Leitfaden zur Artikelserie
Teil 1: Herstellung und Reinigung von modRNA
Teil 2: Nachweisverfahren und Studienlage
Nach der Darstellung der Herstellungs- und Reinigungsprozesse im ersten Teil richtet sich der Blick nun auf die analytische Ebene der Diskussion. Denn die Frage, ob und in welcher Menge DNA-Rückstände in modRNA-basierten Impfstoffen vorhanden sind, hängt entscheidend davon ab, mit welchen Methoden diese überhaupt untersucht werden.
Der Nachweis residualer Nukleinsäuren ist methodisch anspruchsvoll. Unterschiedliche Aufschlussverfahren, Extraktionsmethoden und Analyseplattformen können zu deutlich abweichenden Ergebnissen führen. Damit wird nicht nur die Interpretation einzelner Studien komplex, sondern bereits die grundlegende Frage, was eine Messung unter den jeweiligen Bedingungen tatsächlich erfasst.
Der zweite Teil widmet sich daher den wichtigsten analytischen Nachweisverfahren für DNA und RNA, ihren methodischen Grenzen sowie den bisher veröffentlichten unabhängigen Untersuchungen zu DNA-Rückständen in modRNA-basierten Impfstoffen. Dabei stehen insbesondere die Aussagekraft der verwendeten Methoden und die Vergleichbarkeit der Ergebnisse im Mittelpunkt.
Hinweis
Im allgemeinen Sprachgebrauch ist der Begriff „mRNA-Impfstoff“ gebräuchlich. Fachlich handelt es sich bei den derzeit zugelassenen Präparaten um nukleosid-modifizierte mRNA (modRNA). Aus Gründen der Verständlichkeit wird in dieser Arbeit dennoch der etablierte Begriff „mRNA-Impfstoff“ verwendet.
Inhaltsverzeichnis
2. Methoden zur Messung von DNA und RNA
3. Richtlinien zur Begrenzung von Rest-DNA in Impfstoffen
4. Studien zu DNA-Rückständen in modRNA-basierten Impfstoffen
5. Schlussbetrachtung und Ausblick

2. Methoden zur Messung von RNA und DNA
Die analytische Erfassung von RNA-Konzentration und potenziellen DNA-Rückständen stellt einen zentralen Bestandteil der Qualitätskontrolle mRNA-basierter Arzneimittel dar. Während der Herstellungs- und Reinigungsprozesse entstehen komplexe Nukleinsäuregemische, deren Zusammensetzung durch geeignete Messverfahren charakterisiert und überwacht werden muss. Ziel ist es, sowohl die Integrität und Menge der gewünschten mRNA als auch mögliche Restbestandteile wie DNA-Fragmente zuverlässig zu erfassen.
Dabei existiert kein einzelnes Verfahren, das alle analytischen Fragestellungen gleichermaßen abdeckt. Vielmehr unterscheiden sich die verfügbaren Methoden hinsichtlich Sensitivität, Spezifität, Quantifizierbarkeit und struktureller Auflösung. Einige Verfahren ermöglichen eine schnelle Bestimmung der Gesamtnukleinsäurekonzentration, andere erlauben die selektive Detektion spezifischer DNA-Sequenzen oder sogar die direkte Sequenzanalyse einzelner Moleküle.
Vor diesem Hintergrund werden im Folgenden verschiedene methodische Ansätze systematisch dargestellt – beginnend mit grundlegenden spektralphotometrischen Verfahren, über fluoreszenzbasierte Quantifizierungsmethoden und amplifikationsgestützte Nachweisverfahren bis hin zu sequenzierungsbasierten Technologien. Die Darstellung folgt dabei einer zunehmenden analytischen Auflösung: von der globalen Konzentrationsmessung hin zur sequenzspezifischen Identifikation einzelner Nukleinsäurebestandteile.
Neben dem Funktionsprinzip der jeweiligen Methode werden auch deren Nachweisgrenzen und methodische Einschränkungen berücksichtigt, da diese für die Einordnung möglicher Rest-DNA-Befunde von entscheidender Bedeutung sind.
2.1. Spektralphotometrische Verfahren
2.2. Fluoreszenzbasierte Quantifizierung
2.3. Amplifikationsbasierte Methoden
2.4. Sequenzierungsbasierte Verfahren
2.5. Elektrophoretische Fragmentanalyse
2.6. Vergleich zentraler Nachweisverfahren für Nukleinsäuren
2.1. Spektralphotometrische Verfahren
Spektralphotometrische Verfahren beruhen auf der Wechselwirkung von Licht mit Materie. Gemessen wird die Lichtintensität – vor und nach dem Durchgang durch eine Probe – in Abhängigkeit von der Wellenlänge. Aus der gemessenen Absorption lässt sich unter definierten Bedingungen auf die Konzentration der enthaltenen Substanzen schließen.
Die UV/Vis-Spektrophotometrie stellt eine etablierte Methode zur Bestimmung der Gesamtnukleinsäurekonzentration dar und wird in mehreren Phasen der mRNA-Herstellung eingesetzt. Sie zeichnet sich durch eine schnelle Durchführung, geringen Probenverbrauch und vergleichsweise niedrige Kosten aus.
Wann wird die Photometrie eingesetzt?
Im Herstellungsprozess lassen sich typischerweise drei relevante Messzeitpunkte unterscheiden:
Nach der in-vitro-Transkription: zur Abschätzung, ob die Transkriptionsreaktion erfolgreich war und welche Gesamtausbeute an Nukleinsäure erzielt wurde.
Nach der Aufreinigung: als rascher Kontrollschritt zur Überprüfung, ob die mRNA-Konzentration im erwarteten Bereich liegt.
Vor der Formulierung: Vor der Verkapselung in Lipid-Nanopartikel wird die mRNA-Konzentration erneut bestimmt, da das Mischungsverhältnis zwischen Lipiden und mRNA entscheidend für die Partikelbildung ist.
Es ist zu beachten, dass die Methode nicht zwischen RNA und DNA unterscheiden kann und daher primär eine orientierende Gesamtkonzentrationsbestimmung darstellt.
Typischer Ablauf im Herstellungsprozess
Nach der Aufreinigung der mRNA und vor der LNP-Formulierung wird die mRNA-Wirkstoffcharge einer Konzentrationsbestimmung unterzogen. Hierzu wird unter kontrollierten Bedingungen eine kleine Probe aus dem Prozessbehälter entnommen.
Für die Messung stehen zwei Gerätetypen zur Verfügung, die sich in ihrer Handhabung unterscheiden:
UV/Vis-Spektralphotometer: Die Probe wird in eine Küvette gefüllt – ein kleines, meist rechteckiges Gefäß aus UV-durchlässigem Material (z. B. Quarzglas). Die Küvette wird in das Gerät gestellt, wo die Messung stattfindet.
Mikrovolumen-Spektralphotometer (z. B. NanoDrop): Ein winziger Tropfen (0,5–2,0 µl) wird direkt auf einen Messsockel pipettiert. Ein zweiter (umgeklappter) Sockel bildet durch Oberflächenspannung eine definierte Flüssigkeitssäule zwischen den beiden Messflächen. Der große Vorteil: Es werden weder Küvetten noch nennenswerte Probenmengen benötigt. Diese Geräte werden häufig für DNA- und RNA-Messungen eingesetzt.
Unabhängig vom Gerätetyp dauert die eigentliche spektralphotometrische Analyse nur wenige Sekunden. Unmittelbar nach dem Messvorgang wird das Ergebnis auf dem Bildschirm angezeigt.

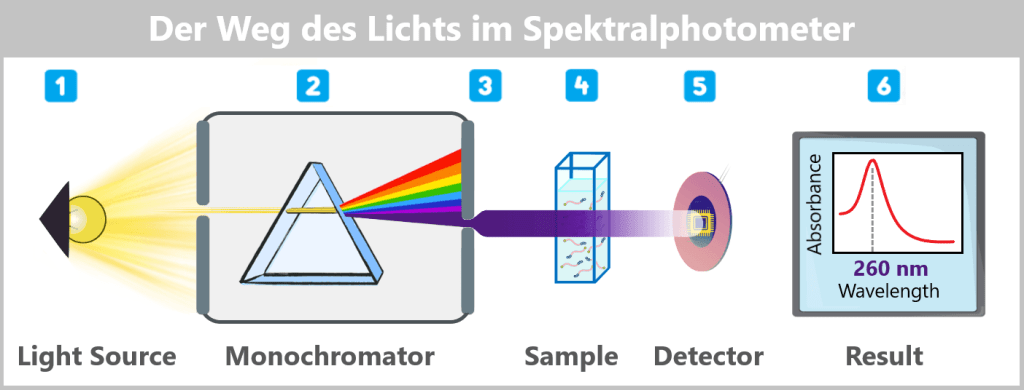
Der Weg des Lichts im Spektralphotometer
1️⃣ Lichterzeugung: Als Lichtquelle dient eine breitbandige UV/Vis-Lampe, beispielsweise eine Xenon-Blitzlampe, die kurze, intensive Lichtblitze aussendet und das gesamte UV- und sichtbare Spektrum abdeckt.
2️⃣ Wellenlängenselektion: Das Licht wird durch den Eintrittsspalt in den Monochromator geleitet und dort durch ein bewegliches Beugungsgitter in seine spektralen Bestandteile zerlegt. Um die Konzentration der mRNA zu bestimmen, wird das Gitter so eingestellt, dass Licht mit einer Wellenlänge von 260 nm – dem Absorptionsmaximum der Nukleinsäuren – auf die Probe trifft.
3️⃣ Lichtführung zur Probe: Über ein System von Spiegeln und Linsen wird der monochromatische Lichtstrahl gebündelt und präzise auf die Probe gerichtet – entweder auf die Küvette oder direkt auf den hängenden Probentropfen.
4️⃣ Wechselwirkung mit der Probe: Ein Teil des Lichts wird von den Nukleinsäuremolekülen absorbiert (verschluckt), der Rest transmittiert (durchdringt) die Probe.
5️⃣ Detektion: Das transmittierte Licht trifft auf einen Detektor (z. B. eine Photodiode oder ein CCD-Sensor), der die ankommende Lichtintensität misst.
6️⃣ Berechnung: Das Gerät vergleicht die gemessene Intensität mit der Intensität des ursprünglich eingestrahlten Lichts (Referenzmessung ohne Probe). Aus dem Verhältnis von eingestrahlter zu transmittierter Lichtintensität berechnet das Gerät gemäß dem Lambert-Beerschen Gesetz die Absorbanz, aus der sich bei bekannter Schichtdicke die Konzentration der Nukleinsäuren in der Probe bestimmen lässt.
Warum absorbieren RNA und DNA bei 260 nm?
Die Absorption von UV-Licht bei 260 nm ist eine charakteristische Eigenschaft der Basen – jener „Buchstaben“, aus denen der genetische Code besteht. Da diese Basen in RNA und DNA nahezu identisch sind, weisen beide Moleküle ein sehr ähnliches Absorptionsverhalten auf.
Das Geheimnis liegt in ihrer chemischen Struktur: Die Basen enthalten sogenannte aromatische Ringe. In diesen Ringen sitzen die Elektronen nicht an festen Positionen zwischen zwei Atomen, sondern sind über das gesamte Ringsystem verteilt. Man kann sich diese „delokalisierten Elektronen“ wie eine Art Elektronenwolke vorstellen, die über dem Molekül schwebt.
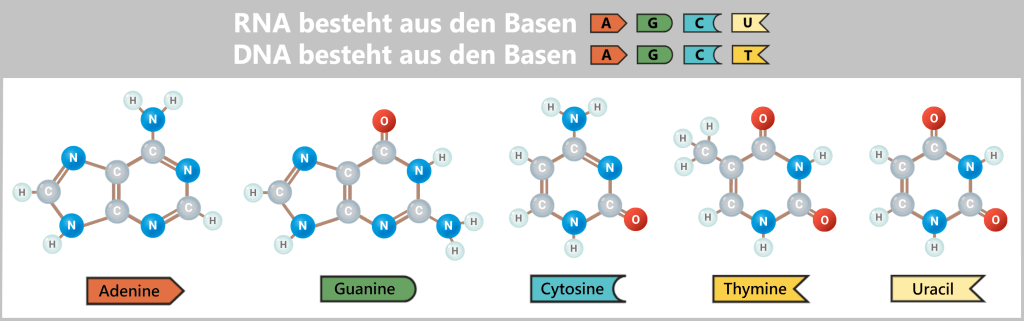
Die Darstellung zeigt die zwei Typen von Basen: die größeren Doppelringe (Purine: A und G) und die kleineren Einzelringe (Pyrimidine: C, T und U). Trotz ihrer unterschiedlichen Größe besitzen beide Typen ein System aus abwechselnden Doppelbindungen. Dieses sorgt dafür, dass die Elektronen über das gesamte Ringsystem verteilt sind und so eine gemeinsame Wolke bilden.
Wenn nun ein Photon (ein Lichtteilchen) mit einer Wellenlänge von etwa 260 nm auf diese Wolke trifft, passt dessen Energie exakt zu den energetischen Eigenschaften der Elektronen in diesen Ringen. Ein Elektron nimmt die Energie des Photons vollständig auf und springt dadurch auf ein höheres Energieniveau – den sogenannten angeregten Zustand. Die Energie des Photons wird dabei vollständig auf das Elektron übertragen, sodass es nicht mehr als Lichtteilchen weiterexistiert – man spricht von Absorption.
Da sowohl RNA als auch DNA aus denselben Basentypen bestehen (mit der Ausnahme von Uracil statt Thymin), zeigen beide Molekülarten ein nahezu identisches Verhalten im UV-Licht. Die Spektrophotometrie erlaubt daher keine direkte Unterscheidung zwischen RNA und DNA.
Bedeutung für die Bewertung von DNA-Rückständen
Die dargestellten physikalischen Grundlagen erklären zugleich eine zentrale Limitation der UV-Spektrophotometrie: Da die Absorption bei etwa 260 nm auf den gemeinsamen Basenstrukturen von RNA und DNA beruht, erfasst die Methode ausschließlich die Gesamtmenge an Nukleinsäuren in einer Probe. Eine Differenzierung zwischen therapeutischer mRNA und eventuell verbliebenen DNA-Restfragmenten ist auf dieser Grundlage nicht möglich.
Für die quantitative Bestimmung spezifischer DNA-Rückstände sind daher analytische Verfahren erforderlich, die entweder selektiv doppelsträngige DNA erfassen oder definierte Zielsequenzen amplifizieren. In der Praxis stehen hierbei insbesondere fluorometrische DNA-Methoden (z. B. Qubit-Technologien) sowie die quantitative Polymerase-Kettenreaktion (qPCR) im Mittelpunkt.
Die Diskussion um geeignete Nachweismethoden für DNA-Rückstände konzentriert sich daher weniger auf photometrische Verfahren, sondern primär auf molekularbiologische und fluoreszenzbasierte Techniken.
2.2. Fluoreszenzbasierte Quantifizierung
2.2.1. DNA-/RNA-spezifische Farbstoffe
a) Wirkmechanismus von PicoGreen
b) Warum PicoGreen bevorzugt doppelsträngige DNA erfasst
c) Grenzen der PicoGreen-Methode
2.2.2. Qubit-Systeme
Im Gegensatz zur UV-Spektrophotometrie, bei der die Abschwächung eines eingestrahlten Lichtstrahls gemessen wird, beruhen fluoreszenzbasierte Verfahren auf der gezielten Anregung spezifischer Moleküle und der anschließenden Messung des von ihnen ausgesandten Lichts. (Oder einfacher formuliert: Während die UV-Spektrophotometrie feststellt, wie viel Licht „verschwindet“, misst die Fluoreszenz nicht den Schatten, sondern das Leuchten.)
Dabei wird der Probe ein Fluoreszenzfarbstoff zugesetzt, der selbst kaum oder gar nicht fluoresziert. Erst nach Bindung an eine Zielstruktur – beispielsweise doppelsträngige DNA oder RNA – ändert sich seine räumliche Anordnung, sodass er nach Anregung mit Licht einer bestimmten Wellenlänge ein charakteristisches Fluoreszenzsignal aussendet. Die Intensität dieses emittierten Lichts ist proportional zur Menge der gebundenen Nukleinsäure.
Das entscheidende Merkmal fluoreszenzbasierter Verfahren ist ihre Selektivität: Durch geeignete Farbstoffe lassen sich gezielt bestimmte Nukleinsäuretypen erfassen, während andere Bestandteile der Probe weitgehend unbeachtet bleiben. Dadurch wird eine deutlich spezifischere Quantifizierung möglich als mit der unspezifischen UV-Absorption bei 260 nm.
Insbesondere bei niedrigen Konzentrationen – etwa bei der Bestimmung von DNA-Rückständen in mRNA-Präparationen – bieten fluoreszenzbasierte Methoden eine wesentlich höhere Sensitivität.
2.2.1. DNA-/RNA-spezifische Farbstoffe
Die Grundlage fluoreszenzbasierter Quantifizierung bilden Nukleinsäure-bindende Farbstoffe, die selektiv mit DNA oder RNA interagieren.
Doppelstrang-DNA-spezifische Farbstoffe
Ein häufig eingesetzter DNA-spezifischer Farbstoff ist PicoGreen. Dieser bindet bevorzugt an doppelsträngige DNA. In freier Lösung zeigt PicoGreen nur eine sehr geringe Eigenfluoreszenz. Erst nach Bindung an die DNA steigt seine Fluoreszenzintensität stark an. Dadurch wird die Quantifizierung selbst sehr geringer DNA-Mengen möglich.
RNA-spezifische Farbstoffe
Für RNA wird häufig RiboGreen verwendet. Auch dieser Farbstoff zeigt erst nach Bindung an RNA ein deutliches Fluoreszenzsignal.
Die unterschiedliche Bindungsaffinität der Farbstoffe erlaubt eine weitgehend selektive Erfassung der jeweiligen Nukleinsäureart.
Das Messprinzip folgt dabei stets demselben Schema:
- Zugabe eines spezifischen Farbstoffs zur Probe
- Bindung des Farbstoffs an die Zielnukleinsäure
- Anregung mit Licht definierter Wellenlänge (λEX – Excitation)
- Messung der emittierten Fluoreszenz (λEM – Emission)
- Vergleich mit einer Kalibrationsreihe bekannter Konzentrationen
Da nur gebundener Farbstoff ein starkes Signal erzeugt, bleibt der Hintergrund gering. Dies erklärt die hohe Empfindlichkeit dieser Methode im Vergleich zur UV-Spektrophotometrie.
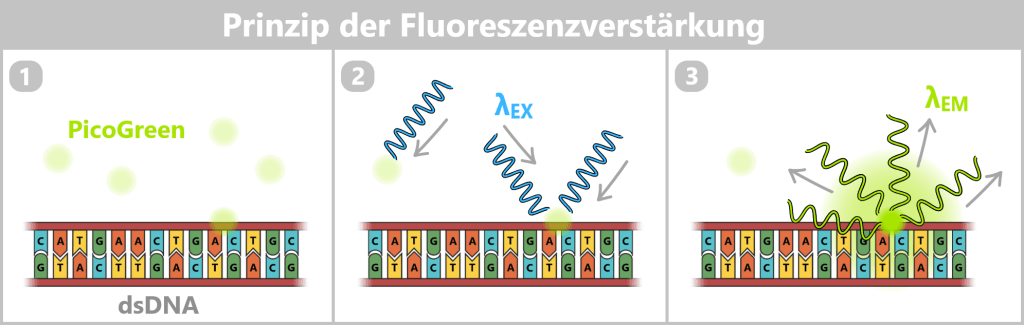
1) Nach Zugabe des Fluoreszenzfarbstoffs (z. B. PicoGreen) erfolgt eine bevorzugte Bindung an doppelsträngige DNA (dsDNA).
2) Die Probe wird mit kurzwelligem, energiereichem Licht einer definierten Wellenlänge (λEX ≈ 480 nm, blau) bestrahlt, wodurch die Farbstoffmoleküle in einen angeregten Zustand versetzt werden.
3) Bei der Rückkehr in den Grundzustand emittiert der gebundene Farbstoff längerwelliges Licht (λEM ≈ 520 nm, grün). Die gemessene Fluoreszenzintensität ist im linearen Messbereich proportional zur vorhandenen DNA-Konzentration in der Probe. Da freier Farbstoff nur eine sehr geringe Eigenfluoreszenz aufweist, entsteht ein deutliches Signal hauptsächlich durch DNA-gebundene Moleküle.
Die Quantifizierung erfolgt über den Vergleich der Signalintensität mit einer Standardreihe bekannter DNA-Konzentrationen.
a) Wirkmechanismus von PicoGreen
PicoGreen ist ein Fluoreszenzfarbstoff mit hoher Affinität zu doppelsträngiger DNA (dsDNA). Seine besondere Empfindlichkeit beruht darauf, dass er im freien Zustand fast nicht leuchtet – nach Bindung an DNA jedoch sehr stark fluoresziert.
Der Unterschied zwischen diesen beiden Zuständen (frei/gebunden) ist entscheidend für die analytische Anwendung.
Freies PicoGreen in Lösung
In wässriger Lösung ist das Molekül beweglich:
- Es kann sich drehen und biegen.
- Seine aromatischen Ringsysteme sind gegeneinander beweglich.
- Es stößt ständig mit Wassermolekülen zusammen.
Wird das Molekül durch Licht angeregt, nimmt es Energie auf. Im freien Zustand geht diese Energie jedoch größtenteils nicht als Licht verloren, sondern wird durch molekulare Bewegungen und Wechselwirkungen mit dem Wasser in Wärme umgewandelt. Die Folge: Die Eigenfluoreszenz ist sehr gering. Freies PicoGreen bleibt nahezu „dunkel“.
Annäherung an die doppelsträngige DNA
PicoGreen trägt positive Ladungen. Das Rückgrat der DNA ist aufgrund seiner Phosphatgruppen negativ geladen. Dadurch entsteht eine elektrostatische Anziehung zwischen Farbstoff und DNA. Das Molekül lagert sich bevorzugt an doppelsträngige DNA (dsDNA) an – insbesondere an Bereiche, in denen zwei DNA-Stränge eng beieinanderliegen. Diese erste Annäherung ist die Voraussetzung für eine stabilere Bindung.
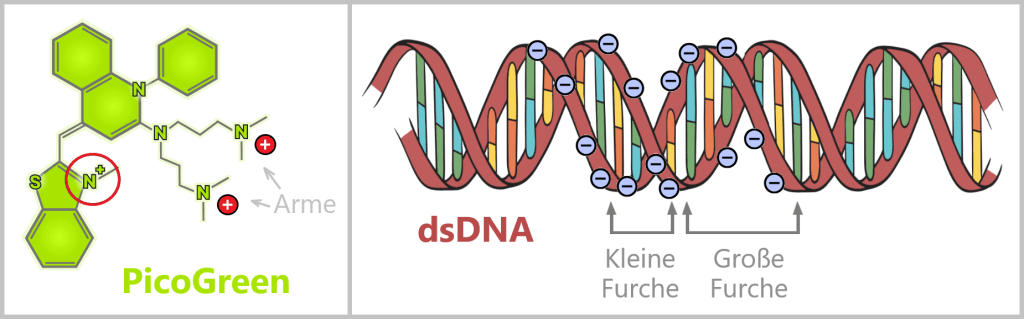
(nicht maßstabsgetreu)
Links ist die vereinfachte Struktur des Fluoreszenzfarbstoffs PicoGreen dargestellt. Die sechseckigen und fünfeckigen Ringe in der Struktur stehen für die aromatischen Ringsysteme – das sind besonders stabile, flache Molekülbausteine, die UV-Licht einfangen können. Zudem besitzt der Farbstoff positiv geladene Seitenarme.
Rechts ist die doppelsträngige DNA mit negativ geladenem Zucker-Phosphat-Rückgrat gezeigt. Die elektrostatische Anziehung zwischen den positiven Seitenarmen des Farbstoffs und dem negativen Rückgrat der DNA bringt den Farbstoff zunächst in die Nähe der DNA.
Bindung und Fixierung
Nach der Bindung wird das Molekül in seiner Beweglichkeit stark eingeschränkt:
- Es kann sich kaum noch drehen oder verbiegen.
- Teile des Moleküls liegen geschützt zwischen oder nahe den Basenpaaren.
- Der direkte Kontakt mit Wasser wird teilweise reduziert.
Die DNA wirkt dabei gewissermaßen wie eine Halterung, die das Molekül in einer festen Position stabilisiert.
Warum die Fluoreszenz nun stark zunimmt
Durch diese Fixierung verändern sich die Energieverluste des Moleküls: Im freien Zustand geht ein Großteil der aufgenommenen Energie durch Bewegung verloren. Im gebundenen Zustand sind diese Bewegungen stark eingeschränkt.
Dadurch steigt die Wahrscheinlichkeit, dass die aufgenommene Energie als Licht abgegeben wird – also als Fluoreszenz. Das Ergebnis ist ein deutlich stärkeres Signal.
Der Unterschied zwischen freiem und DNA-gebundenem PicoGreen ist so groß, dass selbst sehr geringe Mengen doppelsträngiger DNA zuverlässig nachweisbar sind.
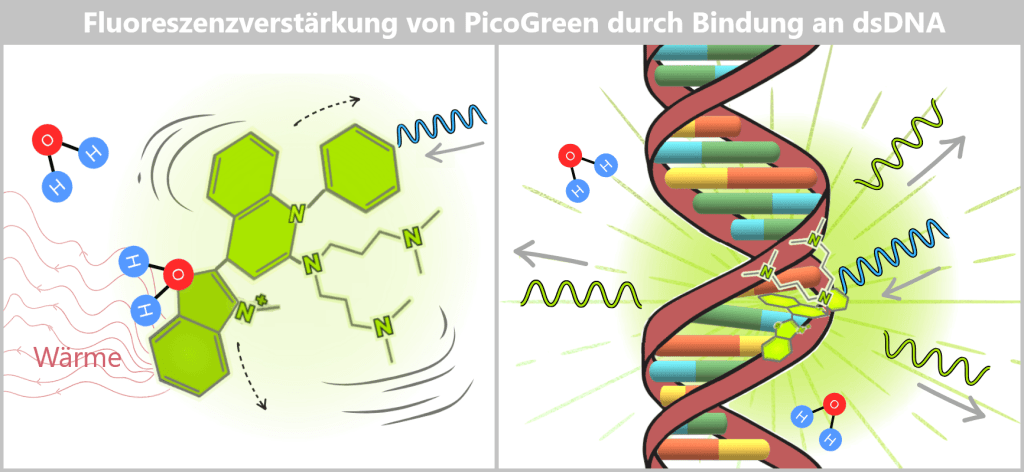
Links: PicoGreen in wässriger Lösung (ohne DNA)
In wässriger Lösung ist das Farbstoffmolekül ständig in Bewegung: Es rotiert und führt innermolekulare Schwingungen aus. Wird es mit blauem Licht angeregt, gibt es die aufgenommene Energie überwiegend über diese Bewegungen an das umgebende Wasser als Wärme ab. Der Farbstoff zeigt daher nur eine sehr schwache Eigenfluoreszenz.
Rechts: PicoGreen gebunden an dsDNA
Nach Bindung an doppelsträngige DNA lagert sich PicoGreen in die Kleine Furche ein und wird zusätzlich durch Stapelwechselwirkungen mit den Basen fixiert. Dadurch ist das Molekül in seiner Beweglichkeit stark eingeschränkt. Die aufgenommene Energie kann nun deutlich weniger über Bewegungsprozesse verloren gehen und wird überwiegend als grünes Licht emittiert. Die Fluoreszenzintensität steigt daher stark an.
Warum diese Eigenschaft analytisch so wertvoll ist
- Freier Farbstoff erzeugt kaum Hintergrundsignal.
- Nur gebundener Farbstoff leuchtet stark.
- Doppelsträngige DNA wird bevorzugt erfasst.
Gerade bei der Bestimmung von DNA-Rückständen in mRNA-Präparationen – auch bei sehr niedrigen Konzentrationen – ist dieser starke Kontrast zwischen „dunkel“ und „leuchtend“ entscheidend für die Sensitivität der Methode.
b) Warum PicoGreen bevorzugt doppelsträngige DNA erfasst
Die Selektivität von PicoGreen beruht nicht darauf, dass der Farbstoff „weiß“, was DNA ist, sondern auf strukturellen Eigenschaften der doppelsträngigen DNA.
Doppelsträngige DNA besitzt:
- einen regelmäßig aufgebauten Basenstapel
- dicht gepackte, planare Basenpaare
- klar definierte große und kleine Furchen
- eine stabile, räumlich geordnete Struktur
Diese geordnete Architektur bietet dem Farbstoff geeignete Bindungsstellen. Er kann sich zwischen oder nahe den gestapelten Basen anlagern und wird dabei mechanisch fixiert. Genau diese Fixierung ist entscheidend für die starke Fluoreszenzzunahme.
Einzelsträngige DNA oder RNA besitzen eine deutlich flexiblere Struktur. Ihnen fehlt der regelmäßig aufgebaute Basenstapel der Doppelhelix. Zwar kann es auch hier zu Wechselwirkungen kommen, doch:
- die Bindung ist schwächer
- die Fixierung weniger stabil
- die Fluoreszenzverstärkung deutlich geringer
Deshalb reagiert PicoGreen besonders empfindlich auf doppelsträngige DNA – und wesentlich schwächer auf RNA oder einzelsträngige Nukleinsäuren.
Für die Analyse von DNA-Rückständen ist genau diese Eigenschaft entscheidend: Restliche Plasmid-DNA oder DNA-Fragmente liegen typischerweise doppelsträngig vor und erzeugen daher ein starkes Signal.
Obwohl DNA-spezifische Fluoreszenzfarbstoffe wie PicoGreen eine hohe Präferenz für doppelsträngige DNA aufweisen, ist ihre Selektivität nicht absolut. Insbesondere in Proben mit sehr hohen RNA-Konzentrationen kann es zu unspezifischen Signalbeiträgen kommen. Diese entstehen durch schwache Bindung an RNA, sekundäre RNA-Strukturen oder schlichte Masseneffekte. Ohne geeignete Kontrollmaßnahmen – etwa eine RNase-Behandlung – kann dies zu einer Überschätzung der DNA-Menge führen.
c) Grenzen der PicoGreen-Methode
Trotz ihrer hohen Empfindlichkeit bleibt die Methode eine quantitative Summenbestimmung von dsDNA. Sie liefert keine Informationen über:
- die Sequenz der DNA
- die genaue Herkunft
- die biologische Funktionalität
- die Fragmentzusammensetzung
Einfluss der Fragmentlänge
Die Signalstärke hängt unter anderem davon ab, wie viele Bindungsstellen zur Verfügung stehen. Sehr kurze DNA-Fragmente können weniger Farbstoff binden als lange, intakte Moleküle.
Das bedeutet: Zwei Proben mit identischer Masse an DNA können – abhängig von der Fragmentierung – leicht unterschiedliche Fluoreszenzsignale erzeugen.
In der Praxis wird dies durch Kalibrationsstandards berücksichtigt, vollständig eliminieren lässt sich dieser Effekt jedoch nicht.
Matrixeffekte
Bestandteile der Probe können das Messergebnis beeinflussen, etwa:
- hohe Salzkonzentrationen
- Restproteine
- Tenside oder Lipidbestandteile
- Pufferzusammensetzung
Solche Faktoren können die Bindungseffizienz oder die Fluoreszenzintensität verändern. Daher werden Messbedingungen standardisiert und validiert.
✧ ✧ ✧
Zwischenfazit
Fluoreszenzbasierte Verfahren wie PicoGreen ermöglichen eine sehr empfindliche Quantifizierung doppelsträngiger DNA und sind grundsätzlich gut geeignet, um geringe DNA-Mengen auch in komplexen Proben nachzuweisen.
Die Methode beruht jedoch nicht auf einer sequenzspezifischen Erkennung, sondern auf strukturellen Eigenschaften der Nukleinsäuren. Das gemessene Signal kann daher durch Faktoren wie RNA-Reste, Fragmentlänge, Sekundärstrukturen oder Bestandteile der Probenmatrix beeinflusst werden. Ohne geeignete Kontrollmaßnahmen – insbesondere RNase-Behandlung und standardisierte Probenaufbereitung – besteht das Risiko einer Über- oder Unterschätzung der tatsächlichen DNA-Menge.
PicoGreen beantwortet damit vor allem die Frage, wie viel fluoreszenzaktive doppelsträngige DNA in einer Probe vorhanden ist. Aussagen über Sequenz, Herkunft, Integrität oder biologische Funktionalität der DNA sind mit dieser Methode allein nicht möglich.
Für solche Fragestellungen sind ergänzende sequenz- oder amplifikationsbasierte Verfahren erforderlich, die in den folgenden Kapitel behandelt werden.
2.2.2. Qubit-Systeme
Während DNA- und RNA-spezifische Farbstoffe wie PicoGreen oder RiboGreen die eigentlichen fluoreszierenden Werkzeuge zur Quantifizierung von Nukleinsäuren darstellen, bezeichnet das Qubit-System das darauf abgestimmte Gesamtsystem zur präzisen Konzentrationsbestimmung. Das Qubit-System ist ein kompaktes Fluorometer, das für die hochsensitive und spezifische Quantifizierung von DNA, RNA und Proteinen von Thermo Fisher Scientific entwickelt wurde. Es kombiniert:
- spezifische Fluoreszenzfarbstoffe = molekulare Sensoren
- optimierte Reagenzienlösungen = definierte Umgebung (Puffer)
- standardisierte Assay-Protokolle = standardisierte Bedienungsanleitungen
Ziel dieses Systems ist eine selektive und empfindliche Quantifizierung von DNA oder RNA in biologischen Proben.
Prinzip des Qubit-Systems
Das zugrunde liegende Messprinzip entspricht dem zuvor beschriebenen Verfahren (im Kapitel 2.2.1): Ein selektiver Fluoreszenzfarbstoff bindet an die Zielnukleinsäure, wird mit Licht definierter Wellenlänge angeregt und emittiert ein messbares Fluoreszenzsignal. Die Intensität dieses Signals ist proportional zur Menge der gebundenen DNA oder RNA. Durch den Vergleich mit Standardlösungen bekannter Konzentration kann daraus die Konzentration der Probe bestimmt werden.
Der Ablauf einer Messung
Die Handhabung ist bewusst einfach gehalten und folgt einem standardisierten Protokoll:
1️⃣ Arbeitslösung ansetzen: Der fluoreszierende Farbstoff (aus dem Assay-Kit) wird mit dem mitgelieferten Puffer im empfohlenen Verhältnis gemischt.
Beispiele für verfügbare Assay-Kits:
| Assay-Kit | Anwendung für |
| dsDNA BR (Broad Range) | höhere DNA-Konzentrationen |
| dsDNA HS (High Sensitivity) | niedrige DNA-Konzentrationen |
| RNA BR (Broad Range) | höhere RNA-Konzentrationen |
| RNA HS (High Sensitivity) | niedrige RNA-Konzentrationen |
| microRNA Assay | kleine RNA-Moleküle wie miRNA und siRNA |
| Protein Assay | Quantifizierung von Proteinen |
2️⃣ Probe zugeben: Ein kleines Volumen der Probe (meist 1–20 µl) wird zur Arbeitslösung gegeben.
3️⃣ Bindung abwarten: Nach einer kurzen Inkubationszeit von nur 2 Minuten (bei DNA/RNA-Assays) bindet der Farbstoff spezifisch an sein Zielmolekül.
4️⃣ Kalibrieren mit Standards: Es werden ein oder zwei mitgelieferte Standards mit bekannter Konzentration gemessen. Daraus erstellt das Gerät automatisch eine Standardkurve.
5️⃣ Probe in das Gerät stellen:
Anregung: Das Gerät bestrahlt die Probe mit der passenden Anregungswellenlänge: z. B. blaues Licht bei ca. 470 nm zur Quantifizierung von DNA.
Emission: Die gebundenen Farbstoffmoleküle absorbieren dieses Licht und strahlen es mit einer längeren Wellenlänge (niedrigerer Energie) wieder ab (z. B. grünes Licht bei ca. 520 nm).
Filterung: Ein optischer Filter im Gerät blockiert das blaue Anregungslicht, sodass der Sensor nur das grüne Leuchten der Probe registriert. Dies erklärt die enorme Genauigkeit: Das Gerät „sieht“ nur das Zielobjekt, störende Anregungsstrahlung wird ausgeblendet.
6️⃣ Messen und Ergebnis: Das Gerät misst das abgestrahlte, längerwellige Licht (z. B. grün). Innerhalb von Sekunden berechnet ein Algorithmus unter Berücksichtigung der Standardkurve die exakte Konzentration. Das Ergebnis wird auf dem Bildschirm angezeigt.
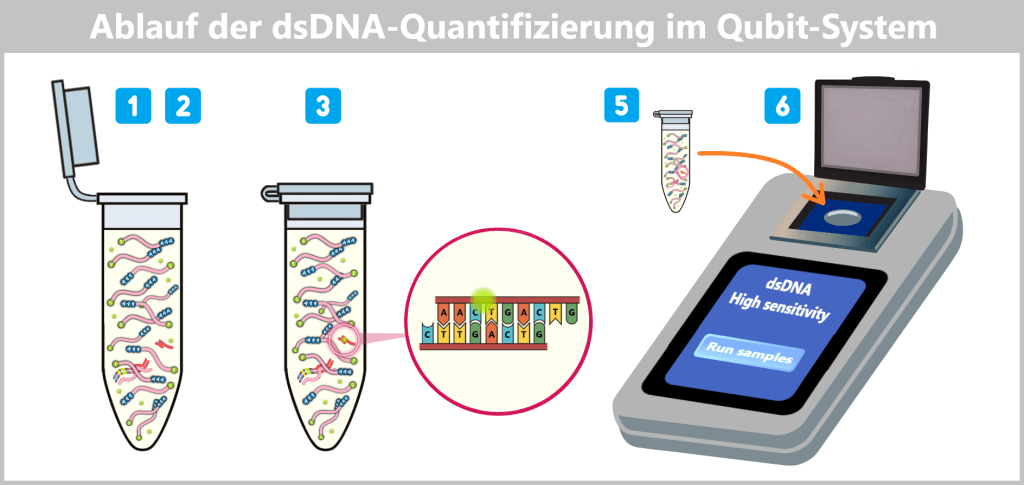
Die Abbildung zeigt den Ablauf der dsDNA-Quantifizierung mit dem Qubit-System. Nach Zugabe des spezifischen Fluoreszenzfarbstoffs zur Probe (1–2) bindet dieser selektiv an doppelsträngige DNA (3). Anschließend wird die Probe in das Qubit-Fluorometer eingesetzt (5), welches die Fluoreszenzintensität misst und die DNA-Konzentration berechnet (6).
Vorteile des Qubit-Systems
- Hohe Spezifität und Selektivität: Die verwendeten Fluoreszenzfarbstoffe besitzen eine starke Präferenz für ihre jeweiligen Zielmoleküle. Dadurch kann beispielsweise doppelsträngige DNA (dsDNA) auch in Gegenwart größerer RNA-Mengen vergleichsweise selektiv quantifiziert werden. Unspezifische Signalbeiträge sind jedoch insbesondere bei sehr hohen RNA-Konzentrationen oder komplexen Probenmatrices nicht vollständig ausgeschlossen.
- Hohe Sensitivität: Im Vergleich zur UV-Photometrie ist die Methode deutlich empfindlicher und erlaubt die Quantifizierung sehr geringer Nukleinsäurekonzentrationen.
- Geringer Probenverbrauch: Für die Messung werden nur kleine Probenvolumina benötigt (typischerweise 1–20 µl), was insbesondere bei limitiertem oder wertvollem Probenmaterial vorteilhaft ist.
- Relative Robustheit gegenüber Verunreinigungen: Viele Substanzen, die UV-spektroskopische Verfahren beeinflussen können – etwa freie Nukleotide, Salze oder Proteine, stören die fluorometrische Messung deutlich weniger. Bestimmte Matrixbestandteile können die Fluoreszenz jedoch weiterhin beeinflussen.
- Einfache Handhabung und schnelle Ergebnisse: Das System ist standardisiert aufgebaut und ermöglicht reproduzierbare Messungen mit vergleichsweise geringem Arbeitsaufwand.
Grenzen des Qubit-Systems
- Keine Aussage über DNA-Integrität: Das Verfahren unterscheidet nicht zwischen intakter und fragmentierter DNA.
- Keine Sequenzinformation: Die Messung liefert keine Informationen über Sequenz, Herkunft oder biologische Funktion der Nukleinsäuren.
- Einfluss der Fragmentlänge: Unterschiedliche Fragmentlängen können die Signalstärke beeinflussen. Sehr kurze oder stark degradierte Fragmente erzeugen unter Umständen ein reduziertes Fluoreszenzsignal.
- Restunspezifität bei hohen RNA-Mengen: Sehr hohe RNA-Konzentrationen können trotz der hohen Farbstoffspezifität zu unspezifischen Signalbeiträgen führen.
