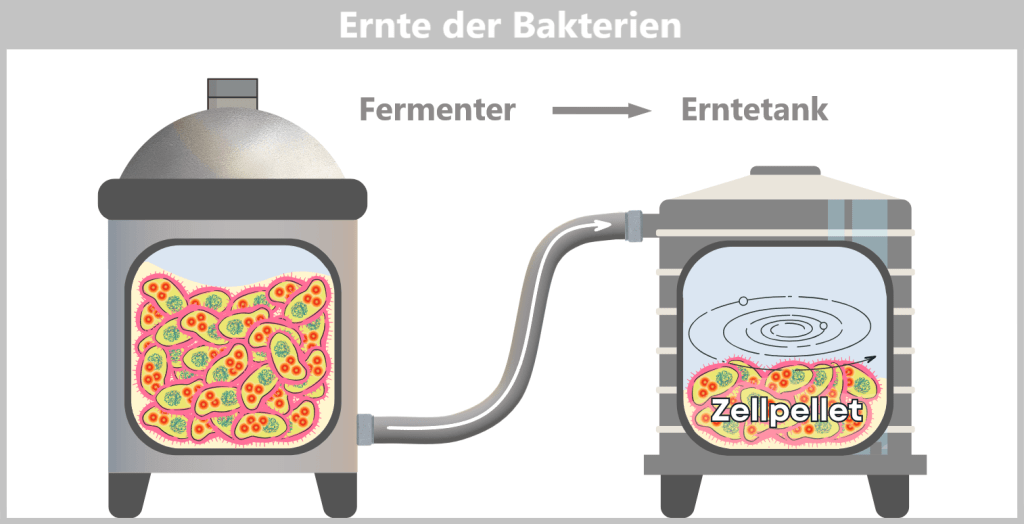
Abb. 1.1.2.2-H1: Überführung des Fermenterinhalts in den Erntetank und Gewinnung eines Zellpellets durch Zentrifugation oder Filtration Der Zellpellet – nun von der Nährlösung getrennt – wird in eine Aufarbeitungsanlage überführt. Entweder wird er hierfür in einer Flüssigkeit fein verteilt, bis ein gleichmäßiger, pumpfähiger „Zellbrei“ entsteht oder er wird als fester Pellet automatisch weitergeleitet. In modernen Anlagen geschieht das alles in einem geschlossenen System, ohne dass die Biomasse offen mit der Umgebung in Kontakt kommt.
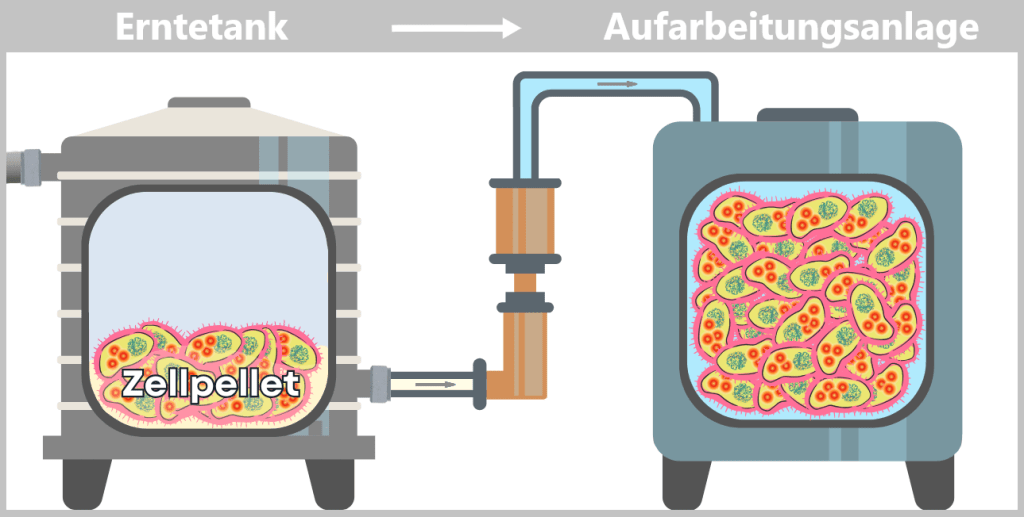
Abb. 1.1.2.2-H2: Überführung der Ernte in die Aufarbeitungsanlage i) Isolierung der modifizierten Plasmide
Nach der Ernte der Bakterienkulturen folgt ein entscheidender Trennungsprozess: Die E. coli-Zellen werden in speziellen Aufarbeitungsanlagen gezielt lysiert, das heißt kontrolliert aufgebrochen.
Die Zugabe von Natronlauge (NaOH) und dem Detergens SDS (einem speziellenSeifenmolekül) löst die fetthaltige Zellhülle auf, vergleichbar mit der Wirkung von Spülmittel auf Fett.
Dabei wird der gesamte Zellinhalt freigesetzt – ein komplexes Gemisch aus den gewünschten Plasmiden, chromosomaler DNA, Proteinen, Membranbestandteilen und zahlreichen weiteren zellulären Komponenten.
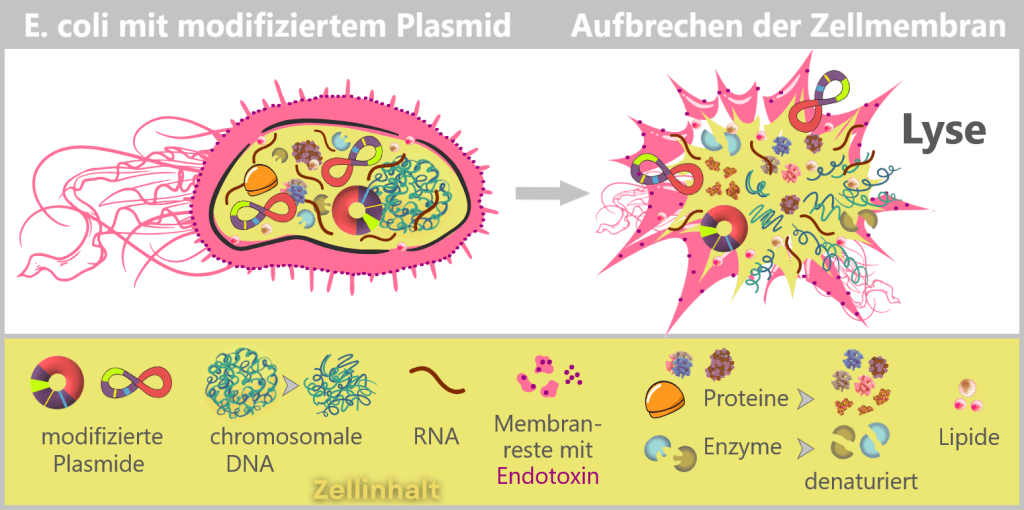
Abb. 1.1.2.2-I1: Lyse der Bakterien und Freisetzung des Zellinhalts Links: Schematischer Querschnitt durch ein E. coli mit modifiziertem Plasmid. Die äußere Membran ist mit eingelagerten Endotoxin-Molekülen (lila) dargestellt.
Rechts: Nach dem Aufbrechen der Zellmembran (Lyse) treten die verschiedenen Zellbestandteile – modifizierte Plasmide, chromosomale DNA (Bakterienchromosom), Proteine, Enzyme und Lipide – aus. Die äußere Membran liegt in Fragmenten vor, die das strukturell gebundene Endotoxin (LPS) enthalten.DNA-Formen in E. coli: Plasmid und Chromosom
Plasmide in Escherichia coli liegen als kleine, ringförmige DNA-Moleküle vor. In der Zelle befinden sie sich überwiegend in einer supercoiled Form. Diese Topologie (räumliche Anordnung) ist biologisch bevorzugt, da sie eine hohe mechanische Stabilität und Kompaktheit verleiht. Der geschlossene DNA-Ring ist dabei zusätzlich verdrillt, wodurch das Molekül platzsparend organisiert ist und gegenüber physikalischen Belastungen relativ widerstandsfähig bleibt.
Der Unterschied zwischen supercoiled (superhelikalen) und relaxed (entspannten, ringförmigen) Plasmiden liegt ausschließlich in ihrer Topologie. Beide Formen bestehen aus identischer genetischer Information, zeigen jedoch deutlich unterschiedliche physikalische Eigenschaften.
In der supercoiled Form steht das DNA-Molekül unter torsionaler Spannung und windet sich eng um sich selbst. Die relaxed Form entsteht typischerweise, wenn einer der beiden DNA-Stränge einen Einzelstrangbruch (Nick) erleidet. Durch diesen Nick kann die Spannung entweichen, und das Plasmid geht in eine offenere, weniger kompakte Ringstruktur über.
Das bakterielle Chromosom (genomische DNA, gDNA) ist ebenfalls ringförmig, aber um ein Vielfaches größer. Es ist hochgradig organisiert, mit Proteinen verknüpft und in viele Domänen gefaltet.
Ergänzende Erläuterungen zur Wirkung der alkalischen Lyse
Zerstörung der Genom-DNA, Erhalt der Plasmide
Während der alkalischen Lyse werden die Zellmembran und Zellwände aufgelöst. Die doppelsträngige DNA beider Molekülarten (Genom-DNA, Plasmid) wird kurzzeitig denaturiert und durch mechanische und chemische Scherkräfte belastet.
Dabei geschieht Folgendes:
- Die lange, faserige genomische DNA wird durch Scherkräfte zerrissen und linearisiert, da sie zu groß und empfindlich ist, um intakt zu bleiben.
- Die kompakte, superhelikale Plasmid-DNA bleibt hingegen weitgehend intakt.
Beim Neutralisieren der zuvor alkalischen Lösung kann sich die kleine, ringförmige Plasmid-DNA wieder korrekt renaturieren, d. h. ihre beiden Einzelstränge finden zueinander zurück und bilden erneut eine stabile Doppelhelix. Dadurch bleibt sie in Lösung.
Die wesentlich längere genomische DNA kann unter diesen Bedingungen nicht mehr vollständig renaturieren. Sie fällt gemeinsam mit Zellresten und Proteinen als unlöslicher Niederschlag aus.
Zerstörung von Proteinstrukturen
Die alkalische Lyse wirkt sich nicht nur auf Nukleinsäuren aus, sondern auch auf Proteine. Unter den stark basischen Bedingungen verlieren Proteine ihre dreidimensionale Faltung und damit ihre biologische Funktion. Auch große Komplexe wie die Ribosomen, die aus ribosomaler RNA und zahlreichen Proteinen bestehen, werden dabei vollständig zerstört und zerfallen in ihre Bestandteile. Während der anschließenden Neutralisation aggregieren (verklumpen) diese zerstörten Proteine und RNA-Fragmente – ähnlich wie Eiweiß beim Kochen fest wird – und bilden einen Niederschlag, der zusammen mit den restlichen Zelltrümmern leicht entfernt werden kann.
Warum nach der bakteriellen Lyse auch kleine RNA-Fragmente auftreten
Beim Aufbrechen der E. coli-Zellen gelangen nicht nur die Plasmide ins Lysat, sondern auch große Mengen bakterieller RNA. Diese besteht aus einer Vielzahl unterschiedlicher Moleküle, die für den Stoffwechsel der Bakterien zuständig waren – darunter viele kurze, stabile RNA-Typen. Zusätzlich werden beim Lysevorgang auch bakterielle Enzyme freigesetzt, die RNA abbauen (RNasen). Aktive RNasen können vorhandene RNA schnell in kleinere Stücke zerlegen. Auch mechanische Scherkräfte während der Lyse tragen zur Fragmentierung bei. Das Ergebnis ist ein komplexes Gemisch aus kurzen bakteriellen RNA-Fragmenten.
Natürliche RNA der Bakterienzelle
Die Bakterienzelle (E. coli) enthält bereits eine enorme Menge ihrer eigenen, natürlichen RNA – deutlich mehr als DNA. Beim Aufbrechen der Zellen (Lyse) gelangt diese gesamte zelluläre RNA mit in das Lysat und stellt eine bedeutende Verunreinigung dar. Dazu gehören:
- rRNA (ribosomale RNA): Macht den Hauptanteil (~80–90 %) der bakteriellen RNA aus und liegt in großen Mengen vor.
- tRNA (transfer-RNA): Sehr kleine, stabile Moleküle in vielen verschiedenen Varianten.
- Bakterielle mRNA: Ist in Bakterien von Natur aus kurzlebig und liegt oft bereits fragmentiert vor.
- Regulatorische kleine RNAs (sRNA): Umfassen natürliche RNA-Spezies von wenigen Dutzend Nukleotiden Länge.
Zusammenfassend bildet diese Gesamtheit an ribosomaler, transfer- und messenger-RNA den natürlichen zellulären RNA-Pool von E. coli.
Zerstörung der Membran und Freisetzung von Endotoxinen
Endotoxine (Lipopolysaccharide, LPS) sind ein natürlicher Bestandteil der äußeren Membran von Bakterien mit einer doppellagigen Zellhülle wie E. coli. In der Fachsprache werden solche Bakterien als ‚gramnegativ‘ bezeichnet. Sie sorgen für einen strukturellen Halt und helfen dem Bakterium bei Angriffen von außen. Bei der Lyse wird diese Membran zerstört, wodurch große Mengen LPS freigesetzt werden. Während Endotoxine für das Bakterium als „Rüstung“ fungieren, sind sie für den Menschen hochgiftig. Der biologisch aktive Bestandteil – Lipid A – kann beim Menschen starke Entzündungsreaktionen auslösen, weshalb Endotoxine zu den kritischsten Verunreinigungen in der biotechnologischen Produktion gehören.
Die Kunst besteht nun darin, die begehrten Plasmide aus dieser biochemischen Vielfalt zu isolieren und von allen unerwünschten Begleitstoffen zu befreien. Hierbei kommt eine mehrstufige Aufreinigung zum Einsatz, die wir später im Abschnitt Reinigungsverfahren im Herstellungsprozess detaillierter betrachten.
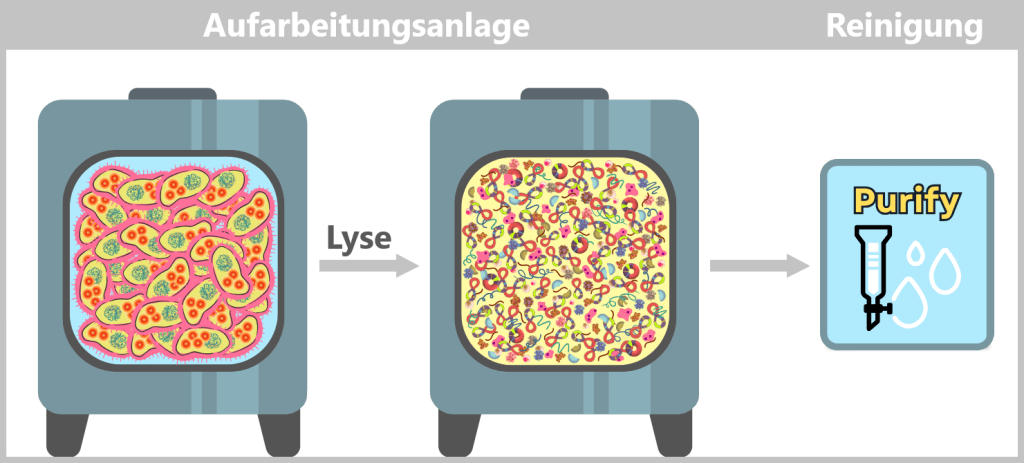
Abb. 1.1.2.2-I2: Lyse in der Aufarbeitungsanlage und Beginn der Plasmid-Reinigung In der Aufarbeitungsanlage werden die geernteten Bakterienzellen lysiert. Dabei entsteht ein „Zellbrei“, der alle zellulären Bestandteile enthält. Anschließend beginnt die Reinigung, bei der die gewünschten Plasmide mit Hilfe spezieller Filter- und Chromatografie-Verfahren aus dem Gemisch herausgetrennt werden.
Nach der Aufreinigung liegen die ringförmigen Plasmide in hoher Reinheit vor.
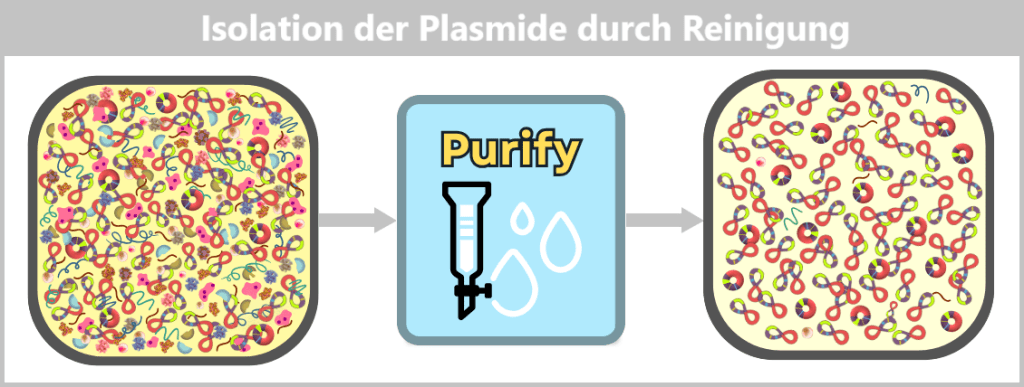
Abb. 1.1.2.2-I3: Ergebnis der Plasmid-Isolation Nach der Aufreinigung liegt die Plasmid-DNA in hoher Reinheit vor. Neben der dominanten supercoiled Konformation ist ein geringer Anteil an relaxed Plasmiden sichtbar. Letztere entstehen durch Einzelstrangbrüche (nicks) während der Zelllyse oder Aufarbeitung.
j) Linearisierung der Spike-Protein-DNA
Die gereinigten ringförmigen Plasmide enthalten zwar bereits den Bauplan für das Spike-Protein, sind für den nächsten Schritt jedoch noch ungeeignet. Für die Transkription muss die kreisförmige Plasmid-DNA in eine lineare Form überführt werden.
Dazu werden die Plasmide gezielt geöffnet: Mithilfe eines spezifischen Restriktionsenzyms erfolgt ein präziser Schnitt an einer definierten Stelle, üblicherweise nach dem Ende des Spike-Protein-Gens. Auf diese Weise entstehen klare DNA-Enden, die das Ablesen der Sequenz erleichtern.
Durch diesen Schnitt wird das gesamte Plasmid von seiner ringförmigen in eine lineare, offene DNA-Kette überführt. Diese enthält nicht nur das Spike-Gen, sondern auch alle übrigen Plasmidbestandteile, wie den Ursprung der Replikation (ORI), Selektionsmarker oder regulatorische Elemente. Eine besonders wichtige dieser Sequenzen ist der T7-Promotor, dies wird im nächsten Schritt deutlich.
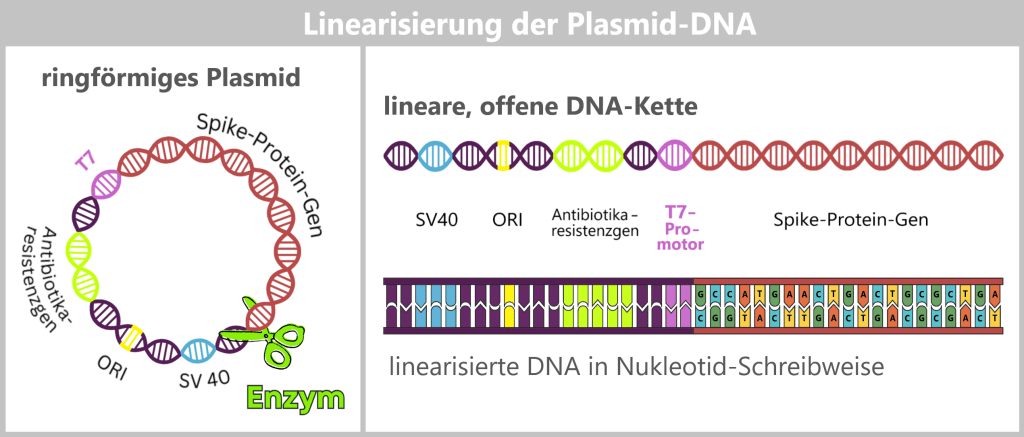
Abb. 1.1.2.2-J1: Linearisierung der Plasmid-DNA Das ringförmige Plasmid wird mit einem Restriktionsenzym aufgeschnitten und in eine lineare DNA-Kette überführt. Diese enthält verschiedene Abschnitte: den Ursprung der Replikation (ORI, gelb), Selektionsmarker (hellgrün), regulatorische Sequenzen wie SV40 (blau) und den T7-Promotor (lila), sowie das eigentliche Spike-Gen (rot). In der detaillierten Darstellung unten ist die Nukleotidfolge des Spike-Gens angedeutet. Die gesamte Nukleotidfolge ist stark gekürzt dargestellt.
Weitere Reinigung nach der Linearisierung
Die Linearisierung der Plasmid-DNA führt zur Entstehung zusätzlicher Reaktionsbestandteile und Nebenprodukte, die vor der in-vitro-Transkription entfernt werden müssen. Hierzu zählen insbesondere:
- Restriktionsenzyme: Die für den gezielten Schnitt eingesetzten Endonukleasen dürfen nicht im weiteren Herstellungsprozess verbleiben.
- DNA-Nebenprodukte: Hierzu gehören unvollständig linearisierte Plasmide (z. B. residuale supercoiled oder relaxed Formen) sowie kurze DNA-Fragmente, die durch unspezifische Brüche oder Nebenreaktionen entstehen können.
- Salze und Pufferreste: Die Linearisierungsreaktion erfolgt in spezifischen Puffersystemen, deren Ionen und Zusatzstoffe die nachfolgende Prozessschritte beeinträchtigen könnten.
Durch geeignete Reinigungsverfahren wird eine hochreine, lineare DNA-Vorlage gewonnen, die als Template für den nächsten Prozessschritt dient.
Der Begriff hochrein beschreibt in diesem Kontext keinen absoluten Zustand, sondern die weitgehende Entfernung prozessrelevanter Verunreinigungen auf ein regulatorisch akzeptiertes Minimum. Geringe Restmengen nicht-linearer Plasmidformen können verbleiben und werden in nachfolgenden Prozessschritten weiter reduziert.
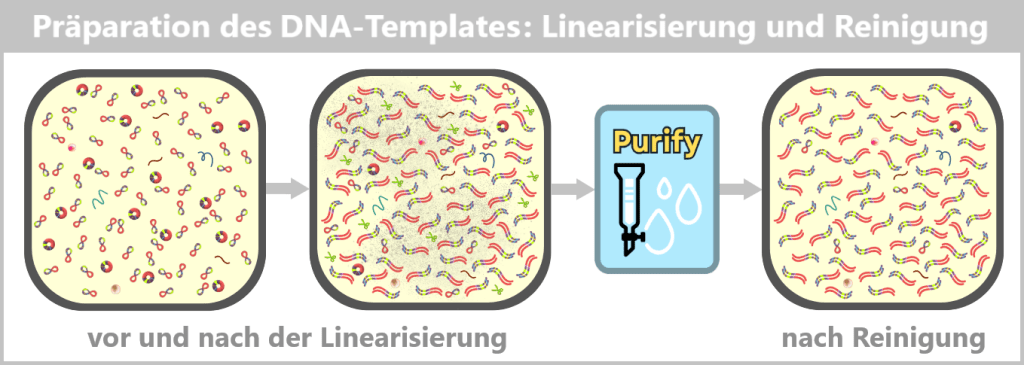
Abb. 1.1.2.2-J2: Vom Plasmid zur DNA-Vorlage: Herstellung des linearen DNA-Templates Links: Die Plasmid-DNA-Lösung vor der Linearisierung. Sie enthält ein Gemisch aus supercoiled (überwiegend) und relaxeden (offenen) Ringformen.
Mitte: Die Lösung nach der Linearisierung durch ein Restriktionsenzym. Die ringförmigen Plasmide wurden an einer definierten Stelle geschnitten und liegen nun als lineare, doppelsträngige DNA-Moleküle vor. Die Lösung enthält aber auch Reste des Enzyms, Puffersalze und mögliche Nebenprodukte.
Rechts: Die Lösung nach der Reinigung. Störende Bestandteile wie das Restriktionsenzym, Pufferkomponenten und unerwünschte DNA-Fragmente wurden entfernt.Schritt 3: In-Vitro-Transkription zur Herstellung der mRNA
Nachdem die Plasmid-DNA linearisiert wurde, dient sie nun als Vorlage für die in-vitro-Transkription (IVT). Ziel dieses Schrittes ist es, aus der DNA das RNA-Molekül herzustellen, das später als mRNA-Impfstoff eingesetzt wird.
Transkription – das „Umschreiben“ der DNA in RNA
Die Transkription erfolgt in einem separaten Bioreaktor unter kontrollierten Bedingungen (z. B. spezifischer pH-Wert, Temperatur, Ionenstärke), die speziell für die RNA-Synthese optimiert sind. Dafür sind drei Dinge notwendig:
1) Das DNA-Template, das das Spike-Gen enthält.
2) Die RNA-Bausteine, sogenannte Nukleotide.
3) Ein Enzym – die T7-RNA-Polymerase, das spezifisch den T7-Promotor erkennt. Der T7-Promotor wurde im Plasmid direkt vor die Spike-Gen-Sequenz eingefügt und markiert den Startpunkt der Transkription.Ablauf der Transkription
Initiation (Start): Die T7-RNA-Polymerase bindet an den T7-Promotor. Sobald das Enzym dort verankert ist, öffnet es die DNA-Doppelhelix um die Transkriptionsstartstelle und legt einen Strang als DNA-Template frei.
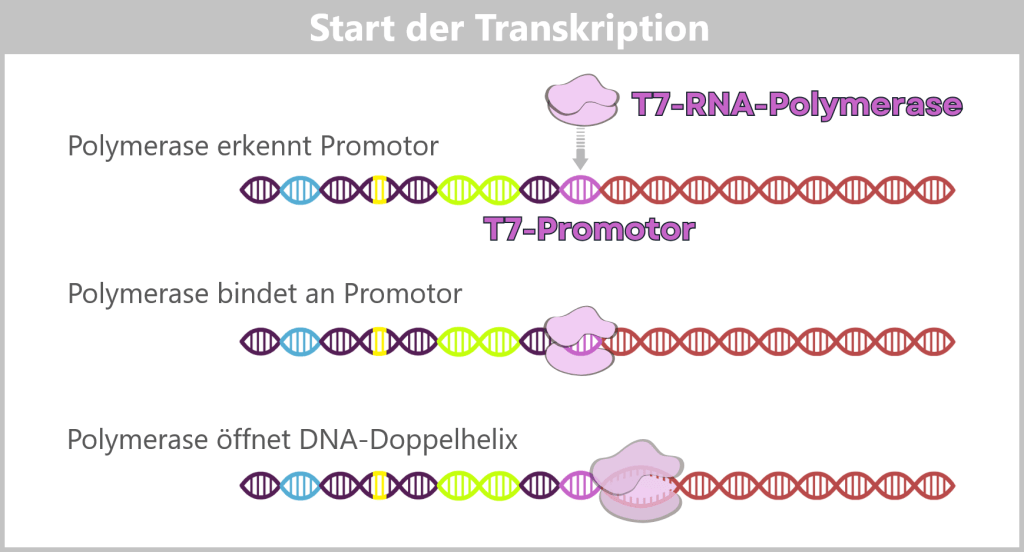
Abb. 1.1.3-A: Schematische Darstellung wie die T7-RNA-Polymerase an die DNA bindet. Die DNA enthält verschiedene funktionelle Abschnitte, darunter den Ursprung der Replikation (gelb), Selektionsmarker (hellgrün), regulatorische Elemente (blau), den T7-Promotor sowie die Spike-Gen-Sequenz (rot). Die Polymerase erkennt den T7-Promotor und setzt genau an dieser Stelle auf. Anschließend öffnet sie den DNA-Doppelstrang und beginnt mit der Synthese des RNA-Strangs entlang des Spike-Gens.
Elongation (Verlängerung): Die Polymerase wandert am DNA-Templatestrang in 3′ → 5′-Richtung entlang und synthetisiert dabei den komplementären RNA-Strang durch schrittweise Anlagerung von Nukleotiden in 5′ → 3′-Richtung. Dabei gilt die komplementäre Basenpaarung:
- DNA-A (Adenin) → RNA-U (Uracil bzw. m¹Ψ)
- DNA-T (Thymin) → RNA-A (Adenin)
- DNA-C (Cytosin) → RNA-G (Guanin)
- DNA-G (Guanin) → RNA-C (Cytosin)
Hinter der Polymerase schließt sich die DNA wieder.
Termination (Ende): Am Ende des Spike-Gens befindet sich eine Terminator-Sequenz. Sobald diese erreicht ist, stoppt die Polymerase, löst sich von der DNA, und die fertige RNA wird freigesetzt.
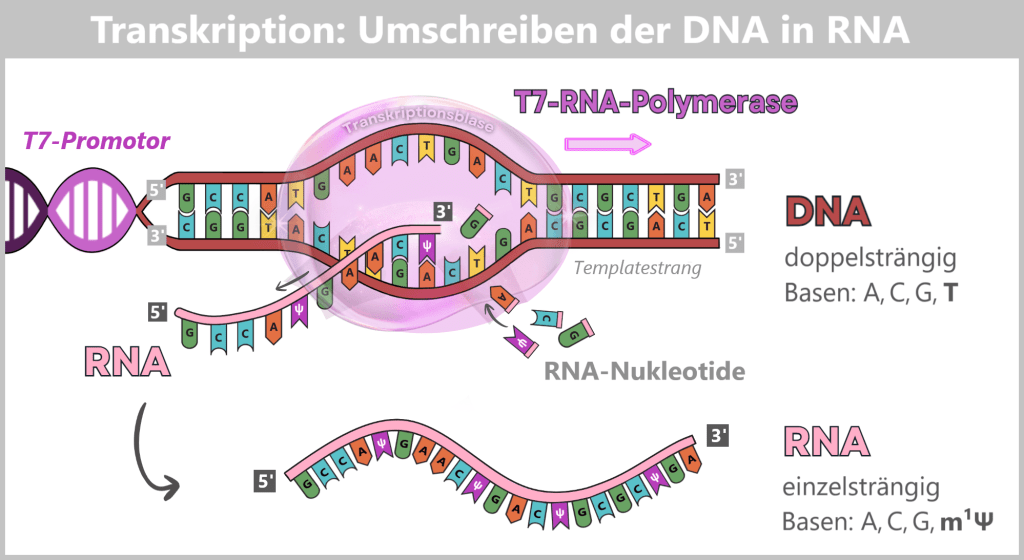
Abb. 1.1.3-B: Die Abbildung veranschaulicht den molekularen Mechanismus der Transkription. Die T7-RNA-Polymerase lagert sich an die DNA an und trennt die beiden Stränge. Entlang des Templatstrangs werden passende RNA-Nukleotide eingebaut. Schrittweise entsteht so ein einzelsträngiger RNA-Strang, der die Information des Spike-Gens trägt.
Die RNA, die auf diese Weise entsteht, ist einzelsträngig und entspricht in ihrer Basenfolge dem Spike-Gen.
Besonderheit der mRNA-Herstellung
Um die Stabilität und Verträglichkeit für den Einsatz im Impfstoff zu verbessern, wird ein modifizierter Baustein verwendet:
Anstelle von Uridin (U) wird N¹-Methyl-Pseudouridin (m¹Ψ) eingebaut.
Diese Modifikation macht die RNA stabiler, schützt sie vor zu schnellem Abbau und reduziert unerwünschte Immunreaktionen.
Sowohl die mRNA-Impfstoffe von Pfizer als auch von Moderna enthalten N¹-Methyl-Pseudouridin (m¹Ψ) anstelle von Uridin. [spektrum]
Vergleich DNA, mRNA und modRNA
Aus der doppelsträngigen DNA entsteht im Verlauf der in-vitro-Transkription eine einzelsträngige RNA. DNA und RNA ähneln sich in ihrer chemischen Grundstruktur, unterscheiden sich jedoch in entscheidenden Eigenschaften wie Stabilität, Lebensdauer und biologischer Funktion. Für mRNA-Impfstoffe kommt zudem keine natürliche mRNA zum Einsatz, sondern eine gezielt chemisch modifizierte Form (modRNA). Diese unterscheidet sich von natürlicher mRNA in mehreren wesentlichen Punkten.
Die folgende Tabelle stellt die Eigenschaften von DNA, natürlicher mRNA und synthetischer modRNA gegenüber.
Eigenschaft DNA Natürliche mRNA Synthetische modRNA Definition Master-Archiv: dauerhafter Speicher der gesamten Erbinformation Abschrift eines einzelnen Gens: enthält die Bauanleitung für ein körpereigenes Protein Kopie eines Gens mit „Tarnkappe“: im Labor hergestellte (synthetische) Form der mRNA, die gezielt chemisch verändert wurde. Sie enthält den Bauplan für ein „nicht körpereigenes“ Protein. Vorkommen Universell: Zellkern (bei Eukaryoten); zusätzlich mitochondriale DNA. Zellspezifisch: Wird bedarfsgerecht nur dort hergestellt, wo das entsprechende Protein benötigt wird. Unspezifisch: kann mithilfe von Lipid-Nanopartikeln in viele Zelltypen des Körpers aufgenommen werden. Struktur doppelsträngig einzelsträngig einzelsträngig Zucker Desoxyribose Ribose Ribose Basen A – Adenin
C – Cytosin
G – Guanin
T – ThyminA – Adenin
C – Cytosin
G – Guanin
U – UracilA – Adenin
C – Cytosin
G – Guanin
m¹Ψ – N1-MethylpseudouridinLebensdauer und Abbau-Geschwindigkeit Sehr stabil: bleibt durch Kernmembran und Reparatursysteme geschützt; wird im Normalfall nicht abgebaut, sondern nur bei Zelltod oder gezieltem DNA-Abbau. Kurzlebig: Minuten bis Stunden. Die Protein-produktion ist flexibel an den aktuellen Stoff-wechselbedarf angepasst. Verlängert: Stunden bis Tage. Durch den Austausch von Uridin durch N1-Methylpseudouridin wird die modRNA weniger stark von RNA-Sensoren des angeborenen Immunsystems erkannt und langsamer abgebaut. Abbau-Enzyme DNasen
(Desoxyribonukleasen)RNasen
(Ribonukleasen)RNasen
(verminderte Erkennung und verlangsamter Abbau)
IVT-Nebenprodukte und Verunreinigungen
Nach der in-vitro-Transkription (IVT) liegt kein reines Produkt vor, sondern ein komplexes Gemisch. Neben der gewünschten mRNA entstehen prozessbedingt verschiedene Nebenprodukte und Verunreinigungen, darunter kurze oder lange einzelsträngige RNA (ssRNA), doppelsträngige RNA (dsRNA) sowie RNA:DNA-Hybride. Diese Begleitprodukte müssen gezielt entfernt werden, da sie andernfalls die Stabilität, Wirksamkeit und Verträglichkeit des Impfstoffs beeinträchtigen können. Die hierfür eingesetzten Reinigungsmethoden werden im Kapitel 1.3. näher erläutert.
Schritt 4: RNA-Prozessierung – Reifung der mRNA
Die RNA-Prozessierung umfasst eine Reihe von Veränderungen, die während oder nach der Transkription stattfinden, um aus der RNA eine reife, funktionale mRNA zu erzeugen.
Bei der Herstellung der mRNA für Impfstoffe wird versucht, die natürlichen Prozesse so weit wie möglich nachzuahmen, die normalerweise auch in menschlichen Zellen stattfinden. Die synthetisch hergestellte mRNA wird so gestaltet, dass sie bestimmte Eigenschaften von natürlich vorkommender mRNA imitiert und dadurch stabil ist sowie effizient in das gewünschte Protein übersetzt werden kann.
Eine funktionelle Impfstoff-mRNA braucht wie eine normale menschliche mRNA:
- eine Schutzkappe (5′-Cap) am vorderen Ende der RNA
- einen Stabilisierungsschwanz (Poly-A-Schwanz) am hinteren Ende
Der 5‘-Cap: Ein „Sicherheitssiegel“ für die Zelle
Damit eine künstlich hergestellte mRNA im Körper funktioniert, muss sie am sogenannten 5′-Ende eine spezielle chemische Schutzstruktur tragen – die Cap-1-Struktur. Diese Kappe ist ein zentrales Erkennungsmerkmal für die Zelle und entscheidet darüber, ob die mRNA stabil bleibt, effizient übersetzt und nicht als fremd erkannt wird.
Die Bauteile der Cap-1-Struktur (m⁷GpppN¹m)
Die Cap-1-Struktur ist kein einfacher „Deckel“, sondern ein präzise aufgebautes Molekül aus drei funktionellen Komponenten:
Das Erkennungsmerkmal: 7-Methylguanosin (m⁷G)
Ein spezieller Guanosin-Baustein mit einer Methylgruppe. Diese Markierung wirkt wie ein molekularer Ausweis: Nur mRNAs mit dieser Struktur werden von der Zelle als korrekt und vertrauenswürdig erkannt.Die Verbindung: Triphosphatbrücke (ppp)
Drei Phosphatgruppen verbinden die Kappe in einer ungewöhnlichen 5′-5′-Verknüpfung mit der mRNA. Diese spezielle Bindung schützt das RNA-Ende effektiv vor enzymatischem Abbau.Die Tarnung: modifiziertes erstes Nukleotid (N¹m)
Das erste Nukleotid der mRNA ist zusätzlich 2′-O-methyliert. Diese Modifikation ist entscheidend, um die mRNA vor der Erkennung durch zelluläre RNA-Sensoren zu schützen.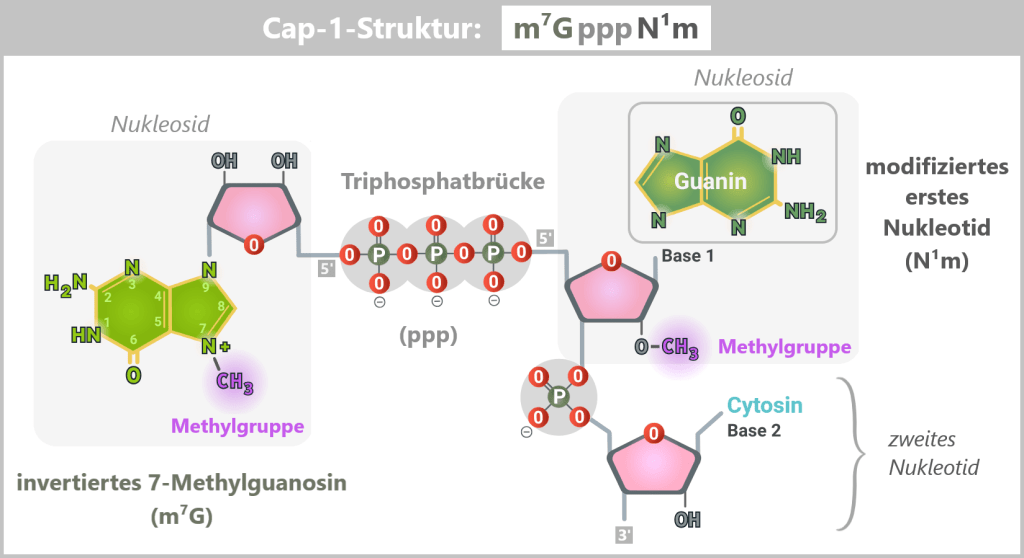
Abb. 1.1.4-A: Schematische Darstellung der Cap-1-Struktur (m⁷GpppN¹m) Das 5′-Ende der mRNA ist durch ein invertiert verknüpftes, am N7-Atom methyliertes Guanosin (m⁷G) über eine 5′–5′-Triphosphatbrücke (ppp) mit dem ersten Nukleotid (in diesem Beispiel Guanin) der mRNA verbunden. Zusätzlich trägt dieses erste Nukleotid eine 2′-O-Methylierung (N¹m), die die Cap-1-Struktur definiert und zur Stabilität, Translationseffizienz und Immuntarnung der mRNA beiträgt.
Bei der natürlichen Genexpression in menschlichen Zellen wird mRNA bereits während ihrer Entstehung im Zellkern prozessiert. Insbesondere erhalten alle durch die RNA-Polymerase II synthetisierten mRNA-Moleküle sehr früh während der Transkription eine 5′-Cap-Struktur sowie begleitende chemische Modifikationen.
Ungecappte mRNA ist in eukaryotischen Zellen unter physiologischen Bedingungen nicht funktionsfähig: Sie ist instabil, wird rasch abgebaut und gelangt nicht in das Zytoplasma. Dies liegt daran, dass ihr 5′-Ende als 5′-Triphosphat (5′-ppp) vorliegt – eine Struktur, die von zellulären Qualitätskontrollsystemen als Anomalie erkannt und beseitigt wird (siehe untere Abbildung).
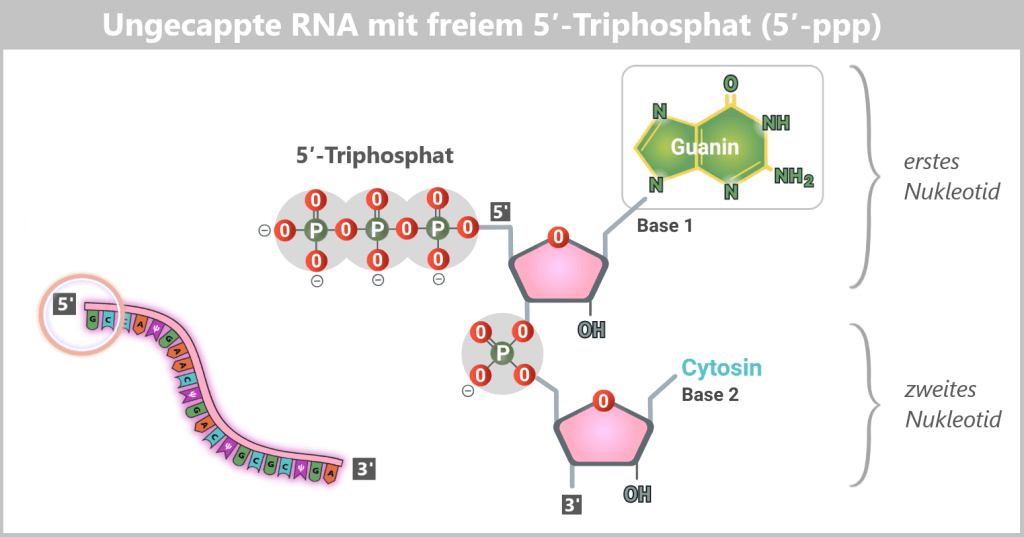
Abb. 1.1.4-B: Ungecappte RNA – das freie 5′-Triphosphat ist ein zentrales Erkennungsmuster für zytosolische Immunsensoren. Warum ist Cap-1 so wichtig?
Die Cap-1-Struktur erfüllt drei zentrale Funktionen:Tarnung (Immunschutz): Zelluläre Mustererkennungsrezeptoren wie RIG-I reagieren empfindlich auf RNA mit freien 5′-Triphosphaten oder unmodifizierten Enden. Die Cap-1-Struktur der modRNA ist chemisch identisch zur Cap-1-Struktur körpereigener mRNA. Da zelluläre RNA-Sensoren diese Struktur als „selbst“ erkennen, wird unter physiologischen Bedingungen keine ausgeprägte angeborene Immunantwort ausgelöst.
Produktionsstart: Das Cap dient als Andockstelle für cap-bindende Proteine und markiert den Startpunkt für die Proteinsynthese durch Ribosomen.
Stabilität: Die Kappe schützt das 5′-Ende der mRNA vor raschem enzymatischem Abbau und verlängert so ihre funktionelle Lebensdauer in der Zelle.
Der Poly-A-Schwanz: Das Schutz- und Kontrollzentrum am Ende
Am anderen Ende der mRNA (dem sogenannten 3′-Ende) befindet sich eine lange Kette, die ausschließlich aus Adenin-Bausteinen besteht – der Poly-A-Schwanz. In der Impfstoffherstellung wird dieser meist aus 100 bis 150 „A“-Gliedern gefertigt.
Wofür ist dieser Schwanz gut?
Die „Sanduhr“ der mRNA: Die Zelle besitzt Enzyme, die mRNA-Moleküle von hinten nach vorne langsam „anknabbern“. Der Poly-A-Schwanz wirkt hier wie ein Puffer oder ein Schutzstück am Ende. Er wird zuerst abgebaut, bevor die eigentliche genetische Information angegriffen wird. Je länger der Schwanz, desto länger überlebt die mRNA in der Zelle und desto mehr Protein kann hergestellt werden.
Übersetzungsbeschleuniger: Spezifische Proteine binden gleichzeitig an das 5′-Cap und den Poly-A-Schwanz und bilden den sogenannten closed-loop-Komplex – eine effiziente „Rundlaufstrecke“. Zum einen signalisiert er dem Ribosom (der Proteinfabrik), dass die mRNA vollständig und für die Translation bereit ist. Zum anderen kann das Ribosom nach Beendigung der Synthese eines Proteins direkt am 5′-Ende erneut mit der Translation beginnen. Dieser Mechanismus steigert die Geschwindigkeit und Ausbeute der Proteinproduktion erheblich.
Zusammenfassend: Während das 5′-Cap die mRNA legitimiert, tarnt und für die Translation verfügbar macht, bestimmt der Poly-A-Schwanz, wie lange und wie oft die Zelle die Information nutzt. Beide Modifikationen sind daher entscheidend für die Lebensdauer der mRNA und für ihre effiziente Übersetzung in Protein.
In den folgenden schematischen Darstellungen wird die Cap-1-Struktur als funktionelle 5′-Einheit vor dem RNA-Strang dargestellt, obwohl sie chemisch eine modifizierte Erweiterung des ersten Nukleotids darstellt. Der Poly-A-Schwanz wird aus didaktischen Gründen durch drei Adeninreste symbolisiert.
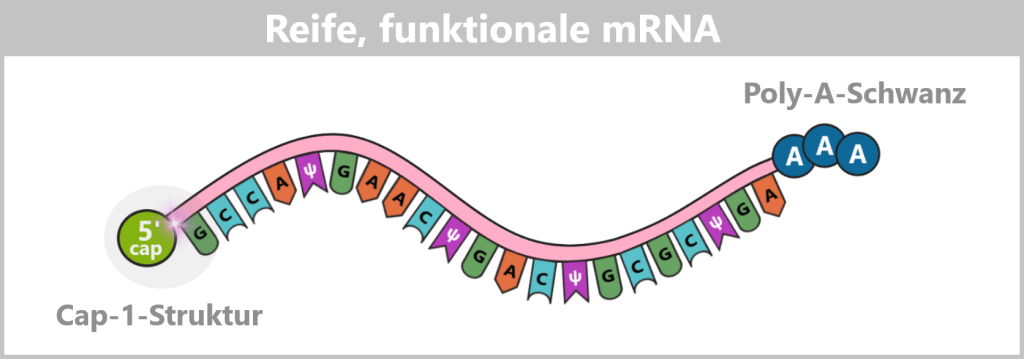
Abb. 1.1.4-C: Schematische Darstellung einer reifen mRNA mit 5′-Cap und Poly-A-Schwanz Die Abbildung ist stark vereinfacht; die tatsächliche mRNA ist deutlich länger und umfasst beim COVID-19-Impfstoff ca. 4200 Nukleotide.
In der biotechnologischen Herstellung haben sich zwei Methoden etabliert, um diese essenziellen Strukturen zu erzeugen:
1. Co-transkriptionelle Modifikation (Die „All-in-One“-Methode)
Hierbei werden Cap und Poly-A-Schwanz bereits während der in-vitro-Transkription (IVT) synthetisiert.
mRNA-Capping: Der Reaktion werden bereits fertige Cap-Bausteine (Cap-Analoga) zugegeben. Die T7-RNA-Polymerase baut diese Cap-Struktur automatisch als erstes Element an das entstehende mRNA-Molekül an.
Polyadenylierung: Das DNA-Template enthält bereits eine Sequenz, die als Matrize für einen definiert langen Poly-A-Schwanz dient. Die Polymerase synthetisiert ihn somit direkt im Anschluss an die kodierende Sequenz.
Vorteil: Der Prozess ist schnell, skalierbar und findet in einem einzigen Reaktionsgefäß statt. Durch die Verwendung moderner Cap-Analoga (z. B. ARCA oder CleanCap) werden Capping-Effizienzen von >95 % erreicht.
In der industriellen Praxis ist die co-transkriptionelle Methode der vorherrschende Standard.
2. Enzymatische Modifikation (Die “Step-by-Step”-Methode)
Bei diesem traditionelleren, mehrstufigen Verfahren werden Cap und Poly-A-Schwanz in separaten Reaktionsschritten nach der IVT angefügt.
Polyadenylierung: Zunächst wird die RNA mit dem Enzym Poly(A)-Polymerase behandelt. Dieses Enzym ist darauf spezialisiert, am 3′-Ende der RNA eine lange Adenin-Kette anzubauen. Die mRNA erhält also nachträglich ihren Poly(A)-Schwanz.
mRNA-Capping: Im Anschluss wird in einer weiteren Reaktion die 5′-Cap-Struktur schrittweise aufgebaut.
Vorteil: Erzielt eine sehr hohe und saubere Capping-Effizienz, ist jedoch aufwändiger.
Unabhängig vom gewählten Verfahren schließt sich eine umfassende Reinigung an, um eine homogene und hochreine mRNA zu erhalten.
Die wesentlichen Schritte – von der reinen DNA-Vorlage bis zum formulierungsfertigen Produkt – sind in der folgenden Übersicht dargestellt:
Abb. 1.1.4-D: Im Prozess: Von der gereinigten DNA zur formulierungsfertigen mRNA Nach der IVT liegt eine komplexe Mischung aus mRNA, Template-DNA, Enzymen, Nebenprodukten und Reaktionskomponenten vor. Durch nachfolgende Reinigungsschritte wird diese Mischung auf eine überwiegend reine mRNA-Lösung reduziert, die als Ausgangsmaterial für die LNP-Formulierung dient.
Schritt 5: Verpackung der mRNA in Lipid-Nanopartikel (LNPs)
Nachdem die mRNA vollständig hergestellt und gereinigt wurde, muss sie noch geschützt und transportfähig gemacht werden. Genau das übernimmt die Verpackung in Lipid-Nanopartikel (LNPs).
Warum braucht die mRNA eine Verpackung?
mRNA ist ein sehr empfindliches Molekül. Ohne Schutz würde sie im Körper sofort abgebaut werden. LNPs übernehmen mehrere Aufgaben gleichzeitig:
- sie schützen die mRNA vor Abbau
- sie transportieren sie in die Körperzellen (durch die Zellmembran)
- sie ermöglichen die kontrollierte Freisetzung der mRNA in der Zelle
Ohne LNPs wäre der Impfstoff nicht funktionsfähig.
Wie entsteht ein LNP? – Der Grundmechanismus
Die Formulierung passiert typischerweise in einem Gerät wie einem Microfluidizer oder Nanopartikel-Mischer. Dabei werden zwei Flüssigkeiten extrem schnell miteinander vermischt:
1) Eine ethanolische Lipid-Lösung
Sie enthält vier verschiedene Lipide:
- ionisierbares kationisches Lipid (bindet die mRNA und ermöglicht Zellaufnahme)
- Phospholipid (stabilisiert die Struktur – ähnlich wie in Zellmembranen)
- PEG-Lipid (sorgt für die richtige Größe und verhindert Aggregation)
- Cholesterin (macht das Partikel flexibel und stabil)
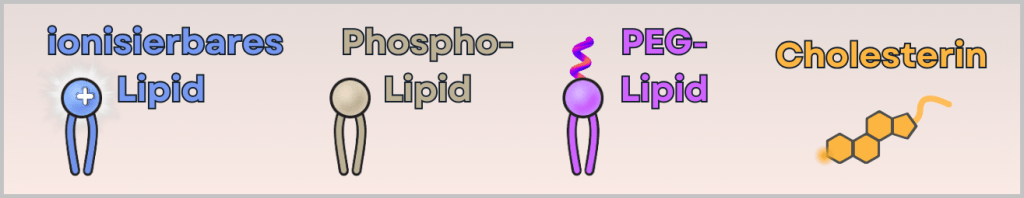
2) Eine wässrige mRNA-Lösung
Diese enthält nur:
- gereinigte mRNA
- einen milden Puffer
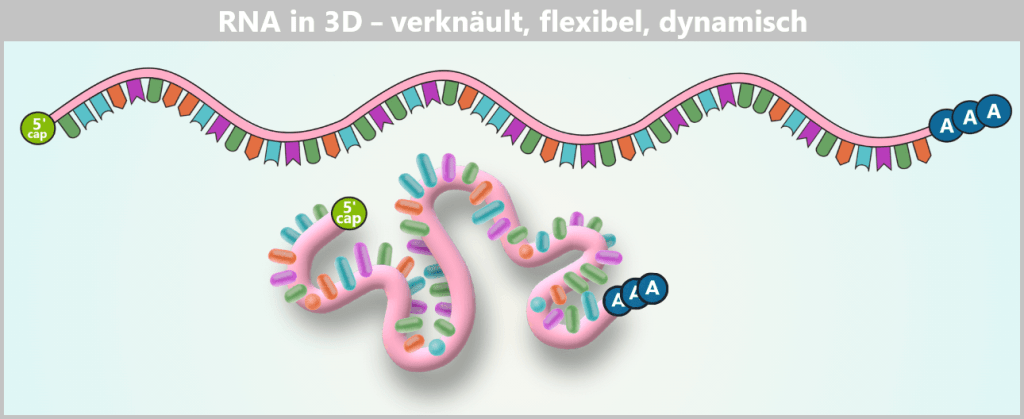
Abb. 1.1.5-A: Schematische Darstellung der RNA in Lösung – linear vs. 3D Oben ist die RNA als linearer Informationsstrang dargestellt – so liest man die Basensequenz. Unten ist gezeigt, wie derselbe Strang in wässriger Lösung tatsächlich existiert: als flexibles, ständig bewegtes 3D-Knäuel, in dem einzelne Abschnitte nur lose oder temporär miteinander wechselwirken.
Was passiert beim Mischen – ein Selbstorganisationseffekt
Wenn die beiden Lösungen in Sekundenbruchteilen aufeinandertreffen, ändern sich:
- pH-Wert
- Löslichkeit der Lipide
- Ladungszustände
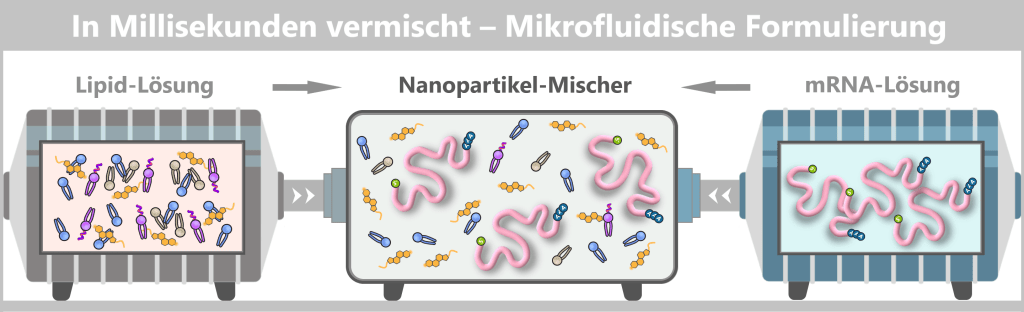
Abb. 1.1.5-B: Das Mikrofluidik-Prinzip: Präzisions-Mischen im Millisekunden-Takt Die Abbildung zeigt schematisch das Prinzip beim mikrofluidischen Mischen. Links und rechts strömen zwei getrennte Flüssigkeiten in den Mischer ein: eine Lipid-Lösung (links) mit den verschiedenen Lipidtypen und eine mRNA-Lösung (rechts) mit frei gelösten mRNA-Strängen. Im zentralen Mikrofluidik-Kanal treffen beide Ströme aufeinander und werden intensiv verwirbelt.
Dadurch passiert ein spontaner, hochpräziser Prozess:
- Das ionisierbare Lipid wird positiv geladen → es zieht die negativ geladene mRNA an.
- Die mRNA wird „eingewickelt“ und eingeschlossen.
- Die anderen Lipide ordnen sich drumherum zu einer stabilen Hülle an.
Es entsteht automatisch ein Nanopartikel mit typischer Größe von 60–100 nm. Es ist also kein „manuelles Verpacken“, sondern eine biophysikalische Selbstorganisation – die Moleküle finden von selbst die richtige Struktur.
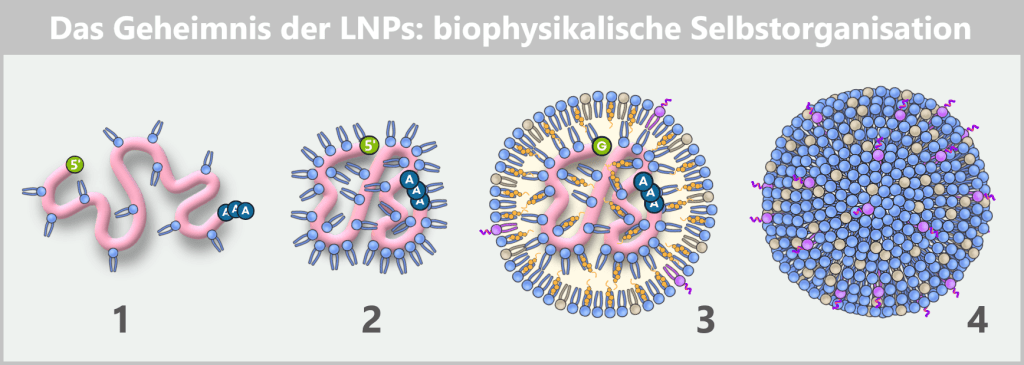
Abb. 1.1.5-C: Wie aus zwei Flüssigkeiten ein Nanopartikel wird: Mikrofluidische Formulierung Die Abbildung zeigt in vier Schritten, wie sich mRNA und Lipide während der Formulierung ganz von selbst zu einem Lipid-Nanopartikel organisieren.
1) Sobald mRNA und ionisierbare Lipide aufeinandertreffen, binden die positiv geladenen Lipidköpfchen sofort an die negativ geladenen Phosphate der mRNA und beginnen, den Strang eng zu umhüllen.
2) Dadurch zieht sich die mRNA lokal zusammen, wird kompakter und verdichtet sich weiter.
3) Auf diese Weise entsteht ein erster „Kern“ aus mRNA-Lipid-Komplexen. Darum herum lagern sich weitere ionisierbare Lipide an – zusätzlich auch Phospholipide, Cholesterin und PEG-Lipide. Zusammen stabilisieren sie die entstehende Struktur, während die mRNA zunehmend dichter eingeschlossen wird.
4) Schließlich bildet sich ein dicht gepackter, nahezu kugelförmiger Nanopartikel. Seine Form entsteht nicht durch äußere Steuerung, sondern allein aus den physikalisch-chemischen Eigenschaften der Moleküle: ein Beispiel für spontane Selbstorganisation auf der Nanoskala.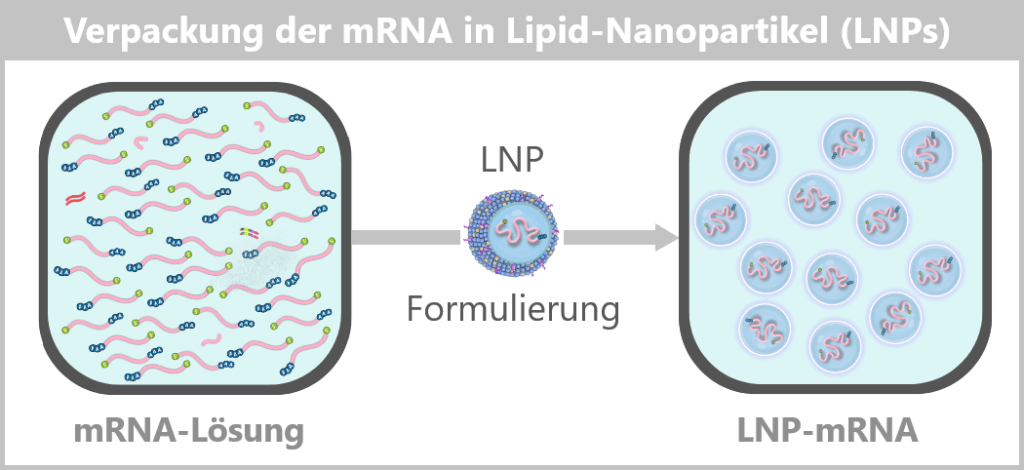
Abb. 1.1.5-D: Formulierungsschritt: Herstellung der LNP-mRNA-Dispersion Die so formulierten LNPs bilden nun das fertige Wirkstoffkonzentrat, das im nächsten Schritt steril abgefüllt wird.
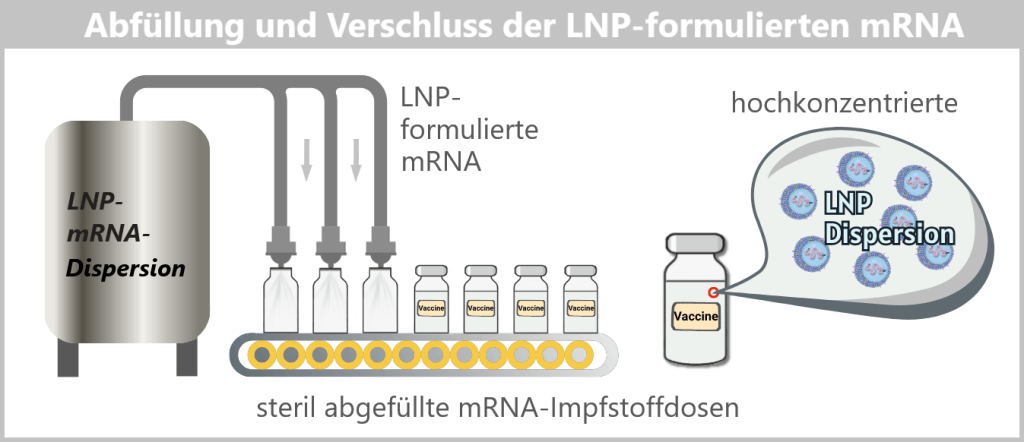
Abb. 1.1.5-E: Schematische Darstellung der sterilen Abfüllung des mRNA-Wirkstoffkonzentrats Ein Impfstoff-Vial enthält eine sehr große Anzahl einzelner Lipid-Nanopartikel, typischerweise im Bereich von 10¹³–10¹⁴ Partikeln pro Fläschchen.
1.2. Verunreinigungen und Nebenprodukte
Die Herstellung einer funktionsfähigen mRNA umfasst mehrere aufeinanderfolgende Prozessschritte – von der Vervielfältigung der DNA-Vorlage bis zur Prozessierung und Formulierung der RNA. In nahezu jedem dieser Schritte entstehen neben dem gewünschten Produkt auch Nebenprodukte, Verunreinigungen oder Reststoffe.
1.2.1. Typische Verunreinigungen bei der bakteriellen Herstellung
1.2.2. Typische Verunreinigungen und Nebenprodukte nach der IVTFür die Sicherheit und Wirksamkeit des Therapeutikums ist deren Identifizierung und spätere Abtrennung im Downstream Prozess entscheidend.
In diesem Abschnitt werfen wir einen Blick auf die wichtigsten potenziellen Verunreinigungen und Nebenprodukte und ihre bekannten biologischen Aktivitäten.
1.2.1. Typische Verunreinigungen bei der bakteriellen Herstellung
Nach der Lyse der Bakterienzellen liegt die Plasmid-DNA in einer komplexen Reaktionsmischung vor. Neben der gewünschten Plasmid-DNA enthält die Lösung chromosomale DNA, bakterielle RNA, Proteine, Enzyme (einschließlich RNasen), Endotoxine sowie Zellwandfragmente.
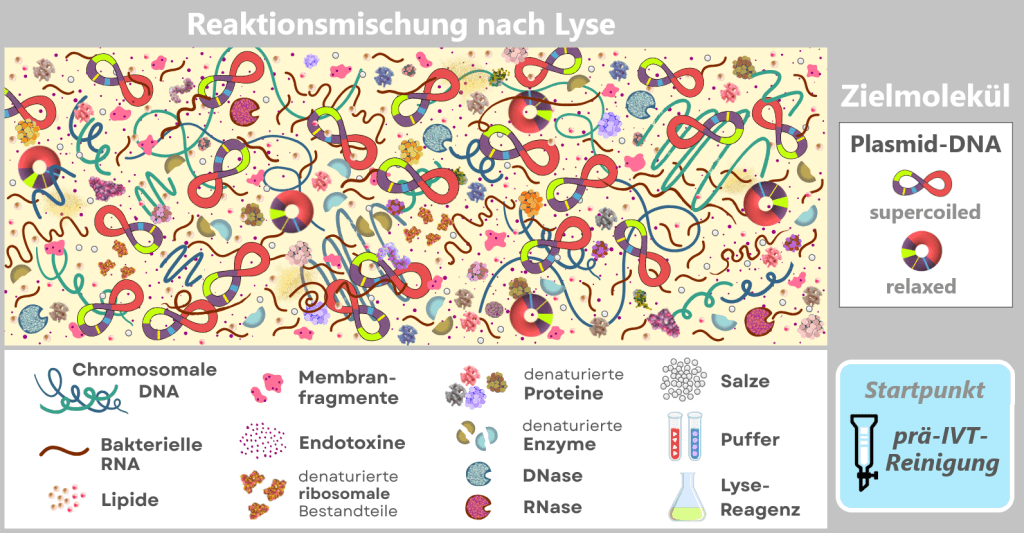
Abb. 1.2.1: Reaktionsmischung nach der bakteriellen Lyse (nicht maßstabsgetreu) Die Abbildung zeigt schematisch die Zusammensetzung der Lösung nach dem Aufschluss der E.-coli-Zellen, in denen das Plasmid vervielfältigt wurde. Neben der gewünschten Plasmid-DNA, die die spätere Expressionskassette für die mRNA enthält, sind zahlreiche bakterielle Bestandteile und Prozesshilfsstoffe vorhanden. Die dargestellten Komponenten sind in eine proteinreiche zytosolische Matrix eingebettet, die aus dem bakteriellen Zytoplasma stammt (gelber Hintergrund).
Diese komplexe und heterogene Mischung verdeutlicht, dass die Plasmid-DNA zunächst aus einer stark verunreinigten biologischen Umgebung isoliert werden muss, bevor sie als Vorlage für die in-vitro-Transkription eingesetzt werden kann.
1.2.2. Typische Verunreinigungen und Nebenprodukte nach der In-vitro-Transkription
Obwohl die in-vitro-Transkription (IVT) eine effiziente Methode zur enzymatischen Herstellung von mRNA darstellt, liefert die Reaktion kein reines Endprodukt. Im Anschluss an die Synthese liegt ein komplexes Gemisch vor, das neben der Ziel-mRNA verschiedene prozessbedingte Verunreinigungen und Nebenprodukte enthält.
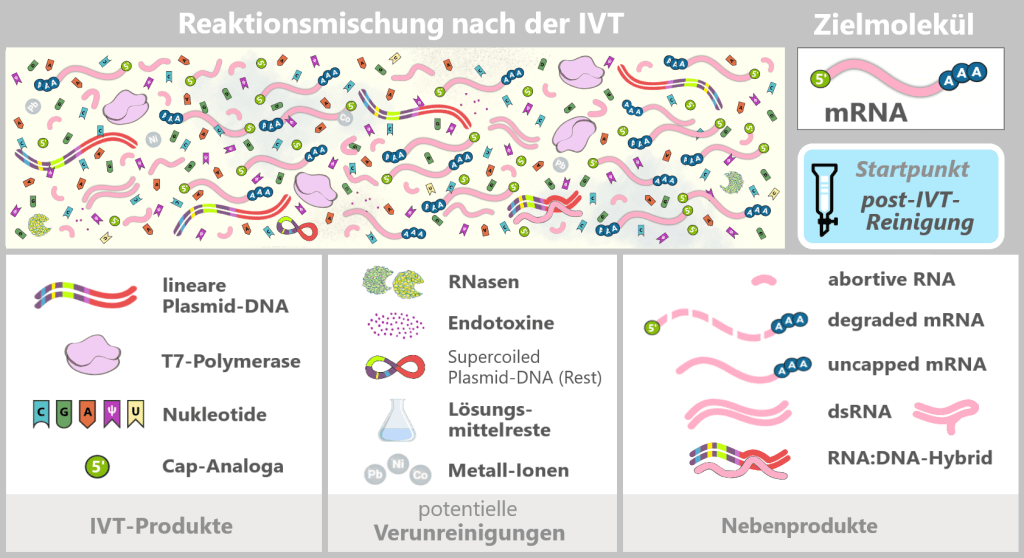
Abb. 1.2.2: Reaktionsmischung nach der in-vitro-Transkription (nicht maßstabsgetreu) Die Abbildung zeigt schematisch die komplexe Zusammensetzung der IVT-Reaktionslösung. Neben der gewünschten, vollständig prozessierten mRNA (Zielmolekül) enthält die Mischung verschiedene RNA-Nebenprodukte, wie uncapped, degradierte oder abortive RNA, doppelsträngige RNA sowie RNA:DNA-Hybride. Weitere Bestandteile:
IVT-Produkte
Lineare Plasmid-DNA: DNA-Vorlage mit der Expressionskassette für das Spike-Protein.
T7-Polymerase: Enzym, das den T7-Promotor erkennt und die mRNA synthetisiert.
Nukleotide: Bausteine der mRNA-Synthese – Cytosin (C), Guanin (G), Adenin (A) und N1-Methylpseudouridin (m¹Ψ) anstelle von Uridin (U). Uridin ist aufgeführt, da geringe U-Anteile aus der Herstellung der modifizierten Nukleotide stammen können.
Cap-Analoga: Synthetische Cap-Analoga, die während der IVT zur Ausbildung der Cap-1-Struktur eingesetzt werden und nach der Reaktion teilweise im Überschuss vorliegen.Potentielle Verunreinigungen
RNasen: Enzyme, die RNA spalten und abbauen können; sie können unbeabsichtigt über Rohstoffe oder bakterielle Reststoffe eingeschleppt werden.
Endotoxine: Bestandteile der äußeren Zellmembran von Gram-negativer Bakterien wie E. coli.
Lösungsmittelreste: Rückstände aus der Herstellung und Reinigung von Ausgangsmaterialien.
Metallionen: Spurenverunreinigungen aus Rohstoffen, Prozesswasser oder Produktionsanlagen.Übersicht: Typische RNA-Nebenprodukte nach der IVT (vor Reinigung)
a) Abortive mRNA
b) Degraded mRNA
c) Uncapped mRNA
d) Doppelsträngige RNA (dsRNA)
e) RNA:DNA-Hybride
a) Abortive mRNA
Zu Beginn der Transkription bindet die T7-RNA-Polymerase fest an den Promotor und bildet einen stabilen Initiationskomplex. Der Übergang von dieser Initiationsphase in eine produktive Elongation ist jedoch mechanisch instabil.
Die Polymerase entwindet lokal die DNA um die Transkriptionsstartstelle und beginnt mit der Synthese eines kurzen RNA-Strangs. Dabei zieht sie die DNA in das Enzym hinein, ohne sich selbst entlang der DNA vorwärtszubewegen. Dieses sogenannte Scrunching führt zur Akkumulation mechanischer Spannung im Transkriptionskomplex, da das Enzym gleichzeitig am Promotor festhält und die DNA weiter einzieht.
Gelingt es der Polymerase nicht, diese Spannung durch das Lösen vom Promotor und den Übergang in die Elongationsphase abzubauen, schnellt die DNA in ihre ursprüngliche Form zurück. Das bereits gebildete kurze RNA-Fragment wird ausgeworfen – ein abortives Transkript entsteht.
Dieser Vorgang kann sich mehrfach wiederholen, sodass eine einzelne Polymerase zahlreiche abortive RNA-Fragmente produziert, bevor eine vollständige Transkription gelingt. Erst wenn eine RNA-Länge von typischerweise etwa 8–14 Nukleotiden erreicht ist, löst sich die Polymerase vom Promotor und bildet einen stabilen Elongationskomplex.
Erscheinungsbild
Abortive RNAs sind sehr kurze RNA-Fragmente, meist nur 2–10 Nukleotide (nt) lang, die während der instabilen Initiationsphase der Transkription entstehen. Trotz der Verwendung co-transkriptioneller Capping-Strategien, bei denen das Cap-Analogon als Initiator dient, erreichen die meisten abortiven Transkripte nicht die für einen stabilen Cap-Einbau erforderliche Länge. Daher liegen sie mehrheitlich als uncapped Fragmente mit einem 5′-Triphosphat-Ende vor. Nur in Einzelfällen – bei längeren abortiven Transkripten im Bereich von etwa 10–12 Nukleotiden – kann formal ein Cap-Analogon eingebaut sein.
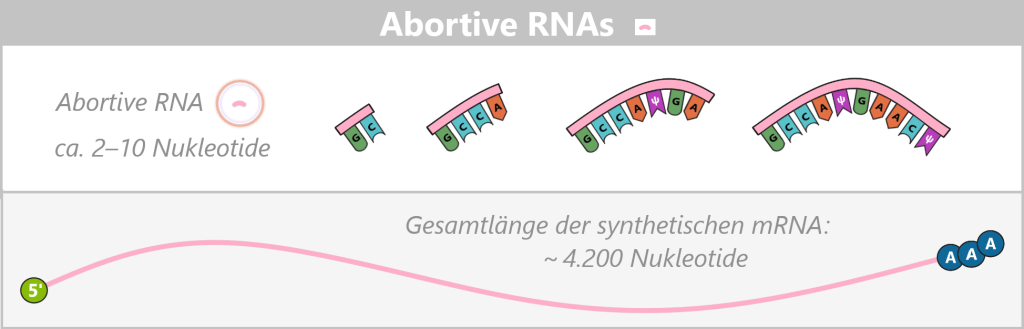
Abb. 1.2.2-A: Abortive RNAs sind schematisch mit freiem 5′-Triphosphat (ppp) dargestellt und erreichen aufgrund der instabilen Initiationsphase keine cap-tragende Elongationsform. Biologische Auswirkung
Die biologischen Auswirkungen abortiver RNA-Fragmente sind bislang nicht vollständig erforscht. Einzelne, isolierte Fragmente von nur wenigen Nukleotiden sind in der Regel zu kurz, um bekannte RNA-Sensoren des angeborenen Immunsystems zu aktivieren, und werden nicht in Proteine übersetzt.
Das potenzielle Risiko liegt daher weniger in den einzelnen 2–10-nt-Fragmenten selbst, sondern in möglichen Sekundäreffekten:
Doppelstrangbildung: Kurze RNA-Fragmente könnten als Primer dienen oder zur Bildung kurzer doppelsträngiger RNA beitragen.
Komplexbildung: Hohe Mengen solcher Fragmente könnten sich zu komplexeren Strukturen zusammenlagern oder die Effizienz nachfolgender Reinigungsschritte beeinträchtigen.
Unspezifischer Belastungseffekt: Eine hohe Konzentration kurzer RNA-Fragmente könnte kurzfristig zellulären Stress erhöhen, auch wenn sie rasch abgebaut werden.BioNTech weist darauf hin, dass abortive Nebenprodukte im Zytosol (dem flüssigen Zellinneren) transfizierter Zellen (Zellen, in die die mRNA eingebracht wurde) potenziell unbekannte Wechselwirkungen mit zelleigenen RNAs oder Mustererkennungsrezeptoren (PRRs) eingehen könnten, was den Bedarf an weiterer Forschung unterstreicht. [Die Auswirkungen von In-vitro-Transkriptionsnebenprodukten und Verunreinigungen verstehen]
Ungefährer Anteil (vor Reinigung)
BioNTech berichtet, dass etwa 44 % der T7-Polymerasen abortive Transkripte produzieren, bevor eine vollständige Transkription gelingt. Daher können sie zahlenmäßig die häufigste RNA-Spezies im Rohansatz darstellen. Jedoch machen sie unter Standard-IVT-Bedingungen weniger als 1 % der gesamten RNA-Masse aus. Der genaue Anteil abortiver RNAs hängt stark von der Sequenz der Vorlage und den Reaktionsbedingungen ab.
b) Degraded mRNA
Im Gegensatz zu abortiver mRNA handelt es sich bei degradierter mRNA um ehemals vollständige oder lange mRNA-Stränge, die durch äußere Einflüsse oder enzymatische Prozesse teilweise oder vollständig zerstört wurden. Der Abbau kann über verschiedene Mechanismen erfolgen:
Enzymatisch durch RNasen: Dies ist die häufigste Ursache für den Abbau während und nach der IVT. RNasen (Ribonukleasen) sind extrem stabile Enzyme, die nahezu überall vorkommen (z. B. auf der Haut, im Staub oder in Rohstoffen). Sie spalten RNA an spezifischen oder strukturell bevorzugten Stellen. Bereits geringste Verunreinigungen im Reaktionsgemisch können dazu führen, dass frisch synthetisierte mRNA in Fragmente zerfällt.
Chemisch/physikalisch: RNA ist aufgrund ihrer chemischen Struktur deutlich instabiler als DNA. Erhöhte pH-Werte, hohe Temperaturen oder bestimmte Metallionen fördern die hydrolytische Spaltung des RNA-Rückgrats und führen zu Kettenbrüchen.
Mechanisch: Wird die Lösung hohen Scherkräften ausgesetzt – etwa durch starkes Rühren oder das Pumpen durch enge Leitungen – können lange mRNA-Stränge physisch zerreißen. Dabei entstehen eher große Bruchstücke als die feinen Fragmente, die durch RNasen verursacht werden.
Abbruch während der Elongation: Streng genommen handelt es sich hierbei nicht um klassische Degradation, sondern um unvollständige mRNA, die bereits während der Synthese entsteht. Fällt die Polymerase während der Elongation vorzeitig vom DNA-Template ab – etwa durch starke Sekundärstrukturen oder limitierte Nukleotidverfügbarkeit – entsteht eine verkürzte mRNA, der das 3′-Ende und damit der Poly-A-Schwanz fehlt.
Erscheinungsbild
Degradierte mRNA liegt als Gemisch von Fragmenten unterschiedlicher Länge vor. Diese Fragmente können einzelne Strangbrüche enthalten oder vollständig zerfallen sein und tragen häufig kein vollständiges 5′-Cap und/oder keinen Poly-A-Schwanz.

Abb. 1.2.2-B: Diese schematische Darstellung zeigt degradierte mRNA … als ein heterogenes Gemisch von Fragmenten, das während oder nach der in-vitro-Transkription entstehen kann. Im Kontrast zur intakten, vollständigen mRNA (unten als Referenz) sind die Abbauprodukte unterschiedlich lang und weisen charakteristische Schäden auf: verlorene oder beschädigte 5′-Cap-Strukturen, freigelegte 5′-Triphosphate (ppp) – die bei Verlust des Caps exponiert werden, verkürzte Poly-A-Schwänze und interne Strangbrüche. Die Fragmente liegen nicht als einheitliche Spezies vor, sondern als komplexes Gemisch.
Biologische Auswirkung
Degradierte mRNA ist keine einheitliche Substanz, sondern ein heterogenes Gemisch. Nicht alle Fragmente sind biologisch problematisch. Das größte Risiko geht von Fragmenten aus, die spezifische immunologische Gefahrensignale tragen, darunter:
- freiliegende 5′-Triphosphate, die von zytosolischen RNA-Sensoren wie RIG-I erkannt werden,
- kurze doppelsträngige RNA-Strukturen, die durch Sekundärstruktur oder Hybridisierung entstehen und Rezeptoren wie MDA5 oder TLR3 aktivieren können,
- ungewöhnliche Endstrukturen (fehlendes Cap oder Poly-A-Schwanz), die als „FREMD“ erkannt werden.
Solche Fragmente können angeborene Immunantworten auslösen und die Verträglichkeit der mRNA-Formulierung beeinträchtigen.
Ungefährer Anteil (vor Reinigung)
Der Anteil degradierter mRNA variiert stark mit der Prozessführung. Typischerweise liegt er vor der Reinigung im Bereich von etwa 1–10 %. In gut optimierten IVT-Prozessen wird dieser Anteil jedoch gezielt auf ein Minimum reduziert.
c) Uncapped mRNA
In modernen IVT-Prozessen wird die 5′-Cap-Struktur häufig co-transkriptionell mithilfe sogenannter CleanCap-Analoga eingeführt. Dabei handelt es sich um synthetische Initiator-Oligonukleotide, die bereits die vollständige 5′-Cap-Struktur einschließlich des ersten transkribierten Nukleotids tragen.
Die T7-RNA-Polymerase nutzt CleanCap gezielt zur Initiation der Transkription; ein direkter Wettbewerb mit GTP (normales Nukleotid), wie er bei klassischen Cap-Analoga auftritt, findet dabei nicht statt. Dadurch entstehen überwiegend korrekt gecappte mRNA-Moleküle, während der Anteil uncapped mRNA stark reduziert wird.
Erscheinungsbild
Uncapped mRNA liegt als vollständige RNA-Sequenz vor, unterscheidet sich jedoch durch das Fehlen der 5′-Cap-Struktur am 5′-Ende.

