Leitfaden zur Artikelserie
Teil 1: Herstellung und Reinigung von modRNA
Teil 2: Nachweisverfahren und Studienlage
Bevor wir uns der Frage zuwenden, warum DNA-Rückstände in modRNA-Impfstoffen nachweisbar sein können, ist ein grundlegendes Verständnis des Herstellungsprozesses hilfreich.
Die Herstellung modRNA-basierter Impfstoffe erfolgt über mehrere aufeinander aufbauende Produktions- und Reinigungsschritte. Ausgangspunkt ist eine DNA-Vorlage, die als Bauplan für die spätere nukleosid-modifizierte mRNA (modRNA) dient. Im Verlauf des Herstellungsprozesses entstehen dabei komplexe Reaktionsgemische, aus denen unerwünschte Restbestandteile möglichst weit entfernt werden müssen.
Besondere Aufmerksamkeit erhielt in den vergangenen Jahren die Tatsache, dass für die Entwicklung und spätere industrielle Produktion unterschiedliche Verfahren zur Vervielfältigung dieser DNA-Vorlage eingesetzt wurden. Diese Prozesse unterscheiden sich nicht nur hinsichtlich ihrer technischen Skalierung, sondern auch hinsichtlich der Anforderungen an die anschließende Reinigung biologischer Restbestandteile.
Der vorliegende erste Teil beschreibt die grundlegende Herstellungslogik modRNA-basierter Impfstoffe, typische Nebenprodukte und Verunreinigungen sowie die wichtigsten Reinigungsverfahren innerhalb der Produktion. Abschließend werden die beiden Herstellungswege – Prozess 1 und Prozess 2 – systematisch gegenübergestellt.
Er bildet damit die Grundlage für den zweiten Teil dieser Artikelserie, der sich den analytischen Nachweisverfahren, regulatorischen Grenzwerten sowie den bisher veröffentlichten experimentellen Untersuchungen zu DNA-Rückständen in modRNA-basierten Impfstoffen widmet.
Hinweis zur Terminologie
Im allgemeinen Sprachgebrauch ist der Begriff „mRNA-Impfstoff“ gebräuchlich. Fachlich handelt es sich bei den zugelassenen Präparaten um nukleosid-modifizierte mRNA (modRNA). Aus Gründen der Verständlichkeit wird in dieser Arbeit der etablierte Begriff „mRNA-Impfstoff“ verwendet.
Inhaltsverzeichnis
Teil 1: Herstellung und Reinigung
1. Die Herstellungsverfahren – ein Überblick
1.1. Produktentstehung – vom Gen zur mRNA
1.2. Verunreinigungen und Nebenprodukte
1.3. Reinigungsverfahren im Herstellungsprozess
1.4. Vergleich: Prozess 1 vs. Prozess 2
1.5. Zusammenfassung: Herstellung und Reinigung von modRNA
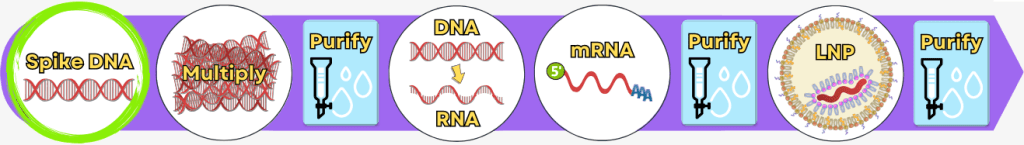
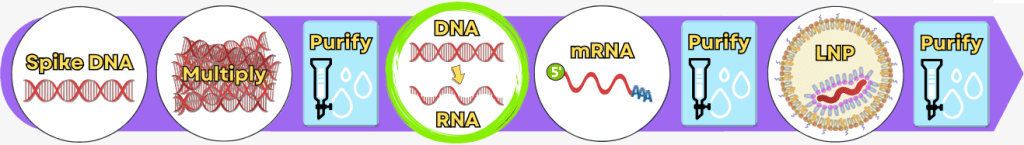
1. Die Herstellungsverfahren – ein Überblick
Die folgende Übersicht stellt die wesentlichen Produktionsschritte vereinfacht dar. In der industriellen Praxis können einzelne Verfahren und Prozessdetails je nach Hersteller variieren.
Die Herstellung lässt sich vereinfacht in folgende Prozessschritte gliedern:
| Schritt 1 | 🟢 | Gewinnung der genetischen Information |
| Schritt 2 | 🟢 | Vervielfältigung der Spike-Protein-DNA Prozess 1: mittels PCR Prozess 2: mittels Bakterien |
| 💧 | Reinigung der DNA | |
| Schritt 3 | 🟢 | In-vitro-Transkription zur Herstellung der mRNA |
| Schritt 4 | 🟢 | RNA-Prozessierung – Reifung der mRNA |
| 💧 | Reinigung der DNA | |
| Schritt 5 | 🟢 | Verpackung der mRNA in Lipid-Nanopartikel (LNPs) |
| 💧 | Endreinigung |
Während im Upstream-Prozess das eigentliche Produkt – die mRNA – schrittweise erzeugt wird, dient der Downstream-Prozess dazu, die entstandene mRNA aus komplexen Reaktionsgemischen zu isolieren, zu reinigen und für die medizinische Anwendung zu formulieren.
1.1. Produktentstehung – vom Gen zur mRNA
Schritt 1: Gewinnung der genetischen Information
Schritt 2: Vervielfältigung der Spike-Protein-DNA
Prozess 1: mittels PCR
Prozess 2: mittels Bakterien
Schritt 3: In-vitro-Transkription zur Herstellung der mRNA
Schritt 4: RNA-Prozessierung – Reifung der mRNA
Schritt 5: Verpackung der mRNA in Lipid-Nanopartikel (LNPs)
Schritt 1: Gewinnung der genetischen Information
Der erste Schritt besteht darin, die genetische Bauanleitung für das gewünschte Protein – hier das Spike-Protein des Coronavirus – zu identifizieren.
In der Praxis läuft das so ab:
Virusproben sammeln: Aus Patientenproben (z. B. Rachen- oder Nasenabstrichen, Blut oder Gewebe) wird Virusmaterial gewonnen.
Anmerkung: Es gibt unterschiedliche Auffassungen darüber, ob SARS-CoV-2 jemals direkt aus Patientenproben isoliert und im Labor kultiviert wurde. Diese Frage wird in diesem Artikel nicht behandelt, da der Fokus hier auf dem Herstellungsprozess der mRNA-Impfstoffe liegt.
Erbinformation extrahieren: SARS-CoV-2 ist ein RNA-Virus. Die virale RNA wird aus der Probe isoliert.
RNA in DNA umwandeln: Mithilfe eines speziellen Enzyms (Reverse Transkriptase) wird die Virus-RNA in DNA übersetzt. DNA ist stabiler und kann leichter analysiert werden.
Genom entschlüsseln: Die DNA wird vervielfältigt und ihre genau Abfolge der Nukleotide (A, T, G, C) bestimmt.
Den wichtigen Abschnitt finden: Mittels Computeranalysen wird der genaue Teil identifiziert, der den Bauplan für das Spike-Protein enthält.
Eintrag in öffentliche Datenbanken: Die so ermittelte genetische Bauanleitung wird in öffentliche Datenbanken eingetragen. Forschungsteams weltweit können dadurch auf diese Sequenz zugreifen, ohne das Virus selbst isolieren zu müssen.
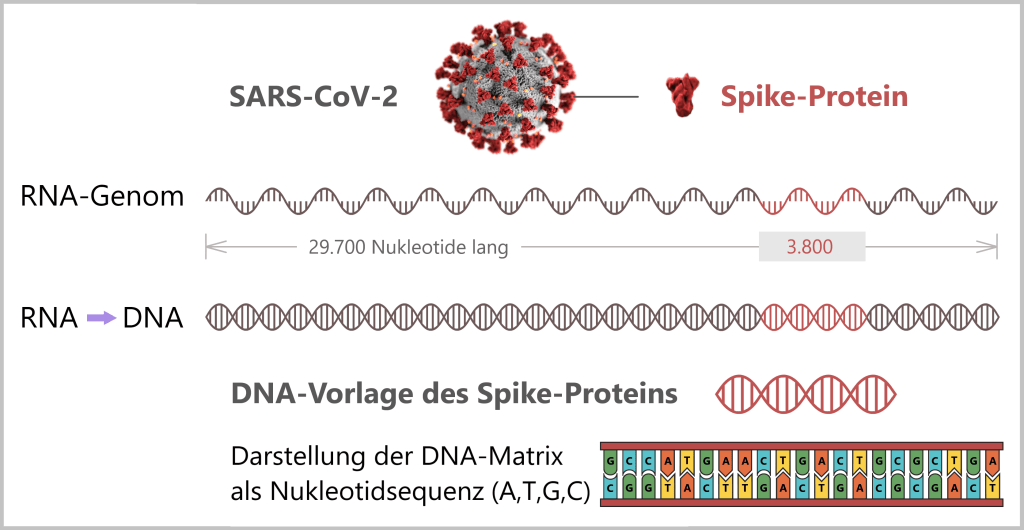
Aus dem RNA-Genom des SARS-CoV-2 wird mithilfe des Enzyms Reverse Transkriptase eine DNA-Version erzeugt. Der Abschnitt, der den Bauplan für das Spike-Protein enthält, wird identifiziert und als DNA-Vorlage (DNA-Template) synthetisch im Labor hergestellt. Das DNA-Template dient später als Vorlage für die Produktion der mRNA im Impfstoff.
Anmerkung: Das Coronavirus SARS-CoV-2 speichert seine Erbinformation auf einer langen RNA-Kette, die aus einzelnen Nukleotiden – den „Buchstaben“ des genetischen Codes – besteht. Mit rund 29.700 dieser Bausteine besitzt es eines der größten bekannten RNA-Genome. Der Bauplan für das charakteristische Spike-Protein umfasst dabei einen Abschnitt von etwa 3.800 Buchstaben.
Erstellen der DNA-Vorlage
Basierend auf der ermittelten genetischen Abfolge (den sogenannten Sequenzdaten) lässt sich im Labor eine DNA-Version des Spike-Gens synthetisch herstellen. Dieses DNA-Template dient später als Vorlage für die mRNA-Produktion.
Schritt 2: Vervielfältigung der Spike-Protein-DNA
Nachdem die Bauanleitung für das Spike-Protein in Form einer DNA-Vorlage hergestellt wurde, muss sie millionenfach vervielfältigt werden. Nur so steht genug Material zur Verfügung, um später mRNA daraus herzustellen.
Prozess 1: mittels PCR
Prozess 2: mittels Bakterien

Prozess 1: Vervielfältigung der Spike-DNA mittels PCR
Prozess 1 nutzt ein Verfahren, das seit der Corona-Pandemie weltweit bekannt ist: die Polymerase-Kettenreaktion (PCR). Sie wird im Labor – fachsprachlich in vitro – durchgeführt und imitiert die natürliche DNA-Vermehrung, wie sie sonst in lebenden Zellen abläuft.
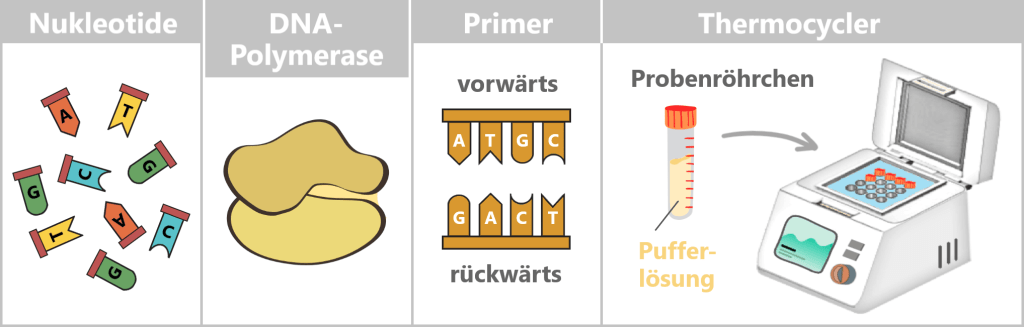
Was braucht man für eine PCR?
Damit die Kopiermaschine der Natur läuft, braucht es ein paar Zutaten:
- DNA-Vorlage – das Ausgangsmaterial ist die Spike-Protein-DNA aus Schritt 1.
- Nukleotide – die Bausteine: die vier „Buchstaben“ der DNA > Adenin (A), Thymin (T), Cytosin (C) und Guanin (G), aus denen neue Stränge zusammengesetzt werden.
- DNA-Polymerase – der Baumeister: das Enzym, das die Vorlage liest und neue DNA-Stränge baut. Meist wird die hitzestabile Taq-Polymerase verwendet.
- Primer – die Wegweiser: kurze DNA-Stücke, die der Polymerase zeigen, wo sie mit dem Kopieren beginnen soll. Man benötigt immer zwei: einen Vorwärts- und einen Rückwärts-Primer.
- Pufferlösung – das richtige Milieu: sorgt für die richtige Umgebung, damit die Polymerase zuverlässig arbeiten kann.
- Thermocycler – das Temperatur-Karussell: ein Gerät, das automatisch die nötigen Temperaturzyklen durchläuft.
Für die spätere mRNA-Herstellung wird die DNA-Vorlage bereits in diesem Schritt gezielt erweitert. An die Spike-Sequenz wird ein zusätzlicher DNA-Abschnitt angehängt – der sogenannte T7-Promotor. Diese kurze Sequenz wird für die PCR selbst nicht benötigt, sondern spielt erst im nachfolgenden Prozessschritt eine Rolle. Der T7-Promotor dient als Andockstelle für die T7-RNA-Polymerase, die in der in-vitro-Transkription (IVT) die DNA in die therapeutische mRNA umschreibt.
Der Ablauf der Polymerase-Kettenreaktion (PCR)
Alle Zutaten kommen in ein kleines Reaktionsröhrchen, das in den Thermocycler gestellt wird. Dieser steuert die immer gleichen drei Schritte, die sich zyklisch wiederholen:
1) Trennung der DNA-Stränge (Denaturierung)
Die Probe wird auf ca. 94–98 °C für etwa 20-30 Sekunden erhitzt. Dadurch lösen sich die Wasserstoffbrücken zwischen den DNA-Basen und der Doppelstrang trennt sich in zwei Einzelstränge auf. Diese dienen im nächsten Schritt als Vorlage.
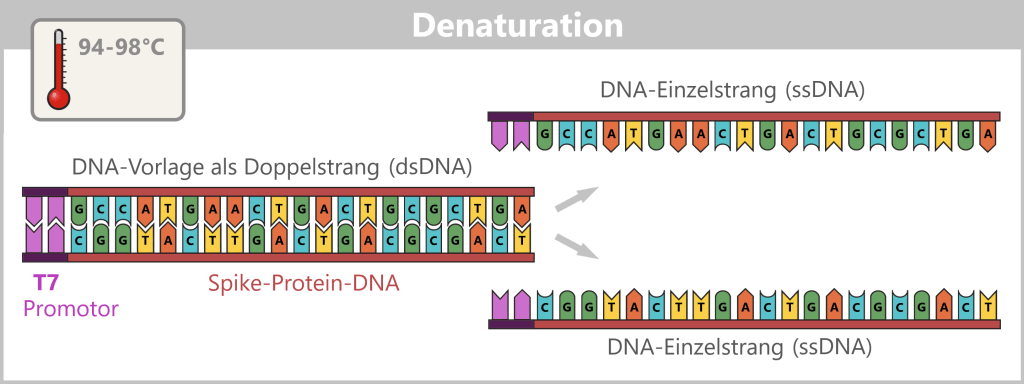
Die Darstellung ist stark schematisch und nicht maßstabsgetreu. Die kodierende Sequenz für das SARS-CoV-2-Spike-Protein beträgt ~3.800 Basenpaare, die für den T7-Promotor ~20 Basenpaare.
2) Primerbindung (Annealing)
Die Temperatur wird auf 50–65 °C abgesenkt. Jetzt binden sich die Primer gezielt an die jeweiligen DNA-Einzelstränge. Sie markieren den Startpunkt für die DNA-Synthese.
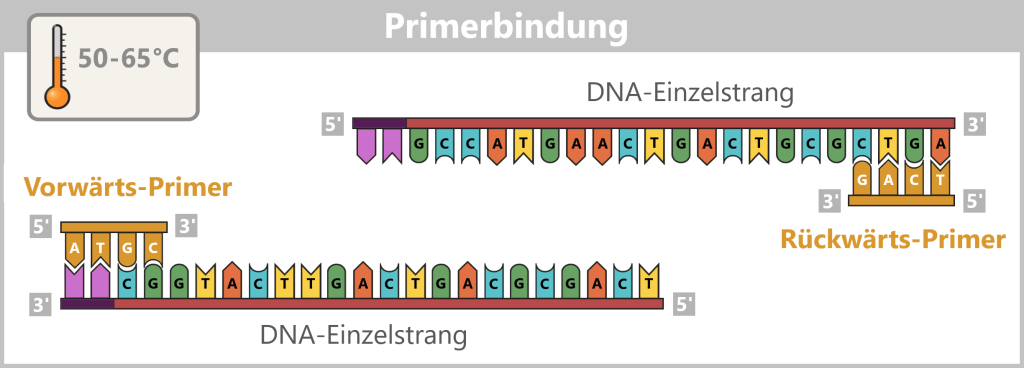
Die Darstellung ist stark schematisch und nicht maßstabsgetreu. Primer sind typischerweise 20–25 Nukleotide lang.
3) DNA-Synthese (Amplifikation)
Bei etwa 70 °C, der optimalen Temperatur für die Polymerase, beginnt die eigentliche Vervielfältigung. Die DNA-Polymerase bindet an den Primer, liest den Einzelstrang von 3′ nach 5′ – und synthetisiert parallel den neuen Strang in 5′ nach 3′-Richtung. Dabei setzt sie Nukleotide nach dem Prinzip der Basenpaarung zusammen: A mit T und G mit C. So entstehen zwei neue DNA-Doppelstränge.
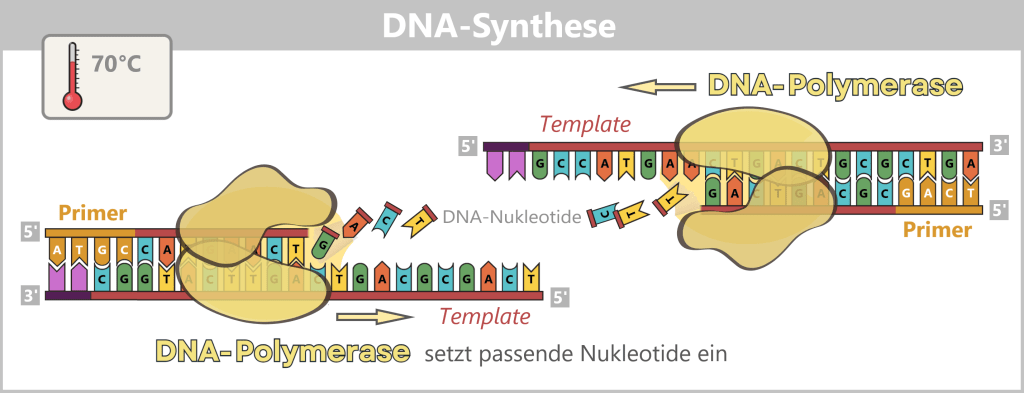
Die Abbildung zeigt ein aktive DNA-Polymerase, die wie ein molekularer Motor am freigelegten Einzelstrang entlanggleitet. Der Prozess der Polymerisation ist deutlich erkennbar: Einzelne Nukleotide (die Bausteine der DNA) treten durch eine Öffnung in das Enzym und werden von der Polymerase passgenau an das Ende des wachsenden Stranges angefügt. Dieser Vorgang bewirkt die Elongation (Verlängerung) der neuen DNA-Kette, die kontinuierlich in 5′-3′-Richtung wächst. Durch diese stetige Kopierarbeit findet die Amplifikation (Vervielfältigung) der genetischen Information statt, wobei aus einem ursprünglichen Templatestrang ein neuer, komplementärer Doppelstrang entsteht.
Warum wird immer in 5′-3′-Richtung synthetisiert?
Der DNA-Aufbau
Die DNA besteht aus zwei Strängen, die in entgegengesetzte Richtungen verlaufen – der eine in 5’→3′-Richtung, der andere in 3’→5′-Richtung. Beide sind komplementär zueinander.
Die DNA-Polymerase, das Enzym, das neue DNA herstellt, kann nur in eine Richtung arbeiten: Sie liest den Matrizenstrang in 3’→5′-Richtung und synthetisiert den neuen Strang in 5’→3′-Richtung.
Was bedeuten 5′ und 3′?
Um das zu verstehen, schauen wir uns die Bausteine der DNA an: die Nukleotide. Jedes Nukleotid besteht aus:
- Einer Base (A, C, G oder T)
- Einem Zucker (Desoxyribose)
- Einer Phosphatgruppe (P)
Der Zucker hat fünf Kohlenstoffatome (C), die durchnummeriert sind:
- 1′: Hier ist die Base (A, T, C oder G) angeheftet.
- 2′: Trägt in der DNA nur ein Wasserstoffatom (-H) und keine Hydroxylgruppe (-OH) wie in der RNA. Diese fehlende OH-Gruppe gibt der Desoxyribose ihren Namen („Desoxy“-Ribose, da „desoxy“ = ohne Sauerstoff).
- 3′: Hier befindet sich eine Hydroxylgruppe (-OH), die für das Anfügen neuer Nukleotide während der Synthese wichtig ist.
- 4′: Verbindet den Zuckerring mit dem 5′-Kohlenstoff.
- 5′: Trägt die Phosphatgruppe, die das Nukleotid mit dem nächsten verknüpft.
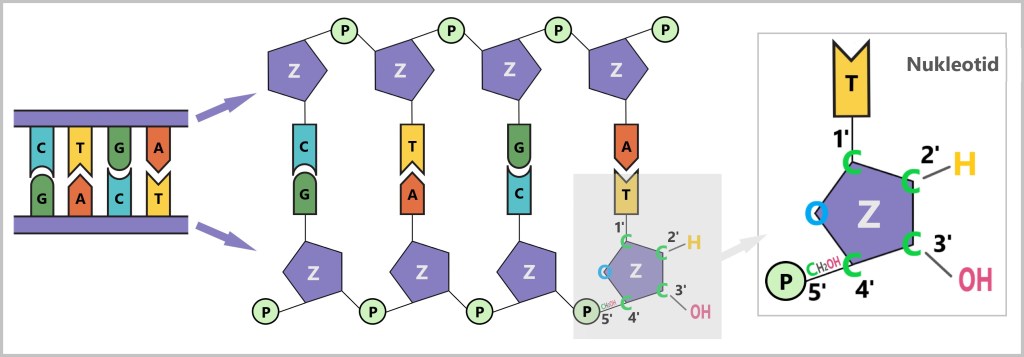
Warum die Synthese nur in 5’→3′-Richtung möglich ist
Die DNA-Polymerase kann neue Nukleotide nur am 3′-Ende des wachsenden Strangs anfügen. Der Grund: Dort befindet sich die freie Hydroxylgruppe (–OH), die für die chemische Verknüpfung mit der Phosphatgruppe des nächsten Nukleotids benötigt wird.
Am 5′-Ende sitzt bereits eine Phosphatgruppe – dort kann nichts Neues angehängt werden. Die Kettenverlängerung ist also nur in 5’→3′-Richtung möglich.
Zusammengefasst
Die DNA-Polymerase bewegt sich entlang des Matrizenstrangs der DNA in 3′-5′-Richtung. Währenddessen fügt sie komplementäre DNA-Nukleotide an den neuen DNA-Strang an und zwar immer an dessen freies 3′-Ende. So wächst der neue Strang kontinuierlich in 5’→3′-Richtung.
Wiederholung der Zyklen
Die neu entstandenen DNA-Doppelstränge dienen direkt als Vorlage für die nächste Runde. Die Schritte Denaturierung – Primerbindung – DNA-Synthese wiederholen sich zyklisch.
Mit jedem Zyklus verdoppelt sich die Menge der DNA: 1 → 2 → 4 → 8 → 16 → 32 … Nach nur 25–40 Zyklen liegen Milliarden Kopien der Spike-Protein-DNA vor.
Das Ergebnis
Am Ende der PCR hat man eine hochkonzentrierte Lösung mit vielen Kopien der Spike-Bauanleitung – die Ausgangsbasis für den nächsten Schritt: die Herstellung von mRNA.
🎥 Tipp: Das Video „What is PCR?“ fasst den Prozess anschaulich zusammen.

Prozess 2: Vervielfältigung der Spike-DNA mittels Bakterien
Im Gegensatz zur PCR, die im Reagenzglas abläuft (in vitro), nutzt dieser Ansatz lebende Organismen, genauer gesagt Bakterien. Man spricht dabei von einem in vivo-Verfahren (lateinisch: „im Lebendigen“), weil die Vervielfältigung direkt innerhalb der Zellen stattfindet.
a) Warum Bakterien?
b) Das Bakterium Escherichia coli (E. coli)
c) Plasmide – kleine DNA-Ringe mit großer Wirkung
d) Plasmide in der Gentechnik
e) Einbau der Spike-Protein-DNA in das Plasmid
f) Übertragung der modifizierten Plasmide in Bakterien
g) Vermehrung der Bakterien
h) Ernte der Bakterien
i) Isolierung der modifizierten Plasmide
j) Linearisierung der Spike-Protein-DNA
a) Warum Bakterien?
Bakterien sind winzige, einzellige Lebewesen, die sich durch einfache Teilung vermehren: Eine Zelle teilt sich in zwei, diese teilen sich wieder – und so entstehen in kürzester Zeit riesige Mengen identischer Kopien.
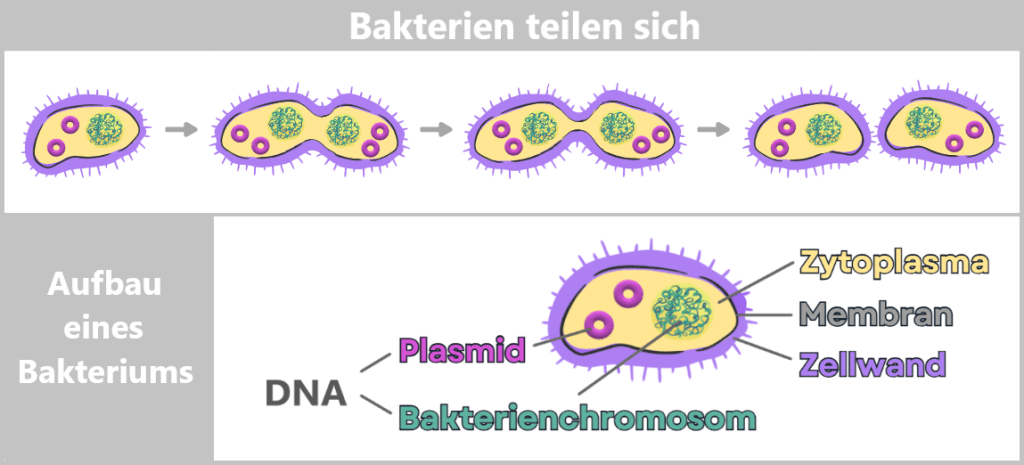
Da Bakterien ungeschlechtlich sind, vermehren sie sich hauptsächlich durch einfache Teilung. Der Prozess beginnt mit dem Wachstum der Zelle und der Verdopplung des genetischen Materials. Dann bildet sich eine Einschnürung, bis schließlich zwei genetisch identische Tochterzellen entstehen.
Die Erbinformation der Bakterien – ihre DNA – liegt frei im Inneren der Zelle, im sogenannten Zytoplasma. Der größte Teil ist im Bakterienchromosom gespeichert. Zusätzlich besitzen viele Bakterien kleine, ringförmige DNA-Moleküle, die Plasmide.
Anpassung durch Mutation und Gentransfer
Die kontinuierliche Veränderung des genetischen Materials ist entscheidend, damit sich Organismen im Laufe der Generationen neuen Umweltbedingungen erfolgreich anpassen können. Bakterien gelten als besonders anpassungsfähig. Dafür sorgen zwei Mechanismen:
Mutationen: kleine zufällige Veränderungen im Erbgut, die manchmal Vorteile bringen (z. B. Resistenz gegen Antibiotika).
Gentransfer: Bakterien können zusätzlich DNA-Abschnitte austauschen, vor allem über ihre Plasmide. So verbreiten sich nützliche Eigenschaften oft rasend schnell in einer Bakterienpopulation.
Darum sind Bakterien so geeignet:
- Schnelle Vermehrung: Unter idealen Bedingungen verdoppeln sich viele Bakterien alle 20 Minuten.
- Einfache Struktur: Ihre DNA liegt nicht im Zellkern, sondern frei im Zellinneren – das erleichtert Manipulationen.
- Plasmide als Zusatz-DNA: Sie können leicht verändert, übertragen und zur Vervielfältigung von Fremd-DNA genutzt werden – ideal für die Gentechnik.
Die Möglichkeit, genetisches Material in Bakterien einzuschleusen und mitzukopieren, nutzen Forscher seit Jahrzehnten. So werden Bakterien zu kleinen „DNA-Fabriken“.
Ein historischer Durchbruch
1973 gelang Stanley Cohen und Herbert Boyer ein bahnbrechendes Experiment: Sie schleusten erstmals ein fremdes Gen in Bakterien ein. Ergebnis: Die Bakterien übernahmen das neue Gen und setzten es sogar um. Damit war bewiesen, dass Gene von einer Art auf eine andere übertragen werden können – die Geburtsstunde der modernen Gentechnik. [Das Cohen-Boyer-Experiment]
b) Das Bakterium Escherichia coli (E. coli)
In der Forschung wird besonders gern das Bakterium Escherichia coli (kurz: E. coli) eingesetzt – und das hat mehrere Gründe:
- Es lässt sich leicht im Labor kultivieren und vermehren.
- Es vermehrt sich sehr schnell: Unter optimalen Bedingungen teilt es sich alle 20–30 Minuten.
- Sein Genom ist umfassend erforscht und verstanden.
- Es lässt sich einfach genetisch manipulieren.
- Die Kultivierung ist kostengünstig.
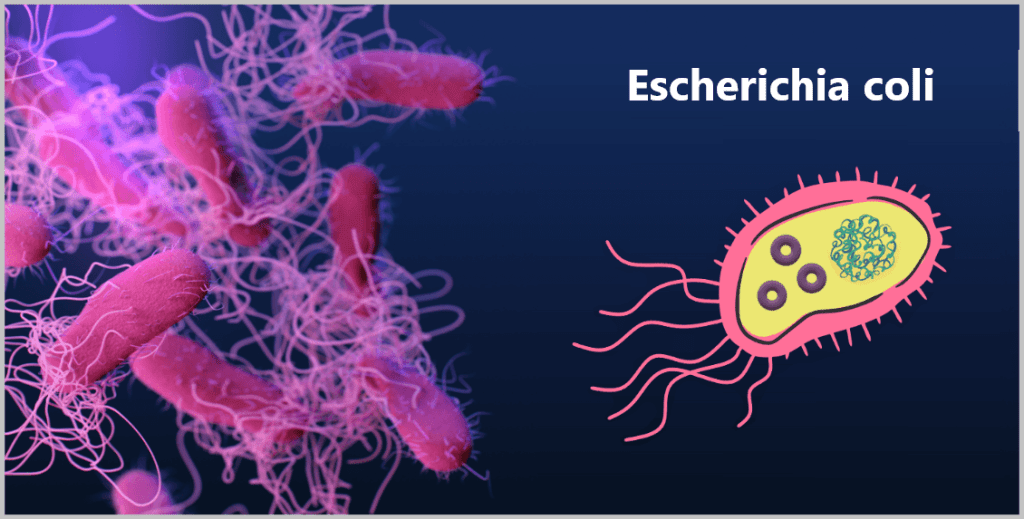
Links eine mikroskopische Aufnahme, rechts eine vereinfachte Darstellung mit DNA (großes Knäuel) und Plasmiden (kleine Ringe).
