Guide to the Article Series
Part 1: Production and Purification of modRNA
Part 2: Detection Methods and Current Evidence
Before we turn to the question of why residual DNA may be detectable in mRNA vaccines, it is helpful to have a basic understanding of the manufacturing process.
The production of modRNA-based vaccines involves multiple sequential manufacturing and purification steps. The starting point is a DNA template that serves as the blueprint for the later nucleoside-modified mRNA (modRNA). During the manufacturing process, complex reaction mixtures are generated, from which unwanted residual components must be removed as thoroughly as possible.
Particular attention in recent years has focused on the fact that different methods were used to amplify this DNA template during development and later industrial production. These processes differ not only in terms of their technical scalability, but also with regard to the requirements for the subsequent purification of biological residual components.
This first part describes the fundamental manufacturing logic of modRNA-based vaccines, typical by-products and impurities, as well as the most important purification methods used during production. Finally, the two manufacturing pathways – Process 1 and Process 2 – are systematically compared.
It thus forms the basis for the second part of this series of articles, which focuses on analytical detection methods, regulatory limits and the experimental studies published to date on residual DNA in mRNA-based vaccines.
Note on terminology
In general usage, the term „mRNA vaccine” is common. From a technical perspective, however, the approved products consist of nucleoside-modified mRNA (modRNA). For reasons of clarity and readability, this work primarily uses the established term „mRNA vaccine”.
Table of contents
1. Manufacturing Processes – An Overview
1.1. Product Formation – From Gene to mRNA
1.2. Impurities and Byproducts
1.3. Purification Methods in the Manufacturing Process
1.4. Comparison: Process 1 vs. Process 2
1.5. Summary: Production and Purification of modRNA
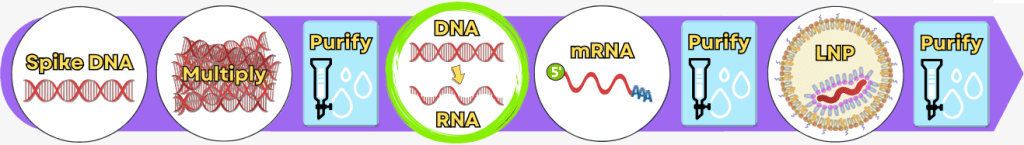
1. Manufacturing Processes – An Overview
The following overview presents the key production steps in a simplified form. In industrial practice, individual procedures and process details may vary depending on the manufacturer.
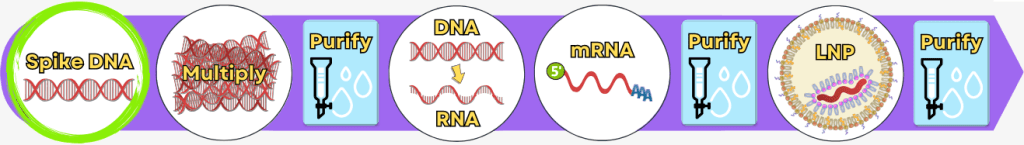
The manufacturing process can be broadly divided into the following steps:
| Step 1 | 🟢 | Extraction of genetic information |
| Step 2 | 🟢 | Amplification of the spike protein DNA Process 1: using PCR Process 2: using bacteria |
| 💧 | Purification of the DNA | |
| Step 3 | 🟢 | In vitro transcription for the production of mRNA |
| Step 4 | 🟢 | RNA processing – maturation of the mRNA |
| 💧 | Purification of the DNA | |
| Step 5 | 🟢 | Encapsulation of the mRNA in lipid nanoparticles (LNPs) |
| 💧 | Final purification |
While the upstream process generates the actual product – the mRNA – step by step, the downstream process serves to isolate the resulting mRNA from complex reaction mixtures, purify it, and formulate it for medical use.
1.1. Product Formation – From Gene to mRNA
Step 1: Extraction of genetic information
Step 2: Amplification of the spike protein DNA
Process 1: using PCR
Process 2: using bacteria
Step 3: In vitro transcription for the production of mRNA
Step 4: RNA processing – maturation of the mRNA
Step 5: Encapsulation of the mRNA in lipid nanoparticles (LNPs)
Step 1: Extraction of genetic information
The first step is to identify the genetic blueprint for the desired protein – in this case, the spike protein of the coronavirus.
In practice, the process unfolds as follows:
Collecting virus samples: Viral material is extracted from patient samples (e.g., throat or nasal swabs, blood, or tissue).
Note: There are differing views on whether SARS-CoV-2 was ever directly isolated from patient samples and cultured in a laboratory. This question is not addressed in this article, as the focus here is on the manufacturing process of mRNA vaccines.
Extracting genetic information: SARS-CoV-2 is an RNA virus. The viral RNA is isolated from the sample.
Converting RNA into DNA: Using a specific enzyme (reverse transcriptase), the viral RNA is converted into DNA. DNA is more stable and can be analyzed more easily.
Decoding the genome: The DNA is amplified, and the exact sequence of nucleotides (A, T, G, C) is determined.
Identifying the relevant section: Using computational analysis, the exact segment containing the blueprint for the spike protein is identified.
Entry into public databases: The resulting genetic blueprint is entered into public databases. This allows research teams worldwide to access the sequence without needing to isolate the virus itself.
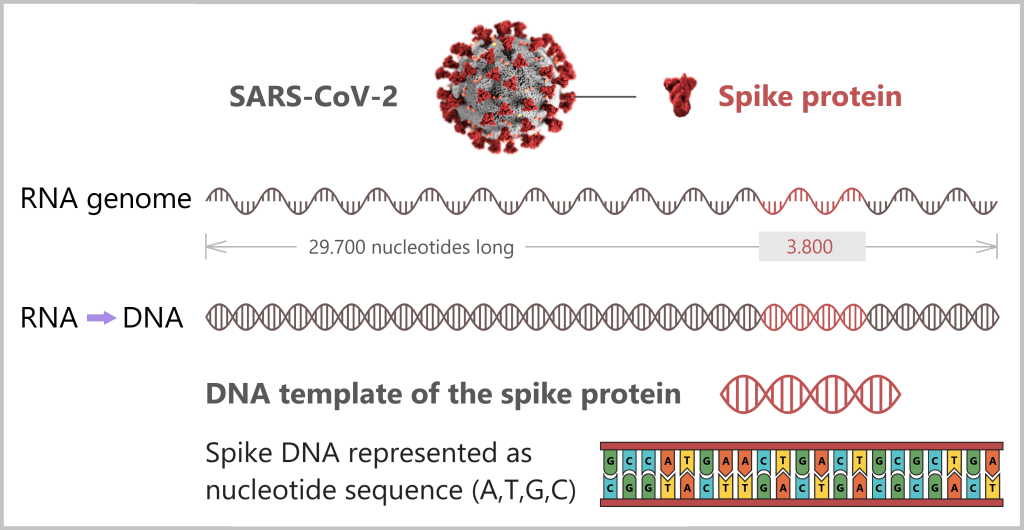
Using the enzyme reverse transcriptase, a DNA version is generated from the RNA genome of SARS-CoV-2. The section containing the blueprint for the spike protein is identified and synthetically produced in the laboratory as a DNA template. This DNA template later serves as the basis for producing the mRNA used in the vaccine.
Note: The coronavirus SARS-CoV-2 stores its genetic information on a long RNA strand composed of individual nucleotides – the „letters” of the genetic code. With around 29,700 of these building blocks, it possesses one of the largest known RNA genomes. The blueprint for the characteristic spike protein comprises a segment of approximately 3,800 letters.
Creating the DNA template
Based on the determined genetic sequence (known as sequence data), a synthetic version of the spike gene can be produced in the laboratory. This DNA template later serves as a template for mRNA production.
Step 2: Amplification of the spike protein DNA
Once the blueprint for the spike protein has been produced in the form of a DNA template, it must be amplified millions of times. Only then is enough material available to subsequently produce mRNA from it.
Process 1: using PCR
Process 2: using bacteria

Process 1: Amplification of spike DNA using PCR
Process 1 uses a method that became widely known during the COVID-19 pandemic: the polymerase chain reaction (PCR). It is carried out in the laboratory – technically known as in vitro – and mimics natural DNA replication as it normally occurs in living cells.
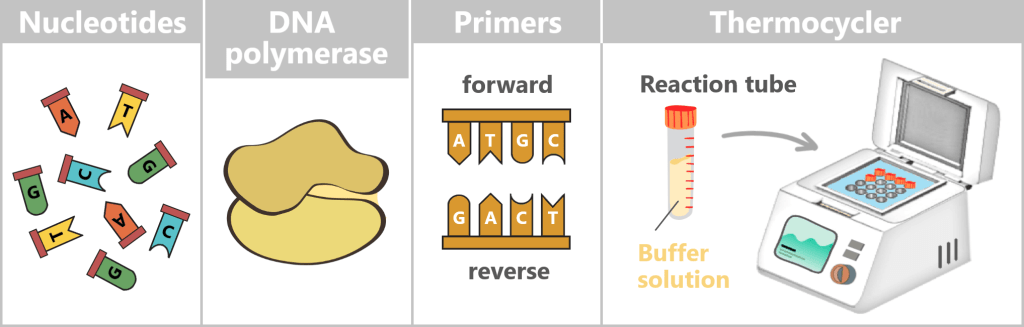
What is needed for PCR?
To get nature’s copying machine running, a few ingredients are required:
- DNA template – the starting material is the spike protein DNA from step 1.
- Nucleotides – the building blocks: the four „letters” of DNA – adenine (A), thymine (T), cytosine (C), and guanine (G) – from which new strands are assembled.
- DNA polymerase – the builder: the enzyme that reads the template and constructs new DNA strands. Most commonly, the heat-stable Taq polymerase is used.
- Primers – the guide markers: short DNA fragments that indicate where the polymerase should start copying. Two are always required: a forward and a reverse primer.
- Buffer solution – the right environment: ensures the proper conditions for the polymerase to function reliably.
- Thermocycler – the temperature carousel: a device that automatically runs through the required temperature cycles.
For the subsequent mRNA production, the DNA template is already deliberately extended in this step. An additional DNA segment – the so-called T7 promoter – is attached to the spike sequence. This short sequence is not required for PCR itself, but only becomes relevant in the following process step. The T7 promoter serves as a binding site for T7 RNA polymerase, which, during in vitro transcription (IVT), transcribes the DNA into therapeutic mRNA.
The Polymerase Chain Reaction (PCR) Process
All ingredients are placed into a small reaction tube, which is then inserted into the thermocycler. The device controls the same three steps, which are repeated cyclically:
1) Separation of the DNA strands (denaturation): The sample is heated to approximately 94–98 °C for about 20-30 seconds. This breaks the hydrogen bonds between the DNA bases, causing the double strand to separate into two single strands. These then serve as templates in the next step.
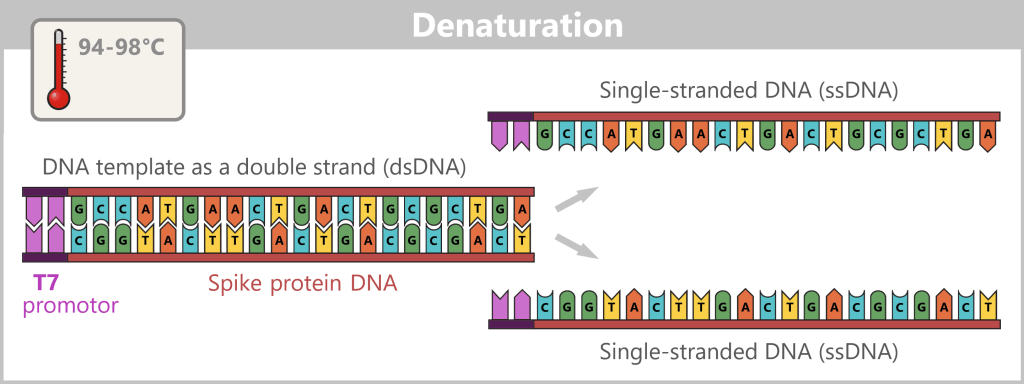
The illustration is highly schematic and not to scale. The coding sequence for the SARS-CoV-2 spike protein comprises approximately 3,800 base pairs, while the T7 promoter consists of approximately 20 base pairs.