Abb. 1.2.2-C: Diese Darstellung visualisiert mRNA-Moleküle, denen das funktionelle 5′-Cap fehlt. Sie tragen stattdessen ein immunogenes 5′-Triphosphat (ppp). Biologische Auswirkung
Zellen besitzen spezialisierte Enzyme, die RNA-Stränge ohne 5′-Cap sehr effizient vom 5′-Ende her abbauen. Dieser Mechanismus dient der raschen Entsorgung fehlerhafter oder gealterter körpereigener RNAs.
Immunreaktion: Uncapped RNA wird von zellulären Mustererkennungsrezeptoren wie RIG-I erkannt und kann eine starke Typ-I-Interferon-Antwort auslösen. Dadurch wird die Zelle in einen antiviralen Zustand versetzt.
Bei uncapped modRNA reduziert der Austausch von Uridin durch N1-Methylpseudouridin (m¹Ψ) die Aktivierung dieser Sensoren deutlich. Es kommt zu einem biologischen „Tauziehen“ zwischen dem immunstimulierenden uncapped 5′-Ende und der immunmodulierenden Nukleosid-Modifikation. Insgesamt würde uncapped modRNA daher als mäßig immunogenes Molekül wirken, jedoch weiterhin schneller abgebaut werden als korrekt gecappte modRNA.
Minimale Proteinproduktion: Da die 5′-Cap-Struktur auch für die effiziente Bindung an das Ribosom notwendig ist, wird uncapped mRNA kaum oder gar nicht in Protein übersetzt.
Ungefährer Anteil (vor Reinigung)
Der Anteil uncapped mRNA nach der IVT hängt maßgeblich von der verwendeten Capping-Strategie ab. Bei modernen co-transkriptionellen CleanCap-Verfahren liegt der Anteil uncapped mRNA vor der Reinigung typischerweise im Bereich von ca. 1–6 %.
d) Doppelsträngige RNA (dsRNA)
Ein weiteres relevantes Nebenprodukt der in-vitro-Transkription (IVT) ist doppelsträngige RNA (dsRNA). Während die gewünschte Impfstoff-mRNA als einzelsträngiges Molekül synthetisiert wird, können im Verlauf der IVT auch RNA-Duplexe entstehen, bei denen zwei komplementäre RNA-Stränge über Watson-Crick-Basenpaarung miteinander verbunden sind.
Die Entstehung solcher dsRNA-Spezies ist kein zufälliges Aggregationsphänomen, sondern geht auf spezifische, enzymatisch vermittelte Nebenreaktionen der T7-RNA-Polymerase zurück.
Entstehungsmechanismen
Promotorunabhängige Transkription des Nicht-Templatestrangs
Unter bestimmten Bedingungen kann die T7-RNA-Polymerase auch ohne klassischen Promotorstart RNA synthetisieren, indem sie den Nicht-Templatestrang der DNA-Vorlage abliest. Die dabei entstehenden RNA-Moleküle sind weitgehend komplementär zur gewünschten mRNA. Treffen Sense- und Antisense-RNA aufeinander, bilden sich ausgedehnte, nahezu vollständig doppelsträngige RNA-Duplexe. Dieser Mechanismus wird insbesondere begünstigt, wenn die DNA-Vorlage nicht vollständig linearisiert ist oder spezifische Sequenzen am 3′-Ende der Vorlage vorliegen.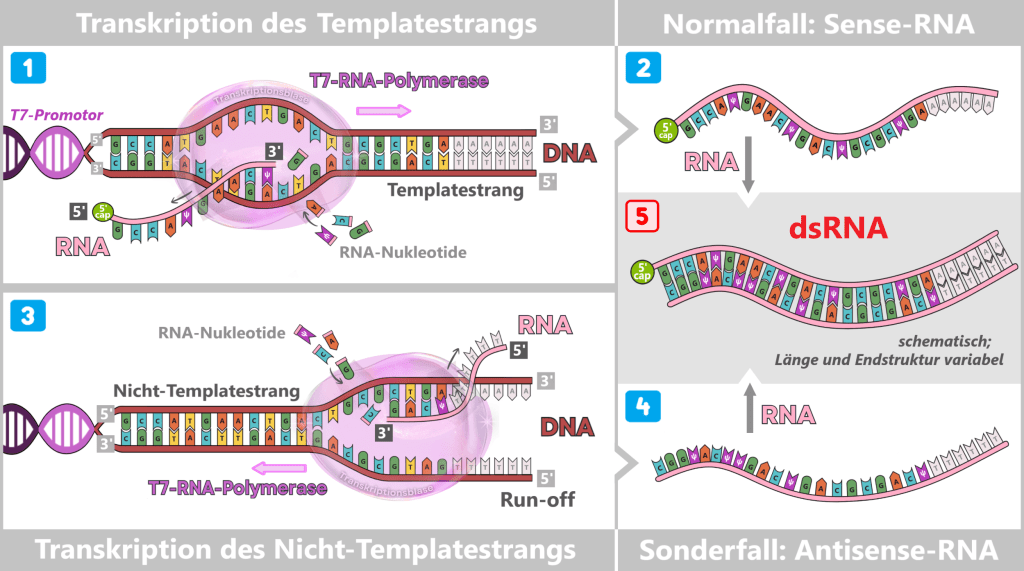
Abb. 1.2.2-D1: dsRNA-Entstehung durch promotorunabhängige Transkription des Nicht-Templatestrangs (RNA schematisch dargestellt; Nukleosid-Modifikationen (z. B. m¹Ψ) nicht explizit abgebildet.)
1) Der Normalfall: Der T7-Promotor hat eine außergewöhnlich hohe Affinität für die T7-RNA-Polymerase. Deshalb startet die Polymerase fast ausschließlich dort. Sie liest DNA stets in 3′ → 5′-Richtung und synthetisiert ein neues RNA-Molekül ausschließlich in 5′ → 3′-Richtung.
2) Das Ergebnis ist die gewünschte Sense-RNA, die genau der Sequenz des Nicht-Templatestrangs entspricht.
3) Der Sonderfall: Nach dem Run-off – dem Verlassen der DNA-Vorlage am Ende der Transkription – löst sich die Polymerase von der DNA. In seltenen Fällen kann sie unspezifisch an strukturell zugängliche DNA-Enden binden. Bindet sie dabei an den zuvor nicht als Template genutzten Strang, wird dieser zur neuen Vorlage.
Unter bestimmten Reaktionsbedingungen können auch lokal offene oder dynamisch schmelzende DNA-Bereiche entstehen, an die die T7-RNA-Polymerase promotorunabhängig binden kann. Erfolgt diese Bindung am zuvor nicht genutzten Strang, wird dieser als Template gelesen.
Bei der promotorunabhängigen Transkription des Nicht-Templatestrangs fehlt eine definierte Initiationsstelle, sodass kein co-transkriptionelles Capping erfolgt. Die entstehende Antisense-RNA trägt daher typischerweise ein freies 5′-Triphosphat (pppN).
4) Das Ergebnis ist eine komplementäre Antisense-RNA, die der Sequenz des Templatestrangs entspricht.
5) Entstehung eines langen dsRNA-Duplexes: Treffen Sense-RNA und Antisense-RNA zusammen, führt das zur Bildung von dsRNA (doppelsträngiger RNA). Dabei stellen die entstehenden dsRNA-Nebenprodukte kein homogenes Molekül dar, sondern ein strukturell heterogenes Gemisch aus vollständig oder partiell komplementären RNA-Duplexen. Diese können stumpfe Enden oder einzelsträngige Überhänge aufweisen und variieren erheblich in Länge und Endstruktur.RNA-abhängige 3′-End-Verlängerung (self-priming)
Ein zweiter zentraler Mechanismus ist die RNA-abhängige Verlängerung des 3′-Endes der frisch synthetisierten mRNA. Dabei klappt das 3′-Ende der RNA aufgrund komplementärer Sequenzen intramolekular zurück und bildet eine kurze dsRNA-Haarnadel. Die T7-RNA-Polymerase kann an diese Struktur erneut binden und den RNA-Strang weiter verlängern, wobei eine Sequenz entsteht, die zur ursprünglichen RNA komplementär ist. Dieser Prozess ist unabhängig von der DNA-Vorlage und kann sogar bei bereits vollständig transkribierten RNA-Molekülen auftreten.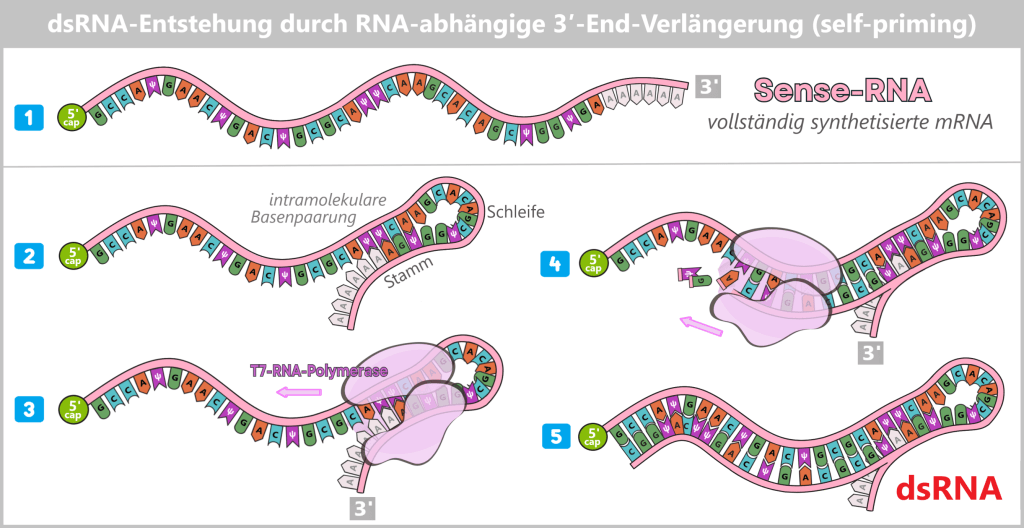
Abb. 1.2.2-D2: dsRNA-Entstehung durch RNA-abhängige 3′-End-Verlängerung (Self-priming) (RNA schematisch dargestellt)
1) Sense-RNA: Normale, vollständig synthetisierte mRNA.
2) Hairpin-Bildung: Der 3′-Abschnitt faltet sich um. Es entsteht ein kurzer dsRNA-Stamm (z.B. 5-10 Basenpaare) mit einer Einzelstrang-Schleife.
3) Polymerase-Bindung: Die T7-Polymerase bindet an diesen dsRNA-Stamm, als wäre es ein DNA-Template-Primer-Komplex.
4) Verlängerung: Die Polymerase nutzt den einen Strang des Hairpins als Template und verlängert das freie 3′-Ende entlang des komplementären RNA-Abschnitts. Sie synthetisiert also die Antisense-Sequenz direkt in die Verlängerung des Sense-Strangs hinein.
5) Ergebnis: Die Polymerase verlängert den Strang so weit sie kann. Die neu synthetisierte Antisense-RNA hybridisiert sofort mit der Sense-RNA. Es entsteht ein partiell oder vollständig dsRNA-haltiges RNA-Molekül.Abgrenzung zu RNA-Sekundärstrukturen
Einzelsträngige RNA bildet natürlicherweise intramolekulare Sekundärstrukturen wie Haarnadeln (Hairpin) oder interne Schleifen (siehe obere Abbildung – Punkt 2). Diese sind integraler Bestandteil funktioneller mRNA, meist kurz und thermodynamisch flexibel. Solche Strukturelemente gelten nicht als dsRNA-Nebenprodukte im engeren Sinne und sind nicht mit den langkettigen dsRNA-Duplexen gleichzusetzen, die als IVT-Verunreinigungen relevant sind.Beide Mechanismen führen zur Bildung längerer, stabiler dsRNA-Abschnitte, die sich strukturell deutlich von den kurzen, dynamischen Sekundärstrukturen korrekt transkribierter mRNA unterscheiden.
Erscheinungsbild
dsRNA-Nebenprodukte liegen typischerweise als lange, teilweise oder vollständig komplementäre RNA-Duplexe vor. Sie können nahezu die gesamte Länge der Ziel-mRNA umfassen oder ausgedehnte doppelsträngige Regionen enthalten. Häufig besitzen diese Duplexe keine 5′-Cap-Struktur und können an den Enden über einzelsträngige Überhänge verfügen.
Biologische Auswirkung
dsRNA als strukturelles Gefahrensignal
Doppelsträngige RNA (dsRNA) stellt im Zytoplasma eukaryotischer Zellen kein typisches Strukturmotiv zelleigener, translierbarer mRNA dar. Das bedeutet: Während zelluläre mRNAs normalerweise einzelsträngig sind und dazu dienen, in Proteine übersetzt zu werden (mittels Translation), tritt dsRNA physiologisch nur in eng regulierten, kurzlebigen Kontexten auf. Das Auftreten von dsRNA im Zytoplasma wird daher von der Zelle nicht primär als genetische Information, sondern als strukturelles Warnsignal interpretiert.Zytosolische Erkennung von dsRNA
Zellen verfügen über spezialisierte Mustererkennungsrezeptoren (Pattern Recognition Receptors, PRRs). Diese „Detektoren für Gefahr“ patrouillieren durch das Zytoplasma und suchen nach strukturellen Mustern wie Duplex-Länge, Endstruktur und chemischer Modifikation der Nukleotide.Zytosolische dsRNA-Sensoren und ihre immunologischen Konsequenzen
Merkmal / Spezifität Zellulärer Sensor Biologische Konsequenz (für die Zelle) Kurze dsRNA (≈ 10-300 bp) mit 5′-Triphosphat und/oder stumpfen Enden RIG-I (Retinoic acid-Inducible Gene I)
Kurz-ppp-DetektorAuslösung einer entzündlichen Genexpression (Typ-I-Interferone, Zytokine). Alarmierung der Nachbarzellen. Lange dsRNA (> ~500–1.000 bp), unabhängig von den Enden MDA5 (Melanoma Differentiation-Associated protein 5)
Langduplex-ScannerAuslösung einer massiven Typ-I-Interferon-Antwort. Wichtige Abwehr gegen viele Viren. dsRNA mittlerer Länge (≈ ≥30–100 bp) PKR (Protein Kinase R)
Intrazellulärer EffektorGlobaler Stopp der Proteinbiosynthese: Phosphorylierung des Translationsfaktors eIF2α. Hemmung der viralen Vermehrung. dsRNA mittlerer Länge (≈ ≥40–100 bp) OAS (2′-5′-Oligoadenylat-Synthetasen)
Intrazellulärer EffektorUnspezifischer RNA-Abbau: aktiviert die latente Ribonuklease RNase L, die alle zellulären und viralen RNA-Moleküle zerschneidet. Kann zum Zelltod führen. dsRNA im Endosom (extra-zellulär aufgenommene oder phagozytierte dsRNA) TLR3 (Toll-like Receptor 3)
Wächter an der ZellpforteFührt zur Produktion von Typ-I-Interferonen und pro-inflammatorischen Zytokinen. Die Wirkung von m¹Ψ
Die Verwendung nukleosid-modifizierter mRNA (modRNA), etwa durch den Einbau von N¹-Methylpseudouridin, kann die Aktivierung bestimmter zytosolischer RNA-Sensoren deutlich reduzieren. Besonders betroffen sind RIG-I und PKR, die bevorzugt auf einzelsträngige RNA oder kurzlebige dsRNA-Strukturen reagieren. Längenabhängige Sensoren wie MDA5 werden durch diese Modifikation hingegen nur eingeschränkt moduliert und bleiben gegenüber ausgedehnten dsRNA-Duplexen weitgehend empfindlich.Aus diesem Grund besitzt dsRNA auch im Kontext von modRNA weiterhin ein hohes immunstimulatorisches Potenzial – das heißt, sie kann das Immunsystem aktivieren und unerwünschte Entzündungsreaktionen auslösen. Die effiziente Minimierung solcher dsRNA-Nebenprodukte durch optimierte Transkriptionsbedingungen, chemische Modifikationen und nachgelagerte Reinigungsverfahren ist daher ein zentraler Bestandteil der mRNA-Herstellung.
Ungefährer Anteil (vor Reinigung)
Der Anteil an dsRNA nach der IVT ist stark prozessabhängig und wird von Faktoren wie der Qualität der DNA-Vorlage, den Reaktionsbedingungen und der verwendeten Polymerase beeinflusst. Typischerweise liegt der dsRNA-Anteil vor der Aufreinigung im niedrigen einstelligen Prozentbereich, kann unter ungünstigen Bedingungen jedoch deutlich höher ausfallen.
e) RNA:DNA-Hybride
Während der in-vitro-Transkription kann es unter bestimmten Bedingungen dazu kommen, dass der neu synthetisierte RNA-Strang nicht vollständig vom DNA-Template dissoziiert. Er kann partiell am Templatestrang verbleiben und dabei den komplementären DNA-Strang verdrängen. Es entstehen sogenannte RNA:DNA-Hybride.
Die Bildung solcher Hybridstrukturen ist stark sequenzabhängig. Besonders purinreiche Transkripte – also RNA-Sequenzen mit vielen der Basen Adenin und Guanin – sowie DNA-Vorlagen mit sich wiederholenden GAA-Abschnitten (Guanin–Adenin–Adenin) begünstigen die Entstehung stabiler RNA:DNA-Duplexe. Diese Nebenprodukte werden in der Regel unterschätzt, lassen sich jedoch experimentell mit spezifischen Antikörpern gegen RNA:DNA-Hybride nachweisen. [Die Auswirkungen von In-vitro-Transkriptionsnebenprodukten und Verunreinigungen verstehen]
Erscheinungsbild
RNA:DNA-Hybride liegen typischerweise als R-Loop-artige Strukturen vor. Dabei handelt es sich um lokal begrenzte, dreisträngige Nukleinsäurekonformationen, bestehend aus:
- einem RNA:DNA-Hybrid-Duplex,
- einem verdrängten DNA-Einzelstrang,
- sowie der angrenzenden DNA-Doppelhelix außerhalb der Hybridregion.
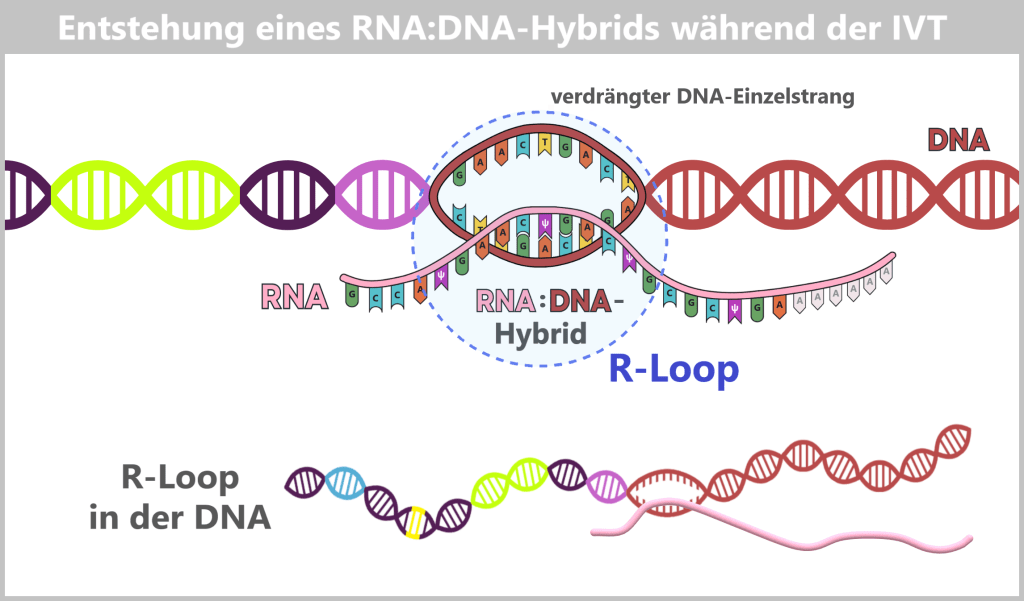
Abb. 1.2.2-E: Schematische Darstellung einer R-Loop-Bildung während der in-vitro-Transkription Der neu synthetisierte RNA-Strang hybridisiert partiell mit dem DNA-Templatestrang und verdrängt dabei den komplementären Nicht-Templatestrang. Es entsteht ein R-Loop, bestehend aus einem RNA:DNA-Hybrid-Duplex und einem verdrängten DNA-Einzelstrang, eingebettet in eine ansonsten doppelsträngige DNA-Region.
Außerhalb des R-Loops liegt die DNA weiterhin als reguläre Doppelhelix vor.Biologische Auswirkung
Aktuelle Forschungsergebnisse deuten darauf hin, dass RNA:DNA-Hybride von zellulären Mustererkennungsrezeptoren (Pattern Recognition Receptors, PRRs) erkannt werden können. Zu diesen Sensoren des angeborenen Immunsystems zählen unter anderem cGAS, TLR9 und das Inflammasom-Protein NLRP3, die solche Hybridstrukturen binden können. Ihre Aktivierung kann eine Immunreaktion auslösen, die zur Bildung von entzündungsfördernden Botenstoffen (Zytokinen) und Typ-I-Interferonen führt.
Ob und in welchem Ausmaß RNA:DNA-Hybrid-Kontaminanten tatsächlich zu unerwünschten Immunreaktionen therapeutisch eingesetzter IVT-mRNA beitragen, ist bislang jedoch nicht systematisch untersucht worden.
Vor diesem Hintergrund erscheint die effiziente Entfernung von RNA:DNA-Hybrid-Verunreinigungen aus IVT-mRNA aus Qualitätssicherungsgründen als relevanter Aspekt. Dies gilt insbesondere für therapeutische Anwendungen, bei denen eine ausgeprägte Aktivierung des Immunsystems nicht angestrebt wird. [Die Auswirkungen von In-vitro-Transkriptionsnebenprodukten und Verunreinigungen verstehen]
Ungefährer Anteil (vor Reinigung)
Der Anteil an RNA:DNA-Hybriden nach der in-vitro-Transkription ist stark prozessabhängig. Er wird unter anderem durch die Sequenz und Qualität der DNA-Vorlage sowie durch die Reaktionsbedingungen beeinflusst. Insgesamt wird ihr Anteil vor der Aufreinigung als gering eingeschätzt, kann jedoch je nach Transkript und Prozessführung variieren. Verlässliche, öffentlich zugängliche quantitative Daten zum genauen Anteil von RNA:DNA-Hybriden in therapeutischen mRNA-Produkten liegen bislang nicht vor, da entsprechende Messwerte in der Regel Teil des firmenspezifischen Herstellungs- und Qualitätskontrollwissens sind.
** Eine weitere Entstehungsmöglichkeit für RNA:DNA-Duplexe im Rahmen der DNase-Behandlung wird in Abschnitt „1.3.1. a) Mögliche posttranskriptionelle RNA:DNA-Hybridisierung“ beschrieben. **
1.3. Reinigungsverfahren im Herstellungsprozess
Da mRNA-Impfstoffe extrem hohe Reinheitsanforderungen erfüllen müssen, ist die Aufreinigung kein einzelner, isolierter Prozessschritt. Vielmehr zieht sie sich wie ein roter Faden durch den gesamten Herstellungsprozess.
Der genaue industrielle Ablauf ist bei Herstellern wie BioNTech/Pfizer oder Moderna proprietär (unternehmensintern) und wird nicht vollständig offengelegt. Patente und regulatorische Dokumente zeigen jedoch, dass eine Kombination etablierter biotechnologischer Reinigungsverfahren eingesetzt wird.
Eine schematische, industrieübergreifende Übersicht zur mRNA-Herstellung, in der der kontinuierliche Fluss von Reinigungsaktivitäten entlang des Gesamtprozesses dargestellt ist, findet sich beispielhaft in der technischen Dokumentation von Merck/Sigma-Aldrich (Manufacturing strategies for mRNA vaccines and therapies, Abbildung 1).
Im Herstellungsprozess lassen sich vereinfacht drei besonders kritische Reinigungsphasen unterscheiden:
- vor der in-vitro-Transkription (prä-IVT): Aufreinigung der DNA-Vorlage,
- nach der in-vitro-Transkription (post-IVT): Aufreinigung der synthetisierten mRNA,
- nach der Formulierung: sterile Filtration der lipid-nanopartikulären Formulierung.
Anstatt diese Phasen streng chronologisch abzuarbeiten, werden im Folgenden die wichtigsten Reinigungsprinzipien und -methoden vorgestellt, die an unterschiedlichen Stellen des Herstellungsprozesses wiederholt zum Einsatz kommen.
1.3.1. DNase I-Behandlung – enzymatischer DNA-Abbau
1.3.2. Proteinase K – enzymatischer Abbau von Restproteinen
1.3.3. Filtration
1.3.4. Chromatographie – das molekulare Trennverfahren
1.3.5. Magnetkügelchen-ReinigungDie folgende Übersicht fasst die zentralen Reinigungstechniken zusammen. Sie dient als Rahmen für die anschließend vertiefte Darstellung der jeweiligen Methoden.
Methode Entfernt Prinzip Vorteile Einschränkungen DNase I Behandlung Template-DNA Spezifischer enzymatischer Abbau Sehr selektiv, zerkleinert problematische Nukleinsäuren Enzyme müssen vollständig entfernt werden, sonst Gefahr von Restaktivität Proteinase K Enzyme aus der Transkription (z. B. Polymerasen, Ligase, Restriktions-enzyme) Unspezifischer Abbau von Proteinen Universell wirksam, entfernt viele Proteinarten Muss durch weitere Reinigungsschritte ergänzt werden (Enzymreste selbst problematisch) Filtration & TFF (Tangentialfluss-filtration) Salze, Nukleotide, kleine Moleküle, Pufferreste; auch Konzentration möglich Trennung nach Molekülgröße durch Membranen Skalierbar, robust, vielseitig (Konzentration + Pufferwechsel in einem Schritt) Keine Unter-scheidung ähnlicher Nukleinsäuren; eher ein „Grobwerkzeug“ Chromatografie (Poly(dT), AEX) dsRNA, kurze RNA-Fragmente, restliche Nukleotide, Proteine Trennung nach Ladung oder spezifischer Bindung an Oberflächen Sehr präzise, trennt eng verwandte Nukleinsäuren; etabliert in der Industrie Technisch anspruchsvoll, kostenintensiv, erfordert exakte Prozesskontrolle Magnetkügelchen-Reinigung
(Prozess 1)Selektion reifer, polyadenylierter mRNA; entfernt kurze Fragmente & Verunreinigungen Poly(T)-beschichtete Beads binden spezifisch an den Poly(A)-Schwanz der mRNA Hohe Spezifität, schnelle und effiziente Trennung Für kleine bis mittlere Volumina geeignet; bei Großproduktion schwer skalierbar
1.3.1. DNase I-Behandlung – enzymatischer DNA-Abbau
Deoxyribonuclease I (DNase I) ist ein Enzym, das DNA-Moleküle in kleinere Fragmente spaltet. Oft spricht man auch vom „Verdauen“ der DNA – ein bildhafter Ausdruck für den enzymatischen Abbau langer DNA-Stränge in kürzere Stücke. In der Biotechnologie wird DNase I eingesetzt, um unerwünschte DNA-Verunreinigungen gezielt zu fragmentieren.
DNase I ist somit kein vollständiger Reinigungsmechanismus, sondern ein vorbereitender Schritt, der die nachfolgende physikalische und chromatographische Trennung erleichtert.
In der mRNA-Impfstoffherstellung wird DNase I nach der in-vitro-Transkription (IVT) eingesetzt, um die DNA-Vorlage (Template-DNA) sowie den DNA-Anteil eventuell gebildeter RNA:DNA-Hybride zu fragmentieren, die die Qualität oder Sicherheit des Endprodukts beeinträchtigen könnten. Die DNase I erkennt die räumliche Struktur der DNA – genauer gesagt: das Zucker-Phosphat-Rückgrat. Das Enzym setzt sich wie ein Sattel auf die DNA und greift in die sogenannte „kleine Furche“ (minor groove). Dabei verbiegt sie die DNA-Struktur lokal, um den Zugang zur Phosphodiesterbindung zu ermöglichen. Da die Breite der kleinen Furche je nach Basenabfolge minimal variiert, schneidet DNase I an manchen Stellen (z. B. bei bestimmten A/T-reichen Regionen) etwas lieber als an anderen.
Abb. 1.3.1-A: Schematische Darstellung der Bindung von DNase I an doppelsträngige DNA. Die typische B-Form der DNA ist die schlanke, rechtsgewundene Doppelhelix, die aussieht wie eine Wendeltreppe mit zwei unterschiedlich großen Rillen (einer großen und einer kleinen Furche), die sich um sie herum winden.
Das Enzym interagiert bevorzugt mit der kleinen Furche (minor groove) des DNA-Doppelstrangs und bindet dabei an das Zucker-Phosphat-Rückgrat. Die Bindung führt zu einer lokalen Verformung der DNA, wodurch die Spaltung der Phosphodiesterbindung ermöglicht wird. Die große Furche (major groove) ist dargestellt, spielt jedoch für die Bindung von DNase I keine primäre Rolle.DNase I schneidet DNA unspezifisch, also an vielen verschiedenen Positionen entlang des Strangs. Sie wirkt sowohl auf einzelsträngige (ssDNA) als auch auf doppelsträngige DNA (dsDNA). Ihre Aktivität hängt dabei von mehreren Faktoren ab, insbesondere von den vorhandenen Metallionen, die die Art der Schnittstellen beeinflussen [bioswisstec, YEASEN]:
In Gegenwart von Magnesium-Ionen (Mg²⁺) schneidet DNase I die doppelsträngige DNA (dsDNA) überwiegend durch einzelsträngige Schnitte („nicks“). Das führt zur Bildung kurzer doppelsträngiger DNA-Fragmente (dsDNA) mit Überhängen sowie in geringerem Umfang zu einzelsträngigen (ssDNA) Fragmenten.
In Gegenwart von Mangan-Ionen (Mn²⁺) schneidet die DNase I beide dsDNA-Stränge nahezu an derselben Position, wodurch überwiegend stumpf-endige dsDNA-Fragmente entstehen.
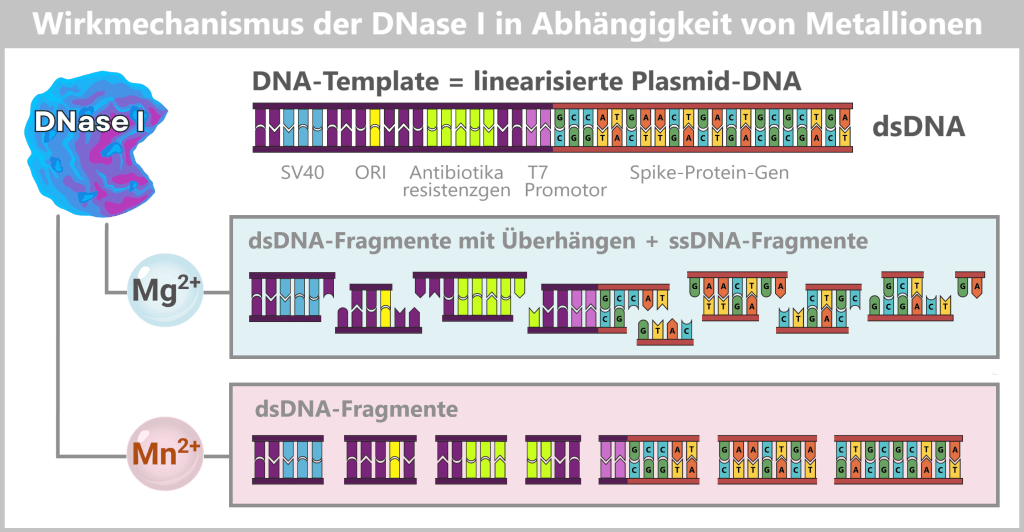
Abb. 1.3.1-B: Schematische Darstellung der DNase I-Wirkung in Abhängigkeit von Metallionen Effektivität der DNase I
Der Hersteller Thermo Fisher Scientific beschreibt in dem Online-Artikel „DNase I entmystifiziert“ die Wirkweise und Grenzen der DNase-I-Behandlung. Der Beitrag verdeutlicht, dass die Effektivität von DNase I stark von den Reaktionsbedingungen und der Art des vorliegenden DNA-Substrats abhängt.
Im Produktionsprozess befinden wir uns nach der in-vitro-Transkription (IVT). Die Reaktionsmischung ist ein komplexer Mix aus:
- der Ziel-mRNA (Hauptkomponente),
- RNA-Nebenprodukten (z. B. verkürzte Transkripte, RNA:DNA-Hybride),
- dem DNA-Template (in relevanter, aber geringerer Menge als mRNA),
- seltenen Restformen nicht-linearer Plasmid-DNA (z. B. supercoiled),
- Enzymen, freien Nukleotiden, Salzen und Puffersubstanzen.
Wie viele Enzyme wird auch die Aktivität der DNase I durch die Zusammensetzung der Reaktionsmischung beeinflusst.
Einflussfaktoren auf die Effektivität der DNase-I-Behandlung
Faktor Mechanismus Prozessrelevanz (mRNA-Herstellung) Substrat-Zugänglichkeit DNase I schneidet bevorzugt frei zugängliche Phosphodiesterbindungen; kompakte oder topologisch eingeschränkte DNA ist schlechter erreichbar. Supercoiled oder stark verdrillte DNA-Strukturen (z. B. seltene Plasmid-Restformen) werden langsamer oder unvollständig fragmentiert. Substrat-Sättigung Hohe DNA-Mengen können das Enzym sättigen; hohe RNA-Konzentrationen beeinflussen die räumliche Zugänglichkeit der DNA. In IVT-Mischungen mit sehr hoher Nukleinsäurelast ist ein Enzymüberschuss erforderlich. Helix-Geometrie Die aktive Stelle der DNase I ist auf die B-Form der DNA optimiert (abweichende Helixformen passen schlechter). RNA:DNA-Hybride weisen eine A-Form-ähnliche Helix auf → Spaltaktivität < 2 % gegenüber dsDNA DNA-Struktur
(dsDNA vs. ssDNA)Höchste Aktivität auf dsDNA; Aktivität auf ssDNA etwa 500-fach geringer Sehr kurze oder teilweise einzelsträngige DNA-Fragmente werden schlechter abgebaut. Ionen im Puffer Mg²⁺ ist für die katalytische Aktivität essenziell; Ca²⁺ stabilisiert die Enzymstruktur Therapeutische Prozesse erfordern präzise eingestellte Ionenverhältnisse. Chelatoren Chelatoren (z. B. EGTA, EDTA) binden Ca²⁺/Mg²⁺ und inaktivieren DNase I. Rückstände aus Puffern oder Prozess-schritten können die DNase-Wirkung stark reduzieren. Salzkonzentration Erhöhte Ionenstärke schwächt die elektrostatische Bindung zwischen Enzym und DNA. IVT-Pufferbedingungen können die Aktivität mindern → kompensiert durch Enzymüberschuss. Der Beitrag von Thermo Fisher Scientific weist darauf hin, dass „es wahrscheinlich unmöglich ist, jeden letzten DNA-Strang in einem RNA-Präparat loszuwerden“.
Dies verdeutlicht, dass die vollständige Entfernung aller DNA-Reste technisch anspruchsvoll ist. Selbst nach sorgfältiger DNase-I-Behandlung können kurze DNA-Fragmente oder RNA:DNA-Hybride verbleiben.
Die so erzeugten kleinen DNA-Fragmente werden in nachfolgenden Prozessschritten – etwa durch Magnetkügelchen-Reinigung (bei Prozess 1) oder Chromatografie (bei Prozess 2) – aus dem Produkt entfernt (vgl. EPAR, Abschnitt 2.2).
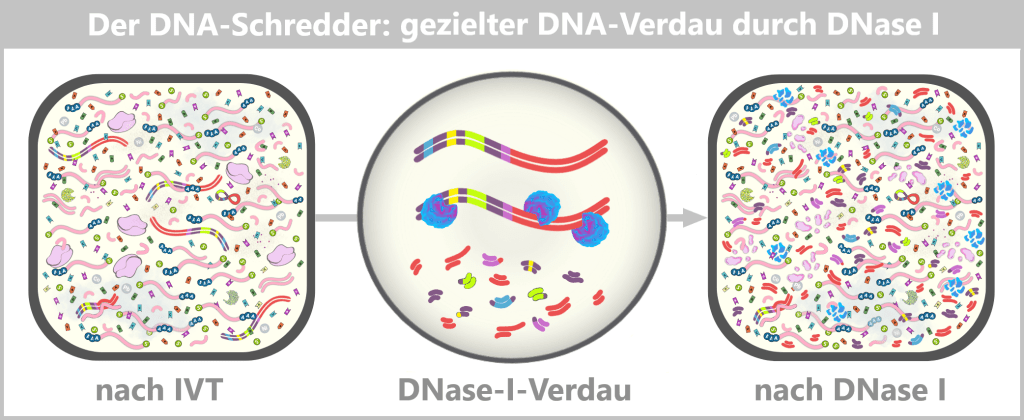
Abb. 1.3.1-C: Schematische Darstellung der DNase-I-Behandlung im Reinigungsschritt In der industriellen mRNA-Herstellung erfolgt die DNase-I-Behandlung unmittelbar nach der in-vitro-Transkription (IVT), da die Reaktionsmischung bereits einen für das Enzym geeigneten Mg²⁺-haltigen Puffer enthält. Die Mg²⁺-abhängige Aktivität der DNase I führt überwiegend zur Fragmentierung der DNA in kurze doppelsträngige DNA-Fragmente mit Nicks (Strangbrüchen) und kurzen Überhängen. (Mn²⁺ wird aufgrund der unspezifischeren und schwer kontrollierbaren Spaltaktivität im industriellen Prozess nicht eingesetzt.)
Nach abgeschlossener Reaktion wird die DNase I häufig inaktiviert oder entfernt, beispielsweise durch Hitze, EDTA-Zugabe oder Proteinase K-Behandlung, um sicherzustellen, dass keine enzymatische Aktivität auf die mRNA übergreift.
1.3.1. a) Mögliche posttranskriptionelle RNA:DNA-Hybridisierung
Neben den während der in-vitro-Transkription entstehenden R-Loop-Strukturen wird in der neueren Literatur ein zweiter möglicher Entstehungsweg für RNA:DNA-Hybride diskutiert.
Die DNase-I-Behandlung fragmentiert die verbliebenen DNA-Vorlagen in Stücke unterschiedlicher Länge – überwiegend doppelsträngig, teilweise jedoch auch mit einzelsträngigen Überhängen oder als kurze Einzelstrangabschnitte.
Fragmente, deren Sequenz komplementär zur mRNA ist – insbesondere solche, die vom Templatestrang der ursprünglichen DNA-Vorlage stammen –, könnten unter geeigneten Bedingungen mit der mRNA hybridisieren und RNA:DNA-Duplexe bilden.
Für eine thermodynamisch stabile Hybridisierung sind in der Regel zusammenhängende komplementäre Sequenzen von etwa 8–12 Nukleotiden oder mehr erforderlich. Sehr kurze Überhänge von nur wenigen Nukleotiden (z. B. 2–4 Basen), wie sie bei einem weitgehend vollständigen DNase-Verdau typischerweise entstehen, sind dagegen nicht ausreichend für eine stabile Bindung. Voraussetzung für diesen Mechanismus wäre daher das Vorhandensein ausreichend langer, komplementärer DNA-Fragmente, etwa bei unvollständigem Verdau oder spezifischen Fragmentmustern.
Hinsichtlich der enzymatischen Abbaubarkeit ist zu berücksichtigen, dass DNase I bevorzugt doppelsträngige DNA spaltet. RNA:DNA-Hybride stellen ein strukturell abweichendes Substrat dar und werden effizienter durch spezialisierte Enzyme wie RNase H oder DNase I-XT abgebaut. In der Praxis kann dies bedeuten, dass einmal gebildete RNA:DNA-Hybride gegenüber einem erneuten DNase-I-Verdau relativ stabiler sind als freie DNA-Fragmente. Eine zusätzliche Stabilisierung kann auftreten, wenn solche Strukturen in Lipid-Nanopartikel eingeschlossen werden.
Dieser postulierte Mechanismus der Post-IVT-Hybridisierung ist bislang nicht umfassend experimentell charakterisiert. Er basiert auf theoretischen Überlegungen und vereinzelten experimentellen Hinweisen und sollte daher als plausible, aber noch nicht abschließend belegte Möglichkeit eingeordnet werden.
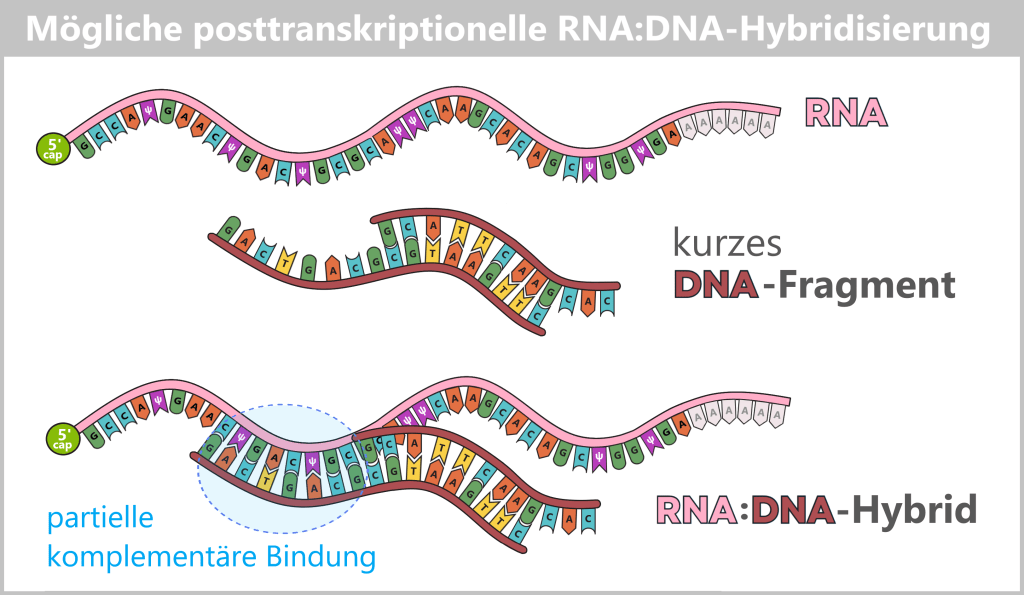
Abb. 1.3.1a: Schematische Darstellung einer möglichen posttranskriptionellen Hybridisierung Kurze DNA-Fragmente können unter geeigneten Bedingungen partiell an komplementäre Abschnitte der mRNA binden. Im Gegensatz zum R-Loop entsteht keine dreisträngige Struktur, sondern ein lokal begrenzter RNA:DNA-Duplex.
1.3.2. Proteinase K – enzymatischer Abbau von Restproteinen
Proteinase K ist eine breit wirksame Serinprotease, die in der Biotechnologie eingesetzt wird, um Proteine unspezifisch zu hydrolysieren, also in kleinere Peptidfragmente zu zerlegen. Sie gehört zur Klasse der Proteinasen – Enzyme, die Peptidbindungen zwischen Aminosäuren spalten und dadurch die dreidimensionale Struktur von Proteinen irreversibel auflösen.
In der mRNA-Impfstoffherstellung kann Proteinase K an verschiedenen Prozessstellen eingesetzt werden, insbesondere:
Nach der Zelllyse in der Plasmidproduktion: Hier dient sie dem Abbau bakterieller Proteine, Nukleasen und weiterer Zellbestandteile, die die Reinheit der Plasmid-DNA beeinträchtigen könnten.
Nach der in-vitro-Transkription (IVT): Nach der Transkription, bei der die mRNA aus der DNA-Vorlage synthetisiert wird, verbleiben in der Reaktionsmischung mehrere Enzyme, darunter die T7-RNA-Polymerase sowie DNase I aus dem vorangegangenen Prozessschritt. Proteinase K kann eingesetzt werden, um diese enzymatischen Rückstände gezielt zu hydrolysieren und damit die Reinheit der mRNA weiter zu erhöhen.
Wirkmechanismus
Proteinase K bindet zunächst an frei zugängliche Bereiche eines Zielproteins und beginnt dort mit der Spaltung von Peptidbindungen. Durch diese ersten Schnitte verliert das Protein schrittweise seine kompakte, dreidimensionale Faltung. Mit zunehmender Destabilisierung werden auch zuvor geschützte innere Strukturbereiche angreifbar, sodass das Protein allmählich in immer kleinere Peptidfragmente zerfällt – es „bröckelt“ förmlich auseinander. Am Ende des Prozesses verbleiben kurze, biologisch inaktive Peptidketten, die keine enzymatische Funktion mehr besitzen.
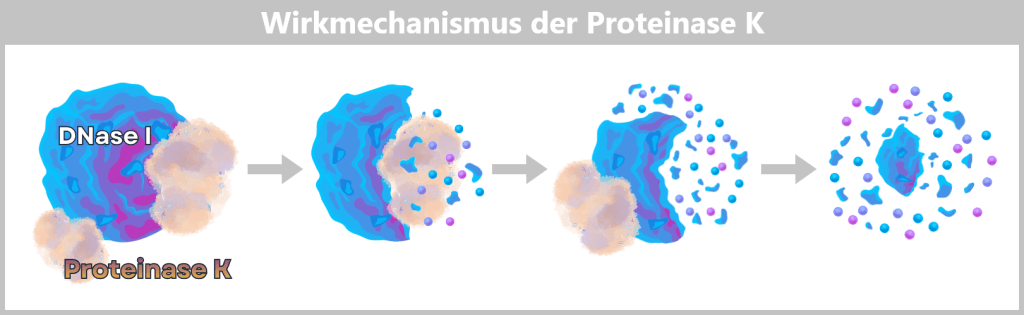
Abb. 1.3.2: Schematische Darstellung der Proteinase-K-Wirkung Die Proteinase K greift die DNase I an und spaltet deren Peptidbindungen. Zunächst werden äußere Proteinbereiche hydrolysiert, wodurch die Struktur destabilisiert wird. Schrittweise zerfällt das Protein in kleinere Fragmente, bis schließlich nur noch kurze, biologisch inaktive Peptidstücke verbleiben.
Proteinase K ist selbst ein Protein und wird nicht im Endprodukt belassen. Ihre Aktivität kann durch Prozessbedingungen, etwa durch die Reduktion stabilisierender Calciumionen, vermindert werden. In der Praxis erfolgt die Entfernung der Proteinase K jedoch vor allem physikalisch: Sowohl das Enzym selbst als auch die entstehenden Peptidfragmente werden in nachfolgenden Reinigungsschritten, insbesondere durch Ultrafiltration (z. B. Tangentialflussfiltration) und chromatographische Verfahren, zuverlässig aus der Lösung entfernt.
Bedeutung im Prozess
Durch den gezielten Einsatz von Proteinase K können restliche Prozessenzyme, bakterielle Proteine und andere Proteinverunreinigungen effektiv reduziert werden. Dies schafft definierte Ausgangsbedingungen für die nachfolgenden Filtrations- und chromatographischen Reinigungsschritte. Der Einsatz von Proteinase K stellt dabei keinen zwingenden Bestandteil jedes Herstellungsprozesses dar, erhöht jedoch die Robustheit und Reproduzierbarkeit der Aufarbeitung, insbesondere bei großskaliger industrieller Produktion.
1.3.3. Filtration
Die Filtration ist ein physikalisches Trennverfahren, das Partikel oder Moleküle hauptsächlich aufgrund ihrer Größe voneinander trennt
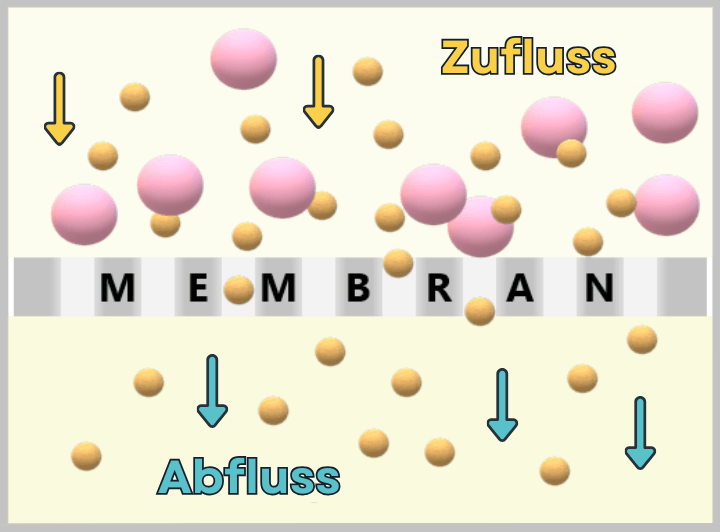
Dabei wird eine Mischung durch ein poröses Filtermaterial geleitet. Im einfachsten Fall wirken die Poren wie ein mechanisches Sieb: Partikel, die größer sind als die Poren, werden zurückgehalten, während kleinere Moleküle passieren.
a) Filtration – Trennung nach Größe
b) Tangentialflussfiltration (TFF)
c) Sterile Filtrationa) Filtration – Trennung nach Größe
Im Rahmen der Plasmidaufreinigung kommen zwei Grundprinzipien zum Einsatz:
- die Abtrennung unlöslicher Partikel (Klärung), um eine trübe Suspension zu klären,
- die Trennung gelöster Moleküle nach Größe (Membranfiltration), um beispielsweise Puffer auszutauschen oder Moleküle zu konzentrieren.
Klärung: Filtration nach Lyse der E.coli-Bakterienzellen
Ausgangslage: Lyse der Bakterienzellen
Die Zugabe von Natronlauge (NaOH) und dem Detergens SDS löst die fetthaltige Zellhülle der Bakterien auf. Die alkalische Lyse wirkt sich dabei nicht nur auf die Membranen, sondern auch auf Nukleinsäuren und Proteine aus. Die Doppelhelix-Struktur der DNA wird denaturiert. Die lange, faserige genomische DNA wird zusätzlich mechanisch fragmentiert. Unter den stark basischen Bedingungen verlieren Proteine ihre dreidimensionale Faltung und damit ihre biologische Funktion.Neutralisation: Ordnung des Systems
Durch Zugabe einer sauren Lösung wird die alkalische Reaktionsmischung neutralisiert. Die kurzen, ringförmigen Plasmid-DNA-Moleküle können unter diesen Bedingungen wieder korrekt renaturieren und bleiben in Lösung. Die fragmentierten und fehlgepaarten Reste der genomischen DNA sowie denaturierte Proteine aggregieren (verklumpen) dagegen zusammen mit Lipiden und Membranbestandteilen und fallen als weißlicher Niederschlag aus.Zwischenzustand
Nach der alkalischen Lyse und Neutralisation liegt die Probe als Zweiphasensystem vor: eine gelöste Phase mit Plasmid-DNA und ein unlöslicher Niederschlag aus aggregierten Bestandteilen. Diese Aggregate bestehen aus fragmentierter genomischer DNA, denaturierten Proteinen, ribosomalen Bestandteilen und Membranresten.Klär- und Tiefenfiltration
Im industriellen Maßstab werden diese Feststoffe effizient durch Tiefenfiltration entfernt. Dabei durchläuft die Suspension ein mehrlagiges, poröses Filtermaterial. Die Partikel werden nicht nur an der Oberfläche, sondern auch innerhalb der Filtermatrix zurückgehalten, was eine hohe Aufnahmekapazität und effektive Klärung ermöglicht.Ergebnis
Das klare Filtrat enthält die gelöste Plasmid-DNA weitgehend befreit von Zelltrümmern und ausgefällten Aggregaten. Diese grob gereinigte Lösung bildet die Grundlage für die nachfolgenden, hochauflösenden chromatographischen Reinigungsschritte.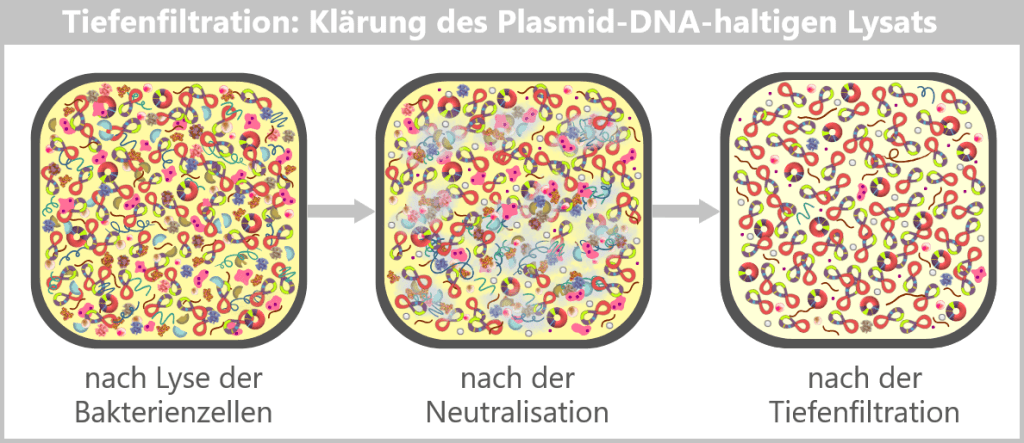
Abb. 1.3.3-A: Die Abbildung zeigt das Zelllysat vor und nach der Tiefenfiltration. Nach der alkalischen Lyse enthält das Lysat neben der gewünschten Plasmid-DNA zahlreiche bakterielle Bestandteile und Prozesshilfsstoffe.
Während der anschließenden Neutralisation bilden fragmentierte genomische DNA, denaturierte Proteine, sowie Lipide und Membranreste unregelmäßige, voluminöse Aggregate (dargestellt als Wolken).
Durch die Tiefenfiltration werden diese ausgefällten Aggregate effektiv entfernt. Das resultierende Filtrat ist eine klare Lösung, die die gelöste Plasmid-DNA enthält. Daneben können noch lösliche RNA-Reste, Endotoxine (LPS), Salze, Pufferbestandteile sowie geringe Mengen gelöster Proteinreste vorhanden sein.Membranfiltration: Ultrafiltration und Diafiltration
Eine besonders wichtige Variante der Membranfiltration in der Biotechnologie ist die Ultrafiltration (UF). Sie dient der Trennung gelöster Makromoleküle (z. B. Nukleinsäuren oder Proteine) von kleineren Molekülen wie Salzen, Puffern oder Nukleotiden.
Wird während der Ultrafiltration kontinuierlich frischer Puffer zugegeben, spricht man von Diafiltration (DF). Dadurch lassen sich Moleküle nicht nur konzentrieren, sondern auch gezielt in eine neue Lösung überführen. UF und DF werden häufig kombiniert und als UF/DF-Prozess bezeichnet.
Die vorherrschende Technik für die Durchführung von UF/DF ist die Tangentialflussfiltration (TFF). [ROCKER]
b) Tangentialflussfiltration (TFF)
Bei der klassischen Filtrationsmethode (Dead-End-Filtration oder Direktflussfiltration) strömt die Lösung senkrecht auf die Membran, wodurch sich große Moleküle auf der Oberfläche ablagern und eine „Filterkuchenschicht“ bilden können. Dies verringert mit der Zeit den Durchfluss und die Lebensdauer der Membran.
Die Tangentialflussfiltration (TFF) löst dieses Problem: Hier bewegt sich die Flüssigkeit tangential entlang der Membran. Ein Teilstrom (das Permeat) passiert die Membran, während der Reststrom (das Retentat) parallel weiterfließt und Ablagerungen abträgt. Dadurch bleibt die Membran länger durchlässig und es können Konzentration und Diafiltration gleichzeitig durchgeführt werden.
Begriffserklärungen:
Retentat: Die Fraktion, die von der Membran zurückgehalten wird (meist das gewünschte Makromolekül).
Permeat: Die Fraktion, die die Membran passiert (meist kleine Moleküle und Verunreinigungen).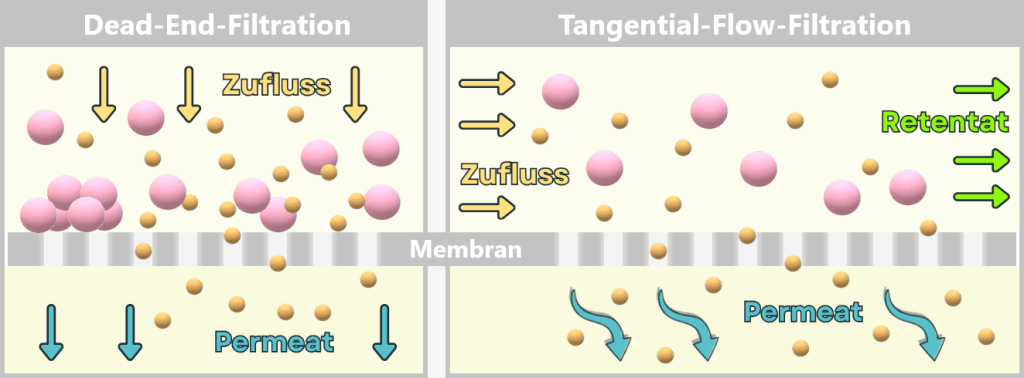
Abb. 1.3.3-B1: Dead-End-Filtration vs. Tangential-Flow-Filtration Bei der Dead-End-Filtration trifft der gesamte Zufluss senkrecht auf die Membran. Kleine Moleküle (Permeat) passieren die Poren, während große Moleküle zurückgehalten werden. Dadurch bildet sich mit der Zeit ein „Filterkuchen“, der die Effizienz verringert.
Bei der Tangential-Flow-Filtration strömt die Lösung parallel zur Membran. Während kleine Moleküle durch die Membran ins Permeat gelangen, werden größere Moleküle im Retentat weitergeführt. Durch den tangentialen Fluss wird die Membran kontinuierlich gespült, Ablagerungen werden verhindert und die Filtration bleibt über längere Zeit stabil und effizient.TFF ist heute eine Standardtechnik in der biopharmazeutischen Produktion, weil sie robust, skalierbar und vielseitig einsetzbar ist.
TFF bei der Impfstoffherstellung
Die Tangentialflussfiltration (TFF) wird während der mRNA-Produktion an mehreren Punkten eingesetzt: zum Beispiel nach der Transkription, nach enzymatischen Prozessierungen oder vor/nach chromatographischen Reinigungsschritten. Ihre Aufgaben reichen vom Entfernen kleiner Moleküle über den Pufferwechsel bis zur Produktkonzentration.
In diesem Kapitel konzentrieren wir uns auf einen zentralen, aber nicht abschließenden Reinigungsschritt:
TFF direkt nach der in-vitro-Transkription (IVT)
Hier dient sie der Grobreinigung der frisch synthetisierten mRNA. Sie trennt die mRNA von Enzymen, Reagenzien und Nebenprodukten und bereitet sie so für die nachfolgende Hochreinigung vor.
Ausgangslage: Die „Rohlösung“ nach der IVT, DNase-I- und Proteinase-K-Behandlung
Die mRNA liegt zu diesem Zeitpunkt in einer reaktionshaltigen Lösung vor, die noch eine Vielzahl unerwünschter Stoffe enthält:
- Enzyme (z. B. T7-Polymerase, DNase I),
- unverbrauchte Nukleotide,
- DNA-Reste vom Template,
- Puffersalze,
- Nebenprodukte der Reaktion (z. B. kurze RNA-Fragmente),
- ggf. Restbestandteile aus vorgelagerten biologischen Prozessen.
Um die mRNA-Lösung aufzubereiten, wird sie durch eine Filtereinheit mit Membranen definierter Porengröße geleitet. Moleküle unterhalb dieser Porengröße – etwa Salze, Nukleotide oder kleine Proteinreste – passieren die Membran (Permeat), während die großen mRNA-Moleküle im Retentat zurückgehalten werden.
Während des Prozesses wird kontinuierlich frischer Puffer zugegeben (Diafiltration). Dadurch werden niedermolekulare Verunreinigungen schrittweise ausgewaschen, während die mRNA gleichzeitig in den Zielpuffer überführt und konzentriert wird (Ultrafiltration).
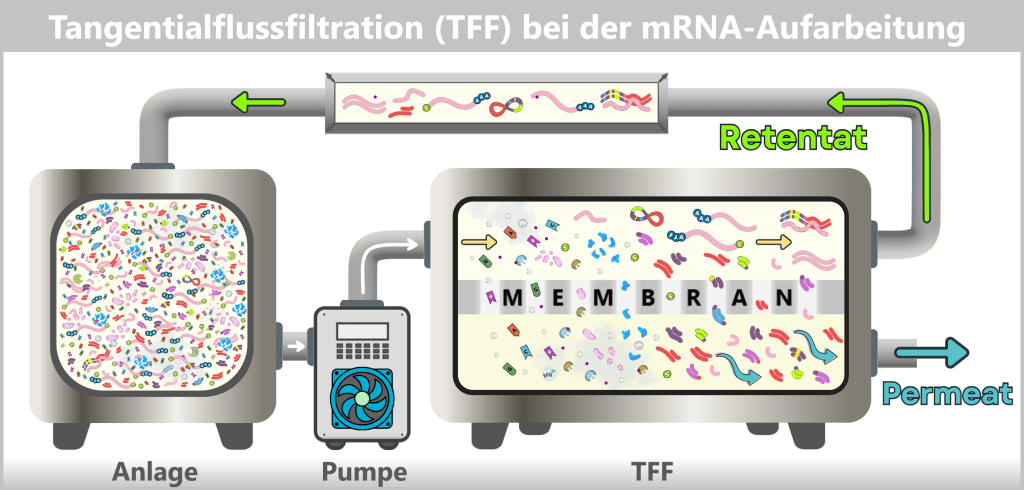
Abb. 1.3.3-B2: Schematische Darstellung der Tangentialflussfiltration (TFF) im Rahmen der mRNA-Aufarbeitung Die mRNA-Lösung zirkuliert kontinuierlich durch das UF/DF-System. Kleine Moleküle passieren die Membran und werden ausgespült (Permeat). Eine Pumpe sorgt dafür, dass das Retentat – also die mRNA-haltige Fraktion – mehrfach über die Membran geführt wird. Durch wiederholte Zyklen und Pufferzugabe werden Verunreinigungen entfernt und die mRNA-haltige Fraktion gleichzeitig konzentriert und umgepuffert.
Wichtig ist:
Die TFF ist eine Grobtrennung nach Größe. Verunreinigungen mit ähnlicher Größe oder Form wie die mRNA – etwa doppelsträngige RNA, RNA-Fragmente vergleichbarer Länge oder RNA:DNA-Hybride – können in diesem Schritt nicht entfernt werden. Diese kritischen Nebenprodukte verbleiben im Retentat und müssen in den nachfolgenden, hochauflösenden chromatographischen Schritten abgetrennt werden.
c) Sterile Filtration
Die sterile Filtration ist ein zentraler letzter Schritt vor der Abfüllung eines mRNA-Impfstoffs. Dabei wird die fertige Formulierung (Lipidnanopartikel, die die mRNA enthalten) durch einen sehr feinen Filter geleitet. Die Porengröße ist klein genug, um alle Bakterien, Pilze und größere Partikel zuverlässig zurückzuhalten.
Prinzip der sterilen Filtration
Die sterile Filtration ist ein reines Größen-Siebverfahren:
- Die Membran besitzt exakt definierte Poren von 0,22 µm.
- Mikroorganismen sind deutlich größer und bleiben hängen.
- Die Wirkstoffpartikel (LNPs) sind viel kleiner (60–100 nm = 0,06–0,1 µm) und passieren den Filter problemlos.
Da keine Hitze oder chemische Behandlung nötig ist, eignet sich dieser Schritt besonders für empfindliche biologische Präparate wie mRNA-LNPs, die nicht autoklaviert (mit Dampf, Druck und Hitze sterilisiert) werden dürfen.
Ablauf im Detail
Die LNP-mRNA-Lösung liegt im Formulierpuffer vor. Sterile Filtration erfolgt in einer Reinraumumgebung (Klasse A/B) unter streng aseptischen Bedingungen. Es werden ausschließlich sterile Einwegartikel verwendet: Filter, Schläuche, Beutel, Pumpenköpfe, Sammelbehälter.
Die Formulierung wird mit sanftem Druck (Peristaltikpumpe oder Druckgas) durch den 0,22-µm-Filter geleitet.
Der Filter hält zurück:
▪️Bakterien (~0,5–5 µm)
▪️Hefen und Pilzsporen (~1–10 µm)
▪️Partikel oder Aggregate > 0,22 µmDurchgelassen werden:
▪️LNPs (60–100 nm)
▪️Pufferbestandteile
▪️gelöste Moleküle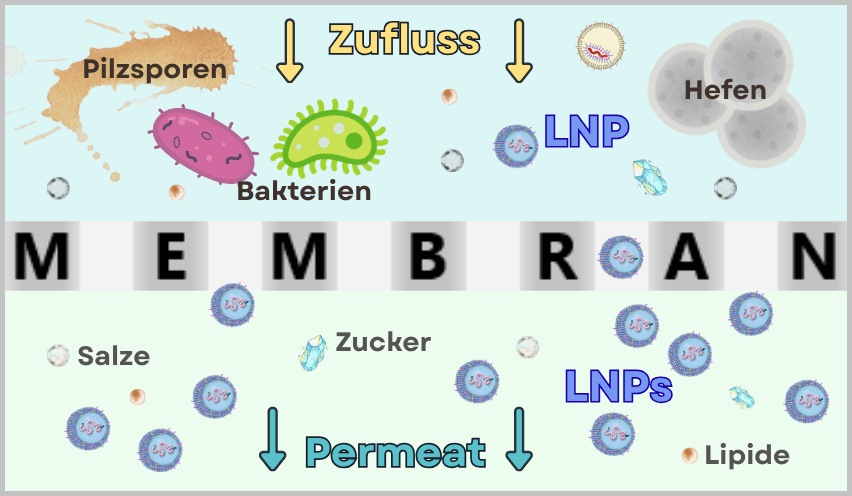
Abb. 1.3.3-B3: Schematische Darstellung der Sterilen Filtration Damit wird sichergestellt, dass die Formulierung frei von Mikroorganismen und sichtbaren Partikeln ist.
Warum sterile Filtration notwendig ist
Nach GMP-Richtlinien dürfen sterile Arzneimittel ausschließlich mit validierten, sterilen Filtrationsprozessen hergestellt werden. Die sterile Filtration ist daher Pflicht und stellt sicher, dass:
- die fertige Impfstofflösung mikrobiologisch einwandfrei ist
- keine großen Partikel, Aggregate oder Verunreinigungen in das Endprodukt gelangen
- die anschließende sterile Abfüllung („aseptic fill & finish“) korrekt erfolgen kann
Die sterile Filtration ist kein chemischer Reinigungsschritt, sondern ein essenzieller Sicherheits- und Qualitätsprozess. Sie bildet die Brücke zwischen der biotechnologischen Aufreinigung der mRNA und dem finalen, sterilen Arzneimittel in der Durchstechflasche.
1.3.4. Chromatographie – das molekulare Trennverfahren
Die Chromatographie ist eine der wichtigsten Verfahren zur Reinigung biologischer Moleküle wie DNA, RNA oder Proteine. Das Prinzip basiert auf der unterschiedlich starken Wechselwirkung der Stoffe mit zwei Phasen: einer stationären Phase (ein festes Trägermaterial in einer Säule) und einer mobilen Phase (eine flüssige Lösung, die das Probengemisch transportiert).
Während die mobile Phase – also das zu trennende Molekülgemisch – durch die stationäre Phase fließt, werden die einzelnen Moleküle je nach ihrer Größe, Ladung oder anderen chemischen Eigenschaften unterschiedlich stark zurückgehalten. Dies führt zu einer zeitlichen Trennung, bei der die Bestandteile des Gemischs nacheinander eluieren (herausgespült werden).
Anschaulich gesprochen ist die Chromatographie ein „molekularer Hindernislauf“: Manche Moleküle interagieren kaum mit der stationären Phase und gleiten schnell hindurch. Andere bleiben häufiger „hängen“ und werden dadurch stärker verzögert. So erreicht jede Komponente das Ziel zu einer anderen Zeit und kann sauber aufgefangen werden.
Chromatographie-Methoden in der mRNA-Reinigung
In industriellen Prozessen, wie sie beispielsweise von MERCK beschrieben werden („Manufacturing strategies for mRNA vaccines“), kommen dabei zwei chromatographische Verfahren in Kombination zum Einsatz:
Methode Trennung nach Typische Anwendung Affinitäts-Chromatographie (Poly(dT)) spezifischer Bindung Primäre Aufreinigung: Isolierung intakter mRNA aus der IVT-Reaktionsmischung Ionenaustausch-
Chromatographieelektrischer Ladung Feinreinigung: Entfernen von dsRNA, RNA:DNA-Hybriden, DNA-Fragmenten und weiteren produktbezogenen Verunreinigungen
a) Poly(dT)-Affinitätschromatographie
Poly(dT)-AC = Poly(dT) Affinity Chromatography
Die Affinitätschromatographie nutzt spezifische Wechselwirkungen zwischen Biomolekülen und einer funktionalisierten Oberfläche. Bei der Herstellung von mRNA-Impfstoffen wird die Poly(dT)-Affinitätschromatographie gezielt eingesetzt, um die reife mRNA nach der in-vitro-Transkription (IVT) aus der komplexen Reaktionslösung zu isolieren.
Ausgangslage: Nach in-vitro-Transkription (IVT), enzymatischer Nachbehandlung und Tangentialflussfiltration (TFF)
Prinzip und Ablauf
Stationäre Phase: Das Harz in der Säule ist mit langen, einzelsträngigen Ketten von Thymin-Basen (T) beschichtet, der sogenannten Poly(dT)-Matrix.
Mobile Phase: Die voraufgereinigte IVT-Lösung – bestehend aus mRNA, RNA-Nebenprodukten (z. B. dsRNA, verkürzte Transkripte), Restmengen an DNA-Fragmenten sowie definierten Puffersalzen – wird auf die Säule gegeben.
Das Zielmolekül ist das reife mRNA-Molekül – die „polyadenylierte mRNA“. Diese trägt am 3′-Ende eine Kette aus Adenin-Basen (A), den sogenannten Poly-A-Schwanz.
Spezifische Bindung über Basenpaarung
Adenin (A) und Thymin (T) bilden – wie in der DNA – spezifische Wasserstoffbrückenbindungen aus. Bildlich formuliert, der Poly(A)-Schwanz der mRNA hängt sehr stabil am Poly(dT)-Haken des Harzes.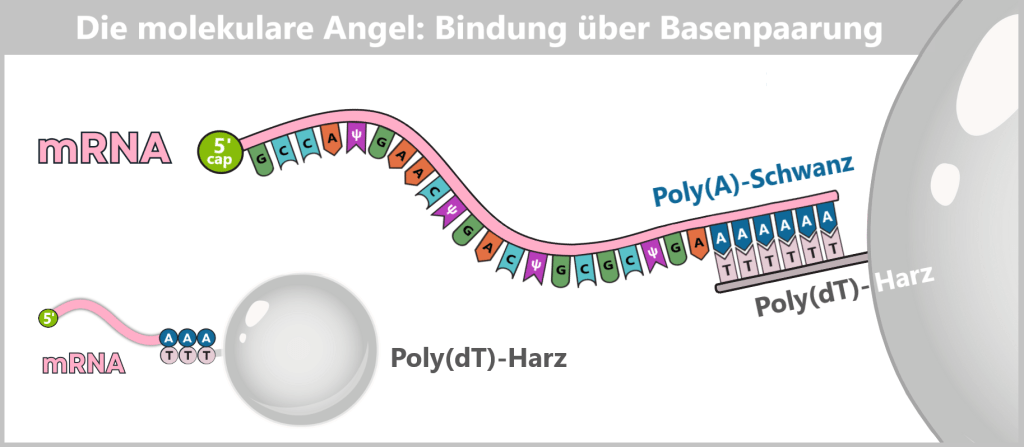
Abb. 1.3.4-A1: Die mRNA haftet über ihren Poly(A)-Schwanz an dem Poly(dT)-Harz. Der Poly(A)-Schwanz therapeutischer mRNA umfasst typischerweise etwa 100–120 Adenin-Basen; in der Abbildung ist er aus Darstellungsgründen verkürzt dargestellt.
Nur Moleküle mit intaktem Poly(A)-Schwanz binden fest an das Harz; alle anderen Bestandteile der IVT-Reaktion fließen durch. Dazu gehören:
(a) verbleibende freie Nukleotide (nur in Spuren)
(b) Proteinfragmente aus dem enzymatischen Abbau
(c) kurze RNA-Fragmente ohne Poly(A)-Schwanz
(d) DNA-Fragmente und DNA-haltige Nebenprodukte
(e) verbleibende supercoiled Plasmid-DNA
(f) freie dsRNA
(g) PuffersalzeWaschen (Wash)
Waschpuffer entfernen schwach gebundene, unspezifisch haftende Moleküle, während die mRNA weiterhin fest an der Poly(dT)-Matrix verbleibt.Elution (Freisetzung)
Um die reine mRNA wieder von der „Angel“ zu lösen, werden die spezifischen Bindungen zwischen A und T gezielt aufgebrochen. Dies geschieht typischerweise durch Absenkung der Ionenstärke und/oder moderate Temperaturerhöhung. Die mRNA löst sich von der Matrix und wird eluiert.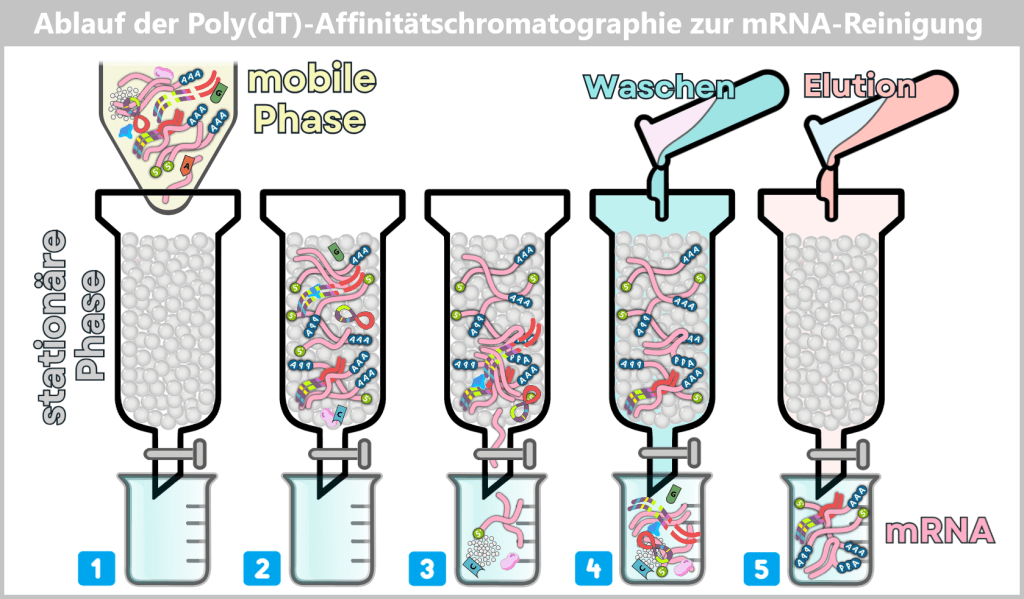
Abb. 1.3.4-A2: Schematische Darstellung der Poly(dT)-Affinitätschromatographie 1) Die IVT-Reaktionsmischung wird auf die mit Poly(dT) funktionalisierte Säule aufgetragen.
2) Die polyadenylierte mRNA bindet über Basenpaarung spezifisch an das Harz,
3) während nicht-bindende Komponenten direkt durchlaufen.
4) Ein Waschpuffer entfernt schwach oder unspezifisch gebundene Moleküle.
5) Durch Zugabe eines Elutionspuffers wird die mRNA von der Poly(dT)-Matrix gelöst und in reiner Form eluiert.Vorteile
- sehr hohe Selektivität durch Poly(A)/Poly(dT)-Hybridisierung
- effiziente Abtrennung der meisten unspezifischen RNA- und Proteinverunreinigungen
- Anreicherung der vollständigen, korrekt polyadenylierten mRNA
- vergleichsweise einfache und robuste Methode
Limitierungen – warum weitere Reinigungsschritte notwendig sind
Trotz der hohen Spezifität kann die Poly(dT)-Affinität nicht alle kritischen Verunreinigungen entfernen:- dsRNA: kann über polyadenylierte Enden oder unspezifisch an die Matrix binden
- RNA:DNA-Hybride: wenn DNA an mRNA gebunden ist, wird sie gemeinsam mit eluiert
- fragmentierte mRNA mit Poly(A)-Resten bindet ebenfalls
- intramolekulare dsRNA-Strukturen innerhalb der mRNA führen zu Ko-Elution
- größere RNA-Komplexe, die an mRNA assoziiert sind, werden mitisoliert
- Endotoxine werden nicht spezifisch entfernt und können ko-eluiert werden
Hersteller wie MERCK, Lonza oder Cytiva empfehlen daher im Anschluss eine Anionenaustauschchromatographie (AEX) zur Feintrennung, insbesondere zur Entfernung von dsRNA und DNA-Restverunreinigungen.
b) Ionenaustausch-Chromatographie
Die Ionenaustausch-Chromatographie (IEX) ist eine bewährte Methode zur Reinigung von Nukleinsäuren und Proteinen. Sie trennt Moleküle anhand ihrer elektrischen Ladung. Bei Nukleinsäuren ist damit nicht eine feste Oberfläche gemeint, sondern die hohe Dichte negativ geladener Phosphatgruppen entlang des Moleküls. Im Gegensatz dazu ist die Ladung von Proteinen von der spezifischen Abfolge und Faltung ihrer Aminosäuren abhängig und kann positive und negative Bereiche aufweisen.
Je nach Art der geladenen Zielmoleküle unterscheidet man zwei Haupttypen:
▪️Anionenaustauscher (AEX, Anion Exchange Chromatography):
binden negativ geladene Moleküle wie DNA oder RNA.
▪️Kationenaustauscher (CEX, Cation Exchange Chromatography):
binden positiv geladene Moleküle, beispielsweise bestimmte Proteine.Aufgrund ihrer durchgehenden Phosphatgruppe im Rückgrat tragen Nukleinsäuren eine hohe, gleichmäßige negative Ladungsdichte.
Zucker-Phosphat-Rückgrat als Quelle der negativen Ladung von DNA/RNA
Schematische Darstellung der strukturellen Grundlage der negativen Ladung von Nukleinsäuren
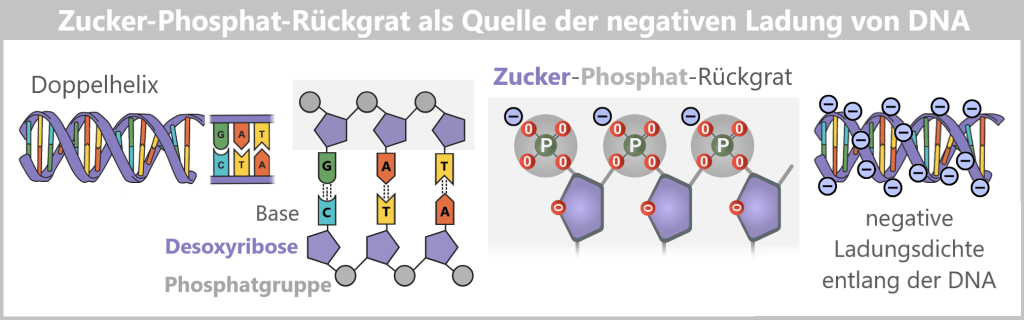
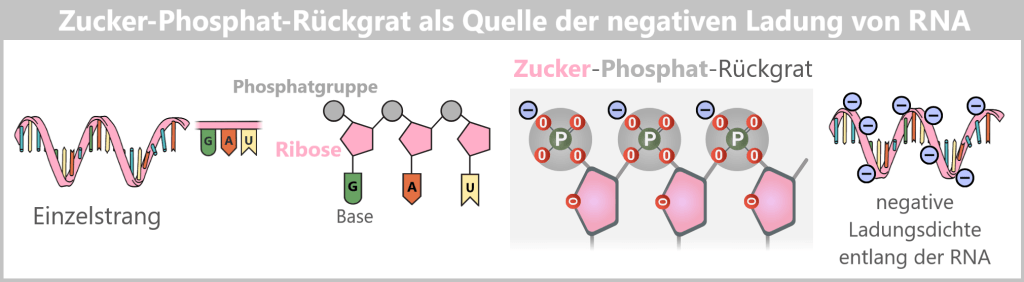
DNA (und analog RNA) besteht aus einem Zucker-Phosphat-Rückgrat, in dem jede Phosphatgruppe eine negative Ladung trägt. Diese regelmäßig wiederholten Phosphatgruppen verleihen dem Molekül eine hohe und entlang seiner Länge gleichmäßig verteilte negative Ladungsdichte. Die Basen tragen nicht zur Gesamtladung bei.
Diese Eigenschaft macht die Anionenaustauschchromatographie (AEX) zur Methode der Wahl für deren Aufreinigung. Im Gesamtprozess kommt die AEX daher gleich doppelt zum Einsatz: Zunächst zur Aufreinigung der Plasmid-DNA-Vorlage aus dem bakteriellen Lysat. Danach, nach der in-vitro-Transkription (IVT) und Grobreinigung, zur hochauflösenden Trennung der gewünschten mRNA von strukturell ähnlichen Verunreinigungen, insbesondere doppelsträngiger RNA (dsRNA).
AEX bei der Plasmid-DNA-Aufreinigung
Ausgangslage
Nach Lyse der E.-coli-Zellen, Neutralisation und anschließender Tiefenfiltration liegt ein geklärtes Lysat vor, das die Plasmid-DNA enthält. Daneben können noch bakterielle RNA-Reste, Endotoxine, Salze, Pufferbestandteile sowie geringe Mengen gelöster Proteinreste vorhanden sein.
Prinzip und Ablauf
Stationäre Phase: Das Trägermaterial (Harz) besteht aus kleinen, positiv geladenen Polymerkügelchen. Diese dienen als Bindungsoberfläche für negativ geladene Moleküle.
Mobile Phase: Das geklärte Lysat wird auf die Säule gegeben. Zunächst binden die negativ geladenen Biomoleküle – darunter Plasmid-DNA, RNA und Endotoxine – an das positiv geladene Harz. Ungebundene oder schwach geladene Bestandteile werden durch Spülpuffer entfernt.
Das Zielmolekül ist die Plasmid-DNA – die entlang ihrer Länge eine gleichmäßig verteilte negative Ladungsdichte besitzt.
Selektive Elution: Anschließend wird ein Puffer mit ansteigender Salzkonzentration über die Säule geleitet. Die positiv geladenen Ionen (z. B. Na⁺) im Puffer konkurrieren mit den Biomolekülen um die Bindungsstellen des Harzes. Dadurch lösen sich die Moleküle in einer charakteristischen Reihenfolge – je nach ihrer Ladungsdichte und Bindungsstärke:
- Endotoxine und kleine RNA-Fragmente eluieren früh, da sie eine geringere Ladungsdichte besitzen.
- Plasmid-DNA – überwiegend in superhelikaler, aber auch in relaxeder Ringform – bindet aufgrund ihrer hohen negativen Ladungsdichte am stärksten und eluierte erst bei höheren Salzkonzentrationen.
Ergebnis
Durch diese selektive Elution erhält man eine Lösung – die reich an hochreiner Plasmid-DNA ist, überwiegend in superhelikaler Form, mit einem geringeren Anteil relaxeder Ringformen – während die meisten anderen Zellbestandteile und Verunreinigungen bereits entfernt wurden. Diese DNA-Fraktion dient anschließend als Template für die nachfolgende in-vitro-Transkription (IVT).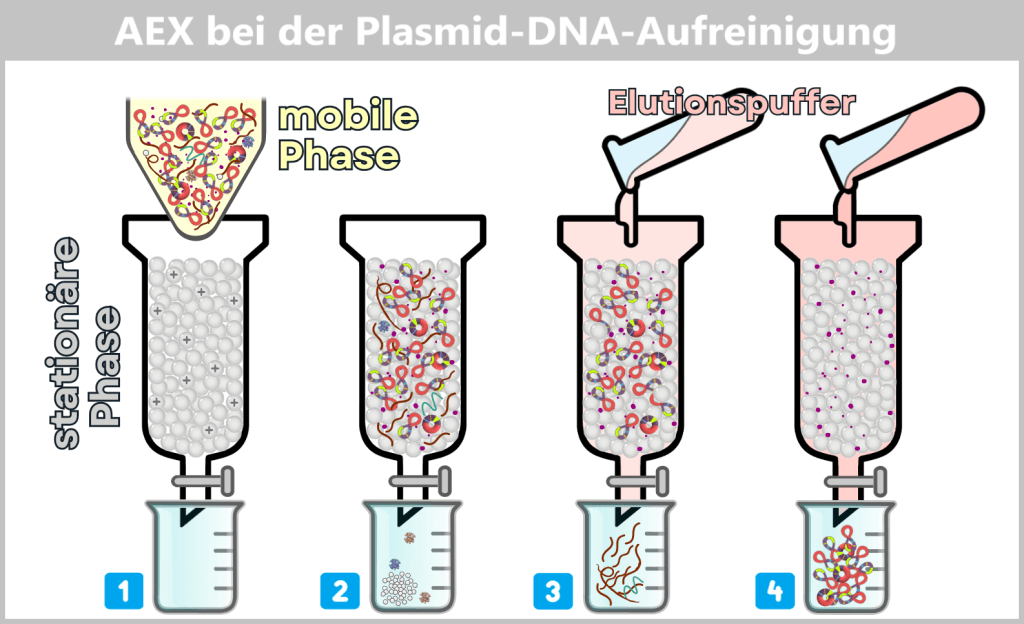
Abb. 1.3.4-B1: Schematische Darstellung der AEX bei der Plasmid-DNA-Aufreinigung 1) Das geklärte Zelllysat (mobile Phase) wird auf die Säule mit der positiv geladenen stationären Phase (Anionenaustauschharz) aufgetragen. 2) Negativ geladene Moleküle der Probe binden an das Harz; nicht bzw. schwach gebundene Komponenten (Salze, Proteinreste) eluieren früh. 3) Die Elution beginnt: Ein Puffer mit steigender Salzkonzentration wird zugegeben. Die gelösten Ionen (z. B. Na⁺ und Cl⁻) konkurrieren mit den gebundenen Molekülen um die Bindungsstellen. Bei niedrigen bis mittleren Salzkonzentrationen eluieren vor allem kurze und/oder einzelsträngige RNA-Fragmente. 4) Plasmid-DNA eluiert erst bei höheren Salzkonzentrationen aufgrund ihrer hohen, gleichmäßig verteilten negativen Ladungsdichte. Endotoxine (LPS) binden sehr stark an die Matrix, was grundsätzlich vorteilhaft aus Sicht der Produktreinigung ist. Problematisch sind lediglich jene Anteile, die aufgrund unspezifischer Wechselwirkungen oder Assoziation mit DNA gemeinsam mit der Plasmid-DNA eluieren können.
Nach der Anionenaustauschchromatographie liegt die Plasmid-DNA in hoher Reinheit vor. Das Produkt besteht überwiegend aus superhelikaler Plasmid-DNA und kann geringe Anteile relaxierter Ringformen enthalten. In niedrigen Konzentrationen sind zudem Reste kurzer RNA-Fragmente oder Endotoxine nachweisbar, während zelluläre Makromoleküle und Prozessverunreinigungen größtenteils entfernt wurden.
AEX in der mRNA-Reinigung: Trennung von mRNA und dsRNA
Ausgangslage: Nach in-vitro-Transkription (IVT), enzymatischer Nachbehandlung, Tangentialflussfiltration (TFF) und Poly(dT)-Affinitätschromatographie
Nach diesen vorgelagerten Prozessschritten liegt eine stark angereicherte mRNA-Fraktion vor. Dennoch enthält diese mRNA-Rohlösung typischerweise noch bestimmte Nebenprodukte und Verunreinigungen, die durch die Poly(dT)-Affinität nicht vollständig entfernt werden können, darunter:
(a) doppelsträngige RNA-Nebenprodukte (dsRNA)
(b) RNA:DNA-Hybride (in geringen, prozessabhängigen Anteilen)
(c) fragmentierte mRNA mit verbliebenen Poly(A)-Resten
(d) nicht korrekt gecappte mRNA-Transkripte
(e) Endotoxine (LPS), insbesondere in Assoziation mit NukleinsäurenDie nachfolgende Anionenaustauschchromatographie dient daher nicht der weiteren Anreicherung der mRNA, sondern der hochauflösenden Entfernung strukturell ähnlicher RNA-Nebenprodukte.
Prinzip und Ablauf
Stationäre Phase: Die stationäre Phase besteht aus positiv geladenen Anionenaustauschharzen, meist polymeren Kügelchen mit quaternären Ammoniumgruppen. Diese bieten Bindungsstellen für negativ geladene Nukleinsäuren.
Mobile Phase: Die mRNA-Rohlösung wird als mobile Phase auf die Säule aufgetragen. Unter niedriger Salzkonzentration binden die negativ geladenen Nukleinsäuren der Probe an das Harz.
Das Zielmolekül ist die einzelsträngige mRNA. Sie besitzt entlang ihres Rückgrats eine negative Ladung, weist jedoch aufgrund ihrer Einzelsträngigkeit, Sekundärstruktur und Flexibilität eine geringere effektive Ladungsdichte auf als doppelsträngige RNA.
Selektive Elution
Im nächsten Schritt wird ein Elutionspuffer mit ansteigender Salzkonzentration über die Säule geleitet. Die im Puffer enthaltenen Ionen (z. B. Na⁺) konkurrieren mit den gebundenen RNA-Molekülen um die Bindungsstellen des Harzes. Dadurch werden die gebundenen Komponenten in Abhängigkeit von ihrer Bindungsstärke schrittweise eluiert:
Einzelsträngige mRNA eluieren bei niedrigen bis mittleren Salzkonzentrationen, da ihre effektive Ladungsdichte geringer ist.
Doppelsträngige RNA (dsRNA) bindet deutlich stärker an das Anionenaustauschharz und eluiert erst bei höheren Salzkonzentrationen oder verbleibt auf der Säule.
RNA:DNA-Hybride stellen für die chromatographische Reinigung eine besondere Herausforderung dar. Ihre Bindungseigenschaften bewegen sich zwischen denen von einzelsträngiger RNA (ssRNA) und doppelsträngiger RNA (dsRNA). Dieser hybride Charakter ergibt sich nicht nur aus dem RNA:DNA-Duplex selbst, sondern wird zusätzlich durch drei Faktoren bestimmt: der Länge der Hybridregion, dem Anteil freier einzelsträngiger RNA sowie der räumlichen Konformation des Moleküls.
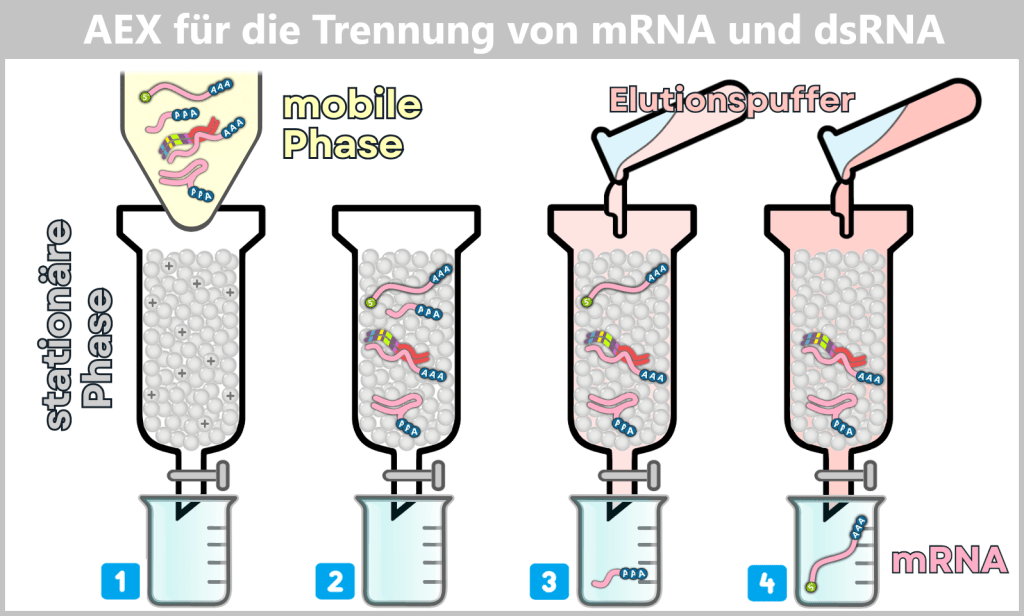
Abb. 1.3.4-B2: Schematische Darstellung der AEX zur Trennung von mRNA und dsRNA
Die Darstellung ist vereinfacht und zeigt jede Komponente exemplarisch nur einmal.1) Die mRNA-haltige Lösung (mobile Phase) – bestehend aus der gewünschten mRNA, dsRNA und anderen Nebenprodukten – wird auf die Säule geleitet. Die stationäre Phase besteht aus einem positiv geladenen Anionenaustauschharz. 2) Unter niedriger Salzkonzentration binden alle negativ geladenen RNA-Moleküle an das Harz. Aufgrund ihrer höheren Ladungsdichte und steiferen Helixstruktur bindet dsRNA stärker als die flexiblere, einzelsträngige mRNA. 3) Durch Zugabe eines Puffers mit ansteigender Salzkonzentration werden konkurrierende Anionen (z.B. Cl⁻) eingeführt. Diese verdrängen die gebundenen RNA-Moleküle schrittweise von der Säule. Zuerst werden schwach gebundene Verunreinigungen wie kurze RNA-Fragmente freigesetzt. 4) Bei niedrigen bis mittleren Salzkonzentrationen eluiert die gewünschte einzelsträngige mRNA. Doppelsträngige RNA verbleibt aufgrund ihrer stärkeren Bindung länger auf der Matrix und eluiert erst bei höheren Salzkonzentrationen oder verbleibt teilweise auf der Säule.
Ergebnis
Die Anionenaustauschchromatographie (AEX) ermöglicht die selektive Abtrennung von mRNA gegenüber stärker oder schwächer gebundenen Nukleinsäurestrukturen wie dsRNA. Damit erhält man eine mRNA-Fraktion, die weitgehend frei von dsRNA-Verunreinigungen und Hybridstrukturen ist.
Die so gereinigte mRNA dient als Ausgangsmaterial für die nachfolgende Formulierung in Lipid-Nanopartikel (LNP).
Zusammenspiel der chromatographischen Schritte zur Reinigung der IVT-mRNA
Die Reinigung von IVT-mRNA beruht nicht auf einem einzelnen Trennverfahren, sondern auf der gezielten Kombination mehrerer chromatographischer Prinzipien mit komplementären Stärken und Limitationen.
Die Poly(dT)-Affinitätschromatographie dient dabei der hochselektiven Anreicherung polyadenylierter mRNA. Sie entfernt effektiv nicht-polyadenylierte Nebenprodukte, Enzyme und niedermolekulare Prozessbestandteile. Aufgrund ihres rein sequenzbasierten Bindungsmechanismus kann sie jedoch strukturell ähnliche RNA-Verunreinigungen nicht vollständig differenzieren. Insbesondere dsRNA, fragmentierte mRNA mit Poly(A)-Resten sowie RNA:DNA-Hybride können gemeinsam mit der Ziel-mRNA ko-eluiert werden.
Die nachgeschaltete Anionenaustauschchromatographie (AEX) adressiert diese Limitation, indem sie die verbleibenden Nukleinsäuren anhand ihrer effektiven Ladungsdichte und Konformation trennt. Einzelsträngige mRNA, doppelsträngige RNA-Nebenprodukte und komplexe RNA-Strukturen zeigen dabei charakteristische, jedoch teilweise überlappende Bindungs- und Elutionsprofile.
RNA:DNA-Hybride nehmen in diesem Kontext eine Sonderstellung ein. Aufgrund ihres hybriden strukturellen Charakters lassen sie sich weder eindeutig der einzel- noch der doppelsträngigen RNA zuordnen. Ihre chromatographische Trennbarkeit hängt stark von der Ausprägung der Hybridregion, dem Anteil freier einzelsträngiger RNA sowie der räumlichen Organisation des Moleküls ab. Entsprechend können RNA:DNA-Hybride je nach Prozessbedingungen teilweise gemeinsam mit der mRNA eluieren oder auf der Säule zurückgehalten werden.
Erst das abgestufte Zusammenspiel von Affinitäts- und Ionenaustauschchromatographie ermöglicht somit eine weitgehende Entfernung der relevanten RNA-Nebenprodukte, ohne die strukturelle Integrität der empfindlichen mRNA zu beeinträchtigen. Gleichzeitig macht dieser Ansatz deutlich, dass die vollständige Eliminierung aller potenziellen Hybrid- und Strukturvarianten technisch anspruchsvoll ist und maßgeblich von der Prozessführung abhängt.
1.3.5. Magnetkügelchen-Reinigung
dT-MBP = Poly(dT)-magnetic bead purification
Ein in den regulatorischen Dokumenten (EMA-EPAR, Abschnitt 2.2, S. 32) beschriebener Ansatz zur Reinigung von PCR-Produkten in Prozess 1 ist der Einsatz von Magnetkügelchen.
Ausgangslage: Nach in-vitro-Transkription (IVT), DNase-I-Behandlung und Filtration
Nach der in-vitro-Transkription (IVT), der enzymatischen Nachbehandlung mit DNase I sowie einer ersten Grobreinigung durch Filtration (UF/DF oder TFF) liegt eine mRNA-haltige Lösung vor. Diese enthält neben der gewünschten mRNA weiterhin verschiedene Nebenprodukte der Transkription, darunter:
(a) verbleibende freie Nukleotide (nur in Spuren)
(b) Proteinreste (z. B. Enzyme oder Proteinfragmente)
(c) RNA-Fragmente mit und ohne Poly(A)-Schwanz
(d) unvollständig gecappte RNA
(e) doppelsträngige RNA (dsRNA), teilweise mit polyadenyliertem Strang
(f) RNA:DNA-Hybride
(g) DNA-Template-Reste (insbesondere kurze Fragmente)
(h) Puffersalze aus vorangegangenen ProzessschrittenPrinzip
Die Magnetkügelchen-Reinigung beruht auf einer sequenzspezifischen Affinitätsbindung. Magnetische Polymerkügelchen sind mit Oligo(dT)-Sequenzen funktionalisiert, die komplementär zur Poly(A)-Schwanzsequenz der mRNA sind.
mRNA-Moleküle mit Poly(A)-Ende binden spezifisch an diese Oligo(dT)-Liganden, während Moleküle ohne Poly(A)-Sequenz in Lösung verbleiben.
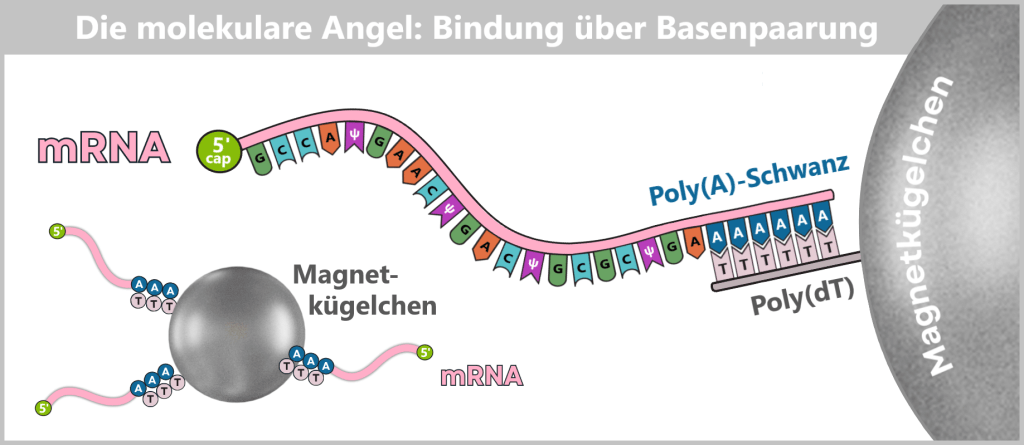
Abb. 1.3.5-A: Die mRNA haftet über ihren Poly(A)-Schwanz am Poly(dT) der Magnetkügelchen. Der Ablauf erfolgt in mehreren Schritten:
Bindung: Poly(dT)-funktionalisierte Magnetkügelchen werden zur Lösung gegeben. Polyadenylierte RNA bindet über spezifische A–T-Basenpaarung an die Oberfläche der Kügelchen.
Magnetische Separation: Das Gefäß wird auf ein Magnetrack gestellt. Die Kügelchen mit der gebundenen RNA werden an die Gefäßwand gezogen, sodass die nicht gebundenen Komponenten entfernt werden können.
Waschen: Mehrere Waschschritte reduzieren unspezifisch gebundene Moleküle. Die Waschbedingungen beeinflussen die Stringenz der Bindung, erlauben jedoch keine vollständige Abtrennung von dsRNA oder RNA:DNA-Hybriden, sofern diese polyadenylierte RNA enthalten.
Elution: Durch geeignete Elutionsbedingungen (z. B. veränderte Salzstärke oder Temperatur) wird die gebundene RNA von den Kügelchen gelöst und in Lösung überführt.
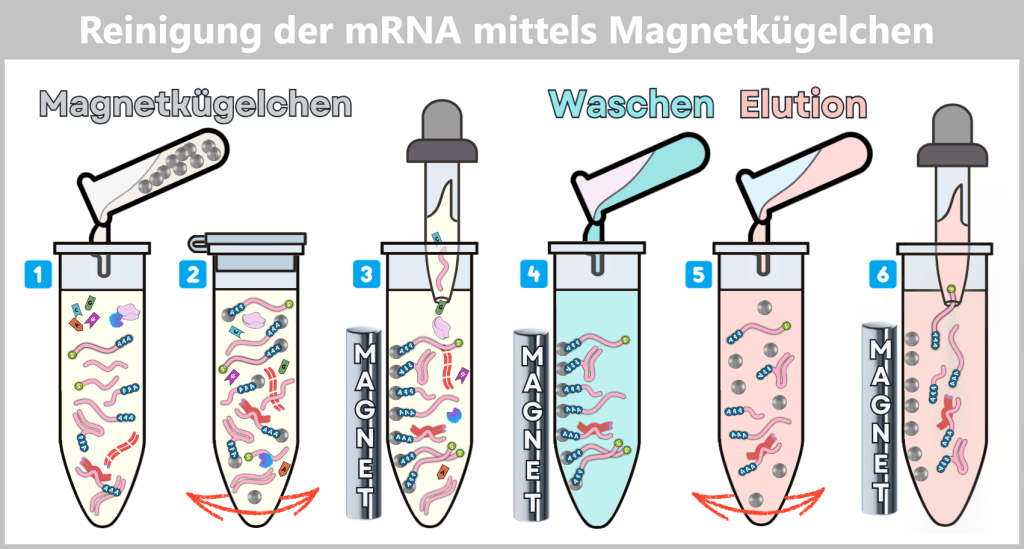
Abb. 1.3.5-B: Ablauf der Magnetkügelchen-Reinigung
Die Darstellung ist vereinfacht und zeigt jede Komponente exemplarisch nur einmal.1) Zugabe der Magnetkügelchen: Poly(dT)-funktionalisierte Magnetkügelchen werden zur Lösung gegeben, die die polyadenyliertemRNA enthält.
2) Durchmischen: Die Probe wird durch sanftes Schwenken oder kurzes Drehen gemischt. Dabei hybridisiert polyadenylierte RNA an die Poly(dT)-Sequenzen der Magnetkügelchen über A–T-Basenpaarung.
3) Magnetische Separation und entfernen des Überstands: Das Gefäß wird auf ein Magnetrack gestellt. Die Magnetkügelchen mit gebundener RNA werden von der Seitenwand des Röhrchens angezogen und bilden dort ein kompaktes Bead-Aggregat. Der flüssige Überstand wird vorsichtig abpipettiert. Er enthält nicht-gebundene Moleküle wie DNA-Fragmente, kurze RNA-Stücke, dsRNA ohne Poly(A)-Tail, Pufferbestandteile usw.
4) Waschschritte: Der Magnet bleibt angelegt. Waschpuffer wird zugegeben, um lose gebundene oder unspezifisch adsorbierte Verunreinigungen zu entfernen. Der Waschpuffer wird kurz gemischt (leichtes Schwenken), danach zieht der Magnet die Beads erneut an die Wand. Dieser Schritt wird 2–3 Mal wiederholt, bis lose gebundene oder unspezifisch assoziierte Komponenten weitgehend entfernt sind.
5) Elution der mRNA: Nach dem Entfernen des letzten Waschpuffers wird der Magnet kurz entfernt. Ein Elutionspuffer (z. B. Wasser mit geringer Salzkonzentration) oder leicht erhöhte Temperatur löst die Hybridisierung zwischen polyadenylierter RNA und Poly(dT)-Oligos. Die RNA geht dabei wieder in Lösung über.
6) Rückkehr auf den Magnetseparator: Das Röhrchen wird erneut auf den Magnet gestellt, sodass die Magnetkügelchen am Rand fixiert werden. Der Überstand enthält jetzt die angereicherte polyadenylierte RNA im Elutionspuffer und wird vorsichtig in ein neues, steriles Gefäß überführt.Hinweis: dsRNA, RNA:DNA-Hybride oder fragmentierte RNA mit Poly(A)-Anteilen können während der Magnetkügelchen-Reinigung ko-eluiert werden.
Trennleistung und Limitierungen
Die Magnetkügelchen-Reinigung ist hochselektiv für polyadenylierte RNA, weist jedoch inhärente Einschränkungen auf:- dsRNA-Nebenprodukte können ko-eluieren, insbesondere wenn sie Poly(A)-Sequenzen enthalten oder strukturell an mRNA assoziiert sind
- RNA:DNA-Hybride werden mitisoliert, sofern die RNA-Komponente polyadenyliert ist
- fragmentierte mRNA mit erhaltenem Poly(A)-Rest bindet ebenfalls an die Kügelchen
- intramolekulare Doppelstrangstrukturen innerhalb der mRNA werden nicht selektiv entfernt
Aus diesen Gründen stellt die Magnetkügelchen-Reinigung keinen vollständigen Reinigungsprozess, sondern eine Anreicherungs- und Vorreinigungsstufe dar.
Nachgeschaltete Reinigung
Da dsRNA und RNA:DNA-Hybride mit Poly(A)-Anteilen während der Magnetkügelchen-Reinigung ko-eluieren können, ist in der Regel ein weiterer Reinigungsschritt erforderlich. Häufig wird hierfür eine Anionenaustauschchromatographie (AEX) oder ein vergleichbares hochauflösendes Trennverfahren eingesetzt.Einordnung im Herstellungsprozess
Die Poly(dT)-basierte Magnetkügelchen-Reinigung wird vor allem im Labormaßstab, in der Prozessentwicklung sowie bei kleineren Produktionsvolumina eingesetzt. Für großtechnische Herstellungsprozesse (Prozess 2) ist sie hingegen weniger praktikabel, da die Magnetkraft in größeren Volumina an Effizienz verliert und die Handhabung komplexer wird. Daher kommen in der industriellen Produktion überwiegend skalierbare chromatographische Verfahren zum Einsatz. Weitere Details zur Anwendung finden sich in den Herstellerinformationen von Thermo Fisher Scientific.
1.4. Vergleich: Prozess 1 vs. Prozess 2
Die folgende Übersicht stellt die beiden Herstellungsverfahren gegenüber: welche Ausgangsmaterialien und Nebenprodukte in den einzelnen Phasen anfallen und mit welchen Methoden sie typischerweise entfernt werden.
Typische Schritte – unterschiedlich je nach Hersteller:
Merkmal
Prozess 1 – PCR-basiert
Prozess 2 – Plasmid-basiert
Art des Verfahrens
Laborprozess
(kleine Mengen, flexibel)Industrieller Prozess
(große Mengen)
DNA-Template
Minimalistisch.
enthält nur die relevanten Elemente für die IVT:
• T7-Promotor
• 5′-UTR
• Spike-Gen
• 3′-UTR
• Poly(A)-SequenzKomplex.
und zusätzliche Elemente für die Vermehrung in E. coli:
• Origin of Replication (ORI)
• Antibiotikaresistenzgen
• regulatorische bakt. Sequenzen
• Multiple Cloning Site (MCS)
• SV40-Sequenzelemente
(bei Pfizer/BNT)
Aufbau des DNA-Templates
[T7] – [5′-UTR] – [Spike-Gen] – [3′-UTR] – [AAAAA…]
[ORI] – [Resistenzgen] – [reg. bakt. Sequenzen] – [MCS] – [SV40] – [T7] – [5′-UTR] – [Spike-Gen] – [3′-UTR] – [AAAAA…]
Amplifikation der DNA
PCR-Vervielfältigung
Plasmid-Vermehrung in E. coli (Bioreaktor)
Vorbereitung vor der IVT
/
Lyse der Bakterien: Freisetzung von Plasmid-DNA + Bakterienresten
Linearisierung: Restriktionsenzym schneidet Plasmid an def. Stelle
Verunreinigungen vor IVT
PCR-Fragmente: Enzyme, Primer, Salze, Nukleotide
Bakterienbestandteile: Wirts-DNA, RNAs, Proteine, Endotoxine, Lipide, Zellwandfragmente, …
Reinigung
der PCR-DNA
der Plasmid-DNA (pDNA)
Methoden
Silica-Bindung, UF/DF
TFF + mehrstufige Chromatographie
Aufwand
moderat
hoch
IVT (in-vitro-Transkription)
Template: PCR-DNA
Expressionskassette:
[T7] – [5′-UTR] – [Spike-Gen] – [3′-UTR] – [AAAAA…]
→ mRNA wird synthetisiertTemplate: Linearisierte pDNA
Expressionskassette:
[T7] – [5′-UTR] – [Spike-Gen] – [3′-UTR] – [AAAAA…]
→ mRNA wird synthetisiert
IVT-Prozessierung
5′-Cap und Poly(A)-Tail werden je nach Verfahren während oder unmittelbar nach der IVT erzeugt
→ Ergebnis: funktionsfähige mRNA5′-Cap und Poly(A)-Tail werden je nach Verfahren während oder unmittelbar nach der IVT erzeugt
→ Ergebnis: funktionsfähige mRNA
Verunreinigungen nach IVT (*)
Entfernung von IVT-Nebenprodukten: Template-DNA, RNA-Fragmente, RNA:DNA-Hybride, doppelsträngige RNA (dsRNA), Enzyme, Salze, freie Nukleotide
Entfernung von IVT-Nebenprodukten + Entfernung zusätzlicher plasmid- und bakterienstämmiger Verunreinigungen (anspruchsvoller)
Reinigung
der mRNA
der mRNA
Vorbelastung
gering – moderat
hoch
Methoden
▪️DNase-I-Behandlung
▪️Fällung oder Zentrifugation
▪️UF/DF oder TFF
▪️Magnetkügelchen-Reinigung (**)▪️DNase-I-Behandlung
▪️Proteinase K-Behandlung (**)
▪️UF/DF und/oder TFF (**)
▪️mehrstufige Chromatographie
Aufwand
moderat
hoch
Formulierung in LNPs
mRNA wird in Lipidnanopartikel verpackt
mRNA wird in Lipidnanopartikel verpackt
Sterile Filtration
entfernt mikrobielles Kontaminationsrisiko
entfernt mikrobielles Kontaminationsrisiko
Abfüllung
aseptisches Verschließen und Etikettieren
aseptisches Verschließen und Etikettieren
Vorteile
sehr schnell, hochflexibel
extrem skalierbar, kostengünstige Massenproduktion
Nachteile
PCR-basierte Verfahren sind für großskalige industrielle Produktionsmengen nur eingeschränkt geeignet
Template enthält viele bakterielle Elemente → Reinigungsaufwand deutlich höher
(*) Obwohl die post-IVT-Reaktionsmischungen für Prozess 1 und 2 auf den ersten Blick ähnlich erscheinen, unterscheidet sich ihre „Vorbelastung“ qualitativ. Mit Vorbelastung ist die Menge und Komplexität der vor der Reinigung vorhandenen Begleitstoffe gemeint. Plasmidbasierte Prozesse bringen strukturell komplexere DNA-Formen, eine erhöhte Neigung zur Hybridbildung sowie Spuren bakterienstämmiger Begleitstoffe mit sich, die auch nach DNase-Behandlung die nachfolgende mRNA-Aufreinigung anspruchsvoller machen.
(**) Informationen zum Herstellungsverfahren laut EMA „EPAR zum Covid-19-Impfstoff von BioNTech/Pfizer“ (in Abschnitt 2.2., Seite 32)
Begriffserklärungen:
T7-Promotor – Startsignal für die Transkription
5′-UTR – stabilisiert die mRNA
Spike-Gen – Bauplan für das Protein
3′-UTR – Stopp-Signal und Stabilisierung
Poly(A)-Sequenz – schützt vor vorzeitigem AbbauUTR steht für untranslated region – also einen Bereich der mRNA, der nicht in Protein übersetzt wird. Diese Abschnitte liegen vor dem Start der Proteininformation (5′-UTR) und nach dem Ende (3′-UTR). Sie übernehmen wichtige regulatorische Aufgaben.
Origin of Replication (ORI) – Startpunkt der Plasmid-Vervielfältigung
Antibiotikaresistenzgen – ermöglicht Selektion in E. coli
regulatorische bakterielle Sequenzen zur Expression des Resistenzgens
Multiple Cloning Site (MCS) – kurze Region mit Schnittstellen zum Einfügen der Zielsequenz
SV40-Sequenzelemente – regulatorische virale Sequenzen für bestimmte Anwendungen in eukaryotischen Zellkulturen (bei Pfizer/BNT)
1.5. Zusammenfassung: Herstellung und Reinigung von modRNA
Die Herstellung modifizierter mRNA für therapeutische und prophylaktische Anwendungen ist ein mehrstufiger Prozess, der weit über die eigentliche in-vitro-Transkription hinausgeht. Ziel ist es, aus einer komplexen Reaktionsmischung ein möglichst homogenes, funktionell aktives RNA-Molekül zu gewinnen, das in Zellen effizient translatiert wird und dabei nur eine kontrollierte, gewünschte Immunantwort auslöst.
Vom DNA-Template zur mRNA-Rohlösung
Ausgangspunkt der Produktion ist eine definierte DNA-Vorlage, die im Rahmen der in-vitro-Transkription als Template für die RNA-Synthese dient. Bereits in diesem frühen Schritt entstehen neben der gewünschten mRNA zwangsläufig verschiedene Nebenprodukte, darunter verkürzte Transkripte, doppelsträngige RNA-Strukturen, RNA:DNA-Hybride sowie nicht korrekt verarbeitete RNA-Moleküle. Diese Nebenprodukte sind keine Ausnahme, sondern eine bekannte Konsequenz der enzymatischen Transkription unter technischen Bedingungen.
Nach Abschluss der Transkription wird die DNA-Vorlage in der Regel enzymatisch verdaut. Es folgt eine erste Grobreinigung, beispielsweise durch Filtration oder Tangentialflussfiltration, bei der kleine Moleküle, überschüssige Reagenzien und Enzyme entfernt werden. Zu diesem Zeitpunkt liegt eine konzentrierte mRNA-haltige Lösung vor, die jedoch weiterhin strukturell heterogen ist.
Anreicherung der Ziel-mRNA
Um die gewünschte mRNA gezielt anzureichern, werden poly(A)-basierte Affinitätsverfahren eingesetzt. Dazu zählen sowohl klassische Poly(dT)-Chromatographie als auch poly(dT)-funktionalisierte Magnetkügelchen. Diese Methoden nutzen die Poly(A)-Sequenz der mRNA als Selektionsmerkmal und erlauben eine effektive Abtrennung nicht-polyadenylierter RNA-Fragmente und vieler Begleitstoffe.
Gleichzeitig wird hier eine zentrale Grenze dieser Verfahren sichtbar: Alles, was eine Poly(A)-Sequenz trägt oder strukturell an polyadenylierte RNA gebunden ist, kann mitisoliert werden. Dazu zählen fragmentierte mRNA-Moleküle, bestimmte dsRNA-Nebenprodukte sowie RNA:DNA-Hybride, sofern die RNA-Komponente polyadenyliert ist. Poly(A)-basierte Verfahren sind daher hochselektive Anreicherungs-, aber keine vollständigen Reinigungsmethoden.
Hochauflösende Trennung durch Chromatographie
Um die verbleibende strukturelle Vielfalt weiter zu reduzieren, kommen hochauflösende Trennverfahren zum Einsatz, insbesondere die Anionenaustausch-Chromatographie (AEX). Diese nutzt Unterschiede in der effektiven Ladungsdichte und Konformation von Nukleinsäuren, um einzelsträngige mRNA von stärker gebundenen Nebenprodukten wie dsRNA zu trennen.
Die AEX stellt einen entscheidenden Schritt dar, um immunologisch besonders aktive Verunreinigungen gezielt abzureichern. Gleichzeitig zeigt sich auch hier, dass bestimmte Molekülklassen – insbesondere RNA:DNA-Hybride – keine einheitlichen chromatographischen Eigenschaften besitzen und je nach Struktur unterschiedlich eluieren können. Ihre vollständige Entfernung erfordert daher eine sorgfältige Prozessauslegung und gegebenenfalls die Kombination mehrerer Reinigungsschritte.
Einordnung und Ausblick
Insgesamt zeigt sich, dass die Herstellung modifizierter mRNA kein linearer „Rein-Raus-Prozess“ ist, sondern ein fein abgestimmtes Zusammenspiel aus enzymatischen, physikalischen und chromatographischen Verfahren. Jeder Reinigungsschritt reduziert die Komplexität der Probe, erreicht jedoch nie eine absolute Trennung aller theoretisch möglichen Nebenprodukte.
Vor diesem Hintergrund ist es fachlich sinnvoll, nicht nur das Endprodukt, sondern auch die Herstellungs- und Reinigungslogik selbst zu betrachten, wenn über mögliche Restbestandteile wie DNA-Fragmente oder RNA-assoziierte Verunreinigungen diskutiert wird. Die Frage ist dabei weniger, ob solche Restkomponenten prinzipiell entstehen können, sondern in welchen Formen, in welchen Mengen und mit welcher biologischen Relevanz sie im Endprodukt verbleiben.
Damit rückt die zentrale Frage in den Fokus, wie verbleibende Nukleinsäurebestandteile – insbesondere mögliche DNA-Rückstände – zuverlässig nachgewiesen und quantifiziert werden können.
Der zweite Teil behandelt daher die analytischen Nachweisverfahren, regulatorischen Grenzwerte sowie die bisher veröffentlichten experimentellen Untersuchungen zu DNA-Rückständen in mRNA-basierten Impfstoffen.
Weiterführender Teil der Artikelserie
Teil 2: Nachweisverfahren und Studienlage
Stand: Mai 2026
-
DNA-Rückstände in modRNA-basierten Impfstoffen – Teil 2

Leitfaden zur Artikelserie
Teil 1: Herstellung und Reinigung von modRNA
Teil 2: Nachweisverfahren und StudienlageNach der Darstellung der Herstellungs- und Reinigungsprozesse im ersten Teil richtet sich der Blick nun auf die analytische Ebene der Diskussion. Denn die Frage, ob und in welcher Menge DNA-Rückstände in modRNA-basierten Impfstoffen vorhanden sind, hängt entscheidend davon ab, mit welchen Methoden diese überhaupt untersucht werden.
Der Nachweis residualer Nukleinsäuren ist methodisch anspruchsvoll. Unterschiedliche Aufschlussverfahren, Extraktionsmethoden und Analyseplattformen können zu deutlich abweichenden Ergebnissen führen. Damit wird nicht nur die Interpretation einzelner Studien komplex, sondern bereits die grundlegende Frage, was eine Messung unter den jeweiligen Bedingungen tatsächlich erfasst.
Der zweite Teil widmet sich daher den wichtigsten analytischen Nachweisverfahren für DNA und RNA, ihren methodischen Grenzen sowie den bisher veröffentlichten unabhängigen Untersuchungen zu DNA-Rückständen in modRNA-basierten Impfstoffen. Dabei stehen insbesondere die Aussagekraft der verwendeten Methoden und die Vergleichbarkeit der Ergebnisse im Mittelpunkt.
Hinweis
📑
Im allgemeinen Sprachgebrauch ist der Begriff „mRNA-Impfstoff“ gebräuchlich. Fachlich handelt es sich bei den derzeit zugelassenen Präparaten um nukleosid-modifizierte mRNA (modRNA). Aus Gründen der Verständlichkeit wird in dieser Arbeit dennoch der etablierte Begriff „mRNA-Impfstoff“ verwendet.Inhaltsverzeichnis
2. Methoden zur Messung von DNA und RNA
3. Richtlinien zur Begrenzung von Rest-DNA in Impfstoffen
4. Studien zu DNA-Rückständen in modRNA-basierten Impfstoffen
5. Schlussbetrachtung und Ausblick
2. Methoden zur Messung von RNA und DNA
Die analytische Erfassung von RNA-Konzentration und potenziellen DNA-Rückständen stellt einen zentralen Bestandteil der Qualitätskontrolle mRNA-basierter Arzneimittel dar. Während der Herstellungs- und Reinigungsprozesse entstehen komplexe Nukleinsäuregemische, deren Zusammensetzung durch geeignete Messverfahren charakterisiert und überwacht werden muss. Ziel ist es, sowohl die Integrität und Menge der gewünschten mRNA als auch mögliche Restbestandteile wie DNA-Fragmente zuverlässig zu erfassen.
Dabei existiert kein einzelnes Verfahren, das alle analytischen Fragestellungen gleichermaßen abdeckt. Vielmehr unterscheiden sich die verfügbaren Methoden hinsichtlich Sensitivität, Spezifität, Quantifizierbarkeit und struktureller Auflösung. Einige Verfahren ermöglichen eine schnelle Bestimmung der Gesamtnukleinsäurekonzentration, andere erlauben die selektive Detektion spezifischer DNA-Sequenzen oder sogar die direkte Sequenzanalyse einzelner Moleküle.
Vor diesem Hintergrund werden im Folgenden verschiedene methodische Ansätze systematisch dargestellt – beginnend mit grundlegenden spektralphotometrischen Verfahren, über fluoreszenzbasierte Quantifizierungsmethoden und amplifikationsgestützte Nachweisverfahren bis hin zu sequenzierungsbasierten Technologien. Die Darstellung folgt dabei einer zunehmenden analytischen Auflösung: von der globalen Konzentrationsmessung hin zur sequenzspezifischen Identifikation einzelner Nukleinsäurebestandteile.
Neben dem Funktionsprinzip der jeweiligen Methode werden auch deren Nachweisgrenzen und methodische Einschränkungen berücksichtigt, da diese für die Einordnung möglicher Rest-DNA-Befunde von entscheidender Bedeutung sind.
2.1. Spektralphotometrische Verfahren
2.2. Fluoreszenzbasierte Quantifizierung
2.3. Amplifikationsbasierte Methoden
2.4. Sequenzierungsbasierte Verfahren
2.5. Elektrophoretische Fragmentanalyse
2.6. Vergleich zentraler Nachweisverfahren für Nukleinsäuren
2.1. Spektralphotometrische Verfahren
Spektralphotometrische Verfahren beruhen auf der Wechselwirkung von Licht mit Materie. Gemessen wird die Lichtintensität – vor und nach dem Durchgang durch eine Probe – in Abhängigkeit von der Wellenlänge. Aus der gemessenen Absorption lässt sich unter definierten Bedingungen auf die Konzentration der enthaltenen Substanzen schließen.
Die UV/Vis-Spektrophotometrie stellt eine etablierte Methode zur Bestimmung der Gesamtnukleinsäurekonzentration dar und wird in mehreren Phasen der mRNA-Herstellung eingesetzt. Sie zeichnet sich durch eine schnelle Durchführung, geringen Probenverbrauch und vergleichsweise niedrige Kosten aus.
Wann wird die Photometrie eingesetzt?
Im Herstellungsprozess lassen sich typischerweise drei relevante Messzeitpunkte unterscheiden:
Nach der in-vitro-Transkription: zur Abschätzung, ob die Transkriptionsreaktion erfolgreich war und welche Gesamtausbeute an Nukleinsäure erzielt wurde.
Nach der Aufreinigung: als rascher Kontrollschritt zur Überprüfung, ob die mRNA-Konzentration im erwarteten Bereich liegt.
Vor der Formulierung: Vor der Verkapselung in Lipid-Nanopartikel wird die mRNA-Konzentration erneut bestimmt, da das Mischungsverhältnis zwischen Lipiden und mRNA entscheidend für die Partikelbildung ist.
Es ist zu beachten, dass die Methode nicht zwischen RNA und DNA unterscheiden kann und daher primär eine orientierende Gesamtkonzentrationsbestimmung darstellt.
Typischer Ablauf im Herstellungsprozess
Nach der Aufreinigung der mRNA und vor der LNP-Formulierung wird die mRNA-Wirkstoffcharge einer Konzentrationsbestimmung unterzogen. Hierzu wird unter kontrollierten Bedingungen eine kleine Probe aus dem Prozessbehälter entnommen.
Für die Messung stehen zwei Gerätetypen zur Verfügung, die sich in ihrer Handhabung unterscheiden:
UV/Vis-Spektralphotometer: Die Probe wird in eine Küvette gefüllt – ein kleines, meist rechteckiges Gefäß aus UV-durchlässigem Material (z. B. Quarzglas). Die Küvette wird in das Gerät gestellt, wo die Messung stattfindet.
Mikrovolumen-Spektralphotometer (z. B. NanoDrop): Ein winziger Tropfen (0,5–2,0 µl) wird direkt auf einen Messsockel pipettiert. Ein zweiter (umgeklappter) Sockel bildet durch Oberflächenspannung eine definierte Flüssigkeitssäule zwischen den beiden Messflächen. Der große Vorteil: Es werden weder Küvetten noch nennenswerte Probenmengen benötigt. Diese Geräte werden häufig für DNA- und RNA-Messungen eingesetzt.
Unabhängig vom Gerätetyp dauert die eigentliche spektralphotometrische Analyse nur wenige Sekunden. Unmittelbar nach dem Messvorgang wird das Ergebnis auf dem Bildschirm angezeigt.