- Begrenzter linearer Messbereich: Die Quantifizierung ist auf den linearen Bereich des jeweiligen Assays beschränkt. Hochkonzentrierte Proben müssen daher entsprechend verdünnt werden.
- Eingeschränkte Aussagen zur Probenreinheit: Aussagen über Reinheit, Zusammensetzung oder mögliche Kontaminationen der Probe sind nur begrenzt möglich.
✧ ✧ ✧
Zusammenfassung
Die Qubit-Fluorometrie gilt als empfindliche und vergleichsweise selektive Methode zur Quantifizierung von DNA und RNA. Im Unterschied zur UV-Spektroskopie basiert sie nicht auf einer unspezifischen Lichtabsorption, sondern auf fluoreszierenden Farbstoffen mit starker Präferenz für bestimmte Nukleinsäurestrukturen. Dadurch sind auch Messungen geringer Konzentrationen in komplexen Proben möglich.
Die Methode bleibt jedoch eine strukturabhängige Summenmessung: Sie liefert Informationen über die Menge bestimmter Nukleinsäuretypen, nicht jedoch über deren Sequenz, Herkunft oder biologische Eigenschaften. Für solche Fragestellungen sind sequenz- oder amplifikationsbasierte Verfahren erforderlich, die in den folgenden Kapitel behandelt werden.
2.3. Amplifikationsbasierte Methoden
Die bisher beschriebenen Verfahren (UV-Spektroskopie und Fluorometrie) ermöglichen die Bestimmung der Gesamtmenge an Nukleinsäuren – also von RNA und/oder DNA. Für den gezielten Nachweis spezifischer DNA- oder RNA-Sequenzen sind jedoch Methoden erforderlich, die auf einer Vervielfältigung der Zielsequenz beruhen. Dieses Prinzip wird als Amplifikation bezeichnet.
Im Herstellungsprozess kommen amplifikationsbasierte Methoden insbesondere zum Einsatz, um verbleibende DNA-Rückstände nachzuweisen.
Grundlage dieser Verfahren ist die Polymerase-Kettenreaktion (PCR). Dabei wird ein zuvor definierter DNA-Abschnitt durch wiederholte enzymatische Synthesezyklen exponentiell vervielfältigt, das heißt, die Menge des Zielabschnitts verdoppelt sich idealerweise in jedem Zyklus. Bereits wenige Ausgangskopien können so in eine messbare Menge überführt werden. Auf diesem Prinzip basieren sowohl die quantitative PCR (qPCR) als auch die digitale PCR (ddPCR).
2.3.1. quantitative PCR (qPCR)
2.3.2. digitale PCR (ddPCR)
2.3.3. Vergleich der PCR-basierten Quantifizierungsmethoden
⚬ ⚬ ⚬
2.3.1. Quantitative Polymerase-Kettenreaktion (qPCR)
Die quantitative PCR, auch Real-Time-PCR genannt, ist eine Weiterentwicklung der klassischen PCR. Sie ermöglicht die gleichzeitige Amplifikation und Quantifizierung eines spezifischen DNA-Abschnitts in Echtzeit.
Die grundlegenden Reaktionsschritte entsprechen denen der konventionellen PCR, wie sie im ersten Teil (Vervielfältigung der Spike-DNA mittels PCR) beschrieben wurden. Der entscheidende Unterschied besteht darin, dass während jedes Amplifikationszyklus die entstehende DNA-Menge durch ein Fluoreszenzsignal gemessen wird.
In der qPCR wird hierzu ein Fluorophor eingesetzt, das als „Lichtsignal“ dient. Grundsätzlich unterscheidet man zwei Strategien:
a) Farbstoffbasierte qPCR
b) Sondenbasierte qPCR
◦ ◦ ◦
a) Farbstoffbasierte qPCR (z.B. mit SYBR® Green)
Bei der Analyse wird der Probe eine Reaktionsmischung zugesetzt, bestehend aus:
- freien Nukleotiden
- einem spezifischen Primerpaar
- einer thermostabilen DNA-Polymerase
- einem Puffer mit Mg²⁺-Ionen (für die Aktivität der Polymerase)
- einem fluoreszierenden Farbstoff (z. B. SYBR® Green)
SYBR® Green ist ein Fluorophor, der selektiv an doppelsträngige DNA bindet. Erst nach Bindung an dsDNA entfaltet der Farbstoff seine starke Fluoreszenz.
Diese Mischung wird in ein qPCR-Gerät – einen sogenannten Real-Time-PCR-Thermocycler – gestellt.
Prinzip der Methode
1️⃣ Denaturierung: Die Reaktionsmischung wird auf etwa 94–98 °C erhitzt. Dabei lösen sich die Wasserstoffbrückenbindungen zwischen den Basenpaaren, und der DNA-Doppelstrang wird in zwei Einzelstränge getrennt.
2️⃣ Primerbindung: Die Temperatur wird auf etwa 50–65 °C abgesenkt. Die spezifischen Primer binden an ihre komplementären Zielsequenzen auf den Einzelsträngen. Sie definieren den Startpunkt der DNA-Synthese.
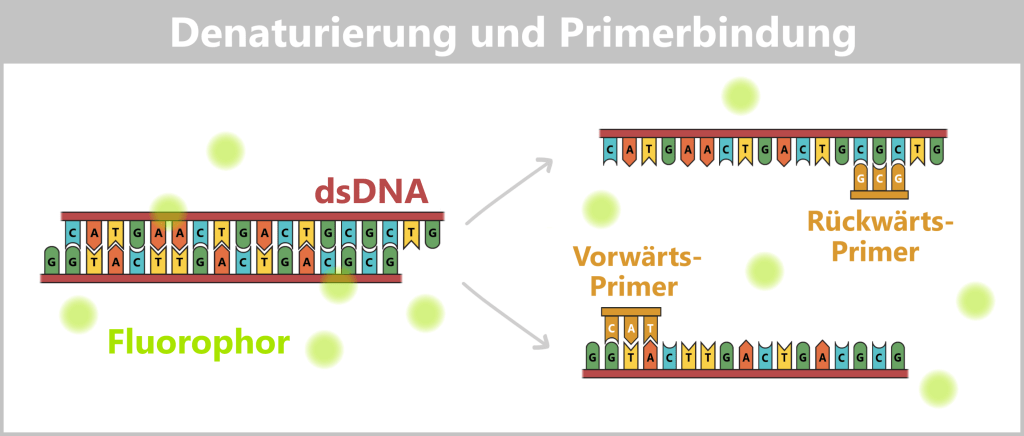
Links: Der Farbstoff SYBR® Green wird vor Beginn der PCR zur Reaktionsmischung gegeben. Er bindet selektiv an doppelsträngige DNA und zeigt in gebundener Form eine starke Fluoreszenz.
Rechts: Während der Denaturierung trennt sich die doppelsträngige DNA in Einzelstränge. Der gebundene Farbstoff wird dabei freigesetzt und verliert weitgehend seine Fluoreszenz. Anschließend binden die spezifischen Primer an ihre komplementären Zielsequenzen. (Hinweis: Die Primer sind zur besseren Übersichtlichkeit verkürzt mit nur drei Nukleotiden dargestellt; in der Praxis sind sie deutlich länger.)
3️⃣ DNA-Synthese (Amplifikation): Bei etwa 72 °C synthetisiert die DNA-Polymerase den komplementären Strang. Dabei entsteht erneut doppelsträngige DNA. Der Farbstoff SYBR® Green bindet an diese neu gebildete dsDNA.
4️⃣ Anregung und Emission: Die Reaktionsmischung wird mit blauem Licht (ca. 490 nm) bestrahlt. Die gebundenen Farbstoffmoleküle absorbieren diese Energie und emittieren grünes Licht (ca. 520 nm). Der gebundene Farbstoff erzeugt ein messbares Fluoreszenzsignal.
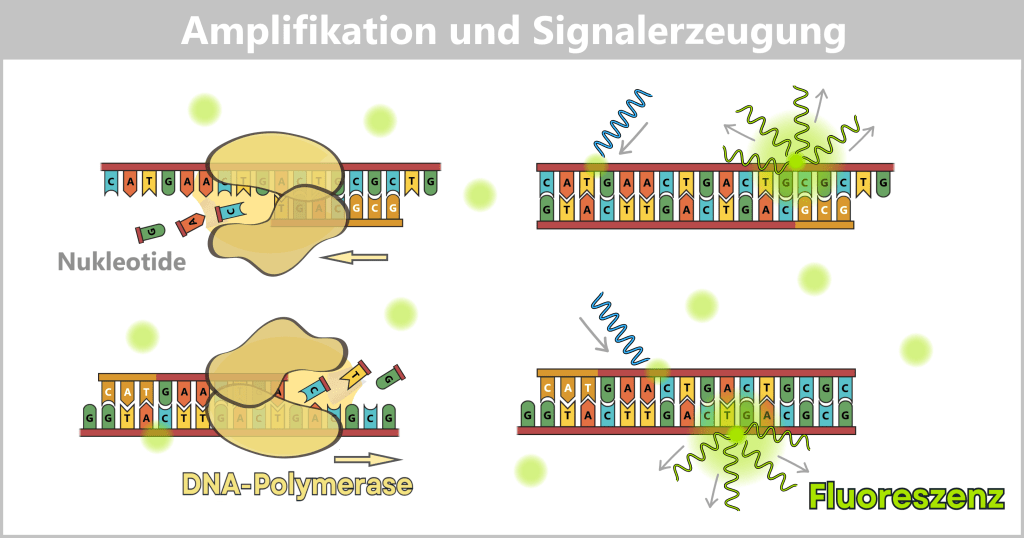
Links: Die DNA-Polymerase synthetisiert neue komplementäre Stränge. Dabei entsteht erneut doppelsträngige DNA, an die SYBR® Green unmittelbar bindet.
Rechts: Am Ende des Zyklus wird die Reaktionsmischung mit Licht geeigneter Wellenlänge angeregt. Die an doppelsträngige DNA gebundenen Farbstoffmoleküle emittieren grünes Fluoreszenzlicht, dessen Intensität proportional zur gebildeten DNA-Menge ist.
5️⃣ Quantifizierung: Die Fluoreszenz wird am Ende jedes Zyklus gemessen. Mit zunehmender DNA-Menge steigt das Signal an. Daraus entsteht eine charakteristische Amplifikationskurve.
Der zentrale Messwert ist der sogenannte Ct-Wert (Cycle threshold). Er bezeichnet den Zyklus, bei dem das Fluoreszenzsignal erstmals eine definierte Schwelle überschreitet.
- Niedriger Ct-Wert → hohe Ausgangsmenge an Ziel-DNA
- Hoher Ct-Wert → geringe Ausgangsmenge an Ziel-DNA
Für eine absolute Quantifizierung wird üblicherweise eine Standardkurve mit bekannten Referenzkonzentrationen erstellt.
Bedeutung der Amplikon-Länge für die Detektion
Als Amplikon bezeichnet man den DNA-Abschnitt, der während der PCR zwischen den beiden Primerbindestellen vervielfältigt wird – einschließlich der Primer selbst.
Die spezifische Detektion der Zielsequenz setzt voraus, dass ein vollständiges Amplikon gebildet werden kann. Das ist nur möglich, wenn beide Primer – Vorwärts- und Rückwärts-Primer – auf demselben DNA-Fragment binden können. Ist das Fragment kürzer als der Abstand zwischen den beiden Primerbindestellen, kann einer der Primer nicht binden. Eine effiziente exponentielle Amplifikation findet nicht statt, es entsteht kein relevantes Fluoreszenzsignal.
Daraus folgt eine wichtige methodische Konsequenz: Je länger das gewählte Amplikon – also der Abstand zwischen den beiden Primerbindestellen, desto mehr kurze Fragmente bleiben unerkannt – nicht weil sie nicht vorhanden sind, sondern weil sie für diesen Assay zu kurz sind. Die Wahl der Amplikon-Länge beeinflusst damit direkt, welcher Anteil der tatsächlich vorhandenen DNA erfasst wird.
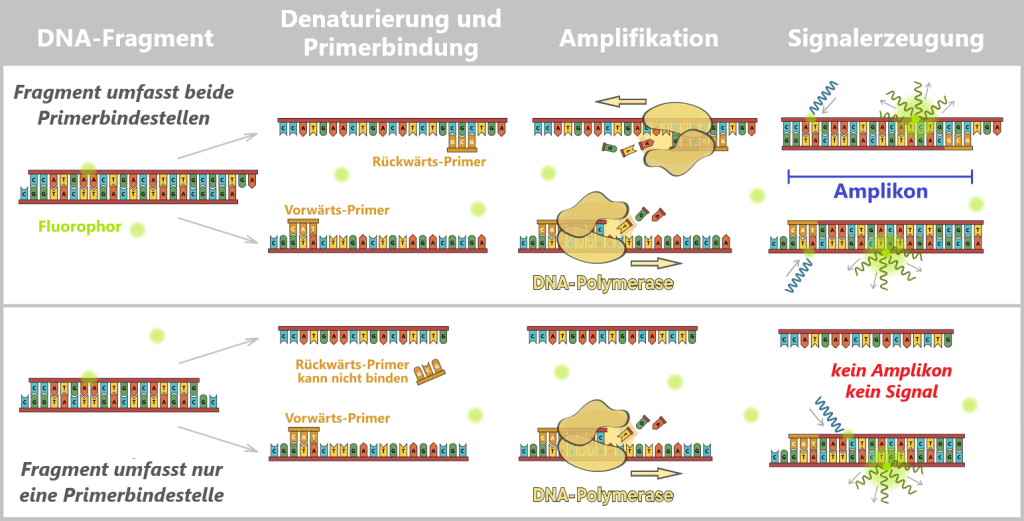
Hinweis: Die Primer sind zur besseren Übersichtlichkeit verkürzt mit nur drei Nukleotiden dargestellt; in der Praxis sind sie deutlich länger. Auch die schematische Darstellung der DNA-Fragmente ist verkürzt.
Oben: Fragment umfasst beide Primerbindestellen
Das DNA-Fragment ist lang genug, dass beide Primer (Vorwärts- und Rückwärts-Primer) an ihre jeweiligen Bindestellen binden können. Die DNA-Polymerase kann den Bereich zwischen den Primern vollständig kopieren. Es entsteht ein spezifisches Amplikon definierter Länge. Im nächsten Zyklus kann dieses Produkt wieder effizient amplifiziert werden. Als Ergebnis entsteht ein starkes Fluoreszenzsignal mit SYBR Green (proportional zur Menge spezifischer dsDNA).
Unten: Fragment umfasst nur eine Primerbindestelle
Das DNA-Fragment ist kürzer als der Abstand zwischen den beiden Primer-Bindestellen. Es kann nur ein Primer binden (der andere hat keine komplementäre Sequenz). Es entsteht kein vollständiges Amplikon, somit ist keine exponentielle Amplifikation möglich. Als Ergebnis entsteht kein oder nur sehr schwaches Fluoreszenzsignal mit SYBR Green.
Vorteile der farbstoffbasierten qPCR
- Geringe Kosten & Aufwand: Die Methode ist vergleichsweise kostengünstig und technisch unkompliziert.
- Einfaches Primer-Design: Es werden nur sequenzspezifische Primer benötigt.
- Hohe Sensitivität: Bereits sehr geringe Mengen an Ziel-DNA lassen sich zuverlässig detektieren.
- Schnelle Durchführung: Die Methode ist breit etabliert und ermöglicht eine zügige Analyse auch größerer Probenzahlen. Die Quantifizierung erfolgt direkt über die Fluoreszenzentwicklung während der Amplifikation.
- Hohe Flexibilität: Neue Zielsequenzen können vergleichsweise schnell durch Anpassung der Primer analysiert werden.
- Schmelzkurvenanalyse als Qualitätskontrolle: Nach der Amplifikation kann die Spezifität der PCR-Produkte anhand ihrer charakteristischen Schmelztemperatur überprüft werden. Dadurch lassen sich unspezifische Produkte oder Primer-Dimere häufig erkennen.
Nachteile der farbstoffbasierten qPCR
- Unspezifische Farbstoffbindung: Der Fluoreszenzfarbstoff bindet an jede doppelsträngige DNA – auch an unspezifische Amplifikationsprodukte wie Primer-Dimere.
- Abhängigkeit vom Primer-Design: Die Spezifität der Analyse hängt stark von der Qualität und Selektivität der verwendeten Primer ab.
- Einfluss der Zielregion: Die Wahl der Zielsequenz und insbesondere die Länge des amplifizierten Abschnitts (Amplikon) beeinflussen das Ergebnis erheblich.
- Einfluss der Fragmentlänge: Stark fragmentierte DNA kann mit längeren Amplikons unvollständig erfasst werden, da häufig nicht mehr beide Primerbindestellen auf demselben Fragment vorhanden sind.
- Einfluss der Amplikonlänge: Kürzere Amplikons erfassen fragmentierte DNA in der Regel effizienter, erhöhen jedoch unter Umständen die Wahrscheinlichkeit unspezifischer Signale.
- Abhängigkeit von Effizienz und Kalibration: Die Genauigkeit der Quantifizierung wird wesentlich durch die PCR-Effizienz sowie die Qualität der verwendeten Standardkurve beeinflusst.
b) Sondenbasierte qPCR (z. B. TaqMan®)
Bei der Analyse wird der Probe eine Reaktionsmischung zugesetzt, bestehend aus:
- freien Nukleotiden
- einem spezifischen Primerpaar
- einer thermostabilen DNA-Polymerase mit „5’→3′-Exonuklease-Aktivität“ (diese Polymerase kann im Weg liegende Hindernisse aktiv abbauen)
- einem Puffer mit Mg²⁺-Ionen (für die Aktivität der Polymerase)
- einer spezifischen Sonde (z. B. TaqMan®-Sonde)
Aufbau und Funktionsweise der TaqMan®-Sonde
Eine TaqMan®-Sonde ist eine kurze, synthetisch hergestellte DNA-Sequenz, die komplementär zu einem Abschnitt der Zielsequenz passt. Die Sonde bindet an einen der beiden DNA-Stränge, und zwar zwischen den beiden Primerbindestellen.
Die Sonde trägt:
• am 5′-Ende einen Fluoreszenzfarbstoff (Reporter)
• am 3′-Ende einen Quencher
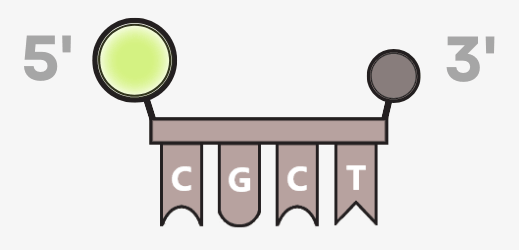
Der Reporter kann unterschiedliche Fluorophore tragen, wodurch auch Multiplex-Analysen – der gleichzeitige Nachweis mehrerer Zielsequenzen – möglich sind.
Solange Reporter und Quencher räumlich nahe beieinander liegen, wird das Fluoreszenzsignal unterdrückt. Das Quenching funktioniert nur, solange die Sonde intakt ist: Durch die räumliche Nähe von Reporter und Quencher kommt es zu einem Fluorescence Resonance Energy Transfer (FRET). Dabei wird die vom Reporter absorbierte Energie auf den Quencher übertragen und als Wärme abgegeben, anstatt als Licht emittiert zu werden.
Die Reaktion wird in einem Real-Time-PCR-Thermocycler durchgeführt, der neben den Temperaturzyklen auch die Fluoreszenz während der Amplifikation misst.
Prinzip der Methode
1️⃣ Denaturierung: Die Reaktionsmischung wird auf etwa 94–98 °C erhitzt. Die Wasserstoffbrückenbindungen brechen auf, wodurch die doppelsträngige DNA in zwei Einzelstränge getrennt wird.
2️⃣ Hybridisierung (Primer- & Sondenbindung): Die Temperatur wird auf etwa 55–60 °C abgesenkt. Neben den spezifischen Primern bindet auch die TaqMan-Sonde an ihre Zielsequenz zwischen den Primern. In diesem Zustand bleibt die Fluoreszenz des Reporters durch den nahen Quencher unterdrückt.
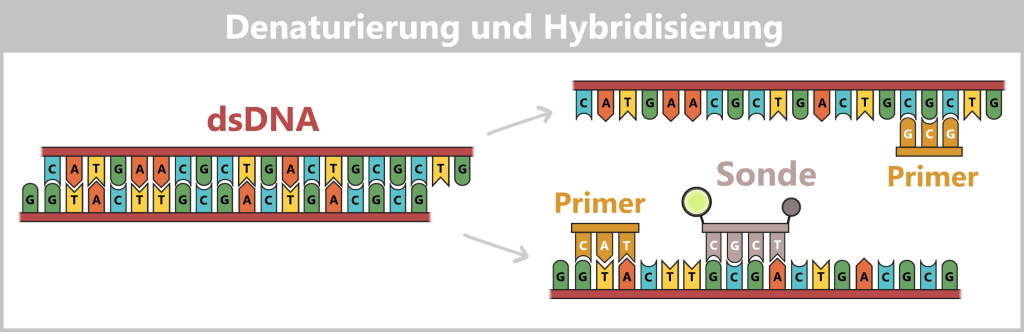
Sobald die Temperatur von 95 °C auf 60 °C sinkt, bindet die Sonde als Erstes an ihre Zielsequenz. Die Sonde hat eine höhere Schmelztemperatur Tm (meist 8–10 °C über der der Primer), damit sie bei der gewählten Arbeitstemperatur bereits fest sitzt, während die Primer gerade erst anfangen zu binden.
Die Sonde ist ein einzelsträngiges DNA-Molekül von etwa 20–30 Nukleotiden Länge. Sie ist so konzipiert, dass sie spezifisch an eine definierte Zielsequenz bindet – einen Abschnitt, der charakteristisch für die gesuchte DNA ist.
3️⃣ DNA-Synthese & Hydrolyse (Amplifikation): Bei etwa 60 °C beginnt die DNA-Polymerase mit der Synthese des neuen Strangs. In vielen modernen qPCR-Protokollen erfolgen Annealing (Primer- & Sondenbindung) und Elongation (DNA-Synthese) bei derselben Temperatur.
Trifft die Polymerase während der Strangsynthese auf die gebundene Sonde, nutzt sie ihre 5’→3′-Exonuklease-Aktivität, um die Sonde zu hydrolysieren (in einzelne Nukleotide zu zerlegen). Bereits der erste Schnitt trennt den Reporter-Farbstoff vom Quencher.
Dadurch wird der Reporter dauerhaft von der quenched Situation befreit.
4️⃣ Anregung und Emission: Während der Messphase wird die Reaktionsmischung mit Licht einer spezifischen Wellenlänge angeregt. Die freigesetzten Reporter-Moleküle absorbieren diese Energie und emittieren Licht einer längeren Wellenlänge (z. B. grün bei FAM oder gelb bei VIC).
Das Fluoreszenzsignal entsteht hier nicht durch Bindung eines Farbstoffs an doppelsträngige DNA (wie bei SYBR Green), sondern durch die enzymatische Spaltung der Sonde während der Amplifikation.
Die Sonde ist nicht an der DNA-Amplifikation beteiligt. Die Amplifikation wird ausschließlich durch den Vorwärts- und den Rückwärtsprimer gesteuert, während die Sonde lediglich als sequenzspezifischer Fluoreszenzmarker dient.
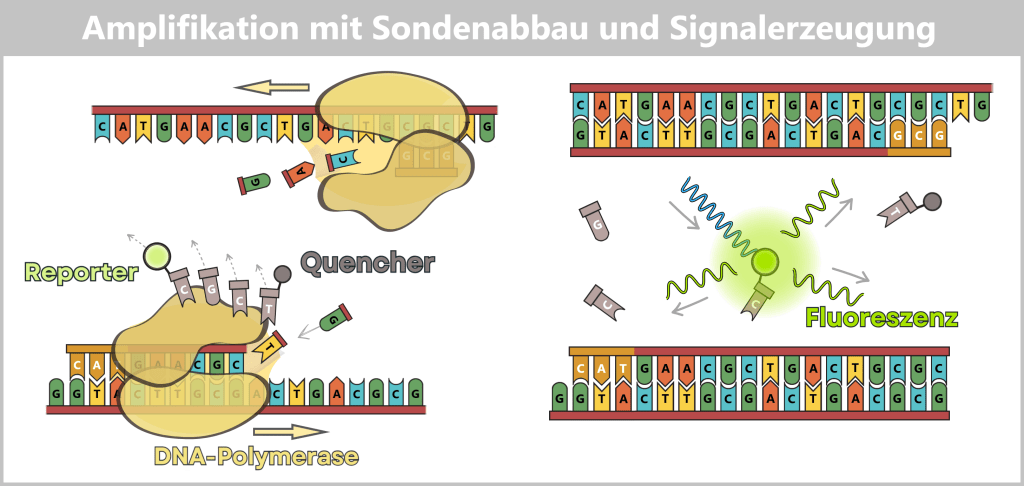
Die Polymerase verlängert vom Primer aus und erreicht die Sonde. Während sie den neuen DNA-Strang synthetisiert, räumt sie die im Weg liegende Sonde weg. Dadurch wird der Reporter vom Quencher getrennt, was das messbare Fluoreszenzsignal erzeugt.
5️⃣ Quantifizierung: Die Fluoreszenz wird nach jedem Zyklus gemessen. Pro neu synthetisiertem DNA-Strang, der eine intakte Sondenbindestelle enthält, wird eine Sonde hydrolysiert. Die Signalintensität steigt daher proportional zur Anzahl korrekt amplifizierter Zielmoleküle.
Der zentrale Messwert ist der Ct-Wert (Cycle threshold):
- Niedriger Ct-Wert → hohe Ausgangsmenge an Ziel-DNA
- Hoher Ct-Wert → geringe Ausgangsmenge an Ziel-DNA
Vorteile der sondenbasierten qPCR
- Hohe Spezifität: Neben den beiden Primern bindet zusätzlich eine sequenzspezifische Fluoreszenzsonde. Ein Signal entsteht nur, wenn die gewünschte Zielsequenz tatsächlich amplifiziert wird. Unspezifische Amplifikationsprodukte wie Primer-Dimere erzeugen daher in der Regel kein relevantes Signal.
- Geeignet für Multiplex-Analysen: Durch unterschiedliche Reporter-Farbstoffe können mehrere Zielsequenzen gleichzeitig in einer Reaktion nachgewiesen werden.
- Verbesserte analytische Robustheit: Die zusätzliche Sondenebene erhöht die Spezifität und verbessert die Zuverlässigkeit der Quantifizierung, insbesondere in komplexen Proben mit hohem Hintergrundsignal.
- Geringere Anfälligkeit für unspezifische Amplifikation: Im Vergleich zu farbstoffbasierten Verfahren sind falsch-positive Signale durch unspezifische PCR-Produkte deutlich reduziert.
Nachteile der sondenbasierten qPCR
- Höhere Kosten: Für jede Zielsequenz muss zusätzlich zu den Primern eine spezifische, fluoreszenzmarkierte Sonde synthetisiert werden.
- Komplexeres Assay-Design: Neben den Primern muss auch die Sonde hinsichtlich Sequenz, Schmelztemperatur, Position und möglicher Sekundärstrukturen optimiert werden.
- Abhängigkeit von der Zielregion: Fragmentierungen, Mutationen oder Schäden innerhalb der Primer- oder Sondenbindestellen können die Amplifikation und Signalgenerierung beeinträchtigen oder verhindern.
- Einfluss der Fragmentlänge: Wie bei anderen PCR-basierten Verfahren hängt die Nachweisbarkeit davon ab, ob ein DNA-Fragment alle erforderlichen Bindestellen für Primer und Sonde enthält. Stark fragmentierte DNA kann daher – insbesondere bei längeren Zielregionen – unvollständig erfasst werden.
- Indirekte Quantifizierung: Die absolute Quantifizierung erfolgt meist über Standardkurven und bleibt abhängig von Amplifikationseffizienz, Assay-Design und Probenvorbereitung.
✧ ✧ ✧
Detaillierte Informationen zur Funktionsweise der beiden qPCR-Methoden sind hier zu finden.
Sowohl die farbstoffbasierte als auch die sondenbasierte qPCR ermöglichen eine sensitive und spezifische Detektion definierter DNA-Sequenzen. Die Quantifizierung erfolgt jedoch indirekt über den Ct-Wert und erfordert in der Regel eine Standardkurve mit bekannten Referenzkonzentrationen. Die Genauigkeit hängt somit von der Amplifikationseffizienz und der Qualität der Referenzstandards ab.
Um diese Abhängigkeit zu reduzieren und eine absolute Quantifizierung ohne Standardkurve zu ermöglichen, wurde die digitale PCR (ddPCR) entwickelt.
2.3.2. Digitale PCR (ddPCR)
ddPCR steht für Droplet Digital PCR (digitale Tröpfchen-PCR).
Die digitale PCR ist eine Weiterentwicklung der PCR-Technik, die eine absolute quantitative Bestimmung von DNA- oder RNA-Molekülen ermöglicht – ohne Standardkurve und mit sehr hoher Präzision.
Im Gegensatz zur qPCR misst sie kein kontinuierlich ansteigendes Fluoreszenzsignal, sondern zählt einzelne, positive Reaktionseinheiten. Das Ergebnis ist binär – „positiv“ oder „negativ“ – daher der Begriff digital.
Grundprinzip von ddPCR
Während bei der qPCR die gesamte Reaktion in einem einzigen Reaktionsvolumen abläuft, wird sie bei der ddPCR in Tausende mikroskopisch kleine, voneinander getrennte Reaktionsräume aufgeteilt – sogenannte Droplets (Tröpfchen). Jedes Droplet stellt eine eigenständige PCR-Reaktion dar.
Ablauf der Methode (am Beispiel DNA):
1️⃣ Probenvorbereitung: Die DNA-Probe wird mit Primern, einer sequenzspezifischen Sonde (meist TaqMan®-ähnlich), Nukleotiden, DNA-Polymerase und Puffer gemischt – analog zur sondenbasierten qPCR.
2️⃣ Partitionierung (Droplet Generator): Die Reaktionsmischung wird in einen Droplet Generator überführt. Dort wird sie zusammen mit einem speziellen Öl durch mikrofluidische Kanäle geleitet.
Durch die Strömungsdynamik entstehen daraus bis zu 20.000 nanoliter-große Droplets. Das Öl stabilisiert die Tröpfchen und bildet eine stabile Emulsion, sodass jedes Droplet ein abgeschlossener Reaktionsraum ist. Die fertigen Tröpfchen landen in einem Well (einer Vertiefung) auf einer 96-Well-Platte.
Die Verteilung der DNA-Moleküle auf die Droplets erfolgt zufällig.
Dabei entstehen:
- viele leere Droplets
- einige Droplets mit einem Zielmolekül
- einige Droplets mit mehreren Molekülen
Diese Verteilung folgt der Poisson-Statistik.
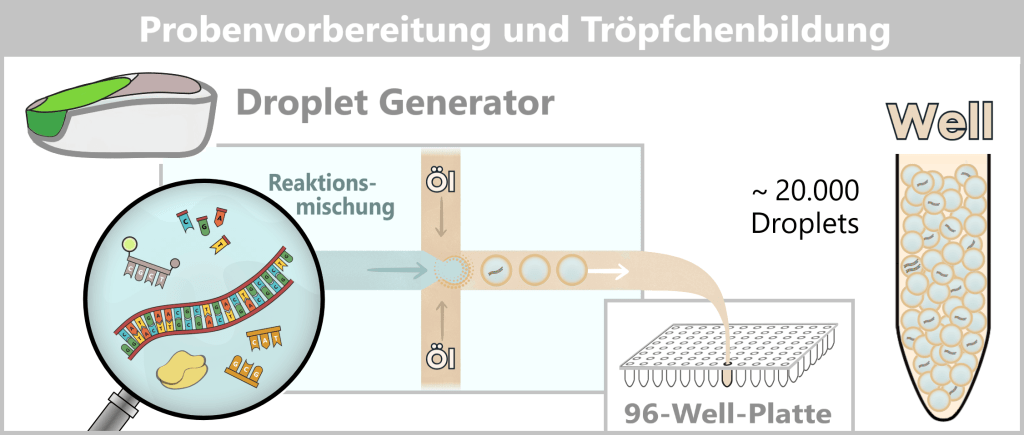
3️⃣ Amplifikation (PCR): Die versiegelte 96-Well-Platte wird in einen Thermocycler überführt.
Die Droplets durchlaufen die üblichen PCR-Zyklen (Denaturierung, Annealing, Elongation), typischerweise 40–45 Zyklen.
Während der Amplifikation wird keine Fluoreszenz gemessen.
Enthält ein Droplet mindestens ein Ziel-DNA-Molekül, wird dieses amplifiziert. Die Sonde wird hydrolysiert und das Droplet entwickelt ein starkes Fluoreszenzsignal.
Droplets ohne Ziel-DNA bleiben dunkel.
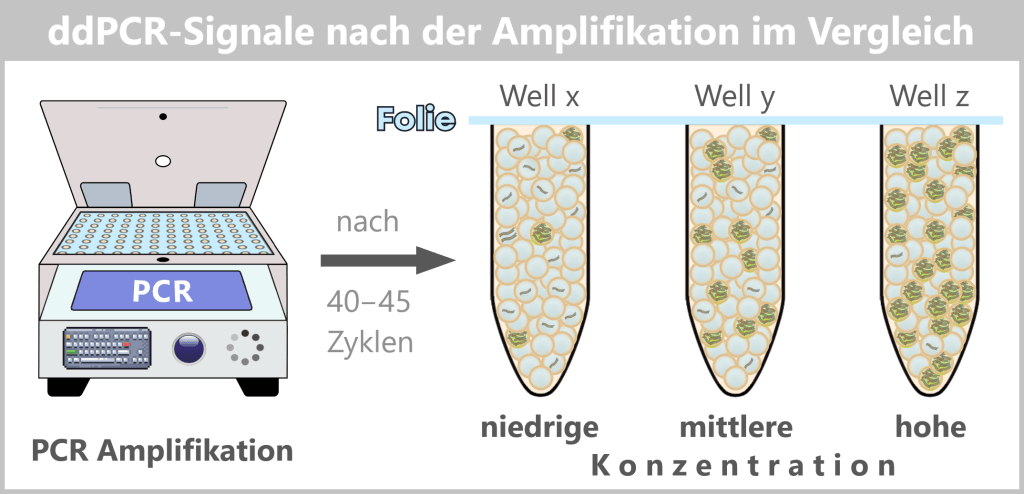
Die Abbildung zeigt schematisch das Ergebnis einer droplet digital PCR (ddPCR) nach Abschluss der Amplifikation.
Die zuvor generierten und versiegelten Droplets durchlaufen in einem Thermocycler 40–45 PCR-Zyklen. Während der Amplifikation erfolgt keine kontinuierliche Fluoreszenzmessung.
Enthält ein Droplet mindestens ein Ziel-DNA-Molekül, kommt es zur Amplifikation der Zielsequenz. Dabei wird die sequenzspezifische Sonde durch die 5′→3′-Exonuklease-Aktivität der DNA-Polymerase hydrolysiert, wodurch der Reporter vom Quencher getrennt wird und ein starkes Fluoreszenzsignal entsteht. Droplets ohne Ziel-DNA bleiben ohne Signal („negativ“).
Mit zunehmender Ausgangskonzentration der Ziel-DNA steigt die Anzahl fluoreszenter (positiver) Droplets, während die Intensität einzelner positiver Droplets vergleichbar bleibt.
4️⃣ Auslesung (Droplet Reader): Nach Abschluss der PCR wird die 96-Well-Platte in einen DropletReader eingesetzt.
Das Gerät:
- saugt die Emulsion aus einem Well an
- leitet die Droplets einzeln durch eine schmale Kapillare
- bestrahlt jedes Droplet mit einem Laser
- misst die Fluoreszenzintensität
Jedes Droplet wird einzeln klassifiziert als:
• positiv (Fluoreszenzsignal vorhanden)
• negativ (kein Signal)
Das Ergebnis ist binär – 0 oder 1.
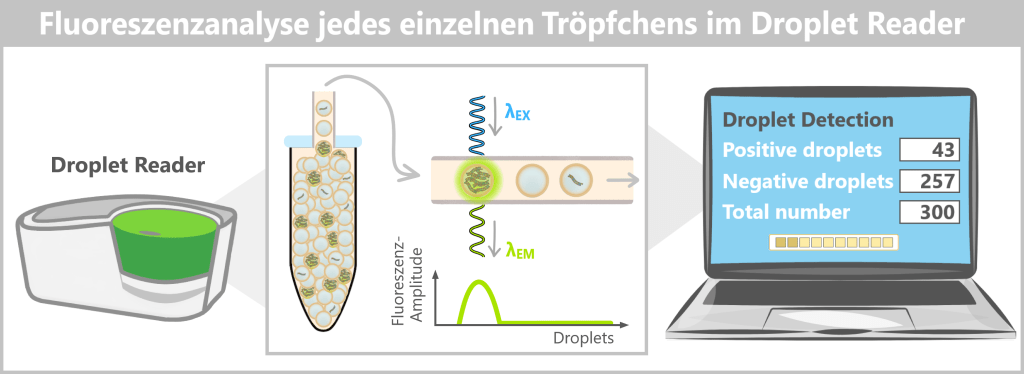
Nach Abschluss der PCR wird die 96-Well-Platte mit der Tröpfchen-Emulsion in den Droplet Reader eingesetzt. Das Gerät saugt die Emulsion aus einem Well an und führt die Tröpfchen einzeln durch eine schmale Kapillare. Jedes Tröpfchen wird dabei mit einem Laser bestrahlt; die gemessene Fluoreszenzintensität wird als Amplitudenwert erfasst. In der Analysesoftware werden diese Werte in einem 1D-Amplitudenplot dargestellt: Die X‑Achse nummeriert die Tröpfchen fortlaufend, die Y‑Achse zeigt die Fluoreszenz-Amplitude. Durch eine softwarebasierte Schwellenwertsetzung werden negative Tröpfchen (kein Signal) von positiven Tröpfchen (Fluoreszenzsignal) getrennt. Die Anzahl der positiven Droplets wird anschließend mittels Poisson-Statistik in die absolute Ziel-DNA-Konzentration der Ausgangsprobe umgerechnet.
5️⃣ Quantifizierung: Da bekannt ist:
- wie viele Droplets insgesamt analysiert wurden
- wie viele davon positiv sind
kann über die Poisson-Verteilung die absolute Anzahl der ursprünglichen DNA-Moleküle berechnet werden.
📍Eine Standardkurve ist nicht erforderlich.
📍Die Quantifizierung ist unabhängig von der Amplifikationseffizienz innerhalb einzelner Zyklen.
🎥 Tipp: Wie das Ganze in der Praxis aussieht, zeigt das Video „Digital PCR Using the Bio-Rad QX100™ ddPCR™ System“.
Vorteile der ddPCR
- Absolute Quantifizierung ohne Standardkurve: Die Anzahl der Zielmoleküle wird direkt aus dem Verhältnis positiver zu negativer Droplets mithilfe der Poisson-Statistik berechnet. Eine externe Standardkurve ist in der Regel nicht erforderlich.
- Hohe Präzision und Reproduzierbarkeit: Durch die Aufteilung der Probe in tausende unabhängige Reaktionsräume wird die Quantifizierung weniger anfällig für Schwankungen der Amplifikationseffizienz als bei klassischen qPCR-Verfahren.
- Hohe Sensitivität bei niedrigen Konzentrationen: Auch sehr geringe Mengen an Ziel-DNA können zuverlässig detektiert werden, da einzelne Moleküle in separaten Droplets amplifiziert werden.
- Relative Robustheit gegenüber PCR-Inhibitoren: Inhibitoren verteilen sich ebenfalls auf die einzelnen Droplets. Dadurch fällt ihre hemmende Wirkung oft geringer aus als bei einer einzelnen Gesamtreaktion.
- Endpunktmessung statt Ct-Interpretation: Die Analyse basiert auf der Anwesenheit oder Abwesenheit eines Signals am Ende der PCR und ist daher weniger abhängig von der Interpretation von Amplifikationskurven.
- Besonders geeignet für seltene Zielsequenzen: Die Methode wird häufig für den Nachweis seltener Mutationen, niedriger Viruslasten oder sehr geringer DNA-Restmengen eingesetzt.
Nachteile der ddPCR
- Aufwendigere Technik und höhere Kosten: Zusätzlich zum Thermocycler werden spezielle Geräte zur Droplet-Erzeugung und -Auslesung benötigt.
- Komplexerer Workflow: Die Partitionierung der Probe sowie die anschließende Droplet-Analyse erfordern zusätzliche Arbeitsschritte und erhöhen den technischen Aufwand.
- Begrenzter Dynamikbereich pro Ansatz: Bei sehr hohen Zielkonzentrationen können zu viele Droplets positiv werden. Dadurch verliert die statistische Auswertung an Genauigkeit (Sättigungseffekt), sodass Verdünnungsreihen erforderlich sein können.