2) Primer binding (annealing): The temperature is lowered to 50–65 °C. The primers now bind specifically to the respective single DNA strands. They mark the starting point for DNA synthesis.
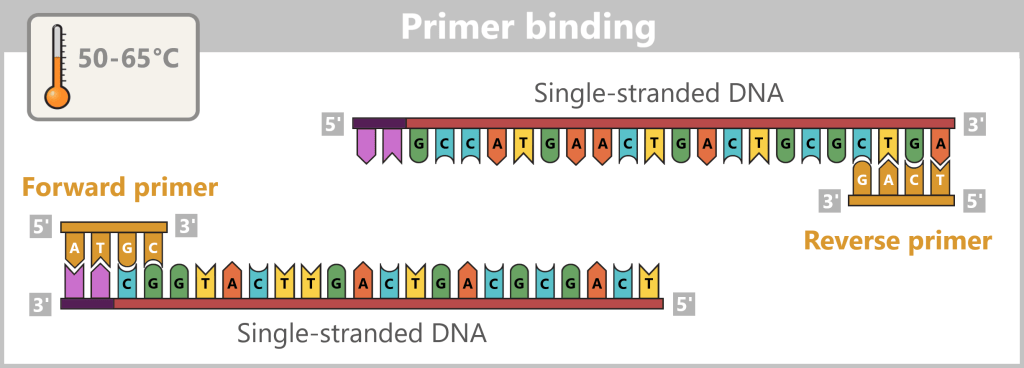
The illustration is highly schematic and not to scale. Primers are typically 20–25 nucleotides long.
3) DNA synthesis (amplification): At around 70 °C, the optimal temperature for the polymerase, the actual amplification begins. The DNA polymerase binds to the primer, reads the single strand in the 3′→5′ direction, and simultaneously synthesizes the new strand in the 5′→3′ direction. In doing so, it assembles nucleotides according to the principle of base pairing: A with T and G with C. In this way, two new double-stranded DNA molecules are produced.
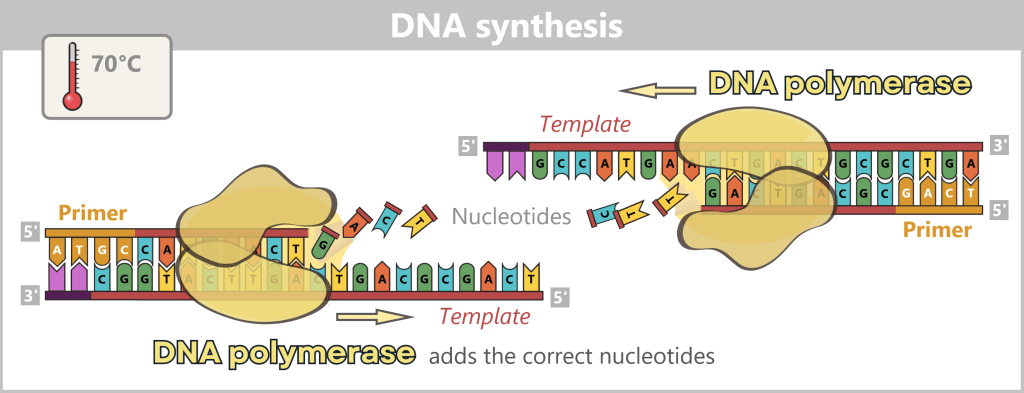
The illustration shows an active DNA polymerase moving along the exposed single strand like a molecular motor. The process of polymerization is clearly visible: individual nucleotides (the building blocks of DNA) enter the enzyme through an opening and are precisely added by the polymerase to the end of the growing strand. This process leads to elongation (extension) of the new DNA strand, which continuously grows in the 5′→3′ direction. Through this ongoing copying activity, amplification (replication) of the genetic information takes place, resulting in a new complementary double strand from the original template strand.
Why is synthesis always carried out in the 5′→3′ direction?
The structure of DNA
DNA consists of two strands that run in opposite directions – one in the 5’→3′ direction and the other in the 3’→5′ direction. The two strands are complementary to each other.
DNA polymerase, the enzyme that synthesizes new DNA, can work in only one direction: it reads the template strand in the 3’→5′ direction and synthesizes the new strand in the 5’→3′ direction.
What do 5′ and 3′ mean?
To understand this, we need to look at the building blocks of DNA: the nucleotides. Each nucleotide consists of:
- A base (A, C, G, or T)
- A sugar (deoxyribose)
- A phosphate group (P)
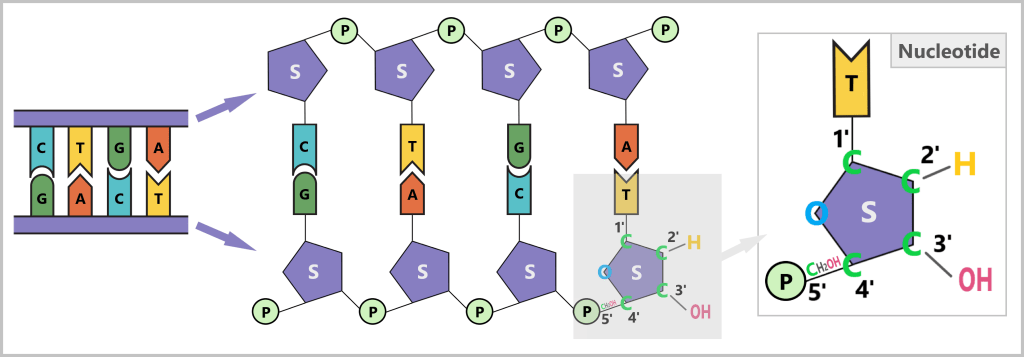
The sugar has five carbon atoms (C), which are numbered as follows:
- 1′: The base (A, T, C, or G) is attached here.
- 2′: In DNA, this position carries only a hydrogen atom (-H) and no hydroxyl group (-OH), unlike RNA. This missing OH group gives deoxyribose its name („deoxy” ribose, where „deoxy” means „without oxygen”).
- 3′: This is where a hydroxyl group (-OH) is located, which is essential for attaching new nucleotides during synthesis.
- 4′: Connects the sugar ring to the 5′ carbon.
- 5′: Carries the phosphate group, which links the nucleotide to the next one.
Why synthesis is only possible in the 5’→3′ direction
DNA polymerase can only add new nucleotides to the 3′ end of the growing strand. The reason is that this end carries a free hydroxyl group (–OH), which is required for forming the phosphodiester bond with the phosphate group of the incoming nucleotide.
At the 5′ end, a phosphate group is already present – no further nucleotide can be added there. Therefore, strand elongation is only possible in the 5′→3′ direction.
In summary
DNA polymerase moves along the DNA template strand in the 3’→5′ direction. At the same time, it adds complementary DNA nucleotides to the new DNA strand, always at its free 3′ end. In this way, the new strand grows continuously in the 5’→3′ direction.
Cycle repetition
The newly formed DNA double strands serve directly as templates for the next round. The steps denaturation – primer binding – DNA synthesis are repeated cyclically.
With each cycle, the amount of DNA doubles: 1 → 2 → 4 → 8 → 16 → 32 …
After only 25–40 cycles, billions of copies of the spike protein DNA are present.
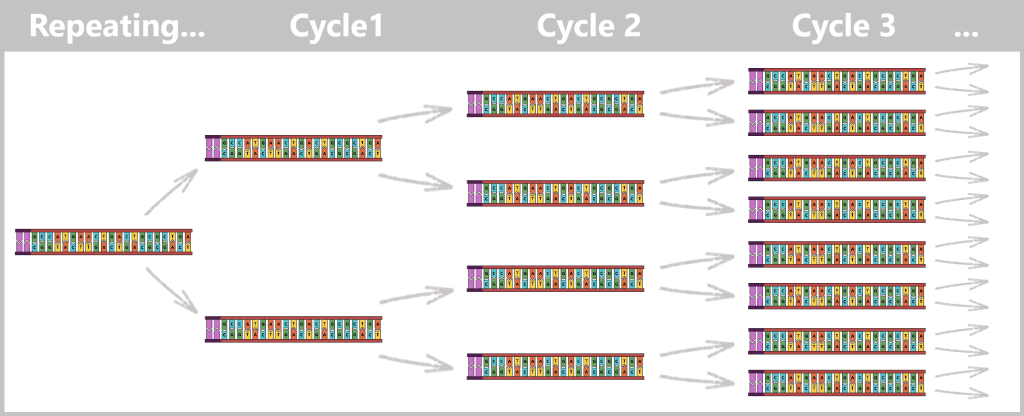
The result
At the end of PCR, you obtain a highly concentrated solution containing many copies of the spike blueprint – the starting material for the next step: the production of mRNA.
🎥 Tip: The video „What is PCR? Polymerase Chain Reaction” provides a clear visual summary of the process.

Process 2: Amplification of spike DNA using bacteria
In contrast to PCR, which takes place in a test tube (in vitro), this approach uses living organisms, specifically bacteria. This is referred to as an in vivo process (Latin for „within the living”), because amplification occurs directly inside the cells.
a) Why bacteria?
b) The bacterium Escherichia coli (E. coli)
c) Plasmids – small DNA rings with a big impact
d) Plasmids in genetic engineering
e) Insertion of spike protein DNA into a plasmid
f) Transfer of modified plasmids into bacteria
g) Bacterial multiplication
h) Harvesting the bacteria
i) Isolation of modified plasmids
j) Linearization of spike protein DNA
a) Why bacteria?
Bacteria are tiny, single-celled organisms that reproduce through simple cell division: one cell divides into two, those divide again – and in a very short time, this produces vast numbers of identical copies.
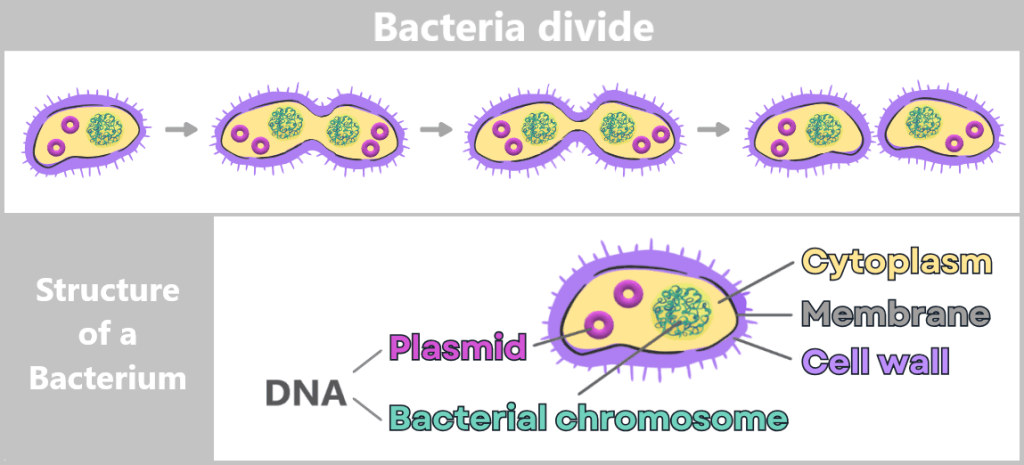
Since bacteria are asexual, they reproduce primarily through simple division. The process begins with cell growth and duplication of the genetic material. A constriction then forms, until two genetically identical daughter cells are ultimately produced.
The genetic information of bacteria – their DNA – is freely located inside the cell in the so-called cytoplasm. Most of it is stored in the bacterial chromosome. In addition, many bacteria contain small, circular DNA molecules called plasmids.
Adaptation through mutation and gene transfer
The continuous alteration of genetic material is essential for organisms to successfully adapt to new environmental conditions over generations. Bacteria are considered particularly adaptable. This is ensured by two mechanisms:
Mutations: small random changes in the genetic material that can sometimes provide advantages (e.g., antibiotic resistance).
Gene transfer: bacteria can additionally exchange DNA segments, especially via their plasmids. In this way, beneficial traits can spread very rapidly through a bacterial population.