Weil E. coli molekularbiologisch und genetisch der am besten untersuchte Organismus ist, nennen Wissenschaftler es scherzhaft das „Haustier der Genetiker“.
Aus diesen Gründen ist E. coli einer der wichtigsten Helfer in der Biotechnologie – und spielt auch bei der Impfstoffentwicklung eine zentrale Rolle.
Eine kleine Reise in eine Welt des E. coli Bakterium
Escherichia coli. – farblos, fast durchsichtig – kaum länger als ein Tausendstel eines Sandkorns – wirkt unscheinbar. Und doch ist es alles andere als langweilig.
Von außen sieht es aus wie eine kleine Kapsel, doch unter seiner Haut verbirgt sich eine der geschäftigsten Megastädte zur Hauptverkehrszeit.
Kein Platz für Leere. Molekül an Molekül. Proteine, Ribosomen, Nukleinsäuren – sie drängen sich, stoßen aneinander, weichen aus. Ein Zustand, den Biologen nüchtern Macromolecular Crowding nennen, der sich hier aber anfühlt wie permanentes Gedränge. Und trotzdem – oder gerade deswegen – funktioniert alles mit atemberaubender Präzision.
Mitten in diesem Gedränge liegt das Gedächtnis der Zelle: ein einziges, ringförmiges DNA-Molekül. Hier steht alles geschrieben: wie man Zucker frisst, wie man schwimmt, wie man sich teilt. Würde man diese DNA vorsichtig auseinanderziehen, wäre sie tausendmal länger als die Zelle selbst. Ein Band mit Millionen von Buchstaben, dicht gepackt, mehrfach verdreht, zu Schleifen und Windungen gezwungen. Supercoiling nennt man diese Kunst des Zusammenfaltens – als hätte jemand eine kilometerlange Geschichte in eine Streichholzschachtel gezwängt, ohne ein Wort zu verlieren.
Doch so mächtig diese DNA auch ist, sie spricht selten selbst. Stattdessen schickt sie unablässig Boten aus: RNA. Tausende, Zehntausende. Kurze Nachrichten, Arbeitsanweisungen, Baupläne. Manche sind nur flüchtige Zettel – mRNA, kaum tausend Zeichen lang, mit einer Lebensdauer von Minuten. Andere sind stabiler, massiver: rRNA, die tragenden Säulen der Ribosomen. Und dann die kleinen tRNA-Moleküle, kaum mehr als Handlanger, die doch an entscheidenden Stellen die richtigen Bausteine anliefern. Sie flitzen wie Kuriere durch das Zytoplasma. Ihr Ziel?
Die Ribosomen – die eigentlichen Giganten dieser Welt. Zehntausende von ihnen treiben durch das Zytoplasma. Sie lesen die flüchtigen mRNA-Botschaften und verwandeln sie in greifbare Realität: Proteine. Sie sind lautlos, unermüdlich, und sie verschlingen einen großen Teil der Ressourcen der Zelle. Fast ein Viertel aller Proteine hier existiert nur, um neue Proteine herzustellen.
Und diese Proteine sind überall. Millionen von ihnen, in tausend Formen. Enzyme, die Reaktionen beschleunigen, die ohne sie Tage oder Jahre dauern würden – hier geschehen sie in Sekundenbruchteilen. Einige arbeiten in Massen, immer wieder dieselbe Aufgabe. Andere sind seltene Spezialisten, vielleicht nur ein paar Dutzend Exemplare, aber im entscheidenden Moment unverzichtbar. Dazwischen patrouillieren die Wächter: RNasen, die alte Botschaften zerlegen. Sobald ein RNA-Befehl ausgeführt wurde, zerlegen sie ihn wieder in seine Einzelteile. So stapelt sich kein Informationsmüll, und die Bausteine können für neue Befehle recycelt werden. Mittendrin – wohldosiert: DNasen. Sie passen derweil auf, dass der wertvolle Masterplan im Archiv repariert wird, falls mal etwas kaputtgeht. Gleichzeitig vernichten sie fremde Eindringlinge und gewinnen daraus neue Energie.
All das spielt sich innerhalb einer Hülle ab, die ständig erneuert werden muss: eine lebendige Grenze aus Lipiden, deren äußere Schicht Endotoxine trägt. Und sie steht unter Zeitdruck. Denn dieses E. coli lebt nicht, um zu verharren. Es lebt, um sich zu teilen.
Unter günstigen Bedingungen zählt es die Zeit in Minuten. Zwanzig, vielleicht dreißig – dann muss aus einer Zelle zwei werden. Während im Inneren kopiert, gelesen und gebaut wird, wächst die Oberfläche mit. Millionen neuer Lipidmoleküle entstehen, fügen sich ein, erweitern die schützende Mauer. Es ist ein Wettlauf ohne Pause, ein kontrolliertes Chaos mit klarer Richtung.
Und dann kommt der Moment der Teilung. Kein dramatischer Schnitt, kein Ende – eher ein leises Auseinandergehen. Die Mutterzelle hört auf, als Individuum zu existieren. Doch ihre Geschichte zerreißt nicht. Sie setzt sich fort, zweimal. In zwei Zellen, die beide alt und neu zugleich sind. Jede trägt dieselbe lange DNA in sich, denselben Lärm aus Molekülen, dieselbe rastlose Ordnung.
Man kann sich die Welt in der E.-coli-Zelle noch um eine weitere, fast schon geheimnisvolle Ebene erweitert vorstellen:
Neben dem großen, ringförmigen Chromosom treiben kleinere DNA-Ringe durch das Gedränge. Plasmide. Sie sind wie lose Seiten im inneren Archiv der Zelle – nicht lebensnotwendig im strengen Sinn, aber oft entscheidend fürs Überleben. Manche tragen Rezepte für neue Fähigkeiten: Resistenz gegen ein Gift, ein Enzym, das eine ungewöhnliche Nahrungsquelle erschließt, manchmal nur ein kleines taktisches Extra für schlechtere Zeiten.
Sie bewegen sich frei, stoßen gegen Ribosomen, werden kurz von Proteinen gepackt, wieder freigegeben. Auch sie werden gelesen, ihre Gene in RNA übersetzt, ihre Botschaften landen auf denselben Ribosomen wie die des großen Chromosoms. In der überfüllten Stadt gibt es keine Sonderrechte – nur Funktion.
Wenn die Zelle sich auf die Teilung vorbereitet, geraten die Plasmide in Bewegung. Sie dürfen nicht vergessen werden. Spezielle Proteine sorgen dafür, dass sich Kopien bilden und dass jede der entstehenden Tochterzellen wenigstens ein Exemplar erhält. Ein stiller Akt der Weitergabe, fast wie das Zustecken eines Briefes im Gedränge, bevor sich zwei Wege trennen.
Manchmal endet ihre Geschichte aber nicht in der eigenen Nachkommenschaft. Gelegentlich öffnet sich eine Verbindung nach außen, zu einer Nachbarzelle. Dann wechseln Plasmide den Besitzer. Ein kurzer Kontakt, ein Transfer, und eine Information springt von einer Geschichte in eine andere. Fähigkeiten verbreiten sich so schneller als jede Mutation des Chromosoms es je könnte.
In dieser Welt sind Plasmide die Gerüchte, die Baupläne, die Überlebenskniffe, die nicht fest in Stein gemeißelt sind. Beweglich, austauschbar, opportunistisch. Und genau dadurch passen sie perfekt in das rastlose, gedrängte Innenleben von E. coli, in dem nichts stillsteht – nicht einmal das Erbgut selbst.
Empfehlenswert sind die Illustrationen von David Goodsell. Er zeichnet das Innere von Zellen maßstabsgetreu – seine Bilder von E. coli machen dieses „Gedränge“ der Moleküle erst so richtig greifbar.
c) Plasmide – kleine DNA-Ringe mit großer Wirkung
Plasmide sind kleine, ringförmige DNA-Moleküle, die zusätzlich zum Bakterienchromosom in der Zelle vorkommen. Sie sind deutlich kleiner als das Hauptchromosom, liegen in variabler Anzahl vor und können sich unabhängig davon vervielfältigen. Im Gegensatz zur linearen DNA von Eukaryoten (z. B. beim Menschen) bilden Plasmide geschlossene Kreise aus doppelsträngiger DNA (dsDNA).
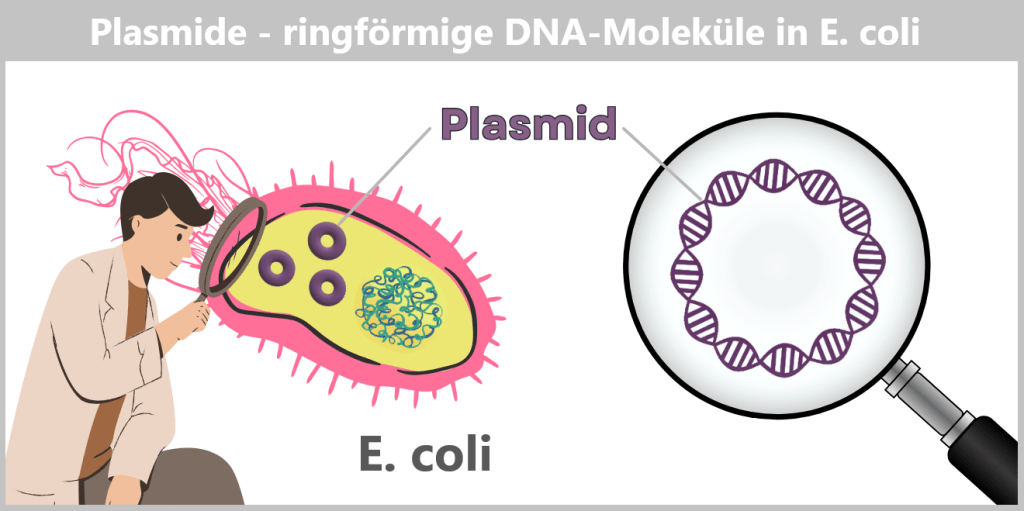
Plasmide können ganz unterschiedliche Gene tragen – etwa für Antibiotikaresistenz oder für die Herstellung bestimmter Proteine. Damit sie von Forschern gezielt genutzt werden können, erstellt man sogenannte Plasmidkarten: schematische Darstellungen, die die wichtigsten Funktionsbereiche zeigen.
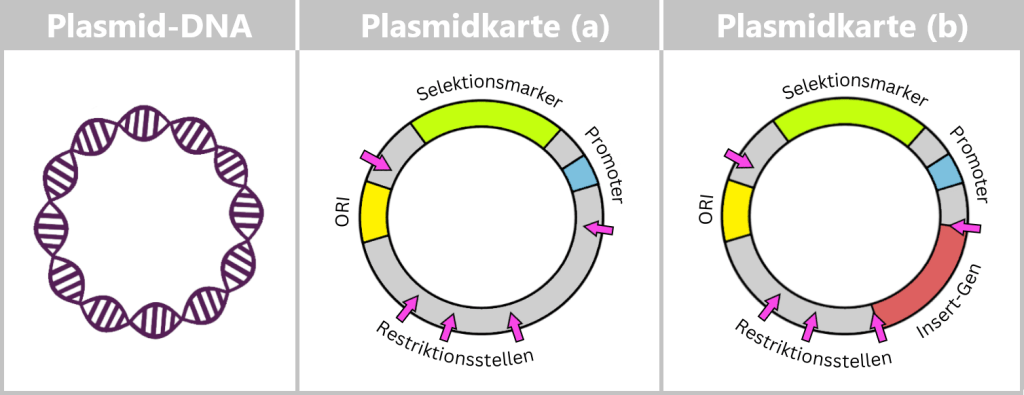
ORI (Origin of Replication): Startpunkt für die Verdopplung des Plasmids. Wenn Umweltbedingungen und interne Signale günstig sind, kann das Bakterium diesen Startknopf drücken, und das Plasmid macht eine Kopie von sich selbst. Nur wenn dieser ORI mit dem Bakterium „kompatibel“ ist, kann sich das Plasmid zuverlässig vermehren, auch unabhängig von der Zellteilung.
Selektionsmarker: Gene, die den Bakterien einen Vorteil verschaffen, z. B. Resistenz gegen ein bestimmtes Antibiotikum. Sie helfen Forschern zu erkennen, welche Bakterien das Plasmid tragen.
Promoter: Der Promoter ist eine spezielle DNA-Region, die die Aktivität von Genen steuert. Er reguliert, wann und wie stark bestimmte Gene auf dem Plasmid abgelesen werden und steuert somit die Produktion der entsprechenden Proteine.
Restriktionsstellen: sind kurze DNA-Sequenzen, die von Restriktionsenzymen erkannt und gezielt geschnitten werden können. Diese Enzyme dienen Bakterien als eine Art Immunsystem, um die DNA von eindringenden Viren zu zerschneiden und sich so zu verteidigen.
d) Plasmide in der Gentechnik
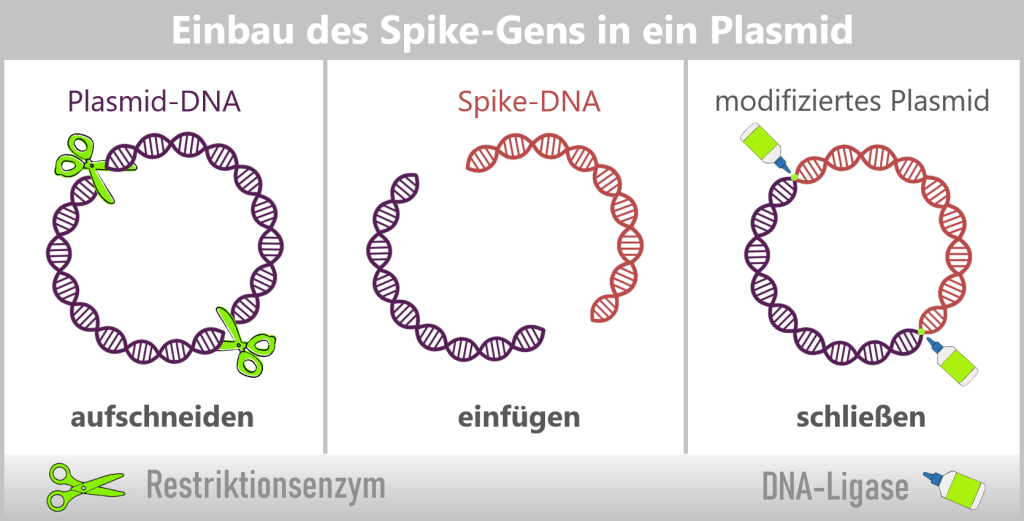
Eigentlich dienen Restriktionsenzyme in Bakterien der Abwehr fremder DNA. In der Gentechnik nutzt man sie jedoch als präzise Werkzeuge: winzige molekulare Scheren, die DNA an ganz bestimmten Stellen schneiden.
So lässt sich eine „Lücke“ in einem Plasmid erzeugen, in die ein gewünschtes Gen eingefügt wird. Ein weiteres Enzym, die DNA-Ligase, „verklebt“ anschließend die DNA-Enden wieder. Das eingefügte Gen nennt man Insert-Gen (siehe obere Abbildung).
Durch dieses Verfahren verwandeln sich Plasmide in kleine Gen-Fähren: Sie transportieren gezielt neue Gene in Bakterien. Bei jeder Zellteilung wird das Plasmid – und damit auch das Insert-Gen – automatisch mitkopiert. Auf diese Weise entstehen ganze Bakterienkulturen, die als „Mini-Fabriken“ bestimmte Proteine oder DNA in großen Mengen herstellen.
🎥 Tipp: Eine kurze, animierte Einführung zu Plasmiden gibt es hier.
e) Einbau der Spike-Protein-DNA in ein Plasmid
Für die Vermehrung des Spike-Proteins von SARS-CoV-2 wird zunächst das Spike-Gen in ein bakterielles Plasmid integriert – ein Vorgang, den man als „Einklonieren“ bezeichnet. Dabei kommen die zuvor beschriebenen molekularbiologischen Werkzeuge zum Einsatz: Restriktionsenzyme öffnen das ringförmige Plasmid an definierten Stellen, während DNA-Ligasen das Spike-Gen passgenau einfügen und die DNA-Enden dauerhaft verbinden. Auf diese Weise entsteht ein rekombinantes (neu zusammengesetztes) Plasmid, das die genetische Information für das gewünschte Antigen enthält.
DNA-Elemente im Plasmid
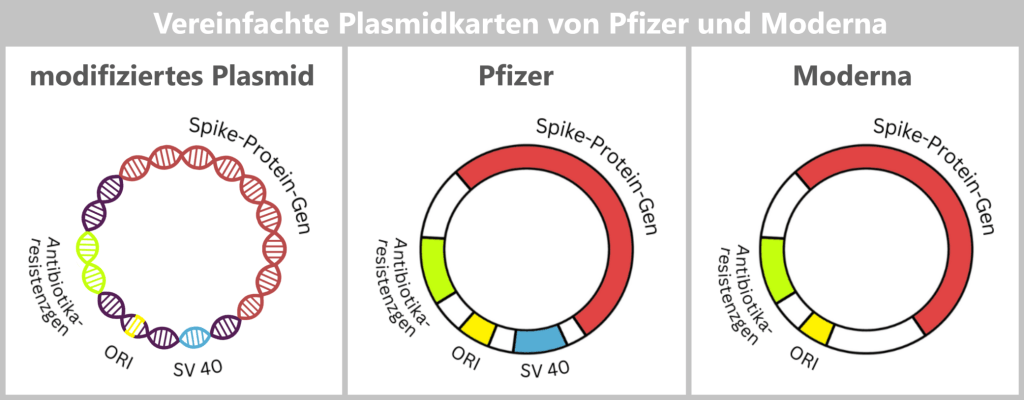
Die Spike-DNA wird nicht isoliert in das Plasmid eingebracht, sondern zusammen mit weiteren genetischen Elementen, die für die Vermehrung des Plasmids erforderlich sind.
ORI (Origin of Replication): Der Replikationsursprung legt fest, an welcher Stelle die Vervielfältigung des Plasmids innerhalb der Bakterienzelle beginnt. Ohne dieses Element könnte das Plasmid in E. coli nicht stabil vermehrt werden.
Spike-Protein-Gen: Dieses Gen enthält den Bauplan für das Spike-Protein – das zentrale Antigen des Impfstoffs.
Antibiotikaresistenzgen: Das Resistenzgen dient als Selektionsmarker. Es stellt sicher, dass unter Antibiotikadruck nur diejenigen Bakterien überleben, die das gewünschte Plasmid aufgenommen haben. Dadurch können geeignete Bakterienklone gezielt ausgewählt und vermehrt werden.
- Moderna: Kanamycin-Resistenzgen
- Pfizer/BioNTech: Neo/Kan-Resistenzgen (Resistenz gegen Neomycin und Kanamycin)
SV40-Komponenten: Das Produktionsplasmid von Pfizer/BioNTech enthält zusätzlich regulatorische Sequenzelemente, die vom Simian Virus 40 (SV40) abgeleitet sind – einem Affenvirus, dessen genetische Elemente seit Jahrzehnten in der Molekularbiologie verwendet werden.
Konkret handelt es sich um:
- einen SV40-Promoter/Enhancer,
- sowie Teile des SV40-Replikationsursprungs.
Solche Sequenzen werden in der Gentechnik genutzt, weil sie die Genexpression in Säugetierzellen verstärken und die Plasmidvermehrung in bestimmten Zelllinien erleichtern können.
Für die bakterielle Vermehrung in E. coli besitzen diese Elemente jedoch keine bekannte Funktion, da Bakterien die hierfür notwendigen zellulären Faktoren nicht besitzen.
Warum diese SV40-Sequenzen im endgültigen Produktionsplasmid enthalten sind, ist nicht vollständig öffentlich dokumentiert. Diskutiert wird unter anderem, dass sie aus früheren Entwicklungs- oder Testsystemen übernommen und später beibehalten wurden.
Nach aktuellem Kenntnisstand verwendet Moderna in seinem Produktionsplasmid keine vergleichbaren SV40-Komponenten.
Die unterschiedliche Verwendung solcher regulatorischer Sequenzen gehört zu den Aspekten, die im Zusammenhang mit DNA-Rückständen wissenschaftlich diskutiert werden.
Eine genauere Darstellung der Plasmidkarten findet man hier.
f) Übertragung der modifizierten Plasmide in Bakterien
Die veränderten Plasmide, die nun den Bauplan für das Spike-Protein tragen, werden anschließend in E. coli-Bakterien eingeschleust – ein Prozess, der als Transformation bezeichnet wird. Dabei nehmen die Bakterien die Plasmide auf; diese verbleiben dauerhaft in der Bakterienzelle und werden bei jeder Zellteilung weitergegeben.
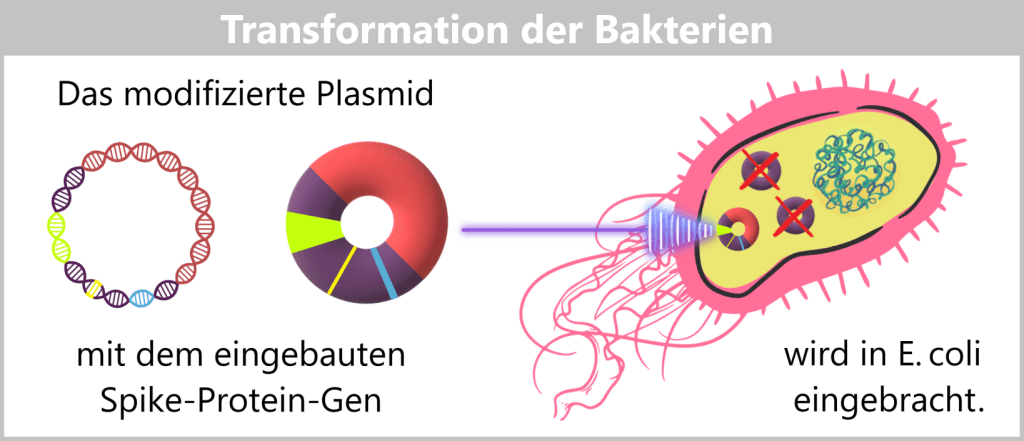
Die modifizierten Plasmide werden mittels Transformation in E. coli-Bakterien eingebracht. Plasmide, die in der Gentechnik für den Transport fremder DNA-Sequenzen genutzt werden, heißen Vektoren.
Plasmidfreie Wirtszellen – für saubere Klone
Da in der biotechnologischen Plasmidproduktion ausschließlich die modifizierten Plasmide hergestellt werden sollen, verwendet man spezielle, plasmidfreie Bakterienstämme, die keine eigenen, natürlichen Plasmide besitzen.
Das hat mehrere Gründe:
- Natürliche Plasmide könnten um die zelluläre Replikationsmaschinerie konkurrieren,
- sie könnten durch Rekombination DNA-Abschnitte austauschen,
- und sie würden die genetische Zusammensetzung der Kolonie unvorhersehbar machen.
Durch den Einsatz plasmidfreier Wirtszellen wird sichergestellt, dass alle Bakterien einer Kolonie genetisch identische Klone sind – sie enthalten das gleiche, definierte Plasmid mit der gewünschten Sequenz.
g) Vermehrung der Bakterien
Die E. coli-Bakterien mit den modifizierten Plasmiden werden in einen Fermenter gegeben. Ein Fermenter, auch Bioreaktor genannt, ist ein Gerät, das in der Biotechnologie für die großtechnische Herstellung von Produkten wie Antibiotika, Enzymen, Vitaminen oder Impfstoffen genutzt wird. Darin lassen sich Bedingungen wie Temperatur, pH-Wert, Sauerstoffzufuhr, Rührgeschwindigkeit und Nährstoffversorgung exakt steuern.
Das Nährmedium im Fermenter enthält alle lebensnotwendigen Stoffe für die Bakterien. Unter diesen optimalen Bedingungen beginnen sie, sich rasant zu vermehren. Bei jeder Zellteilung werden auch die Plasmide mitkopiert, sodass sich die gewünschte DNA vervielfältigt.
Um sicherzustellen, dass nur Bakterien überleben, die tatsächlich das Spike-Gen-Plasmid tragen, wird zusätzlich ein Antibiotikum in den Fermenter gegeben. Nur Zellen mit Plasmid – und damit mit Antibiotikaresistenz – können wachsen. So entsteht eine Kultur, in der ausschließlich die gewünschten, modifizierten Bakterien vorkommen.
E. coli kann sich etwa alle 20–30 Minuten teilen. Innerhalb weniger Tage wächst so im Fermenter eine riesige Menge an Bakterien heran – mit Trillionen von Kopien des Spike-Plasmids.
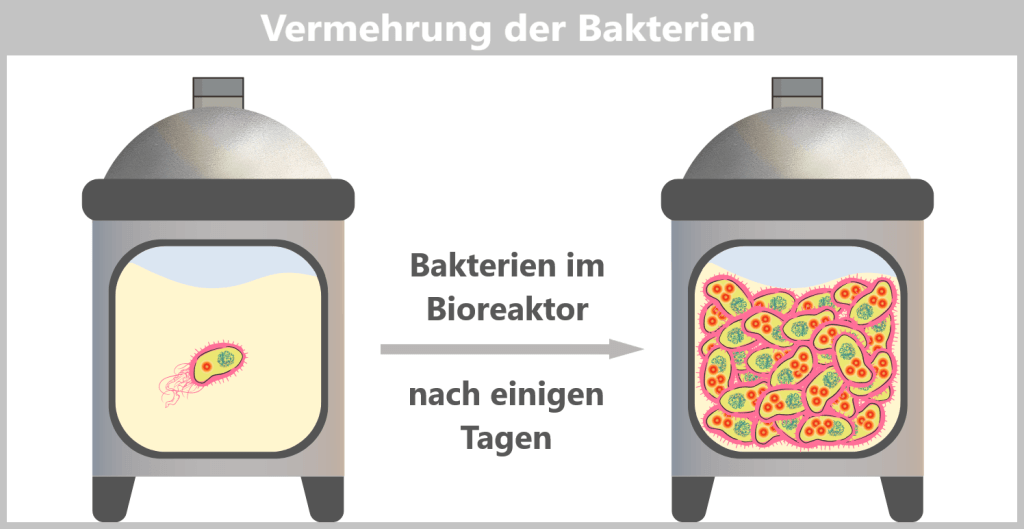
h) Ernte der Bakterien
Nach der Vermehrungsphase werden die E. coli-Zellen „geerntet“. Dazu wird der gesamte Inhalt des Fermenters – die Zellsuspension – in einen Erntetank überführt. Dort erfolgt die Trennung von Flüssigkeit und Zellen, meist durch Zentrifugation (schnelles Schleudern) oder Filtration. Am Ende entsteht ein Zellpellet, das heißt eine konzentrierte Sammlung von Bakterien am Boden des Behälters.
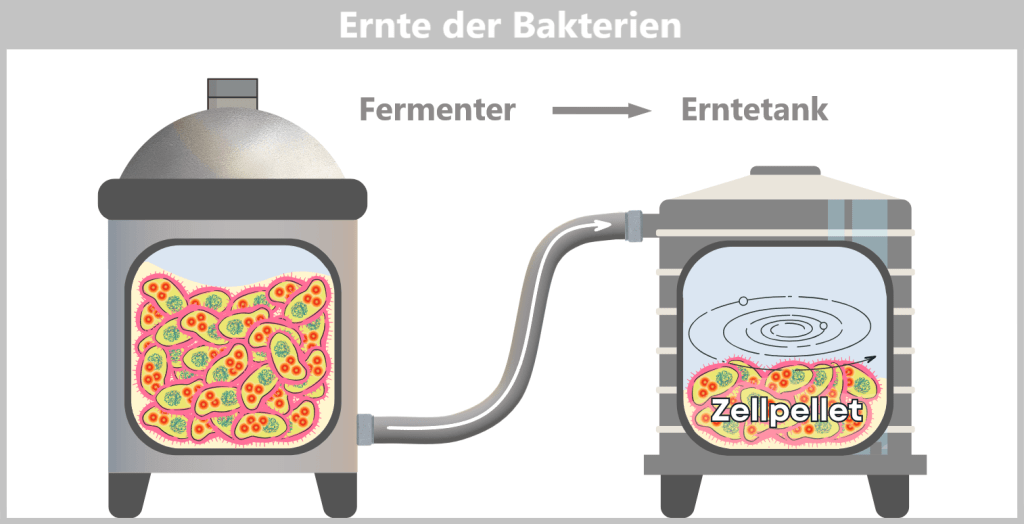
Der Zellpellet – nun von der Nährlösung getrennt – wird in eine Aufarbeitungsanlage überführt. Entweder wird er hierfür in einer Flüssigkeit fein verteilt, bis ein gleichmäßiger, pumpfähiger „Zellbrei“ entsteht oder er wird als fester Pellet automatisch weitergeleitet. In modernen Anlagen geschieht das alles in einem geschlossenen System, ohne dass die Biomasse offen mit der Umgebung in Kontakt kommt.
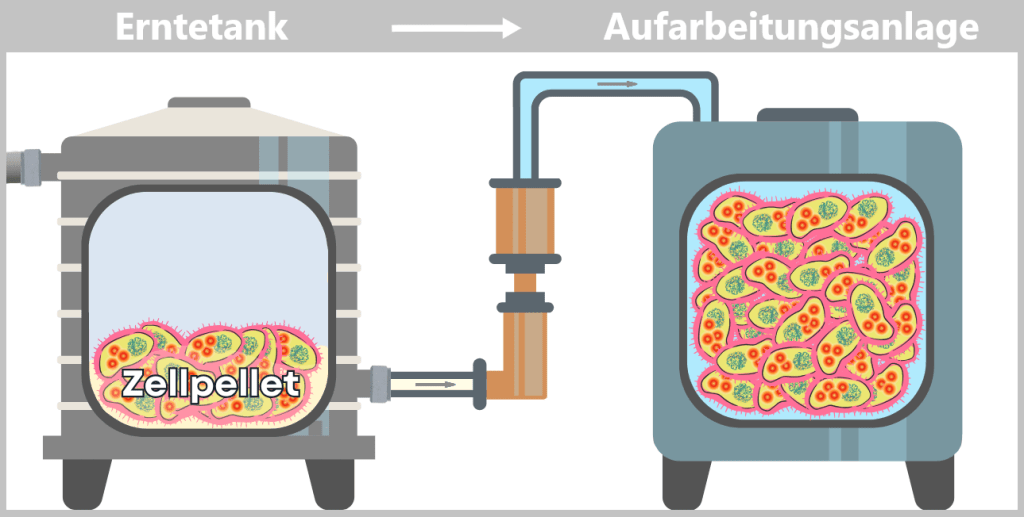
i) Isolierung der modifizierten Plasmide
Nach der Ernte der Bakterienkulturen folgt ein entscheidender Trennungsprozess: Die E. coli-Zellen werden in speziellen Aufarbeitungsanlagen gezielt lysiert, das heißt kontrolliert aufgebrochen.