- Geringerer Probendurchsatz: Im Vergleich zur qPCR ist die Analyse meist zeitaufwendiger und weniger für sehr hohe Probendurchsätze geeignet.
- Weiterhin sequenz- und assayabhängig: Wie bei der qPCR werden nur DNA-Fragmente erfasst, die sämtliche erforderlichen Primer- und gegebenenfalls Sondenbindestellen enthalten.
- Einfluss der Fragmentierung: Stark fragmentierte DNA kann auch bei der ddPCR unvollständig erfasst werden, insbesondere wenn die Zielregion relativ lang ist oder Primerbindestellen fehlen.
- Abhängigkeit von der Probenvorbereitung: Extraktionsverluste, unvollständiger LNP-Aufschluss oder selektive Fragmentverluste können das Ergebnis weiterhin beeinflussen.
2.3.3. Vergleich der PCR-basierten Quantifizierungsmethoden
| Merkmal | Farbstoffbasierte qPCR | Sondenbasierte qPCR | Digitale PCR (ddPCR) |
| Messprinzip | Fluoreszenzbindung an jede dsDNA | Sequenzspezifische Fluoreszenzsonde | Zählung positiver Einzelreaktionen |
| Signalentstehung | Farbstoff bindet an dsDNA | Sonde wird während der Amplifikation hydrolysiert | Endpunktmessung positiver Droplets |
| Quantifizierung | Relativ (über Ct-Wert und Standardkurve) | Relativ (über Ct-Wert und Standardkurve) | Absolut (über Poisson-Verteilung) |
| Standardkurve erforderlich | Ja | Ja | Nein |
| Spezifität | Mittel (primerabhängig) | Hoch (Primer + Sonde) | Hoch (Primer + Sonde) |
| Messzeitpunkt | Während der Amplifikation (Real-Time) | Während der Amplifikation (Real-Time) | Nach Endpunkt-PCR |
| Reaktionsraum | 1 Reaktionsvolumen | 1 Reaktionsvolumen | ~20.000 Droplets pro Well |
| Kosten | Niedrig | Mittel | Hoch |
| Geräteaufwand | Real-Time-PCR-Gerät | Real-Time-PCR-Gerät | Droplet Generator + Thermocycler + Droplet Reader |
2.4. Sequenzierungsbasierte Verfahren
2.4.1. Oxford Nanopore-Technologie
2.4.2. Illumina-Sequenzierung
Während die bisher beschriebenen Methoden (qPCR, ddPCR) gezielt einzelne DNA-Sequenzen nachweisen und quantifizieren können, gehen sequenzierungsbasierte Verfahren einen Schritt weiter: Sie lesen die exakte Abfolge der Basen eines DNA- oder RNA-Moleküls – Stück für Stück, Buchstabe für Buchstabe.
Ein Vertreter dieser Technologien ist die Nanoporen-Sequenzierung. In einer Studie zeigen Gunter et al. (Nature Communications 2023), dass Long-Read-Nanopore-Sequenzierung zur umfassenden Analyse von mRNA-Impfstoffen verwendet werden kann. Methoden wie „VAX-seq“ können Schlüsselqualitätsattribute erfassen – darunter Sequenzidentität, Integrität, Länge und Reinheit – und gleichzeitig mRNA sowie DNA-Rückstände bewerten. Dies macht die Nanopore-Sequenzierung zu einem vielversprechenden analytischen Ansatz für Herstellungs- und Qualitätskontrollprozesse.
Insbesondere die Oxford Nanopore-Technologie (ONT) hat sich in den letzten Jahren als vielversprechendes Werkzeug für die Qualitätskontrolle von mRNA-basierten Wirkstoffen etabliert. Sie liefert direkte Sequenzinformationen einzelner Moleküle und kann sowohl kurze als auch sehr lange Fragmente analysieren – ein entscheidender Vorteil für die Charakterisierung von DNA-Rückständen.
⚬ ⚬ ⚬
2.4.1. Oxford Nanopore-Technologie
Wenn heute von Nanopore-Sequenzierung die Rede ist, ist in der Regel die von Oxford Nanopore Technologies (ONT) entwickelte Plattform gemeint. Obwohl alternative Konzepte zur nanoporenbasierten Sequenzierung existieren, ist ONT derzeit die einzige Technologie, die kommerziell breit verfügbar und praktisch etabliert ist. Seit der Markteinführung im Jahr 2015 hat sie sich als Vertreterin der sogenannten Third-Generation-Sequencing-Verfahren etabliert.
Das Besondere an dieser Methode ist die direkte Analyse einzelner DNA- oder RNA-Moleküle in Echtzeit. Im Gegensatz zu klassischen Sequenzierverfahren sind weder eine zyklische Amplifikation noch chemische Markierungen der Nukleotide erforderlich. Die Sequenzinformation wird unmittelbar aus physikalischen Messsignalen abgeleitet.
Funktionsprinzip
Die ONT-Sequenzierung basiert auf dem Zusammenspiel mehrerer zentraler Komponenten:
Nanoporen – fungieren als winzige molekulare „Lesegeräte“. Wandert ein einzelner DNA- oder RNA-Strang durch die Pore, erzeugt dies charakteristische elektrische Signale – vergleichbar mit einem molekularen „Fingerabdruck“.
Membran – dient als Filter und Barriere. Sie sorgt dafür, dass nur Ionen und Nukleinsäuren die Nanoporen passieren, während unerwünschte Moleküle ausgeschlossen bleiben. Dadurch entsteht ein definierter Messraum für die Signalerfassung.
Chip (Flow Cell) – ist die Basis des Systems. Der Chip enthält die Membran mit zahlreichen Nanoporen sowie die integrierte Elektronik zur Detektion der elektrischen Signale. Er stellt die funktionelle Einheit dar, in der die Sequenzierung stattfindet.
Aufbau des Systems
Der Chip ist in zwei Kammern unterteilt:
| Obere Kammer (cis) | Hier wird die DNA-Probe eingebracht. |
| Untere Kammer (trans) | Nimmt den Strang nach dem Durchtritt durch die Nanopore auf. |
Beide Kammern sind mit einer ionenhaltigen Pufferlösung gefüllt, die elektrischen Strom leitet. Die Membran trennt die Kammern elektrisch voneinander – Strom kann nur an den Stellen fließen, an denen Nanoporen eingebettet sind. Diese Poren sind die einzigen „Tunnel“, durch die Ionen und Nukleinsäuren passieren können.
Durch eine zwischen den Kammern angelegte konstante elektrische Spannung entsteht ein messbarer Ionenstrom durch die Nanoporen. Sobald ein DNA- oder RNA-Strang die Pore durchquert, wird dieser Stromfluss charakteristisch verändert. Die dabei entstehenden Strommodulationen werden kontinuierlich erfasst und als elektrische Rohsignale aufgezeichnet.
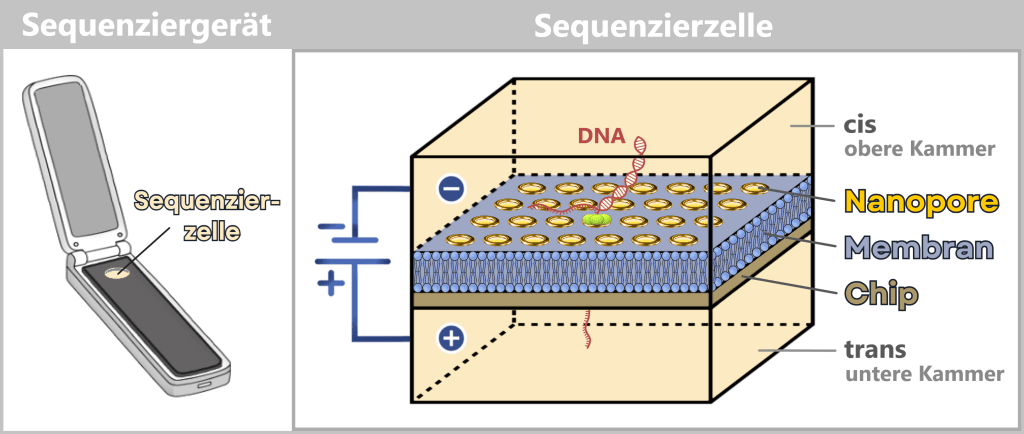
Links ist ein tragbares Nanopore-Sequenziergerät dargestellt, das in seiner Größe in etwa einem etwas breiteren USB-Stick entspricht. Es enthält eine austauschbare Sequenzierzelle (Flow Cell), die für die eigentliche Analyse eingesetzt wird.
Rechts: Die vergrößerte Darstellung zeigt den schematischen Aufbau dieser Sequenzierzelle: Sie besteht aus einem Chip, auf dem eine elektrisch isolierende Membran aufliegt. In diese Membran sind zahlreiche Nanoporen eingebettet, die als molekulare Sensoren dienen. Die Zelle ist in eine obere cis– und eine untere trans-Kammer unterteilt. Durch Anlegen einer elektrischen Spannung wird (hier im Bild) DNA durch die Nanopore gezogen; die dabei entstehenden Stromänderungen werden gemessen und zur Sequenzbestimmung genutzt.
Der Weg der DNA durch die Nanopore
1️⃣ Library Preparation (Bibliotheksvorbereitung)
Bevor die Sequenzierung beginnen kann, muss die in der Probe enthaltene Nukleinsäure (DNA und/oder RNA) für die Nanopore-Technologie vorbereitet werden. Dieser Prozess wird als Library Preparation bezeichnet.
Im Kontext der Qualitätskontrolle therapeutischer mRNA – etwa direkt nach der in-vitro-Transkription oder vor der Formulierung – sind dabei folgende Schritte relevant:
a) Aufreinigung
Nach der Transkription enthält das Reaktionsgemisch neben der gewünschten mRNA oder DNA noch Enzyme (z. B. Polymerasen), freie Nukleotide und Pufferbestandteile.
Diese Komponenten müssen entfernt werden, da sie:
- die Effizienz der Adapter-Ligation beeinträchtigen
- die Nanoporen blockieren könnten
- das elektrische Signal stören
In frühen Prozessschritten ist daher eine gründliche Aufreinigung erforderlich. Kurz vor der finalen Formulierung liegt das Produkt in der Regel bereits gereinigt vor, sodass keine zusätzliche Grundaufreinigung mehr notwendig ist.
b) End-Repair & dA-Tailing (bei DNA)
Soll DNA sequenziert werden – etwa zur Analyse von DNA-Rückständen – müssen die Fragmentenden vorbereitet werden.
Zunächst erfolgt eine End-Reparatur: Überhängende („sticky“) Enden werden enzymatisch aufgefüllt oder zurückgeschnitten, sodass glatte, doppelsträngige Enden entstehen.
Anschließend wird häufig ein sogenanntes dA-Tailing durchgeführt. Dabei hängt eine Polymerase gezielt ein einzelnes Adenosin (A) an jedes 3′-Ende der DNA an. Es entsteht ein definierter 3′-A-Überhang, der für die nachfolgende Adapter-Ligation erforderlich ist.
c) Adapter-Ligation – der entscheidende Schritt
Damit die Nukleinsäuren von der Nanopore erkannt und kontrolliert durch die Pore geschleust (transloziert) werden können, müssen spezielle Adapter-Moleküle an die Enden angefügt (ligiert) werden.
Diese Adapter sind funktionelle Module und enthalten mehrere entscheidende Komponenten:
Motorprotein
Das Motorprotein wirkt als molekulare Bremse. Ohne diese Kontrolle würde die DNA mit extrem hoher Geschwindigkeit durch die Pore „schießen“ – viel zu schnell für eine präzise Signalauflösung.
Das Motorprotein:
- reguliert die Translokationsgeschwindigkeit (≈ 400–500 Basen pro Sekunde)
- sorgt für einen gleichmäßigen Vorschub
- entwindet doppelsträngige DNA bei der Passage
Oxford Nanopore verwendet unterschiedliche Motorproteine (Helicasen) für:
- doppelsträngige DNA
- cDNA
- native (direct) RNA-Sequenzierung
Tether
Der Tether kann als molekularer „Einpark-Assistent“ beschrieben werden. Er enthält meist eine cholesterinartige Struktur, die sich in die Lipidmembran der Flow Cell einlagert. Dadurch wird die Nukleinsäure gezielt in die Nähe der Membranoberfläche gebracht – also genau dorthin, wo sich die Nanoporen befinden.
So erhöht sich die Wahrscheinlichkeit erheblich, dass das Molekül in das elektrische Feld einer Pore gerät und hineingezogen wird.
In manchen Protokollen wird zusätzlich ein sogenannter Tether-Buffer eingesetzt, um die Membranoberfläche mit diesen Ankerstrukturen anzureichern.
Barcode-Sequenzen (optional)
Barcode-Sequenzen ermöglichen die parallele Analyse mehrerer Proben in einem Sequenzierlauf (Multiplexing).
Für die reine Qualitätskontrolle einzelner Chargen sind sie nicht zwingend erforderlich und werden daher im Folgenden nicht weiter berücksichtigt.
⬦ ⬦ ⬦
Adapter-Design bei ONT
Oxford Nanopore stellt die Sequenzier-Adapter bereits vorbeladen mit Motorprotein und Tether bereit.
Bei der Ligation wird somit der gesamte Adapter-Komplex an das Nukleinsäuremolekül gekoppelt.
Bei DNA:
- Der Adapter wird an die Enden der doppelsträngigen DNA ligiert.
- Die Adapter besitzen einen überstehenden Thymin-(T)-Rest, der komplementär zum zuvor erzeugten 3′-A-Überhang der DNA ist.
- Dadurch entsteht eine effiziente und gerichtete Ligation.
Bei RNA (Direct RNA Sequencing):
- Der Adapter wird am 3′-Ende der RNA ligiert, meist über den vorhandenen Poly(A)-Tail.
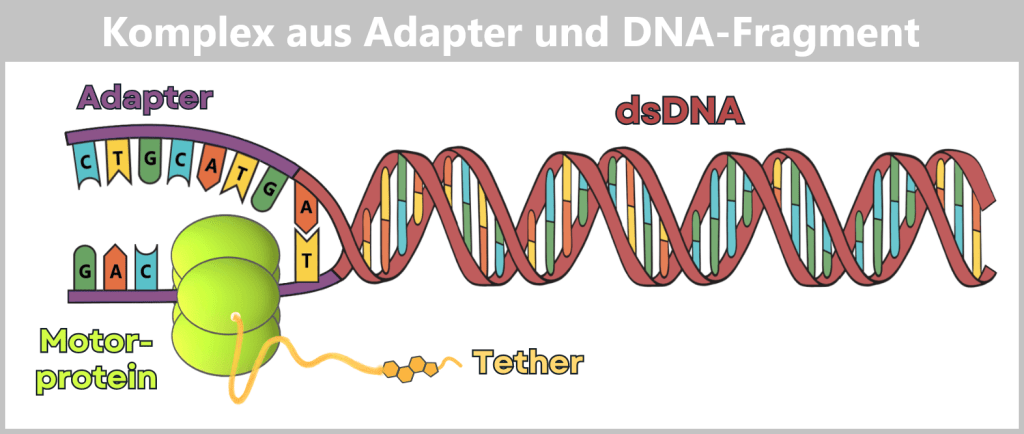
Der Sequenzier-Adapter enthält neben einer doppelsträngigen DNA-Region (meist etwa 30 bis 50 Basenpaare lang) auch zwei kurze einzelsträngige Sequenzen (oft ca. 20 bis 40 Nukleotide). Zur besseren Übersicht zeigt die Abbildung eine stark verkürzte Darstellung.
Ein Einzelstrang ist bereits im Inneren des Motorproteins gebunden, während einige Basen des zweiten Einzelstranges außerhalb sichtbar sind.
Direkt daran befindet sich der Übergang zur eigentlichen Probe. Ein spezifischer Überhang am Ende des Adapters (T) hat sich mit dem komplementären Ende der Proben-DNA (A) verbunden. Diese „Ligation“ verknüpft die zu untersuchende Doppelstrang-DNA (dsDNA) stabil mit dem Sequenzier-System.
Seitlich am Motorprotein ist der Tether befestigt. An seinem freien Ende befindet sich ein Cholesterin-Molekül. Dieses dient als chemischer „Dübel“, der den gesamten Komplex in der Lipidmembran der Pore fixiert, um die Wahrscheinlichkeit zu erhöhen, dass die DNA die Nanopore findet.
Nach der Ligation sind die Adapter stabil mit der Nukleinsäure verbunden und die Bibliothek ist sequenzierbereit.
Besonderheit: Direct RNA Sequencing (z. B. im Rahmen von VAX-seq)
Eine Besonderheit der Nanopore-Technologie ist die Möglichkeit, native RNA direkt zu sequenzieren, ohne vorherige Umschreibung in cDNA.
Für die Qualitätskontrolle therapeutischer mRNA ist das von großer Bedeutung, da:
- chemische Modifikationen (z. B. Pseudouridin) direkt erfasst werden können
- keine Reverse-Transkriptionsartefakte entstehen
- strukturelle Besonderheiten der RNA erhalten bleiben
Damit erlaubt die Technologie nicht nur eine Mengenbestimmung, sondern eine strukturelle und funktionelle Charakterisierung auf Molekülebene.
2️⃣ Anlegen einer elektrischen Spannung
In beiden Kammern der Sequenzierzelle befinden sich geladene Teilchen (Ionen). Sobald zwischen der oberen cis-Kammer und der unteren trans-Kammer eine elektrische Spannung angelegt wird, entsteht ein elektrisches Feld über der Membran. Dieses Feld treibt die Ionen durch die Nanoporen, wodurch ein messbarer elektrischer Strom entsteht.
Solange sich keine DNA in der Pore befindet, bleibt dieser Ionenfluss konstant. Das Gerät registriert in diesem Zustand einen stabilen Grundstrom, der als Referenzsignal dient (siehe folgende Abbildung).
3️⃣ Die DNA wird in die Sequenzierzelle gegeben
Die vorbereitete DNA-Probe wird in die obere cis-Kammer der Sequenzierzelle pipettiert. An den Enden der DNA befinden sich bereits die Sequenzier-Adapter, die mit einem Motorprotein und einem Tether ausgestattet sind.
Der Tether trägt an seinem Ende eine hydrophobe (wasserabweisende) Cholesterin-Gruppe. Sie fungiert als molekularer Anker:
Durch Diffusion bewegen sich die DNA-Stränge in der cis-Kammer ständig umher. Sobald das wasserabweisende Cholesterin dabei zufällig die Membran berührt, bleibt es dort sofort haften. Für das Cholesterin ist es energetisch weitaus günstiger, in der Lipidschicht zu verweilen als im wasserbasierten Puffer.
Dieser Mechanismus bietet zwei wesentliche Vorteile:
Konzentration an der Oberfläche: Die DNA-Moleküle sammeln sich bevorzugt in der Nähe der Membran. Dadurch steigt die Wahrscheinlichkeit, dass das Motorprotein eine Nanopore findet und dort andocken kann.
Laterale Mobilität: Das Cholesterin ist in der Membran nicht starr fixiert, sondern bleibt lateral (seitlich) beweglich. Die DNA kann daher entlang der Membranoberfläche „gleiten“, bis sie vom elektrischen Feld einer Pore erfasst und schließlich eingezogen wird.
4️⃣ Andocken an die Nanopore
Sobald ein einzelsträngiger Abschnitt der DNA in die Nanopore eintritt, zieht das elektrische Feld den negativ geladenen DNA-Strang in Richtung der trans-Kammer. Dadurch wird das Motorprotein gegen die Porenöffnung positioniert und dockt stabil auf der Pore an.
Der elektrische Zug auf den DNA-Strang wirkt dabei wie ein mechanischer Startimpuls für das Motorprotein. Es beginnt nun, seine Helicase-Aktivität auszuführen:
- Es entwindet die doppelsträngige DNA (dsDNA).
- Ein Strang wird schrittweise und mit kontrollierter Geschwindigkeit durch die Nanopore geführt.
- Der zweite Strang bleibt außerhalb der Pore.
Damit startet der eigentliche Sequenzierprozess.
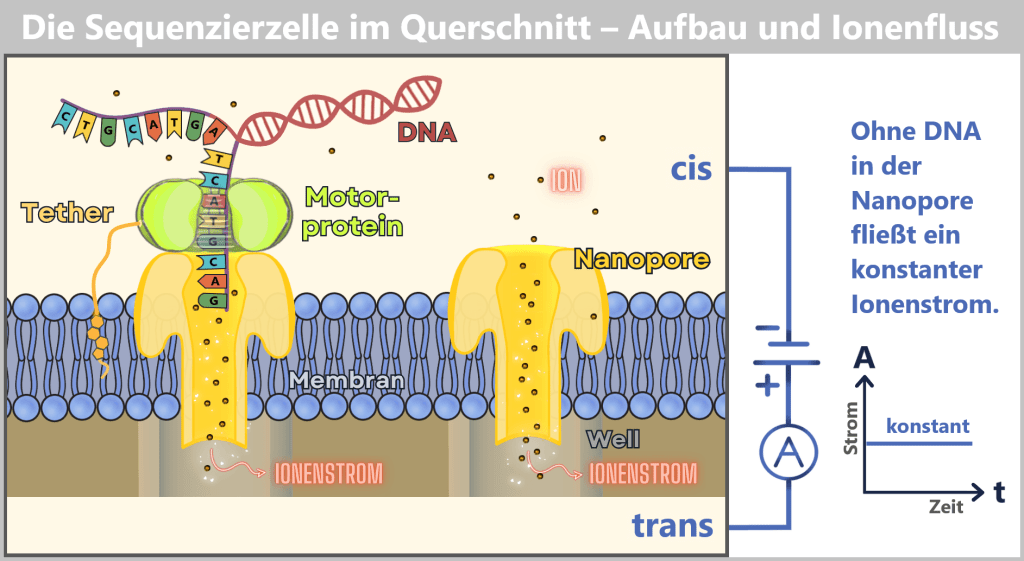
(nicht maßstabsgetreu)
Die Sequenzierzelle besteht aus zwei Kammern: der cis-Kammer (oben) und der trans-Kammer (unten), die durch eine Membran mit eingebetteten Nanoporen getrennt sind. Links hat ein DNA-Molekül mithilfe eines Motorproteins an eine Nanopore angedockt. Rechts ist eine leere Nanopore dargestellt, durch die ein konstanter Ionenstrom fließt.
Die angelegte Spannung zwischen der negativen Elektrode in der cis-Kammer und der positiven Elektrode in der trans-Kammer treibt den Ionenfluss durch die Pore an. Der Ionenstrom wird im Well (einem kleinen Kanal im Chip unterhalb der Pore) gemessen, wie im Strom-Zeit-Diagramm dargestellt. Solange keine DNA durch die Pore wandert, bleibt der Strom konstant.
Das Motorprotein – der Taktgeber der Sequenzierung
Die vorbereitete DNA trägt an ihrem Ende einen Adapter mit einem Motorprotein (einer Helikase). Sobald dieses Molekül in die Nähe der Nanopore gelangt, setzt sich das Protein wie ein passgenauer Deckel auf den Rand der Pore („Docking“). Die elektrische Spannung im System wirkt dabei wie ein unsichtbarer Sog, der die DNA ins Innere der Pore zieht.
Der elektrische Zug unterstützt dabei die Aktivität des Motorproteins. Dieses wirkt wie ein Keil, der den Doppelstrang behutsam auseinanderhebelt. Die Wasserstoffbrücken zwischen den Basenpaaren – die „Sprossen“ der DNA-Leiter – brechen unter diesem Druck auf, da sie deutlich schwächer sind als die stabilisierenden Bindungskräfte innerhalb des Proteins. So wird die doppelsträngige DNA (dsDNA) wie ein Reißverschluss aufgezippt, ohne dass das chemische Rückgrat der Stränge beschädigt wird.
An diesem Punkt beginnt die präzise Kontrolle: Im Inneren des Motorproteins greifen vier bis sechs DNA-bindende Schleifen (sogenannte Hairpins oder Loops) nach dem nun freigelegten Einzelstrang (ssDNA). Diese Greifarme sind spiralig angeordnet, vergleichbar mit den Stufen einer Wendeltreppe oder dem Gewinde einer Schraube.
Man kann sich das Protein als eine „atmende Schraube“ vorstellen: Durch die Zufuhr von Energie (ATP) verändert das Protein rhythmisch seine Form. Es zieht sich zusammen und dehnt sich wieder aus. Bei jeder dieser Bewegungen lösen die obersten Greifarme ihren Griff, rücken ein Stück vor und packen die DNA weiter oben neu, während die unteren Arme den Strang sicher festhalten und kontrolliert nach unten in die Pore schieben.
Diese treppenförmige Anordnung sorgt dafür, dass die DNA nicht unkontrolliert durch die Pore schießt, sondern Schritt für Schritt – also Base für Base – hindurchgeführt wird. Es ist dieser exakte, mechanische Takt des Motorproteins, der es der Nanopore ermöglicht, das elektrische Signal jeder einzelnen Base in aller Ruhe zu registrieren.
5️⃣ Die DNA passiert die Nanopore – das Signal entsteht
DNA besitzt aufgrund ihrer Phosphatgruppen eine negative elektrische Ladung. Durch die angelegte Spannung zwischen der negativ geladenen cis-Kammer und der positiv geladenen trans-Kammer wird der Einzelstrang durch die Nanopore gezogen.
Hier übernimmt das Motorprotein eine entscheidende Funktion:
- Es bewegt die DNA schrittweise durch die Pore.
- Dadurch wird die Geschwindigkeit stark reduziert.
- Die Bewegung erfolgt langsam, gleichmäßig und kontrolliert.
Ohne diese Kontrolle würde die DNA mit einer Geschwindigkeit von Millionen Basen pro Sekunde durch die Nanopore schießen. Das Motorprotein bremst diese Bewegung auf etwa 300–450 Basen pro Sekunde. Erst dadurch wird eine zuverlässige Signalmessung möglich.
Während der DNA-Strang durch die Pore wandert, beeinflusst er den Ionenstrom. Jede Base besitzt eine leicht unterschiedliche Größe und chemische Struktur. Diese Unterschiede verändern den Stromfluss auf charakteristische Weise.
Die Stromänderungen werden von Elektroden gemessen, die mit den einzelnen Wells im Chip verbunden sind. Jeder Well enthält eine Nanopore und besitzt eine eigene Messeinheit. Dadurch kann die Software die Signale eindeutig einer bestimmten Pore zuordnen.
6️⃣ Das elektrische Signal wird aufgezeichnet
Während der Sequenzierung befinden sich typischerweise etwa fünf bis sechs Basen gleichzeitig im engsten Bereich der Nanopore. Dieses kurze DNA-Segment wird als k-mer bezeichnet. Es wirkt im Porenkanal wie ein elektrischer Widerstand: Unterschiedliche Basenkombinationen beeinflussen den Ionenfluss jeweils auf charakteristische Weise und erzeugen dadurch unterschiedliche Stromsignale.
Die DNA-Sequenz lässt sich rekonstruieren, weil sich die DNA Schritt für Schritt durch die Pore bewegt und jedes k-mer ein charakteristisches elektrisches Signal erzeugt. Spezialisierte Software analysiert diese Abfolge von Stromsignalen mithilfe maschinellen Lernens, das die komplexen Signalmuster den jeweiligen Basenabfolgen zuordnet. Dieser Prozess wird Basecalling genannt.
Auf diese Weise entsteht aus den elektrischen Messwerten Schritt für Schritt die vollständige Nukleotidsequenz.
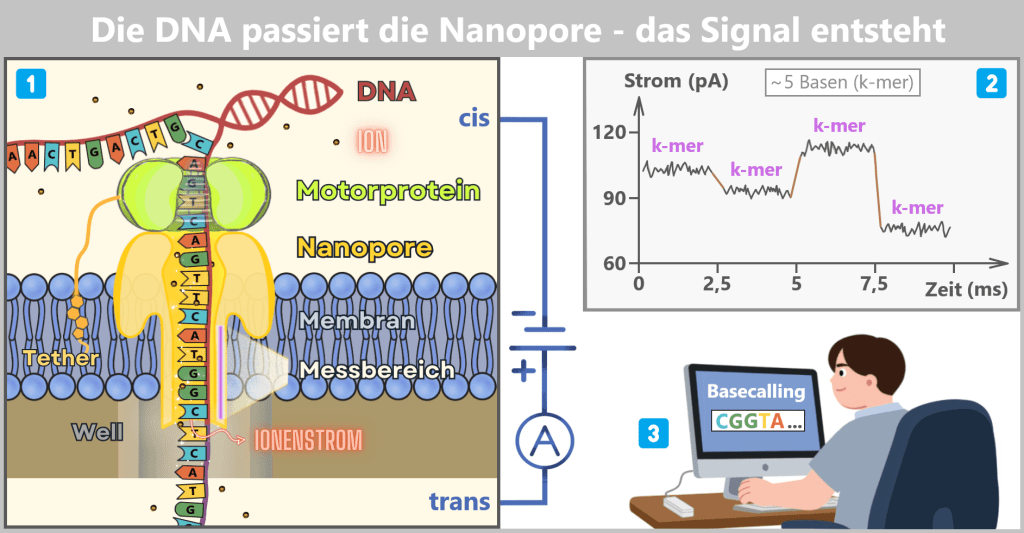
1) Die Sequenzierzelle im Querschnitt
Ein DNA-Einzelstrang wird aufgrund der angelegten Spannung von der cis-Seite zur trans-Seite gezogen. Das Motorprotein sitzt oberhalb auf der Nanopore. Es wirkt dabei wie eine molekulare Bremse und kontrolliert den Vorschub der DNA. Dadurch wird der Einzelstrang mit einer Geschwindigkeit von etwa 400 Basen pro Sekunde durch die Pore bewegt, sodass jedes Signal präzise gemessen werden kann.
In der engsten Stelle der Pore (hier als Messbereich gekennzeichnet) befinden sich gleichzeitig mehrere Basen – typischerweise etwa fünf Nukleotide, ein sogenanntes k-mer. Dieses kurze DNA-Segment verengt den Porenkanal.
Die Ionen (kleine rote Punkte) versuchen weiterhin durch die Pore zu strömen. Da das k-mer den verfügbaren Raum einschränkt, können nur noch weniger Ionen passieren. Dadurch entsteht ein charakteristisches Stromsignal.
2) Die Daten im Strom-Zeit-Diagramm
Die Y-Achse zeigt den Ionenstrom in Picoampere (pA), die X-Achse die Zeit in Millisekunden (ms).
Ein k-mer befindet sich typischerweise für etwa 2 bis 3 Millisekunden im engsten Bereich der Pore. Während dieser Zeit werden mehrere einzelne Strommesswerte für genau diese Basenkombination aufgezeichnet.
Dabei entsteht im Diagramm ein leicht schwankendes Plateau, das durch elektrisches Rauschen verursacht wird. Die braunen Verbindungslinien zwischen den Plateaus markieren den Moment, in dem das Motorprotein die DNA um eine Base weitertransportiert und dadurch ein neues k-mer in den Messbereich gelangt.
3) Basecalling – Umwandlung der Stromsignale in Basen
Die charakteristischen Stromsignale – die Plateaus im Strom-Zeit-Diagramm – entsprechen jeweils dem elektrischen Signal eines bestimmten k-mers, also einer kurzen Basenkombination, die sich für wenige Millisekunden im Messbereich der Pore befindet.
Basecalling-Algorithmen, die häufig auf neuronalen Netzen basieren, analysieren diese Abfolge von Stromsignalen. Aus den charakteristischen Mustern der Signale rekonstruieren sie die zugrunde liegende DNA-Basensequenz.
🎥 Tipp: Das Video „How nanopore sequencing works“ fasst den Prozess anschaulich zusammen.
⬦ ⬦ ⬦
Ergebnis der Nanopore-Sequenzierung
Die Nanopore-Sequenzierung liefert keinen einzelnen Messwert, sondern einen umfangreichen Datensatz aus vielen individuellen Sequenzreads. Jeder dieser Reads entspricht der Analyse eines einzelnen DNA- oder RNA-Moleküls. Aus der Gesamtheit dieser Einzelmessungen ergibt sich ein detailliertes Bild der in der Probe enthaltenen Nukleinsäuren.
Dabei können grundsätzlich alle in der Probe vorhandenen DNA- oder RNA-Moleküle erfasst werden, ohne dass eine spezifische Zielsequenz vorgegeben werden muss. Die Methode ist somit nicht zielgerichtet, sondern explorativ.
◈ ◈ ◈
Zwei zentrale Informationsebenen: Identität und Quantität
Die gewonnenen Sequenzdaten enthalten zwei komplementäre Informationsebenen:
Identität (Sequenzinformation)
Für jedes einzelne Molekül kann die Nukleotidsequenz bestimmt werden. Daraus ergeben sich Informationen über:
- Effizienz der Bibliotheksvorbereitung
- Moleküllänge
- Sequenzeigenschaften
- mögliche Sequenzvarianten
- sowie – mit methodischen Einschränkungen – chemische Modifikationen
Auf dieser Grundlage lässt sich bestimmen, welche Nukleinsäuren in einer Probe vorhanden sind.
Quantität (relative Häufigkeit)
Zusätzlich kann über die Anzahl der Reads abgeschätzt werden, wie häufig bestimmte Sequenzen oder Molekültypen in der Probe vertreten sind. Dabei handelt es sich jedoch nicht um eine absolute Quantifizierung, sondern um eine relative Annäherung, die von verschiedenen Faktoren beeinflusst wird, etwa:
- Effizienz der Bibliotheksvorbereitung
- Moleküllänge
- Sequenzeigenschaften
Die quantitative Aussagekraft der Methode ist daher stets im Kontext dieser Einflussgrößen zu interpretieren.
✧ ✧ ✧
Parallele Einzelmolekülmessung in Echtzeit
Die Datenerzeugung erfolgt durch eine große Anzahl parallel arbeitender Nanoporen innerhalb der sogenannten Flow Cell. Geräte wie das MinION verfügen über mehrere hundert aktive Messkanäle, die gleichzeitig und unabhängig voneinander arbeiten.
Jede Nanopore analysiert jeweils ein einzelnes Molekül. Sobald ein Sequenzvorgang abgeschlossen ist, steht die Pore unmittelbar für das nächste Molekül zur Verfügung. Auf diese Weise entsteht kontinuierlich eine große Zahl individueller Reads.
Die Messung erfolgt in Echtzeit, wobei die Software die Signale vieler einzelner Poren gleichzeitig erfasst und verarbeitet.
✧ ✧ ✧
Charakter der Daten: Verteilung statt Einzelwert
Ein wesentliches Merkmal der Nanopore-Sequenzierung ist, dass die Ergebnisse nicht als einheitlicher Messwert vorliegen, sondern als Verteilung vieler einzelner Sequenzreads. Diese Reads können sich deutlich unterscheiden in:
- Länge
- Sequenz
- Qualität
Insbesondere in komplexen Proben entstehen typischerweise:
- kurze Fragmente
- längere zusammenhängende Moleküle
- unterschiedliche Molekülpopulationen
Die Analyse besteht daher nicht in der Betrachtung einzelner Reads, sondern in der Auswertung der Gesamtheit dieser Verteilung.
✧ ✧ ✧
Besonderheit der Methode: Long Reads
Ein charakteristisches Merkmal der Nanopore-Technologie ist die Fähigkeit, sehr lange Nukleinsäuremoleküle kontinuierlich zu sequenzieren. Im Gegensatz zu Short-Read-Verfahren (wie der Illumina-Sequenzierung), bei denen DNA zunächst in kurze Fragmente zerlegt und anschließend bioinformatisch rekonstruiert wird, können hier zusammenhängende Moleküle direkt erfasst werden.
Die Länge eines Reads ist dabei prinzipiell nicht durch die Technologie begrenzt, sondern wird hauptsächlich durch die Integrität des Ausgangsmoleküls bestimmt.
Dies ermöglicht insbesondere:
- die direkte Analyse vollständiger Moleküle
- die Unterscheidung zwischen intakten Sequenzen und Fragmenten
- die Untersuchung struktureller Zusammenhänge innerhalb einzelner Moleküle
✧ ✧ ✧
Grenzen und methodische Einflüsse
Trotz dieser Vorteile unterliegt die Nanopore-Sequenzierung verschiedenen methodischen Einschränkungen:
Fehlerrate einzelner Reads
Die Genauigkeit einzelner Sequenzreads ist im Vergleich zu klassischen Short-Read-Verfahren geringer. Ursache hierfür ist die indirekte Bestimmung der Sequenz über elektrische Stromsignale. Bestimmte Sequenzmotive, insbesondere homopolymere Bereiche (z. B. lange Wiederholungen derselben Base), können dabei schwieriger aufgelöst werden.
Selektions- und Prozessbias
Nicht alle Moleküle werden mit gleicher Wahrscheinlichkeit erfasst. Unterschiede in Länge, Struktur oder Adapterbindung können dazu führen, dass bestimmte Sequenzen bevorzugt oder seltener sequenziert werden.
Insbesondere die Bibliotheksvorbereitung kann die Zusammensetzung der später sequenzierten Moleküle beeinflussen. Reinigungsschritte mit magnetischen Beads oder andere größenabhängige Prozessschritte können sehr kurze Fragmente teilweise entfernen oder unterrepräsentieren. Gleichzeitig werden längere Moleküle bei der Nanopore-Sequenzierung häufig effizienter erfasst, da sie länger durch die Pore laufen und dadurch stabilere Signale erzeugen.
Die resultierende Read-Verteilung spiegelt daher nicht zwangsläufig exakt die ursprüngliche Fragmentverteilung der Probe wider.
Abhängigkeit von der Probenqualität
Die Qualität und Integrität der Ausgangsnukleinsäuren beeinflussen die resultierenden Reads erheblich. Insbesondere lange Moleküle sind anfällig für Fragmentierung während der Probenvorbereitung.
✧ ✧ ✧
Konsensusbildung und bioinformatische Auswertung
Um die Genauigkeit der Ergebnisse zu erhöhen, werden die Daten nicht auf Basis einzelner Reads interpretiert. Stattdessen werden viele Sequenzreads desselben Molekültyps miteinander verglichen und zu einer Konsensussequenz zusammengeführt.
Da Sequenzierfehler überwiegend zufällig verteilt sind, können sie durch diese Mehrfachanalyse weitgehend reduziert werden. Moderne Basecalling- und Auswertungsverfahren, die häufig auf maschinellem Lernen basieren, tragen zusätzlich zur Verbesserung der Sequenzgenauigkeit bei.
Auf diese Weise lässt sich aus einer Vielzahl einzelner Messungen ein konsistentes und belastbares Gesamtbild ableiten.
✧ ✧ ✧
Einordnung für die Analyse von Nukleinsäuren
Die Nanopore-Sequenzierung ermöglicht somit eine detaillierte Charakterisierung von Nukleinsäuren hinsichtlich ihrer Sequenz, Länge und relativen Häufigkeit. Gleichzeitig erfordert die Interpretation der Daten eine Berücksichtigung methodischer Einflüsse, insbesondere im Hinblick auf Quantifizierung und Repräsentativität der erfassten Moleküle.
2.4.2. Illumina-Sequenzierung
Die Nanopore-Sequenzierung ermöglicht die Analyse langer, zusammenhängender Moleküle in Echtzeit – ihre besondere Stärke liegt in der Erfassung vollständiger Sequenzen, ohne dass diese zuvor zwingend in kurze Standardfragmente zerlegt werden müssen. Eine andere Herangehensweise verfolgt die Illumina-Sequenzierung: Sie arbeitet nicht mit einzelnen langen Molekülen, sondern mit einer sehr großen Zahl kurzer Fragmente, die parallel und hochpräzise gelesen werden. Beide Technologien ergänzen sich – sie beantworten unterschiedliche Fragen mit unterschiedlichen Mitteln.
Die Illumina-Sequenzierung ist ein Short-Read-Verfahren und gehört zur Gruppe der sogenannten Next Generation Sequencing (NGS)-Technologien. Sie ist derzeit das weltweit am weitesten verbreitete Sequenzierverfahren.
Das Grundprinzip unterscheidet sich deutlich von der Nanopore-Technologie: Während ONT einzelne Moleküle in Echtzeit durch eine Pore führt und dabei elektrische Signale misst, basiert Illumina auf einem optischen Verfahren – der sequenzierenden Synthese mit fluoreszenzmarkierten Nukleotiden („Sequencing by Synthesis“). Dabei wird die DNA nicht direkt gelesen, sondern während einer kontrollierten Synthesereaktion schrittweise entschlüsselt.