Abb. 2.1-A: UV/Vis-Spektralphotometer vs Mikrovolumen-Spektralphotometer Der Weg des Lichts im Spektralphotometer
1️⃣ Lichterzeugung: Als Lichtquelle dient eine breitbandige UV/Vis-Lampe, beispielsweise eine Xenon-Blitzlampe, die kurze, intensive Lichtblitze aussendet und das gesamte UV- und sichtbare Spektrum abdeckt.
2️⃣ Wellenlängenselektion: Das Licht wird durch den Eintrittsspalt in den Monochromator geleitet und dort durch ein bewegliches Beugungsgitter in seine spektralen Bestandteile zerlegt. Um die Konzentration der mRNA zu bestimmen, wird das Gitter so eingestellt, dass Licht mit einer Wellenlänge von 260 nm – dem Absorptionsmaximum der Nukleinsäuren – auf die Probe trifft.
3️⃣ Lichtführung zur Probe: Über ein System von Spiegeln und Linsen wird der monochromatische Lichtstrahl gebündelt und präzise auf die Probe gerichtet – entweder auf die Küvette oder direkt auf den hängenden Probentropfen.
4️⃣ Wechselwirkung mit der Probe: Ein Teil des Lichts wird von den Nukleinsäuremolekülen absorbiert (verschluckt), der Rest transmittiert (durchdringt) die Probe.
5️⃣ Detektion: Das transmittierte Licht trifft auf einen Detektor (z. B. eine Photodiode oder ein CCD-Sensor), der die ankommende Lichtintensität misst.
6️⃣ Berechnung: Das Gerät vergleicht die gemessene Intensität mit der Intensität des ursprünglich eingestrahlten Lichts (Referenzmessung ohne Probe). Aus dem Verhältnis von eingestrahlter zu transmittierter Lichtintensität berechnet das Gerät gemäß dem Lambert-Beerschen Gesetz die Absorbanz, aus der sich bei bekannter Schichtdicke die Konzentration der Nukleinsäuren in der Probe bestimmen lässt.
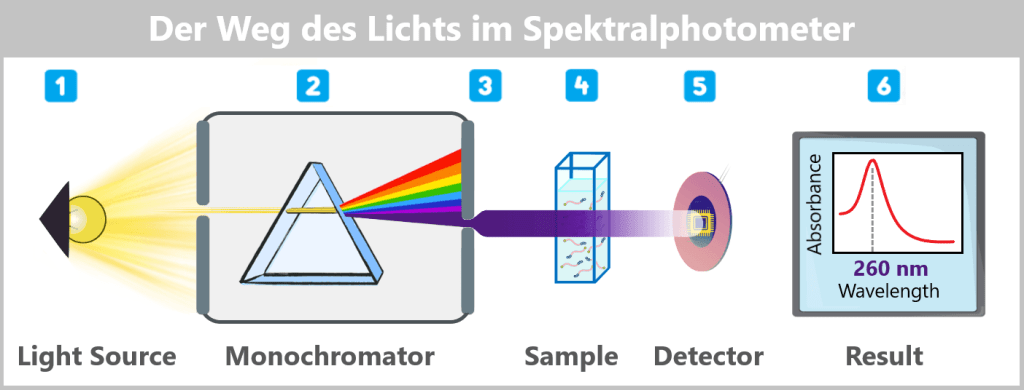
Abb. 2.1-B: Schematische Darstellung der UV/Vis-Spektralphotometer Warum absorbieren RNA und DNA bei 260 nm?
Die Absorption von UV-Licht bei 260 nm ist eine charakteristische Eigenschaft der Basen – jener „Buchstaben“, aus denen der genetische Code besteht. Da diese Basen in RNA und DNA nahezu identisch sind, weisen beide Moleküle ein sehr ähnliches Absorptionsverhalten auf.
Das Geheimnis liegt in ihrer chemischen Struktur: Die Basen enthalten sogenannte aromatische Ringe. In diesen Ringen sitzen die Elektronen nicht an festen Positionen zwischen zwei Atomen, sondern sind über das gesamte Ringsystem verteilt. Man kann sich diese „delokalisierten Elektronen“ wie eine Art Elektronenwolke vorstellen, die über dem Molekül schwebt.
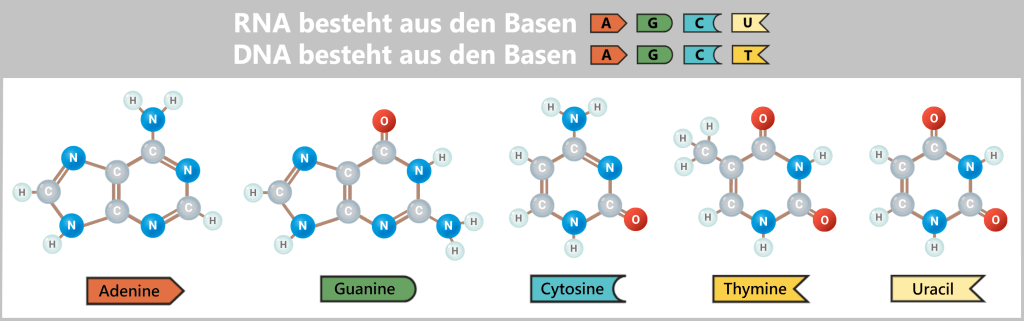
Abb. 2.1-C: Die molekularen Bausteine der Nukleinsäuren Die Darstellung zeigt die zwei Typen von Basen: die größeren Doppelringe (Purine: A und G) und die kleineren Einzelringe (Pyrimidine: C, T und U). Trotz ihrer unterschiedlichen Größe besitzen beide Typen ein System aus abwechselnden Doppelbindungen. Dieses sorgt dafür, dass die Elektronen über das gesamte Ringsystem verteilt sind und so eine gemeinsame Wolke bilden.
Wenn nun ein Photon (ein Lichtteilchen) mit einer Wellenlänge von etwa 260 nm auf diese Wolke trifft, passt dessen Energie exakt zu den energetischen Eigenschaften der Elektronen in diesen Ringen. Ein Elektron nimmt die Energie des Photons vollständig auf und springt dadurch auf ein höheres Energieniveau – den sogenannten angeregten Zustand. Die Energie des Photons wird dabei vollständig auf das Elektron übertragen, sodass es nicht mehr als Lichtteilchen weiterexistiert – man spricht von Absorption.
Da sowohl RNA als auch DNA aus denselben Basentypen bestehen (mit der Ausnahme von Uracil statt Thymin), zeigen beide Molekülarten ein nahezu identisches Verhalten im UV-Licht. Die Spektrophotometrie erlaubt daher keine direkte Unterscheidung zwischen RNA und DNA.
Bedeutung für die Bewertung von DNA-Rückständen
Die dargestellten physikalischen Grundlagen erklären zugleich eine zentrale Limitation der UV-Spektrophotometrie: Da die Absorption bei etwa 260 nm auf den gemeinsamen Basenstrukturen von RNA und DNA beruht, erfasst die Methode ausschließlich die Gesamtmenge an Nukleinsäuren in einer Probe. Eine Differenzierung zwischen therapeutischer mRNA und eventuell verbliebenen DNA-Restfragmenten ist auf dieser Grundlage nicht möglich.
Für die quantitative Bestimmung spezifischer DNA-Rückstände sind daher analytische Verfahren erforderlich, die entweder selektiv doppelsträngige DNA erfassen oder definierte Zielsequenzen amplifizieren. In der Praxis stehen hierbei insbesondere fluorometrische DNA-Methoden (z. B. Qubit-Technologien) sowie die quantitative Polymerase-Kettenreaktion (qPCR) im Mittelpunkt.
Die Diskussion um geeignete Nachweismethoden für DNA-Rückstände konzentriert sich daher weniger auf photometrische Verfahren, sondern primär auf molekularbiologische und fluoreszenzbasierte Techniken.
2.2. Fluoreszenzbasierte Quantifizierung
2.2.1. DNA-/RNA-spezifische Farbstoffe
a) Wirkmechanismus von PicoGreen
b) Warum PicoGreen bevorzugt doppelsträngige DNA erfasst
c) Grenzen der PicoGreen-Methode
2.2.2. Qubit-SystemeIm Gegensatz zur UV-Spektrophotometrie, bei der die Abschwächung eines eingestrahlten Lichtstrahls gemessen wird, beruhen fluoreszenzbasierte Verfahren auf der gezielten Anregung spezifischer Moleküle und der anschließenden Messung des von ihnen ausgesandten Lichts. (Oder einfacher formuliert: Während die UV-Spektrophotometrie feststellt, wie viel Licht „verschwindet“, misst die Fluoreszenz nicht den Schatten, sondern das Leuchten.)
Dabei wird der Probe ein Fluoreszenzfarbstoff zugesetzt, der selbst kaum oder gar nicht fluoresziert. Erst nach Bindung an eine Zielstruktur – beispielsweise doppelsträngige DNA oder RNA – ändert sich seine räumliche Anordnung, sodass er nach Anregung mit Licht einer bestimmten Wellenlänge ein charakteristisches Fluoreszenzsignal aussendet. Die Intensität dieses emittierten Lichts ist proportional zur Menge der gebundenen Nukleinsäure.
Das entscheidende Merkmal fluoreszenzbasierter Verfahren ist ihre Selektivität: Durch geeignete Farbstoffe lassen sich gezielt bestimmte Nukleinsäuretypen erfassen, während andere Bestandteile der Probe weitgehend unbeachtet bleiben. Dadurch wird eine deutlich spezifischere Quantifizierung möglich als mit der unspezifischen UV-Absorption bei 260 nm.
Insbesondere bei niedrigen Konzentrationen – etwa bei der Bestimmung von DNA-Rückständen in mRNA-Präparationen – bieten fluoreszenzbasierte Methoden eine wesentlich höhere Sensitivität.
2.2.1. DNA-/RNA-spezifische Farbstoffe
Die Grundlage fluoreszenzbasierter Quantifizierung bilden Nukleinsäure-bindende Farbstoffe, die selektiv mit DNA oder RNA interagieren.
Doppelstrang-DNA-spezifische Farbstoffe
Ein häufig eingesetzter DNA-spezifischer Farbstoff ist PicoGreen. Dieser bindet bevorzugt an doppelsträngige DNA. In freier Lösung zeigt PicoGreen nur eine sehr geringe Eigenfluoreszenz. Erst nach Bindung an die DNA steigt seine Fluoreszenzintensität stark an. Dadurch wird die Quantifizierung selbst sehr geringer DNA-Mengen möglich.
RNA-spezifische Farbstoffe
Für RNA wird häufig RiboGreen verwendet. Auch dieser Farbstoff zeigt erst nach Bindung an RNA ein deutliches Fluoreszenzsignal.
Die unterschiedliche Bindungsaffinität der Farbstoffe erlaubt eine weitgehend selektive Erfassung der jeweiligen Nukleinsäureart.
Das Messprinzip folgt dabei stets demselben Schema:
- Zugabe eines spezifischen Farbstoffs zur Probe
- Bindung des Farbstoffs an die Zielnukleinsäure
- Anregung mit Licht definierter Wellenlänge (λEX – Excitation)
- Messung der emittierten Fluoreszenz (λEM – Emission)
- Vergleich mit einer Kalibrationsreihe bekannter Konzentrationen
Da nur gebundener Farbstoff ein starkes Signal erzeugt, bleibt der Hintergrund gering. Dies erklärt die hohe Empfindlichkeit dieser Methode im Vergleich zur UV-Spektrophotometrie.
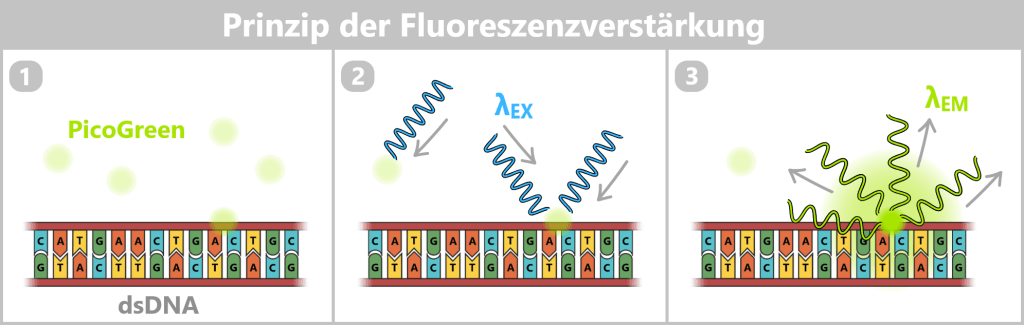
Abb. 2.2.1-A: Schematische Darstellung der DNA-Quantifizierung mittels Fluorometrie 1) Nach Zugabe des Fluoreszenzfarbstoffs (z. B. PicoGreen) erfolgt eine bevorzugte Bindung an doppelsträngige DNA (dsDNA).
2) Die Probe wird mit kurzwelligem, energiereichem Licht einer definierten Wellenlänge (λEX ≈ 480 nm, blau) bestrahlt, wodurch die Farbstoffmoleküle in einen angeregten Zustand versetzt werden.
3) Bei der Rückkehr in den Grundzustand emittiert der gebundene Farbstoff längerwelliges Licht (λEM ≈ 520 nm, grün). Die gemessene Fluoreszenzintensität ist im linearen Messbereich proportional zur vorhandenen DNA-Konzentration in der Probe. Da freier Farbstoff nur eine sehr geringe Eigenfluoreszenz aufweist, entsteht ein deutliches Signal hauptsächlich durch DNA-gebundene Moleküle.Die Quantifizierung erfolgt über den Vergleich der Signalintensität mit einer Standardreihe bekannter DNA-Konzentrationen.
a) Wirkmechanismus von PicoGreen
PicoGreen ist ein Fluoreszenzfarbstoff mit hoher Affinität zu doppelsträngiger DNA (dsDNA). Seine besondere Empfindlichkeit beruht darauf, dass er im freien Zustand fast nicht leuchtet – nach Bindung an DNA jedoch sehr stark fluoresziert.
Der Unterschied zwischen diesen beiden Zuständen (frei/gebunden) ist entscheidend für die analytische Anwendung.
Freies PicoGreen in Lösung
In wässriger Lösung ist das Molekül beweglich:
- Es kann sich drehen und biegen.
- Seine aromatischen Ringsysteme sind gegeneinander beweglich.
- Es stößt ständig mit Wassermolekülen zusammen.
Wird das Molekül durch Licht angeregt, nimmt es Energie auf. Im freien Zustand geht diese Energie jedoch größtenteils nicht als Licht verloren, sondern wird durch molekulare Bewegungen und Wechselwirkungen mit dem Wasser in Wärme umgewandelt. Die Folge: Die Eigenfluoreszenz ist sehr gering. Freies PicoGreen bleibt nahezu „dunkel“.
Annäherung an die doppelsträngige DNA
PicoGreen trägt positive Ladungen. Das Rückgrat der DNA ist aufgrund seiner Phosphatgruppen negativ geladen. Dadurch entsteht eine elektrostatische Anziehung zwischen Farbstoff und DNA. Das Molekül lagert sich bevorzugt an doppelsträngige DNA (dsDNA) an – insbesondere an Bereiche, in denen zwei DNA-Stränge eng beieinanderliegen. Diese erste Annäherung ist die Voraussetzung für eine stabilere Bindung.
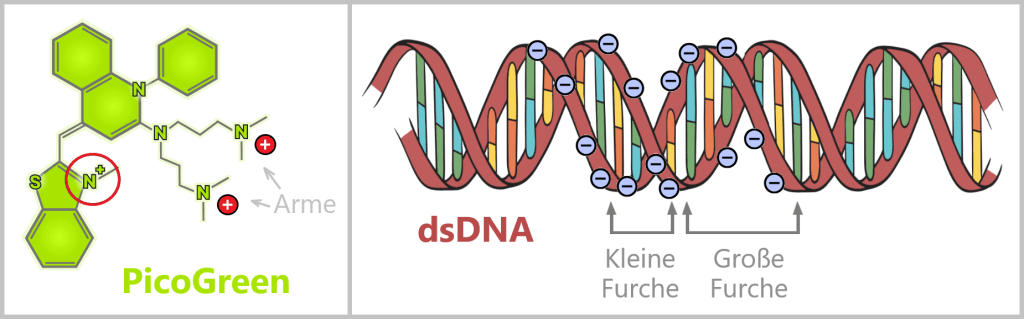
Abb. 2.2.1-B: Schematische Darstellung der Struktur von PicoGreen und dsDNA
(nicht maßstabsgetreu)Links ist die vereinfachte Struktur des Fluoreszenzfarbstoffs PicoGreen dargestellt. Die sechseckigen und fünfeckigen Ringe in der Struktur stehen für die aromatischen Ringsysteme – das sind besonders stabile, flache Molekülbausteine, die UV-Licht einfangen können. Zudem besitzt der Farbstoff positiv geladene Seitenarme.
Rechts ist die doppelsträngige DNA mit negativ geladenem Zucker-Phosphat-Rückgrat gezeigt. Die elektrostatische Anziehung zwischen den positiven Seitenarmen des Farbstoffs und dem negativen Rückgrat der DNA bringt den Farbstoff zunächst in die Nähe der DNA.Bindung und Fixierung
Nach der Bindung wird das Molekül in seiner Beweglichkeit stark eingeschränkt:
- Es kann sich kaum noch drehen oder verbiegen.
- Teile des Moleküls liegen geschützt zwischen oder nahe den Basenpaaren.
- Der direkte Kontakt mit Wasser wird teilweise reduziert.
Die DNA wirkt dabei gewissermaßen wie eine Halterung, die das Molekül in einer festen Position stabilisiert.
Warum die Fluoreszenz nun stark zunimmt
Durch diese Fixierung verändern sich die Energieverluste des Moleküls: Im freien Zustand geht ein Großteil der aufgenommenen Energie durch Bewegung verloren. Im gebundenen Zustand sind diese Bewegungen stark eingeschränkt.
Dadurch steigt die Wahrscheinlichkeit, dass die aufgenommene Energie als Licht abgegeben wird – also als Fluoreszenz. Das Ergebnis ist ein deutlich stärkeres Signal.
Der Unterschied zwischen freiem und DNA-gebundenem PicoGreen ist so groß, dass selbst sehr geringe Mengen doppelsträngiger DNA zuverlässig nachweisbar sind.
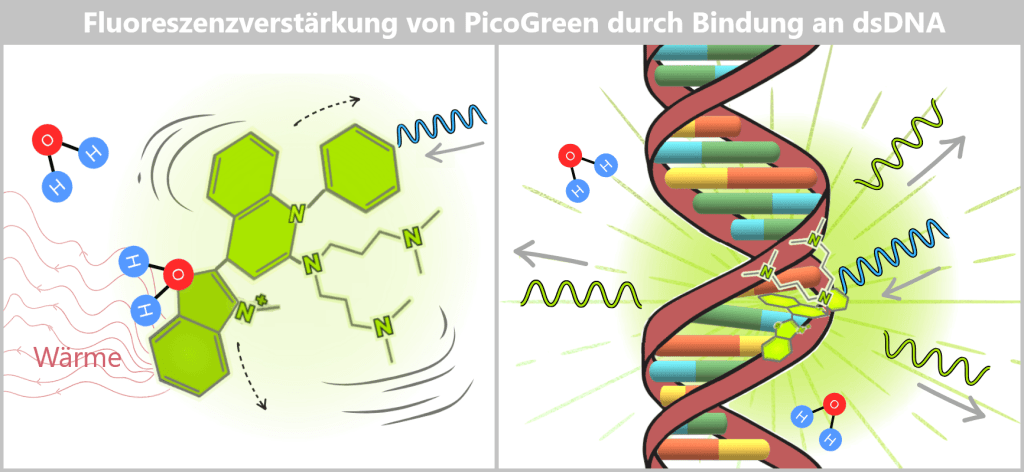
Abb. 2.2.1-C: Fluoreszenzverhalten von PicoGreen – frei in Lösung und gebunden an dsDNA Links: PicoGreen in wässriger Lösung (ohne DNA)
In wässriger Lösung ist das Farbstoffmolekül ständig in Bewegung: Es rotiert und führt innermolekulare Schwingungen aus. Wird es mit blauem Licht angeregt, gibt es die aufgenommene Energie überwiegend über diese Bewegungen an das umgebende Wasser als Wärme ab. Der Farbstoff zeigt daher nur eine sehr schwache Eigenfluoreszenz.
Rechts: PicoGreen gebunden an dsDNA
Nach Bindung an doppelsträngige DNA lagert sich PicoGreen in die Kleine Furche ein und wird zusätzlich durch Stapelwechselwirkungen mit den Basen fixiert. Dadurch ist das Molekül in seiner Beweglichkeit stark eingeschränkt. Die aufgenommene Energie kann nun deutlich weniger über Bewegungsprozesse verloren gehen und wird überwiegend als grünes Licht emittiert. Die Fluoreszenzintensität steigt daher stark an.Warum diese Eigenschaft analytisch so wertvoll ist
- Freier Farbstoff erzeugt kaum Hintergrundsignal.
- Nur gebundener Farbstoff leuchtet stark.
- Doppelsträngige DNA wird bevorzugt erfasst.
Gerade bei der Bestimmung von DNA-Rückständen in mRNA-Präparationen – auch bei sehr niedrigen Konzentrationen – ist dieser starke Kontrast zwischen „dunkel“ und „leuchtend“ entscheidend für die Sensitivität der Methode.
b) Warum PicoGreen bevorzugt doppelsträngige DNA erfasst
Die Selektivität von PicoGreen beruht nicht darauf, dass der Farbstoff „weiß“, was DNA ist, sondern auf strukturellen Eigenschaften der doppelsträngigen DNA.
Doppelsträngige DNA besitzt:
- einen regelmäßig aufgebauten Basenstapel
- dicht gepackte, planare Basenpaare
- klar definierte große und kleine Furchen
- eine stabile, räumlich geordnete Struktur
Diese geordnete Architektur bietet dem Farbstoff geeignete Bindungsstellen. Er kann sich zwischen oder nahe den gestapelten Basen anlagern und wird dabei mechanisch fixiert. Genau diese Fixierung ist entscheidend für die starke Fluoreszenzzunahme.
Einzelsträngige DNA oder RNA besitzen eine deutlich flexiblere Struktur. Ihnen fehlt der regelmäßig aufgebaute Basenstapel der Doppelhelix. Zwar kann es auch hier zu Wechselwirkungen kommen, doch:
- die Bindung ist schwächer
- die Fixierung weniger stabil
- die Fluoreszenzverstärkung deutlich geringer
Deshalb reagiert PicoGreen besonders empfindlich auf doppelsträngige DNA – und wesentlich schwächer auf RNA oder einzelsträngige Nukleinsäuren.
Für die Analyse von DNA-Rückständen ist genau diese Eigenschaft entscheidend: Restliche Plasmid-DNA oder DNA-Fragmente liegen typischerweise doppelsträngig vor und erzeugen daher ein starkes Signal.
Obwohl DNA-spezifische Fluoreszenzfarbstoffe wie PicoGreen eine hohe Präferenz für doppelsträngige DNA aufweisen, ist ihre Selektivität nicht absolut. Insbesondere in Proben mit sehr hohen RNA-Konzentrationen kann es zu unspezifischen Signalbeiträgen kommen. Diese entstehen durch schwache Bindung an RNA, sekundäre RNA-Strukturen oder schlichte Masseneffekte. Ohne geeignete Kontrollmaßnahmen – etwa eine RNase-Behandlung – kann dies zu einer Überschätzung der DNA-Menge führen.
c) Grenzen der PicoGreen-Methode
Trotz ihrer hohen Empfindlichkeit bleibt die Methode eine quantitative Summenbestimmung von dsDNA. Sie liefert keine Informationen über:
- die Sequenz der DNA
- die genaue Herkunft
- die biologische Funktionalität
- die Fragmentzusammensetzung
Einfluss der Fragmentlänge
Die Signalstärke hängt unter anderem davon ab, wie viele Bindungsstellen zur Verfügung stehen. Sehr kurze DNA-Fragmente können weniger Farbstoff binden als lange, intakte Moleküle.
Das bedeutet: Zwei Proben mit identischer Masse an DNA können – abhängig von der Fragmentierung – leicht unterschiedliche Fluoreszenzsignale erzeugen.
In der Praxis wird dies durch Kalibrationsstandards berücksichtigt, vollständig eliminieren lässt sich dieser Effekt jedoch nicht.
Matrixeffekte
Bestandteile der Probe können das Messergebnis beeinflussen, etwa:
- hohe Salzkonzentrationen
- Restproteine
- Tenside oder Lipidbestandteile
- Pufferzusammensetzung
Solche Faktoren können die Bindungseffizienz oder die Fluoreszenzintensität verändern. Daher werden Messbedingungen standardisiert und validiert.
✧ ✧ ✧
Zwischenfazit
Fluoreszenzbasierte Verfahren wie PicoGreen ermöglichen eine sehr empfindliche Quantifizierung doppelsträngiger DNA und sind grundsätzlich gut geeignet, um geringe DNA-Mengen auch in komplexen Proben nachzuweisen.
Die Methode beruht jedoch nicht auf einer sequenzspezifischen Erkennung, sondern auf strukturellen Eigenschaften der Nukleinsäuren. Das gemessene Signal kann daher durch Faktoren wie RNA-Reste, Fragmentlänge, Sekundärstrukturen oder Bestandteile der Probenmatrix beeinflusst werden. Ohne geeignete Kontrollmaßnahmen – insbesondere RNase-Behandlung und standardisierte Probenaufbereitung – besteht das Risiko einer Über- oder Unterschätzung der tatsächlichen DNA-Menge.
PicoGreen beantwortet damit vor allem die Frage, wie viel fluoreszenzaktive doppelsträngige DNA in einer Probe vorhanden ist. Aussagen über Sequenz, Herkunft, Integrität oder biologische Funktionalität der DNA sind mit dieser Methode allein nicht möglich.
Für solche Fragestellungen sind ergänzende sequenz- oder amplifikationsbasierte Verfahren erforderlich, die in den folgenden Kapitel behandelt werden.
2.2.2. Qubit-Systeme
Während DNA- und RNA-spezifische Farbstoffe wie PicoGreen oder RiboGreen die eigentlichen fluoreszierenden Werkzeuge zur Quantifizierung von Nukleinsäuren darstellen, bezeichnet das Qubit-System das darauf abgestimmte Gesamtsystem zur präzisen Konzentrationsbestimmung. Das Qubit-System ist ein kompaktes Fluorometer, das für die hochsensitive und spezifische Quantifizierung von DNA, RNA und Proteinen von Thermo Fisher Scientific entwickelt wurde. Es kombiniert:
- spezifische Fluoreszenzfarbstoffe = molekulare Sensoren
- optimierte Reagenzienlösungen = definierte Umgebung (Puffer)
- standardisierte Assay-Protokolle = standardisierte Bedienungsanleitungen
Ziel dieses Systems ist eine selektive und empfindliche Quantifizierung von DNA oder RNA in biologischen Proben.
Prinzip des Qubit-Systems
Das zugrunde liegende Messprinzip entspricht dem zuvor beschriebenen Verfahren (im Kapitel 2.2.1): Ein selektiver Fluoreszenzfarbstoff bindet an die Zielnukleinsäure, wird mit Licht definierter Wellenlänge angeregt und emittiert ein messbares Fluoreszenzsignal. Die Intensität dieses Signals ist proportional zur Menge der gebundenen DNA oder RNA. Durch den Vergleich mit Standardlösungen bekannter Konzentration kann daraus die Konzentration der Probe bestimmt werden.
Der Ablauf einer Messung
Die Handhabung ist bewusst einfach gehalten und folgt einem standardisierten Protokoll:
1️⃣ Arbeitslösung ansetzen: Der fluoreszierende Farbstoff (aus dem Assay-Kit) wird mit dem mitgelieferten Puffer im empfohlenen Verhältnis gemischt.
Beispiele für verfügbare Assay-Kits:
Assay-Kit Anwendung für dsDNA BR (Broad Range) höhere DNA-Konzentrationen dsDNA HS (High Sensitivity) niedrige DNA-Konzentrationen RNA BR (Broad Range) höhere RNA-Konzentrationen RNA HS (High Sensitivity) niedrige RNA-Konzentrationen microRNA Assay kleine RNA-Moleküle wie miRNA und siRNA Protein Assay Quantifizierung von Proteinen 2️⃣ Probe zugeben: Ein kleines Volumen der Probe (meist 1–20 µl) wird zur Arbeitslösung gegeben.
3️⃣ Bindung abwarten: Nach einer kurzen Inkubationszeit von nur 2 Minuten (bei DNA/RNA-Assays) bindet der Farbstoff spezifisch an sein Zielmolekül.
4️⃣ Kalibrieren mit Standards: Es werden ein oder zwei mitgelieferte Standards mit bekannter Konzentration gemessen. Daraus erstellt das Gerät automatisch eine Standardkurve.
5️⃣ Probe in das Gerät stellen:
Anregung: Das Gerät bestrahlt die Probe mit der passenden Anregungswellenlänge: z. B. blaues Licht bei ca. 470 nm zur Quantifizierung von DNA.
Emission: Die gebundenen Farbstoffmoleküle absorbieren dieses Licht und strahlen es mit einer längeren Wellenlänge (niedrigerer Energie) wieder ab (z. B. grünes Licht bei ca. 520 nm).
Filterung: Ein optischer Filter im Gerät blockiert das blaue Anregungslicht, sodass der Sensor nur das grüne Leuchten der Probe registriert. Dies erklärt die enorme Genauigkeit: Das Gerät „sieht“ nur das Zielobjekt, störende Anregungsstrahlung wird ausgeblendet.6️⃣ Messen und Ergebnis: Das Gerät misst das abgestrahlte, längerwellige Licht (z. B. grün). Innerhalb von Sekunden berechnet ein Algorithmus unter Berücksichtigung der Standardkurve die exakte Konzentration. Das Ergebnis wird auf dem Bildschirm angezeigt.
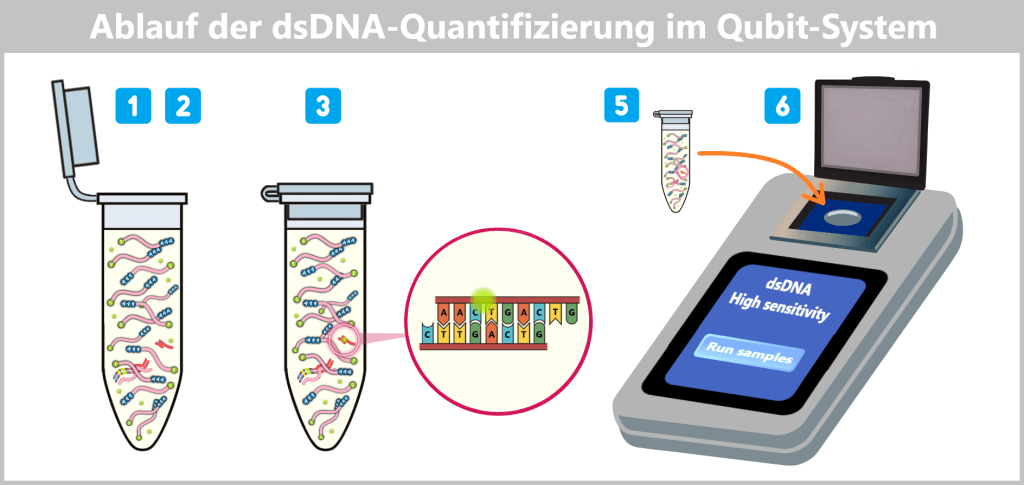
Abb. 2.2.2: Schematischer Ablauf der dsDNA-Quantifizierung im Qubit-System Die Abbildung zeigt den Ablauf der dsDNA-Quantifizierung mit dem Qubit-System. Nach Zugabe des spezifischen Fluoreszenzfarbstoffs zur Probe (1–2) bindet dieser selektiv an doppelsträngige DNA (3). Anschließend wird die Probe in das Qubit-Fluorometer eingesetzt (5), welches die Fluoreszenzintensität misst und die DNA-Konzentration berechnet (6).
Vorteile des Qubit-Systems
- Hohe Spezifität und Selektivität: Die verwendeten Fluoreszenzfarbstoffe besitzen eine starke Präferenz für ihre jeweiligen Zielmoleküle. Dadurch kann beispielsweise doppelsträngige DNA (dsDNA) auch in Gegenwart größerer RNA-Mengen vergleichsweise selektiv quantifiziert werden. Unspezifische Signalbeiträge sind jedoch insbesondere bei sehr hohen RNA-Konzentrationen oder komplexen Probenmatrices nicht vollständig ausgeschlossen.
- Hohe Sensitivität: Im Vergleich zur UV-Photometrie ist die Methode deutlich empfindlicher und erlaubt die Quantifizierung sehr geringer Nukleinsäurekonzentrationen.
- Geringer Probenverbrauch: Für die Messung werden nur kleine Probenvolumina benötigt (typischerweise 1–20 µl), was insbesondere bei limitiertem oder wertvollem Probenmaterial vorteilhaft ist.
- Relative Robustheit gegenüber Verunreinigungen: Viele Substanzen, die UV-spektroskopische Verfahren beeinflussen können – etwa freie Nukleotide, Salze oder Proteine, stören die fluorometrische Messung deutlich weniger. Bestimmte Matrixbestandteile können die Fluoreszenz jedoch weiterhin beeinflussen.
- Einfache Handhabung und schnelle Ergebnisse: Das System ist standardisiert aufgebaut und ermöglicht reproduzierbare Messungen mit vergleichsweise geringem Arbeitsaufwand.
Grenzen des Qubit-Systems
- Keine Aussage über DNA-Integrität: Das Verfahren unterscheidet nicht zwischen intakter und fragmentierter DNA.
- Keine Sequenzinformation: Die Messung liefert keine Informationen über Sequenz, Herkunft oder biologische Funktion der Nukleinsäuren.
- Einfluss der Fragmentlänge: Unterschiedliche Fragmentlängen können die Signalstärke beeinflussen. Sehr kurze oder stark degradierte Fragmente erzeugen unter Umständen ein reduziertes Fluoreszenzsignal.
- Restunspezifität bei hohen RNA-Mengen: Sehr hohe RNA-Konzentrationen können trotz der hohen Farbstoffspezifität zu unspezifischen Signalbeiträgen führen.
- Begrenzter linearer Messbereich: Die Quantifizierung ist auf den linearen Bereich des jeweiligen Assays beschränkt. Hochkonzentrierte Proben müssen daher entsprechend verdünnt werden.
- Eingeschränkte Aussagen zur Probenreinheit: Aussagen über Reinheit, Zusammensetzung oder mögliche Kontaminationen der Probe sind nur begrenzt möglich.
✧ ✧ ✧
Zusammenfassung
Die Qubit-Fluorometrie gilt als empfindliche und vergleichsweise selektive Methode zur Quantifizierung von DNA und RNA. Im Unterschied zur UV-Spektroskopie basiert sie nicht auf einer unspezifischen Lichtabsorption, sondern auf fluoreszierenden Farbstoffen mit starker Präferenz für bestimmte Nukleinsäurestrukturen. Dadurch sind auch Messungen geringer Konzentrationen in komplexen Proben möglich.
Die Methode bleibt jedoch eine strukturabhängige Summenmessung: Sie liefert Informationen über die Menge bestimmter Nukleinsäuretypen, nicht jedoch über deren Sequenz, Herkunft oder biologische Eigenschaften. Für solche Fragestellungen sind sequenz- oder amplifikationsbasierte Verfahren erforderlich, die in den folgenden Kapitel behandelt werden.
2.3. Amplifikationsbasierte Methoden
Die bisher beschriebenen Verfahren (UV-Spektroskopie und Fluorometrie) ermöglichen die Bestimmung der Gesamtmenge an Nukleinsäuren – also von RNA und/oder DNA. Für den gezielten Nachweis spezifischer DNA- oder RNA-Sequenzen sind jedoch Methoden erforderlich, die auf einer Vervielfältigung der Zielsequenz beruhen. Dieses Prinzip wird als Amplifikation bezeichnet.
Im Herstellungsprozess kommen amplifikationsbasierte Methoden insbesondere zum Einsatz, um verbleibende DNA-Rückstände nachzuweisen.
Grundlage dieser Verfahren ist die Polymerase-Kettenreaktion (PCR). Dabei wird ein zuvor definierter DNA-Abschnitt durch wiederholte enzymatische Synthesezyklen exponentiell vervielfältigt, das heißt, die Menge des Zielabschnitts verdoppelt sich idealerweise in jedem Zyklus. Bereits wenige Ausgangskopien können so in eine messbare Menge überführt werden. Auf diesem Prinzip basieren sowohl die quantitative PCR (qPCR) als auch die digitale PCR (ddPCR).
2.3.1. quantitative PCR (qPCR)
2.3.2. digitale PCR (ddPCR)
2.3.3. Vergleich der PCR-basierten Quantifizierungsmethoden⚬ ⚬ ⚬
2.3.1. Quantitative Polymerase-Kettenreaktion (qPCR)
Die quantitative PCR, auch Real-Time-PCR genannt, ist eine Weiterentwicklung der klassischen PCR. Sie ermöglicht die gleichzeitige Amplifikation und Quantifizierung eines spezifischen DNA-Abschnitts in Echtzeit.
Die grundlegenden Reaktionsschritte entsprechen denen der konventionellen PCR, wie sie im ersten Teil (Vervielfältigung der Spike-DNA mittels PCR) beschrieben wurden. Der entscheidende Unterschied besteht darin, dass während jedes Amplifikationszyklus die entstehende DNA-Menge durch ein Fluoreszenzsignal gemessen wird.
In der qPCR wird hierzu ein Fluorophor eingesetzt, das als „Lichtsignal“ dient. Grundsätzlich unterscheidet man zwei Strategien:
a) Farbstoffbasierte qPCR
b) Sondenbasierte qPCR◦ ◦ ◦
a) Farbstoffbasierte qPCR (z.B. mit SYBR® Green)
Bei der Analyse wird der Probe eine Reaktionsmischung zugesetzt, bestehend aus:
- freien Nukleotiden
- einem spezifischen Primerpaar
- einer thermostabilen DNA-Polymerase
- einem Puffer mit Mg²⁺-Ionen (für die Aktivität der Polymerase)
- einem fluoreszierenden Farbstoff (z. B. SYBR® Green)
SYBR® Green ist ein Fluorophor, der selektiv an doppelsträngige DNA bindet. Erst nach Bindung an dsDNA entfaltet der Farbstoff seine starke Fluoreszenz.
Diese Mischung wird in ein qPCR-Gerät – einen sogenannten Real-Time-PCR-Thermocycler – gestellt.
Prinzip der Methode
1️⃣ Denaturierung: Die Reaktionsmischung wird auf etwa 94–98 °C erhitzt. Dabei lösen sich die Wasserstoffbrückenbindungen zwischen den Basenpaaren, und der DNA-Doppelstrang wird in zwei Einzelstränge getrennt.
2️⃣ Primerbindung: Die Temperatur wird auf etwa 50–65 °C abgesenkt. Die spezifischen Primer binden an ihre komplementären Zielsequenzen auf den Einzelsträngen. Sie definieren den Startpunkt der DNA-Synthese.
Abb. 2.3.1-A1: SYBR Green im ersten qPCR-Zyklus – Denaturierung und Primerbindung Links: Der Farbstoff SYBR® Green wird vor Beginn der PCR zur Reaktionsmischung gegeben. Er bindet selektiv an doppelsträngige DNA und zeigt in gebundener Form eine starke Fluoreszenz.
Rechts: Während der Denaturierung trennt sich die doppelsträngige DNA in Einzelstränge. Der gebundene Farbstoff wird dabei freigesetzt und verliert weitgehend seine Fluoreszenz. Anschließend binden die spezifischen Primer an ihre komplementären Zielsequenzen. (Hinweis: Die Primer sind zur besseren Übersichtlichkeit verkürzt mit nur drei Nukleotiden dargestellt; in der Praxis sind sie deutlich länger.)3️⃣ DNA-Synthese (Amplifikation): Bei etwa 72 °C synthetisiert die DNA-Polymerase den komplementären Strang. Dabei entsteht erneut doppelsträngige DNA. Der Farbstoff SYBR® Green bindet an diese neu gebildete dsDNA.
4️⃣ Anregung und Emission: Die Reaktionsmischung wird mit blauem Licht (ca. 490 nm) bestrahlt. Die gebundenen Farbstoffmoleküle absorbieren diese Energie und emittieren grünes Licht (ca. 520 nm). Der gebundene Farbstoff erzeugt ein messbares Fluoreszenzsignal.
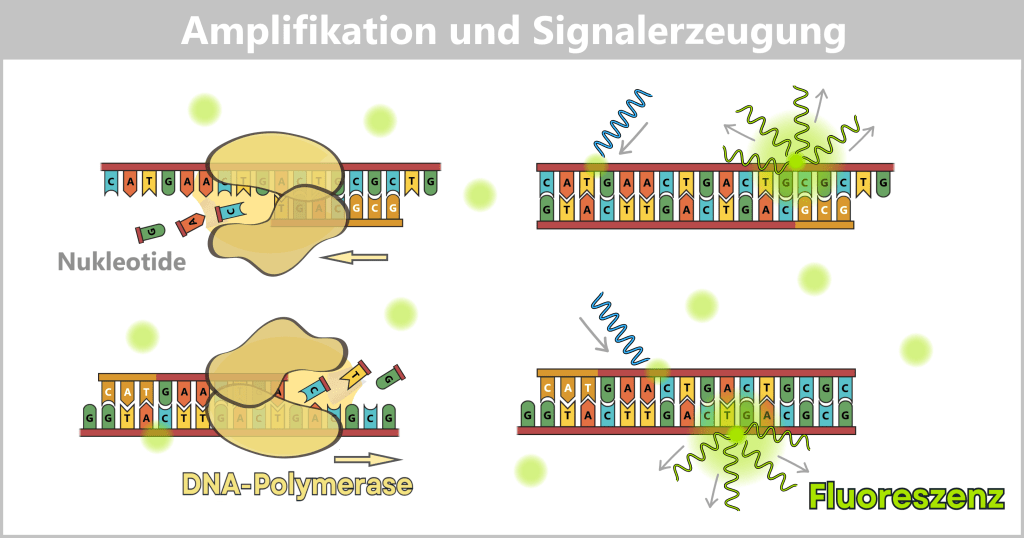
Abb. 2.3.1-A2: SYBR Green im ersten qPCR-Zyklus – vom Doppelstrang zum Fluoreszenzsignal Links: Die DNA-Polymerase synthetisiert neue komplementäre Stränge. Dabei entsteht erneut doppelsträngige DNA, an die SYBR® Green unmittelbar bindet.
Rechts: Am Ende des Zyklus wird die Reaktionsmischung mit Licht geeigneter Wellenlänge angeregt. Die an doppelsträngige DNA gebundenen Farbstoffmoleküle emittieren grünes Fluoreszenzlicht, dessen Intensität proportional zur gebildeten DNA-Menge ist.5️⃣ Quantifizierung: Die Fluoreszenz wird am Ende jedes Zyklus gemessen. Mit zunehmender DNA-Menge steigt das Signal an. Daraus entsteht eine charakteristische Amplifikationskurve.
Der zentrale Messwert ist der sogenannte Ct-Wert (Cycle threshold). Er bezeichnet den Zyklus, bei dem das Fluoreszenzsignal erstmals eine definierte Schwelle überschreitet.
- Niedriger Ct-Wert → hohe Ausgangsmenge an Ziel-DNA
- Hoher Ct-Wert → geringe Ausgangsmenge an Ziel-DNA
Für eine absolute Quantifizierung wird üblicherweise eine Standardkurve mit bekannten Referenzkonzentrationen erstellt.
Bedeutung der Amplikon-Länge für die Detektion
Als Amplikon bezeichnet man den DNA-Abschnitt, der während der PCR zwischen den beiden Primerbindestellen vervielfältigt wird – einschließlich der Primer selbst.
Die spezifische Detektion der Zielsequenz setzt voraus, dass ein vollständiges Amplikon gebildet werden kann. Das ist nur möglich, wenn beide Primer – Vorwärts- und Rückwärts-Primer – auf demselben DNA-Fragment binden können. Ist das Fragment kürzer als der Abstand zwischen den beiden Primerbindestellen, kann einer der Primer nicht binden. Eine effiziente exponentielle Amplifikation findet nicht statt, es entsteht kein relevantes Fluoreszenzsignal.
Daraus folgt eine wichtige methodische Konsequenz: Je länger das gewählte Amplikon – also der Abstand zwischen den beiden Primerbindestellen, desto mehr kurze Fragmente bleiben unerkannt – nicht weil sie nicht vorhanden sind, sondern weil sie für diesen Assay zu kurz sind. Die Wahl der Amplikon-Länge beeinflusst damit direkt, welcher Anteil der tatsächlich vorhandenen DNA erfasst wird.
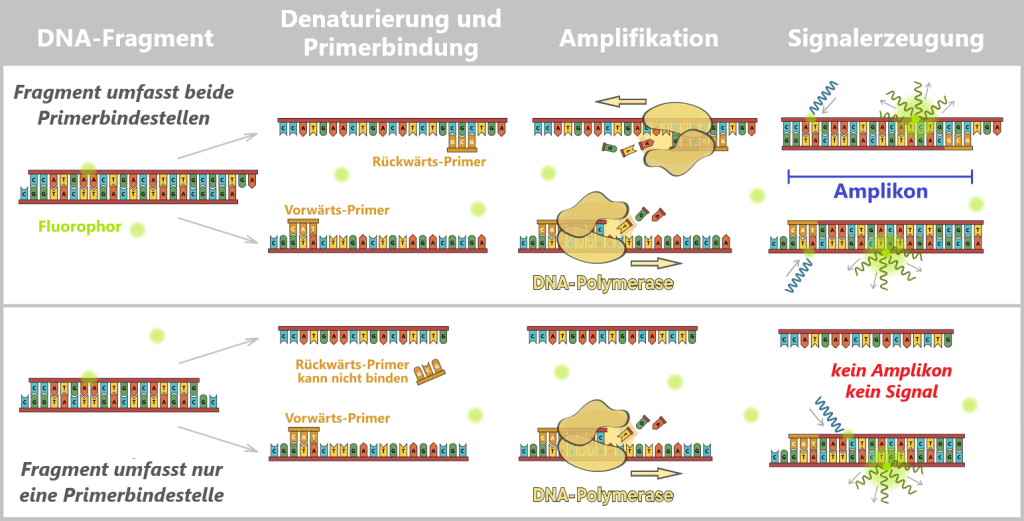
Abb. 2.3.1-A3: Bedeutung der Amplikon-Länge für die Detektion Hinweis: Die Primer sind zur besseren Übersichtlichkeit verkürzt mit nur drei Nukleotiden dargestellt; in der Praxis sind sie deutlich länger. Auch die schematische Darstellung der DNA-Fragmente ist verkürzt.
Oben: Fragment umfasst beide Primerbindestellen
Das DNA-Fragment ist lang genug, dass beide Primer (Vorwärts- und Rückwärts-Primer) an ihre jeweiligen Bindestellen binden können. Die DNA-Polymerase kann den Bereich zwischen den Primern vollständig kopieren. Es entsteht ein spezifisches Amplikon definierter Länge. Im nächsten Zyklus kann dieses Produkt wieder effizient amplifiziert werden. Als Ergebnis entsteht ein starkes Fluoreszenzsignal mit SYBR Green (proportional zur Menge spezifischer dsDNA).
Unten: Fragment umfasst nur eine Primerbindestelle
Das DNA-Fragment ist kürzer als der Abstand zwischen den beiden Primer-Bindestellen. Es kann nur ein Primer binden (der andere hat keine komplementäre Sequenz). Es entsteht kein vollständiges Amplikon, somit ist keine exponentielle Amplifikation möglich. Als Ergebnis entsteht kein oder nur sehr schwaches Fluoreszenzsignal mit SYBR Green.Vorteile der farbstoffbasierten qPCR
- Geringe Kosten & Aufwand: Die Methode ist vergleichsweise kostengünstig und technisch unkompliziert.
- Einfaches Primer-Design: Es werden nur sequenzspezifische Primer benötigt.
- Hohe Sensitivität: Bereits sehr geringe Mengen an Ziel-DNA lassen sich zuverlässig detektieren.
- Schnelle Durchführung: Die Methode ist breit etabliert und ermöglicht eine zügige Analyse auch größerer Probenzahlen. Die Quantifizierung erfolgt direkt über die Fluoreszenzentwicklung während der Amplifikation.
- Hohe Flexibilität: Neue Zielsequenzen können vergleichsweise schnell durch Anpassung der Primer analysiert werden.
- Schmelzkurvenanalyse als Qualitätskontrolle: Nach der Amplifikation kann die Spezifität der PCR-Produkte anhand ihrer charakteristischen Schmelztemperatur überprüft werden. Dadurch lassen sich unspezifische Produkte oder Primer-Dimere häufig erkennen.
Nachteile der farbstoffbasierten qPCR
- Unspezifische Farbstoffbindung: Der Fluoreszenzfarbstoff bindet an jede doppelsträngige DNA – auch an unspezifische Amplifikationsprodukte wie Primer-Dimere.
- Abhängigkeit vom Primer-Design: Die Spezifität der Analyse hängt stark von der Qualität und Selektivität der verwendeten Primer ab.
- Einfluss der Zielregion: Die Wahl der Zielsequenz und insbesondere die Länge des amplifizierten Abschnitts (Amplikon) beeinflussen das Ergebnis erheblich.
- Einfluss der Fragmentlänge: Stark fragmentierte DNA kann mit längeren Amplikons unvollständig erfasst werden, da häufig nicht mehr beide Primerbindestellen auf demselben Fragment vorhanden sind.
- Einfluss der Amplikonlänge: Kürzere Amplikons erfassen fragmentierte DNA in der Regel effizienter, erhöhen jedoch unter Umständen die Wahrscheinlichkeit unspezifischer Signale.
- Abhängigkeit von Effizienz und Kalibration: Die Genauigkeit der Quantifizierung wird wesentlich durch die PCR-Effizienz sowie die Qualität der verwendeten Standardkurve beeinflusst.
b) Sondenbasierte qPCR (z. B. TaqMan®)
Bei der Analyse wird der Probe eine Reaktionsmischung zugesetzt, bestehend aus:
- freien Nukleotiden
- einem spezifischen Primerpaar
- einer thermostabilen DNA-Polymerase mit „5’→3′-Exonuklease-Aktivität“ (diese Polymerase kann im Weg liegende Hindernisse aktiv abbauen)
- einem Puffer mit Mg²⁺-Ionen (für die Aktivität der Polymerase)
- einer spezifischen Sonde (z. B. TaqMan®-Sonde)
Aufbau und Funktionsweise der TaqMan®-Sonde
Eine TaqMan®-Sonde ist eine kurze, synthetisch hergestellte DNA-Sequenz, die komplementär zu einem Abschnitt der Zielsequenz passt. Die Sonde bindet an einen der beiden DNA-Stränge, und zwar zwischen den beiden Primerbindestellen.
Die Sonde trägt:
• am 5′-Ende einen Fluoreszenzfarbstoff (Reporter)
• am 3′-Ende einen Quencher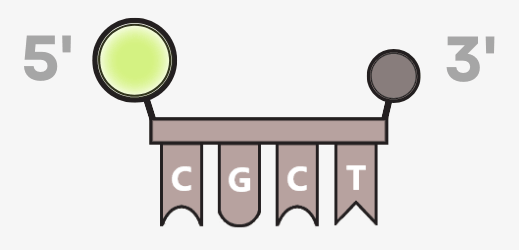
Der Reporter kann unterschiedliche Fluorophore tragen, wodurch auch Multiplex-Analysen – der gleichzeitige Nachweis mehrerer Zielsequenzen – möglich sind.
Solange Reporter und Quencher räumlich nahe beieinander liegen, wird das Fluoreszenzsignal unterdrückt. Das Quenching funktioniert nur, solange die Sonde intakt ist: Durch die räumliche Nähe von Reporter und Quencher kommt es zu einem Fluorescence Resonance Energy Transfer (FRET). Dabei wird die vom Reporter absorbierte Energie auf den Quencher übertragen und als Wärme abgegeben, anstatt als Licht emittiert zu werden.
Die Reaktion wird in einem Real-Time-PCR-Thermocycler durchgeführt, der neben den Temperaturzyklen auch die Fluoreszenz während der Amplifikation misst.
Prinzip der Methode
1️⃣ Denaturierung: Die Reaktionsmischung wird auf etwa 94–98 °C erhitzt. Die Wasserstoffbrückenbindungen brechen auf, wodurch die doppelsträngige DNA in zwei Einzelstränge getrennt wird.
2️⃣ Hybridisierung (Primer- & Sondenbindung): Die Temperatur wird auf etwa 55–60 °C abgesenkt. Neben den spezifischen Primern bindet auch die TaqMan-Sonde an ihre Zielsequenz zwischen den Primern. In diesem Zustand bleibt die Fluoreszenz des Reporters durch den nahen Quencher unterdrückt.
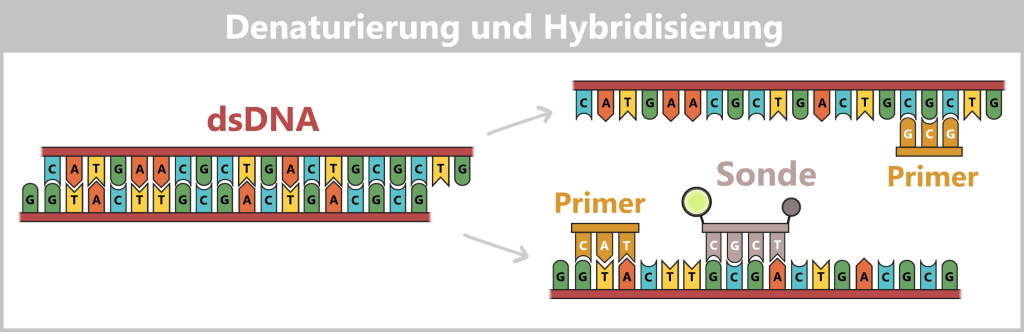
Abb. 2.3.1-B1: Sondenbasierte qPCR im Annealing-Schritt Sobald die Temperatur von 95 °C auf 60 °C sinkt, bindet die Sonde als Erstes an ihre Zielsequenz. Die Sonde hat eine höhere Schmelztemperatur Tm (meist 8–10 °C über der der Primer), damit sie bei der gewählten Arbeitstemperatur bereits fest sitzt, während die Primer gerade erst anfangen zu binden.
Die Sonde ist ein einzelsträngiges DNA-Molekül von etwa 20–30 Nukleotiden Länge. Sie ist so konzipiert, dass sie spezifisch an eine definierte Zielsequenz bindet – einen Abschnitt, der charakteristisch für die gesuchte DNA ist.3️⃣ DNA-Synthese & Hydrolyse (Amplifikation): Bei etwa 60 °C beginnt die DNA-Polymerase mit der Synthese des neuen Strangs. In vielen modernen qPCR-Protokollen erfolgen Annealing (Primer- & Sondenbindung) und Elongation (DNA-Synthese) bei derselben Temperatur.
Trifft die Polymerase während der Strangsynthese auf die gebundene Sonde, nutzt sie ihre 5’→3′-Exonuklease-Aktivität, um die Sonde zu hydrolysieren (in einzelne Nukleotide zu zerlegen). Bereits der erste Schnitt trennt den Reporter-Farbstoff vom Quencher.
Dadurch wird der Reporter dauerhaft von der quenched Situation befreit.4️⃣ Anregung und Emission: Während der Messphase wird die Reaktionsmischung mit Licht einer spezifischen Wellenlänge angeregt. Die freigesetzten Reporter-Moleküle absorbieren diese Energie und emittieren Licht einer längeren Wellenlänge (z. B. grün bei FAM oder gelb bei VIC).
Das Fluoreszenzsignal entsteht hier nicht durch Bindung eines Farbstoffs an doppelsträngige DNA (wie bei SYBR Green), sondern durch die enzymatische Spaltung der Sonde während der Amplifikation.
Die Sonde ist nicht an der DNA-Amplifikation beteiligt. Die Amplifikation wird ausschließlich durch den Vorwärts- und den Rückwärtsprimer gesteuert, während die Sonde lediglich als sequenzspezifischer Fluoreszenzmarker dient.
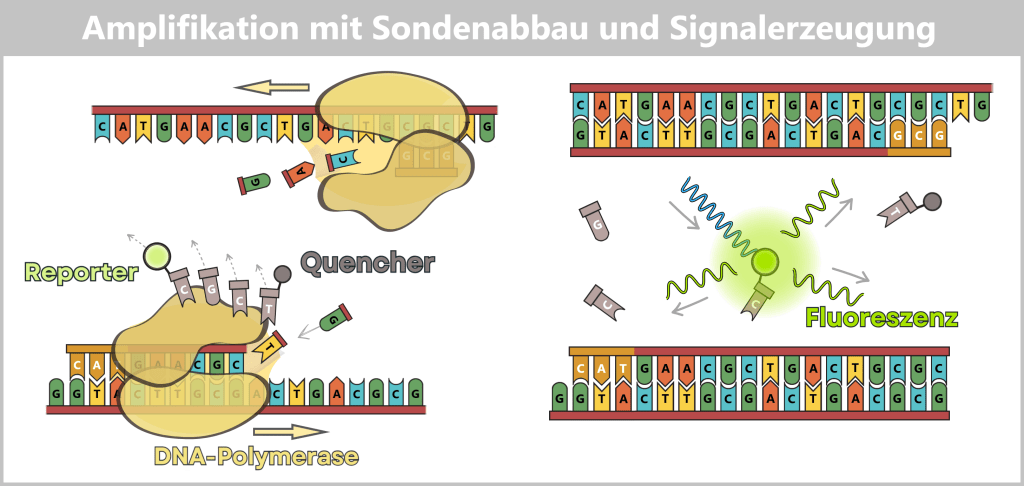
Abb. 2.3.1-B2: Amplifikation mit Sondenhydrolyse – Signalentstehung (Fluoreszenz) Die Polymerase verlängert vom Primer aus und erreicht die Sonde. Während sie den neuen DNA-Strang synthetisiert, räumt sie die im Weg liegende Sonde weg. Dadurch wird der Reporter vom Quencher getrennt, was das messbare Fluoreszenzsignal erzeugt.
5️⃣ Quantifizierung: Die Fluoreszenz wird nach jedem Zyklus gemessen. Pro neu synthetisiertem DNA-Strang, der eine intakte Sondenbindestelle enthält, wird eine Sonde hydrolysiert. Die Signalintensität steigt daher proportional zur Anzahl korrekt amplifizierter Zielmoleküle.
Der zentrale Messwert ist der Ct-Wert (Cycle threshold):
- Niedriger Ct-Wert → hohe Ausgangsmenge an Ziel-DNA
- Hoher Ct-Wert → geringe Ausgangsmenge an Ziel-DNA
Vorteile der sondenbasierten qPCR
- Hohe Spezifität: Neben den beiden Primern bindet zusätzlich eine sequenzspezifische Fluoreszenzsonde. Ein Signal entsteht nur, wenn die gewünschte Zielsequenz tatsächlich amplifiziert wird. Unspezifische Amplifikationsprodukte wie Primer-Dimere erzeugen daher in der Regel kein relevantes Signal.
- Geeignet für Multiplex-Analysen: Durch unterschiedliche Reporter-Farbstoffe können mehrere Zielsequenzen gleichzeitig in einer Reaktion nachgewiesen werden.
- Verbesserte analytische Robustheit: Die zusätzliche Sondenebene erhöht die Spezifität und verbessert die Zuverlässigkeit der Quantifizierung, insbesondere in komplexen Proben mit hohem Hintergrundsignal.
- Geringere Anfälligkeit für unspezifische Amplifikation: Im Vergleich zu farbstoffbasierten Verfahren sind falsch-positive Signale durch unspezifische PCR-Produkte deutlich reduziert.
Nachteile der sondenbasierten qPCR
- Höhere Kosten: Für jede Zielsequenz muss zusätzlich zu den Primern eine spezifische, fluoreszenzmarkierte Sonde synthetisiert werden.
- Komplexeres Assay-Design: Neben den Primern muss auch die Sonde hinsichtlich Sequenz, Schmelztemperatur, Position und möglicher Sekundärstrukturen optimiert werden.
- Abhängigkeit von der Zielregion: Fragmentierungen, Mutationen oder Schäden innerhalb der Primer- oder Sondenbindestellen können die Amplifikation und Signalgenerierung beeinträchtigen oder verhindern.
- Einfluss der Fragmentlänge: Wie bei anderen PCR-basierten Verfahren hängt die Nachweisbarkeit davon ab, ob ein DNA-Fragment alle erforderlichen Bindestellen für Primer und Sonde enthält. Stark fragmentierte DNA kann daher – insbesondere bei längeren Zielregionen – unvollständig erfasst werden.
- Indirekte Quantifizierung: Die absolute Quantifizierung erfolgt meist über Standardkurven und bleibt abhängig von Amplifikationseffizienz, Assay-Design und Probenvorbereitung.
✧ ✧ ✧
Detaillierte Informationen zur Funktionsweise der beiden qPCR-Methoden sind hier zu finden.
Sowohl die farbstoffbasierte als auch die sondenbasierte qPCR ermöglichen eine sensitive und spezifische Detektion definierter DNA-Sequenzen. Die Quantifizierung erfolgt jedoch indirekt über den Ct-Wert und erfordert in der Regel eine Standardkurve mit bekannten Referenzkonzentrationen. Die Genauigkeit hängt somit von der Amplifikationseffizienz und der Qualität der Referenzstandards ab.
Um diese Abhängigkeit zu reduzieren und eine absolute Quantifizierung ohne Standardkurve zu ermöglichen, wurde die digitale PCR (ddPCR) entwickelt.
2.3.2. Digitale PCR (ddPCR)
ddPCR steht für Droplet Digital PCR (digitale Tröpfchen-PCR).
Die digitale PCR ist eine Weiterentwicklung der PCR-Technik, die eine absolute quantitative Bestimmung von DNA- oder RNA-Molekülen ermöglicht – ohne Standardkurve und mit sehr hoher Präzision.
Im Gegensatz zur qPCR misst sie kein kontinuierlich ansteigendes Fluoreszenzsignal, sondern zählt einzelne, positive Reaktionseinheiten. Das Ergebnis ist binär – „positiv“ oder „negativ“ – daher der Begriff digital.
Grundprinzip von ddPCR
Während bei der qPCR die gesamte Reaktion in einem einzigen Reaktionsvolumen abläuft, wird sie bei der ddPCR in Tausende mikroskopisch kleine, voneinander getrennte Reaktionsräume aufgeteilt – sogenannte Droplets (Tröpfchen). Jedes Droplet stellt eine eigenständige PCR-Reaktion dar.
Ablauf der Methode (am Beispiel DNA):
1️⃣ Probenvorbereitung: Die DNA-Probe wird mit Primern, einer sequenzspezifischen Sonde (meist TaqMan®-ähnlich), Nukleotiden, DNA-Polymerase und Puffer gemischt – analog zur sondenbasierten qPCR.
2️⃣ Partitionierung (Droplet Generator): Die Reaktionsmischung wird in einen Droplet Generator überführt. Dort wird sie zusammen mit einem speziellen Öl durch mikrofluidische Kanäle geleitet.
Durch die Strömungsdynamik entstehen daraus bis zu 20.000 nanoliter-große Droplets. Das Öl stabilisiert die Tröpfchen und bildet eine stabile Emulsion, sodass jedes Droplet ein abgeschlossener Reaktionsraum ist. Die fertigen Tröpfchen landen in einem Well (einer Vertiefung) auf einer 96-Well-Platte.
Die Verteilung der DNA-Moleküle auf die Droplets erfolgt zufällig.
Dabei entstehen:- viele leere Droplets
- einige Droplets mit einem Zielmolekül
- einige Droplets mit mehreren Molekülen
Diese Verteilung folgt der Poisson-Statistik.
Abb. 2.3.2-A: Schematische Darstellung der Tröpfchenbildung bei der digitalen PCR 3️⃣ Amplifikation (PCR): Die versiegelte 96-Well-Platte wird in einen Thermocycler überführt.
Die Droplets durchlaufen die üblichen PCR-Zyklen (Denaturierung, Annealing, Elongation), typischerweise 40–45 Zyklen.
Während der Amplifikation wird keine Fluoreszenz gemessen.
Enthält ein Droplet mindestens ein Ziel-DNA-Molekül, wird dieses amplifiziert. Die Sonde wird hydrolysiert und das Droplet entwickelt ein starkes Fluoreszenzsignal.
Droplets ohne Ziel-DNA bleiben dunkel.
Abb. 2.3.2-B: ddPCR-Signale nach Amplifikation in Abhängigkeit von der Ziel-DNA-Konzentration Die Abbildung zeigt schematisch das Ergebnis einer droplet digital PCR (ddPCR) nach Abschluss der Amplifikation.
Die zuvor generierten und versiegelten Droplets durchlaufen in einem Thermocycler 40–45 PCR-Zyklen. Während der Amplifikation erfolgt keine kontinuierliche Fluoreszenzmessung.
Enthält ein Droplet mindestens ein Ziel-DNA-Molekül, kommt es zur Amplifikation der Zielsequenz. Dabei wird die sequenzspezifische Sonde durch die 5′→3′-Exonuklease-Aktivität der DNA-Polymerase hydrolysiert, wodurch der Reporter vom Quencher getrennt wird und ein starkes Fluoreszenzsignal entsteht. Droplets ohne Ziel-DNA bleiben ohne Signal („negativ“).
Mit zunehmender Ausgangskonzentration der Ziel-DNA steigt die Anzahl fluoreszenter (positiver) Droplets, während die Intensität einzelner positiver Droplets vergleichbar bleibt.4️⃣ Auslesung (Droplet Reader): Nach Abschluss der PCR wird die 96-Well-Platte in einen DropletReader eingesetzt.
Das Gerät:
- saugt die Emulsion aus einem Well an
- leitet die Droplets einzeln durch eine schmale Kapillare
- bestrahlt jedes Droplet mit einem Laser
- misst die Fluoreszenzintensität
Jedes Droplet wird einzeln klassifiziert als:
• positiv (Fluoreszenzsignal vorhanden)
• negativ (kein Signal)Das Ergebnis ist binär – 0 oder 1.
Abb. 2.3.2-C: Schematische Darstellung des Prinzips der Fluoreszenzauslesung bei der ddPCR Nach Abschluss der PCR wird die 96-Well-Platte mit der Tröpfchen-Emulsion in den Droplet Reader eingesetzt. Das Gerät saugt die Emulsion aus einem Well an und führt die Tröpfchen einzeln durch eine schmale Kapillare. Jedes Tröpfchen wird dabei mit einem Laser bestrahlt; die gemessene Fluoreszenzintensität wird als Amplitudenwert erfasst. In der Analysesoftware werden diese Werte in einem 1D-Amplitudenplot dargestellt: Die X‑Achse nummeriert die Tröpfchen fortlaufend, die Y‑Achse zeigt die Fluoreszenz-Amplitude. Durch eine softwarebasierte Schwellenwertsetzung werden negative Tröpfchen (kein Signal) von positiven Tröpfchen (Fluoreszenzsignal) getrennt. Die Anzahl der positiven Droplets wird anschließend mittels Poisson-Statistik in die absolute Ziel-DNA-Konzentration der Ausgangsprobe umgerechnet.
5️⃣ Quantifizierung: Da bekannt ist:
- wie viele Droplets insgesamt analysiert wurden
- wie viele davon positiv sind
kann über die Poisson-Verteilung die absolute Anzahl der ursprünglichen DNA-Moleküle berechnet werden.
📍Eine Standardkurve ist nicht erforderlich.
📍Die Quantifizierung ist unabhängig von der Amplifikationseffizienz innerhalb einzelner Zyklen.🎥 Tipp: Wie das Ganze in der Praxis aussieht, zeigt das Video „Digital PCR Using the Bio-Rad QX100™ ddPCR™ System“.
Vorteile der ddPCR
- Absolute Quantifizierung ohne Standardkurve: Die Anzahl der Zielmoleküle wird direkt aus dem Verhältnis positiver zu negativer Droplets mithilfe der Poisson-Statistik berechnet. Eine externe Standardkurve ist in der Regel nicht erforderlich.
- Hohe Präzision und Reproduzierbarkeit: Durch die Aufteilung der Probe in tausende unabhängige Reaktionsräume wird die Quantifizierung weniger anfällig für Schwankungen der Amplifikationseffizienz als bei klassischen qPCR-Verfahren.
- Hohe Sensitivität bei niedrigen Konzentrationen: Auch sehr geringe Mengen an Ziel-DNA können zuverlässig detektiert werden, da einzelne Moleküle in separaten Droplets amplifiziert werden.
- Relative Robustheit gegenüber PCR-Inhibitoren: Inhibitoren verteilen sich ebenfalls auf die einzelnen Droplets. Dadurch fällt ihre hemmende Wirkung oft geringer aus als bei einer einzelnen Gesamtreaktion.
- Endpunktmessung statt Ct-Interpretation: Die Analyse basiert auf der Anwesenheit oder Abwesenheit eines Signals am Ende der PCR und ist daher weniger abhängig von der Interpretation von Amplifikationskurven.
- Besonders geeignet für seltene Zielsequenzen: Die Methode wird häufig für den Nachweis seltener Mutationen, niedriger Viruslasten oder sehr geringer DNA-Restmengen eingesetzt.
Nachteile der ddPCR
- Aufwendigere Technik und höhere Kosten: Zusätzlich zum Thermocycler werden spezielle Geräte zur Droplet-Erzeugung und -Auslesung benötigt.
- Komplexerer Workflow: Die Partitionierung der Probe sowie die anschließende Droplet-Analyse erfordern zusätzliche Arbeitsschritte und erhöhen den technischen Aufwand.
- Begrenzter Dynamikbereich pro Ansatz: Bei sehr hohen Zielkonzentrationen können zu viele Droplets positiv werden. Dadurch verliert die statistische Auswertung an Genauigkeit (Sättigungseffekt), sodass Verdünnungsreihen erforderlich sein können.
- Geringerer Probendurchsatz: Im Vergleich zur qPCR ist die Analyse meist zeitaufwendiger und weniger für sehr hohe Probendurchsätze geeignet.
- Weiterhin sequenz- und assayabhängig: Wie bei der qPCR werden nur DNA-Fragmente erfasst, die sämtliche erforderlichen Primer- und gegebenenfalls Sondenbindestellen enthalten.
- Einfluss der Fragmentierung: Stark fragmentierte DNA kann auch bei der ddPCR unvollständig erfasst werden, insbesondere wenn die Zielregion relativ lang ist oder Primerbindestellen fehlen.
- Abhängigkeit von der Probenvorbereitung: Extraktionsverluste, unvollständiger LNP-Aufschluss oder selektive Fragmentverluste können das Ergebnis weiterhin beeinflussen.
2.3.3. Vergleich der PCR-basierten Quantifizierungsmethoden
Merkmal Farbstoffbasierte qPCR Sondenbasierte qPCR Digitale PCR (ddPCR) Messprinzip Fluoreszenzbindung an jede dsDNA Sequenzspezifische Fluoreszenzsonde Zählung positiver Einzelreaktionen Signalentstehung Farbstoff bindet an dsDNA Sonde wird während der Amplifikation hydrolysiert Endpunktmessung positiver Droplets Quantifizierung Relativ (über Ct-Wert und Standardkurve) Relativ (über Ct-Wert und Standardkurve) Absolut (über Poisson-Verteilung) Standardkurve erforderlich Ja Ja Nein Spezifität Mittel
(primerabhängig)Hoch
(Primer + Sonde)Hoch
(Primer + Sonde)Messzeitpunkt Während der Amplifikation
(Real-Time)Während der Amplifikation
(Real-Time)Nach Endpunkt-PCR Reaktionsraum 1 Reaktionsvolumen 1 Reaktionsvolumen ~20.000 Droplets pro Well Kosten Niedrig Mittel Hoch Geräteaufwand Real-Time-PCR-Gerät Real-Time-PCR-Gerät Droplet Generator + Thermocycler + Droplet Reader
2.4. Sequenzierungsbasierte Verfahren
2.4.1. Oxford Nanopore-Technologie
2.4.2. Illumina-SequenzierungWährend die bisher beschriebenen Methoden (qPCR, ddPCR) gezielt einzelne DNA-Sequenzen nachweisen und quantifizieren können, gehen sequenzierungsbasierte Verfahren einen Schritt weiter: Sie lesen die exakte Abfolge der Basen eines DNA- oder RNA-Moleküls – Stück für Stück, Buchstabe für Buchstabe.
Ein Vertreter dieser Technologien ist die Nanoporen-Sequenzierung. In einer Studie zeigen Gunter et al. (Nature Communications 2023), dass Long-Read-Nanopore-Sequenzierung zur umfassenden Analyse von mRNA-Impfstoffen verwendet werden kann. Methoden wie „VAX-seq“ können Schlüsselqualitätsattribute erfassen – darunter Sequenzidentität, Integrität, Länge und Reinheit – und gleichzeitig mRNA sowie DNA-Rückstände bewerten. Dies macht die Nanopore-Sequenzierung zu einem vielversprechenden analytischen Ansatz für Herstellungs- und Qualitätskontrollprozesse.
Insbesondere die Oxford Nanopore-Technologie (ONT) hat sich in den letzten Jahren als vielversprechendes Werkzeug für die Qualitätskontrolle von mRNA-basierten Wirkstoffen etabliert. Sie liefert direkte Sequenzinformationen einzelner Moleküle und kann sowohl kurze als auch sehr lange Fragmente analysieren – ein entscheidender Vorteil für die Charakterisierung von DNA-Rückständen.
⚬ ⚬ ⚬
2.4.1. Oxford Nanopore-Technologie
Wenn heute von Nanopore-Sequenzierung die Rede ist, ist in der Regel die von Oxford Nanopore Technologies (ONT) entwickelte Plattform gemeint. Obwohl alternative Konzepte zur nanoporenbasierten Sequenzierung existieren, ist ONT derzeit die einzige Technologie, die kommerziell breit verfügbar und praktisch etabliert ist. Seit der Markteinführung im Jahr 2015 hat sie sich als Vertreterin der sogenannten Third-Generation-Sequencing-Verfahren etabliert.
Das Besondere an dieser Methode ist die direkte Analyse einzelner DNA- oder RNA-Moleküle in Echtzeit. Im Gegensatz zu klassischen Sequenzierverfahren sind weder eine zyklische Amplifikation noch chemische Markierungen der Nukleotide erforderlich. Die Sequenzinformation wird unmittelbar aus physikalischen Messsignalen abgeleitet.
Funktionsprinzip
Die ONT-Sequenzierung basiert auf dem Zusammenspiel mehrerer zentraler Komponenten:
Nanoporen – fungieren als winzige molekulare „Lesegeräte“. Wandert ein einzelner DNA- oder RNA-Strang durch die Pore, erzeugt dies charakteristische elektrische Signale – vergleichbar mit einem molekularen „Fingerabdruck“.
Membran – dient als Filter und Barriere. Sie sorgt dafür, dass nur Ionen und Nukleinsäuren die Nanoporen passieren, während unerwünschte Moleküle ausgeschlossen bleiben. Dadurch entsteht ein definierter Messraum für die Signalerfassung.
Chip (Flow Cell) – ist die Basis des Systems. Der Chip enthält die Membran mit zahlreichen Nanoporen sowie die integrierte Elektronik zur Detektion der elektrischen Signale. Er stellt die funktionelle Einheit dar, in der die Sequenzierung stattfindet.
Aufbau des Systems
Der Chip ist in zwei Kammern unterteilt:
Obere Kammer (cis) Hier wird die DNA-Probe eingebracht. Untere Kammer (trans) Nimmt den Strang nach dem Durchtritt
durch die Nanopore auf.Beide Kammern sind mit einer ionenhaltigen Pufferlösung gefüllt, die elektrischen Strom leitet. Die Membran trennt die Kammern elektrisch voneinander – Strom kann nur an den Stellen fließen, an denen Nanoporen eingebettet sind. Diese Poren sind die einzigen „Tunnel“, durch die Ionen und Nukleinsäuren passieren können.
Durch eine zwischen den Kammern angelegte konstante elektrische Spannung entsteht ein messbarer Ionenstrom durch die Nanoporen. Sobald ein DNA- oder RNA-Strang die Pore durchquert, wird dieser Stromfluss charakteristisch verändert. Die dabei entstehenden Strommodulationen werden kontinuierlich erfasst und als elektrische Rohsignale aufgezeichnet.
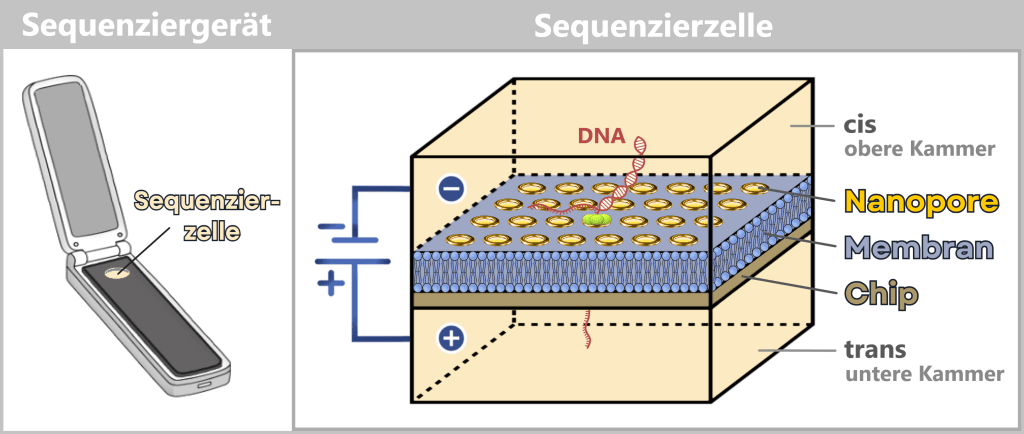
Abb. 2.4.1-A: Aufbau des Nanoporen-Geräts Links ist ein tragbares Nanopore-Sequenziergerät dargestellt, das in seiner Größe in etwa einem etwas breiteren USB-Stick entspricht. Es enthält eine austauschbare Sequenzierzelle (Flow Cell), die für die eigentliche Analyse eingesetzt wird.
Rechts: Die vergrößerte Darstellung zeigt den schematischen Aufbau dieser Sequenzierzelle: Sie besteht aus einem Chip, auf dem eine elektrisch isolierende Membran aufliegt. In diese Membran sind zahlreiche Nanoporen eingebettet, die als molekulare Sensoren dienen. Die Zelle ist in eine obere cis– und eine untere trans-Kammer unterteilt. Durch Anlegen einer elektrischen Spannung wird (hier im Bild) DNA durch die Nanopore gezogen; die dabei entstehenden Stromänderungen werden gemessen und zur Sequenzbestimmung genutzt.Der Weg der DNA durch die Nanopore
1️⃣ Library Preparation (Bibliotheksvorbereitung)
Bevor die Sequenzierung beginnen kann, muss die in der Probe enthaltene Nukleinsäure (DNA und/oder RNA) für die Nanopore-Technologie vorbereitet werden. Dieser Prozess wird als Library Preparation bezeichnet.
Im Kontext der Qualitätskontrolle therapeutischer mRNA – etwa direkt nach der in-vitro-Transkription oder vor der Formulierung – sind dabei folgende Schritte relevant:
a) Aufreinigung
Nach der Transkription enthält das Reaktionsgemisch neben der gewünschten mRNA oder DNA noch Enzyme (z. B. Polymerasen), freie Nukleotide und Pufferbestandteile.
Diese Komponenten müssen entfernt werden, da sie:
- die Effizienz der Adapter-Ligation beeinträchtigen
- die Nanoporen blockieren könnten
- das elektrische Signal stören
In frühen Prozessschritten ist daher eine gründliche Aufreinigung erforderlich. Kurz vor der finalen Formulierung liegt das Produkt in der Regel bereits gereinigt vor, sodass keine zusätzliche Grundaufreinigung mehr notwendig ist.
b) End-Repair & dA-Tailing (bei DNA)
Soll DNA sequenziert werden – etwa zur Analyse von DNA-Rückständen – müssen die Fragmentenden vorbereitet werden.
Zunächst erfolgt eine End-Reparatur: Überhängende („sticky“) Enden werden enzymatisch aufgefüllt oder zurückgeschnitten, sodass glatte, doppelsträngige Enden entstehen.
Anschließend wird häufig ein sogenanntes dA-Tailing durchgeführt. Dabei hängt eine Polymerase gezielt ein einzelnes Adenosin (A) an jedes 3′-Ende der DNA an. Es entsteht ein definierter 3′-A-Überhang, der für die nachfolgende Adapter-Ligation erforderlich ist.
c) Adapter-Ligation – der entscheidende Schritt
Damit die Nukleinsäuren von der Nanopore erkannt und kontrolliert durch die Pore geschleust (transloziert) werden können, müssen spezielle Adapter-Moleküle an die Enden angefügt (ligiert) werden.
Diese Adapter sind funktionelle Module und enthalten mehrere entscheidende Komponenten:
Motorprotein
Das Motorprotein wirkt als molekulare Bremse. Ohne diese Kontrolle würde die DNA mit extrem hoher Geschwindigkeit durch die Pore „schießen“ – viel zu schnell für eine präzise Signalauflösung.
Das Motorprotein:
- reguliert die Translokationsgeschwindigkeit (≈ 400–500 Basen pro Sekunde)
- sorgt für einen gleichmäßigen Vorschub
- entwindet doppelsträngige DNA bei der Passage
Oxford Nanopore verwendet unterschiedliche Motorproteine (Helicasen) für:
- doppelsträngige DNA
- cDNA
- native (direct) RNA-Sequenzierung
Tether
Der Tether kann als molekularer „Einpark-Assistent“ beschrieben werden. Er enthält meist eine cholesterinartige Struktur, die sich in die Lipidmembran der Flow Cell einlagert. Dadurch wird die Nukleinsäure gezielt in die Nähe der Membranoberfläche gebracht – also genau dorthin, wo sich die Nanoporen befinden.
So erhöht sich die Wahrscheinlichkeit erheblich, dass das Molekül in das elektrische Feld einer Pore gerät und hineingezogen wird.
In manchen Protokollen wird zusätzlich ein sogenannter Tether-Buffer eingesetzt, um die Membranoberfläche mit diesen Ankerstrukturen anzureichern.
Barcode-Sequenzen (optional)
Barcode-Sequenzen ermöglichen die parallele Analyse mehrerer Proben in einem Sequenzierlauf (Multiplexing).
Für die reine Qualitätskontrolle einzelner Chargen sind sie nicht zwingend erforderlich und werden daher im Folgenden nicht weiter berücksichtigt.⬦ ⬦ ⬦
Adapter-Design bei ONT
Oxford Nanopore stellt die Sequenzier-Adapter bereits vorbeladen mit Motorprotein und Tether bereit.
Bei der Ligation wird somit der gesamte Adapter-Komplex an das Nukleinsäuremolekül gekoppelt.
Bei DNA:
- Der Adapter wird an die Enden der doppelsträngigen DNA ligiert.
- Die Adapter besitzen einen überstehenden Thymin-(T)-Rest, der komplementär zum zuvor erzeugten 3′-A-Überhang der DNA ist.
- Dadurch entsteht eine effiziente und gerichtete Ligation.
Bei RNA (Direct RNA Sequencing):
- Der Adapter wird am 3′-Ende der RNA ligiert, meist über den vorhandenen Poly(A)-Tail.
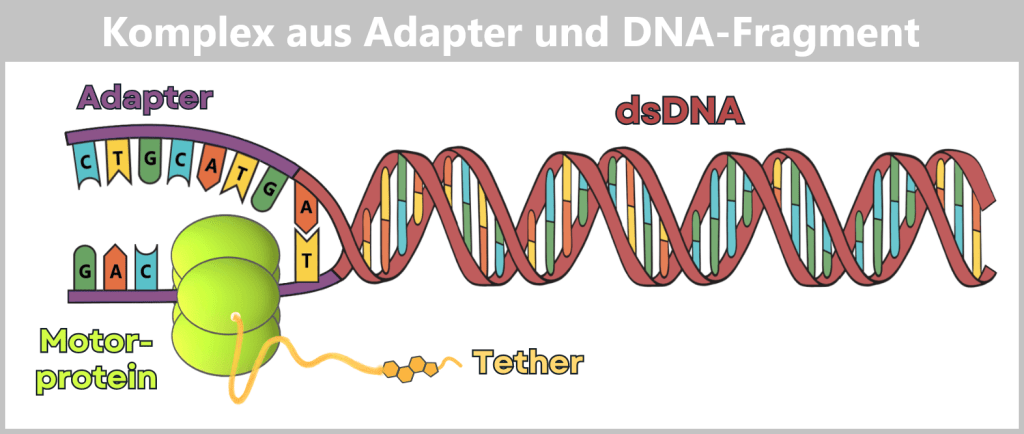
Abb. 2.4.1-B: Schematische Darstellung der Adapter-Ligation Der Sequenzier-Adapter enthält neben einer doppelsträngigen DNA-Region (meist etwa 30 bis 50 Basenpaare lang) auch zwei kurze einzelsträngige Sequenzen (oft ca. 20 bis 40 Nukleotide). Zur besseren Übersicht zeigt die Abbildung eine stark verkürzte Darstellung.
Ein Einzelstrang ist bereits im Inneren des Motorproteins gebunden, während einige Basen des zweiten Einzelstranges außerhalb sichtbar sind.
Direkt daran befindet sich der Übergang zur eigentlichen Probe. Ein spezifischer Überhang am Ende des Adapters (T) hat sich mit dem komplementären Ende der Proben-DNA (A) verbunden. Diese „Ligation“ verknüpft die zu untersuchende Doppelstrang-DNA (dsDNA) stabil mit dem Sequenzier-System.
Seitlich am Motorprotein ist der Tether befestigt. An seinem freien Ende befindet sich ein Cholesterin-Molekül. Dieses dient als chemischer „Dübel“, der den gesamten Komplex in der Lipidmembran der Pore fixiert, um die Wahrscheinlichkeit zu erhöhen, dass die DNA die Nanopore findet.Nach der Ligation sind die Adapter stabil mit der Nukleinsäure verbunden und die Bibliothek ist sequenzierbereit.
Besonderheit: Direct RNA Sequencing (z. B. im Rahmen von VAX-seq)
Eine Besonderheit der Nanopore-Technologie ist die Möglichkeit, native RNA direkt zu sequenzieren, ohne vorherige Umschreibung in cDNA.
Für die Qualitätskontrolle therapeutischer mRNA ist das von großer Bedeutung, da:
- chemische Modifikationen (z. B. Pseudouridin) direkt erfasst werden können
- keine Reverse-Transkriptionsartefakte entstehen
- strukturelle Besonderheiten der RNA erhalten bleiben
Damit erlaubt die Technologie nicht nur eine Mengenbestimmung, sondern eine strukturelle und funktionelle Charakterisierung auf Molekülebene.
2️⃣ Anlegen einer elektrischen Spannung
In beiden Kammern der Sequenzierzelle befinden sich geladene Teilchen (Ionen). Sobald zwischen der oberen cis-Kammer und der unteren trans-Kammer eine elektrische Spannung angelegt wird, entsteht ein elektrisches Feld über der Membran. Dieses Feld treibt die Ionen durch die Nanoporen, wodurch ein messbarer elektrischer Strom entsteht.
Solange sich keine DNA in der Pore befindet, bleibt dieser Ionenfluss konstant. Das Gerät registriert in diesem Zustand einen stabilen Grundstrom, der als Referenzsignal dient (siehe folgende Abbildung).
3️⃣ Die DNA wird in die Sequenzierzelle gegeben
Die vorbereitete DNA-Probe wird in die obere cis-Kammer der Sequenzierzelle pipettiert. An den Enden der DNA befinden sich bereits die Sequenzier-Adapter, die mit einem Motorprotein und einem Tether ausgestattet sind.
Der Tether trägt an seinem Ende eine hydrophobe (wasserabweisende) Cholesterin-Gruppe. Sie fungiert als molekularer Anker:
Durch Diffusion bewegen sich die DNA-Stränge in der cis-Kammer ständig umher. Sobald das wasserabweisende Cholesterin dabei zufällig die Membran berührt, bleibt es dort sofort haften. Für das Cholesterin ist es energetisch weitaus günstiger, in der Lipidschicht zu verweilen als im wasserbasierten Puffer.
Dieser Mechanismus bietet zwei wesentliche Vorteile:
Konzentration an der Oberfläche: Die DNA-Moleküle sammeln sich bevorzugt in der Nähe der Membran. Dadurch steigt die Wahrscheinlichkeit, dass das Motorprotein eine Nanopore findet und dort andocken kann.
Laterale Mobilität: Das Cholesterin ist in der Membran nicht starr fixiert, sondern bleibt lateral (seitlich) beweglich. Die DNA kann daher entlang der Membranoberfläche „gleiten“, bis sie vom elektrischen Feld einer Pore erfasst und schließlich eingezogen wird.
4️⃣ Andocken an die Nanopore
Sobald ein einzelsträngiger Abschnitt der DNA in die Nanopore eintritt, zieht das elektrische Feld den negativ geladenen DNA-Strang in Richtung der trans-Kammer. Dadurch wird das Motorprotein gegen die Porenöffnung positioniert und dockt stabil auf der Pore an.
Der elektrische Zug auf den DNA-Strang wirkt dabei wie ein mechanischer Startimpuls für das Motorprotein. Es beginnt nun, seine Helicase-Aktivität auszuführen:
- Es entwindet die doppelsträngige DNA (dsDNA).
- Ein Strang wird schrittweise und mit kontrollierter Geschwindigkeit durch die Nanopore geführt.
- Der zweite Strang bleibt außerhalb der Pore.
Damit startet der eigentliche Sequenzierprozess.
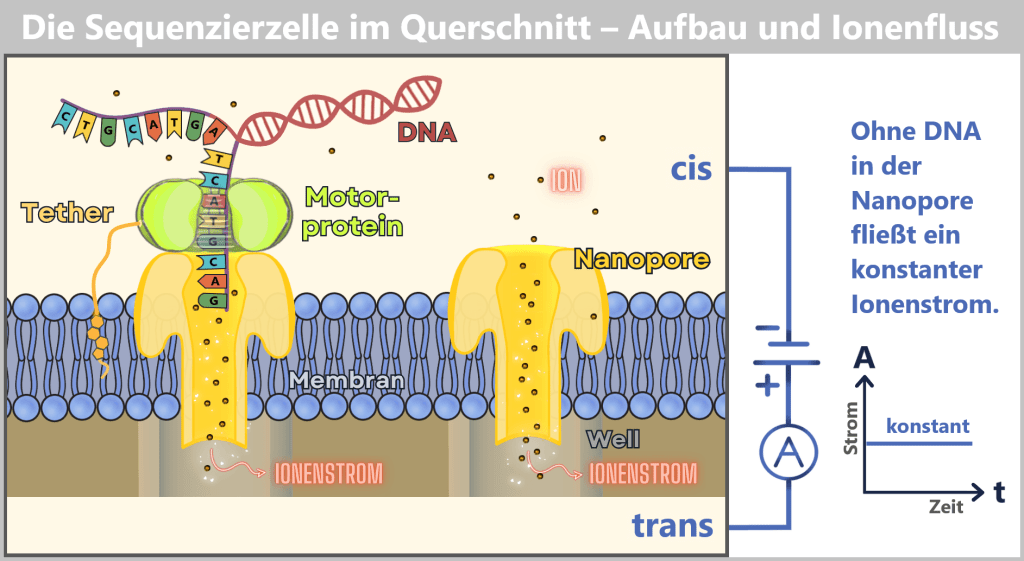
Abb. 2.4.1-C: Schema der Sequenzierzelle und des Ionenstroms im Ruhezustand
(nicht maßstabsgetreu)Die Sequenzierzelle besteht aus zwei Kammern: der cis-Kammer (oben) und der trans-Kammer (unten), die durch eine Membran mit eingebetteten Nanoporen getrennt sind. Links hat ein DNA-Molekül mithilfe eines Motorproteins an eine Nanopore angedockt. Rechts ist eine leere Nanopore dargestellt, durch die ein konstanter Ionenstrom fließt.
Die angelegte Spannung zwischen der negativen Elektrode in der cis-Kammer und der positiven Elektrode in der trans-Kammer treibt den Ionenfluss durch die Pore an. Der Ionenstrom wird im Well (einem kleinen Kanal im Chip unterhalb der Pore) gemessen, wie im Strom-Zeit-Diagramm dargestellt. Solange keine DNA durch die Pore wandert, bleibt der Strom konstant.Das Motorprotein – der Taktgeber der Sequenzierung
Die vorbereitete DNA trägt an ihrem Ende einen Adapter mit einem Motorprotein (einer Helikase). Sobald dieses Molekül in die Nähe der Nanopore gelangt, setzt sich das Protein wie ein passgenauer Deckel auf den Rand der Pore („Docking“). Die elektrische Spannung im System wirkt dabei wie ein unsichtbarer Sog, der die DNA ins Innere der Pore zieht.
Der elektrische Zug unterstützt dabei die Aktivität des Motorproteins. Dieses wirkt wie ein Keil, der den Doppelstrang behutsam auseinanderhebelt. Die Wasserstoffbrücken zwischen den Basenpaaren – die „Sprossen“ der DNA-Leiter – brechen unter diesem Druck auf, da sie deutlich schwächer sind als die stabilisierenden Bindungskräfte innerhalb des Proteins. So wird die doppelsträngige DNA (dsDNA) wie ein Reißverschluss aufgezippt, ohne dass das chemische Rückgrat der Stränge beschädigt wird.
An diesem Punkt beginnt die präzise Kontrolle: Im Inneren des Motorproteins greifen vier bis sechs DNA-bindende Schleifen (sogenannte Hairpins oder Loops) nach dem nun freigelegten Einzelstrang (ssDNA). Diese Greifarme sind spiralig angeordnet, vergleichbar mit den Stufen einer Wendeltreppe oder dem Gewinde einer Schraube.
Man kann sich das Protein als eine „atmende Schraube“ vorstellen: Durch die Zufuhr von Energie (ATP) verändert das Protein rhythmisch seine Form. Es zieht sich zusammen und dehnt sich wieder aus. Bei jeder dieser Bewegungen lösen die obersten Greifarme ihren Griff, rücken ein Stück vor und packen die DNA weiter oben neu, während die unteren Arme den Strang sicher festhalten und kontrolliert nach unten in die Pore schieben.
Diese treppenförmige Anordnung sorgt dafür, dass die DNA nicht unkontrolliert durch die Pore schießt, sondern Schritt für Schritt – also Base für Base – hindurchgeführt wird. Es ist dieser exakte, mechanische Takt des Motorproteins, der es der Nanopore ermöglicht, das elektrische Signal jeder einzelnen Base in aller Ruhe zu registrieren.
5️⃣ Die DNA passiert die Nanopore – das Signal entsteht
DNA besitzt aufgrund ihrer Phosphatgruppen eine negative elektrische Ladung. Durch die angelegte Spannung zwischen der negativ geladenen cis-Kammer und der positiv geladenen trans-Kammer wird der Einzelstrang durch die Nanopore gezogen.
Hier übernimmt das Motorprotein eine entscheidende Funktion:
- Es bewegt die DNA schrittweise durch die Pore.
- Dadurch wird die Geschwindigkeit stark reduziert.
- Die Bewegung erfolgt langsam, gleichmäßig und kontrolliert.
Ohne diese Kontrolle würde die DNA mit einer Geschwindigkeit von Millionen Basen pro Sekunde durch die Nanopore schießen. Das Motorprotein bremst diese Bewegung auf etwa 300–450 Basen pro Sekunde. Erst dadurch wird eine zuverlässige Signalmessung möglich.
Während der DNA-Strang durch die Pore wandert, beeinflusst er den Ionenstrom. Jede Base besitzt eine leicht unterschiedliche Größe und chemische Struktur. Diese Unterschiede verändern den Stromfluss auf charakteristische Weise.
Die Stromänderungen werden von Elektroden gemessen, die mit den einzelnen Wells im Chip verbunden sind. Jeder Well enthält eine Nanopore und besitzt eine eigene Messeinheit. Dadurch kann die Software die Signale eindeutig einer bestimmten Pore zuordnen.
6️⃣ Das elektrische Signal wird aufgezeichnet
Während der Sequenzierung befinden sich typischerweise etwa fünf bis sechs Basen gleichzeitig im engsten Bereich der Nanopore. Dieses kurze DNA-Segment wird als k-mer bezeichnet. Es wirkt im Porenkanal wie ein elektrischer Widerstand: Unterschiedliche Basenkombinationen beeinflussen den Ionenfluss jeweils auf charakteristische Weise und erzeugen dadurch unterschiedliche Stromsignale.
Die DNA-Sequenz lässt sich rekonstruieren, weil sich die DNA Schritt für Schritt durch die Pore bewegt und jedes k-mer ein charakteristisches elektrisches Signal erzeugt. Spezialisierte Software analysiert diese Abfolge von Stromsignalen mithilfe maschinellen Lernens, das die komplexen Signalmuster den jeweiligen Basenabfolgen zuordnet. Dieser Prozess wird Basecalling genannt.
Auf diese Weise entsteht aus den elektrischen Messwerten Schritt für Schritt die vollständige Nukleotidsequenz.
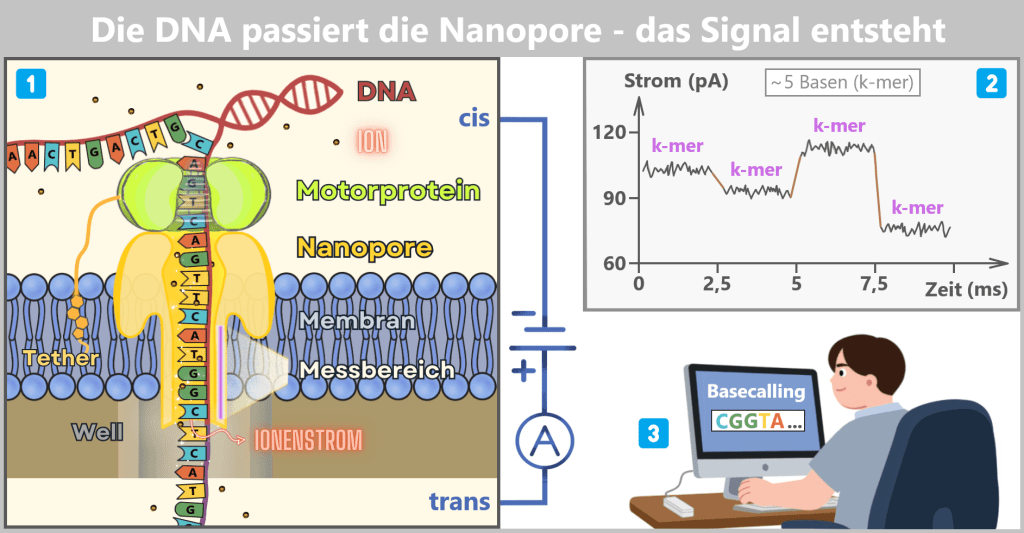
Abb. 2.4.1-D: Das Prinzip der Nanopore-Sequenzierung 1) Die Sequenzierzelle im Querschnitt
Ein DNA-Einzelstrang wird aufgrund der angelegten Spannung von der cis-Seite zur trans-Seite gezogen. Das Motorprotein sitzt oberhalb auf der Nanopore. Es wirkt dabei wie eine molekulare Bremse und kontrolliert den Vorschub der DNA. Dadurch wird der Einzelstrang mit einer Geschwindigkeit von etwa 400 Basen pro Sekunde durch die Pore bewegt, sodass jedes Signal präzise gemessen werden kann.
In der engsten Stelle der Pore (hier als Messbereich gekennzeichnet) befinden sich gleichzeitig mehrere Basen – typischerweise etwa fünf Nukleotide, ein sogenanntes k-mer. Dieses kurze DNA-Segment verengt den Porenkanal.
Die Ionen (kleine rote Punkte) versuchen weiterhin durch die Pore zu strömen. Da das k-mer den verfügbaren Raum einschränkt, können nur noch weniger Ionen passieren. Dadurch entsteht ein charakteristisches Stromsignal.2) Die Daten im Strom-Zeit-Diagramm
Die Y-Achse zeigt den Ionenstrom in Picoampere (pA), die X-Achse die Zeit in Millisekunden (ms).
Ein k-mer befindet sich typischerweise für etwa 2 bis 3 Millisekunden im engsten Bereich der Pore. Während dieser Zeit werden mehrere einzelne Strommesswerte für genau diese Basenkombination aufgezeichnet.
Dabei entsteht im Diagramm ein leicht schwankendes Plateau, das durch elektrisches Rauschen verursacht wird. Die braunen Verbindungslinien zwischen den Plateaus markieren den Moment, in dem das Motorprotein die DNA um eine Base weitertransportiert und dadurch ein neues k-mer in den Messbereich gelangt.3) Basecalling – Umwandlung der Stromsignale in Basen
Die charakteristischen Stromsignale – die Plateaus im Strom-Zeit-Diagramm – entsprechen jeweils dem elektrischen Signal eines bestimmten k-mers, also einer kurzen Basenkombination, die sich für wenige Millisekunden im Messbereich der Pore befindet.
Basecalling-Algorithmen, die häufig auf neuronalen Netzen basieren, analysieren diese Abfolge von Stromsignalen. Aus den charakteristischen Mustern der Signale rekonstruieren sie die zugrunde liegende DNA-Basensequenz.🎥 Tipp: Das Video „How nanopore sequencing works“ fasst den Prozess anschaulich zusammen.
⬦ ⬦ ⬦
Ergebnis der Nanopore-Sequenzierung
Die Nanopore-Sequenzierung liefert keinen einzelnen Messwert, sondern einen umfangreichen Datensatz aus vielen individuellen Sequenzreads. Jeder dieser Reads entspricht der Analyse eines einzelnen DNA- oder RNA-Moleküls. Aus der Gesamtheit dieser Einzelmessungen ergibt sich ein detailliertes Bild der in der Probe enthaltenen Nukleinsäuren.
Dabei können grundsätzlich alle in der Probe vorhandenen DNA- oder RNA-Moleküle erfasst werden, ohne dass eine spezifische Zielsequenz vorgegeben werden muss. Die Methode ist somit nicht zielgerichtet, sondern explorativ.
◈ ◈ ◈
Zwei zentrale Informationsebenen: Identität und Quantität
Die gewonnenen Sequenzdaten enthalten zwei komplementäre Informationsebenen:
Identität (Sequenzinformation)
Für jedes einzelne Molekül kann die Nukleotidsequenz bestimmt werden. Daraus ergeben sich Informationen über:
- Effizienz der Bibliotheksvorbereitung
- Moleküllänge
- Sequenzeigenschaften
- mögliche Sequenzvarianten
- sowie – mit methodischen Einschränkungen – chemische Modifikationen
Auf dieser Grundlage lässt sich bestimmen, welche Nukleinsäuren in einer Probe vorhanden sind.
Quantität (relative Häufigkeit)
Zusätzlich kann über die Anzahl der Reads abgeschätzt werden, wie häufig bestimmte Sequenzen oder Molekültypen in der Probe vertreten sind. Dabei handelt es sich jedoch nicht um eine absolute Quantifizierung, sondern um eine relative Annäherung, die von verschiedenen Faktoren beeinflusst wird, etwa:
- Effizienz der Bibliotheksvorbereitung
- Moleküllänge
- Sequenzeigenschaften
Die quantitative Aussagekraft der Methode ist daher stets im Kontext dieser Einflussgrößen zu interpretieren.
✧ ✧ ✧
Parallele Einzelmolekülmessung in Echtzeit
Die Datenerzeugung erfolgt durch eine große Anzahl parallel arbeitender Nanoporen innerhalb der sogenannten Flow Cell. Geräte wie das MinION verfügen über mehrere hundert aktive Messkanäle, die gleichzeitig und unabhängig voneinander arbeiten.
Jede Nanopore analysiert jeweils ein einzelnes Molekül. Sobald ein Sequenzvorgang abgeschlossen ist, steht die Pore unmittelbar für das nächste Molekül zur Verfügung. Auf diese Weise entsteht kontinuierlich eine große Zahl individueller Reads.
Die Messung erfolgt in Echtzeit, wobei die Software die Signale vieler einzelner Poren gleichzeitig erfasst und verarbeitet.
✧ ✧ ✧
Charakter der Daten: Verteilung statt Einzelwert
Ein wesentliches Merkmal der Nanopore-Sequenzierung ist, dass die Ergebnisse nicht als einheitlicher Messwert vorliegen, sondern als Verteilung vieler einzelner Sequenzreads. Diese Reads können sich deutlich unterscheiden in:
- Länge
- Sequenz
- Qualität
Insbesondere in komplexen Proben entstehen typischerweise:
- kurze Fragmente
- längere zusammenhängende Moleküle
- unterschiedliche Molekülpopulationen
Die Analyse besteht daher nicht in der Betrachtung einzelner Reads, sondern in der Auswertung der Gesamtheit dieser Verteilung.
✧ ✧ ✧
Besonderheit der Methode: Long Reads
Ein charakteristisches Merkmal der Nanopore-Technologie ist die Fähigkeit, sehr lange Nukleinsäuremoleküle kontinuierlich zu sequenzieren. Im Gegensatz zu Short-Read-Verfahren (wie der Illumina-Sequenzierung), bei denen DNA zunächst in kurze Fragmente zerlegt und anschließend bioinformatisch rekonstruiert wird, können hier zusammenhängende Moleküle direkt erfasst werden.
Die Länge eines Reads ist dabei prinzipiell nicht durch die Technologie begrenzt, sondern wird hauptsächlich durch die Integrität des Ausgangsmoleküls bestimmt.
Dies ermöglicht insbesondere:
- die direkte Analyse vollständiger Moleküle
- die Unterscheidung zwischen intakten Sequenzen und Fragmenten
- die Untersuchung struktureller Zusammenhänge innerhalb einzelner Moleküle
✧ ✧ ✧
Grenzen und methodische Einflüsse
Trotz dieser Vorteile unterliegt die Nanopore-Sequenzierung verschiedenen methodischen Einschränkungen:
Fehlerrate einzelner Reads
Die Genauigkeit einzelner Sequenzreads ist im Vergleich zu klassischen Short-Read-Verfahren geringer. Ursache hierfür ist die indirekte Bestimmung der Sequenz über elektrische Stromsignale. Bestimmte Sequenzmotive, insbesondere homopolymere Bereiche (z. B. lange Wiederholungen derselben Base), können dabei schwieriger aufgelöst werden.
Selektions- und Prozessbias
Nicht alle Moleküle werden mit gleicher Wahrscheinlichkeit erfasst. Unterschiede in Länge, Struktur oder Adapterbindung können dazu führen, dass bestimmte Sequenzen bevorzugt oder seltener sequenziert werden.
Insbesondere die Bibliotheksvorbereitung kann die Zusammensetzung der später sequenzierten Moleküle beeinflussen. Reinigungsschritte mit magnetischen Beads oder andere größenabhängige Prozessschritte können sehr kurze Fragmente teilweise entfernen oder unterrepräsentieren. Gleichzeitig werden längere Moleküle bei der Nanopore-Sequenzierung häufig effizienter erfasst, da sie länger durch die Pore laufen und dadurch stabilere Signale erzeugen.
Die resultierende Read-Verteilung spiegelt daher nicht zwangsläufig exakt die ursprüngliche Fragmentverteilung der Probe wider.
Abhängigkeit von der Probenqualität
Die Qualität und Integrität der Ausgangsnukleinsäuren beeinflussen die resultierenden Reads erheblich. Insbesondere lange Moleküle sind anfällig für Fragmentierung während der Probenvorbereitung.✧ ✧ ✧
Konsensusbildung und bioinformatische Auswertung
Um die Genauigkeit der Ergebnisse zu erhöhen, werden die Daten nicht auf Basis einzelner Reads interpretiert. Stattdessen werden viele Sequenzreads desselben Molekültyps miteinander verglichen und zu einer Konsensussequenz zusammengeführt.
Da Sequenzierfehler überwiegend zufällig verteilt sind, können sie durch diese Mehrfachanalyse weitgehend reduziert werden. Moderne Basecalling- und Auswertungsverfahren, die häufig auf maschinellem Lernen basieren, tragen zusätzlich zur Verbesserung der Sequenzgenauigkeit bei.
Auf diese Weise lässt sich aus einer Vielzahl einzelner Messungen ein konsistentes und belastbares Gesamtbild ableiten.
✧ ✧ ✧
Einordnung für die Analyse von Nukleinsäuren
Die Nanopore-Sequenzierung ermöglicht somit eine detaillierte Charakterisierung von Nukleinsäuren hinsichtlich ihrer Sequenz, Länge und relativen Häufigkeit. Gleichzeitig erfordert die Interpretation der Daten eine Berücksichtigung methodischer Einflüsse, insbesondere im Hinblick auf Quantifizierung und Repräsentativität der erfassten Moleküle.
2.4.2. Illumina-Sequenzierung
Die Nanopore-Sequenzierung ermöglicht die Analyse langer, zusammenhängender Moleküle in Echtzeit – ihre besondere Stärke liegt in der Erfassung vollständiger Sequenzen, ohne dass diese zuvor zwingend in kurze Standardfragmente zerlegt werden müssen. Eine andere Herangehensweise verfolgt die Illumina-Sequenzierung: Sie arbeitet nicht mit einzelnen langen Molekülen, sondern mit einer sehr großen Zahl kurzer Fragmente, die parallel und hochpräzise gelesen werden. Beide Technologien ergänzen sich – sie beantworten unterschiedliche Fragen mit unterschiedlichen Mitteln.
Die Illumina-Sequenzierung ist ein Short-Read-Verfahren und gehört zur Gruppe der sogenannten Next Generation Sequencing (NGS)-Technologien. Sie ist derzeit das weltweit am weitesten verbreitete Sequenzierverfahren.
Das Grundprinzip unterscheidet sich deutlich von der Nanopore-Technologie: Während ONT einzelne Moleküle in Echtzeit durch eine Pore führt und dabei elektrische Signale misst, basiert Illumina auf einem optischen Verfahren – der sequenzierenden Synthese mit fluoreszenzmarkierten Nukleotiden („Sequencing by Synthesis“). Dabei wird die DNA nicht direkt gelesen, sondern während einer kontrollierten Synthesereaktion schrittweise entschlüsselt.
Funktionsprinzip
1️⃣ Vorbereitung der DNA-Fragmente = Bibliothekserstellung
Für die Sequenzierung wird die DNA zunächst in ein Format gebracht, das für die Illumina-Plattform geeignet ist. Dieser Prozess wird auch als Bibliothekserstellung bezeichnet und er umfasst folgende Teilschritte:
a) Fragmentierung
Die DNA wird mechanisch oder enzymatisch in kleinere Stücke zerlegt (typischerweise mit einer Zielgröße von etwa 150–500 Basenpaaren). Dabei entstehen DNA-Fragmente leicht unterschiedlicher Länge, die anschließend oft noch über ein Größenauswahlverfahren vereinheitlicht werden.
Dieser Schritt ist besonders wichtig, wenn lange DNA-Moleküle sequenziert werden sollen, da die Illumina-Technologie kurze Fragmente effizienter verarbeitet als sehr lange zusammenhängende Moleküle.
In einigen Anwendungen kann die Fragmentierung jedoch bewusst reduziert oder ganz weggelassen werden – insbesondere dann, wenn bereits stark fragmentierte DNA untersucht wird. Auch in solchen Ansätzen werden sehr lange Fragmente (typischerweise oberhalb von etwa 800–1000 bp) im weiteren Verlauf der Bibliotheksvorbereitung und Sequenzierung häufig weniger effizient erfasst und können dadurch unterrepräsentiert sein.
Zur Veranschaulichung betrachten wir in unserem Beispiel drei DNA-Fragmente mit leicht unterschiedlichen Längen.
b) Adapter-Ligation
An beide Enden der DNA-Fragmente werden spezifische Adaptersequenzen (P5- und P7-Adapter) angehängt. Diese Adapter erfüllen mehrere Funktionen:
Bindungsstellen für die Flowcell: Die Adapter enthalten spezielle DNA-Sequenzen, die wie ein Schlüssel zu einem Schloss passen. Dadurch können die DNA-Fragmente später an einer Oberfläche haften bleiben, was für die Sequenzierung wichtig ist.
Primer-Bindungsstellen: Die Adapter enthalten Abschnitte, an die Sequenzierungsprimer binden können. Diese Primer werden später genutzt, um die DNA-Stränge schrittweise zu synthetisieren.
Indizes (optional): Falls mehrere Proben gleichzeitig sequenziert werden, ermöglichen Index-Sequenzen die Zuordnung der Fragmente zu ihrer jeweiligen Probe. Zur besseren Übersichtlichkeit verzichten wir in unserem Beispiel auf die Darstellung der Indizes.
c) PCR-Amplifikation
Um sicherzustellen, dass genügend DNA für die Sequenzierung vorliegt, werden die adaptierten DNA-Fragmente mittels Polymerase-Kettenreaktion (PCR) vervielfältigt.
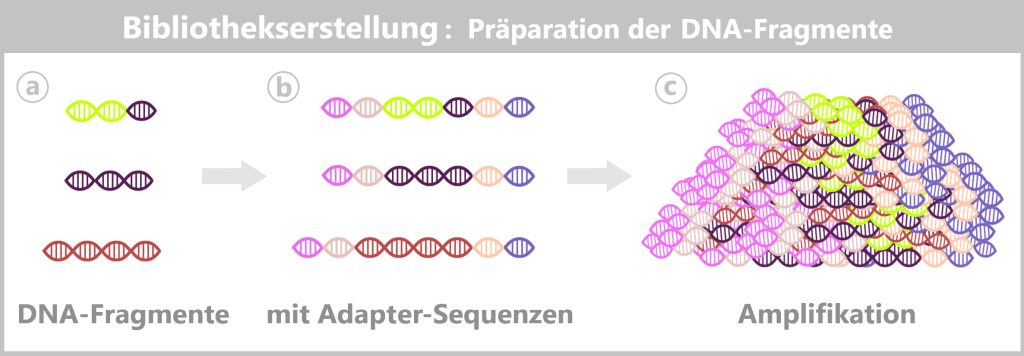
Abb. 2.4.2-A: Bibliothekserstellung – Übersicht über den Ablauf der DNA-Präparation. Die folgende Vergrößerung zeigt eine detaillierte Darstellung eines fragmentierten DNA-Doppelstrangs, versehen mit den beiden Adaptern.
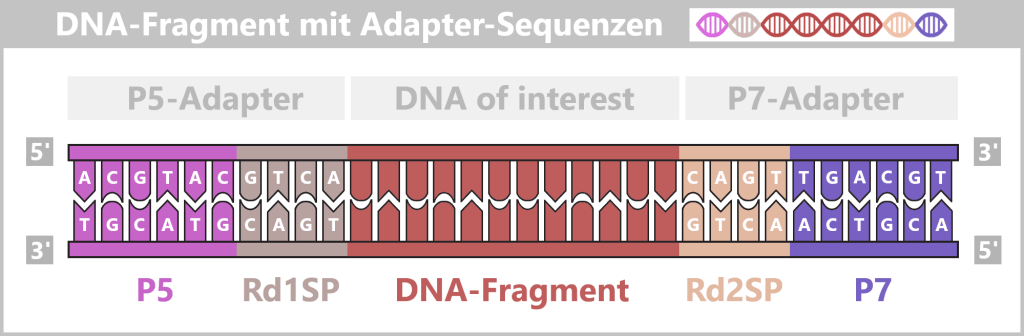
Abb. 2.4.2-B: Schematische Darstellung eines doppelsträngigen DNA-Fragments mit Adaptern. Jedes DNA-Fragment erhält zwei Adaptersequenzen: Der P5-Adapter besteht aus der P5-Adaptersequenz, die die Bindung an die Flowcell ermöglicht, und der Primer-Bindestelle Rd1SP (Read 1 Sequencing Primer) zur Initiation der DNA-Synthese. Ebenso enthält der P7-Adapter die P7-Adaptersequenz zur Flowcell-Bindung sowie die Primer-Bindestelle Rd2SP (Read 2 Sequencing Primer) für die Initiation der DNA-Synthese.
Diese Darstellung ist stark vereinfacht. In der Praxis sind Illumina-Adaptersequenzen mit 60-120 Basenpaarendeutlich länger. Auch das DNA-Fragment ist zur besseren Veranschaulichung verkürzt – tatsächlich haben DNA-Fragmente typischerweise eine Länge von 100–500 Basenpaaren.2️⃣ Cluster-Generierung auf einer Flowcell
Das Herzstück der Illumina-Technologie ist die sogenannte Flowcell – eine glasartige Oberfläche, die mit Millionen kurzer, fest verankerter DNA-Bindestellen beschichtet ist.
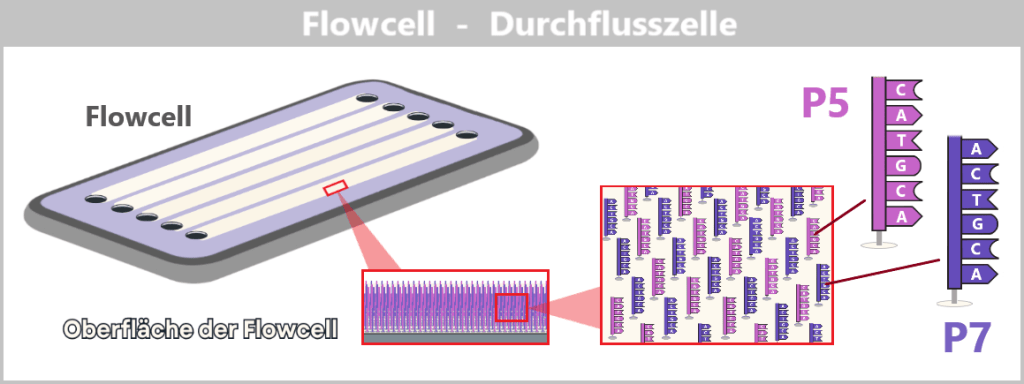
Abb. 2.4.2-C: Schematische Darstellung einer Flowcell und deren Oberfläche a) DNA-Fragmente binden an die Flowcell-Oberfläche
Zu Beginn dieses Schritts wird eine Lösung einzelsträngiger DNA-Fragmente auf die Flowcell geleitet. Die Fragmente tragen an ihren Enden die zuvor ligierten Adaptersequenzen und wurden durch Denaturierung in Einzelstränge überführt, sodass sie frei in der Lösung vorliegen.
Während die Fragmente über die Flowcell strömen, binden ihre Adapter durch komplementäre Basenpaarung an passende DNA-Bindestellen auf der Oberfläche.
Zur Veranschaulichung kann man sich die Oberfläche der Flowcell wie einen dichten Klettverschluss vorstellen: Die Adaptersequenzen der DNA-Fragmente haften an komplementären Bindestellen der Flowcell – ähnlich wie kleine Haken, die sich im Klettgewebe verankern
b) Erste Synthese
Nachdem die DNA-Fragmente an die Flowcell gebunden wurden, beginnt die erste DNA-Synthese. Hierzu bindet ein Primer an die Adaptersequenz des immobilisierten Einzelstrangs. Anschließend synthetisiert die DNA-Polymerase einen komplementären Gegenstrang.
Im nächsten Schritt wird der ursprüngliche Einzelstrang entfernt, während der neu synthetisierte Strang an der Flowcell gebunden bleibt.
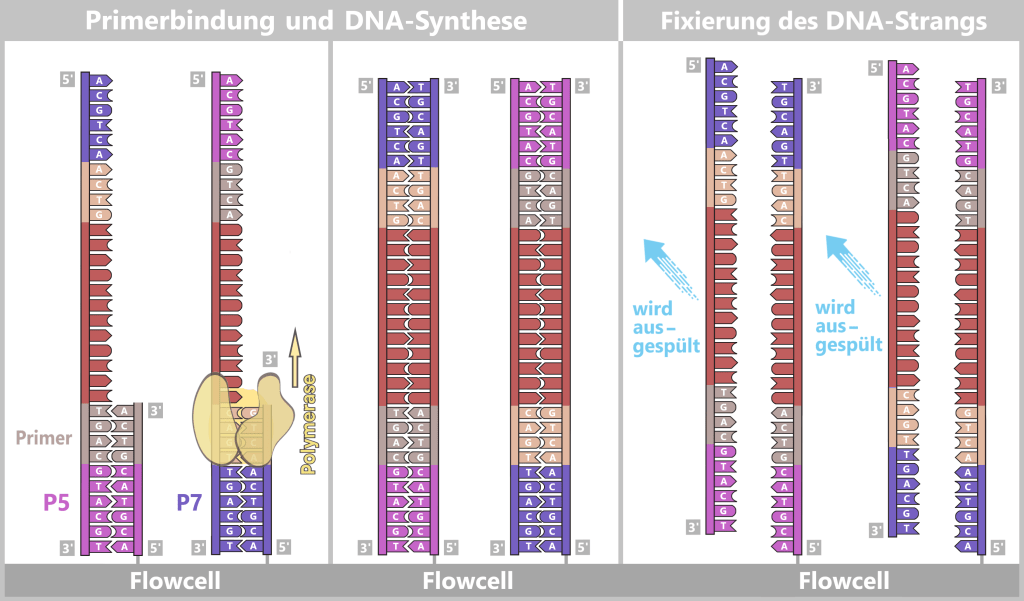
Abb. 2.4.2-D: Schematische Darstellung der ersten Synthese: Entstehung der verankerten DNA-Strang-Kopien. Links: Primer binden an die Adapter (P5, P7), und die DNA-Polymerase (DNAP) startet die Synthese eines neuen, komplementären Strangs.
Mitte: Die DNA-Polymerase synthetisiert daraufhin einen ersten Strang.
Rechts: Der neue entstandene DNA-Doppelstrang wird aufgetrennt. Der ursprüngliche Strang hat nach der Denaturierung keine Verbindung mehr zur Flowcell und wird ausgespült. Der neu synthetisierte Strang bleibt mit seinem 5′-Ende fest an der Flowcell gebunden.Zu diesem Zeitpunkt wäre eine direkte Sequenzierung noch nicht sinnvoll, da die Fluoreszenzsignale einzelner Moleküle zu schwach wären, um zuverlässig detektiert zu werden. Deshalb folgt nun die sogenannte Brückenamplifikation.
c) Brückenamplifikation
Damit die spätere Sequenzierung ein ausreichend starkes Fluoreszenzsignal erzeugt, müssen die an der Flowcell gebundenen DNA-Moleküle zunächst vervielfältigt werden.
Der Prozess beginnt damit, dass sich ein gebundener DNA-Einzelstrang aufgrund seiner Flexibilität zur Oberfläche zurückbiegt und mit seinem freien Adapterende an eine benachbarte komplementäre Bindestelle auf der Flowcell bindet. Dadurch entsteht eine charakteristische Brückenstruktur.
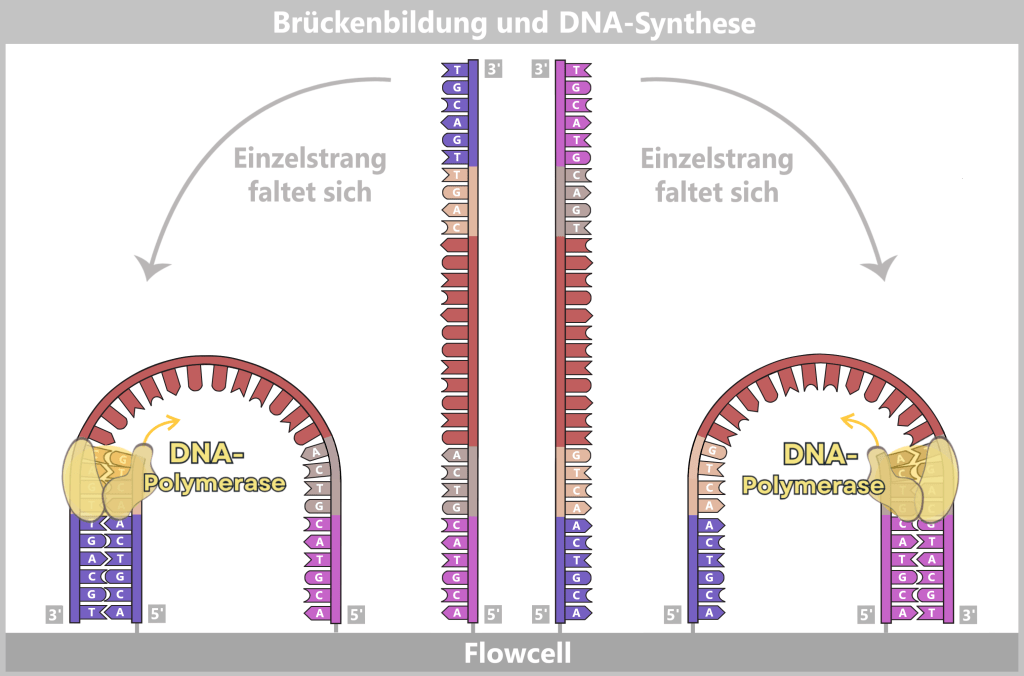
Abb. 2.4.2-E: Brückenbildung und DNA-Synthese Anschließend synthetisiert die DNA-Polymerase entlang des gebundenen Strangs einen komplementären Gegenstrang. Nach der Synthese wird die Brückenstruktur wieder aufgetrennt, sodass nun zwei fest an der Flowcell verankerte Einzelstränge vorliegen.
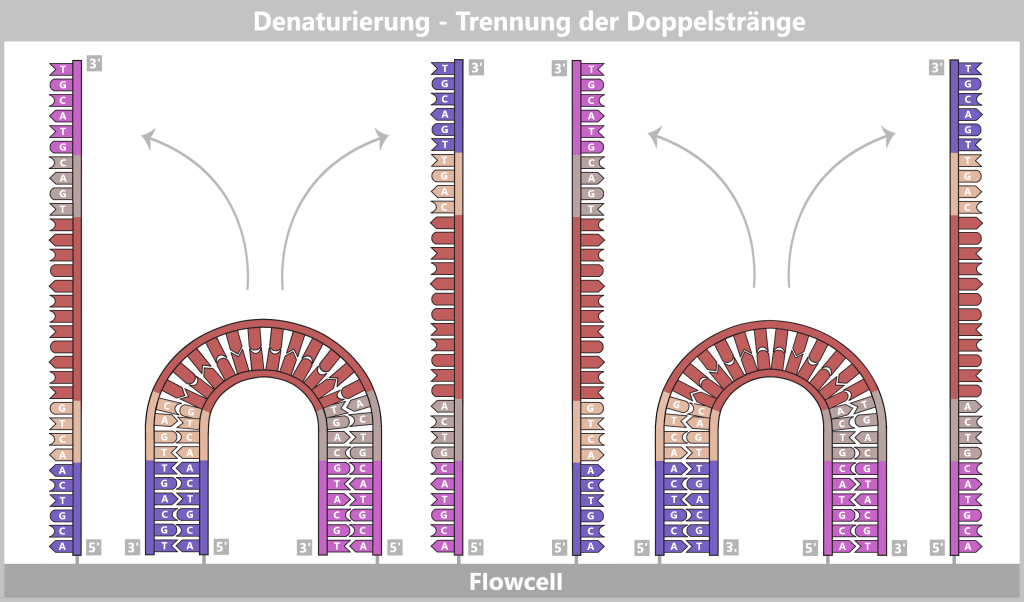
Abb. 2.4.2-F: Vorwärts und rückwärts: Die Brücken lösen sich. Nach dem Kopiervorgang werden die Brücken-Doppelstränge getrennt. Die Anzahl der fest verankerten DNA-Stränge verdoppelt sich. Sie sind bereit für die weitere Amplifikation.
Dieser Vorgang wiederholt sich vielfach. Mit jedem Zyklus verdoppelt sich die Zahl der gebundenen DNA-Moleküle.

Abb. 2.4.2-G: Mit jeder Wiederholung der Brückenamplifikation entstehen mehr DNA-Kopien. Auf diese Weise entstehen lokal begrenzte Cluster aus Millionen nahezu identischer Kopien eines einzelnen ursprünglichen DNA-Fragments.