Die Zugabe von Natronlauge (NaOH) und dem Detergens SDS (einem speziellenSeifenmolekül) löst die fetthaltige Zellhülle auf, vergleichbar mit der Wirkung von Spülmittel auf Fett.
Dabei wird der gesamte Zellinhalt freigesetzt – ein komplexes Gemisch aus den gewünschten Plasmiden, chromosomaler DNA, Proteinen, Membranbestandteilen und zahlreichen weiteren zellulären Komponenten.
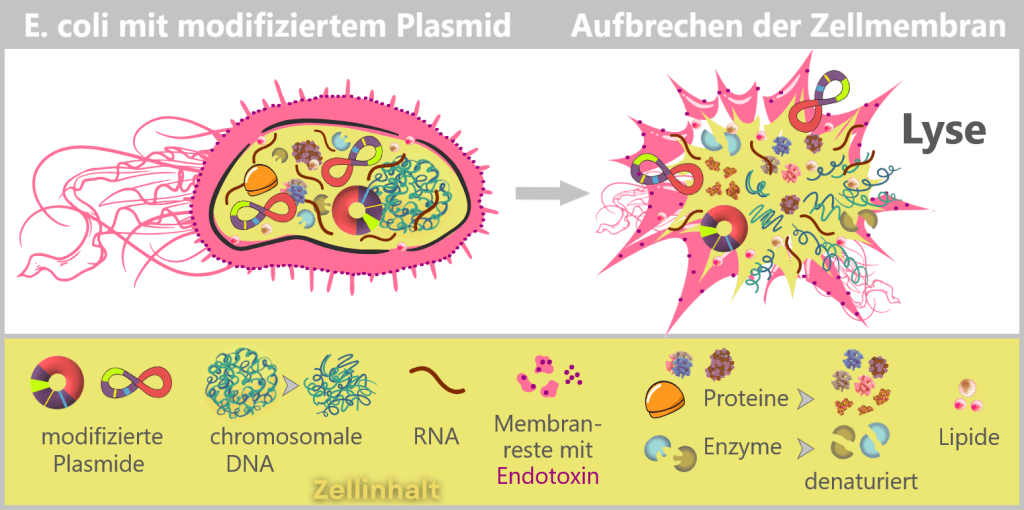
Links: Schematischer Querschnitt durch ein E. coli mit modifiziertem Plasmid. Die äußere Membran ist mit eingelagerten Endotoxin-Molekülen (lila) dargestellt.
Rechts: Nach dem Aufbrechen der Zellmembran (Lyse) treten die verschiedenen Zellbestandteile – modifizierte Plasmide, chromosomale DNA (Bakterienchromosom), Proteine, Enzyme und Lipide – aus. Die äußere Membran liegt in Fragmenten vor, die das strukturell gebundene Endotoxin (LPS) enthalten.
DNA-Formen in E. coli: Plasmid und Chromosom
Plasmide in Escherichia coli liegen als kleine, ringförmige DNA-Moleküle vor. In der Zelle befinden sie sich überwiegend in einer supercoiled Form. Diese Topologie (räumliche Anordnung) ist biologisch bevorzugt, da sie eine hohe mechanische Stabilität und Kompaktheit verleiht. Der geschlossene DNA-Ring ist dabei zusätzlich verdrillt, wodurch das Molekül platzsparend organisiert ist und gegenüber physikalischen Belastungen relativ widerstandsfähig bleibt.

Der Unterschied zwischen supercoiled (superhelikalen) und relaxed (entspannten, ringförmigen) Plasmiden liegt ausschließlich in ihrer Topologie. Beide Formen bestehen aus identischer genetischer Information, zeigen jedoch deutlich unterschiedliche physikalische Eigenschaften.
In der supercoiled Form steht das DNA-Molekül unter torsionaler Spannung und windet sich eng um sich selbst. Die relaxed Form entsteht typischerweise, wenn einer der beiden DNA-Stränge einen Einzelstrangbruch (Nick) erleidet. Durch diesen Nick kann die Spannung entweichen, und das Plasmid geht in eine offenere, weniger kompakte Ringstruktur über.
Das bakterielle Chromosom (genomische DNA, gDNA) ist ebenfalls ringförmig, aber um ein Vielfaches größer. Es ist hochgradig organisiert, mit Proteinen verknüpft und in viele Domänen gefaltet.
Ergänzende Erläuterungen zur Wirkung der alkalischen Lyse
Zerstörung der Genom-DNA, Erhalt der Plasmide
Während der alkalischen Lyse werden die Zellmembran und Zellwände aufgelöst. Die doppelsträngige DNA beider Molekülarten (Genom-DNA, Plasmid) wird kurzzeitig denaturiert und durch mechanische und chemische Scherkräfte belastet.
Dabei geschieht Folgendes:
- Die lange, faserige genomische DNA wird durch Scherkräfte zerrissen und linearisiert, da sie zu groß und empfindlich ist, um intakt zu bleiben.
- Die kompakte, superhelikale Plasmid-DNA bleibt hingegen weitgehend intakt.
Beim Neutralisieren der zuvor alkalischen Lösung kann sich die kleine, ringförmige Plasmid-DNA wieder korrekt renaturieren, d. h. ihre beiden Einzelstränge finden zueinander zurück und bilden erneut eine stabile Doppelhelix. Dadurch bleibt sie in Lösung.
Die wesentlich längere genomische DNA kann unter diesen Bedingungen nicht mehr vollständig renaturieren. Sie fällt gemeinsam mit Zellresten und Proteinen als unlöslicher Niederschlag aus.
Zerstörung von Proteinstrukturen
Die alkalische Lyse wirkt sich nicht nur auf Nukleinsäuren aus, sondern auch auf Proteine. Unter den stark basischen Bedingungen verlieren Proteine ihre dreidimensionale Faltung und damit ihre biologische Funktion. Auch große Komplexe wie die Ribosomen, die aus ribosomaler RNA und zahlreichen Proteinen bestehen, werden dabei vollständig zerstört und zerfallen in ihre Bestandteile. Während der anschließenden Neutralisation aggregieren (verklumpen) diese zerstörten Proteine und RNA-Fragmente – ähnlich wie Eiweiß beim Kochen fest wird – und bilden einen Niederschlag, der zusammen mit den restlichen Zelltrümmern leicht entfernt werden kann.
Warum nach der bakteriellen Lyse auch kleine RNA-Fragmente auftreten
Beim Aufbrechen der E. coli-Zellen gelangen nicht nur die Plasmide ins Lysat, sondern auch große Mengen bakterieller RNA. Diese besteht aus einer Vielzahl unterschiedlicher Moleküle, die für den Stoffwechsel der Bakterien zuständig waren – darunter viele kurze, stabile RNA-Typen. Zusätzlich werden beim Lysevorgang auch bakterielle Enzyme freigesetzt, die RNA abbauen (RNasen). Aktive RNasen können vorhandene RNA schnell in kleinere Stücke zerlegen. Auch mechanische Scherkräfte während der Lyse tragen zur Fragmentierung bei. Das Ergebnis ist ein komplexes Gemisch aus kurzen bakteriellen RNA-Fragmenten.
Natürliche RNA der Bakterienzelle
Die Bakterienzelle (E. coli) enthält bereits eine enorme Menge ihrer eigenen, natürlichen RNA – deutlich mehr als DNA. Beim Aufbrechen der Zellen (Lyse) gelangt diese gesamte zelluläre RNA mit in das Lysat und stellt eine bedeutende Verunreinigung dar. Dazu gehören:
- rRNA (ribosomale RNA): Macht den Hauptanteil (~80–90 %) der bakteriellen RNA aus und liegt in großen Mengen vor.
- tRNA (transfer-RNA): Sehr kleine, stabile Moleküle in vielen verschiedenen Varianten.
- Bakterielle mRNA: Ist in Bakterien von Natur aus kurzlebig und liegt oft bereits fragmentiert vor.
- Regulatorische kleine RNAs (sRNA): Umfassen natürliche RNA-Spezies von wenigen Dutzend Nukleotiden Länge.
Zusammenfassend bildet diese Gesamtheit an ribosomaler, transfer- und messenger-RNA den natürlichen zellulären RNA-Pool von E. coli.
Zerstörung der Membran und Freisetzung von Endotoxinen
Endotoxine (Lipopolysaccharide, LPS) sind ein natürlicher Bestandteil der äußeren Membran von Bakterien mit einer doppellagigen Zellhülle wie E. coli. In der Fachsprache werden solche Bakterien als ‚gramnegativ‘ bezeichnet. Sie sorgen für einen strukturellen Halt und helfen dem Bakterium bei Angriffen von außen. Bei der Lyse wird diese Membran zerstört, wodurch große Mengen LPS freigesetzt werden. Während Endotoxine für das Bakterium als „Rüstung“ fungieren, sind sie für den Menschen hochgiftig. Der biologisch aktive Bestandteil – Lipid A – kann beim Menschen starke Entzündungsreaktionen auslösen, weshalb Endotoxine zu den kritischsten Verunreinigungen in der biotechnologischen Produktion gehören.
Die Kunst besteht nun darin, die begehrten Plasmide aus dieser biochemischen Vielfalt zu isolieren und von allen unerwünschten Begleitstoffen zu befreien. Hierbei kommt eine mehrstufige Aufreinigung zum Einsatz, die wir später im Abschnitt Reinigungsverfahren im Herstellungsprozess detaillierter betrachten.
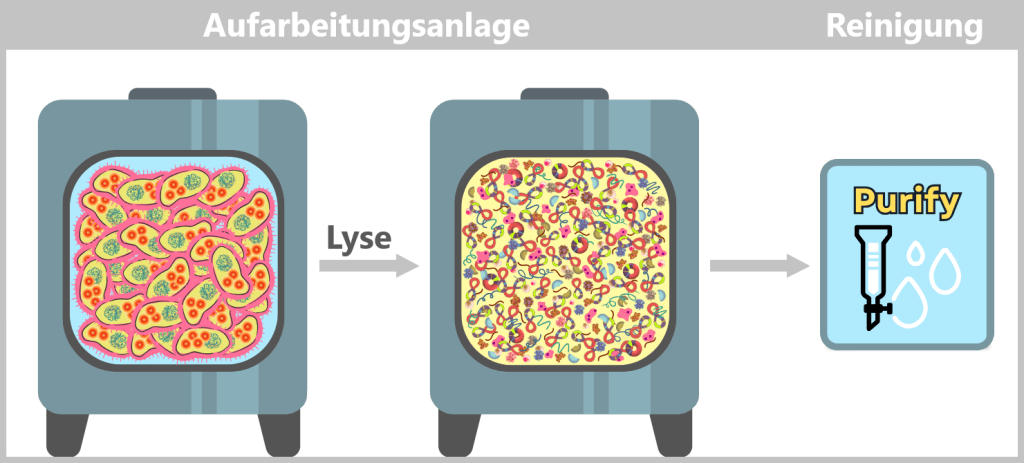
In der Aufarbeitungsanlage werden die geernteten Bakterienzellen lysiert. Dabei entsteht ein „Zellbrei“, der alle zellulären Bestandteile enthält. Anschließend beginnt die Reinigung, bei der die gewünschten Plasmide mit Hilfe spezieller Filter- und Chromatografie-Verfahren aus dem Gemisch herausgetrennt werden.
Nach der Aufreinigung liegen die ringförmigen Plasmide in hoher Reinheit vor.
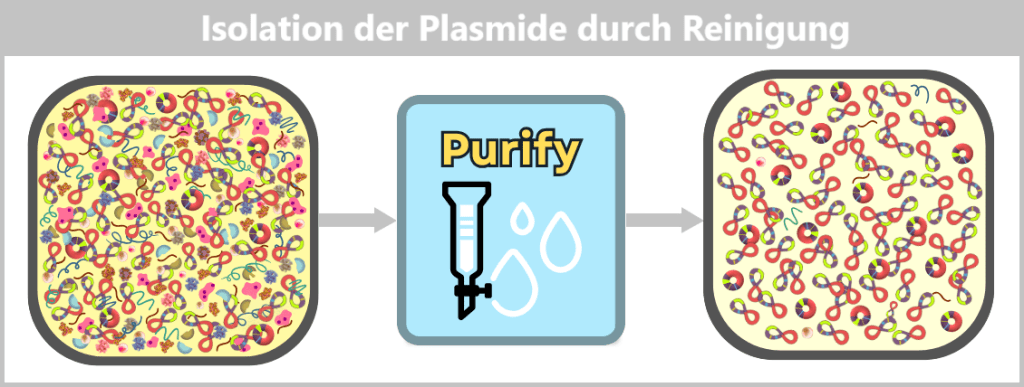
Nach der Aufreinigung liegt die Plasmid-DNA in hoher Reinheit vor. Neben der dominanten supercoiled Konformation ist ein geringer Anteil an relaxed Plasmiden sichtbar. Letztere entstehen durch Einzelstrangbrüche (nicks) während der Zelllyse oder Aufarbeitung.
j) Linearisierung der Spike-Protein-DNA
Die gereinigten ringförmigen Plasmide enthalten zwar bereits den Bauplan für das Spike-Protein, sind für den nächsten Schritt jedoch noch ungeeignet. Für die Transkription muss die kreisförmige Plasmid-DNA in eine lineare Form überführt werden.
Dazu werden die Plasmide gezielt geöffnet: Mithilfe eines spezifischen Restriktionsenzyms erfolgt ein präziser Schnitt an einer definierten Stelle, üblicherweise nach dem Ende des Spike-Protein-Gens. Auf diese Weise entstehen klare DNA-Enden, die das Ablesen der Sequenz erleichtern.
Durch diesen Schnitt wird das gesamte Plasmid von seiner ringförmigen in eine lineare, offene DNA-Kette überführt. Diese enthält nicht nur das Spike-Gen, sondern auch alle übrigen Plasmidbestandteile, wie den Ursprung der Replikation (ORI), Selektionsmarker oder regulatorische Elemente. Eine besonders wichtige dieser Sequenzen ist der T7-Promotor, dies wird im nächsten Schritt deutlich.
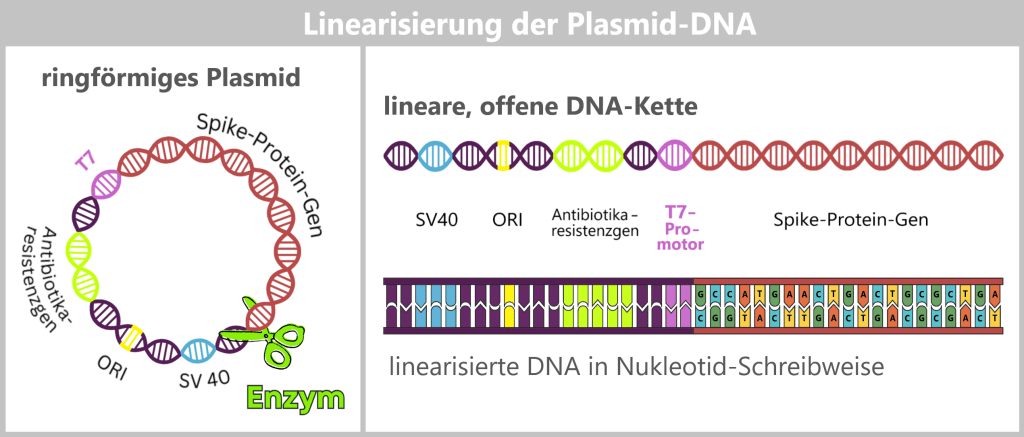
Das ringförmige Plasmid wird mit einem Restriktionsenzym aufgeschnitten und in eine lineare DNA-Kette überführt. Diese enthält verschiedene Abschnitte: den Ursprung der Replikation (ORI, gelb), Selektionsmarker (hellgrün), regulatorische Sequenzen wie SV40 (blau) und den T7-Promotor (lila), sowie das eigentliche Spike-Gen (rot). In der detaillierten Darstellung unten ist die Nukleotidfolge des Spike-Gens angedeutet. Die gesamte Nukleotidfolge ist stark gekürzt dargestellt.
Weitere Reinigung nach der Linearisierung
Die Linearisierung der Plasmid-DNA führt zur Entstehung zusätzlicher Reaktionsbestandteile und Nebenprodukte, die vor der in-vitro-Transkription entfernt werden müssen. Hierzu zählen insbesondere:
- Restriktionsenzyme: Die für den gezielten Schnitt eingesetzten Endonukleasen dürfen nicht im weiteren Herstellungsprozess verbleiben.
- DNA-Nebenprodukte: Hierzu gehören unvollständig linearisierte Plasmide (z. B. residuale supercoiled oder relaxed Formen) sowie kurze DNA-Fragmente, die durch unspezifische Brüche oder Nebenreaktionen entstehen können.
- Salze und Pufferreste: Die Linearisierungsreaktion erfolgt in spezifischen Puffersystemen, deren Ionen und Zusatzstoffe die nachfolgende Prozessschritte beeinträchtigen könnten.
Durch geeignete Reinigungsverfahren wird eine hochreine, lineare DNA-Vorlage gewonnen, die als Template für den nächsten Prozessschritt dient.
Der Begriff hochrein beschreibt in diesem Kontext keinen absoluten Zustand, sondern die weitgehende Entfernung prozessrelevanter Verunreinigungen auf ein regulatorisch akzeptiertes Minimum. Geringe Restmengen nicht-linearer Plasmidformen können verbleiben und werden in nachfolgenden Prozessschritten weiter reduziert.
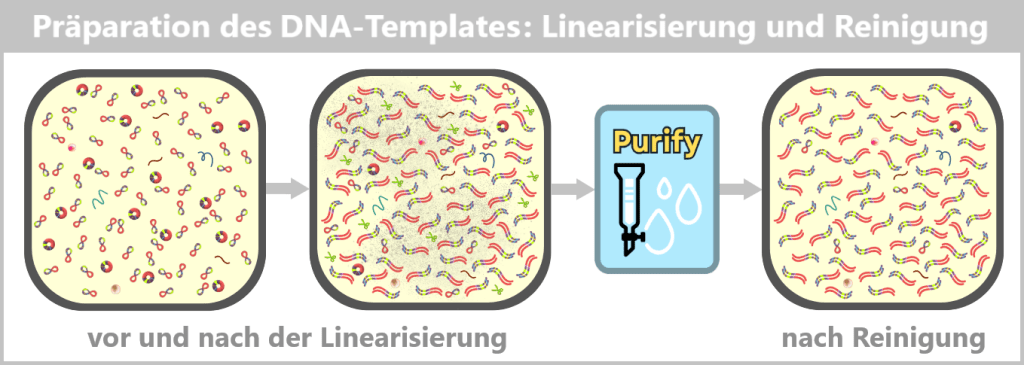
Links: Die Plasmid-DNA-Lösung vor der Linearisierung. Sie enthält ein Gemisch aus supercoiled (überwiegend) und relaxeden (offenen) Ringformen.
Mitte: Die Lösung nach der Linearisierung durch ein Restriktionsenzym. Die ringförmigen Plasmide wurden an einer definierten Stelle geschnitten und liegen nun als lineare, doppelsträngige DNA-Moleküle vor. Die Lösung enthält aber auch Reste des Enzyms, Puffersalze und mögliche Nebenprodukte.
Rechts: Die Lösung nach der Reinigung. Störende Bestandteile wie das Restriktionsenzym, Pufferkomponenten und unerwünschte DNA-Fragmente wurden entfernt.
Schritt 3: In-Vitro-Transkription zur Herstellung der mRNA
Nachdem die Plasmid-DNA linearisiert wurde, dient sie nun als Vorlage für die in-vitro-Transkription (IVT). Ziel dieses Schrittes ist es, aus der DNA das RNA-Molekül herzustellen, das später als mRNA-Impfstoff eingesetzt wird.
Transkription – das „Umschreiben“ der DNA in RNA
Die Transkription erfolgt in einem separaten Bioreaktor unter kontrollierten Bedingungen (z. B. spezifischer pH-Wert, Temperatur, Ionenstärke), die speziell für die RNA-Synthese optimiert sind. Dafür sind drei Dinge notwendig:
1) Das DNA-Template, das das Spike-Gen enthält.
2) Die RNA-Bausteine, sogenannte Nukleotide.
3) Ein Enzym – die T7-RNA-Polymerase, das spezifisch den T7-Promotor erkennt. Der T7-Promotor wurde im Plasmid direkt vor die Spike-Gen-Sequenz eingefügt und markiert den Startpunkt der Transkription.
Ablauf der Transkription
Initiation (Start): Die T7-RNA-Polymerase bindet an den T7-Promotor. Sobald das Enzym dort verankert ist, öffnet es die DNA-Doppelhelix um die Transkriptionsstartstelle und legt einen Strang als DNA-Template frei.
Die DNA enthält verschiedene funktionelle Abschnitte, darunter den Ursprung der Replikation (gelb), Selektionsmarker (hellgrün), regulatorische Elemente (blau), den T7-Promotor sowie die Spike-Gen-Sequenz (rot). Die Polymerase erkennt den T7-Promotor und setzt genau an dieser Stelle auf. Anschließend öffnet sie den DNA-Doppelstrang und beginnt mit der Synthese des RNA-Strangs entlang des Spike-Gens.
Elongation (Verlängerung): Die Polymerase wandert am DNA-Templatestrang in 3′ → 5′-Richtung entlang und synthetisiert dabei den komplementären RNA-Strang durch schrittweise Anlagerung von Nukleotiden in 5′ → 3′-Richtung. Dabei gilt die komplementäre Basenpaarung:
- DNA-A (Adenin) → RNA-U (Uracil bzw. m¹Ψ)
- DNA-T (Thymin) → RNA-A (Adenin)
- DNA-C (Cytosin) → RNA-G (Guanin)
- DNA-G (Guanin) → RNA-C (Cytosin)
Hinter der Polymerase schließt sich die DNA wieder.
Termination (Ende): Am Ende des Spike-Gens befindet sich eine Terminator-Sequenz. Sobald diese erreicht ist, stoppt die Polymerase, löst sich von der DNA, und die fertige RNA wird freigesetzt.
Die T7-RNA-Polymerase lagert sich an die DNA an und trennt die beiden Stränge. Entlang des Templatstrangs werden passende RNA-Nukleotide eingebaut. Schrittweise entsteht so ein einzelsträngiger RNA-Strang, der die Information des Spike-Gens trägt.
Die RNA, die auf diese Weise entsteht, ist einzelsträngig und entspricht in ihrer Basenfolge dem Spike-Gen.
Besonderheit der mRNA-Herstellung
Um die Stabilität und Verträglichkeit für den Einsatz im Impfstoff zu verbessern, wird ein modifizierter Baustein verwendet:
Anstelle von Uridin (U) wird N¹-Methyl-Pseudouridin (m¹Ψ) eingebaut.
Diese Modifikation macht die RNA stabiler, schützt sie vor zu schnellem Abbau und reduziert unerwünschte Immunreaktionen.
Sowohl die mRNA-Impfstoffe von Pfizer als auch von Moderna enthalten N¹-Methyl-Pseudouridin (m¹Ψ) anstelle von Uridin. [spektrum]
Vergleich DNA, mRNA und modRNA
Aus der doppelsträngigen DNA entsteht im Verlauf der in-vitro-Transkription eine einzelsträngige RNA. DNA und RNA ähneln sich in ihrer chemischen Grundstruktur, unterscheiden sich jedoch in entscheidenden Eigenschaften wie Stabilität, Lebensdauer und biologischer Funktion. Für mRNA-Impfstoffe kommt zudem keine natürliche mRNA zum Einsatz, sondern eine gezielt chemisch modifizierte Form (modRNA). Diese unterscheidet sich von natürlicher mRNA in mehreren wesentlichen Punkten.
Die folgende Tabelle stellt die Eigenschaften von DNA, natürlicher mRNA und synthetischer modRNA gegenüber.
| Eigenschaft | DNA | Natürliche mRNA | Synthetische modRNA |
| Definition | Master-Archiv: dauerhafter Speicher der gesamten Erbinformation | Abschrift eines einzelnen Gens: enthält die Bauanleitung für ein körpereigenes Protein | Kopie eines Gens mit „Tarnkappe“: im Labor hergestellte (synthetische) Form der mRNA, die gezielt chemisch verändert wurde. Sie enthält den Bauplan für ein „nicht körpereigenes“ Protein. |
| Vorkommen | Universell: Zellkern (bei Eukaryoten); zusätzlich mitochondriale DNA. | Zellspezifisch: Wird bedarfsgerecht nur dort hergestellt, wo das entsprechende Protein benötigt wird. | Unspezifisch: kann mithilfe von Lipid-Nanopartikeln in viele Zelltypen des Körpers aufgenommen werden. |
| Struktur | doppelsträngig | einzelsträngig | einzelsträngig |
| Zucker | Desoxyribose | Ribose | Ribose |
| Basen | A – Adenin C – Cytosin G – Guanin T – Thymin | A – Adenin C – Cytosin G – Guanin U – Uracil | A – Adenin C – Cytosin G – Guanin m¹Ψ – N1-Methylpseudouridin |
| Lebensdauer und Abbau-Geschwindigkeit | Sehr stabil: bleibt durch Kernmembran und Reparatursysteme geschützt; wird im Normalfall nicht abgebaut, sondern nur bei Zelltod oder gezieltem DNA-Abbau. | Kurzlebig: Minuten bis Stunden. Die Protein-produktion ist flexibel an den aktuellen Stoff-wechselbedarf angepasst. | Verlängert: Stunden bis Tage. Durch den Austausch von Uridin durch N1-Methylpseudouridin wird die modRNA weniger stark von RNA-Sensoren des angeborenen Immunsystems erkannt und langsamer abgebaut. |
| Abbau-Enzyme | DNasen (Desoxyribonukleasen) | RNasen (Ribonukleasen) | RNasen (verminderte Erkennung und verlangsamter Abbau) |
IVT-Nebenprodukte und Verunreinigungen
Nach der in-vitro-Transkription (IVT) liegt kein reines Produkt vor, sondern ein komplexes Gemisch. Neben der gewünschten mRNA entstehen prozessbedingt verschiedene Nebenprodukte und Verunreinigungen, darunter kurze oder lange einzelsträngige RNA (ssRNA), doppelsträngige RNA (dsRNA) sowie RNA:DNA-Hybride. Diese Begleitprodukte müssen gezielt entfernt werden, da sie andernfalls die Stabilität, Wirksamkeit und Verträglichkeit des Impfstoffs beeinträchtigen können. Die hierfür eingesetzten Reinigungsmethoden werden im Kapitel 1.3. näher erläutert.
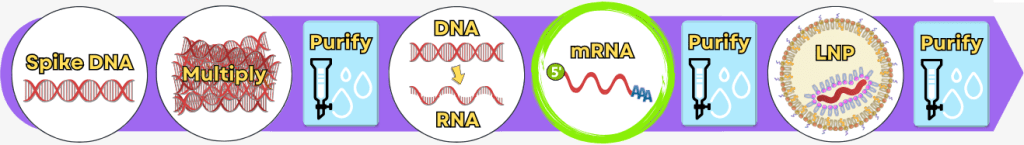
Schritt 4: RNA-Prozessierung – Reifung der mRNA
Die RNA-Prozessierung umfasst eine Reihe von Veränderungen, die während oder nach der Transkription stattfinden, um aus der RNA eine reife, funktionale mRNA zu erzeugen.
Bei der Herstellung der mRNA für Impfstoffe wird versucht, die natürlichen Prozesse so weit wie möglich nachzuahmen, die normalerweise auch in menschlichen Zellen stattfinden. Die synthetisch hergestellte mRNA wird so gestaltet, dass sie bestimmte Eigenschaften von natürlich vorkommender mRNA imitiert und dadurch stabil ist sowie effizient in das gewünschte Protein übersetzt werden kann.
Eine funktionelle Impfstoff-mRNA braucht wie eine normale menschliche mRNA:
- eine Schutzkappe (5′-Cap) am vorderen Ende der RNA
- einen Stabilisierungsschwanz (Poly-A-Schwanz) am hinteren Ende
Der 5‘-Cap: Ein „Sicherheitssiegel“ für die Zelle
Damit eine künstlich hergestellte mRNA im Körper funktioniert, muss sie am sogenannten 5′-Ende eine spezielle chemische Schutzstruktur tragen – die Cap-1-Struktur. Diese Kappe ist ein zentrales Erkennungsmerkmal für die Zelle und entscheidet darüber, ob die mRNA stabil bleibt, effizient übersetzt und nicht als fremd erkannt wird.
Die Bauteile der Cap-1-Struktur (m⁷GpppN¹m)
Die Cap-1-Struktur ist kein einfacher „Deckel“, sondern ein präzise aufgebautes Molekül aus drei funktionellen Komponenten:
Das Erkennungsmerkmal: 7-Methylguanosin (m⁷G)
Ein spezieller Guanosin-Baustein mit einer Methylgruppe. Diese Markierung wirkt wie ein molekularer Ausweis: Nur mRNAs mit dieser Struktur werden von der Zelle als korrekt und vertrauenswürdig erkannt.
Die Verbindung: Triphosphatbrücke (ppp)
Drei Phosphatgruppen verbinden die Kappe in einer ungewöhnlichen 5′-5′-Verknüpfung mit der mRNA. Diese spezielle Bindung schützt das RNA-Ende effektiv vor enzymatischem Abbau.
Die Tarnung: modifiziertes erstes Nukleotid (N¹m)
Das erste Nukleotid der mRNA ist zusätzlich 2′-O-methyliert. Diese Modifikation ist entscheidend, um die mRNA vor der Erkennung durch zelluläre RNA-Sensoren zu schützen.
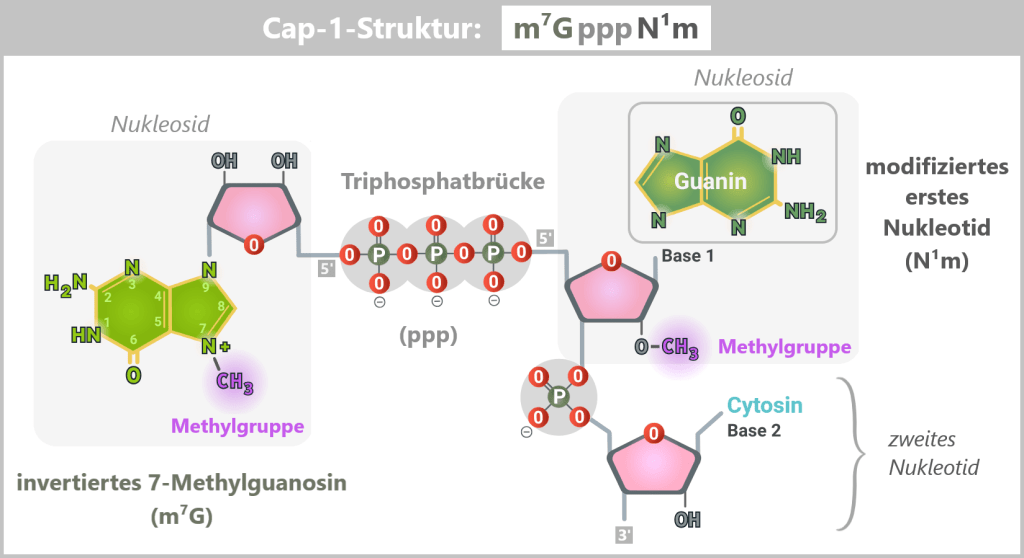
Das 5′-Ende der mRNA ist durch ein invertiert verknüpftes, am N7-Atom methyliertes Guanosin (m⁷G) über eine 5′–5′-Triphosphatbrücke (ppp) mit dem ersten Nukleotid (in diesem Beispiel Guanin) der mRNA verbunden. Zusätzlich trägt dieses erste Nukleotid eine 2′-O-Methylierung (N¹m), die die Cap-1-Struktur definiert und zur Stabilität, Translationseffizienz und Immuntarnung der mRNA beiträgt.