This is why bacteria are so suitable:
- Rapid reproduction: Under ideal conditions, many bacteria double every 20 minutes.
- Simple structure: Their DNA is not enclosed in a cell nucleus but lies freely within the cell, which facilitates manipulation.
- Plasmids as additional DNA: These can be easily modified, transferred, and used for the amplification of foreign DNA – ideal for genetic engineering.
The ability to introduce genetic material into bacteria and have it replicated has been used by researchers for decades. In this way, bacteria become small „DNA factories”.
A historical breakthrough
In 1973, Stanley Cohen and Herbert Boyer achieved a groundbreaking experiment: they were the first to insert a foreign gene into bacteria. The result showed that the bacteria adopted the new gene and even expressed it. This demonstrated that genes can be transferred from one organism to another – marking the birth of modern genetic engineering. [The Cohen–Boyer experiment]
b) The bacterium Escherichia coli (E. coli)
In research, the bacterium Escherichia coli (E. coli for short) is particularly widely used – for several reasons:
- It is easy to culture and grow in the laboratory.
- It reproduces very quickly: under optimal conditions, it divides every 20–30 minutes.
- Its genome has been extensively studied and well understood.
- It can be easily genetically manipulated.
- Its cultivation is inexpensive.
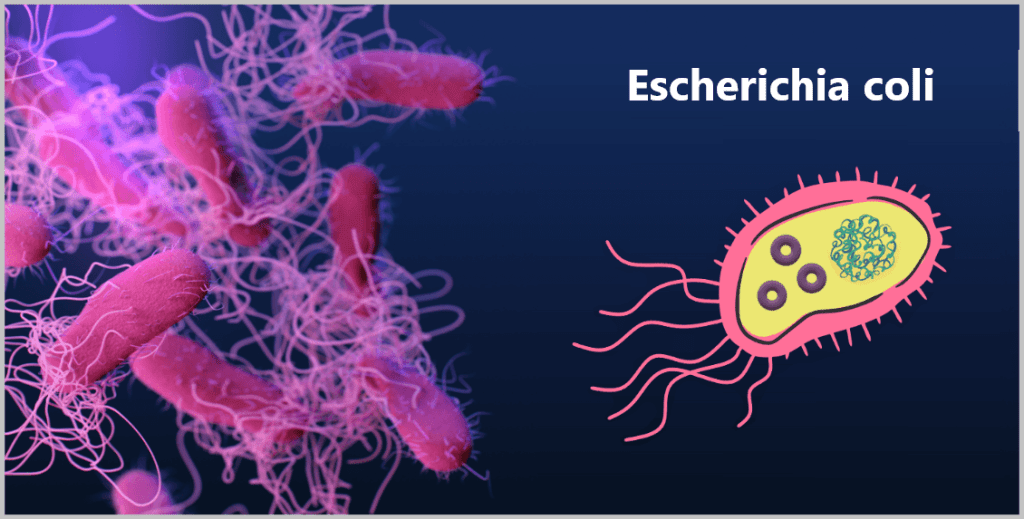
On the left, a microscopic image; on the right, a simplified illustration showing DNA (large tangle) and plasmids (small rings).
Because E. coli is the most extensively studied organism in molecular biology and genetics, scientists jokingly refer to it as the „pet of geneticists“.
For these reasons, E. coli is one of the most important tools in biotechnology – and it also plays a central role in vaccine development.
A brief journey into the world of the E. coli bacterium
Escherichia coli – colorless, almost transparent – scarcely longer than one thousandth of a grain of sand – appears inconspicuous. And yet, it is anything but boring.
From the outside, it looks like a small capsule, but beneath its surface lies one of the busiest megacities at rush hour.
No space for emptiness. Molecule by molecule. Proteins, ribosomes, nucleic acids – they crowd together, collide, and make way for one another. A state that biologists soberly call macromolecular crowding, but which here feels like permanent congestion. And still – or precisely because of it – everything functions with breathtaking precision.
In the midst of this crowded space lies the cell’s memory: a single, circular DNA molecule. Everything is written here – how to consume sugars, how to swim, how to divide. If one were to gently stretch out this DNA, it would be a thousand times longer than the cell itself. A strand of millions of letters, tightly packed, repeatedly twisted, forced into loops and coils. This folding art is called supercoiling – as if someone had crammed a kilometer-long story into a matchbox without losing a single word.
Yet for all its power, this DNA rarely speaks directly. Instead, it continuously sends out messengers: RNA. Thousands, tens of thousands. Short messages, work instructions, construction plans. Some are only fleeting notes – mRNA, barely a thousand characters long, with a lifespan of minutes. Others are more stable and substantial: rRNA, the structural backbone of ribosomes. And then there are the small tRNA molecules, little more than assistants, yet essential in delivering the right building blocks at precisely the right moment. They dart through the cytoplasm like couriers. Their destination?
The ribosomes – the true giants of this world. Tens of thousands of them drift through the cytoplasm. They read the fleeting mRNA messages and turn them into tangible reality: proteins. They are silent, tireless, and they consume a large portion of the cell’s resources. Almost a quarter of all proteins here exist solely to produce new proteins.
And these proteins are everywhere. Millions of them, in thousands of forms. Enzymes that accelerate reactions which would otherwise take days or years – here they occur in fractions of a second. Some operate in bulk, performing the same task repeatedly. Others are rare specialists, perhaps only a few dozen copies, yet indispensable at the decisive moment. Among them patrol the guardians: RNases, which break down old messages. Once an RNA instruction has been executed, they disassemble it back into its basic components. This prevents the buildup of informational waste and allows the building blocks to be recycled for new instructions. Interspersed – in carefully regulated amounts – are DNases. They ensure that the valuable master plan in the archive is repaired if something goes wrong. At the same time, they eliminate foreign intruders and even extract energy from them.
All of this takes place within a shell that must be constantly renewed: a living boundary made of lipids, whose outer layer carries endotoxins. And it is under time pressure. Because this E. coli does not live to remain static – it lives to divide.
Under favourable conditions, it measures time in minutes. Twenty, perhaps thirty – then one cell must become two. While copying, reading, and building take place inside, the surface expands in parallel. Millions of new lipid molecules are produced, integrating themselves and extending the protective wall. It is a race without pause – a controlled chaos with a clear direction.
And then comes the moment of division. No dramatic cut, no ending – more a quiet drifting apart. The mother cell ceases to exist as an individual. Yet its story is not torn apart; it continues, twice. In two cells that are both old and new at the same time. Each carries the same long DNA within it, the same noise of molecules, the same restless order.
One can imagine the world inside the E. coli cell expanding to yet another, almost mysterious layer:
Alongside the large circular chromosome, smaller DNA rings drift through the crowded interior: plasmids. They are like loose pages in the cell’s internal archive – not strictly essential for survival, but often crucial in practice. Some carry instructions for new capabilities: resistance to a toxin, an enzyme that enables the use of an unusual nutrient source, sometimes only a small tactical advantage for difficult times.
They move freely, collide with ribosomes, are briefly bound by proteins, and released again. They, too, are read; their genes are transcribed into RNA, and their messages land on the same ribosomes as those of the main chromosome. In this overcrowded city, there are no special privileges – only function.
When the cell prepares for division, the plasmids begin to move. They must not be lost. Special proteins ensure that copies are made and that each of the resulting daughter cells receives at least one copy. A silent act of transmission – almost like slipping a letter into someone’s hand in a crowd before two paths separate.
Sometimes, however, their story does not end within their own lineage. Occasionally, a connection opens to the outside, to a neighbouring cell. In that moment, plasmids change hands. A brief contact, a transfer, and information jumps from one cellular history into another. In this way, traits can spread faster than any chromosomal mutation could ever achieve.
In this world, plasmids are the rumours, the blueprints, the survival tricks that are not carved in stone. Mobile, exchangeable, opportunistic. And precisely because of this, they fit perfectly into the restless, crowded interior of E. coli, where nothing ever stands still – not even the genetic material itself.
The illustrations by David Goodsell are highly recommended. He depicts the interior of cells true to scale – his images of E. coli make this molecular „crowding” truly tangible.
c) Plasmids – small DNA rings with a big impact
Plasmids are small, circular DNA molecules that occur in addition to the bacterial chromosome within the cell. They are much smaller than the main chromosome, may be present in varying copy numbers, and can replicate independently of it. Unlike the linear DNA of eukaryotes (e.g., humans), plasmids form closed circles of double-stranded DNA (dsDNA).
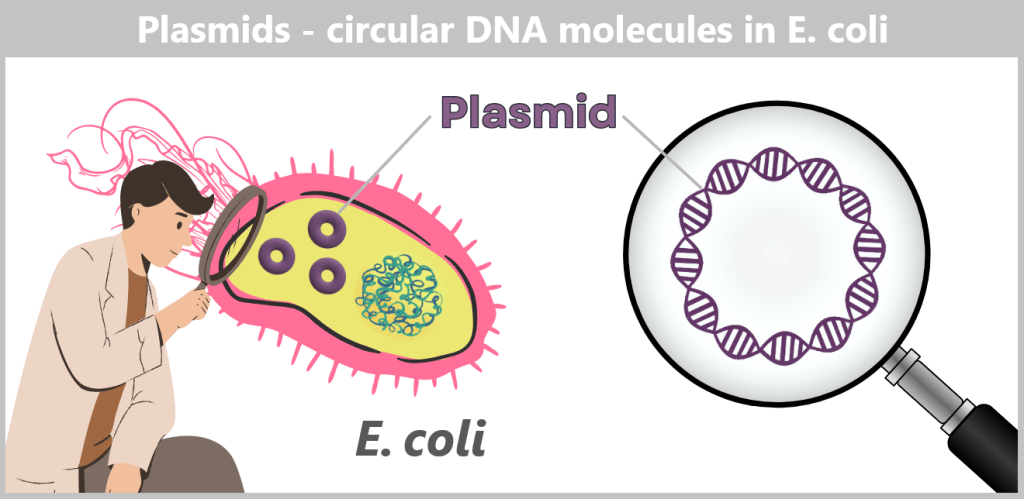
Plasmids can carry a wide variety of genes – for example, genes conferring antibiotic resistance or encoding the production of specific proteins. To enable their targeted use by researchers, so-called plasmid maps are created: schematic representations showing the most important functional regions.
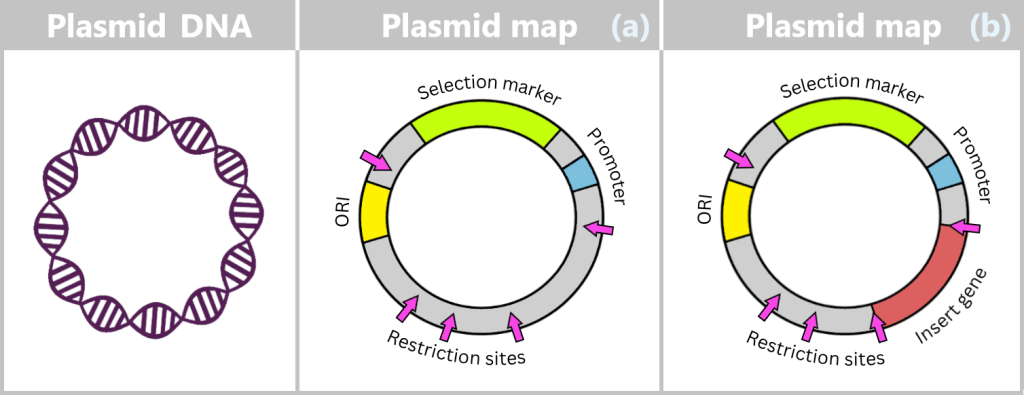
ORI (Origin of Replication): Starting point for the duplication of the plasmid. When environmental conditions and internal signals are favourable, the bacterium can „press this start button”, and the plasmid makes a copy of itself. Only if this ORI is „compatible” with the bacterium can the plasmid reliably replicate, even independently of cell division.
Selection markers: Genes that confer an advantage to bacteria, such as resistance to a specific antibiotic. They help researchers identify which bacteria carry the plasmid.