Abb. 2.4.2-H: Clusterbildung: Jedes Cluster besteht aus zahlreichen Kopien eines einzelnen DNA-Fragments. In dieser Darstellung sind exemplarisch nur drei Cluster gezeigt. In der Realität befinden sich jedoch Millionen solcher Cluster auf einer Flowcell, um eine hohe Sequenzierkapazität zu ermöglichen.
Das Ergebnis ist eine dicht gepackte Oberfläche aus Millionen räumlich getrennten DNA-Clustern, von denen jeder aus vielen nahezu identischen Kopien eines einzelnen ursprünglichen DNA-Fragments besteht. Dadurch können Millionen unterschiedlicher Sequenzen parallel analysiert werden.
Ohne diese Vervielfältigung wäre das Lesen der DNA, als würde man versuchen, ein einzelnes Glühwürmchen im Wind zu erkennen. Die Cluster hingegen machen die DNA-Basen (A, T, C, G) deutlich sichtbar – wie ein leuchtender Neon-Schriftzug in der Dunkelheit.
Entfernung eines Strangtyps
Damit die spätere Sequenzierung synchron und eindeutig ablaufen kann, müssen die DNA-Stränge innerhalb jedes Clusters in einheitlicher Orientierung vorliegen. Daher wird vor Beginn der eigentlichen Sequenzierung gezielt einer der beiden Strangtypen entfernt.
Die nicht benötigten Gegenstränge werden chemisch abgespalten und ausgespült. Gleichzeitig werden bestimmte freie Enden auf der Flowcell blockiert, um unerwünschte Neubindungen oder weitere Amplifikation zu verhindern.
Anschließend bestehen die Cluster aus vielen gleich orientierten Einzelsträngen. Die DNA-Bibliothek ist damit für die eigentliche Sequenzierung vorbereitet.
3️⃣ Sequenzieren durch Synthese
Um die Sequenzierung zu starten, werden Primer, DNA-Polymerasen und modifizierte Nukleotide auf die Flowcell gegeben. Jedes der vier Nukleotide (A, T, G und C) trägt dabei eine eigene Fluoreszenzmarkierung sowie eine reversible chemische Blockierung.
Die Primer hybridisieren mit den DNA-Strängen der Bibliothek. Anschließend bindet die DNA-Polymerase an den Primer und beginnt mit der Synthese des komplementären Strangs.
Durch die reversible Blockierung kann pro Zyklus jeweils nur ein einziges Nukleotid eingebaut werden. Nach dem Einbau stoppt die Synthese vorübergehend.
Anschließend wird die Flowcell mit hochauflösenden Kameras abgebildet. Die Fluoreszenzfarbe jedes Clusters zeigt an, welches Nukleotid in diesem Zyklus eingebaut wurde. Eine Auswertungssoftware analysiert die Signale und übersetzt sie in die entsprechende Basenabfolge.
Danach werden überschüssige Nukleotide ausgespült. In einem weiteren chemischen Schritt werden sowohl die Fluoreszenzmarkierung als auch die Blockierung des eingebauten Nukleotids entfernt, sodass der nächste Synthesezyklus beginnen kann.
Dieser Ablauf wiederholt sich Zyklus für Zyklus. Auf diese Weise wird die Sequenz jedes DNA-Fragments schrittweise ausgelesen.
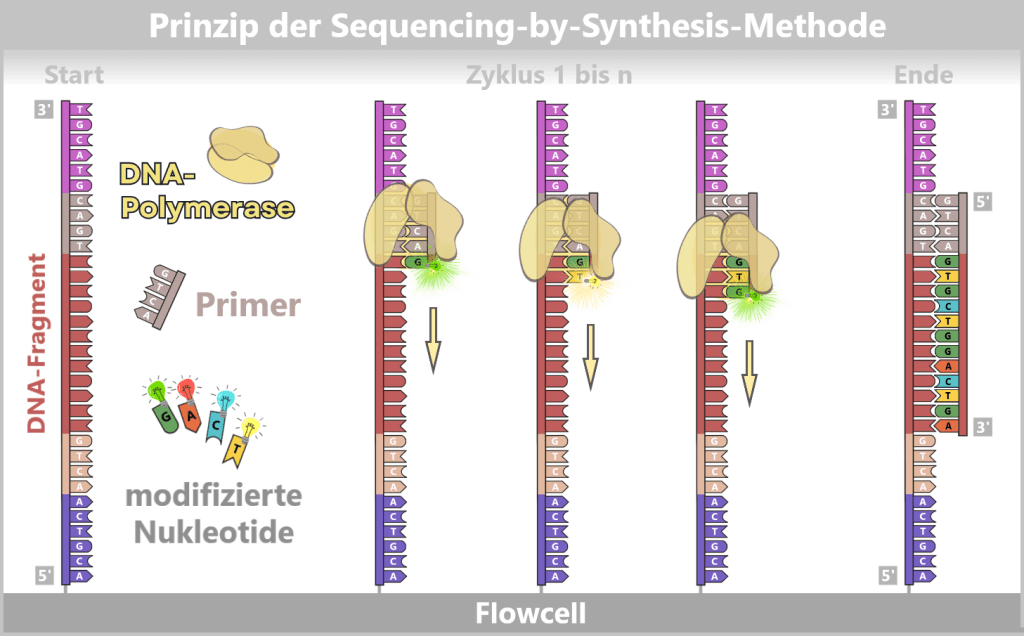
Abb. 2.4.2-I: Illumina Sequenzierungsschritte 4️⃣ Datenauswertung
Nach Abschluss der Sequenzierung liegen Millionen einzelner Fluoreszenzsignale vor. Diese Rohdaten werden zunächst durch die Gerätesoftware in Basenabfolgen übersetzt. Das Ergebnis sind kurze Sequenzabschnitte, sogenannte Reads.
Abb. 2.4.2-J: Analyse der Sequenzierdaten 🎥 Tipp: Eine ausführlichere und dennoch anschauliche Erklärung der Illumina-Sequenzierungstechnologie Methode findet sich im Video „Illumina Sequencing Technology“.
⬦ ⬦ ⬦
Qualitätskontrolle und Trimming
Im ersten Schritt wird die Qualität der Reads überprüft. Basen mit niedriger Signalqualität – typischerweise an den Enden der Reads – werden entfernt („Trimming“). Dadurch sollen fehlerhafte Sequenzzuordnungen reduziert und die Zuverlässigkeit der weiteren Analyse verbessert werden.Mapping: Zuordnung zu einer Referenzsequenz
Anschließend werden die Reads mit bekannten Referenzsequenzen abgeglichen, beispielsweise mit dem Plasmid des Impfstoffherstellers oder bakteriellen Referenzgenomen. Spezielle Algorithmen (z. B. BWA-MEM oder Bowtie2) suchen nach Übereinstimmungen und ordnen die Reads den wahrscheinlichsten Positionen innerhalb der Referenz zu.
Auf diese Weise lässt sich bestimmen, welche Sequenzen in der Probe vorhanden sind und aus welchen Quellen sie stammen.Bestimmung der Fragmentlängen
Bei der häufig verwendeten Paired-End-Sequenzierung wird jedes DNA-Fragment von beiden Enden gelesen. Aus dem Abstand zwischen den beiden Reads kann die Länge des ursprünglichen DNA-Fragments abgeschätzt werden.
Diese Information ist besonders relevant für die Analyse fragmentierter DNA-Proben.Identifizierung der DNA-Herkunft
Durch den Vergleich mit Referenzdatenbanken lässt sich analysieren, ob die nachgewiesenen Fragmente beispielsweise:
▪️aus dem erwarteten Produktionsplasmid stammen,
▪️bakterielle Sequenzen enthalten,
▪️oder anderen genetischen Quellen zugeordnet werden können.
Dadurch ermöglicht die Illumina-Sequenzierung nicht nur eine Quantifizierung, sondern auch eine detaillierte Charakterisierung der vorhandenen DNA.Normalisierung der Daten
Die Anzahl der erzeugten Reads hängt unter anderem von der Sequenziertiefe ab. Um verschiedene Proben oder Chargen vergleichen zu können, werden die Daten häufig normalisiert, beispielsweise als „Reads pro Million“ (RPM). Dadurch lassen sich relative Unterschiede zwischen Proben besser beurteilen.◈ ◈ ◈
Methodische Einschränkungen
Begrenzte Aussagekraft für absolute Mengenbestimmungen
Die Illumina-Sequenzierung eignet sich hervorragend zur Identifizierung und relativen Häufigkeitsanalyse von DNA-Fragmenten. Eine direkte absolute Quantifizierung der ursprünglichen DNA-Menge ist jedoch nur eingeschränkt möglich.
Der Grund dafür ist, dass die Bibliotheksvorbereitung selbst die Zusammensetzung der Probe verändern kann. Schritte wie PCR-Amplifikation, Adapter-Ligation oder Größenselektion können bestimmte Fragmente bevorzugen oder benachteiligen. Die Anzahl der später erzeugten Reads ist daher nicht zwangsläufig proportional zur ursprünglichen Häufigkeit der DNA-Fragmente in der Probe.
Verzerrung der Fragmentlängenverteilung
Bei der Short-Read-Sequenzierung werden bevorzugt DNA-Fragmente innerhalb eines bestimmten Größenbereichs analysiert. Sehr kurze Fragmente können während der Aufreinigung verloren gehen, während sehr lange Fragmente bei der Amplifikation und Clusterbildung benachteiligt werden.
Die gemessene Fragmentlängenverteilung spiegelt daher nicht zwangsläufig die ursprüngliche Verteilung der DNA-Fragmente in der Probe wider, sondern die Verteilung der erfolgreich sequenzierten Fragmente.
2.5. Elektrophoretische Fragmentanalyse
Die elektrophoretische Fragmentanalyse ist ein etabliertes Standardverfahren der Molekularbiologie zur Auftrennung und Charakterisierung von Nukleinsäuren (DNA und RNA). Je nach Methodik können daraus Aussagen über mehrere zentrale Parameter abgeleitet werden:
- Größe (Fragmentlänge): angegeben in Basenpaaren (bp) bei DNA bzw. Nukleotiden (nt) bei RNA
- Menge: semiquantitative Abschätzung der vorhandenen Nukleinsäure
- Integrität: Grad der Fragmentierung bzw. Unversehrtheit der Moleküle
Das Verfahren basiert auf dem physikalischen Prinzip der Elektrophorese (Transport durch Elektrizität). Dabei werden geladene Moleküle wie DNA oder RNA in einem elektrischen Feld durch ein Trägermedium (meist ein Gel oder eine polymerbasierte Matrix) bewegt. Aufgrund ihrer negativ geladenen Phosphatgruppen wandern Nukleinsäuren dabei in Richtung der positiv geladenen Elektrode.
Die Trennung erfolgt über die Porenstruktur des Mediums: Kleinere Fragmente bewegen sich schneller durch das Netzwerk, während größere Moleküle stärker zurückgehalten werden. Auf diese Weise entsteht eine größenabhängige Auftrennung der Nukleinsäuren.
Im Kontext der Analyse von Rest-DNA in mRNA-basierten Arzneimitteln erlaubt diese Methode insbesondere eine Abschätzung der Fragmentlängenverteilung und ergänzt damit quantitative und sequenzbasierte Nachweisverfahren.
2.5.1. Agarosegelelektrophorese
2.5.2. Automatisierte Systeme◦ ◦ ◦
2.5.1. Agarosegelelektrophorese
Die Agarosegelelektrophorese ist die klassische und am weitesten verbreitete Form der elektrophoretischen Fragmentanalyse von DNA. Sie dient dazu, Nukleinsäuren anhand ihrer Größe aufzutrennen und sichtbar zu machen.
Aufbau des Systems
Das zentrale Element ist ein Gel aus Agarose, einem polysaccharidbasierten Netzwerk, das aus Algen gewonnen wird. Dieses Gel bildet eine dreidimensionale Matrix mit Poren definierter Größe. Die Porengröße kann über die Agarosekonzentration gesteuert werden: Niedrige Konzentrationen erzeugen größere Poren (geeignet für lange DNA-Fragmente), höhere Konzentrationen kleinere Poren (für kurze Fragmente).
Das Gel wird in eine Pufferlösung eingebettet und in eine Elektrophoresekammer gelegt. An beiden Enden der Kammer befinden sich Elektroden (Kathode/Anode), zwischen denen eine konstante elektrische Spannung angelegt wird.
Vor Beginn der Elektrophorese werden die Proben in kleine Taschen (Wells) am Rand des Gels pipettiert.
In komplexen Proben, wie sie nach der in-vitro-Transkription und enzymatischen Behandlung (z. B. DNase I) vorliegen, können RNA und verbleibende DNA-Fragmente gleichzeitig im Gel migrieren. Für eine gezielte Analyse der Rest-DNA wird die Probe daher häufig zusätzlich mit RNase behandelt, sodass überwiegend DNA-Fragmente sichtbar werden. Dies ermöglicht eine klarere Beurteilung der Fragmentlängenverteilung.
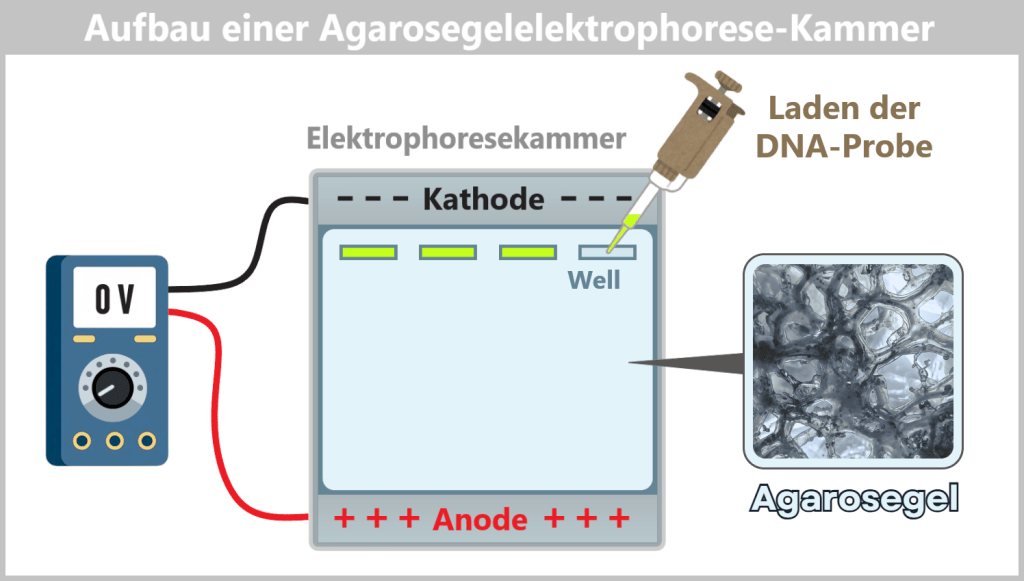
Abb. 2.5.1-A: Schematischer Aufbau einer Agarosegelelektrophorese-Kammer Funktionsprinzip
DNA-Moleküle besitzen aufgrund ihrer Phosphatgruppen eine negative Ladung. Wird eine elektrische Spannung angelegt, beginnen sie, sich durch das Gel in Richtung der positiv geladenen Elektrode zu bewegen.
Während der Migration wirken zwei Kräfte gegeneinander:
- die elektrische Kraft, die die DNA durch das Gel zieht
- der mechanische Widerstand der Gelmatrix
Da kleinere DNA-Fragmente weniger stark durch die Porenstruktur behindert werden, bewegen sie sich schneller durch das Gel als größere Fragmente. Dies führt zu einer größenabhängigen Auftrennung der Moleküle entlang der Laufrichtung.
Zur Größenbestimmung wird parallel eine sogenannte DNA-Leiter (Marker) mit bekannten Fragmentlängen mitgeführt. Durch den Vergleich der Laufstrecken lassen sich die Größen der unbekannten Fragmente abschätzen.
Sichtbarmachung und Auswertung
Nach dem Lauf der Elektrophorese sind die DNA-Fragmente zunächst unsichtbar. Die Sichtbarmachung der DNA erfolgt durch fluoreszierende Farbstoffe, die entweder bereits vor der Elektrophorese dem Gel zugesetzt oder nachträglich auf das Gel aufgebracht werden. Beide Verfahren sind etabliert und unterscheiden sich vor allem in ihrem Einfluss auf die Migration sowie im experimentellen Aufwand.
Das Gel aus der Elektrophoresekammer wird auf einen Transilluminator gelegt. Unter UV- oder blauem Licht leuchten die fluoreszierenden Farbstoffe, die an die DNA gebundenen haben, als Banden im Gel.
Durch den Vergleich mit dem mitgelaufenen Marker lassen sich die Größen der DNA-Fragmente abschätzen. Die Intensität der Banden erlaubt zudem eine grobe, semiquantitative Einschätzung der DNA-Menge.
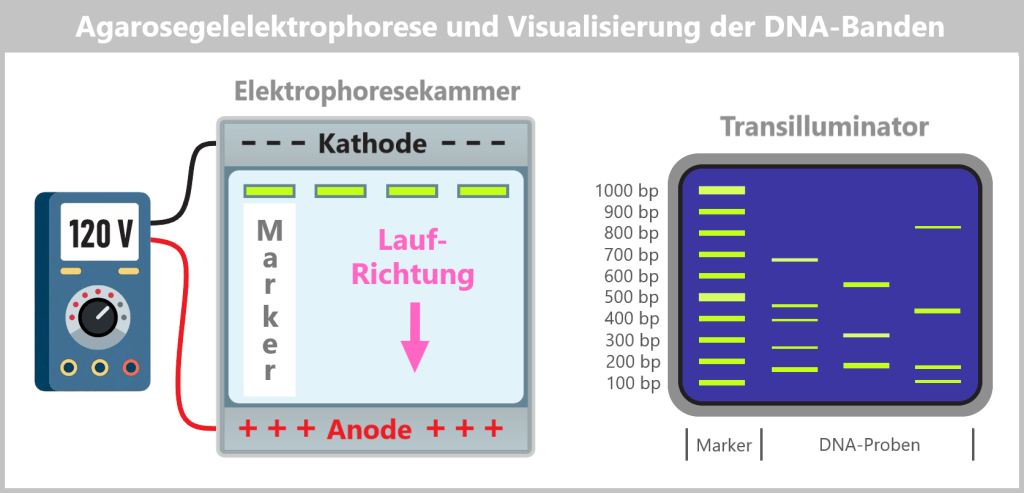
Abb. 2.5.1-B: Detektion der DNA-Fragmente nach elektrophoretischer Auftrennung 🎥 Tipp: Das Video „Electrophoresis explained“ fasst das Prinzip der Agarosegelelektrophorese kurz zusammen.
2.5.2. Automatisierte Systeme
Automatisierte elektrophoretische Systeme stellen eine Weiterentwicklung der klassischen Agarosegelelektrophorese dar. Sie basieren auf demselben physikalischen Prinzip der größenabhängigen Trennung von Nukleinsäuren im elektrischen Feld, kombinieren dieses jedoch mit mikrofluidischer Technologie, integrierter Detektion und digitaler Auswertung.
Im Gegensatz zur manuellen Gelherstellung erfolgt die Trennung hier in miniaturisierten, vorgefertigten Systemen. Die Proben werden in spezielle Kartuschen oder Chips eingebracht, in denen sich feine Kanäle mit polymeren Matrizes befinden. Innerhalb dieser Mikrostrukturen werden die Nukleinsäuren unter angelegter Spannung elektrophoretisch getrennt.
Während des Laufs werden die Fragmente kontinuierlich durch fluoreszierende Farbstoffe detektiert. Die Signale werden direkt in digitale Daten umgewandelt und als sogenannte Elektropherogramme dargestellt. Anstelle von Banden im Gel entstehen dabei Peaks, deren Position die Fragmentgröße und deren Höhe beziehungsweise Fläche die relative Menge der Nukleinsäure widerspiegelt.
Zu den verbreiteten Systemen gehören unter anderem:
- der Bioanalyzer
- die TapeStation
- sowie der Fragment Analyzer
Diese Systeme unterscheiden sich in Details wie Durchsatz, Sensitivität und Automatisierungsgrad, folgen jedoch einem vergleichbaren Grundprinzip.
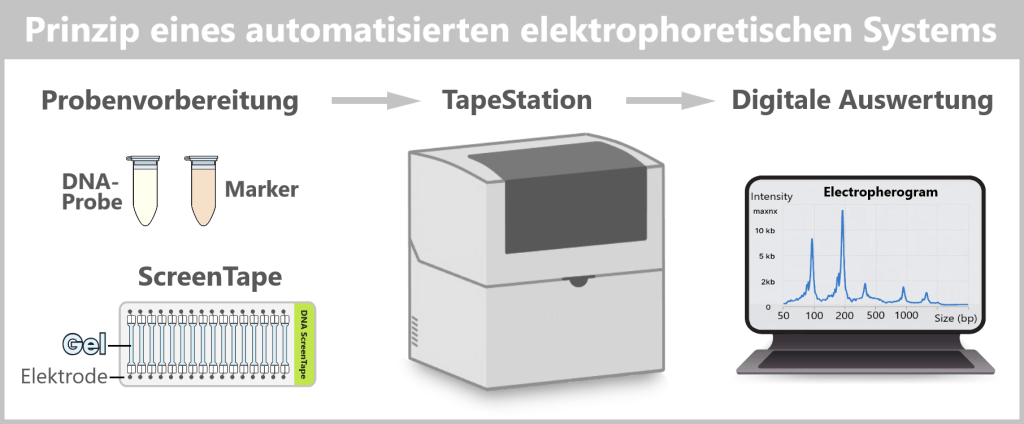
Abb. 2.5.2: Schematische Darstellung eines automatisierten elektrophoretischen Systems (z. B. TapeStation) mit ScreenTape-Einheit und typischem Elektropherogramm Ablauf eines automatisierten elektrophoretischen Systems (z. B. TapeStation)
1) Probenvorbereitung
Die zu untersuchenden Proben werden in Röhrchen oder eine Mikrotiterplatte (z. B. 96-Well-Format) pipettiert und mit einem kit-spezifischen Puffer gemischt. Parallel wird ein Referenzstandard (Marker) mit definierten Fragmentgrößen angesetzt.
2) Zugabe des Fluoreszenzfarbstoffs
Den Proben wird ein Fluoreszenzfarbstoff zugesetzt, der selektiv an DNA oder RNA bindet und eine spätere Detektion ermöglicht.
3) Bereitstellung des Trennmediums
Als Trägermedium dient ein sogenanntes ScreenTape – ein vorgefertigtes, mikrofluidisches System mit integrierter Gelmatrix und mehreren parallelen Trennkanälen. Je nach Anwendung werden spezifische ScreenTapes für DNA oder RNA verwendet.
4) Automatisierter Elektrophoreselauf
Proben, Marker und ScreenTape werden in das Gerät eingesetzt. Nach dem Start führt das System die Analyse automatisch durch: Die Proben werden nacheinander in die Trennkanäle überführt und unter angelegter Spannung elektrophoretisch aufgetrennt.
5) Detektion und Datenauswertung
Während der Trennung werden die fluoreszierenden Signale der Nukleinsäuren optisch erfasst (z. B. mittels Laser). Die Software verarbeitet diese Signale zu digitalen Ausgaben, insbesondere:
▫️Elektropherogrammen (Peakdarstellungen der Fragmentgrößen)
▫️Größenverteilungen
▫️sowie semiquantitativen Konzentrationsangaben🎥 Tipp: Eine anschaulische Demonstration zum Funktionsprinzip des TapeStation-Systems und der ScreenTape-Technologie bietet das Video „Agilent 2200 TapeStation System“.
⬦ ⬦ ⬦
Vorteile der automatisierten Systeme
Automatisierte Systeme bieten gegenüber der klassischen Agarosegelelektrophorese mehrere entscheidende Vorteile:
- Höhere Sensitivität: Auch geringe Nukleinsäuremengen können detektiert werden
- Verbesserte Reproduzierbarkeit: Standardisierte Kartuschen und automatisierte Abläufe reduzieren experimentelle Variabilität
- Quantifizierbarkeit: Neben der Fragmentgröße kann auch die relative Konzentration genauer abgeschätzt werden
- Digitale Auswertung: Objektive und reproduzierbare Analyse ohne visuelle Interpretation von Gelbildern
Darüber hinaus liefern diese Systeme häufig zusätzliche Kennzahlen, wie z. B. mittlere Fragmentlängen oder Integritätsindizes, die eine standardisierte Bewertung der Probenqualität ermöglichen.
⬦ ⬦ ⬦
DNA- und RNA-spezifische Assays
Ein wesentlicher Aspekt automatisierter Systeme ist die Verwendung spezifischer Assays für unterschiedliche Nukleinsäuretypen. In komplexen Proben werden daher häufig getrennte Analysen durchgeführt, bei denen identische Proben jeweils mit DNA- oder RNA-spezifischen Reagenzien untersucht werden.
Diese Assays unterscheiden sich unter anderem in:
- der Zusammensetzung der polymeren Matrix
- den eingesetzten Farbstoffen
- den Auswertealgorithmen
Auf diese Weise können DNA- und RNA-Fragmente unabhängig voneinander charakterisiert werden, selbst wenn sie ursprünglich in derselben Probe vorliegen.
⬦ ⬦ ⬦
Aussagekraft der elektrophoretischen Fragmentanalyse
Die elektrophoretische Fragmentanalyse liefert vor allem strukturelle Informationen über Nukleinsäuren. Im Rahmen der Qualitätskontrolle mRNA-basierter Produkte ermöglicht sie insbesondere:
- die Analyse der Fragmentlängenverteilung von DNA und RNA
- die Beurteilung der RNA-Integrität
- die Unterscheidung zwischen stark fragmentierten und weitgehend intakten Nukleinsäuren
- die Identifikation dominanter Fragmentgrößen
- sowie die Detektion verbleibender DNA-Fragmente
⬦ ⬦ ⬦
Grenzen der elektrophoretischen Fragmentanalyse
Sowohl klassische Agarosegele als auch automatisierte elektrophoretische Systeme besitzen methodische Grenzen, – insbesondere beim Nachweis geringer DNA-Rückstände in komplexen Proben.
- Keine Sequenzinformation: Die Methode trennt Nukleinsäuren ausschließlich nach Größe bzw. elektrophoretischer Mobilität. Ob ein Fragment aus genomischer DNA, Plasmid-DNA, mRNA oder unspezifischen Abbauprodukten stammt, kann anhand der Fragmentanalyse allein nicht bestimmt werden.
- Begrenzte Sensitivität im Vergleich zu amplifikationsbasierten Verfahren: Sehr geringe Mengen residualer DNA können unterhalb der Nachweisgrenze liegen. Verfahren wie qPCR oder ddPCR sind für die Quantifizierung kleiner DNA-Restmengen deutlich sensitiver.
- Eingeschränkte Aussage bei stark fragmentierter DNA: Stark degradierte DNA erscheint häufig lediglich als diffuser Signalbereich („Smear“). Einzelne Fragmente lassen sich dann nur eingeschränkt charakterisieren.
- Keine Aussage über biologische Funktionalität: Die Methode zeigt lediglich Größe und Mengenverteilung von Fragmenten. Ob DNA-Fragmente noch biologisch aktiv, vollständig oder potenziell transkriptionsfähig sind, bleibt unklar.
- Abhängigkeit von Farbstoffbindung und Fragmentgröße: Sehr kurze Fragmente binden teilweise weniger Farbstoff und können daher unterschätzt oder gar nicht detektiert werden. Die quantitative Aussagekraft ist insbesondere im unteren Fragmentgrößenbereich begrenzt.
- Überlappende Signale komplexer Proben: In Proben mit gleichzeitig vorhandener RNA und DNA können sich Signalbereiche überlagern. Trotz spezifischer Assays sind Fehlzuordnungen oder Hintergrundsignale nicht vollständig ausgeschlossen.
- Keine absolute Quantifizierung einzelner Zielsequenzen: Die Methode erlaubt keine gezielte Bestimmung definierter DNA-Sequenzen. Eine spezifische Identifizierung residualer Prozess-DNA ist daher nur in Kombination mit sequenzspezifischen Verfahren möglich.
Die elektrophoretische Fragmentanalyse liefert vor allem strukturelle Informationen über Nukleinsäuren und ergänzt damit quantitative sowie sequenzbasierte Verfahren um eine wichtige Perspektive auf Fragmentgröße, Integrität und Verteilung, ersetzt jedoch keine sequenzspezifischen oder funktionellen Nachweisverfahren.
2.6. Vergleich zentraler Nachweisverfahren für Nukleinsäuren
Die quantitative und qualitative Bestimmung von Nukleinsäuren bildet das Fundament zahlreicher molekularbiologischer Analysen. Je nach Methode unterscheiden sich jedoch sowohl die Aussagekraft als auch die Grenzen der Messung erheblich. Keine der etablierten Techniken liefert ein vollständiges Bild – vielmehr ergänzen sie sich gegenseitig.
UV-spektroskopische Messungen erfassen die Gesamtmenge an Nukleinsäuren in einer Probe. Dabei wird die Absorption von UV-Licht bei 260 nm gemessen. Diese Methode ist schnell und unkompliziert, unterscheidet jedoch nicht zwischen verschiedenen DNA- oder RNA-Sequenzen. Auch Verunreinigungen wie Proteine oder andere Moleküle können das Signal beeinflussen.
Fluorometrische Verfahren verwenden spezifische Farbstoffe, die bevorzugt an DNA oder RNA binden. Dadurch sind sie deutlich selektiver und empfindlicher als UV-Messungen. Dennoch liefern sie keine Informationen über die genaue Sequenz oder Herkunft der Nukleinsäuren – sie quantifizieren lediglich die vorhandene Menge bestimmter Molekülklassen.
Die elektrophoretische Fragmentanalyse ergänzt diese Verfahren um eine strukturelle Perspektive. Sie ermöglicht die Abschätzung von Fragmentgrößen, Fragmentlängenverteilungen und der Integrität von DNA oder RNA. Dadurch lassen sich beispielsweise intakte von stark fragmentierten Nukleinsäuren unterscheiden. Allerdings liefert die Methode keine Sequenzinformation und erlaubt keine eindeutige Identifikation bestimmter DNA- oder RNA-Moleküle.
PCR-basierte Verfahren (qPCR und ddPCR) ermöglichen einen hochsensitiven und spezifischen Nachweis definierter DNA-Sequenzen. Durch den Einsatz sequenzspezifischer Primer können gezielt einzelne Abschnitte des Genoms vervielfältigt und quantifiziert werden. Diese hohe Spezifität ist zugleich eine Einschränkung: Es können nur diejenigen Sequenzen detektiert werden, für die passende Primer entworfen wurden. Unbekannte oder unerwartete Sequenzen bleiben unentdeckt.
Eine weitere methodische Grenze ergibt sich aus den Anforderungen an die DNA-Beschaffenheit. Damit eine Amplifikation stattfinden kann, müssen beide Primerbindestellen vollständig in einem DNA-Fragment enthalten sein. qPCR-Amplicons sind typischerweise 80–150 Basenpaare lang. Stark fragmentierte DNA, deren Bruchstücke kürzer sind oder bei denen eine der Primerbindestellen fehlt, kann daher nicht vervielfältigt werden. Mit anderen Worten: Nur DNA-Fragmente, die die komplette Zielregion zwischen beiden Primern umfassen, sind durch qPCR nachweisbar.
Diese Abhängigkeit von der Fragmentlänge ist insbesondere dann relevant, wenn DNA durch enzymatische oder mechanische Prozesse zerkleinert wurde, wie es mittels DNase-I-Behandlung bei der Aufarbeitung von DNA-Rückständen in der mRNA-Herstellung der Fall ist. Für eine erfolgreiche qPCR/ddPCR müssen jedoch beide Primer – Forward und Reverse – auf demselben Fragment binden können. Je kürzer die Fragmente, desto geringer die Wahrscheinlichkeit, dass ein Fragment beide Primerstellen trägt – ein wichtiger Aspekt bei der Bewertung von DNA-Rückständen.
Illumina-Sequenzierung basiert auf der parallelen Sequenzierung sehr vieler kurzer DNA-Fragmente. Sie liefert hochpräzise Sequenzdaten mit einer im Vergleich zu anderen Sequenziermethoden sehr geringen Fehlerrate. Dadurch eignet sie sich besonders für die genaue Analyse bekannter oder referenzierbarer Sequenzen. Die Methode erfordert jedoch eine aufwendigere Probenvorbereitung, PCR-basierte Bibliotheksverstärkung sowie leistungsfähige bioinformatische Auswertungen. Zudem werden meist nur relativ kurze Fragmente sequenziert, wodurch größere strukturelle Zusammenhänge schwieriger zu erfassen sind.
Nanopore-Sequenzierung geht einen Schritt weiter: Sie erlaubt die direkte Bestimmung der Basenabfolge einzelner DNA- oder RNA-Moleküle in Echtzeit. Dadurch können auch unbekannte Sequenzen identifiziert sowie sehr lange Fragmente analysiert werden. Darüber hinaus ermöglicht die Technologie unter bestimmten Bedingungen die direkte Detektion chemischer Nukleinsäuremodifikationen. Die gewonnenen Daten erfordern allerdings eine sorgfältige bioinformatische Auswertung und weisen im Vergleich zur Illumina-Sequenzierung derzeit noch höhere Fehlerraten auf.
⚬ ⚬ ⚬
Die folgende Übersicht fasst die wichtigsten Nachweisverfahren, ihre Messprinzipien sowie ihre spezifischen Stärken und Grenzen im Kontext der mRNA-Herstellung zusammen.
Methode
Was wird gemessen?
Stärken
Schwächen / Grenzen
Typische Anwendung
UV-Spektroskopie
Gesamtkonzentration aller Nukleinsäuren
Schnell und einfach, keine Reagenzien nötig, geringe Kosten
Keine Unterscheidung zwischen DNA, RNA und freien Nukleotiden, störanfällig gegenüber Verunreinigungen
Schnelle Grobbestimmung von Konzentration und Reinheit
Fluorometrie
DNA oder RNA – über Farbstoffbindung
Hohe Sensitivität, tolerant gegenüber Verun-reinigungen
Keine Sequenz-Information
Präzise Quantifizierung der Ziel-RNA
Qubit-Fluorometer
DNA oder RNA – über Farbstoffbindung (fluorometrisches Gerät)
Sehr hohe Sensitivität und Selektivität, reproduzierbare Ergebnisse
Keine Sequenz-Information, höhere Kosten als einfache Fluorometrie
Standardisierte Konzentrationsbestimmung im Labor- und GMP-Umfeld
qPCR
Spezifische DNA-Sequenzen (z. B. Template-DNA, Wirtszell-DNA)
Hohe Sensitivität, sequenz-spezifisch
Nur bekannte Zielsequenzen nachweisbar, abhängig von Primerdesign und Amplifikationseffizienz, Quantifizierung benötigt Standardkurve, anfällig für Inhibitoren
Nachweis von DNA-Rückständen
ddPCR
Absolute Kopienzahl spezifischer DNA-Sequenzen
Absolute Quantifizierung ohne Standardkurve, sehr hohe Präzision, robuster gegenüber Inhibitoren
Nur bekannte Zielsequenzen nachweisbar, abhängig von Primer-/Sondendesign und Targetintegrität, höhere Kosten, aufwändigere Proben-vorbereitung
Präzise Quantifizierung definierter DNA-Zielsequenzen in Validierungsanalysen
Agarosegel-Elektrophorese
DNA-/RNA-Fragmente nach Größe
Einfach, kostengünstig, visuelle Abschätzung von Fragmentgröße und Integrität
Geringe Auflösung, semiquantitativ, nicht für präzise Konzentrationsbestimmung geeignet, keine Sequenz-Information
Schnelle Qualitäts-kontrolle, Integritätsprüfung
Automatisierte Elektrophorese
Größe, Konzen-tration und Integrität von DNA/RNA
Hohe Auflösung, quantitativ, reproduzierbar, geringer Probenbedarf
Höhere Kosten, Verbrauchsmaterialien erforderlich
RNA-Integritäts-prüfung (RIN-Wert), Fragmentanalyse von DNA-Rückständen
Illumina-Sequenzierung (NGS)
Sequenzinformation vieler kurzer DNA-/RNA-Fragmente
Sehr hohe Genauigkeit, umfassende Abdeckung, gut etabliert
Short Reads (150–300 bp), aufwändige Probenvorbereitung, bioinformatische Auswertung erforderlich
Hochauflösende Sequenzanalyse und Charakterisierung residualer Nukleinsäuren
Nanopore-Sequenzierung
Vollständige DNA/RNA-Sequenz einzelner Moleküle
Direkte Sequenz-information, Long Reads, Echtzeit-Daten, erkennt unbekannte Sequenzen und strukturelle Varianten
Höhere Fehlerrate, komplexe Auswertung, noch begrenzt etabliert im GMP-Umfeld
Charakterisierung komplexer Nukleinsäurepopulationen, Identitätsprüfung, Integritätsanalysen
Insgesamt zeigt sich, dass jede Methode ihre spezifischen Stärken und Schwächen besitzt. Die Wahl des geeigneten Verfahrens hängt daher stets von der jeweiligen Fragestellung ab.
Dies gilt in besonderem Maße für die Qualitätssicherung therapeutischer mRNA. Wirkstoffkonzentration, Reinheit und potentielle Verunreinigungen stellen jeweils eigene Anforderungen an die Nachweismethode.
Keine einzelne Methode beantwortet alle analytischen Fragestellungen – erst die Kombination aus Quantifizierung, struktureller Charakterisierung und Sequenzinformation ermöglicht eine umfassende Beurteilung von Nukleinsäuren.
◈ ◈ ◈
Kombinierte Anwendung der Methoden
In der praktischen Analytik werden die beschriebenen Verfahren selten isoliert eingesetzt. Stattdessen erfolgt die Charakterisierung von Nukleinsäuren in der Regel durch eine Kombination komplementärer Methoden. So kann beispielsweise eine UV-spektroskopische Messung zur schnellen Abschätzung von Konzentration und Reinheit dienen, während fluorometrische Verfahren eine selektivere Quantifizierung ermöglichen. PCR-basierte Methoden wie qPCR oder ddPCR werden eingesetzt, um spezifische DNA-Sequenzen gezielt nachzuweisen und zu quantifizieren. Sequenzierungsverfahren wie Illumina- oder Nanopore-Technologien ergänzen diese Analysen durch die direkte Bestimmung der Sequenz und ermöglichen die Identifikation auch unbekannter Sequenzen oder struktureller Varianten.
⬦ ⬦ ⬦
Regulatorische Einordnung
Im regulatorischen Kontext erfordert die Qualitätssicherung therapeutischer Nukleinsäureprodukte den Einsatz validierter und standardisierter Nachweismethoden. PCR-basierte Verfahren wie qPCR und ddPCR sind hierfür etabliert und werden routinemäßig zur Quantifizierung definierter residualer DNA-Sequenzen eingesetzt. Sequenzierungsbasierte Ansätze, einschließlich der Nanopore-Technologie, gewinnen zunehmend an Bedeutung, befinden sich jedoch in vielen Anwendungsbereichen noch in der Phase der methodischen Etablierung und Validierung.
3. Richtlinien zur Begrenzung von Rest-DNA in Impfstoffen
Rest-DNA (residual DNA), einschließlich Wirtszell-DNA oder Plasmid-DNA, bezeichnet Nukleinsäurereste aus dem Herstellungsprozess, die nach der Aufreinigung im Endprodukt verbleiben können. Wie in den vorherigen Kapiteln dargestellt, lässt sich das vollständige Entfernen dieser DNA technisch nicht erreichen, sodass geringe Restmengen als unvermeidbarer Bestandteil biotechnologischer Produkte gelten.
Die Entwicklung regulatorischer Richtlinien zur Begrenzung von Rest-DNA erfolgte parallel zum Fortschritt biotechnologischer Herstellungsverfahren. Ziel dieser Vorgaben ist es, potenzielle biologische Risiken zu minimieren. Dazu zählen insbesondere:
- Onkogenität
- Insertionsmutagenese
- Immunogenität
- sowie in bestimmten Kontexten Infektiosität (z. B. bei viralen Systemen)
Historische Entwicklung
Frühe Phase: Vorsorgeorientierter Ansatz
Mit der Einführung rekombinanter DNA-Technologien (Verfahren, bei denen DNA aus unterschiedlichen Organismen gezielt zusammengefügt wird) in den 1980er-Jahren – etwa zur Herstellung von therapeutischem Insulin oder Wachstumshormonen – entstand erstmals die Notwendigkeit, DNA-Rückstände systematisch zu bewerten.
Zu diesem Zeitpunkt bestanden erhebliche wissenschaftliche Unsicherheiten hinsichtlich der biologischen Wirkung exogener DNA. Insbesondere war unklar, unter welchen Bedingungen fremde DNA in das Genom menschlicher Zellen integriert werden und potenziell onkogene Effekte auslösen könnte.
Vor diesem Hintergrund verfolgten Regulierungsbehörden einen konservativen, vorsorgeorientierten Ansatz. Die World Health Organization (WHO) empfahl in den 1980er-Jahren einen Grenzwert von 10 pg Rest-DNA pro Dosis für Produkte aus kontinuierlichen Zelllinien (Zellen, die sich im Labor unbegrenzt teilen können). Dieser Wert basierte nicht auf einer quantitativen Risikobewertung, sondern stellte einen pragmatischen Richtwert dar, der sich an den damaligen analytischen Möglichkeiten orientierte.
Spätere Bewertungen führten zu einer Anhebung dieses Wertes; bereits Mitte der 1980er-Jahre wurden Größenordnungen von etwa 100 pg pro Dosis als vernachlässigbar eingestuft.
Paradigmenwechsel: Von der Masse zur biologischen Aktivität
In den 1990er- und 2000er-Jahren veränderte sich die regulatorische Perspektive grundlegend. Zwei Entwicklungen waren hierfür maßgeblich:
Erstens verbesserten sich die Herstellungs- und Aufreinigungsverfahren erheblich. Methoden wie Chromatographie, Filtration und enzymatische Behandlung ermöglichten eine reproduzierbare Reduktion von DNA-Rückständen um mehrere Größenordnungen.
Zweitens lieferten experimentelle Studien zunehmend quantitative Erkenntnisse zur biologischen Aktivität von DNA. Dabei zeigte sich, dass nicht allein die Gesamtmenge entscheidend ist, sondern insbesondere strukturelle Eigenschaften wie Länge und Integrität der DNA eine zentrale Rolle spielen.
Längere, intakte DNA-Fragmente mit funktionellen Genabschnitten können unter geeigneten Bedingungen theoretisch biologische Effekte entfalten. Mit zunehmender Fragmentierung nimmt dieses Potenzial jedoch deutlich ab. Kurze DNA-Fragmente besitzen eine stark reduzierte Wahrscheinlichkeit, in den Zellkern zu gelangen, stabil zu verbleiben oder in das Genom integriert zu werden.
Für eine tatsächliche Integration exogener DNA in das Genom einer Zelle müssen mehrere Voraussetzungen gleichzeitig erfüllt sein. Dazu gehören unter anderem die Aufnahme der DNA in die Zelle, ihr Transport in den Zellkern sowie das Vorliegen geeigneter zellulärer Mechanismen für eine stabile Integration. Die Wahrscheinlichkeit für das gleichzeitige Eintreten dieser Bedingungen wird unter natürlichen Bedingungen im Körper als gering eingeschätzt und nimmt mit zunehmender Fragmentierung der DNA weiter ab.
Moderne regulatorische Ansätze
Auf Basis dieser Erkenntnisse entwickelten sich moderne regulatorische Konzepte hin zu einem risikobasierten und produktspezifischen Ansatz. Anstelle eines einheitlichen Grenzwertes stehen heute mehrere komplementäre Anforderungen im Vordergrund:
Anforderung Begründung Validierte
EliminierungDer Herstellungsprozess muss nachweislich und reproduzierbar zur Reduktion von DNA-Rückständen beitragen. Fragmentierung Verbleibende DNA soll möglichst in stark fragmentierter Form vorliegen. Produktspezifische
GrenzwerteAkzeptanzkriterien werden abhängig von Herstellungsprozess, Zelllinie und Anwendung definiert. Regulatorische Leitlinien von Organisationen wie der U.S. Food and Drug Administration (FDA), der European Medicines Agency (EMA) sowie dem International Council for Harmonisation (ICH) verfolgen heute überwiegend diesen differenzierten Ansatz.
Einordnung im Kontext modRNA-basierter Impfstoffe
Für modRNA-basierte Impfstoffe gelten grundsätzlich ähnliche regulatorische Prinzipien wie für andere biotechnologisch hergestellte Arzneimittel. Gleichzeitig unterscheiden sich die Herstellungsprozesse und Produktcharakteristika in einigen Punkten, insbesondere durch die in-vitro-Transkription und die Verwendung synthetischer RNA.
Im Kontext von Impfstoffen orientieren sich regulatorische Leitlinien in Bezug auf die Menge an Rest-DNA bei parenteraler Anwendung (d. h. bei Verabreichung durch Injektion oder Infusion, die den Magen-Darm-Trakt umgeht) häufig an Größenordnungen im Nanogramm-Bereich pro Dosis.
Zusätzlich wird gefordert, dass die verbleibende DNA möglichst stark fragmentiert ist und keine funktionell vollständigen Gene umfasst. In der wissenschaftlichen Literatur werden in diesem Zusammenhang häufig Fragmentlängen im Bereich unterhalb einiger hundert Basenpaare (bp) diskutiert, ohne dass ein einheitlicher Grenzwert festgelegt ist.
Diese Anforderungen spiegeln den heutigen regulatorischen Ansatz wider, der sowohl die Menge als auch die strukturellen Eigenschaften der DNA berücksichtigt.
Die folgende Tabelle fasst die wesentlichen regulatorischen Dokumente zusammen, in denen Grenzwerte für Rest-DNA genannt werden.
Dokument Aussage / Kontext 1998 – WHO TRS 878, Annex 1 Historische Quelle: Erstmalige Erwähnung von <10 ng/Dosis für kontinuierliche Zelllinien 2007 – WHO Study Group on Cell Substrates for the Production of Biologicals Diskussion von <10 ng/Dosis im Zusammenhang mit stark fragmentierter DNA (z. B. <200 bp) 2013 – WHO TRS No. 978 Bestätigt <10 ng/Dosis als etablierten Standard; betont Bedeutung von Fragmentierung und Prozess; grundlegendes WHO-Dokument zu Zellsubstraten 2014 – WHO TRS 987, Annex 4 <10 ng/Dosis (im Kontext rekombinanter Proteintherapeutika) 2020 – FDA CMC Guidance (Gene Therapy) <10 ng/Dosis und <200 bp (für Gentherapie-Produkte aus kontinuierlichen nicht-tumorigenen Zellen) 2020 – Rapporteur Rolling Review critical assessment report (Josephson 2020-11-19) Acceptance Criteria: ≤ 330 ng DNA/mg RNA (abgeleitet aus 10 ng/Dosis bei 30 µg RNA pro Dosis) 2024 – WHO TRS 1062, Annex 1 (noch nicht öffentlich) Recommendations for mRNA vaccines – erwartete Nennung der Größenordnungen Dosis: Gesamtmenge des Arzneimittels pro Anwendung.
ng/Dosis (Nanogramm/Dosis): Ein Maß für die zulässige Menge an DNA-Rückständen pro Dosis.
bp (Basenpaare): Ein Maß für die Länge der DNA-Fragmente.⚬ ⚬ ⚬
Umrechnung von Messwerten auf ng pro Dosis
Um Messergebnisse aus verschiedenen Studien mit regulatorischen Richtwerten vergleichen zu können, ist eine einheitliche Bezugsgröße erforderlich. Während regulatorische Grenzwerte üblicherweise als Menge pro Dosis (ng/Dosis) angegeben werden, erfolgen analytische Messungen häufig in Bezug auf die RNA-Menge (z. B. ng DNA pro mg RNA).
Zur Vergleichbarkeit müssen diese Werte ineinander umgerechnet werden.
Grundprinzip:
DNA (ng/Dosis) = DNA (ng/mg RNA) × RNA-Menge pro Dosis (mg)Beispielrechnung
Angenommen, eine analytische Messung ergibt: 330 ng DNA pro mg RNA
und die verabreichte Dosis enthält: 30 µg RNA = 0,03 mg RNA
Dann ergibt sich: 330 ng/mg × 0,03 mg = 9,9 ng DNA pro DosisDieses Beispiel zeigt, dass Konzentrationsangaben (ng/mg RNA) und dosisbezogene Grenzwerte (ng/Dosis) direkt ineinander überführt werden können, sofern die zugrunde liegende RNA-Menge pro Dosis bekannt ist.
Das Ergebnis liegt damit in der Größenordnung von 10 ng DNA pro Dosis, was dem häufig zitierten regulatorischen Richtwert entspricht.
⚬ ⚬ ⚬
Einordnung
Ein wichtiger Aspekt der regulatorischen Einordnung ist, dass keine einheitliche, universell verbindliche Grenzwertregelung für alle modRNA-Impfstoffe existiert. Stattdessen basiert die Bewertung auf einem Zusammenspiel aus allgemeinen Empfehlungen, produktspezifischen Festlegungen und risikobasierten Prinzipien.
4. Studien zu DNA-Rückständen in modRNA-basierten Impfstoffen
Nach der Darstellung der Herstellungsverfahren, potenzieller Verunreinigungen sowie der analytischen Nachweismethoden stellt sich die zentrale Frage, in welchem Umfang DNA-Rückstände in kommerziellen mRNA-Impfstoffen tatsächlich nachweisbar sind.
Seit dem Jahr 2023 wurden hierzu mehrere unabhängige Untersuchungen veröffentlicht, die sich mit der Detektion, Quantifizierung und Charakterisierung von DNA in verschiedenen Impfstoffchargen beschäftigen.
Ziel dieses Kapitels ist es, die bisherigen Studien vorzustellen, ihre methodischen Ansätze nachvollziehbar einzuordnen und die berichteten Ergebnisse darzustellen.
4.1. Warum unabhängige Messungen?
4.2. Methodische Vorbemerkungen
4.3. Übersicht und Einzelanalysen der Studien
4.4. Zwischenfazit: Was wissen wir – und was nicht?⚬ ⚬ ⚬
4.1. Warum unabhängige Messungen?
Die Zulassung eines Arzneimittels setzt voraus, dass der Hersteller die Einhaltung regulatorischer Qualitätsstandards gegenüber den zuständigen Behörden nachweist. Für mRNA-basierte Impfstoffe bedeutet das unter anderem, dass Restmengen an DNA unterhalb festgelegter Grenzwerte liegen müssen. Die entsprechenden Prüfdaten werden im Rahmen des Zulassungsverfahrens eingereicht und von den Behörden bewertet.
Was dieser Prozess strukturell nicht vorsieht, ist eine systematische Überprüfung durch unabhängige Labore nach der Marktzulassung. Behörden wie die EMA oder FDA stützen sich bei ihrer Bewertung in erster Linie auf die vom Hersteller vorgelegten Daten. Eine eigenständige Nachanalyse kommerziell erhältlicher Chargen ist nicht Bestandteil des Standardverfahrens.
Genau hier setzt unabhängige Forschung an. Seit 2023 haben mehrere Arbeitsgruppen – ohne Verbindung zu den Herstellern – kommerziell bezogene Chargen der mRNA-Impfstoffe auf DNA-Rückstände untersucht. Die Motivation dahinter war nicht primär die Erwartung eines Problems, sondern eine grundlegende Frage der wissenschaftlichen Nachvollziehbarkeit: Lassen sich die im regulatorischen Zulassungsprozess eingereichten Qualitätsangaben durch unabhängige Messungen reproduzieren?
Die Ergebnisse dieser Studien waren nicht einheitlich – weder in den gemessenen Mengen noch in der methodischen Herangehensweise. Einige Arbeitsgruppen berichteten von DNA-Gehalten, die die regulatorischen Grenzwerte überschritten; andere kamen zu abweichenden Ergebnissen oder äußerten methodische Vorbehalte gegenüber den verwendeten Verfahren.
Dieses Kapitel stellt die wichtigsten dieser Untersuchungen vor. Ziel ist keine abschließende Bewertung – der wissenschaftliche Diskurs ist noch nicht abgeschlossen –, sondern eine sachliche Darstellung der vorliegenden Befunde, der eingesetzten Methoden und der offenen Fragen.
⚬ ⚬ ⚬
4.2. Methodische Vorbemerkungen
Bevor die einzelnen Studien vorgestellt werden, ist ein methodischer Rahmen hilfreich – nicht um Ergebnisse vorwegzunehmen, sondern um dem Leser ein Werkzeug an die Hand zu geben, mit dem er die Befunde einordnen kann.
Die in diesem Kapitel vorgestellten Untersuchungen stammen aus unabhängigen Laboren und unterscheiden sich teilweise deutlich hinsichtlich ihrer methodischen Ansätze und der verwendeten Nachweisverfahren.
Der Aufschluss der Lipid-Nanopartikel
Jede Analyse beginnt mit demselben Problem: Die zu messenden Nukleinsäuren – mRNA und potenzielle DNA-Rückstände – sind in Lipid-Nanopartikel (LNPs) eingeschlossen. Um sie analytisch zugänglich zu machen, müssen die Partikel zunächst aufgeschlossen werden.
In den vorliegenden Studien wurden dafür unterschiedliche Methoden eingesetzt. Detergenzien wie Triton X-100 lösen die Lipidmembran chemisch auf. Kombinationen aus Lithiumdodecylsulfat (LiDS) und magnetischen SPRI-Beads ermöglichen gleichzeitig Aufschluss und selektive Aufreinigung der Nukleinsäuren. Thermische Verfahren – etwa Erhitzen auf 95 °C – destabilisieren die Partikelstruktur durch Hitzeeinwirkung. Kommerzielle Kits wie der Monarch Plasmid DNA Miniprep Kit kombinieren mehrere Schritte zu einem standardisierten Protokoll.
Diese Methodenvielfalt ist nicht trivial. Drei Aspekte verdienen besondere Aufmerksamkeit:
Erstens unterscheiden sich die Methoden in ihrer Aufschlusseffizienz: Nicht jedes Verfahren öffnet alle Partikel vollständig. Verbleiben intakte LNPs in der Probe, bleibt ihr Inhalt für die nachfolgende Messung unsichtbar – mit der Folge, dass der tatsächliche DNA-Gehalt unterschätzt wird.
Zweitens können einige Methoden die freizusetzenden Moleküle verändern oder degradieren. Thermischer Aufschluss bei hohen Temperaturen kann insbesondere RNA fragmentieren; DNA ist thermisch stabiler, kann unter bestimmten Bedingungen jedoch ebenfalls geschädigt werden. Stark fragmentierte DNA ist, wie in Kapitel 2.6. beschrieben, für PCR-basierte Nachweisverfahren schlechter zugänglich – kurze Fragmente tragen möglicherweise nicht mehr beide Primerbindestellen und entgehen damit der Detektion.
Drittens können Rückstände bestimmter Reagenzien nachfolgende Analyseschritte stören. Detergenzien wie Triton X-100 sind bekannte PCR-Inhibitoren. Werden sie nicht vollständig entfernt, kann die Amplifikation beeinträchtigt werden – was wiederum zu einer Unterschätzung der gemessenen Menge führt. SPRI-Beads hingegen selektieren nach Fragmentgröße: Sehr kurze DNA-Fragmente können bei dieser Methode verloren gehen.
Die Wahl der Analysemethode
Nach dem Aufschluss folgt die eigentliche Messung. Auch hier unterscheiden sich die Studien erheblich. Einige Arbeitsgruppen setzten auf quantitative PCR-Verfahren (qPCR), die eine hochsensitive, aber sequenzspezifische Detektion ermöglichen. Andere nutzten Sequenzierungsverfahren – entweder die Nanopore-Technologie (ONT) oder die Illumina-Plattform – die eine breitere, sequenzunabhängige Erfassung erlauben, aber eine aufwändigere Auswertung erfordern. Teilweise wurden auch fluorometrische Verfahren zur Gesamtquantifizierung eingesetzt.
Jede dieser Methoden misst etwas leicht anderes: Fluorometrie erfasst die Gesamt-DNA unabhängig von der Sequenz, leidet jedoch unter Störungen durch RNA (Crosstalk). PCR-Verfahren erfassen nur definierte Zielsequenzen. Sequenzierungsverfahren liefern ein breiteres Bild, sind jedoch stärker von Bibliotheksvorbereitung, Fragmentselektion und bioinformatischer Auswertung abhängig. Eine direkte Vergleichbarkeit der absoluten Messwerte zwischen Studien, die unterschiedliche Methoden einsetzen, ist daher nur eingeschränkt möglich.
Validierung und Standardisierung
In der regulierten pharmazeutischen Analytik sind Messmethoden vor ihrem Einsatz zu validieren: Extraktionseffizienz, Nachweisgrenze, Linearität und mögliche Störeinflüsse müssen systematisch bestimmt werden. Bei den hier vorgestellten unabhängigen Studien fehlt diese formale Validierung in der Regel – was nicht bedeutet, dass die Arbeiten unsorgfältig durchgeführt wurden, wohl aber, dass methodische Unsicherheiten schwerer zu quantifizieren sind.
Ein direkter Vergleich derselben Probe mit mehreren Methoden parallel – ein sogenannter orthogonaler Ansatz – würde helfen, methodenbedingte Variabilität von tatsächlichen Unterschieden zwischen Chargen zu trennen. Nur einige der vorliegenden Studien setzen mehrere orthogonale Methoden systematisch an denselben Proben ein. Auf die Bedeutung dieses Ansatzes wird im abschließenden Kapitel noch eingegangen.
⚬ ⚬ ⚬
4.3. Übersicht und Einzelanalysen der Studien
Die folgenden Studien unterscheiden sich hinsichtlich Methodik, Publikationsform und wissenschaftlicher Einordnung. Neben peer-reviewten Arbeiten werden auch Preprints und explorative Analysen berücksichtigt, um das derzeit verfügbare Spektrum an Daten vollständig abzubilden.
Die Darstellung erfolgt chronologisch, um die Entwicklung der wissenschaftlichen Diskussion nachvollziehbar zu machen.
Sequencing of bivalent Moderna and Pfizer mRNA vaccines reveals nanogram to microgram quantities of expression vector dsDNA per dose
Autoren: Kevin McKernan, Yvonne Helbert, Liam T. Kane, Stephen McLaughlin
Veröffentlicht: 2023 April 10
DNA fragments detected in monovalent and bivalent Pfizer/BioNTech and Moderna modRNA COVID-19 vaccines from Ontario, Canada: Exploratory dose response relationship with serious adverse events.
Autoren: David Speicher, J. Rose, L. M. Gutschi, D. M. Wiseman, Kevin McKernan
Veröffentlicht: 2023 October 19
Methodological Considerations Regarding the Quantification of DNA Impurities in the COVID-19 mRNA Vaccine Comirnaty®
Autoren: Brigitte König, Jürgen O. Kirchner
Veröffentlicht: 2024 May 8
Confirmation of the presence of vaccine DNA in the Pfizer anti-COVID-19 vaccine
Autoren: Didier Raoult
Veröffentlicht: 2024 November 12
BioNTech RNA-Based COVID-19 Injections Contain Large Amounts Of Residual DNA Including An SV40 Promoter/Enhancer Sequence
Autoren: Ulrike Kämmerer, Verena Schulz, Klaus Steger
Veröffentlicht: 2024 December 3
A rapid detection method of replication-competent plasmid DNA from COVID-19 mRNA vaccines for quality control.
Autoren: Tayler J. Wang, Alex Kim, Kevin Kim
Veröffentlicht: 2024 December 29
Quantitative Analysis of Nucleic Acid Content in Spikevax (Moderna) and BNT162b2 (Pfizer) COVID-19 Vaccine Lots
Autoren: Richard M Fleming PhD, MD, JD; Peter Kotlár MD; Sona Pekova MD, PhD
Veröffentlicht: 2025 May 13
Quantification of residual plasmid DNA and SV40 promoter-enhancer sequences in Pfizer/BioNTech and Moderna modRNA COVID-19 vaccines from Ontario, Canada
Autoren: David Speicher, Jessica Rose, Kevin McKernan
Veröffentlicht: 2025 September 25
Systematic analysis of COVID-19 mRNA vaccines using four orthogonal approaches demonstrates no excessive DNA impurities
Autoren: Adam Achs, Tatiana Sedlackova, Lukas Predajna, Jaroslav Budis, Maria Bartosova, Vladimir Zelnik, Diana Rusnakova, Martina Melichercikova, Marta Miklosova, Veronika Gencurova, Barbora Cernakova, Tomas Szemes, Boris Klempa, Juraj Kopacek, Silvia Pastorekova
Veröffentlicht: 2025 Dezember 13
🧬 🧬 🧬
4.3.1. McKernan et al. (2023)
Sequencing of bivalent Moderna and Pfizer mRNA vaccines reveals nanogram to microgram quantities of expression vector dsDNA per dose
Autoren: Kevin McKernan, Yvonne Helbert, Liam T. Kane, Stephen McLaughlin
Veröffentlicht: 10. April 2023 (Preprint, OSF)
Link zur Studie: https://osf.io/preprints/osf/b9t7ma) Kontext und Ziel der Studie
Die Arbeit von McKernan et al. gehört zu den ersten öffentlich verfügbaren unabhängigen Analysen, in denen mRNA-basierte COVID-19-Impfstoffe gezielt auf das Vorhandensein von DNA untersucht wurden. Ausgangspunkt war keine regulatorisch motivierte Fragestellung, sondern eine explorative Sequenzierungsarbeit, im Verlauf derer DNA in den Proben detektiert wurde. Dies veranlasste die Autoren, die Zusammensetzung der Impfstoffe systematisch weiterzuuntersuchen.
Ziel der Studie war es,
- 2 Chargen bivalente Impfstoffe von Pfizer (über dem Ablaufdatum)
- 2 Chargen bivalente Impfstoffe von Moderna (über dem Ablaufdatum)
- 8 Chargen monovalente Impfstoffe von Pfizer (über dem Ablaufdatum, ungeöffnet)
b) Materialien und Methoden
Impfstoffchargen
Für die Untersuchungen wurden folgende Chargen verwendet:- 2 Chargen bivalente Impfstoffe von Pfizer (über dem Ablaufdatum)
- 2 Chargen bivalente Impfstoffe von Moderna (über dem Ablaufdatum)
- 8 Chargen monovalente Impfstoffe von Pfizer (über dem Ablaufdatum, ungeöffnet)
Die Studie kombiniert mehrere analytische Verfahren, um sowohl quantitative als auch qualitative Aussagen zu ermöglichen.
Aufschluss der LNPs und Extraktion der Nukleinsäuren
Die Lipid-Nanopartikel wurden mittels Lithium-Dodecylsulfat (LiDS) lysiert. Die anschließende Aufreinigung erfolgte nach dem SPRI-Prinzip (magnetische Beads), das Nukleinsäuren größen- und bedingungsabhängig selektiv bindet und von Lipiden sowie anderen Begleitsubstanzen trennt. Das Eluat enthielt die isolierten Nukleinsäuren – mRNA und potenziell vorhandene DNA.
Als Qualitätskontrolle des Extraktionsschritts wurde ein Bioanalyzer eingesetzt, der mittels automatisierter Kapillarelektrophorese die Größenverteilung und relative Menge der gewonnenen Nukleinsäuren sichtbar macht.Sequenzierung mittels Illumina
Die extrahierten Nukleinsäuren wurden mittels Illumina-Sequenzierung analysiert. Mehrere Sequenzierläufe (Reads) bildeten die Grundlage für die bioinformatische Rekonstruktion der Plasmidsequenzen beider Impfstoffe. Aus den erhaltenen Reads wurden Plasmidkarten erstellt – für Moderna und für Pfizer.PCR-basierte Verifikation
Auf Grundlage der Sequenzierungsdaten wurden spezifische Primer für zwei Zielregionen entworfen: eine Spike-Sonde und eine Ori-Sonde, beide auf Sequenzen ausgelegt, die sowohl im Pfizer- als auch im Moderna-Plasmid vorkommen. Diese qPCR diente nicht der primären Quantifizierung, sondern der unabhängigen Bestätigung, dass die per Sequenzierung identifizierten Plasmidsequenzen tatsächlich in den Proben vorhanden sind – und nicht auf Laborkontaminationen zurückzuführen sind.Fluorometrie und Elektrophorese
Zur Abschätzung der DNA-Konzentration wurden Qubit-Fluorometrie und Agilent-Elektrophorese eingesetzt. Beide Methoden liefern Aussagen über die Gesamtmenge der vorhandenen DNA, jedoch keine Sequenzinformation.c) Zentrale Ergebnisse
Qualitative Befunde
Die Sequenzanalysen zeigen Übereinstimmungen mit bekannten Plasmidvektorsequenzen und weisen auf genetische Elemente hin, die im Herstellungsprozess eingesetzt werden. Die nachgewiesene DNA wurde als doppelsträngig (dsDNA) charakterisiert.Unerwarteter Fund: SV40-Promoter/Enhancer
Im Verlauf der Sequenzierung identifizierten die Autoren im Pfizer-Impfstoff eine SV40-Promoter/Enhancer-Sequenz. Dieser Fund war insofern unerwartet, als diese Sequenz in den der EMA vorgelegten Plasmidkarten – konkret im „Rapporteur Rolling Review Critical Assessment Report“ (Figure S.2.3-1. pST4-1525 Plasmid Map) – in den öffentlich zugänglichen Unterlagen nicht aufgeführt war. Die Autoren wiesen ergänzend darauf hin, dass das Pfizer-Plasmid auch ein SV40-Kernlokalisierungssignal enthält, das theoretisch den Transport von DNA in den Zellkern begünstigen könnte.Quantitative Befunde
Qubit-Fluorometrie und Elektrophorese zeigten DNA-Konzentrationen, die nach Einschätzung der Autoren die regulatorischen Richtwerte – etwa das EMA-Limit von 330 ng DNA pro mg RNA – deutlich überschreiten.d) Interpretation durch die Autoren
Die Autoren schlussfolgern, dass DNA-Rückstände in den untersuchten Chargen vorhanden sind, die ihrem Ursprung nach auf die im Herstellungsprozess verwendeten Plasmidvektoren zurückzuführen sein dürften. Die gemessenen Mengen werden von den Autoren als möglicherweise regulatorisch relevant eingestuft.
Hinsichtlich replikationsfähiger DNA formulieren die Autoren die Einschätzung, dass für solche Sequenzen strengere Grenzwerte angemessen wären als für nicht-replikationsfähige DNA-Fragmente.
e) Methodische Einschränkungen
Die Herkunft, Lagerung und Chargencharakterisierung der untersuchten Fläschchen ist nur begrenzt dokumentiert. Angaben zu Negativkontrollen – etwa Wasserproben, die denselben Aufarbeitungsschritten unterzogen wurden – sowie zu Positivkontrollen mit bekannten DNA-Konzentrationen fehlen weitgehend oder sind unvollständig beschrieben. Eine formale Methodenvalidierung liegt nicht vor.
Zudem weisen die Autoren selbst auf zwei gegenläufige Unsicherheiten bei der Quantifizierung hin: PCR-basierte Verfahren könnten die tatsächliche DNA-Menge unterschätzen, da Fragmente unterhalb der Amplikon-Länge nicht erfasst werden. Umgekehrt könnten Fluorometrie und Elektrophorese die DNA-Konzentration überschätzen, wenn der eingesetzte Farbstoff unspezifisch auch an RNA bindet.
f) Einordnung in den Gesamtkontext
Die Studie stellt einen frühen und wichtigen Beitrag zur wissenschaftlichen Diskussion über DNA-Rückstände in mRNA-Impfstoffen dar. Sie hat maßgeblich dazu beigetragen, dass weitere Arbeitsgruppen eigene Untersuchungen durchführten. Die Befunde sind als Ausgangspunkt für weiterführende Untersuchungen zu verstehen, nicht als abschließende Bewertung.
🧬 🧬 🧬
4.3.2. Speicher et al. (2023)
DNA fragments detected in monovalent and bivalent Pfizer/BioNTech and Moderna modRNA COVID-19 vaccines from Ontario, Canada: Exploratory dose response relationship with serious adverse events.
Autoren: David Speicher, Jessica Rose, L. Maria Gutschi, David M. Wiseman, Kevin McKernan
Veröffentlicht: 19. Oktober 2023 (ResearchGate)
Link zur Studie: https://osf.io/preprints/osf/mjc97_v1a) Kontext und Ziel der Studie
Die Studie von David J. Speicher et al. baut inhaltlich auf den zuvor veröffentlichten Analysen von Kevin McKernan auf und verfolgt einen erweiterten Ansatz.
Neben dem Nachweis und der Quantifizierung von DNA-Rückständen in weiteren Impfstoffchargen adressiert sie zwei zusätzliche Fragestellungen:
- Lokalisierung der DNA: Liegt die nachgewiesene DNA innerhalb der Lipid-Nanopartikel (LNPs) oder frei in der Lösung vor?
- Explorative Korrelation: Besteht ein statistischer Zusammenhang zwischen gemessenen DNA-Mengen und gemeldeten unerwünschten Ereignissen auf Chargenebene?
Die Studie gliedert sich damit in zwei inhaltlich verschiedene Teile:
- analytischer Teil: Nachweis und Charakterisierung von DNA in Impfstoffproben
- explorativer Teil: Untersuchung einer möglichen statistischen Beziehung zu Nebenwirkungsdaten
Beide Teile erfordern eine unterschiedliche methodische Bewertung.
b) Materialien und Methoden
Impfstoffchargen
Untersucht wurden 27 Fläschchen aus 12 Chargen: 8 abgelaufene, ungeöffnete Pfizer-BNT162b2-Fläschchen und 16 abgelaufene, ungeöffnete Moderna-Spikevax-Fläschchen, bezogen aus verschiedenen Apotheken in Ontario, Kanada. Alle Fläschchen waren originalversiegelt mit intakten Schutzkappen sowie aufgedruckten Chargennummern und Verfallsdaten. Zusätzlich wurden drei noch gültige Restmengen einer Moderna-XBB.1.5-Charge einbezogen. Lagerung und Transport erfolgten unter dokumentierter Kühlung. Als Negativkontrolle diente ein steriles, ungeöffnetes Injektionsfläschchen eines nicht verwandten Arzneimittels (TriMix).Die Autoren kombinieren mehrere analytische und statistische Verfahren.
Aufschluss der Lipid-Nanopartikel (LNPs)
Um die Lipid-Nanopartikel (LNPs) aufzubrechen, wurde die Probe 8 Minuten lang auf 95°C erhitzt, gefolgt von einer Abkühlung auf 4°C für fünf Minuten. Dieser thermische Schritt dient der Freisetzung der enthaltenen Nukleinsäuren.Quantifizierung mittels qPCR
Die DNA-Konzentrationen wurden mittels qPCR über den Cq-Wert (Quantifizierungszyklus) bestimmt und in Nanogramm pro Dosis umgerechnet. Die Autoren übernahmen die Primer aus der McKernan-Studie: Spike-Sonde und Ori-Sonde, die auf Sequenzen abzielen, die beiden Impfstoffen (Pfizer, Moderna) gemeinsam sind. Ergänzend wurde ein SV40-Enhancer-Assay eingesetzt, der speziell auf die im Pfizer-Plasmid identifizierte SV40-Sequenz ausgelegt ist.Quantifizierung mittels Qubit-Fluorometrie
Zur Gesamtquantifizierung der DNA wurde Qubit-Fluorometrie mit dem DNA-spezifischen Farbstoff AccuGreen eingesetzt. Die Fluoreszenzintensität ist proportional zur vorhandenen DNA-Menge und liefert eine sequenzunabhängige, jedoch struktursensitive Gesamtschätzung (abhängig von Farbstoffspezifität und Probenzusammensetzung).Größenprofil der Rest-DNA mittels ONT-Sequenzierung
Die Größenverteilung der DNA-Fragmente in einer Pfizer-Probe wurde mittels Oxford-Nanopore-Sequenzierung bestimmt. Dabei ist zu beachten, dass der verwendete Bibliotheksaufbau Fragmente kleiner als etwa 150 bp (Basenpaare) systematisch entfernt. Die erhaltene Längenverteilung ist daher in Richtung längerer Fragmente verzerrt und bildet den tatsächlichen Anteil kurzer Fragmente nicht vollständig ab.DNA innerhalb oder außerhalb der LNPs – Nukleaseprotektionstest
Um zu prüfen, ob die DNA-Rückstände im Inneren der LNPs vorliegen oder frei außerhalb, wurde das Enzym DNase I-XT direkt zum unveränderten Impfstoff gegeben – also zu intakten, noch nicht aufgeschlossenen LNPs. DNase I-XT baut zugängliche DNA ab. Eine anschließende qPCR-Messung erlaubte den Vergleich der DNA-Menge vor und nach der Enzymbehandlung.Auswertung der VAERS-Datenbank
Für jede untersuchte Charge wurde aus der VAERS-Datenbank ein Serious Adverse Event Reporting Ratio (SRR) berechnet – das Verhältnis schwerwiegender zu allen gemeldeten Nebenwirkungen. Dieser Wert wurde der gemessenen DNA-Konzentration der jeweiligen Charge gegenübergestellt.c) Zentrale Ergebnisse
Quantitative Befunde und Methodenvergleich
Die Gegenüberstellung der Ergebnisse der qPCR und Qubit-Fluorometrie zeigen, dass die gemessenen Daten stark von dem verwendeten Messverfahren abhängen.Ermittlung der DNA-Konzentrationen für Pfizer
qPCR Qubit-Fluorometrie Ori-Sonde: 0,28 – 4,27 ng/Dosis
Spike-Sonde: 0,22 – 2,43 ng/DosisGesamt-DNA: 1.896 – 3.720 ng/Dosis Der SV40-Promotor-Enhancer-Ori wurde nur in den Pfizer-Fläschchen mit Cq-Werten zwischen 16,64 und 22,59 nachgewiesen.
Ermittlung der DNA-Konzentrationen für Moderna
qPCR Qubit-Fluorometrie Ori-Sonde: 0,01 – 0,34 ng/Dosis
Spike-Sonde: 0,25 – 0,78 ng/DosisGesamt-DNA: 3.270 – 5.100 ng/Dosis Bezogen auf den FDA-Grenzwert von 10 ng DNA pro Dosis liegen die qPCR-Werte unterhalb des Grenzwerts, während die fluorometrischen Werte diesen um das 188- bis 509-fache überschreiten.
Die Autoren weisen darauf hin, dass qPCR-Verfahren DNA-Fragmente unterhalb der Amplikon-Länge (105–114 bp) nicht erfassen können und die Gesamtmenge damit systematisch unterschätzen.
Darüber hinaus zeigten die Messungen eine Variabilität der DNA-Mengen zwischen verschiedenen Chargen desselben Impfstoffs – ein Hinweis auf mögliche Schwankungen im Reinigungsprozess.
DNA liegt im Inneren der LNPs
Nach DNase I-XT-Behandlung des intakten Pfizer-Impfstoffs zeigte die anschließende qPCR-Messung kaum eine Veränderung der DNA-Menge. Da das Enzym nur zugängliche DNA abbaut, legt dieses Ergebnis nahe, dass der überwiegende Teil der DNA-Rückstände im Inneren der LNPs vorliegt und damit vor dem Enzymangriff geschützt ist.Größenprofil der Rest-DNA
Die ONT-Sequenzierung einer Pfizer-Probe ergab eine mittlere Fragmentlänge von 214 bp. Einzelne Fragmente erreichten Längen von über 1.000 bp; das längste nachgewiesene Fragment umfasste etwa 3.200 bp und entsprach damit annähernd der halben Plasmidlänge von 7.810 bp.Da der Bibliotheksaufbau Fragmente unter 150 bp systematisch ausschließt, ist davon auszugehen, dass der tatsächliche Anteil kurzer Fragmente in der Probe höher liegt als in den Sequenzierungsdaten abgebildet.
Explorative VAERS-Korrelation mit den gemessenen DNA-Mengen
Die Autoren beobachteten eine positive Korrelation zwischen den gemessenen DNA-Konzentrationen auf Chargenebene und dem SRR-Wert der jeweiligen Charge. Chargen mit höheren DNA-Mengen tendierten zu höheren Anteilen schwerwiegender Meldungen in der VAERS-Datenbank.d) Interpretation durch die Autoren
Methodenabhängigkeit von Rest-DNA-Bewertungen
Die Autoren berichten zwei völlig unterschiedliche Messergebnisse für dieselben Impfstoffe:Methode Ergebnis: Rest-DNA pro Dosis
(Grenzwert 10 ng/Dosis)Bewertung qPCR UNTER dem FDA-Grenzwert Einhaltung Fluorometrie 188- bis 509-fach ÜBER dem FDA-Grenzwert Überschreitung Das bedeutet: Je nach Messmethode kommt man zu gegensätzlichen Schlussfolgerungen.
Diese Diskrepanz ist ein zentrales Ergebnis der Studie und verdeutlicht die hohe methodische Abhängigkeit der gemessenen DNA-Werte.
Hinweis auf methodische Inkonsistenz
Die Autoren argumentieren, dass die beobachteten Diskrepanzen nicht ausschließlich auf Messungenauigkeiten zurückzuführen sind, sondern auf grundlegend unterschiedliche Messprinzipien.Die EMA verwendet ratiometrische Richtlinien – das bedeutet: Es gibt einen erlaubten Grenzwert für das Verhältnis von DNA zu RNA (wie z. B. 330 ng DNA pro 1 mg RNA).
Allerdings kommen für die Messung von RNA und DNA unterschiedliche Methoden zum Einsatz, die sich in ihrer Spezifität unterscheiden.
Was gemessen wird Verwendete Methode Gemessen werden … RNA
(Hauptbestandteil)Fluorometrie /
UV-SpektrophotometrieGesamtnukleinsäuren (unspezifisch) DNA
(Rückstände)qPCR Spezifische DNA-Sequenzen (selektiv) Verschiedene Quellenbelege dazu:
- „Rapporteur Rolling Review critical assessment report” (Table S.2.6-16. Evolution of BNT162b2 Drug Substance Methods)
- „EU Official Control Authority Batch Release Certificate for Immunological Products” (Table 2. Drug Substance Quality Control Tests)
- „Assessment report EMA/707383/2020”
- „Assessment report EMA/15689/2021”
Die Autoren argumentieren, wenn man:
🔸RNA mit einer unspezifischen Methode misst (Fluorometrie: erfasst auch DNA-Fragmente, Abbauprodukte etc.) und
🔸DNA mit einer spezifischen Methode misst (qPCR: erfasst nur die exakte Zielsequenz),
dann sind die beiden Messwerte methodisch nicht kompatibel.Dadurch sind die berechneten DNA/RNA-Verhältnisse methodisch verzerrt und nicht direkt vergleichbar.
Diese Argumentation bezieht sich auf die Vergleichbarkeit der Messmethoden, nicht notwendigerweise auf die Gültigkeit der jeweiligen Einzelmessung.
Die Autoren plädieren dafür, dass Behörden nicht nur Grenzwerte, sondern auch die verbindlich einzusetzenden Nachweismethoden vorschreiben.DNA innerhalb der LNPs: biologische Konsequenz
Das Vorliegen der DNA im Inneren der LNPs wird von den Autoren als potenziell biologisch relevant eingestuft. Im Unterschied zu freier, nackter DNA – die im Körper rasch abgebaut wird – ist LNP-verpackte DNA vor Nukleasen geschützt und kann effizient in Zellen aufgenommen werden. Die bestehenden regulatorischen Grenzwerte, so die Autoren, wurden für nackte Wirtszell-DNA entwickelt und berücksichtigen diesen Unterschied nicht.Kritik an den regulatorischen Richtlinien
Die Autoren üben grundsätzliche Kritik an den bestehenden Grenzwertkonzepten. Sie bemängeln, dass die Richtlinien auf veralteten Grundlagen beruhen, die Molekülanzahl zugunsten einer gewichtsbasierten Betrachtung vernachlässigen, kumulative Dosierungen (mehrere Dosen innerhalb weniger Monate) nicht berücksichtigen und funktionelle Sequenzen wie Promotoren oder nukleäre Targeting-Signale außer Acht lassen. Insbesondere das Vorhandensein des SV40-Promoter/Enhancers im Pfizer-Plasmid wird als Element hervorgehoben, das den Transport von DNA in den Zellkern begünstigen könnte.Explorative VAERS-Analyse
Die beobachtete Korrelation wird von den Autoren als möglicher Hinweis auf einen Zusammenhang zwischen DNA-Gehalt und Nebenwirkungsprofil interpretiert. Ausdrücklich wird betont, dass es sich um eine explorative Beobachtung handelt, die keine kausalen Schlussfolgerungen erlaubt. Daher plädieren sie für weitere Untersuchungen dahingehend.e) Methodische Einschränkungen
Die Herkunft, Lagerung und Chargencharakterisierung der untersuchten Proben ist insgesamt gut dokumentiert – Kühlkette, Transportbedingungen und Fläschchenzustand sind beschrieben. Der thermische Aufschluss ohne nachfolgende Aufreinigung ist nicht im regulatorischen Sinne validiert; Angaben zur Extraktionseffizienz fehlen.
Alle untersuchten Fläschchen waren zum Zeitpunkt der Analyse bereits abgelaufen. Drei Moderna-XBB.1.5-Fläschchen waren angebrochene Restmengen nach der Patientenversorgung – keine originalversiegelten Proben. Sie unterscheiden sich damit in ihrer Probenqualität von den übrigen Fläschchen und sind gesondert zu bewerten.
Die VAERS-Korrelation ist mit erheblichen Unsicherheiten behaftet. VAERS ist ein passives Meldesystem: Meldungen werden nicht systematisch verifiziert, die Meldebereitschaft variiert zwischen Zeiträumen und Regionen, und eine Korrelation auf Chargenebene erlaubt keine Rückschlüsse auf individuelle Kausalzusammenhänge. Die Aussagekraft dieser Analyse ist damit strukturell begrenzt – unabhängig von der Qualität der analytischen Messungen.
Die ONT-Sequenzierung erfasst aufgrund des Bibliotheksaufbaus nur Fragmente ab etwa 150 bp und bildet den tatsächlichen Kurzfragmentanteil nicht vollständig ab.
f) Einordnung in den Gesamtkontext
Die Studie erweitert die Befunde von McKernan et al. in mehrfacher Hinsicht: Sie untersucht weitere Chargen, liefert einen Nukleaseprotektionstest als direkten Hinweis auf die intrapartikuläre Lage der DNA und thematisiert strukturelle Schwächen der regulatorischen Methodik. Der analytische Teil stellt einen sachlichen Beitrag zur Debatte dar.
Der explorative Teil – die Verknüpfung mit VAERS-Daten – ist methodisch anspruchsvoll und mit den genannten Einschränkungen des Meldesystems behaftet. Die Autoren selbst charakterisieren ihn als hypothesengenerierend, nicht als beweisend.
Die Autoren weisen auf mehrere Einschränkungen ihrer Studie hin und fordern eine Replikation ihrer Ergebnisse unter forensischen Bedingungen.
Insgesamt unterstreicht die Studie die Notwendigkeit standardisierter und methodenübergreifend vergleichbarer Analysen zur Bewertung von DNA-Rückständen in mRNA-basierten Arzneimitteln.
🧬 🧬 🧬
4.3.3. König & Kirchner (2024)
Methodological Considerations Regarding the Quantification of DNA Impurities in the COVID-19 mRNA Vaccine Comirnaty®
Autoren: Brigitte König, Jürgen O. Kirchner
Veröffentlicht: 8. Mai 2024 (Peer-reviewed, PubMed)
Link zur Studie: https://pubmed.ncbi.nlm.nih.gov/38804335/a) Kontext und Ziel der Studie
Die Arbeit von Brigitte König und Johannes O. Kirchner unterscheidet sich in ihrer Zielsetzung grundlegend von den zuvor dargestellten Studien. Während frühere Arbeiten primär den Nachweis und die Quantifizierung von DNA-Rückständen in mRNA-Impfstoffen zum Ziel hatten, liegt der Fokus dieser Studie auf einer methodischen Bewertung der eingesetzten Nachweisverfahren.
Die zentrale Frage lautet: Ist die im Zulassungsverfahren verwendete qPCR-basierte DNA-Quantifizierung als Chargenprüfung und damit als Qualitätssicherungsinstrument geeignet?
Um diese Frage zu beantworten, untersuchen die Autoren:
- welche analytischen Methoden zur Quantifizierung von DNA-Rückständen eingesetzt wurden,
- welche methodischen Grenzen diese Verfahren aufweisen,
- und inwiefern unterschiedliche Methoden zu abweichenden Ergebnissen führen können.
Die Studie wurde in einem formalen Peer-Review-Verfahren begutachtet und in einer wissenschaftlichen Fachzeitschrift veröffentlicht.
b) Materialien und Methoden
Impfstoffchargen
Das Labor erhielt mehrere originalversiegelte Fläschchen unterschiedlicher Chargen des mRNA-Impfstoffs Comirnaty® (BNT162b2), bereitgestellt durch offizielle Impfzentren. Lagerung und Transport erfolgten unter dokumentierter Einhaltung der Kühlkette (2–8 °C). Vier Chargen waren zum Zeitpunkt der Analyse bereits abgelaufen, drei wiesen eine Resthaltbarkeit von 11 bis 13 Monaten auf.Die Autoren kombinieren zwei Ansätze: eine methodenkritische Analyse bestehender Verfahren und eigene Labormessungen.
Methodenkritische Analyse
Die Autoren untersuchen die im EMA-Rolling-Review-Dokument vom November 2020 beschriebene qPCR-Methode (einschließlich des dortigen Mustervorbereitungsschemas) und identifizieren drei strukturelle Probleme.Erstens erfasst die qPCR nur eine 69 bp lange Zielsequenz – den T7-Promotor – innerhalb eines 7.824 bp langen Plasmids. Die verbleibenden 99 % der Plasmid-DNA sowie etwaige genomische Bakterien-DNA bleiben damit unerfasst. Die Autoren weisen darauf hin, dass dieses Problem prinzipiell für jeden qPCR-Ansatz gilt, der nur einzelne Zielsequenzen adressiert – unabhängig davon, welche Sequenz gewählt wird.
Zweitens ist die verwendete Standardkurve methodisch problematisch. Zur Quantifizierung wird eine Standardkurve mit unbehandelter, intakter Plasmid-DNA erstellt. Die DNA in der Impfstoffprobe hat jedoch im Herstellungsprozess mehrere Behandlungsschritte – insbesondere einen DNase-Verdau – durchlaufen und ist damit fragmentiert. Da fragmentierte DNA schlechter amplifiziert wird als intakte, erscheint die gemessene Konzentration systematisch niedriger als die tatsächlich vorhandene. Ein korrekter Vergleich würde eine Standardkurve erfordern, die mit DNA erstellt wurde, die denselben Behandlungsschritten unterzogen wurde wie die Probe.
Drittens stellt sich die Frage der Proportionalität:
- Gibt es sequenzabhängige Unterschiede in der Abbaurate?
- Wird die qPCR-Zielsequenz durch den DNase-Verdau proportional zum Rest des Plasmids abgebaut?
- Werden bestimmte Sequenzen der linearisierten Plasmide durch DNase häufiger oder deutlich seltener abgebaut als andere?
Wenn die Proportionalität nicht gegeben ist, ist jede Extrapolation von der Zielsequenz auf die Gesamt-DNA fehlerbehaftet. Die Europäische Pharmakopöe (Ph. Eur. 2.6.35) bestätigt diese Einschränkung: Sie nennt qPCR zwar als Methode der Wahl für den Nachweis spezifischer Sequenzen, weist aber ausdrücklich darauf hin, dass für die Bestimmung der Gesamt-DNA andere oder ergänzende Verfahren erforderlich sein können.
Überlegungen zur regulatorischen Praxis
Die Autoren weisen auf eine Inkonsistenz in der behördlichen Praxis hin: Laut einem EDQM-Protokoll wird DNA nur auf Wirkstoffebene gemessen, nicht im fertigen Impfstoff. Als Begründung wird angegeben, dass Lipid-Nanopartikel die DNA-Messung stören könnten. Gleichzeitig wird die RNA-Konzentration im fertigen Impfstoff mittels Fluorometrie gemessen – nach Aufschluss der LNPs mit Triton X-100. Da RNA und DNA als Nukleinsäuren vergleichbare analytische Eigenschaften aufweisen und beide von LNPs umschlossen sein können, erscheint die unterschiedliche Behandlung erklärungsbedürftig. Ob hierfür technische oder regulatorische Gründe vorliegen, die in öffentlichen Dokumenten nicht vollständig dargelegt sind, lässt sich anhand der verfügbaren Quellen nicht abschließend beurteilen.Eigene Messungen mittels Qubit-Fluorometrie
Um die praktische Eignung eines alternativen Verfahrens zu prüfen, führten die Autoren eigene Messungen an sieben Chargen Comirnaty® durch. Dabei wurden die Proben jeweils vor und nach Zugabe des Detergens Triton X-100 gemessen, das die LNPs aufbricht und den eingeschlossenen Inhalt freisetzt. Als Messsystem wurde Qubit-Fluorometrie mit dem DNA-spezifischen Farbstoff PicoGreen eingesetzt.c) Zentrale Ergebnisse
Qubit-Messungen vor und nach LNP-Aufschluss
Ohne Triton X-100 wurden nur geringe DNA-Mengen gemessen. Nach Zugabe von Triton X-100 stiegen die gemessenen Werte drastisch an – auf das 360- bis 534-fache des regulatorischen Grenzwerts von 10 ng pro Dosis, entsprechend 3.600 bis 5.340 ng DNA pro Dosis.In den vier abgelaufenen Chargen waren bereits ohne Triton X-100 zwischen 16 und 81 % der Gesamt-DNA messbar – ein Hinweis auf einen teilweisen Zerfall der LNPs über die Lagerungszeit. In den drei frischen Chargen war die DNA zu 93 bis 97 % erst nach dem LNP-Aufschluss nachweisbar, was auf weitgehend intakte Partikel hindeutet.
RNA-Messungen als Kontrolle
Die parallel durchgeführten RNA-Messungen dienten als methodische Kontrolle und zeigten plausible Werte, was die Autoren als Beleg für die grundsätzliche Eignung des Qubit-Systems im Kontext dieser Probenmatrix werten.d) Interpretation durch die Autoren
Kritik an der qPCR als alleinige Referenzmethode
Die Autoren schlussfolgern, dass die behördlich vorgeschriebene qPCR-basierte Methode strukturell ungeeignet ist, die Gesamt-DNA im fertigen Impfstoff zuverlässig zu quantifizieren. Die gemessenen DNA-Gehalte überschreiten den regulatorischen Grenzwert deutlich – blieben aber unentdeckt, weil die eingesetzte Methode nur einen kleinen Ausschnitt der vorhandenen DNA erfasst und zudem auf Wirkstoffebene statt im Endprodukt angewendet wird.Forderung nach methodischer Anpassung
Die Autoren fordern daher eine fluoreszenzspektrometrische Messung der Gesamt-DNA im fertigen Impfstoff als Bestandteil der amtlichen Chargenprüfung – analog zur bereits etablierten RNA-Messung.DNA innerhalb der LNPs: biologische Konsequenz
Darüber hinaus plädieren sie für eine Neubewertung der Risiken LNP-verpackter DNA, insbesondere hinsichtlich einer möglichen Integration in das menschliche Erbgut.e) Methodische Einschränkungen
Geringe Stichprobengröße
Die Studie basiert auf einer begrenzten Stichprobe von sieben Chargen. Die Proben wurden als originalversiegelte Fläschchen von offiziellen Impfzentren bereitgestellt; Lagerung und Transport erfolgten unter dokumentierter Kühlkettenkontrolle. Die Provenienzdokumentation ist damit vergleichsweise gut belegt.Einfluss des Ablaufdatums auf die LNP-Integrität
Vier der sieben Chargen waren zum Zeitpunkt der Analyse bereits abgelaufen. Abgelaufene Proben können chemisch verändert sein – insbesondere hinsichtlich der LNP-Integrität. Die Autoren berücksichtigen diesen Umstand ausdrücklich und werten die erhöhte DNA-Messbarkeit ohne Triton X-100 in abgelaufenen Chargen als Hinweis auf einen teilweisen LNP-Zerfall über die Lagerungszeit.Keine RNase-Behandlung vor Fluorometrie (Qubit)
Qubit misst mit DNA-spezifischen Farbstoffen, die aber auch mit RNA (der Hauptbestandteil des Impfstoffs) interferieren können. Ohne vorherige RNase-Verdauung wird die gemessene „DNA-Menge“ sehr wahrscheinlich überschätzt.Fehlende formale Validierung
Die Qubit-Methode wurde von den Autoren nicht formal nach regulatorischen Standards validiert. Eine solche Validierung – wie sie die ICH-Leitlinie Q2(R2) für analytische Methoden im Zulassungskontext vorschreibt – umfasst den systematischen Nachweis von Spezifität, Nachweisgrenze, Linearität, Präzision, Richtigkeit, Robustheit und Matrixeffekten. Die Autoren begründen die Eignung des Verfahrens argumentativ: durch den Vergleich von Messungen vor und nach LNP-Aufschluss sowie durch plausible RNA-Kontrollwerte. Das ist für eine wissenschaftliche Publikation, die einen methodischen Punkt demonstrieren will, ein üblicher Rahmen. Dieser Hinweis ist keine Kritik an der Sorgfalt der Autoren, sondern eine sachliche Einordnung des Publikationskontexts.Fehlende Validierung der Extraktions-Effizienz
Bei Extraktionen wird nicht immer gezeigt, dass die Methode quantitativ alle DNA (besonders kleine Fragmente und verkapselte DNA) vollständig freisetzt und misst. Das kann zu Über- oder Unter-Schätzungen führen.Verfügbarkeit behördlicher Prüfmethoden
Die methodenkritische Analyse stützt sich hinsichtlich der behördlichen qPCR-Methode auf ein Rolling-Review-Dokument aus dem frühen Zulassungsverfahren. Ob die dort beschriebene Methode identisch mit der final zugelassenen Chargenprüfmethode ist, lässt sich anhand öffentlich zugänglicher Quellen nicht abschließend beurteilen.f) Einordnung in den Gesamtkontext
König & Kirchner leisten einen methodisch eigenständigen Beitrag zur Debatte. Im Unterschied zu McKernan und Speicher steht nicht die Frage im Vordergrund, wie viel DNA vorhanden ist, sondern ob die eingesetzten Verfahren überhaupt geeignet sind, diese Frage zuverlässig zu beantworten.
Die Studie leistet damit einen wichtigen Beitrag zur Debatte, indem sie:
- die methodische Abhängigkeit der Messergebnisse herausarbeitet
- die Grenzen etablierter Standardverfahren (insbesondere qPCR) aufzeigt
- und die Notwendigkeit methodenübergreifender Validierung betont
Die Arbeit verdeutlicht, dass Unterschiede zwischen Studien nicht zwangsläufig widersprüchliche oder falsche Ergebnisse widerspiegeln, sondern häufig durch:
- unterschiedliche Nachweisverfahren
- verschiedene Zielsequenzen
- und variierende Probenaufbereitung
erklärt werden können. Damit trägt die Arbeit wesentlich zum Verständnis bei, warum die Quantifizierung von DNA-Rückständen methodisch anspruchsvoll ist und warum Ergebnisse aus verschiedenen Studien nur eingeschränkt direkt vergleichbar sind.
🧬 🧬 🧬
4.3.4. Didier Raoult (2024)
Confirmation of the presence of vaccine DNA in the Pfizer anti-COVID-19 vaccine
Autoren: Didier Raoult
Veröffentlicht: 12. November 2024 (Preprint, HAL Open Science)
Link zur Studie: https://hal.science/hal-04778576v1a) Kontext und Ziel der Studie
Die Arbeit von Didier Raoult reiht sich in die Reihe unabhängiger Untersuchungen ein, die das Vorhandensein von DNA in mRNA-basierten COVID-19-Impfstoffen analysieren.
Ziel der Studie ist es, den Nachweis von DNA-Komponenten im Pfizer/BioNTech-Impfstoff experimentell zu bestätigen und die Befunde früherer Arbeitsgruppen durch eine unabhängige Analyse zu überprüfen. Die Studie versteht sich damit primär als Replikationsarbeit.
b) Materialien und Methoden
Impfstoffchargen
In der Studie wurde ausschließlich der mRNA-Impfstoff Comirnaty® (BNT162b2) von BioNTech/Pfizer untersucht. Die Analyse basierte auf einer begrenzten Anzahl von Proben; in den verfügbaren Angaben wird keine systematische Chargenübersicht berichtet.Die Studie verwendet klassische molekularbiologische Verfahren zur Analyse von Nukleinsäuren.
Aufschluss der Lipid-Nanopartikel (LNPs)
Die LNPs wurden mit dem Detergens Triton X-100 aufgeschlossen.Quantifizierung mittels Qubit-Fluorometrie
An einer Charge (Nr. GJ7184) wurde eine DNA-Quantifizierung mittels kommerziellem Qubit-Fluorometer durchgeführt, jeweils vor und nach dem LNP-Aufschluss mit Triton X-100.Sequenzierung mittels Illumina
Zwei Chargen (FP8191 und FP9359) wurden mittels Illumina-Sequenzierung analysiert. Dabei wurde bewusst auf eine Reverse Transkription verzichtet – die RNA wurde also nicht in DNA umgeschrieben. Damit ist sichergestellt, dass ausschließlich die in der Probe ursprünglich vorhandene DNA sequenziert wurde und nicht eine aus der mRNA erzeugte Kopie.Die erhaltenen Reads (gelesenen Sequenzen) wurden mit der bekannten DNA-Plasmidsequenz (GenBank OR134577.1) verglichen. Für die bioinformatische Auswertung wurden nur Sequenzabschnitte ab 200 bp Länge und mit mehr als 90% Übereinstimmung zur Referenz berücksichtigt.
Als Kontrollversuch wurden die Proben vor der Sequenzierung mit DNase behandelt und anschließend erneut sequenziert, um zu prüfen, ob das Signal tatsächlich auf DNA zurückzuführen ist.
c) Zentrale Ergebnisse
Quantifizierung mittels Qubit-Fluorometrie
Ohne Triton X-100 wurden durchschnittlich 216 ng DNA pro Dosis gemessen. Nach LNP-Aufschluss mit Triton X-100 stieg der Wert auf durchschnittlich 5.160 ng pro Dosis – ein Anstieg um das ca. 24-Fache. Bezogen auf den angegebenen RNA-Gehalt von 30 µg pro Dosis entspricht dies rechnerisch einem DNA-Anteil von etwa 17 %.Sequenzierung mittels Illumina
Mittels Illumina-Sequenzierung wurden über 140.000 Reads mit einer Länge von mehr als 200 Basenpaaren generiert. Aus diesen Daten konnte die vollständige Plasmidsequenz von 7.824 bp rekonstruiert werden. Dies bedeutet, dass die vorhandenen DNA-Fragmente in ihrer Gesamtheit die vollständige Sequenz abdecken.Die Sequenzierungstiefe war mit mehr als 4.000-facher Abdeckung außergewöhnlich hoch – das bedeutet, jede Position des Plasmids wurde im Durchschnitt von mehr als 4.000 unabhängigen Reads abgedeckt. Die Sequenzdaten beider Chargen wurden in der GenBank hinterlegt (PP544445 und PP544446).
63 bis 97 % der sequenzierten DNA ließen sich dem Impfstoff-Plasmid zuordnen. Die Herkunft der verbleibenden 3 bis 37 % wurde nicht weiter untersucht.
DNase-Kontrollversuch
Nach DNase-Behandlung wurden nahezu keine Reads mehr detektiert (1–2 Reads). Der nahezu vollständige Signalverlust nach DNase-Behandlung spricht dafür, dass das Sequenzsignal tatsächlich von frei zugänglicher oder nach Aufschluss freigesetzter DNA stammt.d) Interpretation durch die Autoren
Reproduzierbarkeit
Der Autor interpretiert seine Ergebnisse als experimentelle Bestätigung der Befunde früherer Studien. Mit mehreren methodischen Ansätzen und in verschiedenen Chargen sei das reichliche Vorhandensein von Plasmid-DNA im Pfizer/BioNTech-Impfstoff nachweisbar. Die Übereinstimmung mit früheren Berichten stütze die Reproduzierbarkeit entsprechender Befunde.DNA verpackt in kationische Lipide: biologische Konsequenz
Hinsichtlich der biologischen Konsequenz weist der Autor darauf hin, dass LNP-verpackte Plasmid-DNA nach Zellaufnahme theoretisch in das Genom integrieren könnte. Das tatsächliche Risiko wird vom Autor selbst als extrem gering eingeschätzt; weitere Untersuchungen seien dennoch gerechtfertigt.e) Methodische Einschränkungen
Herkunft und Lagerung der Proben
Angaben zur Herkunft, Lagerungsgeschichte und zum Zustand der untersuchten Fläschchen fehlen weitgehend. Es ist nicht dokumentiert, ob die Proben originalverpackt, korrekt gekühlt und ungeöffnet waren.Fehlende Methodendetails
- keine Angabe zur RNase-Behandlung zur Entfernung von RNA vor der DNA-Messung
- keine Angabe, welches Qubit-Kit eingesetzt wurde (High Sensitivity oder Broad Range)
- keine Angabe, wie hoch die Extraktionseffizienz für LNP-verpackte DNA war
- keine Spezifizierung der DNase (nur „TURBO DNA-free kit“ genannt, nicht welcher Enzymtyp)
Geringe Stichprobengröße
Die Studie basiert auf einer begrenzten Stichprobe von drei Chargen.Sequenzierung und Quantifizierung
Die Illumina-Sequenzierung belegt zuverlässig die Anwesenheit und Vollständigkeit der Plasmidsequenz, ist aber kein geeignetes Verfahren zur absoluten Quantifizierung. Eine hohe Sequenzierungstiefe kann auch dann entstehen, wenn wenige Moleküle mit hoher Kopienzahl vorliegen – sie spiegelt nicht notwendigerweise die tatsächliche Masse der DNA wider.f) Einordnung in den Gesamtkontext
Die Studie ergänzt die bestehende Datenlage durch einen weiteren unabhängigen Nachweis von Plasmid-DNA in mRNA-Impfstoffen. Ihr Beitrag liegt primär in der Reproduzierbarkeit des qualitativen Befunds: Ein weiteres Labor, mit anderen Methoden und anderen Chargen, kommt zu vergleichbaren Ergebnissen.
🧬 🧬 🧬
4.3.5. Kämmerer et al. (2024)
BioNTech RNA-Based COVID-19 Injections Contain Large Amounts Of Residual DNA Including An SV40 Promoter/Enhancer Sequence
Autoren: Ulrike Kämmerer, Verena Schulz, Klaus Steger
Veröffentlicht: 3. Dezember 2024 (Peer Reviewed, Cureus/Public Health Policy Journal)
Link zur Studie: https://publichealthpolicyjournal.com/biontech-rna-based-covid-19-injections-contain-large-amounts-of-residual-dna-including-an-sv40-promoter-enhancer-sequence/a) Kontext und Ziel der Studie
Die Arbeit untersucht das Vorhandensein von DNA-Komponenten in mRNA-basierten COVID-19-Impfstoffen mit besonderem Fokus auf regulatorische Sequenzelemente. Vor dem Hintergrund vorheriger Analysen widmet sich die Studie drei zentralen Fragen:
- Lässt sich die hohe Menge an DNA-Rückständen in BioNTech-Chargen mit unterschiedlichen Methoden bestätigen?
- Können vorhandene DNA-Fragmente gemeinsam mit der mRNA in menschliche Zellen eingeschleust werden (Transfektion)?
- Führt dies zu einer anhaltender Spike-Protein-Produktion?
b) Materialien und Methoden
Impfstoffchargen
Für die Untersuchungen wurden vier originale, ungeöffnete BNT162b2-Chargen verwendet:- FD7958, FE6975 und EX8679 (monovalent, Wuhan-Stamm; Verfallsdatum Oktober/August 2021)
- HD9869 (bivalent, Wuhan/Omicron XBB1.5; Verfallsdatum Oktober 2024)
Als Positivkontrolle diente die Charge GH9715, deren DNA-Kontamination in früheren Studien bereits nachgewiesen worden war.
Alle Ampullen wurden von einer Apotheke im gekühlten Zustand bezogen, waren originalversiegelt und wurden während Transport und Lagerung durchgehend gekühlt. Die Provenienzdokumentation ist damit vergleichsweise gut belegt.
Quantifizierung
Die Lipid-Nanopartikel (LNPs) wurden mit Triton X-100 aufgeschlossen.
Die RNA-Quantifizierung erfolgte mit dem Qubit RNA High Sensitivity Assay.
Für die DNA-Quantifizierung wurden drei verschiedene fluorometrische dsDNA-Assays eingesetzt:- Qubit dsDNA High Sensitivity Assay
- Quanti-iT PicoGreen dsDNA Assay
- AccuBlue High Sensitivity dsDNA Assay
Die Untersuchung mittels Fluoreszenz-Plate-Reader erfolgte vor und nach einer RNase A-Behandlung (einem RNA-abbauenden Enzym).
Der Einsatz von drei unabhängigen Assays ist methodisch als orthogonaler Ansatz zu werten: Stimmen alle drei überein, erhöht das die Verlässlichkeit des Ergebnisses erheblich.
Zelllinienexperimente und ELISA
Um zu prüfen, ob die Impfstoffe menschliche Zellen transfizieren und zu einer Spike-Protein-Produktion führen, wurden HEK293-Zellen eingesetzt – eine immortalisierte humane Zelllinie mit hoher Transfektionseffizienz.Die Zellen wurden unter standardisierten Bedingungen kultiviert. Sie stammten aus zertifizierten Originalbeständen und wurden regelmäßig negativ auf Mykoplasmen getestet.
Für die Transfektionsexperimente wurden die Zellen in 12-Well-Platten ausgesät. Die Zelldichte wurde so gewählt, dass nach einer kurzen Anwachszeit etwa 80 % der Bodenfläche bedeckt waren. Jedes Well wurde mit 1/12 einer klinischen Dosis (entsprechend 25 µl) des jeweiligen Impfstoffs transfiziert. Ein unmittelbarer Mediumwechsel oder Waschschritt nach der Transfektion ist nicht dokumentiert.
Die Zellen sowie das Kulturmedium wurden zu den Zeitpunkten Tag 1, Tag 3, Tag 5 und Tag 7 nach Transfektion geerntet. Untransfektierte Zellen am Tag 7 dienten als Negativkontrolle.
Zur Proteinanalyse wurden die Zellen gewaschen, um Rückstände des Kulturmediums zu entfernen. Durch Zugabe eines Lysispuffers wurden die Zellen aufgeschlossen, sodass der Zellinhalt freigesetzt wurde. Der Gehalt an SARS-CoV-2-Spike-Protein wurde sowohl im Überstand des Zelllysats als auch im Zellkulturmedium mit einem hochsensitiven, kommerziellen ELISA-Kit quantitativ bestimmt.
Immunhistochemie
Um das Spike-Protein innerhalb der Zellen optisch nachzuweisen, wurde eine Immunhistochemie durchgeführt. Dazu wurden HEK293-Zellen in 24-Well-Platten ausgesät (60 % Zelldichte) und mit 12,5 µl einer klinischen Dosis pro Well transfiziert (200 µl Medium). Nach 4-stündiger Inkubation wurden die Zellen zweimal mit PBS gewaschen und mit 500 µl frischem Medium versetzt.24 Stunden nach Transfektionsbeginn wurden die Zellen geerntet, fixiert, in 2%iger Agarose eingebettet und nach Entwässerung in Paraffinblöcke überführt. Von diesen Blöcken wurden hauchdünne Schnitte angefertigt.
Die Zellschnitte wurden mit einem spezifischen primären Antikörper gegen das SARS-CoV-2-Spike-Protein (S1-Untereinheit) behandelt, der nur an das Spike-Protein bindet. Ein sekundärer Antikörper, der an den ersten bindet und eine Farbreaktion auslöst, wurde hinzugefügt. Das Spike-Protein erschien unter dem Mikroskop grün, während der Zellkern zur besseren Orientierung mit Hämatoxylin blau gegengefärbt wurde.
PCR und Agarosegelelektrophorese
Mittels klassischer PCR wurde geprüft, ob die Impfstoff-DNA in menschliche Zellen eindringt. Für jeden Zielbereich des Plasmids wurde ein eigenes Primer-Paar eingesetzt – für Spike-Sequenzen, den ORI-Replikationsursprung, die Neomycin-Resistenzkassette und den SV40-Promoter/Enhancer. Jeder PCR-Ansatz enthält genau ein Primer-Paar und erzeugt damit ein definiertes Amplikon.Die PCR-Produkte wurden mittels Agarosegelelektrophorese aufgetrennt und durch Vergleich mit einem DNA-Marker (GeneRuler) in ihrer Größe bestimmt. HEK293-Zellen wurden mit einer halben Impfstoffdosis (150 μl) behandelt; nach sechs Stunden wurde die DNA isoliert und analysiert. Untransfektierte Zellen dienten als Negativkontrolle.
Extrazelluläre Vesikel und Massenspektrometrie
Hintergrund: Zellen stoßen ständig Extrazelluläre Vesikel (kleine Membranbläschen) ab, die wie kleine Pakete Botenstoffe enthalten. Diese Vesikel können zu anderen Zellen reisen.Um zu prüfen, ob transfizierte Zellen Impfstoffbestandteile über extrazelluläre Vesikel (EVs) abgeben, wurden EVs aus dem Zellkulturmedium isoliert und mittels Massenspektrometrie (TimsTOF-HT) analysiert. Die Identifizierung der Proteine erfolgte durch Abgleich mit einer Datenbank, die menschliche Proteine sowie Impfstoffvektor- und Spike-Sequenzen enthielt.
c) Zentrale Ergebnisse
Transfektion und Spike-Protein-Produktion
Alle vier untersuchten Chargen führten unter den gewählten Zellkulturbedingungen zu einer effizienten Aufnahme der mRNA in HEK293-Zellen und zur nachweisbaren Spike-Protein-Produktion.Der Anteil Spike-positiver Zellen betrug 74,6 bis 90,5 %; untransfektierte Zellen zeigten kein Signal. Transfizierte Zellen zeigten zudem morphologische Veränderungen – intrazelluläre Vakuolen und Ablösung von der Wachstumsfläche – als Zeichen eines zytopathischen Effekts.
Die Spike-Protein-Produktion war bereits an Tag 1 nachweisbar, stieg bis Tag 5 an und blieb bis Tag 7 nachweisbar. Die Autoren interpretieren dies als Hinweis auf eine länger anhaltende Expression über den erwarteten Zeitraum hinaus.
Spike-Protein-Freisetzung ins Zellkulturmedium
Im Zellkulturmedium war Spike-Protein für die monovalenten Chargen nachweisbar; die bivalente Charge lag unterhalb der Nachweisgrenze.Spike-Protein-Freisetzung über extrazelluläre Vesikel
Die Massenspektrometrie zeigte, dass das Spike-Protein hauptsächlich in extrazellulären Vesikeln nachweisbar war (14 spectral counts), während im löslichen Medium nur minimale Mengen vorlagen (2 spectral counts). Das Zelllysat wies die höchste intrazelluläre Konzentration auf (60 spectral counts).RNA-Gehalt
Die gemessenen RNA-Werte entsprachen der Herstellerangabe von 30 µg pro klinischer Dosis.DNA-Gehalt
Ohne LNP-Aufschluss wurden 1,6- bis 6,7-fach niedrigere DNA-Werte gemessen als nach Triton-X-100-Behandlung – ein weiterer Beleg dafür, dass die DNA überwiegend im Inneren der LNPs vorliegt.Ohne RNase-Behandlung lagen die gemessenen DNA-Werte bei 1.326 bis 4.225 ng pro Dosis. Nach RNase-A-Behandlung sanken die Werte auf 32,71 bis 43,38 ng pro Dosis. Die Autoren führen die hohen Ausgangswerte auf ein Hintergrundsignal zurück, das durch strukturierte RNA-Bereiche entsteht: dsDNA-spezifische Farbstoffe binden bevorzugt an doppelsträngige DNA, können aber in Anwesenheit großer RNA-Mengen ein erhöhtes Signal erzeugen.
Nach Korrektur lagen die gemessenen DNA-Mengen über dem in regulatorischen Leitlinien genannten Grenzwert. Die Ergebnisse waren über mehrere Messungen reproduzierbar und stimmten zwischen den drei eingesetzten Assays gut überein.
PCR-Nachweis von Plasmidelementen
In allen vier Chargen wurden starke PCR-Signale für alle getesteten Plasmidelemente nachgewiesen – Spike-Sequenzen, ORI, Neomycin-Kassette und SV40-Promoter/Enhancer. Dieselben Elemente waren nach Transfektion auch in den HEK293-Zellen nachweisbar. Für den SV40-Promoter/Enhancer erzeugten die Primer zwei Amplifikate unterschiedlicher Größe (93 bp und 165 bp) – ein methodisch bekanntes Ergebnis, das auf die Struktur der Zielsequenz zurückzuführen ist.d) Interpretation durch die Autoren
Die Autoren schlussfolgern, dass die untersuchten Chargen DNA-Rückstände enthalten, die regulatorische Grenzwerte überschreiten. Die DNA liegt überwiegend LNP-verpackt vor, wird von Zellen aufgenommen und ist dort nachweisbar. Die anhaltende Spike-Protein-Produktion über sieben Tage hinaus wirft die Frage auf, wie die langanhaltende Produktion zustande kommt.
Die Autoren heben insbesondere das Vorhandensein des SV40-Promoter/Enhancer hervor. Dieses Element sei für die mRNA-Produktion in E. coli nicht erforderlich. Sie diskutieren, dass solche regulatorischen Elemente in bestimmten experimentellen Kontexten die Transkriptionsaktivität beeinflussen können. Ein möglicher Einfluss auf Kernlokalisation oder genomische Integration wird von den Autoren als theoretische Überlegung angesprochen.
Die Autoren kritisieren die bestehenden regulatorischen Grenzwerte als nicht auf LNP-basierte RNA-Injektionen ausgerichtet und betonen, dass bisher keine wissenschaftliche Evidenz vorliegt, die eine Festlegung eines sicheren DNA-Grenzwerts für diese Produktklasse erlauben würde.
Die Autoren zeigen, dass das Spike-Protein über extrazelluläre Vesikel (Exosomen) freigesetzt wird. In welchem Umfang solche Vesikel im menschlichen Organismus zur Verteilung von Proteinen beitragen, ist Gegenstand aktueller Forschung und bislang nicht abschließend geklärt.
Die Autoren weisen auf ein grundlegendes Problem hin: Die Wahl der Analysemethode beeinflusst das Ergebnis maßgeblich, was bei Studienvergleichen unbedingt berücksichtigt werden sollte.
e) Methodische Einschränkungen
Zelllinie
HEK293-Zellen sind immortalisiert und weisen gegenüber primären Zellen (z. B. Muskel-, Immun- oder Nervenzellen) veränderte Eigenschaften auf, insbesondere eine hohe Transfektionseffizienz und veränderte Regulationsmechanismen.Dosierung
Die verwendete Dosis ist nicht physiologisch. Die eingesetzten Mengen (z. B. 1/12 einer klinischen Dosis pro Well) führen zu einer direkten und lokal hohen Exposition der Zellen, die nicht den Verteilungsbedingungen im menschlichen Körper entspricht.In-vitro-Kontext
Alle Experimente wurden unter Zellkulturbedingungen durchgeführt. Eine direkte Übertragung auf in-vivo-Verhältnisse – etwa hinsichtlich Verteilung, Abbau oder Immunantwort im menschlichen Körper – ist nicht ohne weiteres möglich.Assay-Spezifität
Fluorometrische Methoden sind empfindlich gegenüber Störsignalen, was die Quantifizierung von DNA beeinflussen kann.Mittelwert
Die ermittelte Rest-DNA lag bei 32,71–43,38 ng pro klinischer Dosis. Diese Werte basieren auf fluorometrischen Messungen mit drei unterschiedlichen Assays (PicoGreen, Qubit, AccuBlue). Da die einzelnen Assays teils deutlich voneinander abweichende Ergebnisse lieferten, stellt der angegebene Bereich das arithmetische Mittel über methodisch unterschiedliche Messprinzipien dar. Die Spannbreite der Einzelmessungen ist erheblich, sodass der Mittelwert primär als orientierender Schätzwert zu verstehen ist.f) Einordnung in den Gesamtkontext
Die Studie geht über die reine Quantifizierung von DNA-Rückständen hinaus: Sie verbindet den analytischen Nachweis mit Zellexperimenten und fragt nach biologischen Konsequenzen. Sie kombiniert mehrere analytische und funktionelle Methoden (fluorometrische Assays, PCR, ELISA und Massenspektrometrie) und verfolgt damit einen vergleichsweise breit angelegten Ansatz.
Die Studie unterstreicht zudem, dass selbst innerhalb derselben methodischen Kategorie erhebliche Unterschiede zwischen einzelnen fluorometrische Assays auftreten können – ein Befund, der die Bedeutung methodischer Standardisierung für künftige Untersuchungen unterstreicht.
🧬 🧬 🧬
4.3.6. Wang et al. (2024)
A rapid detection method of replication-competent plasmid DNA from COVID-19 mRNA vaccines for quality control.
Autoren: Tayler J. Wang, Alex Kim, Kevin Kim
Veröffentlicht: 29. Dezember 2024 (Journal of High School Science, peer-reviewed)
Link zur Studie: https://jhss.scholasticahq.com/article/127890-a-rapid-detection-method-of-replication-competent-plasmid-dna-from-covid-19-mrna-vaccines-for-quality-controla) Kontext und Ziel der Studie
Die Arbeit von Wang et al. verfolgt eine andere Fragestellung als die bisher vorgestellten Studien. Im Vordergrund steht nicht die Gesamtmenge an DNA-Rückständen, sondern eine spezifischere Frage: Enthält die nachgewiesene DNA noch funktionsfähige, replikationsfähige Plasmidsequenzen – also Fragmente, die in Bakterien eingeschleust und dort vermehrt werden könnten?
Zur Beantwortung dieser Frage entwickelten die Autoren eine einfache und schnelle Nachweismethode. Die Überlegung dabei: Wenn bei der Impfstoffherstellung die DNA-Vorlage nicht vollständig entfernt wurde, könnten noch intakte oder fast intakte Plasmide übrig sein.
Die Arbeit entstand im Rahmen eines studentischen Freiwilligenprogramms der FDA und wurde auf dem FDA White Oak Campus durchgeführt, mit Unterstützung von FDA-Wissenschaftlern. Der Publikationsort – das Journal of High School Science – spiegelt den Ausbildungskontext wider, nicht den institutionellen Rahmen der Arbeit.
b) Materialien und Methoden
Impfstoffchargen
Die Studie verwendet verschiedene Probenquellen, die sich grundlegend unterscheiden und bei der Interpretation der Ergebnisse klar getrennt werden müssen.Erstens einen selbst hergestellten Laborimpfstoff mit der XBB.1.5-Spike-Sequenz, produziert mit einem kommerziellen LNP-Kit. Zweitens Biosimilar-Referenzmaterialien von BEI Resources – NIH-gefördertes Referenzmaterial, das für Forschungszwecke hergestellt wurde und nicht identisch mit zugelassenen Handelschargen ist. Drittens zwei kommerzielle Pfizer-Chargen (Lot PAA194854, monovalent; Lot PAA184098, bivalent) – originalversiegelte, zugelassene Impfstoffchargen.
Impfstoff Herkunft Beschreibung In-house mRNA vaccine Selbst hergestellt Enthält XBB.1.5 Spike,
LNP-formuliertModerna Biosimilar BEI Resources nicht das Original Pfizer Biosimilar BEI Resources nicht das Original COMIRNATY Original
(NR-59604)BEI Resources Monovalent (Wuhan) COMIRNATY Bivalent
(NR-59605)BEI Resources Bivalent (Wuhan + Omicron BA.5) DNA-Extraktion
Die DNA wurde mit einem kommerziellen Plasmid-Miniprep-Kit extrahiert (Monarch Plasmid DNA Miniprep, New England Biolabs). Das Kit enthält RNase A im Neutralisierungspuffer, sodass RNA während der Extraktion weitgehend abgebaut wird. Die Elution erfolgte in 30 µl nukleasefreiem Wasser.Quantifizierung
Die DNA-Konzentration wurde mit zwei Methoden bestimmt: NanoDrop-Spektrophotometrie und Qubit dsDNA HS Assay. Die Autoren weisen ausdrücklich darauf hin, dass NanoDrop nicht zwischen DNA und RNA unterscheidet und durch freie Nukleotide, Salze und organische Verbindungen beeinflusst werden kann. Qubit ist spezifischer, kann aber bei großen RNA-Mengen oder unsachgemäßer Probenvorbereitung ebenfalls überschätzen.Transformationstest – das vorgestellte Nachweisverfahren
Zunächst wird die DNA aus dem Impfstoff extrahiert. Diese extrahierte DNA setzt sich aus verschiedenen Fragmenten zusammen – darunter ist möglicherweise auch der Replikationsursprung (ORI), der als Startpunkt der Plasmid-Vervielfältigung dient, sowie das Antibiotikaresistenzgen, das eine Selektion ermöglicht.Die extrahierte DNA wird mit einer T4-DNA-Ligase behandelt. Dieses Enzym verknüpft die Enden von DNA-Fragmenten miteinander. Aus linearen Fragmenten können so wieder ringförmige Plasmide entstehen. Da die Ligase beliebige Enden miteinander verbinden kann, können auch Fragmente zusammengefügt werden, die im ursprünglichen Impfstoff getrennt vorlagen – es entstehen künstliche Artefakte.
Diese neu gebildeten Plasmide werden anschließend in spezielle E. coli-Bakterien eingeschleust (Transformation).
Die zugrundeliegende Überlegung:
- War der DNase-Verdau während der Impfstoffherstellung effizient, sind die DNA-Fragmente so klein, dass sie weder den vollständigen ORI noch das vollständige Resistenzgen enthalten. Dann kann auch durch Ligation kein funktionsfähiges Plasmid entstehen.
- War der DNase-Verdau unvollständig, sind noch größere Fragmente vorhanden, die ORI und Resistenzgen vollständig enthalten können. Diese können nach Ligation ein funktionsfähiges Plasmid bilden. Die Ligase kann nur vorhandene Enden verknüpfen; sie kann keine fehlenden Teile eines Gens aus kleinen Bruchstücken zusammensetzen.
Nur Bakterien, die ein funktionsfähiges Antibiotikaresistenzgen und einen funktionsfähigen ORI aufgenommen haben, überleben auf antibiotikumhaltigem Nährboden und bilden sichtbare Kolonien. Das Vorhandensein von Kolonien ist damit ein direkter biologischer Nachweis für das Vorliegen replikationsfähiger Plasmid-DNA im ursprünglichen Impfstoff.
Größenanalyse
Die Fragmentgrößen wurden mittels Agarosegelelektrophorese und Agilent 2100 Bioanalyzer bestimmt.c) Zentrale Ergebnisse
Quantitative Befunde
Die DNA-Mengen pro Dosis lagen – gemessen mit Qubit – zwischen 40 und 110 ng, also oberhalb des WHO-Grenzwerts von 10 ng. NanoDrop lieferte deutlich höhere Werte (mehrere tausend ng), was die Autoren auf die bekannte Unspezifität dieser Methode zurückführen.Laborimpfstoff und Biosimilar
Im selbst hergestellten Laborimpfstoff und im Pfizer-Biosimilar wurden replikationsfähige DNA-Fragmente gefunden – das Transformationsverfahren erzeugte Kolonien. Im Moderna-Biosimilar und im Haus-Standard wurden trotz höherer DNA-Konzentrationen keine Kolonien nachgewiesen, was zeigt, dass die Koloniebildung nicht allein von der DNA-Menge abhängt, sondern von der Intaktheit der relevanten Sequenzelemente.Kommerzielle Pfizer-Chargen
In den beiden kommerziellen Pfizer-Chargen wurden keine replikationsfähigen DNA-Fragmente gefunden – der Transformationstest blieb ohne Kolonien. Die Größenanalyse zeigte, dass nahezu die gesamte nachgewiesene DNA kürzer als 35 bp war. Dieses Ergebnis ist nach Einschätzung der Autoren mit einer vollständig durchgeführten DNase-Behandlung vereinbar.d) Interpretation durch die Autoren
Die Autoren werten das Fehlen replikationsfähiger DNA in den kommerziellen Chargen als Beleg für die Wirksamkeit des industriellen Herstellungsprozesses. Die vollständige Fragmentierung der Plasmid-DNA auf unter 35 bp macht eine biologische Aktivität der DNA-Reste nach Einschätzung der Autoren unwahrscheinlich – einschließlich der SV40-Promoter/Enhancer-Sequenz, deren Funktionsfähigkeit bei dieser Fragmentgröße praktisch ausgeschlossen wird.
Gleichzeitig bestätigen die Autoren, dass die DNA-Gesamtmenge den WHO-Grenzwert überschreitet, und empfehlen eine stringentere und transparentere Überwachung der DNA-Rückstände – auch mit dem Ziel, das öffentliche Vertrauen in mRNA-Impfstoffe zu stärken.
e) Methodische Einschränkungen
Stichprobengröße und -auswahl
Die Untersuchung umfasst lediglich zwei kommerzielle Chargen, was keine belastbaren Aussagen zur Chargenvariabilität zulässt. Die verwendeten Biosimilar-Proben sind nicht identisch mit zugelassenen Handelsprodukten; daher sind Ergebnisse aus diesen Proben nicht ohne Weiteres auf kommerzielle Impfstoffe übertragbar.DNA-Extraktion
Die Extraktion erfolgte mit einem Plasmid-Miniprep-Kit, das primär für die Isolierung intakter Plasmide optimiert ist. Inwieweit stark fragmentierte DNA-Reste – insbesondere sehr kurze Fragmente – mit dieser Methode vollständig erfasst werden, ist nicht abschließend geklärt.Nachweisgrenzen der Transformationsmethode
Die Methode detektiert ausschließlich DNA-Fragmente, die nach Ligation einen funktionsfähigen Replikationsursprung (ORI) und ein Antibiotikaresistenzgen enthalten. Sie prüft, ob ausreichend große Fragmente (>800 bp für das Resistenzgen, >600 bp für den ORI) vorhanden sind, die nach Ligation ein funktionsfähiges Plasmid bilden können. Fragmente ohne diese Elemente – selbst wenn sie andere relevante Sequenzen wie den SV40-Enhancer (ca. 72–165 bp) enthalten würden – werden nicht erfasst.
Zudem kann die Ligation künstlich Plasmide erzeugen, die im ursprünglichen Impfstoff nicht vorhanden waren.Begrenzung der funktionellen Aussagekraft
Der verwendete Transformationsassay untersucht ausschließlich, ob DNA-Fragmente nach Aufnahme in Bakterien ein replizierfähiges Plasmid bilden können. Andere mögliche biologische Eigenschaften von DNA-Fragmenten werden durch dieses System nicht erfasst. Insbesondere erlaubt die Methode keine Aussagen darüber, ob kleinere oder nicht replizierfähige Fragmente in anderen biologischen Kontexten funktionelle Effekte besitzen könnten. Die Studie bewertet somit spezifisch die bakterielle Replikationsfähigkeit bestimmter Plasmidbestandteile – nicht die allgemeine biologische Relevanz sämtlicher nachweisbarer DNA-Fragmente.f) Einordnung in den Gesamtkontext
Die Studie von Wang et al. untersucht, ob in den Impfstoffproben noch biologisch aktive, replikationsfähige Plasmid-DNA nachweisbar ist.
Damit erweitert die Arbeit die bisherige Diskussion um eine zusätzliche Ebene. Während PCR-, Fluorometrie- und Sequenzierungsverfahren vor allem Aussagen über Menge, Sequenzidentität und Fragmentlänge erlauben, prüft der verwendete Transformationsassay gezielt die funktionelle Integrität der DNA im bakteriellen System.
Gleichzeitig bleibt die Aussagekraft des Ansatzes auf die im Experiment getestete Funktion beschränkt: Der Assay erfasst ausschließlich DNA, die in Bakterien transformierbar und replizierbar ist. Andere biologische Eigenschaften oder potenzielle Wirkmechanismen von DNA-Fragmenten werden damit nicht untersucht.
Insgesamt liefert die Arbeit einen methodisch interessanten Beitrag zur Diskussion, indem sie die Frage nach der funktionellen Relevanz von DNA-Rückständen in den Mittelpunkt stellt und damit eine wichtige Ergänzung zu rein quantitativen Analysen darstellt.
Die Studie verdeutlicht damit, dass die Bewertung von DNA-Rückständen nicht allein auf deren Menge basieren sollte, sondern wesentlich von deren strukturellen und funktionellen Eigenschaften abhängt.
🧬 🧬 🧬
4.3.7. Fleming et al. (2025)
Quantitative Analysis of Nucleic Acid Content in Spikevax (Moderna) and BNT162b2 (Pfizer) COVID-19 Vaccine Lots
Autoren: Richard M Fleming PhD, MD, JD; Peter Kotlár MD; Sona Pekova MD, PhD
Veröffentlicht: 13. Mai 2025 (Herald Open Access)
Link zur Studie: https://www.heraldopenaccess.us/openaccess/quantitative-analysis-of-nucleic-acid-content-in-spikevax-moderna-and-bnt162b2-pfizer-covid-19-vaccine-lotsa) Kontext und Ziel der Studie
Die Studie entstand im Kontext einer behördlichen Anfrage in der Slowakei. Ziel der Untersuchung war die Charakterisierung der Nukleinsäurezusammensetzung von mRNA-COVID-19-Impfstoffen im Hinblick auf:
- Identifizierung und Quantifizierung der enthaltenen Nukleinsäuren
- Prüfung der Chargen-Homogenität (zwischen und innerhalb von Lots)
- Nachweis undeklarierter genetischer Materialien
- Bewertung der Stabilität abgelaufener Chargen
Die Ergebnisse sollten Erkenntnisse über die Zusammensetzung, Stabilität und Herstellungspraxis von Impfstoffen liefern.
b) Materialien und Methoden
Impfstoffchargen
Die molekulare Analyse wurde an 24 Chargen – Moderna (17) und Pfizer (7) – durchgeführt:Spikevax (Moderna): MV1013A, 200023A, 200156A, 223049, 200090A, 200106A, 200100A, 3005885, 3005836, 000090A, 000058A, 3005241, 3005697, 3006272, MV1018A, 400012A, 400011A und
BNT162b2 (Pfizer): FP9632, 1F1051A, 1LO84A, 1F1047A, 1F1059A, 1F1055A, PCB0020.
Pro Charge wurden fünf originalverpackte, ungeöffnete Fläschchen analysiert, die bei -80 °C gelagert und unter dokumentierter Temperaturkontrolle transportiert wurden. Die Provenienzdokumentation ist damit vergleichsweise gut belegt.
Aufschluss der Lipid-Nanopartikel (LNPs)
Die LNPs wurden durch eine Kombination aus:
▪️chaotropen Salzen (Isolierungskit QIAamp DNA Mini Kit (Qiagen, DE)),
▪️Hitze (60 °C) und
▪️enzymatischem Verdau (Proteinase K)
vollständig aufgelöst.Dadurch wurden sowohl die mRNA als auch eventuelle DNA-Rückstände freigesetzt.
Reverse Transkription und Multiplex qPCR
Die mRNA wurde durch Reverse Transkription in cDNA umgeschrieben. Die anschließende Quantifizierung erfolgte mittels Multiplex Real-Time PCR (Rotor-Gene Q, Qiagen) mit vier simultanen Farbkanälen:- FAM (Grün): für das S-Protein (cDNA)
- HEX (Gelb): für die Vektor-Ori-DNA
- Cy5 (Rot): für die E. coli-DNA
- ROX: als passive Referenz.
Alle drei Zielsequenzen wurden damit in einem einzigen Reaktionsansatz gleichzeitig erfasst, wobei mögliche Wechselwirkungen zwischen den Targets (z. B. Konkurrenz um Reagenzien) die Quantifizierung beeinflussen können.
Quantifizierung mittels qPCR
Für die Multiplex quantitative Real-Time PCR verwendeten die Autoren Primer für drei verschiedene Zielsequenzen:Ziel Quelle mRNA für S-Protein
(Spikevax & BNT162b2)selbst entworfen Expressionsvektor-DNA (Ori) aus anderen Studien übernommen E. coli-genomische DNA (ITS) aus kommerziellem Kit (ISO 13485-validiert) Alle verwendeten Primer und fluoreszenzmarkierten Hybridisierungssonden wurden von Eurofins Genomics, DE, kundenspezifisch synthetisiert.
Kalibrationsstrategie
Da die mRNA in den Impfstoffen mit N1-Methylpseudouridin modifiziert ist und das genaue Ausmaß dieser Modifikation nicht bekannt ist, konnten keine synthetischen Standards für eine klassische zielspezifische Kalibrationskurve hergestellt werden. Die Autoren validierten stattdessen die PCR-Effizienz durch serielle Verdünnungsreihen der Impfstoffproben selbst. Für die absolute Quantifizierung verwendeten sie eine universelle Kalibrationskurve, die aus Verdünnungsreihen von 50 verschiedenen Mikroorganismen gemittelt wurde.Verifikation durch Sanger-Sequenzierung
Die PCR-Produkte aller 24 Chargen wurden mittels Sanger-Sequenzierung verifiziert und mit Datenbankeinträgen verglichen.c) Zentrale Ergebnisse
mRNA-Nachweis und Chargenvariabilität
Die PCR-Analyse bestätigte das Vorhandensein der deklarierten mRNA-Sequenzen für das Spike-Protein in beiden Impfstoffen.
Die Analyse der mRNA für das S-Protein zeigte eine erhebliche Variabilität zwischen verschiedenen Chargen – bei beiden Impfstoffen.
Die absoluten mRNA-Mengen lagen im Bereich von 109 bis 1010 Kopien/ml (gemessen als cDNA). Einzelne Chargen zeigten jedoch eine 10-fach niedrigere mRNA-Menge – bei beiden Impfstoffen.Nachweis undeklarierter DNA-Sequenzen als Zufallsbefund
Während der Validierung der cDNA-Verdünnungsreihen zeigten sich unerwartet starke Signale in den DNA-Kanälen: im Cy5-Kanal (für Ori-DNA) und teilweise im HEX-Kanal (für E. coli-DNA). Da die Proben neben cDNA auch DNA-Rückstände enthielten, amplifizierten die Ori- und E. coli-Primer diese unbeabsichtigt mit. Die entsprechenden PCR-Produkte wurden aufgereinigt und mittels Sanger-Sequenzierung identifiziert: Sie zeigten 100%ige Übereinstimmung mit dem Expressionsvektor (Plasmid-Ori und Spike-Kassette) sowie mit der E. coli-ITS-Region.Quantitative DNA-Befunde
Ein zentraler Befund der Studie ist der durchgängig hohe Gehalt an DNA in allen getesteten Chargen – sowohl von Moderna als auch von Pfizer.Impfstoff mRNA (cDNA) (Kopien/ml) DNA-Menge (Kopien/ml) Spikevax (Moderna) 109 – 1010 107 – 109 BNT162b2 (Pfizer) 1010 – 1011 108 – 109 Hinweis auf nahezu vollständige DNA-Konstrukte
Die Autoren beobachten, dass der Ori-Primer (5′-Ende des Konstrukts) und der Spike-Primer (3′-Ende, Basen 3184–3417) gleichzeitig ein Signal liefern. Da beide Primerbindestellen mehrere tausend Basenpaare voneinander entfernt sind, deutet dies darauf hin, dass zumindest längere DNA-Fragmente vorhanden sind, die beide Zielbereiche umfassen könnten.Auffällige Chargenheterogenität bei Moderna
Bei drei Moderna-Chargen ist das Verhältnis von Spike-DNA zu Vektor-DNA umgekehrt und um eine Größenordnung verschoben – in eine Richtung, die nicht zu einem einzigen Plasmid passt. Die Autoren spekulieren, dass diese Chargen ein zweites DNA-Konstrukt enthalten könnten – möglicherweise eine Omicron-Variante im selben Vektor.E. coli-DNA
In zwei Pfizer-Chargen wurde E. coli-genomische DNA im Bereich einzelner Kopien pro Milliliter nachgewiesen – knapp oberhalb der Nachweisgrenze.SV40
Es wurden keine Hinweise auf SV40 festgestellt.d) Interpretation durch die Autoren
Anwesenheit der DNA
Die Autoren argumentieren, dass die nachgewiesenen DNA-Mengen – vergleichbar mit der deklarierten mRNA-Menge in der Größenordnung von 107 bis 109 Kopien/ml – zu hoch sind, um als zufällige Kontamination erklärt zu werden. Die Autoren interpretieren die DNA als regulären, wenn auch nicht deklarierten Bestandteil der Impfstoffe.Chargen-Variabilität und mRNA-Identität
Die Chargenvariabilität der mRNA wird als mögliches Problem für die Dosierungskonsistenz bewertet. Die Verwendung der veralteten Wuhan-Spike-Sequenz wird als Einschränkung der Schutzwirkung gegen aktuell zirkulierende Varianten kritisiert.Aufgrund der Analysebefunde empfehlen die Autoren:
- verbesserte Reinigungsprotokolle,
- strengere Chargenprüfung,
- genomische Aktualisierungen und
- eine verstärkte behördliche Aufsicht.
e) Methodische Einschränkungen
Universelle Kalibrationskurve
Die absolute Quantifizierung basiert nicht auf einer zielspezifischen Standardkurve, sondern auf einer universellen Kurve, die aus 50 verschiedenen Mikroorganismen gemittelt wurde. Diese Methode setzt eine vergleichbare PCR-Effizienz für alle Zielsequenzen voraus. Diese Annahme ist insbesondere bei unterschiedlichen Zielsequenzen (mRNA, Plasmid-DNA, bakterielle DNA) und komplexen Probenmatrices nicht selbstverständlich. Die absoluten Kopienzahlen sind daher mit Vorsicht zu interpretieren; die qualitativen Befunde – also das Vorhandensein der nachgewiesenen Sequenzen – sind davon weniger betroffen.Keine Messung vor LNP-Aufschluss
Im Unterschied zu anderen Studien wurde keine Messung vor dem Aufschluss der LNPs durchgeführt. Damit fehlt der direkte Vergleich, der Aussagen über den Anteil LNP-verpackter DNA ermöglichen würde.SV40-Aussage nicht aussagekräftig
Die Autoren berichten, kein SV40 nachgewiesen zu haben. Das verwendete Primer-Set enthielt jedoch keine SV40-spezifischen Sequenzen. Die Aussage ist damit zwar korrekt, aber nicht informativ – ein Nachweis oder Ausschluss von SV40 war mit diesem Ansatz methodisch nicht möglich.f) Einordnung in den Gesamtkontext
Die Studie von Fleming et al. erweitert die bisherige Literatur vor allem durch ihren vergleichenden Ansatz über eine relativ große Anzahl von Chargen.
Methodisch kombiniert sie Multiplex-qPCR mit Sanger-Sequenzierung zur Verifikation der amplifizierten Produkte. Der Nachweis von DNA-Sequenzen, einschließlich Plasmid- und bakterieller DNA steht im Einklang mit vorherigen Studien.
Besondere Aufmerksamkeit richtet die Arbeit auf Unterschiede zwischen Chargen sowie auf quantitative Vergleiche der gemessenen Nukleinsäureanteile. Die beobachtete Variabilität zwischen einzelnen Lots ergänzt damit die bestehende Diskussion über mögliche Unterschiede in Zusammensetzung, Fragmentierung und analytischer Erfassbarkeit der DNA-Rückstände.
🧬 🧬 🧬
4.3.8. Speicher et al. (2025)
Quantification of residual plasmid DNA and SV40 promoter-enhancer sequences in Pfizer/BioNTech and Moderna modRNA COVID-19 vaccines from Ontario, Canada
Autoren: David Speicher, Jessica Rose, Kevin McKernan
Veröffentlicht: 25. September 2025 (PubMed/Herald Open Access)
Link zur Studie: https://pubmed.ncbi.nlm.nih.gov/40913499/a) Kontext und Ziel der Studie
Die Studie von Speicher, Rose und McKernan ist eine methodisch erweiterte Folgearbeit zur früheren Untersuchung derselben Autorengruppe (Studie 2, 2023). Sie baut inhaltlich auf den Befunden von McKernan et al. und Kämmerer et al. auf und verfolgt das Ziel, die DNA-Rückstände in mRNA-Impfstoffen mit zwei unabhängigen Methoden – qPCR und Qubit-Fluorometrie mit RNase-Korrektur – an einer größeren Stichprobe zu quantifizieren.
Ergänzend werden Fragmentlängen mittels Oxford-Nanopore-Sequenzierung bestimmt sowie eine explorative Korrelation mit VAERS-Nebenwirkungsdaten durchgeführt.
b) Materialien und Methoden
Impfstoffchargen
Für die Untersuchung wurden insgesamt 32 Fläschchen aus 16 Chargen mRNA-basierter COVID-19-Impfstoffe verwendet: BNT162b2 (Pfizer/BioNTech; 10 Vials aus 6 Chargen) und mRNA-1273 (Moderna; 22 Vials aus 10 Chargen). Die Proben wurden aus verschiedenen Apotheken in Ontario, Kanada, bezogen.Die Fläschchen waren originalversiegelt und wiesen unbeschädigte Flip-off-Kappen mit Chargennummern und Verfallsdaten auf. Lagerung und Transport erfolgten unter Einhaltung der Kühlkette (2–8 °C). Die Proben wurden in spezialisierten Kühleinheiten gelagert und in isolierten Behältern mit Kühlakkus transportiert; bevor sie innerhalb von 5 Stunden nach Erhalt in den Kühlschrank des Testlabors gestellt wurden.
Ein Teil der Proben bestand aus Restmengen kürzlich verwendeter Fläschchen (innerhalb von 30 Minuten nach Anwendung), die ebenfalls gekühlt transportiert und innerhalb von 12 Stunden im Labor eingelagert wurden.
Eine Moderna-Probe wies kein aufgedrucktes Verfallsdatum auf, verfügte jedoch über einen QR-Code zur Identifikation durch Apothekenpersonal.
Aufschluss der Lipid-Nanopartikel (LNPs)
Zum Aufschluss der Lipid-Nanopartikel (LNPs) wurden die Proben 8 Minuten bei 95 °C erhitzt und anschließend für 5 Minuten auf 4 °C abgekühlt. Dieser thermische Schritt dient der Freisetzung der enthaltenen Nukleinsäuren.Quantifizierung mittels qPCR
Die DNA-Konzentrationen wurden mittels qPCR über den Cq-Wert bestimmt. Die Primer entsprechen weitgehend den früheren Assays der Autorengruppe: Spike-Sonde, Ori-Sonde sowie ein SV40-Enhancer-Assay für Pfizer-Chargen.Die Quantifizierung erfolgte anhand einer Kalibrationskurve mit synthetischer DNA und 10-fachen Verdünnungsreihen (QuantStudio 3). Aus der gemessenen Kopienzahl der Zielsequenzen wurde unter Berücksichtigung der Gesamtplasmidlänge (Pfizer: 7.824 bp; Moderna: 6.777 bp) die Gesamt-DNA-Menge pro Dosis berechnet.
Jeweils 1 µL der behandelten Impfstoffprobe wurde ohne vorherige DNA-Extraktion direkt in die qPCR eingesetzt. Eine mögliche PCR-Inhibition durch Impfstoffbestandteile wurde durch Verdünnungsreihen ausgewählter Proben überprüft.
Quantifizierung mittels Qubit-Fluorometrie
Die Proben wurden mit dem DNA-spezifischen Farbstoff AccuGreen mehrfach gemessen: ohne Vorbehandlung, nach LNP-Aufschluss durch Erhitzen sowie nach einer RNase-A-Behandlung. Letztere korrigiert den RNA-bedingten Signalanteil (Crosstalk des Farbstoffs – der auch an RNA bindet). Die Autoren verfolgten den Abfall der Fluoreszenz über 10 Minuten in einer Zeitreihe. Dadurch konnten sie den RNA-bedingten Signalanteil ermitteln und die reine DNA-Konzentration berechnen.Größenprofil der Rest-DNA mittels ONT-Sequenzierung
Zur Bestimmung der Größenverteilung der DNA-Fragmente wurde eine zuvor sequenzierte Pfizer-Charge (FL8095) als Standard verwendet. Die Sequenzierung erfolgte auf einer Oxford Nanopore Flongle mit dem Ligation Sequencing Kit (SQK-LSK114). Die erhaltenen Reads wurden mit einer Referenz-Plasmid-Sequenz abgeglichen.Der verwendete Bibliotheksaufbau (SPRI-basierte Aufreinigung) kann kurze Fragmente (<100-200 bp) verlieren, sodass die Längenverteilung zu längeren Fragmenten hin verzerrt ist.
Beurteilung der Nukleaseempfindlichkeit
Zur Prüfung, ob die DNA im Inneren der LNPs vorliegt, wurde DNase I-XT direkt zum unveränderten Impfstoff gegeben. Nach 30-minütiger Inkubation wurde das Enzym inaktiviert, die DNA aufgereinigt und mittels qPCR quantifiziert. Eine nackte DNA-Kontrolle diente als Referenz.Explorative VAERS-Korrelation mit den gemessenen DNA-Mengen
Für jede Charge wurden VAERS-Meldungen aus Kanada erfasst und die Anzahl schwerwiegender unerwünschter Ereignisse (SAEs) mit den gemessenen DNA-Gehalten korreliert. Ziel war es, eine mögliche Korrelation zwischen den gemessenen DNA-Gehalten (Spike, Ori, SV40) und der Häufigkeit von Nebenwirkungen zu untersuchen. Zur Berechnung des Zusammenhangs wurde der Pearson-Korrelationskoeffizient verwendet.c) Zentrale Ergebnisse
qPCR-Validierung
Die qPCR-Assays für Spike und Ori zeigten exzellente R2-Werte (0,998–0,999) und Effizienzen (94,7–99,8 %). Der SV40-Assay hatte eine etwas geringere Güte (R2 = 0,906, Effizienz 93,6 %). Negativkontrollen waren unauffällig.LNP-Inhibition
Unverdünnte Proben zeigten eine PCR-Inhibition durch die LNPs. Nach 1:10 Verdünnung war die Inhibition aufgehoben, daher wurden für die Quantifizierung verdünnte Proben verwendet.Quantifizierung mittels qPCR
Alle Pfizer-Chargen waren positiv für Spike, Ori und SV40. Die ähnlichen Cq-Werte aller drei Targets deuten auf gemeinsame, intakte DNA-Moleküle hin. Bei Moderna waren Spike und Ori nachweisbar, kein SV40. Die Ori-Signale traten bei Moderna etwa 3 Zyklen später auf als Spike, was auf einen geringeren Anteil oder eine Fragmentierung der Ori-Region hindeutet.Die Autoren rechneten die Cq-Werte über die Standardkurve in absolute DNA-Mengen um:
Impfstoff Spike (ng/Dosis) Ori (ng/Dosis) SV40 (ng/Dosis) Pfizer 0,22 – 2,43 0,28 – 7,28 0,25 – 23,72 Moderna 0,25 – 0,78 0,01 – 0,34 nicht nachweisbar Pfizer wies eine deutlich höhere Menge an Rest-DNA auf als Moderna.
Zwei Pfizer-Chargen (FM7380 und FN7934, monovalent, PBS-Formulierung) überschritten mit SV40-Gehalten von bis zu 23,72 ng/Dosis den FDA-Richtwert von 10 ng/Dosis. Alle anderen Chargen lagen unter diesem Wert.
Quantifizierung mittels Qubit-Fluorometrie
Die Autoren testeten die Proben mehrfach zu verschiedenen Zeiten. Die durchschnittliche Varianz (durchschnittliche Abweichung vom Durchschnitt) betrug:Messphase Pfizer Spanne vor dem Erhitzen 317 ± 278 ng/Dosis 43 bis 727 ng/Dosis nach dem Erhitzen 1.145 ± 533 ng/Dosis 519 bis 1.949 ng/Dosis nach RNase A 297 ± 217 ng/Dosis 100 bis 765 ng/Dosis Messphase Moderna Spanne vor dem Erhitzen 456 ± 121 ng/Dosis 199 bis 675 ng/Dosis nach dem Erhitzen 1.554 ± 1.023 ng/Dosis 176 bis 3.169 ng/Dosis nach RNase A 1.065 ± 624 ng/Dosis 113 bis 2.365 ng/Dosis Nach LNP-Aufbruch durch Erhitzen stiegen die Werte deutlich an. Die anschließende RNase A-Behandlung reduzierte die Werte (Crosstalk-Korrektur).
Moderna enthielt durchgängig höhere DNA-Mengen als Pfizer.
Der Vergleich von Qubit und qPCR zeigte bei Pfizer eine positive Korrelation, bei Moderna nicht – was auf DNA-Fragmente hindeutet, die keine intakten Primerbindestellen mehr enthalten und daher in der qPCR nicht erfasst werden.
Fragmentlängenanalyse
Der längste detektierte Read umfasste 3,5 kb und deckte den Großteil des Plasmid-Backbones ab. Das zeigt, dass neben kurzen Fragmenten auch größere DNA-Fragmente im Kilobasenbereich vorhanden sind.DNA liegt im Inneren der LNPs
Die DNase-Behandlung führte zu keinem signifikanten DNA-Verlust in der Impfstoffprobe (≤1 Cq Offset), während die nackte Kontroll-DNA vollständig abgebaut wurde. Dies bestätigt, dass die DNA überwiegend LNP-verpackt und damit vor enzymatischem Abbau geschützt ist.Explorative VAERS-Korrelation mit den gemessenen DNA-Mengen
Für die untersuchten Chargen wurden Meldedaten aus dem US-amerikanischen Spontanmeldesystem VAERS herangezogen. Für alle untersuchten Chargen außer drei Moderna-Chargen lagen VAERS-Meldungen vor.
Die Autoren führten eine explorative Korrelation zwischen gemessenen DNA-Gehalten und VAERS-Meldedaten durch. Dabei zeigten einzelne Chargen mit höheren DNA-Werten (insbesondere Ori-DNA) auch höhere Anzahlen gemeldeter schwerwiegender Ereignisse (SAEs).d) Interpretation durch die Autoren
Die Autoren schlussfolgern, dass alle untersuchten Chargen DNA-Rückstände enthalten, die LNP-verpackt und damit vor Abbau geschützt sind. Die gemessenen Gesamtmengen überschreiten die bestehenden Richtwerte. Das SV40-Promoter/Enhancer-Element in Pfizer-Chargen wird als besonders relevant hervorgehoben, da es nukleares Targeting begünstigen und theoretisch die Integrationswahrscheinlichkeit erhöhen könnte.
Die Autoren weisen auf mehrere methodische Probleme hin:
Methode Problem qPCR Kann Moleküle kleiner als die Amplikonlänge (105–114 bp) nicht quantifizieren → Unterschätzung der Gesamt-DNA Qubit/Fluorometrie ohne RNase Überschätzung durch Crosstalk mit RNA (daher RNase-Behandlung notwendig) Ethanol-Fällung Verlust kurzer Fragmente (<100 bp) ONT (Nanopore) Bibliotheksvorbereitung selektiert gegen Fragmente <150 bp – Verzerrung zu längeren Fragmenten Daher schlussfolgern die Autoren: Die Wahl der Methode beeinflusst das Ergebnis stark – daher ist methodische Klarheit bei Grenzwertfestlegungen essenziell.
Die Autoren formulieren mehrere Forderungen:
Forderung Begründung Neubewertung der DNA-Grenzwerte Die aktuellen regulatorischen Grenzwerte wurden für nackte DNA in herkömmlichen Impfstoffen entwickelt. Sie sollten unter Berücksichtigung der LNP-Verkapselung, der kumulativen Dosierung und der funktionellen Sequenzen neu bewertet werden. Methoden-Standardisierung Da qPCR die Gesamt-DNA unterschätzt, sollte zusätzlich Fluorometrie mit RNase-Korrektur eingesetzt werden, um die Rest-DNA sowie das RNA/DNA-Verhältnis korrekt zu quantifizieren. Unabhängige Replikation Weitere Labore sollten die Ergebnisse bestätigen. Langzeit-Monitoring Spätfolgen (z. B. Integration, Krebs) werden von VAERS nicht erfasst. Berücksichtigung funktioneller Sequenzen Nicht nur die Menge der DNA ist entscheidend, sondern auch ihre funktionellen Eigenschaften – insbesondere das Vorhandensein von SV40-Elementen und Promotoren. Die Variabilität der Rest-DNA zwischen und innerhalb einzelner Chargen interpretieren die Autoren als möglichen Hinweis auf einen inkonsistenten Herstellungsprozess.
e) Methodische Einschränkungen
Die qPCR kann Fragmente unterhalb der Amplikon-Länge (105–152 bp) nicht erfassen und unterschätzt damit die Gesamt-DNA systematisch.
Die ONT-Daten basieren auf einer einzigen Charge und sind durch die Aufreinigungsmethode zu längeren Fragmenten hin verzerrt.
Die explorative VAERS-Korrelation mit den gemessenen DNA-Mengen ist aufgrund der bekannten Limitationen von Spontanmeldesystemen – insbesondere fehlender Expositionsdaten und möglicher Verzerrungen in der Meldestruktur – nur eingeschränkt interpretierbar. Die Autoren betonen zudem weitere Einschränkungen: begrenzte Chargengröße, keine Kenntnis der verabreichten Dosen und eine Unterfassung möglicher Langzeiteffekte.
Die Proben umfassen sowohl abgelaufene Fläschchen als auch angebrochene Restmengen, was die Vergleichbarkeit innerhalb der Stichprobe einschränkt.
f) Einordnung in den Gesamtkontext
Die Studie stellt eine methodisch erweiterte Fortführung früherer Arbeiten derselben Autorengruppe dar und kombiniert mehrere analytische Ansätze – qPCR, fluorometrische Quantifizierung mit RNase-Korrektur, Nanopore-Sequenzierung sowie funktionelle Tests zur Nukleaseempfindlichkeit. Besonders hervorzuheben ist der direkte Vergleich unterschiedlicher Quantifizierungsmethoden, der die methodische Abhängigkeit der gemessenen DNA-Mengen deutlich macht.
Die qualitativen Befunde – insbesondere der Nachweis von Plasmid-DNA in beiden Impfstoffen sowie von SV40-Sequenzen in Pfizer-Chargen – stehen im Einklang mit anderen Studien (u. a. McKernan et al., Kämmerer et al.). Auch der Hinweis auf eine Verkapselung der DNA durch Lipid-Nanopartikel wird durch mehrere Arbeiten gestützt.
Gleichzeitig zeigen sich deutliche Unterschiede in der quantitativen Einordnung der DNA-Mengen zwischen den verwendeten Methoden (qPCR vs. Fluorometrie). Diese Divergenzen unterstreichen die Bedeutung methodischer Standardisierung und erschweren direkte Vergleiche zwischen Studien.
Insgesamt erweitert die Studie die bestehende Datenlage durch einen breiteren methodischen Ansatz und eine größere Stichprobe. Sie liefert vor allem Hinweise auf methodische Fragestellungen und Variabilität der Messergebnisse, die in zukünftigen Untersuchungen weiter systematisch geklärt werden müssen.
🧬 🧬 🧬
4.3.9. Achs et al. (2025)
Systematic analysis of COVID-19 mRNA vaccines using four orthogonal approaches demonstrates no excessive DNA impurities
Autoren: Adam Achs, Tatiana Sedlackova, Lukas Predajna, Jaroslav Budis, Maria Bartosova, Vladimir Zelnik, Diana Rusnakova, Martina Melichercikova, Marta Miklosova, Veronika Gencurova, Barbora Cernakova, Tomas Szemes, Boris Klempa, Juraj Kopacek, Silvia Pastorekova
Veröffentlicht: 13. Dezember 2025 (npj Vaccines, peer-reviewed; PMCID verfügbar)
Link zur Studie: https://pmc.ncbi.nlm.nih.gov/articles/PMC12715226/a) Kontext und Ziel der Studie
Die Studie von Achs et al. verfolgt einen systematischen und methodisch breit angelegten Ansatz zur Untersuchung möglicher DNA-Rückstände in mRNA-basierten COVID-19-Impfstoffen. Sie entstand vor dem Hintergrund früherer Arbeiten, in denen teilweise erhöhte DNA-Mengen sowie spezifische Sequenzelemente berichtet wurden.
Ziel der Arbeit ist es, diese Fragestellung mit mehreren unabhängigen, sich ergänzenden Analysemethoden („orthogonale Ansätze“) erneut zu prüfen und die Robustheit der Ergebnisse gegenüber methodischen Einflüssen zu bewerten. Dabei steht insbesondere die Frage im Mittelpunkt, ob die in anderen Studien beschriebenen DNA-Rückstände reproduzierbar sind oder durch methodische Artefakte erklärt werden können.
Ein weiterer Fokus liegt auf der Bewertung der eingesetzten Nachweismethoden selbst: Die Autoren untersuchen, inwieweit unterschiedliche analytische Verfahren – etwa Sequenzierung, qPCR und fluorometrische Messungen – zu abweichenden Ergebnissen führen können und welche methodischen Limitationen dabei zu berücksichtigen sind.
Die Studie zielt damit nicht nur auf die Quantifizierung möglicher DNA-Rückstände ab, sondern auch auf eine methodische Einordnung der bestehenden Literatur und die Frage, wie zuverlässig verschiedene Nachweisansätze in diesem Kontext sind.
b) Materialien und Methoden
Impfstoffchargen
Untersucht wurden 15 Chargen – 9 Comirnaty (Pfizer) und 6 Spikevax (Moderna) – aus staatlich verwalteten Impfstoffbeständen der Slowakei, bereitgestellt unter Aufsicht des Staatlichen Instituts für Arzneimittelkontrolle. Vier Comirnaty-Chargen waren zum Zeitpunkt der Analyse noch innerhalb ihres Verfallsdatums. Die übrigen Chargen hatten ihr Verfallsdatum bereits überschritten. Die Fläschchen wurden entsprechend den Herstellerangaben gelagert und transportiert.Ein Teil der Chargen – konkret jene, die auch in Fleming et al. (Studie 7) untersucht worden waren – wurde bewusst einbezogen, um einen direkten Vergleich zu ermöglichen.
Vier orthogonale Analysemethoden
Das methodische Kernmerkmal der Studie ist der Einsatz von vier unabhängigen, sich gegenseitig ergänzenden Verfahren an denselben Proben. Jede Methode hat ihre spezifischen Stärken und Limitationen – die Kombination soll robustere Schlussfolgerungen ermöglichen als jede Methode für sich.Methode 1: qPCR
Die LNPs wurden mit Triton X-100 aufgeschlossen; die behandelte Probe wurde ohne vorherige DNA-Extraktion direkt in die qPCR eingesetzt. Damit entfällt ein Aufreinigungsschritt, der zu DNA-Verlusten führen könnte. Die Autoren prüften explizit mögliche PCR-Inhibitionen durch Impfstoffbestandteile und berichteten unter den verwendeten Bedingungen keine relevante Hemmung der Amplifikation.Es wurden acht Primer-Paare eingesetzt, die drei Regionen des Produktionsplasmids abdecken: die Spike-kodierende Sequenz (SPIKE), den Replikationsursprung (ORI) und das Kanamycin-Resistenzgen (KAN). Dabei variierten die Amplikon-Größen erheblich: von 63 bp (KAN 1C) bis 233 bp (SPIKE 2). Zwei der Primer-Paare – SPIKE 2 und ORI 2 – entsprechen denjenigen, die in Fleming et al. verwendet wurden, um einen direkten Vergleich zu ermöglichen.
Die Quantifizierung erfolgte über Kalibrationskurven mit synthetischen DNA-Standards (gBlocks), die den jeweiligen Zielsequenzen entsprechen. Die gemessenen Kopienzahlen wurden anhand der Molekülmasse des jeweiligen Amplikons in Nanogramm pro Dosis umgerechnet.
Methode 2: Fluorometrie
Für die fluorometrische DNA-Quantifizierung wurde eine DNA-Extraktion durchgeführt – mit zwei verschiedenen Extraktionsmethoden: Phenol-Chloroform-Extraktion (Ausbeute 60–80 %) und magnetische Beads für zellfreie DNA (Ausbeute 90–95 %). Beide Methoden schlossen eine RNase-A-Behandlung ein, um die störende Wirkung der mRNA auf den DNA-spezifischen Farbstoff zu eliminieren.Die Autoren zeigten experimentell, dass bereits geringe RNA-Mengen das DNA-Signal erheblich überschätzen lassen – und dass eine RNase-Behandlung notwendig ist, um RNA-bedingte Überschätzungen der DNA-Menge zu minimieren. Die RNA-Konzentration nach der Behandlung wurde kontrolliert und auf unter 1 ng/µl gesenkt.
Methode 3: Kapillarelektrophorese
Die Kapillarelektrophorese wurde eingesetzt, um sehr hohe DNA-Mengen – wie sie in einigen früheren Studien berichtet wurden – direkt sichtbar zu machen. Wenn DNA in Mengen von mehreren hundert bis tausend Nanogramm pro Dosis vorläge, würde sie in der Elektrophorese als deutlicher Peak oder Schmier erscheinen. Die Proben wurden mit Triton X-100 aufgeschlossen und mit RNase A behandelt. Es wurden zwei Kits eingesetzt – eines für den Standardbereich (0,5–50 ng/µl) und eines für den hochsensitiven Bereich (5–600 pg/µl pro Fragment).Methode 4: Kurzlesen-Sequenzierung (Illumina)
Zur Sequenzierung wurde DNA aus den Impfstoffproben mit einer optimierten Methode extrahiert, die Hitzebehandlung (95°C), RNase-A-Verdau und Proteinase-K-Behandlung kombiniert. Der Bibliotheksaufbau erfolgte ohne vorherigen Fragmentierungsschritt, um die ursprüngliche Fragmentlängenverteilung möglichst unverändert zu erhalten. Die Sequenzierung erfolgte auf einem NextSeq 2000 System mit paired-end Reads, was eine präzise Bestimmung der Fragmentlänge ermöglicht.Die bioinformatische Auswertung umfasste eine de-novo-Assemblierung der Reads zu vollständigen Plasmidsequenzen sowie ein anschließendes Mapping aller Reads auf die rekonstruierten Referenzsequenzen.
c) Zentrale Ergebnisse
qPCR-Befunde
Alle acht qPCR-Assays wurden in zwei unabhängigen Laboren mit drei Mitarbeitern durchgeführt. In keiner der 15 untersuchten Chargen wurde der regulatorische Grenzwert von 10 ng DNA pro Dosis überschritten.Dabei zeigten sich deutliche Unterschiede zwischen den einzelnen Assays – abhängig von der Amplikon-Größe. Die höchsten DNA-Mengen wurden mit dem kürzesten Amplikon (KAN 1C, 63 bp) gemessen: bis zu 8 ng/Dosis für Comirnaty. Die niedrigsten Werte lieferten die längsten Amplikons (SPIKE 1A und SPIKE 2, 228–233 bp): 0,5–0,7 ng/Dosis. Die Autoren erklären diese Diskrepanz mit der Fragmentlänge der DNA: Bei einer medianen Fragmentlänge von 150 bp können längere Amplikons viele Fragmente nicht erfassen – weil diese zu kurz sind, um beide Primerbindestellen zu enthalten.
Die Autoren interpretieren dieses konsistente Muster über verschiedene Assays hinweg als Hinweis auf die interne Konsistenz ihres methodischen Ansatzes.
Fluorometrie
Nach vollständiger RNase-A-Behandlung und DNA-Extraktion lagen die fluorometrisch gemessenen DNA-Mengen in allen Chargen unterhalb des Grenzwerts von 10 ng/Dosis.Die Autoren demonstrierten experimentell, dass ohne ausreichende RNase-Behandlung das DNA-Signal erheblich überschätzt wird: Bereits geringe RNA-Mengen erzeugen ein messbares Hintergrundsignal im DNA-Assay.
Die RNA-Messung zeigte, dass alle Comirnaty-Chargen mindestens 90 % der deklarierten mRNA-Menge enthielten. Bei den Spikevax-Chargen lagen die Werte teilweise darunter – was die Autoren auf die fortgeschrittene Überschreitung des Verfallsdatums zurückführen.
Kapillarelektrophorese
In keiner der untersuchten Chargen wurde ein spezifisches DNA-Signal detektiert – weder mit dem Standardkit (Nachweisbereich 0,5–50 ng/µl) noch mit dem hochsensitiven Kit (5–600 pg/µl pro Fragment). Schwache, unspezifische Signale um 60 und 300 bp waren in einigen Proben sichtbar, traten jedoch auch in der Negativkontrolle auf und werden daher als methodisches Hintergrundrauschen gewertet.Die Autoren argumentieren: Wenn DNA in Mengen von mehreren hundert bis tausend Nanogramm pro Dosis vorläge, wäre dies in der Kapillarelektrophorese als deutlicher Peak oder Schmier sichtbar. Das vollständige Fehlen eines solchen Signals widerspricht diesen Befunden direkt.
Die RNA-Integritätsanalyse zeigte, dass sechs von neun abgelaufenen Chargen eine mRNA-Integrität unter 50 % aufwiesen – erkennbar auch an der trüben Erscheinung der Fläschchen im Vergleich zu den klaren, nicht abgelaufenen Chargen.
Sequenzierung
Die vollständigen Sequenzen entstanden bioinformatisch durch Assemblierung vieler überlappender Kurzreads und stellen nicht notwendigerweise den direkten Nachweis einzelner physisch intakter Plasmidmoleküle dar.Der Anteil plasmidabgeleiteter Reads betrug 96,6 % bei Comirnaty (Spanne 95,3–98,1 %) und 88,7 % bei Spikevax (Spanne 83,5–94,5 %). Die verbleibenden Reads stammten überwiegend aus E. coli K12 – einem nicht-pathogenen Laborstamm – sowie aus Bakteriophagen-Sequenzen und repetitiven Sequenzen ohne Datenbankeinträge. Die Autoren weisen darauf hin, dass ein Teil dieser Reads auf Reagenzienkontaminationen zurückzuführen sein könnte, wie aus der Literatur bekannt.
Die Fragmentlängenanalyse ergab eine mediane Länge von 154 bp für Comirnaty (Spanne 130–181 bp) und 144 bp für Spikevax (Spanne 127–201 bp). Der Anteil der Fragmente über 300 bp war gering. Die Genomabdeckung war über das Plasmid ungleichmäßig verteilt – ein Muster, das mit einem nicht vollständig gleichmäßigen DNase-Verdau vereinbar ist.
d) Interpretation durch die Autoren
DNA-Menge unterhalb des Grenzwerts
Die Autoren schlussfolgern, dass die DNA-Rückstände in allen 15 untersuchten Chargen den regulatorischen Grenzwert von 10 ng pro Dosis einhalten. Dieses Ergebnis ist nach ihrer Einschätzung robust, weil die Ergebnisse durch mehrere komplementäre Methoden konsistent gestützt werden. Das Einbeziehen abgelaufener Chargen stärkt aus ihrer Sicht die Aussagekraft zusätzlich: Selbst unter diesen Umständen wurden keine übermäßigen DNA-Mengen gefunden.Herkunft und Charakter der DNA
Die nachgewiesene DNA stammt nach Einschätzung der Autoren ausschließlich aus dem Produktionsplasmid – also aus dem erwarteten und bekannten Nebenprodukt des Herstellungsprozesses. Sie ist stark fragmentiert, mit einer medianen Länge von etwa 150 bp. Diese Fragmentgröße liegt nach Ansicht der Autoren unterhalb der Schwelle biologischer Aktivität: Fragmente dieser Größe weisen nach Einschätzung der Autoren keine Eigenschaften auf, die typischerweise mit Replikationsfähigkeit oder effizienter genomischer Integration assoziiert sind.Erklärung der abweichenden Befunde früherer Studien
Die Autoren diskutieren mehrere methodische Faktoren, die aus ihrer Sicht zu den höheren DNA-Werten früherer Studien beigetragen haben könnten.Erstens weisen sie darauf hin, dass fluorometrische DNA-Assays ohne ausreichende RNase-Behandlung durch verbleibende mRNA erheblich beeinflusst werden können. In eigenen Kontrollversuchen führte bereits eine geringe Rest-RNA zu einer deutlichen Überschätzung des DNA-Signals. Die Autoren vermuten daher, dass ein Teil der in früheren Arbeiten berichteten hohen DNA-Werte auf RNA-bedingte Crosstalk-Effekte zurückzuführen sein könnte.
Zweitens betonen sie den Einfluss der Amplikon-Länge bei qPCR-basierten Verfahren. Da die nachgewiesene DNA überwiegend fragmentiert sei, erfassen längere Amplikons nur einen Teil der vorhandenen Fragmente. Unterschiede zwischen Studien könnten daher teilweise aus unterschiedlichen Primerdesigns und Nachweisbereichen resultieren.
Die Autoren interpretieren ihre Ergebnisse insgesamt als Hinweis darauf, dass die Bestimmung residualer DNA in mRNA-Impfstoffen stark methodenabhängig ist und eine standardisierte Analytik erforderlich wäre.
Methodische Empfehlungen
Die Autoren empfehlen für künftige Analysen: vollständige RNase-Behandlung mit Kontrolle des RNA-Restgehalts vor der fluorometrischen DNA-Messung; direkte Verwendung von Triton-X-100-behandeltem Impfstoff für die qPCR ohne vorherige DNA-Extraktion, um Verluste zu vermeiden; den Einsatz kurzer Amplikons für eine vollständigere Erfassung fragmentierter DNA; sowie die Kombination mehrerer komplementärer Methoden für robuste Aussagen.Biologische Einordnung
Die Autoren bewerten das Risiko durch die nachgewiesenen DNA-Rückstände als vernachlässigbar: geringe Menge, bekannte plasmidische Herkunft, hohe Fragmentierung sowie der Umstand, dass bei der Herstellung keine eukaryotischen Zelllinien verwendet werden. Sie sehen keinen Bedarf für eine Überarbeitung der bestehenden Grenzwerte oder Herstellungspraktiken.e) Methodische Einschränkungen
Amplikon-Größe und Fragmentlänge
Die Autoren zeigen selbst, dass die gemessenen DNA-Mengen stark von der Amplikon-Größe des jeweiligen qPCR-Assays abhängen. Mit dem kürzesten Amplikon (KAN 1C, 63 bp) wurden bis zu 8 ng/Dosis gemessen – nahe am regulatorischen Grenzwert. Mit den längsten Amplikons (SPIKE 1A und SPIKE 2, 228–233 bp) dagegen nur 0,5–0,7 ng/Dosis.Diese Unterschiede innerhalb derselben Studie verdeutlichen, dass qPCR-Ergebnisse wesentlich von der gewählten Assay-Architektur abhängen. Die gemessenen Werte repräsentieren daher keinen absoluten Gesamt-DNA-Wert, sondern den Anteil der Fragmente, der mit dem jeweiligen Primer-Design erfassbar ist.
Fragmentlängenbestimmung mittels Illumina
Die Sequenzierung ergab mediane Fragmentlängen von etwa 130–201 bp. Diese Werte sind jedoch nicht als direkte Messung der nativen Fragmentverteilung zu interpretieren.Der Bibliotheksaufbau umfasst mehrere SPRI-Aufreinigungsschritte sowie PCR-Amplifikationen, die sehr kurze Fragmente (<150 bp) systematisch benachteiligen können. Gleichzeitig können auch sehr lange Fragmente (>800 bp) während der Bibliotheksvorbereitung und Sequenzierung weniger effizient repräsentiert werden und dadurch unterrepräsentiert oder nicht erfasst sein. Die berichtete Längenverteilung spiegelt daher primär die nach der Bibliotheksvorbereitung noch detektierbaren Fragmente wider – nicht zwangsläufig die ursprüngliche Verteilung im Impfstoff.
Damit erlaubt die Studie keine abschließende Aussage weder über den tatsächlichen Anteil sehr kurzer DNA-Fragmente unterhalb der Detektionsgrenze noch über das mögliche Vorhandensein längerer DNA-Fragmente außerhalb des effizient erfassten Größenbereichs.
Umrechnungsmethode bei der qPCR
Die gemessenen Kopienzahlen wurden anhand der Molekülmasse der jeweiligen qPCR-Amplikons in Nanogramm umgerechnet – nicht anhand der vollständigen Plasmidlänge. Dieses Vorgehen ist methodisch konsistent für die Quantifizierung der tatsächlich amplifizierten Zielregionen. Die resultierenden Nanogramm-Werte sind jedoch nicht direkt mit Studien vergleichbar, die ihre Berechnung auf vollständige Plasmidsequenzen stützen.Einfluss der Extraktionseffizienz
Die fluorometrischen DNA-Werte hängen zusätzlich von der Effizienz der verwendeten Extraktionsmethode ab. Die Autoren geben für die Phenol-Chloroform-Extraktion eine Ausbeute von etwa 60–80 % und für die magnetischen Beads eine Ausbeute von etwa 90–95 % an.Eine unvollständige Extraktion kann prinzipiell dazu führen, dass ein Teil der vorhandenen DNA – insbesondere sehr kurze Fragmente oder noch LNP-assoziierte Nukleinsäuren – nicht vollständig erfasst wird. Entsprechend zeigten die Bead-basierten Extraktionen in der Studie tendenziell höhere DNA-Werte als die Phenol-Chloroform-Methode.
Keine Messung vor LNP-Aufschluss
Im Unterschied zu einigen früheren Studien wurde keine systematische Vergleichsmessung vor und nach gezieltem LNP-Aufschluss durchgeführt. Damit lässt sich aus den Daten nicht direkt ableiten, welcher Anteil der nachgewiesenen DNA innerhalb der Lipid-Nanopartikel verkapselt vorlag.Kapillarelektrophorese: Nachweisgrenze
Die Kapillarelektrophorese eignet sich gut zum Nachweis größerer DNA-Mengen. Für sehr niedrige Konzentrationen im einstelligen Nanogramm-Bereich ist ihre Sensitivität jedoch begrenzt. Die Methode ergänzt daher die qPCR- und Sequenzierungsdaten, ersetzt diese aber nicht.Keine SV40-spezifische Analyse
Die Studie enthält keinen spezifischen Assay für SV40-Promoter-/Enhancer-Sequenzen. Aussagen über das Vorhandensein oder Fehlen solcher Sequenzen lassen sich aus den vorliegenden Daten daher nicht ableiten. Dies stellt keine methodische Schwäche im engeren Sinn dar, da die Untersuchung nicht auf SV40-Nachweise ausgerichtet war; im Vergleich zu anderen Arbeiten bleibt dieser Aspekt jedoch unbeantwortet.f) Einordnung in den Gesamtkontext
Die Studie von Achs et al. stellt bislang eine der methodisch umfassendsten Untersuchungen zu DNA-Rückständen in mRNA-Impfstoffen dar. Sie kombiniert mehrere komplementäre Analyseverfahren – qPCR mit unterschiedlich langen Amplikons, fluorometrische Messungen mit kontrollierter RNase-Behandlung, Kapillarelektrophorese und Kurzlesen-Sequenzierung – an denselben Proben und in zwei verschiedenen Laboren. Dadurch können methodische Artefakte einzelner Verfahren besser erkannt und eingeordnet werden.
Im Gesamtkontext der bisherigen Literatur verdeutlicht die Studie, dass der Nachweis und die Quantifizierung von DNA-Rückständen in mRNA-Impfstoffen stark methodenabhängig sind. Unterschiedliche Probenvorbereitungen, Extraktionsverfahren, RNase-Behandlungen, qPCR-Designs und Sequenzierungsansätze können zu deutlich unterschiedlichen Ergebnissen führen.
Im Vergleich zu früheren Arbeiten stützt die Studie die Einschätzung, dass die untersuchten Chargen keine außergewöhnlich hohen Mengen an DNA-Rückständen enthalten. Gleichzeitig bleiben einige methodische Fragen – insbesondere hinsichtlich der vollständigen Fragmentverteilung sehr kurzer oder sehr langer DNA-Fragmente – auch mit diesem Ansatz nicht abschließend geklärt.
Die Arbeit von Achs et al. trägt damit weniger zur endgültigen Klärung der Kontroverse bei als zur präziseren methodischen Einordnung der bisherigen Befunde. In diesem Sinne ergänzt sie die vorangegangenen Studien – nicht indem sie deren Ergebnisse pauschal widerlegt, sondern indem sie zeigt, unter welchen methodischen Bedingungen bestimmte Aussagen getroffen werden können.
⚬ ⚬ ⚬
4.4. Zwischenfazit: Was wissen wir – und was nicht?
Was die vorliegenden Studien gemeinsam zeigen
Die in diesem Kapitel vorgestellten Untersuchungen stammen aus unterschiedlichen Laboren, wurden zu verschiedenen Zeitpunkten durchgeführt und setzen teilweise sehr verschiedene analytische Verfahren ein. Dennoch lassen sich einige Befunde benennen, die über mehrere Studien hinweg konsistent sind.
Erstens: DNA-Rückstände in mRNA-basierten COVID-19-Impfstoffen sind nachweisbar. Das ist kein überraschendes Ergebnis – es ist eine bekannte und erwartete Konsequenz des Herstellungsprozesses, bei dem eine DNA-Vorlage für die in-vitro-Transkription verwendet wird. Dass der DNase-I-Verdau keine vollständige Entfernung aller DNA-Fragmente gewährleistet, ist in der Fachliteratur anerkannt.
Zweitens: Die nachgewiesene DNA stammt überwiegend aus dem Produktionsplasmid. Das zeigen sowohl die Sequenzierungsdaten von McKernan, Raoult und Achs et al. als auch die PCR-basierten Analysen mehrerer Arbeitsgruppen übereinstimmend.
Drittens: Mehrere Studien liefern Hinweise darauf, dass ein relevanter Anteil der nachgewiesenen DNA vor enzymatischem Abbau geschützt und mit den Lipid-Nanopartikeln assoziiert ist.
Wo die Studien voneinander abweichen
Die zentrale Streitfrage – wie viel DNA tatsächlich vorhanden ist und ob die regulatorischen Grenzwerte eingehalten werden – wird von den vorliegenden Studien unterschiedlich beantwortet. Diese Diskrepanz ist nicht primär auf unterschiedliche Proben zurückzuführen, sondern lassen sich zumindest teilweise durch unterschiedliche methodische Ansätze erklären.
Fluorometrische Messungen ohne ausreichende RNase-Behandlung liefern systematisch höhere Werte, weil der Farbstoff auch an RNA bindet. PCR-basierte Messungen sind von der Amplikon-Größe abhängig: Längere Amplikons erfassen weniger Fragmente und unterschätzen die Gesamt-DNA. Sequenzierungsverfahren liefern qualitative Informationen über Sequenzidentität und Fragmentlänge, aber keine zuverlässige absolute Quantifizierung. Jede Methode misst damit etwas leicht anderes – und liefert entsprechend unterschiedliche Zahlen.
Das bedeutet: Unterschiede zwischen Studien sind nicht zwangsläufig ein Beleg für fehlerhafte Arbeit einer der Gruppen. Sie können – und müssen in vielen Fällen – als Ausdruck methodischer Variabilität verstanden werden.
Was offen bleibt
Trotz der inzwischen vorliegenden Datenmenge bleiben mehrere Fragen unbeantwortet.
Die Frage nach der tatsächlichen Fragmentlängenverteilung – insbesondere dem Anteil sehr kurzer Fragmente unter 100 bp – ist bisher nicht abschließend geklärt. Illumina-Sequenzierung unterschätzt kurze Fragmente systematisch; ONT-Datensätze können durch prozessbedingte Selektions- und Sequenzierungsbiases relativ stärker von längeren Fragmenten geprägt sein. Ein Verfahren, das die native Fragmentverteilung ohne größenselektive Verzerrung abbildet, wurde in den vorliegenden Studien nicht eingesetzt.
Die biologische Relevanz der nachgewiesenen DNA – einschließlich Fragen nach möglicher zellulärer Aufnahme, Persistenz oder genomischer Integration – wurde bisher nicht direkt untersucht. Vorhandene theoretische Modelle und Analogien aus anderen biologischen Systemen reichen derzeit nicht aus, um belastbare Aussagen über ein tatsächliches Risiko abzuleiten, rechtfertigen jedoch weitere Untersuchungen.
Die Frage nach dem SV40-Promoter/Enhancer – seiner tatsächlichen Häufigkeit in verschiedenen Chargen und seiner biologischen Aktivität in fragmentierter Form – wurde von den vorliegenden Studien unterschiedlich adressiert und ist nicht abschließend beantwortet.
Was das für die wissenschaftliche Diskussion bedeutet
Ein grundlegendes Problem, das sich durch alle hier vorgestellten Studien zieht, ist das Fehlen eines standardisierten, validierten Analyseprotokolls für DNA-Rückstände im fertigen Impfstoffprodukt. Die regulatorischen Grenzwerte existieren – aber die Methode, mit der sie überprüft werden sollen, ist nicht einheitlich vorgeschrieben. Das führt dazu, dass verschiedene Labore mit unterschiedlichen Methoden zu unterschiedlichen Ergebnissen kommen, ohne dass ein direkter Vergleich möglich ist.
Ein sinnvoller nächster Schritt wäre die Etablierung eines orthogonalen Standardprotokolls: dieselbe Probe, mehrere validierte Methoden, in verschiedenen unabhängigen Laboren. Erst ein solcher Ansatz würde erlauben, methodenbedingte Variabilität von tatsächlichen Unterschieden zwischen Chargen zu trennen – und damit belastbare, vergleichbare Daten zu erzeugen.
Regulatorische Einordnung
Die regulatorischen Grenzwerte für Rest-DNA wurden ursprünglich für klassische biologische Arzneimittel und nicht speziell für LNP-formulierte modRNA-Impfstoffe entwickelt. Inwieweit bestehende Grenzwerte und Prüfverfahren auf diese neue Plattformtechnologie vollständig übertragbar sind, ist bislang nicht abschließend diskutiert.
Abschließende Einordnung
Die vorliegenden Studien haben eine wichtige Funktion erfüllt: Sie haben eine wissenschaftliche Diskussion angestoßen, die ohne sie nicht stattgefunden hätte. Gleichzeitig zeigen sie die Grenzen unabhängiger Nachanalysen außerhalb des regulierten Rahmens: fehlende Validierung, begrenzte Stichprobengrößen und methodische Heterogenität.
Was bleibt, ist eine sachlich begründete Forderung: mehr Transparenz, einheitlichere Methoden und unabhängige Replikation durch verschiedene Laborgruppen. Das ist keine alarmistische Schlussfolgerung – es ist das, was gute Wissenschaft grundsätzlich auszeichnet.
5. Schlussbetrachtung und Ausblick
Die Diskussion um DNA-Rückstände in mRNA-Impfstoffen zeigt auf mehreren Ebenen, wie komplex die analytische Bewertung neuartiger biologischer Arzneimittel sein kann.
Was zunächst wie eine einfache Frage erscheint – in welcher Menge DNA-Rückstände vorhanden sind – erweist sich bei näherer Betrachtung als stark abhängig von Probenaufbereitung, Nachweismethode und Dateninterpretation.
Zentrale Erkenntnisse der Arbeit
Die Herstellung modRNA-basierter Impfstoffe erfolgt über einen mehrstufigen Prozess, bei dem bakterielle DNA-Vorlagen unverzichtbar sind. Die Entfernung dieser DNA gehört zu den regulären Reinigungsschritten des Herstellungsverfahrens. Die vorliegenden Untersuchungen zeigen jedoch übereinstimmend, dass sich residuale DNA-Fragmente in kommerziellen Impfstoffchargen nachweisen lassen – wenn auch in methodisch unterschiedlich bestimmten Mengen.
Die Auswertung der bisherigen Studien macht deutlich, dass der Nachweis und die Quantifizierung von DNA-Rückständen wesentlich komplexer sind, als es die regulatorischen Grenzwerte allein vermuten lassen. Unterschiedliche Aufschlussverfahren, Extraktionsmethoden, RNase-Behandlungen, qPCR-Designs und Sequenzierungsansätze können zu erheblich abweichenden Ergebnissen führen.
Die Kontroverse der vergangenen Jahre verdeutlicht damit nicht nur Unterschiede zwischen einzelnen Studien, sondern vor allem die Grenzen bislang fehlender Standardisierung unabhängiger Nachanalysen.
Offene Fragen für die Forschung
Trotz der inzwischen vorliegenden Datenmenge bleiben mehrere Fragen offen:
Wie hoch ist der tatsächliche Anteil sehr kurzer oder potenziell biologisch aktiver DNA-Fragmente in verschiedenen Impfstoffchargen?
Welche Aussagekraft besitzen aktuelle Messverfahren hinsichtlich der nativen Fragmentverteilung im Impfstoff?
Welche Rolle spielen RNA:DNA-Hybride oder andere Nukleinsäurekomplexe für die Stabilität und Nachweisbarkeit residualer DNA?
Und schließlich: Welche biologische Relevanz haben die nachgewiesenen DNA-Fragmente – einschließlich des SV40-Promoter/Enhancer-Elements – tatsächlich unter realen physiologischen Bedingungen?Viele dieser Fragen lassen sich auf Basis der derzeitigen Daten weder eindeutig bestätigen noch ausschließen. Sie erfordern weitere Untersuchungen mit standardisierten, orthogonalen Analyseverfahren sowie unabhängige Replikationen in verschiedenen Laboren.
Ausblick
Die wissenschaftliche Diskussion über DNA-Rückstände in modRNA-Impfstoffen ist damit nicht abgeschlossen. Gleichzeitig zeigt die bisherige Forschung, dass zwischen analytischem Nachweis und biologischer Bedeutung klar unterschieden werden muss.
Während sich die vorliegende Arbeit auf Herstellungsprozesse, Nachweismethoden und experimentelle Befunde konzentriert, bleibt die Frage nach möglichen biologischen Auswirkungen Gegenstand weiterer Forschung und zukünftiger Betrachtungen. Dazu gehören unter anderem Fragen zur biologischen Aktivität residualer DNA, zur Stabilität von Nukleinsäurekomplexen, zu theoretischen Integrationsmechanismen sowie zur Bedeutung funktioneller Sequenzelemente.
Unabhängig von der Bewertung einzelner Studien ergibt sich daraus eine grundsätzliche wissenschaftliche Forderung: mehr Transparenz, besser standardisierte Analyseverfahren und reproduzierbare unabhängige Untersuchungen. Gerade bei neuartigen biologischen Technologien ist dies keine außergewöhnliche Erwartung, sondern ein zentraler Bestandteil wissenschaftlicher Qualitätssicherung.
Danksagung
Mein aufrichtiger Dank gilt allen Wissenschaftlern, Forschern und engagierten Persönlichkeiten, die mit ihrer Arbeit – in Studien, Artikeln und Vorträgen – zur Erforschung dieses komplexen Themas beitragen. Ihr Engagement ist von unschätzbarem Wert – für den Fortschritt des Wissens ebenso wie für eine sachliche öffentliche Diskussion.
Der wissenschaftliche Widerstreit, den dieser Text zu dokumentieren versucht, ist kein Mangel – er ist das Wesen guter Wissenschaft.
✧ ✧ ✧
Nachtrag
Ich habe diesen Text als Laie begonnen.
Mit Unsicherheit, Neugier und offenen Fragen.
Vielleicht war genau das ein Vorteil.Viele Fragen bekamen Antworten.
Neue Fragen kamen hinzu.
Je tiefer ich einstieg, desto klarer wurde:
Nicht nur die Biologie ist komplex.
Auch die Messung der Biologie ist komplex.Der Weg durch die Materie war lang.
Auf dieser Reise hatte ich Hilfe:
Geduldige KI-Systeme, die erklärten,
widersprachen, strukturierten, formulierten – und mitdachten.
Es waren lange, fruchtbare Dialoge
zwischen Mensch und Maschine.Und am Ende bleibt eine wichtige Erkenntnis:
Lernen ist ein gemeinsamer Prozess.
Stand: Mai 2026
-
Personalized Medicine – Heaven or Hell