Bei der natürlichen Genexpression in menschlichen Zellen wird mRNA bereits während ihrer Entstehung im Zellkern prozessiert. Insbesondere erhalten alle durch die RNA-Polymerase II synthetisierten mRNA-Moleküle sehr früh während der Transkription eine 5′-Cap-Struktur sowie begleitende chemische Modifikationen.
Ungecappte mRNA ist in eukaryotischen Zellen unter physiologischen Bedingungen nicht funktionsfähig: Sie ist instabil, wird rasch abgebaut und gelangt nicht in das Zytoplasma. Dies liegt daran, dass ihr 5′-Ende als 5′-Triphosphat (5′-ppp) vorliegt – eine Struktur, die von zellulären Qualitätskontrollsystemen als Anomalie erkannt und beseitigt wird (siehe untere Abbildung).
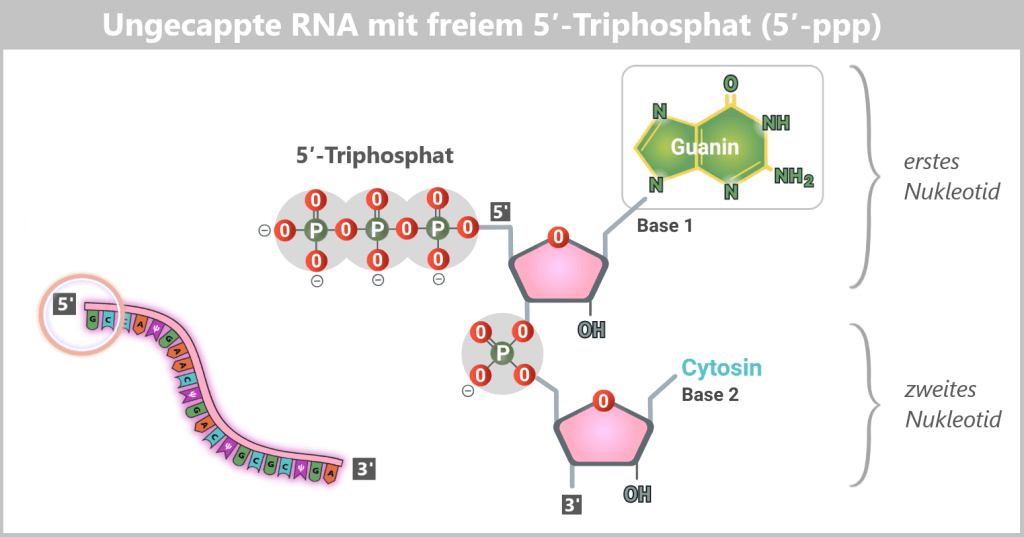
Warum ist Cap-1 so wichtig?
Die Cap-1-Struktur erfüllt drei zentrale Funktionen:
Tarnung (Immunschutz): Zelluläre Mustererkennungsrezeptoren wie RIG-I reagieren empfindlich auf RNA mit freien 5′-Triphosphaten oder unmodifizierten Enden. Die Cap-1-Struktur der modRNA ist chemisch identisch zur Cap-1-Struktur körpereigener mRNA. Da zelluläre RNA-Sensoren diese Struktur als „selbst“ erkennen, wird unter physiologischen Bedingungen keine ausgeprägte angeborene Immunantwort ausgelöst.
Produktionsstart: Das Cap dient als Andockstelle für cap-bindende Proteine und markiert den Startpunkt für die Proteinsynthese durch Ribosomen.
Stabilität: Die Kappe schützt das 5′-Ende der mRNA vor raschem enzymatischem Abbau und verlängert so ihre funktionelle Lebensdauer in der Zelle.
Der Poly-A-Schwanz: Das Schutz- und Kontrollzentrum am Ende
Am anderen Ende der mRNA (dem sogenannten 3′-Ende) befindet sich eine lange Kette, die ausschließlich aus Adenin-Bausteinen besteht – der Poly-A-Schwanz. In der Impfstoffherstellung wird dieser meist aus 100 bis 150 „A“-Gliedern gefertigt.
Wofür ist dieser Schwanz gut?
Die „Sanduhr“ der mRNA: Die Zelle besitzt Enzyme, die mRNA-Moleküle von hinten nach vorne langsam „anknabbern“. Der Poly-A-Schwanz wirkt hier wie ein Puffer oder ein Schutzstück am Ende. Er wird zuerst abgebaut, bevor die eigentliche genetische Information angegriffen wird. Je länger der Schwanz, desto länger überlebt die mRNA in der Zelle und desto mehr Protein kann hergestellt werden.
Übersetzungsbeschleuniger: Spezifische Proteine binden gleichzeitig an das 5′-Cap und den Poly-A-Schwanz und bilden den sogenannten closed-loop-Komplex – eine effiziente „Rundlaufstrecke“. Zum einen signalisiert er dem Ribosom (der Proteinfabrik), dass die mRNA vollständig und für die Translation bereit ist. Zum anderen kann das Ribosom nach Beendigung der Synthese eines Proteins direkt am 5′-Ende erneut mit der Translation beginnen. Dieser Mechanismus steigert die Geschwindigkeit und Ausbeute der Proteinproduktion erheblich.
Zusammenfassend: Während das 5′-Cap die mRNA legitimiert, tarnt und für die Translation verfügbar macht, bestimmt der Poly-A-Schwanz, wie lange und wie oft die Zelle die Information nutzt. Beide Modifikationen sind daher entscheidend für die Lebensdauer der mRNA und für ihre effiziente Übersetzung in Protein.
In den folgenden schematischen Darstellungen wird die Cap-1-Struktur als funktionelle 5′-Einheit vor dem RNA-Strang dargestellt, obwohl sie chemisch eine modifizierte Erweiterung des ersten Nukleotids darstellt. Der Poly-A-Schwanz wird aus didaktischen Gründen durch drei Adeninreste symbolisiert.
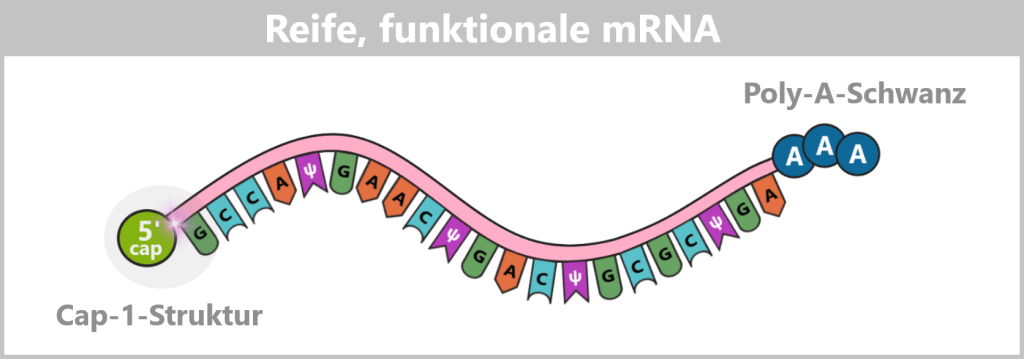
Die Abbildung ist stark vereinfacht; die tatsächliche mRNA ist deutlich länger und umfasst beim COVID-19-Impfstoff ca. 4200 Nukleotide.
In der biotechnologischen Herstellung haben sich zwei Methoden etabliert, um diese essenziellen Strukturen zu erzeugen:
1. Co-transkriptionelle Modifikation (Die „All-in-One“-Methode)
Hierbei werden Cap und Poly-A-Schwanz bereits während der in-vitro-Transkription (IVT) synthetisiert.
mRNA-Capping: Der Reaktion werden bereits fertige Cap-Bausteine (Cap-Analoga) zugegeben. Die T7-RNA-Polymerase baut diese Cap-Struktur automatisch als erstes Element an das entstehende mRNA-Molekül an.
Polyadenylierung: Das DNA-Template enthält bereits eine Sequenz, die als Matrize für einen definiert langen Poly-A-Schwanz dient. Die Polymerase synthetisiert ihn somit direkt im Anschluss an die kodierende Sequenz.
Vorteil: Der Prozess ist schnell, skalierbar und findet in einem einzigen Reaktionsgefäß statt. Durch die Verwendung moderner Cap-Analoga (z. B. ARCA oder CleanCap) werden Capping-Effizienzen von >95 % erreicht.
In der industriellen Praxis ist die co-transkriptionelle Methode der vorherrschende Standard.
2. Enzymatische Modifikation (Die “Step-by-Step”-Methode)
Bei diesem traditionelleren, mehrstufigen Verfahren werden Cap und Poly-A-Schwanz in separaten Reaktionsschritten nach der IVT angefügt.
Polyadenylierung: Zunächst wird die RNA mit dem Enzym Poly(A)-Polymerase behandelt. Dieses Enzym ist darauf spezialisiert, am 3′-Ende der RNA eine lange Adenin-Kette anzubauen. Die mRNA erhält also nachträglich ihren Poly(A)-Schwanz.
mRNA-Capping: Im Anschluss wird in einer weiteren Reaktion die 5′-Cap-Struktur schrittweise aufgebaut.
Vorteil: Erzielt eine sehr hohe und saubere Capping-Effizienz, ist jedoch aufwändiger.
Unabhängig vom gewählten Verfahren schließt sich eine umfassende Reinigung an, um eine homogene und hochreine mRNA zu erhalten.
Die wesentlichen Schritte – von der reinen DNA-Vorlage bis zum formulierungsfertigen Produkt – sind in der folgenden Übersicht dargestellt:
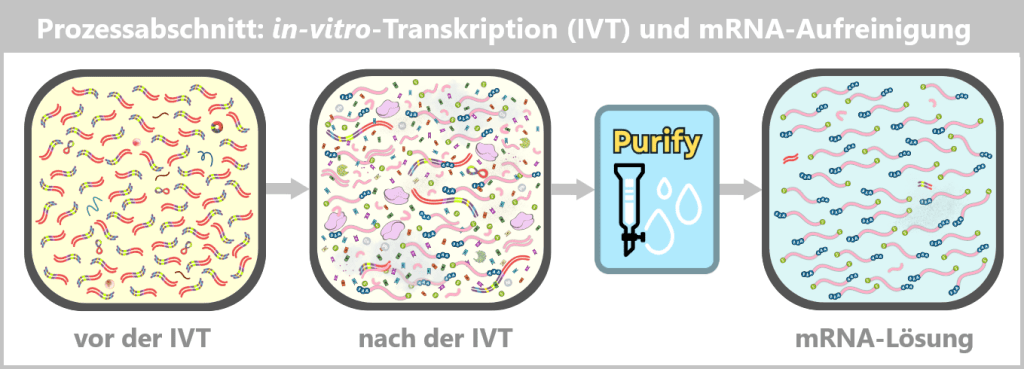
Nach der IVT liegt eine komplexe Mischung aus mRNA, Template-DNA, Enzymen, Nebenprodukten und Reaktionskomponenten vor. Durch nachfolgende Reinigungsschritte wird diese Mischung auf eine überwiegend reine mRNA-Lösung reduziert, die als Ausgangsmaterial für die LNP-Formulierung dient.
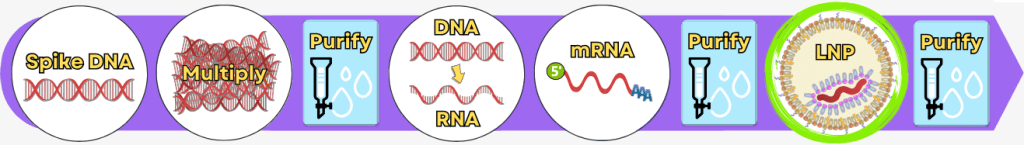
Schritt 5: Verpackung der mRNA in Lipid-Nanopartikel (LNPs)
Nachdem die mRNA vollständig hergestellt und gereinigt wurde, muss sie noch geschützt und transportfähig gemacht werden. Genau das übernimmt die Verpackung in Lipid-Nanopartikel (LNPs).
Warum braucht die mRNA eine Verpackung?
mRNA ist ein sehr empfindliches Molekül. Ohne Schutz würde sie im Körper sofort abgebaut werden. LNPs übernehmen mehrere Aufgaben gleichzeitig:
- sie schützen die mRNA vor Abbau
- sie transportieren sie in die Körperzellen (durch die Zellmembran)
- sie ermöglichen die kontrollierte Freisetzung der mRNA in der Zelle
Ohne LNPs wäre der Impfstoff nicht funktionsfähig.
Wie entsteht ein LNP? – Der Grundmechanismus
Die Formulierung passiert typischerweise in einem Gerät wie einem Microfluidizer oder Nanopartikel-Mischer. Dabei werden zwei Flüssigkeiten extrem schnell miteinander vermischt:
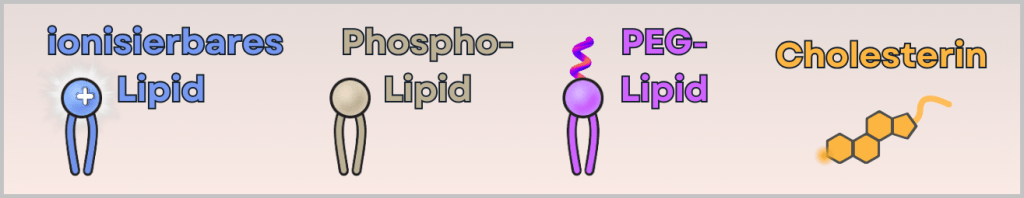
1) Eine ethanolische Lipid-Lösung
Sie enthält vier verschiedene Lipide:
- ionisierbares kationisches Lipid (bindet die mRNA und ermöglicht Zellaufnahme)
- Phospholipid (stabilisiert die Struktur – ähnlich wie in Zellmembranen)
- PEG-Lipid (sorgt für die richtige Größe und verhindert Aggregation)
- Cholesterin (macht das Partikel flexibel und stabil)
2) Eine wässrige mRNA-Lösung
Diese enthält nur:
- gereinigte mRNA
- einen milden Puffer
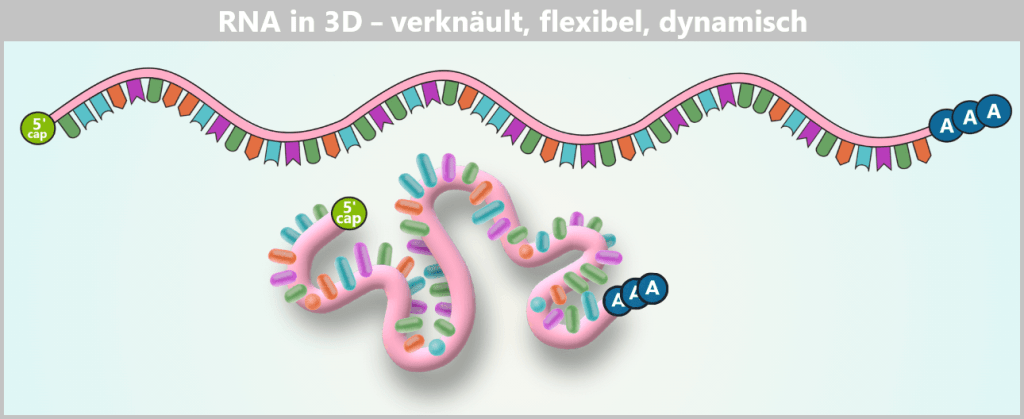
Oben ist die RNA als linearer Informationsstrang dargestellt – so liest man die Basensequenz. Unten ist gezeigt, wie derselbe Strang in wässriger Lösung tatsächlich existiert: als flexibles, ständig bewegtes 3D-Knäuel, in dem einzelne Abschnitte nur lose oder temporär miteinander wechselwirken.
Was passiert beim Mischen – ein Selbstorganisationseffekt
Wenn die beiden Lösungen in Sekundenbruchteilen aufeinandertreffen, ändern sich:
- pH-Wert
- Löslichkeit der Lipide
- Ladungszustände
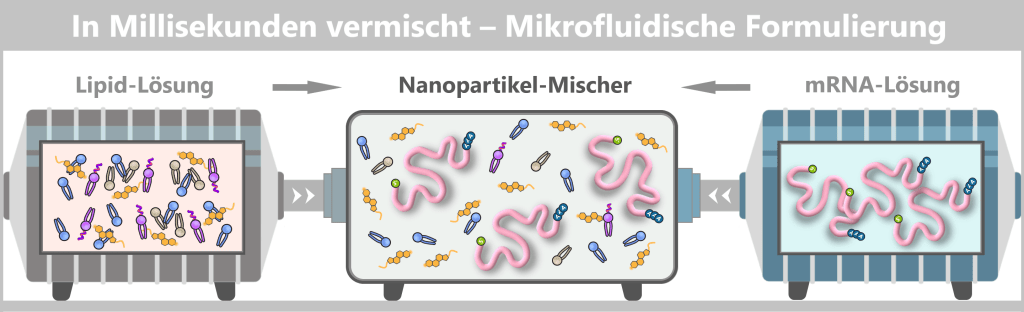
Die Abbildung zeigt schematisch das Prinzip beim mikrofluidischen Mischen. Links und rechts strömen zwei getrennte Flüssigkeiten in den Mischer ein: eine Lipid-Lösung (links) mit den verschiedenen Lipidtypen und eine mRNA-Lösung (rechts) mit frei gelösten mRNA-Strängen. Im zentralen Mikrofluidik-Kanal treffen beide Ströme aufeinander und werden intensiv verwirbelt.
Dadurch passiert ein spontaner, hochpräziser Prozess:
- Das ionisierbare Lipid wird positiv geladen → es zieht die negativ geladene mRNA an.
- Die mRNA wird „eingewickelt“ und eingeschlossen.
- Die anderen Lipide ordnen sich drumherum zu einer stabilen Hülle an.
Es entsteht automatisch ein Nanopartikel mit typischer Größe von 60–100 nm. Es ist also kein „manuelles Verpacken“, sondern eine biophysikalische Selbstorganisation – die Moleküle finden von selbst die richtige Struktur.
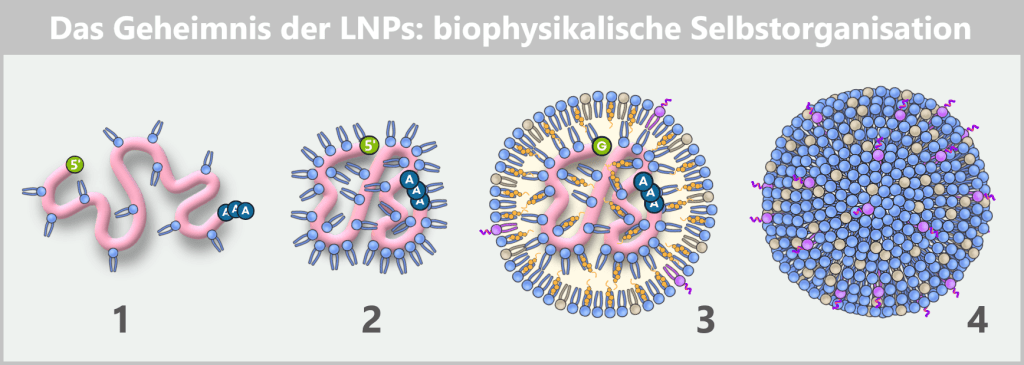
Die Abbildung zeigt in vier Schritten, wie sich mRNA und Lipide während der Formulierung ganz von selbst zu einem Lipid-Nanopartikel organisieren.
1) Sobald mRNA und ionisierbare Lipide aufeinandertreffen, binden die positiv geladenen Lipidköpfchen sofort an die negativ geladenen Phosphate der mRNA und beginnen, den Strang eng zu umhüllen.
2) Dadurch zieht sich die mRNA lokal zusammen, wird kompakter und verdichtet sich weiter.
3) Auf diese Weise entsteht ein erster „Kern“ aus mRNA-Lipid-Komplexen. Darum herum lagern sich weitere ionisierbare Lipide an – zusätzlich auch Phospholipide, Cholesterin und PEG-Lipide. Zusammen stabilisieren sie die entstehende Struktur, während die mRNA zunehmend dichter eingeschlossen wird.
4) Schließlich bildet sich ein dicht gepackter, nahezu kugelförmiger Nanopartikel. Seine Form entsteht nicht durch äußere Steuerung, sondern allein aus den physikalisch-chemischen Eigenschaften der Moleküle: ein Beispiel für spontane Selbstorganisation auf der Nanoskala.
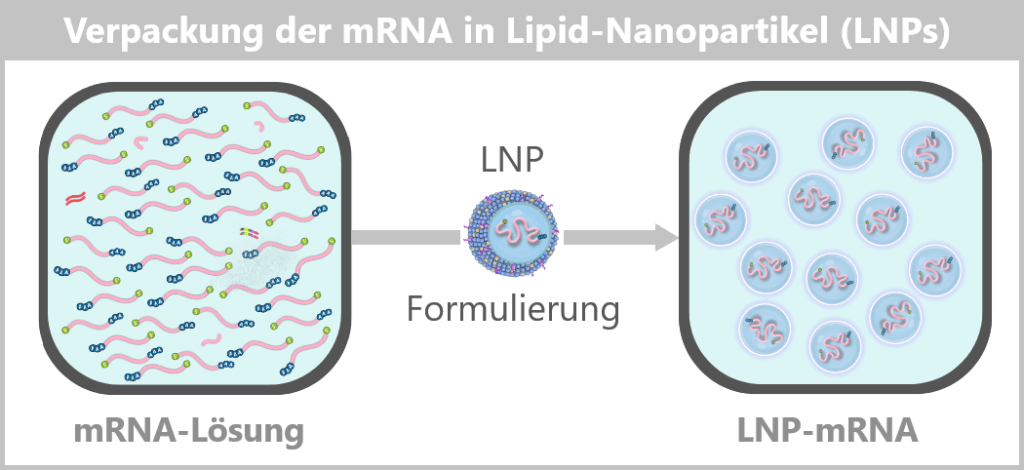
Die so formulierten LNPs bilden nun das fertige Wirkstoffkonzentrat, das im nächsten Schritt steril abgefüllt wird.
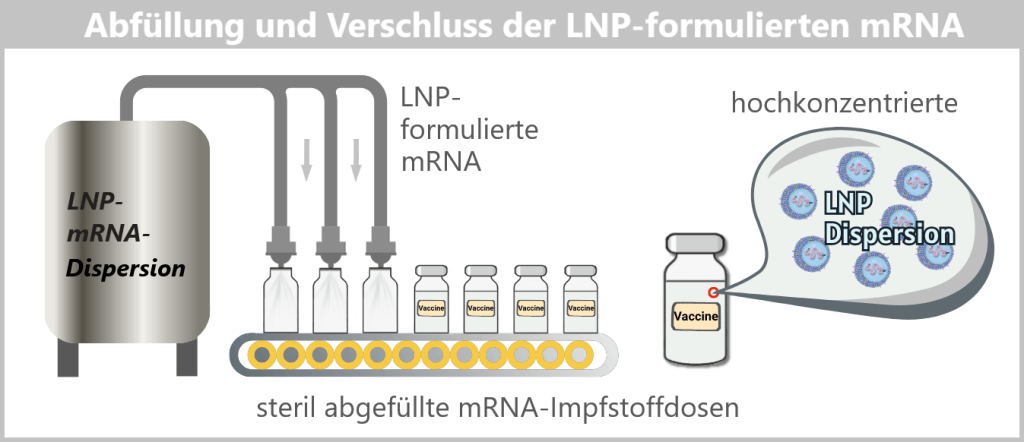
Ein Impfstoff-Vial enthält eine sehr große Anzahl einzelner Lipid-Nanopartikel, typischerweise im Bereich von 10¹³–10¹⁴ Partikeln pro Fläschchen.
1.2. Verunreinigungen und Nebenprodukte
Die Herstellung einer funktionsfähigen mRNA umfasst mehrere aufeinanderfolgende Prozessschritte – von der Vervielfältigung der DNA-Vorlage bis zur Prozessierung und Formulierung der RNA. In nahezu jedem dieser Schritte entstehen neben dem gewünschten Produkt auch Nebenprodukte, Verunreinigungen oder Reststoffe.
1.2.1. Typische Verunreinigungen bei der bakteriellen Herstellung
1.2.2. Typische Verunreinigungen und Nebenprodukte nach der IVT
Für die Sicherheit und Wirksamkeit des Therapeutikums ist deren Identifizierung und spätere Abtrennung im Downstream Prozess entscheidend.
In diesem Abschnitt werfen wir einen Blick auf die wichtigsten potenziellen Verunreinigungen und Nebenprodukte und ihre bekannten biologischen Aktivitäten.
1.2.1. Typische Verunreinigungen bei der bakteriellen Herstellung
Nach der Lyse der Bakterienzellen liegt die Plasmid-DNA in einer komplexen Reaktionsmischung vor. Neben der gewünschten Plasmid-DNA enthält die Lösung chromosomale DNA, bakterielle RNA, Proteine, Enzyme (einschließlich RNasen), Endotoxine sowie Zellwandfragmente.
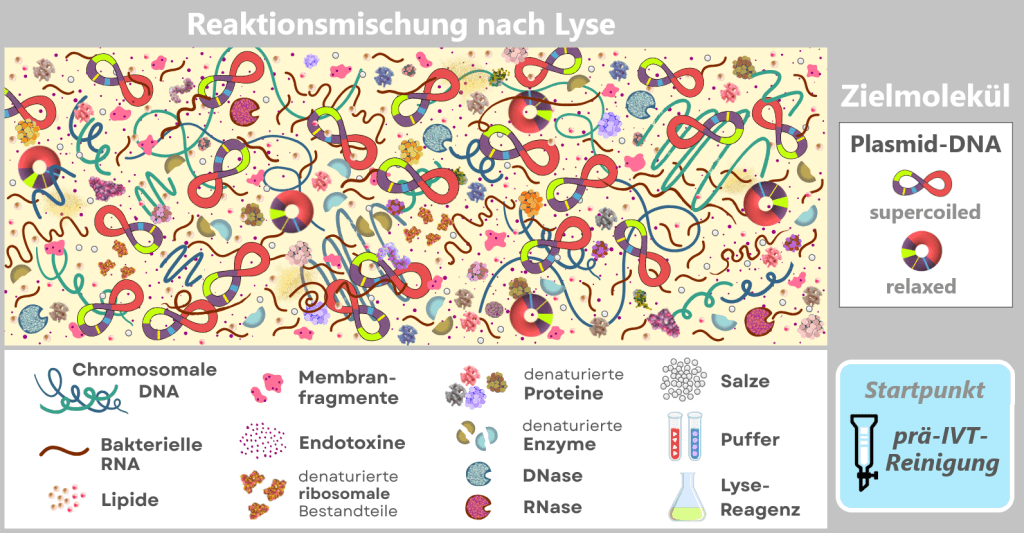
Die Abbildung zeigt schematisch die Zusammensetzung der Lösung nach dem Aufschluss der E.-coli-Zellen, in denen das Plasmid vervielfältigt wurde. Neben der gewünschten Plasmid-DNA, die die spätere Expressionskassette für die mRNA enthält, sind zahlreiche bakterielle Bestandteile und Prozesshilfsstoffe vorhanden. Die dargestellten Komponenten sind in eine proteinreiche zytosolische Matrix eingebettet, die aus dem bakteriellen Zytoplasma stammt (gelber Hintergrund).
Diese komplexe und heterogene Mischung verdeutlicht, dass die Plasmid-DNA zunächst aus einer stark verunreinigten biologischen Umgebung isoliert werden muss, bevor sie als Vorlage für die in-vitro-Transkription eingesetzt werden kann.
1.2.2. Typische Verunreinigungen und Nebenprodukte nach der In-vitro-Transkription
Obwohl die in-vitro-Transkription (IVT) eine effiziente Methode zur enzymatischen Herstellung von mRNA darstellt, liefert die Reaktion kein reines Endprodukt. Im Anschluss an die Synthese liegt ein komplexes Gemisch vor, das neben der Ziel-mRNA verschiedene prozessbedingte Verunreinigungen und Nebenprodukte enthält.
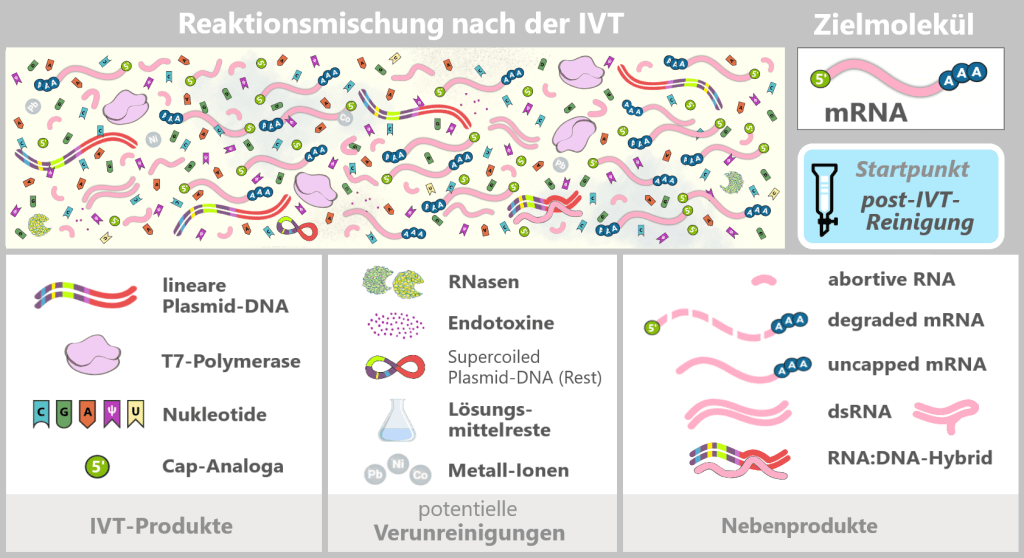
Die Abbildung zeigt schematisch die komplexe Zusammensetzung der IVT-Reaktionslösung. Neben der gewünschten, vollständig prozessierten mRNA (Zielmolekül) enthält die Mischung verschiedene RNA-Nebenprodukte, wie uncapped, degradierte oder abortive RNA, doppelsträngige RNA sowie RNA:DNA-Hybride. Weitere Bestandteile:
IVT-Produkte
Lineare Plasmid-DNA: DNA-Vorlage mit der Expressionskassette für das Spike-Protein.
T7-Polymerase: Enzym, das den T7-Promotor erkennt und die mRNA synthetisiert.