Funktionsprinzip
1️⃣ Vorbereitung der DNA-Fragmente = Bibliothekserstellung
Für die Sequenzierung wird die DNA zunächst in ein Format gebracht, das für die Illumina-Plattform geeignet ist. Dieser Prozess wird auch als Bibliothekserstellung bezeichnet und er umfasst folgende Teilschritte:
a) Fragmentierung
Die DNA wird mechanisch oder enzymatisch in kleinere Stücke zerlegt (typischerweise mit einer Zielgröße von etwa 150–500 Basenpaaren). Dabei entstehen DNA-Fragmente leicht unterschiedlicher Länge, die anschließend oft noch über ein Größenauswahlverfahren vereinheitlicht werden.
Dieser Schritt ist besonders wichtig, wenn lange DNA-Moleküle sequenziert werden sollen, da die Illumina-Technologie kurze Fragmente effizienter verarbeitet als sehr lange zusammenhängende Moleküle.
In einigen Anwendungen kann die Fragmentierung jedoch bewusst reduziert oder ganz weggelassen werden – insbesondere dann, wenn bereits stark fragmentierte DNA untersucht wird. Auch in solchen Ansätzen werden sehr lange Fragmente (typischerweise oberhalb von etwa 800–1000 bp) im weiteren Verlauf der Bibliotheksvorbereitung und Sequenzierung häufig weniger effizient erfasst und können dadurch unterrepräsentiert sein.
Zur Veranschaulichung betrachten wir in unserem Beispiel drei DNA-Fragmente mit leicht unterschiedlichen Längen.
b) Adapter-Ligation
An beide Enden der DNA-Fragmente werden spezifische Adaptersequenzen (P5- und P7-Adapter) angehängt. Diese Adapter erfüllen mehrere Funktionen:
Bindungsstellen für die Flowcell: Die Adapter enthalten spezielle DNA-Sequenzen, die wie ein Schlüssel zu einem Schloss passen. Dadurch können die DNA-Fragmente später an einer Oberfläche haften bleiben, was für die Sequenzierung wichtig ist.
Primer-Bindungsstellen: Die Adapter enthalten Abschnitte, an die Sequenzierungsprimer binden können. Diese Primer werden später genutzt, um die DNA-Stränge schrittweise zu synthetisieren.
Indizes (optional): Falls mehrere Proben gleichzeitig sequenziert werden, ermöglichen Index-Sequenzen die Zuordnung der Fragmente zu ihrer jeweiligen Probe. Zur besseren Übersichtlichkeit verzichten wir in unserem Beispiel auf die Darstellung der Indizes.
c) PCR-Amplifikation
Um sicherzustellen, dass genügend DNA für die Sequenzierung vorliegt, werden die adaptierten DNA-Fragmente mittels Polymerase-Kettenreaktion (PCR) vervielfältigt.
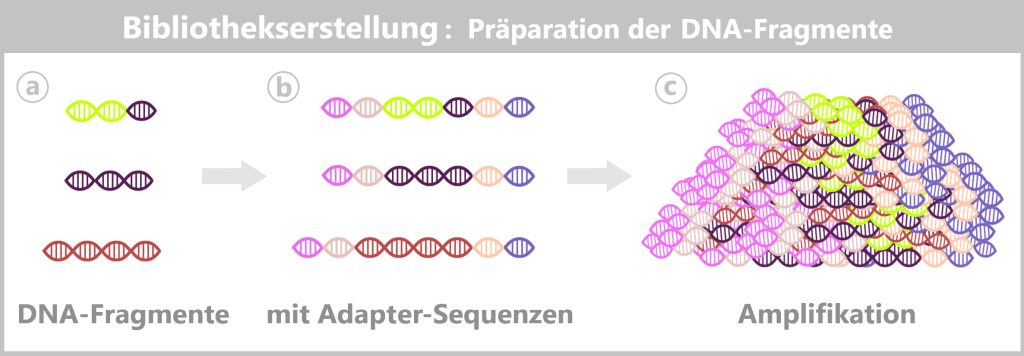
Die folgende Vergrößerung zeigt eine detaillierte Darstellung eines fragmentierten DNA-Doppelstrangs, versehen mit den beiden Adaptern.
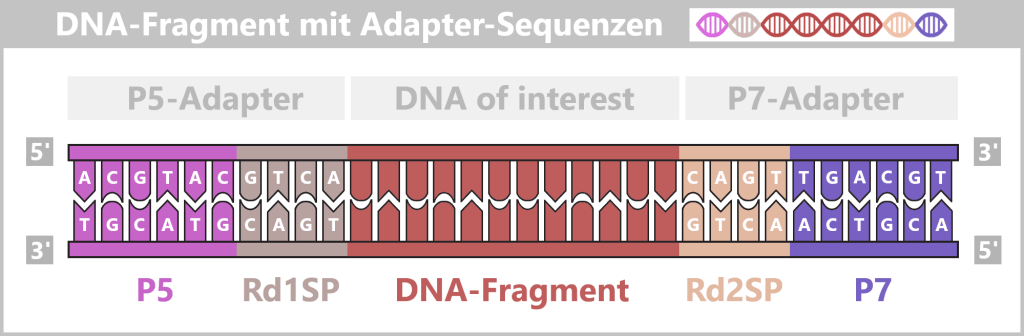
Jedes DNA-Fragment erhält zwei Adaptersequenzen: Der P5-Adapter besteht aus der P5-Adaptersequenz, die die Bindung an die Flowcell ermöglicht, und der Primer-Bindestelle Rd1SP (Read 1 Sequencing Primer) zur Initiation der DNA-Synthese. Ebenso enthält der P7-Adapter die P7-Adaptersequenz zur Flowcell-Bindung sowie die Primer-Bindestelle Rd2SP (Read 2 Sequencing Primer) für die Initiation der DNA-Synthese.
Diese Darstellung ist stark vereinfacht. In der Praxis sind Illumina-Adaptersequenzen mit 60-120 Basenpaarendeutlich länger. Auch das DNA-Fragment ist zur besseren Veranschaulichung verkürzt – tatsächlich haben DNA-Fragmente typischerweise eine Länge von 100–500 Basenpaaren.
2️⃣ Cluster-Generierung auf einer Flowcell
Das Herzstück der Illumina-Technologie ist die sogenannte Flowcell – eine glasartige Oberfläche, die mit Millionen kurzer, fest verankerter DNA-Bindestellen beschichtet ist.
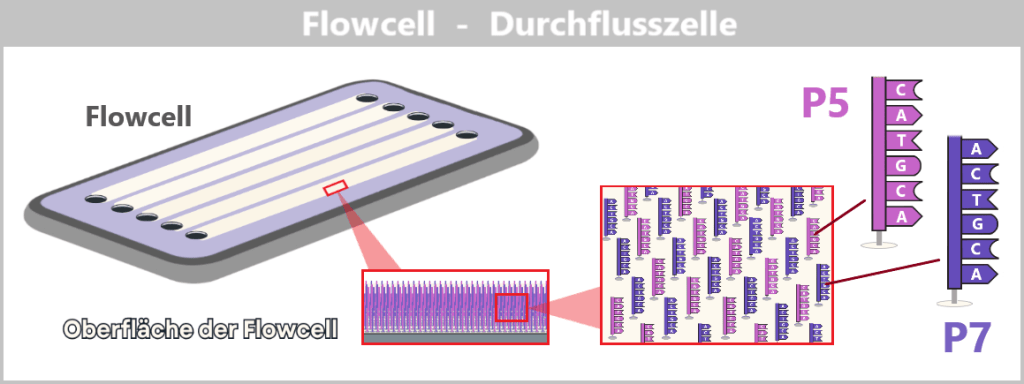
a) DNA-Fragmente binden an die Flowcell-Oberfläche
Zu Beginn dieses Schritts wird eine Lösung einzelsträngiger DNA-Fragmente auf die Flowcell geleitet. Die Fragmente tragen an ihren Enden die zuvor ligierten Adaptersequenzen und wurden durch Denaturierung in Einzelstränge überführt, sodass sie frei in der Lösung vorliegen.
Während die Fragmente über die Flowcell strömen, binden ihre Adapter durch komplementäre Basenpaarung an passende DNA-Bindestellen auf der Oberfläche.
Zur Veranschaulichung kann man sich die Oberfläche der Flowcell wie einen dichten Klettverschluss vorstellen: Die Adaptersequenzen der DNA-Fragmente haften an komplementären Bindestellen der Flowcell – ähnlich wie kleine Haken, die sich im Klettgewebe verankern
b) Erste Synthese
Nachdem die DNA-Fragmente an die Flowcell gebunden wurden, beginnt die erste DNA-Synthese. Hierzu bindet ein Primer an die Adaptersequenz des immobilisierten Einzelstrangs. Anschließend synthetisiert die DNA-Polymerase einen komplementären Gegenstrang.
Im nächsten Schritt wird der ursprüngliche Einzelstrang entfernt, während der neu synthetisierte Strang an der Flowcell gebunden bleibt.
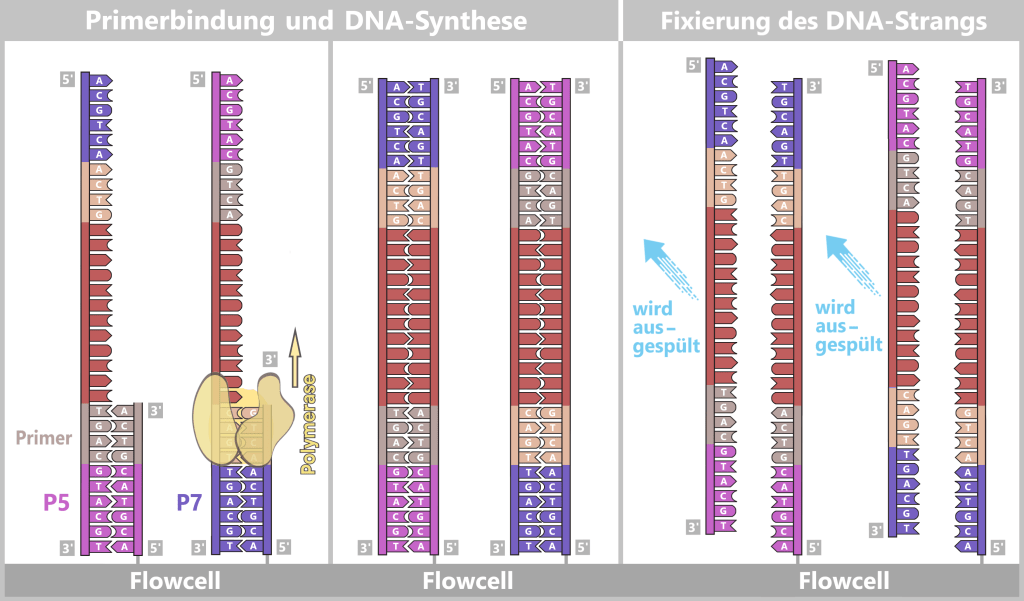
Links: Primer binden an die Adapter (P5, P7), und die DNA-Polymerase (DNAP) startet die Synthese eines neuen, komplementären Strangs.
Mitte: Die DNA-Polymerase synthetisiert daraufhin einen ersten Strang.
Rechts: Der neue entstandene DNA-Doppelstrang wird aufgetrennt. Der ursprüngliche Strang hat nach der Denaturierung keine Verbindung mehr zur Flowcell und wird ausgespült. Der neu synthetisierte Strang bleibt mit seinem 5′-Ende fest an der Flowcell gebunden.
Zu diesem Zeitpunkt wäre eine direkte Sequenzierung noch nicht sinnvoll, da die Fluoreszenzsignale einzelner Moleküle zu schwach wären, um zuverlässig detektiert zu werden. Deshalb folgt nun die sogenannte Brückenamplifikation.
c) Brückenamplifikation
Damit die spätere Sequenzierung ein ausreichend starkes Fluoreszenzsignal erzeugt, müssen die an der Flowcell gebundenen DNA-Moleküle zunächst vervielfältigt werden.
Der Prozess beginnt damit, dass sich ein gebundener DNA-Einzelstrang aufgrund seiner Flexibilität zur Oberfläche zurückbiegt und mit seinem freien Adapterende an eine benachbarte komplementäre Bindestelle auf der Flowcell bindet. Dadurch entsteht eine charakteristische Brückenstruktur.
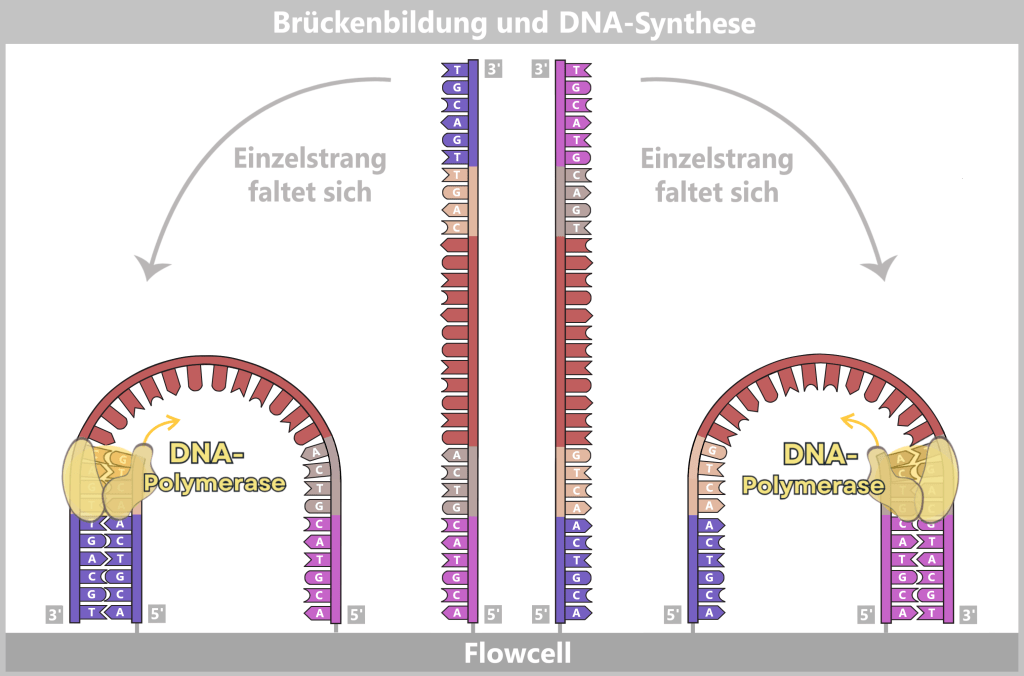
Anschließend synthetisiert die DNA-Polymerase entlang des gebundenen Strangs einen komplementären Gegenstrang. Nach der Synthese wird die Brückenstruktur wieder aufgetrennt, sodass nun zwei fest an der Flowcell verankerte Einzelstränge vorliegen.
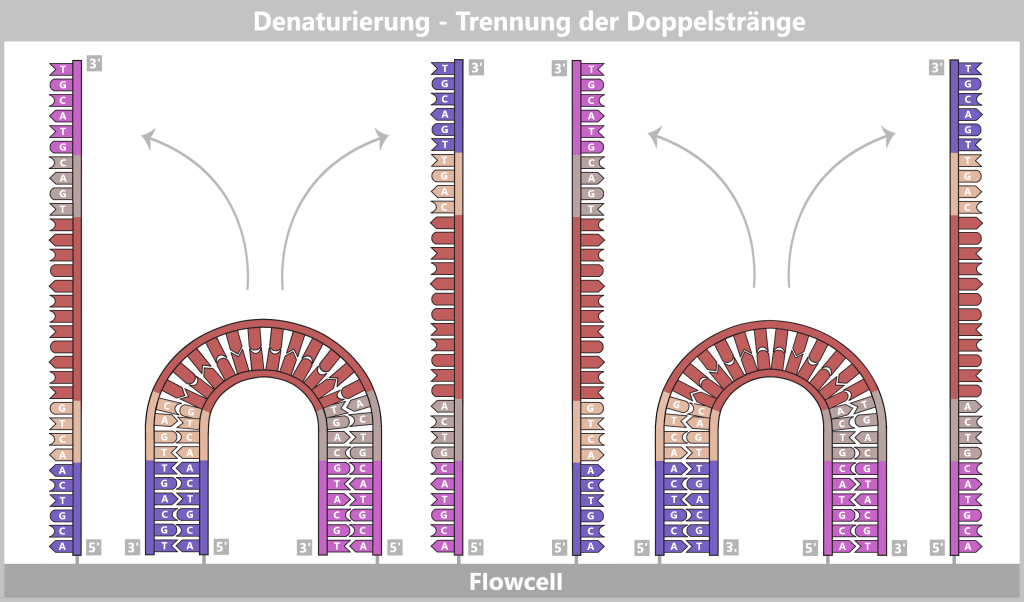
Nach dem Kopiervorgang werden die Brücken-Doppelstränge getrennt. Die Anzahl der fest verankerten DNA-Stränge verdoppelt sich. Sie sind bereit für die weitere Amplifikation.
Dieser Vorgang wiederholt sich vielfach. Mit jedem Zyklus verdoppelt sich die Zahl der gebundenen DNA-Moleküle.

Auf diese Weise entstehen lokal begrenzte Cluster aus Millionen nahezu identischer Kopien eines einzelnen ursprünglichen DNA-Fragments.

In dieser Darstellung sind exemplarisch nur drei Cluster gezeigt. In der Realität befinden sich jedoch Millionen solcher Cluster auf einer Flowcell, um eine hohe Sequenzierkapazität zu ermöglichen.
Das Ergebnis ist eine dicht gepackte Oberfläche aus Millionen räumlich getrennten DNA-Clustern, von denen jeder aus vielen nahezu identischen Kopien eines einzelnen ursprünglichen DNA-Fragments besteht. Dadurch können Millionen unterschiedlicher Sequenzen parallel analysiert werden.
Ohne diese Vervielfältigung wäre das Lesen der DNA, als würde man versuchen, ein einzelnes Glühwürmchen im Wind zu erkennen. Die Cluster hingegen machen die DNA-Basen (A, T, C, G) deutlich sichtbar – wie ein leuchtender Neon-Schriftzug in der Dunkelheit.
Entfernung eines Strangtyps
Damit die spätere Sequenzierung synchron und eindeutig ablaufen kann, müssen die DNA-Stränge innerhalb jedes Clusters in einheitlicher Orientierung vorliegen. Daher wird vor Beginn der eigentlichen Sequenzierung gezielt einer der beiden Strangtypen entfernt.
Die nicht benötigten Gegenstränge werden chemisch abgespalten und ausgespült. Gleichzeitig werden bestimmte freie Enden auf der Flowcell blockiert, um unerwünschte Neubindungen oder weitere Amplifikation zu verhindern.
Anschließend bestehen die Cluster aus vielen gleich orientierten Einzelsträngen. Die DNA-Bibliothek ist damit für die eigentliche Sequenzierung vorbereitet.
3️⃣ Sequenzieren durch Synthese
Um die Sequenzierung zu starten, werden Primer, DNA-Polymerasen und modifizierte Nukleotide auf die Flowcell gegeben. Jedes der vier Nukleotide (A, T, G und C) trägt dabei eine eigene Fluoreszenzmarkierung sowie eine reversible chemische Blockierung.
Die Primer hybridisieren mit den DNA-Strängen der Bibliothek. Anschließend bindet die DNA-Polymerase an den Primer und beginnt mit der Synthese des komplementären Strangs.
Durch die reversible Blockierung kann pro Zyklus jeweils nur ein einziges Nukleotid eingebaut werden. Nach dem Einbau stoppt die Synthese vorübergehend.
Anschließend wird die Flowcell mit hochauflösenden Kameras abgebildet. Die Fluoreszenzfarbe jedes Clusters zeigt an, welches Nukleotid in diesem Zyklus eingebaut wurde. Eine Auswertungssoftware analysiert die Signale und übersetzt sie in die entsprechende Basenabfolge.
Danach werden überschüssige Nukleotide ausgespült. In einem weiteren chemischen Schritt werden sowohl die Fluoreszenzmarkierung als auch die Blockierung des eingebauten Nukleotids entfernt, sodass der nächste Synthesezyklus beginnen kann.
Dieser Ablauf wiederholt sich Zyklus für Zyklus. Auf diese Weise wird die Sequenz jedes DNA-Fragments schrittweise ausgelesen.
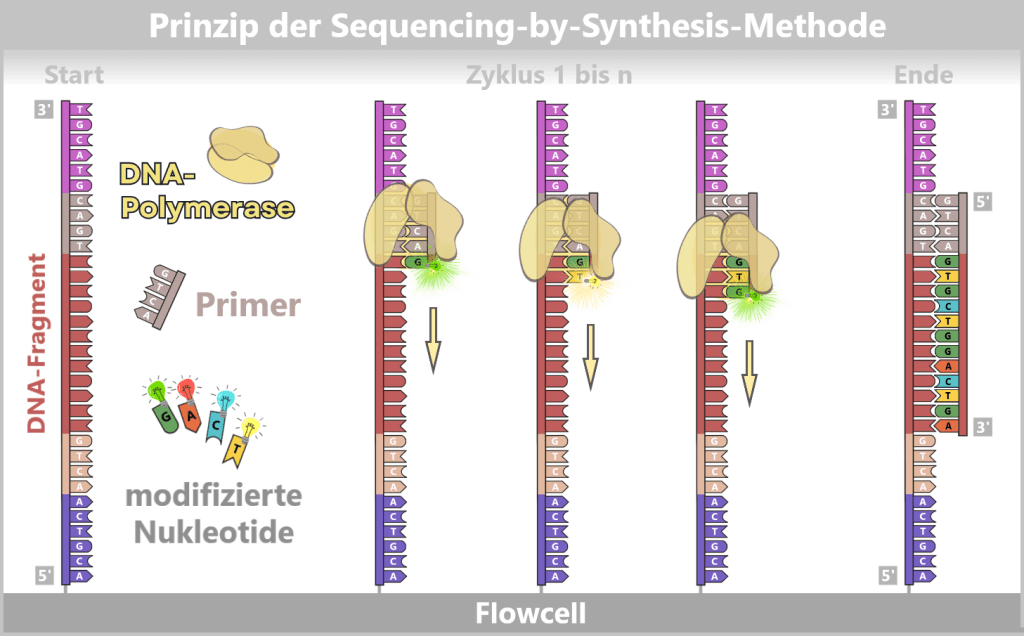
4️⃣ Datenauswertung
Nach Abschluss der Sequenzierung liegen Millionen einzelner Fluoreszenzsignale vor. Diese Rohdaten werden zunächst durch die Gerätesoftware in Basenabfolgen übersetzt. Das Ergebnis sind kurze Sequenzabschnitte, sogenannte Reads.
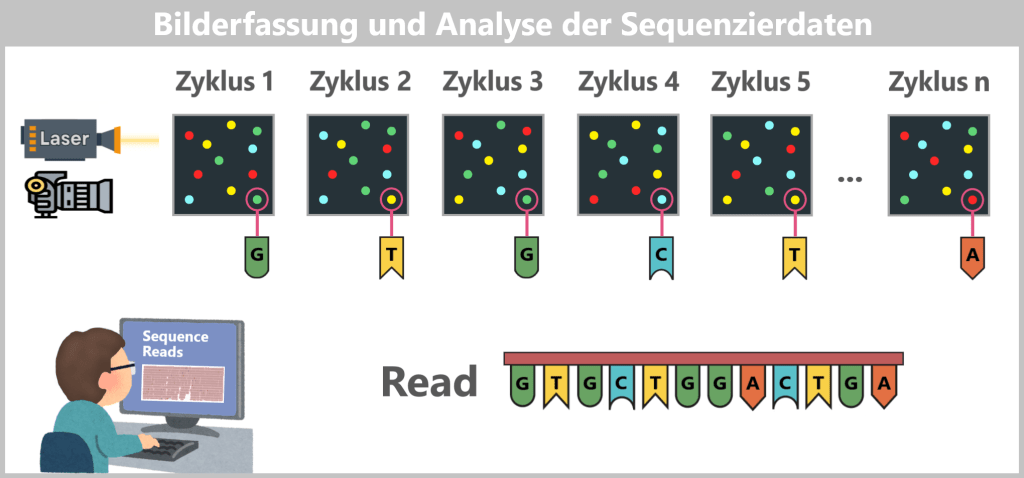
🎥 Tipp: Eine ausführlichere und dennoch anschauliche Erklärung der Illumina-Sequenzierungstechnologie Methode findet sich im Video „Illumina Sequencing Technology“.
⬦ ⬦ ⬦
Qualitätskontrolle und Trimming
Im ersten Schritt wird die Qualität der Reads überprüft. Basen mit niedriger Signalqualität – typischerweise an den Enden der Reads – werden entfernt („Trimming“). Dadurch sollen fehlerhafte Sequenzzuordnungen reduziert und die Zuverlässigkeit der weiteren Analyse verbessert werden.
Mapping: Zuordnung zu einer Referenzsequenz
Anschließend werden die Reads mit bekannten Referenzsequenzen abgeglichen, beispielsweise mit dem Plasmid des Impfstoffherstellers oder bakteriellen Referenzgenomen. Spezielle Algorithmen (z. B. BWA-MEM oder Bowtie2) suchen nach Übereinstimmungen und ordnen die Reads den wahrscheinlichsten Positionen innerhalb der Referenz zu.
Auf diese Weise lässt sich bestimmen, welche Sequenzen in der Probe vorhanden sind und aus welchen Quellen sie stammen.
Bestimmung der Fragmentlängen
Bei der häufig verwendeten Paired-End-Sequenzierung wird jedes DNA-Fragment von beiden Enden gelesen. Aus dem Abstand zwischen den beiden Reads kann die Länge des ursprünglichen DNA-Fragments abgeschätzt werden.
Diese Information ist besonders relevant für die Analyse fragmentierter DNA-Proben.
Identifizierung der DNA-Herkunft
Durch den Vergleich mit Referenzdatenbanken lässt sich analysieren, ob die nachgewiesenen Fragmente beispielsweise:
▪️aus dem erwarteten Produktionsplasmid stammen,
▪️bakterielle Sequenzen enthalten,
▪️oder anderen genetischen Quellen zugeordnet werden können.
Dadurch ermöglicht die Illumina-Sequenzierung nicht nur eine Quantifizierung, sondern auch eine detaillierte Charakterisierung der vorhandenen DNA.
Normalisierung der Daten
Die Anzahl der erzeugten Reads hängt unter anderem von der Sequenziertiefe ab. Um verschiedene Proben oder Chargen vergleichen zu können, werden die Daten häufig normalisiert, beispielsweise als „Reads pro Million“ (RPM). Dadurch lassen sich relative Unterschiede zwischen Proben besser beurteilen.
◈ ◈ ◈
Methodische Einschränkungen
Begrenzte Aussagekraft für absolute Mengenbestimmungen
Die Illumina-Sequenzierung eignet sich hervorragend zur Identifizierung und relativen Häufigkeitsanalyse von DNA-Fragmenten. Eine direkte absolute Quantifizierung der ursprünglichen DNA-Menge ist jedoch nur eingeschränkt möglich.
Der Grund dafür ist, dass die Bibliotheksvorbereitung selbst die Zusammensetzung der Probe verändern kann. Schritte wie PCR-Amplifikation, Adapter-Ligation oder Größenselektion können bestimmte Fragmente bevorzugen oder benachteiligen. Die Anzahl der später erzeugten Reads ist daher nicht zwangsläufig proportional zur ursprünglichen Häufigkeit der DNA-Fragmente in der Probe.
Verzerrung der Fragmentlängenverteilung
Bei der Short-Read-Sequenzierung werden bevorzugt DNA-Fragmente innerhalb eines bestimmten Größenbereichs analysiert. Sehr kurze Fragmente können während der Aufreinigung verloren gehen, während sehr lange Fragmente bei der Amplifikation und Clusterbildung benachteiligt werden.
Die gemessene Fragmentlängenverteilung spiegelt daher nicht zwangsläufig die ursprüngliche Verteilung der DNA-Fragmente in der Probe wider, sondern die Verteilung der erfolgreich sequenzierten Fragmente.
2.5. Elektrophoretische Fragmentanalyse
Die elektrophoretische Fragmentanalyse ist ein etabliertes Standardverfahren der Molekularbiologie zur Auftrennung und Charakterisierung von Nukleinsäuren (DNA und RNA). Je nach Methodik können daraus Aussagen über mehrere zentrale Parameter abgeleitet werden:
- Größe (Fragmentlänge): angegeben in Basenpaaren (bp) bei DNA bzw. Nukleotiden (nt) bei RNA
- Menge: semiquantitative Abschätzung der vorhandenen Nukleinsäure
- Integrität: Grad der Fragmentierung bzw. Unversehrtheit der Moleküle
Das Verfahren basiert auf dem physikalischen Prinzip der Elektrophorese (Transport durch Elektrizität). Dabei werden geladene Moleküle wie DNA oder RNA in einem elektrischen Feld durch ein Trägermedium (meist ein Gel oder eine polymerbasierte Matrix) bewegt. Aufgrund ihrer negativ geladenen Phosphatgruppen wandern Nukleinsäuren dabei in Richtung der positiv geladenen Elektrode.
Die Trennung erfolgt über die Porenstruktur des Mediums: Kleinere Fragmente bewegen sich schneller durch das Netzwerk, während größere Moleküle stärker zurückgehalten werden. Auf diese Weise entsteht eine größenabhängige Auftrennung der Nukleinsäuren.
Im Kontext der Analyse von Rest-DNA in mRNA-basierten Arzneimitteln erlaubt diese Methode insbesondere eine Abschätzung der Fragmentlängenverteilung und ergänzt damit quantitative und sequenzbasierte Nachweisverfahren.
2.5.1. Agarosegelelektrophorese
2.5.2. Automatisierte Systeme
◦ ◦ ◦
2.5.1. Agarosegelelektrophorese
Die Agarosegelelektrophorese ist die klassische und am weitesten verbreitete Form der elektrophoretischen Fragmentanalyse von DNA. Sie dient dazu, Nukleinsäuren anhand ihrer Größe aufzutrennen und sichtbar zu machen.
Aufbau des Systems
Das zentrale Element ist ein Gel aus Agarose, einem polysaccharidbasierten Netzwerk, das aus Algen gewonnen wird. Dieses Gel bildet eine dreidimensionale Matrix mit Poren definierter Größe. Die Porengröße kann über die Agarosekonzentration gesteuert werden: Niedrige Konzentrationen erzeugen größere Poren (geeignet für lange DNA-Fragmente), höhere Konzentrationen kleinere Poren (für kurze Fragmente).
Das Gel wird in eine Pufferlösung eingebettet und in eine Elektrophoresekammer gelegt. An beiden Enden der Kammer befinden sich Elektroden (Kathode/Anode), zwischen denen eine konstante elektrische Spannung angelegt wird.
Vor Beginn der Elektrophorese werden die Proben in kleine Taschen (Wells) am Rand des Gels pipettiert.
In komplexen Proben, wie sie nach der in-vitro-Transkription und enzymatischen Behandlung (z. B. DNase I) vorliegen, können RNA und verbleibende DNA-Fragmente gleichzeitig im Gel migrieren. Für eine gezielte Analyse der Rest-DNA wird die Probe daher häufig zusätzlich mit RNase behandelt, sodass überwiegend DNA-Fragmente sichtbar werden. Dies ermöglicht eine klarere Beurteilung der Fragmentlängenverteilung.
Funktionsprinzip
DNA-Moleküle besitzen aufgrund ihrer Phosphatgruppen eine negative Ladung. Wird eine elektrische Spannung angelegt, beginnen sie, sich durch das Gel in Richtung der positiv geladenen Elektrode zu bewegen.
Während der Migration wirken zwei Kräfte gegeneinander:
- die elektrische Kraft, die die DNA durch das Gel zieht
- der mechanische Widerstand der Gelmatrix
Da kleinere DNA-Fragmente weniger stark durch die Porenstruktur behindert werden, bewegen sie sich schneller durch das Gel als größere Fragmente. Dies führt zu einer größenabhängigen Auftrennung der Moleküle entlang der Laufrichtung.
Zur Größenbestimmung wird parallel eine sogenannte DNA-Leiter (Marker) mit bekannten Fragmentlängen mitgeführt. Durch den Vergleich der Laufstrecken lassen sich die Größen der unbekannten Fragmente abschätzen.
Sichtbarmachung und Auswertung
Nach dem Lauf der Elektrophorese sind die DNA-Fragmente zunächst unsichtbar. Die Sichtbarmachung der DNA erfolgt durch fluoreszierende Farbstoffe, die entweder bereits vor der Elektrophorese dem Gel zugesetzt oder nachträglich auf das Gel aufgebracht werden. Beide Verfahren sind etabliert und unterscheiden sich vor allem in ihrem Einfluss auf die Migration sowie im experimentellen Aufwand.
Das Gel aus der Elektrophoresekammer wird auf einen Transilluminator gelegt. Unter UV- oder blauem Licht leuchten die fluoreszierenden Farbstoffe, die an die DNA gebundenen haben, als Banden im Gel.
Durch den Vergleich mit dem mitgelaufenen Marker lassen sich die Größen der DNA-Fragmente abschätzen. Die Intensität der Banden erlaubt zudem eine grobe, semiquantitative Einschätzung der DNA-Menge.
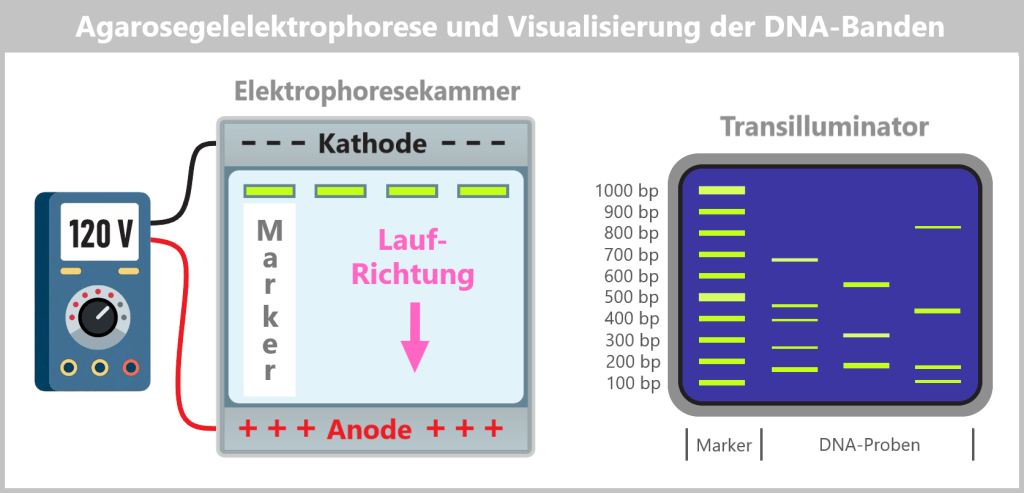
🎥 Tipp: Das Video „Electrophoresis explained“ fasst das Prinzip der Agarosegelelektrophorese kurz zusammen.
2.5.2. Automatisierte Systeme
Automatisierte elektrophoretische Systeme stellen eine Weiterentwicklung der klassischen Agarosegelelektrophorese dar. Sie basieren auf demselben physikalischen Prinzip der größenabhängigen Trennung von Nukleinsäuren im elektrischen Feld, kombinieren dieses jedoch mit mikrofluidischer Technologie, integrierter Detektion und digitaler Auswertung.
Im Gegensatz zur manuellen Gelherstellung erfolgt die Trennung hier in miniaturisierten, vorgefertigten Systemen. Die Proben werden in spezielle Kartuschen oder Chips eingebracht, in denen sich feine Kanäle mit polymeren Matrizes befinden. Innerhalb dieser Mikrostrukturen werden die Nukleinsäuren unter angelegter Spannung elektrophoretisch getrennt.
Während des Laufs werden die Fragmente kontinuierlich durch fluoreszierende Farbstoffe detektiert. Die Signale werden direkt in digitale Daten umgewandelt und als sogenannte Elektropherogramme dargestellt. Anstelle von Banden im Gel entstehen dabei Peaks, deren Position die Fragmentgröße und deren Höhe beziehungsweise Fläche die relative Menge der Nukleinsäure widerspiegelt.
Zu den verbreiteten Systemen gehören unter anderem:
- der Bioanalyzer
- die TapeStation
- sowie der Fragment Analyzer
Diese Systeme unterscheiden sich in Details wie Durchsatz, Sensitivität und Automatisierungsgrad, folgen jedoch einem vergleichbaren Grundprinzip.
Ablauf eines automatisierten elektrophoretischen Systems (z. B. TapeStation)
1) Probenvorbereitung
Die zu untersuchenden Proben werden in Röhrchen oder eine Mikrotiterplatte (z. B. 96-Well-Format) pipettiert und mit einem kit-spezifischen Puffer gemischt. Parallel wird ein Referenzstandard (Marker) mit definierten Fragmentgrößen angesetzt.
2) Zugabe des Fluoreszenzfarbstoffs
Den Proben wird ein Fluoreszenzfarbstoff zugesetzt, der selektiv an DNA oder RNA bindet und eine spätere Detektion ermöglicht.
3) Bereitstellung des Trennmediums
Als Trägermedium dient ein sogenanntes ScreenTape – ein vorgefertigtes, mikrofluidisches System mit integrierter Gelmatrix und mehreren parallelen Trennkanälen. Je nach Anwendung werden spezifische ScreenTapes für DNA oder RNA verwendet.
4) Automatisierter Elektrophoreselauf
Proben, Marker und ScreenTape werden in das Gerät eingesetzt. Nach dem Start führt das System die Analyse automatisch durch: Die Proben werden nacheinander in die Trennkanäle überführt und unter angelegter Spannung elektrophoretisch aufgetrennt.
5) Detektion und Datenauswertung
Während der Trennung werden die fluoreszierenden Signale der Nukleinsäuren optisch erfasst (z. B. mittels Laser). Die Software verarbeitet diese Signale zu digitalen Ausgaben, insbesondere:
▫️Elektropherogrammen (Peakdarstellungen der Fragmentgrößen)
▫️Größenverteilungen
▫️sowie semiquantitativen Konzentrationsangaben
🎥 Tipp: Eine anschaulische Demonstration zum Funktionsprinzip des TapeStation-Systems und der ScreenTape-Technologie bietet das Video „Agilent 2200 TapeStation System“.
⬦ ⬦ ⬦
Vorteile der automatisierten Systeme
Automatisierte Systeme bieten gegenüber der klassischen Agarosegelelektrophorese mehrere entscheidende Vorteile:
- Höhere Sensitivität: Auch geringe Nukleinsäuremengen können detektiert werden
- Verbesserte Reproduzierbarkeit: Standardisierte Kartuschen und automatisierte Abläufe reduzieren experimentelle Variabilität
- Quantifizierbarkeit: Neben der Fragmentgröße kann auch die relative Konzentration genauer abgeschätzt werden
- Digitale Auswertung: Objektive und reproduzierbare Analyse ohne visuelle Interpretation von Gelbildern
Darüber hinaus liefern diese Systeme häufig zusätzliche Kennzahlen, wie z. B. mittlere Fragmentlängen oder Integritätsindizes, die eine standardisierte Bewertung der Probenqualität ermöglichen.
⬦ ⬦ ⬦
DNA- und RNA-spezifische Assays
Ein wesentlicher Aspekt automatisierter Systeme ist die Verwendung spezifischer Assays für unterschiedliche Nukleinsäuretypen. In komplexen Proben werden daher häufig getrennte Analysen durchgeführt, bei denen identische Proben jeweils mit DNA- oder RNA-spezifischen Reagenzien untersucht werden.
Diese Assays unterscheiden sich unter anderem in:
- der Zusammensetzung der polymeren Matrix
- den eingesetzten Farbstoffen
- den Auswertealgorithmen
Auf diese Weise können DNA- und RNA-Fragmente unabhängig voneinander charakterisiert werden, selbst wenn sie ursprünglich in derselben Probe vorliegen.
⬦ ⬦ ⬦
Aussagekraft der elektrophoretischen Fragmentanalyse
Die elektrophoretische Fragmentanalyse liefert vor allem strukturelle Informationen über Nukleinsäuren. Im Rahmen der Qualitätskontrolle mRNA-basierter Produkte ermöglicht sie insbesondere:
- die Analyse der Fragmentlängenverteilung von DNA und RNA
- die Beurteilung der RNA-Integrität
- die Unterscheidung zwischen stark fragmentierten und weitgehend intakten Nukleinsäuren
- die Identifikation dominanter Fragmentgrößen
- sowie die Detektion verbleibender DNA-Fragmente
⬦ ⬦ ⬦
Grenzen der elektrophoretischen Fragmentanalyse
Sowohl klassische Agarosegele als auch automatisierte elektrophoretische Systeme besitzen methodische Grenzen, – insbesondere beim Nachweis geringer DNA-Rückstände in komplexen Proben.
- Keine Sequenzinformation: Die Methode trennt Nukleinsäuren ausschließlich nach Größe bzw. elektrophoretischer Mobilität. Ob ein Fragment aus genomischer DNA, Plasmid-DNA, mRNA oder unspezifischen Abbauprodukten stammt, kann anhand der Fragmentanalyse allein nicht bestimmt werden.