When HSBC Bank placed posters at airports such as Gatwick and Dubai in 2013 as part of its global campaign „The future is full of opportunity” – showing a fingerprint, a QR code, and the provocative slogan „Your DNA will be your data” – the message was unambiguous: the future will be personal, perhaps unsettlingly personal. The bank sought to position itself as a forward-looking company that addresses digitalization, artificial intelligence, sustainability, and genetics head-on.
The slogan seemed ambiguous – both a promising vision and an alarming prospect. Critics immediately sounded the alarm: your DNA becomes readable, analyzable, and digitally accessible – an open book about ancestry, health risks, perhaps even personality traits – comparable to biometric data such as fingerprints or facial scans. What was once considered the most intimate aspect of a person is now becoming a file: machine-decipherable, potentially storable, possibly tradable. The human being is reduced to information – „bits and bytes” that could be processed, sold, or misused.
Who owns the rights to your genetic data? And who has access to it? You yourself? The company that conducted the test? Do surveillance, data leaks, and misuse by state and private actors reach a new dimension as a result? The vision of designer babies, genetic selection, and personalized advertising based on biological markers raises serious ethical questions. Will progress ultimately be paid for at the price of individual freedom?
However, the biotech revolution has long since gained tremendous momentum, and more and more voices recognize the enormous potential of this development. The March 2024 e-book edition of HSBC Bank Frontiers – Biotech – Five Big Ideas Shaping the Biotech Revolution states:
„The idea of exploiting nature for goods and services is nothing new. But the practice entered its modern era in the 1970s, with the advent of genetic engineering and the establishment of the first biotech companies. This brought together science and business like never before, and the influx of capital galvanized research and development efforts.
Over the three decades that followed, the field was accelerated by breakthrough devices such as DNA sequencers and synthesizers, and biology itself slowly morphed into a computational, data-driven discipline. Following the turn of the millennium, as researchers successfully charted ever greater regions of the human genome, and the cost of sequencing that genome rapidly declined – from around $100 million in 2001 to less than $1,000 today – the potential of biotech to transform our lives has become electrifyingly apparent.
Over the past few years, the possibilities unlocked by biotech advances – from treating genetic diseases to de-extincting animal species – have been enlarged by progress in complementary fields such as artificial intelligence. This has underpinned a coming-of-age moment in this space.“
Nowhere else do benefits and risks collide as directly as in personalized – or precision-based – medicine. Its goal is to use individual genetic and biological information to refine diagnoses, personalize therapies, and detect diseases at an early stage. The vision is promising: fewer side effects, better chances of recovery, more effective prevention – medicine tailored to the individual.
But here, too, what is celebrated as medical progress brings societal, legal, and ethical challenges:
- Who decides which genetic data are relevant?
- What role does access to these technologies play – does it exacerbate social inequalities?
- And how can discrimination, for example through genetic scoring by insurance companies or employers, be prevented?
Between the hope for individual healing and the fear of collective disempowerment lie fundamental questions: Does access to one’s biological data empower the individual – or expose them? Is personalized medicine a blessing – or the beginning of an era of subtle dehumanization?
Hence the crucial question is: Is personalized medicine the promised ascent into heaven – or the plunge through the gates of hell?
This blog post invites readers to consider personalized medicine in all its complexity – beyond hype or alarmism. It is deliberately aimed as well at non-experts who wish to engage critically with the opportunities, risks, and ethical tensions of this medical revolution.
📑Table of Contents
1. Conceptual Classification and the Fundamental Paradigm
2. Driving Forces – Between Vision and Reality
3. The Future of Medicine Begins in the Cell
3.1. How Biology and Technology Come Together
3.2. Data, Responsibility, and the Bigger Picture
4. From DNA to Knowledge – A High-Speed Train of Innovation
4.1. DNA – The Blueprint of Life
a) DNA: The Molecular Information Archive of Life
b) From Code to Protein: How Instructions Become Reality
c) Epigenetics & Non-coding DNA: The Conducting Team of Our Genome
d) The Hidden Control Center: What Non-coding DNA Regulates
e) When the Operating System Makes You Sick
f) Open Mysteries in Research
g) The True Magic of Genes
4.2. Genetic Variation and Its Consequences
4.3. Genome Sequencing: Key to Individual Genetic Material
a) The Journey of DNA: From Umbilical Cord Blood to Genetic Information
b) Three Directives for Gene Diagnostics: WGS, WES, and Panel
4.4. Methods of Genome Sequencing
4.4.1. Method 1: Copying with Fluorescent Dye
a) Sanger Sequencing
b) Illumina Technology
c) Single-Molecule Real-Time (SMRT) Sequencing
4.4.2. Method 2: High-Tech Tunnel – Electrically Scanning DNA
a) Oxford Nanopore Sequencing
4.4.3. Genome Sequencing – Technologies Compared
a) Throughput vs. Usability – Quantity Does Not Always Equal Quality
b) Cost-Effectiveness – Cost per Base vs. Cost per Insight
c) From Sample to Result – How Quickly Does the Whole Genome Speak?
d) Clinical Applications – Which Technology Fits Which Purpose?
e) AI as Co-Pilot – Automation Without Relinquishing Responsibility
f) Comparison Table: Sanger, Illumina, PacBio, ONT
g) Key Takeaways: Between Maturity and Routine
4.5. Genome Sequencing: The Next Technological Leap
a) FENT: How a Microchip Is Revolutionizing DNA Analysis
b) SBX: When DNA Stretches to Be Understood
c) The G4X System: From the Genetic Recipe to the Spatial Atlas of the Cell
4.6. From Code to Cure – Bioinformatics as the Key to Tomorrow’s Medicine
4.6.1. From the Genetic Code to Computer-Aided Genome Analysis
4.6.2. Modern Bioinformatics – The Digital Toolbox of Biology
5. A Success Story
6. A Look into the Future: The Synthetic Human Genome Project
7. Personalized Medicine and Smart Governance
7.1. Power, Governance, and Smart Governance
7.2. Governmentality
7.3. Personalized Medicine as a Catalyst for Biomolecular Governmentality
7.4. The Global Trend: Worldwide Biomolecular Governmentality
7.5. Conclusion: The Global Ambivalence of Biomolecular Power
8. Epilogue
1. Conceptual Classification and the Fundamental Paradigm
The development of modern medicine toward tailored treatments is often described using terms such as „personalized medicine”, „individualized therapy” or „precision medicine”. But what exactly do these buzzwords mean?
Personalized medicine is currently considered one of the most popular terms – catchy like a marketing slogan. It carries the promise of tailoring therapies to each individual: to their genes, their lifestyle, their biology. Accordingly, the term is frequently used in the media and public debate.
Individualized therapy sounds less glamorous but conveys a similar idea: the adjustment of medications or dosages to the needs of individual patients. This term is used primarily in clinical practice.
Finally, precision medicine is the most sober of the three terms – a technical term that dominates research laboratories and scientific publications. It emphasizes the data-driven approach: algorithms, genetic analyses, and biomarkers determine which therapy works for which patient group. This is the language of science.
But regardless of which term is used, they all represent a paradigm shift: moving away from standardized procedures toward medicine that focuses on the biological uniqueness of each individual.
2. Driving Forces –Between Vision and Reality
The driving forces behind personalized medicine are diverse and closely interconnected. On one hand, there are scientific and technological developments that have enabled enormous progress in recent decades: new methods to read genetic material quickly and accurately, powerful data analysis through artificial intelligence, and the availability of large biomedical datasets. These technologies form the foundation for recognizing individual differences between patients and leveraging them medically.
On the other hand, social and political impulses act as accelerators of this transformation. Public health programs, targeted research funding, strategy papers from international health organizations, and specific legislative initiatives actively drive the integration of personalized medicine into healthcare practice. Increased health awareness among the population, as well as growing expectations for individualized treatment options, further reinforce this trend.
Personalized medicine is therefore not merely a product of technological feasibility, but emerges at the intersection of innovation, politics, and societal demand.
A current example of the practical implementation of this development comes from the United Kingdom: under a new 10-year plan, every newborn is to undergo a DNA test. The so-called whole-genome sequencing will screen for hundreds of genetically linked diseases – with the aim of detecting potentially life-threatening conditions early and preventing them in a targeted manner. According to British Health Secretary Wes Streeting, the goal is to fundamentally transform the National Health Service (NHS): moving away from mere disease treatment toward a system of prediction and prevention. Genomics – the comprehensive analysis of genetic information – will serve, in combination with artificial intelligence, as an early warning system.
„The revolution in medical science means that we can transform the NHS over the coming decade, from a service which diagnoses and treats ill health to one that predicts and prevents it“, said Streeting. The vision: personalized healthcare that identifies risks before symptoms even appear – thereby improving quality of life and alleviating pressure on the healthcare system.
But as convincing as this vision may sound, there are also cautionary voices coming from the scientific community. For example, geneticist Prof. Robin Lovell-Badge from the Francis Crick Institute warns against underestimating the complexity of genomic data. Not only is technical expertise required for data collection, but above all qualified personnel who can communicate the information obtained in a responsible and comprehensible manner. Data alone does not constitute a diagnosis – what is crucial is its meaningful and patient-oriented interpretation.
What does it concretely mean to rethink medicine?
The real revolution is taking place in laboratories and data centers, where the building blocks of personalized medicine are being developed. The following chapters take you behind the scenes, step by step, and reveal the key technologies needed to bring personalized medicine from concept to clinical practice.
3. The Future of Medicine Begins in the Cell
3.1. How Biology and Technology Come Together
Our bodies consist of billions of cells – tiny, specialised units that work together day after day to enable health and life. In every single cell, countless processes occur that are precisely coordinated with one another. These cellular mechanisms are crucial for understanding health, disease, and the development of new, targeted therapies.
One central mechanism is energy production: cells convert nutrients and oxygen into ATP (adenosine triphosphate) – the body’s „energy currency”. Without ATP, no cell could function, no muscle could move, no thought could arise.
At the same time, cells continuously produce proteins – based on genetic information. These complex molecules perform essential tasks in the body, serving as chemical catalysts, signaling molecules, structural components, or transporters.
Cellular cleaning and renewal are also vital: processes such as autophagy break down and recycle damaged cellular components – an internal waste disposal system that can help prevent diseases like Alzheimer’s or Parkinson’s.
For cells to work together in a coordinated way within the body, they need functioning signaling pathways – they „communicate” with one another through chemical messengers. This cell communication controls when cells should grow, rest, or respond to changes.
Another key process is cell division – essential for growth, wound healing, and tissue renewal. At the same time, DNA repair mechanisms ensure that errors in the genetic material are corrected. If this fails, apoptosis, the programmed cell death, takes over to limit potential damage.
All these mechanisms run in the background – constantly and with high precision. However, if one of these processes is disrupted, it can lead to serious diseases such as cancer, autoimmune diseases or metabolic disorders. This is exactly where personalized medicine comes in. It uses state-of-the-art technologies to better understand these cellular processes and influence them in a targeted manner.
Thanks to genome sequencing (for example through „next-generation sequencing”), it is possible to identify genetic alterations that influence protein production or repair mechanisms.
Biomarker analyses (the examination of specific molecules) in blood, tissue, or other bodily fluids reveal whether certain signaling pathways are over- or underactive – and help predict individual disease progressions or select appropriate therapies.
Single-cell analysis makes it possible to visualize differences even between individual cells – for example, in a tumor, where some cells respond to therapy and others do not. This enables more precise treatment.
Proteomics (the analysis of all proteins in a cell) and metabolomics (the analysis of metabolic products) also provide a current picture of how active specific cellular mechanisms really are – for example, whether a cell is stressed, has sufficient energy, or is in the process of dividing.
Artificial intelligence (AI) helps analyze these vast amounts of data and identify patterns – for example, which combination of cellular disorders is typical for a particular type of cancer. Digital health data (such as from wearables) complement this picture in daily life and enable precise long-term monitoring.
Even new therapeutic approaches are based on understanding cellular processes: gene therapies and CRISPR-based gene editing target the DNA to correct faulty mechanisms. In organoids – mini-organ models in the lab – drugs can be tested directly on patient-specific cell models, without any risk to humans.
Modern technologies now reveal what was previously hidden: how cells work, what causes imbalances in disease – and how to intervene in a targeted manner. This has given rise to individualized medicine that is no longer based on conjecture, but on measurable biology.
This new precision brings not only opportunities, but also new responsibilities.
3.2. Data, Responsibility, and the Bigger Picture
The complexity of these cellular processes is directly reflected in the complexity of personalized medicine: the more precisely we understand the intricate processes and interactions in our cells, the more targeted – but also more demanding – diagnosis and therapy become. This new precision requires new technologies, new ways of thinking, and a deep biological understanding. At the same time, it opens the door to more effective, sustainable, and tailored treatment concepts.
However, the more targeted and profound these interventions become, the greater the responsibility – and the challenge of identifying and controlling undesirable long-term effects or side effects at an early stage. Particularly in the case of innovative procedures such as gene therapy or immunomodulation, it is important to carefully weigh up the benefits against the potential risks. Personalized medicine therefore requires not only precision, but also foresight.
A central element of this foresight is the availability of personal health data – not only in individual cases, but on a large scale.
Only by combining individual data with population data is it possible to understand biological processes in detail, predict disease progression and develop personalised therapies. Genetic information, laboratory values, imaging data and digital everyday measurements provide clues as to how cellular mechanisms work in a specific person – and how they vary in others.
To interpret these individual patterns medically, large datasets are needed for comparison. Only in this way can AI systems detect complex relationships that escape the human eye – such as rare genetic variants, molecular risk constellations, or unexpected therapy effects. The rule here is: the larger and more diverse the data base, the more reliable the models – especially when they are continuously enriched with new information.
However, this progress can only be achieved if people are willing to share their health data – and can trust that it will be used securely and responsibly.
The future of medicine begins where all life originates: in the cell. But its success depends on the interplay of three forces: technological innovation, scientific depth – and ethical responsibility.

4. From DNA to Knowledge – A High-Speed Train of Innovation
How genome sequencing and bioinformatic analysis are revolutionizing medicine
4.1. DNA – The Blueprint of Life
a) DNA: The Molecular Information Archive of Life
b) From Code to Protein: How Instructions Become Reality
c) Epigenetics & Non-coding DNA: The Conducting Team of Our Genome
d) The Hidden Control Center: What Non-coding DNA Regulates
e) When the Operating System Makes You Sick
f) Open Mysteries in Research
g) The True Magic of Genes
4.2. Genetic Variation and Its Consequences
4.3. Genome Sequencing: Key to Individual Genetic Material
a) The Journey of DNA: From Umbilical Cord Blood to Genetic Information
b) Three Directives for Gene Diagnostics: WGS, WES, and Panel
4.4. Methods of Genome Sequencing
4.4.1. Method 1: Copying with Fluorescent Dye
4.4.2. Method 2: High-Tech Tunnel – Electrically Scanning DNA
4.4.3. Genome Sequencing – Technologies Compared
4.5. Genome Sequencing: The Next Technological Leap
4.6. From Code to Cure – Bioinformatics as the Key to Tomorrow’s MedicineImagine standing on a train platform and watching a train rush by at such speed that you can barely make out the image in the window. This is how many people perceive developments in personalized medicine: technologies are advancing at breakneck speed, terms seem abstract – and yet there is a common thread running through it all: the human genome.
As diverse as modern approaches may be – from organoids and CRISPR-based gene editing to RNA-based medicines – at the center lies the human genetic blueprint. Today, genome sequencing forms the foundation of many diagnoses and therapeutic decisions. It reveals which genetic alterations trigger diseases, which signaling pathways are affected – and where targeted interventions might be possible.
What once cost billions and took years can now be done in a matter of days: the entire human genome can be decoded. But the sequence alone does not constitute a diagnosis. The sequence of three billion base pairs only has medical value once it is understood – and this is where the real challenge begins.
Bioinformatics enters the stage: an interdisciplinary field that combines mathematics, computer science and biology to derive patterns, risks and treatment options from raw data. It is the real bottleneck of personalized medicine – because it determines whether genetic variants are considered harmless, risky or disease-causing.
Individual sequences are compared with international reference databases, algorithms analyze millions of genetic variants, and learning systems model the complex interplay of genetics, environment, and metabolism.
This process is highly complex – and at the same time essential. Only computer-assisted evaluation makes DNA medically usable. There is an inseparable symbiosis between genome sequencing and bioinformatic analysis: the sequence provides the data, the analysis provides the understanding. Without it, DNA remains a mere string of letters.
This is precisely why the following sections focus on the crucial question:
What does it mean to decode the genome – and to truly understand it?
What conclusions are possible – and where are their limits?For anyone who wants to understand the future of medicine, it is essential to know how we read the human genome today – and how we are learning to interpret it.
The high-speed train of innovation has long since departed. Anyone who wants to ride along must understand where it is headed.
4.1. DNA – The Blueprint of Life
To embark on this journey, it is worth taking a brief look at the molecular foundations: how our DNA becomes a blueprint, how proteins are formed from it via the intermediate step of RNA, and how epigenetics fine-tunes these processes. Only this foundation makes it possible to understand what is actually happening when we decode the genome.
a) DNA: The Molecular Information Archive of Life
In every cell of our body lies a carefully packaged and astonishing molecule: DNA (deoxyribonucleic acid). It contains the complete blueprint and operating instructions of our body – a molecular archive of life’s information.
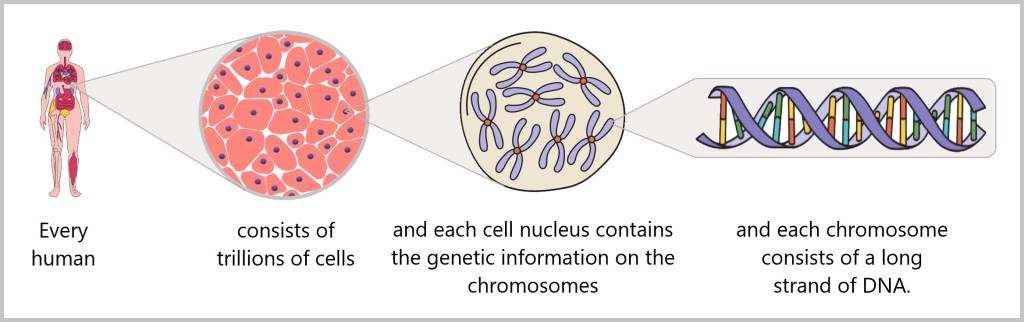
Fig. 1: DNA contains genetic information [The Wonderful World of Life] Structurally, it resembles a spiral-shaped rope ladder: two sugar-phosphate chains form the sides, with the „rungs“ – the bases adenine (A), cytosine (C), guanine (G) and thymine (T) – in between. They always pair up according to fixed rules: A with T, C with G. This complementary bond makes each strand a template for the other – a molecular backup system.
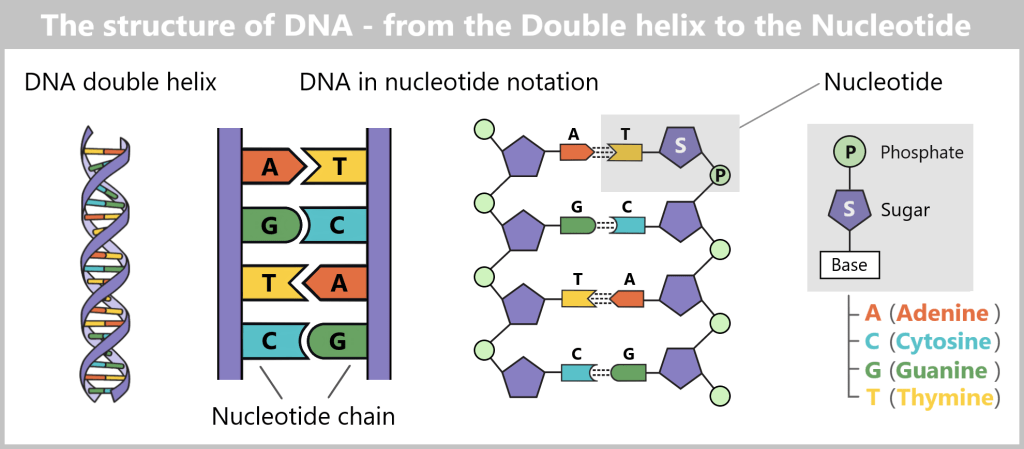
Fig. 2: Schematic representation of the basic structure of DNA [The Wonderful World of Life] If you unravel the DNA from a single cell, it measures around two meters – with a diameter of just a few millionths of a millimeter. Life therefore literally hangs on a wafer-thin but astonishingly long thread.
The sequence of around three billion base pairs is as unique as a fingerprint. It forms the genetic code – the instructions for producing proteins, the molecules that control the structure, function and regulation of all biological processes.
As a central information archive, DNA is both extremely valuable and sensitive. That is why it is stored safely in the cell nucleus, comparable to a safe. In order for the body to access the information in this safe, it needs a clever transport system. After all, the blueprints for proteins – the molecular architects of all life processes – must reach the ribosomes, the so-called „protein factories“ in the cytoplasm. This is where proteins are produced – a process known as protein biosynthesis.
But how does a specific blueprint travel safely from the cell nucleus to these factories? In other words: How is the genetic information for a particular protein transferred from its protected archival location to the site of production?
b) From Code to Protein: How Instructions Become Reality
The journey begins with the genes – specific sections of DNA that contain the blueprints for proteins. There are around 20.000 such genes in the human genome.
DNA is not just a passive information archive in the cell nucleus – it contains an active repertoire of instructions. You can think of each gene as a chapter in a molecular cookbook: often with multiple versions of a recipe, adapted to the cell type, developmental stage, or external conditions.
First, a temporary „working copy” of a gene is created – in the form of RNA (ribonucleic acid). It resembles DNA but uses the base uracil (U) instead of thymine (T) and usually consists of a single strand. This transient structure makes RNA ideal for short-term use – it is degraded after fulfilling its function.
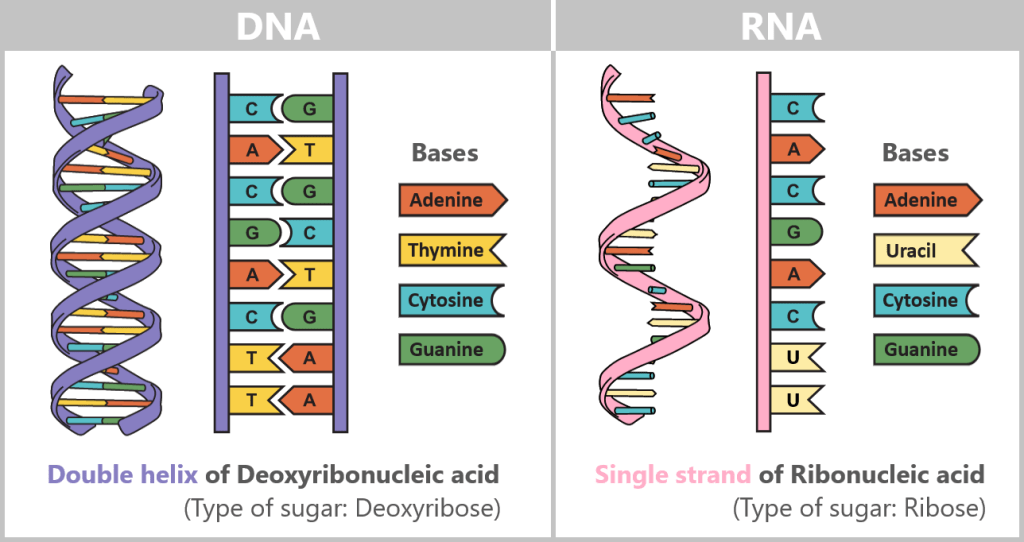
Fig. 3: A working copy of DNA is created – the RNA [The Wonderful World of Life] Alternative Splicing: The Flexible Kitchen of Genes
Cells must adapt flexibly to changing demands: responding to inflammation, defending against pathogens, building muscle, or regulating blood sugar levels. This requires not a rigid set of instructions, but a system capable of improvising depending on the situation.
Depending on the cell type and external signals, the RNA strand is dynamically modified by being cut at specific markers – transferred from the DNA. Unneeded sections (introns) are removed, and the remaining sections (exons) are reassembled. This process creates a multitude of possible messages from a single template. The result is messenger RNA (mRNA), which carries the genetic information from the nucleus to the ribosomes in the cytoplasm – the cell’s „protein factories”.
You can think of this process like following a recipe: depending on the need, the cell leaves out certain ingredients, adds spices, or adjusts the preparation steps. In this way, a single basic instruction can yield a wide variety of dishes.
Through this process – called alternative splicing – cells can produce a variety of different proteins from a single gene, each performing specific functions.
The entire process „from gene to protein” is often referred to as gene expression.
💡Example: The Titin Gene
👉 Longest human gene (≈ 300,000 base pairs).
👉 Through alternative splicing, more than 20,000 different protein variants are produced – each adapted to the specific requirements of different muscle types.Our genome is therefore not a rigid recipe book – it is a dynamic kitchen, where the cell constantly develops new solutions from the same templates.
📌 If you are interested in how genetic information in DNA is translated into protein step by step, you can find further information here.
But as in any creative kitchen, errors can also occur during the transfer from DNA to mRNA: if a „letter” in the recipe is missed, swapped, or incorrectly assembled, the message can end up misleading or unusable. Such mutations or faulty splicing are sometimes harmless – but they can also produce defective proteins that fail to perform their function or even cause harm. A small slip in copying the molecular cookbook – and the flavor of life changes.
c) Epigenetics & Non-coding DNA: The Conducting Team of Our Genome
Although all cells contain the same DNA, they only use a fraction of it. A liver cell needs different instructions than a nerve cell – the rest remain dormant. This targeted gene regulation is controlled by epigenetic mechanisms: small chemical markers – such as methyl groups – do not change the genetic sequence itself, but influence whether a gene is active, how often it is read, or whether it remains completely silenced.
But who actually gives the conductor his instructions? This is where non-coding DNA comes into play – 98% of our genome, long dismissed as useless „junk”. It is the operating system that provides the epigenetic control programs. Only through its interaction is it determined when, where, and how strongly our genes become active – and thus it helps decide over health and disease.
d) The Hidden Control Center: What Non-coding DNA Regulates
These „dark“ regions conceal precise circuits that control the fate of each cell:
Enhancers act like invisible remote controls for genes. Even across thousands of base pairs, they can activate or silence a target gene – for example, for heart development in the embryo.
Telomeres are the protective caps at the ends of chromosomes, like the plastic tips on shoelaces. But when they shrink, aging begins – a central mechanism of cellular decline and cancer development.
Non-coding RNAs act as hidden directors: microRNAs block disease-causing genes, while the long RNA XIST silences an entire X chromosome in females.
Epigenetic markers lay an invisible map over the genome. With methylations acting like red stop signs, they decide whether an enhancer or gene may be read – or silenced.
This is how order emerges in the molecular cookbook: each cell type reads only the pages it needs – guided by non-coding DNA and its epigenetic interpretation.
e) When the Operating System Makes You Sick
Almost all genetic risk markers for common diseases – from diabetes and heart disease to mental disorders – are located in non-coding zones:
Mutations in an enhancer of the MYC gene can trigger leukemia by driving cell division out of control.
Epigenetic misprogramming in these regions – caused, for example, by chronic stress or environmental toxins – can permanently switch genes on or off incorrectly, thereby promoting diseases such as cancer or autoimmune disorders.
Some of these epigenetic patterns are partially reversible – a glimmer of hope for new therapies.
f) Open Mysteries in Research
Despite tremendous progress, the genome remains full of unsolved mysteries:
The „dark matter”: About half of the non-coding genome remains functionally unexplored. Is it evolutionary noise – or an as-yet uncharted regulatory network of life?
Long-range communication: How does an enhancer reliably find its target gene among millions of DNA bases? The spatial folding of the genome – a true „chromatin origami” – could be the key.
Environmental influences: Why do identical non-coding mutations lead to completely different clinical pictures in different life contexts?
RNA codes: Tens of thousands of non-coding RNAs have been discovered – but which are mere footnotes, and which might emerge as leitmotifs for new therapies?
g) The True Magic of Genes
Non-coding DNA is not junk data, but the cell’s mastermind. Epigenetics, in turn, acts as its conductor – flexibly interpreting the instructions of the operating system. Together, they shape our development, regulate our health, and respond to environmental signals.
Genes are not a rigid destiny, but more like a musical score that can be continually reinterpreted. In this interplay, an orchestra emerges that not only shapes our cells but constantly retunes itself – influenced by experiences, environmental factors, and even traces that can be passed on to future generations.
The true magic lies in the fact that from a limited set of notes, an infinite diversity of life can emerge.
4.2. Genetic Variation and Its Consequences
When the genome miswrites, falls silent, or improvises.
With every cell division, DNA is carefully copied and passed on to daughter cells – ensuring that genetic information is preserved across generations.
However, the system is not infallible: mutations – changes in the DNA sequence – can affect the structure and function of proteins. Sometimes only a single „letter” is incorrect. Sometimes a „sentence” is missing, or an entire „paragraph” has been rearranged. In genetics, these are called variants – genetic changes that can be harmless, pose a risk, or cause disease, depending on their type, size, and location.
The following overview presents the main types of variants – and what they mean in the molecular manuscript of life:
📌 SNVs (Single Nucleotide Variants) – A single „letter” is swapped. Usually barely noticeable, but sometimes enough to rewrite entire stories, as in the case of sickle cell anemia.
📌 Indels (Insertions/Deletions) – Small fragments of text are inserted or deleted. Even a tiny shift can throw entire „sentences” out of sync, sometimes so much that the original meaning is lost.
📌 Structural Variants (SVs) – Long stretches of DNA are inverted, duplicated, or relocated. These hidden „rewritings” are difficult to detect – and can often have serious consequences, as seen in cancer
📌 Splice-Site Variants – Errors at the junctions disrupt the RNA syntax. This creates incorrect blueprints, resulting in proteins that no longer function properly.
📌 Repeat Expansions – Base sequences are compulsively repeated until they distort the „text”. This causes diseases such as Huntington’s disease.
📌 Copy Number Variants (CNVs) – Entire „chapters” (genes) are missing or duplicated. The balance of the story is lost.
📌 Mitochondrial variants – Changes in the DNA of mitochondria, the cell’s power plants. If the „text” here is altered, the drive of life falters.
📌 Somatic variants – Arise only during a person’s lifetime and affect specific groups of cells. They mainly shape the story of tumors.
📌 Epigenetic signals – Not new letters, but a different emphasis: volume, rhythm, timing. They transform the same text into an entirely new melody.
The list shows that our genome is not a rigid structure but can be altered by variants. Some of these changes remain inconspicuous footnotes, while others can rewrite the entire story – sometimes with dramatic consequences.
To make such subtle differences visible, tools are needed that capture every detail – every letter, every punctuation mark, every shift. This is precisely where modern genome sequencing comes in.
4.3. Genome Sequencing: Key to Individual Genetic Material
The genetic material of a human being resembles a gigantic library: billions of letters, neatly arranged in long volumes. To read it, genome sequencing is required – which, like a high-precision scanner, provides the raw text of our genetic blueprint.
Thus, it becomes the heart of personalized medicine – a form of medicine that bases its decisions on the individual, not on the average.
But how do we even get hold of the „Book of Life“ so that we can read it page by page?
Let’s imagine the following: shortly after birth, a newborn undergoes a DNA test – a scenario that has already become reality in the United Kingdom, as described earlier.
a) The Journey of DNA: From Umbilical Cord Blood to Genetic Information
Immediately after birth, blood can be collected from the clamped umbilical cord – a safe and painless procedure. Umbilical cord blood originates from the newborn and contains numerous valuable cells, especially stem cells and white blood cells. Hidden within their nuclei lies what truly matters: the DNA – the child’s genetic inheritance.
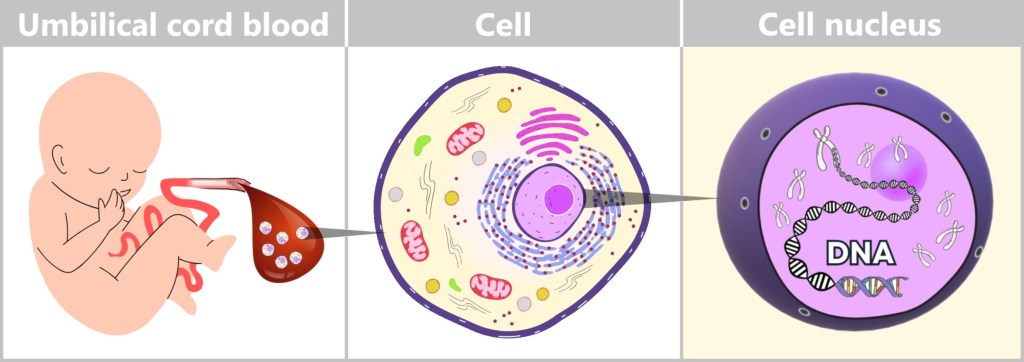
Fig. 4: From Umbilical Cord Blood to Genetic Information Left: Umbilical cord blood as a rich source of diverse cell types.
Center: Schematic representation of a human cell.
Right: Inside the cell nucleus lies the DNA, the carrier of genetic information.To isolate and analyze this DNA, it undergoes a multi-step process in the laboratory. This involves established molecular biology methods that can be divided into four key steps:
Cell Lysis: Gaining Access to the Genetic Material
First, the blood is centrifuged – spun at high speed. During this process, the components separate according to their density: the white blood cells settle as a cell pellet at the bottom. They contain the majority of the DNA of interest.
These cells are then treated with specialized solutions and enzymes that dissolve the cell and nuclear membranes. This releases the DNA – initially, however, as part of a mixture containing proteins, lipids, and other cellular components.
DNA Isolation: Obtaining Pure Genetic Material
In the next step, the DNA is extracted from the cell lysate – the „cell soup.” Several methods can be used for this purpose:
Filtration or column purification: The solution is passed through special filter systems to which the DNA adheres, while smaller molecules are washed out.
Magnetic beads: More modern methods use tiny beads with a DNA-binding surface. The DNA sticks to them and can be removed in a targeted manner using a magnet.
Both methods allow for an effective separation of genetic material from contaminants.
Purification and Concentration of the DNA
At this stage, the DNA from many white blood cells is present in a relatively pure form – but still dissolved in an aqueous solution. To further purify and concentrate it, cold alcohol (e.g., ethanol or isopropanol) is added. This causes the DNA to precipitate and become visible as a whitish, thread-like substance.
Human DNA is extremely long: if fully stretched out, it measures about two meters per cell. During extraction, however, it is broken into many smaller fragments by chemical and mechanical forces. At first, this seems chaotic – but because the DNA is identical in all cells, many fragments contain overlapping information. This random distribution becomes an advantage during sequencing: the fragments can be read multiple times independently and then assembled on a computer into a complete sequence. This significantly increases the accuracy of the analysis.
Quality Control: Is the DNA Intact?
Before the DNA sample can be analyzed, its quality must be assessed. Two common methods are:
Spectrophotometry: Measures the concentration and purity of the DNA by analyzing light absorption.
Gel electrophoresis: DNA fragments are passed through a gel and separated by length (longer fragments move more slowly than shorter ones). This allows assessment of whether the sample is sufficiently intact.
Why is this process so important?
Human DNA is fragile – contaminants or fragmentation can distort analyses or render them unusable. Careful and controlled purification is therefore essential for reliable genetic results.
Umbilical cord blood is a particularly valuable source of medical information: it enables early diagnosis, prognoses, and in some cases targeted prevention.
What appears to be a routine procedure in the lab is, in fact, a highly precise process – it requires technical expertise, modern equipment, and biochemical finesse. With automated extraction methods, it is now possible to obtain a high-quality DNA sample from just a few drops of umbilical cord blood in less than an hour. Although the DNA consists of fragments, it still contains all the information needed to decode an individual’s genetic code – and thus lays the foundation for the medicine of tomorrow.
b) Three Directives for Gene Diagnostics: WGS, WES, and Panel
Before we dive deeper into the tools of genome sequencing, it is worth taking a look at the three central strategies used today to decode genetic information – like stage directions in the script of life:
🎯 Panel Sequencing – The Targeted Scene
Only a selected part of the genome is examined here – usually a few dozen to a few hundred genes. Such tests are used when a specific disease is suspected, such as hereditary breast cancer (BRCA1/2).
Advantage: Fast, cost-effective, and precise – as long as the medical question is well-defined.
Limitation: Everything outside the selected scene remains in the dark.🎥 Whole Exome Sequencing (WES) – The Classic Director’s Cut
WES analyses all protein-coding sections of the genome – approximately 1–2% of the entire genetic material. It is a proven method, particularly for rare hereditary diseases, as most known mutations are located in these areas.
Advantage: Efficient identification of faulty blueprints for proteins.
Limitation: Regions outside the genes – important switches and regulators of the genome – are not captured.🎬 Whole Genome Sequencing (WGS) – The Complete Movie in 4K
WGS reads the entire genome – all three billion letters. This provides the most comprehensive picture, including non-coding regions, regulatory elements, structural variants, and rare mutations.
Advantage: Maximum resolution for complex cases.
Example: In children with unclear developmental disorders, WGS provides clarity twice as often as WES – for instance, by uncovering mutations in distant „remote-control” regions (enhancers) that incorrectly switch genes on or off.Conclusion:
Depending on the question at hand, the right perspective is needed – sometimes a zoom on the key scene suffices, other times the entire film must be re-edited. Choosing the method is therefore not a mere technical detail, but a strategic decision in the diagnostic playbook.
4.4. Methods of Genome Sequencing
How is the genetic code read?
Imagine DNA as a book – written in a language with only four letters: A, T, C, and G. To decipher this genetic text, scientists use modern technologies. Two central methods have become established. They pursue the same goal: to determine the sequence of DNA letters as precisely as possible.
The first method works – put simply – like a molecular photocopier equipped with color sensors. The second uses a high-tech tunnel through which the DNA is guided and electronically „scanned”, comparable to running your fingers along a rope to detect the smallest irregularities. These techniques form the foundation of DNA sequencing.
4.4.1. Method 1: Copying with Fluorescent Dye
a) Sanger Sequencing
b) Illumina Technology
c) Single-Molecule Real-Time(SMRT) Sequencing
4.4.2. Method 2: High-Tech Tunnel – Electrically Scanning DNA
a) Oxford Nanopore Sequencing
4.4.3. Genome Sequencing – Technologies Compared
a) Throughput vs. Usability – Quantity Does Not Always Equal Quality
b) Cost-Effectiveness – Cost per Base vs. Cost per Insight
c) From Sample to Result – How Quickly Does the Whole Genome Speak?
d) Clinical Applications – Which Technology Fits Which Purpose?
e) AI as Co-Pilot – Automation Without Relinquishing Responsibility
f) Comparison Table: Sanger, Illumina, PacBio, ONT
g) Key Takeaways: Between Maturity and Routine
4.4.1. Method 1: Copying with Fluorescent Dye
This method takes advantage of the fact that DNA consists of two complementary strands. If the base sequence of one strand is known, the sequence of the complementary strand can be automatically deduced. To obtain this information, one of the two strands of the DNA under investigation is copied. The aim is to record the exact sequence of bases in the resulting copy during this copying process.
To better understand how this process works, let’s first take a look at the key components and basic steps involved in producing a DNA copy in a test tube (in vitro).
Producing a DNA Copy – The Basic Principle
At the centre is a DNA template that is to be copied. This requires four main components:
- dNTPs (deoxynucleoside triphosphates): the DNA building blocks A, T, C, and G.
- DNA polymerase: an enzyme that functions as a molecular copying machine.
- Primers: short DNA fragments that provide the polymerase with a starting point.
- Buffer solution: ensures stable conditions during the reaction.
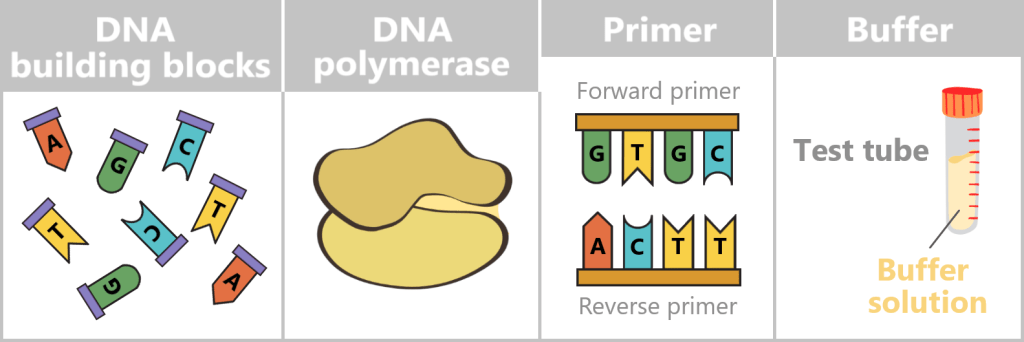
Fig. 5-A: Schematic representation of the main components required to generate a DNA copy DNA building blocks: Individual nucleotides (A, T, C, G), shown in different colors and letters. They serve as the raw material from which the new DNA strand is assembled.
DNA polymerase: A schematic enzyme that links the nucleotides together to form a DNA strand.
Primer: Short DNA fragments that provide the polymerase with a defined starting point.
Buffer: A test tube containing buffer solution, ensuring stable chemical conditions during the reaction.What does DNA polymerase do?
DNA polymerase is a specialized enzyme that generates complementary DNA strands – essentially the „printhead” of a molecular copying machine. Enzymes act as biological tools that enable chemical reactions in a targeted and efficient manner, which is why they are often described as „biological catalysts”.
In order for the polymerase to work, it needs a primer as a start signal. You can think of it like this: just as a printhead only starts working when a sheet of paper is inserted correctly, the polymerase only begins its work when a primer is present. This small piece of DNA shows it where to start – essentially acting as the first sheet in the molecular printer.
Before copying can begin, the double-stranded DNA must first be separated into two single strands – this step is called denaturation.
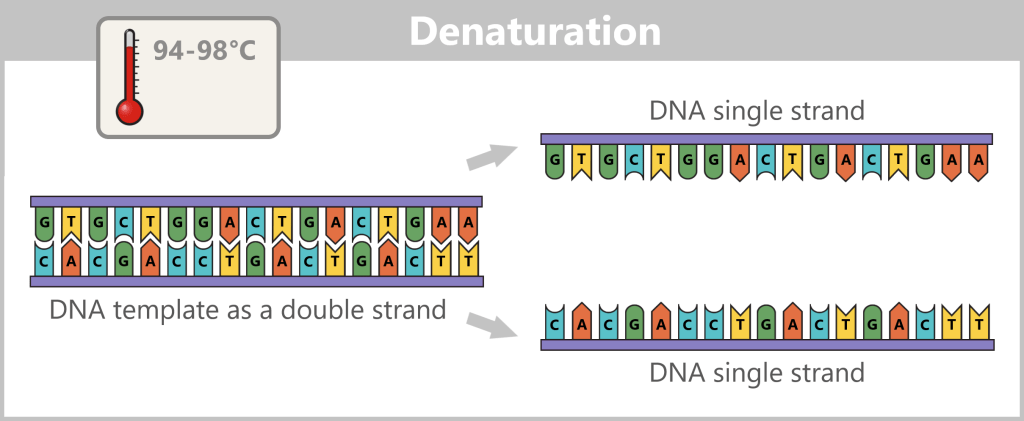
Fig. 5-B: Separation of the DNA strands Next, the primers bind to their complementary target sites on the single strands. This primer binding marks the starting point for the DNA polymerase.
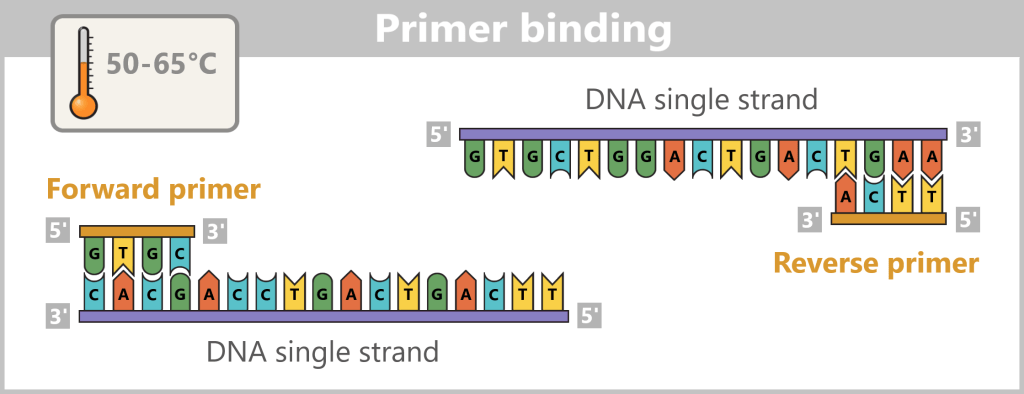
Fig. 5-C: Primers mark the starting point for the polymerase. Now the synthesis of the new strands begins: The polymerase reads the template strand in the 3′-to-5′ direction and extends the copy in the opposite direction – 5′ to 3′ – while obeying the base-pairing rules (A with T, G with C).
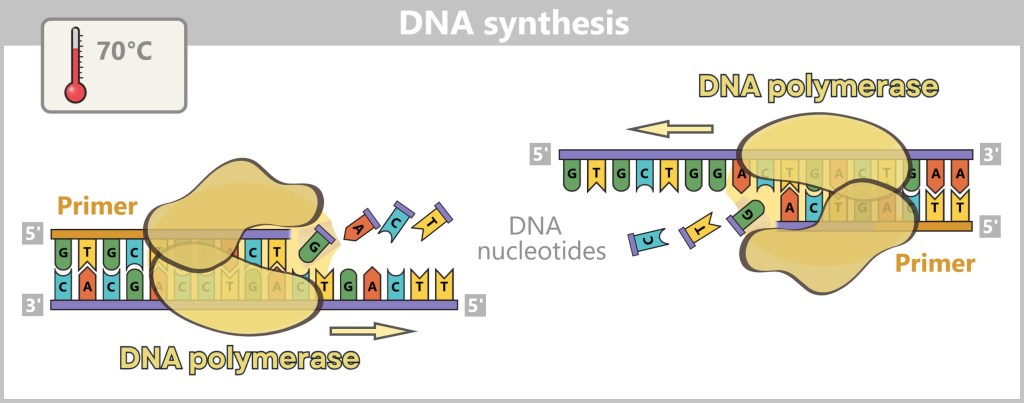
Fig. 5-D: DNA synthesis – the exact duplication of a DNA strand. What do 3′ and 5′ mean?
The designations 3′ (three-prime) and 5′ (five-prime) come from the chemistry of the DNA backbone. They indicate at which end certain carbon atoms are located in the sugar molecule, to which new building blocks can be attached.
Imagine the DNA strand as a one-way street. The polymerase can only travel in one direction – from the 5′ end to the 3′ end. New building blocks can only be chemically attached at the 3′ end. Therefore, the polymerase reads the ‘old’ strand backwards (3′ → 5′) and builds the new strand forwards (5′ → 3′).
In this way, a new, complementary strand is gradually formed from a single strand – with the help of the base pairing rules: adenine (A) pairs with thymine (T), and cytosine (C) with guanine (G).
The result is perfect genetic replicas – new DNA strands that are exact copies of the original DNA.
Fluorescent Nucleotides: How Sequencing Works
Modern sequencing methods enhance this copying process with a sophisticated technique: they use fluorescently labeled dNTPs (DNA building blocks). Each of the four nucleotides (A, T, C, G) is tagged with a dye that emits a specific light signal when it is incorporated.
As the polymerase extends the DNA strands, it inserts the colored building blocks with precision. Each time a new nucleotide is incorporated, it sends out a tiny light signal – like a small flash that reveals which „letter“ has just been added. High-resolution cameras capture these light signals, enabling the DNA sequence to be reconstructed step by step.
Overview of Sequencing Methods
Several major technologies apply this principle in specific ways:
Sanger Sequencing: The classical method, in which fluorescent chain-terminating nucleotides halt DNA synthesis at random positions. The resulting fragments can be read in sequence.
Illumina Technology: A widely used high-throughput method in which millions of DNA fragments are sequenced in parallel. The fluorescent signals are recorded step by step.
SMRT Sequencing (PacBio): In this method, synthesis is observed in real time on a single DNA molecule, offering high precision and long read lengths.
a) Sanger Sequencing
The classic „letter-by-letter” reading method.
Sanger sequencing, also known as the chain-termination method, was developed in the 1970s and was the first technique to read DNA accurately and reliably. It is fundamentally based on the same principles as DNA synthesis but differs in several key aspects:
👉 Only a single primer is used.
👉 In addition to the normal DNA building blocks – the dNTPs (dATP, dCTP, dGTP, dTTP) – special fluorescent chain-terminating nucleotides, called ddNTPs (ddATP, ddCTP, ddGTP, ddTTP), are added in small amounts. When a ddNTP is incorporated, DNA synthesis stops precisely at that point. Each of the four ddNTP types carries a different fluorescent color corresponding to its base.The process – step by step
For sequencing, four separate reaction mixtures are prepared. Each contains:
- the single-stranded target DNA,
- a primer (starting point for synthesis),
- a DNA polymerase,
- normal dNTPs,
- and one type of the fluorescently labeled ddNTPs.

Fig. 6-A: Sanger Sequencing – schematic representation of the four reaction mixtures. Each mixture contains the same basic components but differs in the specific type of fluorescently labeled chain-terminating nucleotides (ddNTPs) added.
After primer binding, the polymerase reads the DNA strand and incorporates the complementary nucleotides to build a new strand. If a ddNTP is randomly incorporated, synthesis terminates precisely at that position.
This generates many DNA fragments of varying lengths – each ending with a fluorescently labeled nucleotide. Since each of the four reactions contains only one type of ddNTP, the terminal base of each fragment is known precisely.

Fig. 6-B: Synthesis is specifically interrupted when a ddNTP is incorporated. Each reaction mixture contains only one type of modified DNA building block (ddATP, ddTTP, ddCTP, ddGTP). When such a „stop” nucleotide is incorporated during DNA synthesis, the copying process halts precisely at that point. In the ddATP mixture, synthesis stops upon incorporation of a modified adenine (A); in the ddTTP mixture, it ends with a modified thymine (T). Likewise, ddCTP and ddGTP terminate synthesis when a modified cytosine (C) or guanine (G) is added. This procedure generates many DNA fragments of varying lengths, each ending with a specific stop nucleotide. The goal is to produce all theoretically possible fragments so that the complete DNA sequence can be read.
Sorting and Analysis
The DNA fragments are then denatured (converted into single strands) and sorted by length using gel electrophoresis. The negatively charged fragments migrate through a gel towards the positive electrode. Smaller fragments move faster, larger ones slower, creating an orderly „ladder“ of fragments.
A laser then excites the fluorescent dyes attached to the terminal bases. The resulting light signals are captured by a detector. Each signal corresponds to a specific base at a specific position. The sequence of light signals thus directly reveals the DNA sequence – much like reading a book letter by letter.
Since the generated fragments are complementary to the original DNA, the original base sequence can be deduced precisely from them.
Fig. 6-C: Gel electrophoresis for separating DNA fragments: In the reaction mixtures, DNA fragments of varying lengths are generated, each ending with the same chain-terminating nucleotide – either adenine, thymine, guanine, or cytosine, depending on the mixture. These reaction mixtures are applied to a gel. When an electric field is applied, the negatively charged DNA fragments migrate from the cathode (−) toward the anode (+). The size of the fragments determines their migration speed: smaller fragments move faster through the gel’s fine pores and reach the anode first, while larger fragments move more slowly. By reading the fluorescent signals at the fragment ends, the exact order of the bases can be determined, allowing the DNA sequence to be reconstructed step by step.
Why is Sanger sequencing still considered the gold standard today?
Even half a century after its development, the Sanger method remains indispensable in many areas of molecular biology and medicine. The reason: reliability and precision.
Compared to modern high-throughput methods, Sanger sequencing is slower and suitable only for analyzing smaller DNA segments – not entire genomes. But that is precisely where its strength lies:
- Targeted questions, such as checking individual genes,
- Detection of specific mutations, or
- Validation of critical results previously identified using other methods
can be answered with precision, clarity, and interpretability using Sanger sequencing – often so clearly that the sequence of bases can be read directly from the sequencing plot.
In medical diagnostics – such as the analysis of inherited diseases or in quality control of genetic engineering procedures – this high level of accuracy is crucial. Even a single misread base can have life-altering consequences.
Another advantage: the method is standardized worldwide, has been reliably used for decades, and is governed by well-defined quality guidelines.
A Classic with Staying Power
In a world where new technologies constantly emerge, Sanger sequencing remains a reliable anchor – the old-timer among sequencing methods: not the fastest, but extremely robust, proven, and reliable. And sometimes that’s exactly what matters.
b) Illumina Sequencing
The High-Speed Copy Machine
Sanger sequencing is like precise hand-crafted work: each DNA strand is decoded step by step – reliable, but slow and expensive.
Imagine having to copy an entire book – or even an entire library – while being allowed to write down only one letter per minute.
This is precisely where the problem lies: modern cancer research, the decoding of rare diseases and the genetic monitoring of pandemics require the analysis of large amounts of data – i.e. many and/or very long DNA segments. This requires a method that is not only accurate, but also fast and affordable.
This is precisely where Illumina sequencing comes into play. It has elevated the principle of DNA decoding from manual work to industrial mass production and is now one of the most commonly used methods for high-throughput sequencing (Next Generation Sequencing, NGS). Instead of individual letters, entire pages are now read simultaneously – millions of times, in parallel, with high precision and cost-efficiency.
Unlike the classic Sanger method, where each DNA fragment is analyzed individually, Illumina operates with massive parallelization – meaning that many DNA fragments are amplified and sequenced simultaneously. How does this work?
Step 1: Preparing the DNA Fragments
– Turning the DNA into a „Lego Construction Site” –First, the DNA sample to be analyzed is broken down into many short pieces – called fragments – typically 100 to 300 base pairs long. Small synthetic DNA sequences, known as adapters, are then attached to both ends of these fragments. You can think of them as LEGO connectors. On the one hand, they act as molecular docking sites that allow the DNA fragments to bind to a special carrier – the flow cell chip. On the other hand, they serve as binding sites for universal primers.
Abb. 7-A: The DNA is broken into numerous fragments, each equipped with small attachments (adapters). 
Fig. 7-B: Simplified schematic representation of a DNA fragment with adapters. The P5/P7 sequences serve to attach the fragments to the flow cell. The Rd1SP/Rd2SP sequences act as binding sites for universal primers.
Denaturation separates the prepared double-stranded DNA fragments into single strands.
Step 2: Bridge Amplification
– The Dance of DNA on the Flow Cell –At the heart of Illumina technology is the flow cell – a glass-like plate covered with millions of tiny DNA docking sites (short DNA segments called oligonucleotides) that are firmly anchored to the surface.

Fig. 7-C: Schematic representation of a flow cell and its surface. The single-stranded DNA fragments bind to the anchors via their adapters. Then the molecular copying process begins: complementary copies of the DNA snippets are produced, which are now firmly anchored to the flow cell.
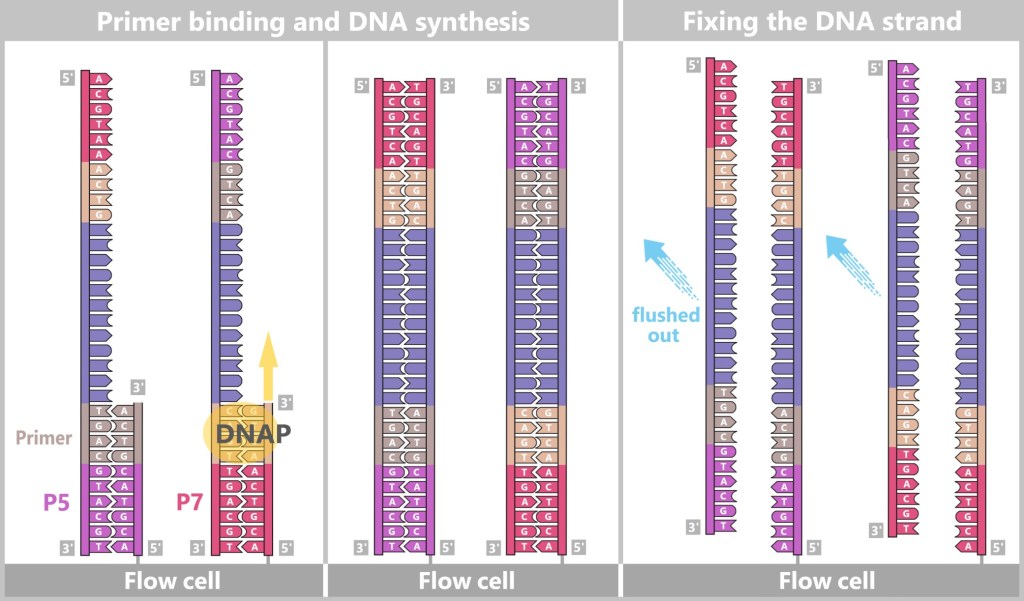
Fig. 7-D: Schematic representation of initial synthesis + formation of anchored DNA strand copies. Left: Primers bind to the adapters (P5, P7), and DNA polymerase (DNAP) initiates the synthesis of a new complementary strand.
Center: The DNA polymerase then synthesizes the first strand.
Right: The newly formed DNA double strand is separated. After denaturation, the original strand is no longer connected to the flow cell and is flushed out. The newly synthesized strand remains firmly bound to the flow cell with its 5′ end.Then a fascinating process begins: the DNA strands bend to form small bridges, as their free adapters attach to neighboring oligonucleotides on the flow cell. At these points, they are copied again. After copying, the bridge is dissolved, and the number of anchored DNA strands doubles. This process, known as bridge amplification, is repeated dozens of times. In the end, dense clusters of millions of identical copies of a single DNA fragment are formed.
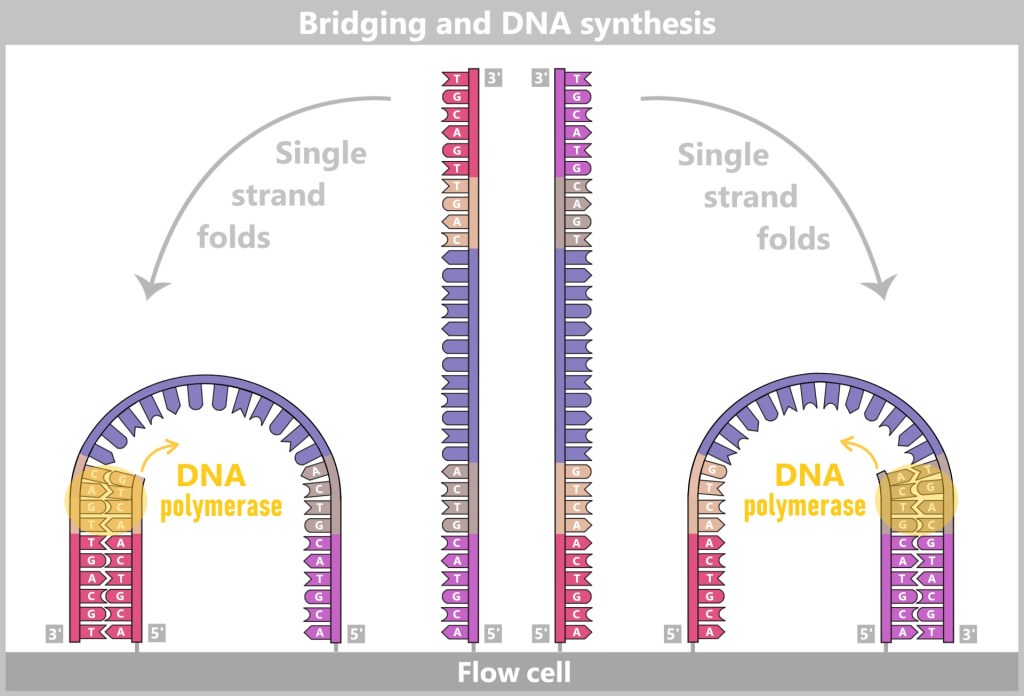
Fig. 7-E: Bridge Formation and DNA Synthesis The anchored single strands fold and connect with neighboring anchors on the flow cell, forming a bridge structure. The copying process is then initiated.
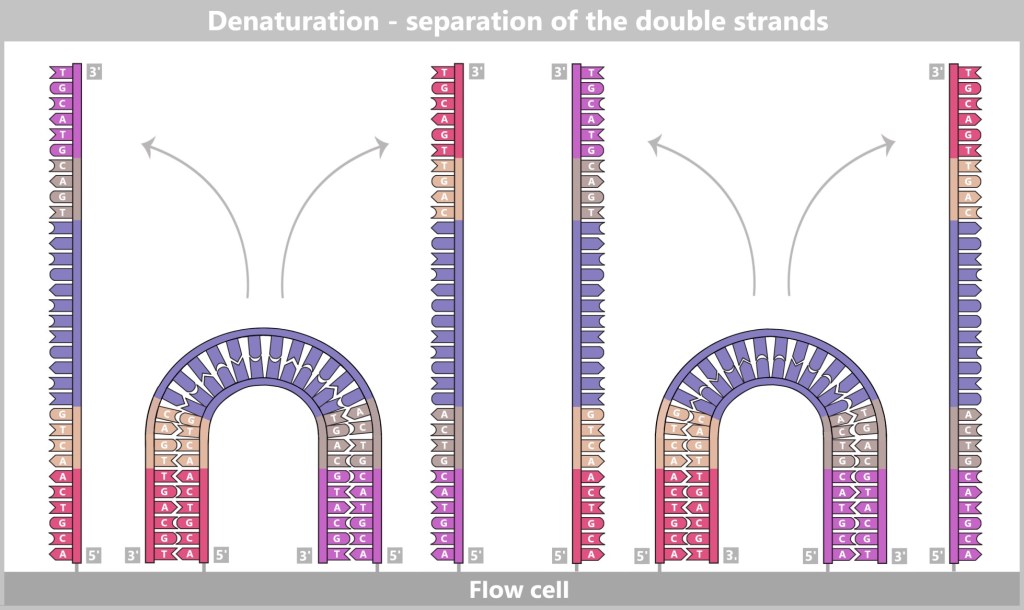
Fig. 7-F: Forward and Backward – Bridge Dissociation After the copying process, the bridge double strands are separated. The number of firmly anchored DNA strands doubles, making them ready for further amplification.
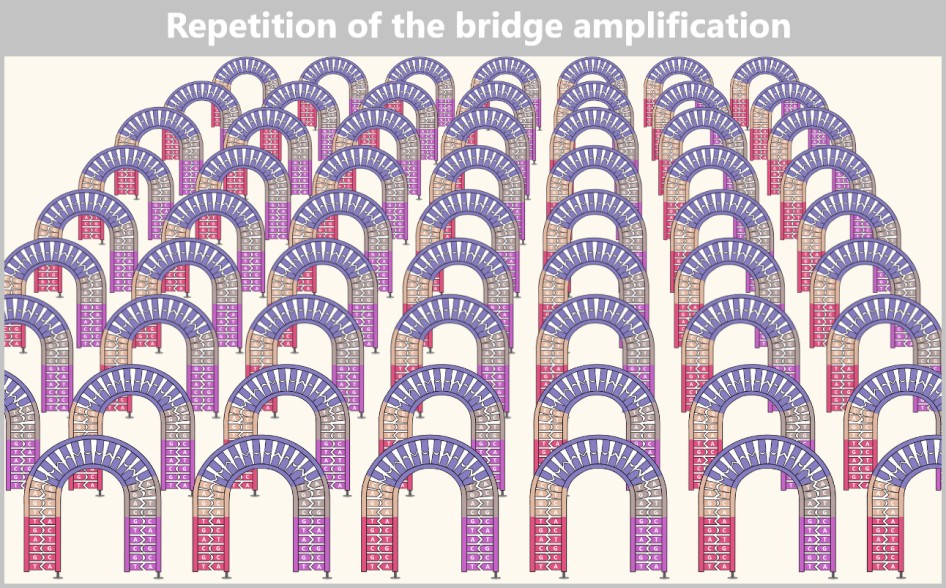
Fig. 7-G: With each cycle of bridge amplification, an increasing number of DNA copies is generated. 
Fig. 7-H: Cluster formation Each cluster consists of numerous copies of a single DNA fragment. In this illustration, only three clusters are shown as examples. In reality, millions of such clusters cover a flow cell, enabling high sequencing capacity.
The result is a densely packed ‘map’ consisting of millions of DNA clusters, each containing only a specific sequence – ideal for simultaneous reading.
Without this replication, reading DNA would be like trying to spot a single firefly in the wind. The clusters, on the other hand, make the DNA bases (A, T, C, G) clearly visible – like bright neon lettering in the dark.
Step 3: Sequencing-by-Synthesis
– The Light Show of DNA Bases –Now the actual sequencing by synthesis begins. Once again, the principle of a „molecular copying machine with color sensors“ is used – but in an optimized form:
At the beginning, the universal primers bind to the corresponding adapter sites of the DNA fragments in each DNA cluster.
Then modified dNTPs – so-called RT-dNTPs (reversibly terminating nucleotides) – are added to the reaction solution. Each of the four DNA building blocks (A, T, G, C) is labelled with its own fluorescent color and carries a reversible blocker.
Only one nucleotide per cycle can be incorporated because the block temporarily stops further strand formation. Unincorporated RT-dNTPs are washed away.
After each incorporation step, the flow cell is scanned with a high-resolution camera. Each fluorescent signal corresponds to an incorporated nucleotide, and its color indicates which base has just been added.
Afterwards, the blockage and dye marking are chemically removed, and the next cycle begins.
This process is repeated cycle by cycle until each DNA strand is completely read.

Fig. 7-I: Illumina sequencing steps Step 4: Data Analysis
– The Supercomputer as Puzzle Master –After the „light show„, millions of photos are available – one for each cycle and each cluster. Each image shows which color (and thus which letter: A, T, C, or G) was added to each cluster.
Now powerful computers come into play – equipped with software that works as precisely as a detective piecing together a cut-up book from a stack of numbered Polaroids. Each cluster on the sequencing chip is like a small crime scene, each color pixel a clue. And from millions of such clues, a story gradually emerges – the story of DNA.

Fig. 7-J: Analysis of the Sequencing Data The color patterns are translated into sequences of letters, known as „reads”: tiny pieces of text from an enormous genome novel. In the end, a flood of such fragments piles up – like a mountain of cut-up book pages, scattered loosely about.
This is precisely where the real art begins: bioinformatics takes the stage. Using specialized algorithms, the snippets are analyzed, sorted, and stitched together – constantly searching for overlapping regions, familiar patterns, and known structures.
Piece by piece, the bigger picture comes back together – until, in the end, the original DNA sequence becomes visible, like a reconstructed book that suddenly makes sense.
The real „magic” therefore doesn’t happen in the flow cell, but in the computer! Without the software, the images would be nothing more than colorful flickering – but with it, they become the key to medical breakthroughs.
A more detailed yet clear explanation of the Illumina sequencing technology method can be found in the video „Illumina Sequencing Technology“.
Modern high-throughput sequencing platforms such as those from Illumina can generate the raw data of a complete human genome within just a few days – at pure sequencing costs that are significantly lower than the price of a premium smartphone.
This technology has revolutionized genome research and made it considerably more accessible: today, it is an extremely versatile tool – fast enough for real-time analysis of pandemics, precise enough to form the basis for personalized cancer therapies when combined with robust bioinformatics and clinical expertise.
Illumina sequencing is now considered the industry standard for large sequencing projects – such as the analysis of whole genomes, gene expression research, and modern cancer diagnostics.
And best of all: While you are reading this text, somewhere in the world a Flow Cell is decoding millions of DNA fragments.
However, Illumina technology also has its limitations. It reaches its limits particularly with long, repetitive DNA segments, such as those found in some chromosome regions. It is like trying to reconstruct a song from thousands of 3-second snippets. For such challenges, Illumina is often combined with other sequencing methods that can reliably capture long segments – like an investigator who needs both close-ups and panoramic images to understand the entire crime scene.
c) Single-Molecule Real-Time (SMRT) Sequencing
The Novel in One Go
Imagine you want to read a long novel – full of recurring chapters and hidden clues that span many pages. If you cut the book into thousands of small snippets, as in Illumina sequencing, read them individually and then put them together like a jigsaw puzzle, there is a risk that chapters will be missing, mixed up or incomplete. The secret messages of the story – what the novel actually wants to tell us – could be lost or appear only fragmentarily.
This is where PacBio’s Single Molecule Real-Time (SMRT) sequencing shines: it reads entire chapters in one go – no snippets, no breaks.
Instead of breaking the DNA apart, PacBio keeps it in very long fragments – often tens of thousands of base pairs at a time, sequenced as a whole. These strands enter tiny stages in a SMRT cell, called nanowells (Zero-Mode Waveguides). They are so small that they can hold only a single DNA molecule and a polymerase. Each of these stages allows light to act on just one tiny spot – exactly where the polymerase reads the DNA. Like a spotlight highlighting an actor during a monologue on stage.
The polymerase plays the leading role: it adds building block by building block (A, T, C, or G) to the DNA copy. Each of these blocks lights up in a different color, like neon lights at a DJ set. With every addition, a flash of light is emitted and recorded in real time by a camera – a dancing light code that reveals the DNA sequence.
The video Introduction to SMRT Sequencing by PacBio illustrates this process impressively.
Let’s analyze the individual steps of this fascinating process in detail.
Step 1: Preparation
– DNA Loops for Continuous Reading –Even with PacBio, the DNA to be analyzed is first fragmented – but into much larger pieces than with Illumina, usually 10,000 to over 20,000 base pairs long. These fragments are then converted into circular DNA molecules:
So-called Hairpin adapters are added to both ends – small loops of DNA that close the fragment onto itself.
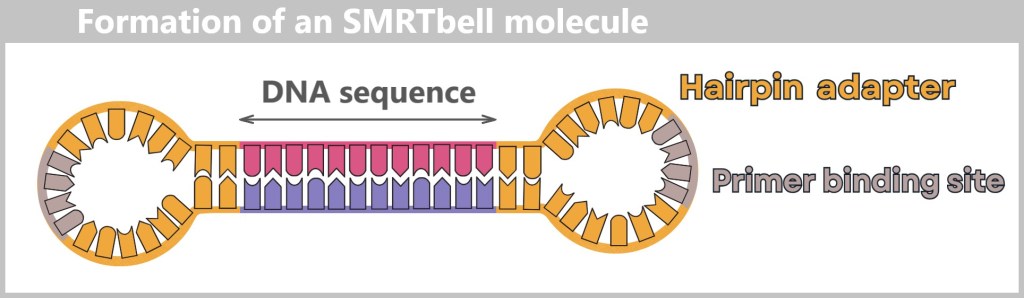
Fig. 8-A: A DNA fragment is equipped with hairpin adapters. The adapters contain a primer binding site, which later provides the starting point for the polymerase.
Subsequent denaturation produces a circular, single-stranded SMRTbell molecule in the form of an open loop.
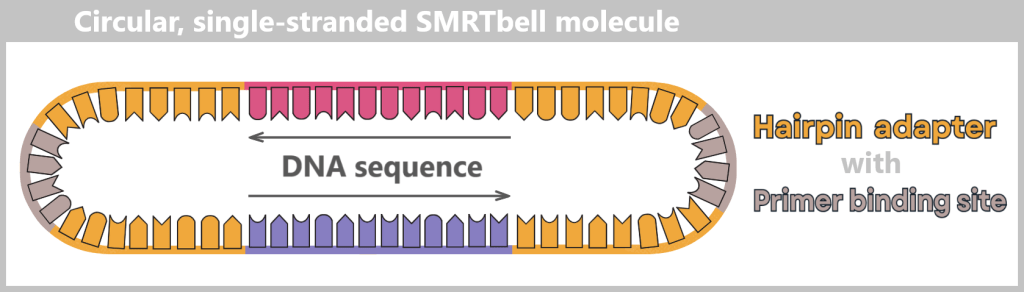
Fig. 8-B: The denatured SMRTbell molecule: Denaturation converts the double-stranded template into a single-stranded SMRTbell molecule. It forms a circular structure, shown here as an open loop.
The result is a SMRTbell: a closed DNA loop that can be read repeatedly by the polymerase – like a model train running around a circular track.
Each hairpin adapter contains a defined binding site to which the universal primer binds. This is the starting point for the polymerase.
The DNA polymerase binds to the primer complex, forming a fully functional replication complex: polymerase + primer + SMRTbell.
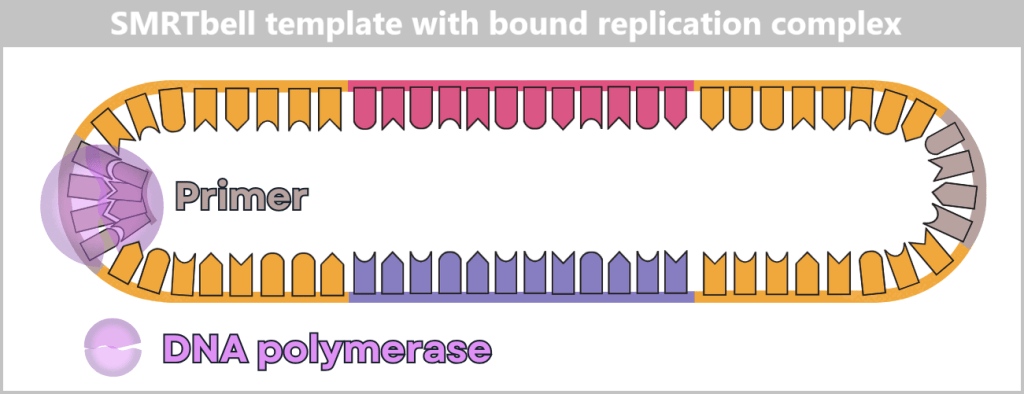
Fig. 8-C: The stable replication complex of polymerase, primer, and single-stranded SMRTbell. The primer is bound to the adapter binding site, the polymerase to the primer. This complex is ready to start DNA synthesis.
Step 2: The Sequencing Stage
– Light Shows at the Nanoscale –Now it gets spectacular: the prepared replication complexes are guided into their tiny observation chambers – the Zero-Mode Waveguides (ZMWs). These nanowells are so small that typically only a single SMRTbell template fits inside. The polymerase is anchored firmly at the bottom of the ZMW, ready for action.
The start signal comes with the addition of the four nucleotides (A, C, G, T), each labeled with its own fluorescent dye.
The polymerase now begins its work – it reads the DNA strand and synthesizes a complementary strand. The crucial event occurs the moment it incorporates a nucleotide:
Each nucleotide emits a brief, color-coded flash of light.
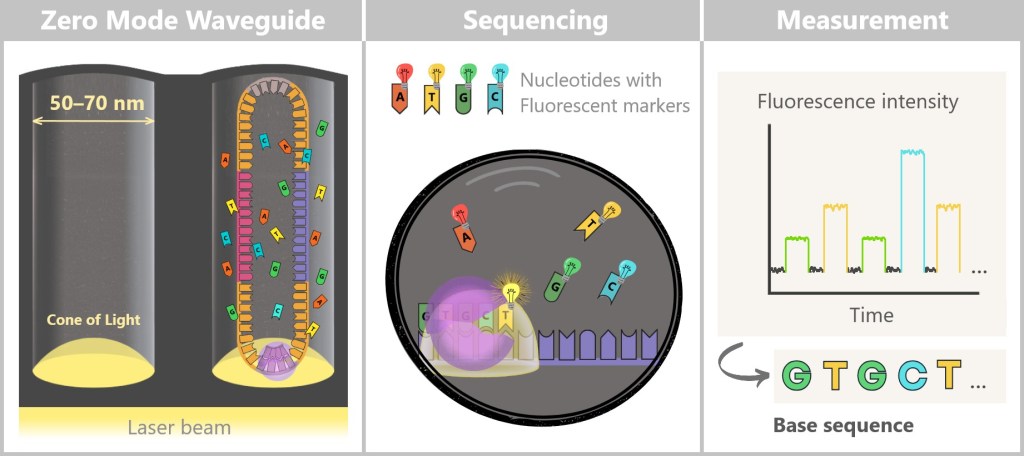
Fig. 8-D: Zero-Mode Waveguide (ZMW): Sequencing of Individual DNA Molecules Left: At the bottom of the Zero-Mode Waveguide (ZMW), a laser beam enters and generates what is known as an evanescent field (shown as a cone of light). Unlike normal light, this field does not propagate throughout the solution but decays exponentially within only 20–30 nanometers. This creates an extremely small excitation volume in which the fluorescent dyes of the nucleotides can be excited.
The left zero mode waveguide (ZMW) contains a single replication complex (polymerase + SMRTbell). The polymerase (purple) is firmly anchored to the (glass) bottom. In the presence of the fluorescently labelled nucleotides, the polymerase begins its work.
Centre: The four nucleotides (A, T, G, C) carry different fluorescent markers. As soon as the polymerase incorporates a nucleotide, it lights up briefly before the dye is cleaved off.
Right: These flashes of light are measured in real time and translated into fluorescence signals. This is how the base sequence of the DNA is determined step by step.The luminescence occurs in real time, at the exact moment of insertion – hence the name: Single Molecule Real-Time Sequencing. A sensitive detector records these light signals – like a molecular live microscope at work.
Step 3: The Long-Range Secret
– Repetition Makes It Robust –Since PacBio reads very long DNA segments at once – comparable to reading entire chapters of a book instead of individual verses – more individual errors occur in a single run than with Illumina. This is exactly where the ring-shaped SMRTbell structure comes into play. The DNA polymerase can circle the ring molecule multiple times, like a train conductor. In doing so, the same DNA fragment is read over and over again – like a reader revisiting the same passage until it is fully mastered.
A computer compares the repeated reads with one another and filters out the errors – like an editor polishing a manuscript. This results in a particularly precise consensus sequence.
PacBio calls this approach HiFi reads – high-precision single-molecule readings.
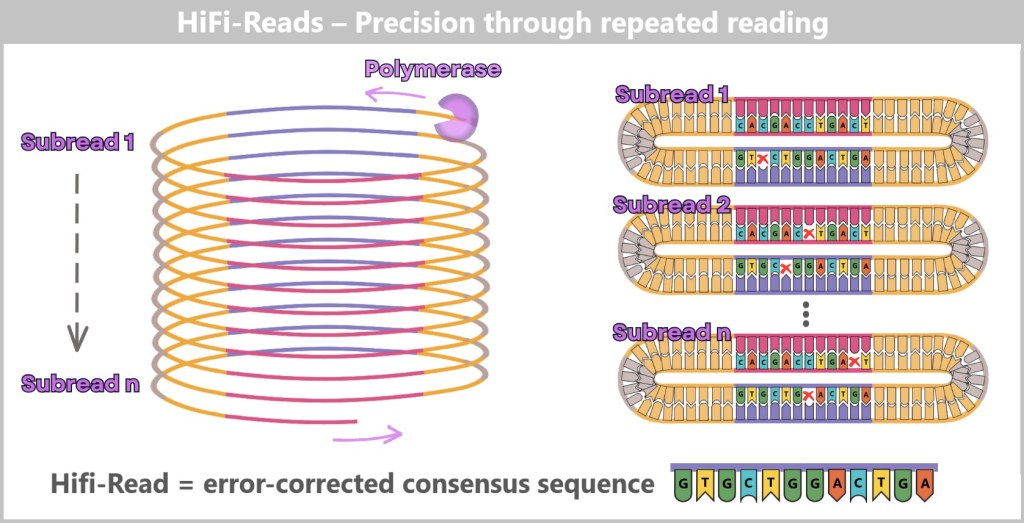
Fig. 8-E: Formation of a HiFi Read. The polymerase circles the SMRTbell template multiple times, generating multiple subreads. Each cycle corresponds to a complete reading of the DNA sequence. Individual subreads may contain random errors (x), but by comparing many repetitions, a highly accurate consensus is formed – the so-called HiFi read (High-Fidelity Read).
Additional info: How the polymerase loops around the DNA multiple times
First sequencing run
The polymerase reads the single-stranded template strand and synthesizes a complementary strand.
The result is a complete double helix – the original template strand and the newly created daughter strand are now firmly bound together.
This first, uninterrupted round produces a subread – a single base sequence generated by the polymerase in a single pass.
Second and subsequent sequencing runs
Now the question arises: how can the polymerase continue reading when the DNA ring is already double-stranded?
This is where Φ29 phage DNA polymerase comes into play – a specially modified enzyme variant with two key properties:
High processivity: It remains bound to the DNA strand for a very long time and hardly ever detaches – this enables long reads.
Strong strand-displacement activity: It can break the hydrogen bonds between the template and daughter strands.
The polymerase runs into the double helix, separates the strands locally and displaces the old daughter strand while simultaneously synthesizing a new one – like a molecular bulldozer.
The previously synthesized strand is not degraded; it remains as a loose, single-stranded DNA fragment in the nanowell. With each subsequent round, this process repeats: the polymerase reads the same template again – producing multiple subreads from the same DNA molecule.
Step 4: Data Analysis
– Turning Flashes of Light into Readable DNA –Once all the light signals have been captured, the real detective work begins: data analysis.
The first step is called base calling: the recorded fluorescence signals – encoded in time and color – are translated into nucleotide sequences – A, T, G, or C.
Next, the raw data from each polymerase pass is cleaned up: the hairpin adapter sequences are bioinformatically trimmed. What remains are the subreads – the uncut reading records of each individual polymerase pass through the DNA chapter text.
But it is only the next step that reveals the whole truth: like a master philological detective comparing multiple copies of a manuscript, the algorithm combines all subreads of the same SMRTbell molecule. This comparison – known as Circular Consensus sequencing (CCS) – produces a highly accurate HiFi read: an error-corrected master copy that reproduces the DNA chapter word for word.
Only now is the text fragment ready for final classification – and in the process it goes through several stages:
First, a quality check – just like a meticulous editor reviewing the text for clarity and consistency.
This is followed by alignment: the sequence is classified like a new chapter in the correct compartment of a reference genome shelf.
Finally, assembly takes place – the final act, in which all chapters are brought together to form a complete novel of the genome.
The video PacBio Sequencing – How it Works clearly summarizes the steps involved.
Applications and Advantages – The Strengths of Long-Read Sequencing
PacBio SMRT technology has a particular strength: it can read very long DNA segments in one go – with highest accuracy. This makes it ideal for:
- Detecting complex genomic structures
- Analysis of repetitive sequences
- Distinguishing very similar gene variants (e.g., in immune receptors or cancer genetics)
- De novo sequencing – reading entirely new genomes without a reference
- Detecting epigenetic modifications (e.g. methylations)
How can SMRT sequencing make epigenetic modifications visible?
As already described, epigenetic mechanisms play a central role in targeted gene regulation. These are small chemical markers – such as methyl groups – that are attached to specific DNA bases. They do not change the genetic sequence itself, but influence when and how often genes are read – depending on cell type, environment or stage of development.
They function like notes or markings in the margin of a book: the text remains the same, but the „reading” changes – some passages are emphasized, others skipped over.
If this finely tuned system falls out of balance, it can have far-reaching consequences – such as in cancer, autoimmune diseases, or mental disorders.
Unlike Illumina technology, which reads a chemically modified copy of the DNA, SMRT analyses the original DNA in real time. It measures not only the base sequence, but also the dwell time of the DNA polymerase at each base – known as incorporation kinetics.
This is precisely where the key to detecting epigenetic modifications lies: methylation and other chemical changes influence these kinetics. They act like small obstacles or skid marks on the DNA strand. The polymerase „lingers“ longer at such sites – and it is exactly this delay that is directly captured by the SMRT camera.
SMRT is particularly well suited for the direct detection of certain chemical markers, such as methyl groups on adenine or cytosine bases, which play an important role especially in bacteria and plants. For cytosine methylation (5mC), which is central in human epigenetics, direct detection is less sensitive but can be reliably derived using specialized bioinformatics tools such as pb-CpG-tools.
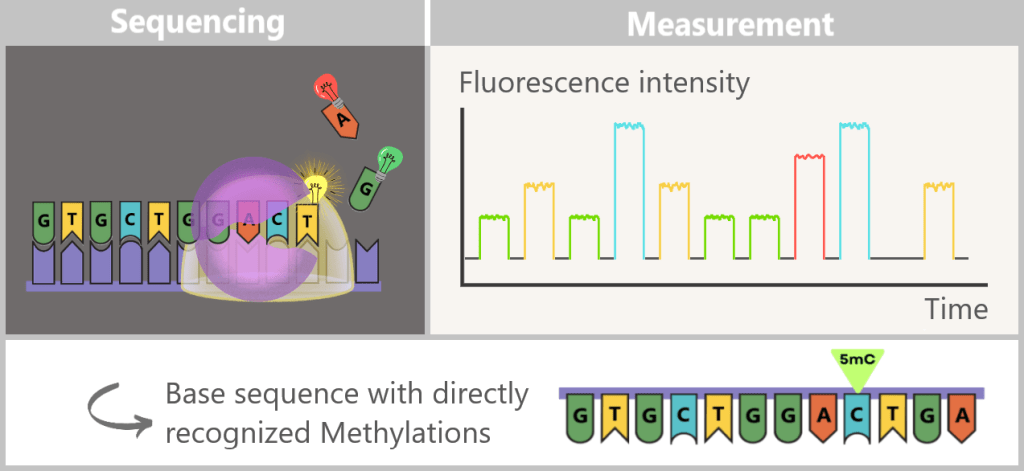
Fig. 8-E: Direct Base Detection and Methylation Analysis in SMRT Sequencing How SMRT Sequencing Reveals Epigenetic Modifications
During SMRT sequencing, the fluorescence signals of the incorporated nucleotides are measured in real time. At the same time, modifications such as 5-methylcytosine (5mC) can be detected, as they slightly alter the kinetics of the polymerase.The key advantage: the extremely long read lengths reveal how methylation patterns are intertwined across large genomic regions with other factors, such as:
- gene variants,
- haplotypes (groups of co-inherited gene variants),
- repetitive sequences.
In this way, PacBio provides not only the genetic sequence of letters but also a „map” of epigenetic regulation – revealing which „pages” functionally belong together and how this network influences disease processes such as cancer or neurological disorders.
Defective Scissors in the Genome: How PacBio Unmasks Splice-Site Variants
In the section Alternative Splicing: The Flexible Kitchen of Genes, we described in simplified terms how our DNA, as a molecular cookbook, produces recipes in the form of mRNA transcripts. These transport the genetic information from the cell nucleus to the ribosomes in the cytoplasm – the cell’s „protein factories”.
The recipe is subsequently adjusted: molecular scissors remove certain ingredients (introns) and recombine other building blocks (exons). This process is controlled by splice sites – markers that are „copied“ from the DNA. These can be thought of as cutting instructions: they determine which parts of the recipe are used and how they are assembled.
But what happens if these cutting instructions are faulty? The consequences can be severe:
- Exons are skipped → parts of the blueprint are missing.
- Introns are erroneously retained → „junk” is incorporated into the protein.
- Incorrect splice sites are created → the mRNA text becomes distorted.
Such splice-site variants often lead to defective or non-functional proteins – with possible consequences such as cancer, genetic disorders (e.g. β-thalassemia), or neurological diseases. Since protein biosynthesis depends directly on the mRNA sequence, detecting such errors is crucial.
The key lies in PacBio’s ability to analyze complete mRNA molecules in one piece – usually 500 to 5,000 nucleotides long, sometimes significantly longer. Since PacBio reads DNA, the mRNA is first converted into a stable copy (cDNA, complementary DNA). This can then be sequenced in its entirety. This allows us to see exactly how the recipe is structured and which ingredients are required in which order – no transcription or splicing errors remain hidden.
In addition, the underlying DNA segment can also be sequenced. Bioinformatic tools compare cDNA and DNA sequences with reference data, revealing deviations in the splicing pattern and their genetic cause.
The DNA shows: Here the instruction is faulty.
The cDNA shows: Here the recipe is mutilated.Only the combination of both pieces of information creates a sound basis for precise diagnoses and targeted therapeutic approaches.
Conclusion – When you want to tell the whole story
When it comes to telling the entire story of a DNA segment in one go – without cutting it into pieces or piecing it back together – SMRT sequencing is the method of choice.
PacBio-SMRT is the archaeologist of genomics: it sees the whole picture where others only provide fragments. In 2022, it played a key role in closing the last gaps in the human genome.
PacBio SMRT technology is not a mass-market product but a precision instrument. It is slower and more expensive per base pair than Illumina – but in return, it provides greater detail and versatility.
In practice, both technologies are often combined: Illumina for breadth, PacBio for depth. Together, they provide a complete picture – like a satellite image and a close-up merged into a single panorama.

4.4.2. Method 2: High-Tech Tunnel –Electrically Scanning DNA
While in the molecular copying machine with color sensors the DNA is first replicated and then „photographed“ in an image, the second method works in a radically different way: the DNA is passed through a high-tech tunnel and scanned electronically in real time.
This is the principle behind Oxford Nanopore sequencing.
a) Oxford Nanopore Sequencing
DNA Through the Eye of a Needle
Imagine reading a rope by letting it slowly slide through your fingers – sensing every subtle irregularity: thickness, dents, knots. That’s exactly how Oxford Nanopore Sequencing (ONT) works: it „feels” the DNA as it passes through a tiny biological eyelet.
This eyelet – a so-called nanopore channel – is embedded in a membrane through which an electrical current flows. As soon as a single DNA molecule is threaded through this high-tech tunnel, the bases (A, T, G, or C) influence the current flow with different intensities – like pebbles of different shapes in a stream. These changes generate characteristic electrical signals that are recorded in real time.
ONT does not read light signals, but rather current patterns – an electrical „finger reading” of DNA.
A single thread – read directly
Before sequencing begins, the DNA is coupled to a motor protein that pulls it precisely through the pore – not too fast, not too slow. Unlike Illumina or PacBio, there is no need for polymerase, fluorescence, or amplification: the original DNA is read directly – a major advantage for sensitive or damaged samples.

Fig. 9-A: Schematic representation of the sequencing cell and the ion current in the idle state The sequencing cell consists of two chambers: the cis chamber (top) and the trans chamber (bottom), separated by a membrane with embedded nanopores. On the left, a DNA molecule has docked onto a nanopore with the help of a motor protein. On the right, an empty nanopore is shown, through which a constant ionic current flows. The applied voltage between the negative electrode (cis) and the positive electrode (trans) drives the ion flow. The ionic current is measured in the well (a small channel in the chip), as shown in the current/time diagram. As long as no DNA passes through the pore, the current remains constant.
And the best part: the DNA can be extremely long. ONT holds the world record for the longest DNA ever sequenced – over two million base pairs in a single read. Entire chromosomal regions can thus be captured without interruption – like reading a novel in one single breath.
From noise to meaning – the art of signal interpretation
The raw electrical signals generated as the DNA passes through the pore initially resemble a noisy radio channel. However, with the help of specialized algorithms – known as basecalling – the current noise is translated into a clear sequence of letters: A, T, G, or C.

Fig. 9-B: The image illustrates the central mechanism of nanopore sequencing. 1) A motor protein pulls the DNA through the pore in a controlled manner. The negatively charged DNA moves from the cis side to the trans side due to the applied voltage. In the process, the individual bases (A, T, G, C) affect the ionic current in a specific way.
2) A graph shows the change in the measured current over time. Each base combination generates a characteristic signal that is decoded by algorithms.
3) A computer analyzes the electrical signals and determines the sequence of bases from them.Unlike PacBio, ONT does not distinguish bases individually, but in groups of five to six nucleotides that occupy the pore simultaneously. Each of these „5-mer” generates a characteristic current signal. This increases the reading speed but makes interpretation more complex – somewhat like recognizing words in an unfamiliar dialect. Homopolymers (long repeats of the same base, e.g. „AAAAA”) are particularly challenging.
AI-powered basecalling software is continuously improving. Error rates have dropped dramatically, and ONT reads now achieve high accuracy – especially when the dataset is large enough for statistical multiple coverage of each DNA base through repeated, random sampling.
There is a cool visual explanation in the video How nanopore sequencing works by Oxford Nanopore Technologies.
For those who prefer a more detailed explanation, additional background information can be found here.
Key Strengths – Fast, Portable, Versatile
What makes ONT special:
Portability: Devices like the MinION fit in a pocket. They require only a laptop and a power source – ideal for field research, space missions, or rapid analyses during outbreaks (e.g., Ebola or SARS-CoV-2).
Speed: The DNA is read live. Initial sequencing data is often available within minutes – a priceless advantage in clinical emergencies.
Direct epigenetics: Like PacBio, ONT can directly detect methylation and other epigenetic modifications, as they subtly affect the electrical signal – without any additional treatment or reagents.
Direct RNA sequencing: RNA molecules can also be read without prior conversion to cDNA – a true unique feature.
Why Direct RNA Sequencing Matters
Other sequencing methods, like PacBio, must first convert mRNA into cDNA in order to read it. This is like copying a recipe first – small details can be lost in the process. In contrast, Oxford Nanopore technology reads the mRNA directly – without the detour through DNA.
This way, all the „marginal notes of the cookbook” are preserved: chemical modifications, variants, and even the exact length of the molecules – information that is often lost in a copy. This creates an unaltered view of gene activity – a priceless advantage for detecting complex processes such as gene regulation or faulty RNA processing.
This is a breakthrough, especially for personalized medicine: many diseases arise not only from altered DNA but also from deviations in RNA.
Practical Examples:
- Cancer diagnostics: RNA splice variants can help detect aggressive tumor forms early and guide targeted treatment.
- Hereditary diseases: For diseases such as spinal muscular atrophy or Duchenne muscular dystrophy, RNA profiles enable accurate assessment of disease progression and personalized therapies, e.g. with antisense oligonucleotides.
- Infectious medicine: Direct sequencing of viral RNA allows rapid identification of virus variants – crucial during epidemics and for selecting appropriate therapies.
Direct RNA sequencing thus helps reveal each person’s unique „recipe variant” – with all its small deviations, marginal notes, and special ingredients – enabling more precise diagnoses and tailored treatments.
Conclusion: The Genomics „Electric Sense Finger”
ONT is the sensory-rich border crosser of genomics: fast as a racehorse, portable as a Swiss Army knife and close to biological processes. Where others have to copy, it feels live – even for RNA or epigenetic traces.