Promoter: The promoter is a specific DNA region that controls gene activity. It regulates when and how strongly certain genes on the plasmid are transcribed, thereby controlling the production of the corresponding proteins.
Restriction sites: These are short DNA sequences that can be recognized and specifically cleaved by restriction enzymes. These enzymes serve as a kind of immune system for bacteria, enabling them to cut the DNA of invading viruses and thus defend themselves.
d) Plasmids in genetic engineering
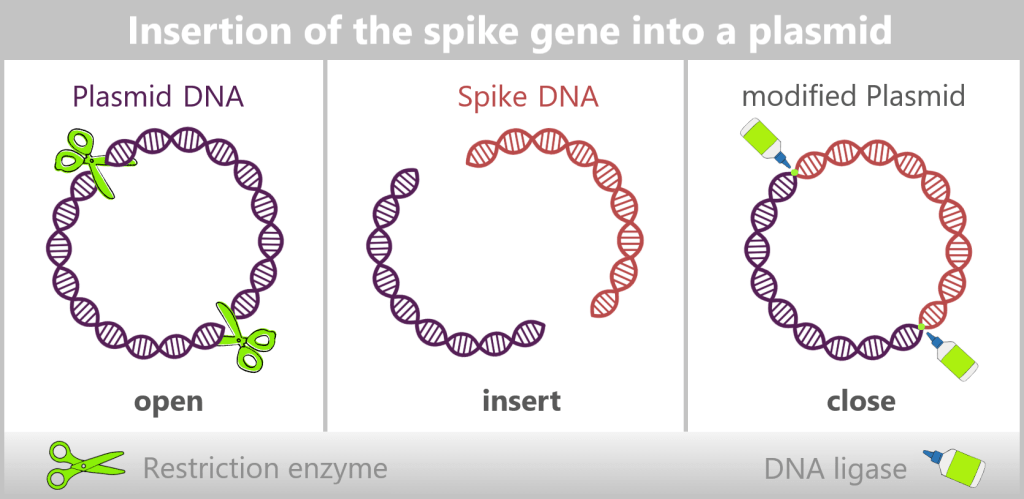
In bacteria, restriction enzymes actually serve to defend against foreign DNA. In genetic engineering, however, they are used as precise tools: tiny molecular scissors that cut DNA at very specific sites.
This creates a „gap” in a plasmid into which a desired gene can be inserted. Another enzyme, DNA ligase, then „glues” the DNA ends back together. The inserted gene is called the insert gene (see upper figure).
Through this process, plasmids are transformed into small gene shuttles: they specifically transport new genes into bacteria. With each cell division, the plasmid – and thus the insert gene – is automatically copied as well. In this way, entire bacterial cultures are created that act as „mini-factories”, producing specific proteins or DNA in large quantities.
🎥 Tip: A short animated introduction to plasmids can be found here.
e) Insertion of spike protein DNA into a plasmid
For the amplification of the SARS-CoV-2 spike protein sequence, the spike gene is first integrated into a bacterial plasmid – a process referred to as „cloning”. In this step, the molecular biology tools described earlier are used: restriction enzymes open the circular plasmid at defined sites, while DNA ligases precisely insert the spike gene and permanently join the DNA ends. In this way, a recombinant (newly assembled) plasmid is created that contains the genetic information for the desired antigen.
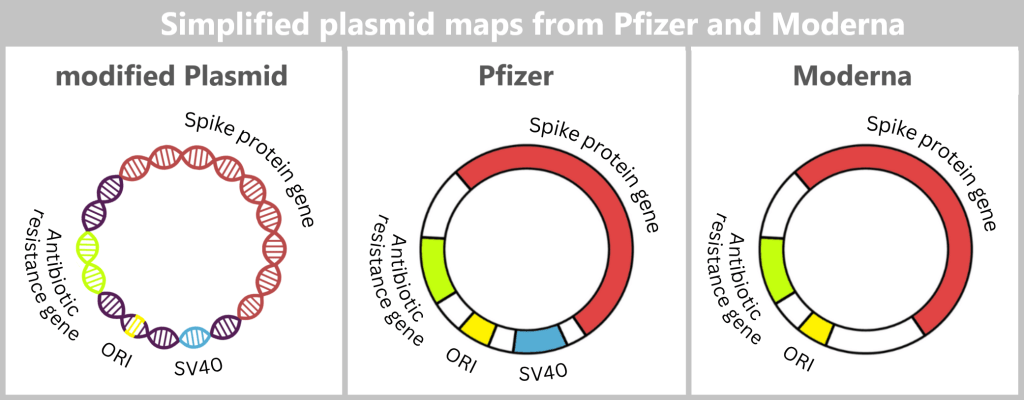
DNA elements within the plasmid
The spike DNA is not inserted into the plasmid in isolation, but together with additional genetic elements required for plasmid replication.
ORI (Origin of Replication): The origin of replication determines the site at which plasmid replication begins within the bacterial cell. Without this element, the plasmid could not be stably amplified in E. coli.
Spike protein gene: This gene contains the blueprint for the spike protein – the central antigen of the vaccine.
Antibiotic resistance gene: The resistance gene serves as a selection marker. It ensures that, under antibiotic pressure, only those bacteria survive that have taken up the desired plasmid. This allows suitable bacterial clones to be specifically selected and amplified.
- Moderna: kanamycin resistance gene
- Pfizer/BioNTech: Neo/Kan resistance gene (conferring resistance to neomycin and kanamycin)
SV40 components (Pfizer/BioNTech): The production plasmid used by Pfizer/BioNTech additionally contains regulatory sequence elements derived from Simian Virus 40 (SV40) – a monkey virus whose genetic components have been used in molecular biology for decades.
Specifically, these include:
- an SV40 promoter/enhancer,
- as well as parts of the SV40 origin of replication.
Such sequences are used in genetic engineering because they can enhance gene expression in mammalian cells and may facilitate plasmid replication in certain cell lines.
However, for bacterial amplification in E. coli, these elements have no known functional role, since bacteria do not possess the necessary cellular factors required for their activity.
Why these SV40 sequences are included in the final production plasmid is not fully documented in publicly available sources. It is discussed, among other things, that they may have been carried over from earlier development or testing systems and later retained.
According to current knowledge, Moderna does not use comparable SV40 components in its production plasmid.
The varying use of such regulatory sequences is among the issues currently being debated in scientific circles in connection with residual DNA.
A more detailed representation of the plasmid maps can be found here.
f) Transfer of modified plasmids into bacteria
The modified plasmids, which now carry the blueprint for the spike protein, are then introduced into E. coli bacteria – a process known as transformation. During this step, the bacteria take up the plasmids; these remain permanently within the bacterial cell and are passed on during each cell division.
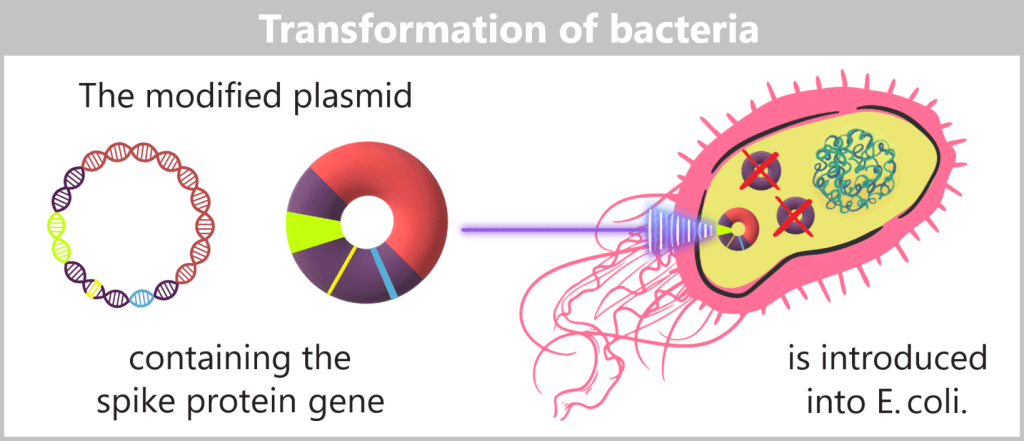
The modified plasmids are introduced into E. coli bacteria via transformation. Plasmids used in genetic engineering to transport foreign DNA sequences are called vectors.
Plasmid-free host cells – for clean clones
In biotechnological plasmid production, only the modified plasmids are intended to be produced. Therefore, special plasmid-free bacterial strains are used that do not carry any of their own natural plasmids.
This has several reasons:
- Natural plasmids could compete for the cell’s replication machinery,
- they could exchange DNA segments through recombination,
- and they would make the genetic composition of the colony unpredictable.
By using plasmid-free host cells, it is ensured that all bacteria in a colony are genetically identical clones – each containing the same, defined plasmid with the desired sequence.
g) Bacterial multiplication
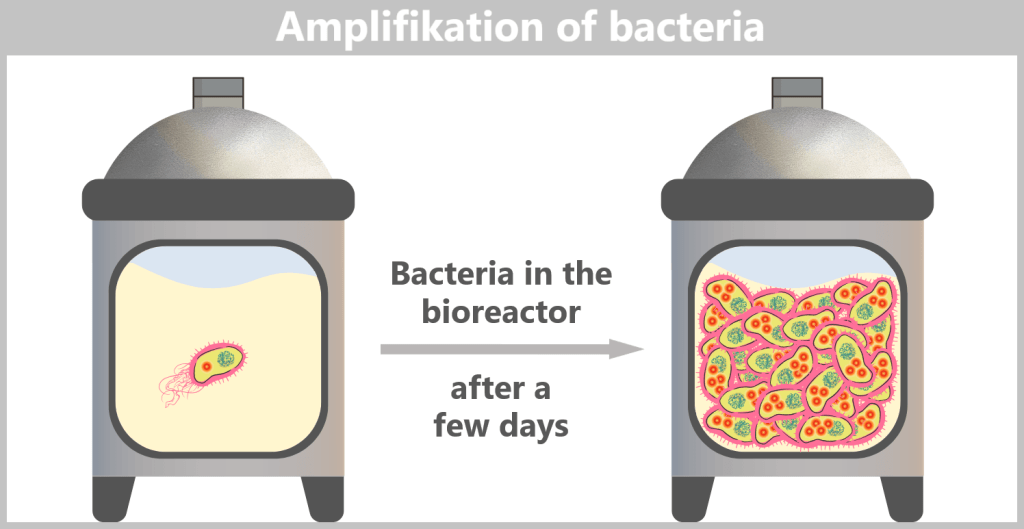
The E. coli bacteria carrying the modified plasmids are transferred into a fermenter. A fermenter, also called a bioreactor, is a device used in biotechnology for the large-scale production of products such as antibiotics, enzymes, vitamins, or vaccines. It allows precise control of conditions such as temperature, pH, oxygen supply, stirring speed, and nutrient availability.
The nutrient medium in the fermenter contains all essential substances required for bacterial growth. Under these optimal conditions, the bacteria begin to multiply rapidly. With each cell division, the plasmids are also copied, so that the desired DNA is amplified as well.
To ensure that only bacteria survive which actually carry the spike gene plasmid, an antibiotic is additionally added to the fermenter. Only cells containing the plasmid – and therefore the antibiotic resistance gene – can grow. This results in a culture composed exclusively of the desired, modified bacteria.
E. coli can divide approximately every 20–30 minutes. Within just a few days, this leads to an enormous bacterial population in the fermenter – containing trillions of copies of the spike plasmid.