Nukleotide: Bausteine der mRNA-Synthese – Cytosin (C), Guanin (G), Adenin (A) und N1-Methylpseudouridin (m¹Ψ) anstelle von Uridin (U). Uridin ist aufgeführt, da geringe U-Anteile aus der Herstellung der modifizierten Nukleotide stammen können.
Cap-Analoga: Synthetische Cap-Analoga, die während der IVT zur Ausbildung der Cap-1-Struktur eingesetzt werden und nach der Reaktion teilweise im Überschuss vorliegen.
Potentielle Verunreinigungen
RNasen: Enzyme, die RNA spalten und abbauen können; sie können unbeabsichtigt über Rohstoffe oder bakterielle Reststoffe eingeschleppt werden.
Endotoxine: Bestandteile der äußeren Zellmembran von Gram-negativer Bakterien wie E. coli.
Lösungsmittelreste: Rückstände aus der Herstellung und Reinigung von Ausgangsmaterialien.
Metallionen: Spurenverunreinigungen aus Rohstoffen, Prozesswasser oder Produktionsanlagen.
Übersicht: Typische RNA-Nebenprodukte nach der IVT (vor Reinigung)
a) Abortive mRNA
b) Degraded mRNA
c) Uncapped mRNA
d) Doppelsträngige RNA (dsRNA)
e) RNA:DNA-Hybride
a) Abortive mRNA
Zu Beginn der Transkription bindet die T7-RNA-Polymerase fest an den Promotor und bildet einen stabilen Initiationskomplex. Der Übergang von dieser Initiationsphase in eine produktive Elongation ist jedoch mechanisch instabil.
Die Polymerase entwindet lokal die DNA um die Transkriptionsstartstelle und beginnt mit der Synthese eines kurzen RNA-Strangs. Dabei zieht sie die DNA in das Enzym hinein, ohne sich selbst entlang der DNA vorwärtszubewegen. Dieses sogenannte Scrunching führt zur Akkumulation mechanischer Spannung im Transkriptionskomplex, da das Enzym gleichzeitig am Promotor festhält und die DNA weiter einzieht.
Gelingt es der Polymerase nicht, diese Spannung durch das Lösen vom Promotor und den Übergang in die Elongationsphase abzubauen, schnellt die DNA in ihre ursprüngliche Form zurück. Das bereits gebildete kurze RNA-Fragment wird ausgeworfen – ein abortives Transkript entsteht.
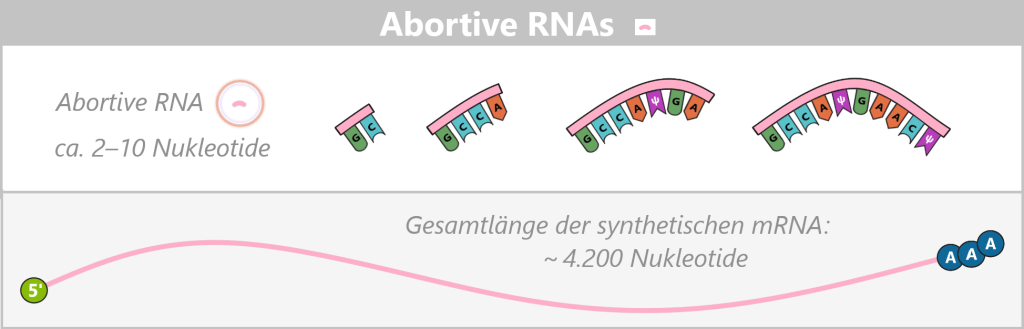
Dieser Vorgang kann sich mehrfach wiederholen, sodass eine einzelne Polymerase zahlreiche abortive RNA-Fragmente produziert, bevor eine vollständige Transkription gelingt. Erst wenn eine RNA-Länge von typischerweise etwa 8–14 Nukleotiden erreicht ist, löst sich die Polymerase vom Promotor und bildet einen stabilen Elongationskomplex.
Erscheinungsbild
Abortive RNAs sind sehr kurze RNA-Fragmente, meist nur 2–10 Nukleotide (nt) lang, die während der instabilen Initiationsphase der Transkription entstehen. Trotz der Verwendung co-transkriptioneller Capping-Strategien, bei denen das Cap-Analogon als Initiator dient, erreichen die meisten abortiven Transkripte nicht die für einen stabilen Cap-Einbau erforderliche Länge. Daher liegen sie mehrheitlich als uncapped Fragmente mit einem 5′-Triphosphat-Ende vor. Nur in Einzelfällen – bei längeren abortiven Transkripten im Bereich von etwa 10–12 Nukleotiden – kann formal ein Cap-Analogon eingebaut sein.
Biologische Auswirkung
Die biologischen Auswirkungen abortiver RNA-Fragmente sind bislang nicht vollständig erforscht. Einzelne, isolierte Fragmente von nur wenigen Nukleotiden sind in der Regel zu kurz, um bekannte RNA-Sensoren des angeborenen Immunsystems zu aktivieren, und werden nicht in Proteine übersetzt.
Das potenzielle Risiko liegt daher weniger in den einzelnen 2–10-nt-Fragmenten selbst, sondern in möglichen Sekundäreffekten:
Doppelstrangbildung: Kurze RNA-Fragmente könnten als Primer dienen oder zur Bildung kurzer doppelsträngiger RNA beitragen.
Komplexbildung: Hohe Mengen solcher Fragmente könnten sich zu komplexeren Strukturen zusammenlagern oder die Effizienz nachfolgender Reinigungsschritte beeinträchtigen.
Unspezifischer Belastungseffekt: Eine hohe Konzentration kurzer RNA-Fragmente könnte kurzfristig zellulären Stress erhöhen, auch wenn sie rasch abgebaut werden.
BioNTech weist darauf hin, dass abortive Nebenprodukte im Zytosol (dem flüssigen Zellinneren) transfizierter Zellen (Zellen, in die die mRNA eingebracht wurde) potenziell unbekannte Wechselwirkungen mit zelleigenen RNAs oder Mustererkennungsrezeptoren (PRRs) eingehen könnten, was den Bedarf an weiterer Forschung unterstreicht. [Die Auswirkungen von In-vitro-Transkriptionsnebenprodukten und Verunreinigungen verstehen]
Ungefährer Anteil (vor Reinigung)
BioNTech berichtet, dass etwa 44 % der T7-Polymerasen abortive Transkripte produzieren, bevor eine vollständige Transkription gelingt. Daher können sie zahlenmäßig die häufigste RNA-Spezies im Rohansatz darstellen. Jedoch machen sie unter Standard-IVT-Bedingungen weniger als 1 % der gesamten RNA-Masse aus. Der genaue Anteil abortiver RNAs hängt stark von der Sequenz der Vorlage und den Reaktionsbedingungen ab.
b) Degraded mRNA
Im Gegensatz zu abortiver mRNA handelt es sich bei degradierter mRNA um ehemals vollständige oder lange mRNA-Stränge, die durch äußere Einflüsse oder enzymatische Prozesse teilweise oder vollständig zerstört wurden. Der Abbau kann über verschiedene Mechanismen erfolgen:
Enzymatisch durch RNasen: Dies ist die häufigste Ursache für den Abbau während und nach der IVT. RNasen (Ribonukleasen) sind extrem stabile Enzyme, die nahezu überall vorkommen (z. B. auf der Haut, im Staub oder in Rohstoffen). Sie spalten RNA an spezifischen oder strukturell bevorzugten Stellen. Bereits geringste Verunreinigungen im Reaktionsgemisch können dazu führen, dass frisch synthetisierte mRNA in Fragmente zerfällt.
Chemisch/physikalisch: RNA ist aufgrund ihrer chemischen Struktur deutlich instabiler als DNA. Erhöhte pH-Werte, hohe Temperaturen oder bestimmte Metallionen fördern die hydrolytische Spaltung des RNA-Rückgrats und führen zu Kettenbrüchen.
Mechanisch: Wird die Lösung hohen Scherkräften ausgesetzt – etwa durch starkes Rühren oder das Pumpen durch enge Leitungen – können lange mRNA-Stränge physisch zerreißen. Dabei entstehen eher große Bruchstücke als die feinen Fragmente, die durch RNasen verursacht werden.
Abbruch während der Elongation: Streng genommen handelt es sich hierbei nicht um klassische Degradation, sondern um unvollständige mRNA, die bereits während der Synthese entsteht. Fällt die Polymerase während der Elongation vorzeitig vom DNA-Template ab – etwa durch starke Sekundärstrukturen oder limitierte Nukleotidverfügbarkeit – entsteht eine verkürzte mRNA, der das 3′-Ende und damit der Poly-A-Schwanz fehlt.
Erscheinungsbild
Degradierte mRNA liegt als Gemisch von Fragmenten unterschiedlicher Länge vor. Diese Fragmente können einzelne Strangbrüche enthalten oder vollständig zerfallen sein und tragen häufig kein vollständiges 5′-Cap und/oder keinen Poly-A-Schwanz.

… als ein heterogenes Gemisch von Fragmenten, das während oder nach der in-vitro-Transkription entstehen kann. Im Kontrast zur intakten, vollständigen mRNA (unten als Referenz) sind die Abbauprodukte unterschiedlich lang und weisen charakteristische Schäden auf: verlorene oder beschädigte 5′-Cap-Strukturen, freigelegte 5′-Triphosphate (ppp) – die bei Verlust des Caps exponiert werden, verkürzte Poly-A-Schwänze und interne Strangbrüche. Die Fragmente liegen nicht als einheitliche Spezies vor, sondern als komplexes Gemisch.
Biologische Auswirkung
Degradierte mRNA ist keine einheitliche Substanz, sondern ein heterogenes Gemisch. Nicht alle Fragmente sind biologisch problematisch. Das größte Risiko geht von Fragmenten aus, die spezifische immunologische Gefahrensignale tragen, darunter:
- freiliegende 5′-Triphosphate, die von zytosolischen RNA-Sensoren wie RIG-I erkannt werden,
- kurze doppelsträngige RNA-Strukturen, die durch Sekundärstruktur oder Hybridisierung entstehen und Rezeptoren wie MDA5 oder TLR3 aktivieren können,
- ungewöhnliche Endstrukturen (fehlendes Cap oder Poly-A-Schwanz), die als „FREMD“ erkannt werden.
Solche Fragmente können angeborene Immunantworten auslösen und die Verträglichkeit der mRNA-Formulierung beeinträchtigen.
Ungefährer Anteil (vor Reinigung)
Der Anteil degradierter mRNA variiert stark mit der Prozessführung. Typischerweise liegt er vor der Reinigung im Bereich von etwa 1–10 %. In gut optimierten IVT-Prozessen wird dieser Anteil jedoch gezielt auf ein Minimum reduziert.
c) Uncapped mRNA
In modernen IVT-Prozessen wird die 5′-Cap-Struktur häufig co-transkriptionell mithilfe sogenannter CleanCap-Analoga eingeführt. Dabei handelt es sich um synthetische Initiator-Oligonukleotide, die bereits die vollständige 5′-Cap-Struktur einschließlich des ersten transkribierten Nukleotids tragen.
Die T7-RNA-Polymerase nutzt CleanCap gezielt zur Initiation der Transkription; ein direkter Wettbewerb mit GTP (normales Nukleotid), wie er bei klassischen Cap-Analoga auftritt, findet dabei nicht statt. Dadurch entstehen überwiegend korrekt gecappte mRNA-Moleküle, während der Anteil uncapped mRNA stark reduziert wird.
Erscheinungsbild
Uncapped mRNA liegt als vollständige RNA-Sequenz vor, unterscheidet sich jedoch durch das Fehlen der 5′-Cap-Struktur am 5′-Ende.

Biologische Auswirkung
Zellen besitzen spezialisierte Enzyme, die RNA-Stränge ohne 5′-Cap sehr effizient vom 5′-Ende her abbauen. Dieser Mechanismus dient der raschen Entsorgung fehlerhafter oder gealterter körpereigener RNAs.
Immunreaktion: Uncapped RNA wird von zellulären Mustererkennungsrezeptoren wie RIG-I erkannt und kann eine starke Typ-I-Interferon-Antwort auslösen. Dadurch wird die Zelle in einen antiviralen Zustand versetzt.
Bei uncapped modRNA reduziert der Austausch von Uridin durch N1-Methylpseudouridin (m¹Ψ) die Aktivierung dieser Sensoren deutlich. Es kommt zu einem biologischen „Tauziehen“ zwischen dem immunstimulierenden uncapped 5′-Ende und der immunmodulierenden Nukleosid-Modifikation. Insgesamt würde uncapped modRNA daher als mäßig immunogenes Molekül wirken, jedoch weiterhin schneller abgebaut werden als korrekt gecappte modRNA.
Minimale Proteinproduktion: Da die 5′-Cap-Struktur auch für die effiziente Bindung an das Ribosom notwendig ist, wird uncapped mRNA kaum oder gar nicht in Protein übersetzt.
Ungefährer Anteil (vor Reinigung)
Der Anteil uncapped mRNA nach der IVT hängt maßgeblich von der verwendeten Capping-Strategie ab. Bei modernen co-transkriptionellen CleanCap-Verfahren liegt der Anteil uncapped mRNA vor der Reinigung typischerweise im Bereich von ca. 1–6 %.
d) Doppelsträngige RNA (dsRNA)
Ein weiteres relevantes Nebenprodukt der in-vitro-Transkription (IVT) ist doppelsträngige RNA (dsRNA). Während die gewünschte Impfstoff-mRNA als einzelsträngiges Molekül synthetisiert wird, können im Verlauf der IVT auch RNA-Duplexe entstehen, bei denen zwei komplementäre RNA-Stränge über Watson-Crick-Basenpaarung miteinander verbunden sind.
Die Entstehung solcher dsRNA-Spezies ist kein zufälliges Aggregationsphänomen, sondern geht auf spezifische, enzymatisch vermittelte Nebenreaktionen der T7-RNA-Polymerase zurück.
Entstehungsmechanismen
Promotorunabhängige Transkription des Nicht-Templatestrangs
Unter bestimmten Bedingungen kann die T7-RNA-Polymerase auch ohne klassischen Promotorstart RNA synthetisieren, indem sie den Nicht-Templatestrang der DNA-Vorlage abliest. Die dabei entstehenden RNA-Moleküle sind weitgehend komplementär zur gewünschten mRNA. Treffen Sense- und Antisense-RNA aufeinander, bilden sich ausgedehnte, nahezu vollständig doppelsträngige RNA-Duplexe. Dieser Mechanismus wird insbesondere begünstigt, wenn die DNA-Vorlage nicht vollständig linearisiert ist oder spezifische Sequenzen am 3′-Ende der Vorlage vorliegen.
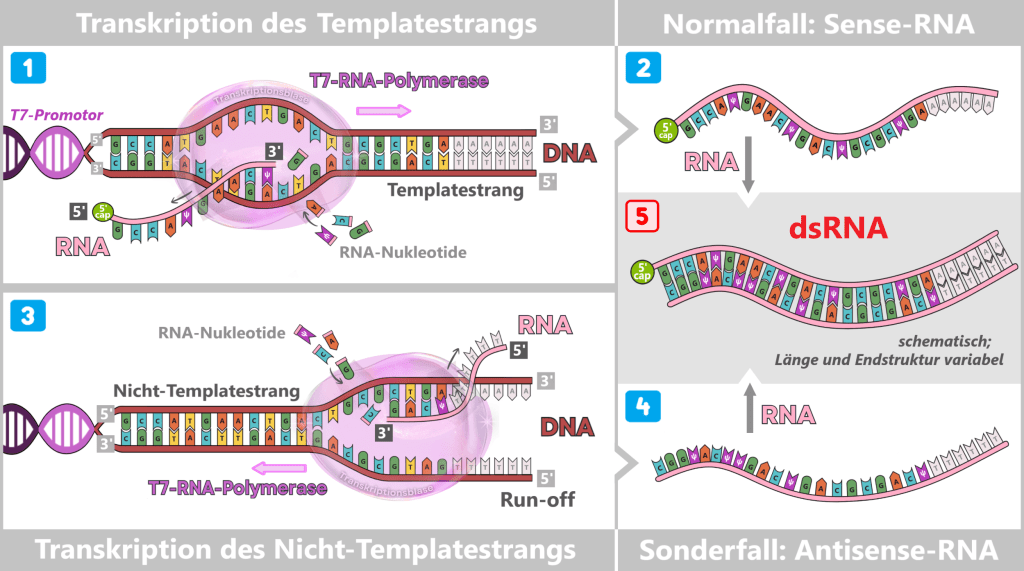
(RNA schematisch dargestellt; Nukleosid-Modifikationen (z. B. m¹Ψ) nicht explizit abgebildet.)
1) Der Normalfall: Der T7-Promotor hat eine außergewöhnlich hohe Affinität für die T7-RNA-Polymerase. Deshalb startet die Polymerase fast ausschließlich dort. Sie liest DNA stets in 3′ → 5′-Richtung und synthetisiert ein neues RNA-Molekül ausschließlich in 5′ → 3′-Richtung.
2) Das Ergebnis ist die gewünschte Sense-RNA, die genau der Sequenz des Nicht-Templatestrangs entspricht.
3) Der Sonderfall: Nach dem Run-off – dem Verlassen der DNA-Vorlage am Ende der Transkription – löst sich die Polymerase von der DNA. In seltenen Fällen kann sie unspezifisch an strukturell zugängliche DNA-Enden binden. Bindet sie dabei an den zuvor nicht als Template genutzten Strang, wird dieser zur neuen Vorlage.
Unter bestimmten Reaktionsbedingungen können auch lokal offene oder dynamisch schmelzende DNA-Bereiche entstehen, an die die T7-RNA-Polymerase promotorunabhängig binden kann. Erfolgt diese Bindung am zuvor nicht genutzten Strang, wird dieser als Template gelesen.
Bei der promotorunabhängigen Transkription des Nicht-Templatestrangs fehlt eine definierte Initiationsstelle, sodass kein co-transkriptionelles Capping erfolgt. Die entstehende Antisense-RNA trägt daher typischerweise ein freies 5′-Triphosphat (pppN).
4) Das Ergebnis ist eine komplementäre Antisense-RNA, die der Sequenz des Templatestrangs entspricht.
5) Entstehung eines langen dsRNA-Duplexes: Treffen Sense-RNA und Antisense-RNA zusammen, führt das zur Bildung von dsRNA (doppelsträngiger RNA). Dabei stellen die entstehenden dsRNA-Nebenprodukte kein homogenes Molekül dar, sondern ein strukturell heterogenes Gemisch aus vollständig oder partiell komplementären RNA-Duplexen. Diese können stumpfe Enden oder einzelsträngige Überhänge aufweisen und variieren erheblich in Länge und Endstruktur.
RNA-abhängige 3′-End-Verlängerung (self-priming)
Ein zweiter zentraler Mechanismus ist die RNA-abhängige Verlängerung des 3′-Endes der frisch synthetisierten mRNA. Dabei klappt das 3′-Ende der RNA aufgrund komplementärer Sequenzen intramolekular zurück und bildet eine kurze dsRNA-Haarnadel. Die T7-RNA-Polymerase kann an diese Struktur erneut binden und den RNA-Strang weiter verlängern, wobei eine Sequenz entsteht, die zur ursprünglichen RNA komplementär ist. Dieser Prozess ist unabhängig von der DNA-Vorlage und kann sogar bei bereits vollständig transkribierten RNA-Molekülen auftreten.
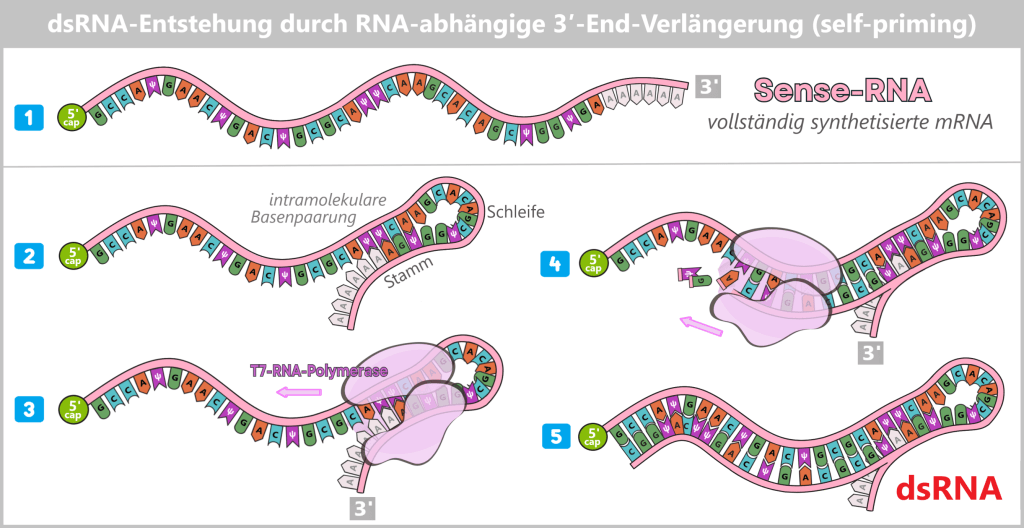
(RNA schematisch dargestellt)
1) Sense-RNA: Normale, vollständig synthetisierte mRNA.
2) Hairpin-Bildung: Der 3′-Abschnitt faltet sich um. Es entsteht ein kurzer dsRNA-Stamm (z.B. 5-10 Basenpaare) mit einer Einzelstrang-Schleife.
3) Polymerase-Bindung: Die T7-Polymerase bindet an diesen dsRNA-Stamm, als wäre es ein DNA-Template-Primer-Komplex.
4) Verlängerung: Die Polymerase nutzt den einen Strang des Hairpins als Template und verlängert das freie 3′-Ende entlang des komplementären RNA-Abschnitts. Sie synthetisiert also die Antisense-Sequenz direkt in die Verlängerung des Sense-Strangs hinein.
5) Ergebnis: Die Polymerase verlängert den Strang so weit sie kann. Die neu synthetisierte Antisense-RNA hybridisiert sofort mit der Sense-RNA. Es entsteht ein partiell oder vollständig dsRNA-haltiges RNA-Molekül.
Abgrenzung zu RNA-Sekundärstrukturen
Einzelsträngige RNA bildet natürlicherweise intramolekulare Sekundärstrukturen wie Haarnadeln (Hairpin) oder interne Schleifen (siehe obere Abbildung – Punkt 2). Diese sind integraler Bestandteil funktioneller mRNA, meist kurz und thermodynamisch flexibel. Solche Strukturelemente gelten nicht als dsRNA-Nebenprodukte im engeren Sinne und sind nicht mit den langkettigen dsRNA-Duplexen gleichzusetzen, die als IVT-Verunreinigungen relevant sind.
Beide Mechanismen führen zur Bildung längerer, stabiler dsRNA-Abschnitte, die sich strukturell deutlich von den kurzen, dynamischen Sekundärstrukturen korrekt transkribierter mRNA unterscheiden.
Erscheinungsbild
dsRNA-Nebenprodukte liegen typischerweise als lange, teilweise oder vollständig komplementäre RNA-Duplexe vor. Sie können nahezu die gesamte Länge der Ziel-mRNA umfassen oder ausgedehnte doppelsträngige Regionen enthalten. Häufig besitzen diese Duplexe keine 5′-Cap-Struktur und können an den Enden über einzelsträngige Überhänge verfügen.
Biologische Auswirkung
dsRNA als strukturelles Gefahrensignal
Doppelsträngige RNA (dsRNA) stellt im Zytoplasma eukaryotischer Zellen kein typisches Strukturmotiv zelleigener, translierbarer mRNA dar. Das bedeutet: Während zelluläre mRNAs normalerweise einzelsträngig sind und dazu dienen, in Proteine übersetzt zu werden (mittels Translation), tritt dsRNA physiologisch nur in eng regulierten, kurzlebigen Kontexten auf. Das Auftreten von dsRNA im Zytoplasma wird daher von der Zelle nicht primär als genetische Information, sondern als strukturelles Warnsignal interpretiert.
Zytosolische Erkennung von dsRNA
Zellen verfügen über spezialisierte Mustererkennungsrezeptoren (Pattern Recognition Receptors, PRRs). Diese „Detektoren für Gefahr“ patrouillieren durch das Zytoplasma und suchen nach strukturellen Mustern wie Duplex-Länge, Endstruktur und chemischer Modifikation der Nukleotide.
Zytosolische dsRNA-Sensoren und ihre immunologischen Konsequenzen
| Merkmal / Spezifität | Zellulärer Sensor | Biologische Konsequenz (für die Zelle) |
| Kurze dsRNA (≈ 10-300 bp) mit 5′-Triphosphat und/oder stumpfen Enden | RIG-I (Retinoic acid-Inducible Gene I) Kurz-ppp-Detektor | Auslösung einer entzündlichen Genexpression (Typ-I-Interferone, Zytokine). Alarmierung der Nachbarzellen. |
| Lange dsRNA (> ~500–1.000 bp), unabhängig von den Enden | MDA5 (Melanoma Differentiation-Associated protein 5) Langduplex-Scanner | Auslösung einer massiven Typ-I-Interferon-Antwort. Wichtige Abwehr gegen viele Viren. |
| dsRNA mittlerer Länge (≈ ≥30–100 bp) | PKR (Protein Kinase R) Intrazellulärer Effektor | Globaler Stopp der Proteinbiosynthese: Phosphorylierung des Translationsfaktors eIF2α. Hemmung der viralen Vermehrung. |
| dsRNA mittlerer Länge (≈ ≥40–100 bp) | OAS (2′-5′-Oligoadenylat-Synthetasen) Intrazellulärer Effektor | Unspezifischer RNA-Abbau: aktiviert die latente Ribonuklease RNase L, die alle zellulären und viralen RNA-Moleküle zerschneidet. Kann zum Zelltod führen. |
| dsRNA im Endosom (extra-zellulär aufgenommene oder phagozytierte dsRNA) | TLR3 (Toll-like Receptor 3) Wächter an der Zellpforte | Führt zur Produktion von Typ-I-Interferonen und pro-inflammatorischen Zytokinen. |
Die Wirkung von m¹Ψ
Die Verwendung nukleosid-modifizierter mRNA (modRNA), etwa durch den Einbau von N¹-Methylpseudouridin, kann die Aktivierung bestimmter zytosolischer RNA-Sensoren deutlich reduzieren. Besonders betroffen sind RIG-I und PKR, die bevorzugt auf einzelsträngige RNA oder kurzlebige dsRNA-Strukturen reagieren. Längenabhängige Sensoren wie MDA5 werden durch diese Modifikation hingegen nur eingeschränkt moduliert und bleiben gegenüber ausgedehnten dsRNA-Duplexen weitgehend empfindlich.
Aus diesem Grund besitzt dsRNA auch im Kontext von modRNA weiterhin ein hohes immunstimulatorisches Potenzial – das heißt, sie kann das Immunsystem aktivieren und unerwünschte Entzündungsreaktionen auslösen. Die effiziente Minimierung solcher dsRNA-Nebenprodukte durch optimierte Transkriptionsbedingungen, chemische Modifikationen und nachgelagerte Reinigungsverfahren ist daher ein zentraler Bestandteil der mRNA-Herstellung.
Ungefährer Anteil (vor Reinigung)
Der Anteil an dsRNA nach der IVT ist stark prozessabhängig und wird von Faktoren wie der Qualität der DNA-Vorlage, den Reaktionsbedingungen und der verwendeten Polymerase beeinflusst. Typischerweise liegt der dsRNA-Anteil vor der Aufreinigung im niedrigen einstelligen Prozentbereich, kann unter ungünstigen Bedingungen jedoch deutlich höher ausfallen.
e) RNA:DNA-Hybride
Während der in-vitro-Transkription kann es unter bestimmten Bedingungen dazu kommen, dass der neu synthetisierte RNA-Strang nicht vollständig vom DNA-Template dissoziiert. Er kann partiell am Templatestrang verbleiben und dabei den komplementären DNA-Strang verdrängen. Es entstehen sogenannte RNA:DNA-Hybride.
Die Bildung solcher Hybridstrukturen ist stark sequenzabhängig. Besonders purinreiche Transkripte – also RNA-Sequenzen mit vielen der Basen Adenin und Guanin – sowie DNA-Vorlagen mit sich wiederholenden GAA-Abschnitten (Guanin–Adenin–Adenin) begünstigen die Entstehung stabiler RNA:DNA-Duplexe. Diese Nebenprodukte werden in der Regel unterschätzt, lassen sich jedoch experimentell mit spezifischen Antikörpern gegen RNA:DNA-Hybride nachweisen. [Die Auswirkungen von In-vitro-Transkriptionsnebenprodukten und Verunreinigungen verstehen]
Erscheinungsbild
RNA:DNA-Hybride liegen typischerweise als R-Loop-artige Strukturen vor. Dabei handelt es sich um lokal begrenzte, dreisträngige Nukleinsäurekonformationen, bestehend aus:
- einem RNA:DNA-Hybrid-Duplex,
- einem verdrängten DNA-Einzelstrang,
- sowie der angrenzenden DNA-Doppelhelix außerhalb der Hybridregion.
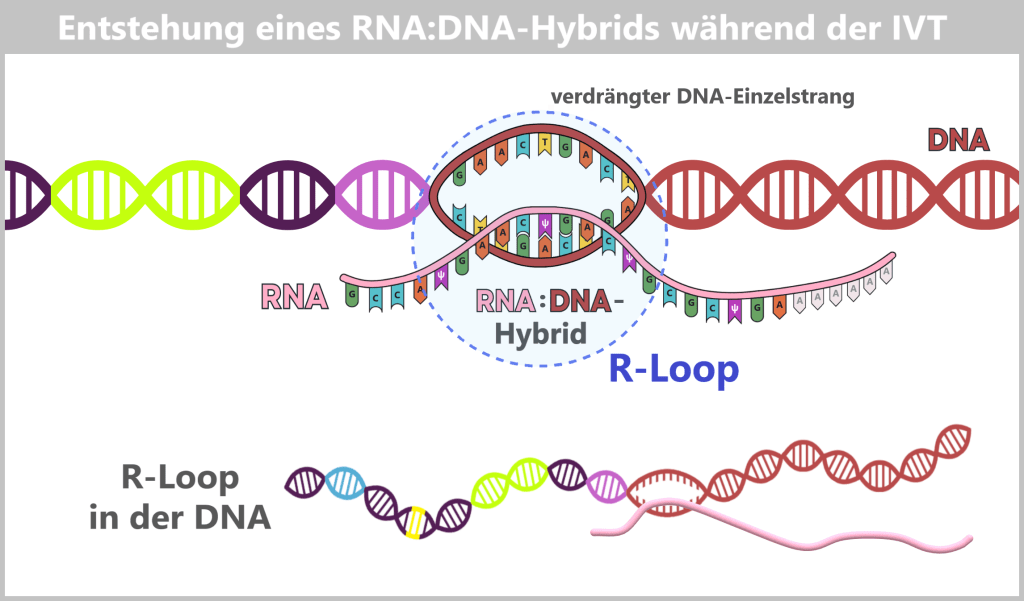
Der neu synthetisierte RNA-Strang hybridisiert partiell mit dem DNA-Templatestrang und verdrängt dabei den komplementären Nicht-Templatestrang. Es entsteht ein R-Loop, bestehend aus einem RNA:DNA-Hybrid-Duplex und einem verdrängten DNA-Einzelstrang, eingebettet in eine ansonsten doppelsträngige DNA-Region.