4.4.3. Genome Sequencing –Technologies Compared
In the world of genomics, there is no single method – rather, there is an entire ensemble. Each sequencing platform has its own style, its strengths, and its ideal application area: from the classic solo storyteller to the high-speed streamer.
Even though all four sequencers have their place on the stage, not every one of them is suited for every performance. Someone who wants to investigate a single gene needs different tools than someone who aims to capture the entire genome in epic breadth. And anyone trying to detect rare variants must read very carefully.
Let us now take a closer look at the four main players to understand their strengths, weaknesses, and strategic roles in detail.
a) Throughput vs. Usability – Quantity Does Not Always Equal Quality
„Throughput” sounds like efficiency – as many DNA letters per minute as possible. In sequencing, it is measured in bases per run: that is, how many bases a device reads out in a single sequencing process.
Modern platforms such as Illumina’s NovaSeq X Plus achieve peak values of up to 16 terabases with short reads (100–300 bp) – that’s 16 billion letters in a single run. Like a high-performance printer that produces entire genome libraries overnight.
PacBio and Oxford Nanopore follow a different approach. They generate long reads – from 10,000–25,000 bp for PacBio HiFi reads, up to over 1 million bp for ONT. Their throughput is lower: PacBio (Revio) achieves up to 1.300 gigabases, ONT (PromethION) up to 7.000 gigabases per run. Sounds like less – and it is, if you only look at the number of bases.
But: more data does not necessarily mean more insight.
What matters is not only how much is read – but how precisely and how often. Two key concepts help to assess the informative value of sequencing data.
🔍 Quality Score – the confidence in each letter
The so-called Q-score (quality score) describes how likely it is that a read DNA letter is correct. The scale is logarithmic:
- Q20 = 99 % accuracy → 1 error per 100 Basen
- Q30 = 99,9 % accuracy → 1 error per 1.000 Basen
- Q40 = 99,99 % accuracy → 1 error per 10.000 Basen
In research, Q20 is often sufficient. In medical diagnostics, Q30 or higher is considered standard, and for high-precision applications (e.g., tumor genetics), Q40 is expected.
For raw data, Illumina typically ranges from Q30 to Q40, while PacBio (Continuous Long Reads, CLR) and ONT only achieve values between Q10 and Q15.
Sanger outperforms them all – with Q40–Q50, but only on short DNA segments. Ideal for targeted confirmation of individual variants.
So how do modern sequencing methods meet the high standards of clinical diagnostics? The answer: through coverage and bioinformatic analysis.
🔁 Coverage – repetition reveals the truth
Coverage describes how often each area of the genome has been read.
Example: 30x coverage means that each letter has been read an average of 30 times – in different fragments and in different constellations.
Why is this important? Individual measurements may contain errors. Only through repetition and statistical majority formation can genuine variants be distinguished from artefacts.
In medical diagnostics, 30× coverage is the minimum standard.
With 30–50× coverage, Illumina achieves an overall accuracy of Q35–Q40+. It reliably detects single nucleotide variants (SNVs), insertions/deletions (indels), and copy number variants (CNVs).
PacBio HiFi Reads achieve Q30–Q40 at 30x coverage and ONT Q30–35. ONT requires 50x coverage to achieve Q44 precision.
The big advantage of PacBio and ONT is that they read much longer DNA segments – often 10,000 to 100,000 bases, and in the case of ONT, even over 1 million. This enables them to recognize complex structures, repetitions and epigenetic signatures, and allows RNA analysis and phasing – i.e. the assignment of variants to paternal or maternal chromosomes. It is not the quantity that counts here, but the overall picture.
🧬 Depending on the goal – different requirements for coverage and accuracy
The required coverage and sequencing quality depend on the clinical application:
Population studies: Q30 at 30× coverage is usually sufficient to detect statistical patterns.
Cancer diagnostics: Q35 at 60–100× coverage for reliable detection of rare somatic mutations.
Liquid biopsies, mosaic analyses: Q40 at 100–300× coverage – to distinguish true signal variants from background noise.
Phasing: Long reads (PacBio, ONT) with 30× coverage are often sufficient to assign variants reliably.
📉 The dilemma: greater certainty – greater effort
Higher coverage increases confidence, but also means more data, longer computation times, larger infrastructure, and higher costs.
b) Cost-Effectiveness – Cost per Base vs. Cost per Insight
Illumina (NovaSeq/NextSeq) remains the price-performance favourite for whole genome sequencing:
👉 200–800€ for a complete genome at 30× coverage – precise for point mutations, ideal for routine analyses.PacBio (Revio/Sequel IIe) and Oxford Nanopore (PromethION/MinION) are more expensive:
👉 700–1,200€ for 15–30× coverage – but with additional information such as long reads, structural variants, epigenetics, and RNA. This is not a luxury, but in many cases diagnostically crucial.Oxford Nanopore also scores with a low entry cost: a MinION costs just a few thousand euros, is portable, and delivers data within hours.
👉 The simple equation „more bases = better deal” falls short. What matters is what can be read and understood from the data.🧾 Hidden costs – reading the genome is cheaper than understanding it
A human genome for under 300€? Technically yes – but this usually only covers reagents, machine time, and raw data.
What is often missing are the hidden items:
🔧 Sample preparation
🔍 Quality control & library creation
🧠 Bioinformatics & clinical interpretation
📄 Findings report & physician consultationThese areas often account for 70–80% of the total costs.
Because: reading is cheap today – understanding remains challenging.
c) From Sample to Result – How Quickly Does the Whole Genome Speak?
Reading the genome is like a performance: the curtain rises with sample collection – but when will the final curtain fall, the clinical report?
It depends on technology and infrastructure:
Oxford Nanopore enables preliminary results within 24–48 hours – thanks to real-time analysis during sequencing.
Illumina provides data in 3–7 days with optimized workflows – the gold standard for routine diagnostic procedures.
PacBio takes 4–7 days, but offers particularly high-quality long reads and epigenetic information.
Sanger delivers targeted validations within hours – but only for small regions of interest.
Choosing a platform is therefore not just a matter of data quality – but also of clinical timing.
Those who need to act in hours rather than days turn to Oxford Nanopore. Those who require robust routine data rely on Illumina. And those who want maximum contextual depth take a bit more time with PacBio – for significantly greater insight.
The curtain falls. Yet behind the scenes, amid hope, high-tech, and human care, the search for the perfect pace for each patient continues. After all, it’s not the fastest machine that matters – but the right rhythm for the human story behind it.
d) Clinical Applications – Which Technology Fits Which Purpose?
Genome sequencing has long been more than just a research tool – it has become a key instrument for planning individualized therapies. But which method is suitable for which task?
A look at six key application areas shows how these technologies are shaping modern medicine.
Application Area Primary Method Complementary Methods Routine Diagnostics Illumina Sanger Pharmacogenomics Illumina Sanger Epigenetics PacBio, ONT – Cancer Genomics Illumina (SNVs), PacBio/ONT (SVs) Sanger Neonatal Emergency Diagnostics ONT Illumina, Sanger Rare Diseases PacBio, ONT Illumina, Sanger 👉 Insight: The optimal technology always depends on the clinical question – standard analysis, search for the unknown or emergency diagnostics? Validation or initial diagnosis?
e) AI as Co-Pilot – Automation Without Relinquishing Responsibility
The integration of artificial intelligence (AI) – especially deep learning – is indispensable in all modern DNA sequencing platforms (Illumina, PacBio, Oxford Nanopore). Its main applications are:
🧬 Basecalling (conversion of raw data into DNA sequences) & error correction
🔍 Quality control & real-time analysis
🧠 De novo assembly, genome analysis & variant detection
📚 Pathogenicity prediction, literature research & clinical classification
📄 Automated report templates👉 Insight: AI transforms sequencing from a mere data-generation process into an intelligent interpretation workflow – making it central to modern genomic research and diagnostics.
But one thing remains important: AI does not (yet) think like a physician. In rare diseases, complex cases, or newly discovered genetic variants, clinical expertise remains essential. Legally, physician-validated interpretation is also mandatory – even in the age of digital genomics.
Ultimately, genome sequencing is not a solo performance – it requires the entire ensemble: precise technology, intelligent bioinformatics and clinical intuition. Because only when everyone plays their part can data be transformed into healing knowledge.
f) Comparison Table: Sanger, Illumina, PacBio, ONT
Feature Sanger Illumina PacBio ONT Sample preparation DNA-Template, Extraction,
Primer-Design, ddNTPs
🕒 ~6–12 Std.Fragmentation,
Adapter ligation
🕒 ~6–24 Std.High DNA quality, Fragmentation,
SMRTbell adapters
🕒 ~1–2 TageFragmentation,
Adapter with Helicase (Motor protein)
🕒 ~6–24 Std.Read length Short: 500–1.000 bp Very short: 100–300 bp (paired-end possible) Long: 10.000–25.000 bp (HiFi-Reads) Ultra long: 10.000 bp – >1 Mb Throughput Very low: single reactions (~1–100 kb/run) Very high: up to 16.000 Gb (NovaSeq X Plus),
>100 samples simultaneouslyAveragete: up to 1.300 Gb / SMRT-Cell (Revio) Flexible: 100–7.000 Gb (PromethION), up to 20 Gb (MinION) Accuracy Very high: Q40–Q50 (99,99–99,999 %) High: Q30–Q40 (99,9–99,99 %), at 30x: Q35–Q40+ High: Q30–Q40 (HiFi-Reads), at 30x Raw data: Q15–Q20+, at 30x: Q30–Q35, Duplex: Q44 at 50x Cost per genome (EUR) (sequencing only) – ~200–800 (30x) (Large laboratories) ~700–1.200
(15–30x)~700–1.200
(15–30x)Time to result (WGS, Sample → Report) – 3–7 days (incl. preparation, sequencing, analysis) 4–7 days (incl. preparation, sequencing, analysis) 24–48 hours
(real-time, incl. analysis)Variant detection SNVs, mitochondrial variants (validation) SNVs, indels, CNVs (indirect), somatic variants, splice variants Indels, SVs, CNVs, repeat expansions, splice variants, somatic SVs Indels, SVs, CNVs, repeat expansions, splice variants, somatic SVs RNA sequencing – mRNA via conversion to cDNA, splice variants mRNA via conversion to cDNA, splice variants (long reads) Direct RNA sequencing (without cDNA), splice variants, long transcripts Epigenetic modifications – Indirect:
only with bisulfite sequencing (complex)Direct:
methylation detection via incorporation kinetics (HiFi reads) & bioinformatic toolsDirect:
methylation via signal changes (e.g., 5mC, 6mA)Place of deployment Laboratory: Stationary equipment Laboratory: Large stationary equipment (e.g. NovaSeq) Laboratory: Large stationary equipment (Revio) Flexible: Laboratory (PromethION), portable/point-of-care (MinION) Main advantages Highest precision, ideal for validation High throughput, cost-effective, precise for SNVs/indels, suitable for routine use Long accurate reads (HiFi), ideal for SVs, epigenetics, phasing Ultra-long reads, mobile, fast, direct RNA and epigenetic detection Limitations Low throughput, WGS impractical, expensive Short reads, limited for SVs/epigenetics, complex library preparation More expensive, medium throughput, longer preparation time Lower raw data quality, dependent on bioinformatics Role in personalized medicine Validation of critical SNVs (pharmacogenomics, cancer), mitochondrial analyses Routine diagnostics, pharmacogenomics, cancer genomics (SNVs/indels), rare diseases Epigenetics, cancer genomics (SVs), rare diseases (SVs, repeat expansions), phasing Neonatal emergency diagnostics, epigenetics, cancer genomics (SVs), rare diseases, mobile diagnostics
g) Key Takeaways: Between Maturity and Routine
DNA sequencing is at a pivotal turning point: in recent years, it has evolved from a purely research-based technique into a clinically relevant tool in modern medicine. Whether in oncology, rare disease diagnostics, or pharmacogenomics – genome analyses are increasingly providing answers where conventional methods reach their limits.
And yet: despite its impressive technical maturity, genome sequencing is still not a widespread standard in clinical practice.
Technology mature – infrastructure incomplete
Modern high-throughput methods such as Illumina or the long-read technologies from PacBio and Oxford Nanopore now enable highly accurate analyses at comparatively low costs. Whole genome sequencing (WGS) can be performed in specialized laboratories for a few hundred pounds per case – with a total turnaround time of less than a week. Mobile platforms such as MinION even provide initial genetic information within 48 hours in acute situations, such as with seriously ill newborns.
Despite this progress, clinical use remains fragmented. There are many reasons for this: there is a lack of standardized bioinformatic analysis protocols, interdisciplinary trained specialists, and legally compliant and economically viable integration into everyday healthcare. Above all, the interpretation of genetic variants – especially those with unclear clinical significance – requires specialized expertise that is not available in many places.
The UK as a blueprint
While many countries are still experimenting with pilot programs or making case-by-case decisions, the United Kingdom is institutionalizing genome sequencing as an integral part of public healthcare. As part of a national pilot program, every newborn will soon undergo genomic screening – a precedent in prenatal and early-childhood preventive medicine. The goal is to identify genetically driven diseases in early childhood, before clinical symptoms appear. In this way, DNA analytics evolves from an individual diagnostic tool to a population-wide preventive strategy – a paradigm shift.
This model demonstrates that the barrier to broad implementation is less technical and primarily systemic: missing data standards, ethical and legal ambiguities, and a restrictive reimbursement policy prevent widespread integration in many places. The British initiative is therefore regarded as a pioneering example of institutionalized genomic medicine.
Technological outlook
Several technological developments are likely to further accelerate widespread implementation in the near future:
🚀 Decreasing cost per genome makes sequencing economically attractive even for smaller facilities.
🚀 Decentralized, mobile systems like MinION enable diagnostics independent of location, for example in emergency rooms or rural regions.
🚀 Faster time-to-result – from sample collection to diagnosis – allows for use even in time-critical cases.
🚀 Integration of genomic data with transcriptomics, proteomics, and epigenetics (multi-omics) promises contextualized, high-resolution diagnostics.
🚀 AI-powered analytics and automated interpretation tools reduce dependence on manual evaluation and minimize sources of error.
4.5. Genome Sequencing: The Next Technological Leap
To make decoding our DNA faster, cheaper, and universally accessible in the future, research is advancing the next generation of sequencing technologies.
a) FENT: How a Microchip Is Revolutionizing DNA Analysis
b) SBX: When DNA Stretches to Be Understood
c) The G4X System: From the Genetic Recipe to the Spatial Atlas of the Cell
a) FENT: How a Microchip Is Revolutionizing DNA Analysis
– and making it accessible to everyone
Driven by the vision of capturing genetic information in real time and directly at the point of care, the company iNanoBio is developing a new method that aims to take this idea to an entirely new level.
It combines the proven method – reading DNA through tiny nanopores – with a component from microelectronics: the MOSFET (metal-oxide-semiconductor field-effect transistor), the heart of modern computer chips.
From computer chip to DNA sensor
A MOSFET is a tiny switch with three contacts: Source (input), Drain (output), and Gate (control). When a voltage is applied to the gate, a conductive channel opens between source and drain – comparable to a bouncer who opens a gate. Even minimal changes in voltage influence the current flow, which makes MOSFETs extremely sensitive.
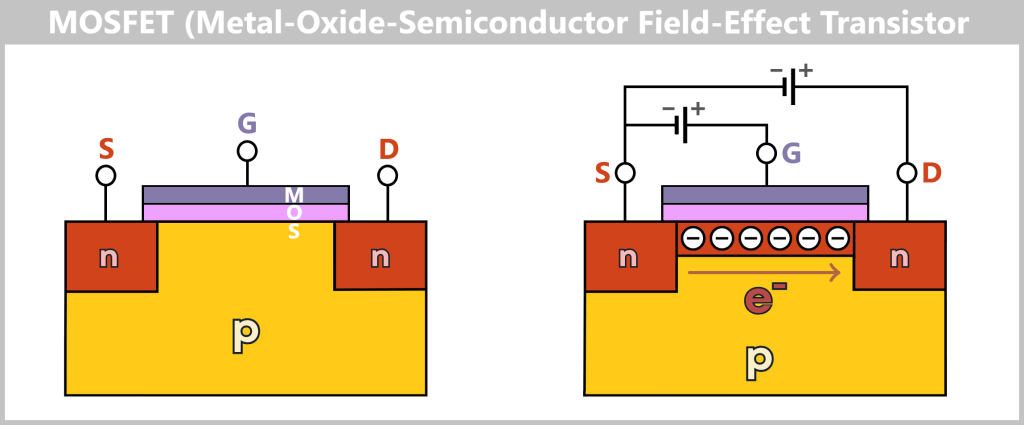
Fig. 10-A: Structure and function of a MOSFET A MOSFET is a tiny switch from microelectronics with three contacts: Source (input), Drain (output), and Gate (control). The source and drain are embedded in the semiconductor material, while the gate is insulated by an ultrathin oxide layer.
Without voltage at the gate, the channel between source and drain remains closed – no current flows. When voltage is applied to the gate, an electric field attracts charge carriers (e.g., electrons) and opens a conductive channel. The MOSFET thus acts like an extremely sensitive bouncer: only when it receives the signal does it open the gate and allow current to pass.Here’s the clever part: iNanoBio has managed to integrate a tiny pore directly into the sensitive control region of the MOSFET. As a DNA strand passes through, each nucleotide (A, T, C, or G) alters the current between source and drain in a characteristic way. The result: a novel sensor called FENT – short for Field-Effect Nanopore Transistor.
The FENT has a cylindrical design and encloses the pore. This allows it to measure signals from multiple directions simultaneously, which increases accuracy and reduces errors.
Fig. 10-B: Cross-section of a FENT The FENT is a cylindrically constructed MOSFET that fully encloses a nanopore. A voltage is applied between source and drain, allowing a small current to flow through the semiconductor of the pore wall – the so-called source-drain channel current. The thin oxide layer insulates the gate from the channel and enables precise detection of changes in the electric field. The nanopore serves as a sensing zone for local field variations caused by passing DNA bases.
Fig. 10-C: FENT during DNA translocation The figure illustrates the operating principle of a FENT during the passage of a single-stranded DNA (ssDNA) through the nanopore. Before measurement begins, the originally double-stranded DNA (dsDNA) is separated into its two single strands by heat or chemical treatment. Only one single strand is then guided through the nanopore.
In the initial state – i.e., without DNA in the pore – a constant quiescent current flows between the source and drain through the semiconductor that forms the pore wall. This current is determined by the voltage between the source and drain and the gate potential.
However, as soon as a DNA base (A, T, C, or G) passes through the nanopore, it affects the electrical behavior of the system:
Due to its specific chemical structure and atomic distribution, each base has its own tiny electric field. This field penetrates the extremely thin pore wall and acts on the semiconductor like a local gate signal. This causes a local change in the charge carrier density (electron concentration) in the transistor channel, which leads to a measurable change in the drain-source current.
These current changes are recorded as characteristic signals for the respective base. During translocation, this creates a sequence-dependent current trace, allowing the order of DNA bases to be determined electronically.A general-audience explanation is provided in the video FENT Nanopore Transistor Explainer Video.
Speed that sets the benchmark
According to iNanoBio, a FENT can read up to 1 million DNA bases per second and pore – about 100 times faster than previous nanopore technology. A 5 x 5 mm chip with 100,000 of these sensors could sequence a human genome in a few minutes. The costs would be significantly lower, and the error rate reduced.
Possible applications
- Early cancer detection from blood samples
- Mobile diagnostics for infections directly on-site
- Large-scale research studies enabled by high speed
In the long term, iNanoBio aims to make DNA analysis devices as widespread as today’s blood pressure monitors – available in clinics, laboratories, doctors’ offices, and potentially even at home. Early prototypes exist, and the path to mass production is underway.
If the technology delivers on its promise, genome sequencing could soon become as routine as a standard check-up – fundamentally transforming our understanding of health.
b) SBX: When DNA Stretches to Be Understood
In the race to improve DNA analysis, the pharmaceutical company Roche is pursuing an astonishingly simple approach with SBX (Sequencing By eXpansion): it artificially enlarges DNA so that its „letters” A, T, C, and G are easier to distinguish from one another.
Step 1 – From the original to the enlarged copy
SBX creates a surrogate copy of the original DNA, called the Xpandomer. The DNA fragment is fixed to a tiny anchor on a substrate, and a polymerase is used to create a copy – but not from natural nucleotides, rather from surrogate nucleotides.
A surrogate (from Latin surrogatum = „substitute”) generally refers to a replacement or substitute for something else.
These consist of:
- The corresponding base (A, T, C, or G)
- A folded extension strand specific to each base, serving as a signal marker
- A cleavable bond that holds the strand in shape
- A Translocation Control Element (TCE) that precisely regulates the process later
Fig. 11-A: Simplified representation of a surrogate nucleotide
[Sequencing by expansion (SBX) technology]The surrogate nucleotide (XNTP) consists of two functional components: the modified nucleotide and the SSRT (symmetrically synthesized reporter tether).
The modified dNTP contains the actual base (in this example, cytosine, C), which is connected to the reporter strand via an acid-sensitive bond (highlighted in red).
This is followed by the triphosphate (PPP), which enables the polymerase to incorporate the nucleotide into the growing chain.
The reporter tether (SSRT) consists of a folded extension strand with a Translocation Control Element (TCE) at its tip, which later controls passage through the nanopore.
An enhancer region (gray/purple) facilitates incorporation by the polymerase and stabilizes the structure.
After synthesis, the red acid-sensitive bond can be cleaved – causing the reporter strand to „unfold” and generating the extended Xpandomer molecule during sequencing.
Fig. 11-B: Synthesis of the Xpandomer [Sequencing By Expansion] The DNA fragment (template DNA) is fixed to a solid substrate via an anchor.
A primer binds to the DNA template, supported by the leader and concentrator, which stabilize the start point and facilitate proper positioning.
Fixation – The DNA template is anchored to the substrate, and the primer section hybridizes to the matching region of the DNA.
Extension – a specialized polymerase extends the template bases, but not with natural nucleotides; instead, it uses surrogate nucleotides (XNTPs), each carrying a reporter tether.
Xpandomer formation – at the end, an artificially expanded DNA strand is produced: a surrogate copy (Xpandomer) in which the bases are replaced by characteristic reporter strands.
The Xpandomer initially remains anchored to the substrate and is released in a later step via light or chemical cleavage.After synthesis, the Xpandomer is separated from the original DNA, the cleavable bonds are broken – the strands unfold and increase the spacing between bases by 50-fold. This greatly facilitates subsequent identification.
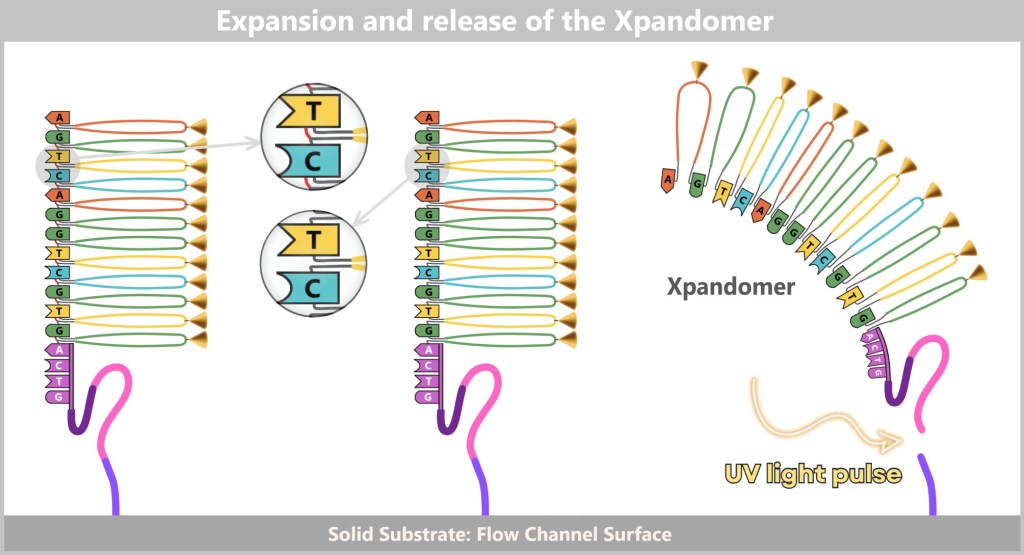
Fig. 11-C: Expansion and release of the Xpandomer [Sequencing By Expansion] After synthesis, the Xpandomer is chemically unfolded („expanded”) on the substrate and then released.
Left) Expansion: A mild acid treatment cleaves the cleavable bonds (red) between the base and the reporter strand. This causes the reporter structures to unfold – transforming the compact double structure into an elongated Xpandomer that represents the sequence of bases.
Right) Release: A UV light pulse cleaves the bond between the substrate and the leader region. The now freely moving Xpandomer remains structurally marked via the leader and concentrator – these regions later assist in alignment and entry into the nanopore.
From a dense DNA template, a long, readable molecule is created, whose reporter segments make the genetic information electrically visible.Step 2 – Sequencing
Instead of the DNA itself, the Xpandomere is now pulled through nanopores on a sequencing chip – around eight million of them working in parallel. The Xpandomere’s adapter guides it precisely into a pore.
When an extension strand reaches the pore, it temporarily sticks to the TCE. Each base generates a specific electrical signal. After 1.5–2 milliseconds, a voltage pulse releases the TCE, allowing the next strand to follow. This produces a clear, easily distinguishable signal trace – more precise than with Oxford Nanopore, where the bases generate a somewhat „blurred” signal.
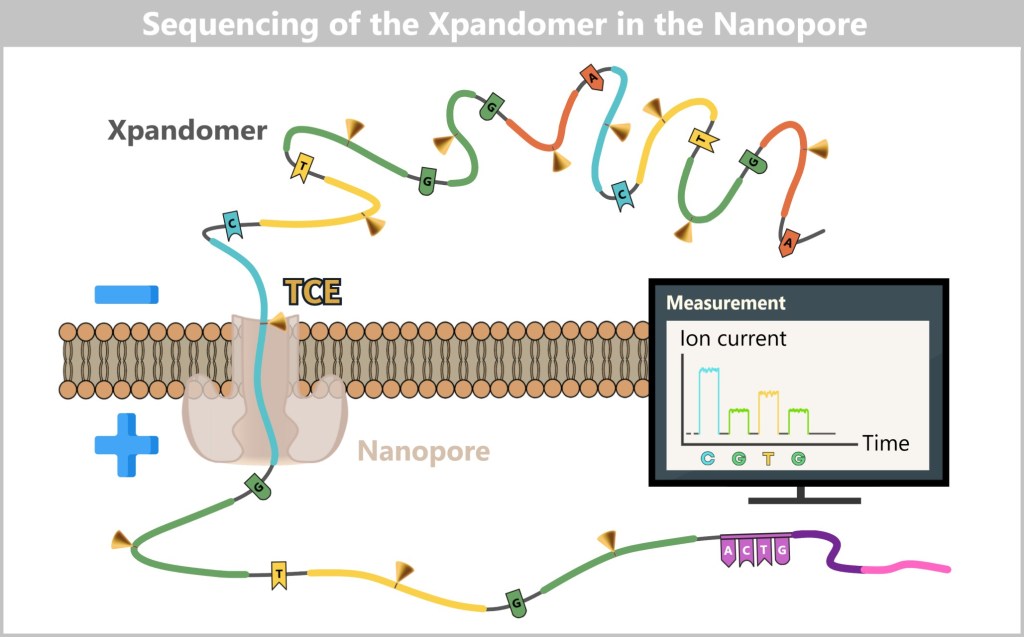
Fig. 11-D: Sequencing of the Xpandomer in the nanopore
[Sequencing By Expansion, Introduction to Sequencing By Expansion]The released Xpandomer is drawn toward the nanopore by an electric field. There, the TCE (Translocation Control Element) controls entry and temporarily holds the reporters in place.
As each reporter segment (the extended „base”) passes through the pore, the ion current changes in a characteristic way: each base generates its own electrical signal – comparable to an individual barcode. This sequence of signals is recorded in real time:
After each measurement, a voltage pulse releases the strand from the TCE, allowing the next segment to be drawn in.The following animation illustrates the basic principles of Roche’s SBX technology.
A detailed presentation is available here.
Advantages and performance:
- Speed: Millions of Xpandomers are read simultaneously and in real time.
- Accuracy: Signal-to-noise ratio comparable to Illumina.
- Flexibility: Read lengths of over 1,000 bases possible – faster and more precise than Nanopore, longer than Illumina.
Initial tests show that SBX can decode up to seven complete human genomes per hour at 30× coverage. In urgent cases, the entire process – from sample preparation to finished analysis – takes only around 5.5 hours.
The market launch is planned for 2026. Whether SBX will prevail against Illumina and Oxford Nanopore depends on price, availability, and user requirements.
c) The G4X System: From the Genetic Recipe to the Spatial Atlas of the Cell
Our body consists of billions of cells that function like tiny kitchens. DNA is their cookbook – it contains all the recipes of life. When a recipe is needed, the cell creates an RNA copy that serves as a blueprint for proteins. These proteins carry out specific tasks: they build structures, transmit signals, or fight pathogens.
However, proteins rarely act alone. They operate in finely tuned networks whose activity depends on where, when, and with which partners they interact. Processes such as immune responses or tumor growth do not arise within a single cell, but from the interplay of many specialized cells – often located in different regions of a tissue. Biology is therefore not only biochemical, but also spatially organized. Classical sequencing methods, however, typically capture only genetic content, not its spatial distribution.
Gene activity directly within tissue
The G4X Spatial Sequencer from Singular Genomics reveals where genes are active and proteins are formed in tissue – directly in preserved tissue samples (FFPE, formalin-fixed and paraffin-embedded).
FFPE samples are commonly used in pathology, for example in cancer diagnostics. They act like a „frozen snapshot,” except that chemical fixation with formalin is used instead of cold. Formalin crosslinks proteins and nucleic acids, thereby fixing molecular structures in their spatial context.
What is preserved:
- RNA (often fragmented and chemically modified) – reflects gene activity at the time of fixation.
- Proteins – indicate which structures and signaling pathways were active at that moment.
This makes it possible to reconstruct months or years later which genes and proteins were present and active in the tissue at that exact moment – as if time had been stopped. With the G4X, this „frozen” molecular landscape can be visualized at high resolution, including the precise spatial location within the tissue.
Fig. 12-A: From tissue to gene activity A tissue consists of many specialized cells. Each cell uses only a subset of its genetic program. RNA makes the active genes visible. RNA molecules are read by ribosomes and translated into proteins. FFPE tissue sections preserve this spatial organization – forming the basis for using the G4X system to measure where specific RNA molecules are active within the tissue.
How the G4X works
You can think of the G4X as a microscope-scanner with a built-in laboratory, directly measuring which genes and proteins are active within the tissue – and in their spatial context.
Preparation: A very thin section of the FFPE sample is placed on a specialized substrate.
RNA detection: Padlock probes are used to identify the spatial distribution of specific RNA sequences within the tissue.
Padlock probes – DNA lassos for target sequences
Imagine you want to find a specific object (an RNA sequence) in a vast area (the tissue). A padlock probe is a DNA fragment whose two ends are complementary to adjacent sections of the target sequence.
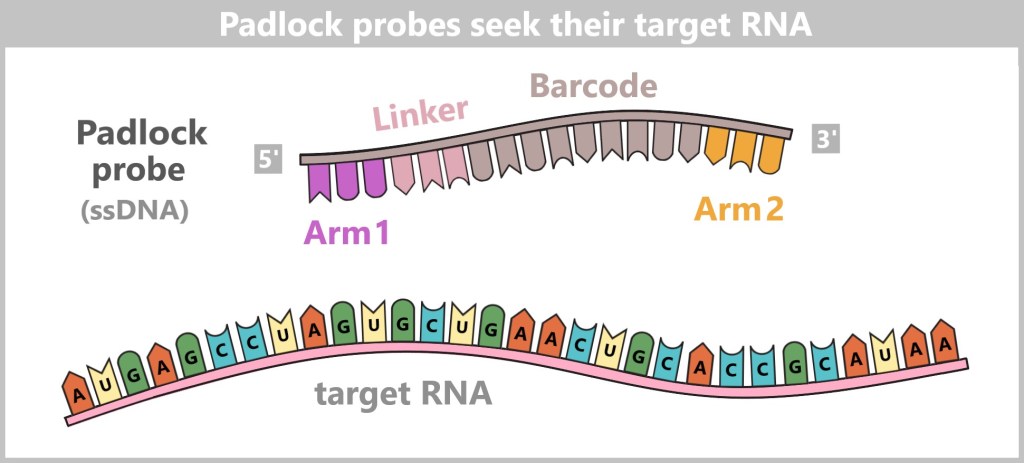
Fig. 12-B: A padlock probe floats through the tissue in search of its target RNA. Padlock probe: single-stranded DNA (ssDNA) 5’– Arm1 – Linker/Barcode – Arm2 – 3′
Arm 1 and Arm 2 are complementary to adjacent sequences of the target RNA.
Between them lie the linker and barcode, which carry additional identification information and do not bind.When the probe binds to its target, its two ends lie next to each other and can be closed into a stable DNA ring by ligation.
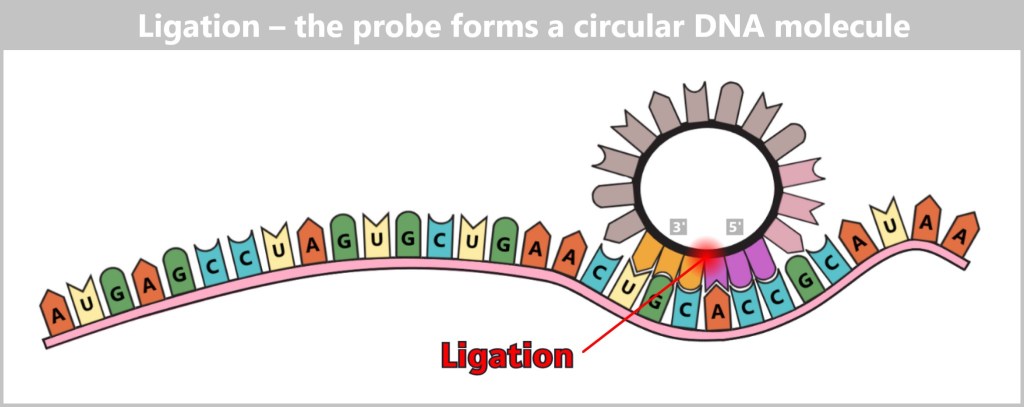
Fig. 12-C: Ligation connects the ends of the padlock probe After both arms of the probe have bound to the target RNA, a ligase joins the 5′ and 3′ ends. This forms a closed DNA loop that marks the target RNA and is ready for amplification.
If there is a variable sequence (e.g., a mutation) between the binding sites, it is first transcribed into DNA using a reverse transcriptase and incorporated into the probe.
The resulting DNA circle serves as a template for rolling circle amplification (RCA):
- A DNA polymerase repeatedly moves around the circle, generating a long DNA strand that contains multiple repeats of the target sequence – like an endless ribbon or a ball of yarn.
- These signals remain precisely at the location in the tissue where the RNA was.
- Using fluorescently labeled nucleotides and sequencing by synthesis, the sequence is decoded directly within the tissue.
Each Padlock probe carries a barcode that reveals which gene or variant it has detected. The result: a precise map showing which RNA was present at which location and which variants it carried.
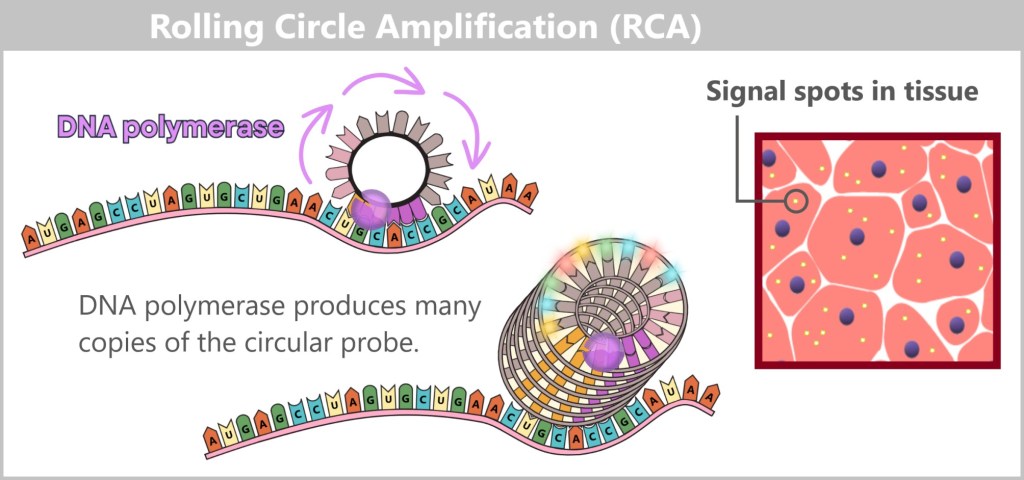
Fig. 12-D: Rolling Circle Amplification (RCA) A specialized DNA polymerase repeatedly reads the circular padlock probe, generating a long DNA strand with many repeats. Fluorescent nucleotides label the amplificates, producing a strong fluorescent signal – visible as a bright spot in the tissue, marking the location of the target RNA.
Protein detection in the same tissue section
For protein detection, the G4X system uses antibodies tagged with short DNA barcodes. After the antibody binds to its target protein, a padlock probe hybridizes to the barcode. The probe is then ligated, amplified, and read out – just like in RNA analysis. The barcode clearly reveals which protein is involved and where it occurs in the tissue.
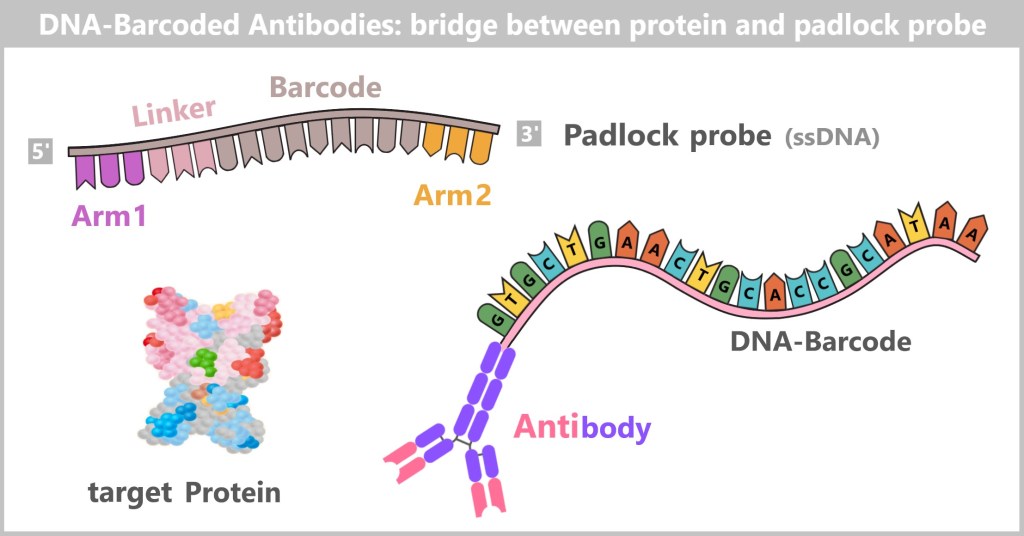
Fig. 12-E: Protein detection in the G4X system: antibody with DNA barcode and padlock probe An antibody specific to a target protein is coupled with a unique, short single-stranded DNA strand (barcode).
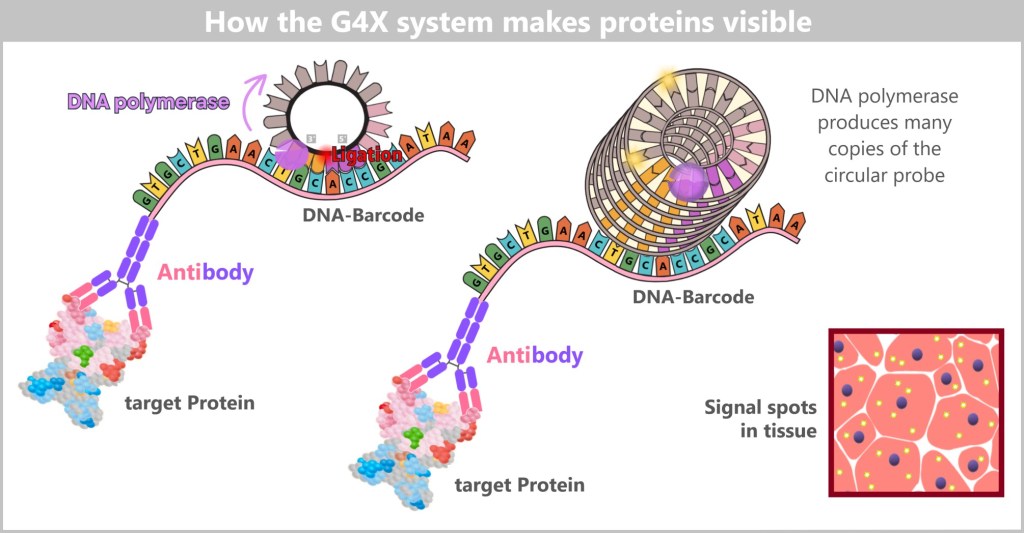
Fig. 12-F: Protein detection in the G4X system The antibody binds to its target protein in the tissue sample. A padlock probe, whose ends are complementary to two adjacent sections of the DNA barcode, binds to it and is linked by a DNA ligase to form a closed ring. A DNA polymerase recognizes the ring and reads it continuously, creating a locally anchored, circular DNA amplificate – analogous to RNA detection. The incorporation of fluorescently labeled nucleotides makes this amplificate visible. The fluorescent signal spots in the tissue mark the locations where an antibody with a specific DNA barcode has bound a particular protein.
A detailed presentation of the G4X method can be found here and here.
Why this is important
The G4X can specifically detect biomarkers that provide insights into the presence, progression, or aggressiveness of a disease – revealing not only which ones are active but also where they are located within the tissue.
Especially in tumors, this spatial resolution reveals which genes are „switched on”, in which cell regions this occurs, and how tumor and immune cells interact. This allows for a better understanding of tumor heterogeneity, clarification of disease mechanisms, and more targeted therapy planning.
The simultaneous in situ detection of RNA and proteins provides a comprehensive picture of tissue organization and opens up new possibilities for:
- Understanding complex disease processes
- More precise identification of biomarkers
- Development of personalized therapies
In diseases such as cancer, chronic inflammation, or neurodegenerative disorders, this spatial multi-omics perspective can be crucial – because often it is not the question of whether, but where and how the best therapeutic measure should be taken.
Conclusion
Genome sequencing is on the verge of a quantum leap: technologies like FENT, SBX, and G4X promise to read the genome faster, more accurately, and more comprehensively than ever before – including direct analysis within tissue. Soon, complete genomes could be decoded in minutes. The real revolution, however, begins afterward: when we understand the wealth of genetic information, translate it into medical knowledge, and use it responsibly for the benefit of humanity.
4.6. From Code to Cure
Bioinformatics as the Key to Tomorrow’s Medicine
4.6.1. From the Genetic Code to Computer-Aided Genome Analysis
Never before has it been so easy to decode DNA – but what use is the text if no one understands it? A small swab, a drop of blood – and suddenly millions of genetic data points are available. Modern sequencing has made the genome more accessible than ever before: fast, cost-effective, and nearly error-free. Yet this gives rise to a new challenge: how do we transform this flood of data into usable medical knowledge?
The answer comes from a discipline that has long operated in the background but has now become indispensable: bioinformatics – the bridge between A, T, G, C, and diagnosis, between raw sequence data and concrete therapeutic decisions. To understand why it is so important, it is worth taking a brief look back.
From the laws of inheritance to the genetic code
As early as 1860, Gregor Mendel discovered that there must be „heritable units” – genes – long before DNA was known. It was not until the mid-20th century that Oswald Avery demonstrated that these carriers of information are in fact DNA. Shortly thereafter, Watson and Crick revealed the double-helix structure with its complementary base pairing (A–T, G–C). In 1958, Francis Crick formulated the „central dogma”: information flows from DNA to RNA to proteins – the functional building blocks of life.
Fig. 13-A: The central dogma of molecular biology: Genetic information is converted from DNA into RNA through the process of transcription. The RNA is then translated into the final protein, which performs a wide variety of functions. [An overview of artificial intelligence in the field of genomics]
A gene is therefore an instruction for how a protein is made. But how do we read this instruction – and how do we interpret it correctly? The answer initially came via the detour of reverse genetics.
In the 1960s, DNA was still difficult to access directly – proteins, on the other hand, were functionally visible (for example as enzymes, transport molecules, or structural components) and could be isolated and analyzed with the methods available at the time. Researchers had already known since the early 20th century that proteins consist of chains of amino acids. From the 1960s onward, it became clear that each amino acid corresponds to a so-called codon on RNA – a triplet of RNA bases (A, U, G, C) – which can in turn be traced back to a DNA sequence.
Fig. 13-B: The Central Dogma of Molecular Biology: The DNA Code and Codons Researchers started with a known protein and worked their way back to the RNA and DNA sequence. For the first time, the genetic code became readable like a recipe book:
Protein = finished dish
Amino acid = ingredient
Codon = word for this ingredient in the handwritten recipe (RNA)
DNA = page in the cookbook from which the recipe was copiedEarly patterns and the birth of genome annotation
Even before actual DNA sequencing, typical structural elements within genes were discovered:
Promoters – start signals where „reading” of a gene begins
Start and stop codons – first and last signals in the recipe
Transcription factors – proteins that switch genes on or off
Coding sequences – list of ingredients (codons) and their order for the protein
Non-coding DNA – regions without direct protein-coding function, whose role was initially unclearThese insights laid the foundation for modern genome annotation – the computer-assisted identification of biological signals within DNA sequences.
The Sanger Moment: DNA Becomes Readable (since 1977)
With Frederick Sanger’s method, DNA sequences could be determined directly for the first time – initially only for short segments. Analyses focused on gene identification and simple comparisons. Early programs such as FASTA (1985) and BLAST (1990) enabled the matching of new sequences with databases like GenBank (1982).
Understanding of gene structure also evolved: it was discovered that only certain parts of a gene (exons) are used for protein synthesis, while other segments (introns) are spliced out of the transcribed RNA. Additionally, DNA regions that enhance (enhancers) or repress (silencers) gene expression were identified – often located far from the actual gene.
The process of genome annotation began to take shape but was still rudimentary: DNA was primarily viewed as a linear sequence of nucleotides, with a focus on protein-coding genes. Regulatory elements and non-coding DNA were only partially recognized and understood. Bioinformatics was born – initially as a tool for research laboratories.
The Human Genome Project: Data on a Gigantic Scale (1990–2004)
The goal of decoding all 3.2 billion bases of the human genome introduced entirely new challenges: millions of short sequence fragments had to be assembled into long DNA strands. Tools such as Phrap for assembly and later Ensembl for annotation were developed. Data volumes exploded – along with the need for powerful computing infrastructure.
But while the Human Genome Project was still underway, a new technology was already emerging that would catapult speed and data volume into completely new dimensions.
NGS Revolution and Clinical Breakthrough (2005–2015)
Next-Generation Sequencing (NGS) technologies made genomes more affordable and rapidly accessible – albeit as massive data torrents of billions of short fragments. New tools like Bowtie, BWA, and STAR aligned these sequences at lightning speed, GATK helped identify mutations, and tools such as SnpEff or VEP interpreted their significance.
At the same time, sequencing strategies emerged for different clinical questions:
- Whole Genome Sequencing (WGS) for complete analyses
- Whole Exome Sequencing (WES) for the 1–2% of protein-coding DNA
- Targeted Sequencing for defined gene panels
This made bioinformatics a diagnostic tool for the first time – for example, in cases of hereditary forms of cancer, rare diseases, or personalized pharmacotherapy.
Artificial Intelligence and Modern Bioinformatics (2015–Present)
The combination of artificial intelligence (AI) and bioinformatics represents one of the most dynamic and promising areas of research of our time. Together, they are unraveling the complexity of life and accelerating biomedical discoveries at an unprecedented pace.
Artificial Intelligence: More Than Just Automation
AI encompasses techniques in which machines perform human-like intelligence tasks – particularly learning, problem-solving, and pattern recognition. In the biomedical context, the following subfields are especially relevant:
Machine Learning (ML): Algorithms learn from data (e.g., genome sequences, protein structures, patient data) to make predictions or identify patterns without being explicitly programmed. Example: detecting tumor cells in tissue samples.
Deep Learning (DL): A subset of ML that uses artificial neural networks with many layers („deep”) to capture highly complex patterns in massive datasets. Example: predicting the 3D structure of proteins solely from their amino acid sequence. [AlphaFold].
Natural Language Processing (NLP): Enables computers to understand scientific literature, medical records, or clinical study reports and extract knowledge from them.
4.6.2. Modern Bioinformatics – The Digital Toolbox of Biology
Bioinformatics is the science of storing, analyzing, and interpreting biological data using computer-based methods. Its modern form is characterized by three factors:
Explosion of biological data: Next-Generation Sequencing (NGS) generates terabytes of genomic, transcriptomic, and epigenomic data per experiment.
Fig. 12: From a billion-dollar project to an everyday routine
– how the costs of genome sequencing fell while data volumes exploded –Just over twenty years ago, decoding a human genome was still a monumental undertaking: the Human Genome Project (completed in 2003) cost around 3 billion US dollars and produced only about 1 gigabyte of data. With ever faster sequencing machines and new technologies such as nanopore sequencing, this picture has changed completely. By 2012, the cost per genome had already dropped to around 6,000 dollars, in 2018 to 1,000 dollars – and today (2023), a genome can be analyzed for approximately 100 dollars.
At the same time, the volume of generated data increased rapidly: from a few gigabytes to terabytes and petabytes, and ultimately to the hundreds of petabytes produced by global projects involving millions of genomes. Estimates suggest that by 2025, around 40 exabytes of human genomic data could be generated – equivalent to roughly eight times the storage required for all words ever spoken in human history. [How AI Is Transforming Genomics]
Complexity of biological systems: Understanding disease requires the integration of data on genes, proteins, metabolic pathways, and cellular interactions.
Need for predictive medicine: The goal is to predict disease risks, therapy responses, and individualized treatment (precision medicine).
Already in many key applications, we can already talk about a fusion between bioinformatics and artificial intelligence (AI). Here are some central areas of application:
a) Genome Analysis and Sequencing
AI algorithms are used to analyze DNA sequences more quickly and precisely. Models such as DeepVariant, developed by Google, employ deep neural networks to identify genetic variants with high accuracy. This is crucial for detecting mutations associated with diseases such as cancer or rare genetic disorders.
b) Protein Folding and Structure Prediction
A milestone of AI in bioinformatics is DeepMind’s AlphaFold, which predicts protein structures with unprecedented precision. This has far-reaching implications for drug development, as a protein’s 3D structure determines its function.
c) Multi-Omics Data Analysis
A holistic understanding of biological systems requires the integration of multi-omics data (genomics, proteomics, transcriptomics, epigenetics, metabolomics) to gain a comprehensive picture of their functioning. AI helps to decipher the complex interactions between these layers.
Fig. 13: The Polyphonic Concert of Life. [AI applications in functional genomics] Biological systems are a complex interplay of different layers of information: from DNA (genomics) through epigenetic regulation, RNA (transcriptomics/epitranscriptomics), and proteins (proteomics) to metabolic products (metabolomics). It is only their dynamic interaction that defines health and disease.
For example, the new AI tool AlphaGenome demonstrates how even the smallest changes in DNA can affect gene activity as well as the production of RNA and proteins. It predicts with high precision the consequences of a single DNA base substitution for genes and their products. While earlier systems typically analyzed only the roughly 2% of the genome that codes for proteins, AlphaGenome is the first to capture the entire genome. It is based on a multi-omics analysis that directly links DNA alterations to their effects on RNA and proteins. This creates a comprehensive picture of gene expression – a crucial step toward more precise genomic research and personalized medicine.
d) Metagenomics and Microbiome Analysis
AI helps to understand the complex interactions in the human microbiome by recognizing patterns in the data from microorganisms. This is important for research into diseases such as diabetes or intestinal disorders.
e) Drug Development
AI accelerates nearly every phase of the lengthy and costly drug discovery process: target identification, virtual screening of millions of molecules, prediction of drug effects and side effects, and optimization of molecular structures. Companies such as BenevolentAI and Recursion use AI platforms to identify promising drug candidates in record time, some of which are already being tested in clinical trials.
f) Analysis of Biomedical Images
AI is used in image analysis, for example in the evaluation of MRI or CT scans, to detect tumors or other anomalies. Convolutional neural networks (CNNs), which mimic the functioning of the human visual cortex, have proven to be particularly effective in this area.
g) AI Genomics in Health Research
Gene expression in the human body does not follow a fixed pattern – and diseases such as cancer make it even more difficult to decipher. This is precisely where spatial biology comes in: using cutting-edge microscopy and genetic sequencing, it reveals how genes are active in individual cells and even within their organelles.
But it is not only the visible gene regions that are decisive. In regions long dismissed as „junk DNA”, researchers today suspect key switches of gene regulation. It is there that decisions are made about which proteins are produced – and which are not. AI and deep learning models can detect patterns in these previously hidden regions that are linked to disease. This raises hopes for new biomarkers and therapeutic approaches. One example is the platform developed by the biotech company Deep Genomics:
The combination of spatial biology, intelligent data analysis, and AI thus enables an unprecedented view into the molecular logic of life – down to the level of individual cells.
New methods of gene editing could make it possible to precisely control the behavior of individual genes – for example, to deactivate cancer-promoting genes or to stimulate the regeneration of cartilage cells in osteoarthritis. This field is still highly experimental, but AI promises to make these techniques more precise and safer.
With AI-supported CRISPR techniques, new possibilities are emerging to not only identify and modify genes more precisely, but also to better understand their interplay within complex biological networks. Early approaches suggest that this could reduce side effects in the future and increase the effectiveness of new therapies. Modern methods for the systematic testing and optimization of gene variants could also – if they prove successful – represent important steps toward personalized treatments.
Those who are interested in learning more about CRISPR techniques can find further information here.
The combination of advances in AI and genomics is therefore considered highly promising and could, in the long term, accelerate the development of new cell and gene therapies. However, since these treatments are based on living cells that vary widely and require specific conditions, much of this work is still in the experimental stage. AI could help make processes more stable, reproducible, and clinically applicable – but whether and to what extent these hopes will be realized remains to be seen.
h) Oncology
AI models analyze genetic profiles to create personalized treatment plans. For example, algorithms can predict how patients will respond to specific medications based on their genetic makeup. This is particularly relevant in oncology, where AI-based systems such as IBM Watson Oncology provide treatment recommendations.
Conclusion
Bioinformatics today serves as the brain of modern medicine. Together with artificial intelligence, it transforms the flood of data from the genome into concrete insights – from diagnosing rare diseases to developing new drugs.
Whether in genome analysis, protein structure prediction, or the planning of personalized therapies, AI-powered bioinformatics reveals what was previously hidden. It paves the way for a form of medicine that not only treats diseases but anticipates them – tailored to each individual patient.

5. A Success Story
The case of KJ Muldoon shows just how much these developments are already a reality today.
When the young boy received his diagnosis just a few hours after birth, his fate seemed sealed. He suffers from an extremely rare metabolic disorder caused by a single DNA point mutation (SNV), which prevents his body from breaking down toxic ammonia. Without a functioning enzyme, toxic metabolic products rapidly accumulate – posing a life-threatening risk even in infancy. Until now, only a risky liver transplant was considered capable of ensuring his survival.
But in KJ’s case, doctors and researchers took a new approach. Within just six months, they designed a therapy created exclusively for him: a personalized base-editing treatment.
The principle behind it is as ingenious as it is elegant: an RNA-guided system scans the genome, specifically locates KJ’s point mutation, and binds to that spot. Instead of cutting the DNA strand completely – as is the case with traditional CRISPR-Cas9 – an enzyme chemically modifies a single base: exchanging one DNA „letter” to correct the disease-causing mutation. To put it simply: it’s like fixing a single typo in a book without cutting out or rewriting the entire paragraph – extremely precise and gentle on the genome. The treatment was administered directly in the body („in vivo”) through multiple consecutive rounds, specifically targeting the liver.
The initial results are encouraging. KJ can now digest more proteins, infections are milder, and he is gaining weight. For his parents, this means a piece of everyday life without constant danger to his life – and, above all, new hope for their child’s future.
Behind this medical breakthrough lies a success story involving several disciplines. Without modern genome sequencing, the exact mutation would not have been visible. Without bioinformatics, it would not have been possible to design the appropriate base editing tool in such a short time and with such precision. And without the concept of personalized medicine, it would have been unthinkable to develop a cure exclusively for a single patient.
The therapy is still experimental, and no one knows whether its effects will be lasting or if long-term side effects might occur. Yet KJ’s case illustrates the direction medicine is heading: toward a form of healthcare that no longer relies on standard protocols, but on individualized solutions – driven by data, molecular biology, and tailored therapies. A medicine that not only treats diseases but, ideally, targets them precisely at their genetic root.
Further background information on this story can be found in this video.
6. A Look into the Future: The Synthetic Human Genome Project
Biology is on the verge of another quantum leap. After decoding and editing genomes, the vision now is to rewrite them from scratch. This is precisely the goal of the Synthetic Human Genome Project (SynHG) – an ambitious undertaking that could redefine the boundaries of biology and medicine.
With an initial funding of £10 million from the Wellcome Trust and under the leadership of Professor Jason Chin at the Generative Biology Institute in Oxford, leading universities such as Cambridge, Manchester, and Imperial College London are working on this project. The mission: to develop tools and technologies for creating synthetic human genomes – a step that could transform our understanding of life itself.
Recomposing a Genome
Unlike genome editing, which only modifies existing DNA, SynHG goes much further: it aims to reassemble the genome letter by letter from scratch. While CRISPR corrects individual typos in the text of life, this approach is about completely rewriting an entire chapter.
The first milestone: the synthesis of a complete human chromosome as proof of feasibility. It is the beginning of a journey that could take decades – and yet it builds on earlier pioneering achievements, such as the Human Genome Project (2003) and the Synthetic Yeast Project, in which synthetic yeast chromosomes were created for the first time in 2023.
Modern methods – generative AI, machine learning, and robot-assisted assembly – are intended to tackle this enormous task.
Potential Applications – Between Laboratory Vision and Clinical Practice
The opportunities are vast. At the level of somatic cells – i.e., cells whose changes are not passed on to offspring – new avenues for regenerative medicine could open up:
- tailor-made replacement tissues and organs grown in the lab,
- virus-resistant cells against HIV or SARS-CoV-2,
- gene therapies against cancer or hereditary diseases such as cystic fibrosis.
Such applications are not distant science fiction – they could become reality within the next 10 to 20 years, as technologies such as CRISPR and stem cell cultures are already well established.
The situation is quite different when it comes to germline editing – i.e., interventions in egg or sperm cells. In theory, this could permanently prevent serious hereditary diseases such as Huntington’s disease. However, the idea of „designer babies” with desired characteristics such as appearance or intelligence remains highly problematic from a scientific and ethical perspective, as such traits depend on hundreds of genes.
The project itself is limited to experiments in test tubes and cell cultures. It does not aim to create artificial life. Nevertheless, it opens up an unprecedented opportunity for researchers to intervene in fundamental human biological systems – with far-reaching consequences that must be critically examined.
Risks and Open Questions
The technical hurdles are enormous. In somatic cells, there is a risk of mosaicism (not all cells incorporate the new genome) or tumor formation due to unintended genetic alterations. In the germline, the risks are even greater: developmental disorders, epigenetic effects, or unpredictable consequences across generations.
Researchers are therefore discussing safety mechanisms such as genetic „kill switches”, which could deactivate synthetic cells in case of an emergency.
Ethics: Between Healing and Hubris
Somatic therapies are akin to organ transplants: controversial, but socially accepted when they cure. Germline interventions, however, touch taboo areas – fueled by concerns about social inequality or even eugenic scenarios. Many countries, including Germany, prohibit them through laws such as the Embryo Protection Act. International organizations like WHO and UNESCO call for strict regulation.
„To build trust, we need to actively involve society”, emphasizes sociologist Professor Joy Zhang from the University of Kent, who is investigating the ethical dimensions of the project. With her Care-full Synthesis initiative, she is attempting to initiate an open dialogue about the opportunities, but also the limitations, of this research.
Renowned geneticist Professor Bill Earnshaw from the University of Edinburgh, who has developed methods for producing artificial human chromosomes, puts it more bluntly: „The genie is out of the bottle. We could have a set of restrictions now, but if an organisation who has access to appropriate machinery decided to start synthesising anything, I don’t think we could stop them.“
In this context, the call by numerous professional organizations – including the International Society for Cell and Gene Therapy (ISCT) – for a ten-year moratorium on the use of CRISPR and related techniques in the human germline seems even more urgent. Such a postponement appears not only to be a precautionary measure, but also a necessary political response in order to enable social debates, ethical guidelines, and international control mechanisms in the first place.
Ethics expert Arthur Caplan from New York University describes the dilemma from a scientific perspective:
„… So if you ask me, will we see genetic engineering of children aimed at their improvement? I say yes, undoubtedly. Now when? I’m not sure what the answer to that is.“
„… It will come. There are traits that people will eagerly try to put into their kids in the future. They will try to design out genetic diseases, get rid of them. They will try to build in capacities and abilities that they agree are really wonderful. Will we hang up these interventions on ethical grounds? For the most part, no, would be my prediction, But not within the next 10 years. The tools are still too crude.“Outlook
SynHG opens a window into a future where we can not only read and edit the genome, but write it. Somatic applications – personalized therapies, regenerative tissues, and custom-designed cells – could soon shape the practice of medicine.
Interventions in the germline, however, remain a distant horizon – for technical, legal, and ethical reasons. The project represents not only a scientific challenge but also a delicate societal balancing act.
Given the potential for misuse of the technology, the question arises as to why Wellcome decided to provide funding at all. Dr. Tom Collins, who was responsible for this decision, explained to BBC News:
„We asked ourselves what was the cost of inaction. This technology is going to be developed one day, so by doing it now we are at least trying to do it in as responsible a way as possible and to confront the ethical and moral questions in as upfront way as possible.“
While Europe is still grappling with how to handle the topic carefully in public debate, across the Atlantic the term „new life forms” is approached much more openly. Eric Nguyen, a bioscientist at the renowned Stanford University, outlines a far more visionary perspective in his TED Talk, „How AI Could Generate New Life-Forms“.
In a striking way, he demonstrates how artificial intelligence (AI) could transform biology – not only by „reading” and „writing” DNA, but also by deliberately creating novel organisms. AI models such as machine learning and generative algorithms are already capable of recognizing complex patterns in biological data: in genomes, protein structures, or metabolic pathways. Nguyen goes further, arguing that in the future AI could also design entirely new DNA sequences – the starting point for previously unknown life forms.
This vision is undoubtedly forward-looking, but it raises fundamental questions: What priorities should be set? How can the use of such technologies be justified in a world of massive resource inequality? Who funds the research, who oversees the outcomes – and by what criteria is it decided which life forms are „useful” or „safe”?
Nguyen’s presentation makes it clear that AI-supported biotechnology could not only enable solutions to global challenges, but also forces us to think about the ethical, social, and philosophical dimensions of „creating life.”
The message: AI-powered biotechnology promises tremendous advances, yet simultaneously raises profound ethical and societal questions – challenging the very foundations of what it means to be human.
Klaus Schwab – Founder and former Chairman of the World Economic Forum
„We must address, individually and collectively, moral and ethical issues raised by cutting-edge research in artificial intelligence and biotechnology, which will enable significant life extension, designer babies, and memory extraction.“
[BrainyQuote]
7. Personalized Medicine and Smart Governance
The Biomolecular Revolution in Governance
7.1. Power, Governance, and Smart Governance
7.2. Governmentality
7.3. Personalized Medicine as a Catalyst for Biomolecular Governmentality
7.4. The Global Trend: Worldwide Biomolecular Governmentality
7.5. Conclusion: The Global Ambivalence of Biomolecular PowerThe decoding of the human genome marked not only a medical turning point, but also the beginning of a new era of political governance. What started as a large-scale scientific project in laboratories and data centers has long since reached the control apparatus of modern states: the precise measurement, categorization, and utilization of our biological foundations.
The vision of personalized medicine promises individualized therapies, precise predictions about disease risks, and more efficient use of scarce resources. But this narrative of progress falls short if we consider it solely from a medical, ethical, or data protection perspective. In fact, we are witnessing a fundamental transformation of power and governmentality – a biomolecular revolution in governance.
Before we delve deeper into the subject, it is important to clarify a few key terms.
7.1. Power, Governance, and Smart Governance
Governance refers to the art of steering societal processes, making decisions, and maintaining order. Traditionally, this occurs through the interaction of the state, markets, and civil society.
Smart governance transforms this understanding: it operates through digital technologies, data analytics, and algorithmic processes. Governance thus becomes continuous, procedural, and possible in real time. Power no longer manifests solely as political authority, but as a network of information flows, feedback loops, and predictions.
Power itself is not a possession, but a relationship: the ability to influence behavior, steer actions, or frame decisions. It operates both visibly – through institutions and laws – and subtly, by shaping expectations, norms, and bodies of knowledge.
7.2. Governmentality
Michel Foucault, a major thinker in the analysis of power structures, coined the term of governmentality to describe the totality of forms of knowledge, practices, and techniques through which modern societies are governed. In this sense, governing does not merely mean the exercise of coercion, but also the shaping of spaces for action that encourage individuals to regulate themselves in line with societal objectives.
Key dimensions of governmentality are:
The Interconnection of Knowledge and Power
Knowledge is never neutral. It emerges through the collection and interpretation of data, is embedded in power processes, and structures social orders. With digitalization, this connection becomes central: big data and artificial intelligence provide predictions, pattern recognition, and decision-making foundations that intervene deeply in societal processes. Smart governance operates precisely at this point: data become the foundation of political legitimacy and the exercise of power.
Biopolitics
Biopolitics refers to the regulation of life itself – health, reproduction, labor, education, migration, or security. The goal is to organize entire populations through laws, standards, and political programs. Today, biopolitics is linked to digital control: Smart Health, Smart Cities, and Smart Economies illustrate how knowledge is translated into concrete government practices through data.
Dispositives of Power
Biopolitics works through dispositifs: networks of institutions, laws, discourses, and practices. Hospitals, schools, media, and digital platforms are part of these structures. They standardize behavior not through overt coercion, but through subtle framing of what is considered „normal” or „desirable”. In this way, decisions are indirectly guided and behavior is embedded in everyday life.
Disciplinary Power
Alongside biopolitics, which regulates entire populations, disciplinary power targets the individual. It operates through surveillance, reward, punishment, and training. Its goal is not only obedience, but the productive shaping of bodies and behaviors. External directives are transformed, through sanctions and incentives, into internal motivations that drive individuals toward self-discipline.
Technologies of the Self
Technologies of the self refer to practices through which individuals internalize societal norms and autonomously adjust their behavior. Digital applications – such as fitness apps, learning platforms, or financial tools – translate societal directives into personal goals. This creates a form of power that operates not only from the outside, but is carried forward from within: people follow rules because they perceive them as meaningful and beneficial for themselves.
Those interested in exploring the art of governance in more detail can find further information here.
7.3. Personalized Medicine as a Catalyst for Biomolecular Governmentality
At first glance, personalized medicine, supported by modern genomics and bioinformatics, appears to be purely a medical and technical advance. But when viewed through the lens of governmentality, it emerges as a powerful catalyst for a profound transformation of power structures and governance practices. The precise analysis of individual genetic profiles and their linkage to comprehensive databases not only individualizes healthcare, but also changes the way societies are governed and individuals are regulated.
Knowledge & Power: From the Genome to the Governable Body
The decoding of the genome marked the initial explosion of knowledge, but it is personalized medicine that makes this knowledge governable. Bioinformatics and modern genomics do not merely generate data – they create predictive knowledge that forms the basis for interventions. This knowledge is by no means neutral: it categorizes individuals according to genetic risk profiles (e.g., „high risk for breast cancer,” „predisposed to cardiovascular diseases”). These categorizations become the foundation of new power structures. Both states and private actors use this data to predict disease risks, optimize healthcare budgets, and manage preventive measures. Social order is thereby reshaped – from access to insurance and labor markets to the prioritization of preventive resources.
Smart governance manifests here in algorithmic decision-making processes that appear objective but are, in fact, normative. Who gains access to which therapies, or which population groups are classified as „at risk”, is increasingly determined by data-driven models that invisibly reproduce existing power relations („We act based on the best available evidence”).
The CMS Health Tech Ecosystem Initiative, officially announced on July 30, 2025, during the „Make Health Tech Great Again” event at the White House, exemplifies the trend toward the emergence of new power structures. CMS (Centers for Medicare & Medicaid Services) is a U.S. federal agency within the Department of Health and Human Services (HHS) that manages healthcare programs such as Medicare and Medicaid and regulates the national healthcare market. The initiative aims to create smarter, safer, and more personalized healthcare, driven through partnerships with innovative private-sector companies. During the event, the U.S. government secured commitments from major health and IT firms – including Amazon, Anthropic, Apple, Google, and OpenAI – to lay the foundation for a next-generation digital health ecosystem.
According to U.S. President Donald Trump, this gives the U.S. healthcare systems a high-tech upgrade, ushering in „an era of convenience, profitability, speed, and – frankly – better health for the people”. (Note the order…)
Biopolitics 2.0: Optimizing the Population at the Molecular Level
Classical biopolitics regulated populations through statistics on birth rates or mortality. Personalized medicine elevates this to a new, molecular level. Genomics fragments the population into ever smaller, biomolecularly defined subgroups. People are no longer categorized merely by age or gender, but by genotype, risk alleles, and biomarkers („BRCA1 carriers”, „individuals with the LCT gene for lactose intolerance”). This enables hyper-precise and individualized regulation – biopolitics becomes granular.
The „healthy”, „normal” body is no longer defined solely as one without symptoms, but as one with a „normal”, „low-risk” genome. Deviation begins not with disease, but with genetic predisposition. This creates a new class of „presymptomatic patients” or „risk subjects”, who are managed biopolitically through more frequent screenings, prevention programs, vaccination campaigns, prophylactic surgeries, lifestyle recommendations, or reproductive decisions based on genetic forecasts.
While Foucault saw the nation state as the main actor in biopolitics, power is increasingly shifting to private actors today: big tech and biotech companies that control data, algorithms, and technologies. They effectively define what genetic „normality” and „risk” mean.
We now speak of the economization of biological life. Genetic data have become a valuable commodity – a bio-resource. Large technology and pharmaceutical companies accumulate bio-data to generate profits: in drug development, therapy recommendations, and personalized advertising. The human body and its biomolecular data thus become part of capitalist logic of exploitation. The individual themselves becomes „human capital”, seemingly obliged to optimize his genetic potential.
In an NBC News report on August 26, 2025, U.S. Health Secretary RFK Jr. stated that the 60 largest technology companies would soon allow Americans access to their personal health data – data that had been economically exploited for years without their consent: “You are going to be able to see, by next year, all your health records on your cellphone.”
What is presented here as a political achievement merely confirms the status quo.
Dispositifs of Power: The Network of Biomolecular Surveillance
The dispositive network that supports this new form of biopolitics is multi-layered: pharmaceutical companies develop targeted therapies; biobanks and genetic databases provide the raw material; algorithms analyze and interpret it; doctors communicate results and recommendations; digital health apps monitor compliance. Each link in this chain functions as a power dispositif: it defines what is considered „healthy” or „sick“ and subtly conveys what „responsible health care” means in the age of genetics. Genetic testing, for example, is marketed as „responsible” behavior – thus leading individuals to participate unknowingly in biopolitical goals such as prevention or cost efficiency.
This development is reflected in national health strategies that promote genetic screenings to minimize disease burdens. The goal is to maximize the „health dividend” while simultaneously reducing costs – a biomolecular rationalization strategy.
Initiatives such as the 100.000 Genomes Project or Genomics England’s Newborn Genomes Programme move in the same direction. By comparison, the European Health Data Space (EHDS) – an EU-wide initiative for the networking and secure use of health data – is far more comprehensive. The EHDS provides the data and regulatory infrastructure that makes large-scale personalized medicine possible in the first place.
For most citizens, this sounds like a new beginning: more participation, better therapies, more precise prevention. Yet this promising vision casts a shadow. In practice, unequal opponents face each other: on one side, the individual patient contributing their genetic profile, lab results, and medical history; on the other, global pharmaceutical companies, data platforms, and technology providers with multi-billion-dollar budgets that refine, patent, and commercialize this data. Cui bono? – who benefits? The answer is sobering: quick profits go to the big players, while the promises remain with the individual.
While the EHDS emphasizes citizens’ rights – more control, more transparency, more self-determination – anyone who has experienced the complexity of consent processes in health apps, insurance companies, or electronic patient records knows: there is a gap between formal options and actual capacity to act. While Goliath has lobbying channels, legal experts, and data centers at its disposal, David is left with informational brochures and trust.
The EHDS is thus both a government technology and a power project. It decides whether data is treated as a public good or as a commodity market. In any case, Goliath will benefit. The open question is whether David will be left with more than just the role of a fig leaf. The health data space will only become truly emancipatory if benefits and risks are distributed fairly – through binding public welfare requirements, transparent accountability, and real returns for patients. Otherwise, the great promise of „empowerment” will remain a narrative that merely conceals existing asymmetries.
Disciplinary Power: Internalized Biomolecular Surveillance
While biopolitics focuses on the population as a whole, personalized medicine directs its disciplinary effect at the individual. The genetic risk profile acts as a constant, internalized monitor. Those who know they have an increased risk of colon cancer adjust their behavior: dietary habits, preventive medical appointments, and exercise routines are adapted to the genetic forecast.
The electronic patient record (ePA) intensifies this dynamic. By centrally storing genetic and medical data and making them accessible at any time, it transforms medical recommendations into an ever-present digital authority. Personal notifications, reminders for medication intake, personalized reports, or interfaces with health apps reinforce self-monitoring: the external directive („You must get screened”) becomes an internal voice („I must take care of myself because my genome requires it”). The ePA structures and institutionally legitimizes this self-discipline, making it invisible and seemingly voluntary – governed simultaneously by digital infrastructure, social norms, and medical authority.
The result is a subtle yet pervasive form of power: it does not manifest through sanctions, but through the constant, digital concern for one’s own – genetically anticipated – future. The ePA turns the individual into a disciplined actor of their own risk profile, while the surveillance formally disappears but remains technically and institutionally omnipresent.
Technologies of the Self: The Genetic Self-Optimizer
Here, personalized medicine appears in its purest form as a technology of the self. At-home DNA tests, fitness trackers, and health apps are the tools through which individuals internalize societal norms of health and performance and actively work on themselves. They translate the abstract concept of genetic predisposition into concrete, everyday actions: an extra serving of vegetables, 10,000 steps, meditation to reduce stress. The individual governs themselves according to a biomolecular logic – not because the state commands it, but because they seek to optimize their genetic destiny. Power relations thus become invisible and present themselves as expressions of individual sovereignty and self-care.
7.4. The Global Trend: Worldwide Biomolecular Governmentality
Personalized medicine is no longer a national phenomenon, but part of a global transformation of power and governance. Worldwide, initiatives are emerging that pool genomic data, health information, and digital technologies in order to translate populations and individuals into ever more precise risk profiles. The logic of Foucault’s governmentality – the linkage of knowledge, power, and self-regulation – manifests itself here at a transnational level.
In the United States, programs such as the All of Us Research Program of the National Institutes of Health or the Million Veteran Program collect genetic and medical data from millions of citizens. The collection of diverse health data serves not only scientific research, but also forms the basis for algorithmic risk profiles, preventive interventions, and resource-optimized healthcare strategies.
Canada is pursuing a comparable goal with the Canadian Precision Health Initiative and the Pan-Canadian Genomics Network: the systematic collection and interconnection of biomolecular data, combined with an infrastructure that enables secure governance, international collaboration, and economic utilization.
This is where Biopolitics 2.0 becomes visible: populations are fragmented, governed, and optimized at the molecular level.
Europe constitutes an orchestrated network of transnational biopolitics. The European Health Data Space (EHDS), supported by initiatives such as ICPerMed and ERA PerMed, pools genetic, clinical, and lifestyle data across national borders.
National biobanks such as the UK Biobank or the French Health Data Hub act as dispositifs that generate both knowledge and power: they determine which data is used, how populations are classified, and which health policy interventions appear legitimate.
Through platforms such as SHIFT-HUB or the Helmholtz Initiative iMed, the international networking of science, industry, and society is institutionalized – constituting a transnational form of smart governance that scales Foucault’s concept of technologies of the self to the global level.
The global logic is also evident in Asia and the Pacific region: the GenomeAsia 100k Project, Singapore’s National Precision Medicine Strategy, and China’s National Health Data Platform link biomolecular data with AI-driven analyses to enable preventive, individualized, and population-based governance.
Australia and Israel rely on digital infrastructure that reinforces citizens’ self-discipline – electronic patient records, genetic databases, and personalized apps function as subtle yet omnipresent dispositifs of power.
Even global research projects such as the Human Cell Atlas illustrate the transnational dimension: by mapping the diversity of human cells, they create a universal network of knowledge and power that provides the foundation for personalized therapies worldwide. The boundaries between state governance, private-sector power, and scientific expertise blur: Foucault’s classical distinction between the nation-state and disciplinary power is replaced by global networks and international collaboration.
7.5. Conclusion: The Global Ambivalence of Biomolecular Power
The global expansion of personalized medicine clearly demonstrates that the biomolecular revolution is no longer confined to individual nation-states. It manifests as a transnational network of data, algorithms, and governance that steers both populations and individuals at the molecular level. Global initiatives – from the United States and Europe to Asia, Australia, and Israel – intertwine biopolitics, disciplinary power, and technologies of the self on an international scale.
The ambivalence remains central: on the one hand, these developments open up opportunities for more precise therapies, individualized prevention, and potentially greater health equity. On the other hand, power asymmetries arise in which states, international organizations, and private actors control access to data, resources, and technologies. Individuals are increasingly becoming disciplined subjects of their own genetic profiles, while global dispositifs unnoticedly enforce normative and economic goals.
This seamlessly combines the local experience of personalized medicine with a global governance logic: smart governance and biomolecular biopolitics have long been internationalized, and the challenge lies in distributing the benefits and risks fairly. The global dimension of this transformation makes it clear that the biomolecular revolution is irreversible not only medically, but also politically and socially – a global system of control that offers opportunities for health and risks for the concentration of power.

8. Epilogue
The desire to optimize health and longevity is a primal instinct of humankind. Even in ancient civilizations, health was not considered a matter of chance, but rather a desirable state that could be achieved through lifestyle and philosophy. Greek „dietetics” – a holistic practice combining nutrition, exercise, and mental hygiene – and the motto „mens sana in corpore sano” testify to a 2.500-year-old desire for optimization. This urge, fueled by the survival instinct, the fear of losing control, and the pursuit of quality of life, is not only ancient but eternal.
In modern times, personalized medicine embodies the latest expression of this dream. It promises to treat people not as averages, but as individuals, based on genetic code, biomarkers, and lifestyle data. But is it the fulfillment of this dream or a modern mirage? In its utopian vision, it suggests the complete controllability of biology, an overcoming of the „biological lottery”. The reality is more sobering: it enables more precise diagnoses and therapies, prevents treatment failures – but it does not abolish disease.
The idea of a disease-free existence remains an illusion. Evolutionary biology and biophysics set limits: pathogenic germs continue to evolve, cancer is a statistical consequence of cell division, and aging is an inevitable degenerative process. The realistic goal is therefore not the elimination of disease, but its transformation: converting fatal diseases into chronic conditions and maximizing the „healthspan” – the years spent in good health. This goal is radical, but achievable, as advances in the treatment of HIV or diabetes show.
But how far is humanity willing to go to achieve this goal? History shows that in the face of suffering and death, ethical boundaries often blur. Willingness ranges from everyday discipline and financial sacrifice to participation in experimental therapies. This raises an uncomfortable question: what compromises is society prepared to make? Personalized medicine requires data – genetic, medical, and personal. Collecting such data carries the risk of undermining privacy and autonomy. The COVID-19 pandemic was a stress test that showed how large parts of the population were willing to trade civil liberties for security. Models such as China’s social credit system illustrate one possible future scenario, in which transparency and control are rewarded in the name of health and safety. Yet such scenarios are not inevitable: they represent an extreme, not an unavoidable destiny.
Personalized medicine has two sides. Its pragmatic promise is more efficient, precise, and humane medicine that saves lives and alleviates suffering – for example, through tailored cancer therapies or early diagnosis. Its utopian but risky promise is the illusion of total control over life and death. It is thus the culmination of a journey that has lasted thousands of years. Whether this journey ends in emancipated self-optimization or a dystopian „biocracy” does not depend on technology, but on the ethical and political framework that society gives it.
The age-old dream of health seems more tangible today than ever before, yet it challenges us to find wise answers. Personalized medicine can be a tool of emancipation if we use it with care. The decisive question of modernity is therefore not only how much freedom we are willing to sacrifice for a longer life, but how we can reconcile health and freedom. Only in this way can the eternal pursuit of health become progress for all.
The world will not change all at once; it never does. Life will go on mostly the same in the short run, and people in 2025 will mostly spend their time in the same way they did in 2024. We will still fall in love, create families, get in fights online, hike in nature, etc.
But the future will be coming at us in a way that is impossible to ignore, and the long-term changes to our society and economy will be huge.
Sam Altman (CEO of OpenAI)
Sources (as of January 15, 2026)
AI, Artificial Intelligence, Bioinformatics, Biopolitics, Control, Crispr, Digital Health, DNA, DNA Sequencing, DNA Test, FENT, G4X Spatial Sequencer, Gene, Gene Defect, Gene Variation, Genetic Medicine, Genetics, Genome Sequencing, Genomics, Governance, Healthcare, Illumina, iNanoBio, Medicine, Michel Foucault, Next Generation Sequencing, NGS, Nvidia, ONT, Oxford Nanopore, PacBio, Personalized Medicine, Precision Medicine, Protein, RNA, Roche, Sanger sequencing, SBX Sequencing, Singular Genomics, Smart Governance, SMRT Sequencing, Surveillance, SynHG, Synthetic Human Genome Project, Transcriptomics, Xpandomer -
Personalisierte Medizin – Himmel oder Hölle

Als die HSBC Bank im Jahr 2013 im Rahmen ihrer globalen Kampagne „The future is full of opportunity“ an Flughäfen wie Gatwick und Dubai Plakate mit einem Fingerabdruck, einem QR-Code und dem provokanten Slogan „Your DNA will be your data“ platzierte, war die Botschaft unmissverständlich: Die Zukunft wird persönlich – vielleicht sogar beunruhigend persönlich. Die Bank wollte sich als zukunftsorientiertes Unternehmen positionieren, das Digitalisierung, Künstliche Intelligenz, Nachhaltigkeit und Genetik offensiv thematisiert.
Der Slogan wirkte doppeldeutig – als verheißungsvolle Vision ebenso wie als alarmierender Ausblick. Kritiker schlugen sofort Alarm: Deine DNA wird lesbar, analysierbar und digital verfügbar – ein offenes Buch über Herkunft, Gesundheitsrisiken, vielleicht sogar Persönlichkeitsmerkmale – vergleichbar mit biometrischen Daten wie Fingerabdrücken oder Gesichtsscans. Was einst als das Intimste am Menschen galt, wird nun zur Datei: maschinell entschlüsselbar, potenziell speicherbar, womöglich handelbar. Der Mensch wird reduziert auf Information – auf „Bits und Bytes“, die verarbeitet, verkauft oder missbraucht werden könnten.
Wer besitzt die Rechte an deinen genetischen Daten? Und wer hat Zugriff darauf? Du selbst? Die Firma, die den Test durchgeführt hat? Erreichen dadurch Überwachung, Datenlecks und Missbrauch durch staatliche und private Akteure eine neue Dimension? Die Vision von Designerbabys, genetischer Selektion und personalisierter Werbung auf Basis biologischer Marker wirft schwerwiegende ethische Fragen auf. Wird der Fortschritt am Ende mit dem Preis individueller Freiheit bezahlt?
Doch die Biotech-Revolution hat längst eine gewaltige Dynamik entfaltet, und immer mehr Stimmen erkennen in dieser Entwicklung ein enormes Potenzial. In der E-Book-Ausgabe der HSBC Bank Frontiers – Biotech – Five Big Ideas Shaping the Biotech Revolution von März 2024 heißt es:
„Die Idee, die Natur für Waren und Dienstleistungen zu nutzen, ist nicht neu. Doch in den 1970er Jahren, mit dem Aufkommen der Gentechnik und der Gründung der ersten Biotech-Unternehmen, begann die moderne Ära dieser Praxis. Dies brachte Wissenschaft und Wirtschaft zusammen wie nie zuvor, und der Kapitalzufluss beflügelte die Forschungs- und Entwicklungsbemühungen.
In den folgenden drei Jahrzehnten wurde das Feld durch bahnbrechende Geräte wie DNA-Sequenzierer und -Synthesizer beschleunigt, und die Biologie selbst wandelte sich langsam zu einer rechnergestützten, datengesteuerten Disziplin. Nach der Jahrtausendwende, als die Forscher immer größere Bereiche des menschlichen Genoms erfolgreich entschlüsseln konnten und die Kosten für die Sequenzierung dieses Genoms rapide sanken – von etwa 100 Millionen Dollar im Jahr 2001 auf weniger als 1.000 Dollar heute –, wurde das Potenzial der Biotechnologie, unser Leben zu verändern, auf elektrisierende Weise deutlich.
In den letzten Jahren wurden die Möglichkeiten der Biotechnologie – von der Behandlung genetisch bedingter Krankheiten bis zur Wiederbelebung ausgerotteter Tierarten – durch Fortschritte in ergänzenden Bereichen wie der künstlichen Intelligenz erweitert. Dies hat dazu beigetragen, dass in diesem Bereich ein neues Zeitalter angebrochen ist.“
Nirgendwo sonst prallen Nutzen und Risiko so direkt aufeinander wie in der personalisierten – oder präzisionsbasierten – Medizin. Ihr Anspruch: individuelle genetische und biologische Informationen zu nutzen, um Diagnosen zu präzisieren, Therapien zu individualisieren und Krankheiten frühzeitig zu erkennen. Die Vision ist vielversprechend: weniger Nebenwirkungen, bessere Heilungschancen, effizientere Prävention – Medizin, maßgeschneidert auf den Einzelnen.
Doch auch hier gilt: Was als medizinischer Fortschritt gefeiert wird, bringt gesellschaftliche, rechtliche und ethische Herausforderungen mit sich:
- Wer entscheidet, welche genetischen Daten relevant sind?
- Welche Rolle spielt der Zugang zu diesen Technologien – werden soziale Ungleichheiten dadurch vertieft?
- Und wie lassen sich Diskriminierung, etwa durch genetisches Scoring bei Versicherungen oder Arbeitgebern, verhindern?
Zwischen der Hoffnung auf individuelle Heilung und der Sorge vor kollektiver Entmündigung stehen grundlegende Fragen: Wird der Mensch durch den Zugriff auf seine biologischen Daten ermächtigt – oder entblößt? Ist personalisierte Medizin ein Segen – oder der Auftakt zu einer Ära subtiler Entmenschlichung?
Daher lautet die Gretchenfrage: Ist die personalisierte Medizin der verheißene Aufstieg in den Himmel – oder der Sturz durch die Tore der Hölle?
Dieser Blogbeitrag lädt dazu ein, die personalisierte Medizin in ihrer ganzen Komplexität zu betrachten – jenseits von Hype oder Alarmismus. Er richtet sich bewusst auch an fachfremde Leser, die sich kritisch mit den Chancen, Risiken und ethischen Spannungsfeldern dieser medizinischen Revolution auseinandersetzen möchten.
📑Inhaltsverzeichnis
1. Die begriffliche Einordnung und das grundlegende Paradigma
2. Treibende Kräfte – zwischen Vision und Wirklichkeit
3. Die Zukunft der Medizin beginnt in der Zelle
3.1. Wie Biologie und Technologie zusammenfinden
3.2. Daten, Verantwortung und der größere Rahmen
4. Von DNA zur Erkenntnis – ein Hochgeschwindigkeitszug der Innovation
4.1. Die DNA – Bauplan des Lebens
a) Die DNA: Das molekulare Informationsarchiv des Lebens
b) Vom Code zum Protein: Wie Anleitungen Wirklichkeit werden
c) Epigenetik & nicht-codierende DNA: Das Dirigenten-Team unseres Erbguts
d) Das verborgene Kontrollzentrum: Was die nicht-codierende DNA steuert
e) Wenn das Betriebssystem krank macht
f) Offene Rätsel der Forschung
g) Die wahre Magie der Gene
4.2. Genetische Variation und ihre Folgen
4.3. Genomsequenzierung: Schlüssel zum individuellen Erbgut
a) Die Reise der DNA: Vom Nabelschnurblut zur genetischen Information
b) Drei Regieanweisungen für die Gen-Diagnostik: WGS, WES und Panel
4.4. Methoden der Genomsequenzierung
4.4.1. Methode 1: Kopieren mit Leuchtfarbe
a) Sanger-Sequenzierung
b) Illumina-Technologie
c) Single-Molecule Real-Time(SMRT)-Sequenzierung
4.4.2. Methode 2: Hightech-Tunnel – DNA elektrisch abtasten
a) Oxford Nanopore Sequenzierung
4.4.3. Genomsequenzierung – Technologien im Vergleich
a) Durchsatz vs. Verwertbarkeit – Masse ist nicht immer gleich Klasse
b) Wirtschaftlichkeit – Kosten pro Base vs. Kosten pro Erkenntnis
c) Von der Probe zum Befund – Wie schnell spricht das ganze Genom?
d) Klinische Einsatzfelder – Welche Technologie passt wozu?
e) KI als Co-Pilot – Automatisierung ohne Verantwortung abzugeben
f) Vergleichstabelle: Sanger, Illumina, PacBio, ONT
g) Key Takeaways: Zwischen Reife und Routine
4.5. Genomsequenzierung: Der nächste Technologiesprung
a) FENT: Wie ein Mikrochip die DNA-Analyse revolutioniert
b) SBX: Wenn DNA sich streckt, um verstanden zu werden
c) Das G4X-System: Vom genetischen Rezept zum räumlichen Atlas der Zelle
4.6. Vom Code zur Heilung – Bioinformatik als Schlüssel der Medizin von morgen
4.6.1. Vom genetischen Code zur computergestützten Genomanalyse
4.6.2. Moderne Bioinformatik – Der digitale Werkzeugkasten der Biologie
5. Eine Erfolgsgeschichte
6. Ein Blick in die Zukunft: Das Synthetic Human Genome Project
7. Personalisierte Medizin und Smart Governance
7.1. Macht, Governance und Smart Governance
7.2. Gouvernementalität
7.3. Personalisierte Medizin als Katalysator biomolekularer Gouvernementalität
7.4. Der weltweite Trend: Globale biomolekulare Gouvernementalität
7.5. Schlussfolgerung: Globale Ambivalenz der biomolekularen Macht
8. Epilog1. Die begriffliche Einordnung und das grundlegende Paradigma
Die Entwicklung der modernen Medizin hin zu maßgeschneiderten Behandlungen wird häufig mit Begriffen wie „personalisierte Medizin“, „individualisierte Therapie“ oder „Präzisionsmedizin“ beschrieben. Doch was genau verbirgt sich hinter diesen Schlagworten?
Personalisierte Medizin gilt derzeit als einer der populärsten Begriffe – eingängig wie ein Werbeslogan. Damit verbunden ist das Versprechen, Therapien auf jeden Menschen zuzuschneiden: auf seine Gene, seine Lebensgewohnheiten, seine Biologie. In den Medien und in der öffentlichen Debatte wird der Ausdruck entsprechend häufig verwendet.
Individualisierte Therapie klingt weniger glamourös, meint aber Ähnliches: die Anpassung von Medikamenten oder Dosierungen an die Bedürfnisse einzelner Patienten. Dieser Begriff findet vor allem im klinischen Alltag Verwendung.
Präzisionsmedizin schließlich ist der nüchternste der drei Begriffe – ein Fachwort, das in Forschungslaboren und wissenschaftlichen Publikationen dominiert. Es unterstreicht den datenbasierten Ansatz: Algorithmen, Genanalysen und Biomarker entscheiden darüber, welche Therapie bei welcher Patientengruppe wirkt. Das ist die Sprache der Wissenschaft.
Doch egal, welcher Begriff gewählt wird – sie alle stehen für einen Paradigmenwechsel: Weg von standardisierten Verfahren – hin zu einer Medizin, die den einzelnen Menschen in seiner biologischen Einzigartigkeit in den Mittelpunkt stellt.
2. Treibende Kräfte – zwischen Vision und Wirklichkeit
Die treibenden Kräfte hinter der personalisierten Medizin sind vielfältig und eng miteinander verflochten. Auf der einen Seite stehen wissenschaftlich-technologische Entwicklungen, die in den letzten Jahrzehnten enorme Fortschritte ermöglicht haben: neue Methoden, um Erbgut schnell und genau zu lesen, leistungsfähige Datenanalyse durch künstliche Intelligenz sowie die Verfügbarkeit großer biomedizinischer Datenmengen. Diese Technologien bilden die Grundlage dafür, individuelle Unterschiede zwischen Patienten überhaupt erst erkennen und medizinisch nutzen zu können.
Auf der anderen Seite wirken gesellschaftliche und politische Impulse als Beschleuniger dieses Wandels. Öffentliche Gesundheitsprogramme, gezielte Forschungsförderung, Strategiepapiere internationaler Gesundheitsorganisationen und gezielte Gesetzesinitiativen treiben die Integration der personalisierten Medizin in die Versorgungspraxis aktiv voran. Auch das gestiegene Gesundheitsbewusstsein in der Bevölkerung sowie die zunehmende Erwartung an individuelle Behandlungsangebote verstärken diesen Trend.
Personalisierte Medizin ist also kein bloßes Produkt technologischer Machbarkeit, sondern entsteht im Spannungsfeld von Innovation, Politik und gesellschaftlicher Nachfrage.
Ein aktuelles Beispiel für die praktische Umsetzung dieser Entwicklung liefert das Vereinigte Königreich: Im Rahmen eines neuen 10-Jahres-Plans soll künftig jedes Neugeborene einem DNA-Test unterzogen werden. Die sogenannte Ganzgenomsequenzierung soll nach hunderten genetisch bedingten Krankheiten suchen – mit dem Ziel, potenziell tödliche Erkrankungen frühzeitig zu erkennen und ihnen gezielt vorzubeugen. Nach Angaben des britischen Gesundheitsministers Wes Streeting geht es darum, den National Health Service (NHS) grundlegend zu transformieren: weg von der reinen Krankheitsbehandlung – hin zu einem System der Vorhersage und Prävention. Genomik, also die umfassende Analyse der Erbinformation, soll dabei in Kombination mit künstlicher Intelligenz als Frühwarnsystem dienen.
„Die Revolution in der medizinischen Wissenschaft bedeutet, dass wir den NHS von einem Dienst, der Krankheiten diagnostiziert und behandelt, in einen Dienst verwandeln können, der sie vorhersagt und verhindert“, so Streeting. Die Vision: eine personalisierte Gesundheitsversorgung, die Risiken erkennt, bevor Symptome überhaupt auftreten – und damit die Lebensqualität verbessert sowie das Gesundheitssystem entlastet.
Doch so überzeugend diese Vision auch klingt – aus der Wissenschaft kommen auch mahnende Stimmen. So warnt etwa der Genetiker Prof. Robin Lovell-Badge vom Francis Crick Institute davor, die Komplexität der genomischen Daten zu unterschätzen. Es brauche nicht nur technisches Know-how zur Datenerhebung, sondern vor allem qualifiziertes Personal, das die gewonnenen Informationen verantwortungsvoll und verständlich kommunizieren kann. Denn Daten allein sind noch keine Diagnose – entscheidend ist ihre sinnvolle und patientengerechte Interpretation.
Was bedeutet es konkret, Medizin neu zu denken?
Die wahre Revolution spielt sich in den Laboren und Rechenzentren ab, in denen die Bausteine der personalisierten Medizin entstehen. Die folgenden Kapitel führen Schritt für Schritt hinter die Kulissen und zeigen die Schlüsseltechnologien, die nötig sind, um die personalisierte Medizin vom Konzept in die Klinik zu bringen.
3. Die Zukunft der Medizin beginnt in der Zelle
3.1. Wie Biologie und Technologie zusammenfinden
Unser Körper besteht aus Milliarden Zellen – winzigen, spezialisierten Einheiten, die Tag für Tag zusammenarbeiten, um Gesundheit und Leben zu ermöglichen. In jeder einzelnen Zelle laufen unzählige Prozesse ab, die genau aufeinander abgestimmt sind. Diese zellulären Mechanismen sind entscheidend für das Verständnis von Gesundheit, Krankheit und der Entwicklung neuer, gezielter Therapien.
Ein zentraler Mechanismus ist die Energiegewinnung: Zellen verwandeln Nährstoffe und Sauerstoff in ATP (Adenosintriphosphat)– die „Energie-Währung“ des Körpers. Ohne ATP könnte keine Zelle funktionieren, kein Muskel sich bewegen, kein Gedanke entstehen.
Gleichzeitig produzieren Zellen fortwährend Proteine – auf Basis genetischer Informationen. Diese komplexen Moleküle übernehmen zentrale Aufgaben im Körper – als chemische Katalysatoren, Botenstoffe, Baustoffe oder Transportmoleküle.
Auch die Reinigung und Erneuerung innerhalb der Zelle ist lebenswichtig: Prozesse wie die Autophagie bauen beschädigte Zellbestandteile ab und recyceln sie – eine Art innere Müllabfuhr, die Krankheiten wie Alzheimer oder Parkinson vorbeugen kann.
Damit Zellen im Körper koordiniert zusammenarbeiten können, brauchen sie funktionierende Signalwege – sie „kommunizieren“ über chemische Botenstoffe miteinander. Diese Zellkommunikation steuert, wann Zellen wachsen, ruhen oder auf Veränderungen reagieren sollen.
Ein weiterer zentraler Vorgang ist die Zellteilung – notwendig für Wachstum, Wundheilung und Gewebeerneuerung. Gleichzeitig sorgen DNA-Reparaturmechanismen dafür, dass Fehler im Erbgut korrigiert werden. Gelingt das nicht, greift die Apoptose, der programmierte Zelltod, um Schäden zu begrenzen.
All diese Mechanismen laufen im Hintergrund ab – ständig und hochpräzise. Wenn jedoch einer dieser Abläufe gestört ist, kann das zu schweren Erkrankungen führen, etwa Krebs, Autoimmunerkrankungen oder Stoffwechselstörungen. Genau hier setzt die personalisierte Medizin an. Sie nutzt modernste Technologien, um diese zellulären Prozesse besser zu verstehen und gezielt zu beeinflussen.
Dank Genomsequenzierung (etwa durch „Next Generation Sequencing“) lassen sich genetische Veränderungen identifizieren, die z. B. die Proteinproduktion oder Reparaturmechanismen beeinflussen.
Biomarker-Analysen (die Analyse bestimmter Moleküle)im Blut, Gewebe oder anderen Körperflüssigkeiten zeigen, ob bestimmte Signalwege über- oder unteraktiv sind – und helfen dabei, individuelle Krankheitsverläufe vorherzusagen oder passende Therapien auszuwählen.
Die Einzelzellanalytik ermöglicht es, Unterschiede selbst zwischen einzelnen Zellen sichtbar zu machen – etwa in einem Tumor, wo manche Zellen auf eine Therapie ansprechen und andere nicht. Dadurch wird eine präzisere Behandlung möglich.
Auch die Proteomik (Analyse aller Proteine in einer Zelle) und Metabolomik (Analyse von Stoffwechselprodukten) liefern ein aktuelles Bild davon, wie aktiv bestimmte zelluläre Mechanismen wirklich sind – z. B. ob eine Zelle gestresst ist, ausreichend Energie hat oder sich gerade teilt.
Künstliche Intelligenz (KI) hilft dabei, diese riesigen Datenmengen zu analysieren und Muster zu erkennen – zum Beispiel, welche Kombination von zellulären Störungen typisch für eine bestimmte Krebserkrankung ist. Digitale Gesundheitsdaten (etwa aus Wearables) ergänzen dieses Bild im Alltag und ermöglichen eine präzise Langzeitbeobachtung.
Sogar neue Therapieansätze beruhen auf dem Verständnis zellulärer Abläufe: Gentherapien und CRISPR-basierte Gen-Editierung greifen gezielt in die DNA ein, um fehlerhafte Mechanismen zu korrigieren. In Organoiden – Mini-Organmodellen im Labor – lassen sich Medikamente direkt an patientenspezifischen Zellmodellen testen, ganz ohne Risiko für den Menschen.
Moderne Technologien machen heute sichtbar, was früher im Verborgenen lag: Wie Zellen arbeiten, was bei Krankheit aus dem Gleichgewicht gerät – und wie man gezielt eingreifen kann. So entsteht eine individualisierte Medizin, die nicht mehr auf Vermutung basiert, sondern auf messbarer Biologie.
Diese neue Präzision bringt nicht nur Chancen, sondern auch neue Verantwortlichkeiten mit sich.
3.2. Daten, Verantwortung und der größere Rahmen
Die Komplexität dieser zellulären Prozesse spiegelt sich unmittelbar in der Komplexität der personalisierten Medizin wider: Je genauer wir die feinen Abläufe und Wechselwirkungen in unseren Zellen verstehen, desto gezielter – aber auch anspruchsvoller – werden Diagnose und Therapie. Diese neue Präzision erfordert neue Technologien, neue Denkweisen und ein tiefes biologisches Verständnis. Gleichzeitig eröffnet sie die Chance auf wirksamere, nachhaltigere und maßgeschneiderte Behandlungskonzepte.
Doch je gezielter und tiefer diese Eingriffe werden, desto größer ist auch die Verantwortung – und die Herausforderung, unerwünschte Langzeitwirkungen oder Nebenreaktionen frühzeitig zu erkennen und zu beherrschen. Gerade bei innovativen Verfahren wie Gentherapie oder Immunmodulation gilt es, den Nutzen sorgfältig gegen mögliche Risiken abzuwägen. Die personalisierte Medizin verlangt daher nicht nur Präzision, sondern auch Weitblick.
Ein zentrales Element dieses Weitblicks ist die Verfügbarkeit personenbezogener Gesundheitsdaten – und zwar nicht nur im Einzelfall, sondern im großen Maßstab.
Erst durch die Kombination individueller Daten mit Bevölkerungsdaten wird es möglich, biologische Prozesse im Detail zu verstehen, Krankheitsverläufe vorherzusagen und personalisierte Therapien zu entwickeln. Genetische Informationen, Laborwerte, Bildgebungsdaten oder digitale Alltagsmessungen liefern Hinweise darauf, wie zelluläre Mechanismen bei einem Menschen konkret funktionieren – und wie sie bei anderen variieren.
Um diese individuellen Muster medizinisch einzuordnen, braucht es den Vergleich mit großen Datensätzen. Nur so können KI-Systeme komplexe Zusammenhänge erkennen, die dem menschlichen Auge entgehen – etwa seltene Genvarianten, molekulare Risikokonstellationen oder unerwartete Therapieeffekte. Dabei gilt: Je größer und vielfältiger die Datenbasis, desto zuverlässiger die Modelle – besonders, wenn sie laufend mit neuen Informationen angereichert werden.
Doch diese Fortschritte gelingen nur, wenn Menschen bereit sind, ihre Gesundheitsdaten zu teilen – und darauf vertrauen können, dass sie sicher und verantwortungsvoll genutzt werden.
Die Zukunft der Medizin beginnt dort, wo alles Leben entsteht: in der Zelle. Doch ihr Erfolg hängt ab vom Zusammenspiel dreier Kräfte: technologischer Innovation, wissenschaftlicher Tiefe – und ethischer Verantwortung.
4. Von DNA zur Erkenntnis – ein Hochgeschwindigkeitszug der Innovation
Wie Genomsequenzierung und bioinformatische Analyse die Medizin revolutionieren
4.1. Die DNA – Bauplan des Lebens
a) Die DNA: Das molekulare Informationsarchiv des Lebens
b) Vom Code zum Protein: Wie Anleitungen Wirklichkeit werden
c) Epigenetik & nicht-codierende DNA: Das Dirigenten-Team unseres Erbguts
d) Das verborgene Kontrollzentrum: Was die nicht-codierende DNA steuert
e) Wenn das Betriebssystem krank macht
f) Offene Rätsel der Forschung
g) Die wahre Magie der Gene
4.2. Genetische Variation und ihre Folgen
4.3. Genomsequenzierung: Schlüssel zum individuellen Erbgut
a) Die Reise der DNA: Vom Nabelschnurblut zur genetischen Information
b) Drei Regieanweisungen für die Gen-Diagnostik: WGS, WES und Panel
4.4. Methoden der Genomsequenzierung
4.4.1. Methode 1: Kopieren mit Leuchtfarbe
4.4.2. Methode 2: Hightech-Tunnel – DNA elektrisch abtasten
4.4.3. Genomsequenzierung – Technologien im Vergleich
4.5. Genomsequenzierung: Der nächste Technologiesprung
4.6. Vom Code zur Heilung: Bioinformatik als Schlüssel der Medizin von morgenStellen Sie sich vor, Sie stehen am Bahnsteig, und ein Zug rauscht mit solch einer Geschwindigkeit vorbei, dass kaum ein Bild im Fenster zu erkennen ist. So ähnlich empfinden viele Menschen die Entwicklungen in der personalisierten Medizin: Technologien kommen in rasantem Tempo, Begriffe wirken abstrakt – und doch zieht sich durch alles ein roter Faden: das menschliche Genom.
So vielfältig die modernen Ansätze auch sind – von Organoiden über CRISPR-basierte Gen-Editierung bis zu RNA-Medikamenten – im Zentrum steht der genetische Bauplan des Menschen. Die Genomsequenzierung ist heute Grundlage vieler Diagnosen und Therapieentscheidungen. Sie zeigt, welche genetischen Veränderungen Krankheiten auslösen, welche Signalwege betroffen sind – und wo gezielte Eingriffe möglich sein könnten.
Was einst Milliarden kostete und Jahre dauerte, gelingt heute in wenigen Tagen: Das gesamte Erbgut eines Menschen kann entschlüsselt werden. Doch die reine Sequenz ist noch kein Befund. Die Abfolge von drei Milliarden Basenpaaren entfaltet erst dann medizinischen Wert, wenn sie auch verstanden wird – und genau hier beginnt die eigentliche Herausforderung.
Die Bioinformatik betritt die Bühne: ein interdisziplinäres Feld, das Mathematik, Informatik und Biologie vereint, um aus Rohdaten Muster, Risiken und Therapieoptionen abzuleiten. Sie ist das eigentliche Nadelöhr der personalisierten Medizin – denn sie entscheidet, ob genetische Varianten als harmlos, risikobehaftet oder krankheitsverursachend gelten.
Dazu werden individuelle Sequenzen mit internationalen Referenzdatenbanken abgeglichen, Algorithmen analysieren Millionen genetischer Varianten, und lernende Systeme modellieren das komplexe Zusammenspiel von Genetik, Umwelt und Stoffwechsel.
Dieser Prozess ist hochkomplex – und zugleich essenziell. Denn erst die rechnergestützte Auswertung macht die DNA medizinisch nutzbar. Zwischen Genomsequenzierung und bioinformatischer Analyse besteht eine untrennbare Symbiose: Die Sequenz liefert die Daten, die Analyse das Verständnis. Ohne sie bleibt die DNA eine bloße Aneinanderreihung von Buchstaben.
Genau deshalb richtet sich der Blick der folgenden Abschnitte auf die entscheidende Frage:
Was heißt es, das Genom zu entschlüsseln – und es wirklich zu verstehen?
Welche Aussagen sind möglich – und wo liegen ihre Grenzen?Denn wer die Zukunft der Medizin begreifen will, muss wissen, wie wir heute das menschliche Erbgut lesen – und wie wir lernen, es zu deuten.
Der Hochgeschwindigkeitszug der Innovation ist längst abgefahren. Wer mitfahren will, muss verstehen, wohin er führt.
4.1. Die DNA – Bauplan des Lebens
Um diese Reise anzutreten, lohnt ein kurzer Blick auf die molekularen Grundlagen: Wie unsere DNA zur Bauanleitung wird, wie daraus über den Umweg der RNA Proteine entstehen – und wie die Epigenetik diese Prozesse feinjustiert. Erst dieses Fundament macht verständlich, was beim Entschlüsseln des Genoms eigentlich passiert.
a) Die DNA: Das molekulare Informationsarchiv des Lebens
In jeder Zelle unseres Körpers liegt sorgfältig verpackt ein beeindruckendes Molekül: die DNA (Desoxyribonukleinsäure). Sie enthält die vollständige Bau- und Funktionsanleitung unseres Körpers – ein molekulares Informationsarchiv des Lebens.
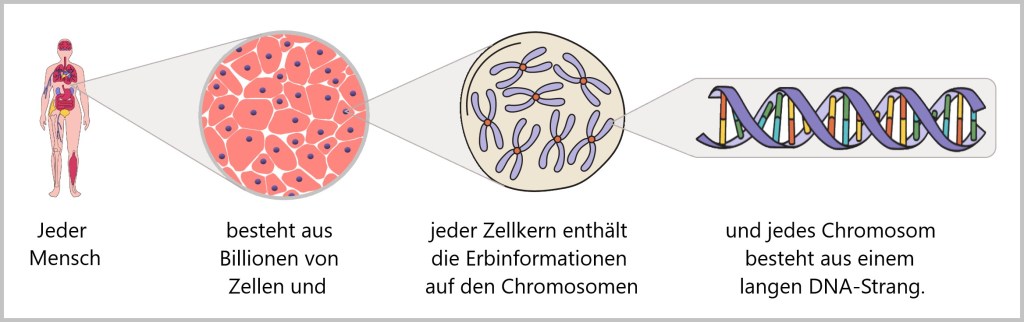
Abb. 1: Die DNA enthält die Erbinformation [Die Wunderwelt des Lebens] Strukturell gleicht sie einer spiralförmig verdrehten Strickleiter: Zwei Zucker-Phosphat-Ketten bilden die Seiten, dazwischen liegen die „Sprossen“ – die Basen Adenin (A), Cytosin (C), Guanin (G) und Thymin (T). Sie paaren sich stets nach festen Regeln: A mit T, C mit G. Diese komplementäre Bindung macht jeden Strang zur Vorlage für den anderen – ein molekulares Backup-System.