- Begrenzte Sensitivität im Vergleich zu amplifikationsbasierten Verfahren: Sehr geringe Mengen residualer DNA können unterhalb der Nachweisgrenze liegen. Verfahren wie qPCR oder ddPCR sind für die Quantifizierung kleiner DNA-Restmengen deutlich sensitiver.
- Eingeschränkte Aussage bei stark fragmentierter DNA: Stark degradierte DNA erscheint häufig lediglich als diffuser Signalbereich („Smear“). Einzelne Fragmente lassen sich dann nur eingeschränkt charakterisieren.
- Keine Aussage über biologische Funktionalität: Die Methode zeigt lediglich Größe und Mengenverteilung von Fragmenten. Ob DNA-Fragmente noch biologisch aktiv, vollständig oder potenziell transkriptionsfähig sind, bleibt unklar.
- Abhängigkeit von Farbstoffbindung und Fragmentgröße: Sehr kurze Fragmente binden teilweise weniger Farbstoff und können daher unterschätzt oder gar nicht detektiert werden. Die quantitative Aussagekraft ist insbesondere im unteren Fragmentgrößenbereich begrenzt.
- Überlappende Signale komplexer Proben: In Proben mit gleichzeitig vorhandener RNA und DNA können sich Signalbereiche überlagern. Trotz spezifischer Assays sind Fehlzuordnungen oder Hintergrundsignale nicht vollständig ausgeschlossen.
- Keine absolute Quantifizierung einzelner Zielsequenzen: Die Methode erlaubt keine gezielte Bestimmung definierter DNA-Sequenzen. Eine spezifische Identifizierung residualer Prozess-DNA ist daher nur in Kombination mit sequenzspezifischen Verfahren möglich.
Die elektrophoretische Fragmentanalyse liefert vor allem strukturelle Informationen über Nukleinsäuren und ergänzt damit quantitative sowie sequenzbasierte Verfahren um eine wichtige Perspektive auf Fragmentgröße, Integrität und Verteilung, ersetzt jedoch keine sequenzspezifischen oder funktionellen Nachweisverfahren.
2.6. Vergleich zentraler Nachweisverfahren für Nukleinsäuren
Die quantitative und qualitative Bestimmung von Nukleinsäuren bildet das Fundament zahlreicher molekularbiologischer Analysen. Je nach Methode unterscheiden sich jedoch sowohl die Aussagekraft als auch die Grenzen der Messung erheblich. Keine der etablierten Techniken liefert ein vollständiges Bild – vielmehr ergänzen sie sich gegenseitig.
UV-spektroskopische Messungen erfassen die Gesamtmenge an Nukleinsäuren in einer Probe. Dabei wird die Absorption von UV-Licht bei 260 nm gemessen. Diese Methode ist schnell und unkompliziert, unterscheidet jedoch nicht zwischen verschiedenen DNA- oder RNA-Sequenzen. Auch Verunreinigungen wie Proteine oder andere Moleküle können das Signal beeinflussen.
Fluorometrische Verfahren verwenden spezifische Farbstoffe, die bevorzugt an DNA oder RNA binden. Dadurch sind sie deutlich selektiver und empfindlicher als UV-Messungen. Dennoch liefern sie keine Informationen über die genaue Sequenz oder Herkunft der Nukleinsäuren – sie quantifizieren lediglich die vorhandene Menge bestimmter Molekülklassen.
Die elektrophoretische Fragmentanalyse ergänzt diese Verfahren um eine strukturelle Perspektive. Sie ermöglicht die Abschätzung von Fragmentgrößen, Fragmentlängenverteilungen und der Integrität von DNA oder RNA. Dadurch lassen sich beispielsweise intakte von stark fragmentierten Nukleinsäuren unterscheiden. Allerdings liefert die Methode keine Sequenzinformation und erlaubt keine eindeutige Identifikation bestimmter DNA- oder RNA-Moleküle.
PCR-basierte Verfahren (qPCR und ddPCR) ermöglichen einen hochsensitiven und spezifischen Nachweis definierter DNA-Sequenzen. Durch den Einsatz sequenzspezifischer Primer können gezielt einzelne Abschnitte des Genoms vervielfältigt und quantifiziert werden. Diese hohe Spezifität ist zugleich eine Einschränkung: Es können nur diejenigen Sequenzen detektiert werden, für die passende Primer entworfen wurden. Unbekannte oder unerwartete Sequenzen bleiben unentdeckt.
Eine weitere methodische Grenze ergibt sich aus den Anforderungen an die DNA-Beschaffenheit. Damit eine Amplifikation stattfinden kann, müssen beide Primerbindestellen vollständig in einem DNA-Fragment enthalten sein. qPCR-Amplicons sind typischerweise 80–150 Basenpaare lang. Stark fragmentierte DNA, deren Bruchstücke kürzer sind oder bei denen eine der Primerbindestellen fehlt, kann daher nicht vervielfältigt werden. Mit anderen Worten: Nur DNA-Fragmente, die die komplette Zielregion zwischen beiden Primern umfassen, sind durch qPCR nachweisbar.
Diese Abhängigkeit von der Fragmentlänge ist insbesondere dann relevant, wenn DNA durch enzymatische oder mechanische Prozesse zerkleinert wurde, wie es mittels DNase-I-Behandlung bei der Aufarbeitung von DNA-Rückständen in der mRNA-Herstellung der Fall ist. Für eine erfolgreiche qPCR/ddPCR müssen jedoch beide Primer – Forward und Reverse – auf demselben Fragment binden können. Je kürzer die Fragmente, desto geringer die Wahrscheinlichkeit, dass ein Fragment beide Primerstellen trägt – ein wichtiger Aspekt bei der Bewertung von DNA-Rückständen.
Illumina-Sequenzierung basiert auf der parallelen Sequenzierung sehr vieler kurzer DNA-Fragmente. Sie liefert hochpräzise Sequenzdaten mit einer im Vergleich zu anderen Sequenziermethoden sehr geringen Fehlerrate. Dadurch eignet sie sich besonders für die genaue Analyse bekannter oder referenzierbarer Sequenzen. Die Methode erfordert jedoch eine aufwendigere Probenvorbereitung, PCR-basierte Bibliotheksverstärkung sowie leistungsfähige bioinformatische Auswertungen. Zudem werden meist nur relativ kurze Fragmente sequenziert, wodurch größere strukturelle Zusammenhänge schwieriger zu erfassen sind.
Nanopore-Sequenzierung geht einen Schritt weiter: Sie erlaubt die direkte Bestimmung der Basenabfolge einzelner DNA- oder RNA-Moleküle in Echtzeit. Dadurch können auch unbekannte Sequenzen identifiziert sowie sehr lange Fragmente analysiert werden. Darüber hinaus ermöglicht die Technologie unter bestimmten Bedingungen die direkte Detektion chemischer Nukleinsäuremodifikationen. Die gewonnenen Daten erfordern allerdings eine sorgfältige bioinformatische Auswertung und weisen im Vergleich zur Illumina-Sequenzierung derzeit noch höhere Fehlerraten auf.
⚬ ⚬ ⚬
Die folgende Übersicht fasst die wichtigsten Nachweisverfahren, ihre Messprinzipien sowie ihre spezifischen Stärken und Grenzen im Kontext der mRNA-Herstellung zusammen.
Methode
Was wird gemessen?
Stärken
Schwächen / Grenzen
Typische Anwendung
UV-Spektroskopie
Gesamtkonzentration aller Nukleinsäuren
Schnell und einfach, keine Reagenzien nötig, geringe Kosten
Keine Unterscheidung zwischen DNA, RNA und freien Nukleotiden, störanfällig gegenüber Verunreinigungen
Schnelle Grobbestimmung von Konzentration und Reinheit
Fluorometrie
DNA oder RNA – über Farbstoffbindung
Hohe Sensitivität, tolerant gegenüber Verun-reinigungen
Keine Sequenz-Information
Präzise Quantifizierung der Ziel-RNA
Qubit-Fluorometer
DNA oder RNA – über Farbstoffbindung (fluorometrisches Gerät)
Sehr hohe Sensitivität und Selektivität, reproduzierbare Ergebnisse
Keine Sequenz-Information, höhere Kosten als einfache Fluorometrie
Standardisierte Konzentrationsbestimmung im Labor- und GMP-Umfeld
qPCR
Spezifische DNA-Sequenzen (z. B. Template-DNA, Wirtszell-DNA)
Hohe Sensitivität, sequenz-spezifisch
Nur bekannte Zielsequenzen nachweisbar, abhängig von Primerdesign und Amplifikationseffizienz, Quantifizierung benötigt Standardkurve, anfällig für Inhibitoren
Nachweis von DNA-Rückständen
ddPCR
Absolute Kopienzahl spezifischer DNA-Sequenzen
Absolute Quantifizierung ohne Standardkurve, sehr hohe Präzision, robuster gegenüber Inhibitoren
Nur bekannte Zielsequenzen nachweisbar, abhängig von Primer-/Sondendesign und Targetintegrität, höhere Kosten, aufwändigere Proben-vorbereitung
Präzise Quantifizierung definierter DNA-Zielsequenzen in Validierungsanalysen
Agarosegel-Elektrophorese
DNA-/RNA-Fragmente nach Größe
Einfach, kostengünstig, visuelle Abschätzung von Fragmentgröße und Integrität
Geringe Auflösung, semiquantitativ, nicht für präzise Konzentrationsbestimmung geeignet, keine Sequenz-Information
Schnelle Qualitäts-kontrolle, Integritätsprüfung
Automatisierte Elektrophorese
Größe, Konzen-tration und Integrität von DNA/RNA
Hohe Auflösung, quantitativ, reproduzierbar, geringer Probenbedarf
Höhere Kosten, Verbrauchsmaterialien erforderlich
RNA-Integritäts-prüfung (RIN-Wert), Fragmentanalyse von DNA-Rückständen
Illumina-Sequenzierung (NGS)
Sequenzinformation vieler kurzer DNA-/RNA-Fragmente
Sehr hohe Genauigkeit, umfassende Abdeckung, gut etabliert
Short Reads (150–300 bp), aufwändige Probenvorbereitung, bioinformatische Auswertung erforderlich
Hochauflösende Sequenzanalyse und Charakterisierung residualer Nukleinsäuren
Nanopore-Sequenzierung
Vollständige DNA/RNA-Sequenz einzelner Moleküle
Direkte Sequenz-information, Long Reads, Echtzeit-Daten, erkennt unbekannte Sequenzen und strukturelle Varianten
Höhere Fehlerrate, komplexe Auswertung, noch begrenzt etabliert im GMP-Umfeld
Charakterisierung komplexer Nukleinsäurepopulationen, Identitätsprüfung, Integritätsanalysen
Insgesamt zeigt sich, dass jede Methode ihre spezifischen Stärken und Schwächen besitzt. Die Wahl des geeigneten Verfahrens hängt daher stets von der jeweiligen Fragestellung ab.
Dies gilt in besonderem Maße für die Qualitätssicherung therapeutischer mRNA. Wirkstoffkonzentration, Reinheit und potentielle Verunreinigungen stellen jeweils eigene Anforderungen an die Nachweismethode.
Keine einzelne Methode beantwortet alle analytischen Fragestellungen – erst die Kombination aus Quantifizierung, struktureller Charakterisierung und Sequenzinformation ermöglicht eine umfassende Beurteilung von Nukleinsäuren.
◈ ◈ ◈
Kombinierte Anwendung der Methoden
In der praktischen Analytik werden die beschriebenen Verfahren selten isoliert eingesetzt. Stattdessen erfolgt die Charakterisierung von Nukleinsäuren in der Regel durch eine Kombination komplementärer Methoden. So kann beispielsweise eine UV-spektroskopische Messung zur schnellen Abschätzung von Konzentration und Reinheit dienen, während fluorometrische Verfahren eine selektivere Quantifizierung ermöglichen. PCR-basierte Methoden wie qPCR oder ddPCR werden eingesetzt, um spezifische DNA-Sequenzen gezielt nachzuweisen und zu quantifizieren. Sequenzierungsverfahren wie Illumina- oder Nanopore-Technologien ergänzen diese Analysen durch die direkte Bestimmung der Sequenz und ermöglichen die Identifikation auch unbekannter Sequenzen oder struktureller Varianten.
⬦ ⬦ ⬦
Regulatorische Einordnung
Im regulatorischen Kontext erfordert die Qualitätssicherung therapeutischer Nukleinsäureprodukte den Einsatz validierter und standardisierter Nachweismethoden. PCR-basierte Verfahren wie qPCR und ddPCR sind hierfür etabliert und werden routinemäßig zur Quantifizierung definierter residualer DNA-Sequenzen eingesetzt. Sequenzierungsbasierte Ansätze, einschließlich der Nanopore-Technologie, gewinnen zunehmend an Bedeutung, befinden sich jedoch in vielen Anwendungsbereichen noch in der Phase der methodischen Etablierung und Validierung.
3. Richtlinien zur Begrenzung von Rest-DNA in Impfstoffen
Rest-DNA (residual DNA), einschließlich Wirtszell-DNA oder Plasmid-DNA, bezeichnet Nukleinsäurereste aus dem Herstellungsprozess, die nach der Aufreinigung im Endprodukt verbleiben können. Wie in den vorherigen Kapiteln dargestellt, lässt sich das vollständige Entfernen dieser DNA technisch nicht erreichen, sodass geringe Restmengen als unvermeidbarer Bestandteil biotechnologischer Produkte gelten.
Die Entwicklung regulatorischer Richtlinien zur Begrenzung von Rest-DNA erfolgte parallel zum Fortschritt biotechnologischer Herstellungsverfahren. Ziel dieser Vorgaben ist es, potenzielle biologische Risiken zu minimieren. Dazu zählen insbesondere:
- Onkogenität
- Insertionsmutagenese
- Immunogenität
- sowie in bestimmten Kontexten Infektiosität (z. B. bei viralen Systemen)
Historische Entwicklung
Frühe Phase: Vorsorgeorientierter Ansatz
Mit der Einführung rekombinanter DNA-Technologien (Verfahren, bei denen DNA aus unterschiedlichen Organismen gezielt zusammengefügt wird) in den 1980er-Jahren – etwa zur Herstellung von therapeutischem Insulin oder Wachstumshormonen – entstand erstmals die Notwendigkeit, DNA-Rückstände systematisch zu bewerten.
Zu diesem Zeitpunkt bestanden erhebliche wissenschaftliche Unsicherheiten hinsichtlich der biologischen Wirkung exogener DNA. Insbesondere war unklar, unter welchen Bedingungen fremde DNA in das Genom menschlicher Zellen integriert werden und potenziell onkogene Effekte auslösen könnte.
Vor diesem Hintergrund verfolgten Regulierungsbehörden einen konservativen, vorsorgeorientierten Ansatz. Die World Health Organization (WHO) empfahl in den 1980er-Jahren einen Grenzwert von 10 pg Rest-DNA pro Dosis für Produkte aus kontinuierlichen Zelllinien (Zellen, die sich im Labor unbegrenzt teilen können). Dieser Wert basierte nicht auf einer quantitativen Risikobewertung, sondern stellte einen pragmatischen Richtwert dar, der sich an den damaligen analytischen Möglichkeiten orientierte.
Spätere Bewertungen führten zu einer Anhebung dieses Wertes; bereits Mitte der 1980er-Jahre wurden Größenordnungen von etwa 100 pg pro Dosis als vernachlässigbar eingestuft.
Paradigmenwechsel: Von der Masse zur biologischen Aktivität
In den 1990er- und 2000er-Jahren veränderte sich die regulatorische Perspektive grundlegend. Zwei Entwicklungen waren hierfür maßgeblich:
Erstens verbesserten sich die Herstellungs- und Aufreinigungsverfahren erheblich. Methoden wie Chromatographie, Filtration und enzymatische Behandlung ermöglichten eine reproduzierbare Reduktion von DNA-Rückständen um mehrere Größenordnungen.
Zweitens lieferten experimentelle Studien zunehmend quantitative Erkenntnisse zur biologischen Aktivität von DNA. Dabei zeigte sich, dass nicht allein die Gesamtmenge entscheidend ist, sondern insbesondere strukturelle Eigenschaften wie Länge und Integrität der DNA eine zentrale Rolle spielen.
Längere, intakte DNA-Fragmente mit funktionellen Genabschnitten können unter geeigneten Bedingungen theoretisch biologische Effekte entfalten. Mit zunehmender Fragmentierung nimmt dieses Potenzial jedoch deutlich ab. Kurze DNA-Fragmente besitzen eine stark reduzierte Wahrscheinlichkeit, in den Zellkern zu gelangen, stabil zu verbleiben oder in das Genom integriert zu werden.
Für eine tatsächliche Integration exogener DNA in das Genom einer Zelle müssen mehrere Voraussetzungen gleichzeitig erfüllt sein. Dazu gehören unter anderem die Aufnahme der DNA in die Zelle, ihr Transport in den Zellkern sowie das Vorliegen geeigneter zellulärer Mechanismen für eine stabile Integration. Die Wahrscheinlichkeit für das gleichzeitige Eintreten dieser Bedingungen wird unter natürlichen Bedingungen im Körper als gering eingeschätzt und nimmt mit zunehmender Fragmentierung der DNA weiter ab.
Moderne regulatorische Ansätze
Auf Basis dieser Erkenntnisse entwickelten sich moderne regulatorische Konzepte hin zu einem risikobasierten und produktspezifischen Ansatz. Anstelle eines einheitlichen Grenzwertes stehen heute mehrere komplementäre Anforderungen im Vordergrund:
| Anforderung | Begründung |
| Validierte Eliminierung | Der Herstellungsprozess muss nachweislich und reproduzierbar zur Reduktion von DNA-Rückständen beitragen. |
| Fragmentierung | Verbleibende DNA soll möglichst in stark fragmentierter Form vorliegen. |
| Produktspezifische Grenzwerte | Akzeptanzkriterien werden abhängig von Herstellungsprozess, Zelllinie und Anwendung definiert. |
Regulatorische Leitlinien von Organisationen wie der U.S. Food and Drug Administration (FDA), der European Medicines Agency (EMA) sowie dem International Council for Harmonisation (ICH) verfolgen heute überwiegend diesen differenzierten Ansatz.
Einordnung im Kontext modRNA-basierter Impfstoffe
Für modRNA-basierte Impfstoffe gelten grundsätzlich ähnliche regulatorische Prinzipien wie für andere biotechnologisch hergestellte Arzneimittel. Gleichzeitig unterscheiden sich die Herstellungsprozesse und Produktcharakteristika in einigen Punkten, insbesondere durch die in-vitro-Transkription und die Verwendung synthetischer RNA.
Im Kontext von Impfstoffen orientieren sich regulatorische Leitlinien in Bezug auf die Menge an Rest-DNA bei parenteraler Anwendung (d. h. bei Verabreichung durch Injektion oder Infusion, die den Magen-Darm-Trakt umgeht) häufig an Größenordnungen im Nanogramm-Bereich pro Dosis.
Zusätzlich wird gefordert, dass die verbleibende DNA möglichst stark fragmentiert ist und keine funktionell vollständigen Gene umfasst. In der wissenschaftlichen Literatur werden in diesem Zusammenhang häufig Fragmentlängen im Bereich unterhalb einiger hundert Basenpaare (bp) diskutiert, ohne dass ein einheitlicher Grenzwert festgelegt ist.
Diese Anforderungen spiegeln den heutigen regulatorischen Ansatz wider, der sowohl die Menge als auch die strukturellen Eigenschaften der DNA berücksichtigt.
Die folgende Tabelle fasst die wesentlichen regulatorischen Dokumente zusammen, in denen Grenzwerte für Rest-DNA genannt werden.
| Dokument | Aussage / Kontext |
|---|---|
| 1998 – WHO TRS 878, Annex 1 | Historische Quelle: Erstmalige Erwähnung von <10 ng/Dosis für kontinuierliche Zelllinien |
| 2007 – WHO Study Group on Cell Substrates for the Production of Biologicals | Diskussion von <10 ng/Dosis im Zusammenhang mit stark fragmentierter DNA (z. B. <200 bp) |
| 2013 – WHO TRS No. 978 | Bestätigt <10 ng/Dosis als etablierten Standard; betont Bedeutung von Fragmentierung und Prozess; grundlegendes WHO-Dokument zu Zellsubstraten |
| 2014 – WHO TRS 987, Annex 4 | <10 ng/Dosis (im Kontext rekombinanter Proteintherapeutika) |
| 2020 – FDA CMC Guidance (Gene Therapy) | <10 ng/Dosis und <200 bp (für Gentherapie-Produkte aus kontinuierlichen nicht-tumorigenen Zellen) |
| 2020 – Rapporteur Rolling Review critical assessment report (Josephson 2020-11-19) | Acceptance Criteria: ≤ 330 ng DNA/mg RNA (abgeleitet aus 10 ng/Dosis bei 30 µg RNA pro Dosis) |
| 2024 – WHO TRS 1062, Annex 1 (noch nicht öffentlich) | Recommendations for mRNA vaccines – erwartete Nennung der Größenordnungen |
Dosis: Gesamtmenge des Arzneimittels pro Anwendung.
ng/Dosis (Nanogramm/Dosis): Ein Maß für die zulässige Menge an DNA-Rückständen pro Dosis.
bp (Basenpaare): Ein Maß für die Länge der DNA-Fragmente.
⚬ ⚬ ⚬
Umrechnung von Messwerten auf ng pro Dosis
Um Messergebnisse aus verschiedenen Studien mit regulatorischen Richtwerten vergleichen zu können, ist eine einheitliche Bezugsgröße erforderlich. Während regulatorische Grenzwerte üblicherweise als Menge pro Dosis (ng/Dosis) angegeben werden, erfolgen analytische Messungen häufig in Bezug auf die RNA-Menge (z. B. ng DNA pro mg RNA).
Zur Vergleichbarkeit müssen diese Werte ineinander umgerechnet werden.
Grundprinzip:
DNA (ng/Dosis) = DNA (ng/mg RNA) × RNA-Menge pro Dosis (mg)
Beispielrechnung
Angenommen, eine analytische Messung ergibt: 330 ng DNA pro mg RNA
und die verabreichte Dosis enthält: 30 µg RNA = 0,03 mg RNA
Dann ergibt sich: 330 ng/mg × 0,03 mg = 9,9 ng DNA pro Dosis
Dieses Beispiel zeigt, dass Konzentrationsangaben (ng/mg RNA) und dosisbezogene Grenzwerte (ng/Dosis) direkt ineinander überführt werden können, sofern die zugrunde liegende RNA-Menge pro Dosis bekannt ist.
Das Ergebnis liegt damit in der Größenordnung von 10 ng DNA pro Dosis, was dem häufig zitierten regulatorischen Richtwert entspricht.
⚬ ⚬ ⚬
Einordnung
Ein wichtiger Aspekt der regulatorischen Einordnung ist, dass keine einheitliche, universell verbindliche Grenzwertregelung für alle modRNA-Impfstoffe existiert. Stattdessen basiert die Bewertung auf einem Zusammenspiel aus allgemeinen Empfehlungen, produktspezifischen Festlegungen und risikobasierten Prinzipien.

4. Studien zu DNA-Rückständen in modRNA-basierten Impfstoffen
Nach der Darstellung der Herstellungsverfahren, potenzieller Verunreinigungen sowie der analytischen Nachweismethoden stellt sich die zentrale Frage, in welchem Umfang DNA-Rückstände in kommerziellen mRNA-Impfstoffen tatsächlich nachweisbar sind.
Seit dem Jahr 2023 wurden hierzu mehrere unabhängige Untersuchungen veröffentlicht, die sich mit der Detektion, Quantifizierung und Charakterisierung von DNA in verschiedenen Impfstoffchargen beschäftigen.
Ziel dieses Kapitels ist es, die bisherigen Studien vorzustellen, ihre methodischen Ansätze nachvollziehbar einzuordnen und die berichteten Ergebnisse darzustellen.
4.1. Warum unabhängige Messungen?
4.2. Methodische Vorbemerkungen
4.3. Übersicht und Einzelanalysen der Studien
4.4. Zwischenfazit: Was wissen wir – und was nicht?
⚬ ⚬ ⚬
4.1. Warum unabhängige Messungen?
Die Zulassung eines Arzneimittels setzt voraus, dass der Hersteller die Einhaltung regulatorischer Qualitätsstandards gegenüber den zuständigen Behörden nachweist. Für mRNA-basierte Impfstoffe bedeutet das unter anderem, dass Restmengen an DNA unterhalb festgelegter Grenzwerte liegen müssen. Die entsprechenden Prüfdaten werden im Rahmen des Zulassungsverfahrens eingereicht und von den Behörden bewertet.
Was dieser Prozess strukturell nicht vorsieht, ist eine systematische Überprüfung durch unabhängige Labore nach der Marktzulassung. Behörden wie die EMA oder FDA stützen sich bei ihrer Bewertung in erster Linie auf die vom Hersteller vorgelegten Daten. Eine eigenständige Nachanalyse kommerziell erhältlicher Chargen ist nicht Bestandteil des Standardverfahrens.
Genau hier setzt unabhängige Forschung an. Seit 2023 haben mehrere Arbeitsgruppen – ohne Verbindung zu den Herstellern – kommerziell bezogene Chargen der mRNA-Impfstoffe auf DNA-Rückstände untersucht. Die Motivation dahinter war nicht primär die Erwartung eines Problems, sondern eine grundlegende Frage der wissenschaftlichen Nachvollziehbarkeit: Lassen sich die im regulatorischen Zulassungsprozess eingereichten Qualitätsangaben durch unabhängige Messungen reproduzieren?
Die Ergebnisse dieser Studien waren nicht einheitlich – weder in den gemessenen Mengen noch in der methodischen Herangehensweise. Einige Arbeitsgruppen berichteten von DNA-Gehalten, die die regulatorischen Grenzwerte überschritten; andere kamen zu abweichenden Ergebnissen oder äußerten methodische Vorbehalte gegenüber den verwendeten Verfahren.
Dieses Kapitel stellt die wichtigsten dieser Untersuchungen vor. Ziel ist keine abschließende Bewertung – der wissenschaftliche Diskurs ist noch nicht abgeschlossen –, sondern eine sachliche Darstellung der vorliegenden Befunde, der eingesetzten Methoden und der offenen Fragen.
⚬ ⚬ ⚬
4.2. Methodische Vorbemerkungen
Bevor die einzelnen Studien vorgestellt werden, ist ein methodischer Rahmen hilfreich – nicht um Ergebnisse vorwegzunehmen, sondern um dem Leser ein Werkzeug an die Hand zu geben, mit dem er die Befunde einordnen kann.
Die in diesem Kapitel vorgestellten Untersuchungen stammen aus unabhängigen Laboren und unterscheiden sich teilweise deutlich hinsichtlich ihrer methodischen Ansätze und der verwendeten Nachweisverfahren.
Der Aufschluss der Lipid-Nanopartikel
Jede Analyse beginnt mit demselben Problem: Die zu messenden Nukleinsäuren – mRNA und potenzielle DNA-Rückstände – sind in Lipid-Nanopartikel (LNPs) eingeschlossen. Um sie analytisch zugänglich zu machen, müssen die Partikel zunächst aufgeschlossen werden.
In den vorliegenden Studien wurden dafür unterschiedliche Methoden eingesetzt. Detergenzien wie Triton X-100 lösen die Lipidmembran chemisch auf. Kombinationen aus Lithiumdodecylsulfat (LiDS) und magnetischen SPRI-Beads ermöglichen gleichzeitig Aufschluss und selektive Aufreinigung der Nukleinsäuren. Thermische Verfahren – etwa Erhitzen auf 95 °C – destabilisieren die Partikelstruktur durch Hitzeeinwirkung. Kommerzielle Kits wie der Monarch Plasmid DNA Miniprep Kit kombinieren mehrere Schritte zu einem standardisierten Protokoll.
Diese Methodenvielfalt ist nicht trivial. Drei Aspekte verdienen besondere Aufmerksamkeit:
Erstens unterscheiden sich die Methoden in ihrer Aufschlusseffizienz: Nicht jedes Verfahren öffnet alle Partikel vollständig. Verbleiben intakte LNPs in der Probe, bleibt ihr Inhalt für die nachfolgende Messung unsichtbar – mit der Folge, dass der tatsächliche DNA-Gehalt unterschätzt wird.
Zweitens können einige Methoden die freizusetzenden Moleküle verändern oder degradieren. Thermischer Aufschluss bei hohen Temperaturen kann insbesondere RNA fragmentieren; DNA ist thermisch stabiler, kann unter bestimmten Bedingungen jedoch ebenfalls geschädigt werden. Stark fragmentierte DNA ist, wie in Kapitel 2.6. beschrieben, für PCR-basierte Nachweisverfahren schlechter zugänglich – kurze Fragmente tragen möglicherweise nicht mehr beide Primerbindestellen und entgehen damit der Detektion.
Drittens können Rückstände bestimmter Reagenzien nachfolgende Analyseschritte stören. Detergenzien wie Triton X-100 sind bekannte PCR-Inhibitoren. Werden sie nicht vollständig entfernt, kann die Amplifikation beeinträchtigt werden – was wiederum zu einer Unterschätzung der gemessenen Menge führt. SPRI-Beads hingegen selektieren nach Fragmentgröße: Sehr kurze DNA-Fragmente können bei dieser Methode verloren gehen.
Die Wahl der Analysemethode
Nach dem Aufschluss folgt die eigentliche Messung. Auch hier unterscheiden sich die Studien erheblich. Einige Arbeitsgruppen setzten auf quantitative PCR-Verfahren (qPCR), die eine hochsensitive, aber sequenzspezifische Detektion ermöglichen. Andere nutzten Sequenzierungsverfahren – entweder die Nanopore-Technologie (ONT) oder die Illumina-Plattform – die eine breitere, sequenzunabhängige Erfassung erlauben, aber eine aufwändigere Auswertung erfordern. Teilweise wurden auch fluorometrische Verfahren zur Gesamtquantifizierung eingesetzt.
Jede dieser Methoden misst etwas leicht anderes: Fluorometrie erfasst die Gesamt-DNA unabhängig von der Sequenz, leidet jedoch unter Störungen durch RNA (Crosstalk). PCR-Verfahren erfassen nur definierte Zielsequenzen. Sequenzierungsverfahren liefern ein breiteres Bild, sind jedoch stärker von Bibliotheksvorbereitung, Fragmentselektion und bioinformatischer Auswertung abhängig. Eine direkte Vergleichbarkeit der absoluten Messwerte zwischen Studien, die unterschiedliche Methoden einsetzen, ist daher nur eingeschränkt möglich.
Validierung und Standardisierung
In der regulierten pharmazeutischen Analytik sind Messmethoden vor ihrem Einsatz zu validieren: Extraktionseffizienz, Nachweisgrenze, Linearität und mögliche Störeinflüsse müssen systematisch bestimmt werden. Bei den hier vorgestellten unabhängigen Studien fehlt diese formale Validierung in der Regel – was nicht bedeutet, dass die Arbeiten unsorgfältig durchgeführt wurden, wohl aber, dass methodische Unsicherheiten schwerer zu quantifizieren sind.
Ein direkter Vergleich derselben Probe mit mehreren Methoden parallel – ein sogenannter orthogonaler Ansatz – würde helfen, methodenbedingte Variabilität von tatsächlichen Unterschieden zwischen Chargen zu trennen. Nur einige der vorliegenden Studien setzen mehrere orthogonale Methoden systematisch an denselben Proben ein. Auf die Bedeutung dieses Ansatzes wird im abschließenden Kapitel noch eingegangen.
⚬ ⚬ ⚬
4.3. Übersicht und Einzelanalysen der Studien
Die folgenden Studien unterscheiden sich hinsichtlich Methodik, Publikationsform und wissenschaftlicher Einordnung. Neben peer-reviewten Arbeiten werden auch Preprints und explorative Analysen berücksichtigt, um das derzeit verfügbare Spektrum an Daten vollständig abzubilden.
Die Darstellung erfolgt chronologisch, um die Entwicklung der wissenschaftlichen Diskussion nachvollziehbar zu machen.
Sequencing of bivalent Moderna and Pfizer mRNA vaccines reveals nanogram to microgram quantities of expression vector dsDNA per dose
Autoren: Kevin McKernan, Yvonne Helbert, Liam T. Kane, Stephen McLaughlin
Veröffentlicht: 2023 April 10
DNA fragments detected in monovalent and bivalent Pfizer/BioNTech and Moderna modRNA COVID-19 vaccines from Ontario, Canada: Exploratory dose response relationship with serious adverse events.
Autoren: David Speicher, J. Rose, L. M. Gutschi, D. M. Wiseman, Kevin McKernan
Veröffentlicht: 2023 October 19
Methodological Considerations Regarding the Quantification of DNA Impurities in the COVID-19 mRNA Vaccine Comirnaty®
Autoren: Brigitte König, Jürgen O. Kirchner
Veröffentlicht: 2024 May 8
Confirmation of the presence of vaccine DNA in the Pfizer anti-COVID-19 vaccine
Autoren: Didier Raoult
Veröffentlicht: 2024 November 12
BioNTech RNA-Based COVID-19 Injections Contain Large Amounts Of Residual DNA Including An SV40 Promoter/Enhancer Sequence
Autoren: Ulrike Kämmerer, Verena Schulz, Klaus Steger
Veröffentlicht: 2024 December 3
A rapid detection method of replication-competent plasmid DNA from COVID-19 mRNA vaccines for quality control.
Autoren: Tayler J. Wang, Alex Kim, Kevin Kim
Veröffentlicht: 2024 December 29
Quantitative Analysis of Nucleic Acid Content in Spikevax (Moderna) and BNT162b2 (Pfizer) COVID-19 Vaccine Lots
Autoren: Richard M Fleming PhD, MD, JD; Peter Kotlár MD; Sona Pekova MD, PhD
Veröffentlicht: 2025 May 13
Quantification of residual plasmid DNA and SV40 promoter-enhancer sequences in Pfizer/BioNTech and Moderna modRNA COVID-19 vaccines from Ontario, Canada
Autoren: David Speicher, Jessica Rose, Kevin McKernan
Veröffentlicht: 2025 September 25
Systematic analysis of COVID-19 mRNA vaccines using four orthogonal approaches demonstrates no excessive DNA impurities
Autoren: Adam Achs, Tatiana Sedlackova, Lukas Predajna, Jaroslav Budis, Maria Bartosova, Vladimir Zelnik, Diana Rusnakova, Martina Melichercikova, Marta Miklosova, Veronika Gencurova, Barbora Cernakova, Tomas Szemes, Boris Klempa, Juraj Kopacek, Silvia Pastorekova
Veröffentlicht: 2025 Dezember 13
🧬 🧬 🧬
4.3.1. McKernan et al. (2023)
| Sequencing of bivalent Moderna and Pfizer mRNA vaccines reveals nanogram to microgram quantities of expression vector dsDNA per dose Autoren: Kevin McKernan, Yvonne Helbert, Liam T. Kane, Stephen McLaughlin Veröffentlicht: 10. April 2023 (Preprint, OSF) Link zur Studie: https://osf.io/preprints/osf/b9t7m |
a) Kontext und Ziel der Studie
Die Arbeit von McKernan et al. gehört zu den ersten öffentlich verfügbaren unabhängigen Analysen, in denen mRNA-basierte COVID-19-Impfstoffe gezielt auf das Vorhandensein von DNA untersucht wurden. Ausgangspunkt war keine regulatorisch motivierte Fragestellung, sondern eine explorative Sequenzierungsarbeit, im Verlauf derer DNA in den Proben detektiert wurde. Dies veranlasste die Autoren, die Zusammensetzung der Impfstoffe systematisch weiterzuuntersuchen.
Ziel der Studie war es,
- 2 Chargen bivalente Impfstoffe von Pfizer (über dem Ablaufdatum)
- 2 Chargen bivalente Impfstoffe von Moderna (über dem Ablaufdatum)
- 8 Chargen monovalente Impfstoffe von Pfizer (über dem Ablaufdatum, ungeöffnet)
b) Materialien und Methoden
Impfstoffchargen
Für die Untersuchungen wurden folgende Chargen verwendet:
- 2 Chargen bivalente Impfstoffe von Pfizer (über dem Ablaufdatum)
- 2 Chargen bivalente Impfstoffe von Moderna (über dem Ablaufdatum)
- 8 Chargen monovalente Impfstoffe von Pfizer (über dem Ablaufdatum, ungeöffnet)
Die Studie kombiniert mehrere analytische Verfahren, um sowohl quantitative als auch qualitative Aussagen zu ermöglichen.
Aufschluss der LNPs und Extraktion der Nukleinsäuren
Die Lipid-Nanopartikel wurden mittels Lithium-Dodecylsulfat (LiDS) lysiert. Die anschließende Aufreinigung erfolgte nach dem SPRI-Prinzip (magnetische Beads), das Nukleinsäuren größen- und bedingungsabhängig selektiv bindet und von Lipiden sowie anderen Begleitsubstanzen trennt. Das Eluat enthielt die isolierten Nukleinsäuren – mRNA und potenziell vorhandene DNA.
Als Qualitätskontrolle des Extraktionsschritts wurde ein Bioanalyzer eingesetzt, der mittels automatisierter Kapillarelektrophorese die Größenverteilung und relative Menge der gewonnenen Nukleinsäuren sichtbar macht.
Sequenzierung mittels Illumina
Die extrahierten Nukleinsäuren wurden mittels Illumina-Sequenzierung analysiert. Mehrere Sequenzierläufe (Reads) bildeten die Grundlage für die bioinformatische Rekonstruktion der Plasmidsequenzen beider Impfstoffe. Aus den erhaltenen Reads wurden Plasmidkarten erstellt – für Moderna und für Pfizer.
PCR-basierte Verifikation
Auf Grundlage der Sequenzierungsdaten wurden spezifische Primer für zwei Zielregionen entworfen: eine Spike-Sonde und eine Ori-Sonde, beide auf Sequenzen ausgelegt, die sowohl im Pfizer- als auch im Moderna-Plasmid vorkommen. Diese qPCR diente nicht der primären Quantifizierung, sondern der unabhängigen Bestätigung, dass die per Sequenzierung identifizierten Plasmidsequenzen tatsächlich in den Proben vorhanden sind – und nicht auf Laborkontaminationen zurückzuführen sind.
Fluorometrie und Elektrophorese
Zur Abschätzung der DNA-Konzentration wurden Qubit-Fluorometrie und Agilent-Elektrophorese eingesetzt. Beide Methoden liefern Aussagen über die Gesamtmenge der vorhandenen DNA, jedoch keine Sequenzinformation.
c) Zentrale Ergebnisse
Qualitative Befunde
Die Sequenzanalysen zeigen Übereinstimmungen mit bekannten Plasmidvektorsequenzen und weisen auf genetische Elemente hin, die im Herstellungsprozess eingesetzt werden. Die nachgewiesene DNA wurde als doppelsträngig (dsDNA) charakterisiert.
Unerwarteter Fund: SV40-Promoter/Enhancer
Im Verlauf der Sequenzierung identifizierten die Autoren im Pfizer-Impfstoff eine SV40-Promoter/Enhancer-Sequenz. Dieser Fund war insofern unerwartet, als diese Sequenz in den der EMA vorgelegten Plasmidkarten – konkret im „Rapporteur Rolling Review Critical Assessment Report“ (Figure S.2.3-1. pST4-1525 Plasmid Map) – in den öffentlich zugänglichen Unterlagen nicht aufgeführt war. Die Autoren wiesen ergänzend darauf hin, dass das Pfizer-Plasmid auch ein SV40-Kernlokalisierungssignal enthält, das theoretisch den Transport von DNA in den Zellkern begünstigen könnte.
Quantitative Befunde
Qubit-Fluorometrie und Elektrophorese zeigten DNA-Konzentrationen, die nach Einschätzung der Autoren die regulatorischen Richtwerte – etwa das EMA-Limit von 330 ng DNA pro mg RNA – deutlich überschreiten.
d) Interpretation durch die Autoren
Die Autoren schlussfolgern, dass DNA-Rückstände in den untersuchten Chargen vorhanden sind, die ihrem Ursprung nach auf die im Herstellungsprozess verwendeten Plasmidvektoren zurückzuführen sein dürften. Die gemessenen Mengen werden von den Autoren als möglicherweise regulatorisch relevant eingestuft.
Hinsichtlich replikationsfähiger DNA formulieren die Autoren die Einschätzung, dass für solche Sequenzen strengere Grenzwerte angemessen wären als für nicht-replikationsfähige DNA-Fragmente.
e) Methodische Einschränkungen
Die Herkunft, Lagerung und Chargencharakterisierung der untersuchten Fläschchen ist nur begrenzt dokumentiert. Angaben zu Negativkontrollen – etwa Wasserproben, die denselben Aufarbeitungsschritten unterzogen wurden – sowie zu Positivkontrollen mit bekannten DNA-Konzentrationen fehlen weitgehend oder sind unvollständig beschrieben. Eine formale Methodenvalidierung liegt nicht vor.