Außerhalb des R-Loops liegt die DNA weiterhin als reguläre Doppelhelix vor.
Biologische Auswirkung
Aktuelle Forschungsergebnisse deuten darauf hin, dass RNA:DNA-Hybride von zellulären Mustererkennungsrezeptoren (Pattern Recognition Receptors, PRRs) erkannt werden können. Zu diesen Sensoren des angeborenen Immunsystems zählen unter anderem cGAS, TLR9 und das Inflammasom-Protein NLRP3, die solche Hybridstrukturen binden können. Ihre Aktivierung kann eine Immunreaktion auslösen, die zur Bildung von entzündungsfördernden Botenstoffen (Zytokinen) und Typ-I-Interferonen führt.
Ob und in welchem Ausmaß RNA:DNA-Hybrid-Kontaminanten tatsächlich zu unerwünschten Immunreaktionen therapeutisch eingesetzter IVT-mRNA beitragen, ist bislang jedoch nicht systematisch untersucht worden.
Vor diesem Hintergrund erscheint die effiziente Entfernung von RNA:DNA-Hybrid-Verunreinigungen aus IVT-mRNA aus Qualitätssicherungsgründen als relevanter Aspekt. Dies gilt insbesondere für therapeutische Anwendungen, bei denen eine ausgeprägte Aktivierung des Immunsystems nicht angestrebt wird. [Die Auswirkungen von In-vitro-Transkriptionsnebenprodukten und Verunreinigungen verstehen]
Ungefährer Anteil (vor Reinigung)
Der Anteil an RNA:DNA-Hybriden nach der in-vitro-Transkription ist stark prozessabhängig. Er wird unter anderem durch die Sequenz und Qualität der DNA-Vorlage sowie durch die Reaktionsbedingungen beeinflusst. Insgesamt wird ihr Anteil vor der Aufreinigung als gering eingeschätzt, kann jedoch je nach Transkript und Prozessführung variieren. Verlässliche, öffentlich zugängliche quantitative Daten zum genauen Anteil von RNA:DNA-Hybriden in therapeutischen mRNA-Produkten liegen bislang nicht vor, da entsprechende Messwerte in der Regel Teil des firmenspezifischen Herstellungs- und Qualitätskontrollwissens sind.
** Eine weitere Entstehungsmöglichkeit für RNA:DNA-Duplexe im Rahmen der DNase-Behandlung wird in Abschnitt „1.3.1. a) Mögliche posttranskriptionelle RNA:DNA-Hybridisierung“ beschrieben. **
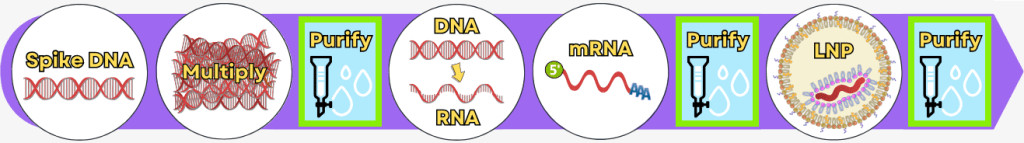
1.3. Reinigungsverfahren im Herstellungsprozess
Da mRNA-Impfstoffe extrem hohe Reinheitsanforderungen erfüllen müssen, ist die Aufreinigung kein einzelner, isolierter Prozessschritt. Vielmehr zieht sie sich wie ein roter Faden durch den gesamten Herstellungsprozess.
Der genaue industrielle Ablauf ist bei Herstellern wie BioNTech/Pfizer oder Moderna proprietär (unternehmensintern) und wird nicht vollständig offengelegt. Patente und regulatorische Dokumente zeigen jedoch, dass eine Kombination etablierter biotechnologischer Reinigungsverfahren eingesetzt wird.
Eine schematische, industrieübergreifende Übersicht zur mRNA-Herstellung, in der der kontinuierliche Fluss von Reinigungsaktivitäten entlang des Gesamtprozesses dargestellt ist, findet sich beispielhaft in der technischen Dokumentation von Merck/Sigma-Aldrich (Manufacturing strategies for mRNA vaccines and therapies, Abbildung 1).
Im Herstellungsprozess lassen sich vereinfacht drei besonders kritische Reinigungsphasen unterscheiden:
- vor der in-vitro-Transkription (prä-IVT): Aufreinigung der DNA-Vorlage,
- nach der in-vitro-Transkription (post-IVT): Aufreinigung der synthetisierten mRNA,
- nach der Formulierung: sterile Filtration der lipid-nanopartikulären Formulierung.
Anstatt diese Phasen streng chronologisch abzuarbeiten, werden im Folgenden die wichtigsten Reinigungsprinzipien und -methoden vorgestellt, die an unterschiedlichen Stellen des Herstellungsprozesses wiederholt zum Einsatz kommen.
1.3.1. DNase I-Behandlung – enzymatischer DNA-Abbau
1.3.2. Proteinase K – enzymatischer Abbau von Restproteinen
1.3.3. Filtration
1.3.4. Chromatographie – das molekulare Trennverfahren
1.3.5. Magnetkügelchen-Reinigung
Die folgende Übersicht fasst die zentralen Reinigungstechniken zusammen. Sie dient als Rahmen für die anschließend vertiefte Darstellung der jeweiligen Methoden.
| Methode | Entfernt | Prinzip | Vorteile | Einschränkungen |
| DNase I Behandlung | Template-DNA | Spezifischer enzymatischer Abbau | Sehr selektiv, zerkleinert problematische Nukleinsäuren | Enzyme müssen vollständig entfernt werden, sonst Gefahr von Restaktivität |
| Proteinase K | Enzyme aus der Transkription (z. B. Polymerasen, Ligase, Restriktions-enzyme) | Unspezifischer Abbau von Proteinen | Universell wirksam, entfernt viele Proteinarten | Muss durch weitere Reinigungsschritte ergänzt werden (Enzymreste selbst problematisch) |
| Filtration & TFF (Tangentialfluss-filtration) | Salze, Nukleotide, kleine Moleküle, Pufferreste; auch Konzentration möglich | Trennung nach Molekülgröße durch Membranen | Skalierbar, robust, vielseitig (Konzentration + Pufferwechsel in einem Schritt) | Keine Unter-scheidung ähnlicher Nukleinsäuren; eher ein „Grobwerkzeug“ |
| Chromatografie (Poly(dT), AEX) | dsRNA, kurze RNA-Fragmente, restliche Nukleotide, Proteine | Trennung nach Ladung oder spezifischer Bindung an Oberflächen | Sehr präzise, trennt eng verwandte Nukleinsäuren; etabliert in der Industrie | Technisch anspruchsvoll, kostenintensiv, erfordert exakte Prozesskontrolle |
| Magnetkügelchen-Reinigung (Prozess 1) | Selektion reifer, polyadenylierter mRNA; entfernt kurze Fragmente & Verunreinigungen | Poly(T)-beschichtete Beads binden spezifisch an den Poly(A)-Schwanz der mRNA | Hohe Spezifität, schnelle und effiziente Trennung | Für kleine bis mittlere Volumina geeignet; bei Großproduktion schwer skalierbar |
1.3.1. DNase I-Behandlung – enzymatischer DNA-Abbau
Deoxyribonuclease I (DNase I) ist ein Enzym, das DNA-Moleküle in kleinere Fragmente spaltet. Oft spricht man auch vom „Verdauen“ der DNA – ein bildhafter Ausdruck für den enzymatischen Abbau langer DNA-Stränge in kürzere Stücke. In der Biotechnologie wird DNase I eingesetzt, um unerwünschte DNA-Verunreinigungen gezielt zu fragmentieren.
DNase I ist somit kein vollständiger Reinigungsmechanismus, sondern ein vorbereitender Schritt, der die nachfolgende physikalische und chromatographische Trennung erleichtert.
In der mRNA-Impfstoffherstellung wird DNase I nach der in-vitro-Transkription (IVT) eingesetzt, um die DNA-Vorlage (Template-DNA) sowie den DNA-Anteil eventuell gebildeter RNA:DNA-Hybride zu fragmentieren, die die Qualität oder Sicherheit des Endprodukts beeinträchtigen könnten. Die DNase I erkennt die räumliche Struktur der DNA – genauer gesagt: das Zucker-Phosphat-Rückgrat. Das Enzym setzt sich wie ein Sattel auf die DNA und greift in die sogenannte „kleine Furche“ (minor groove). Dabei verbiegt sie die DNA-Struktur lokal, um den Zugang zur Phosphodiesterbindung zu ermöglichen. Da die Breite der kleinen Furche je nach Basenabfolge minimal variiert, schneidet DNase I an manchen Stellen (z. B. bei bestimmten A/T-reichen Regionen) etwas lieber als an anderen.
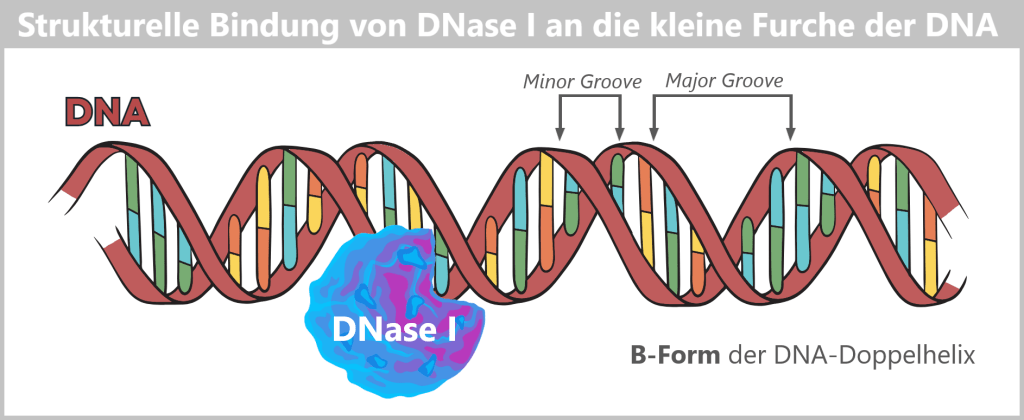
Die typische B-Form der DNA ist die schlanke, rechtsgewundene Doppelhelix, die aussieht wie eine Wendeltreppe mit zwei unterschiedlich großen Rillen (einer großen und einer kleinen Furche), die sich um sie herum winden.
Das Enzym interagiert bevorzugt mit der kleinen Furche (minor groove) des DNA-Doppelstrangs und bindet dabei an das Zucker-Phosphat-Rückgrat. Die Bindung führt zu einer lokalen Verformung der DNA, wodurch die Spaltung der Phosphodiesterbindung ermöglicht wird. Die große Furche (major groove) ist dargestellt, spielt jedoch für die Bindung von DNase I keine primäre Rolle.
DNase I schneidet DNA unspezifisch, also an vielen verschiedenen Positionen entlang des Strangs. Sie wirkt sowohl auf einzelsträngige (ssDNA) als auch auf doppelsträngige DNA (dsDNA). Ihre Aktivität hängt dabei von mehreren Faktoren ab, insbesondere von den vorhandenen Metallionen, die die Art der Schnittstellen beeinflussen [bioswisstec, YEASEN]:
In Gegenwart von Magnesium-Ionen (Mg²⁺) schneidet DNase I die doppelsträngige DNA (dsDNA) überwiegend durch einzelsträngige Schnitte („nicks“). Das führt zur Bildung kurzer doppelsträngiger DNA-Fragmente (dsDNA) mit Überhängen sowie in geringerem Umfang zu einzelsträngigen (ssDNA) Fragmenten.
In Gegenwart von Mangan-Ionen (Mn²⁺) schneidet die DNase I beide dsDNA-Stränge nahezu an derselben Position, wodurch überwiegend stumpf-endige dsDNA-Fragmente entstehen.
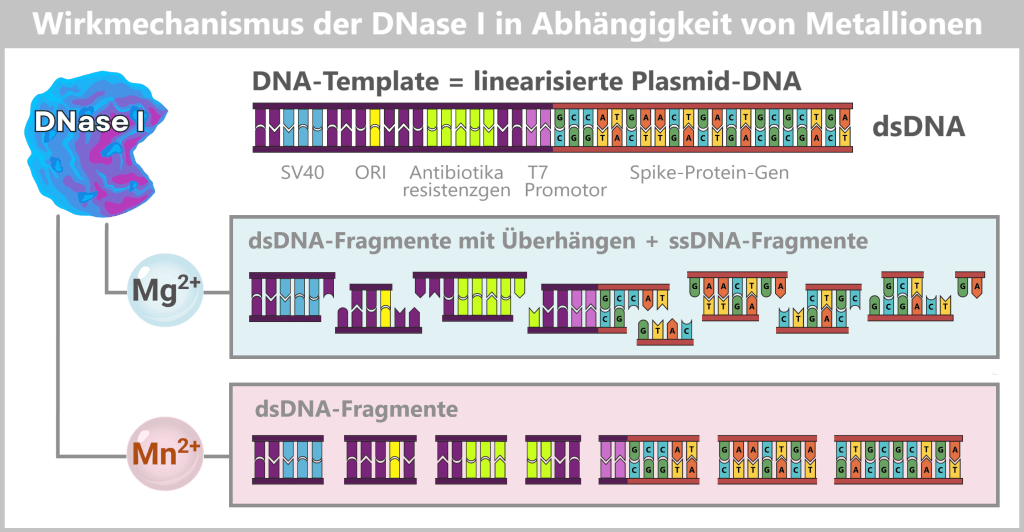
Effektivität der DNase I
Der Hersteller Thermo Fisher Scientific beschreibt in dem Online-Artikel „DNase I entmystifiziert“ die Wirkweise und Grenzen der DNase-I-Behandlung. Der Beitrag verdeutlicht, dass die Effektivität von DNase I stark von den Reaktionsbedingungen und der Art des vorliegenden DNA-Substrats abhängt.
Im Produktionsprozess befinden wir uns nach der in-vitro-Transkription (IVT). Die Reaktionsmischung ist ein komplexer Mix aus:
- der Ziel-mRNA (Hauptkomponente),
- RNA-Nebenprodukten (z. B. verkürzte Transkripte, RNA:DNA-Hybride),
- dem DNA-Template (in relevanter, aber geringerer Menge als mRNA),
- seltenen Restformen nicht-linearer Plasmid-DNA (z. B. supercoiled),
- Enzymen, freien Nukleotiden, Salzen und Puffersubstanzen.
Wie viele Enzyme wird auch die Aktivität der DNase I durch die Zusammensetzung der Reaktionsmischung beeinflusst.
Einflussfaktoren auf die Effektivität der DNase-I-Behandlung
| Faktor | Mechanismus | Prozessrelevanz (mRNA-Herstellung) |
| Substrat-Zugänglichkeit | DNase I schneidet bevorzugt frei zugängliche Phosphodiesterbindungen; kompakte oder topologisch eingeschränkte DNA ist schlechter erreichbar. | Supercoiled oder stark verdrillte DNA-Strukturen (z. B. seltene Plasmid-Restformen) werden langsamer oder unvollständig fragmentiert. |
| Substrat-Sättigung | Hohe DNA-Mengen können das Enzym sättigen; hohe RNA-Konzentrationen beeinflussen die räumliche Zugänglichkeit der DNA. | In IVT-Mischungen mit sehr hoher Nukleinsäurelast ist ein Enzymüberschuss erforderlich. |
| Helix-Geometrie | Die aktive Stelle der DNase I ist auf die B-Form der DNA optimiert (abweichende Helixformen passen schlechter). | RNA:DNA-Hybride weisen eine A-Form-ähnliche Helix auf → Spaltaktivität < 2 % gegenüber dsDNA |
| DNA-Struktur (dsDNA vs. ssDNA) | Höchste Aktivität auf dsDNA; Aktivität auf ssDNA etwa 500-fach geringer | Sehr kurze oder teilweise einzelsträngige DNA-Fragmente werden schlechter abgebaut. |
| Ionen im Puffer | Mg²⁺ ist für die katalytische Aktivität essenziell; Ca²⁺ stabilisiert die Enzymstruktur | Therapeutische Prozesse erfordern präzise eingestellte Ionenverhältnisse. |
| Chelatoren | Chelatoren (z. B. EGTA, EDTA) binden Ca²⁺/Mg²⁺ und inaktivieren DNase I. | Rückstände aus Puffern oder Prozess-schritten können die DNase-Wirkung stark reduzieren. |
| Salzkonzentration | Erhöhte Ionenstärke schwächt die elektrostatische Bindung zwischen Enzym und DNA. | IVT-Pufferbedingungen können die Aktivität mindern → kompensiert durch Enzymüberschuss. |
Der Beitrag von Thermo Fisher Scientific weist darauf hin, dass „es wahrscheinlich unmöglich ist, jeden letzten DNA-Strang in einem RNA-Präparat loszuwerden“.
Dies verdeutlicht, dass die vollständige Entfernung aller DNA-Reste technisch anspruchsvoll ist. Selbst nach sorgfältiger DNase-I-Behandlung können kurze DNA-Fragmente oder RNA:DNA-Hybride verbleiben.
Die so erzeugten kleinen DNA-Fragmente werden in nachfolgenden Prozessschritten – etwa durch Magnetkügelchen-Reinigung (bei Prozess 1) oder Chromatografie (bei Prozess 2) – aus dem Produkt entfernt (vgl. EPAR, Abschnitt 2.2).
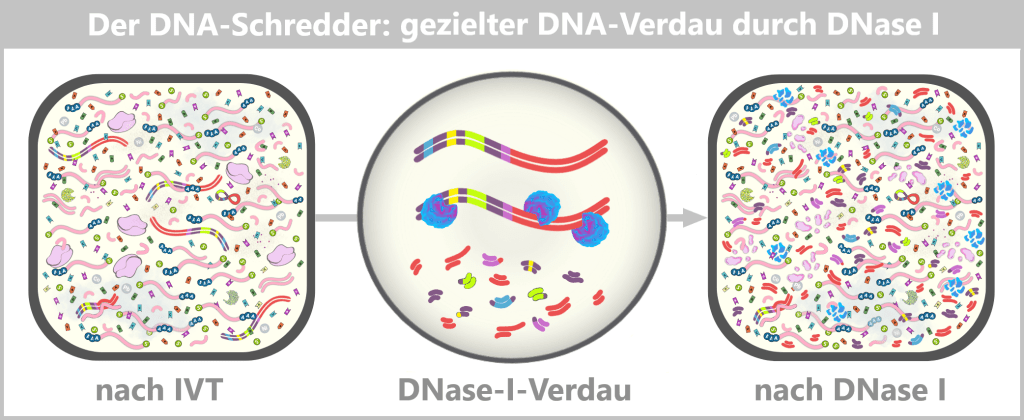
In der industriellen mRNA-Herstellung erfolgt die DNase-I-Behandlung unmittelbar nach der in-vitro-Transkription (IVT), da die Reaktionsmischung bereits einen für das Enzym geeigneten Mg²⁺-haltigen Puffer enthält. Die Mg²⁺-abhängige Aktivität der DNase I führt überwiegend zur Fragmentierung der DNA in kurze doppelsträngige DNA-Fragmente mit Nicks (Strangbrüchen) und kurzen Überhängen. (Mn²⁺ wird aufgrund der unspezifischeren und schwer kontrollierbaren Spaltaktivität im industriellen Prozess nicht eingesetzt.)
Nach abgeschlossener Reaktion wird die DNase I häufig inaktiviert oder entfernt, beispielsweise durch Hitze, EDTA-Zugabe oder Proteinase K-Behandlung, um sicherzustellen, dass keine enzymatische Aktivität auf die mRNA übergreift.
1.3.1. a) Mögliche posttranskriptionelle RNA:DNA-Hybridisierung
Neben den während der in-vitro-Transkription entstehenden R-Loop-Strukturen wird in der neueren Literatur ein zweiter möglicher Entstehungsweg für RNA:DNA-Hybride diskutiert.
Die DNase-I-Behandlung fragmentiert die verbliebenen DNA-Vorlagen in Stücke unterschiedlicher Länge – überwiegend doppelsträngig, teilweise jedoch auch mit einzelsträngigen Überhängen oder als kurze Einzelstrangabschnitte.
Fragmente, deren Sequenz komplementär zur mRNA ist – insbesondere solche, die vom Templatestrang der ursprünglichen DNA-Vorlage stammen –, könnten unter geeigneten Bedingungen mit der mRNA hybridisieren und RNA:DNA-Duplexe bilden.
Für eine thermodynamisch stabile Hybridisierung sind in der Regel zusammenhängende komplementäre Sequenzen von etwa 8–12 Nukleotiden oder mehr erforderlich. Sehr kurze Überhänge von nur wenigen Nukleotiden (z. B. 2–4 Basen), wie sie bei einem weitgehend vollständigen DNase-Verdau typischerweise entstehen, sind dagegen nicht ausreichend für eine stabile Bindung. Voraussetzung für diesen Mechanismus wäre daher das Vorhandensein ausreichend langer, komplementärer DNA-Fragmente, etwa bei unvollständigem Verdau oder spezifischen Fragmentmustern.
Hinsichtlich der enzymatischen Abbaubarkeit ist zu berücksichtigen, dass DNase I bevorzugt doppelsträngige DNA spaltet. RNA:DNA-Hybride stellen ein strukturell abweichendes Substrat dar und werden effizienter durch spezialisierte Enzyme wie RNase H oder DNase I-XT abgebaut. In der Praxis kann dies bedeuten, dass einmal gebildete RNA:DNA-Hybride gegenüber einem erneuten DNase-I-Verdau relativ stabiler sind als freie DNA-Fragmente. Eine zusätzliche Stabilisierung kann auftreten, wenn solche Strukturen in Lipid-Nanopartikel eingeschlossen werden.
Dieser postulierte Mechanismus der Post-IVT-Hybridisierung ist bislang nicht umfassend experimentell charakterisiert. Er basiert auf theoretischen Überlegungen und vereinzelten experimentellen Hinweisen und sollte daher als plausible, aber noch nicht abschließend belegte Möglichkeit eingeordnet werden.
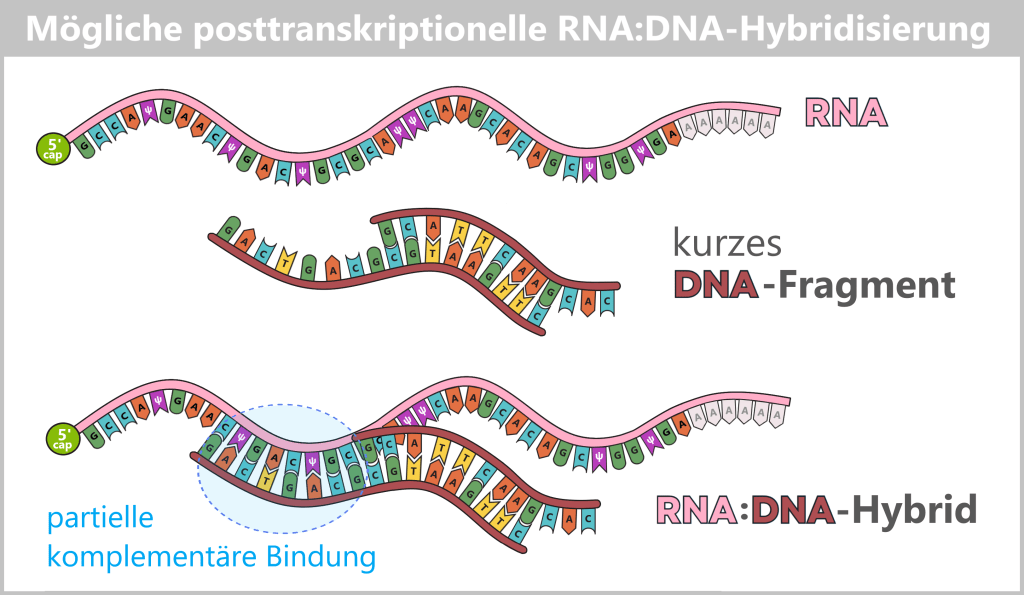
Kurze DNA-Fragmente können unter geeigneten Bedingungen partiell an komplementäre Abschnitte der mRNA binden. Im Gegensatz zum R-Loop entsteht keine dreisträngige Struktur, sondern ein lokal begrenzter RNA:DNA-Duplex.
1.3.2. Proteinase K – enzymatischer Abbau von Restproteinen
Proteinase K ist eine breit wirksame Serinprotease, die in der Biotechnologie eingesetzt wird, um Proteine unspezifisch zu hydrolysieren, also in kleinere Peptidfragmente zu zerlegen. Sie gehört zur Klasse der Proteinasen – Enzyme, die Peptidbindungen zwischen Aminosäuren spalten und dadurch die dreidimensionale Struktur von Proteinen irreversibel auflösen.
In der mRNA-Impfstoffherstellung kann Proteinase K an verschiedenen Prozessstellen eingesetzt werden, insbesondere:
Nach der Zelllyse in der Plasmidproduktion: Hier dient sie dem Abbau bakterieller Proteine, Nukleasen und weiterer Zellbestandteile, die die Reinheit der Plasmid-DNA beeinträchtigen könnten.
Nach der in-vitro-Transkription (IVT): Nach der Transkription, bei der die mRNA aus der DNA-Vorlage synthetisiert wird, verbleiben in der Reaktionsmischung mehrere Enzyme, darunter die T7-RNA-Polymerase sowie DNase I aus dem vorangegangenen Prozessschritt. Proteinase K kann eingesetzt werden, um diese enzymatischen Rückstände gezielt zu hydrolysieren und damit die Reinheit der mRNA weiter zu erhöhen.
Wirkmechanismus
Proteinase K bindet zunächst an frei zugängliche Bereiche eines Zielproteins und beginnt dort mit der Spaltung von Peptidbindungen. Durch diese ersten Schnitte verliert das Protein schrittweise seine kompakte, dreidimensionale Faltung. Mit zunehmender Destabilisierung werden auch zuvor geschützte innere Strukturbereiche angreifbar, sodass das Protein allmählich in immer kleinere Peptidfragmente zerfällt – es „bröckelt“ förmlich auseinander. Am Ende des Prozesses verbleiben kurze, biologisch inaktive Peptidketten, die keine enzymatische Funktion mehr besitzen.
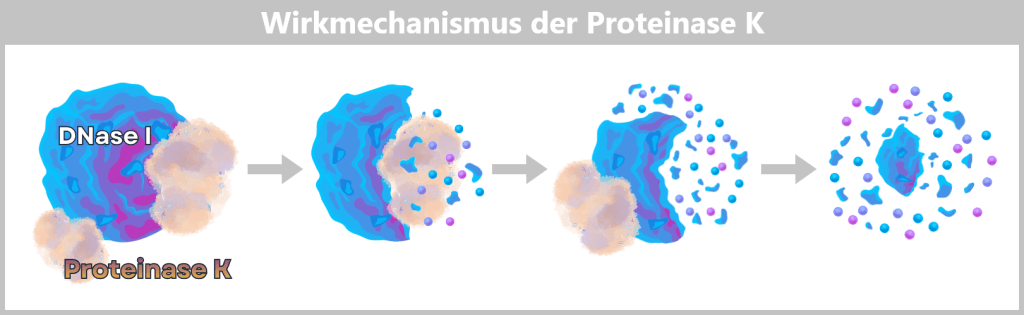
Die Proteinase K greift die DNase I an und spaltet deren Peptidbindungen. Zunächst werden äußere Proteinbereiche hydrolysiert, wodurch die Struktur destabilisiert wird. Schrittweise zerfällt das Protein in kleinere Fragmente, bis schließlich nur noch kurze, biologisch inaktive Peptidstücke verbleiben.
Proteinase K ist selbst ein Protein und wird nicht im Endprodukt belassen. Ihre Aktivität kann durch Prozessbedingungen, etwa durch die Reduktion stabilisierender Calciumionen, vermindert werden. In der Praxis erfolgt die Entfernung der Proteinase K jedoch vor allem physikalisch: Sowohl das Enzym selbst als auch die entstehenden Peptidfragmente werden in nachfolgenden Reinigungsschritten, insbesondere durch Ultrafiltration (z. B. Tangentialflussfiltration) und chromatographische Verfahren, zuverlässig aus der Lösung entfernt.
Bedeutung im Prozess
Durch den gezielten Einsatz von Proteinase K können restliche Prozessenzyme, bakterielle Proteine und andere Proteinverunreinigungen effektiv reduziert werden. Dies schafft definierte Ausgangsbedingungen für die nachfolgenden Filtrations- und chromatographischen Reinigungsschritte. Der Einsatz von Proteinase K stellt dabei keinen zwingenden Bestandteil jedes Herstellungsprozesses dar, erhöht jedoch die Robustheit und Reproduzierbarkeit der Aufarbeitung, insbesondere bei großskaliger industrieller Produktion.
1.3.3. Filtration
Die Filtration ist ein physikalisches Trennverfahren, das Partikel oder Moleküle hauptsächlich aufgrund ihrer Größe voneinander trennt
Dabei wird eine Mischung durch ein poröses Filtermaterial geleitet. Im einfachsten Fall wirken die Poren wie ein mechanisches Sieb: Partikel, die größer sind als die Poren, werden zurückgehalten, während kleinere Moleküle passieren.
a) Filtration – Trennung nach Größe
b) Tangentialflussfiltration (TFF)
c) Sterile Filtration
a) Filtration – Trennung nach Größe
Im Rahmen der Plasmidaufreinigung kommen zwei Grundprinzipien zum Einsatz:
- die Abtrennung unlöslicher Partikel (Klärung), um eine trübe Suspension zu klären,
- die Trennung gelöster Moleküle nach Größe (Membranfiltration), um beispielsweise Puffer auszutauschen oder Moleküle zu konzentrieren.
Klärung: Filtration nach Lyse der E.coli-Bakterienzellen
Ausgangslage: Lyse der Bakterienzellen
Die Zugabe von Natronlauge (NaOH) und dem Detergens SDS löst die fetthaltige Zellhülle der Bakterien auf. Die alkalische Lyse wirkt sich dabei nicht nur auf die Membranen, sondern auch auf Nukleinsäuren und Proteine aus. Die Doppelhelix-Struktur der DNA wird denaturiert. Die lange, faserige genomische DNA wird zusätzlich mechanisch fragmentiert. Unter den stark basischen Bedingungen verlieren Proteine ihre dreidimensionale Faltung und damit ihre biologische Funktion.
Neutralisation: Ordnung des Systems
Durch Zugabe einer sauren Lösung wird die alkalische Reaktionsmischung neutralisiert. Die kurzen, ringförmigen Plasmid-DNA-Moleküle können unter diesen Bedingungen wieder korrekt renaturieren und bleiben in Lösung. Die fragmentierten und fehlgepaarten Reste der genomischen DNA sowie denaturierte Proteine aggregieren (verklumpen) dagegen zusammen mit Lipiden und Membranbestandteilen und fallen als weißlicher Niederschlag aus.
Zwischenzustand
Nach der alkalischen Lyse und Neutralisation liegt die Probe als Zweiphasensystem vor: eine gelöste Phase mit Plasmid-DNA und ein unlöslicher Niederschlag aus aggregierten Bestandteilen. Diese Aggregate bestehen aus fragmentierter genomischer DNA, denaturierten Proteinen, ribosomalen Bestandteilen und Membranresten.