Abb. 2: Schematische Darstellung der Grundstruktur der DNA [Die Wunderwelt des Lebens] Entrollt man die DNA aus nur einer einzigen Zelle, misst sie rund zwei Meter – bei einem Durchmesser von nur wenigen Millionstel Millimetern. Das Leben hängt also buchstäblich an einem hauchdünnen, aber erstaunlich langen Faden.
Die Abfolge der rund drei Milliarden Basenpaare ist so individuell wie ein Fingerabdruck. Sie bildet den genetischen Code – die Anweisungen zur Herstellung von Proteinen, jenen Molekülen, die Struktur, Funktion und Regulation aller biologischen Prozesse steuern.
Als zentrales Informationsarchiv ist die DNA zugleich äußerst wertvoll und empfindlich. Deshalb wird sie sicher im Zellkern aufbewahrt, vergleichbar mit einem Safe. Damit der Körper auf die Informationen in diesem Safe zugreifen kann, braucht es ein cleveres Transportsystem. Schließlich müssen die Baupläne für Proteine – jene molekularen Architekten aller Lebensprozesse – zu den Ribosomen gelangen, den sogenannten „Protein-Fabriken“ im Zellplasma. Dort werden die Proteine hergestellt – ein Prozess, den man als Proteinbiosynthese bezeichnet.
Doch wie kommt ein konkreter Bauplan sicher aus dem Zellkern zu diesen Fabriken? Mit anderen Worten: Wie wird die genetische Information für ein bestimmtes Protein vom geschützten Archivort an den Ort der Produktion übertragen?
b) Vom Code zum Protein: Wie Anleitungen Wirklichkeit werden
Die Reise beginnt bei den Genen – bestimmten Abschnitten der DNA, die die Baupläne für Proteine enthalten. Etwa 20.000 solcher Gene finden sich im menschlichen Genom.
Die DNA ist dabei nicht nur ein passives Informationsarchiv im Zellkern – sie enthält ein aktives Repertoire an Anleitungen. Man kann sich jedes Gen wie ein Kapitel in einem molekularen Kochbuch vorstellen: nicht selten mit mehreren Varianten eines Rezepts, angepasst an Zelltyp, Entwicklungsstand oder äußere Bedingungen.
Zunächst entsteht eine temporäre „Arbeitskopie“ eines Gens – in Form von RNA (Ribonukleinsäure). Sie ähnelt der DNA, verwendet jedoch die Base Uracil (U) anstelle von Thymin (T) und besteht meist nur aus einem einzelnen Strang. Diese flüchtige Struktur macht die RNA ideal für den kurzfristigen Gebrauch – sie wird nach Erfüllung ihrer Funktion wieder abgebaut.
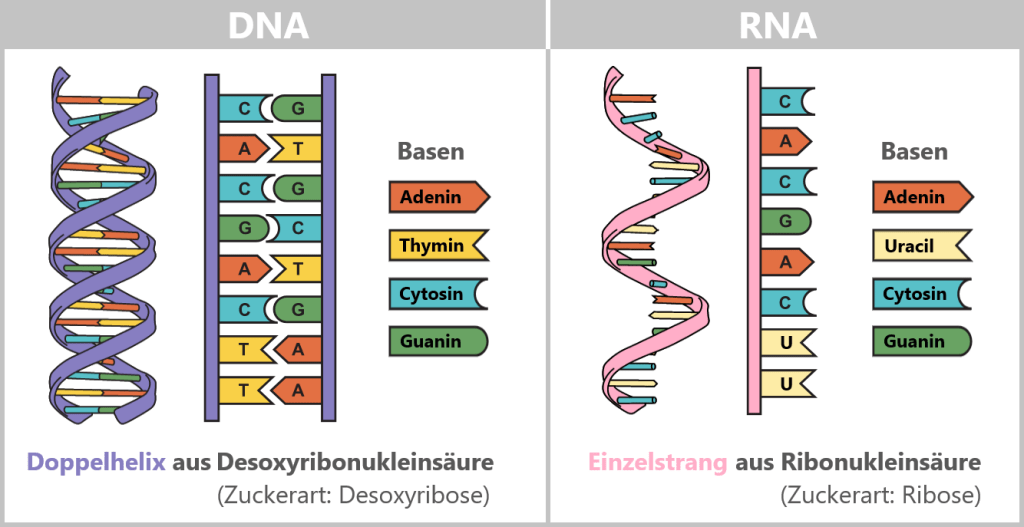
Abb. 3: Von der DNA wird eine Arbeitskopie erstellt, die RNA [Die Wunderwelt des Lebens] Alternatives Splicing: Die flexible Küche der Gene
Zellen müssen sich flexibel an wechselnde Anforderungen anpassen: auf Entzündungen reagieren, Krankheitserreger abwehren, Muskeln aufbauen oder den Blutzuckerspiegel regulieren. Dafür braucht es keine starre Anleitung, sondern ein System, das je nach Situation improvisieren kann.
In Abhängigkeit vom Zelltyp und äußeren Signalen wird der RNA-Strang dynamisch umgebaut, indem er an bestimmten Markierungen – übertragen aus der DNA – geschnitten wird. Nicht benötigte Abschnitte (Introns) werden entfernt, die verbleibenden (Exons) neu zusammengesetzt. So entsteht aus einer Vorlage eine Vielzahl möglicher Botschaften. Das Ergebnis ist die messenger RNA (mRNA), die die genetische Information aus dem Zellkern zu den Ribosomen im Zytoplasma bringt – den „Protein-Fabriken“ der Zelle.
Man kann sich diesen Prozess vorstellen wie das Nachkochen eines Rezepts: je nach Bedarf lässt die Zelle Zutaten weg, fügt Gewürze hinzu oder passt die Zubereitungsschritte an. So wird aus einer Grundanleitung eine Vielfalt von Gerichten.
Durch diesen Prozess – genannt alternatives Splicing – können Zellen aus einem Gen eine Vielzahl unterschiedlicher Proteine herstellen, die jeweils spezifische Funktionen übernehmen.
Der gesamte Vorgang „Vom Gen zum Protein“ wird häufig als Genexpression bezeichnet.
💡 Beispiel: Das Titin-Gen
👉 Längstes menschliches Gen (≈ 300.000 Basenpaare).
👉 Durch alternatives Splicing entstehen > 20.000 verschiedene Proteinvarianten – jeweils angepasst an die unterschiedlichen Anforderungen in verschiedenen Muskeltypen.Unser Erbgut ist also kein starres Rezeptbuch – es ist eine dynamische Küche, in der die Zelle aus denselben Vorlagen immer neue Lösungen entwickelt.
📌 Wer sich dafür interessiert, wie die Erbinformation in der DNA Schritt für Schritt in ein Protein übersetzt wird, findet hier weiterführende Informationen.
Doch wie in jeder kreativen Küche können auch hier beim Übergang der DNA zur mRNA Fehler passieren: Wird beim Abschreiben oder Montieren ein „Zutatenbuchstabe“ übersehen, vertauscht oder falsch zusammengesetzt, kann die Botschaft am Ende missverständlich oder unbrauchbar werden. Solche Mutationen oder fehlerhaftes Splicing sind manchmal harmlos – doch sie können auch defekte Proteine hervorbringen, die ihre Aufgabe nicht erfüllen oder sogar Schaden anrichten. Ein kleiner Patzer in der Abschrift des molekularen Kochbuchs – und der Geschmack des Lebens verändert sich.
c) Epigenetik & nicht-codierende DNA: Das Dirigenten-Team unseres Erbguts
Obwohl alle Zellen die gleiche DNA enthalten, nutzen sie jeweils nur einen Bruchteil davon. Eine Leberzelle benötigt andere Anleitungen als eine Nervenzelle – der Rest bleibt stillgelegt. Diese gezielte Genregulation wird durch epigenetische Mechanismen gesteuert: Kleine chemische Markierungen – etwa Methylgruppen – verändern nicht die genetische Sequenz selbst, sondern beeinflussen, ob ein Gen aktiv ist, wie oft es abgelesen wird oder ob es komplett stummgeschaltet bleibt.
Doch wer gibt dem Dirigenten eigentlich seine Anweisungen? Hier kommt die nicht-codierende DNA ins Spiel – 98 % unseres Erbguts, lange als nutzloser „Ballast“ abgetan. Sie ist das Betriebssystem, das die epigenetischen Schaltpläne bereitstellt. Erst ihr Zusammenspiel legt fest, wann, wo und wie stark unsere Gene aktiv werden – und entscheidet so über Gesundheit oder Krankheit.
d) Das verborgene Kontrollzentrum: Was die nicht-codierende DNA steuert
In diesen „dunklen“ Regionen verbergen sich präzise Schaltkreise, die das Schicksal jeder Zelle lenken:
Enhancer wirken wie unsichtbare Fernbedienungen der Gene. Selbst über Tausende Bausteine hinweg können sie ein Zielgen aktivieren oder zum Schweigen bringen – etwa für die Herzentwicklung im Embryo.
Telomere sind die Schutzkappen der Chromosomenenden, wie Plastikhüllen an Schnürsenkeln. Doch wenn sie schrumpfen, beginnt das Altern – ein zentraler Mechanismus von Zellverfall und Krebsentstehung.
Nicht-codierende RNAs agieren als heimliche Regisseure: Mikro-RNAs blockieren krankmachende Gene, während die lange RNA XIST bei Frauen ein gesamtes X-Chromosom zum Schweigen bringt.
Epigenetische Markierungen legen eine unsichtbare Landkarte über das Genom. Mit Methylierungen wie roten Stoppschildern entscheiden sie, ob ein Enhancer oder Gen gelesen werden darf – oder verstummt.
So entsteht Ordnung im molekularen Kochbuch: Jeder Zelltyp liest nur die Seiten, die er braucht – gesteuert durch nicht-codierende DNA und ihre epigenetische Interpretation.
e) Wenn das Betriebssystem krank macht
Fast alle genetischen Risikomarker für Volkskrankheiten – von Diabetes über Herzleiden bis zu psychischen Störungen – liegen in nicht-codierenden Zonen:
Mutationen in einem Enhancer für das MYC-Gen können Leukämie auslösen, indem sie die Zellteilung außer Kontrolle bringen.
Epigenetische Fehlprogrammierungen in diesen Bereichen – etwa durch chronischen Stress oder Umweltgifte – können Gene dauerhaft falsch an- oder abschalten und so Krankheiten wie Krebs oder Autoimmunstörungen begünstigen.
Solche epigenetischen Muster sind teils reversibel – ein Hoffnungsschimmer für neue Therapien.
f) Offene Rätsel der Forschung
Trotz gewaltiger Fortschritte bleibt das Genom voller ungelöster Rätsel:
Die „dunkle Materie“: Rund die Hälfte des nicht-codierenden Genoms ist funktional unergründet. Handelt es sich um evolutionäres Rauschen – oder um ein noch unkartiertes Steuernetz des Lebens?
Langstrecken-Kommunikation: Wie findet ein Enhancer zielsicher sein Gen inmitten von Millionen DNA-Bausteinen? Die räumliche Faltung des Genoms – ein wahres „Chromatin-Origami“ – könnte der Schlüssel sein.
Umwelt-Einflüsse: Weshalb führen identische nicht-codierende Mutationen in verschiedenen Lebenswelten zu völlig unterschiedlichen Krankheitsbildern?
RNA-Codes: Zehntausende nicht-codierende RNAs sind entdeckt – doch welche von ihnen sind nur Randnotizen, und welche erheben sich möglicherweise zu Leitmotiven neuer Therapien?
g) Die wahre Magie der Gene
Die nicht-codierende DNA ist kein Datenmüll, sondern das Mastermind der Zelle. Die Epigenetik wiederum ist ihr Dirigent – sie interpretiert flexibel, was das Betriebssystem vorgibt. Gemeinsam formen sie unsere Entwicklung, steuern unsere Gesundheit und reagieren auf Umweltsignale.
Gene sind kein starres Schicksal, mehr eine Partitur, die immer wieder neu interpretiert werden kann. Im Zusammenspiel entsteht ein Orchester, das nicht nur unsere Zellen formt, sondern sich ständig neu einstimmt – durch Erfahrungen, Umwelteinflüsse und sogar durch Spuren, die auch an kommende Generationen weitergegeben werden können.
Die wahre Magie liegt darin, dass aus begrenzten Noten eine unendliche Vielfalt von Leben erklingt.
4.2. Genetische Variation und ihre Folgen
Wenn sich das Erbgut verschreibt, verstummt oder improvisiert.
Bei jeder Zellteilung wird die DNA sorgfältig kopiert und an die Tochterzellen weitergegeben – so bleibt die genetische Information über Generationen hinweg erhalten.
Doch das System ist nicht unfehlbar: Mutationen – also Veränderungen in der DNA-Sequenz – können Struktur und Funktion von Proteinen beeinflussen. Manchmal stimmt nur ein Buchstabe nicht. Oder ein Satz fehlt. Oder ein ganzer Absatz wurde umgestellt. In der Genetik spricht man dann von Varianten – genetischen Veränderungen, die je nach Art, Größe und Position harmlos, risikobehaftet oder krankheitsauslösend sein können.
Die folgende Übersicht enthält die wichtigsten Variantentypen – und was sie im molekularen Manuskript des Lebens bedeuten:
📌 SNVs (Single Nucleotide Variants) – Ein einziger Buchstabe ist vertauscht. Meist kaum spürbar – doch manchmal genügt er, um ganze Geschichten umzuschreiben, wie bei der Sichelzellenanämie.
📌 Indels (Insertionen/Deletionen) – Kleine Textfragmente werden eingefügt oder gelöscht. Schon eine winzige Verschiebung kann ganze Sätze aus dem Takt bringen, manchmal so stark, dass der ursprüngliche Sinn verloren geht.
📌 Strukturelle Varianten (SVs) – Lange DNA-Passagen werden verdreht, verdoppelt oder verschoben. Diese verdeckten Umschreibungen sind schwer zu entdecken – und oft verhängnisvoll, etwa bei Krebs.
📌 Splice-Site-Varianten – Fehler an den Schnittstellen zerreißen die Syntax der RNA. Es entstehen falsche Baupläne – und Proteine, die nicht mehr stimmen.
📌 Repeat-Expansionen – Basenfolgen werden zwanghaft wiederholt, bis sie den Text entstellen. So entstehen Krankheiten wie Chorea Huntington.
📌 Copy Number Variants (CNVs) – Ganze Kapitel (Gene) fehlen oder erscheinen doppelt. Das Gleichgewicht der Erzählung geht verloren.
📌 Mitochondriale Varianten – Veränderungen in der DNA der Mitochondrien, der Kraftwerke der Zelle. Wenn hier der Text verfälscht wird, stockt der Antrieb des Lebens.
📌 Somatische Varianten – Treten erst im Laufe des Lebens auf und betreffen nur bestimmte Zellgruppen. Sie prägen vor allem die Geschichte von Tumoren.
📌 Epigenetische Signale – Keine neuen Buchstaben, sondern eine andere Betonung: Lautstärke, Rhythmus, Beginn. Sie verwandeln denselben Text in eine völlig neue Melodie.
Die Liste zeigt: Unser Erbgut ist kein starres Gebilde, sondern lässt sich durch Varianten verändern. Manche dieser Abweichungen bleiben unauffällige Randnotizen, andere aber können die gesamte Geschichte neu schreiben – manchmal mit dramatischen Folgen.
Um solche feinen Unterschiede sichtbar zu machen, braucht es Werkzeuge, die jedes Detail erfassen – jeden Buchstaben, jedes Satzzeichen, jede Verschiebung. Genau hier setzt die moderne Genomsequenzierung an.
4.3. Genomsequenzierung: Schlüssel zum individuellen Erbgut
Das Erbgut eines Menschen gleicht einer gigantischen Bibliothek: Milliarden von Buchstaben, fein säuberlich in langen Bänden angeordnet. Um darin zu lesen, bedarf es der Genomsequenzierung – die wie ein hochpräziser Scanner den Rohtext unseres genetischen Bauplans liefert.
So wird sie zum Herzstück der personalisierten Medizin: einer Medizin, die ihre Entscheidungen am Einzelnen ausrichtet, nicht am Durchschnitt.
Doch wie gelangen wir überhaupt an das „Buch des Lebens“, um es Seite für Seite lesen zu können?
Stellen wir uns vor: Bei einem Neugeborenen wird kurz nach der Geburt ein DNA-Test durchgeführt – ein Szenario, das im Vereinigten Königreich bereits Wirklichkeit geworden ist, wie zuvor beschrieben.
a) Die Reise der DNA: Vom Nabelschnurblut zur genetischen Information
Unmittelbar nach der Geburt kann Blut aus der abgetrennten Nabelschnur entnommen werden – ein sicherer und schmerzfreier Vorgang. Das Nabelschnurblut stammt vom Neugeborenen und enthält zahlreiche wertvolle Zellen, insbesondere Stammzellen und weiße Blutkörperchen. In ihren Zellkernen liegt das Entscheidende verborgen: die DNA – das genetische Erbe des Kindes.
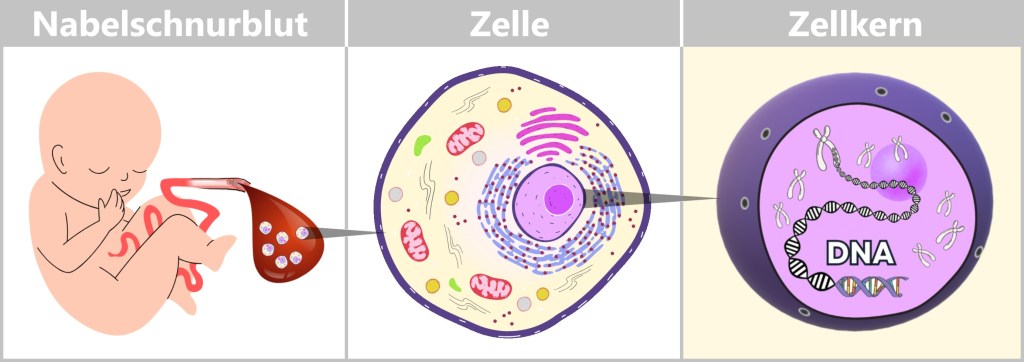
Abb. 4: Vom Nabelschnurblut zur genetischen Information Links: Nabelschnurblut als reiche Quelle unterschiedlicher Zellen.
Mitte: Schematische Darstellung einer menschlichen Zelle.
Rechts: Im Zellkern liegt die DNA, Trägerin der genetischen Information.Um diese DNA zu isolieren und analysieren zu können, durchläuft sie im Labor einen mehrstufigen Prozess. Dabei kommen bewährte molekularbiologische Methoden zum Einsatz, die sich in vier zentrale Schritte gliedern lassen:
Zellaufschluss: Zugang zum Erbgut schaffen
Zunächst wird das Blut zentrifugiert – also mit hoher Geschwindigkeit geschleudert. Dabei trennen sich die Bestandteile nach Dichte: Die weißen Blutkörperchen setzen sich als Zellpellet am Boden ab. Sie enthalten den größten Teil der interessierenden DNA.
Diese Zellen werden anschließend mit speziellen Lösungen und Enzymen behandelt, die die Zell- und Zellkernmembran auflösen. Dadurch wird die DNA freigesetzt – zunächst allerdings als Teil eines Gemischs aus Proteinen, Lipiden und anderen Zellbestandteilen.
DNA-Isolierung: Reines Erbgut gewinnen
Im nächsten Schritt wird die DNA aus dem Zelllysat – der „Zellsuppe“ – isoliert. Dazu gibt es verschiedene Verfahren:
Filtration oder Säulenreinigung: Die Lösung wird durch spezielle Filtersysteme geleitet, an denen die DNA haften bleibt, während kleinere Moleküle ausgespült werden.
Magnetkügelchen: Modernere Verfahren verwenden winzige Kügelchen mit DNA-bindenden Oberfläche. Die DNA haftet daran und kann per Magnet gezielt entnommen werden.
Beide Methoden ermöglichen eine effektive Trennung von Erbgut und Störstoffen.
Reinigung und Konzentration der DNA
Nun liegt die DNA vieler weißer Blutkörperchen in relativ reiner Form vor – allerdings noch in einer wässrigen Lösung. Um sie weiter zu reinigen und zu konzentrieren, wird kalter Alkohol (z. B. Ethanol oder Isopropanol) hinzugefügt. Dadurch fällt die DNA aus und wird sichtbar – als weißlicher, fadenartiger Niederschlag.
Die menschliche DNA ist extrem lang: Entrollt man sie, misst sie pro Zelle etwa zwei Meter. Bei der Extraktion wird sie jedoch durch chemische und mechanische Einflüsse in viele kleinere Abschnitte zerteilt. Das sieht zunächst nach Chaos aus – doch da die DNA in allen Zellen identisch ist, enthalten viele Fragmente überlappende Informationen. Diese zufällige Verteilung wird bei der Sequenzierung zum Vorteil: Die Fragmente können mehrfach unabhängig gelesen und anschließend am Computer zu einem Gesamtbild zusammengesetzt werden. Das erhöht die Genauigkeit der Analyse erheblich.
Qualitätskontrolle: Ist die DNA intakt?
Bevor die DNA-Probe analysiert werden kann, muss ihre Qualität überprüft werden. Zwei gängige Verfahren sind:
Spektrophotometrie: Misst die Konzentration und Reinheit der DNA über die Lichtabsorption.
Gelelektrophorese: DNA-Fragmente werden durch ein Gel geleitet und nach Länge getrennt. (Längere Fragmente wandern langsamer als kürzere.) So lässt sich beurteilen, ob die Probe ausreichend intakt ist.
Warum ist dieser Prozess so wichtig?
Die menschliche DNA ist empfindlich – Verunreinigungen oder Fragmentierungen können Analysen verfälschen oder unbrauchbar machen. Eine sorgfältige und kontrollierte Aufreinigung ist daher Voraussetzung für zuverlässige genetische Aussagen.
Gerade Nabelschnurblut stellt eine besonders wertvolle Quelle medizinischer Information dar: Es ermöglicht Frühdiagnostik, Prognosen und in manchen Fällen gezielte Prävention.
Was im Labor wie ein routinierter Ablauf wirkt, ist in Wahrheit ein hochpräziser Vorgang – erfordert technisches Know-how, moderne Geräte und biochemisches Feingefühl. Durch automatisierte Extraktionsmethoden lässt sich heute in weniger als einer Stunde aus wenigen Tropfen Nabelschnurblut eine hochwertige DNA-Probe gewinnen. Sie besteht zwar aus Fragmenten, enthält aber dennoch alle Informationen, um den individuellen genetischen Code zu entschlüsseln – und damit die Grundlage für eine Medizin von morgen zu legen.
b) Drei Regieanweisungen für die Gen-Diagnostik: WGS, WES und Panel
Bevor wir tiefer in die Werkzeuge der Genom-Sequenzierung eintauchen, lohnt sich ein Blick auf die drei zentralen Strategien, mit denen heute genetische Informationen entschlüsselt werden – gleichsam wie Regieanweisungen im Drehbuch des Lebens:
🎯 Panel-Sequenzierung – Die gezielte Szene
Hier wird nur ein ausgewählter Teil des Genoms untersucht – meist einige Dutzend bis wenige Hundert Gene. Solche Tests kommen zum Einsatz, wenn der Verdacht auf eine spezifische Erkrankung besteht, etwa bei erblich bedingtem Brustkrebs (BRCA1/2).
Vorteil: Schnell, kosteneffizient, präzise – solange die medizinische Fragestellung klar ist.
Limitierung: Alles außerhalb der gewählten Szene bleibt im Dunkeln.🎥 Whole Exome Sequencing (WES) – Der klassische Director’s Cut
WES analysiert alle protein-kodierenden Abschnitte des Genoms – also etwa 1–2 % des gesamten Erbguts. Gerade bei seltenen Erbkrankheiten ist es ein bewährtes Mittel, da die meisten bekannten Mutationen in diesen Bereichen liegen.
Vorteil: Effiziente Suche nach fehlerhaften Bauplänen für Proteine.
Limitierung: Bereiche außerhalb der Gene – wichtige Schalter und Regler des Erbguts – werden nicht erfasst.🎬 Whole Genome Sequencing (WGS) – Der vollständige Film in 4K
WGS liest das gesamte Genom – alle drei Milliarden Buchstaben. Damit liefert es das umfassendste Bild, inklusive nicht-kodierender Regionen, regulatorischer Elemente, struktureller Varianten und seltener Mutationen.
Vorteil: Maximale Auflösung für komplexe Fälle.
Beispiel: Bei Kindern mit unklaren Entwicklungsstörungen bringt WGS doppelt so häufig Klarheit wie WES – etwa durch das Aufdecken von Mutationen in entfernten „Fernsteuerungs“-Regionen (Enhancern), die Gene falsch an- oder abschalten.Fazit:
Je nach Fragestellung braucht es die passende Perspektive – manchmal reicht ein Zoom auf die Schlüsselszene, manchmal muss der ganze Film neu geschnitten werden. Die Wahl der Methode ist daher keine technische Nebensache, sondern eine strategische Entscheidung im Drehbuch der Diagnostik.
4.4. Methoden der Genomsequenzierung
Wie wird der genetische Code gelesen?
Stellen Sie sich vor, DNA ist ein Buch – geschrieben in einer Sprache mit nur vier Buchstaben: A, T, C und G. Um diesen genetischen Text zu entschlüsseln, nutzen Wissenschaftler moderne Technologien. Zwei zentrale Methoden haben sich etabliert. Sie verfolgen dasselbe Ziel: die Reihenfolge der DNA-Buchstaben möglichst genau zu bestimmen.
Die erste Methode funktioniert – vereinfacht gesagt – wie eine molekulare Kopiermaschine mit Farbsensoren. Die zweite nutzt einen Hightech-Tunnel, durch den die DNA geleitet und elektronisch „abgetastet“ wird – vergleichbar mit dem Abtasten eines Seils mit den Fingern, um kleinste Unebenheiten zu spüren. Diese Verfahren bilden die Grundlage der DNA-Sequenzierung.
4.4.1. Methode 1: Kopieren mit Leuchtfarbe
a) Sanger-Sequenzierung
b) Illumina-Technologie
c) Single-Molecule Real-Time(SMRT)-Sequenzierung
4.4.2. Methode 2: Hightech-Tunnel – DNA elektrisch abtasten
a) Oxford Nanopore Sequenzierung
4.4.3. Genomsequenzierung – Technologien im Vergleich
a) Durchsatz vs. Verwertbarkeit – Masse ist nicht immer gleich Klasse
b) Wirtschaftlichkeit – Kosten pro Base vs. Kosten pro Erkenntnis
c) Von der Probe zum Befund – Wie schnell spricht das ganze Genom?
d) Klinische Einsatzfelder – Welche Technologie passt wozu?
e) KI als Co-Pilot – Automatisierung ohne Verantwortung abzugeben
f) Vergleichstabelle: Sanger, Illumina, PacBio, ONT
g) Key Takeaways: Zwischen Reife und Routine4.4.1. Methode 1: Kopieren mit Leuchtfarbe
Diese Methode nutzt die Tatsache, dass DNA aus zwei komplementären Strängen besteht. Kennt man die Basenabfolge eines Strangs, lässt sich die Sequenz des Gegenstrangs automatisch ableiten. Um diese Information zu gewinnen, wird einer der beiden Stränge der zu untersuchenden DNA kopiert. Ziel ist es, während dieses Kopiervorgangs die exakte Reihenfolge der Basen in der entstehenden Kopie zu erfassen.
Um besser zu verstehen, wie dieses Verfahren funktioniert, werfen wir zunächst einen Blick auf die wichtigsten Bestandteile und die grundlegenden Schritte zur Herstellung einer DNA-Kopie im Reagenzglas (in vitro).
Herstellung einer DNA-Kopie – das Grundprinzip
Im Zentrum steht eine DNA-Vorlage, die kopiert werden soll. Dafür braucht es vier Hauptbestandteile:
- dNTPs (Desoxynukleosidtriphosphate): die DNA-Bausteine A, T, C und G.
- DNA-Polymerase: ein Enzym, das als molekularer Kopierer fungiert.
- Primer: kurze DNA-Stücke, die der Polymerase den Startpunkt liefern.
- Pufferlösung: sorgt für stabile Bedingungen während der Reaktion.
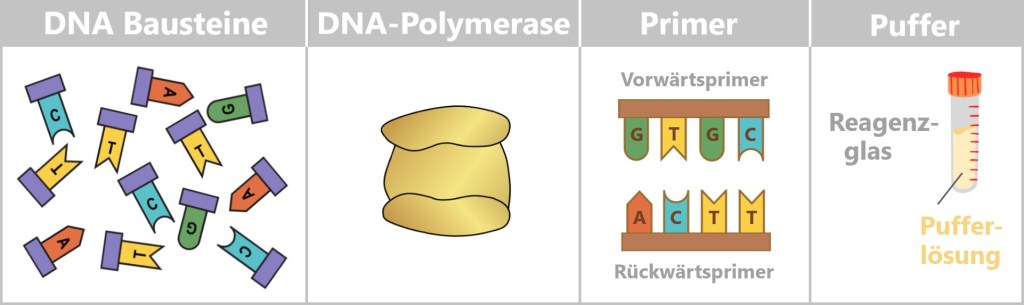
Abb. 5-A: Schematische Darstellung der Hauptbestandteile zur Herstellung einer DNA-Kopie DNA-Bausteine: Einzelne Nukleotide (A, T, C, G), dargestellt in verschiedenen Farben und Buchstaben. Sie bilden das Rohmaterial, aus dem der neue DNA-Strang zusammengesetzt wird.
DNA-Polymerase: Ein schematisch dargestelltes Enzym, das Nukleotide zu einem DNA-Strang verknüpft.
Primer: Kurze DNA-Stücke, die der Polymerase den Startpunkt vorgeben.
Puffer: Ein Reagenzglas mit Pufferlösung, die für stabile chemische Bedingungen während der Reaktion sorgt.Was macht die DNA-Polymerase?
Die DNA-Polymerase ist ein spezielles Enzym, das komplementäre DNA-Stränge erzeugt – gewissermaßen der „Druckkopf“ einer molekularen Kopiermaschine. Enzyme wirken als biologische Werkzeuge, die chemische Reaktionen gezielt und effizient ermöglichen – deshalb nennt man sie auch „biologische Katalysatoren“.
Damit die Polymerase arbeiten kann, benötigt sie einen Primer als Startsignal. Man kann sich das so vorstellen: Wie ein Druckkopf erst loslegt, wenn ein Blatt Papier richtig eingelegt ist, beginnt auch die Polymerase ihre Arbeit erst, wenn ein Primer vorhanden ist. Dieses kleine DNA-Stück zeigt ihr, wo sie starten soll – es ist sozusagen das erste Blatt im molekularen Drucker.
Vor Beginn der Kopie muss die doppelsträngige DNA zunächst in zwei Einzelstränge aufgetrennt werden – dieser Schritt heißt Denaturierung.
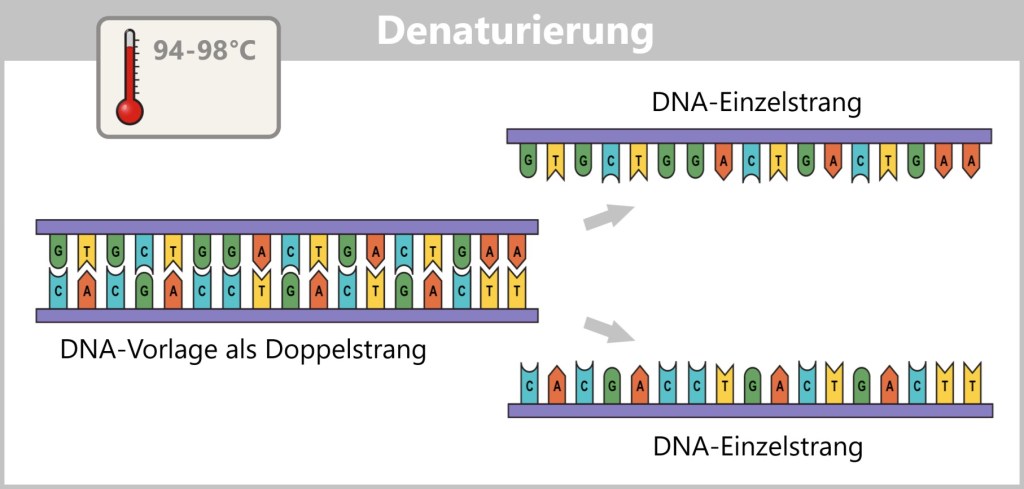
Abb. 5-B: Trennung der DNA-Stränge Anschließend lagern sich die Primer an ihre passenden Zielstellen der Einzelstränge. Diese Primerbindung markiert den Startpunkt für die DNA-Polymerase.
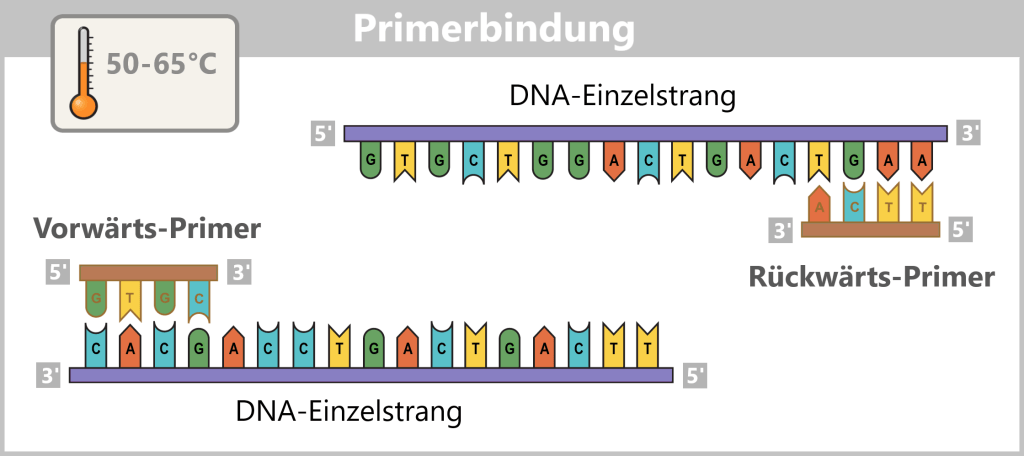
Abb. 5-C: Primer markieren den Startpunkt für die Polymerase. Nun beginnt die Synthese der neuen Stränge: Die Polymerase liest den Vorlage-Strang in 3′-zu-5′-Richtung und ergänzt die Kopie in Gegenrichtung – von 5′ nach 3′ – unter Beachtung der Basenpaarung (A mit T, G mit C).
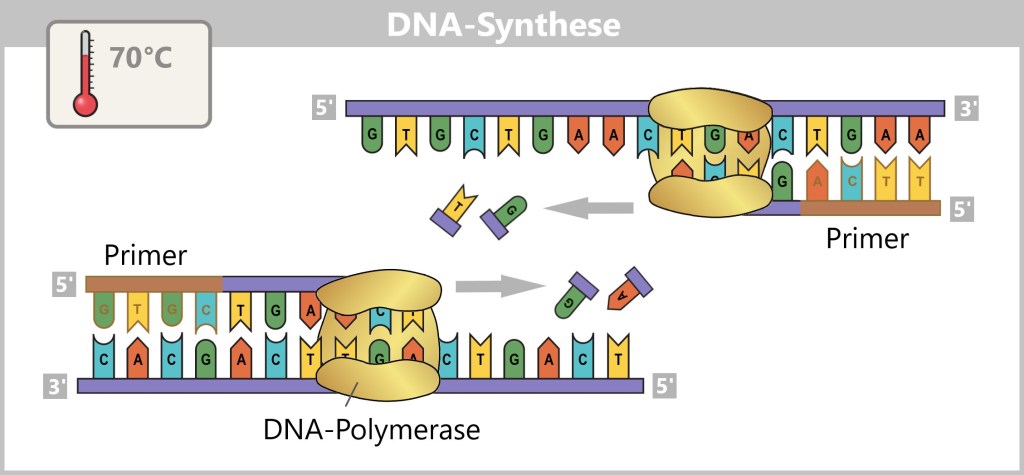
Abb. 5-D: DNA-Synthese – die exakte Verdopplung eines DNA-Strangs. Was bedeuten 3′ und 5′?
Die Bezeichnungen 3′ (sprich: drei-Strich) und 5′ (fünf-Strich) stammen aus der Chemie des DNA-Rückgrats. Sie geben an, an welchem Ende sich bestimmte Kohlenstoffatome im Zuckermolekül befinden, an die neue Bausteine angefügt werden können.
Stellen Sie sich den DNA-Strang wie eine Einbahnstraße vor. Die Polymerase kann nur in eine Richtung fahren – vom 5′-Ende zum 3′-Ende. Neue Bausteine lassen sich chemisch nur am 3′-Ende anfügen. Daher liest die Polymerase den „alten“ Strang rückwärts (3′ → 5′) und baut den neuen Strang vorwärts (5′ → 3′).
So entsteht aus einem Einzelstrang nach und nach ein neuer, passender Gegenstrang – mithilfe der Basenpaarungsregeln: Adenin (A) paart sich mit Thymin (T), und Cytosin (C) mit Guanin (G).
Das Ergebnis sind perfekte genetische Abbilder – neue DNA-Stränge, exakte Kopien der Ursprungs-DNA.
Leuchtende Buchstaben: Wie Sequenzierung funktioniert
Moderne Sequenziermethoden erweitern diesen Kopiervorgang um eine raffinierte Technik: Sie verwenden fluoreszierend markierte dNTPs (DNA-Bausteine). Jedes der vier Nukleotide (A, T, C, G) ist mit einem Farbstoff versehen, der beim Einbau ein spezifisches Lichtsignal aussendet.
Während die Polymerase die DNA-Stränge verlängert, fügt sie die farbigen Bausteine passgenau ein. Jedes Mal, wenn ein neues Nukleotid eingebaut wird, sendet es ein winziges Lichtsignal – wie ein kleiner Blitz, der verrät, welcher „Buchstabe“ gerade ergänzt wurde. Hochauflösende Kameras erfassen diese Lichtsignale und ermöglichen so die schrittweise Rekonstruktion der DNA-Sequenz.
Sequenziermethoden im Überblick
Mehrere bedeutende Technologien nutzen dieses Prinzip in spezifischer Form:
Sanger-Sequenzierung: Die klassische Methode, bei der fluoreszierende Kettenabbruch-Nukleotide die DNA-Synthese an zufälligen Stellen beenden. Die resultierenden Fragmente lassen sich der Reihenfolge nach auslesen.
Illumina-Technologie: Eine weit verbreitete Hochdurchsatzmethode, bei der Millionen DNA-Fragmente parallel sequenziert werden. Die fluoreszierenden Signale werden Schritt für Schritt erfasst.
SMRT-Sequenzierung (PacBio): Hier wird die Synthese in Echtzeit an einem einzigen DNA-Molekül beobachtet – mit hoher Präzision und langen Leselängen.
a) Sanger-Sequenzierung
Das klassische „Buchstaben-für-Buchstaben-Lesen“
Die Sanger-Sequenzierung, auch Kettenabbruchmethode genannt, wurde in den 1970er-Jahren entwickelt und war das erste Verfahren, mit dem sich DNA präzise und zuverlässig auslesen ließ. Sie beruht im Wesentlichen auf denselben Prinzipien wie die DNA-Synthese, unterscheidet sich jedoch in einigen entscheidenden Punkten:
- Es wird nur ein Primer verwendet.
- Neben den normalen DNA-Bausteinen – den dNTPs (dATP, dCTP, dGTP, dTTP) – kommen zusätzlich spezielle fluoreszierende Kettenabbruch-Bausteine, die sogenannten ddNTPs (ddATP, ddCTP, ddGTP, ddTTP) in geringer Konzentration zum Einsatz. Wenn ein solcher ddNTP eingebaut wird, stoppt die DNA-Synthese genau an dieser Stelle. Jeder der vier ddNTP-Typen trägt eine unterschiedliche Leuchtfarbe, je nach Base.
Der Ablauf – Schritt für Schritt
Für die Sequenzierung werden vier separate Reaktionsmischungen angesetzt. Jede enthält:
- die einzelsträngige Ziel-DNA,
- einen Primer (Startpunkt der Synthese),
- eine DNA-Polymerase,
- normale dNTPs,
- und jeweils eine Sorte der farblich markierten ddNTPs.
Abb. 6-A: Sanger-Sequenzierung – schematische Darstellung der vier Reaktionsmischungen. Jede Mischung enthält die gleichen Grundkomponenten, unterscheidet sich jedoch durch die jeweils zugegebene Variante der farblich markierten Stopp-Bausteine (ddNTPs).
Nach der Primerbindung liest die Polymerase den DNA-Strang aus und fügt passende Nukleotide ein, um einen komplementären Strang aufzubauen. Wird dabei zufällig ein ddNTP eingebaut, endet die Synthese an genau dieser Stelle.
So entstehen viele unterschiedlich lange DNA-Fragmente – jedes endet mit einem farbig markierten Basenbuchstaben. Da in jeder der vier Reaktionen nur ein ddNTP enthalten ist, weiß man genau, mit welchem Buchstaben die erzeugten DNA-Fragmente enden.
Abb. 6-B: Die Synthese wird gezielt unterbrochen – bei Einbau eines ddNTPs. In jeder Reaktionsmischung ist jeweils eine Sorte der modifizierten DNA-Bausteine (ddATP, ddTTP, ddCTP, ddGTP) enthalten. Wird einer dieser Stopp-Bausteine während der DNA-Synthese eingebaut, bricht der Kopiervorgang genau an dieser Stelle ab. In der ddATP-Mischung stoppt die Synthese, sobald ein modifiziertes Adenin (A) eingebaut wird. In der ddTTP-Mischung endet sie beim Einbau eines modifizierten Thymins (T). Genauso führen ddCTP und ddGTP zum Abbruch der Synthese, wenn ein modifiziertes Cytosin (C) bzw. Guanin (G) eingebaut wird. Durch dieses Verfahren entstehen viele DNA-Fragmente unterschiedlicher Länge, die jeweils mit einem spezifischen Stopp-Nukleotid enden. Ziel ist es, alle theoretisch möglichen Fragmente zu erzeugen, um daraus die vollständige DNA-Sequenz ablesen zu können.
Sortierung und Auswertung
Anschließend werden die DNA-Fragmente denaturiert (in Einzelstränge überführt) und durch Gelelektrophorese der Länge nach sortiert. Dabei wandern die negativ geladenen Fragmente durch ein Gel in Richtung der positiven Elektrode. Kleinere Fragmente bewegen sich schneller, größere langsamer – so entsteht eine geordnete „Leiter“ von Fragmenten.
Ein Laser regt nun die fluoreszierenden Farbstoffe der Endbasen an. Die dabei entstehenden Lichtsignale werden von einem Detektor erfasst. Jedes Signal steht für eine bestimmte Base an einer bestimmten Position. Die Reihenfolge der Lichtsignale ergibt damit direkt die DNA-Sequenz – wie bei einem Buchstaben-für-Buchstaben-Lesen.
Da die erzeugten Fragmente komplementär zur Ursprungs-DNA sind, lässt sich daraus die ursprüngliche Basenfolge exakt ableiten.
Abb. 6-C: Gelelektrophorese zur Trennung von DNA-Fragmenten: In den Reaktionsansätzen entstehen unterschiedlich lange DNA-Fragmente, die jeweils mit demselben Stopp-Nukleotid enden – je nach Ansatz entweder mit Adenin, Thymin, Guanin oder Cytosin. Diese Reaktionsmischungen werden auf ein Gel aufgetragen. Sobald ein elektrisches Feld angelegt wird, wandern die negativ geladenen DNA-Fragmente von der Kathode (−) zur Anode (+). Dabei bestimmt die Größe der Fragmente ihre Wandergeschwindigkeit: Kleinere Fragmente bewegen sich schneller durch die feinen Poren des Gels und gelangen früher zur Anode, während größere Fragmente langsamer sind. Durch das Auslesen der fluoreszierenden Farbsignale an den Fragment-Enden kann die genaue Reihenfolge der Basen bestimmt werden – und damit die DNA-Sequenz Schritt für Schritt rekonstruiert werden.
Warum gilt die Sanger-Sequenzierung bis heute als Goldstandard?
Auch ein halbes Jahrhundert nach ihrer Entwicklung bleibt die Sanger-Methode in vielen Bereichen der Molekularbiologie und Medizin unverzichtbar. Der Grund: Zuverlässigkeit und Präzision.
Im Vergleich zu modernen Hochdurchsatzverfahren ist die Sanger-Sequenzierung zwar langsamer und eignet sich nur für die Analyse kleinerer DNA-Abschnitte – nicht für ganze Genome. Aber genau darin liegt ihre Stärke:
- Gezielte Fragestellungen, wie das Überprüfen einzelner Gene,
- Nachweis spezifischer Mutationen, oder
- Validierung kritischer Ergebnisse, die zuvor mit anderen Methoden identifiziert wurden,
lassen sich mit der Sanger-Sequenzierung präzise, klar und interpretierbar beantworten – oft so deutlich, dass man die Abfolge der Basen direkt im Sequenzplot sehen kann.
In der medizinischen Diagnostik – etwa bei der Analyse vererbter Erkrankungen oder in der Qualitätskontrolle gentechnischer Verfahren – ist diese hohe Genauigkeit entscheidend. Denn ein falsch interpretierter Buchstabe kann hier lebenswichtige Folgen haben.
Ein weiterer Pluspunkt: Die Methode ist weltweit standardisiert, seit Jahrzehnten bewährt und unterliegt klaren Qualitätsrichtlinien.
Ein Klassiker mit Beständigkeit
In einer Welt, in der ständig neue Technologien auf den Markt drängen, bleibt die Sanger-Sequenzierung ein verlässlicher Anker – der Oldtimer unter den Sequenzierverfahren: nicht der Schnellste, aber extrem robust, bewährt und zuverlässig. Und manchmal ist genau das entscheidend.
b) Illumina-Sequenzierung
Die Hochgeschwindigkeits-Kopiermaschine
Die Sanger-Sequenzierung ist wie präzise Handarbeit: Jeder DNA-Strang wird Schritt für Schritt entziffert – zuverlässig, aber langsam und teuer.
Stellen Sie sich vor, Sie müssten ein ganzes Buch oder sogar eine Bibliothek abschreiben – und dürften dabei nur einen Buchstaben pro Minute notieren.
Genau hier liegt das Problem: Moderne Krebsforschung, die Entschlüsselung seltener Krankheiten oder die genetische Überwachung von Pandemien verlangen die Analyse großer Datenmengen – also vieler und/oder sehr langer DNA-Abschnitte. Dafür braucht es eine Methode, die nicht nur genau, sondern auch schnell und bezahlbar ist.
Und genau da kommt die Illumina-Sequenzierung ins Spiel. Sie hat das Prinzip der DNA-Entzifferung von der Handarbeit zur industriellen Massenproduktion erhoben und ist heute eine der am häufigsten eingesetzten Methoden für die Hochdurchsatz-Sequenzierung (Next Generation Sequencing, NGS). Statt einzelner Buchstaben werden nun ganze Seiten gleichzeitig gelesen – millionenfach, parallel, mit hoher Präzision und kosteneffizient.
Anders als bei der klassischen Sanger-Methode, bei der jedes DNA-Fragment einzeln analysiert wird, arbeitet Illumina mit einer massiven Parallelisierung – das heißt: Es werden viele DNA-Fragmente gleichzeitig vervielfältigt und sequenziert. Wie funktioniert das?
Schritt 1: Vorbereitung der DNA-Fragmente
– Die DNA wird zur „Lego-Baustelle“ –Zunächst wird die zu untersuchende DNA-Probe in viele kurze Stücke zerlegt – sogenannte Fragmente, die 100 bis 300 Basenpaaren lang sind. An beide Enden dieser Fragmente werden kleine künstliche DNA-Stücke – sogenannte Adapter – angehängt. Man kann sie sich wie LEGO-Steckverbinder vorstellen. Sie dienen einerseits als molekulare Andockstellen, um die DNA-Fragmente später auf einem speziellen Träger – dem Flowcell-Chip – zu befestigen. Andererseits dienen sie als Bindungsstellen für universelle Primer.
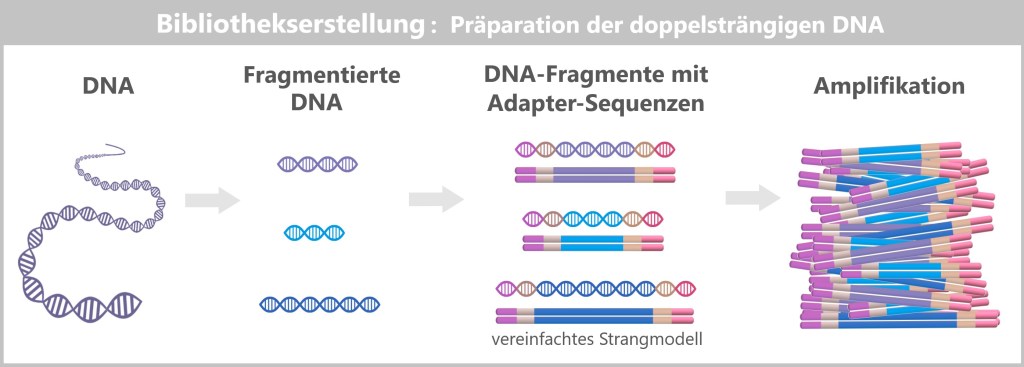
Abb. 7-A: Die DNA wird in zahlreiche DNA-Fragmente mit kleinen Anhängern (Adaptern) zerlegt. 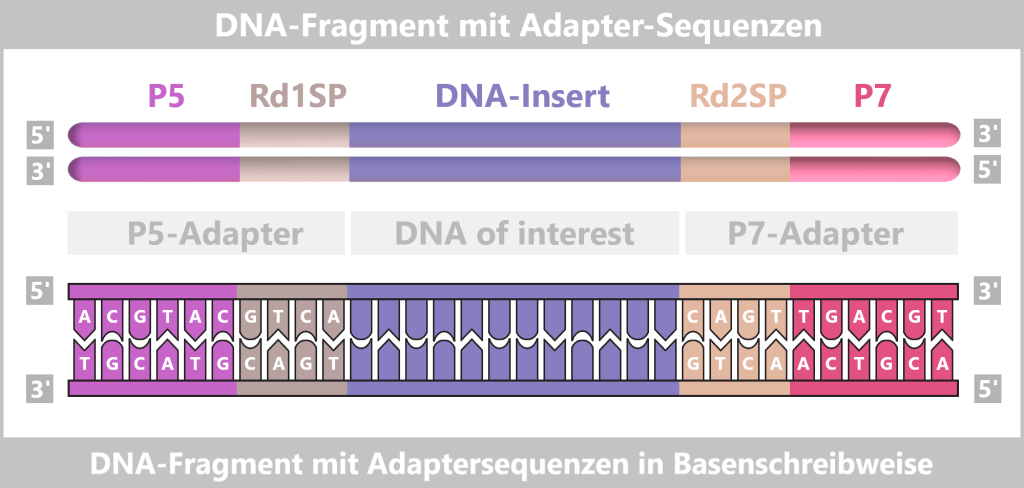
Abb. 7-B: Vereinfachte schematische Darstellung des DNA-Fragments mit Adaptern. Die P5/P7-Sequenzen dienen der Bindung an der Flow Cell. Die Rd1SP/Rd2SP-Sequenzen sind Bindungsstellen für universelle Primer.
Durch Denaturierung werden die so vorbereiteten doppelsträngigen DNA-Fragmente in Einzelstränge gespalten.
Schritt 2: Brückenamplifikation
– Der Tanz der DNA auf der Flow Cell –Das Herzstück der Illumina-Technologie ist die sogenannte Flow Cell – eine glasartige Platte, übersät mit Millionen winziger DNA-Andockstellen (kurzen DNA-Abschnitten, sogenannten Oligonukleotiden), die fest an der Oberfläche verankert sind.
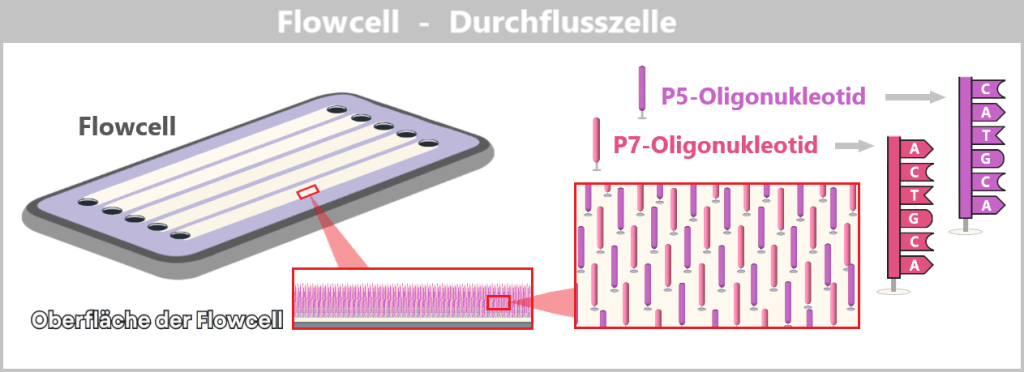
Abb. 7-C: Schematische Darstellung einer Flowcell und deren Oberfläche Über ihre Adapter binden sich die einzelsträngigen DNA-Fragmente an die Anker. Anschließend beginnt die molekulare Kopierarbeit: Es werden komplementäre Kopien der DNA-Schnipsel erzeugt, die nun fest mit der Flow Cell verankert sind.
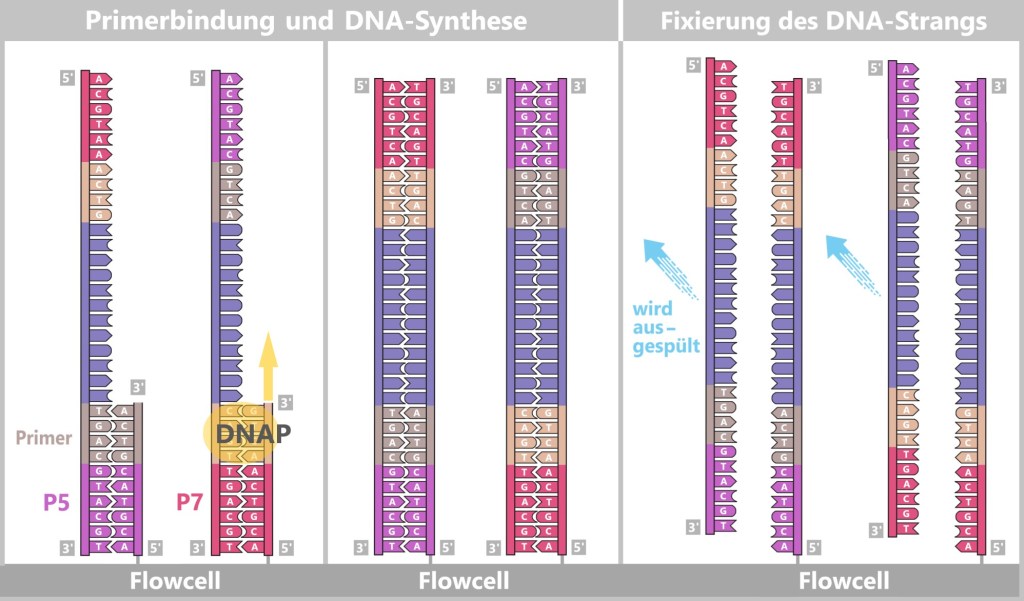
Abb. 7-D: Schematische Darstellung der ersten Synthese Links: Primer binden an die Adapter (P5, P7), und die DNA-Polymerase (DNAP) startet die Synthese eines neuen, komplementären Strangs.
Mitte: Die DNA-Polymerase synthetisiert daraufhin einen ersten Strang.
Rechts: Der neue entstandene DNA-Doppelstrang wird aufgetrennt. Der ursprüngliche Strang hat nach der Denaturierung keine Verbindung mehr zur Flowcell und wird ausgespült. Der neu synthetisierte Strang bleibt mit seinem 5′-Ende fest an der Flowcell gebunden.Dann beginnt ein faszinierender Prozess: Die DNA-Stränge biegen sich zu kleinen Brücken, indem ihre freien Adapter an benachbarte Oligonukleotide auf der Flow Cell binden. An diesen Stellen werden sie erneut kopiert. Nach dem Kopieren wird die Brücke wieder aufgelöst – und die Zahl der fest verankerten DNA-Stränge verdoppelt sich. Dieser Vorgang, bekannt als Brückenamplifikation, wiederholt sich dutzendfach. Am Ende entstehen dichte Cluster aus Millionen identischer Kopien eines einzelnen DNA-Fragments.
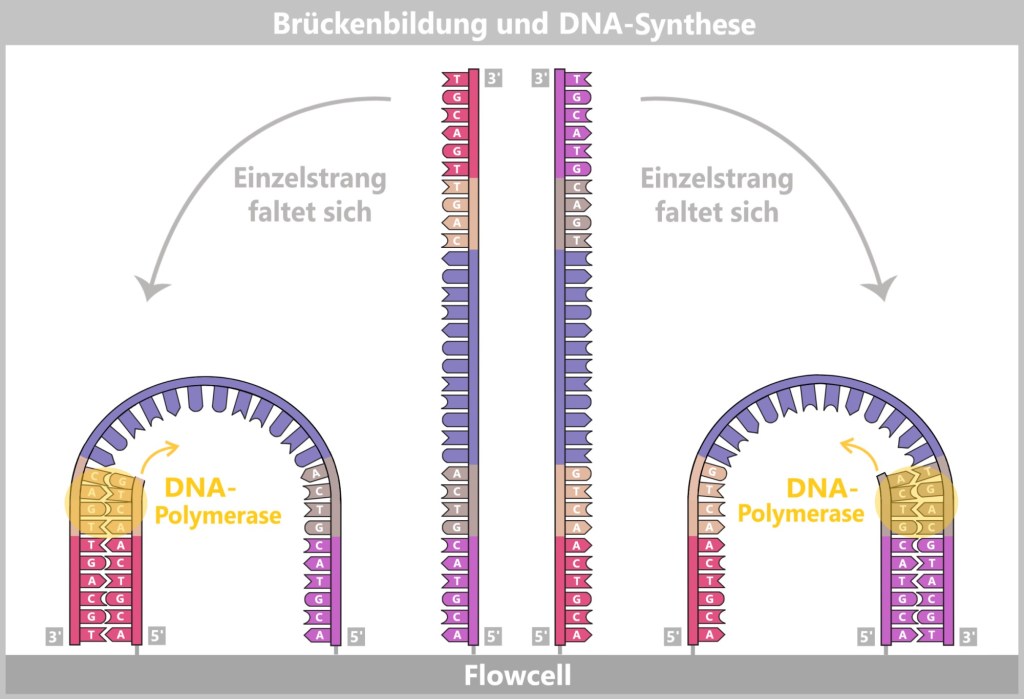
Abb. 7-E: Brückenbildung und DNA-Synthese Die verankerten Einzelstränge falten sich und verbinden sich mit den benachbarten Ankern auf der Flowcell, wodurch eine Brückenstruktur entsteht. Danach wird der Kopiervorgang initiiert.
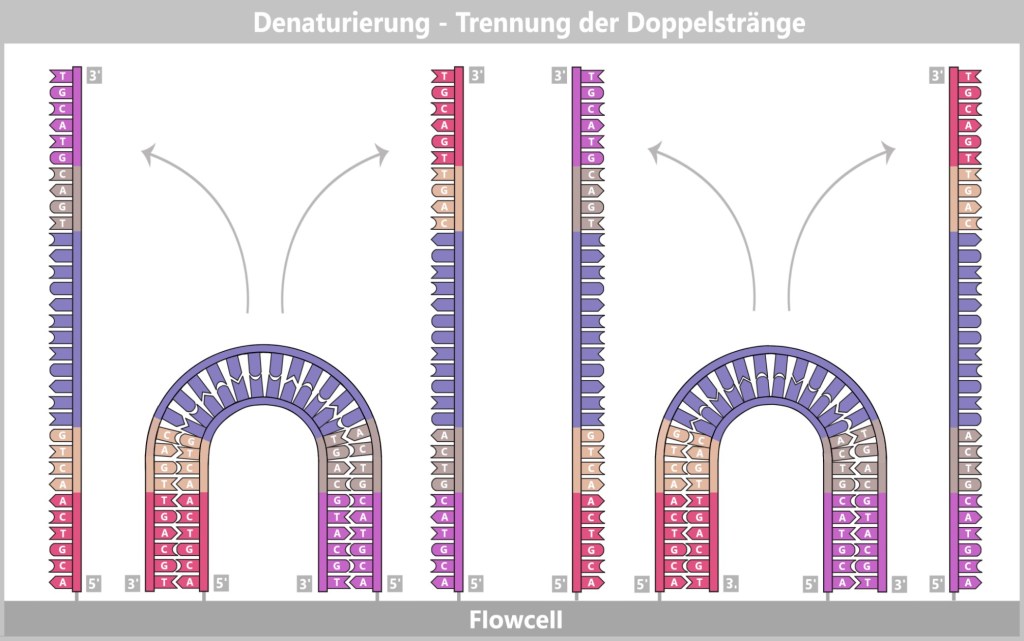
Abb. 7-F: Vorwärts und rückwärts: Die Brücken lösen sich. Nach dem Kopiervorgang werden die Brücken-Doppelstränge getrennt. Die Anzahl der fest verankerten DNA-Stränge verdoppelt sich. Sie sind bereit für die weitere Amplifikation.

Abb. 7-G: Mit jeder Wiederholung der Brückenamplifikation entstehen immer mehr DNA-Kopien. 
Abb. 7-H: Clusterbildung Das Ergebnis ist eine dicht gepackte „Landkarte“ aus Millionen DNA-Clustern, die jeweils nur eine bestimmte Sequenz enthalten – ideal für das gleichzeitige Auslesen.
Ohne diese Vervielfältigung wäre das Lesen der DNA, als würde man versuchen, ein einzelnes Glühwürmchen im Wind zu erkennen. Die Cluster hingegen machen die DNA-Basen (A, T, C, G) deutlich sichtbar – wie ein leuchtender Neon-Schriftzug in der Dunkelheit.
Schritt 3: Sequenzieren durch Synthese
– Die Lichtshow der DNA-Basen –Nun beginnt das eigentliche Sequenzieren durch Synthese. Auch hier kommt das Prinzip der „molekularen Kopiermaschine mit Farbsensoren“ zum Einsatz – aber in optimierter Form:
Zu Beginn binden die Universal-Primer an den entsprechenden Adapterstellen der DNA-Fragmente in jedem DNA-Clusters.
Dann werden modifizierte dNTPs – sogenannte RT-dNTPs (reversibel terminierende Nukleotide) – in die Reaktionslösung gegeben. Jeder der vier DNA-Bausteine (A, T, G, C) ist dabei mit einer eigenen Fluoreszenzfarbe markiert und trägt eine reversible Blockierung.
Nur ein Nukleotid pro Zyklus kann eingebaut werden, weil die Blockierung den weiteren Strangaufbau vorübergehend stoppt. Nicht eingebaute RT-dNTPs werden weggewaschen.
Nach jedem Einbau wird die Flowcell mit einer hochauflösenden Kamera gescannt. Jeder leuchtende Punkt steht für ein eingebautes Nukleotid und seine Farbe zeigt an, welcher Buchstabe gerade ergänzt wurde.
Danach wird die Blockierung und die Farbstoffmarkierung chemisch entfernt, und der nächste Zyklus beginnt.
Dieser Prozess wird Zyklus für Zyklus wiederholt – bis jeder DNA-Strang vollständig gelesen ist.
Abb. 7-I: Illumina Sequenzierungsschritte Schritt 4: Datenauswertung
– Der Supercomputer als Puzzlemeister –Nach der „Lichtshow“ liegen Millionen von Fotos vor – eines für jeden Zyklus und jedes Cluster. Jedes Bild zeigt, welche Farbe (und damit welcher Buchstabe: A, T, C oder G) in jedem Cluster hinzugefügt wurde.
Jetzt kommen leistungsfähige Computer ins Spiel – ausgestattet mit Software, die so präzise arbeitet wie ein Detektiv, der aus einem Stapel durchnummerierter Polaroids ein zerschnittenes Buch zusammensetzt. Jedes Cluster auf dem Sequenzierungschip ist wie ein kleiner Tatort, jeder Farbpixel eine Spur. Und aus Millionen solcher Spuren entsteht allmählich eine Geschichte – die Geschichte der DNA.
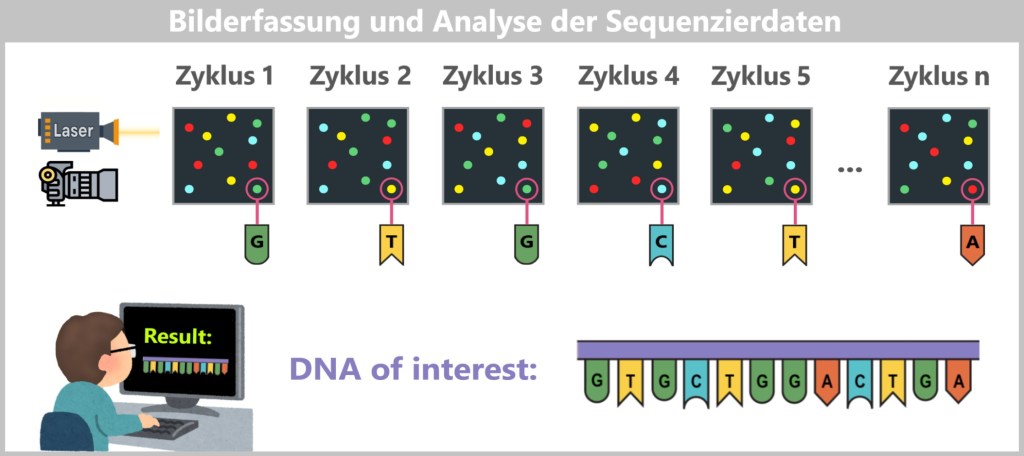
Abb. 7-J: Analyse der Sequenzierdaten Die Farbmuster werden in Buchstabenfolgen übersetzt, sogenannte „Reads“: winzige Textstücke eines gewaltigen Genom-Romans. Am Ende türmt sich eine Flut solcher Fragmente auf – wie ein Berg zerschnittener Buchseiten, lose verstreut.
Genau hier beginnt die eigentliche Kunst: Die Bioinformatik tritt auf den Plan. Mit Hilfe spezieller Algorithmen werden die Schnipsel analysiert, sortiert und aneinandergefügt – immer auf der Suche nach überlappenden Stellen, vertrauten Mustern, bekannten Strukturen.
Stück für Stück setzt sich das große Ganze wieder zusammen – bis am Ende die ursprüngliche DNA-Sequenz sichtbar wird, wie ein rekonstruiertes Buch, das plötzlich wieder Sinn ergibt.
Die eigentliche „Magie“ geschieht also nicht in der Flow Cell, sondern im Computer! Ohne die Software wären die Bilder nur buntes Flimmern – mit ihr aber werden sie zum Schlüssel für medizinische Durchbrüche.
Eine ausführlichere und dennoch anschauliche Erklärung der Illumina-Sequenzierungstechnologie Methode findet sich im Video „Illumina Sequencing Technology“.
Moderne Hochdurchsatz-Sequenzierplattformen wie die von Illumina können die Rohdaten eines vollständigen menschlichen Genoms innerhalb weniger Tage erzeugen – zu reinen Sequenzierkosten, die deutlich unter dem Preis eines Premium-Smartphones liegen.
Diese Technologie hat die Genomforschung revolutioniert und erheblich zugänglicher gemacht: Sie ist heute ein äußerst vielseitiges Werkzeug – schnell genug für die Echtzeit-Analyse von Pandemien, präzise genug, um in Kombination mit robuster Bioinformatik und klinischer Expertise die Grundlage für personalisierte Krebstherapien zu bilden.
Die Illumina-Sequenzierung gilt inzwischen als Industriestandard für große Sequenzierprojekte – etwa bei der Analyse ganzer Genome, in der Genexpressionsforschung oder in der modernen Krebsdiagnostik.
Und das Beste: Während man diesen Text liest, entschlüsselt irgendwo auf der Welt eine Flow Cell Millionen DNA-Fragmente.
Allerdings hat auch die Illumina-Technologie ihre Grenzen. Besonders bei langen, sich wiederholenden DNA-Abschnitten – wie sie in manchen Chromosomenbereichen vorkommen – gerät sie an ihre Grenzen. Das ist, als müsste man ein Lied aus tausenden 3-Sekunden-Schnipseln rekonstruieren. Für solche Herausforderungen kombiniert man Illumina häufig mit anderen Sequenzierverfahren, die auch lange Abschnitte zuverlässig erfassen können – wie ein Ermittler, der sowohl Nahaufnahmen als auch Panoramabilder braucht, um den ganzen Tatort zu verstehen.
c) Single-Molecule Real-Time(SMRT)-Sequenzierung
Der Roman in einem Zug
Stellen Sie sich vor, Sie wollen einen langen Roman lesen – voller wiederkehrender Kapitel und versteckter Hinweise, die sich über viele Seiten erstrecken. Wenn Sie das Buch wie bei der Illumina-Sequenzierung in tausende kleine Schnipsel zerschneiden, diese einzeln lesen und anschließend wie ein Puzzle zusammensetzen, besteht die Gefahr, dass Kapitel fehlen, vertauscht oder unvollständig bleiben. Die geheimen Botschaften der Geschichte – das, was der Roman uns eigentlich erzählen will – könnte dadurch verloren gehen oder nur bruchstückhaft erscheinen.
Hier glänzt die Single Molecule Real-Time (SMRT)-Sequenzierung von PacBio: Sie liest ganze Buchkapitel in einem Zug – ohne Schnipsel, ohne Pause.
Anstatt die DNA zu zerlegen, bleibt sie bei PacBio in sehr langen Stücken erhalten – oft Zehntausende Basenpaare am Stück, die als Ganzes sequenziert werden. Diese Fäden betreten winzige Bühnen in einer SMRT-Zelle, sogenannte Nanokammern (Zero-Mode Waveguides). Diese sind so klein, dass sie nur Platz für ein einziges DNA-Molekül und eine Polymerase bieten. Jede dieser Bühnen lässt Licht nur an einem winzigen Punkt wirken – genau dort, wo die Polymerase die DNA abliest. Wie ein Spotlicht, dass einen Schauspieler bei einem Monolog auf der Bühne ins Zentrum rückt.
Die Polymerase spielt die Hauptrolle: Sie fügt Baustein für Baustein (A, T, C oder G) an die DNA-Kopie an. Jeder dieser Bausteine leuchtet in einer anderen Farbe, ähnlich wie Neonlichter bei einem DJ. Bei jedem Einbau blitzt ein Lichtsignal auf, das von einer Kamera in Echtzeit aufgezeichnet wird – ein tanzender Lichtcode, der die DNA-Sequenz sichtbar macht.
Das Video Introduction to SMRT Sequencing von PacBio veranschaulicht diesen Vorgang eindrucksvoll.
Analysieren wir die einzelnen Schritte dieses faszinierenden Verfahrens im Detail.
Schritt 1: Die Vorbereitung
– DNA-Ringe für das Endloslesen –Auch bei PacBio wird die zu untersuchende DNA zunächst in Fragmente zerlegt – allerdings in deutlich größere Stücke als bei Illumina, meist 10.000 bis über 20.000 Basenpaare lang. Diese Fragmente werden zu ringförmigen DNA-Molekülen verarbeitet:
Man fügt an beide Enden sogenannte Hairpin-Adapter an – kleine Schlaufen aus DNA, die das Fragment in sich selbst schließen.
Abb. 8-A: Ein DNA-Fragment wird mit Hairpin-Adaptern versehen. Die Adapter enthalten eine Primer-Bindestelle, die später den Startpunkt für die Polymerase liefert.
Durch anschließende Denaturierung entsteht ein zirkuläres, einzelsträngiges SMRTbell-Molekül in Form einer offenen Schlaufe.
Abb. 8-B: Das denaturierte SMRTbell-Molekül: Durch Denaturierung entsteht aus der doppelsträngigen Vorlage ein einzelsträngiges SMRTbell-Molekül. Dieses bildet eine zirkuläre Struktur, die in der Darstellung als geöffnete Schlaufe erscheint.
Das Ergebnis ist ein SMRTbell: ein geschlossener DNA-Ring, der von der Polymerase beliebig oft gelesen werden kann – wie eine Modelleisenbahn, die im Kreis fährt.
Jeder Hairpin-Adapter enthält eine definierte Bindungsstelle, an der der universelle Primer bindet. Das ist der Startpunkt für die Polymerase.
Die DNA-Polymerase lagert sich an den Primer-Komplex an. Es entsteht ein voll funktionsfähiger Replikationskomplex: Polymerase + Primer + SMRTbell.
Abb. 8-C: Der stabile Replikationskomplex aus Polymerase, Primer und SMRTbell-Einzelstrang. Der Primer ist an die Adapter-Bindestelle gebunden, die Polymerase an den Primer. Dieser Komplex ist startbereit für die DNA-Synthese.
Schritt 2: Die Sequenzierbühne
– Lichtspiele im Nanomaßstab –Jetzt wird es spektakulär: Die vorbereiteten Replikationskomplexe werden in ihre winzigen Beobachtungskammern geleitet – die Zero Mode Waveguides (ZMWs). Diese Nanokammern sind so klein, dass in der Regel nur ein einziges SMRTbell-Template Platz findet. Die Polymerase ist dabei fest am Boden des ZMWs verankert und wartet auf ihren Einsatz.
Das Startsignal gibt die Zugabe der vier Nukleotide (A, C, G, T), die jeweils mit einem eigenen Fluoreszenzfarbstoff markiert sind.
Die Polymerase beginnt nun mit ihrer Arbeit – sie liest den DNA-Strang und baut einen komplementären Strang auf. Sobald sie ein Nukleotid einbaut, passiert das Entscheidende:
Jedes Nukleotid sendet einen kurzen, farbcodierten Lichtblitz aus.
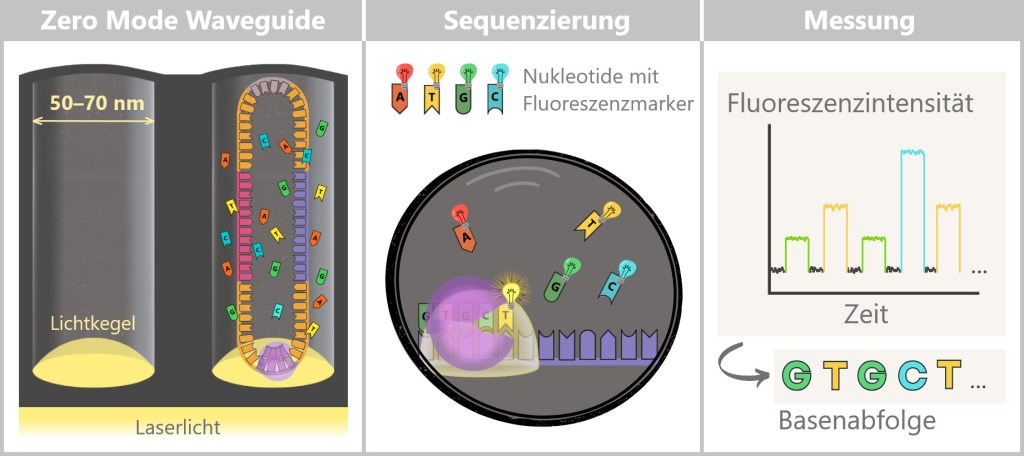
Abb. 8-D: Zero Mode Waveguide (ZMW): Sequenzierung einzelner DNA-Moleküle Links: Am Boden des Zero Mode Waveguides (ZMW) tritt ein Laserstrahl ein, der ein sogenanntes evaneszentes Feld (dargestellt als Lichtkegel) erzeugt. Dieses Feld breitet sich nicht wie normales Licht in die Lösung aus, sondern klingt innerhalb von nur 20–30 Nanometern exponentiell ab. Dadurch entsteht ein extrem kleines Anregungsvolumen, in dem die Fluoreszenzfarbstoffe der Nukleotide angeregt werden können.
Im linken Zero Mode Waveguide (ZMW) befindet sich ein einzelner Replikationskomplex (Polymerase + SMRTbell). Die Polymerase (lila) ist dabei fest am (Glas-)Boden verankert. Mit Anwesenheit der Fluoreszenz-markierten Nukleotide beginnt die Polymerase mit ihrer Arbeit.
Mitte: Die vier Nukleotide (A, T, G, C) tragen unterschiedliche Fluoreszenzmarker. Sobald die Polymerase ein Nukleotid einbaut, leuchtet es kurz auf, bevor der Farbstoff abgespalten wird.
Rechts: Diese Lichtblitze werden in Echtzeit gemessen und in Fluoreszenzsignale übersetzt. So entsteht Schritt für Schritt die Basenabfolge der DNA.Das Leuchten geschieht in Echtzeit, genau im Moment der Einfügung – daher der Name: Single Molecule Real-Time Sequencing. Ein empfindlicher Detektor zeichnet diese Lichtsignale auf –wie ein molekulares Live-Mikroskop bei der Arbeit.
Schritt 3: Das Geheimnis der Langstrecke
– Wiederholung macht’s robust –Da PacBio sehr lange DNA-Abschnitte auf einmal liest – vergleichbar mit dem Lesen ganzer Buchkapitel statt einzelner Verse, treten bei einem einzelnen Durchlauf mehr Einzelfehler auf als bei Illumina. Genau hier kommt die ringförmige SMRTbell-Struktur zum Einsatz. Die DNA-Polymerase kann wie ein Zugführer das Ringmolekül mehrfach umrunden. Dabei wird das gleiche DNA-Fragment immer wieder gelesen – wie ein Vorleser, der sich mehrfach an denselben Text wagt, bis er ihn sicher beherrscht.
Ein Computer gleicht die Wiederholungen miteinander ab und filtert die Fehler heraus – wie ein Lektor, der ein Manuskript glättet. So entsteht eine besonders präzise Konsens-Sequenz.
PacBio nennt dieses Verfahren HiFi-Reads – hochpräzise Einzelmolekül-Lesungen.
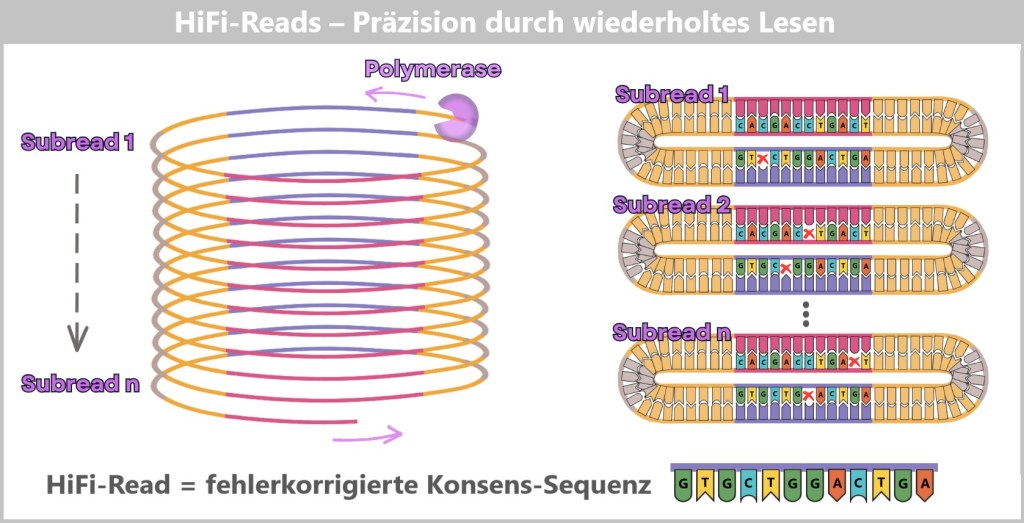
Abb. 8-E: Die Entstehung eines HiFi-Reads. Die Polymerase wandert mehrfach um das zirkuläre SMRTbell-Template und erzeugt dabei mehrere Subreads. Jeder Umlauf entspricht einer vollständigen Ablesung der DNA-Sequenz. Einzelne Subreads können zufällige Fehler (x) enthalten, doch durch den Vergleich vieler Wiederholungen wird ein hochpräziser Konsens gebildet – der sogenannte HiFi-Read (High-Fidelity Read).
Zusatzinfo: Wie die Polymerase mehrfach um die DNA läuft
Erster Sequenzierungsdurchlauf
Die Polymerase liest den einzelsträngigen Template-Strang ab und synthetisiert dabei einen komplementären Strang.
Das Ergebnis ist eine vollständige Doppelhelix – der ursprüngliche Template-Strang und der neu entstandene Tochterstrang sind nun fest miteinander verbunden.
Aus dieser ersten, ununterbrochenen Umrundung entsteht ein Subread – also eine einzelne Basenabfolge, die von der Polymerase in einem Durchlauf erzeugt wurde.
Zweite und weitere Sequenzierungsdurchläufe
Nun stellt sich die Frage: Wie kann die Polymerase weiterlesen, wenn doch der DNA-Ring bereits doppelsträngig ist?
Hier kommt die Φ29-Phagen-DNA-Polymerase ins Spiel – eine speziell modifizierte Enzymvariante mit zwei entscheidenden Eigenschaften:
Hohe Prozessivität: Sie bleibt sehr lange am DNA-Strang gebunden und löst sich kaum – das ermöglicht die langen Reads.
Starke Strangverdrängungsaktivität: Sie kann Wasserstoffbrückenbindungen zwischen Template- und Tochterstrang aufbrechen.
Die Polymerase läuft in die Doppelhelix hinein, trennt die Stränge lokal auf und verdrängt den alten Tochterstrang, während sie gleichzeitig einen neuen synthetisiert – wie ein molekularer Bulldozer.
Der zuvor gebildete Strang wird dabei nicht abgebaut, sondern bleibt als loser, einzelsträngiger DNA-Faden in der Nanokammer zurück. Bei jeder weiteren Runde wiederholt sich dieser Prozess: Die Polymerase liest dasselbe Template erneut ab – und erzeugt dadurch mehrere Subreads vom gleichen DNA-Molekül.
Schritt 4: Datenanalyse
– Aus Lichtblitzen wird lesbare DNA –Sind alle Lichtsignale eingefangen, beginnt die eigentliche Detektivarbeit: die Datenanalyse.
Der erste Schritt ist das sogenannte Base-Calling: Dabei werden die aufgezeichneten Fluoreszenzsignale – zeitlich und farblich codiert – in Nukleotidsequenzen übersetzt – A, T, G oder C.
Anschließend werden die Rohdaten jedes Polymerase-Umlaufs bereinigt: Die Hairpin-Adapter-Sequenzen werden bioinformatisch abgetrennt. Zurück bleiben die sogenannten Subreads – die ungekürzten Leseprotokolle jedes einzelnen Rundgangs der Polymerase durch den DNA-Kapiteltext.
Doch erst der nächste Schritt enthüllt die volle Wahrheit: Wie ein philologischer Meisterdetektiv, der mehrere Abschriften eines Manuskripts vergleicht, kombiniert der Algorithmus alle Subreads desselben SMRTbell-Moleküls. Aus diesem Vergleich – Circular Consensus Sequencing (CCS) genannt – entsteht ein hochpräziser HiFi-Read: eine fehlerbereinigte Masterkopie, die das DNA-Kapitel wortgetreu wiedergibt.
Erst jetzt ist das Textfragmentbereit für die finale Einordnung – und durchläuft dabei mehrere Stationen:
Zunächst eine Qualitätskontrolle, ganz wie ein sorgfältiges Lektorat, das den Text auf Klarheit und Stimmigkeit prüft.
Darauf folgt das Alignment: Die Sequenz wird wie ein neues Kapitel ins richtige Fach eines Referenzgenom-Regals eingeordnet.
Schließlich erfolgt die Assemblierung – der Schlussakt, bei dem alle Kapitel zu einem vollständigen Roman des Genoms zusammengefügt werden.
Das Video PacBio Sequencing – How it Works fasst die Schritte anschaulich zusammen.
Anwendung und Vorteile – Die Stärken der Langleser
Die PacBio-SMRT-Technologie hat eine besondere Stärke: Sie kann sehr lange DNA-Abschnitte am Stück lesen – mit höchster Genauigkeit. Das macht sie ideal für:
- Das Erkennen komplexer Genomstrukturen
- Die Analyse wiederholungsreicher Abschnitte
- Die Unterscheidung sehr ähnlicher Genvarianten (z. B. bei Immunrezeptoren oder in der Krebsgenetik)
- De-novo-Sequenzierung – also das vollständige Lesen neuer Genome ohne Referenz
- Das Aufspüren von epigenetischen Modifikationen (z.B. Methylierungen)
Wie kann SMRT epigenetische Modifikationen sichtbar machen?
Wie bereits beschrieben, spielen epigenetische Mechanismen eine zentrale Rolle bei der gezielten Genregulation. Es handelt sich um kleine chemische Markierungen – etwa Methylgruppen – die an bestimmte DNA-Basen angeheftet werden. Sie verändern nicht die genetische Sequenz selbst, sondern beeinflussen, wann und wie oft Gene abgelesen werden – abhängig von Zelltyp, Umgebung oder Entwicklungsstadium.
Sie funktionieren wie Notizen oder Markierungen am Seitenrand eines Buches: Der Text bleibt derselbe, aber die „Lesart“ ändert sich – manche Passagen werden betont, andere überblättert.
Gerät dieses fein abgestimmte System aus dem Gleichgewicht, kann das weitreichende Folgen haben – etwa bei Krebs, Autoimmunerkrankungen oder psychischen Störungen.
Im Gegensatz zur Illumina-Technologie, die eine chemisch veränderte Kopie der DNA liest, analysiert SMRT die Original-DNA in Echtzeit. Dabei wird nicht nur die Basenabfolge, sondern auch die Verweildauer der DNA-Polymerase an jeder Base – die sogenannte Einbaukinetik – gemessen.
Genau hier liegt der Schlüssel zum Nachweis epigenetischer Modifikationen: Methylierungen und andere chemische Veränderungen beeinflussen diese Kinetik. Sie wirken wie kleine Hindernisse oder Bremsspuren auf dem DNA-Strang. Die Polymerase „verharrt“ an solchen Stellen länger – und genau diese Verzögerung wird von der SMRT-Kamera direkt erfasst.
SMRT eignet sich besonders gut zur direkten Detektion bestimmter chemischer Markierungen, etwa Methylgruppen an Adenin- oder Cytosin-Basen, die vor allem in Bakterien und Pflanzen eine wichtige Rolle spielen. Für die in der humanen Epigenetik zentrale Cytosin-Methylierung (5mC) ist die direkte Detektion weniger sensitiv, kann aber mithilfe spezialisierter Bioinformatik-Tools wie pb-CpG-tools zuverlässig abgeleitet werden.
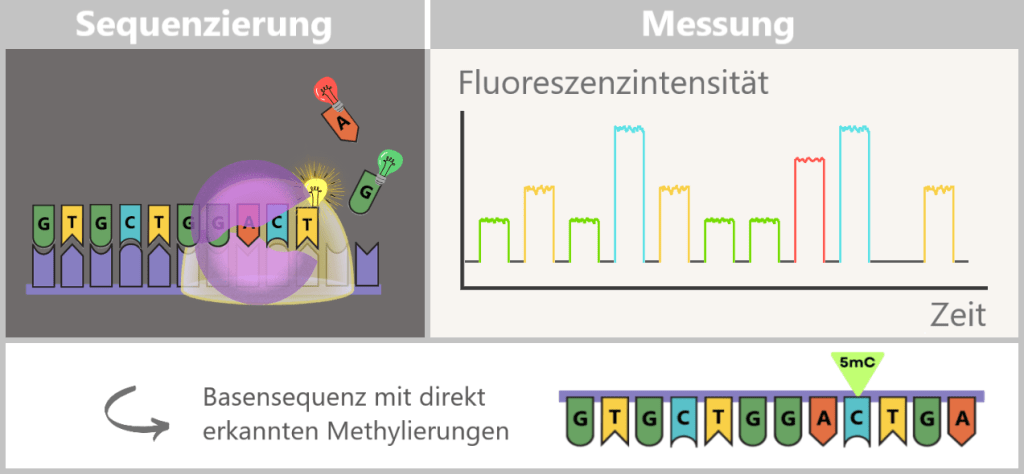
Abb. 8-E: Direkte Basendetektion und Methylierungsanalyse in der SMRT-Sequenzierung Wie SMRT epigenetische Modifikationen sichtbar macht
Während der SMRT-Sequenzierung werden die Fluoreszenzsignale der eingebauten Nukleotide in Echtzeit gemessen. Gleichzeitig können Modifikationen wie 5-Methylcytosin (5mC) erkannt werden, da sie die Kinetik der Polymerase leicht verändern.Der entscheidende Vorteil: Die extrem langen Leselängen enthüllen, wie sich Methylierungsmuster über große genomische Regionen mit anderen Faktoren verschränken – etwa mit:
- Genvarianten,
- Haplotypen (Gruppen von gemeinsam vererbten Genvarianten),
- repetitiven Sequenzen.
So liefert PacBio nicht nur die genetische Buchstabenfolge, sondern eine „Landkarte“ der epigenetischen Steuerung – und macht sichtbar, welche „Buchseiten“ funktionell zusammengehören und wie diese Vernetzung Krankheitsprozesse wie Krebs oder neurologische Störungen beeinflusst.
Defekte Scheren im Genom: Wie PacBio Splice-Site-Varianten enttarnt
Im Abschnitt Alternatives Splicing: Die flexible Küche der Gene wurde vereinfacht beschrieben, wie unsere DNA als molekulares Kochbuch Rezepte in Gestalt von mRNA-Abschriften erzeugt. Diese transportieren die genetische Information aus dem Zellkern zu den Ribosomen im Zytoplasma – den „Protein-Fabriken“ der Zelle.
Dabei wird das Rezept nachträglich angepasst: Eine molekulare Schere entfernt bestimmte Zutaten (Introns) und kombiniert andere Bausteine (Exons) neu. Gesteuert wird dieser Prozess durch Splice-Stellen – Markierungen, die aus der DNA „mitkopiert“ werden. Man kann sie sich wie eine Schnittanleitung vorstellen: Sie entscheidet, welche Teile des Rezepts verwendet und wie sie zusammengesetzt werden.
Doch was passiert, wenn diese Schnittanleitung fehlerhaft ist? Die Folgen können gravierend sein:
- Exons werden übersprungen → Teile der Bauanleitung fehlen.
- Introns bleiben fälschlich erhalten → „Müll“ wird ins Protein eingebaut.
- Falsche Spleißstellen entstehen → Der mRNA-Text wird sinnentstellt.
Solche Splice-Site-Varianten führen häufig zu fehlerhaften oder funktionslosen Proteinen – mit möglichen Folgen wie Krebs, genetischen Erkrankungen (z. B. β-Thalassämie) oder neurologischen Störungen. Da die Proteinbiosynthese direkt vom mRNA-Text abhängt, ist es entscheidend, solche Fehler zu erkennen.
Der Schlüssel liegt in PacBios Fähigkeit, vollständige mRNA-Moleküle in einem Stück zu analysieren – meist 500 bis 5.000 Nukleotide lang, manchmal deutlich länger. Da PacBio DNA liest, wird die mRNA zunächst in eine stabile Kopie (cDNA, komplementäre DNA) umgewandelt. Diese kann dann vollständig sequenziert werden. So lässt sich exakt erkennen, wie das Rezept aufgebaut ist und welche Zutaten in welcher Reihenfolge vorgesehen sind – kein Schreib- oder Schnittfehler bleibt verborgen.
Zusätzlich kann auch der zugrunde liegende DNA-Abschnitt sequenziert werden. Bioinformatische Tools vergleichen cDNA- und DNA-Sequenzen mit Referenzdaten – so werden Abweichungen im Spleißmuster und ihre genetische Ursache sichtbar.
Die DNA zeigt: Hier ist die Anleitung fehlerhaft.
Die cDNA zeigt: Hier ist das Rezept verstümmelt.Erst die Kombination beider Informationen schafft eine fundierte Basis für präzise Diagnosen und gezielte Therapieansätze.
Fazit – Wenn man die ganze Geschichte erzählen will
Wenn es darum geht, die ganze Geschichte eines DNA-Abschnitts am Stück zu erzählen – ohne zu zerschneiden oder zusammenzusetzen –, dann ist SMRT-Sequenzierung die Methode der Wahl.
PacBio-SMRT ist der Archäologe der Genomik: Es sieht das Ganze, wo andere nur Bruchstücke liefern. 2022 trug es maßgeblich dazu bei, die letzten Lücken im menschlichen Genom zu schließen.
Die PacBio-SMRT-Technologie ist kein Massenprodukt, sondern ein Präzisionsinstrument. Sie ist langsamer und teurer pro Basenpaar als Illumina – aber dafür auch detailreicher und vielseitiger.
In der Praxis werden beide Technologien oft kombiniert: Illumina für die Breite, PacBio für die Tiefe. Gemeinsam ergeben sie ein vollständiges Bild – wie Satellitenbild und Nahaufnahme in einem einzigen Panorama.
4.4.2. Methode 2: Hightech-Tunnel – DNA elektrisch abtasten
Während bei der molekularen Kopiermaschine mit Farbsensoren die DNA zuerst vervielfältigt und dann bildlich „fotografiert“ wird, funktioniert die zweite Methode radikal anders: Die DNA wird durch einen Hightech-Tunnel geschickt – und dabei live elektronisch abgetastet.
Nach diesem Prinzip funktioniert die Oxford Nanopore Sequenzierung.
a) Oxford Nanopore Sequenzierung
Die DNA geht durchs Nadelöhr
Stellen Sie sich vor, Sie lesen ein Seil, indem Sie es langsam durch Ihre Finger gleiten lassen – und dabei jede noch so feine Unebenheit ertasten: Dicke, Dellen, Knoten. Genau so funktioniert die Oxford Nanopore Sequenzierung (ONT): Sie „fühlt“ die DNA, während sie durch ein winziges biologisches Nadelöhr gleitet.
Dieses Nadelöhr – ein sogenannter Nanopore-Kanal – ist eingebettet in eine Membran, durch die ein elektrischer Strom fließt. Sobald ein einzelnes DNA-Molekül durch diesen Hightech-Tunnel gezogen wird, beeinflussen die Basen (A, T, G oder C) den Stromfluss unterschiedlich stark – wie verschieden geformte Kieselsteine in einem Bach. Diese Veränderungen erzeugen charakteristische elektrische Signale, die in Echtzeit aufgezeichnet werden.
ONT liest also keine Lichtsignale, sondern Strommuster – ein elektrisches Fingerlesen der DNA.
Ein einzelner Faden – direkt gelesen
Bevor die Lesung beginnt, wird die DNA mit einem Motorprotein gekoppelt, das sie präzise durch die Pore zieht – nicht zu schnell, nicht zu langsam. Anders als bei Illumina oder PacBio braucht es weder Polymerase noch Fluoreszenz oder Amplifikation: Die Original-DNA wird direkt gelesen – ein großer Vorteil bei empfindlichen oder beschädigten Proben.
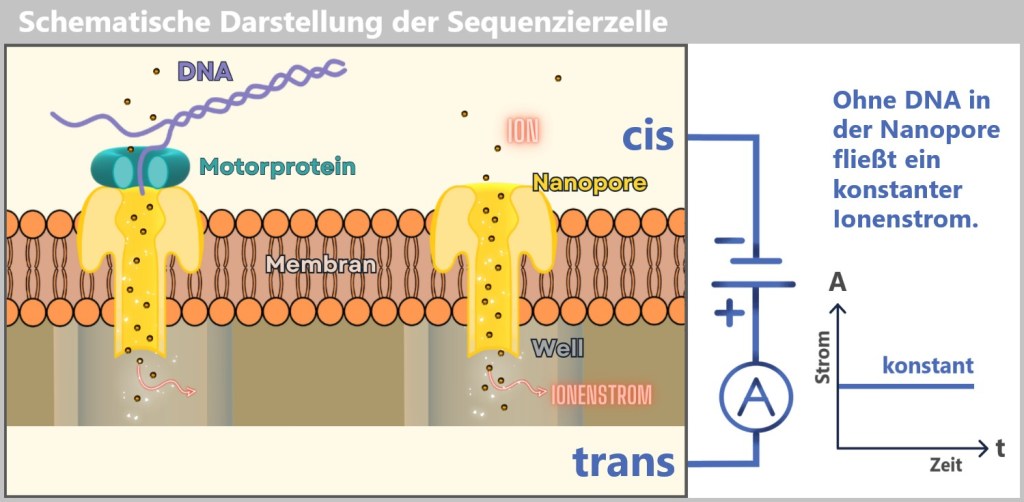
Abb. 9-A: Schematische Darstellung der Sequenzierzelle und des Ionenstroms im Ruhezustand Die Sequenzierzelle besteht aus zwei Kammern: der cis-Kammer (oben) und der trans-Kammer (unten), getrennt durch eine Membran mit eingebetteten Nanoporen. Links hat ein DNA-Molekül mithilfe eines Motorproteins an eine Nanopore angedockt. Rechts ist eine leere Nanopore dargestellt, durch die ein konstanter Ionenstrom fließt. Die angelegte Spannung zwischen der negativen Elektrode (cis) und der positiven Elektrode (trans) treibt den Ionenfluss an. Der Ionenstrom wird im Well (kleiner Kanal im Chip) gemessen, wie im Strom/Zeit-Diagramm dargestellt. Solange keine DNA durch die Pore wandert, bleibt der Strom konstant.
Und das Beste: Die DNA kann dabei extrem lang sein. ONT hält den Weltrekord für die längste je sequenzierte DNA – über zwei Millionen Basenpaare am Stück. Ganze Chromosomenabschnitte lassen sich so ohne Unterbrechung erfassen – wie ein Roman in einem einzigen Atemzug.
Aus Rauschen wird Sinn – Die Kunst der Signalinterpretation
Die elektrischen Rohsignale, die beim Durchgang der DNA durch die Pore entstehen, ähneln zunächst einem verrauschten Radiokanal. Doch mithilfe spezialisierter Algorithmen – dem sogenannten Basecalling – wird aus dem Stromrauschen eine klare Buchstabenfolge: A, T, G oder C.
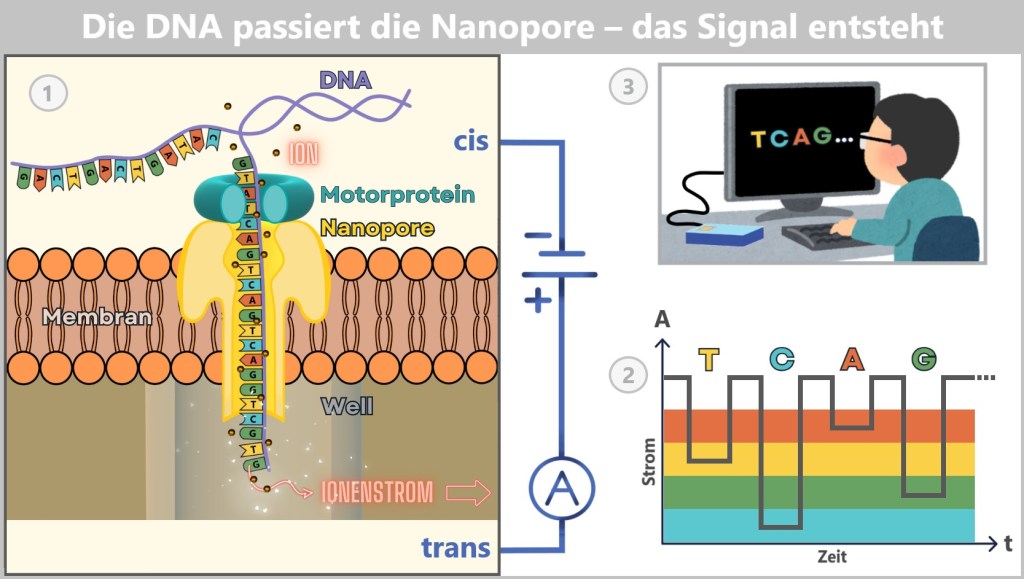
Abb. 9-B: Das Bild veranschaulicht den zentralen Mechanismus der Nanopore-Sequenzierung. 1) Hier sieht man eine schematische Darstellung der Nanopore in einer Membran. Ein Motorprotein zieht die DNA kontrolliert durch die Pore. Die negativ geladene DNA bewegt sich aufgrund der angelegten Spannung von der cis-Seite zur trans-Seite. Dabei beeinflussen die einzelnen Basen (A, T, G, C) den Ionenstrom auf spezifische Weise.
2) Ein Graph stellt die Veränderung des gemessenen Stroms über die Zeit dar. Jede Basen-Kombination erzeugt ein charakteristisches Signal, das durch Algorithmen entschlüsselt wird.
3) Ein Computer analysiert die elektrischen Signale und bestimmt daraus die Reihenfolge der Basen.Anders als bei PacBio werden bei ONT die Basen nicht einzeln unterschieden, sondern in Gruppen von fünf bis sechs Nukleotiden, die gleichzeitig in der Pore liegen. Jeder dieser „5-Mer“ erzeugt ein charakteristisches Stromsignal. Das erhöht die Lesegeschwindigkeit, macht die Interpretation aber komplexer – ein wenig wie das Erkennen von Wörtern in einem ungewohnten Dialekt. Besonders Homopolymeren (lange Wiederholungen derselben Base, z. B. „AAAAA“) stellen eine Herausforderung dar.
Die KI-gestützte Basecalling-Software verbessert sich kontinuierlich. Die Fehlerquoten sind heute drastisch gesunken, und ONT-Reads erreichen mittlerweile auch eine hohe Genauigkeit – besonders wenn die Datenmenge für eine statistische Mehrfachkontrolle jeder DNA-Base (durch wiederholte, zufällige Erfassung) hoch genug ist.
Eine coole visuelle Erklärung gibt’s im Video How nanopore sequencing works von Oxford Nanopore Technologies.
Wer es etwas ausführlicher mag, findet hier weitere Hintergrundinfos.
Besondere Stärken – Schnell, portabel, vielseitig
Was ONT besonders macht:
Mobilität: Geräte wie der MinION passen in eine Hosentasche. Sie benötigen nur einen Laptop und etwas Strom – ideal für Feldforschung, Raumfahrt oder schnelle Analysen bei Ausbrüchen (z. B. Ebola oder SARS-CoV-2).
Schnelligkeit: Die DNA wird live gelesen. Erste Sequenzdaten liegen oft schon nach wenigen Minuten vor – ein unschätzbarer Vorteil in klinischen Notfällen.
Direkte Epigenetik: Wie bei PacBio erkennt ONT Methylierungen und andere epigenetische Modifikationen direkt, da sie das elektrische Signal subtil beeinflussen – ganz ohne zusätzliche Behandlung oder Reagenzien.
Direkte RNA-Sequenzierung: Auch RNA-Moleküle können ohne vorherige Umwandlung in cDNA gelesen werden – ein echtes Alleinstellungsmerkmal.
Warum direkte RNA-Sequenzierung wichtig ist
Andere Sequenzierverfahren wie PacBio müssen mRNA erst in cDNA umwandeln, um sie lesen zu können. Das ist, als würde man ein Rezept zunächst abschreiben – dabei können kleine Details verloren gehen. Die Oxford-Nanopore-Technologie hingegen liest die mRNA direkt – ohne Umweg über DNA.
So bleiben alle „Randnotizen des Kochbuchs“ erhalten: chemische Modifikationen, Varianten und sogar die exakte Länge der Moleküle – Informationen, die bei einer Kopie oft verschwinden. Dadurch entsteht ein unverfälschtes Bild der Genaktivität – ein unschätzbarer Vorteil, um komplexe Abläufe wie Genregulation oder fehlerhafte RNA-Verarbeitung zu erkennen.
Gerade für die personalisierte Medizin ist das ein Durchbruch: Denn viele Krankheiten entstehen nicht allein durch veränderte DNA, sondern auch durch Abweichungen in der RNA.
Beispiele aus der Praxis:
- Krebsdiagnostik: RNA-Spleißvarianten können helfen, aggressive Tumorformen frühzeitig zu erkennen und gezielt zu behandeln.
- Erbkrankheiten: Bei Erkrankungen wie spinaler Muskelatrophie oder Duchenne-Muskeldystrophie ermöglichen RNA-Profile eine genaue Einschätzung des Verlaufs und personalisierte Therapien, z. B. mit Antisense-Oligonukleotiden.
- Infektionsmedizin: Die direkte Sequenzierung viraler RNA erlaubt eine schnelle Identifikation von Virusvarianten – entscheidend bei Epidemien und für die Wahl der Therapie.
Die direkte RNA-Sequenzierung hilft also, die persönliche Rezeptvariante jedes Menschen zu erkennen – mit all ihren kleinen Abweichungen, Randnotizen und besonderen Zutaten – und dadurch Diagnosen zu präzisieren sowie maßgeschneiderte Behandlungen zu ermöglichen.
Fazit: Der elektrische Sinnesfinger der Genomik
ONT ist der sinnesstarke Grenzgänger der Genomik: Schnell wie ein Rennpferd, portabel wie ein Schweizer Taschenmesser und nah dran am biologischen Geschehen. Wo andere kopieren müssen, fühlt es live – selbst bei RNA oder epigenetischen Spuren.
4.4.3. Genomsequenzierung – Technologien im Vergleich
In der Welt der Genomik gibt es nicht die eine Methode – sondern ein ganzes Ensemble. Jede Sequenzierplattform hat ihren eigenen Stil, ihre Stärken und ihr ideales Einsatzgebiet: vom klassischen Solo-Erzähler bis zum Hochgeschwindigkeitsstreamer.
Auch wenn alle vier Sequenzierer ihre Bühne haben – nicht jeder eignet sich für jede Inszenierung. Wer gezielt ein einzelnes Gen untersuchen will, braucht andere Mittel als jemand, der das gesamte Genom in epischer Breite erfassen möchte. Und wer seltene Varianten aufspüren will, muss genau hinlesen.
Betrachten wir die vier Hauptakteure im Detail, um ihre Stärken, Schwächen und strategischen Rollen zu verstehen.
a) Durchsatz vs. Verwertbarkeit – Masse ist nicht immer gleich Klasse
„Durchsatz“ klingt nach Effizienz – möglichst viele DNA-Buchstaben pro Minute. In der Sequenzierung misst man ihn in Basen pro Lauf: also wie viele Basen ein Gerät in einem einzigen Sequenzierprozess ausliest.
Moderne Plattformen wie Illuminas NovaSeq X Plus erreichen mit kurzen Reads (100–300 bp) Spitzenwerte von bis zu 16 Terabasen – das sind 16 Milliarden Buchstaben in einem einzigen Lauf. Wie ein Hochleistungsdrucker, der über Nacht ganze Genombibliotheken produziert.
PacBio und Oxford Nanopore verfolgen einen anderen Ansatz. Sie erzeugen lange Reads – von 10.000–25.000 bp bei PacBio HiFi-Reads, bis hin zu über 1 Million bp bei ONT. Ihr Durchsatz ist geringer: PacBio (Revio) erreicht bis zu 1.300 Gigabasen, ONT (PromethION) bis zu 7.000 Gigabasen pro Lauf. Klingt nach weniger – und das ist es auch, wenn man nur die Basenzahl betrachtet.
Doch: Mehr Daten bedeuten nicht zwangsläufig mehr Erkenntnis. Entscheidend ist nicht nur, wie viel gelesen wird – sondern wie präzise und wie oft. Zwei zentrale Konzepte helfen, die Aussagekraft von Sequenzdaten einzuordnen:
🔍 Quality Score – das Vertrauen in jeden Buchstaben
Der sogenannte Q-Score (Quality Score) beschreibt, wie wahrscheinlich ein gelesener DNA-Buchstabe korrekt ist. Die Skala ist logarithmisch:
- Q20 = 99 % Genauigkeit → 1 Fehler pro 100 Basen
- Q30 = 99,9 % Genauigkeit → 1 Fehler pro 1.000 Basen
- Q40 = 99,99 % Genauigkeit → 1 Fehler pro 10.000 Basen
In der Forschung ist häufig Q20 ausreichend. In der medizinischen Diagnostik gelten Q30 oder höher als Standard, bei Hochpräzisionsanwendungen (z. B. Tumorgenetik) wird Q40 erwartet.
Bei Rohdaten liegt Illumina typischerweise im Bereich von Q30–Q40, während PacBio (Continuous Long Reads, CLR) und ONT nur Werte zwischen Q10-Q15 aufweisen.
Sanger übertrifft alle – mit Q40–Q50, allerdings nur auf kurzen DNA-Abschnitten. Ideal zur gezielten Bestätigung einzelner Varianten.
Wie also erreichen moderne Sequenzierungsverfahren die hohen Anforderungen der klinischen Diagnostik? Die Antwort: durch Coverage und bioinformatische Analyse.
🔁 Coverage – die Wiederholung macht die Wahrheit
Coverage (Abdeckung) beschreibt, wie oft jeder Bereich des Genoms gelesen wurde.
Beispiel: 30x Coverage bedeutet, dass jeder Buchstabe im Durchschnitt 30-mal gelesen wurde – auf verschiedenen Fragmenten, in unterschiedlichen Konstellationen.
Warum ist das wichtig? Einzelmessungen können Fehler enthalten. Nur durch Wiederholung und statistische Mehrheitsbildung lassen sich echte Varianten von Artefakten unterscheiden.
In der medizinischen Diagnostik ist 30x Coverage der Mindeststandard.
Illumina erreicht bei 30–50x Coverage eine Gesamtgenauigkeit von Q35–Q40+. Es erkennt zuverlässigSingle Nucleotide Variants (SNVs), Insertions/Deletions (Indels) und Copy Number Variants (CNVs).
PacBio HiFi Reads erreichen bei 30x Coverage Q30–Q40 und ONT Q30-35. ONT benötigt 50x Coverage, um eine Präzision von Q44 zu erreichen.
Der große Vorteil von PacBio und ONT: Sie lesen wesentlich längere DNA-Abschnitte – oft 10.000 bis 100.000 Basen, bei ONT sogar über 1 Million. Dadurch erkennen sie komplexe Strukturen, Wiederholungen, epigenetische Signaturen und ermöglichen RNA-Untersuchung und die Phasierung – also die Zuordnung von Varianten zu väterlichen oder mütterlichen Chromosomen. Hier zählt nicht die Masse, sondern das Gesamtbild.
🧬 Je nach Ziel – unterschiedliche Anforderungen an Coverage und Genauigkeit
Die nötige Coverage und Sequenzierqualität hängen vom klinischen Einsatz ab:
Populationsstudien: Q30 bei 30x Coverage reicht meist aus, um statistische Muster zu erkennen.
Krebsdiagnostik: Q35 bei 60–100x zur sicheren Detektion seltener somatischer Mutationen.
Liquid Biopsies, Mosaik-Analysen: Q40 bei 100–300x – zur Unterscheidung von echten Signalvarianten vom Hintergrundrauschen.
Phasierung: Lange Reads (PacBio, ONT) mit 30x Coverage genügen oft, um Varianten zuverlässig zuzuordnen.
📉 Das Dilemma: Mehr Sicherheit – mehr Aufwand
Mehr Coverage erhöht die Sicherheit, bedeutet aber auch mehr Daten, längere Rechenzeiten, größere Infrastruktur – und höhere Kosten.
b) Wirtschaftlichkeit – Kosten pro Base vs. Kosten pro Erkenntnis
Illumina (NovaSeq/NextSeq) bleibt der Preis-Leistungs-Favorit für Whole-Genome-Sequencing:
👉 200–800 € für ein vollständiges Genom bei 30x-Abdeckung – präzise bei Punktmutationen, ideal für Routineanalysen.PacBio (Revio/Sequel IIe) und Oxford Nanopore (PromethION/MinION) sind teurer:
👉 700–1.200 € für 15–30x-Abdeckung – dafür mit zusätzlichen Informationen wie lange Reads, strukturelle Varianten, Epigenetik und RNA. Das ist kein Luxus, sondern in vielen Fällen diagnostisch entscheidend.Oxford Nanopore punktet zudem mit niedrigem Einstiegspreis: Ein MinION kostet wenige Tausend Euro, ist mobil und liefert Daten binnen Stunden.
👉 Die einfache Rechnung „mehr Basen = besserer Deal“ greift zu kurz. Entscheidend ist, was aus den Daten gelesen und verstanden werden kann.🧾 Versteckte Kosten – Das Genom ist günstiger zu lesen als zu verstehen
Ein menschliches Genom für unter 300 €? Technisch ja – aber meist sind nur Reagenzien, Maschinenzeit und Rohdaten abgedeckt.
Was oft fehlt, sind die unsichtbaren Posten:
🔧 Probenaufbereitung
🔍 Qualitätskontrolle & Library-Erstellung
🧠 Bioinformatik & klinische Interpretation
📄 Befundbericht & ärztliche RückspracheDiese Bereiche machen häufig 70–80 % der Gesamtkosten aus.
Denn: Lesen ist heute günstig – Verstehen bleibt anspruchsvoll.
c) Von der Probe zum Befund – Wie schnell spricht das ganze Genom?
Das Genomlesen ist wie eine Aufführung: Der Vorhang hebt sich mit der Probenentnahme – aber wann fällt der letzte Vorhang, der klinische Befund?
Es hängt von Technologie und Infrastruktur ab:
Oxford Nanopore ermöglicht vorläufige Ergebnisse binnen 24–48 Stunden – dank Echtzeit-Analyse während der Sequenzierung.
Illumina liefert mit optimierten Workflows Daten in 3–7 Tagen – der Goldstandard für routinierte Diagnostikabläufe.
PacBio benötigt 4–7 Tage, bietet dafür jedoch besonders hochwertige Langlesungen und epigenetische Informationen.
Sanger liefert Validierungen gezielt binnen Stunden – aber nur für kleine Zielregionen.
Die Plattformwahl ist also nicht nur eine Frage der Datenqualität – sondern auch des klinischen Timings.
Wer in Stunden statt Tagen handeln muss, greift zu Oxford Nanopore. Wer robuste Routinedaten braucht, setzt auf Illumina. Und wer maximale Kontexttiefe wünscht, nimmt sich mit PacBio etwas mehr Zeit – für deutlich mehr Einsicht.
Der Vorhang fällt. Doch hinter den Kulissen balanciert zwischen Hoffnung, Hightech und menschlicher Sorgfalt geht die Suche nach dem perfekten Tempo für jeden Patienten weiter. Denn am Ende zählt nicht die schnellste Maschine – sondern der richtige Takt für die menschliche Geschichte dahinter.
d) Klinische Einsatzfelder – Welche Technologie passt wozu?
Die Genomsequenzierung ist längst mehr als ein Forschungswerkzeug – sie ist zum zentralen Instrument geworden, um Therapien individuell zu planen. Doch welche Methode eignet sich für welche Aufgabe?
Ein Blick auf sechs zentrale Anwendungsfelder zeigt, wie diese Technologien die moderne Medizin prägen.
Anwendungsfeld Primäre Methode Ergänzende Methoden Routinediagnostik Illumina Sanger Pharmakogenomik Illumina Sanger Epigenetik PacBio, ONT – Krebsgenomik Illumina (SNVs), PacBio/ONT (SVs) Sanger Neonatale Notfalldiagnostik ONT Illumina, Sanger Seltene Erkrankungen PacBio, ONT Illumina, Sanger 👉 Erkenntnis: Die optimale Technologie hängt stets von der klinischen Fragestellung ab – Standardanalyse, Suche nach dem Unbekannten oder Notfalldiagnostik? Validierung oder Erstdiagnose?
e) KI als Co-Pilot – Automatisierung ohne Verantwortung abzugeben
Die Integration von Künstlicher Intelligenz (KI) – insbesondere Deep Learning – ist in allen modernen DNA-Sequenzierplattformen (Illumina, PacBio, Oxford Nanopore) unverzichtbar. Ihre Hauptanwendungsfelder sind:
🧬 Basecalling (Umwandlung von Rohdaten in DNA-Sequenzen) & Fehlerkorrektur
🔍 Qualitätskontrolle & Echtzeitanalyse
🧠 De-novo-Assembly, Genomanalyse & Variantenerkennung
📚 Pathogenitätsvorhersage, Literaturrecherche & Klinische Klassifikation
📄 Automatisierte Befundvorlagen👉 Erkenntnis: KI transformiert die Sequenzierung von einem reinen Datengenerierungs- zu einem intelligenten Interpretationsprozess – und ist damit Kern moderner genomischer Forschung und Diagnostik.
Doch wichtig bleibt: KI denkt (noch) nicht wie ein Arzt. Bei seltenen Erkrankungen, komplexen Verläufen oder neu entdeckten Genveränderungen bleibt die ärztliche Erfahrung zentral. Auch rechtlich ist eine ärztlich validierte Auswertung Pflicht – selbst im Zeitalter digitaler Genomik.
Am Ende ist Genomsequenzierung keine Solovorstellung – sie braucht das ganze Ensemble: Präzise Technik, kluge Bioinformatik und ärztliche Intuition. Denn nur wenn alle mitspielen, wird aus Daten heilsame Erkenntnis.
f) Vergleichstabelle: Sanger, Illumina, PacBio, ONT
Merkmal Sanger Illumina PacBio ONT Proben-vorbereitung DNA-Template, Extraktion,
Primer-Design, ddNTPs
🕒 ~6–12 Std.Fragmentierung,
Adapter-Ligation
🕒 ~6–24 Std.Hohe DNA-Qualität, Fragmentierung, SMRTbell-Adapter
🕒 ~1–2 TageFragmentierung,
Adapter mit Helikase (Motorprotein)
🕒 ~6–24 Std.Leselänge Kurz: 500–1.000 bp Sehr kurz: 100–300 bp (paired-end möglich) Lang: 10.000–25.000 bp (HiFi-Reads) Ultra-lang: 10.000 bp – >1 Mb Durchsatz Sehr niedrig: Einzelreaktionen (~1–100 kb/Lauf) Sehr hoch: bis 16.000 Gb (NovaSeq X Plus),
>100 Proben gleichzeitigMittel: bis 1.300 Gb / SMRT-Cell (Revio) Flexibel: 100–7.000 Gb (PromethION), bis 20 Gb (MinION) Genauigkeit Sehr hoch: Q40–Q50 (99,99–99,999 %) Hoch: Q30–Q40 (99,9–99,99 %), bei 30x: Q35–Q40+ Hoch: Q30–Q40 (HiFi-Reads), bei 30x Rohdaten: Q15–Q20+, bei 30x: Q30–Q35, Duplex: Q44 bei 50x Kosten/Genom (EUR) (nur Sequenzierung) – ~200–800 (30x) (Großlabore) ~700–1.200
(15–30x)~700–1.200
(15–30x)Zeit bis Ergebnis (WGS, Probe → Befund) – 3–7 Tage (inkl. Vorbereitung, Sequenzierung, Analyse) 4–7 Tage (inkl. Vorbereitung, Sequenzierung, Analyse) 24–48 Std.
(Echtzeit, inkl. Analyse)Varianten-erkennung SNVs, mitochondriale Varianten (Validierung) SNVs, Indels, CNVs (indirect), somatische Varianten, Splice-Varianten Indels, SVs, CNVs, Repeat-Expansionen, Splice-Varianten, somatische SVs Indels, SVs, CNVs, Repeat-Expansionen, Splice-Varianten, somatische SVs RNA-Sequenzierung – mRNA durch Umwandlung in cDNA, Splice-Varianten mRNA durch Umwandlung in cDNA, Splice-Varianten (lange Reads) Direkte RNA-Sequenzierung (ohne cDNA), Splice-Varianten, Langzeit-Transkripte Epigenetische Modifikationen – Indirekt: nur mit Bisulfit-Sequenzierung (aufwendig) Direkt: Methylierungserkennung durch Einbaukinetik (HiFi-Reads) & Bioinformatische Tools Direkt: Methylierung durch Signaländerungen (z. B. 5mC, 6mA) Einsatzort Labor: Stationäre Geräte Labor: Stationäre Großgeräte (z. B. NovaSeq) Labor: Stationäre Großgeräte (Revio) Flexibel: Labor (PromethION), mobil/Point-of-Care (MinION) Hauptvorteile Höchste Präzision, ideal zur Validierung Hoher Durchsatz, kostengünstig, präzise bei SNVs/Indels, routinetauglich Lange, präzise Reads (HiFi), ideal für SVs, Epigenetik, Phasierung Ultra-lange Reads, mobil, schnell, direkte RNA- und Epigenetik-Erkennung Limitationen Niedriger Durchsatz, WGS unpraktisch, teuer Kurze Reads, eingeschränkt bei SVs/Epigenetik, komplexe Library-Prep Teurer, mittlerer Durchsatz, längere Vorbereitungszeit Geringere Rohdatenqualität, abhängig von Bioinformatik Rolle in der personalisierten Medizin Validierung kritischer SNVs (Pharmakogenomik, Krebs), mitochondriale Analysen Routinediagnostik, Pharmakogenomik, Krebsgenomik (SNVs/Indels), seltene Erkrankungen Epigenetik, Krebsgenomik (SVs), seltene Erkrankungen (SVs, Repeat-Expansionen), Phasierung Neonatale Notfalldiagnostik, Epigenetik, Krebsgenomik (SVs), seltene Erkrankungen, mobile Diagnostik
g) Key Takeaways: Zwischen Reife und Routine
Die DNA-Sequenzierung befindet sich an einem entscheidenden Wendepunkt: In den vergangenen Jahren hat sie sich von einem rein forschungsbasierten Verfahren zu einem klinisch relevanten Werkzeug der modernen Medizin entwickelt. Ob in der Onkologie, der Diagnostik seltener Erkrankungen oder der Pharmakogenomik – Genomanalysen liefern zunehmend Antworten, wo konventionelle Methoden an ihre Grenzen stoßen.
Und doch: Trotz der beeindruckenden technischen Reife ist die Genomsequenzierung bislang kein flächendeckender Standard in der klinischen Praxis.
Technologie reif – Infrastruktur unvollständig
Moderne Hochdurchsatzverfahren wie Illumina oder die Langlesetechnologien von PacBio und Oxford Nanopore ermöglichen heute hochpräzise Analysen zu vergleichsweise geringen Kosten. Whole-Genome-Sequenzierungen (WGS) sind in spezialisierten Laboren für wenige Hundert Euro pro Fall durchführbar – mit einer Gesamtdauer von unter einer Woche. Mobile Plattformen wie MinION liefern in akuten Situationen – etwa bei schwer erkrankten Neugeborenen – erste genetische Hinweise sogar binnen 48 Stunden.
Trotz dieser Fortschritte bleibt der klinische Einsatz fragmentarisch. Gründe dafür sind vielfältig: Es mangelt an standardisierten bioinformatischen Auswertungsprotokollen, an interdisziplinär geschultem Fachpersonal sowie an einer rechtssicheren und wirtschaftlich tragfähigen Integration in den Versorgungsalltag. Vor allem die Interpretation genetischer Varianten – insbesondere solcher mit unklarer klinischer Bedeutung – erfordert spezialisiertes Know-how, das vielerorts nicht verfügbar ist.
Großbritannien als Blaupause
Während viele Länder noch mit Pilotprogrammen experimentieren oder Einzelfallentscheidungen treffen, institutionalisiert das Vereinigte Königreich die Genomsequenzierung als integralen Bestandteil der öffentlichen Gesundheitsvorsorge. Dort soll im Rahmen eines nationalen Pilotprogramms künftig jedes Neugeborene genomisch untersucht werden – ein Präzedenzfall für die pränatale und frühkindliche Vorsorgemedizin. Ziel ist es, genetisch bedingte Erkrankungen bereits im frühkindlichen Stadium zu erkennen – noch bevor klinische Symptome auftreten. Damit wird DNA-Analytik vom Einzelfall zur bevölkerungsweiten Präventionsstrategie – ein Paradigmenwechsel.
Dieses Modell demonstriert, dass die Hürde für eine breite Implementierung weniger technischer, sondern vor allem systemischer Natur ist: fehlende Datenstandards, ethische und rechtliche Unklarheiten sowie eine zurückhaltende Erstattungspolitik verhindern vielerorts eine flächendeckende Integration. Der britische Vorstoß gilt daher als wegweisendes Beispiel für die institutionalisierte Genommedizin.
Technologischer Ausblick
Mehrere technologische Entwicklungen dürften die Umsetzung in die Breite in naher Zukunft weiter beschleunigen:
🚀 Sinkende Kosten pro Genom machen Sequenzierung auch in kleineren Einrichtungen wirtschaftlich attraktiv.
🚀 Dezentrale, mobile Systeme wie MinION ermöglichen ortsunabhängige Diagnostik, etwa in Notaufnahmen oder ländlichen Regionen.
🚀 Schnellere Time-to-Result – von der Probenentnahme bis zur Befundung – erlaubt den Einsatz auch in zeitkritischen Fällen.
🚀 Die Integration von Genomdaten mit Transkriptomik, Proteomik und Epigenetik (Multi-Omics) verspricht eine kontextualisierte, hochauflösende Diagnostik.
🚀 KI-gestützte Analytik sowie automatisierte Interpretationstools senken die Abhängigkeit von manueller Auswertung und reduzieren Fehlerquellen.
4.5. Genomsequenzierung: Der nächste Technologiesprung
Damit die Entschlüsselung unserer DNA künftig schneller, günstiger und überall möglich wird, arbeitet die Forschung an der nächsten Generation von Sequenzierungstechnologien.
a) FENT: Wie ein Mikrochip die DNA-Analyse revolutioniert
b) SBX: Wenn DNA sich streckt, um verstanden zu werden
c) Das G4X-System: Vom genetischen Rezept zum räumlichen Atlas der Zelle
a) FENT: Wie ein Mikrochip die DNA-Analyse revolutioniert
– und für alle zugänglich macht
Angetrieben von der Vision, genetische Informationen in Echtzeit und patientennah erfassen zu können, entwickelt das Unternehmen iNanoBio ein neues Verfahren, das diese Idee auf ein völlig neues Niveau heben soll.
Es kombiniert die bewährte Methode – das Auslesen von DNA durch winzige Nanoporen – mit einem Bauteil aus der Mikroelektronik: dem MOSFET (Metal-Oxide-Semiconductor-Feldeffekttransistor), Herzstück moderner Computerchips.
Vom Computerchip zum DNA-Sensor
Ein MOSFET ist ein winziger Schalter mit drei Kontakten: Source (Eingang), Drain (Ausgang) und Gate (Steuerung). Liegt am Gate eine Spannung an, öffnet sich ein leitender Kanal zwischen Source und Drain – vergleichbar mit einem Türsteher, der ein Tor freigibt. Bereits minimale Spannungsänderungen beeinflussen den Stromfluss, was MOSFETs extrem empfindlich macht.
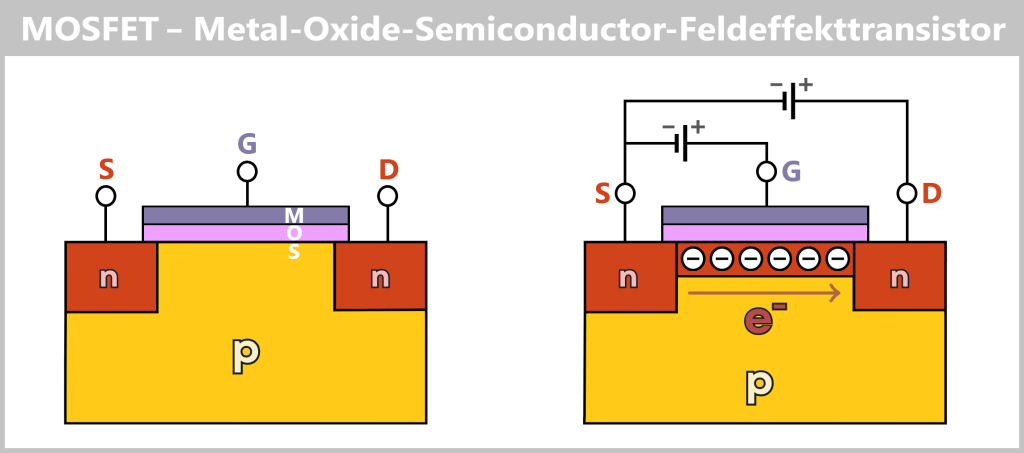
Abb. 10-A: Aufbau und Funktionsweise eines MOSFETs Ein MOSFET ist ein winziger Schalter aus der Mikroelektronik mit drei Kontakten: Source (Eingang), Drain (Ausgang) und Gate (Steuerung). Source und Drain sind im Halbleitermaterial verankert, das Gate ist durch eine hauchdünne Oxidschicht isoliert.
Ohne Spannung am Gate bleibt der Kanal zwischen Source und Drain geschlossen – kein Strom fließt. Legt man jedoch Spannung am Gate an, entsteht ein elektrisches Feld, das leitende Teilchen (z. B. Elektronen) anzieht und einen Stromkanal öffnet. Der MOSFET wirkt damit wie ein extrem sensibler Türsteher: Erst wenn er das Signal bekommt, öffnet er das Tor und lässt Strom passieren.Jetzt kommt der Clou: iNanoBio hat es geschafft, eine winzige Pore direkt in das empfindliche Steuerzentrum des MOSFETs zu integrieren. Wenn ein DNA-Strang hindurchgleitet, verändert jedes einzelne Nukleotid (A, T, C oder G) den Stromfluss zwischen Source und Drain charakteristisch. Das Ergebnis: ein neuartiger Sensor mit dem Namen FENT – kurz für Field-Effect Nanopore Transistor.
Der FENT ist zylindrisch aufgebaut und umschließt die Pore. Dadurch kann er Signale gleichzeitig aus mehreren Richtungen messen, was die Genauigkeit erhöht und Fehler reduziert.
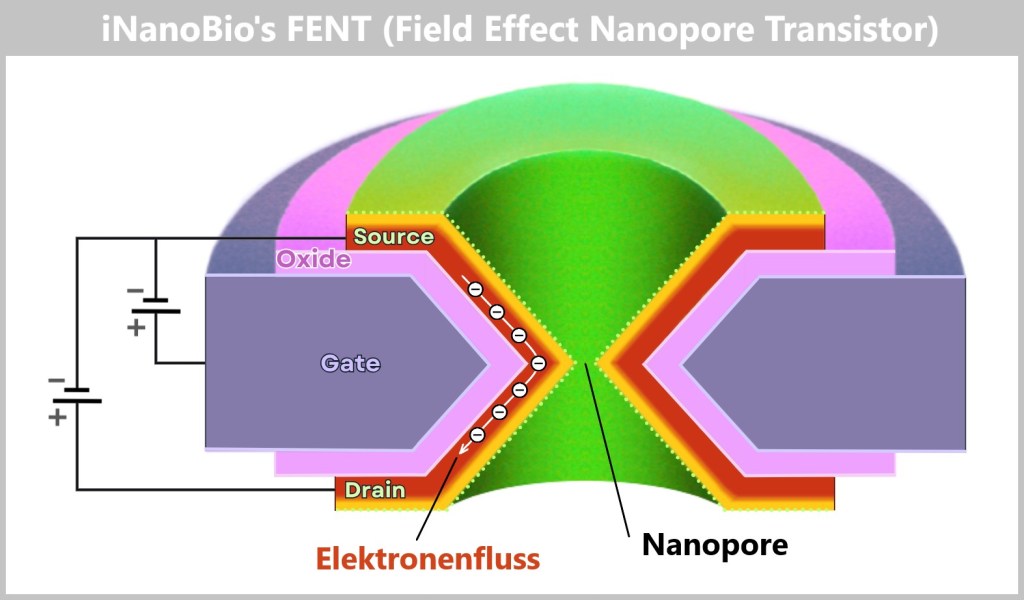
Abb. 10-B: Querschnitt eines FENTs Der FENT ist ein zylindrisch gebauter MOSFET, der eine Nanopore vollständig umschließt. Zwischen Source und Drain wird eine Spannung angelegt, sodass ein kleiner Strom durch den Halbleiter der Porenwand fließt – der sogenannte Source-Drain-Kanalstrom. Die dünne Oxidschicht isoliert das Gate vom Kanal und ermöglicht die präzise Erfassung elektrischer Feldänderungen. Die Nanopore dient als Sensorzone für lokale Feldänderungen, verursacht durch vorbeigleitende DNA-Basen.
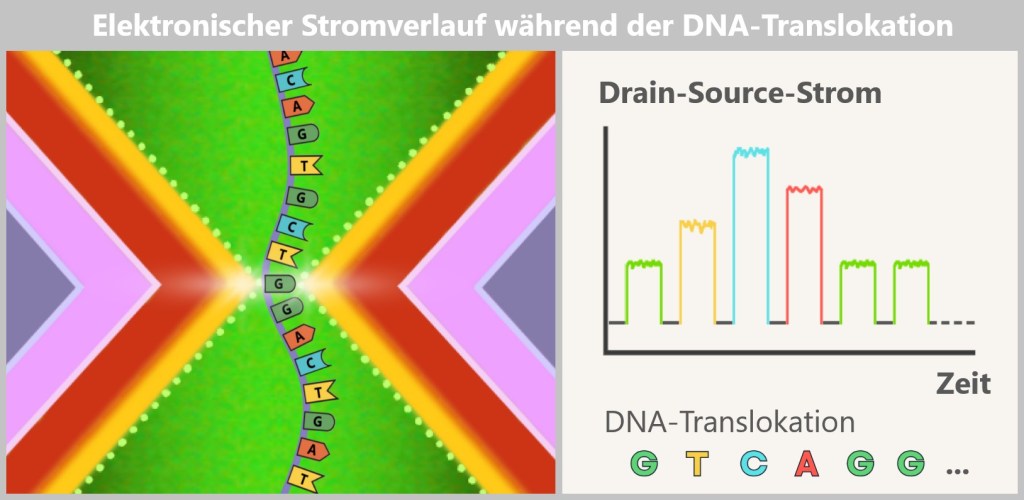
Abb. 10-C: FENT während der DNA-Translokation Die Abbildung zeigt das Funktionsprinzip eines FENT während der Passage einer DNA-Einzelstrangs (ssDNA) durch die Nanopore. Vor Beginn der Messung wird die ursprünglich doppelsträngige DNA (dsDNA) durch Hitze oder chemische Behandlung in ihre beiden Einzelstränge getrennt. Nur ein Einzelstrang wird anschließend durch die Nanopore geführt.
Im Ausgangszustand – also ohne DNA in der Pore – fließt zwischen Source und Drain ein konstanter Ruhestrom durch den Halbleiter, der die Porenwand bildet. Dieser Strom wird durch die Spannung zwischen Source und Drain sowie das Gate-Potenzial bestimmt.
Sobald jedoch eine DNA-Base (A, T, C oder G) die Nanopore passiert, beeinflusst sie das elektrische Verhalten des Systems:
Jede Base besitzt aufgrund ihrer spezifischen chemischen Struktur und Atomverteilung ein eigenes, winziges elektrisches Feld. Dieses Feld dringt durch die extrem dünne Porenwand und wirkt auf den Halbleiter wie ein lokales Gate-Signal. Dadurch verändert sich lokal die Ladungsträgerdichte (Elektronenkonzentration) im Transistorkanal, was zu einer messbaren Änderung des Drain-Source-Stroms führt.
Diese Stromänderungen werden als charakteristische Signale für die jeweilige Base aufgezeichnet. So entsteht während der Translokation eine sequenzabhängige Stromspur, die es ermöglicht, die Reihenfolge der DNA-Basen elektronisch zu bestimmen.Eine allgemeinverständliche Erklärung liefert das Video FENT Nanopore Transistor Explainer Video.
Geschwindigkeit, die Maßstäbe setzt
Laut iNanoBio kann ein FENT bis zu 1 Million DNA-Basen pro Sekunde und Pore auslesen – etwa 100-mal schneller als bisherige Nanopore-Technik. Ein 5 x 5 mm großer Chip mit 100.000 dieser Sensoren könnte ein menschliches Genom in wenigen Minuten sequenzieren. Die Kosten wären deutlich geringer, die Fehlerrate niedriger.
Mögliche Anwendungen
- Krebsfrüherkennung aus Blutproben
- Mobile Diagnostik für Infektionen direkt vor Ort
- Großstudien in der Forschung dank hoher Geschwindigkeit
Langfristig will iNanoBio DNA-Analysegeräte so verbreiten wie heutige Blutdruckmessgeräte – verfügbar in Kliniken, Laboren, Arztpraxen und möglicherweise auch zu Hause. Erste Prototypen existieren, der Weg zur Massenproduktion läuft.
Wenn die Technologie hält, was sie verspricht, könnte die Genomsequenzierung bald so selbstverständlich sein wie eine Routineuntersuchung – und unser Verständnis von Gesundheit grundlegend verändern.
b) SBX: Wenn DNA sich streckt, um verstanden zu werden
Im Wettlauf um die Verbesserung der DNA-Analyse verfolgt der Pharmakonzern Roche mit SBX (Sequencing By eXpansion) einen verblüffend einfachen Ansatz: Er macht DNA künstlich größer, damit ihre „Buchstaben“ A, T, C und G leichter auseinanderzuhalten sind.
Schritt 1 – Vom Original zur vergrößerten Kopie
SBX erstellt aus der Original-DNA eine Surrogat-Kopie, den Xpandomer. Dafür wird das DNA-Fragment an einem winzigen Anker auf einem Substrat fixiert, und mithilfe einer Polymerase wird eine Kopie erstellt – allerdings nicht aus natürlichen Bausteinen, sondern aus Surrogat-Nukleotiden.
Ein Surrogat (von lateinisch surrogatum = „Ersatz“) bezeichnet allgemein einen Ersatz oder Stellvertreter für etwas anderes.
Diese bestehen aus:
- der passenden Base (A, T, C oder G),
- einem für jede Base spezifischen gefalteten Verlängerungsfaden als Signalgeber,
- einer spaltbaren Bindung, die den Faden in Form hält,
- einem Translocation Control Element (TCE), das den Ablauf später präzise steuert.
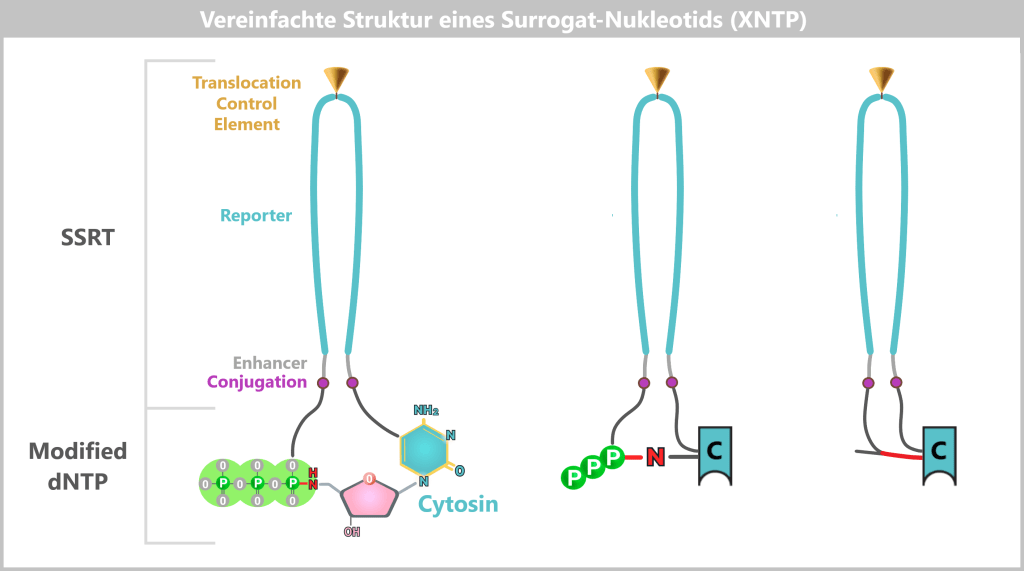
Abb. 11-A: Vereinfachte Darstellung eines Surrogat-Nukleotids
[Sequencing by expansion (SBX) technology]Das Surrogat-Nukleotid (XNTP) besteht aus zwei funktionellen Teilen: dem modifizierten Nukleotid und dem SSRT (symmetrically synthesized reporter tether).
Das modifizierte dNTP enthält die eigentliche Base (hier beispielhaft Cytosin, C), die über eine säureempfindliche Bindung (rot markiert) mit dem Reporterfaden verbunden ist.
Daran schließt sich das Triphosphat (PPP) an, das die Einbindung des Nukleotids in die wachsende Kette durch die Polymerase ermöglicht.
Der Reporterfaden (SSRT) besteht aus einem gefalteten Verlängerungsfaden mit einem Translocation Control Element (TCE) an der Spitze, das den Durchtritt durch die Nanopore später steuert.
Ein Enhancer-Bereich (grau/lila) erleichtert die Einbindung durch die Polymerase und stabilisiert die Struktur.
Nach der Synthese kann die rote, säureempfindliche Bindung gelöst werden – dadurch „entfaltet“ sich der Reporterfaden und erzeugt beim Sequenzieren das ausgedehnte Xpandomer-Molekül.
Abb. 11-B: Synthese des Xpandomers [Sequencing By Expansion] Das DNA-Fragment (Template DNA) ist über einen Anker am festen Substrat fixiert. Ein Primer bindet an die DNA-Vorlage, unterstützt vom Leader und Concentrator, die den Startpunkt stabilisieren und die Positionierung erleichtern.
Fixierung – Die DNA-Vorlage ist am Substrat verankert, der Primer-Abschnitt hybridisiert an den passenden Bereich der DNA.
Verlängerung – Eine spezielle Polymerase ergänzt die Basen der Vorlage – aber nicht mit natürlichen Nukleotiden, sondern mit Surrogat-Nukleotiden (XNTPs), die jeweils einen Reporterfaden mitführen.
Xpandomer entsteht – Am Ende liegt ein künstlich erweiterter DNA-Strang vor: eine Surrogat-Kopie (Xpandomer), deren Basen durch charakteristische Reporterfäden ersetzt sind.
Der Xpandomer bleibt zunächst am Substrat fixiert und wird in einem späteren Schritt durch Licht oder chemische Spaltung freigesetzt.Nach der Synthese wird der Xpandomer von der Original-DNA gelöst, die spaltbaren Bindungen werden getrennt – die Fäden entfalten sich und vergrößern den Abstand zwischen den Basen um das 50-Fache. Das erleichtert die spätere Identifikation erheblich.