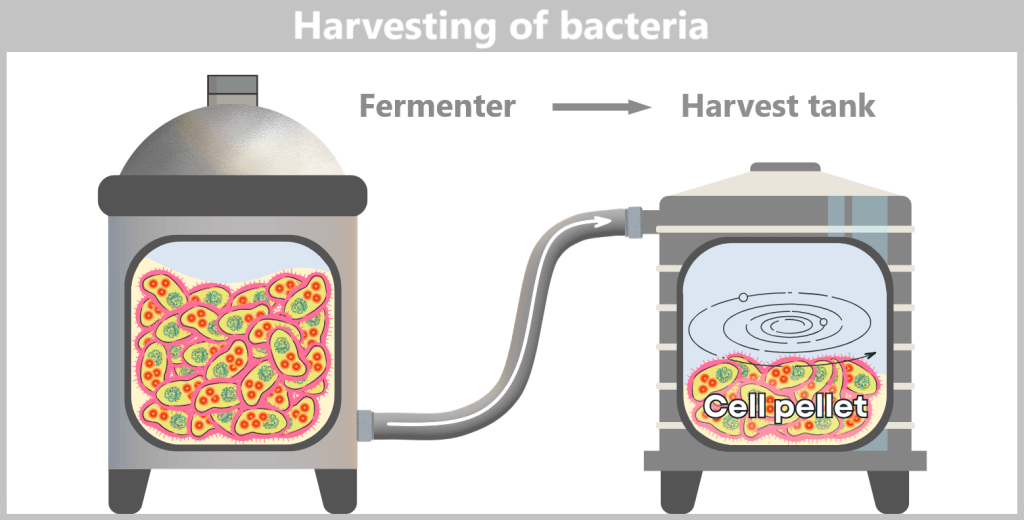
h) Harvesting the bacteria
After the growth phase, the E. coli cells are „harvested”. For this purpose, the entire contents of the fermenter – the cell suspension – are transferred into a harvest tank. There, the separation of liquid and cells takes place, usually by centrifugation (rapid spinning) or filtration. In the end, a cell pellet is formed, meaning a concentrated collection of bacteria at the bottom of the container.
The cell pellet – now separated from the nutrient medium – is transferred to a downstream processing facility. It is either resuspended in a liquid to form a uniform, pumpable „cell slurry”, or it is automatically conveyed as a solid pellet. In modern facilities, all of this takes place in a closed system, without the biomass being exposed to the external environment.
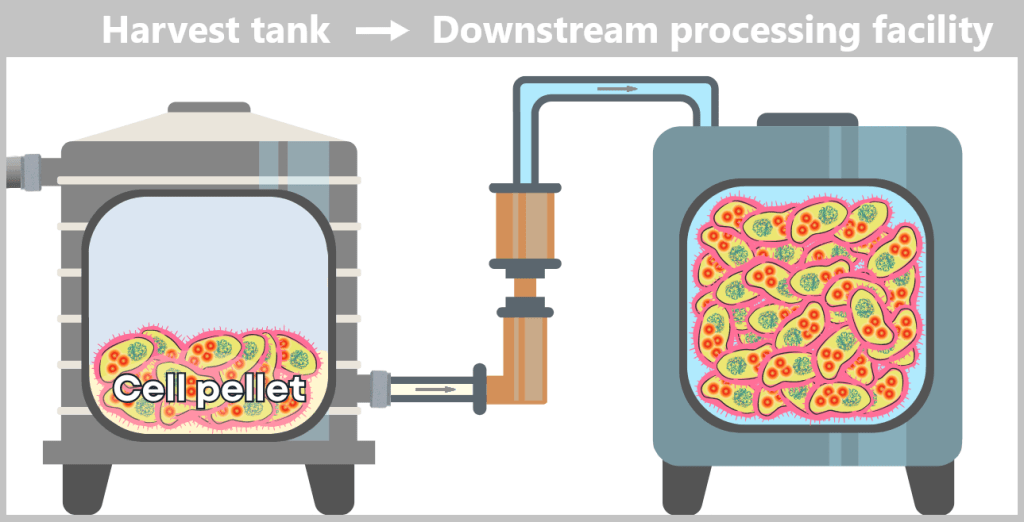
i) Isolation of modified plasmids
After harvesting the bacterial cultures, a crucial separation step follows: the E. coli cells are specifically lysed in dedicated downstream processing systems, meaning they are carefully broken open.
The addition of sodium hydroxide (NaOH) and the detergent SDS (a specialized soap-like molecule) dissolves the lipid-based cell envelope, similar to how dishwashing detergent removes grease.
This releases the entire cellular content – a complex mixture consisting of the desired plasmids, chromosomal DNA, proteins, membrane components, and many other cellular substances.
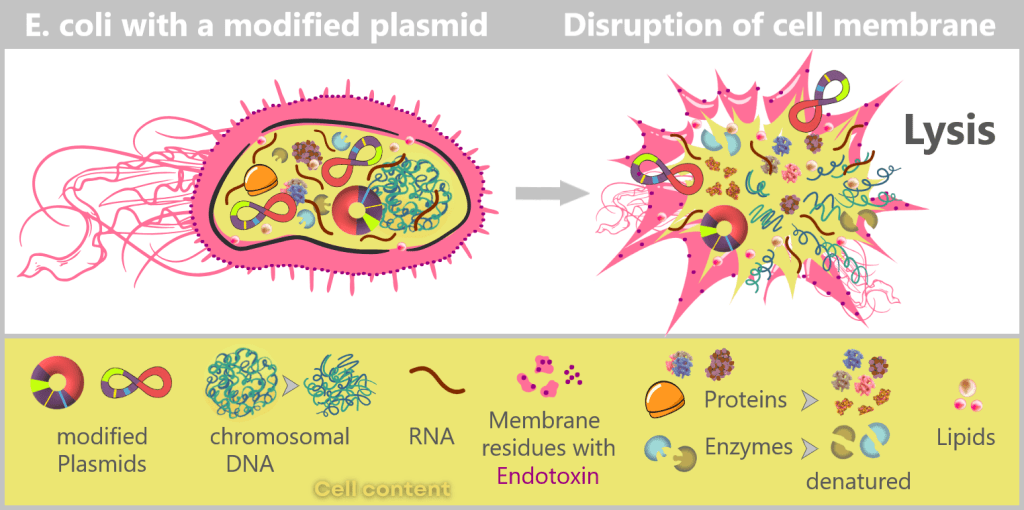
Left: Schematic cross-section of an E. coli cell containing a modified plasmid. The outer membrane is shown with embedded endotoxin molecules (purple).
Right: After disruption of the cell membrane (lysis), the various cellular components are released – modified plasmids, chromosomal DNA (bacterial chromosome), proteins, enzymes, and lipids. The outer membrane appears in fragments containing structurally bound endotoxin (LPS).
DNA forms in E. coli: plasmid and chromosome
Plasmids in Escherichia coli exist as small, circular DNA molecules. Within the cell, they are predominantly found in a supercoiled form. This topology (spatial arrangement) is biologically preferred because it provides high mechanical stability and compactness. The closed DNA ring is additionally twisted, allowing the molecule to be organized in a space-saving manner while remaining relatively resistant to physical stress.

The difference between supercoiled (superhelical) and relaxed (ring-shaped) plasmids lies exclusively in their topology. Both forms contain identical genetic information, but they exhibit markedly different physical properties.
In the supercoiled form, the DNA molecule is under torsional strain and tightly wound around itself. The relaxed form typically arises when one of the two DNA strands acquires a single-strand break (a nick). This nick relieves the torsional stress, and the plasmid adopts a more open, less compact circular structure.
The bacterial chromosome (genomic DNA, gDNA) is also circular, but many times larger. It is highly organized, associated with proteins, and folded into multiple domains.
Supplementary explanation regarding the mechanism of alkaline lysis
Destruction of genomic DNA, preservation of plasmids
During alkaline lysis, the cell membrane and cell wall are disrupted. The double-stranded DNA of both molecular types (genomic DNA and plasmids) is briefly denatured and subjected to mechanical and chemical shear forces.
The following occurs:
- The long, fibrous genomic DNA is broken and linearized by shear forces, as it is too large and fragile to remain intact.
- In contrast, the compact, supercoiled plasmid DNA remains largely intact.
When the previously alkaline solution is neutralized, the small circular plasmid DNA can renature correctly: its two single strands find each other again and re-form a stable double helix. As a result, it remains in solution.
The much longer genomic DNA can no longer fully renature under these conditions. It precipitates together with cell debris and proteins as an insoluble pellet.
Destruction of protein structures
Alkaline lysis affects not only nucleic acids but also proteins. Under strongly basic conditions, proteins lose their three-dimensional folding and thus their biological function. Large complexes such as ribosomes, which consist of ribosomal RNA and numerous proteins, are also completely destroyed and break down into their components. During subsequent neutralization, these denatured proteins and RNA fragments aggregate (clump together) – similar to how egg white solidifies when cooked – forming a precipitate that can be easily removed together with the remaining cell debris.
Why small RNA fragments appear after bacterial lysis
When E. coli cells are broken open, not only plasmids enter the lysate but also large amounts of bacterial RNA. This consists of a wide variety of molecules that were responsible for the bacteria’s metabolism – including many short, stable RNA types. In addition, bacterial enzymes that degrade RNA (RNases) are released during lysis. Active RNases can rapidly break existing RNA down into smaller pieces. Mechanical shear forces during the lysis process also contribute to fragmentation. The result is a complex mixture of short bacterial RNA fragments.
Natural RNA of the bacterial cell
The bacterial cell (E. coli) already contains a vast amount of its own natural RNA – significantly more than DNA. When the cells are disrupted (lysis), all of this cellular RNA enters the lysate and represents a major impurity. This includes:
- rRNA (ribosomal RNA): makes up the majority (~80–90%) of bacterial RNA and is present in large quantities.
- tRNA (transfer RNA): very small, stable molecules in many different variants.
- Bacterial mRNA: naturally short-lived in bacteria and often already partially degraded.
- Regulatory small RNAs (sRNAs): natural RNA species only a few dozen nucleotides in length.
In summary, this collection of ribosomal, transfer, and messenger RNA forms the natural cellular RNA pool of E. coli.
Destruction of the membrane and release of endotoxins
Endotoxins (lipopolysaccharides, LPS) are a natural component of the outer membrane of bacteria with a double-layered cell envelope such as E. coli. In scientific terminology, such bacteria are referred to as ‚Gram-negative‘. They provide structural stability and help the bacterium withstand external stress. During lysis, this membrane is disrupted, releasing large amounts of LPS. While endotoxins act as a kind of „armour” for the bacterium, they are highly toxic to humans. The biologically active component – lipid A – can trigger strong inflammatory responses in humans, which is why endotoxins are among the most critical contaminants in biotechnological production.
The challenge now lies in isolating the desired plasmids from this biochemical complexity and removing all unwanted accompanying substances. This involves a multi-stage purification process, which we will examine in more detail later in the section on Purification Methods in the Manufacturing Process.
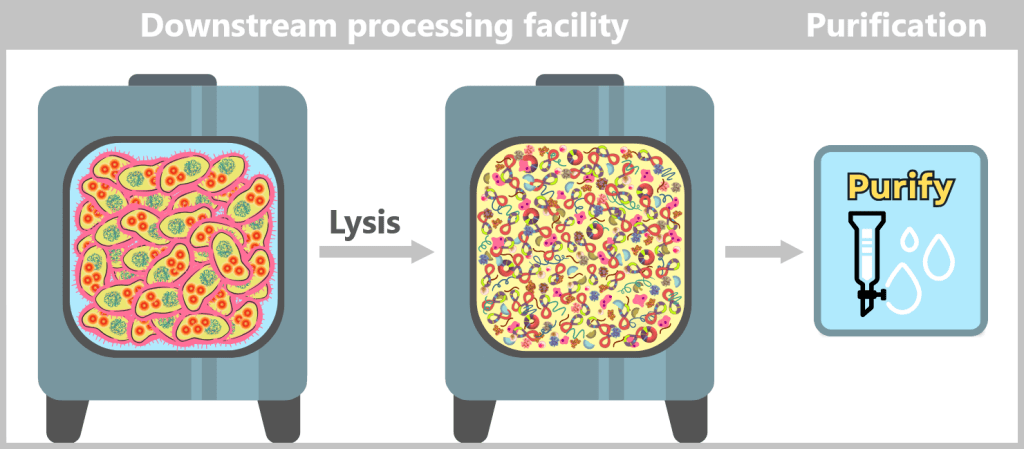
In the downstream processing facility, the harvested bacterial cells are lysed. This produces a „cell slurry” containing all cellular components. The purification process then begins, in which the desired plasmids are separated from the mixture using specific filtration and chromatography techniques.