Zudem weisen die Autoren selbst auf zwei gegenläufige Unsicherheiten bei der Quantifizierung hin: PCR-basierte Verfahren könnten die tatsächliche DNA-Menge unterschätzen, da Fragmente unterhalb der Amplikon-Länge nicht erfasst werden. Umgekehrt könnten Fluorometrie und Elektrophorese die DNA-Konzentration überschätzen, wenn der eingesetzte Farbstoff unspezifisch auch an RNA bindet.
f) Einordnung in den Gesamtkontext
Die Studie stellt einen frühen und wichtigen Beitrag zur wissenschaftlichen Diskussion über DNA-Rückstände in mRNA-Impfstoffen dar. Sie hat maßgeblich dazu beigetragen, dass weitere Arbeitsgruppen eigene Untersuchungen durchführten. Die Befunde sind als Ausgangspunkt für weiterführende Untersuchungen zu verstehen, nicht als abschließende Bewertung.
🧬 🧬 🧬
4.3.2. Speicher et al. (2023)
| DNA fragments detected in monovalent and bivalent Pfizer/BioNTech and Moderna modRNA COVID-19 vaccines from Ontario, Canada: Exploratory dose response relationship with serious adverse events. Autoren: David Speicher, Jessica Rose, L. Maria Gutschi, David M. Wiseman, Kevin McKernan Veröffentlicht: 19. Oktober 2023 (ResearchGate) Link zur Studie: https://osf.io/preprints/osf/mjc97_v1 |
a) Kontext und Ziel der Studie
Die Studie von David J. Speicher et al. baut inhaltlich auf den zuvor veröffentlichten Analysen von Kevin McKernan auf und verfolgt einen erweiterten Ansatz.
Neben dem Nachweis und der Quantifizierung von DNA-Rückständen in weiteren Impfstoffchargen adressiert sie zwei zusätzliche Fragestellungen:
- Lokalisierung der DNA: Liegt die nachgewiesene DNA innerhalb der Lipid-Nanopartikel (LNPs) oder frei in der Lösung vor?
- Explorative Korrelation: Besteht ein statistischer Zusammenhang zwischen gemessenen DNA-Mengen und gemeldeten unerwünschten Ereignissen auf Chargenebene?
Die Studie gliedert sich damit in zwei inhaltlich verschiedene Teile:
- analytischer Teil: Nachweis und Charakterisierung von DNA in Impfstoffproben
- explorativer Teil: Untersuchung einer möglichen statistischen Beziehung zu Nebenwirkungsdaten
Beide Teile erfordern eine unterschiedliche methodische Bewertung.
b) Materialien und Methoden
Impfstoffchargen
Untersucht wurden 27 Fläschchen aus 12 Chargen: 8 abgelaufene, ungeöffnete Pfizer-BNT162b2-Fläschchen und 16 abgelaufene, ungeöffnete Moderna-Spikevax-Fläschchen, bezogen aus verschiedenen Apotheken in Ontario, Kanada. Alle Fläschchen waren originalversiegelt mit intakten Schutzkappen sowie aufgedruckten Chargennummern und Verfallsdaten. Zusätzlich wurden drei noch gültige Restmengen einer Moderna-XBB.1.5-Charge einbezogen. Lagerung und Transport erfolgten unter dokumentierter Kühlung. Als Negativkontrolle diente ein steriles, ungeöffnetes Injektionsfläschchen eines nicht verwandten Arzneimittels (TriMix).
Die Autoren kombinieren mehrere analytische und statistische Verfahren.
Aufschluss der Lipid-Nanopartikel (LNPs)
Um die Lipid-Nanopartikel (LNPs) aufzubrechen, wurde die Probe 8 Minuten lang auf 95°C erhitzt, gefolgt von einer Abkühlung auf 4°C für fünf Minuten. Dieser thermische Schritt dient der Freisetzung der enthaltenen Nukleinsäuren.
Quantifizierung mittels qPCR
Die DNA-Konzentrationen wurden mittels qPCR über den Cq-Wert (Quantifizierungszyklus) bestimmt und in Nanogramm pro Dosis umgerechnet. Die Autoren übernahmen die Primer aus der McKernan-Studie: Spike-Sonde und Ori-Sonde, die auf Sequenzen abzielen, die beiden Impfstoffen (Pfizer, Moderna) gemeinsam sind. Ergänzend wurde ein SV40-Enhancer-Assay eingesetzt, der speziell auf die im Pfizer-Plasmid identifizierte SV40-Sequenz ausgelegt ist.
Quantifizierung mittels Qubit-Fluorometrie
Zur Gesamtquantifizierung der DNA wurde Qubit-Fluorometrie mit dem DNA-spezifischen Farbstoff AccuGreen eingesetzt. Die Fluoreszenzintensität ist proportional zur vorhandenen DNA-Menge und liefert eine sequenzunabhängige, jedoch struktursensitive Gesamtschätzung (abhängig von Farbstoffspezifität und Probenzusammensetzung).
Größenprofil der Rest-DNA mittels ONT-Sequenzierung
Die Größenverteilung der DNA-Fragmente in einer Pfizer-Probe wurde mittels Oxford-Nanopore-Sequenzierung bestimmt. Dabei ist zu beachten, dass der verwendete Bibliotheksaufbau Fragmente kleiner als etwa 150 bp (Basenpaare) systematisch entfernt. Die erhaltene Längenverteilung ist daher in Richtung längerer Fragmente verzerrt und bildet den tatsächlichen Anteil kurzer Fragmente nicht vollständig ab.
DNA innerhalb oder außerhalb der LNPs – Nukleaseprotektionstest
Um zu prüfen, ob die DNA-Rückstände im Inneren der LNPs vorliegen oder frei außerhalb, wurde das Enzym DNase I-XT direkt zum unveränderten Impfstoff gegeben – also zu intakten, noch nicht aufgeschlossenen LNPs. DNase I-XT baut zugängliche DNA ab. Eine anschließende qPCR-Messung erlaubte den Vergleich der DNA-Menge vor und nach der Enzymbehandlung.
Auswertung der VAERS-Datenbank
Für jede untersuchte Charge wurde aus der VAERS-Datenbank ein Serious Adverse Event Reporting Ratio (SRR) berechnet – das Verhältnis schwerwiegender zu allen gemeldeten Nebenwirkungen. Dieser Wert wurde der gemessenen DNA-Konzentration der jeweiligen Charge gegenübergestellt.
c) Zentrale Ergebnisse
Quantitative Befunde und Methodenvergleich
Die Gegenüberstellung der Ergebnisse der qPCR und Qubit-Fluorometrie zeigen, dass die gemessenen Daten stark von dem verwendeten Messverfahren abhängen.
Ermittlung der DNA-Konzentrationen für Pfizer
| qPCR | Qubit-Fluorometrie |
| Ori-Sonde: 0,28 – 4,27 ng/Dosis Spike-Sonde: 0,22 – 2,43 ng/Dosis | Gesamt-DNA: 1.896 – 3.720 ng/Dosis |
Der SV40-Promotor-Enhancer-Ori wurde nur in den Pfizer-Fläschchen mit Cq-Werten zwischen 16,64 und 22,59 nachgewiesen.
Ermittlung der DNA-Konzentrationen für Moderna
| qPCR | Qubit-Fluorometrie |
| Ori-Sonde: 0,01 – 0,34 ng/Dosis Spike-Sonde: 0,25 – 0,78 ng/Dosis | Gesamt-DNA: 3.270 – 5.100 ng/Dosis |
Bezogen auf den FDA-Grenzwert von 10 ng DNA pro Dosis liegen die qPCR-Werte unterhalb des Grenzwerts, während die fluorometrischen Werte diesen um das 188- bis 509-fache überschreiten.
Die Autoren weisen darauf hin, dass qPCR-Verfahren DNA-Fragmente unterhalb der Amplikon-Länge (105–114 bp) nicht erfassen können und die Gesamtmenge damit systematisch unterschätzen.
Darüber hinaus zeigten die Messungen eine Variabilität der DNA-Mengen zwischen verschiedenen Chargen desselben Impfstoffs – ein Hinweis auf mögliche Schwankungen im Reinigungsprozess.
DNA liegt im Inneren der LNPs
Nach DNase I-XT-Behandlung des intakten Pfizer-Impfstoffs zeigte die anschließende qPCR-Messung kaum eine Veränderung der DNA-Menge. Da das Enzym nur zugängliche DNA abbaut, legt dieses Ergebnis nahe, dass der überwiegende Teil der DNA-Rückstände im Inneren der LNPs vorliegt und damit vor dem Enzymangriff geschützt ist.
Größenprofil der Rest-DNA
Die ONT-Sequenzierung einer Pfizer-Probe ergab eine mittlere Fragmentlänge von 214 bp. Einzelne Fragmente erreichten Längen von über 1.000 bp; das längste nachgewiesene Fragment umfasste etwa 3.200 bp und entsprach damit annähernd der halben Plasmidlänge von 7.810 bp.
Da der Bibliotheksaufbau Fragmente unter 150 bp systematisch ausschließt, ist davon auszugehen, dass der tatsächliche Anteil kurzer Fragmente in der Probe höher liegt als in den Sequenzierungsdaten abgebildet.
Explorative VAERS-Korrelation mit den gemessenen DNA-Mengen
Die Autoren beobachteten eine positive Korrelation zwischen den gemessenen DNA-Konzentrationen auf Chargenebene und dem SRR-Wert der jeweiligen Charge. Chargen mit höheren DNA-Mengen tendierten zu höheren Anteilen schwerwiegender Meldungen in der VAERS-Datenbank.
d) Interpretation durch die Autoren
Methodenabhängigkeit von Rest-DNA-Bewertungen
Die Autoren berichten zwei völlig unterschiedliche Messergebnisse für dieselben Impfstoffe:
| Methode | Ergebnis: Rest-DNA pro Dosis (Grenzwert 10 ng/Dosis) | Bewertung |
| qPCR | UNTER dem FDA-Grenzwert | Einhaltung |
| Fluorometrie | 188- bis 509-fach ÜBER dem FDA-Grenzwert | Überschreitung |
Das bedeutet: Je nach Messmethode kommt man zu gegensätzlichen Schlussfolgerungen.
Diese Diskrepanz ist ein zentrales Ergebnis der Studie und verdeutlicht die hohe methodische Abhängigkeit der gemessenen DNA-Werte.
Hinweis auf methodische Inkonsistenz
Die Autoren argumentieren, dass die beobachteten Diskrepanzen nicht ausschließlich auf Messungenauigkeiten zurückzuführen sind, sondern auf grundlegend unterschiedliche Messprinzipien.
Die EMA verwendet ratiometrische Richtlinien – das bedeutet: Es gibt einen erlaubten Grenzwert für das Verhältnis von DNA zu RNA (wie z. B. 330 ng DNA pro 1 mg RNA).
Allerdings kommen für die Messung von RNA und DNA unterschiedliche Methoden zum Einsatz, die sich in ihrer Spezifität unterscheiden.
| Was gemessen wird | Verwendete Methode | Gemessen werden … |
| RNA (Hauptbestandteil) | Fluorometrie / UV-Spektrophotometrie | Gesamtnukleinsäuren (unspezifisch) |
| DNA (Rückstände) | qPCR | Spezifische DNA-Sequenzen (selektiv) |
Verschiedene Quellenbelege dazu:
- „Rapporteur Rolling Review critical assessment report” (Table S.2.6-16. Evolution of BNT162b2 Drug Substance Methods)
- „EU Official Control Authority Batch Release Certificate for Immunological Products” (Table 2. Drug Substance Quality Control Tests)
- „Assessment report EMA/707383/2020”
- „Assessment report EMA/15689/2021”
Die Autoren argumentieren, wenn man:
🔸RNA mit einer unspezifischen Methode misst (Fluorometrie: erfasst auch DNA-Fragmente, Abbauprodukte etc.) und
🔸DNA mit einer spezifischen Methode misst (qPCR: erfasst nur die exakte Zielsequenz),
dann sind die beiden Messwerte methodisch nicht kompatibel.
Dadurch sind die berechneten DNA/RNA-Verhältnisse methodisch verzerrt und nicht direkt vergleichbar.
Diese Argumentation bezieht sich auf die Vergleichbarkeit der Messmethoden, nicht notwendigerweise auf die Gültigkeit der jeweiligen Einzelmessung.
Die Autoren plädieren dafür, dass Behörden nicht nur Grenzwerte, sondern auch die verbindlich einzusetzenden Nachweismethoden vorschreiben.
DNA innerhalb der LNPs: biologische Konsequenz
Das Vorliegen der DNA im Inneren der LNPs wird von den Autoren als potenziell biologisch relevant eingestuft. Im Unterschied zu freier, nackter DNA – die im Körper rasch abgebaut wird – ist LNP-verpackte DNA vor Nukleasen geschützt und kann effizient in Zellen aufgenommen werden. Die bestehenden regulatorischen Grenzwerte, so die Autoren, wurden für nackte Wirtszell-DNA entwickelt und berücksichtigen diesen Unterschied nicht.
Kritik an den regulatorischen Richtlinien
Die Autoren üben grundsätzliche Kritik an den bestehenden Grenzwertkonzepten. Sie bemängeln, dass die Richtlinien auf veralteten Grundlagen beruhen, die Molekülanzahl zugunsten einer gewichtsbasierten Betrachtung vernachlässigen, kumulative Dosierungen (mehrere Dosen innerhalb weniger Monate) nicht berücksichtigen und funktionelle Sequenzen wie Promotoren oder nukleäre Targeting-Signale außer Acht lassen. Insbesondere das Vorhandensein des SV40-Promoter/Enhancers im Pfizer-Plasmid wird als Element hervorgehoben, das den Transport von DNA in den Zellkern begünstigen könnte.
Explorative VAERS-Analyse
Die beobachtete Korrelation wird von den Autoren als möglicher Hinweis auf einen Zusammenhang zwischen DNA-Gehalt und Nebenwirkungsprofil interpretiert. Ausdrücklich wird betont, dass es sich um eine explorative Beobachtung handelt, die keine kausalen Schlussfolgerungen erlaubt. Daher plädieren sie für weitere Untersuchungen dahingehend.
e) Methodische Einschränkungen
Die Herkunft, Lagerung und Chargencharakterisierung der untersuchten Proben ist insgesamt gut dokumentiert – Kühlkette, Transportbedingungen und Fläschchenzustand sind beschrieben. Der thermische Aufschluss ohne nachfolgende Aufreinigung ist nicht im regulatorischen Sinne validiert; Angaben zur Extraktionseffizienz fehlen.
Alle untersuchten Fläschchen waren zum Zeitpunkt der Analyse bereits abgelaufen. Drei Moderna-XBB.1.5-Fläschchen waren angebrochene Restmengen nach der Patientenversorgung – keine originalversiegelten Proben. Sie unterscheiden sich damit in ihrer Probenqualität von den übrigen Fläschchen und sind gesondert zu bewerten.
Die VAERS-Korrelation ist mit erheblichen Unsicherheiten behaftet. VAERS ist ein passives Meldesystem: Meldungen werden nicht systematisch verifiziert, die Meldebereitschaft variiert zwischen Zeiträumen und Regionen, und eine Korrelation auf Chargenebene erlaubt keine Rückschlüsse auf individuelle Kausalzusammenhänge. Die Aussagekraft dieser Analyse ist damit strukturell begrenzt – unabhängig von der Qualität der analytischen Messungen.
Die ONT-Sequenzierung erfasst aufgrund des Bibliotheksaufbaus nur Fragmente ab etwa 150 bp und bildet den tatsächlichen Kurzfragmentanteil nicht vollständig ab.
f) Einordnung in den Gesamtkontext
Die Studie erweitert die Befunde von McKernan et al. in mehrfacher Hinsicht: Sie untersucht weitere Chargen, liefert einen Nukleaseprotektionstest als direkten Hinweis auf die intrapartikuläre Lage der DNA und thematisiert strukturelle Schwächen der regulatorischen Methodik. Der analytische Teil stellt einen sachlichen Beitrag zur Debatte dar.
Der explorative Teil – die Verknüpfung mit VAERS-Daten – ist methodisch anspruchsvoll und mit den genannten Einschränkungen des Meldesystems behaftet. Die Autoren selbst charakterisieren ihn als hypothesengenerierend, nicht als beweisend.
Die Autoren weisen auf mehrere Einschränkungen ihrer Studie hin und fordern eine Replikation ihrer Ergebnisse unter forensischen Bedingungen.
Insgesamt unterstreicht die Studie die Notwendigkeit standardisierter und methodenübergreifend vergleichbarer Analysen zur Bewertung von DNA-Rückständen in mRNA-basierten Arzneimitteln.
🧬 🧬 🧬
4.3.3. König & Kirchner (2024)
| Methodological Considerations Regarding the Quantification of DNA Impurities in the COVID-19 mRNA Vaccine Comirnaty® Autoren: Brigitte König, Jürgen O. Kirchner Veröffentlicht: 8. Mai 2024 (Peer-reviewed, PubMed) Link zur Studie: https://pubmed.ncbi.nlm.nih.gov/38804335/ |
a) Kontext und Ziel der Studie
Die Arbeit von Brigitte König und Johannes O. Kirchner unterscheidet sich in ihrer Zielsetzung grundlegend von den zuvor dargestellten Studien. Während frühere Arbeiten primär den Nachweis und die Quantifizierung von DNA-Rückständen in mRNA-Impfstoffen zum Ziel hatten, liegt der Fokus dieser Studie auf einer methodischen Bewertung der eingesetzten Nachweisverfahren.
Die zentrale Frage lautet: Ist die im Zulassungsverfahren verwendete qPCR-basierte DNA-Quantifizierung als Chargenprüfung und damit als Qualitätssicherungsinstrument geeignet?
Um diese Frage zu beantworten, untersuchen die Autoren:
- welche analytischen Methoden zur Quantifizierung von DNA-Rückständen eingesetzt wurden,
- welche methodischen Grenzen diese Verfahren aufweisen,
- und inwiefern unterschiedliche Methoden zu abweichenden Ergebnissen führen können.
Die Studie wurde in einem formalen Peer-Review-Verfahren begutachtet und in einer wissenschaftlichen Fachzeitschrift veröffentlicht.
b) Materialien und Methoden
Impfstoffchargen
Das Labor erhielt mehrere originalversiegelte Fläschchen unterschiedlicher Chargen des mRNA-Impfstoffs Comirnaty® (BNT162b2), bereitgestellt durch offizielle Impfzentren. Lagerung und Transport erfolgten unter dokumentierter Einhaltung der Kühlkette (2–8 °C). Vier Chargen waren zum Zeitpunkt der Analyse bereits abgelaufen, drei wiesen eine Resthaltbarkeit von 11 bis 13 Monaten auf.
Die Autoren kombinieren zwei Ansätze: eine methodenkritische Analyse bestehender Verfahren und eigene Labormessungen.
Methodenkritische Analyse
Die Autoren untersuchen die im EMA-Rolling-Review-Dokument vom November 2020 beschriebene qPCR-Methode (einschließlich des dortigen Mustervorbereitungsschemas) und identifizieren drei strukturelle Probleme.
Erstens erfasst die qPCR nur eine 69 bp lange Zielsequenz – den T7-Promotor – innerhalb eines 7.824 bp langen Plasmids. Die verbleibenden 99 % der Plasmid-DNA sowie etwaige genomische Bakterien-DNA bleiben damit unerfasst. Die Autoren weisen darauf hin, dass dieses Problem prinzipiell für jeden qPCR-Ansatz gilt, der nur einzelne Zielsequenzen adressiert – unabhängig davon, welche Sequenz gewählt wird.
Zweitens ist die verwendete Standardkurve methodisch problematisch. Zur Quantifizierung wird eine Standardkurve mit unbehandelter, intakter Plasmid-DNA erstellt. Die DNA in der Impfstoffprobe hat jedoch im Herstellungsprozess mehrere Behandlungsschritte – insbesondere einen DNase-Verdau – durchlaufen und ist damit fragmentiert. Da fragmentierte DNA schlechter amplifiziert wird als intakte, erscheint die gemessene Konzentration systematisch niedriger als die tatsächlich vorhandene. Ein korrekter Vergleich würde eine Standardkurve erfordern, die mit DNA erstellt wurde, die denselben Behandlungsschritten unterzogen wurde wie die Probe.
Drittens stellt sich die Frage der Proportionalität:
- Gibt es sequenzabhängige Unterschiede in der Abbaurate?
- Wird die qPCR-Zielsequenz durch den DNase-Verdau proportional zum Rest des Plasmids abgebaut?
- Werden bestimmte Sequenzen der linearisierten Plasmide durch DNase häufiger oder deutlich seltener abgebaut als andere?
Wenn die Proportionalität nicht gegeben ist, ist jede Extrapolation von der Zielsequenz auf die Gesamt-DNA fehlerbehaftet. Die Europäische Pharmakopöe (Ph. Eur. 2.6.35) bestätigt diese Einschränkung: Sie nennt qPCR zwar als Methode der Wahl für den Nachweis spezifischer Sequenzen, weist aber ausdrücklich darauf hin, dass für die Bestimmung der Gesamt-DNA andere oder ergänzende Verfahren erforderlich sein können.
Überlegungen zur regulatorischen Praxis
Die Autoren weisen auf eine Inkonsistenz in der behördlichen Praxis hin: Laut einem EDQM-Protokoll wird DNA nur auf Wirkstoffebene gemessen, nicht im fertigen Impfstoff. Als Begründung wird angegeben, dass Lipid-Nanopartikel die DNA-Messung stören könnten. Gleichzeitig wird die RNA-Konzentration im fertigen Impfstoff mittels Fluorometrie gemessen – nach Aufschluss der LNPs mit Triton X-100. Da RNA und DNA als Nukleinsäuren vergleichbare analytische Eigenschaften aufweisen und beide von LNPs umschlossen sein können, erscheint die unterschiedliche Behandlung erklärungsbedürftig. Ob hierfür technische oder regulatorische Gründe vorliegen, die in öffentlichen Dokumenten nicht vollständig dargelegt sind, lässt sich anhand der verfügbaren Quellen nicht abschließend beurteilen.
Eigene Messungen mittels Qubit-Fluorometrie
Um die praktische Eignung eines alternativen Verfahrens zu prüfen, führten die Autoren eigene Messungen an sieben Chargen Comirnaty® durch. Dabei wurden die Proben jeweils vor und nach Zugabe des Detergens Triton X-100 gemessen, das die LNPs aufbricht und den eingeschlossenen Inhalt freisetzt. Als Messsystem wurde Qubit-Fluorometrie mit dem DNA-spezifischen Farbstoff PicoGreen eingesetzt.
c) Zentrale Ergebnisse
Qubit-Messungen vor und nach LNP-Aufschluss
Ohne Triton X-100 wurden nur geringe DNA-Mengen gemessen. Nach Zugabe von Triton X-100 stiegen die gemessenen Werte drastisch an – auf das 360- bis 534-fache des regulatorischen Grenzwerts von 10 ng pro Dosis, entsprechend 3.600 bis 5.340 ng DNA pro Dosis.
In den vier abgelaufenen Chargen waren bereits ohne Triton X-100 zwischen 16 und 81 % der Gesamt-DNA messbar – ein Hinweis auf einen teilweisen Zerfall der LNPs über die Lagerungszeit. In den drei frischen Chargen war die DNA zu 93 bis 97 % erst nach dem LNP-Aufschluss nachweisbar, was auf weitgehend intakte Partikel hindeutet.
RNA-Messungen als Kontrolle
Die parallel durchgeführten RNA-Messungen dienten als methodische Kontrolle und zeigten plausible Werte, was die Autoren als Beleg für die grundsätzliche Eignung des Qubit-Systems im Kontext dieser Probenmatrix werten.
d) Interpretation durch die Autoren
Kritik an der qPCR als alleinige Referenzmethode
Die Autoren schlussfolgern, dass die behördlich vorgeschriebene qPCR-basierte Methode strukturell ungeeignet ist, die Gesamt-DNA im fertigen Impfstoff zuverlässig zu quantifizieren. Die gemessenen DNA-Gehalte überschreiten den regulatorischen Grenzwert deutlich – blieben aber unentdeckt, weil die eingesetzte Methode nur einen kleinen Ausschnitt der vorhandenen DNA erfasst und zudem auf Wirkstoffebene statt im Endprodukt angewendet wird.
Forderung nach methodischer Anpassung
Die Autoren fordern daher eine fluoreszenzspektrometrische Messung der Gesamt-DNA im fertigen Impfstoff als Bestandteil der amtlichen Chargenprüfung – analog zur bereits etablierten RNA-Messung.
DNA innerhalb der LNPs: biologische Konsequenz
Darüber hinaus plädieren sie für eine Neubewertung der Risiken LNP-verpackter DNA, insbesondere hinsichtlich einer möglichen Integration in das menschliche Erbgut.
e) Methodische Einschränkungen
Geringe Stichprobengröße
Die Studie basiert auf einer begrenzten Stichprobe von sieben Chargen. Die Proben wurden als originalversiegelte Fläschchen von offiziellen Impfzentren bereitgestellt; Lagerung und Transport erfolgten unter dokumentierter Kühlkettenkontrolle. Die Provenienzdokumentation ist damit vergleichsweise gut belegt.
Einfluss des Ablaufdatums auf die LNP-Integrität
Vier der sieben Chargen waren zum Zeitpunkt der Analyse bereits abgelaufen. Abgelaufene Proben können chemisch verändert sein – insbesondere hinsichtlich der LNP-Integrität. Die Autoren berücksichtigen diesen Umstand ausdrücklich und werten die erhöhte DNA-Messbarkeit ohne Triton X-100 in abgelaufenen Chargen als Hinweis auf einen teilweisen LNP-Zerfall über die Lagerungszeit.
Keine RNase-Behandlung vor Fluorometrie (Qubit)
Qubit misst mit DNA-spezifischen Farbstoffen, die aber auch mit RNA (der Hauptbestandteil des Impfstoffs) interferieren können. Ohne vorherige RNase-Verdauung wird die gemessene „DNA-Menge“ sehr wahrscheinlich überschätzt.
Fehlende formale Validierung
Die Qubit-Methode wurde von den Autoren nicht formal nach regulatorischen Standards validiert. Eine solche Validierung – wie sie die ICH-Leitlinie Q2(R2) für analytische Methoden im Zulassungskontext vorschreibt – umfasst den systematischen Nachweis von Spezifität, Nachweisgrenze, Linearität, Präzision, Richtigkeit, Robustheit und Matrixeffekten. Die Autoren begründen die Eignung des Verfahrens argumentativ: durch den Vergleich von Messungen vor und nach LNP-Aufschluss sowie durch plausible RNA-Kontrollwerte. Das ist für eine wissenschaftliche Publikation, die einen methodischen Punkt demonstrieren will, ein üblicher Rahmen. Dieser Hinweis ist keine Kritik an der Sorgfalt der Autoren, sondern eine sachliche Einordnung des Publikationskontexts.
Fehlende Validierung der Extraktions-Effizienz
Bei Extraktionen wird nicht immer gezeigt, dass die Methode quantitativ alle DNA (besonders kleine Fragmente und verkapselte DNA) vollständig freisetzt und misst. Das kann zu Über- oder Unter-Schätzungen führen.
Verfügbarkeit behördlicher Prüfmethoden
Die methodenkritische Analyse stützt sich hinsichtlich der behördlichen qPCR-Methode auf ein Rolling-Review-Dokument aus dem frühen Zulassungsverfahren. Ob die dort beschriebene Methode identisch mit der final zugelassenen Chargenprüfmethode ist, lässt sich anhand öffentlich zugänglicher Quellen nicht abschließend beurteilen.
f) Einordnung in den Gesamtkontext
König & Kirchner leisten einen methodisch eigenständigen Beitrag zur Debatte. Im Unterschied zu McKernan und Speicher steht nicht die Frage im Vordergrund, wie viel DNA vorhanden ist, sondern ob die eingesetzten Verfahren überhaupt geeignet sind, diese Frage zuverlässig zu beantworten.
Die Studie leistet damit einen wichtigen Beitrag zur Debatte, indem sie:
- die methodische Abhängigkeit der Messergebnisse herausarbeitet
- die Grenzen etablierter Standardverfahren (insbesondere qPCR) aufzeigt
- und die Notwendigkeit methodenübergreifender Validierung betont
Die Arbeit verdeutlicht, dass Unterschiede zwischen Studien nicht zwangsläufig widersprüchliche oder falsche Ergebnisse widerspiegeln, sondern häufig durch:
- unterschiedliche Nachweisverfahren
- verschiedene Zielsequenzen
- und variierende Probenaufbereitung
erklärt werden können. Damit trägt die Arbeit wesentlich zum Verständnis bei, warum die Quantifizierung von DNA-Rückständen methodisch anspruchsvoll ist und warum Ergebnisse aus verschiedenen Studien nur eingeschränkt direkt vergleichbar sind.
🧬 🧬 🧬
4.3.4. Didier Raoult (2024)
| Confirmation of the presence of vaccine DNA in the Pfizer anti-COVID-19 vaccine Autoren: Didier Raoult Veröffentlicht: 12. November 2024 (Preprint, HAL Open Science) Link zur Studie: https://hal.science/hal-04778576v1 |
a) Kontext und Ziel der Studie
Die Arbeit von Didier Raoult reiht sich in die Reihe unabhängiger Untersuchungen ein, die das Vorhandensein von DNA in mRNA-basierten COVID-19-Impfstoffen analysieren.
Ziel der Studie ist es, den Nachweis von DNA-Komponenten im Pfizer/BioNTech-Impfstoff experimentell zu bestätigen und die Befunde früherer Arbeitsgruppen durch eine unabhängige Analyse zu überprüfen. Die Studie versteht sich damit primär als Replikationsarbeit.
b) Materialien und Methoden
Impfstoffchargen
In der Studie wurde ausschließlich der mRNA-Impfstoff Comirnaty® (BNT162b2) von BioNTech/Pfizer untersucht. Die Analyse basierte auf einer begrenzten Anzahl von Proben; in den verfügbaren Angaben wird keine systematische Chargenübersicht berichtet.
Die Studie verwendet klassische molekularbiologische Verfahren zur Analyse von Nukleinsäuren.
Aufschluss der Lipid-Nanopartikel (LNPs)
Die LNPs wurden mit dem Detergens Triton X-100 aufgeschlossen.
Quantifizierung mittels Qubit-Fluorometrie
An einer Charge (Nr. GJ7184) wurde eine DNA-Quantifizierung mittels kommerziellem Qubit-Fluorometer durchgeführt, jeweils vor und nach dem LNP-Aufschluss mit Triton X-100.
Sequenzierung mittels Illumina
Zwei Chargen (FP8191 und FP9359) wurden mittels Illumina-Sequenzierung analysiert. Dabei wurde bewusst auf eine Reverse Transkription verzichtet – die RNA wurde also nicht in DNA umgeschrieben. Damit ist sichergestellt, dass ausschließlich die in der Probe ursprünglich vorhandene DNA sequenziert wurde und nicht eine aus der mRNA erzeugte Kopie.
Die erhaltenen Reads (gelesenen Sequenzen) wurden mit der bekannten DNA-Plasmidsequenz (GenBank OR134577.1) verglichen. Für die bioinformatische Auswertung wurden nur Sequenzabschnitte ab 200 bp Länge und mit mehr als 90% Übereinstimmung zur Referenz berücksichtigt.
Als Kontrollversuch wurden die Proben vor der Sequenzierung mit DNase behandelt und anschließend erneut sequenziert, um zu prüfen, ob das Signal tatsächlich auf DNA zurückzuführen ist.
c) Zentrale Ergebnisse
Quantifizierung mittels Qubit-Fluorometrie
Ohne Triton X-100 wurden durchschnittlich 216 ng DNA pro Dosis gemessen. Nach LNP-Aufschluss mit Triton X-100 stieg der Wert auf durchschnittlich 5.160 ng pro Dosis – ein Anstieg um das ca. 24-Fache. Bezogen auf den angegebenen RNA-Gehalt von 30 µg pro Dosis entspricht dies rechnerisch einem DNA-Anteil von etwa 17 %.
Sequenzierung mittels Illumina
Mittels Illumina-Sequenzierung wurden über 140.000 Reads mit einer Länge von mehr als 200 Basenpaaren generiert. Aus diesen Daten konnte die vollständige Plasmidsequenz von 7.824 bp rekonstruiert werden. Dies bedeutet, dass die vorhandenen DNA-Fragmente in ihrer Gesamtheit die vollständige Sequenz abdecken.
Die Sequenzierungstiefe war mit mehr als 4.000-facher Abdeckung außergewöhnlich hoch – das bedeutet, jede Position des Plasmids wurde im Durchschnitt von mehr als 4.000 unabhängigen Reads abgedeckt. Die Sequenzdaten beider Chargen wurden in der GenBank hinterlegt (PP544445 und PP544446).
63 bis 97 % der sequenzierten DNA ließen sich dem Impfstoff-Plasmid zuordnen. Die Herkunft der verbleibenden 3 bis 37 % wurde nicht weiter untersucht.
DNase-Kontrollversuch
Nach DNase-Behandlung wurden nahezu keine Reads mehr detektiert (1–2 Reads). Der nahezu vollständige Signalverlust nach DNase-Behandlung spricht dafür, dass das Sequenzsignal tatsächlich von frei zugänglicher oder nach Aufschluss freigesetzter DNA stammt.
d) Interpretation durch die Autoren
Reproduzierbarkeit
Der Autor interpretiert seine Ergebnisse als experimentelle Bestätigung der Befunde früherer Studien. Mit mehreren methodischen Ansätzen und in verschiedenen Chargen sei das reichliche Vorhandensein von Plasmid-DNA im Pfizer/BioNTech-Impfstoff nachweisbar. Die Übereinstimmung mit früheren Berichten stütze die Reproduzierbarkeit entsprechender Befunde.
DNA verpackt in kationische Lipide: biologische Konsequenz
Hinsichtlich der biologischen Konsequenz weist der Autor darauf hin, dass LNP-verpackte Plasmid-DNA nach Zellaufnahme theoretisch in das Genom integrieren könnte. Das tatsächliche Risiko wird vom Autor selbst als extrem gering eingeschätzt; weitere Untersuchungen seien dennoch gerechtfertigt.
e) Methodische Einschränkungen
Herkunft und Lagerung der Proben
Angaben zur Herkunft, Lagerungsgeschichte und zum Zustand der untersuchten Fläschchen fehlen weitgehend. Es ist nicht dokumentiert, ob die Proben originalverpackt, korrekt gekühlt und ungeöffnet waren.
Fehlende Methodendetails
- keine Angabe zur RNase-Behandlung zur Entfernung von RNA vor der DNA-Messung
- keine Angabe, welches Qubit-Kit eingesetzt wurde (High Sensitivity oder Broad Range)
- keine Angabe, wie hoch die Extraktionseffizienz für LNP-verpackte DNA war
- keine Spezifizierung der DNase (nur „TURBO DNA-free kit“ genannt, nicht welcher Enzymtyp)
Geringe Stichprobengröße
Die Studie basiert auf einer begrenzten Stichprobe von drei Chargen.
Sequenzierung und Quantifizierung
Die Illumina-Sequenzierung belegt zuverlässig die Anwesenheit und Vollständigkeit der Plasmidsequenz, ist aber kein geeignetes Verfahren zur absoluten Quantifizierung. Eine hohe Sequenzierungstiefe kann auch dann entstehen, wenn wenige Moleküle mit hoher Kopienzahl vorliegen – sie spiegelt nicht notwendigerweise die tatsächliche Masse der DNA wider.
f) Einordnung in den Gesamtkontext
Die Studie ergänzt die bestehende Datenlage durch einen weiteren unabhängigen Nachweis von Plasmid-DNA in mRNA-Impfstoffen. Ihr Beitrag liegt primär in der Reproduzierbarkeit des qualitativen Befunds: Ein weiteres Labor, mit anderen Methoden und anderen Chargen, kommt zu vergleichbaren Ergebnissen.
🧬 🧬 🧬
4.3.5. Kämmerer et al. (2024)
| BioNTech RNA-Based COVID-19 Injections Contain Large Amounts Of Residual DNA Including An SV40 Promoter/Enhancer Sequence Autoren: Ulrike Kämmerer, Verena Schulz, Klaus Steger Veröffentlicht: 3. Dezember 2024 (Peer Reviewed, Cureus/Public Health Policy Journal) Link zur Studie: https://publichealthpolicyjournal.com/biontech-rna-based-covid-19-injections-contain-large-amounts-of-residual-dna-including-an-sv40-promoter-enhancer-sequence/ |
a) Kontext und Ziel der Studie
Die Arbeit untersucht das Vorhandensein von DNA-Komponenten in mRNA-basierten COVID-19-Impfstoffen mit besonderem Fokus auf regulatorische Sequenzelemente. Vor dem Hintergrund vorheriger Analysen widmet sich die Studie drei zentralen Fragen:
- Lässt sich die hohe Menge an DNA-Rückständen in BioNTech-Chargen mit unterschiedlichen Methoden bestätigen?
- Können vorhandene DNA-Fragmente gemeinsam mit der mRNA in menschliche Zellen eingeschleust werden (Transfektion)?
- Führt dies zu einer anhaltender Spike-Protein-Produktion?
b) Materialien und Methoden
Impfstoffchargen
Für die Untersuchungen wurden vier originale, ungeöffnete BNT162b2-Chargen verwendet:
- FD7958, FE6975 und EX8679 (monovalent, Wuhan-Stamm; Verfallsdatum Oktober/August 2021)
- HD9869 (bivalent, Wuhan/Omicron XBB1.5; Verfallsdatum Oktober 2024)
Als Positivkontrolle diente die Charge GH9715, deren DNA-Kontamination in früheren Studien bereits nachgewiesen worden war.
Alle Ampullen wurden von einer Apotheke im gekühlten Zustand bezogen, waren originalversiegelt und wurden während Transport und Lagerung durchgehend gekühlt. Die Provenienzdokumentation ist damit vergleichsweise gut belegt.
Quantifizierung
Die Lipid-Nanopartikel (LNPs) wurden mit Triton X-100 aufgeschlossen.
Die RNA-Quantifizierung erfolgte mit dem Qubit RNA High Sensitivity Assay.
Für die DNA-Quantifizierung wurden drei verschiedene fluorometrische dsDNA-Assays eingesetzt:
- Qubit dsDNA High Sensitivity Assay
- Quanti-iT PicoGreen dsDNA Assay
- AccuBlue High Sensitivity dsDNA Assay
Die Untersuchung mittels Fluoreszenz-Plate-Reader erfolgte vor und nach einer RNase A-Behandlung (einem RNA-abbauenden Enzym).
Der Einsatz von drei unabhängigen Assays ist methodisch als orthogonaler Ansatz zu werten: Stimmen alle drei überein, erhöht das die Verlässlichkeit des Ergebnisses erheblich.
Zelllinienexperimente und ELISA
Um zu prüfen, ob die Impfstoffe menschliche Zellen transfizieren und zu einer Spike-Protein-Produktion führen, wurden HEK293-Zellen eingesetzt – eine immortalisierte humane Zelllinie mit hoher Transfektionseffizienz.
Die Zellen wurden unter standardisierten Bedingungen kultiviert. Sie stammten aus zertifizierten Originalbeständen und wurden regelmäßig negativ auf Mykoplasmen getestet.
Für die Transfektionsexperimente wurden die Zellen in 12-Well-Platten ausgesät. Die Zelldichte wurde so gewählt, dass nach einer kurzen Anwachszeit etwa 80 % der Bodenfläche bedeckt waren. Jedes Well wurde mit 1/12 einer klinischen Dosis (entsprechend 25 µl) des jeweiligen Impfstoffs transfiziert. Ein unmittelbarer Mediumwechsel oder Waschschritt nach der Transfektion ist nicht dokumentiert.
Die Zellen sowie das Kulturmedium wurden zu den Zeitpunkten Tag 1, Tag 3, Tag 5 und Tag 7 nach Transfektion geerntet. Untransfektierte Zellen am Tag 7 dienten als Negativkontrolle.
Zur Proteinanalyse wurden die Zellen gewaschen, um Rückstände des Kulturmediums zu entfernen. Durch Zugabe eines Lysispuffers wurden die Zellen aufgeschlossen, sodass der Zellinhalt freigesetzt wurde. Der Gehalt an SARS-CoV-2-Spike-Protein wurde sowohl im Überstand des Zelllysats als auch im Zellkulturmedium mit einem hochsensitiven, kommerziellen ELISA-Kit quantitativ bestimmt.
Immunhistochemie
Um das Spike-Protein innerhalb der Zellen optisch nachzuweisen, wurde eine Immunhistochemie durchgeführt. Dazu wurden HEK293-Zellen in 24-Well-Platten ausgesät (60 % Zelldichte) und mit 12,5 µl einer klinischen Dosis pro Well transfiziert (200 µl Medium). Nach 4-stündiger Inkubation wurden die Zellen zweimal mit PBS gewaschen und mit 500 µl frischem Medium versetzt.