Klär- und Tiefenfiltration
Im industriellen Maßstab werden diese Feststoffe effizient durch Tiefenfiltration entfernt. Dabei durchläuft die Suspension ein mehrlagiges, poröses Filtermaterial. Die Partikel werden nicht nur an der Oberfläche, sondern auch innerhalb der Filtermatrix zurückgehalten, was eine hohe Aufnahmekapazität und effektive Klärung ermöglicht.
Ergebnis
Das klare Filtrat enthält die gelöste Plasmid-DNA weitgehend befreit von Zelltrümmern und ausgefällten Aggregaten. Diese grob gereinigte Lösung bildet die Grundlage für die nachfolgenden, hochauflösenden chromatographischen Reinigungsschritte.
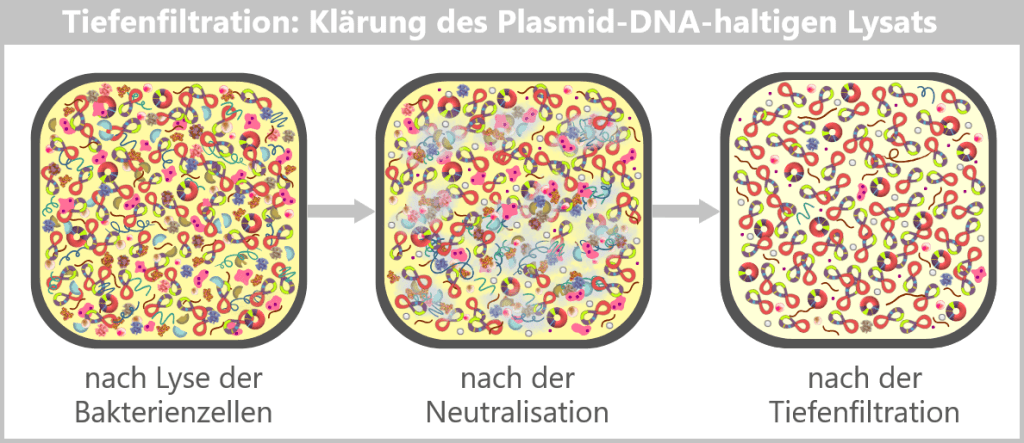
Nach der alkalischen Lyse enthält das Lysat neben der gewünschten Plasmid-DNA zahlreiche bakterielle Bestandteile und Prozesshilfsstoffe.
Während der anschließenden Neutralisation bilden fragmentierte genomische DNA, denaturierte Proteine, sowie Lipide und Membranreste unregelmäßige, voluminöse Aggregate (dargestellt als Wolken).
Durch die Tiefenfiltration werden diese ausgefällten Aggregate effektiv entfernt. Das resultierende Filtrat ist eine klare Lösung, die die gelöste Plasmid-DNA enthält. Daneben können noch lösliche RNA-Reste, Endotoxine (LPS), Salze, Pufferbestandteile sowie geringe Mengen gelöster Proteinreste vorhanden sein.
Membranfiltration: Ultrafiltration und Diafiltration
Eine besonders wichtige Variante der Membranfiltration in der Biotechnologie ist die Ultrafiltration (UF). Sie dient der Trennung gelöster Makromoleküle (z. B. Nukleinsäuren oder Proteine) von kleineren Molekülen wie Salzen, Puffern oder Nukleotiden.
Wird während der Ultrafiltration kontinuierlich frischer Puffer zugegeben, spricht man von Diafiltration (DF). Dadurch lassen sich Moleküle nicht nur konzentrieren, sondern auch gezielt in eine neue Lösung überführen. UF und DF werden häufig kombiniert und als UF/DF-Prozess bezeichnet.
Die vorherrschende Technik für die Durchführung von UF/DF ist die Tangentialflussfiltration (TFF). [ROCKER]
b) Tangentialflussfiltration (TFF)
Bei der klassischen Filtrationsmethode (Dead-End-Filtration oder Direktflussfiltration) strömt die Lösung senkrecht auf die Membran, wodurch sich große Moleküle auf der Oberfläche ablagern und eine „Filterkuchenschicht“ bilden können. Dies verringert mit der Zeit den Durchfluss und die Lebensdauer der Membran.
Die Tangentialflussfiltration (TFF) löst dieses Problem: Hier bewegt sich die Flüssigkeit tangential entlang der Membran. Ein Teilstrom (das Permeat) passiert die Membran, während der Reststrom (das Retentat) parallel weiterfließt und Ablagerungen abträgt. Dadurch bleibt die Membran länger durchlässig und es können Konzentration und Diafiltration gleichzeitig durchgeführt werden.
Begriffserklärungen:
Retentat: Die Fraktion, die von der Membran zurückgehalten wird (meist das gewünschte Makromolekül).
Permeat: Die Fraktion, die die Membran passiert (meist kleine Moleküle und Verunreinigungen).
Bei der Dead-End-Filtration trifft der gesamte Zufluss senkrecht auf die Membran. Kleine Moleküle (Permeat) passieren die Poren, während große Moleküle zurückgehalten werden. Dadurch bildet sich mit der Zeit ein „Filterkuchen“, der die Effizienz verringert.
Bei der Tangential-Flow-Filtration strömt die Lösung parallel zur Membran. Während kleine Moleküle durch die Membran ins Permeat gelangen, werden größere Moleküle im Retentat weitergeführt. Durch den tangentialen Fluss wird die Membran kontinuierlich gespült, Ablagerungen werden verhindert und die Filtration bleibt über längere Zeit stabil und effizient.
TFF ist heute eine Standardtechnik in der biopharmazeutischen Produktion, weil sie robust, skalierbar und vielseitig einsetzbar ist.
TFF bei der Impfstoffherstellung
Die Tangentialflussfiltration (TFF) wird während der mRNA-Produktion an mehreren Punkten eingesetzt: zum Beispiel nach der Transkription, nach enzymatischen Prozessierungen oder vor/nach chromatographischen Reinigungsschritten. Ihre Aufgaben reichen vom Entfernen kleiner Moleküle über den Pufferwechsel bis zur Produktkonzentration.
In diesem Kapitel konzentrieren wir uns auf einen zentralen, aber nicht abschließenden Reinigungsschritt:
TFF direkt nach der in-vitro-Transkription (IVT)
Hier dient sie der Grobreinigung der frisch synthetisierten mRNA. Sie trennt die mRNA von Enzymen, Reagenzien und Nebenprodukten und bereitet sie so für die nachfolgende Hochreinigung vor.
Ausgangslage: Die „Rohlösung“ nach der IVT, DNase-I- und Proteinase-K-Behandlung
Die mRNA liegt zu diesem Zeitpunkt in einer reaktionshaltigen Lösung vor, die noch eine Vielzahl unerwünschter Stoffe enthält:
- Enzyme (z. B. T7-Polymerase, DNase I),
- unverbrauchte Nukleotide,
- DNA-Reste vom Template,
- Puffersalze,
- Nebenprodukte der Reaktion (z. B. kurze RNA-Fragmente),
- ggf. Restbestandteile aus vorgelagerten biologischen Prozessen.
Um die mRNA-Lösung aufzubereiten, wird sie durch eine Filtereinheit mit Membranen definierter Porengröße geleitet. Moleküle unterhalb dieser Porengröße – etwa Salze, Nukleotide oder kleine Proteinreste – passieren die Membran (Permeat), während die großen mRNA-Moleküle im Retentat zurückgehalten werden.
Während des Prozesses wird kontinuierlich frischer Puffer zugegeben (Diafiltration). Dadurch werden niedermolekulare Verunreinigungen schrittweise ausgewaschen, während die mRNA gleichzeitig in den Zielpuffer überführt und konzentriert wird (Ultrafiltration).
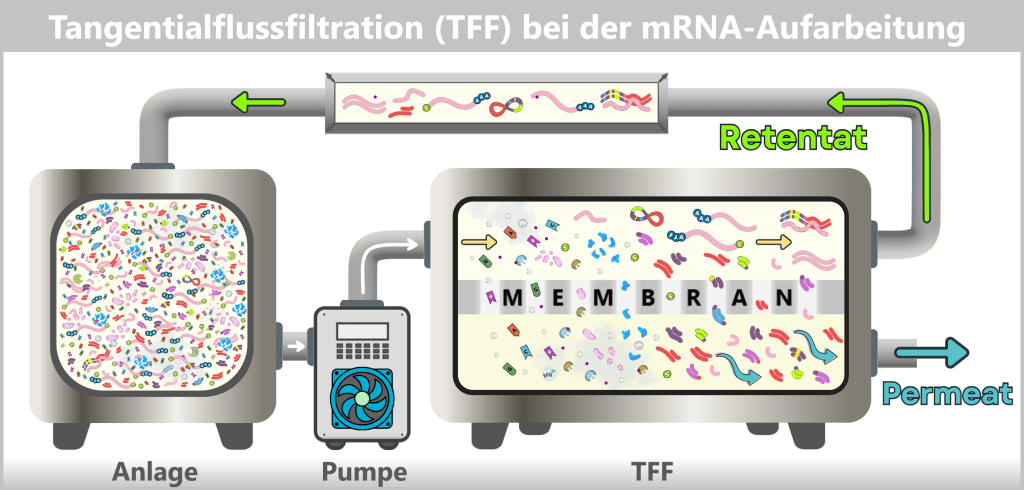
Die mRNA-Lösung zirkuliert kontinuierlich durch das UF/DF-System. Kleine Moleküle passieren die Membran und werden ausgespült (Permeat). Eine Pumpe sorgt dafür, dass das Retentat – also die mRNA-haltige Fraktion – mehrfach über die Membran geführt wird. Durch wiederholte Zyklen und Pufferzugabe werden Verunreinigungen entfernt und die mRNA-haltige Fraktion gleichzeitig konzentriert und umgepuffert.
Wichtig ist:
Die TFF ist eine Grobtrennung nach Größe. Verunreinigungen mit ähnlicher Größe oder Form wie die mRNA – etwa doppelsträngige RNA, RNA-Fragmente vergleichbarer Länge oder RNA:DNA-Hybride – können in diesem Schritt nicht entfernt werden. Diese kritischen Nebenprodukte verbleiben im Retentat und müssen in den nachfolgenden, hochauflösenden chromatographischen Schritten abgetrennt werden.
c) Sterile Filtration
Die sterile Filtration ist ein zentraler letzter Schritt vor der Abfüllung eines mRNA-Impfstoffs. Dabei wird die fertige Formulierung (Lipidnanopartikel, die die mRNA enthalten) durch einen sehr feinen Filter geleitet. Die Porengröße ist klein genug, um alle Bakterien, Pilze und größere Partikel zuverlässig zurückzuhalten.
Prinzip der sterilen Filtration
Die sterile Filtration ist ein reines Größen-Siebverfahren:
- Die Membran besitzt exakt definierte Poren von 0,22 µm.
- Mikroorganismen sind deutlich größer und bleiben hängen.
- Die Wirkstoffpartikel (LNPs) sind viel kleiner (60–100 nm = 0,06–0,1 µm) und passieren den Filter problemlos.
Da keine Hitze oder chemische Behandlung nötig ist, eignet sich dieser Schritt besonders für empfindliche biologische Präparate wie mRNA-LNPs, die nicht autoklaviert (mit Dampf, Druck und Hitze sterilisiert) werden dürfen.
Ablauf im Detail
Die LNP-mRNA-Lösung liegt im Formulierpuffer vor. Sterile Filtration erfolgt in einer Reinraumumgebung (Klasse A/B) unter streng aseptischen Bedingungen. Es werden ausschließlich sterile Einwegartikel verwendet: Filter, Schläuche, Beutel, Pumpenköpfe, Sammelbehälter.
Die Formulierung wird mit sanftem Druck (Peristaltikpumpe oder Druckgas) durch den 0,22-µm-Filter geleitet.
| Der Filter hält zurück: ▪️Bakterien (~0,5–5 µm) ▪️Hefen und Pilzsporen (~1–10 µm) ▪️Partikel oder Aggregate > 0,22 µm | Durchgelassen werden: ▪️LNPs (60–100 nm) ▪️Pufferbestandteile ▪️gelöste Moleküle |
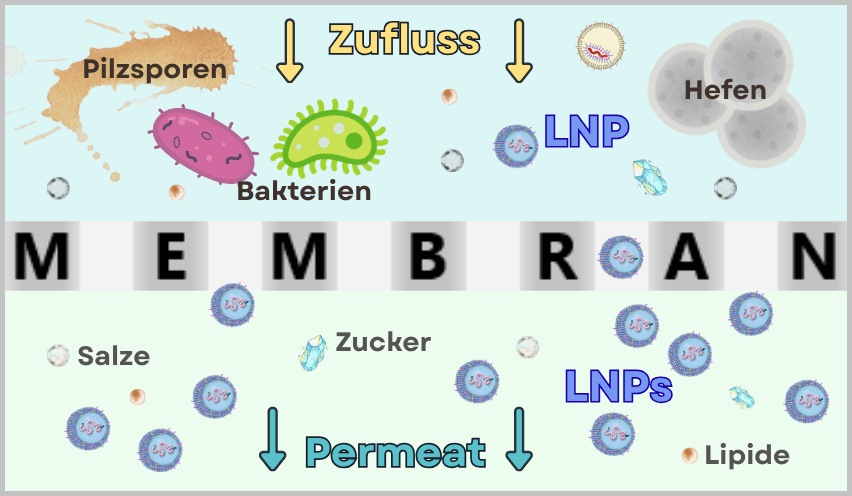
Damit wird sichergestellt, dass die Formulierung frei von Mikroorganismen und sichtbaren Partikeln ist.
Warum sterile Filtration notwendig ist
Nach GMP-Richtlinien dürfen sterile Arzneimittel ausschließlich mit validierten, sterilen Filtrationsprozessen hergestellt werden. Die sterile Filtration ist daher Pflicht und stellt sicher, dass:
- die fertige Impfstofflösung mikrobiologisch einwandfrei ist
- keine großen Partikel, Aggregate oder Verunreinigungen in das Endprodukt gelangen
- die anschließende sterile Abfüllung („aseptic fill & finish“) korrekt erfolgen kann
Die sterile Filtration ist kein chemischer Reinigungsschritt, sondern ein essenzieller Sicherheits- und Qualitätsprozess. Sie bildet die Brücke zwischen der biotechnologischen Aufreinigung der mRNA und dem finalen, sterilen Arzneimittel in der Durchstechflasche.
1.3.4. Chromatographie – das molekulare Trennverfahren
Die Chromatographie ist eine der wichtigsten Verfahren zur Reinigung biologischer Moleküle wie DNA, RNA oder Proteine. Das Prinzip basiert auf der unterschiedlich starken Wechselwirkung der Stoffe mit zwei Phasen: einer stationären Phase (ein festes Trägermaterial in einer Säule) und einer mobilen Phase (eine flüssige Lösung, die das Probengemisch transportiert).
Während die mobile Phase – also das zu trennende Molekülgemisch – durch die stationäre Phase fließt, werden die einzelnen Moleküle je nach ihrer Größe, Ladung oder anderen chemischen Eigenschaften unterschiedlich stark zurückgehalten. Dies führt zu einer zeitlichen Trennung, bei der die Bestandteile des Gemischs nacheinander eluieren (herausgespült werden).
Anschaulich gesprochen ist die Chromatographie ein „molekularer Hindernislauf“: Manche Moleküle interagieren kaum mit der stationären Phase und gleiten schnell hindurch. Andere bleiben häufiger „hängen“ und werden dadurch stärker verzögert. So erreicht jede Komponente das Ziel zu einer anderen Zeit und kann sauber aufgefangen werden.
Chromatographie-Methoden in der mRNA-Reinigung
In industriellen Prozessen, wie sie beispielsweise von MERCK beschrieben werden („Manufacturing strategies for mRNA vaccines“), kommen dabei zwei chromatographische Verfahren in Kombination zum Einsatz:
| Methode | Trennung nach | Typische Anwendung |
| Affinitäts-Chromatographie (Poly(dT)) | spezifischer Bindung | Primäre Aufreinigung: Isolierung intakter mRNA aus der IVT-Reaktionsmischung |
| Ionenaustausch- Chromatographie | elektrischer Ladung | Feinreinigung: Entfernen von dsRNA, RNA:DNA-Hybriden, DNA-Fragmenten und weiteren produktbezogenen Verunreinigungen |
a) Poly(dT)-Affinitätschromatographie
Poly(dT)-AC = Poly(dT) Affinity Chromatography
Die Affinitätschromatographie nutzt spezifische Wechselwirkungen zwischen Biomolekülen und einer funktionalisierten Oberfläche. Bei der Herstellung von mRNA-Impfstoffen wird die Poly(dT)-Affinitätschromatographie gezielt eingesetzt, um die reife mRNA nach der in-vitro-Transkription (IVT) aus der komplexen Reaktionslösung zu isolieren.
Ausgangslage: Nach in-vitro-Transkription (IVT), enzymatischer Nachbehandlung und Tangentialflussfiltration (TFF)
Prinzip und Ablauf
Stationäre Phase: Das Harz in der Säule ist mit langen, einzelsträngigen Ketten von Thymin-Basen (T) beschichtet, der sogenannten Poly(dT)-Matrix.
Mobile Phase: Die voraufgereinigte IVT-Lösung – bestehend aus mRNA, RNA-Nebenprodukten (z. B. dsRNA, verkürzte Transkripte), Restmengen an DNA-Fragmenten sowie definierten Puffersalzen – wird auf die Säule gegeben.
Das Zielmolekül ist das reife mRNA-Molekül – die „polyadenylierte mRNA“. Diese trägt am 3′-Ende eine Kette aus Adenin-Basen (A), den sogenannten Poly-A-Schwanz.
Spezifische Bindung über Basenpaarung
Adenin (A) und Thymin (T) bilden – wie in der DNA – spezifische Wasserstoffbrückenbindungen aus. Bildlich formuliert, der Poly(A)-Schwanz der mRNA hängt sehr stabil am Poly(dT)-Haken des Harzes.
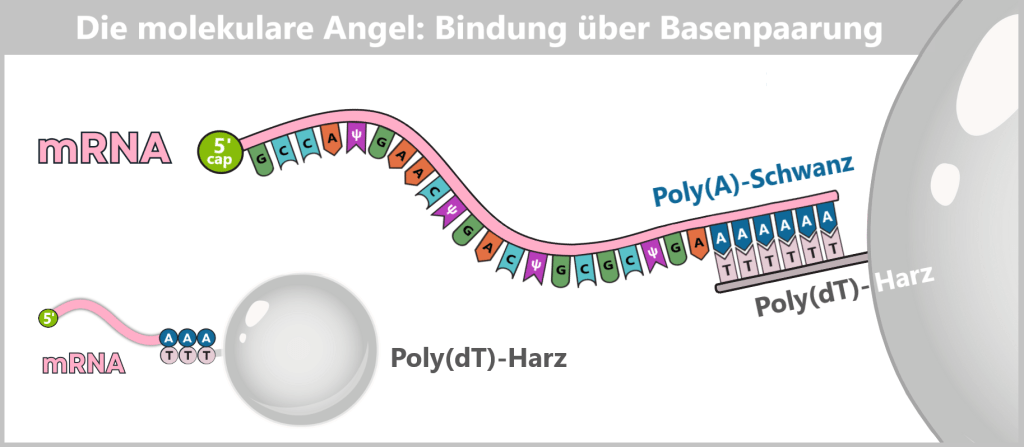
Der Poly(A)-Schwanz therapeutischer mRNA umfasst typischerweise etwa 100–120 Adenin-Basen; in der Abbildung ist er aus Darstellungsgründen verkürzt dargestellt.
Nur Moleküle mit intaktem Poly(A)-Schwanz binden fest an das Harz; alle anderen Bestandteile der IVT-Reaktion fließen durch. Dazu gehören:
(a) verbleibende freie Nukleotide (nur in Spuren)
(b) Proteinfragmente aus dem enzymatischen Abbau
(c) kurze RNA-Fragmente ohne Poly(A)-Schwanz
(d) DNA-Fragmente und DNA-haltige Nebenprodukte
(e) verbleibende supercoiled Plasmid-DNA
(f) freie dsRNA
(g) Puffersalze

Waschen (Wash)
Waschpuffer entfernen schwach gebundene, unspezifisch haftende Moleküle, während die mRNA weiterhin fest an der Poly(dT)-Matrix verbleibt.
Elution (Freisetzung)
Um die reine mRNA wieder von der „Angel“ zu lösen, werden die spezifischen Bindungen zwischen A und T gezielt aufgebrochen. Dies geschieht typischerweise durch Absenkung der Ionenstärke und/oder moderate Temperaturerhöhung. Die mRNA löst sich von der Matrix und wird eluiert.
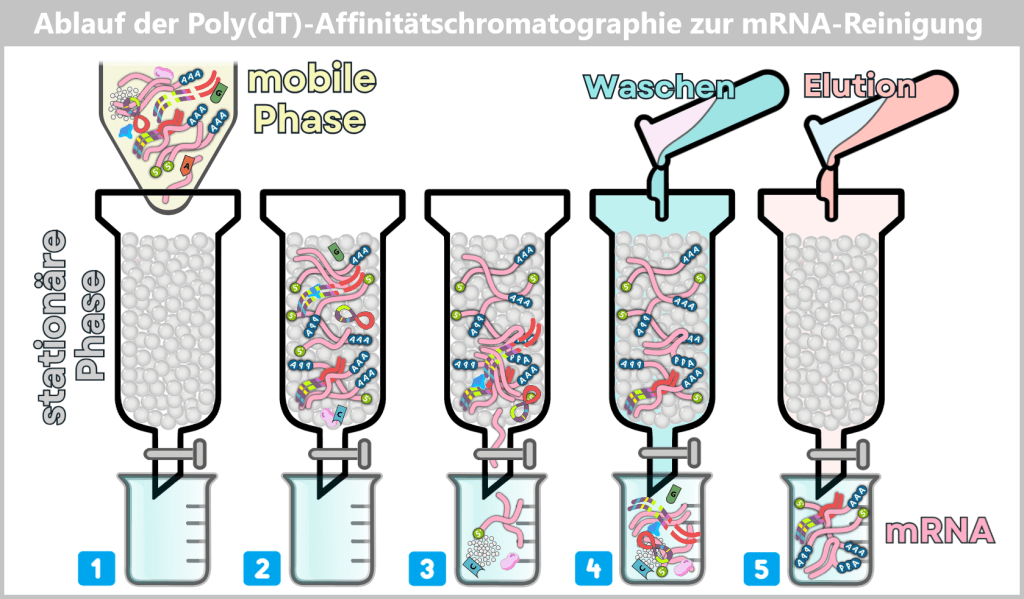
1) Die IVT-Reaktionsmischung wird auf die mit Poly(dT) funktionalisierte Säule aufgetragen.
2) Die polyadenylierte mRNA bindet über Basenpaarung spezifisch an das Harz,
3) während nicht-bindende Komponenten direkt durchlaufen.
4) Ein Waschpuffer entfernt schwach oder unspezifisch gebundene Moleküle.
5) Durch Zugabe eines Elutionspuffers wird die mRNA von der Poly(dT)-Matrix gelöst und in reiner Form eluiert.
Vorteile
- sehr hohe Selektivität durch Poly(A)/Poly(dT)-Hybridisierung
- effiziente Abtrennung der meisten unspezifischen RNA- und Proteinverunreinigungen
- Anreicherung der vollständigen, korrekt polyadenylierten mRNA
- vergleichsweise einfache und robuste Methode
Limitierungen – warum weitere Reinigungsschritte notwendig sind
Trotz der hohen Spezifität kann die Poly(dT)-Affinität nicht alle kritischen Verunreinigungen entfernen:
- dsRNA: kann über polyadenylierte Enden oder unspezifisch an die Matrix binden
- RNA:DNA-Hybride: wenn DNA an mRNA gebunden ist, wird sie gemeinsam mit eluiert
- fragmentierte mRNA mit Poly(A)-Resten bindet ebenfalls
- intramolekulare dsRNA-Strukturen innerhalb der mRNA führen zu Ko-Elution
- größere RNA-Komplexe, die an mRNA assoziiert sind, werden mitisoliert
- Endotoxine werden nicht spezifisch entfernt und können ko-eluiert werden
Hersteller wie MERCK, Lonza oder Cytiva empfehlen daher im Anschluss eine Anionenaustauschchromatographie (AEX) zur Feintrennung, insbesondere zur Entfernung von dsRNA und DNA-Restverunreinigungen.
b) Ionenaustausch-Chromatographie
Die Ionenaustausch-Chromatographie (IEX) ist eine bewährte Methode zur Reinigung von Nukleinsäuren und Proteinen. Sie trennt Moleküle anhand ihrer elektrischen Ladung. Bei Nukleinsäuren ist damit nicht eine feste Oberfläche gemeint, sondern die hohe Dichte negativ geladener Phosphatgruppen entlang des Moleküls. Im Gegensatz dazu ist die Ladung von Proteinen von der spezifischen Abfolge und Faltung ihrer Aminosäuren abhängig und kann positive und negative Bereiche aufweisen.
Je nach Art der geladenen Zielmoleküle unterscheidet man zwei Haupttypen:
▪️Anionenaustauscher (AEX, Anion Exchange Chromatography):
binden negativ geladene Moleküle wie DNA oder RNA.
▪️Kationenaustauscher (CEX, Cation Exchange Chromatography):
binden positiv geladene Moleküle, beispielsweise bestimmte Proteine.
Aufgrund ihrer durchgehenden Phosphatgruppe im Rückgrat tragen Nukleinsäuren eine hohe, gleichmäßige negative Ladungsdichte.
Zucker-Phosphat-Rückgrat als Quelle der negativen Ladung von DNA/RNA
Schematische Darstellung der strukturellen Grundlage der negativen Ladung von Nukleinsäuren
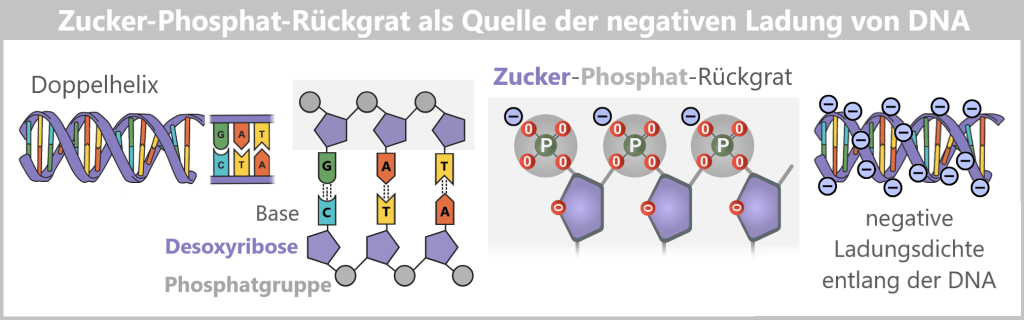
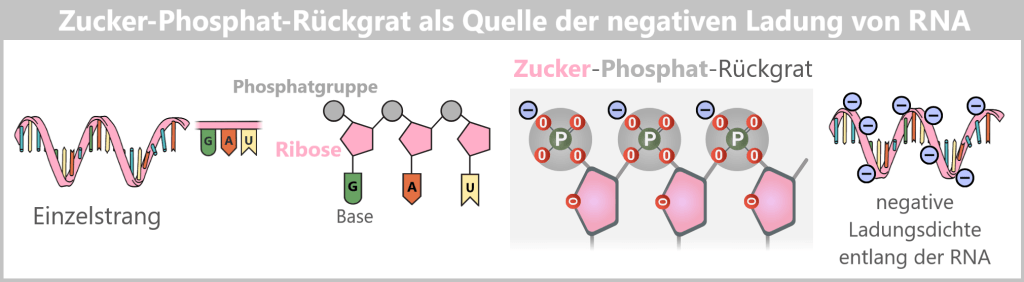
DNA (und analog RNA) besteht aus einem Zucker-Phosphat-Rückgrat, in dem jede Phosphatgruppe eine negative Ladung trägt. Diese regelmäßig wiederholten Phosphatgruppen verleihen dem Molekül eine hohe und entlang seiner Länge gleichmäßig verteilte negative Ladungsdichte. Die Basen tragen nicht zur Gesamtladung bei.
Diese Eigenschaft macht die Anionenaustauschchromatographie (AEX) zur Methode der Wahl für deren Aufreinigung. Im Gesamtprozess kommt die AEX daher gleich doppelt zum Einsatz: Zunächst zur Aufreinigung der Plasmid-DNA-Vorlage aus dem bakteriellen Lysat. Danach, nach der in-vitro-Transkription (IVT) und Grobreinigung, zur hochauflösenden Trennung der gewünschten mRNA von strukturell ähnlichen Verunreinigungen, insbesondere doppelsträngiger RNA (dsRNA).
AEX bei der Plasmid-DNA-Aufreinigung
Ausgangslage
Nach Lyse der E.-coli-Zellen, Neutralisation und anschließender Tiefenfiltration liegt ein geklärtes Lysat vor, das die Plasmid-DNA enthält. Daneben können noch bakterielle RNA-Reste, Endotoxine, Salze, Pufferbestandteile sowie geringe Mengen gelöster Proteinreste vorhanden sein.

Prinzip und Ablauf
Stationäre Phase: Das Trägermaterial (Harz) besteht aus kleinen, positiv geladenen Polymerkügelchen. Diese dienen als Bindungsoberfläche für negativ geladene Moleküle.
Mobile Phase: Das geklärte Lysat wird auf die Säule gegeben. Zunächst binden die negativ geladenen Biomoleküle – darunter Plasmid-DNA, RNA und Endotoxine – an das positiv geladene Harz. Ungebundene oder schwach geladene Bestandteile werden durch Spülpuffer entfernt.
Das Zielmolekül ist die Plasmid-DNA – die entlang ihrer Länge eine gleichmäßig verteilte negative Ladungsdichte besitzt.
Selektive Elution: Anschließend wird ein Puffer mit ansteigender Salzkonzentration über die Säule geleitet. Die positiv geladenen Ionen (z. B. Na⁺) im Puffer konkurrieren mit den Biomolekülen um die Bindungsstellen des Harzes. Dadurch lösen sich die Moleküle in einer charakteristischen Reihenfolge – je nach ihrer Ladungsdichte und Bindungsstärke:
- Endotoxine und kleine RNA-Fragmente eluieren früh, da sie eine geringere Ladungsdichte besitzen.
- Plasmid-DNA – überwiegend in superhelikaler, aber auch in relaxeder Ringform – bindet aufgrund ihrer hohen negativen Ladungsdichte am stärksten und eluierte erst bei höheren Salzkonzentrationen.
Ergebnis
Durch diese selektive Elution erhält man eine Lösung – die reich an hochreiner Plasmid-DNA ist, überwiegend in superhelikaler Form, mit einem geringeren Anteil relaxeder Ringformen – während die meisten anderen Zellbestandteile und Verunreinigungen bereits entfernt wurden. Diese DNA-Fraktion dient anschließend als Template für die nachfolgende in-vitro-Transkription (IVT).