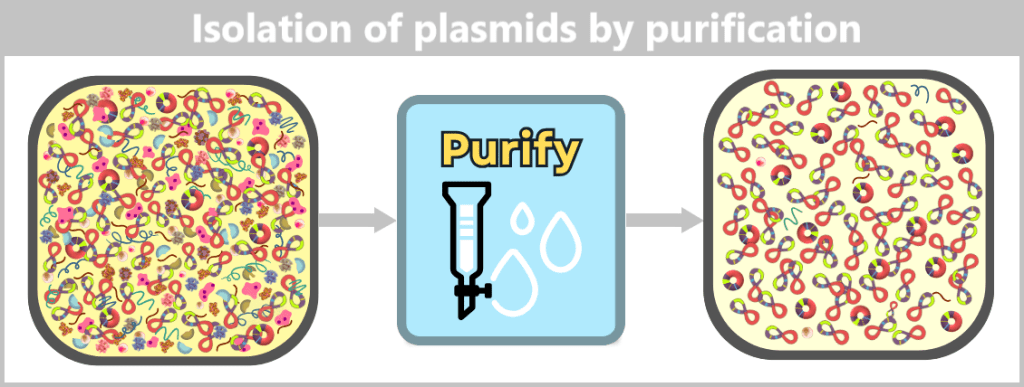
After purification, the plasmid DNA is obtained in high purity. In addition to the dominant supercoiled conformation, a small proportion of relaxed plasmids is visible. The latter arise from single-strand breaks (nicks) during cell lysis or downstream processing.
j) Linearization of spike protein DNA
The purified circular plasmids already contain the blueprint for the spike protein, but they are not yet suitable for the next step. For transcription, the circular plasmid DNA must be converted into a linear form.
To achieve this, the plasmids are specifically opened: a targeted restriction enzyme makes a precise cut at a defined site, typically after the end of the spike protein gene. In this way, clean DNA ends are generated that facilitate the reading of the sequence.
This cut converts the entire plasmid from its circular form into a linear, open DNA strand. This strand contains not only the spike gene, but also all other plasmid components, such as the origin of replication (ORI), selection markers, and regulatory elements. One particularly important of these sequences is the T7 promoter; this will become clear in the next step.
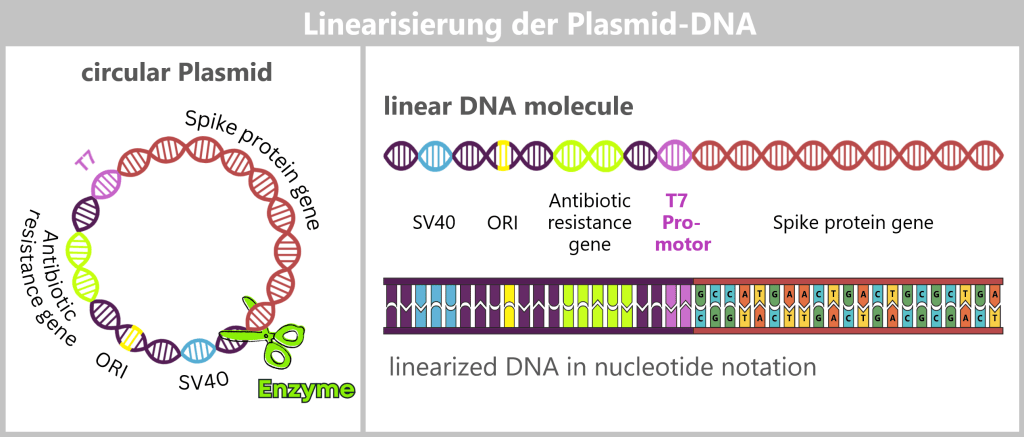
The circular plasmid is cut open with a restriction enzyme, resulting in a linear DNA molecule. This contains different sections: the origin of replication (ORI, yellow), selection markers (light green), regulatory sequences such as SV40 (blue) and the T7 promoter (purple), as well as the actual spike gene (red). In the detailed view below, the nucleotide sequence of the spike gene is indicated. The complete nucleotide sequence is shown in a strongly shortened form.
Further purification after linearization
The linearization of plasmid DNA generates additional reaction components and by-products that must be removed before in vitro transcription. These include, in particular:
- Restriction enzymes: The endonucleases used for the targeted cleavage must not remain in the subsequent manufacturing process.
- DNA by-products: These include incompletely linearized plasmids (e.g., residual supercoiled or relaxed forms) as well as short DNA fragments that may arise from nonspecific breaks or side reactions.
- Salts and buffer residues: The linearization reaction is carried out in specific buffer systems whose ions and additives could interfere with subsequent process steps.
Appropriate purification methods are used to obtain a highly purified linear DNA template, which serves as the template for the next process step.
In this context, the term highly purified does not describe an absolute state, but rather the extensive removal of process-related impurities to a regulatorily acceptable minimum. Small residual amounts of non-linear plasmid forms may remain and are further reduced in subsequent process steps.
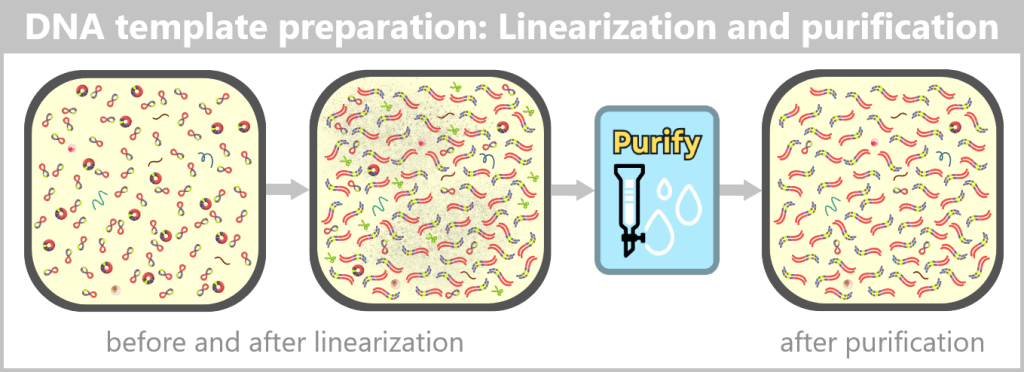
Left: The plasmid DNA solution before linearization. It contains a mixture of supercoiled (predominant) and relaxed (open circular) plasmid forms.
Center: The solution after linearization by a restriction enzyme. The circular plasmids have been cut at a defined site and now exist as linear, double-stranded DNA molecules. However, the solution also contains residual enzyme, buffer salts, and possible by-products.
Right: The solution after purification. Interfering components such as the restriction enzyme, buffer constituents, and unwanted DNA fragments have been removed.
Step 3: In vitro transcription for the production of mRNA
After the plasmid DNA has been linearized, it now serves as the template for in vitro transcription (IVT). The goal of this step is to produce the RNA molecule from the DNA that will later be used as the mRNA vaccine.
Transcription – the „rewriting” of DNA into RNA
Transcription takes place in a separate bioreactor under controlled conditions (e.g., specific pH, temperature, and ionic strength) that are optimized for RNA synthesis. Three components are required for this process:
1) The DNA template containing the spike gene.
2) The RNA building blocks, known as nucleotides.
3) An enzyme – T7 RNA polymerase – which specifically recognizes the T7 promoter. The T7 promoter was inserted into the plasmid directly upstream of the spike gene sequence and marks the starting point of transcription.
Course of transcription
Initiation (start): The T7 RNA polymerase binds to the T7 promoter. Once the enzyme is anchored there, it opens the DNA double helix at the transcription start site and exposes one strand as the DNA template.
The DNA contains various functional regions, including the origin of replication (yellow), selection markers (light green), regulatory elements (blue), the T7 promoter, and the spike gene sequence (red). The polymerase recognizes the T7 promoter and binds precisely at this site. It then unwinds the DNA double helix and begins synthesizing the RNA strand along the spike gene.
Elongation (extension): The polymerase moves along the DNA template strand in the 3′ → 5′ direction and simultaneously synthesizes the complementary RNA strand by stepwise addition of nucleotides in the 5′ → 3′ direction. The following complementary base pairing applies:
- DNA A (adenine) → RNA U (uracil or m¹Ψ)
- DNA T (thymine) → RNA A (adenine)
- DNA C (cytosine) → RNA G (guanine)
- DNA G (guanine) → RNA C (cytosine)
Behind the polymerase, the DNA double helix re-forms.
Termination (end): At the end of the spike gene, there is a terminator sequence. Once this is reached, the polymerase stops, detaches from the DNA, and the completed RNA molecule is released.
T7 RNA polymerase binds to the DNA and separates the two strands. Along the template strand, matching RNA nucleotides are incorporated. In this stepwise process, a single-stranded RNA molecule is formed that carries the information of the spike gene.
The RNA produced in this way is single-stranded and corresponds in its base sequence to the spike gene.
A distinctive feature of mRNA production
To improve stability and tolerability for use in vaccines, a modified building block is used:
Instead of uridine (U), N¹-methyl-pseudouridine (m¹Ψ) is incorporated.
This modification makes the RNA more stable, protects it from rapid degradation, and reduces unwanted immune reactions.
Both Pfizer’s and Moderna’s mRNA vaccines contain N¹-methyl-pseudouridine (m¹Ψ) instead of uridine. [The Critical Contribution of Pseudouridine to mRNA COVID-19 Vaccines]
Comparison of DNA, mRNA, and modRNA
During in vitro transcription, double-stranded DNA is converted into single-stranded RNA. DNA and RNA are similar in their basic chemical structure but differ in key properties such as stability, lifetime, and biological function. In mRNA vaccines, however, natural mRNA is not used; instead, a chemically modified form (modRNA) is employed. This differs from natural mRNA in several essential aspects.
The following table compares the properties of DNA, natural mRNA, and synthetic modRNA.
| Property | DNA | Natural mRNA | Synthetic modRNA |
| Definition | Master archive: permanent storage of all genetic information | Transcript of a single gene: contains the blueprint for a body´s own protein | Gene copy with a „disguise”: laboratory-produced (synthetic) form of mRNA that has been chemically modified. It contains the blueprint for a „non-native” protein. |
| Occurrence | Universal: nucleus (in eukaryotes); also mitochondrial DNA | Cell-specific: produced on demand only where the corresponding protein is needed. | Non-specific: can be taken up by many cell types in the body via lipid nanoparticles |
| Structure | Double-stranded | Single-stranded | Single-stranded |
| Sugar | Desoxyribose | Ribose | Ribose |
| Basen | A – adenine C – cytosine G – guanine T – thymine | A – adenie C – cytosine G – guanine U – uracil | A – adenine C – cytosine G – guanine m¹Ψ – N1-Methylpseudouridine |
| Lifetime and degradation rate | Very stable: protected by the nuclear membrane and repair systems; normally not degraded except during cell death or targeted DNA breakdown. | Short-lived: minutes to hours. Protein production is flexibly adapted to current metabolic needs. | Extended: hours to days. By replacing uridine with N1-methylpseudouridine, modRNA is less strongly recognized by innate immune RNA sensors and is degraded more slowly. |
| Degrading enzymes | DNases (deoxyribonucleases) | RNases (ribonucleases) | RNases (reduced recognition and slower degradation) |
✧ ✧ ✧
IVT by-products and impurities
After in vitro transcription (IVT), the result is not a pure product but a complex mixture. In addition to the desired mRNA, various by-products and impurities are formed as a result of the process, including short or long single-stranded RNA (ssRNA), double-stranded RNA (dsRNA), and RNA:DNA hybrids.
These accompanying products must be selectively removed, as they can otherwise impair the stability, efficacy, and tolerability of the vaccine. The purification methods used for this purpose are explained in more detail in Chapter 1.3.