24 Stunden nach Transfektionsbeginn wurden die Zellen geerntet, fixiert, in 2%iger Agarose eingebettet und nach Entwässerung in Paraffinblöcke überführt. Von diesen Blöcken wurden hauchdünne Schnitte angefertigt.
Die Zellschnitte wurden mit einem spezifischen primären Antikörper gegen das SARS-CoV-2-Spike-Protein (S1-Untereinheit) behandelt, der nur an das Spike-Protein bindet. Ein sekundärer Antikörper, der an den ersten bindet und eine Farbreaktion auslöst, wurde hinzugefügt. Das Spike-Protein erschien unter dem Mikroskop grün, während der Zellkern zur besseren Orientierung mit Hämatoxylin blau gegengefärbt wurde.
PCR und Agarosegelelektrophorese
Mittels klassischer PCR wurde geprüft, ob die Impfstoff-DNA in menschliche Zellen eindringt. Für jeden Zielbereich des Plasmids wurde ein eigenes Primer-Paar eingesetzt – für Spike-Sequenzen, den ORI-Replikationsursprung, die Neomycin-Resistenzkassette und den SV40-Promoter/Enhancer. Jeder PCR-Ansatz enthält genau ein Primer-Paar und erzeugt damit ein definiertes Amplikon.
Die PCR-Produkte wurden mittels Agarosegelelektrophorese aufgetrennt und durch Vergleich mit einem DNA-Marker (GeneRuler) in ihrer Größe bestimmt. HEK293-Zellen wurden mit einer halben Impfstoffdosis (150 μl) behandelt; nach sechs Stunden wurde die DNA isoliert und analysiert. Untransfektierte Zellen dienten als Negativkontrolle.
Extrazelluläre Vesikel und Massenspektrometrie
Hintergrund: Zellen stoßen ständig Extrazelluläre Vesikel (kleine Membranbläschen) ab, die wie kleine Pakete Botenstoffe enthalten. Diese Vesikel können zu anderen Zellen reisen.
Um zu prüfen, ob transfizierte Zellen Impfstoffbestandteile über extrazelluläre Vesikel (EVs) abgeben, wurden EVs aus dem Zellkulturmedium isoliert und mittels Massenspektrometrie (TimsTOF-HT) analysiert. Die Identifizierung der Proteine erfolgte durch Abgleich mit einer Datenbank, die menschliche Proteine sowie Impfstoffvektor- und Spike-Sequenzen enthielt.
c) Zentrale Ergebnisse
Transfektion und Spike-Protein-Produktion
Alle vier untersuchten Chargen führten unter den gewählten Zellkulturbedingungen zu einer effizienten Aufnahme der mRNA in HEK293-Zellen und zur nachweisbaren Spike-Protein-Produktion.
Der Anteil Spike-positiver Zellen betrug 74,6 bis 90,5 %; untransfektierte Zellen zeigten kein Signal. Transfizierte Zellen zeigten zudem morphologische Veränderungen – intrazelluläre Vakuolen und Ablösung von der Wachstumsfläche – als Zeichen eines zytopathischen Effekts.
Die Spike-Protein-Produktion war bereits an Tag 1 nachweisbar, stieg bis Tag 5 an und blieb bis Tag 7 nachweisbar. Die Autoren interpretieren dies als Hinweis auf eine länger anhaltende Expression über den erwarteten Zeitraum hinaus.
Spike-Protein-Freisetzung ins Zellkulturmedium
Im Zellkulturmedium war Spike-Protein für die monovalenten Chargen nachweisbar; die bivalente Charge lag unterhalb der Nachweisgrenze.
Spike-Protein-Freisetzung über extrazelluläre Vesikel
Die Massenspektrometrie zeigte, dass das Spike-Protein hauptsächlich in extrazellulären Vesikeln nachweisbar war (14 spectral counts), während im löslichen Medium nur minimale Mengen vorlagen (2 spectral counts). Das Zelllysat wies die höchste intrazelluläre Konzentration auf (60 spectral counts).
RNA-Gehalt
Die gemessenen RNA-Werte entsprachen der Herstellerangabe von 30 µg pro klinischer Dosis.
DNA-Gehalt
Ohne LNP-Aufschluss wurden 1,6- bis 6,7-fach niedrigere DNA-Werte gemessen als nach Triton-X-100-Behandlung – ein weiterer Beleg dafür, dass die DNA überwiegend im Inneren der LNPs vorliegt.
Ohne RNase-Behandlung lagen die gemessenen DNA-Werte bei 1.326 bis 4.225 ng pro Dosis. Nach RNase-A-Behandlung sanken die Werte auf 32,71 bis 43,38 ng pro Dosis. Die Autoren führen die hohen Ausgangswerte auf ein Hintergrundsignal zurück, das durch strukturierte RNA-Bereiche entsteht: dsDNA-spezifische Farbstoffe binden bevorzugt an doppelsträngige DNA, können aber in Anwesenheit großer RNA-Mengen ein erhöhtes Signal erzeugen.
Nach Korrektur lagen die gemessenen DNA-Mengen über dem in regulatorischen Leitlinien genannten Grenzwert. Die Ergebnisse waren über mehrere Messungen reproduzierbar und stimmten zwischen den drei eingesetzten Assays gut überein.
PCR-Nachweis von Plasmidelementen
In allen vier Chargen wurden starke PCR-Signale für alle getesteten Plasmidelemente nachgewiesen – Spike-Sequenzen, ORI, Neomycin-Kassette und SV40-Promoter/Enhancer. Dieselben Elemente waren nach Transfektion auch in den HEK293-Zellen nachweisbar. Für den SV40-Promoter/Enhancer erzeugten die Primer zwei Amplifikate unterschiedlicher Größe (93 bp und 165 bp) – ein methodisch bekanntes Ergebnis, das auf die Struktur der Zielsequenz zurückzuführen ist.
d) Interpretation durch die Autoren
Die Autoren schlussfolgern, dass die untersuchten Chargen DNA-Rückstände enthalten, die regulatorische Grenzwerte überschreiten. Die DNA liegt überwiegend LNP-verpackt vor, wird von Zellen aufgenommen und ist dort nachweisbar. Die anhaltende Spike-Protein-Produktion über sieben Tage hinaus wirft die Frage auf, wie die langanhaltende Produktion zustande kommt.
Die Autoren heben insbesondere das Vorhandensein des SV40-Promoter/Enhancer hervor. Dieses Element sei für die mRNA-Produktion in E. coli nicht erforderlich. Sie diskutieren, dass solche regulatorischen Elemente in bestimmten experimentellen Kontexten die Transkriptionsaktivität beeinflussen können. Ein möglicher Einfluss auf Kernlokalisation oder genomische Integration wird von den Autoren als theoretische Überlegung angesprochen.
Die Autoren kritisieren die bestehenden regulatorischen Grenzwerte als nicht auf LNP-basierte RNA-Injektionen ausgerichtet und betonen, dass bisher keine wissenschaftliche Evidenz vorliegt, die eine Festlegung eines sicheren DNA-Grenzwerts für diese Produktklasse erlauben würde.
Die Autoren zeigen, dass das Spike-Protein über extrazelluläre Vesikel (Exosomen) freigesetzt wird. In welchem Umfang solche Vesikel im menschlichen Organismus zur Verteilung von Proteinen beitragen, ist Gegenstand aktueller Forschung und bislang nicht abschließend geklärt.
Die Autoren weisen auf ein grundlegendes Problem hin: Die Wahl der Analysemethode beeinflusst das Ergebnis maßgeblich, was bei Studienvergleichen unbedingt berücksichtigt werden sollte.
e) Methodische Einschränkungen
Zelllinie
HEK293-Zellen sind immortalisiert und weisen gegenüber primären Zellen (z. B. Muskel-, Immun- oder Nervenzellen) veränderte Eigenschaften auf, insbesondere eine hohe Transfektionseffizienz und veränderte Regulationsmechanismen.
Dosierung
Die verwendete Dosis ist nicht physiologisch. Die eingesetzten Mengen (z. B. 1/12 einer klinischen Dosis pro Well) führen zu einer direkten und lokal hohen Exposition der Zellen, die nicht den Verteilungsbedingungen im menschlichen Körper entspricht.
In-vitro-Kontext
Alle Experimente wurden unter Zellkulturbedingungen durchgeführt. Eine direkte Übertragung auf in-vivo-Verhältnisse – etwa hinsichtlich Verteilung, Abbau oder Immunantwort im menschlichen Körper – ist nicht ohne weiteres möglich.
Assay-Spezifität
Fluorometrische Methoden sind empfindlich gegenüber Störsignalen, was die Quantifizierung von DNA beeinflussen kann.
Mittelwert
Die ermittelte Rest-DNA lag bei 32,71–43,38 ng pro klinischer Dosis. Diese Werte basieren auf fluorometrischen Messungen mit drei unterschiedlichen Assays (PicoGreen, Qubit, AccuBlue). Da die einzelnen Assays teils deutlich voneinander abweichende Ergebnisse lieferten, stellt der angegebene Bereich das arithmetische Mittel über methodisch unterschiedliche Messprinzipien dar. Die Spannbreite der Einzelmessungen ist erheblich, sodass der Mittelwert primär als orientierender Schätzwert zu verstehen ist.
f) Einordnung in den Gesamtkontext
Die Studie geht über die reine Quantifizierung von DNA-Rückständen hinaus: Sie verbindet den analytischen Nachweis mit Zellexperimenten und fragt nach biologischen Konsequenzen. Sie kombiniert mehrere analytische und funktionelle Methoden (fluorometrische Assays, PCR, ELISA und Massenspektrometrie) und verfolgt damit einen vergleichsweise breit angelegten Ansatz.
Die Studie unterstreicht zudem, dass selbst innerhalb derselben methodischen Kategorie erhebliche Unterschiede zwischen einzelnen fluorometrische Assays auftreten können – ein Befund, der die Bedeutung methodischer Standardisierung für künftige Untersuchungen unterstreicht.
🧬 🧬 🧬
4.3.6. Wang et al. (2024)
| A rapid detection method of replication-competent plasmid DNA from COVID-19 mRNA vaccines for quality control. Autoren: Tayler J. Wang, Alex Kim, Kevin Kim Veröffentlicht: 29. Dezember 2024 (Journal of High School Science, peer-reviewed) Link zur Studie: https://jhss.scholasticahq.com/article/127890-a-rapid-detection-method-of-replication-competent-plasmid-dna-from-covid-19-mrna-vaccines-for-quality-control |
a) Kontext und Ziel der Studie
Die Arbeit von Wang et al. verfolgt eine andere Fragestellung als die bisher vorgestellten Studien. Im Vordergrund steht nicht die Gesamtmenge an DNA-Rückständen, sondern eine spezifischere Frage: Enthält die nachgewiesene DNA noch funktionsfähige, replikationsfähige Plasmidsequenzen – also Fragmente, die in Bakterien eingeschleust und dort vermehrt werden könnten?
Zur Beantwortung dieser Frage entwickelten die Autoren eine einfache und schnelle Nachweismethode. Die Überlegung dabei: Wenn bei der Impfstoffherstellung die DNA-Vorlage nicht vollständig entfernt wurde, könnten noch intakte oder fast intakte Plasmide übrig sein.
Die Arbeit entstand im Rahmen eines studentischen Freiwilligenprogramms der FDA und wurde auf dem FDA White Oak Campus durchgeführt, mit Unterstützung von FDA-Wissenschaftlern. Der Publikationsort – das Journal of High School Science – spiegelt den Ausbildungskontext wider, nicht den institutionellen Rahmen der Arbeit.
b) Materialien und Methoden
Impfstoffchargen
Die Studie verwendet verschiedene Probenquellen, die sich grundlegend unterscheiden und bei der Interpretation der Ergebnisse klar getrennt werden müssen.
Erstens einen selbst hergestellten Laborimpfstoff mit der XBB.1.5-Spike-Sequenz, produziert mit einem kommerziellen LNP-Kit. Zweitens Biosimilar-Referenzmaterialien von BEI Resources – NIH-gefördertes Referenzmaterial, das für Forschungszwecke hergestellt wurde und nicht identisch mit zugelassenen Handelschargen ist. Drittens zwei kommerzielle Pfizer-Chargen (Lot PAA194854, monovalent; Lot PAA184098, bivalent) – originalversiegelte, zugelassene Impfstoffchargen.
| Impfstoff | Herkunft | Beschreibung |
| In-house mRNA vaccine | Selbst hergestellt | Enthält XBB.1.5 Spike, LNP-formuliert |
| Moderna Biosimilar | BEI Resources | nicht das Original |
| Pfizer Biosimilar | BEI Resources | nicht das Original |
| COMIRNATY Original (NR-59604) | BEI Resources | Monovalent (Wuhan) |
| COMIRNATY Bivalent (NR-59605) | BEI Resources | Bivalent (Wuhan + Omicron BA.5) |
DNA-Extraktion
Die DNA wurde mit einem kommerziellen Plasmid-Miniprep-Kit extrahiert (Monarch Plasmid DNA Miniprep, New England Biolabs). Das Kit enthält RNase A im Neutralisierungspuffer, sodass RNA während der Extraktion weitgehend abgebaut wird. Die Elution erfolgte in 30 µl nukleasefreiem Wasser.
Quantifizierung
Die DNA-Konzentration wurde mit zwei Methoden bestimmt: NanoDrop-Spektrophotometrie und Qubit dsDNA HS Assay. Die Autoren weisen ausdrücklich darauf hin, dass NanoDrop nicht zwischen DNA und RNA unterscheidet und durch freie Nukleotide, Salze und organische Verbindungen beeinflusst werden kann. Qubit ist spezifischer, kann aber bei großen RNA-Mengen oder unsachgemäßer Probenvorbereitung ebenfalls überschätzen.
Transformationstest – das vorgestellte Nachweisverfahren
Zunächst wird die DNA aus dem Impfstoff extrahiert. Diese extrahierte DNA setzt sich aus verschiedenen Fragmenten zusammen – darunter ist möglicherweise auch der Replikationsursprung (ORI), der als Startpunkt der Plasmid-Vervielfältigung dient, sowie das Antibiotikaresistenzgen, das eine Selektion ermöglicht.
Die extrahierte DNA wird mit einer T4-DNA-Ligase behandelt. Dieses Enzym verknüpft die Enden von DNA-Fragmenten miteinander. Aus linearen Fragmenten können so wieder ringförmige Plasmide entstehen. Da die Ligase beliebige Enden miteinander verbinden kann, können auch Fragmente zusammengefügt werden, die im ursprünglichen Impfstoff getrennt vorlagen – es entstehen künstliche Artefakte.
Diese neu gebildeten Plasmide werden anschließend in spezielle E. coli-Bakterien eingeschleust (Transformation).
Die zugrundeliegende Überlegung:
- War der DNase-Verdau während der Impfstoffherstellung effizient, sind die DNA-Fragmente so klein, dass sie weder den vollständigen ORI noch das vollständige Resistenzgen enthalten. Dann kann auch durch Ligation kein funktionsfähiges Plasmid entstehen.
- War der DNase-Verdau unvollständig, sind noch größere Fragmente vorhanden, die ORI und Resistenzgen vollständig enthalten können. Diese können nach Ligation ein funktionsfähiges Plasmid bilden. Die Ligase kann nur vorhandene Enden verknüpfen; sie kann keine fehlenden Teile eines Gens aus kleinen Bruchstücken zusammensetzen.
Nur Bakterien, die ein funktionsfähiges Antibiotikaresistenzgen und einen funktionsfähigen ORI aufgenommen haben, überleben auf antibiotikumhaltigem Nährboden und bilden sichtbare Kolonien. Das Vorhandensein von Kolonien ist damit ein direkter biologischer Nachweis für das Vorliegen replikationsfähiger Plasmid-DNA im ursprünglichen Impfstoff.
Größenanalyse
Die Fragmentgrößen wurden mittels Agarosegelelektrophorese und Agilent 2100 Bioanalyzer bestimmt.
c) Zentrale Ergebnisse
Quantitative Befunde
Die DNA-Mengen pro Dosis lagen – gemessen mit Qubit – zwischen 40 und 110 ng, also oberhalb des WHO-Grenzwerts von 10 ng. NanoDrop lieferte deutlich höhere Werte (mehrere tausend ng), was die Autoren auf die bekannte Unspezifität dieser Methode zurückführen.
Laborimpfstoff und Biosimilar
Im selbst hergestellten Laborimpfstoff und im Pfizer-Biosimilar wurden replikationsfähige DNA-Fragmente gefunden – das Transformationsverfahren erzeugte Kolonien. Im Moderna-Biosimilar und im Haus-Standard wurden trotz höherer DNA-Konzentrationen keine Kolonien nachgewiesen, was zeigt, dass die Koloniebildung nicht allein von der DNA-Menge abhängt, sondern von der Intaktheit der relevanten Sequenzelemente.
Kommerzielle Pfizer-Chargen
In den beiden kommerziellen Pfizer-Chargen wurden keine replikationsfähigen DNA-Fragmente gefunden – der Transformationstest blieb ohne Kolonien. Die Größenanalyse zeigte, dass nahezu die gesamte nachgewiesene DNA kürzer als 35 bp war. Dieses Ergebnis ist nach Einschätzung der Autoren mit einer vollständig durchgeführten DNase-Behandlung vereinbar.
d) Interpretation durch die Autoren
Die Autoren werten das Fehlen replikationsfähiger DNA in den kommerziellen Chargen als Beleg für die Wirksamkeit des industriellen Herstellungsprozesses. Die vollständige Fragmentierung der Plasmid-DNA auf unter 35 bp macht eine biologische Aktivität der DNA-Reste nach Einschätzung der Autoren unwahrscheinlich – einschließlich der SV40-Promoter/Enhancer-Sequenz, deren Funktionsfähigkeit bei dieser Fragmentgröße praktisch ausgeschlossen wird.
Gleichzeitig bestätigen die Autoren, dass die DNA-Gesamtmenge den WHO-Grenzwert überschreitet, und empfehlen eine stringentere und transparentere Überwachung der DNA-Rückstände – auch mit dem Ziel, das öffentliche Vertrauen in mRNA-Impfstoffe zu stärken.
e) Methodische Einschränkungen
Stichprobengröße und -auswahl
Die Untersuchung umfasst lediglich zwei kommerzielle Chargen, was keine belastbaren Aussagen zur Chargenvariabilität zulässt. Die verwendeten Biosimilar-Proben sind nicht identisch mit zugelassenen Handelsprodukten; daher sind Ergebnisse aus diesen Proben nicht ohne Weiteres auf kommerzielle Impfstoffe übertragbar.
DNA-Extraktion
Die Extraktion erfolgte mit einem Plasmid-Miniprep-Kit, das primär für die Isolierung intakter Plasmide optimiert ist. Inwieweit stark fragmentierte DNA-Reste – insbesondere sehr kurze Fragmente – mit dieser Methode vollständig erfasst werden, ist nicht abschließend geklärt.
Nachweisgrenzen der Transformationsmethode
Die Methode detektiert ausschließlich DNA-Fragmente, die nach Ligation einen funktionsfähigen Replikationsursprung (ORI) und ein Antibiotikaresistenzgen enthalten. Sie prüft, ob ausreichend große Fragmente (>800 bp für das Resistenzgen, >600 bp für den ORI) vorhanden sind, die nach Ligation ein funktionsfähiges Plasmid bilden können. Fragmente ohne diese Elemente – selbst wenn sie andere relevante Sequenzen wie den SV40-Enhancer (ca. 72–165 bp) enthalten würden – werden nicht erfasst.
Zudem kann die Ligation künstlich Plasmide erzeugen, die im ursprünglichen Impfstoff nicht vorhanden waren.
Begrenzung der funktionellen Aussagekraft
Der verwendete Transformationsassay untersucht ausschließlich, ob DNA-Fragmente nach Aufnahme in Bakterien ein replizierfähiges Plasmid bilden können. Andere mögliche biologische Eigenschaften von DNA-Fragmenten werden durch dieses System nicht erfasst. Insbesondere erlaubt die Methode keine Aussagen darüber, ob kleinere oder nicht replizierfähige Fragmente in anderen biologischen Kontexten funktionelle Effekte besitzen könnten. Die Studie bewertet somit spezifisch die bakterielle Replikationsfähigkeit bestimmter Plasmidbestandteile – nicht die allgemeine biologische Relevanz sämtlicher nachweisbarer DNA-Fragmente.
f) Einordnung in den Gesamtkontext
Die Studie von Wang et al. untersucht, ob in den Impfstoffproben noch biologisch aktive, replikationsfähige Plasmid-DNA nachweisbar ist.
Damit erweitert die Arbeit die bisherige Diskussion um eine zusätzliche Ebene. Während PCR-, Fluorometrie- und Sequenzierungsverfahren vor allem Aussagen über Menge, Sequenzidentität und Fragmentlänge erlauben, prüft der verwendete Transformationsassay gezielt die funktionelle Integrität der DNA im bakteriellen System.
Gleichzeitig bleibt die Aussagekraft des Ansatzes auf die im Experiment getestete Funktion beschränkt: Der Assay erfasst ausschließlich DNA, die in Bakterien transformierbar und replizierbar ist. Andere biologische Eigenschaften oder potenzielle Wirkmechanismen von DNA-Fragmenten werden damit nicht untersucht.
Insgesamt liefert die Arbeit einen methodisch interessanten Beitrag zur Diskussion, indem sie die Frage nach der funktionellen Relevanz von DNA-Rückständen in den Mittelpunkt stellt und damit eine wichtige Ergänzung zu rein quantitativen Analysen darstellt.
Die Studie verdeutlicht damit, dass die Bewertung von DNA-Rückständen nicht allein auf deren Menge basieren sollte, sondern wesentlich von deren strukturellen und funktionellen Eigenschaften abhängt.
🧬 🧬 🧬
4.3.7. Fleming et al. (2025)
| Quantitative Analysis of Nucleic Acid Content in Spikevax (Moderna) and BNT162b2 (Pfizer) COVID-19 Vaccine Lots Autoren: Richard M Fleming PhD, MD, JD; Peter Kotlár MD; Sona Pekova MD, PhD Veröffentlicht: 13. Mai 2025 (Herald Open Access) Link zur Studie: https://www.heraldopenaccess.us/openaccess/quantitative-analysis-of-nucleic-acid-content-in-spikevax-moderna-and-bnt162b2-pfizer-covid-19-vaccine-lots |
a) Kontext und Ziel der Studie
Die Studie entstand im Kontext einer behördlichen Anfrage in der Slowakei. Ziel der Untersuchung war die Charakterisierung der Nukleinsäurezusammensetzung von mRNA-COVID-19-Impfstoffen im Hinblick auf:
- Identifizierung und Quantifizierung der enthaltenen Nukleinsäuren
- Prüfung der Chargen-Homogenität (zwischen und innerhalb von Lots)
- Nachweis undeklarierter genetischer Materialien
- Bewertung der Stabilität abgelaufener Chargen
Die Ergebnisse sollten Erkenntnisse über die Zusammensetzung, Stabilität und Herstellungspraxis von Impfstoffen liefern.
b) Materialien und Methoden
Impfstoffchargen
Die molekulare Analyse wurde an 24 Chargen – Moderna (17) und Pfizer (7) – durchgeführt:
Spikevax (Moderna): MV1013A, 200023A, 200156A, 223049, 200090A, 200106A, 200100A, 3005885, 3005836, 000090A, 000058A, 3005241, 3005697, 3006272, MV1018A, 400012A, 400011A und
BNT162b2 (Pfizer): FP9632, 1F1051A, 1LO84A, 1F1047A, 1F1059A, 1F1055A, PCB0020.
Pro Charge wurden fünf originalverpackte, ungeöffnete Fläschchen analysiert, die bei -80 °C gelagert und unter dokumentierter Temperaturkontrolle transportiert wurden. Die Provenienzdokumentation ist damit vergleichsweise gut belegt.
Aufschluss der Lipid-Nanopartikel (LNPs)
Die LNPs wurden durch eine Kombination aus:
▪️chaotropen Salzen (Isolierungskit QIAamp DNA Mini Kit (Qiagen, DE)),
▪️Hitze (60 °C) und
▪️enzymatischem Verdau (Proteinase K)
vollständig aufgelöst.
Dadurch wurden sowohl die mRNA als auch eventuelle DNA-Rückstände freigesetzt.
Reverse Transkription und Multiplex qPCR
Die mRNA wurde durch Reverse Transkription in cDNA umgeschrieben. Die anschließende Quantifizierung erfolgte mittels Multiplex Real-Time PCR (Rotor-Gene Q, Qiagen) mit vier simultanen Farbkanälen:
- FAM (Grün): für das S-Protein (cDNA)
- HEX (Gelb): für die Vektor-Ori-DNA
- Cy5 (Rot): für die E. coli-DNA
- ROX: als passive Referenz.
Alle drei Zielsequenzen wurden damit in einem einzigen Reaktionsansatz gleichzeitig erfasst, wobei mögliche Wechselwirkungen zwischen den Targets (z. B. Konkurrenz um Reagenzien) die Quantifizierung beeinflussen können.
Quantifizierung mittels qPCR
Für die Multiplex quantitative Real-Time PCR verwendeten die Autoren Primer für drei verschiedene Zielsequenzen:
| Ziel | Quelle |
| mRNA für S-Protein (Spikevax & BNT162b2) | selbst entworfen |
| Expressionsvektor-DNA (Ori) | aus anderen Studien übernommen |
| E. coli-genomische DNA (ITS) | aus kommerziellem Kit (ISO 13485-validiert) |
Alle verwendeten Primer und fluoreszenzmarkierten Hybridisierungssonden wurden von Eurofins Genomics, DE, kundenspezifisch synthetisiert.
Kalibrationsstrategie
Da die mRNA in den Impfstoffen mit N1-Methylpseudouridin modifiziert ist und das genaue Ausmaß dieser Modifikation nicht bekannt ist, konnten keine synthetischen Standards für eine klassische zielspezifische Kalibrationskurve hergestellt werden. Die Autoren validierten stattdessen die PCR-Effizienz durch serielle Verdünnungsreihen der Impfstoffproben selbst. Für die absolute Quantifizierung verwendeten sie eine universelle Kalibrationskurve, die aus Verdünnungsreihen von 50 verschiedenen Mikroorganismen gemittelt wurde.
Verifikation durch Sanger-Sequenzierung
Die PCR-Produkte aller 24 Chargen wurden mittels Sanger-Sequenzierung verifiziert und mit Datenbankeinträgen verglichen.
c) Zentrale Ergebnisse
mRNA-Nachweis und Chargenvariabilität
Die PCR-Analyse bestätigte das Vorhandensein der deklarierten mRNA-Sequenzen für das Spike-Protein in beiden Impfstoffen.
Die Analyse der mRNA für das S-Protein zeigte eine erhebliche Variabilität zwischen verschiedenen Chargen – bei beiden Impfstoffen.
Die absoluten mRNA-Mengen lagen im Bereich von 109 bis 1010 Kopien/ml (gemessen als cDNA). Einzelne Chargen zeigten jedoch eine 10-fach niedrigere mRNA-Menge – bei beiden Impfstoffen.
Nachweis undeklarierter DNA-Sequenzen als Zufallsbefund
Während der Validierung der cDNA-Verdünnungsreihen zeigten sich unerwartet starke Signale in den DNA-Kanälen: im Cy5-Kanal (für Ori-DNA) und teilweise im HEX-Kanal (für E. coli-DNA). Da die Proben neben cDNA auch DNA-Rückstände enthielten, amplifizierten die Ori- und E. coli-Primer diese unbeabsichtigt mit. Die entsprechenden PCR-Produkte wurden aufgereinigt und mittels Sanger-Sequenzierung identifiziert: Sie zeigten 100%ige Übereinstimmung mit dem Expressionsvektor (Plasmid-Ori und Spike-Kassette) sowie mit der E. coli-ITS-Region.
Quantitative DNA-Befunde
Ein zentraler Befund der Studie ist der durchgängig hohe Gehalt an DNA in allen getesteten Chargen – sowohl von Moderna als auch von Pfizer.
| Impfstoff | mRNA (cDNA) (Kopien/ml) | DNA-Menge (Kopien/ml) |
| Spikevax (Moderna) | 109 – 1010 | 107 – 109 |
| BNT162b2 (Pfizer) | 1010 – 1011 | 108 – 109 |
Hinweis auf nahezu vollständige DNA-Konstrukte
Die Autoren beobachten, dass der Ori-Primer (5′-Ende des Konstrukts) und der Spike-Primer (3′-Ende, Basen 3184–3417) gleichzeitig ein Signal liefern. Da beide Primerbindestellen mehrere tausend Basenpaare voneinander entfernt sind, deutet dies darauf hin, dass zumindest längere DNA-Fragmente vorhanden sind, die beide Zielbereiche umfassen könnten.
Auffällige Chargenheterogenität bei Moderna
Bei drei Moderna-Chargen ist das Verhältnis von Spike-DNA zu Vektor-DNA umgekehrt und um eine Größenordnung verschoben – in eine Richtung, die nicht zu einem einzigen Plasmid passt. Die Autoren spekulieren, dass diese Chargen ein zweites DNA-Konstrukt enthalten könnten – möglicherweise eine Omicron-Variante im selben Vektor.
E. coli-DNA
In zwei Pfizer-Chargen wurde E. coli-genomische DNA im Bereich einzelner Kopien pro Milliliter nachgewiesen – knapp oberhalb der Nachweisgrenze.
SV40
Es wurden keine Hinweise auf SV40 festgestellt.
d) Interpretation durch die Autoren
Anwesenheit der DNA
Die Autoren argumentieren, dass die nachgewiesenen DNA-Mengen – vergleichbar mit der deklarierten mRNA-Menge in der Größenordnung von 107 bis 109 Kopien/ml – zu hoch sind, um als zufällige Kontamination erklärt zu werden. Die Autoren interpretieren die DNA als regulären, wenn auch nicht deklarierten Bestandteil der Impfstoffe.
Chargen-Variabilität und mRNA-Identität
Die Chargenvariabilität der mRNA wird als mögliches Problem für die Dosierungskonsistenz bewertet. Die Verwendung der veralteten Wuhan-Spike-Sequenz wird als Einschränkung der Schutzwirkung gegen aktuell zirkulierende Varianten kritisiert.
Aufgrund der Analysebefunde empfehlen die Autoren:
- verbesserte Reinigungsprotokolle,
- strengere Chargenprüfung,
- genomische Aktualisierungen und
- eine verstärkte behördliche Aufsicht.
e) Methodische Einschränkungen
Universelle Kalibrationskurve
Die absolute Quantifizierung basiert nicht auf einer zielspezifischen Standardkurve, sondern auf einer universellen Kurve, die aus 50 verschiedenen Mikroorganismen gemittelt wurde. Diese Methode setzt eine vergleichbare PCR-Effizienz für alle Zielsequenzen voraus. Diese Annahme ist insbesondere bei unterschiedlichen Zielsequenzen (mRNA, Plasmid-DNA, bakterielle DNA) und komplexen Probenmatrices nicht selbstverständlich. Die absoluten Kopienzahlen sind daher mit Vorsicht zu interpretieren; die qualitativen Befunde – also das Vorhandensein der nachgewiesenen Sequenzen – sind davon weniger betroffen.
Keine Messung vor LNP-Aufschluss
Im Unterschied zu anderen Studien wurde keine Messung vor dem Aufschluss der LNPs durchgeführt. Damit fehlt der direkte Vergleich, der Aussagen über den Anteil LNP-verpackter DNA ermöglichen würde.
SV40-Aussage nicht aussagekräftig
Die Autoren berichten, kein SV40 nachgewiesen zu haben. Das verwendete Primer-Set enthielt jedoch keine SV40-spezifischen Sequenzen. Die Aussage ist damit zwar korrekt, aber nicht informativ – ein Nachweis oder Ausschluss von SV40 war mit diesem Ansatz methodisch nicht möglich.
f) Einordnung in den Gesamtkontext
Die Studie von Fleming et al. erweitert die bisherige Literatur vor allem durch ihren vergleichenden Ansatz über eine relativ große Anzahl von Chargen.
Methodisch kombiniert sie Multiplex-qPCR mit Sanger-Sequenzierung zur Verifikation der amplifizierten Produkte. Der Nachweis von DNA-Sequenzen, einschließlich Plasmid- und bakterieller DNA steht im Einklang mit vorherigen Studien.
Besondere Aufmerksamkeit richtet die Arbeit auf Unterschiede zwischen Chargen sowie auf quantitative Vergleiche der gemessenen Nukleinsäureanteile. Die beobachtete Variabilität zwischen einzelnen Lots ergänzt damit die bestehende Diskussion über mögliche Unterschiede in Zusammensetzung, Fragmentierung und analytischer Erfassbarkeit der DNA-Rückstände.
🧬 🧬 🧬
4.3.8. Speicher et al. (2025)
| Quantification of residual plasmid DNA and SV40 promoter-enhancer sequences in Pfizer/BioNTech and Moderna modRNA COVID-19 vaccines from Ontario, Canada Autoren: David Speicher, Jessica Rose, Kevin McKernan Veröffentlicht: 25. September 2025 (PubMed/Herald Open Access) Link zur Studie: https://pubmed.ncbi.nlm.nih.gov/40913499/ |
a) Kontext und Ziel der Studie
Die Studie von Speicher, Rose und McKernan ist eine methodisch erweiterte Folgearbeit zur früheren Untersuchung derselben Autorengruppe (Studie 2, 2023). Sie baut inhaltlich auf den Befunden von McKernan et al. und Kämmerer et al. auf und verfolgt das Ziel, die DNA-Rückstände in mRNA-Impfstoffen mit zwei unabhängigen Methoden – qPCR und Qubit-Fluorometrie mit RNase-Korrektur – an einer größeren Stichprobe zu quantifizieren.
Ergänzend werden Fragmentlängen mittels Oxford-Nanopore-Sequenzierung bestimmt sowie eine explorative Korrelation mit VAERS-Nebenwirkungsdaten durchgeführt.
b) Materialien und Methoden
Impfstoffchargen
Für die Untersuchung wurden insgesamt 32 Fläschchen aus 16 Chargen mRNA-basierter COVID-19-Impfstoffe verwendet: BNT162b2 (Pfizer/BioNTech; 10 Vials aus 6 Chargen) und mRNA-1273 (Moderna; 22 Vials aus 10 Chargen). Die Proben wurden aus verschiedenen Apotheken in Ontario, Kanada, bezogen.
Die Fläschchen waren originalversiegelt und wiesen unbeschädigte Flip-off-Kappen mit Chargennummern und Verfallsdaten auf. Lagerung und Transport erfolgten unter Einhaltung der Kühlkette (2–8 °C). Die Proben wurden in spezialisierten Kühleinheiten gelagert und in isolierten Behältern mit Kühlakkus transportiert; bevor sie innerhalb von 5 Stunden nach Erhalt in den Kühlschrank des Testlabors gestellt wurden.
Ein Teil der Proben bestand aus Restmengen kürzlich verwendeter Fläschchen (innerhalb von 30 Minuten nach Anwendung), die ebenfalls gekühlt transportiert und innerhalb von 12 Stunden im Labor eingelagert wurden.
Eine Moderna-Probe wies kein aufgedrucktes Verfallsdatum auf, verfügte jedoch über einen QR-Code zur Identifikation durch Apothekenpersonal.
Aufschluss der Lipid-Nanopartikel (LNPs)
Zum Aufschluss der Lipid-Nanopartikel (LNPs) wurden die Proben 8 Minuten bei 95 °C erhitzt und anschließend für 5 Minuten auf 4 °C abgekühlt. Dieser thermische Schritt dient der Freisetzung der enthaltenen Nukleinsäuren.
Quantifizierung mittels qPCR
Die DNA-Konzentrationen wurden mittels qPCR über den Cq-Wert bestimmt. Die Primer entsprechen weitgehend den früheren Assays der Autorengruppe: Spike-Sonde, Ori-Sonde sowie ein SV40-Enhancer-Assay für Pfizer-Chargen.
Die Quantifizierung erfolgte anhand einer Kalibrationskurve mit synthetischer DNA und 10-fachen Verdünnungsreihen (QuantStudio 3). Aus der gemessenen Kopienzahl der Zielsequenzen wurde unter Berücksichtigung der Gesamtplasmidlänge (Pfizer: 7.824 bp; Moderna: 6.777 bp) die Gesamt-DNA-Menge pro Dosis berechnet.
Jeweils 1 µL der behandelten Impfstoffprobe wurde ohne vorherige DNA-Extraktion direkt in die qPCR eingesetzt. Eine mögliche PCR-Inhibition durch Impfstoffbestandteile wurde durch Verdünnungsreihen ausgewählter Proben überprüft.
Quantifizierung mittels Qubit-Fluorometrie
Die Proben wurden mit dem DNA-spezifischen Farbstoff AccuGreen mehrfach gemessen: ohne Vorbehandlung, nach LNP-Aufschluss durch Erhitzen sowie nach einer RNase-A-Behandlung. Letztere korrigiert den RNA-bedingten Signalanteil (Crosstalk des Farbstoffs – der auch an RNA bindet). Die Autoren verfolgten den Abfall der Fluoreszenz über 10 Minuten in einer Zeitreihe. Dadurch konnten sie den RNA-bedingten Signalanteil ermitteln und die reine DNA-Konzentration berechnen.
Größenprofil der Rest-DNA mittels ONT-Sequenzierung
Zur Bestimmung der Größenverteilung der DNA-Fragmente wurde eine zuvor sequenzierte Pfizer-Charge (FL8095) als Standard verwendet. Die Sequenzierung erfolgte auf einer Oxford Nanopore Flongle mit dem Ligation Sequencing Kit (SQK-LSK114). Die erhaltenen Reads wurden mit einer Referenz-Plasmid-Sequenz abgeglichen.
Der verwendete Bibliotheksaufbau (SPRI-basierte Aufreinigung) kann kurze Fragmente (<100-200 bp) verlieren, sodass die Längenverteilung zu längeren Fragmenten hin verzerrt ist.
Beurteilung der Nukleaseempfindlichkeit
Zur Prüfung, ob die DNA im Inneren der LNPs vorliegt, wurde DNase I-XT direkt zum unveränderten Impfstoff gegeben. Nach 30-minütiger Inkubation wurde das Enzym inaktiviert, die DNA aufgereinigt und mittels qPCR quantifiziert. Eine nackte DNA-Kontrolle diente als Referenz.
Explorative VAERS-Korrelation mit den gemessenen DNA-Mengen
Für jede Charge wurden VAERS-Meldungen aus Kanada erfasst und die Anzahl schwerwiegender unerwünschter Ereignisse (SAEs) mit den gemessenen DNA-Gehalten korreliert. Ziel war es, eine mögliche Korrelation zwischen den gemessenen DNA-Gehalten (Spike, Ori, SV40) und der Häufigkeit von Nebenwirkungen zu untersuchen. Zur Berechnung des Zusammenhangs wurde der Pearson-Korrelationskoeffizient verwendet.
c) Zentrale Ergebnisse
qPCR-Validierung
Die qPCR-Assays für Spike und Ori zeigten exzellente R2-Werte (0,998–0,999) und Effizienzen (94,7–99,8 %). Der SV40-Assay hatte eine etwas geringere Güte (R2 = 0,906, Effizienz 93,6 %). Negativkontrollen waren unauffällig.
LNP-Inhibition
Unverdünnte Proben zeigten eine PCR-Inhibition durch die LNPs. Nach 1:10 Verdünnung war die Inhibition aufgehoben, daher wurden für die Quantifizierung verdünnte Proben verwendet.
Quantifizierung mittels qPCR
Alle Pfizer-Chargen waren positiv für Spike, Ori und SV40. Die ähnlichen Cq-Werte aller drei Targets deuten auf gemeinsame, intakte DNA-Moleküle hin. Bei Moderna waren Spike und Ori nachweisbar, kein SV40. Die Ori-Signale traten bei Moderna etwa 3 Zyklen später auf als Spike, was auf einen geringeren Anteil oder eine Fragmentierung der Ori-Region hindeutet.
Die Autoren rechneten die Cq-Werte über die Standardkurve in absolute DNA-Mengen um:
| Impfstoff | Spike (ng/Dosis) | Ori (ng/Dosis) | SV40 (ng/Dosis) |
| Pfizer | 0,22 – 2,43 | 0,28 – 7,28 | 0,25 – 23,72 |
| Moderna | 0,25 – 0,78 | 0,01 – 0,34 | nicht nachweisbar |
Pfizer wies eine deutlich höhere Menge an Rest-DNA auf als Moderna.
Zwei Pfizer-Chargen (FM7380 und FN7934, monovalent, PBS-Formulierung) überschritten mit SV40-Gehalten von bis zu 23,72 ng/Dosis den FDA-Richtwert von 10 ng/Dosis. Alle anderen Chargen lagen unter diesem Wert.