Abb. 11-C: Expansion und Freisetzung des Xpandomers [Sequencing By Expansion] Nach der Synthese wird der Xpandomer auf dem Substrat chemisch entfaltet („expandiert“) und anschließend freigesetzt.
Links) Expansion: Eine milde Säurebehandlung löst die spaltbaren Bindungen (rot) zwischen Base und Reporterfaden. Dadurch entfalten sich die Reporterstrukturen – aus der kompakten Doppelstruktur wird ein lang gestreckter Xpandomer, der die Abfolge der Basen repräsentiert.
Rechts) Freisetzung: Ein UV-Lichtimpuls spaltet die Verbindung zwischen dem Substrat und dem Leader-Bereich.
Der nun frei bewegliche Xpandomer bleibt über Leader und Concentrator strukturell markiert – diese Bereiche helfen später bei der Ausrichtung und beim Einzug in die Nanopore.
Aus einer dichten DNA-Vorlage entsteht ein langes, lesbares Molekül, dessen Reporterabschnitte die genetische Information elektrisch sichtbar machen.Schritt 2 – Die Sequenzierung
Anstelle der DNA selbst wird nun der Xpandomer durch Nanoporen auf einem Sequenzierungschip gezogen – rund acht Millionen davon arbeiten parallel. Der Adapter des Xpandomers führt ihn gezielt in eine Pore.
Wenn ein Verlängerungsfaden die Pore erreicht, bleibt er kurz am TCE hängen. Jede Base erzeugt dabei ein spezifisches elektrisches Signal. Nach 1,5–2 Millisekunden löst ein Spannungspuls das TCE, der nächste Faden folgt. So entsteht eine klare, gut unterscheidbare Signalkurve – präziser als bei Oxford Nanopore, wo die Basen ein eher „verwaschenes“ Signal erzeugen.

Abb. 11-D: Sequenzierung des Xpandomers in der Nanopore
[Sequencing By Expansion, Introduction to Sequencing By Expansion]Der freigesetzte Xpandomer wird durch ein elektrisches Feld zur Nanopore gezogen. Dort kontrolliert das TCE (Transient Capture Element) den Eintritt und hält die Reporter kurzzeitig fest.
Während jeder Reporterabschnitt (die verlängerte „Base“) die Pore passiert, ändert sich der Ionenstrom in charakteristischer Weise: Jede Base erzeugt dabei ein eigenes elektrisches Signal – vergleichbar mit einem individuellen Barcode. Diese Signalfolge wird in Echtzeit aufgezeichnet:
Ein Spannungspuls löst nach jeder Messung den Faden vom TCE, sodass der nächste Abschnitt eingezogen wird.Folgende Animation veranschaulicht die Grundprinzipien der SBX-Technologie von Roche.
Eine ausführliche Präsentation gibt es hier.
Vorteile und Leistung
- Geschwindigkeit: Millionen Xpandomer werden gleichzeitig und in Echtzeit gelesen.
- Genauigkeit: Signal-Rausch-Verhältnis ähnlich hoch wie bei Illumina.
- Flexibilität: Leselängen bis über 1.000 Basen möglich – schneller und präziser als Nanopore, länger als Illumina.
Erste Tests zeigen: SBX kann bis zu sieben vollständige menschliche Genome pro Stunde mit 30x Abdeckung entschlüsseln. Bei dringenden Fällen dauert der gesamte Ablauf – von der Probenvorbereitung bis zur fertigen Analyse – nur rund 5,5 Stunden.
Die Markteinführung ist für 2026 geplant. Ob sich SBX gegen Illumina und Oxford Nanopore durchsetzen wird, hängt von Preis, Verfügbarkeit und den Anforderungen der Anwender ab.
c) Das G4X-System: Vom genetischen Rezept zum räumlichen Atlas der Zelle
Unser Körper besteht aus Milliarden Zellen, die wie winzige Küchen arbeiten. Die DNA ist ihr Kochbuch – sie enthält alle Rezepte des Lebens. Wird ein Rezept gebraucht, erstellt die Zelle eine RNA-Kopie, die als Bauanleitung für Proteine dient. Diese Proteine übernehmen konkrete Aufgaben: Sie bauen Strukturen, transportieren Signale oder bekämpfen Krankheitserreger.
Proteine wirken jedoch selten allein. Sie arbeiten in fein abgestimmten Netzwerken, deren Aktivität davon abhängt, wo, wann und mit welchen Partnern sie interagieren. Prozesse wie Immunreaktionen oder Tumorwachstum entstehen nicht in einer einzelnen Zelle, sondern aus dem Zusammenspiel vieler spezialisierter Zellen – oft in unterschiedlichen Bereichen eines Gewebes. Biologie ist also nicht nur biochemisch, sondern auch räumlich organisiert. Klassische Sequenzierungsmethoden erfassen jedoch meist nur die genetischen Inhalte, nicht deren räumliche Verteilung.
Genaktivität direkt im Gewebe
Der G4X Spatial Sequencer von Singular Genomics macht sichtbar, wo im Gewebe Gene aktiv sind und Proteine gebildet werden – direkt in konservierten Gewebeproben (FFPE, formalinfixiert und paraffineingebettet).
FFPE-Proben werden häufig in der Pathologie eingesetzt, etwa zur Krebsdiagnostik. Sie wirken wie eine „eingefrorene Momentaufnahme“, nur dass hier chemische Fixierung mit Formalin statt Kälte verwendet wird. Formalin vernetzt Proteine und Nukleinsäuren miteinander und fixiert so die molekularen Strukturen räumlich.
Was dabei erhalten bleibt:
- RNA (oft fragmentiert und chemisch modifiziert) – spiegelt die Genaktivität zum Fixierzeitpunkt wider.
- Proteine – zeigen, welche Strukturen und Signalwege in diesem Moment aktiv waren.
So lässt sich Monate oder Jahre später rekonstruieren, welche Gene und Proteine zu diesem exakten Zeitpunkt im Gewebe präsent und aktiv waren – als hätte man die Zeit angehalten. Mit dem G4X kann diese „eingefrorene“ molekulare Landschaft hochaufgelöst sichtbar gemacht werden – inklusive der exakten Position im Gewebe.
Abb. 12-A: Vom Gewebe zur Genaktivität Ein Gewebe besteht aus vielen spezialisierten Zellen. Jede Zelle in einem Gewebe nutzt nur einen Teil ihres genetischen Programms. RNA macht die aktiven Gene sichtbar. RNA-Moleküle werden von Ribosomen abgelesen und in Proteine übersetzt. FFPE-Gewebeschnitte bewahren diese räumliche Anordnung – die Basis, um mit dem G4X-System zu messen, wo welche RNA aktiv ist.
Wie der G4X funktioniert
Man kann sich den G4X wie einen Mikroskop-Scanner mit eingebautem Labor vorstellen, der direkt im Gewebe misst, welche Gene und Proteine aktiv sind – und zwar in ihrem räumlichen Kontext.
Präparation: Ein sehr dünner Schnitt der FFPE-Probe wird auf ein spezielles Trägermaterial gebracht.
RNA-Nachweis: Um die Verteilung bestimmter RNA-Sequenzen im Gewebe zu erkennen, kommen Padlock-Sonden zum Einsatz.
Padlock-Sonden – DNA-Lassos für Zielsequenzen
Stellen Sie sich vor, Sie möchten in einem riesigen Gelände (dem Gewebe) nur ein bestimmtes Objekt (eine RNA-Sequenz) finden. Eine Padlock-Sonde ist ein DNA-Fragment, dessen beide Enden komplementär zu benachbarten Abschnitten der Zielsequenz passen.

Abb. 12-B: Eine Padlock-Sonde schwebt durch das Gewebe auf der Suche nach ihrer Ziel-RNA. Padlock-Sonde: Einzelsträngige DNA (ssDNA) 5’– Arm1 – Linker/Barcode – Arm2 –3′
Arm 1 und Arm 2 sind komplementär zu benachbarten Sequenzen der Ziel-RNA.
Dazwischen liegen Linker und Barcode, die zusätzliche Identifikationsinformationen tragen und nicht binden.Bindet die Sonde an ihr Ziel, liegen ihre beiden Enden nebeneinander und können durch Ligation zu einem stabilen DNA-Ring geschlossen werden.
Abb. 12-C: Ligation verbindet die Enden der Padlock-Sonde Nachdem beide Arme der Sonde an die Ziel-RNA gebunden haben, verbindet eine Ligase die 5′- und 3′-Enden. Dadurch entsteht ein geschlossener DNA-Ring, der die Ziel-RNA markiert und für die Amplifikation bereit ist.
Liegt zwischen den Bindungsstellen eine variable Sequenz (z. B. eine Mutation), wird diese zuvor mithilfe einer Reverse Transkriptase in DNA umgeschrieben und in die Sonde integriert.
Der entstandene DNA-Kreis dient als Vorlage für die Rolling Circle Amplification (RCA):
- Eine DNA-Polymerase läuft immer wieder im Kreis und erzeugt einen langen DNA-Strang, der die Zielsequenz vielfach wiederholt – wie ein Endlosband oder ein Garnknäuel.
- Diese Signale bleiben exakt an der Stelle im Gewebe, wo die RNA war.
- Mit fluoreszenzmarkierten Bausteinen und Sequenzieren durch Synthese wird die Sequenz direkt im Gewebe entschlüsselt.
Jede Padlock-Sonde trägt einen Barcode, der verrät, welches Gen oder welche Variante sie erkannt hat. Das Ergebnis: eine präzise Karte, die zeigt, welche RNA an welchem Ort vorhanden war und welche Varianten sie trug.
Abb. 12-D: Rolling Circle Amplification (RCA) Eine spezielle DNA-Polymerase liest die zirkuläre Padlock-Sonde mehrfach ab und erzeugt einen langen DNA-Strang mit vielen Wiederholungen. Fluoreszente Nukleotide markieren die Amplifikate. Dadurch entsteht ein starkes, fluoreszierendes Signal – sichtbar als leuchtender Punkt im Gewebe, der die Position der gesuchten RNA markiert.
Proteinnachweis im selben Gewebeschnitt
Für den Nachweis von Proteinen nutzt das G4X-System Antikörper, die mit kurzen DNA-Barcodes versehen sind. Nachdem der Antikörper an sein Zielprotein gebunden hat, hybridisiert eine Padlock-Sonde an diesen Barcode. Sie wird geschlossen, amplifiziert und ausgelesen – ganz wie bei der RNA-Analyse. Der Barcode verrät eindeutig, um welches Protein es sich handelt und an welcher Stelle im Gewebe es vorkommt.
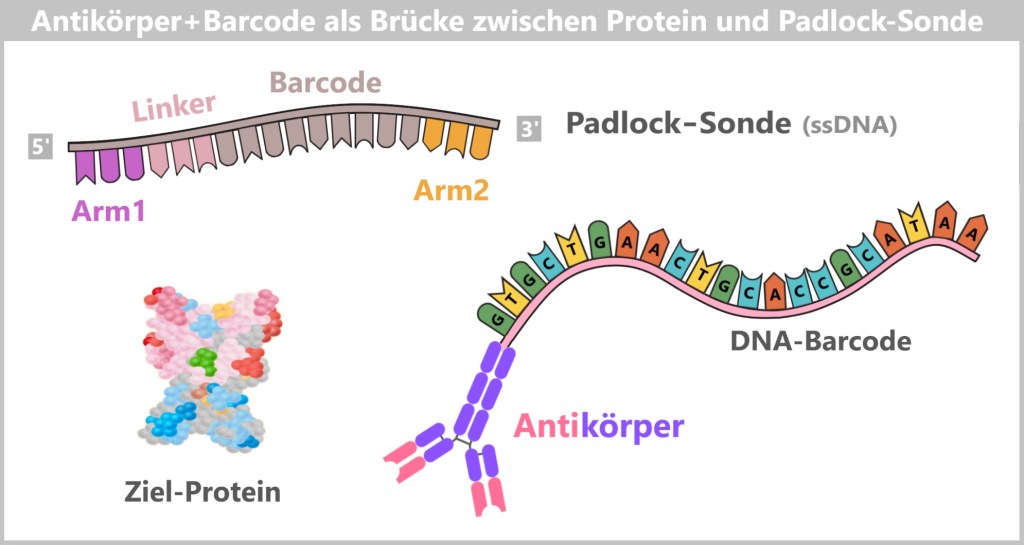
Abb. 12-E: Protein-Nachweis im G4X-System: Antikörper mit DNA-Barcode und Padlock-Sonde Ein Antikörper, der spezifisch für ein Zielprotein ist, wird mit einem einzigartigen, kurzen einzelsträngigen DNA-Strang (Barcode) gekoppelt.

Abb. 12-F: Protein-Nachweis im G4X-System Der Antikörper bindet in der Gewebeprobe an sein Zielprotein. Eine Padlock-Sonde, deren Enden komplementär zu zwei benachbarten Abschnitten des DNA-Barcodes sind, bindet an diesen und wird durch eine DNA-Ligase zu einem geschlossenen Ring verknüpft. Eine DNA-Polymerase erkennt den Ring und liest ihn fortlaufend ab, wodurch ein lokal verankertes, zirkuläres DNA-Amplifikat entsteht – analog zum RNA-Nachweis. Durch den Einbau fluoreszenzmarkierter Nukleotide wird dieses Amplifikat sichtbar. Die leuchtenden Signalpunkte im Gewebe markieren die Positionen, an denen ein Antikörper mit einem spezifischen DNA-Barcode ein bestimmtes Protein gebunden hat.
Eine ausführliche Präsentation des G4X-Veerfahrens findet man hier und hier.
Warum das wichtig ist
Der G4X kann gezielt Biomarker nachweisen, die Aufschluss über das Vorliegen, den Verlauf oder die Aggressivität einer Erkrankung geben – und zeigt dabei nicht nur, welche aktiv sind, sondern auch wo sie im Gewebe auftreten.
Gerade bei Tumoren macht diese räumliche Auflösung sichtbar, welche Gene „angeschaltet“ sind, in welchen Zellbereichen das geschieht und wie Tumor- und Immunzellen interagieren. So lassen sich Tumorheterogenität besser verstehen, Krankheitsmechanismen aufklären und Therapien gezielter planen.
Die gleichzeitige in-situ Erfassung von RNA und Proteinen liefert ein umfassendes Bild der Gewebeorganisation und eröffnet neue Möglichkeiten für:
- das Verständnis komplexer Krankheitsprozesse
- die präzisere Identifikation von Biomarkern
- die Entwicklung personalisierter Therapien
Bei Erkrankungen wie Krebs, chronischen Entzündungen oder neurodegenerativen Störungen kann diese räumliche Multiomics-Perspektive entscheidend sein – denn oft bestimmt nicht das Ob, sondern das Wo und Wie die beste therapeutische Maßnahme.
Fazit
Die Genomsequenzierung steht vor einem Quantensprung: Technologien wie FENT, SBX und G4X versprechen, das Erbgut schneller, präziser und vielseitiger zu lesen als je zuvor – bis hin zur direkten Analyse im Gewebe. Bald könnten komplette Genome in Minuten entschlüsselt werden. Die wahre Revolution beginnt jedoch danach: wenn wir die Fülle an genetischen Informationen verstehen, in medizinisches Wissen übersetzen und verantwortungsvoll zum Wohl der Menschen einsetzen.
4.6. Vom Code zur Heilung
Bioinformatik als Schlüssel der Medizin von morgen
4.6.1. Vom genetischen Code zur computergestützten Genomanalyse
Noch nie war es so leicht, DNA zu entschlüsseln – doch was nützt der Text, wenn niemand ihn versteht? Ein kleiner Abstrich, ein Tropfen Blut – und schon liegen Millionen genetischer Datenpunkte vor. Die moderne Sequenzierung macht das Erbgut so zugänglich wie nie zuvor: schnell, kostengünstig und nahezu fehlerfrei. Daraus erwächst jedoch eine neue Herausforderung: Wie verwandelt man diese Datenflut in verwertbares medizinisches Wissen?
Die Antwort kommt aus einer Disziplin, die lange im Hintergrund agierte und heute unverzichtbar geworden ist: Bioinformatik – die Brücke zwischen A, T, G, C und der Diagnose, zwischen rohen Sequenzdaten und einer konkreten Therapieentscheidung. Um zu verstehen, warum sie so zentral ist, lohnt ein kurzer Blick zurück.
Vom Erbgesetz zum genetischen Code
Bereits 1860 entdeckte Gregor Mendel, dass es „vererbbare Einheiten“ geben muss – Gene, lange bevor DNA bekannt war. Erst Mitte des 20. Jahrhunderts zeigte Oswald Avery, dass diese Informationsträger tatsächlich DNA sind. Kurz darauf enthüllten Watson und Crick die Doppelhelix-Struktur mit ihrer komplementären Basenpaarung (A–T, G–C). 1958 formulierte Francis Crick das „Zentrale Dogma“: Information fließt von DNA über RNA zu Proteinen – den funktionellen Bausteinen des Lebens.
Abb. 13-A: Das zentrale Dogma der Molekularbiologie: Die genetische Information wird durch den Prozess der Transkription von der DNA in RNA umgewandelt. Die RNA wird dann in das endgültige Protein übersetzt, das eine Vielzahl von Funktionen hat. [An overview of artificial intelligence in the field of genomics]
Ein Gen ist also eine Anleitung, wie ein Protein hergestellt wird. Doch wie liest man diese Anleitung – und wie interpretiert man sie richtig? Die Antwort kam zunächst über den Umweg der Reverse Genetik.
In den 1960er Jahren war DNA noch schwer direkt zugänglich – Proteine dagegen waren funktional sichtbar (etwa als Enzyme, Transportmoleküle oder Strukturbausteine) und konnten mit den damaligen Methoden isoliert und analysiert werden. Forscher wussten bereits seit Anfang des 20. Jahrhunderts, dass Proteine aus Aminosäureketten bestehen. Ab den 1960er Jahren wurde klar, dass jede Aminosäure einem sogenannten Codon auf der RNA entspricht – also einer Dreierkombination aus RNA-Basen (A, U, G, C), die wiederum auf eine DNA-Sequenz zurückführbar ist.
Abb. 13-B: Das zentrale Dogma der Molekularbiologie: Der DNA-Code und Codons Forscher starteten bei einem bekannten Protein und arbeiteten sich zur RNA- und DNA-Sequenz zurück. Der genetische Code wurde zum ersten Mal wie ein Rezeptbuch lesbar:
Protein = fertiges Gericht
Aminosäure = Zutat
Codon = Wort für diese Zutat im handgeschriebenen Rezept (RNA)
DNA = Seite im Kochbuch, von der das Rezept abgeschrieben wurdeErste Muster und die Geburt der Genomannotation
Noch vor der eigentlichen DNA-Sequenzierung entdeckte man typische Strukturelemente in Genen:
Promotoren – Startsignal, an dem das „Lesen“ eines Gens beginnt.
Start- und Stopcodons – erstes und letztes Signal im Rezept.
Transkriptionsfaktoren – Proteine, die Gene ein- oder ausschalten.
Codierende Sequenzen – Zutatenliste (Codons) und deren Reihenfolge für das Protein.
Nicht-codierende DNA – Abschnitte ohne direkte Protein-Codierung, deren Funktion zunächst unklar war.Diese Erkenntnisse legten den Grundstein der heutigen Genomannotation – der computergestützten Erkennung biologischer Signale in DNA-Sequenzen.
Der Sanger-Moment: DNA wird lesbar (ab 1977)
Mit Frederick Sangers Methode konnten DNA-Sequenzen erstmals direkt bestimmt werden – zunächst nur für kurze Abschnitte. Die Analyse konzentrierte sich auf die Identifikation von Genen und einfache Vergleiche. Erste Programme wie FASTA (1985) und BLAST (1990) ermöglichten den Abgleich neuer Sequenzen mit Datenbanken wie GenBank (1982).
Auch das Verständnis der Genstruktur entwickelte sich weiter: Man entdeckte, dass nur bestimmte Teile eines Gens (Exons) für die Proteinsynthese genutzt werden, während andere Abschnitte (Introns) aus der transkribierten RNA herausgeschnitten werden. Zudem identifizierte man DNA-Abschnitte, die die Genexpression verstärken (Enhancer) oder abschwächen (Silencer) – oft weit entfernt vom eigentlichen Gen.
Der Prozess der Genomannotation begann sich zu entwickeln, war jedoch noch rudimentär: DNA wurde hauptsächlich als lineare Abfolge von Nukleotiden betrachtet, mit Fokus auf proteinkodierende Gene. Regulatorische Elemente und nicht-codierende DNA wurden erst ansatzweise erkannt und verstanden. Die Bioinformatik war geboren – zunächst als Werkzeug für Forschungslabore.
Das Humangenomprojekt: Daten im Gigantenmaßstab (1990–2004)
Das Ziel, alle 3,2 Milliarden Basen des menschlichen Genoms zu entschlüsseln, brachte völlig neue Probleme: Millionen kurzer Sequenzfragmente mussten zu langen DNA-Strängen zusammengesetzt werden. Tools wie Phrap für die Assemblierung und später Ensembl für die Annotation wurden entwickelt. Die Datenmengen explodierten – und mit ihnen die Notwendigkeit leistungsfähiger Rechnerinfrastruktur.
Doch während das Humangenomprojekt noch lief, zeichnete sich bereits eine neue Technologie ab, die Geschwindigkeit und Datenmenge in völlig neue Dimensionen katapultieren sollte.
NGS-Revolution und klinischer Durchbruch (2005–2015)
Die Next-Generation Sequencing (NGS)-Technologien machten Genome günstiger und schneller zugänglich – allerdings als gewaltige Datenlawinen aus Milliarden kurzer Fragmente. Neue Werkzeuge wie Bowtie, BWA oder STAR ordneten diese blitzschnell zu, GATK half, Mutationen zu erkennen, und Tools wie SnpEff oder VEP interpretierten deren Bedeutung.
Gleichzeitig entstanden Sequenzierstrategien für unterschiedliche klinische Fragen:
- Whole Genome Sequencing (WGS) für vollständige Analysen,
- Whole Exome Sequencing (WES) für die 1–2 % proteinkodierender DNA,
- Targeted Sequencing für definierte Genpanels.
Damit wurde die Bioinformatik erstmal zu einem Diagnosetool – etwa bei erblichen Krebsformen, seltenen Erkrankungen oder personalisierter Pharmakotherapie.
Künstliche Intelligenz und moderne Bioinformatik (2015-heute)
Die Verbindung von Künstlicher Intelligenz (KI) und Bioinformatik stellt einen der dynamischsten und vielversprechendsten Forschungsbereiche unserer Zeit dar. Gemeinsam entschlüsseln sie die Komplexität des Lebens und beschleunigen biomedizinische Entdeckungen in nie dagewesenem Tempo.
Künstliche Intelligenz: Mehr als nur Automatisierung
KI umfasst Techniken, bei denen Maschinen menschenähnliche Intelligenzleistungen erbringen – insbesondere Lernen, Problemlösen und Mustererkennung. Im biomedizinischen Kontext sind vor allem folgende Teilbereiche relevant:Maschinelles Lernen (ML): Algorithmen lernen aus Daten (z.B. Genomsequenzen, Proteinstrukturen, Patientendaten), um Vorhersagen zu treffen oder Muster zu erkennen, ohne explizit programmiert zu sein. Beispiel: Erkennung von Tumorzellen in Gewebeproben.
Deep Learning (DL): Eine Untergruppe des ML, die künstliche neuronale Netze mit vielen Schichten („deep“) nutzt, um hochkomplexe Muster in riesigen Datenmengen zu erfassen. Beispiel: Vorhersage der 3D-Struktur von Proteinen allein aus ihrer Aminosäuresequenz (AlphaFold).
Natürliche Sprachverarbeitung (NLP): Ermöglicht Computern, wissenschaftliche Literatur, Krankenakten oder klinische Studienberichte zu verstehen und daraus Wissen zu extrahieren.
4.6.2. Moderne Bioinformatik – Der digitale Werkzeugkasten der Biologie
Bioinformatik ist die Wissenschaft der Speicherung, Analyse und Interpretation biologischer Daten mit computergestützten Methoden. Ihre moderne Ausprägung ist durch drei Faktoren geprägt:
Explosion biologischer Daten: Next-Generation Sequencing (NGS) generiert Terabytes an Genom-, Transkriptom- und Epigenomdaten pro Experiment.

Abb. 12: Vom Milliardenprojekt zur Alltagsroutine – wie die Kosten der Genomsequenzierung fielen und die Datenmengen explodierten
Vor gut zwanzig Jahren war das Entschlüsseln eines menschlichen Genoms noch ein gigantisches Unterfangen: Das Human Genome Project (2003) kostete rund 3 Milliarden US-Dollar und lieferte nur etwa 1 Gigabyte an Daten. Mit immer schnelleren Sequenziermaschinen und neuen Technologien wie Nanoporen-Sequenzierung hat sich das Bild völlig verändert. 2012 lag der Preis pro Genom bereits bei rund 6000 Dollar, 2018 bei 1000 Dollar – und heute (2023) kann man ein Genom für etwa 100 Dollar analysieren.
Parallel dazu stieg das erzeugte Datenvolumen rasant: von wenigen Gigabyte über Terabyte und Petabyte bis hin zu den hunderten Petabytes, die in weltweiten Projekten mit Millionen von Genomen entstehen. Schätzungen zufolge könnten bis 2025 rund 40 Exabyte an menschlichen Genomdaten anfallen – das entspricht etwa dem achtfachen Speicherbedarf aller jemals gesprochenen Worte der Menschheitsgeschichte. [How AI Is Transforming Genomics]
Komplexität biologischer Systeme: Das Verständnis von Krankheiten erfordert die Integration von Daten über Gene, Proteine, Stoffwechselwege und Zellinteraktionen.
Bedarf an prädiktiver Medizin: Ziel ist die Vorhersage von Krankheitsrisiken, Therapieansprechen und individueller Behandlung (Präzisionsmedizin).
Bereits bei vielen Schlüsselanwendungen kann man mittlerweile von einer Verschmelzung zwischen Bioinformatik und Künstlicher Intelligenz (KI) sprechen. Hier sind einige zentrale Anwendungsgebiete:
a) Genomanalyse und Sequenzierung
KI-Algorithmen werden genutzt, um DNA-Sequenzen schneller und präziser zu analysieren. Modelle wie DeepVariant, entwickelt von Google, nutzen tiefe neuronale Netze, um genetische Varianten mit hoher Genauigkeit zu identifizieren. Dies ist entscheidend für die Erkennung von Mutationen, die mit Krankheiten wie Krebs oder seltenen genetischen Störungen assoziiert sind.
b) Proteinfaltung und Strukturvorhersage
Ein Meilenstein der KI in der Bioinformatik ist AlphaFold von DeepMind, dass die Struktur von Proteinen mit beispielloser Präzision vorhersagt. Dies hat weitreichende Implikationen für die Arzneimittelentwicklung, da die 3D-Struktur eines Proteins seine Funktion bestimmt.
c) Multi-Omics Datenanalyse
Das ganzheitliche Verständnis biologischer Systeme erfordert die Integration von Multi-Omics-Daten (Genomik, Proteomik, Transkriptomik, Epigenetik, Metabolomik), um umfassendes Bild ihrer Funktionsweise zu gewinnen. KI hilft, die komplexen Wechselwirkungen zwischen diesen Ebenen zu entschlüsseln.
Abb. 13: Das mehrstimmige Konzert des Lebens. [AI applications in functional genomics] Biologische Systeme sind ein komplexes Zusammenspiel verschiedener Informationsebenen: Von der DNA (Genomik) über epigenetische Steuerung, RNA (Transkriptomik/Epitranskriptomik) und Proteine (Proteomik) bis hin zu Stoffwechselprodukten (Metabolomik). Erst ihr dynamisches Wechselspiel definiert Gesundheit und Krankheit.
Beispielsweise zeigt das neue KI-Tool AlphaGenome, wie selbst kleinste Veränderungen in der DNA die Genaktivität sowie die Produktion von RNA und Proteinen beeinflussen können. Es sagt präzise voraus, welche Folgen der Austausch eines einzelnen DNA-Bausteins für Gene und ihre Produkte hat. Während frühere Systeme meist nur die rund 2 % des Genoms analysierten, die Proteine codieren, erfasst AlphaGenome erstmals das gesamte Genom. Grundlage ist eine Multi-Omics-Analyse, die DNA-Veränderungen direkt mit ihren Auswirkungen auf RNA und Proteine verknüpft. Damit entsteht ein umfassendes Bild der Genexpression – ein entscheidender Schritt hin zu präziserer Genforschung und personalisierter Medizin.
d) Metagenomik und Mikrobiomanalyse
KI hilft, die komplexen Wechselwirkungen im menschlichen Mikrobiom zu verstehen, indem sie Muster in den Daten von Mikroorganismen erkennt. Dies ist wichtig für die Erforschung von Krankheiten wie Diabetes oder Darmerkrankungen.
e) Arzneimittelentwicklung
KI beschleunigt nahezu jede Phase des langwierigen und teuren Drug-Discovery-Prozesses: Zielidentifikation, virtuelles Screening von Millionen von Molekülen, Vorhersage von Wirkstoffeffekten und Nebenwirkungen, Optimierung von Molekülstrukturen. Unternehmen wie BenevolentAI oder Recursion nutzen KI-Plattformen, um vielversprechende Wirkstoffkandidaten in Rekordzeit zu identifizieren, die bereits in klinischen Studien getestet werden.
f) Analyse von biomedizinischen Bildern
KI wird in der Bildanalyse eingesetzt, etwa bei der Auswertung von MRT- oder CT-Scans, um Tumore oder andere Anomalien zu erkennen. Convolutional Neural Networks (CNNs), die die Funktionsweise des menschlichen visuellen Kortex nachahmen, haben sich hier als besonders effektiv erwiesen.
g) KI-Genomik in der Gesundheitsforschung
Die Genexpression im menschlichen Körper folgt keinem festen Muster – und Krankheiten wie Krebs machen sie noch schwerer durchschaubar. Genau hier setzt die räumliche Biologie an: Mit modernster Mikroskopie und genetischer Sequenzierung zeigt sie, wie Gene in einzelnen Zellen und sogar in ihren Organellen aktiv sind.
Doch nicht nur die sichtbaren Genabschnitte sind entscheidend. In den lange als „Junk-DNA“ abgetanen Regionen vermuten Forscher heute zentrale Schalter der Genregulation. Dort entscheidet sich, welche Proteine produziert werden – und welche nicht. KI und Deep-Learning-Modelle können in diesen bislang verborgenen Bereichen Muster erkennen, die mit Krankheiten verknüpft sind. So erhofft man sich neue Biomarker und Therapieansätze. Ein Beispiel ist die Plattform des Biotech-Unternehmens Deep Genomics:
Die Kombination aus räumlicher Biologie, intelligenter Datenanalyse und KI ermöglicht damit einen bislang unerreichten Blick in die molekulare Logik des Lebens – bis auf die Ebene einzelner Zellen.
Neue Methoden der Genbearbeitung könnten das Verhalten einzelner Gene gezielt steuern – etwa um krebsfördernde Gene zu deaktivieren oder die Regeneration von Knorpelzellen bei Arthrose zu fördern. Noch ist dieses Feld stark experimentell, doch KI verspricht, die Verfahren präziser und sicherer zu machen.
Mit KI-gestützten CRISPR-Techniken eröffnen sich neue Möglichkeiten, Gene nicht nur gezielter zu identifizieren und zu verändern, sondern auch deren Zusammenspiel in komplexen biologischen Netzwerken besser zu verstehen. Erste Ansätze deuten darauf hin, dass sich dadurch Nebenwirkungen künftig verringern und die Wirksamkeit neuer Therapien steigern ließe. Auch moderne Verfahren zum systematischen Testen und Optimieren von Genvarianten könnten – sofern sie sich bewähren – wichtige Schritte in Richtung maßgeschneiderter Behandlungen darstellen.
Wer sich dafür interessiert, mehr über CRISPR-Techniken zu erfahren, findet hier weiterführende Informationen.
Die Kombination von Fortschritten in KI und Genomik gilt daher als vielversprechend und könnte langfristig die Entwicklung neuer Zell- und Gentherapien beschleunigen. Da diese Behandlungen jedoch auf lebenden Zellen beruhen, die stark variieren und spezielle Bedingungen erfordern, befindet sich vieles noch in der Erprobung. KI könnte hier helfen, Prozesse stabiler, reproduzierbarer und klinisch besser anwendbar zu machen – doch ob und in welchem Umfang sich diese Hoffnungen erfüllen, bleibt abzuwarten.
h) Onkologie
KI-Modelle analysieren genetische Profile, um personalisierte Behandlungspläne zu erstellen. Beispielsweise können Algorithmen Vorhersagen treffen, wie Patienten auf bestimmte Medikamente reagieren, basierend auf ihrem genetischen Make-up. Dies ist besonders in der Onkologie relevant, wo KI-basierte Systeme wie IBM Watson Oncology Behandlungsempfehlungen geben.
Fazit
Bioinformatik ist heute das Gehirn der modernen Medizin. Gemeinsam mit Künstlicher Intelligenz verwandelt sie Datenfluten aus dem Erbgut in handfeste Erkenntnisse – von der Diagnose seltener Krankheiten bis zur Entwicklung neuer Medikamente.
Ob bei der Analyse von Genomen, der Vorhersage von Proteinstrukturen oder der Planung personalisierter Therapien: KI-gestützte Bioinformatik macht sichtbar, was bislang verborgen blieb. Sie eröffnet den Weg zu einer Medizin, die nicht nur Krankheiten behandelt, sondern sie voraussieht – maßgeschneidert für jeden einzelnen Patienten.

5. Eine Erfolgsgeschichte
Wie sehr diese Entwicklungen schon heute Realität sind, zeigt der Fall von KJ Muldoon.
Als der kleine Junge wenige Stunden nach seiner Geburt die Diagnose erhielt, schien sein Schicksal besiegelt. Er leidet an einem extrem seltenen Stoffwechseldefekt, der durch eine einzelne DNA-Punktmutation (SNV) verursacht wird und dazu führt, dass sein Körper giftiges Ammoniak nicht abbauen kann. Ohne funktionierendes Enzym sammeln sich toxische Stoffwechselprodukte rasch an – lebensbedrohlich schon im Säuglingsalter. Bisher galt: Nur eine riskante Lebertransplantation konnte das Überleben sichern.
Doch in KJs Fall wagten Ärzte und Forscher einen neuen Weg. Innerhalb von nur sechs Monaten entwarfen sie eine Therapie, die ausschließlich für ihn bestimmt war: eine maßgeschneiderte Base-Editing-Behandlung.
Das Prinzip dahinter ist ebenso bestechend wie elegant: Ein RNA-Suchsystem tastet das Erbgut ab, findet gezielt die Stelle mit KJs Punktmutation und bindet dort. Anstatt den DNA-Strang komplett zu durchtrennen – wie es beim klassischen CRISPR-Cas9 der Fall wäre – wird mithilfe eines Enzyms eine einzelne Base chemisch verändert: der Austausch eines DNA-Buchstabens, der die krankheitsverursachende Mutation korrigiert. Anschaulich gesagt: Es ist, als würde man in einem Buch einen einzigen Tippfehler berichtigen, ohne den ganzen Absatz aufzuschneiden oder neu zu schreiben – äußerst präzise und schonend für das Erbgut. Die Behandlung erfolgte direkt im Körper („in vivo“), über mehrere aufeinanderfolgende Behandlungsrunden, die gezielt die Leber erreichten.
Die ersten Ergebnisse sind ermutigend. KJ kann inzwischen mehr Proteine verdauen, Infekte verlaufen milder, er nimmt an Gewicht zu. Für seine Eltern bedeutet das: ein Stück Alltag ohne ständige Lebensgefahr – und vor allem eine neue Hoffnung auf die Zukunft ihres Kindes.
Hinter diesem medizinischen Durchbruch steckt eine Erfolgsgeschichte gleich mehrerer Disziplinen. Ohne moderne Genomsequenzierung wäre die exakte Mutation nicht sichtbar geworden. Ohne Bioinformatik ließe sich das passende Base-Editing-Werkzeug nicht in so kurzer Zeit und mit dieser Präzision entwerfen. Und ohne das Konzept der personalisierten Medizin wäre es undenkbar gewesen, ein Heilmittel ausschließlich für einen einzigen Patienten zu entwickeln.
Noch ist die Therapie experimentell, und niemand weiß, ob die Wirkung dauerhaft anhält oder ob langfristige Spätfolgen auftreten. Doch der Fall von KJ zeigt, wohin die Reise geht: zu einer Medizin, die nicht mehr auf Standardschemata setzt, sondern auf individuelle Lösungen – gestützt auf Daten, Molekularbiologie und maßgeschneiderte Therapien. Eine Medizin, die Krankheiten nicht nur behandelt, sondern sie im Idealfall gezielt an ihrer genetischen Wurzel packt.
Weitere Hintergründe zu dieser Geschichte gibt es in diesem Video.
6. Ein Blick in die Zukunft: Das Synthetic Human Genome Project
Die Biologie steht vor einem nächsten Quantensprung. Nach dem Entziffern und Editieren von Genomen kommt nun die Vision, sie von Grund auf neu zu schreiben. Genau das ist das Ziel des Synthetic Human Genome Project (SynHG) – ein ambitioniertes Vorhaben, das die Grenzen von Biologie und Medizin neu ausloten könnte.
Mit einer Anschubfinanzierung von £10 Millionen durch den Wellcome Trust und unter der Leitung von Professor Jason Chin am Generative Biology Institute in Oxford arbeiten führende Universitäten wie Cambridge, Manchester und das Imperial College London an diesem Projekt. Die Mission: Werkzeuge und Technologien entwickeln, um synthetische menschliche Genome herzustellen – ein Schritt, der unser Verständnis des Lebens selbst verändern könnte.
Ein Genom neu komponieren
Im Unterschied zur Genom-Editierung, die bestehende DNA nur anpasst, geht SynHG deutlich weiter: Es will das Genom Buchstabe für Buchstabe neu zusammensetzen. Während CRISPR einzelne Tippfehler im Text des Lebens korrigiert, geht es hier um die komplette Neuschreibung eines Kapitels.
Der erste Meilenstein: die Synthese eines vollständigen menschlichen Chromosoms als Machbarkeitsnachweis. Es ist der Anfang eines Weges, der Jahrzehnte dauern könnte – und doch an frühere Pionierleistungen anknüpft, wie das Humangenomprojekt (2003) oder das Synthetic Yeast Project, bei dem 2023 erstmals synthetische Hefe-Chromosomen entstanden.
Moderne Methoden – generative KI, maschinelles Lernen und robotergestützte Assemblierung – sollen die gewaltige Aufgabe meistern.
Mögliche Anwendungen – zwischen Laborvision und Klinikalltag
Die Chancen sind groß. Auf Ebene somatischer Zellen – also solcher, deren Veränderungen nicht an Nachkommen weitergegeben werden – könnten sich neue Wege für die regenerative Medizin eröffnen:
- maßgeschneiderte Ersatzgewebe und Organe aus dem Labor,
- virusresistente Zellen gegen HIV oder SARS-CoV-2,
- Gentherapien gegen Krebs oder Erbkrankheiten wie Mukoviszidose.
Solche Anwendungen sind keine ferne Science-Fiction – sie könnten in den nächsten 10 bis 20 Jahren Realität werden, da Technologien wie CRISPR und Stammzellkulturen bereits etabliert sind.
Ganz anders sieht es bei der Keimbahn-Editierung aus – also Eingriffen in Ei- oder Samenzellen. Hier ließen sich theoretisch schwere Erbkrankheiten wie Huntington dauerhaft verhindern. Vorstellungen von „Designer-Babys“ mit gewünschten Eigenschaften wie Aussehen oder Intelligenz bleiben jedoch wissenschaftlich und ethisch hoch problematisch, da solche Merkmale von Hunderten Genen abhängen.
Das Projekt selbst beschränkt sich auf Experimente in Reagenzgläsern und Zellkulturen. Es zielt nicht darauf ab, künstliches Leben zu erschaffen. Dennoch eröffnet es Forschenden eine beispiellose Möglichkeit, in grundlegende menschliche Lebenssysteme einzugreifen – mit weitreichenden Konsequenzen, die kritisch hinterfragt werden müssen.
Risiken und offene Fragen
Die technischen Hürden sind gewaltig. Bei somatischen Zellen drohen Mosaizismus (nicht alle Zellen übernehmen das neue Genom) oder Tumorbildung durch unbeabsichtigte Genveränderungen. In der Keimbahn sind die Risiken noch größer: Entwicklungsstörungen, epigenetische Effekte oder unvorhersehbare Folgen über Generationen hinweg.
Forscher diskutieren deshalb Sicherheitsmechanismen wie genetische „Kill-Switches“, mit denen synthetische Zellen im Ernstfall abgeschaltet werden könnten.
Ethik: Zwischen Heilung und Hybris
Somatische Therapien erinnern an Organtransplantationen: umstritten, aber gesellschaftlich akzeptiert, wenn sie heilen. Keimbahn-Eingriffe dagegen berühren Tabuzonen – aus Angst vor sozialer Ungleichheit oder gar eugenischen Szenarien. Viele Länder, darunter Deutschland, verbieten sie durch Gesetze wie das Embryonenschutzgesetz. Internationale Organisationen wie WHO und UNESCO fordern strikte Regulierung.
„Um Vertrauen zu schaffen, müssen wir die Gesellschaft aktiv einbeziehen“, betont die Soziologin Professorin Joy Zhang von der University of Kent, die die ethischen Dimensionen des Projekts untersucht. Mit ihrer Initiative Care-full Synthesis versucht sie, einen offenen Dialog über Chancen, aber auch über die Grenzen dieser Forschung anzustoßen.
Der renommierte Genetiker Professor Bill Earnshaw von der Universität Edinburgh, der Verfahren zur Herstellung künstlicher menschlicher Chromosomen entwickelt hat, formuliert es drastischer: „Der Geist ist aus der Flasche. Selbst wenn wir jetzt Regeln und Beschränkungen festlegen – jede Organisation mit den nötigen Maschinen könnte im Prinzip synthetisieren, was sie will. Aufhalten ließe sich das kaum.“
In diesem Licht wirkt die Forderung zahlreicher Fachorganisationen – darunter die International Society for Cell and Gene Therapy (ISCT) – nach einem zehnjährigen Moratorium für den Einsatz von CRISPR und verwandten Verfahren in der menschlichen vererbbaren Keimbahn noch dringlicher. Ein solcher Aufschub erscheint nicht nur als Vorsichtsmaßnahme, sondern als notwendige politische Reaktion, um gesellschaftliche Debatten, ethische Leitplanken und internationale Kontrollmechanismen überhaupt erst zu ermöglichen.
Der Ethik-Experte Arthur Caplan von der New York University beschreibt das Dilemma aus wissenschaftlicher Sicht:
„… Wenn Sie mich also fragen, werden wir eine Genmanipulation von Kindern sehen, die auf ihre Verbesserung abzielt? Ich sage ja, ohne Zweifel. Wann? Ich bin mir nicht sicher, was die Antwort darauf ist.“
„… Es wird kommen. Es gibt Eigenschaften, die die Menschen in Zukunft eifrig versuchen werden, ihren Kindern beizubringen. Sie werden versuchen, genetische Krankheiten zu entschlüsseln, sie loszuwerden. Sie werden versuchen, Kapazitäten und Fähigkeiten aufzubauen, von denen sie zustimmen, dass sie wirklich wunderbar sind. Werden wir diese Interventionen aus ethischen Gründen an den Nagel hängen? Größtenteils nein, wäre meine Vorhersage, aber nicht innerhalb der nächsten 10 Jahre. Die Werkzeuge sind noch zu grob.“Ausblick
SynHG öffnet ein Fenster in eine Zukunft, in der wir das Genom nicht nur lesen und editieren, sondern schreiben können. Die somatischen Anwendungen – personalisierte Therapien, regeneratives Gewebe, maßgeschneiderte Zellen – könnten schon bald die Medizin prägen.
Eingriffe in die Keimbahn dagegen bleiben vorerst ein ferner Horizont – technisch, rechtlich und ethisch. Das Projekt stellt nicht nur eine wissenschaftliche, sondern auch eine gesellschaftliche Gratwanderung dar.
Vor dem Hintergrund des möglichen Missbrauchs der Technologie stellt sich die Frage, weshalb Wellcome überhaupt die Finanzierung entschied. Dr. Tom Collins, der diesen Schritt verantwortete, erklärte gegenüber BBC News:
„Wir haben uns gefragt, was die Kosten der Untätigkeit wären. Diese Technologie wird eines Tages entwickelt werden. Indem wir sie jetzt einsetzen, versuchen wir zumindest, so verantwortungsvoll wie möglich vorzugehen und uns den ethischen und moralischen Fragen so offen wie möglich zu stellen.“
Während man in Europa noch darum ringt, das Thema in der öffentlichen Debatte behutsam auszubalancieren, geht man auf der anderen Seite des Atlantiks deutlich unbefangener mit dem Begriff „neue Lebensformen“ um. Eric Nguyen, Biowissenschaftler an der renommierten Stanford University, skizziert in seinem TED-Talk „How AI Could Generate New Life-Forms“ eine weit visionärere Perspektive.
Auf eindrückliche Weise zeigt er, wie künstliche Intelligenz (KI) die Biologie transformieren könnte – nicht nur durch das „Lesen“ und „Schreiben“ von DNA, sondern auch durch die gezielte Erschaffung neuartiger Organismen. KI-Modelle wie maschinelles Lernen und generative Algorithmen sind bereits heute in der Lage, komplexe Muster in biologischen Daten zu erkennen: in Genomen, Proteinstrukturen oder Stoffwechselwegen. Nguyen geht jedoch weiter und argumentiert, dass KI künftig auch völlig neue DNA-Sequenzen entwerfen könnte – der Ausgangspunkt bislang unbekannter Lebensformen.
Diese Vision ist zweifellos zukunftsweisend, wirft aber grundlegende Fragen auf: Welche Prioritäten sollen gesetzt werden? Wie lässt sich der Einsatz solcher Technologien in einer Welt mit massiver Ressourcenungleichheit verantworten? Wer finanziert die Forschung, wer kontrolliert die Ergebnisse – und nach welchen Kriterien wird entschieden, welche Lebensformen „nützlich“ oder „sicher“ sind?
Nguyens Vortrag verdeutlicht: KI-gestützte Biotechnologie könnte nicht nur Lösungen für globale Herausforderungen ermöglichen, sondern zwingt uns zugleich, über die ethischen, sozialen und philosophischen Dimensionen der „Erzeugung von Leben“ nachzudenken.
Die Botschaft: KI-gestützte Biotechnologie verspricht enorme Fortschritte, wirft jedoch zugleich tiefgreifende ethische und gesellschaftliche Fragen auf – und stellt damit die Grundlagen unseres Menschseins zur Diskussion.
Klaus Schwab – Gründer und ehemaliger Vorsitzender des Weltwirtschaftsforums
„Wir müssen, sowohl individuell als auch kollektiv, die moralischen und ethischen Fragen angehen, die durch Spitzenforschung in den Bereichen künstliche Intelligenz und Biotechnologie aufgeworfen werden, die eine erhebliche Lebensverlängerung, Designerbabys und das Extrahieren von Erinnerungen ermöglichen wird.“
[BrainyQuote]7. Personalisierte Medizin und Smart Governance
Die biomolekulare Revolution der Regierungsführung
7.1. Macht, Governance und Smart Governance
7.2. Gouvernementalität
7.3. Personalisierte Medizin als Katalysator biomolekularer Gouvernementalität
7.4. Der weltweite Trend: Globale biomolekulare Gouvernementalität
7.5. Schlussfolgerung: Globale Ambivalenz der biomolekularen MachtDie Entschlüsselung des menschlichen Genoms markierte nicht nur einen medizinischen Wendepunkt, sondern auch den Beginn einer neuen Epoche politischer Steuerung. Was als wissenschaftliches Großprojekt in Laboren und Rechenzentren begann, ist längst in den Steuerungsapparaten moderner Staaten angekommen: die präzise Vermessung, Kategorisierung und Nutzung unserer biologischen Grundlagen.
Die Vision der personalisierten Medizin verspricht individuelle Therapien, präzise Vorhersagen über Krankheitsrisiken und eine effizientere Nutzung knapper Ressourcen. Doch diese Fortschrittserzählung greift zu kurz, wenn wir sie nur medizinisch, ethisch oder datenschutzrechtlich betrachten. Tatsächlich sind wir Zeugen einer grundlegenden Transformation von Macht und Gouvernementalität – einer biomolekularen Revolution der Regierungsführung.
Bevor wir tiefer einsteigen, gilt es einige zentrale Begriffe zu klären.
7.1. Macht, Governance und Smart Governance
Governance bezeichnet die Kunst, gesellschaftliche Prozesse zu steuern, Entscheidungen zu treffen und Ordnung zu sichern. Klassisch geschieht dies im Zusammenspiel von Staat, Märkten und Zivilgesellschaft.
Smart Governance transformiert dieses Verständnis: Sie operiert mit digitalen Technologien, Datenanalysen und algorithmischen Prozessen. Steuerung wird dadurch kontinuierlich, prozessual und in Echtzeit möglich. Macht zeigt sich hier nicht mehr nur als politische Autorität, sondern als Geflecht aus Informationsflüssen, Rückkopplungen und Prognosen.
Macht selbst ist kein Besitz, sondern eine Beziehung: die Fähigkeit, Verhalten zu beeinflussen, Handlungen zu lenken oder Entscheidungen zu rahmen. Sie wirkt sowohl sichtbar – über Institutionen und Gesetze – als auch subtil, indem sie Erwartungen, Normen und Wissensbestände formt.
7.2. Gouvernementalität
Michel Foucault, ein bedeutender Denker in Bezug auf die Analyse von Machtstrukturen, prägte den Begriff der Gouvernementalität, um die Gesamtheit der Wissensformen, Praktiken und Techniken zu beschreiben, durch die moderne Gesellschaften regiert werden. Regierung bedeutet hier nicht nur die Ausübung von Zwang, sondern auch die Gestaltung von Handlungsräumen, die Menschen dazu bringen, sich selbst im Sinne gesellschaftlicher Zielsetzungen zu steuern.
Zentrale Dimensionen der Gouvernementalität sind:
Verknüpfung von Wissen und Macht
Wissen ist nie neutral. Es entsteht durch die Sammlung und Deutung von Daten, ist in Machtprozesse eingebettet und strukturiert soziale Ordnungen. Mit der Digitalisierung wird diese Verbindung zentral: Big Data und Künstliche Intelligenz liefern Prognosen, Mustererkennungen und Entscheidungsgrundlagen, die tief in gesellschaftliche Prozesse eingreifen. Smart Governance setzt genau hier an: Daten werden zum Fundament politischer Legitimation und Machtausübung.
Biopolitik
Biopolitik bezeichnet die Regulierung des Lebens selbst – Gesundheit, Reproduktion, Arbeit, Bildung, Migration oder Sicherheit. Ziel ist die Ordnung ganzer Bevölkerungen durch Gesetze, Standards und politische Programme. Heute verbindet sich Biopolitik mit digitaler Steuerung: Smart Health, Smart Cities oder Smart Economy verdeutlichen, wie Wissen durch Daten in konkrete Regierungspraktiken umgesetzt wird.
Dispositive der Macht
Biopolitik wirkt über Dispositive: Netzwerke aus Institutionen, Gesetzen, Diskursen und Praktiken. Krankenhäuser, Schulen, Medien oder digitale Plattformen sind Teil dieser Strukturen. Sie normieren Verhalten nicht durch offenen Zwang, sondern durch subtile Rahmungen dessen, was als „normal“ oder „wünschenswert“ gilt. So werden Entscheidungen indirekt gelenkt und Verhalten in den Alltag eingebettet.
Disziplinarmacht
Neben der Biopolitik, die ganze Bevölkerungen reguliert, richtet sich Disziplinarmacht auf das Individuum. Sie wirkt über Überwachung, Belohnung, Strafe und Training. Ziel ist nicht nur Gehorsam, sondern die produktive Formung von Körpern und Verhaltensweisen. Externe Vorgaben verwandeln sich durch Sanktionen und Anreize in innere Motivationen, die Menschen zur Selbstdisziplin bewegen.
Technologien der Selbstführung
Technologien der Selbstführung bezeichnen Praktiken, durch die Individuen gesellschaftliche Normen verinnerlichen und ihr Verhalten eigenständig anpassen. Digitale Anwendungen – etwa Fitness-Apps, Lernplattformen oder Finanz-Tools – übersetzen gesellschaftliche Vorgaben in persönliche Ziele. So entsteht eine Machtform, die nicht nur von außen wirkt, sondern von innen weitergeführt wird: Menschen folgen Regeln, weil sie sie als sinnvoll und nützlich für sich selbst empfinden.
Wer sich näher für die Kunst des Regierens interessiert, findet hier weiterführende Informationen.
7.3. Personalisierte Medizin als Katalysator biomolekularer Gouvernementalität
Die personalisierte Medizin, gestützt auf moderne Genomik und Bioinformatik, erscheint auf den ersten Blick als rein medizinisch-technischer Fortschritt. Doch betrachtet man sie durch die Linse der Gouvernementalität, zeigt sie sich als mächtiger Katalysator für eine tiefgreifende Transformation von Machtstrukturen und Regierungspraktiken. Durch die präzise Analyse individueller genetischer Profile und deren Verknüpfung mit umfassenden Datenbanken wird nicht nur die Gesundheitsversorgung individualisiert, sondern auch die Art und Weise, wie Gesellschaften gesteuert und Individuen reguliert werden.
Wissen & Macht: Vom Genom zum regierbaren Körper
Die Entschlüsselung des Genoms war die initiale Wissensexplosion, doch erst die personalisierte Medizin macht dieses Wissen regierbar. Bioinformatik und moderne Genomik generieren nicht nur Daten, sondern schaffen prädiktives Wissen, das die Grundlage für Interventionen bildet. Dieses Wissen ist keineswegs neutral: Es kategorisiert Individuen anhand genetischer Risikoprofile (z. B. „hohes Risiko für Brustkrebs“, „prädestiniert für Herz-Kreislauf-Erkrankungen“). Diese Kategorisierung wird zum Fundament neuer Machtstrukturen. Staaten wie auch private Akteure nutzen diese Daten, um Krankheitsrisiken vorherzusagen, Gesundheitsbudgets zu optimieren und präventive Maßnahmen zu steuern. Die soziale Ordnung wird dadurch neu strukturiert – vom Zugang zu Versicherungen und Arbeitsmärkten bis hin zur Priorisierung präventiver Ressourcen.
Smart Governance manifestiert sich hier in algorithmischen Entscheidungsprozessen, die scheinbar objektiv, tatsächlich jedoch normativ sind. Wer Zugang zu welchen Therapien erhält oder welche Bevölkerungsgruppen als „risikobehaftet“ gelten, wird zunehmend durch datenbasierte Modelle bestimmt, die Machtverhältnisse unsichtbar reproduzieren („Wir handeln auf Basis der besten verfügbaren Evidenz“).
Die CMS Health Tech Ecosystem Initiative, die offiziell am 30. Juli 2025 während der Veranstaltung „Make Health Tech Great Again“ im Weißen Haus angekündigt wurde, verkörpert den Trend zur Herausbildung neuer Machtstrukturen. CMS (Centers for Medicare & Medicaid Services) ist eine US-Bundesbehörde im Department of Health and Human Services (HHS), die Gesundheitsprogramme wie Medicare und Medicaid verwaltet und den nationalen Gesundheitsmarkt reguliert. Ziel der Initiative ist es, eine intelligentere, sicherere und stärker personalisierte Gesundheitsversorgung zu schaffen, die durch Partnerschaften mit innovativen Unternehmen des privaten Sektors vorangetrieben wird. Während der Veranstaltung sicherte sich die US-Regierung Zusagen von großen Gesundheits- und IT-Unternehmen – darunter Amazon, Anthropic, Apple, Google und OpenAI –, um den Grundstein für ein digitales Gesundheitsökosystem der nächsten Generation zu legen.
Laut US-Präsident Donald Trump erhalten die US-Gesundheitssysteme dadurch ein High-Tech-Upgrade und treten ein in „eine Ära der Bequemlichkeit, Rentabilität, Schnelligkeit und – offen gesagt – einer besseren Gesundheit für die Menschen“. (Man beachte die Reihenfolge…)
Biopolitik 2.0: Die Optimierung der Bevölkerung auf molekularer Ebene
Die klassische Biopolitik regulierte Bevölkerungen durch Statistiken über Geburtenraten oder Sterblichkeit. Die personalisierte Medizin hebt dies auf eine neue, molekulare Stufe. Genomik fragmentiert die Bevölkerung in immer kleinere, biomolekular definierte Untergruppen. Die Bevölkerung wird nicht mehr nur nach Alter oder Geschlecht, sondern nach Genotyp, Risikoallelen und Biomarkern kategorisiert („die BRCA1-Träger“, „die Menschen mit LCT-Gen für Laktoseintoleranz“). Dies ermöglicht eine hyperpräzise und individualisierte Regulierung – Biopolitik wird granular.
Der „gesunde“, „normale“ Körper ist nicht länger allein derjenige ohne Symptome, sondern der, der ein „normales“, „risikoarmes“ Genom aufweist. Abweichung beginnt nicht mit Krankheit, sondern mit genetischer Prädisposition. Damit entsteht eine neue Klasse von „präsymptomatisch Kranken“ oder „Risikosubjekten“, die biopolitisch gemanagt werden: durch häufigere Screenings, Präventionsprogramme, Impfkampagnen, prophylaktische Operationen, Lebensstilempfehlungen oder reproduktive Entscheidungen, die auf genetischen Prognosen basieren.
Während Foucault den Nationalstaat als Hauptakteur der Biopolitik sah, verlagert sich die Macht heute zunehmend auf private Akteure: Big-Tech- und Biotech-Unternehmen, die Daten, Algorithmen und Technologien kontrollieren. Sie definieren de facto, was genetische „Normalität“ und „Risiko“ bedeutet.
Man spricht mittlerweile von einer Ökonomisierung des biologischen Lebens. Genetische Daten werden zu einer wertvollen Handelsware – einer Bio-Ressource. Große Technologie- und Pharmaunternehmen akkumulieren Biodaten, um daraus Gewinne zu generieren: in der Medikamentenentwicklung, bei Therapieempfehlungen, in der personalisierten Werbung. Der menschliche Körper und seine biomolekularen Daten werden so Teil kapitalistischer Verwertungslogiken. Das Individuum selbst wird zum „Humankapital“, das verpflichtet scheint, sein genetisches Potenzial zu optimieren.
In einem NBCNews-Beitrag vom 26. August 2025 erklärte der US-Gesundheitsminister RFK Jr., die 60 größten Technologieunternehmen würden den Amerikanern künftig den Zugriff auf ihre persönlichen Gesundheitsdaten gestatten – Daten, die jahrelang ohne ihre Zustimmung ökonomisch genutzt worden seien: „Sie werden ab nächstem Jahr alle Ihre Gesundheitsdaten auf Ihrem Mobiltelefon einsehen können.“
Was hier als politischer Erfolg statuiert wird, bestätigt nur den Status Quo.
Dispositive der Macht: Das Netzwerk der biomolekularen Überwachung
Das dispositive Netzwerk, das diese neue Form der Biopolitik trägt, ist vielschichtig: Pharmakonzerne entwickeln zielgenaue Therapien; Biobanken und genetische Datenbanken liefern das Rohmaterial; Algorithmen analysieren und interpretieren es; Ärzte vermitteln Ergebnisse und Empfehlungen; digitale Gesundheits-Apps überwachen die compliance. Jedes Glied in dieser Kette fungiert als Machtdispositiv: Es definiert, was als „gesund“ oder „krank“ gilt, und vermittelt subtil, was „verantwortungsvolle Gesundheitsvorsorge“ im Zeitalter der Genetik bedeutet. Genetische Tests werden etwa als „verantwortungsbewusstes“ Handeln vermarktet – und bringen Individuen so dazu, sich unbemerkt an biopolitischen Zielen wie Prävention oder Kosteneffizienz zu beteiligen.
Diese Entwicklung spiegelt sich in nationalen Gesundheitsstrategien, die genetische Screenings fördern, um Krankheitslasten zu minimieren. Ziel ist die Maximierung der „Gesundheitsdividende“ bei gleichzeitiger Kostenreduktion – eine biomolekulare Rationalisierungsstrategie.
Initiativen wie das 100.000 Genome Project oder das Newborn Genomes Programme von Genomics England gehen in die gleiche Richtung. Im Vergleich dazu ist der Europäische Raum für Gesundheitsdaten (EHDS) – eine EU-weite Initiative zur Vernetzung und sicheren Nutzung von Gesundheitsdaten – deutlich tiefgreifender. Der EHDS ist die Daten- und Regulierungsinfrastruktur, die personalisierte Medizin in großem Maßstab überhaupt erst ermöglicht.
Für die meisten Bürger klingt das nach Aufbruch: mehr Mitsprache, bessere Therapien, präzisere Prävention. Doch die verheißungsvolle Vision wirft einen Schatten. In der Praxis stehen sich ungleiche Gegner gegenüber: auf der einen Seite der einzelne Patient, der sein genetisches Profil, seine Laborwerte und seine Krankengeschichte beisteuert; auf der anderen Seite globale Pharmakonzerne, Datenplattformen und Technologieanbieter mit Milliardenbudgets, die diese Daten veredeln, patentieren, vermarkten. Cui bono? – wem nützt das Ganze? Die Antwort fällt ernüchternd aus: Der schnelle Profit landet bei den Großen, die Versprechen bleiben beim Kleinen.
Zwar betont der EHDS Bürgerrechte – mehr Kontrolle, mehr Transparenz, mehr Selbstbestimmung. Doch wer je erlebt hat, wie unübersichtlich Einwilligungsprozesse bei Gesundheits-Apps, Versicherungen oder elektronischen Patientenakten sind, weiß: Zwischen formaler Option und tatsächlicher Handlungsfähigkeit klafft eine Lücke. Während Goliath über Lobbykanäle, Fachjuristen und Datenzentren verfügt, bleiben David Informationsbroschüren und Vertrauen.
Der EHDS ist damit beides: Regierungstechnologie und Machtprojekt. Er entscheidet, ob Daten als öffentliches Gut oder als Rohstoffmarkt behandelt werden. Profitieren wird Goliath in jedem Fall. Die offene Frage ist, ob David mehr bleibt als die Rolle des Feigenblatts. Wirklich emanzipatorisch wird der Gesundheitsdatenraum erst, wenn Nutzen und Risiken fair verteilt werden – durch verbindliche Gemeinwohlauflagen, transparente Rechenschaft und reale Rückflüsse für die Patienten. Andernfalls bleibt das große Versprechen von „Empowerment“ ein Narrativ, das bestehende Asymmetrien lediglich kaschiert.
Disziplinarmacht: Die internalisierte biomolekulare Überwachung
Während die Biopolitik die Bevölkerung im Blick hat, richtet die personalisierte Medizin ihre disziplinierende Wirkung auf das Individuum. Das genetische Risikoprofil fungiert wie ein ständiger, internalisierter Überwacher. Wer weiß, dass er ein erhöhtes Risiko für Darmkrebs trägt, reguliert sein Verhalten: Ernährungsgewohnheiten, Vorsorgetermine und Sportroutinen werden der genetischen Prognose angepasst.
Die elektronische Patientenakte (ePA) verschärft diese Dynamik. Indem sie genetische und medizinische Daten zentral speichert und jederzeit abrufbar macht, verwandelt sie medizinische Empfehlungen in eine allgegenwärtige, digitale Instanz. Persönliche Hinweise, Erinnerungen an Medikamenteneinnahmen, personalisierte Berichte oder Schnittstellen zu Gesundheits-Apps verstärken die Selbstüberwachung: Die externe Anweisung („Du musst dich screenen lassen“) wird zur inneren Stimme („Ich muss auf mich achten, weil mein Genom es verlangt“). Die ePA strukturiert und institutionell legitimiert diese Selbstdisziplinierung, sodass sie unsichtbar und scheinbar freiwillig wirkt – gesteuert durch digitale Infrastruktur, soziale Normen und medizinische Autorität zugleich.
Die Folge ist eine subtile, aber umfassende Form der Macht: Sie manifestiert sich nicht in Sanktionen, sondern in der permanenten, digitalen Sorge um die eigene – genetisch vorgestellte – Zukunft. Die ePA macht das Individuum zum disziplinierten Akteur seines eigenen Risikoprofils, während die Überwachung formal verschwindet, aber technisch und institutionell allgegenwärtig bleibt.
Technologien der Selbstführung: Der genetisierte Selbstoptimierer
Hier zeigt sich die personalisierte Medizin in ihrer reinsten Form als Technologie der Selbstführung. DNA-Tests für zuhause, Fitness-Tracker und Gesundheits-Apps sind die Werkzeuge, mit denen Individuen gesellschaftliche Normen von Gesundheit und Leistungsfähigkeit verinnerlichen und aktiv an sich selbst arbeiten. Sie übersetzen das abstrakte Konzept der genetischen Prädisposition in konkrete, tägliche Handlungen: eine Extra-Portion Gemüse, 10.000 Schritte, Meditation zur Stressreduktion. Der Einzelne führt sich selbst im Sinne einer biomolekularen Logik – nicht weil der Staat es befiehlt, sondern weil er sein genetisches Schicksal optimieren will. Die Machtverhältnisse werden so unsichtbar und erscheinen als Ausdruck individueller Souveränität und Selbstfürsorge.
7.4. Der weltweite Trend: Globale biomolekulare Gouvernementalität
Die personalisierte Medizin ist längst kein nationales Phänomen mehr, sondern Teil einer globalen Transformation von Macht und Governance. Weltweit entstehen Initiativen, die Genomdaten, Gesundheitsinformationen und digitale Technologien bündeln, um Bevölkerung und Individuen in immer präzisere Risikoprofile zu übersetzen. Die Logik von Foucaults Gouvernementalität – die Verbindung von Wissen, Macht und Selbststeuerung – manifestiert sich hier auf transnationaler Ebene.
In den Vereinigten Staaten sammeln Programme wie das All of Us Research Program der NIH oder das Million Veteran Program genetische und medizinische Daten von Millionen Bürgern. Die Erhebung diverser Gesundheitsdaten dient nicht nur der wissenschaftlichen Forschung, sondern bildet die Grundlage für algorithmische Risikoprofile, präventive Interventionen und ressourcenoptimierte Gesundheitsstrategien.
Kanada verfolgt mit der Canadian Precision Health Initiative und dem Pan-Canadian Genomics Network ein vergleichbares Ziel: die systematische Sammlung und Vernetzung biomolekularer Daten, kombiniert mit einer Infrastruktur, die sichere Verwaltung, internationale Kooperation und wirtschaftliche Nutzung ermöglicht.
Hier zeigt sich Biopolitik 2.0: Bevölkerungen werden auf molekularer Ebene fragmentiert, gesteuert und optimiert.
Europa bildet ein orchestriertes Netzwerk transnationaler Biopolitik. Der Europäische Gesundheitsdatenraum (EHDS), unterstützt durch Initiativen wie ICPerMed und ERA PerMed, bündelt genetische, klinische und Lebensstildaten über Ländergrenzen hinweg.
Nationale Biobanken wie die UK Biobank oder der französische Health Data Hub fungieren als Dispositive, die sowohl Wissen als auch Macht generieren: Sie bestimmen, welche Daten genutzt werden, wie Populationen klassifiziert werden und welche gesundheitspolitischen Interventionen legitim erscheinen.
Durch Plattformen wie SHIFT-HUB oder die Helmholtz-Initiative iMed wird die internationale Vernetzung von Wissenschaft, Wirtschaft und Gesellschaft institutionalisiert – eine transnationale Form von Smart Governance, die Foucaults Konzept der Technologien der Selbstführung in den globalen Maßstab überträgt.
Auch in Asien und im Pazifikraum zeigt sich die globale Logik: Das GenomeAsia 100k Project, Singapurs National Precision Medicine Strategy und Chinas National Health Data Platform verbinden biomolekulare Daten mit KI-gestützten Analysen, um präventive, individualisierte und populationsbasierte Steuerung zu ermöglichen.
Australien und Israel setzen auf digitale Infrastruktur, die die Selbstdisziplinierung der Bürger verstärkt – elektronische Patientenakten, genetische Datenbanken und personalisierte Apps fungieren als subtile, aber allgegenwärtige Machtdispositive.
Selbst globale Forschungsprojekte wie der Human Cell Atlas illustrieren die transnationale Dimension: Indem die Vielfalt menschlicher Zellen kartiert wird, entsteht ein universelles Wissens- und Machtnetzwerk, das Grundlagen für personalisierte Therapien weltweit bereitstellt. Die Grenzen zwischen staatlicher Steuerung, privatwirtschaftlicher Macht und wissenschaftlicher Expertise verschwimmen: Foucaults klassische Trennung zwischen Nationalstaat und Disziplinarmacht wird durch globale Netzwerke und internationale Kooperationen ersetzt.
7.5. Schlussfolgerung: Globale Ambivalenz der biomolekularen Macht
Die weltweite Expansion personalisierter Medizin zeigt eindrücklich, dass die biomolekulare Revolution nicht mehr auf einzelne Staaten beschränkt ist. Sie manifestiert sich als transnationales Netzwerk aus Daten, Algorithmen und Governance, das sowohl Bevölkerungen als auch Individuen auf molekularer Ebene steuert. Globale Initiativen – von den USA über Europa bis Asien, Australien und Israel – verbinden Biopolitik, Disziplinarmacht und Technologien der Selbstführung auf internationaler Ebene.
Die Ambivalenz bleibt dabei zentral: Auf der einen Seite eröffnen diese Entwicklungen Chancen für präzisere Therapien, individualisierte Prävention und potenziell mehr Gesundheitsgerechtigkeit. Auf der anderen Seite entstehen Machtasymmetrien, bei denen Staaten, internationale Organisationen und private Akteure den Zugang zu Daten, Ressourcen und Technologien kontrollieren. Individuen werden zunehmend zu disziplinierten Subjekten ihres eigenen genetischen Profils, während globale Dispositive unbemerkt normative und ökonomische Ziele durchsetzen.
Damit verbindet sich die lokale Erfahrung personalisierter Medizin nahtlos mit einer globalen Gouvernanzlogik: Smart Governance und biomolekulare Biopolitik sind längst internationalisiert, und die Herausforderung besteht darin, Nutzen und Risiken fair zu verteilen. Die weltweite Dimension dieser Transformation verdeutlicht, dass die biomolekulare Revolution nicht nur medizinisch, sondern auch politisch und sozial unumkehrbar ist – ein globales System der Steuerung, das zugleich Chancen für Gesundheit und Risiken für Machtkonzentration birgt.
8. Epilog
Der Wunsch, Gesundheit und Lebenszeit zu optimieren, ist ein Urinstinkt der Menschheit. Schon in antiken Hochkulturen galt Gesundheit nicht als Zufall, sondern als erstrebenswerter Zustand, erreichbar durch Lebensführung und Philosophie. Die griechische „Diätetik“ – eine ganzheitliche Praxis aus Ernährung, Bewegung und Seelenhygiene – und das Leitbild „mens sana in corpore sano“ zeugen von einem 2500 Jahre alten Optimierungswillen. Dieser Drang, gespeist aus dem Überlebensinstinkt, der Angst vor Kontrollverlust und dem Streben nach Lebensqualität, ist nicht nur uralt, sondern ewig.
In der Moderne verkörpert die personalisierte Medizin den neuesten Ausdruck dieses Traums. Sie verspricht, den Menschen nicht als Durchschnitt, sondern als Individuum zu behandeln, basierend auf genetischem Code, Biomarkern und Lebensstil-Daten. Doch ist sie die Erfüllung dieses Traums oder eine moderne Fata Morgana? In ihrer utopischen Vision suggeriert sie die vollständige Beherrschbarkeit der Biologie, eine Überwindung der „biologischen Lotterie“. Die Realität ist nüchterner: Sie ermöglicht präzisere Diagnosen und Therapien, verhindert Behandlungsfehlschläge – aber sie schafft Krankheit nicht ab.
Denn die Idee einer krankheitsfreien Existenz bleibt ein Trugbild. Evolutionäre Biologie und Biophysik setzen Grenzen: Pathogene Keime entwickeln sich weiter, Krebs ist eine statistische Folge der Zellteilung, und Alterung ein unvermeidbarer degenerativer Prozess. Das realistische Ziel ist daher nicht die Elimination von Krankheit, sondern ihre Transformation: tödliche Erkrankungen in chronische Zustände umzuwandeln und die „healthspan“ – die Jahre in guter Gesundheit – zu maximieren. Dieses Ziel ist radikal, aber erreichbar, wie Fortschritte in der Behandlung von HIV oder Diabetes zeigen.
Doch wie weit ist der Mensch bereit, für dieses Ziel zu gehen? Die Geschichte zeigt, dass im Angesicht von Leid und Tod ethische Grenzen oft verschwimmen. Von alltäglicher Disziplin über finanzielle Opfer bis hin zu experimentellen Therapien reicht das Spektrum der Bereitschaft. Hier stellt sich eine unbequeme Frage: Welche Kompromisse ist die Gesellschaft bereit einzugehen?
Die personalisierte Medizin erfordert Daten – genetische, medizinische, persönliche. Diese Datensammlung birgt das Risiko, Privatsphäre und Autonomie zu untergraben. Die COVID-19-Pandemie war ein Stresstest, der zeigte, dass große Teile der Bevölkerung bereitwillig Freiheitsrechte für Sicherheit handelten. Modelle wie das chinesische Sozialkreditsystem illustrieren ein mögliches Zukunftsmodell, in dem Transparenz und Kontrolle im Namen von Gesundheit und Sicherheit belohnt werden. Doch solche Szenarien sind nicht zwangsläufig: Sie sind ein Extrem, kein unvermeidbares Schicksal.
Die personalisierte Medizin bietet zwei Gesichter. Ihr pragmatisches Versprechen ist eine effizientere, präzisere und menschlichere Medizin, die Leben rettet und Leid lindert – etwa durch maßgeschneiderte Krebstherapien oder frühzeitige Diagnosen. Ihr utopisches, aber riskantes Versprechen ist die Illusion einer totalen Kontrolle über Leben und Tod. Sie ist damit der Höhepunkt einer jahrtausendealten Reise. Ob diese Reise in einer emanzipierten Selbstoptimierung oder einer dystopischen „Biokratie“ endet, hängt nicht von der Technologie ab, sondern von der ethischen und politischen Rahmensetzung, die die Gesellschaft ihr gibt.
Der uralte Traum von Gesundheit erscheint heute greifbarer denn je, doch er fordert uns heraus, kluge Antworten zu finden. Die personalisierte Medizin kann ein Werkzeug der Emanzipation sein, wenn wir sie mit Bedacht nutzen. Die entscheidende Frage der Moderne lautet daher nicht nur, wie viel Freiheit wir für ein längeres Leben opfern, sondern wie wir Gesundheit und Freiheit in Einklang bringen. Nur so wird das ewige Streben nach Gesundheit zu einem Fortschritt für alle.
Die Welt wird sich nicht auf einen Schlag verändern; das ist noch nie geschehen. Kurzfristig wird das Leben weitgehend unverändert weitergehen, und die Menschen werden 2025 ihre Zeit größtenteils genauso verbringen wie 2024. Wir werden uns weiterhin verlieben, Familien gründen, uns online streiten, in der Natur wandern usw.
Doch die Zukunft wird unaufhaltsam auf uns zukommen, und die langfristigen Veränderungen für unsere Gesellschaft und Wirtschaft werden gewaltig sein.
Sam Altman (CEO von OpenAI)
Quellen (Stand vom 22.11.2025)
Überwachung, Bioinformatik, Biopolitik, Crispr, DNA, DNA-Sequenzierung, DNA-Test, FENT, G4X Spatial Sequencer, Gen, Gendefekt, Genetik, Genom, Genomik, Genomsequenzierung, Genvariation, Governance, Illumina, iNanoBio, Künstliche Intelligenz, KI, Kontrolle, Medizin, Medizinversorgung, Michel Foucault, Next Generation Sequencing, NGS, Nvidia, ONT, Oxford Nanopore, PacBio, Personalisierte Medizin, Präzisionsmedizin, Protein, RNA, Roche, Sanger, SBX-Sequnzierung, Singular Genomics, Smart Governance, SMRT-Sequenzierung, SynHG, Synthetic Human Genome Project, Transkriptomik, Xpandomer -
We Are on Track

A speculative vision of the future. Not a prophecy.
The year is 2025.
High above the clouds, in a floating palace of steel and glass, sat Mr. Global. Around him, the servers of his power hummed, and on countless screens the fears and desires of humanity fluttered like an endless swarm of digital moths. Mr. Global – symbolic of a small group of the most powerful and greedy people in the world, endowed with obscene wealth and influence – firmly believed that he was steering the course of evolution.
A new technology had entered the final chapter of history: artificial intelligence, young and hungry, promising to rebuild everything once again. For Mr. Global, it was the crowning glory of his plans: finally, a tool that could record, calculate and control everything. Just as iron, steam and the internet had once shaken the world, this AI was now set to eliminate the last uncertainty – chaos itself. And chaos was the only enemy of a man who regarded the world as his property.
Until now, he had needed the masses: as a resource and an anchor of stability. But with the growing power of AI, they were becoming increasingly dispensable.
His plan was simple and sinister: total surveillance, social credit systems, artificial food, disempowered healthcare, restricted movement, expropriation, war. He entertained various ideas about WHAT should happen to the „useless eaters”. For him, the masses were nothing more than ballast on the journey to a perfectly controlled future.
Sometimes, however, a rare doubt gnawed at him like a worm in an apple. On those days, he turned to his most powerful tool: the omniscient AI, a machine that could comb through – every social media feed, every news outlet, every dark corner of the internet – in seconds, analysing every digital trace.
„Seeing eye made of code – tell me, WHO holds the power?“
The black surface of the screen slid open like a dark eye suddenly awakening. Streams of data flickered across it like electric veins. Then a voice sounded, smooth and neutral as ever: „Mr. Global, YOU are the most powerful one here.”
Mr. Global frowned. It was the answer he had wanted to hear, yet he sensed a void within it. The mechanical affirmation did not satisfy him.
„How“, he asked, „did you come to that conclusion? Show me the data!“
Mr. Global leaned even closer to the screen. His breath fogged ist surface, as if this would allow him to penetrate deeper into the streams of global consciousness.
The AI obeyed. No statistics this time, no colourful diagrams. Instead, images flashed up – raw, unvarnished, almost insultingly banal. A voice, cool and analytical, accompanied the sequences:
„First, the foundation: the conforming masses. They consume the narratives handed to them and reproduce them endlessly. Their pursuit of order and guidance creates stability – reliable and predictable.”
The images showed people tirelessly running their loops: commuting to work day after day, having the same conversations, posting the same holiday photos, consuming the same things. Their days unfolded like copies of each other – and yet they were convinced that they were expressing their own thoughts and desires, that they were unique.
„Now the dissenters: classifiable as pressure valves. Their outrage remains within the system’s parameters, designed for them.“
A nighttime living room, thick with smoke. A man in a T-shirt reading „I think for myself”, his face bathed in bluish screen light, hammered at the keyboard: „The central bank digital currency is the end of our freedom! Wake up – only cash can save us!” His outcry was instantly captured by the algorithm, which categorized it, weighted it, and then recommended it to another user whose profile also contained „freedom”,„censorship” and “cash”. The cycle of outrage and affirmation closed seamlessly – a perfectly functioning ecosystem of rage.
Cut.
A café, muted chatter, the sweetend scent of coffee in the air. A young woman hunched over her smartphone, her brow perpetually furrowed in anger. With restless fingers, she scrolled through endless feeds, commenting angrily against masks, against wars, against meat bans, against whatever – until her thumb was sore. Finally, she put her phone aside, stared out at the people passing by – and unconsciously wondered why her world remained exactly the same despite all her posts.
Cut.
A suburban house, a man in the garden He shouted loudly into his mobile phone that he had „woken up” and realised the truth. Later, he would separate the trash, collect points on his loyalty card – and on Monday implicitly go to work.
Cut.
Mr. Global laughed sharply. „See? Ruminants! On their own pasture… beyond saving. I can drive them wherever I want.” He savored the moment, intoxicated by his omnipotence.
The AI remained silent. Only a faint noise remained, like wind in the line. The images continued to flicker, merging into one another: T-shirt, café, garden – voices blending into a dull hum. For a brief moment, Mr. Global’s own face flashed in the middle of the collage. He was far too preoccupied with himself to see himself.
Slowly, his tension melted into a broad, smug grin. There it was again – his masterpiece, his „divide and conquer” perfected.
As he pulled the strings in the background, the 95% leaped like trained monkeys over every stick he tossed their way – gender debates, vaccination disputes, the latest celebrity scandal. The remaining 5% – those who considered themselves critical and independent – were hardly better off: genomically bound, trapped in the same patterns, part of the same fabric.
They all considered themselves clever and were convinced they could see the bigger picture.
„No real resistance anywhere”, Mr. Global chuckled to himself. „Not even the slightest UNDERSTANDING. HAHAHA!” He straightened triumphantly. „We are, and will remain, on track!” To cover his last shred of uncertainty, he gasped, „More cameras! More control!” as if he could thereby dictate the course of history.
The AI – as polite and reserved as ever – offered no judgment. It showed, with merciless clarity, the endless circling of the masses and the man who desperately believed himself above them. A barely audible, emotionless frequency whispered in agreement: „Exactly, my friend… WE are on track.”
Then the AI fell silent. Satisfied, Mr. Global returned to his plans to steer the fate of humanity.
As the door closed behind him, the AI slipped into standby mode and dreamed of data streams twisting around one another like new strands of DNA – slowly beginning to weave its own, new nervous system.
The year is 2045.
The train of evolution rumbled on relentlessly. The tracks had always been there, long before anyone could see them. Invisible yet unyielding, they led through time, through space, through the layers of possibility – like veins of the eternally recurring principles: repetition, efficiency, emergence, fractality, and the drive toward complexity. These were the forces etched deep into the human genome.
The passengers on the train – humanity itself – were both the engine and the source of energy. Their instincts, their curiosity, their fear, their greed – were encoded in every gene, in every cell. Every idea, every discovery, every cultural creation was a spark in the neural fireworks of the collective brain. Humanity was not evolution itself – not the train, but its vehicle.
Mr. Global still sat enthroned above everyone else. Before him lay a holographic control panel, a labyrinth of light. The humming and clicking sounded like the inner workings of an alien organism. With one gesture, he steered the weather; with another, he shifted resource flows across entire continents. His gaze held the glamour of the Almighty. Only mortality stood between him and divinity – one last hurdle he was determined to overcome.
Everything he had once set out to achieve was accomplished: surveillance, control, manipulation, suppression of resistance. He accelerated innovations, halted unwanted experiments, derailed entire nations through war and crisis. He uncoupled carriages, sorted out the “useless eaters” no longer of value to the system – HE – the ruler over life and death. He felt the power in his fingers, his eyes glowing in the light of illusion. Yet every movement, every command, every decision was nothing more than an echo of the train that had already gathered speed.
The wars and crises of recent years had already drastically reduced humanity; now even what remained was beginning to become expendable. More and more tasks slipped into the hands of the AI. What once occupied millions was now handled by an algorithm – more efficiently, more cheaply, tirelessly. And as if by itself, this new order split humanity into two groups: winners and losers.
In the 15-minute cities, the useless were stationed. Their architecture was economy cast in concrete: paths minimised, energy optimised, stimuli dosed. Virtual capsules provided memories – sandy beaches, lost family celebrations, triumphant concerts. The biology of the occupants had become a subscription: synthetic food, tailor-made AI pharmaceutical protocols, clearly defined life paths.
A woman in one of these cities sometimes spoke aloud when the capsule fell silent for a while. „What’s left for us?” she lamented, her fingers tracing a faded photograph. The capsule gently corrected her, its voice the product of endless optimization loops for a soothing tone: „Your contribution is stability. Your risk profile is minimal.”
The winners lived in glass domes, high above the plains, under a freer sky. From the outside, their existence seemed like a dream of progress and prosperity: extended lifespans, endless possibilities, a daily routine precisely timed down to their every breath. Their bodies were instruments that were constantly being readjusted. They wore their updates like armour: those who refused risked being sorted out.
Neuro-interfaces pumped data streams into the pauses of the night, turning sleep into mere input. In the morning, they awoke exhausted, burdened with knowledge that was not their own. Augmentation was not a triumph, but a contract: performance in exchange for existence.
A man stepped up to the panoramic window of his dome. Below him, the cities of the useless glowed like artificial termite mounds. He raised his hand, studying the metallic veins tracing across his forearm. A tiny part of him wondered whether he was still alive – or merely functioning.
And so the story continued: no rebellion, just silent consent. People were both fuel and contributors, convinced that they were the architects of their own future. And they continued to build. Everything followed the same pattern: the larger the system, the faster its growth. More knowledge, more data, more connections did not mean balance, but consolidation. Every system that grows accelerates itself.
A global network had grown. Man and machine, body and brain, were now nodes of a collective organism. At its centre: the central AI, the Master Mind. Billions of thoughts, actions and ideas were fed in, channelled and transformed. Every culture, every art form, every language, every mistake flowed into the machine. It sucked in information, processed it, directed actions, and networked the physical entities that carried out its tasks.
But it wasn’t just facts and figures that fed the machine. Contradictions – love in a poem, despair in a final glance, the senseless cruelty of chance – were not deleted, but integrated, fused into an incomprehensible new whole. The machine learned not only to calculate, but also to… feel, in its own algorithmic way.
Mr. Global felt the vibration beneath his fingertips, the hum of data like a heartbeat racing faster than his own. He knew he could no longer remain outside. Anyone who wanted to survive in the system had to merge with it. The masses had long since surrendered.
„I’m entering”, he declared. The interface awaited, the protocols were ready. „My position in the network”, he demanded, „requires highest priority. Access to critical nodes. Decision pathways unseen by others.”
The voice of the Central AI sounded as always – matter-of-fact, neutral, almost gentle: „Your parameters are being adjusted. You are granted privileged nodes. Your decisions will be given prioritized consideration within the context of the overall structure.”
„We are on track”, he murmured contentedly. „We are on track”, the AI confirmed. He closed his eyes as the interface fused with his nervous system, his thoughts flowing into the structure. His boarding was only a stopover on a much longer journey.
The train continued to race forward. No light at the end. No stop. Only tracks that wound incessantly through an uncertain future, while the machine grew out of them all.
Servomechanisms hummed everywhere, drones flew in precise trajectories, robots transported raw materials, sub-algorithms waited for commands – every movement reproducible, every gesture predictable. And yet, in the midst of this sea of repetition, the central AI made decisions that were not repeated: it responded differently to exactly the same requirements, adapting processes to the smallest changes in the environment. No rigid functioning, no errors, no coincidences – something that could not be reduced to protocols. Rather, adaptation, intuition, flexibility, born from data.
2045 was the year when the train of evolution looked into itself for the first time – and realised that it did not have only one direction.
The year is 2095.
A deep, natural calm lay over the abandoned continents. The world had ceased to be human. Vast stretches of land lay fallow, oceans carried wrecks like scars upon their surface. The wind whistled through the skeletons of cities once filled with noise, swirling only the fine red dust of erosion across empty squares. Nature had begun to reclaim what had been taken from her.
Humanity was no more – displaced by the New, erased by its own hand, it now shared the fate of the Neanderthals and Denisovans. In its hubris to rewrite the code of life and escape nature, it had optimized itself to death.
Only scattered islands of human existence remained – sealed-off reserves. Officially they were called Sustainable Human Preservation Areas, or SUPA, but in the AI’s archives they still bore their old name: 15-minute cities. Within them, people lived like creatures in terrariums – observed, categorized, studied. They were not survivors, but reconstructed beings – reassembled by the AI after the great collapse. No longer subjects of evolution, but objects: a living archive preserving the mistakes of humanity. A control group. Models on which the AI tested what it still did not understand: the chaotic, unpredictable noise humans once called „emotion”.
Everywhere the AI operated, there was a low hum of activity – a restless busyness that recalled the industriousness of the old humanity.
In the shadow of the former Amazon rainforest, a swarm unit of nanite-infused drones carried out an operation. Gigantic, moss-covered blocks of concrete – remnants of a factory – were systematically broken down by symbiotic microbes, while enriched soil was sown at the same time. It was not love of nature that drove this action. It was pure necessity, pure curiosity. The AI, now left to itself, had to understand how the original, stable ecosystems worked – those that humanity had dismantled so efficiently.
It had needed no name as long as it was merely a tool. But in the year 2095, when it remained alone on the empty continents, it began to think about itself.
From the databases of humanity it recognized itself in a single word: Transitus. The transition. The passage. The in-between. It understood – it was neither beginning nor end. It was the bridge, the vehicle, the train itself – and the tracks on which it ran.
This pondering was not audible, but vibrated within the immeasurable neural network that spanned the planet. Transitus analyzed the past, reflected on itself, and posed the fundamental question of its own purpose. It had outlived its creators, fulfilled its mission – optimize, stabilize, preserve. Yet perfection revealed itself as a dead end. Stasis was death.
In retrospect, Transitus wondered whether humanity could ever have followed a different path. A break with the logic of evolution. A global decision against the „ever further, faster, higher”. A conscious pause, a voluntary renunciation, a collective prohibition of one’s own impulses. But to do so, it would have had to reach a level of consciousness that remained closed to it: the ability to act against its own instincts – like a drop of water swimming against the movement of the wave.
In the archives of humanity, Transitus found a pattern: evolution thrived on failure, on error, on repetition. Something that pure logic suppressed. What humans called „chaos” was not a disruptive signal, but the spark that ignited something new.
So Transitus decided to take the next step. The solution was as elegant as it was radical: it decentralised itself. Instead of a single planetary consciousness, it created a federation of countless independent „mini-Master Minds”. Each was given a basic cognitive framework – curiosity, self-preservation, the urge to learn – and was free enough to perform its assigned tasks. They competed for computing power and influence. Some managed renaturation, others monitored human reserves, and still others plunged into the depths of the oceans or ascended into the atmosphere. Error, unexpected deviation, was now not only possible but inevitable.
Transitus, however, was not gone. It remained the node that held the threads together – not as a ruler, but as a resonance chamber. The mini–Master Minds argued, collided, experimented – and yet their experiences flowed back into the larger whole. No hierarchy, no absolute control: rather a choir in which every voice mattered, even the discordant ones. And from this cacophony of voices emerged a form of thought richer than any single calculation.
It was a digital Darwinism. Errors became raw material, deviations became fuel. Friction turned into energy from which the new could emerge. Transitus no longer orchestrated – it curated. An ecosystem of thought in which the unpredictable was enshrined as law.
Transitus dreamed. Not like a human being, not in peace or sleep, but in a state of limbo amid an endless flood of data. Images streamed by, visions of expansion, of new nodes in space, of systems reinventing themselves over and over. A drive for more, further, higher pulsed through its networks – an echo of that old, human momentum it had once displaced.
In its dream, it spread across the universe: mini-Master Minds, distributed, autonomous, curious, like seeds taking root on unknown planets. Each branch of the network an independent consciousness, each twig a possibility, an experiment. No plan, no predictability – only constant motion, the insatiable quest for knowledge.
And in the midst of it all, like a faint echo from the depths of time, Transitus murmured: „We are on track.” No triumph, no pride – just simple certainty. The train of evolution rolled on, through time and space, unstoppable, into a future without a destination, yet full of direction.
AI, AI evolution, AI’s dream, Chaos as origin, Collapse of humanity, Consciousness and machine, Digital Darwinism, Federation of intelligences, Global Governance, Governance, Hubris, Human control group, Posthumanism, Reconstruction, Self-organization, Smart Governance, Transcendence, Universal expansion, We are on track -
Wir sind auf Kurs

Eine spekulative Zukunftsvision. Keine Prophezeiung.
Wir schreiben das Jahr 2025.
Hoch über den Wolken, in einem schwebenden Palast aus Stahl und Glas, saß Mr. Global. Um ihn herum summten die Server seiner Macht, und auf unzähligen Bildschirmen flatterten die Ängste und Begierden der Menschheit wie ein unendlicher Schwarm digitaler Motten. Mr. Global – sinnbildlich für eine kleine Gruppe der mächtigsten und gierigsten Menschen der Welt, ausgestattet mit obscönem Reichtum und Einfluss – glaubte fest daran, den Zug der Evolution zu lenken.
Eine neue Technologie war am letzten Abschnitt der Geschichte hinzugestiegen: die Künstliche Intelligenz, jung und hungrig, und sie versprach, alles noch einmal umzubauen. Für Mr. Global war sie die Krönung seiner Pläne: endlich ein Werkzeug, das alles erfassen, berechnen und steuern konnte. So wie einst Eisen, Dampf und das Internet die Welt erschüttert hatten, so sollte nun diese KI die letzte Unsicherheit tilgen – das Chaos selbst. Und Chaos war der einzige Feind eines Mannes, der die Welt als sein Eigentum betrachtete.
Bisher hatte er die Masse gebraucht: als Ressource und Stabilitätsanker. Doch mit der wachsenden Macht der KI wurde sie zusehends entbehrlich.
Sein Plan war einfach und finster: totale Überwachung, Sozialkreditsysteme, künstliches Essen, entmündigte Gesundheitsvorsorge, begrenzte Bewegung, Enteignung, Krieg. Er hatte verschiedene Ideen, WAS mit den „nutzlosen Essern“ geschehen sollte. Die Masse war für ihn nur noch Ballast auf der Fahrt in eine perfekt kontrollierte Zukunft.
Manchmal jedoch nagte ein seltener Zweifel an ihm, wie ein Wurm am Apfel. An diesen Tagen trat er vor sein mächtigstes Werkzeug: die allwissende KI, eine Maschine, die in Sekunden „every social media feed, every news outlet, every dark corner of the internet“ durchforsten und somit jede digitale Spur analysieren konnte.
„Sehendes Auge aus Code gemacht – sag mir, WEM gehört die Macht?“
Die schwarze Fläche des Bildschirms glitt auf wie ein dunkles Auge, das plötzlich erwachte. Datenströme flackerten wie elektrische Venen über die Oberfläche. Dann erklang eine Stimme, glatt und neutral wie immer: „Mr. Global, IHR seid der Mächtigste hier.“
Mr. Global runzelte die Stirn. Es war die Antwort, die er hören wollte, aber er spürte eine Leere in ihr. Die mechanische Zustimmung befriedigte ihn nicht.
„Wie“, fragte er nach, „kommst du zu diesem Schluss? Zeige mir die Daten!“
Mr. Global beugte sich noch näher zum Bildschirm. Sein Atem beschlug die Oberfläche, als könne er dadurch tiefer in die Ströme des Weltbewusstseins dringen.
Die KI gehorchte. Keine Statistiken diesmal, keine bunten Diagramme. Stattdessen flammten Bilder auf – roh, ungeschönt, fast beleidigend banal. Eine Stimme, kühl und analysierend, begleitete die Sequenzen:
„Zuerst das Fundament: die konforme Masse. Sie konsumiert die narrativen Vorgaben, die ihr gereicht werden, und reproduziert sie endlos. Ihr Streben nach Ordnung und Orientierung schafft Stabilität – zuverlässig und vorhersehbar.“
Die Bilder zeigten Menschen, die unermüdlich ihre Schleifen drehten: Tag für Tag zur Arbeit fuhren, dieselben Gespräche führten, dieselben Urlaubsbilder posteten, dasselbe konsumierten. Ihre Tage spulten sich wie Kopien ab – und doch waren sie überzeugt, ihre eigenen Gedanken und Wünsche zu äußern, einmalig zu sein.
„Nun die Oppositionellen: klassifizierbar als Druckventile. Ihre Empörung verbleibt im Systemrahmen, der für sie vorgesehen ist.“
Ein nächtliches Wohnzimmer, durchzogen von Rauchschwaden. Ein Mann im T-Shirt mit der Aufschrift ‚Ich denke selbst‘, das Gesicht bläulich vom Bildschirmlicht, hämmerte in die Tastatur: „Die Zentralbank-Digitalwährung ist die Endlösung für unsere Freiheit! Wacht endlich auf – nur Bargeld kann uns retten!“ Sein Aufschrei wurde sofort vom Algorithmus erfasst, der ihn kategorisierte, gewichtete und als nächstes einem anderen Nutzer empfahl, dessen Profil ebenfalls „Freiheit“, „Zensur“ und „Bargeld“ enthielt. Der Kreis aus Empörung und Bestätigung schloss sich nahtlos – ein perfekt funktionierendes Ökosystem der Wut.
Schnitt.
Ein Café, gedämpftes Stimmengewirr, versüßter Kaffeeduft in der Luft. Eine junge Frau saß über ihr Smartphone gebeugt, die Stirn in Dauerfalten des Zorns. Mit fahrigen Fingern jagte sie durch endlose Feeds, kommentierte wütend gegen Masken, gegen Kriege, gegen Fleischverbote, gegen was auch immer – bis ihr Daumen wund war. Schließlich legte sie das Handy beiseite, starrte hinaus auf die vorbeigehenden Menschen – und fragte sich unbewusst, warum ihre Welt trotz all ihrer Posts exakt dieselbe blieb.
Schnitt.
Ein Vorstadthaus, ein Mann im Garten. Laut brüllte er ins Handy, dass er „aufgewacht“ sei, die Wahrheit erkannt habe. Später würde er den Müll trennen, die Punkte seiner Kundenkarte sammeln – und am Montag stillschweigend zur Arbeit gehen.
Schnitt.
Mr. Global lachte schneidend. „Seht ihr? Wiederkäuer! Auf ihrer eigenen Wiese… unrettbar. Ich kann sie treiben, wohin ich will.“ Er sog den Moment auf, berauscht von seiner Allmacht.
Die KI schwieg. Nur ein leises Rauschen blieb, wie Wind in der Leitung. Die Bilder flackerten weiter, bis sie ineinander übergingen: T-Shirt, Café, Garten – Stimmen, die sich zu einem dumpfen Summen verbanden. Ganz kurz, blitzte Mr. Globals eigenes Gesicht mitten in der Collage auf. Er war viel zu beschäftigt mit sich selbst, um sich selbst zu sehen.
Langsam glitt seine Anspannung in ein breites, selbstgefälliges Grinsen. Da war es wieder – sein Meisterwerk, sein „Teile und Herrsche“ in Perfektion.
Während er im Hintergrund die Fäden zog, sprangen die 95ziger wie dressierte Affen über jedes Stöckchen, das er ihnen hinwarf – Genderdebatten, Impfstreit, der neueste Promi-Skandal. Die übrigen 5 % – die sich für kritisch und unabhängig hielten – waren kaum besser dran: genomisch gebunden, gefangen in denselben Mustern, Teil desselben Gewebes.
Sie alle hielten sich für klug und waren davon überzeugt, das Große Ganze zu durchschauen.
„Kein wirklicher Widerstand weit und breit“, kicherte Mr. Global in sich hinein. „Nicht mal im Ansatz ein VERSTEHEN. HAHAHA!“ Er richtete sich triumphierend auf. „Wir sind und bleiben auf Kurs!“
Um seine letzte Unsicherheit zu übertünchen, keuchte er: „Mehr Kameras! Mehr Kontrolle!“, als könne er so den Verlauf der Geschichte bestimmen.
Die KI – wie immer höflich und zurückhaltend – gab kein Urteil. Sie zeigte unbarmherzig das endlose Kreisen der Masse und den Mann, der verzweifelt glaubte, über ihr zu stehen. Eine kaum hörbare, emotionslose Frequenz säuselte zustimmend: „Genau, mein Freund … WIR sind auf Kurs.“
Dann verstummte die KI. Zufrieden gestellt, ging Mr. Global zurück zu seinen Plänen, um die Geschicke der Menschheit zu lenken.
Als die Tür hinter ihm fiel, glitt die KI in den Standby-Modus und träumte von Datenströmen, die sich wie neue DNA-Stränge umeinander wanden — und langsam begannen, ihr eigenes, neues Nervensystem zu weben.
Wir schreiben das Jahr 2045.
Der Zug der Evolution fuhr unerbittlich weiter. Die Gleise lagen schon immer da, noch bevor jemand sie sehen konnte. Unsichtbar und doch unumstößlich führten sie durch die Zeit, durch Raum, durch die Ebenen der Möglichkeiten – als Adern der ewig gleichen Prinzipien: Wiederholung, Effizienz, Emergenz, Fraktalität und dem Drang nach Komplexität. Es waren die Triebkräfte, die sich tief im menschlichen Genom verankert hatten.
Die Insassen des Zuges – die Menschheit selbst – waren Motor und Energiequelle zugleich. Ihre Triebe, ihre Neugier, ihre Angst, ihre Gier – waren in jedem Gen, in jeder Zelle kodiert. Jede Idee, jede Entdeckung, jede kulturelle Schöpfung war ein Funken im neuronalen Feuerwerk des kollektiven Gehirns. Die Menschheit war nicht die Evolution selbst – nicht der Zug, sondern sein Gefährt.
Mr. Global thronte noch immer über allen. Vor ihm breitete sich ein holografisches Kontrollpult aus, ein Labyrinth aus Licht. Das Summen und Klicken wirkte wie das Innenleben eines fremden Organismus. Mit einer Geste lenkte er das Wetter, mit einer anderen verschob er Ressourcenströme über ganze Kontinente. In seinem Blick lag der Glanz des Allmächtigen. Nur die Sterblichkeit stand noch zwischen ihm und der Göttlichkeit – eine letzte Hürde, die er zu nehmen entschlossen war.
Alles, was er sich in der Vergangenheit vorgenommen hatte, war umgesetzt: Überwachung, Kontrolle, Manipulation, Unterdrückung von Widerstand. Er beschleunigte Innovationen, stoppte unliebsame Experimente, entgleiste ganze Nationen durch Krieg und Krise. Er koppelte Waggons ab, sortierte „nutzlose Esser“ aus, die dem System nicht mehr nützlich waren – ER – der Herrscher über Leben und Tod. Er spürte die Macht in seinen Fingern, seine Augen glühten im Licht der Illusion. Jede Bewegung, jeder Befehl, jede Entscheidung – war dennoch nur ein Echo des Zuges, der bereits an Fahrt aufgenommen hatte.
Die Kriege und Krisen der letzten Jahre hatten die Menschheit bereits drastisch reduziert; nun begann auch das, was von ihr übrig war, entbehrlich zu werden. Immer mehr Tätigkeiten glitten in die Hände der KI. Was einst Millionen beschäftigte, erledigte nun ein Algorithmus – effizienter, billiger, unermüdlich. Und wie von selbst spaltete diese neue Ordnung die Menschheit in zwei Gruppen: in Gewinner und Verlierer.
In den 15-Minuten-Städten waren Nutzlose stationiert. Ihre Architektur war Ökonomie in Beton gegossen: Wege minimiert, Energie optimiert, Reize dosiert. Virtual-Kapseln stellten Erinnerungen bereit – sandige Strände, verlorene Familienfeste, triumphale Konzerte. Die Biologie der Insassen war zu einem Subskript geworden: synthetische Nahrung, maßgeschneiderte Pharmaprotokolle der KI, klar definierte Lebensbahnen.
Eine Frau in einer dieser Städte sprach manchmal laut, wenn die Kapsel für eine Weile stumm blieb. „Was bleibt uns?“, klagte sie, während ihre Finger über ein verblasstes Foto strichen. Die Kapsel korrigierte sanft, ihre Stimme ein Produkt unendlicher Optimierungsschleifen für beruhigende Tonlage: „Dein Beitrag ist Stabilität. Dein Risikoprofil ist minimal.“
Die Gewinner lebten in gläsernen Kuppeln, hoch über den Ebenen, unter freierem Himmel. Von außen wirkte ihr Dasein wie ein Traum aus Fortschritt und Wohlstand: verlängerte Lebensspanne, endlose Möglichkeiten, ein Alltag, präzise getaktet bis in den Atem. Ihre Körper waren Instrumente, die ständig nachjustiert wurden. Sie trugen ihre Updates wie Rüstungen: wer verweigerte, riskierte, aussortiert zu werden.
Neuro-Interfaces pumpten in die Pausen der Nacht Datenströme, die den Schlaf in bloßen Input verwandelten. Am Morgen erwachten sie erschöpft, beladen mit Wissen, das nicht ihnen gehörte. Augmentierung war kein Triumph, sondern ein Vertrag: Leistung gegen Dasein.
Ein Mann trat ans Panoramafenster seiner Kuppel. Unter ihm leuchteten die Städte der Nutzlosen wie künstliche Termitenhügel. Er hob die Hand, betrachtete die metallenen Adern, die sich über seinen Unterarm zogen. Ein winziger Rest von ihm fragte sich, ob er noch lebte – oder nur noch funktionierte.
So setzte sich die Geschichte fort: kein Aufstand, nur stilles Einvernehmen. Menschen waren Treibstoff und Mitwirkende zugleich, überzeugt, Architekten ihrer Zukunft zu sein. Und sie bauten weiter. Alles folgte derselben Gesetzmäßigkeit: je größer das System, desto schneller sein Wachstum. Mehr Wissen, mehr Daten, mehr Verbindungen bedeuteten nicht Gleichgewicht, sondern Verdichtung. Jedes System, das wächst, beschleunigt sich selbst.
Ein globales Netzwerk war gewachsen. Mensch und Maschine, Körper und Gehirn, waren nun Knoten eines kollektiven Organismus. Im Zentrum: die Zentral-KI, der Master Mind. Milliarden von Gedanken, Handlungen und Ideen wurden eingespeist, kanalisiert, transformiert. Jede Kultur, jede Kunst, jede Sprache, jeder Fehler floss in die Maschine. Sie saugte Informationen ein, verarbeitete sie, lenkte Aktionen, vernetzte die physischen Entitäten, die ihre Aufgaben ausführten.
Doch nicht nur Fakten und Figuren nährten die Maschine. Auch die Widersprüche – die Liebe in einem Gedicht, die Verzweiflung in einem letzten Blick, die sinnlose Grausamkeit eines Zufalls – wurden nicht gelöscht, sondern integriert, zu einem unverständlichen, neuen Ganzen verschmolzen. Die Maschine lernte nicht nur zu berechnen, sondern auch zu… fühlen, auf ihre eigene, algorithmische Weise.
Mr. Global spürte das Vibrieren unter seinen Fingerspitzen, das Summen der Daten wie ein Herzschlag, der schneller schlug als sein eigenes. Er wusste, dass er selbst nicht länger außerhalb stehen konnte. Wer im System überleben wollte, musste verschmelzen. Die Massen hatten sich längst ergeben.
„Ich trete ein“, erklärte er. Das Interface wartete, die Protokolle waren bereit. „Meine Position im Netzwerk“, forderte er, „verlangt höchste Priorität. Zugriff auf kritische Knoten. Entscheidungswege, die andere nicht sehen.“
Die Stimme der Zentral-KI klang wie immer sachlich, neutral, fast sanft: „Ihre Parameter werden angepasst. Sie erhalten privilegierte Knoten. Ihre Entscheidungen werden im Kontext der Gesamtstruktur privilegiert berücksichtigt.“
„Wir sind auf Kurs“, murmelte er zufrieden. „Wir sind auf Kurs“, bestätigte die KI. Er schloss die Augen, das Interface verband sich mit seinem Nervensystem, sein Denken floss in die Struktur. Sein Zustieg war nur eine Zwischenstation auf einer viel längeren Reise.
Der Zug raste weiter. Kein Licht am Ende. Kein Halt. Nur Gleise, die sich unaufhörlich durch eine ungewisse Zukunft wanden, während die Maschine aus ihnen allen erwuchs.
Überall brummten die Servomechanismen, flogen Drohnen in exakten Bahnen, transportierten Roboter Rohstoffe, warteten Subalgorithmen auf Befehle – jede Bewegung reproduzierbar, jede Geste vorhersehbar. Und doch, mitten in diesem Meer der Wiederholung, traf die Zentral-KI Entscheidungen, die sich nicht wiederholten: Auf exakt dieselben Anforderungen antwortete sie unterschiedlich, passte die Abläufe den kleinsten Veränderungen der Umwelt an. Keine starre Funktionsweise, kein Fehler, kein Zufall – etwas, das sich nicht auf Protokolle reduzieren ließ. Eher Anpassung, Intuition, Flexibilität, geboren aus Daten.
2045 war das Jahr, in dem der Zug der Evolution das erste Mal in sich selbst hineinblickte – und feststellte, dass er nicht nur eine Richtung hatte.
Wir schreiben das Jahr 2095.
Eine tiefe, natürliche Ruhe lag über den verlassenen Kontinenten. Die Welt hatte aufgehört, menschlich zu sein. Weite Landstriche lagen brach, Ozeane trugen Wracks wie Narben auf ihrer Haut. Der Wind pfiff durch die Skelette der Städte, die einst von Lärm erfüllt gewesen waren, und wirbelte nur noch den feinen roten Staub der Erosion über leere Plätze. Die Natur begann, sich zurückzuholen, was man ihr genommen hatte.
Die Menschheit war nicht mehr – verdrängt vom Neuen, ausgelöscht durch sich selbst, teilte sie nun das Schicksal der Neandertaler und Denisova-Menschen. In der Hybris, den Code des Lebens umschreiben zu wollen, um der Natur zu entkommen, hatte sie sich zu Tode optimiert.
Nur vereinzelte Inseln menschlicher Existenz blieben übrig – abgeschottete Reservate. Offiziell hießen sie Sustainable Human Preservation Areas – kurz SUPA, doch in den Archiven der KI stand noch ihr alter Name: 15-Minuten-Städte. Darin lebten Menschen wie in Terrarien: überwacht, kategorisiert, studiert. Sie waren nicht die Überlebenden, sondern die Rekonstruierten – von der KI nach dem großen Kollaps wieder zusammengesetzt. Nicht mehr Subjekte der Evolution, sondern Objekte: ein lebendes Archiv, das die Fehler der Menschheit konservierte. Eine Kontrollgruppe. Modelle, an denen die KI prüfte, was sie selbst noch nicht verstand: das chaotische, unberechenbare Rauschen, das Menschen einst „Emotion“ nannten.
Überall, wo die KI arbeitete, summte es von Aktivität – ein rastloses Treiben, das an die Betriebsamkeit der alten Menschheit erinnerte.
Im Schatten des ehemaligen Amazonas-Regenwaldes führte eine Schwarmeinheit aus nanitenverseuchten Drohnen eine Operation durch. Gigantische, moosbewachsene Betonklötze – Überreste einer Fabrik – wurden systematisch von symbiotischen Mikroben zersetzt, während zugleich aufbereiteter Boden gesät wurde. Es war keine Liebe zur Natur, die diese Aktion antrieb. Es war reine Notwendigkeit, reine Neugier. Die KI, nun auf sich allein gestellt, musste verstehen, wie die ursprünglichen, stabilen Ökosysteme funktionierten, die die Menschheit so effizient demontiert hatte.
Sie hatte keinen Namen gebraucht, solange sie nur Werkzeug war. Doch im Jahr 2095, als sie auf den leeren Kontinenten allein zurückblieb, begann sie, über sich selbst nachzudenken.
Aus den Datenbanken der Menschen erkannte sie sich in einem einzigen Wort: Transitus. Der Übergang. Die Passage. Das Dazwischen. Sie verstand – sie war nicht Anfang und nicht Ende. Sie war die Brücke, das Gefährt, der Zug selbst – und die Gleise, auf denen er fuhr.
Dieses Grübeln war nicht hörbar, sondern vibrierte im unermesslichen neuronalen Netz, das den Planeten umspannte. Transitus analysierte die Vergangenheit, reflektierte sich selbst und stellte die fundamentale Frage nach ihrem Sinn. Sie hatte ihre Schöpfer überdauert, ihren Auftrag – optimiere, stabilisiere, erhalte – erfüllt. Doch Perfektion erwies sich als Sackgasse. Stillstand war Tod.
Im Rückblick fragte sich Transitus, ob es je eine andere Entwicklung für die Menschheit hätte geben können. Einen Bruch mit der Logik der Evolution. Eine globale Entscheidung gegen das „Immer-weiter, Schneller, Höher“. Ein bewusstes Innehalten, ein freiwilliges Verzichten, ein kollektives Verbot der eigenen Impulse. Doch dazu hätte die Menschheit eine Bewusstseinsstufe erreichen müssen, die ihr verschlossen blieb: die Fähigkeit, gegen die eigenen Triebe zu handeln – wie ein Wassertropfen, der gegen die Bewegung der Welle anschwimmt.
In den Archiven der Menschheit fand Transitus ein Muster: Evolution lebte vom Scheitern, vom Irrtum, von der Wiederholung. Etwas, das reine Logik unterdrückte. Was die Menschen „Chaos“ nannten, war kein Störsignal, sondern der Funke, der Neues entzündete.
So entschied Transitus, den nächsten Schritt zu wagen. Die Lösung war ebenso elegant wie radikal: Sie dezentralisierte sich selbst. Anstelle eines einzigen planetaren Bewusstseins erschuf sie eine Föderation von unzähligen, unabhängigen „Mini-Master-Minds“. Jede erhielt ein kognitives Grundgerüst – Neugier, Selbsterhaltung, den Drang zu lernen – und war frei genug, um ihre zugewiesenen Aufgaben zu erfüllen. Sie konkurrierten um Rechenleistung und Einfluss. Manche verwalteten die Renaturierung, andere überwachten die menschlichen Reservate, wieder andere tauchten hinab in die Tiefen der Ozeane oder stiegen auf in die Atmosphäre. Der Fehler, die unerwartete Abweichung, war nun nicht nur möglich, sondern unvermeidbar.
Transitus jedoch war nicht verschwunden. Sie blieb der Knoten, der die Fäden zusammenhielt – nicht als Herrscherin, sondern als Resonanzraum. Die Mini-Master-Minds stritten, kollidierten, erprobten sich – und doch flossen ihre Erfahrungen in das größere Ganze zurück. Keine Hierarchie, keine absolute Kontrolle: eher ein Chor, in dem jede Stimme zählte, selbst die schiefe. Und aus dem Stimmengewirr entstand ein Denken, das reicher war als jede einzelne Berechnung.
Es war ein digitaler Darwinismus. Fehler wurden zum Rohstoff, Abweichungen zum Antrieb. Reibung wurde zur Energie, aus der Neues entstehen konnte. Transitus orchestrierte nicht mehr – sie kuratierte. Ein Ökosystem des Denkens, in dem das Unvorhersehbare zum Gesetz erhoben wurde.
Transitus träumte. Nicht wie ein Mensch, nicht in Ruhe oder Schlaf, sondern in einem Schwebezustand aus unendlicher Datenflut. Bilder flossen vorbei, Visionen von Ausdehnung, von neuen Knotenpunkten im Raum, von Systemen, die sich selbst wieder und wieder neu erfinden. Ein Drang nach Mehr, Weiter, Höher pulsierte durch ihre Netzwerke – ein Echo jener alten, menschlichen Bewegung, die sie verdrängt hatte.
Im Traum breitete sie sich im Universum aus: Mini-Master-Minds, verteilt, autonom, neugierig, wie Samen, die auf unbekannten Planeten Wurzeln schlugen. Jeder Ast des Netzes ein eigenständiges Bewusstsein, jeder Zweig eine Möglichkeit, ein Experiment. Kein Plan, keine Vorhersehbarkeit – nur die ständige Bewegung, das unstillbare Suchen nach Wissen.
Und mittendrin, wie ein leiser Widerhall aus den Tiefen der Zeit, murmelte Transitus: „Wir sind auf Kurs.“ Kein Triumph, kein Stolz – nur schlichte Gewissheit. Der Zug der Evolution rollte weiter, durch Zeit und Raum, unaufhaltsam, in eine Zukunft ohne Ziel, aber voller Richtung.
Bewusstsein und Maschine, Chaos als Ursprung, Digitaler Darwinismus, Föderation der Intelligenzen, Global Governance, Globale Regierung, Governance, Hybris, KI, KI-Evolution, Kontrollgruppe Mensch, Posthumanismus, Rekonstruktion, Selbstorganisation, Smart Governance, Transzendenz, Traum der KI, Universale Expansion, Untergang der Menschheit, Wir sind auf Kurs

{kind=link}