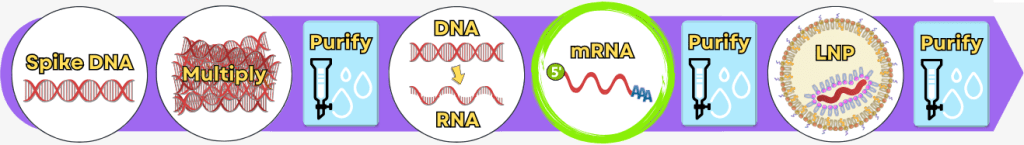
Step 4: RNA processing – maturation of the mRNA
RNA processing comprises a series of modifications that occur during or after transcription in order to produce a mature, functional mRNA from the RNA.
In the production of mRNA for vaccines, efforts are made to mimic natural processes as closely as possible, as they normally occur in human cells. The synthetically produced mRNA is designed to imitate certain properties of naturally occurring mRNA, thereby making it stable and enabling efficient translation into the desired protein.
A functional vaccine mRNA requires, like normal human mRNA:
- a protective cap (5′ cap) at the front end of the RNA
- a stabilizing tail (poly-A tail) at the back end
The 5′ cap: a „security seal” for the cell
For a synthetically produced mRNA to function in the body, it must carry a specific chemical protective structure at its 5′ end – the Cap-1 structure. This cap is a key recognition feature for the cell and determines whether the mRNA remains stable, is efficiently translated, and is not recognized as foreign.
The components of the Cap-1 structure (m⁷GpppN¹m)
The Cap-1 structure is not a simple „cap”, but a precisely built molecule consisting of three functional components:
The recognition element: 7-methylguanosine (m⁷G)
A special guanosine building block with a methyl group. This modification acts like a molecular ID card: only mRNAs with this structure are recognized by the cell as correct and trustworthy.
The linkage: triphosphate bridge (ppp)
Three phosphate groups connect the cap to the mRNA via an unusual 5′-5′ linkage. This special bond effectively protects the RNA end from enzymatic degradation.
The disguise: modified first nucleotide (N¹m)
The first nucleotide of the mRNA is additionally 2′-O-methylated. This modification is crucial for protecting the mRNA from recognition by cellular RNA sensors.
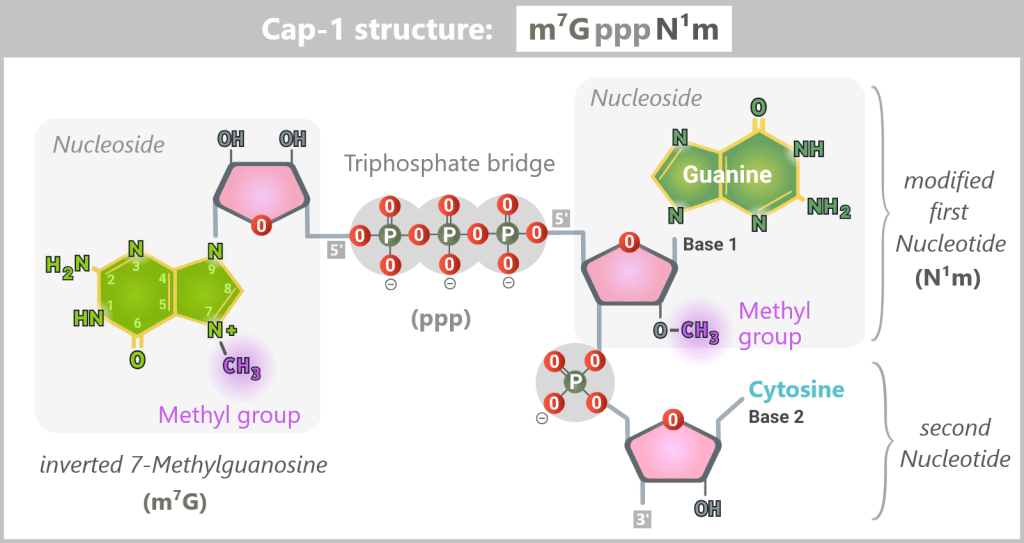
The 5′ end of the mRNA is linked to the first nucleotide (guanine in this example) via a 5′–5′ triphosphate bridge (ppp) and an invertedly linked, N7-methylated guanosine (m⁷G). Additionally, this first nucleotide bears a 2′-O-methylation (N¹m), which defines the Cap-1 structure and contributes to the stability, translational efficiency, and immune evasion of the mRNA.
During natural gene expression in human cells, mRNA is already processed while it is being synthesized in the nucleus. In particular, all mRNA molecules produced by RNA polymerase II receive a 5′ cap structure very early during transcription, along with additional chemical modifications.
Uncapped mRNA is non-functional under physiological conditions in eukaryotic cells: it is unstable, rapidly degraded, and does not reach the cytoplasm. This is because its 5′ end exists as a 5′ triphosphate (5′-ppp) structure, which is recognized and eliminated by cellular quality control systems as an abnormality (see lower figure).
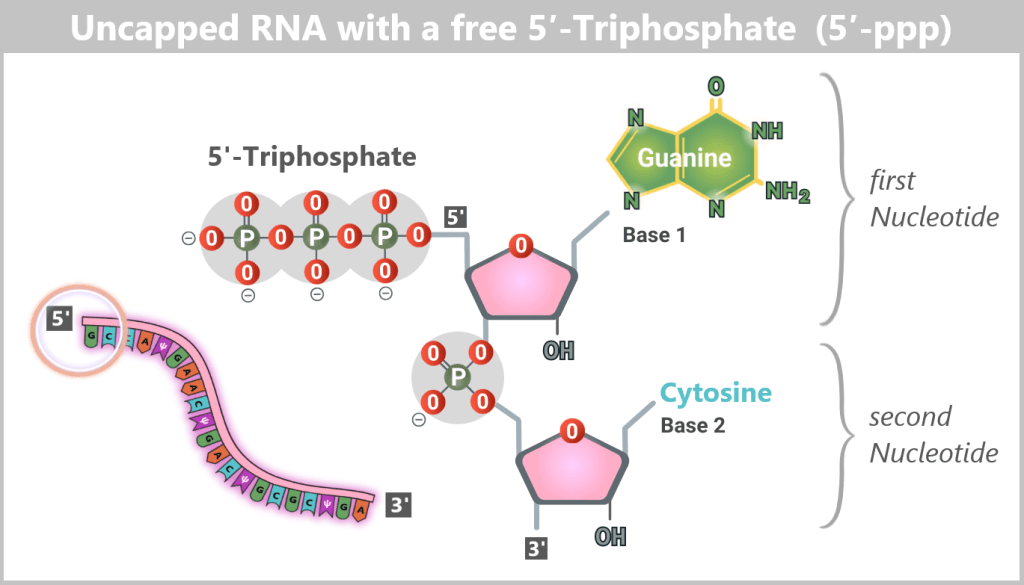
Why is Cap-1 so important?
The Cap-1 structure fulfills three central functions:
Immune evasion (camouflage): Cellular pattern recognition receptors such as RIG-I are highly sensitive to RNA with free 5′ triphosphates or unmodified ends. The Cap-1 structure of modRNA is chemically identical to the Cap-1 structure of the body’s own mRNA. Because cellular RNA sensors recognize this structure as „self”, no strong innate immune response is triggered under physiological conditions.
Initiation of protein production: The cap serves as a docking site for cap-binding proteins and marks the starting point for protein synthesis by ribosomes.
Stability: The cap protects the 5′ end of the mRNA from rapid enzymatic degradation, thereby extending its functional lifetime within the cell.
The poly-A tail: the protection and control center at the end
At the other end of the mRNA (the so-called 3′ end) is a long sequence consisting exclusively of adenine nucleotides – the poly-A tail. In vaccine production, it is usually made up of 100 to 150 ‘A’ units.
What is this tail for?
The „hourglass” of mRNA: The cell contains enzymes that gradually „nibble away” mRNA molecules from back to front. The poly-A tail acts as a buffer or protective extension at the end. It is degraded first, before the actual genetic information is attacked. The longer the tail, the longer the mRNA survives in the cell and the more protein can be produced.
Translation enhancer: Specific proteins bind simultaneously to the 5′ cap and the poly-A tail, forming the so-called closed-loop complex – an efficient „circular track”. On the one hand, this signals to the ribosome (the protein factory) that the mRNA is complete and ready for translation. On the other hand, after finishing the synthesis of one protein, the ribosome can directly restart translation at the 5′ end. This mechanism greatly increases the speed and efficiency of protein production.
✧ ✧ ✧
In summary: while the 5′ cap legitimizes the mRNA, disguises it, and makes it available for translation, the poly-A tail determines how long and how often the cell can use the information. Both modifications are therefore crucial for the lifetime of the mRNA and for its efficient translation into protein.
In the following schematic representations, the Cap-1 structure is shown as a functional 5′ unit positioned in front of the RNA strand, although chemically it represents a modified extension of the first nucleotide. For didactic reasons, the poly-A tail is symbolized by three adenine residues.
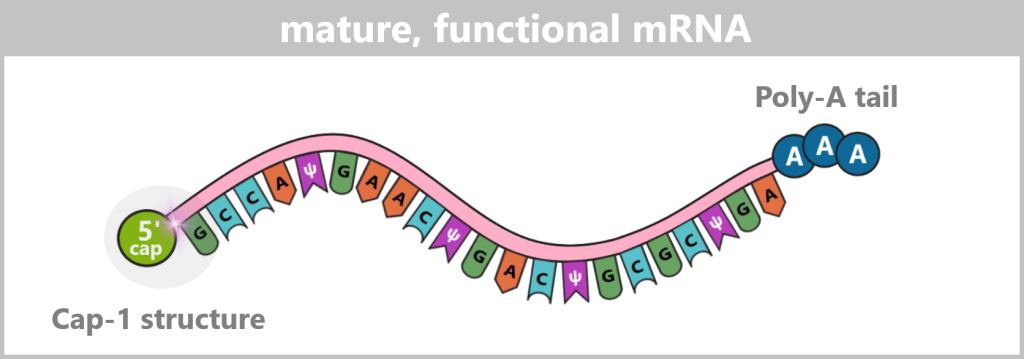
The figure is highly simplified; the actual mRNA is significantly longer and comprises approximately 4,200 nucleotides in the COVID-19 vaccine.
In biotechnological production, two methods have become established for generating these essential structures:
1. Co-transcriptional modification (the „all-in-one” method)
In this approach, the cap and poly-A tail are generated during in vitro transcription (IVT).
mRNA capping: Pre-formed cap building blocks (cap analogs) are added to the reaction. The T7 RNA polymerase automatically incorporates this cap structure as the first element at the beginning of the emerging mRNA molecule.
Polyadenylation: The DNA template already contains a sequence that serves as a template for a defined-length poly-A tail. The polymerase therefore synthesizes it directly following the coding sequence.
Advantage: The process is fast, scalable, and takes place in a single reaction vessel. By using modern cap analogs (e.g. ARCA or CleanCap), capping efficiencies of >95% can be achieved.
In industrial practice, the co-transcriptional method is the predominant standard.
2. Enzymatic modification (the „step-by-step” method)
In this more traditional, multi-step approach, the cap and poly-A tail are added in separate reaction steps after IVT.
Polyadenylation: First, the RNA is treated with the enzyme poly(A) polymerase. This enzyme specifically adds a long sequence of adenine nucleotides to the 3′ end of the RNA. The mRNA thus receives its poly(A) tail after synthesis.
mRNA capping: In a subsequent step, the 5′ cap structure is built up in a separate reaction.
Advantage: This method achieves very high and clean capping efficiency, but it is more labor-intensive.
Regardless of the chosen method, a comprehensive purification step follows in order to obtain a homogeneous and highly pure mRNA product.