Quantifizierung mittels Qubit-Fluorometrie
Die Autoren testeten die Proben mehrfach zu verschiedenen Zeiten. Die durchschnittliche Varianz (durchschnittliche Abweichung vom Durchschnitt) betrug:
| Messphase | Pfizer | Spanne |
| vor dem Erhitzen | 317 ± 278 ng/Dosis | 43 bis 727 ng/Dosis |
| nach dem Erhitzen | 1.145 ± 533 ng/Dosis | 519 bis 1.949 ng/Dosis |
| nach RNase A | 297 ± 217 ng/Dosis | 100 bis 765 ng/Dosis |
| Messphase | Moderna | Spanne |
| vor dem Erhitzen | 456 ± 121 ng/Dosis | 199 bis 675 ng/Dosis |
| nach dem Erhitzen | 1.554 ± 1.023 ng/Dosis | 176 bis 3.169 ng/Dosis |
| nach RNase A | 1.065 ± 624 ng/Dosis | 113 bis 2.365 ng/Dosis |
Nach LNP-Aufbruch durch Erhitzen stiegen die Werte deutlich an. Die anschließende RNase A-Behandlung reduzierte die Werte (Crosstalk-Korrektur).
Moderna enthielt durchgängig höhere DNA-Mengen als Pfizer.
Der Vergleich von Qubit und qPCR zeigte bei Pfizer eine positive Korrelation, bei Moderna nicht – was auf DNA-Fragmente hindeutet, die keine intakten Primerbindestellen mehr enthalten und daher in der qPCR nicht erfasst werden.
Fragmentlängenanalyse
Der längste detektierte Read umfasste 3,5 kb und deckte den Großteil des Plasmid-Backbones ab. Das zeigt, dass neben kurzen Fragmenten auch größere DNA-Fragmente im Kilobasenbereich vorhanden sind.
DNA liegt im Inneren der LNPs
Die DNase-Behandlung führte zu keinem signifikanten DNA-Verlust in der Impfstoffprobe (≤1 Cq Offset), während die nackte Kontroll-DNA vollständig abgebaut wurde. Dies bestätigt, dass die DNA überwiegend LNP-verpackt und damit vor enzymatischem Abbau geschützt ist.
Explorative VAERS-Korrelation mit den gemessenen DNA-Mengen
Für die untersuchten Chargen wurden Meldedaten aus dem US-amerikanischen Spontanmeldesystem VAERS herangezogen. Für alle untersuchten Chargen außer drei Moderna-Chargen lagen VAERS-Meldungen vor.
Die Autoren führten eine explorative Korrelation zwischen gemessenen DNA-Gehalten und VAERS-Meldedaten durch. Dabei zeigten einzelne Chargen mit höheren DNA-Werten (insbesondere Ori-DNA) auch höhere Anzahlen gemeldeter schwerwiegender Ereignisse (SAEs).
d) Interpretation durch die Autoren
Die Autoren schlussfolgern, dass alle untersuchten Chargen DNA-Rückstände enthalten, die LNP-verpackt und damit vor Abbau geschützt sind. Die gemessenen Gesamtmengen überschreiten die bestehenden Richtwerte. Das SV40-Promoter/Enhancer-Element in Pfizer-Chargen wird als besonders relevant hervorgehoben, da es nukleares Targeting begünstigen und theoretisch die Integrationswahrscheinlichkeit erhöhen könnte.
Die Autoren weisen auf mehrere methodische Probleme hin:
| Methode | Problem |
| qPCR | Kann Moleküle kleiner als die Amplikonlänge (105–114 bp) nicht quantifizieren → Unterschätzung der Gesamt-DNA |
| Qubit/Fluorometrie ohne RNase | Überschätzung durch Crosstalk mit RNA (daher RNase-Behandlung notwendig) |
| Ethanol-Fällung | Verlust kurzer Fragmente (<100 bp) |
| ONT (Nanopore) | Bibliotheksvorbereitung selektiert gegen Fragmente <150 bp – Verzerrung zu längeren Fragmenten |
Daher schlussfolgern die Autoren: Die Wahl der Methode beeinflusst das Ergebnis stark – daher ist methodische Klarheit bei Grenzwertfestlegungen essenziell.
Die Autoren formulieren mehrere Forderungen:
| Forderung | Begründung |
| Neubewertung der DNA-Grenzwerte | Die aktuellen regulatorischen Grenzwerte wurden für nackte DNA in herkömmlichen Impfstoffen entwickelt. Sie sollten unter Berücksichtigung der LNP-Verkapselung, der kumulativen Dosierung und der funktionellen Sequenzen neu bewertet werden. |
| Methoden-Standardisierung | Da qPCR die Gesamt-DNA unterschätzt, sollte zusätzlich Fluorometrie mit RNase-Korrektur eingesetzt werden, um die Rest-DNA sowie das RNA/DNA-Verhältnis korrekt zu quantifizieren. |
| Unabhängige Replikation | Weitere Labore sollten die Ergebnisse bestätigen. |
| Langzeit-Monitoring | Spätfolgen (z. B. Integration, Krebs) werden von VAERS nicht erfasst. |
| Berücksichtigung funktioneller Sequenzen | Nicht nur die Menge der DNA ist entscheidend, sondern auch ihre funktionellen Eigenschaften – insbesondere das Vorhandensein von SV40-Elementen und Promotoren. |
Die Variabilität der Rest-DNA zwischen und innerhalb einzelner Chargen interpretieren die Autoren als möglichen Hinweis auf einen inkonsistenten Herstellungsprozess.
e) Methodische Einschränkungen
Die qPCR kann Fragmente unterhalb der Amplikon-Länge (105–152 bp) nicht erfassen und unterschätzt damit die Gesamt-DNA systematisch.
Die ONT-Daten basieren auf einer einzigen Charge und sind durch die Aufreinigungsmethode zu längeren Fragmenten hin verzerrt.
Die explorative VAERS-Korrelation mit den gemessenen DNA-Mengen ist aufgrund der bekannten Limitationen von Spontanmeldesystemen – insbesondere fehlender Expositionsdaten und möglicher Verzerrungen in der Meldestruktur – nur eingeschränkt interpretierbar. Die Autoren betonen zudem weitere Einschränkungen: begrenzte Chargengröße, keine Kenntnis der verabreichten Dosen und eine Unterfassung möglicher Langzeiteffekte.
Die Proben umfassen sowohl abgelaufene Fläschchen als auch angebrochene Restmengen, was die Vergleichbarkeit innerhalb der Stichprobe einschränkt.
f) Einordnung in den Gesamtkontext
Die Studie stellt eine methodisch erweiterte Fortführung früherer Arbeiten derselben Autorengruppe dar und kombiniert mehrere analytische Ansätze – qPCR, fluorometrische Quantifizierung mit RNase-Korrektur, Nanopore-Sequenzierung sowie funktionelle Tests zur Nukleaseempfindlichkeit. Besonders hervorzuheben ist der direkte Vergleich unterschiedlicher Quantifizierungsmethoden, der die methodische Abhängigkeit der gemessenen DNA-Mengen deutlich macht.
Die qualitativen Befunde – insbesondere der Nachweis von Plasmid-DNA in beiden Impfstoffen sowie von SV40-Sequenzen in Pfizer-Chargen – stehen im Einklang mit anderen Studien (u. a. McKernan et al., Kämmerer et al.). Auch der Hinweis auf eine Verkapselung der DNA durch Lipid-Nanopartikel wird durch mehrere Arbeiten gestützt.
Gleichzeitig zeigen sich deutliche Unterschiede in der quantitativen Einordnung der DNA-Mengen zwischen den verwendeten Methoden (qPCR vs. Fluorometrie). Diese Divergenzen unterstreichen die Bedeutung methodischer Standardisierung und erschweren direkte Vergleiche zwischen Studien.
Insgesamt erweitert die Studie die bestehende Datenlage durch einen breiteren methodischen Ansatz und eine größere Stichprobe. Sie liefert vor allem Hinweise auf methodische Fragestellungen und Variabilität der Messergebnisse, die in zukünftigen Untersuchungen weiter systematisch geklärt werden müssen.
🧬 🧬 🧬
4.3.9. Achs et al. (2025)
| Systematic analysis of COVID-19 mRNA vaccines using four orthogonal approaches demonstrates no excessive DNA impurities Autoren: Adam Achs, Tatiana Sedlackova, Lukas Predajna, Jaroslav Budis, Maria Bartosova, Vladimir Zelnik, Diana Rusnakova, Martina Melichercikova, Marta Miklosova, Veronika Gencurova, Barbora Cernakova, Tomas Szemes, Boris Klempa, Juraj Kopacek, Silvia Pastorekova Veröffentlicht: 13. Dezember 2025 (npj Vaccines, peer-reviewed; PMCID verfügbar) Link zur Studie: https://pmc.ncbi.nlm.nih.gov/articles/PMC12715226/ |
a) Kontext und Ziel der Studie
Die Studie von Achs et al. verfolgt einen systematischen und methodisch breit angelegten Ansatz zur Untersuchung möglicher DNA-Rückstände in mRNA-basierten COVID-19-Impfstoffen. Sie entstand vor dem Hintergrund früherer Arbeiten, in denen teilweise erhöhte DNA-Mengen sowie spezifische Sequenzelemente berichtet wurden.
Ziel der Arbeit ist es, diese Fragestellung mit mehreren unabhängigen, sich ergänzenden Analysemethoden („orthogonale Ansätze“) erneut zu prüfen und die Robustheit der Ergebnisse gegenüber methodischen Einflüssen zu bewerten. Dabei steht insbesondere die Frage im Mittelpunkt, ob die in anderen Studien beschriebenen DNA-Rückstände reproduzierbar sind oder durch methodische Artefakte erklärt werden können.
Ein weiterer Fokus liegt auf der Bewertung der eingesetzten Nachweismethoden selbst: Die Autoren untersuchen, inwieweit unterschiedliche analytische Verfahren – etwa Sequenzierung, qPCR und fluorometrische Messungen – zu abweichenden Ergebnissen führen können und welche methodischen Limitationen dabei zu berücksichtigen sind.
Die Studie zielt damit nicht nur auf die Quantifizierung möglicher DNA-Rückstände ab, sondern auch auf eine methodische Einordnung der bestehenden Literatur und die Frage, wie zuverlässig verschiedene Nachweisansätze in diesem Kontext sind.
b) Materialien und Methoden
Impfstoffchargen
Untersucht wurden 15 Chargen – 9 Comirnaty (Pfizer) und 6 Spikevax (Moderna) – aus staatlich verwalteten Impfstoffbeständen der Slowakei, bereitgestellt unter Aufsicht des Staatlichen Instituts für Arzneimittelkontrolle. Vier Comirnaty-Chargen waren zum Zeitpunkt der Analyse noch innerhalb ihres Verfallsdatums. Die übrigen Chargen hatten ihr Verfallsdatum bereits überschritten. Die Fläschchen wurden entsprechend den Herstellerangaben gelagert und transportiert.
Ein Teil der Chargen – konkret jene, die auch in Fleming et al. (Studie 7) untersucht worden waren – wurde bewusst einbezogen, um einen direkten Vergleich zu ermöglichen.
Vier orthogonale Analysemethoden
Das methodische Kernmerkmal der Studie ist der Einsatz von vier unabhängigen, sich gegenseitig ergänzenden Verfahren an denselben Proben. Jede Methode hat ihre spezifischen Stärken und Limitationen – die Kombination soll robustere Schlussfolgerungen ermöglichen als jede Methode für sich.
Methode 1: qPCR
Die LNPs wurden mit Triton X-100 aufgeschlossen; die behandelte Probe wurde ohne vorherige DNA-Extraktion direkt in die qPCR eingesetzt. Damit entfällt ein Aufreinigungsschritt, der zu DNA-Verlusten führen könnte. Die Autoren prüften explizit mögliche PCR-Inhibitionen durch Impfstoffbestandteile und berichteten unter den verwendeten Bedingungen keine relevante Hemmung der Amplifikation.
Es wurden acht Primer-Paare eingesetzt, die drei Regionen des Produktionsplasmids abdecken: die Spike-kodierende Sequenz (SPIKE), den Replikationsursprung (ORI) und das Kanamycin-Resistenzgen (KAN). Dabei variierten die Amplikon-Größen erheblich: von 63 bp (KAN 1C) bis 233 bp (SPIKE 2). Zwei der Primer-Paare – SPIKE 2 und ORI 2 – entsprechen denjenigen, die in Fleming et al. verwendet wurden, um einen direkten Vergleich zu ermöglichen.
Die Quantifizierung erfolgte über Kalibrationskurven mit synthetischen DNA-Standards (gBlocks), die den jeweiligen Zielsequenzen entsprechen. Die gemessenen Kopienzahlen wurden anhand der Molekülmasse des jeweiligen Amplikons in Nanogramm pro Dosis umgerechnet.
Methode 2: Fluorometrie
Für die fluorometrische DNA-Quantifizierung wurde eine DNA-Extraktion durchgeführt – mit zwei verschiedenen Extraktionsmethoden: Phenol-Chloroform-Extraktion (Ausbeute 60–80 %) und magnetische Beads für zellfreie DNA (Ausbeute 90–95 %). Beide Methoden schlossen eine RNase-A-Behandlung ein, um die störende Wirkung der mRNA auf den DNA-spezifischen Farbstoff zu eliminieren.
Die Autoren zeigten experimentell, dass bereits geringe RNA-Mengen das DNA-Signal erheblich überschätzen lassen – und dass eine RNase-Behandlung notwendig ist, um RNA-bedingte Überschätzungen der DNA-Menge zu minimieren. Die RNA-Konzentration nach der Behandlung wurde kontrolliert und auf unter 1 ng/µl gesenkt.
Methode 3: Kapillarelektrophorese
Die Kapillarelektrophorese wurde eingesetzt, um sehr hohe DNA-Mengen – wie sie in einigen früheren Studien berichtet wurden – direkt sichtbar zu machen. Wenn DNA in Mengen von mehreren hundert bis tausend Nanogramm pro Dosis vorläge, würde sie in der Elektrophorese als deutlicher Peak oder Schmier erscheinen. Die Proben wurden mit Triton X-100 aufgeschlossen und mit RNase A behandelt. Es wurden zwei Kits eingesetzt – eines für den Standardbereich (0,5–50 ng/µl) und eines für den hochsensitiven Bereich (5–600 pg/µl pro Fragment).
Methode 4: Kurzlesen-Sequenzierung (Illumina)
Zur Sequenzierung wurde DNA aus den Impfstoffproben mit einer optimierten Methode extrahiert, die Hitzebehandlung (95°C), RNase-A-Verdau und Proteinase-K-Behandlung kombiniert. Der Bibliotheksaufbau erfolgte ohne vorherigen Fragmentierungsschritt, um die ursprüngliche Fragmentlängenverteilung möglichst unverändert zu erhalten. Die Sequenzierung erfolgte auf einem NextSeq 2000 System mit paired-end Reads, was eine präzise Bestimmung der Fragmentlänge ermöglicht.
Die bioinformatische Auswertung umfasste eine de-novo-Assemblierung der Reads zu vollständigen Plasmidsequenzen sowie ein anschließendes Mapping aller Reads auf die rekonstruierten Referenzsequenzen.
c) Zentrale Ergebnisse
qPCR-Befunde
Alle acht qPCR-Assays wurden in zwei unabhängigen Laboren mit drei Mitarbeitern durchgeführt. In keiner der 15 untersuchten Chargen wurde der regulatorische Grenzwert von 10 ng DNA pro Dosis überschritten.
Dabei zeigten sich deutliche Unterschiede zwischen den einzelnen Assays – abhängig von der Amplikon-Größe. Die höchsten DNA-Mengen wurden mit dem kürzesten Amplikon (KAN 1C, 63 bp) gemessen: bis zu 8 ng/Dosis für Comirnaty. Die niedrigsten Werte lieferten die längsten Amplikons (SPIKE 1A und SPIKE 2, 228–233 bp): 0,5–0,7 ng/Dosis. Die Autoren erklären diese Diskrepanz mit der Fragmentlänge der DNA: Bei einer medianen Fragmentlänge von 150 bp können längere Amplikons viele Fragmente nicht erfassen – weil diese zu kurz sind, um beide Primerbindestellen zu enthalten.
Die Autoren interpretieren dieses konsistente Muster über verschiedene Assays hinweg als Hinweis auf die interne Konsistenz ihres methodischen Ansatzes.
Fluorometrie
Nach vollständiger RNase-A-Behandlung und DNA-Extraktion lagen die fluorometrisch gemessenen DNA-Mengen in allen Chargen unterhalb des Grenzwerts von 10 ng/Dosis.
Die Autoren demonstrierten experimentell, dass ohne ausreichende RNase-Behandlung das DNA-Signal erheblich überschätzt wird: Bereits geringe RNA-Mengen erzeugen ein messbares Hintergrundsignal im DNA-Assay.
Die RNA-Messung zeigte, dass alle Comirnaty-Chargen mindestens 90 % der deklarierten mRNA-Menge enthielten. Bei den Spikevax-Chargen lagen die Werte teilweise darunter – was die Autoren auf die fortgeschrittene Überschreitung des Verfallsdatums zurückführen.
Kapillarelektrophorese
In keiner der untersuchten Chargen wurde ein spezifisches DNA-Signal detektiert – weder mit dem Standardkit (Nachweisbereich 0,5–50 ng/µl) noch mit dem hochsensitiven Kit (5–600 pg/µl pro Fragment). Schwache, unspezifische Signale um 60 und 300 bp waren in einigen Proben sichtbar, traten jedoch auch in der Negativkontrolle auf und werden daher als methodisches Hintergrundrauschen gewertet.
Die Autoren argumentieren: Wenn DNA in Mengen von mehreren hundert bis tausend Nanogramm pro Dosis vorläge, wäre dies in der Kapillarelektrophorese als deutlicher Peak oder Schmier sichtbar. Das vollständige Fehlen eines solchen Signals widerspricht diesen Befunden direkt.
Die RNA-Integritätsanalyse zeigte, dass sechs von neun abgelaufenen Chargen eine mRNA-Integrität unter 50 % aufwiesen – erkennbar auch an der trüben Erscheinung der Fläschchen im Vergleich zu den klaren, nicht abgelaufenen Chargen.
Sequenzierung
Die vollständigen Sequenzen entstanden bioinformatisch durch Assemblierung vieler überlappender Kurzreads und stellen nicht notwendigerweise den direkten Nachweis einzelner physisch intakter Plasmidmoleküle dar.
Der Anteil plasmidabgeleiteter Reads betrug 96,6 % bei Comirnaty (Spanne 95,3–98,1 %) und 88,7 % bei Spikevax (Spanne 83,5–94,5 %). Die verbleibenden Reads stammten überwiegend aus E. coli K12 – einem nicht-pathogenen Laborstamm – sowie aus Bakteriophagen-Sequenzen und repetitiven Sequenzen ohne Datenbankeinträge. Die Autoren weisen darauf hin, dass ein Teil dieser Reads auf Reagenzienkontaminationen zurückzuführen sein könnte, wie aus der Literatur bekannt.
Die Fragmentlängenanalyse ergab eine mediane Länge von 154 bp für Comirnaty (Spanne 130–181 bp) und 144 bp für Spikevax (Spanne 127–201 bp). Der Anteil der Fragmente über 300 bp war gering. Die Genomabdeckung war über das Plasmid ungleichmäßig verteilt – ein Muster, das mit einem nicht vollständig gleichmäßigen DNase-Verdau vereinbar ist.
d) Interpretation durch die Autoren
DNA-Menge unterhalb des Grenzwerts
Die Autoren schlussfolgern, dass die DNA-Rückstände in allen 15 untersuchten Chargen den regulatorischen Grenzwert von 10 ng pro Dosis einhalten. Dieses Ergebnis ist nach ihrer Einschätzung robust, weil die Ergebnisse durch mehrere komplementäre Methoden konsistent gestützt werden. Das Einbeziehen abgelaufener Chargen stärkt aus ihrer Sicht die Aussagekraft zusätzlich: Selbst unter diesen Umständen wurden keine übermäßigen DNA-Mengen gefunden.
Herkunft und Charakter der DNA
Die nachgewiesene DNA stammt nach Einschätzung der Autoren ausschließlich aus dem Produktionsplasmid – also aus dem erwarteten und bekannten Nebenprodukt des Herstellungsprozesses. Sie ist stark fragmentiert, mit einer medianen Länge von etwa 150 bp. Diese Fragmentgröße liegt nach Ansicht der Autoren unterhalb der Schwelle biologischer Aktivität: Fragmente dieser Größe weisen nach Einschätzung der Autoren keine Eigenschaften auf, die typischerweise mit Replikationsfähigkeit oder effizienter genomischer Integration assoziiert sind.
Erklärung der abweichenden Befunde früherer Studien
Die Autoren diskutieren mehrere methodische Faktoren, die aus ihrer Sicht zu den höheren DNA-Werten früherer Studien beigetragen haben könnten.
Erstens weisen sie darauf hin, dass fluorometrische DNA-Assays ohne ausreichende RNase-Behandlung durch verbleibende mRNA erheblich beeinflusst werden können. In eigenen Kontrollversuchen führte bereits eine geringe Rest-RNA zu einer deutlichen Überschätzung des DNA-Signals. Die Autoren vermuten daher, dass ein Teil der in früheren Arbeiten berichteten hohen DNA-Werte auf RNA-bedingte Crosstalk-Effekte zurückzuführen sein könnte.
Zweitens betonen sie den Einfluss der Amplikon-Länge bei qPCR-basierten Verfahren. Da die nachgewiesene DNA überwiegend fragmentiert sei, erfassen längere Amplikons nur einen Teil der vorhandenen Fragmente. Unterschiede zwischen Studien könnten daher teilweise aus unterschiedlichen Primerdesigns und Nachweisbereichen resultieren.
Die Autoren interpretieren ihre Ergebnisse insgesamt als Hinweis darauf, dass die Bestimmung residualer DNA in mRNA-Impfstoffen stark methodenabhängig ist und eine standardisierte Analytik erforderlich wäre.
Methodische Empfehlungen
Die Autoren empfehlen für künftige Analysen: vollständige RNase-Behandlung mit Kontrolle des RNA-Restgehalts vor der fluorometrischen DNA-Messung; direkte Verwendung von Triton-X-100-behandeltem Impfstoff für die qPCR ohne vorherige DNA-Extraktion, um Verluste zu vermeiden; den Einsatz kurzer Amplikons für eine vollständigere Erfassung fragmentierter DNA; sowie die Kombination mehrerer komplementärer Methoden für robuste Aussagen.
Biologische Einordnung
Die Autoren bewerten das Risiko durch die nachgewiesenen DNA-Rückstände als vernachlässigbar: geringe Menge, bekannte plasmidische Herkunft, hohe Fragmentierung sowie der Umstand, dass bei der Herstellung keine eukaryotischen Zelllinien verwendet werden. Sie sehen keinen Bedarf für eine Überarbeitung der bestehenden Grenzwerte oder Herstellungspraktiken.
e) Methodische Einschränkungen
Amplikon-Größe und Fragmentlänge
Die Autoren zeigen selbst, dass die gemessenen DNA-Mengen stark von der Amplikon-Größe des jeweiligen qPCR-Assays abhängen. Mit dem kürzesten Amplikon (KAN 1C, 63 bp) wurden bis zu 8 ng/Dosis gemessen – nahe am regulatorischen Grenzwert. Mit den längsten Amplikons (SPIKE 1A und SPIKE 2, 228–233 bp) dagegen nur 0,5–0,7 ng/Dosis.
Diese Unterschiede innerhalb derselben Studie verdeutlichen, dass qPCR-Ergebnisse wesentlich von der gewählten Assay-Architektur abhängen. Die gemessenen Werte repräsentieren daher keinen absoluten Gesamt-DNA-Wert, sondern den Anteil der Fragmente, der mit dem jeweiligen Primer-Design erfassbar ist.
Fragmentlängenbestimmung mittels Illumina
Die Sequenzierung ergab mediane Fragmentlängen von etwa 130–201 bp. Diese Werte sind jedoch nicht als direkte Messung der nativen Fragmentverteilung zu interpretieren.
Der Bibliotheksaufbau umfasst mehrere SPRI-Aufreinigungsschritte sowie PCR-Amplifikationen, die sehr kurze Fragmente (<150 bp) systematisch benachteiligen können. Gleichzeitig können auch sehr lange Fragmente (>800 bp) während der Bibliotheksvorbereitung und Sequenzierung weniger effizient repräsentiert werden und dadurch unterrepräsentiert oder nicht erfasst sein. Die berichtete Längenverteilung spiegelt daher primär die nach der Bibliotheksvorbereitung noch detektierbaren Fragmente wider – nicht zwangsläufig die ursprüngliche Verteilung im Impfstoff.
Damit erlaubt die Studie keine abschließende Aussage weder über den tatsächlichen Anteil sehr kurzer DNA-Fragmente unterhalb der Detektionsgrenze noch über das mögliche Vorhandensein längerer DNA-Fragmente außerhalb des effizient erfassten Größenbereichs.
Umrechnungsmethode bei der qPCR
Die gemessenen Kopienzahlen wurden anhand der Molekülmasse der jeweiligen qPCR-Amplikons in Nanogramm umgerechnet – nicht anhand der vollständigen Plasmidlänge. Dieses Vorgehen ist methodisch konsistent für die Quantifizierung der tatsächlich amplifizierten Zielregionen. Die resultierenden Nanogramm-Werte sind jedoch nicht direkt mit Studien vergleichbar, die ihre Berechnung auf vollständige Plasmidsequenzen stützen.
Einfluss der Extraktionseffizienz
Die fluorometrischen DNA-Werte hängen zusätzlich von der Effizienz der verwendeten Extraktionsmethode ab. Die Autoren geben für die Phenol-Chloroform-Extraktion eine Ausbeute von etwa 60–80 % und für die magnetischen Beads eine Ausbeute von etwa 90–95 % an.
Eine unvollständige Extraktion kann prinzipiell dazu führen, dass ein Teil der vorhandenen DNA – insbesondere sehr kurze Fragmente oder noch LNP-assoziierte Nukleinsäuren – nicht vollständig erfasst wird. Entsprechend zeigten die Bead-basierten Extraktionen in der Studie tendenziell höhere DNA-Werte als die Phenol-Chloroform-Methode.
Keine Messung vor LNP-Aufschluss
Im Unterschied zu einigen früheren Studien wurde keine systematische Vergleichsmessung vor und nach gezieltem LNP-Aufschluss durchgeführt. Damit lässt sich aus den Daten nicht direkt ableiten, welcher Anteil der nachgewiesenen DNA innerhalb der Lipid-Nanopartikel verkapselt vorlag.
Kapillarelektrophorese: Nachweisgrenze
Die Kapillarelektrophorese eignet sich gut zum Nachweis größerer DNA-Mengen. Für sehr niedrige Konzentrationen im einstelligen Nanogramm-Bereich ist ihre Sensitivität jedoch begrenzt. Die Methode ergänzt daher die qPCR- und Sequenzierungsdaten, ersetzt diese aber nicht.
Keine SV40-spezifische Analyse
Die Studie enthält keinen spezifischen Assay für SV40-Promoter-/Enhancer-Sequenzen. Aussagen über das Vorhandensein oder Fehlen solcher Sequenzen lassen sich aus den vorliegenden Daten daher nicht ableiten. Dies stellt keine methodische Schwäche im engeren Sinn dar, da die Untersuchung nicht auf SV40-Nachweise ausgerichtet war; im Vergleich zu anderen Arbeiten bleibt dieser Aspekt jedoch unbeantwortet.
f) Einordnung in den Gesamtkontext
Die Studie von Achs et al. stellt bislang eine der methodisch umfassendsten Untersuchungen zu DNA-Rückständen in mRNA-Impfstoffen dar. Sie kombiniert mehrere komplementäre Analyseverfahren – qPCR mit unterschiedlich langen Amplikons, fluorometrische Messungen mit kontrollierter RNase-Behandlung, Kapillarelektrophorese und Kurzlesen-Sequenzierung – an denselben Proben und in zwei verschiedenen Laboren. Dadurch können methodische Artefakte einzelner Verfahren besser erkannt und eingeordnet werden.
Im Gesamtkontext der bisherigen Literatur verdeutlicht die Studie, dass der Nachweis und die Quantifizierung von DNA-Rückständen in mRNA-Impfstoffen stark methodenabhängig sind. Unterschiedliche Probenvorbereitungen, Extraktionsverfahren, RNase-Behandlungen, qPCR-Designs und Sequenzierungsansätze können zu deutlich unterschiedlichen Ergebnissen führen.
Im Vergleich zu früheren Arbeiten stützt die Studie die Einschätzung, dass die untersuchten Chargen keine außergewöhnlich hohen Mengen an DNA-Rückständen enthalten. Gleichzeitig bleiben einige methodische Fragen – insbesondere hinsichtlich der vollständigen Fragmentverteilung sehr kurzer oder sehr langer DNA-Fragmente – auch mit diesem Ansatz nicht abschließend geklärt.
Die Arbeit von Achs et al. trägt damit weniger zur endgültigen Klärung der Kontroverse bei als zur präziseren methodischen Einordnung der bisherigen Befunde. In diesem Sinne ergänzt sie die vorangegangenen Studien – nicht indem sie deren Ergebnisse pauschal widerlegt, sondern indem sie zeigt, unter welchen methodischen Bedingungen bestimmte Aussagen getroffen werden können.
⚬ ⚬ ⚬
4.4. Zwischenfazit: Was wissen wir – und was nicht?
Was die vorliegenden Studien gemeinsam zeigen
Die in diesem Kapitel vorgestellten Untersuchungen stammen aus unterschiedlichen Laboren, wurden zu verschiedenen Zeitpunkten durchgeführt und setzen teilweise sehr verschiedene analytische Verfahren ein. Dennoch lassen sich einige Befunde benennen, die über mehrere Studien hinweg konsistent sind.
Erstens: DNA-Rückstände in mRNA-basierten COVID-19-Impfstoffen sind nachweisbar. Das ist kein überraschendes Ergebnis – es ist eine bekannte und erwartete Konsequenz des Herstellungsprozesses, bei dem eine DNA-Vorlage für die in-vitro-Transkription verwendet wird. Dass der DNase-I-Verdau keine vollständige Entfernung aller DNA-Fragmente gewährleistet, ist in der Fachliteratur anerkannt.
Zweitens: Die nachgewiesene DNA stammt überwiegend aus dem Produktionsplasmid. Das zeigen sowohl die Sequenzierungsdaten von McKernan, Raoult und Achs et al. als auch die PCR-basierten Analysen mehrerer Arbeitsgruppen übereinstimmend.
Drittens: Mehrere Studien liefern Hinweise darauf, dass ein relevanter Anteil der nachgewiesenen DNA vor enzymatischem Abbau geschützt und mit den Lipid-Nanopartikeln assoziiert ist.
Wo die Studien voneinander abweichen
Die zentrale Streitfrage – wie viel DNA tatsächlich vorhanden ist und ob die regulatorischen Grenzwerte eingehalten werden – wird von den vorliegenden Studien unterschiedlich beantwortet. Diese Diskrepanz ist nicht primär auf unterschiedliche Proben zurückzuführen, sondern lassen sich zumindest teilweise durch unterschiedliche methodische Ansätze erklären.
Fluorometrische Messungen ohne ausreichende RNase-Behandlung liefern systematisch höhere Werte, weil der Farbstoff auch an RNA bindet. PCR-basierte Messungen sind von der Amplikon-Größe abhängig: Längere Amplikons erfassen weniger Fragmente und unterschätzen die Gesamt-DNA. Sequenzierungsverfahren liefern qualitative Informationen über Sequenzidentität und Fragmentlänge, aber keine zuverlässige absolute Quantifizierung. Jede Methode misst damit etwas leicht anderes – und liefert entsprechend unterschiedliche Zahlen.
Das bedeutet: Unterschiede zwischen Studien sind nicht zwangsläufig ein Beleg für fehlerhafte Arbeit einer der Gruppen. Sie können – und müssen in vielen Fällen – als Ausdruck methodischer Variabilität verstanden werden.
Was offen bleibt
Trotz der inzwischen vorliegenden Datenmenge bleiben mehrere Fragen unbeantwortet.
Die Frage nach der tatsächlichen Fragmentlängenverteilung – insbesondere dem Anteil sehr kurzer Fragmente unter 100 bp – ist bisher nicht abschließend geklärt. Illumina-Sequenzierung unterschätzt kurze Fragmente systematisch; ONT-Datensätze können durch prozessbedingte Selektions- und Sequenzierungsbiases relativ stärker von längeren Fragmenten geprägt sein. Ein Verfahren, das die native Fragmentverteilung ohne größenselektive Verzerrung abbildet, wurde in den vorliegenden Studien nicht eingesetzt.
Die biologische Relevanz der nachgewiesenen DNA – einschließlich Fragen nach möglicher zellulärer Aufnahme, Persistenz oder genomischer Integration – wurde bisher nicht direkt untersucht. Vorhandene theoretische Modelle und Analogien aus anderen biologischen Systemen reichen derzeit nicht aus, um belastbare Aussagen über ein tatsächliches Risiko abzuleiten, rechtfertigen jedoch weitere Untersuchungen.
Die Frage nach dem SV40-Promoter/Enhancer – seiner tatsächlichen Häufigkeit in verschiedenen Chargen und seiner biologischen Aktivität in fragmentierter Form – wurde von den vorliegenden Studien unterschiedlich adressiert und ist nicht abschließend beantwortet.
Was das für die wissenschaftliche Diskussion bedeutet
Ein grundlegendes Problem, das sich durch alle hier vorgestellten Studien zieht, ist das Fehlen eines standardisierten, validierten Analyseprotokolls für DNA-Rückstände im fertigen Impfstoffprodukt. Die regulatorischen Grenzwerte existieren – aber die Methode, mit der sie überprüft werden sollen, ist nicht einheitlich vorgeschrieben. Das führt dazu, dass verschiedene Labore mit unterschiedlichen Methoden zu unterschiedlichen Ergebnissen kommen, ohne dass ein direkter Vergleich möglich ist.
Ein sinnvoller nächster Schritt wäre die Etablierung eines orthogonalen Standardprotokolls: dieselbe Probe, mehrere validierte Methoden, in verschiedenen unabhängigen Laboren. Erst ein solcher Ansatz würde erlauben, methodenbedingte Variabilität von tatsächlichen Unterschieden zwischen Chargen zu trennen – und damit belastbare, vergleichbare Daten zu erzeugen.
Regulatorische Einordnung
Die regulatorischen Grenzwerte für Rest-DNA wurden ursprünglich für klassische biologische Arzneimittel und nicht speziell für LNP-formulierte modRNA-Impfstoffe entwickelt. Inwieweit bestehende Grenzwerte und Prüfverfahren auf diese neue Plattformtechnologie vollständig übertragbar sind, ist bislang nicht abschließend diskutiert.
Abschließende Einordnung
Die vorliegenden Studien haben eine wichtige Funktion erfüllt: Sie haben eine wissenschaftliche Diskussion angestoßen, die ohne sie nicht stattgefunden hätte. Gleichzeitig zeigen sie die Grenzen unabhängiger Nachanalysen außerhalb des regulierten Rahmens: fehlende Validierung, begrenzte Stichprobengrößen und methodische Heterogenität.
Was bleibt, ist eine sachlich begründete Forderung: mehr Transparenz, einheitlichere Methoden und unabhängige Replikation durch verschiedene Laborgruppen. Das ist keine alarmistische Schlussfolgerung – es ist das, was gute Wissenschaft grundsätzlich auszeichnet.
5. Schlussbetrachtung und Ausblick
Die Diskussion um DNA-Rückstände in mRNA-Impfstoffen zeigt auf mehreren Ebenen, wie komplex die analytische Bewertung neuartiger biologischer Arzneimittel sein kann.
Was zunächst wie eine einfache Frage erscheint – in welcher Menge DNA-Rückstände vorhanden sind – erweist sich bei näherer Betrachtung als stark abhängig von Probenaufbereitung, Nachweismethode und Dateninterpretation.
Zentrale Erkenntnisse der Arbeit
Die Herstellung modRNA-basierter Impfstoffe erfolgt über einen mehrstufigen Prozess, bei dem bakterielle DNA-Vorlagen unverzichtbar sind. Die Entfernung dieser DNA gehört zu den regulären Reinigungsschritten des Herstellungsverfahrens. Die vorliegenden Untersuchungen zeigen jedoch übereinstimmend, dass sich residuale DNA-Fragmente in kommerziellen Impfstoffchargen nachweisen lassen – wenn auch in methodisch unterschiedlich bestimmten Mengen.
Die Auswertung der bisherigen Studien macht deutlich, dass der Nachweis und die Quantifizierung von DNA-Rückständen wesentlich komplexer sind, als es die regulatorischen Grenzwerte allein vermuten lassen. Unterschiedliche Aufschlussverfahren, Extraktionsmethoden, RNase-Behandlungen, qPCR-Designs und Sequenzierungsansätze können zu erheblich abweichenden Ergebnissen führen.
Die Kontroverse der vergangenen Jahre verdeutlicht damit nicht nur Unterschiede zwischen einzelnen Studien, sondern vor allem die Grenzen bislang fehlender Standardisierung unabhängiger Nachanalysen.
Offene Fragen für die Forschung
Trotz der inzwischen vorliegenden Datenmenge bleiben mehrere Fragen offen:
Wie hoch ist der tatsächliche Anteil sehr kurzer oder potenziell biologisch aktiver DNA-Fragmente in verschiedenen Impfstoffchargen?
Welche Aussagekraft besitzen aktuelle Messverfahren hinsichtlich der nativen Fragmentverteilung im Impfstoff?
Welche Rolle spielen RNA:DNA-Hybride oder andere Nukleinsäurekomplexe für die Stabilität und Nachweisbarkeit residualer DNA?
Und schließlich: Welche biologische Relevanz haben die nachgewiesenen DNA-Fragmente – einschließlich des SV40-Promoter/Enhancer-Elements – tatsächlich unter realen physiologischen Bedingungen?
Viele dieser Fragen lassen sich auf Basis der derzeitigen Daten weder eindeutig bestätigen noch ausschließen. Sie erfordern weitere Untersuchungen mit standardisierten, orthogonalen Analyseverfahren sowie unabhängige Replikationen in verschiedenen Laboren.
Ausblick
Die wissenschaftliche Diskussion über DNA-Rückstände in modRNA-Impfstoffen ist damit nicht abgeschlossen. Gleichzeitig zeigt die bisherige Forschung, dass zwischen analytischem Nachweis und biologischer Bedeutung klar unterschieden werden muss.
Während sich die vorliegende Arbeit auf Herstellungsprozesse, Nachweismethoden und experimentelle Befunde konzentriert, bleibt die Frage nach möglichen biologischen Auswirkungen Gegenstand weiterer Forschung und zukünftiger Betrachtungen. Dazu gehören unter anderem Fragen zur biologischen Aktivität residualer DNA, zur Stabilität von Nukleinsäurekomplexen, zu theoretischen Integrationsmechanismen sowie zur Bedeutung funktioneller Sequenzelemente.
Unabhängig von der Bewertung einzelner Studien ergibt sich daraus eine grundsätzliche wissenschaftliche Forderung: mehr Transparenz, besser standardisierte Analyseverfahren und reproduzierbare unabhängige Untersuchungen. Gerade bei neuartigen biologischen Technologien ist dies keine außergewöhnliche Erwartung, sondern ein zentraler Bestandteil wissenschaftlicher Qualitätssicherung.

Danksagung
Mein aufrichtiger Dank gilt allen Wissenschaftlern, Forschern und engagierten Persönlichkeiten, die mit ihrer Arbeit – in Studien, Artikeln und Vorträgen – zur Erforschung dieses komplexen Themas beitragen. Ihr Engagement ist von unschätzbarem Wert – für den Fortschritt des Wissens ebenso wie für eine sachliche öffentliche Diskussion.
Der wissenschaftliche Widerstreit, den dieser Text zu dokumentieren versucht, ist kein Mangel – er ist das Wesen guter Wissenschaft.
✧ ✧ ✧
Nachtrag
Ich habe diesen Text als Laie begonnen.
Mit Unsicherheit, Neugier und offenen Fragen.
Vielleicht war genau das ein Vorteil.
Viele Fragen bekamen Antworten.
Neue Fragen kamen hinzu.
Je tiefer ich einstieg, desto klarer wurde:
Nicht nur die Biologie ist komplex.
Auch die Messung der Biologie ist komplex.
Der Weg durch die Materie war lang.
Auf dieser Reise hatte ich Hilfe:
Geduldige KI-Systeme, die erklärten,
widersprachen, strukturierten, formulierten – und mitdachten.
Es waren lange, fruchtbare Dialoge
zwischen Mensch und Maschine.
Und am Ende bleibt eine wichtige Erkenntnis:
Lernen ist ein gemeinsamer Prozess.
Stand: Mai 2026