1) Das geklärte Zelllysat (mobile Phase) wird auf die Säule mit der positiv geladenen stationären Phase (Anionenaustauschharz) aufgetragen. 2) Negativ geladene Moleküle der Probe binden an das Harz; nicht bzw. schwach gebundene Komponenten (Salze, Proteinreste) eluieren früh. 3) Die Elution beginnt: Ein Puffer mit steigender Salzkonzentration wird zugegeben. Die gelösten Ionen (z. B. Na⁺ und Cl⁻) konkurrieren mit den gebundenen Molekülen um die Bindungsstellen. Bei niedrigen bis mittleren Salzkonzentrationen eluieren vor allem kurze und/oder einzelsträngige RNA-Fragmente. 4) Plasmid-DNA eluiert erst bei höheren Salzkonzentrationen aufgrund ihrer hohen, gleichmäßig verteilten negativen Ladungsdichte. Endotoxine (LPS) binden sehr stark an die Matrix, was grundsätzlich vorteilhaft aus Sicht der Produktreinigung ist. Problematisch sind lediglich jene Anteile, die aufgrund unspezifischer Wechselwirkungen oder Assoziation mit DNA gemeinsam mit der Plasmid-DNA eluieren können.
Nach der Anionenaustauschchromatographie liegt die Plasmid-DNA in hoher Reinheit vor. Das Produkt besteht überwiegend aus superhelikaler Plasmid-DNA und kann geringe Anteile relaxierter Ringformen enthalten. In niedrigen Konzentrationen sind zudem Reste kurzer RNA-Fragmente oder Endotoxine nachweisbar, während zelluläre Makromoleküle und Prozessverunreinigungen größtenteils entfernt wurden.
AEX in der mRNA-Reinigung: Trennung von mRNA und dsRNA
Ausgangslage: Nach in-vitro-Transkription (IVT), enzymatischer Nachbehandlung, Tangentialflussfiltration (TFF) und Poly(dT)-Affinitätschromatographie
Nach diesen vorgelagerten Prozessschritten liegt eine stark angereicherte mRNA-Fraktion vor. Dennoch enthält diese mRNA-Rohlösung typischerweise noch bestimmte Nebenprodukte und Verunreinigungen, die durch die Poly(dT)-Affinität nicht vollständig entfernt werden können, darunter:
(a) doppelsträngige RNA-Nebenprodukte (dsRNA)
(b) RNA:DNA-Hybride (in geringen, prozessabhängigen Anteilen)
(c) fragmentierte mRNA mit verbliebenen Poly(A)-Resten
(d) nicht korrekt gecappte mRNA-Transkripte
(e) Endotoxine (LPS), insbesondere in Assoziation mit Nukleinsäuren

Die nachfolgende Anionenaustauschchromatographie dient daher nicht der weiteren Anreicherung der mRNA, sondern der hochauflösenden Entfernung strukturell ähnlicher RNA-Nebenprodukte.
Prinzip und Ablauf
Stationäre Phase: Die stationäre Phase besteht aus positiv geladenen Anionenaustauschharzen, meist polymeren Kügelchen mit quaternären Ammoniumgruppen. Diese bieten Bindungsstellen für negativ geladene Nukleinsäuren.
Mobile Phase: Die mRNA-Rohlösung wird als mobile Phase auf die Säule aufgetragen. Unter niedriger Salzkonzentration binden die negativ geladenen Nukleinsäuren der Probe an das Harz.
Das Zielmolekül ist die einzelsträngige mRNA. Sie besitzt entlang ihres Rückgrats eine negative Ladung, weist jedoch aufgrund ihrer Einzelsträngigkeit, Sekundärstruktur und Flexibilität eine geringere effektive Ladungsdichte auf als doppelsträngige RNA.
Selektive Elution
Im nächsten Schritt wird ein Elutionspuffer mit ansteigender Salzkonzentration über die Säule geleitet. Die im Puffer enthaltenen Ionen (z. B. Na⁺) konkurrieren mit den gebundenen RNA-Molekülen um die Bindungsstellen des Harzes. Dadurch werden die gebundenen Komponenten in Abhängigkeit von ihrer Bindungsstärke schrittweise eluiert:
Einzelsträngige mRNA eluieren bei niedrigen bis mittleren Salzkonzentrationen, da ihre effektive Ladungsdichte geringer ist.
Doppelsträngige RNA (dsRNA) bindet deutlich stärker an das Anionenaustauschharz und eluiert erst bei höheren Salzkonzentrationen oder verbleibt auf der Säule.
RNA:DNA-Hybride stellen für die chromatographische Reinigung eine besondere Herausforderung dar. Ihre Bindungseigenschaften bewegen sich zwischen denen von einzelsträngiger RNA (ssRNA) und doppelsträngiger RNA (dsRNA). Dieser hybride Charakter ergibt sich nicht nur aus dem RNA:DNA-Duplex selbst, sondern wird zusätzlich durch drei Faktoren bestimmt: der Länge der Hybridregion, dem Anteil freier einzelsträngiger RNA sowie der räumlichen Konformation des Moleküls.
Die Darstellung ist vereinfacht und zeigt jede Komponente exemplarisch nur einmal.
1) Die mRNA-haltige Lösung (mobile Phase) – bestehend aus der gewünschten mRNA, dsRNA und anderen Nebenprodukten – wird auf die Säule geleitet. Die stationäre Phase besteht aus einem positiv geladenen Anionenaustauschharz. 2) Unter niedriger Salzkonzentration binden alle negativ geladenen RNA-Moleküle an das Harz. Aufgrund ihrer höheren Ladungsdichte und steiferen Helixstruktur bindet dsRNA stärker als die flexiblere, einzelsträngige mRNA. 3) Durch Zugabe eines Puffers mit ansteigender Salzkonzentration werden konkurrierende Anionen (z.B. Cl⁻) eingeführt. Diese verdrängen die gebundenen RNA-Moleküle schrittweise von der Säule. Zuerst werden schwach gebundene Verunreinigungen wie kurze RNA-Fragmente freigesetzt. 4) Bei niedrigen bis mittleren Salzkonzentrationen eluiert die gewünschte einzelsträngige mRNA. Doppelsträngige RNA verbleibt aufgrund ihrer stärkeren Bindung länger auf der Matrix und eluiert erst bei höheren Salzkonzentrationen oder verbleibt teilweise auf der Säule.
Ergebnis
Die Anionenaustauschchromatographie (AEX) ermöglicht die selektive Abtrennung von mRNA gegenüber stärker oder schwächer gebundenen Nukleinsäurestrukturen wie dsRNA. Damit erhält man eine mRNA-Fraktion, die weitgehend frei von dsRNA-Verunreinigungen und Hybridstrukturen ist.
Die so gereinigte mRNA dient als Ausgangsmaterial für die nachfolgende Formulierung in Lipid-Nanopartikel (LNP).
Zusammenspiel der chromatographischen Schritte zur Reinigung der IVT-mRNA
Die Reinigung von IVT-mRNA beruht nicht auf einem einzelnen Trennverfahren, sondern auf der gezielten Kombination mehrerer chromatographischer Prinzipien mit komplementären Stärken und Limitationen.
Die Poly(dT)-Affinitätschromatographie dient dabei der hochselektiven Anreicherung polyadenylierter mRNA. Sie entfernt effektiv nicht-polyadenylierte Nebenprodukte, Enzyme und niedermolekulare Prozessbestandteile. Aufgrund ihres rein sequenzbasierten Bindungsmechanismus kann sie jedoch strukturell ähnliche RNA-Verunreinigungen nicht vollständig differenzieren. Insbesondere dsRNA, fragmentierte mRNA mit Poly(A)-Resten sowie RNA:DNA-Hybride können gemeinsam mit der Ziel-mRNA ko-eluiert werden.
Die nachgeschaltete Anionenaustauschchromatographie (AEX) adressiert diese Limitation, indem sie die verbleibenden Nukleinsäuren anhand ihrer effektiven Ladungsdichte und Konformation trennt. Einzelsträngige mRNA, doppelsträngige RNA-Nebenprodukte und komplexe RNA-Strukturen zeigen dabei charakteristische, jedoch teilweise überlappende Bindungs- und Elutionsprofile.
RNA:DNA-Hybride nehmen in diesem Kontext eine Sonderstellung ein. Aufgrund ihres hybriden strukturellen Charakters lassen sie sich weder eindeutig der einzel- noch der doppelsträngigen RNA zuordnen. Ihre chromatographische Trennbarkeit hängt stark von der Ausprägung der Hybridregion, dem Anteil freier einzelsträngiger RNA sowie der räumlichen Organisation des Moleküls ab. Entsprechend können RNA:DNA-Hybride je nach Prozessbedingungen teilweise gemeinsam mit der mRNA eluieren oder auf der Säule zurückgehalten werden.
Erst das abgestufte Zusammenspiel von Affinitäts- und Ionenaustauschchromatographie ermöglicht somit eine weitgehende Entfernung der relevanten RNA-Nebenprodukte, ohne die strukturelle Integrität der empfindlichen mRNA zu beeinträchtigen. Gleichzeitig macht dieser Ansatz deutlich, dass die vollständige Eliminierung aller potenziellen Hybrid- und Strukturvarianten technisch anspruchsvoll ist und maßgeblich von der Prozessführung abhängt.
1.3.5. Magnetkügelchen-Reinigung
dT-MBP = Poly(dT)-magnetic bead purification
Ein in den regulatorischen Dokumenten (EMA-EPAR, Abschnitt 2.2, S. 32) beschriebener Ansatz zur Reinigung von PCR-Produkten in Prozess 1 ist der Einsatz von Magnetkügelchen.
Ausgangslage: Nach in-vitro-Transkription (IVT), DNase-I-Behandlung und Filtration
Nach der in-vitro-Transkription (IVT), der enzymatischen Nachbehandlung mit DNase I sowie einer ersten Grobreinigung durch Filtration (UF/DF oder TFF) liegt eine mRNA-haltige Lösung vor. Diese enthält neben der gewünschten mRNA weiterhin verschiedene Nebenprodukte der Transkription, darunter:
(a) verbleibende freie Nukleotide (nur in Spuren)
(b) Proteinreste (z. B. Enzyme oder Proteinfragmente)
(c) RNA-Fragmente mit und ohne Poly(A)-Schwanz
(d) unvollständig gecappte RNA
(e) doppelsträngige RNA (dsRNA), teilweise mit polyadenyliertem Strang
(f) RNA:DNA-Hybride
(g) DNA-Template-Reste (insbesondere kurze Fragmente)
(h) Puffersalze aus vorangegangenen Prozessschritten
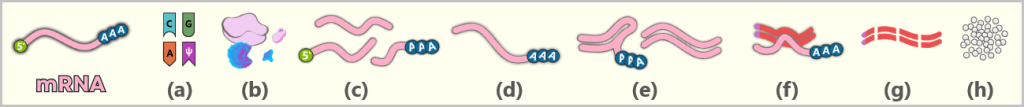
Prinzip
Die Magnetkügelchen-Reinigung beruht auf einer sequenzspezifischen Affinitätsbindung. Magnetische Polymerkügelchen sind mit Oligo(dT)-Sequenzen funktionalisiert, die komplementär zur Poly(A)-Schwanzsequenz der mRNA sind.
mRNA-Moleküle mit Poly(A)-Ende binden spezifisch an diese Oligo(dT)-Liganden, während Moleküle ohne Poly(A)-Sequenz in Lösung verbleiben.
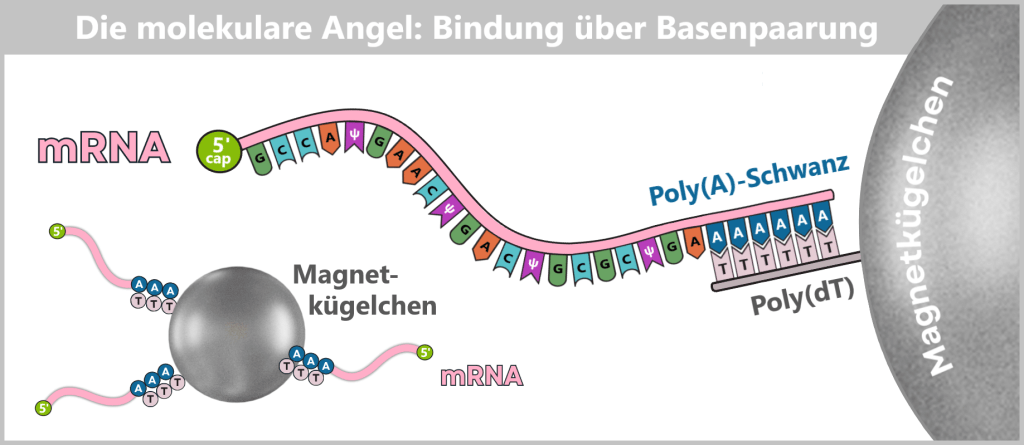
Der Ablauf erfolgt in mehreren Schritten:
Bindung: Poly(dT)-funktionalisierte Magnetkügelchen werden zur Lösung gegeben. Polyadenylierte RNA bindet über spezifische A–T-Basenpaarung an die Oberfläche der Kügelchen.
Magnetische Separation: Das Gefäß wird auf ein Magnetrack gestellt. Die Kügelchen mit der gebundenen RNA werden an die Gefäßwand gezogen, sodass die nicht gebundenen Komponenten entfernt werden können.
Waschen: Mehrere Waschschritte reduzieren unspezifisch gebundene Moleküle. Die Waschbedingungen beeinflussen die Stringenz der Bindung, erlauben jedoch keine vollständige Abtrennung von dsRNA oder RNA:DNA-Hybriden, sofern diese polyadenylierte RNA enthalten.
Elution: Durch geeignete Elutionsbedingungen (z. B. veränderte Salzstärke oder Temperatur) wird die gebundene RNA von den Kügelchen gelöst und in Lösung überführt.
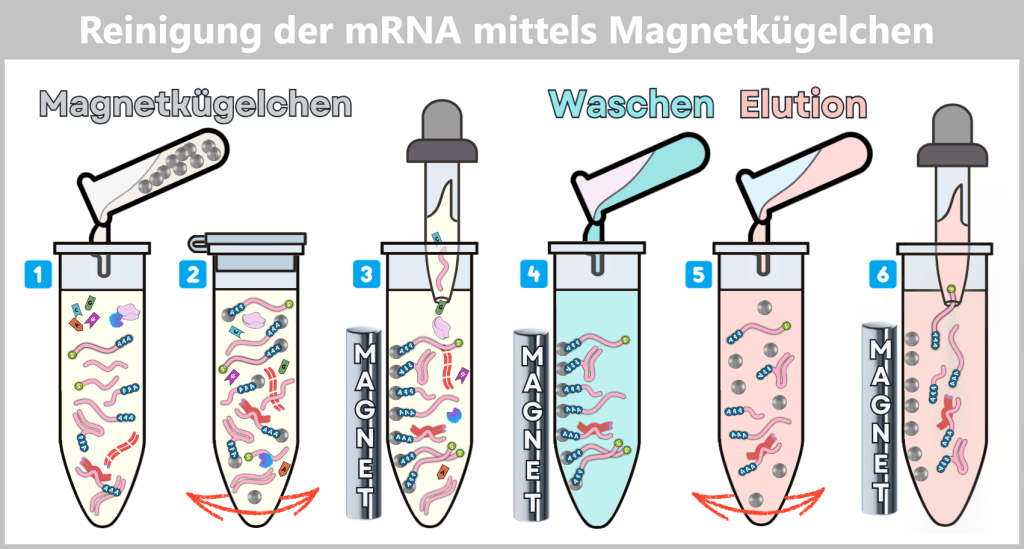
Die Darstellung ist vereinfacht und zeigt jede Komponente exemplarisch nur einmal.
1) Zugabe der Magnetkügelchen: Poly(dT)-funktionalisierte Magnetkügelchen werden zur Lösung gegeben, die die polyadenyliertemRNA enthält.
2) Durchmischen: Die Probe wird durch sanftes Schwenken oder kurzes Drehen gemischt. Dabei hybridisiert polyadenylierte RNA an die Poly(dT)-Sequenzen der Magnetkügelchen über A–T-Basenpaarung.
3) Magnetische Separation und entfernen des Überstands: Das Gefäß wird auf ein Magnetrack gestellt. Die Magnetkügelchen mit gebundener RNA werden von der Seitenwand des Röhrchens angezogen und bilden dort ein kompaktes Bead-Aggregat. Der flüssige Überstand wird vorsichtig abpipettiert. Er enthält nicht-gebundene Moleküle wie DNA-Fragmente, kurze RNA-Stücke, dsRNA ohne Poly(A)-Tail, Pufferbestandteile usw.
4) Waschschritte: Der Magnet bleibt angelegt. Waschpuffer wird zugegeben, um lose gebundene oder unspezifisch adsorbierte Verunreinigungen zu entfernen. Der Waschpuffer wird kurz gemischt (leichtes Schwenken), danach zieht der Magnet die Beads erneut an die Wand. Dieser Schritt wird 2–3 Mal wiederholt, bis lose gebundene oder unspezifisch assoziierte Komponenten weitgehend entfernt sind.
5) Elution der mRNA: Nach dem Entfernen des letzten Waschpuffers wird der Magnet kurz entfernt. Ein Elutionspuffer (z. B. Wasser mit geringer Salzkonzentration) oder leicht erhöhte Temperatur löst die Hybridisierung zwischen polyadenylierter RNA und Poly(dT)-Oligos. Die RNA geht dabei wieder in Lösung über.
6) Rückkehr auf den Magnetseparator: Das Röhrchen wird erneut auf den Magnet gestellt, sodass die Magnetkügelchen am Rand fixiert werden. Der Überstand enthält jetzt die angereicherte polyadenylierte RNA im Elutionspuffer und wird vorsichtig in ein neues, steriles Gefäß überführt.
Hinweis: dsRNA, RNA:DNA-Hybride oder fragmentierte RNA mit Poly(A)-Anteilen können während der Magnetkügelchen-Reinigung ko-eluiert werden.
Trennleistung und Limitierungen
Die Magnetkügelchen-Reinigung ist hochselektiv für polyadenylierte RNA, weist jedoch inhärente Einschränkungen auf:
- dsRNA-Nebenprodukte können ko-eluieren, insbesondere wenn sie Poly(A)-Sequenzen enthalten oder strukturell an mRNA assoziiert sind
- RNA:DNA-Hybride werden mitisoliert, sofern die RNA-Komponente polyadenyliert ist
- fragmentierte mRNA mit erhaltenem Poly(A)-Rest bindet ebenfalls an die Kügelchen
- intramolekulare Doppelstrangstrukturen innerhalb der mRNA werden nicht selektiv entfernt
Aus diesen Gründen stellt die Magnetkügelchen-Reinigung keinen vollständigen Reinigungsprozess, sondern eine Anreicherungs- und Vorreinigungsstufe dar.
Nachgeschaltete Reinigung
Da dsRNA und RNA:DNA-Hybride mit Poly(A)-Anteilen während der Magnetkügelchen-Reinigung ko-eluieren können, ist in der Regel ein weiterer Reinigungsschritt erforderlich. Häufig wird hierfür eine Anionenaustauschchromatographie (AEX) oder ein vergleichbares hochauflösendes Trennverfahren eingesetzt.
Einordnung im Herstellungsprozess
Die Poly(dT)-basierte Magnetkügelchen-Reinigung wird vor allem im Labormaßstab, in der Prozessentwicklung sowie bei kleineren Produktionsvolumina eingesetzt. Für großtechnische Herstellungsprozesse (Prozess 2) ist sie hingegen weniger praktikabel, da die Magnetkraft in größeren Volumina an Effizienz verliert und die Handhabung komplexer wird. Daher kommen in der industriellen Produktion überwiegend skalierbare chromatographische Verfahren zum Einsatz. Weitere Details zur Anwendung finden sich in den Herstellerinformationen von Thermo Fisher Scientific.
1.4. Vergleich: Prozess 1 vs. Prozess 2
Die folgende Übersicht stellt die beiden Herstellungsverfahren gegenüber: welche Ausgangsmaterialien und Nebenprodukte in den einzelnen Phasen anfallen und mit welchen Methoden sie typischerweise entfernt werden.
Typische Schritte – unterschiedlich je nach Hersteller:
Merkmal
Prozess 1 – PCR-basiert
Prozess 2 – Plasmid-basiert
Art des Verfahrens
Laborprozess
(kleine Mengen, flexibel)
Industrieller Prozess
(große Mengen)
DNA-Template
Minimalistisch.
enthält nur die relevanten Elemente für die IVT:
• T7-Promotor
• 5′-UTR
• Spike-Gen
• 3′-UTR
• Poly(A)-Sequenz
Komplex.
und zusätzliche Elemente für die Vermehrung in E. coli:
• Origin of Replication (ORI)
• Antibiotikaresistenzgen
• regulatorische bakt. Sequenzen
• Multiple Cloning Site (MCS)
• SV40-Sequenzelemente
(bei Pfizer/BNT)
Aufbau des DNA-Templates
[T7] – [5′-UTR] – [Spike-Gen] – [3′-UTR] – [AAAAA…]
[ORI] – [Resistenzgen] – [reg. bakt. Sequenzen] – [MCS] – [SV40] – [T7] – [5′-UTR] – [Spike-Gen] – [3′-UTR] – [AAAAA…]
Amplifikation der DNA
PCR-Vervielfältigung
Plasmid-Vermehrung in E. coli (Bioreaktor)
Vorbereitung vor der IVT
/
Lyse der Bakterien: Freisetzung von Plasmid-DNA + Bakterienresten
Linearisierung: Restriktionsenzym schneidet Plasmid an def. Stelle
Verunreinigungen vor IVT
PCR-Fragmente: Enzyme, Primer, Salze, Nukleotide
Bakterienbestandteile: Wirts-DNA, RNAs, Proteine, Endotoxine, Lipide, Zellwandfragmente, …
Reinigung
der PCR-DNA
der Plasmid-DNA (pDNA)
Methoden
Silica-Bindung, UF/DF
TFF + mehrstufige Chromatographie
Aufwand
moderat
hoch
IVT (in-vitro-Transkription)
Template: PCR-DNA
Expressionskassette:
[T7] – [5′-UTR] – [Spike-Gen] – [3′-UTR] – [AAAAA…]
→ mRNA wird synthetisiert
Template: Linearisierte pDNA
Expressionskassette:
[T7] – [5′-UTR] – [Spike-Gen] – [3′-UTR] – [AAAAA…]
→ mRNA wird synthetisiert
IVT-Prozessierung
5′-Cap und Poly(A)-Tail werden je nach Verfahren während oder unmittelbar nach der IVT erzeugt
→ Ergebnis: funktionsfähige mRNA
5′-Cap und Poly(A)-Tail werden je nach Verfahren während oder unmittelbar nach der IVT erzeugt
→ Ergebnis: funktionsfähige mRNA
Verunreinigungen nach IVT (*)
Entfernung von IVT-Nebenprodukten: Template-DNA, RNA-Fragmente, RNA:DNA-Hybride, doppelsträngige RNA (dsRNA), Enzyme, Salze, freie Nukleotide
Entfernung von IVT-Nebenprodukten + Entfernung zusätzlicher plasmid- und bakterienstämmiger Verunreinigungen (anspruchsvoller)
Reinigung
der mRNA
der mRNA
Vorbelastung
gering – moderat
hoch
Methoden
▪️DNase-I-Behandlung
▪️Fällung oder Zentrifugation
▪️UF/DF oder TFF
▪️Magnetkügelchen-Reinigung (**)
▪️DNase-I-Behandlung
▪️Proteinase K-Behandlung (**)
▪️UF/DF und/oder TFF (**)
▪️mehrstufige Chromatographie
Aufwand
moderat
hoch
Formulierung in LNPs
mRNA wird in Lipidnanopartikel verpackt
mRNA wird in Lipidnanopartikel verpackt
Sterile Filtration
entfernt mikrobielles Kontaminationsrisiko
entfernt mikrobielles Kontaminationsrisiko
Abfüllung
aseptisches Verschließen und Etikettieren
aseptisches Verschließen und Etikettieren
Vorteile
sehr schnell, hochflexibel
extrem skalierbar, kostengünstige Massenproduktion
Nachteile
PCR-basierte Verfahren sind für großskalige industrielle Produktionsmengen nur eingeschränkt geeignet
Template enthält viele bakterielle Elemente → Reinigungsaufwand deutlich höher
(*) Obwohl die post-IVT-Reaktionsmischungen für Prozess 1 und 2 auf den ersten Blick ähnlich erscheinen, unterscheidet sich ihre „Vorbelastung“ qualitativ. Mit Vorbelastung ist die Menge und Komplexität der vor der Reinigung vorhandenen Begleitstoffe gemeint. Plasmidbasierte Prozesse bringen strukturell komplexere DNA-Formen, eine erhöhte Neigung zur Hybridbildung sowie Spuren bakterienstämmiger Begleitstoffe mit sich, die auch nach DNase-Behandlung die nachfolgende mRNA-Aufreinigung anspruchsvoller machen.
(**) Informationen zum Herstellungsverfahren laut EMA „EPAR zum Covid-19-Impfstoff von BioNTech/Pfizer“ (in Abschnitt 2.2., Seite 32)
Begriffserklärungen:
T7-Promotor – Startsignal für die Transkription
5′-UTR – stabilisiert die mRNA
Spike-Gen – Bauplan für das Protein
3′-UTR – Stopp-Signal und Stabilisierung
Poly(A)-Sequenz – schützt vor vorzeitigem Abbau
UTR steht für untranslated region – also einen Bereich der mRNA, der nicht in Protein übersetzt wird. Diese Abschnitte liegen vor dem Start der Proteininformation (5′-UTR) und nach dem Ende (3′-UTR). Sie übernehmen wichtige regulatorische Aufgaben.
Origin of Replication (ORI) – Startpunkt der Plasmid-Vervielfältigung
Antibiotikaresistenzgen – ermöglicht Selektion in E. coli
regulatorische bakterielle Sequenzen zur Expression des Resistenzgens
Multiple Cloning Site (MCS) – kurze Region mit Schnittstellen zum Einfügen der Zielsequenz
SV40-Sequenzelemente – regulatorische virale Sequenzen für bestimmte Anwendungen in eukaryotischen Zellkulturen (bei Pfizer/BNT)
1.5. Zusammenfassung: Herstellung und Reinigung von modRNA
Die Herstellung modifizierter mRNA für therapeutische und prophylaktische Anwendungen ist ein mehrstufiger Prozess, der weit über die eigentliche in-vitro-Transkription hinausgeht. Ziel ist es, aus einer komplexen Reaktionsmischung ein möglichst homogenes, funktionell aktives RNA-Molekül zu gewinnen, das in Zellen effizient translatiert wird und dabei nur eine kontrollierte, gewünschte Immunantwort auslöst.
Vom DNA-Template zur mRNA-Rohlösung
Ausgangspunkt der Produktion ist eine definierte DNA-Vorlage, die im Rahmen der in-vitro-Transkription als Template für die RNA-Synthese dient. Bereits in diesem frühen Schritt entstehen neben der gewünschten mRNA zwangsläufig verschiedene Nebenprodukte, darunter verkürzte Transkripte, doppelsträngige RNA-Strukturen, RNA:DNA-Hybride sowie nicht korrekt verarbeitete RNA-Moleküle. Diese Nebenprodukte sind keine Ausnahme, sondern eine bekannte Konsequenz der enzymatischen Transkription unter technischen Bedingungen.
Nach Abschluss der Transkription wird die DNA-Vorlage in der Regel enzymatisch verdaut. Es folgt eine erste Grobreinigung, beispielsweise durch Filtration oder Tangentialflussfiltration, bei der kleine Moleküle, überschüssige Reagenzien und Enzyme entfernt werden. Zu diesem Zeitpunkt liegt eine konzentrierte mRNA-haltige Lösung vor, die jedoch weiterhin strukturell heterogen ist.
Anreicherung der Ziel-mRNA
Um die gewünschte mRNA gezielt anzureichern, werden poly(A)-basierte Affinitätsverfahren eingesetzt. Dazu zählen sowohl klassische Poly(dT)-Chromatographie als auch poly(dT)-funktionalisierte Magnetkügelchen. Diese Methoden nutzen die Poly(A)-Sequenz der mRNA als Selektionsmerkmal und erlauben eine effektive Abtrennung nicht-polyadenylierter RNA-Fragmente und vieler Begleitstoffe.
Gleichzeitig wird hier eine zentrale Grenze dieser Verfahren sichtbar: Alles, was eine Poly(A)-Sequenz trägt oder strukturell an polyadenylierte RNA gebunden ist, kann mitisoliert werden. Dazu zählen fragmentierte mRNA-Moleküle, bestimmte dsRNA-Nebenprodukte sowie RNA:DNA-Hybride, sofern die RNA-Komponente polyadenyliert ist. Poly(A)-basierte Verfahren sind daher hochselektive Anreicherungs-, aber keine vollständigen Reinigungsmethoden.
Hochauflösende Trennung durch Chromatographie
Um die verbleibende strukturelle Vielfalt weiter zu reduzieren, kommen hochauflösende Trennverfahren zum Einsatz, insbesondere die Anionenaustausch-Chromatographie (AEX). Diese nutzt Unterschiede in der effektiven Ladungsdichte und Konformation von Nukleinsäuren, um einzelsträngige mRNA von stärker gebundenen Nebenprodukten wie dsRNA zu trennen.
Die AEX stellt einen entscheidenden Schritt dar, um immunologisch besonders aktive Verunreinigungen gezielt abzureichern. Gleichzeitig zeigt sich auch hier, dass bestimmte Molekülklassen – insbesondere RNA:DNA-Hybride – keine einheitlichen chromatographischen Eigenschaften besitzen und je nach Struktur unterschiedlich eluieren können. Ihre vollständige Entfernung erfordert daher eine sorgfältige Prozessauslegung und gegebenenfalls die Kombination mehrerer Reinigungsschritte.
Einordnung und Ausblick
Insgesamt zeigt sich, dass die Herstellung modifizierter mRNA kein linearer „Rein-Raus-Prozess“ ist, sondern ein fein abgestimmtes Zusammenspiel aus enzymatischen, physikalischen und chromatographischen Verfahren. Jeder Reinigungsschritt reduziert die Komplexität der Probe, erreicht jedoch nie eine absolute Trennung aller theoretisch möglichen Nebenprodukte.
Vor diesem Hintergrund ist es fachlich sinnvoll, nicht nur das Endprodukt, sondern auch die Herstellungs- und Reinigungslogik selbst zu betrachten, wenn über mögliche Restbestandteile wie DNA-Fragmente oder RNA-assoziierte Verunreinigungen diskutiert wird. Die Frage ist dabei weniger, ob solche Restkomponenten prinzipiell entstehen können, sondern in welchen Formen, in welchen Mengen und mit welcher biologischen Relevanz sie im Endprodukt verbleiben.
Damit rückt die zentrale Frage in den Fokus, wie verbleibende Nukleinsäurebestandteile – insbesondere mögliche DNA-Rückstände – zuverlässig nachgewiesen und quantifiziert werden können.
Der zweite Teil behandelt daher die analytischen Nachweisverfahren, regulatorischen Grenzwerte sowie die bisher veröffentlichten experimentellen Untersuchungen zu DNA-Rückständen in mRNA-basierten Impfstoffen.
Weiterführender Teil der Artikelserie
Teil 2: Nachweisverfahren und Studienlage
Stand: Mai 2026

{kind=link}