The key steps – from the purified DNA template to the formulation-ready product – are summarized in the following overview:
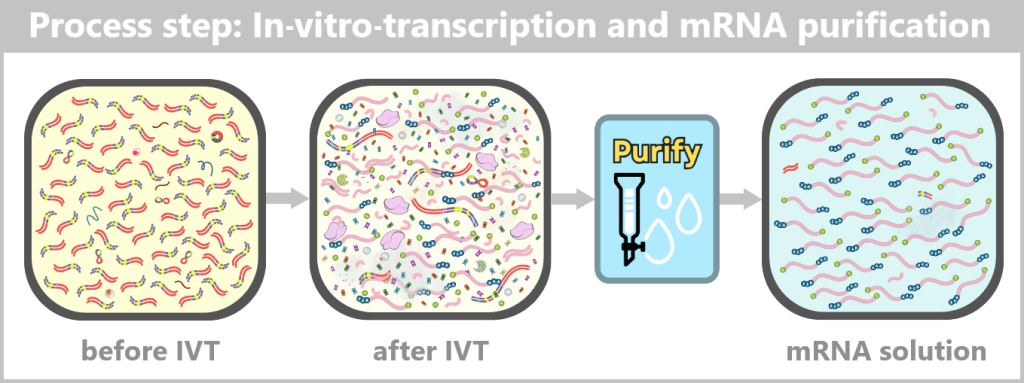
After IVT, a complex mixture of mRNA, template DNA, enzymes, by-products, and reaction components is present. Through subsequent purification steps, this mixture is reduced to a predominantly pure mRNA solution, which serves as the starting material for LNP formulation.
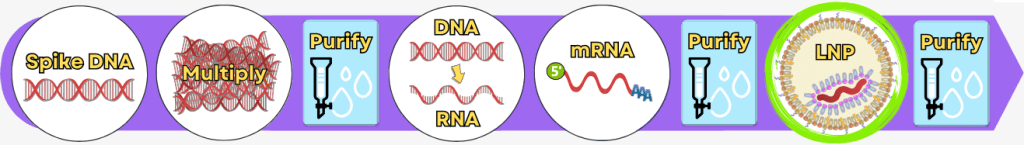
Step 5: Encapsulation of the mRNA in lipid nanoparticles (LNPs)
After the mRNA has been fully produced and purified, it still needs to be protected and made transportable. This is exactly the role of packaging it into lipid nanoparticles (LNPs).
Why does mRNA need packaging?
mRNA is a very fragile molecule. Without protection, it would be rapidly degraded in the body. LNPs fulfill several functions simultaneously:
- they protect the mRNA from degradation
- they transport it into body cells (through the cell membrane)
- they enable controlled release of the mRNA inside the cell
Without LNPs, the vaccine would not be functional.
How does an LNP form? – The basic mechanism
Formulation typically takes place in a device such as a microfluidizer or nanoparticle mixer. In this process, two liquids are rapidly mixed at very high speed:
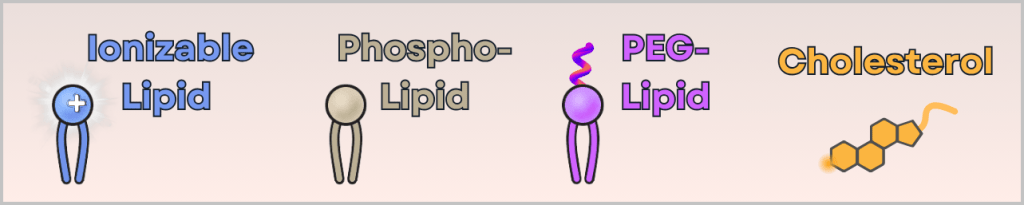
1) An ethanolic lipid solution
This contains four different lipids:
- ionizable cationic lipid (binds the mRNA and enables cellular uptake)
- phospholipid (stabilizes the structure – similar to cell membranes)
- PEG-lipid (controls particle size and prevents aggregation)
- cholesterol (makes the particle flexible and stable)
2) An aqueous mRNA solution
This contains only:
- purified mRNA
- a mild buffer
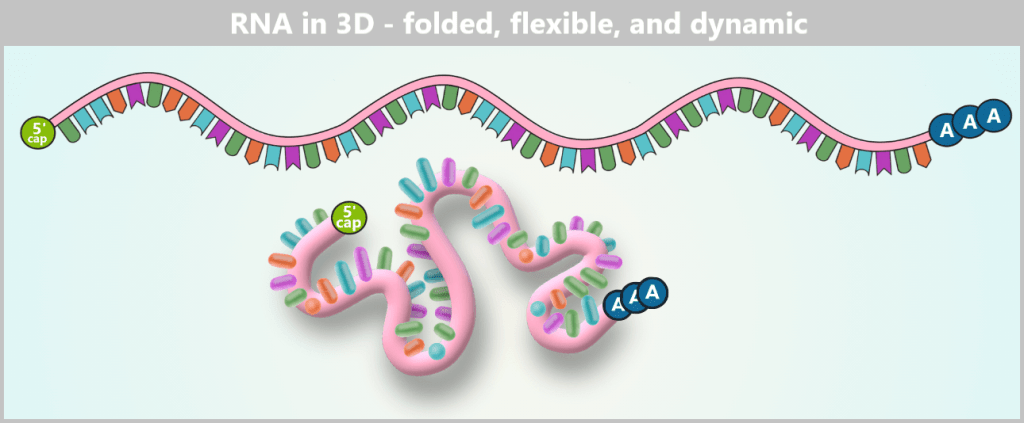
The RNA is shown in its linear form above, representing how the base sequence is read. Below, the same strand is illustrated as it actually exists in aqueous solution: a flexible, constantly moving 3D coil in which individual segments interact only loosely or transiently with each other.
What happens during mixing – a self-assembly effect
When the two solutions meet within fractions of a second, several parameters change:
- pH value
- lipid solubility
- charge states

The figure schematically illustrates the principle of microfluidic mixing. On the left and right, two separate liquids enter the mixer: a lipid solution (left) containing the different lipid types and an mRNA solution (right) containing freely dissolved mRNA strands. In the central microfluidic channel, both streams meet and are intensely mixed.
As a result, a spontaneous and highly precise process occurs:
- The ionizable lipid becomes positively charged → it attracts the negatively charged mRNA.
- The mRNA is „wrapped” and encapsulated.
- The other lipids arrange themselves around it to form a stable outer shell.
This automatically generates a nanoparticle with a typical size of 60–100 nm. It is therefore not a „manual packaging” process, but rather a biophysical self-assembly process – the molecules spontaneously organize into the correct structure.
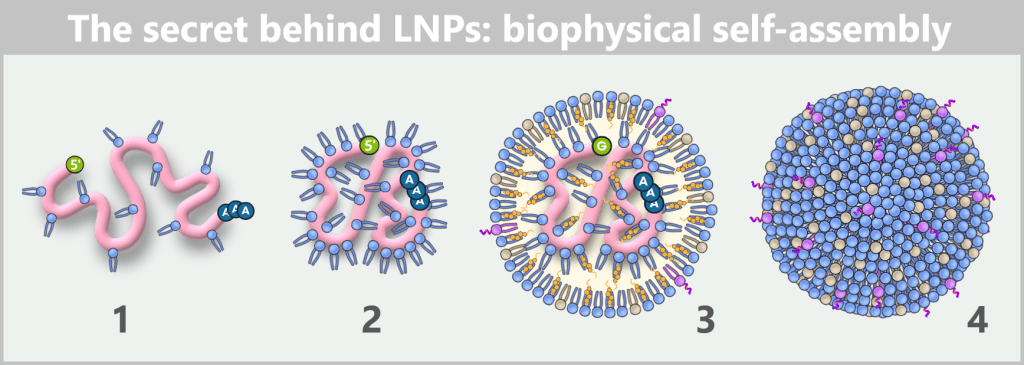
The figure shows in four steps how mRNA and lipids spontaneously organize into a lipid nanoparticle during formulation.
1) As soon as mRNA and ionizable lipids come into contact, the positively charged lipid head groups immediately bind to the negatively charged phosphate backbone of the mRNA and begin to tightly wrap around the strand.
2) As a result, the mRNA contracts locally, becomes more compact and continues to condense.
3) In this way, an initial „core” of mRNA–lipid complexes is formed. Additional ionizable lipids then accumulate around it, along with phospholipids, cholesterol, and PEG-lipids. Together, they stabilize the emerging structure while the mRNA becomes progressively more tightly enclosed.
4) Finally, a densely packed, nearly spherical nanoparticle is formed. Its shape is not determined by external control but solely by the physicochemical properties of the molecules – an example of spontaneous self-organization on the nanoscale.
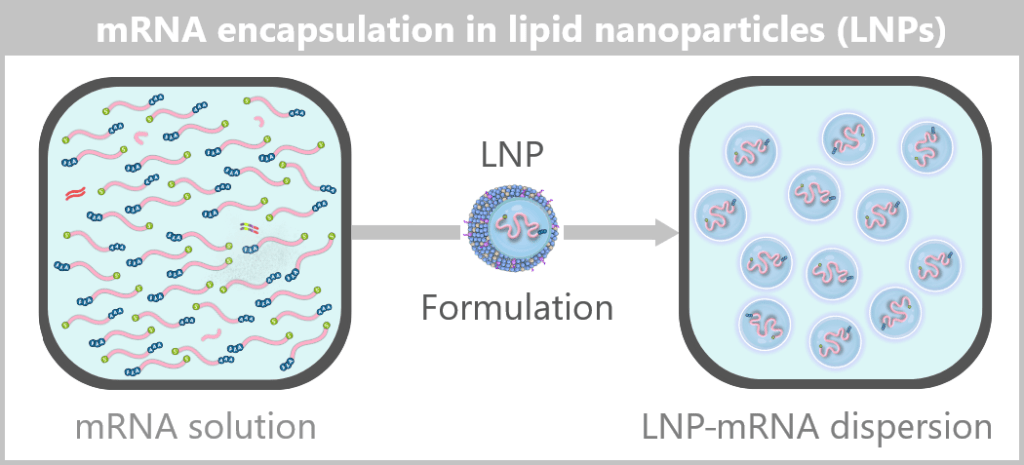
The formulated LNPs now constitute the final drug substance concentrate, which is subsequently filled under sterile conditions in the next step.
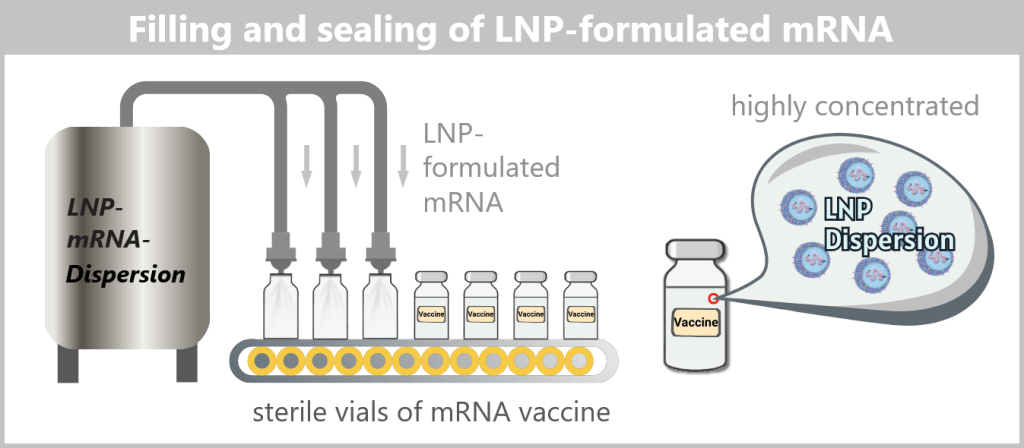
A vaccine vial contains a very large number of individual lipid nanoparticles, typically in the range of 10¹³–10¹⁴ particles per vial.
1.2. Impurities and Byproducts
The production of a functional mRNA involves several sequential process steps – from the amplification of the DNA template to the processing and formulation of the RNA. In nearly every one of these steps, by-products, impurities, or residual substances are also generated alongside the desired product.
1.2.1. Typical impurities in bacterial production
1.2.2. Typical impurities and by-products after in vitro transcription
For the safety and efficacy of the therapeutic product, their identification and subsequent removal in the downstream process is crucial.
In this section, we take a look at the most important potential impurities and by-products and their known biological activities.
✧ ✧ ✧
1.2.1. Typical impurities in bacterial production
After bacterial cell lysis, the plasmid DNA is present in a complex reaction mixture. In addition to the desired plasmid DNA, the solution contains chromosomal DNA, bacterial RNA, proteins, enzymes (including RNases), endotoxins, and fragments of the cell wall.
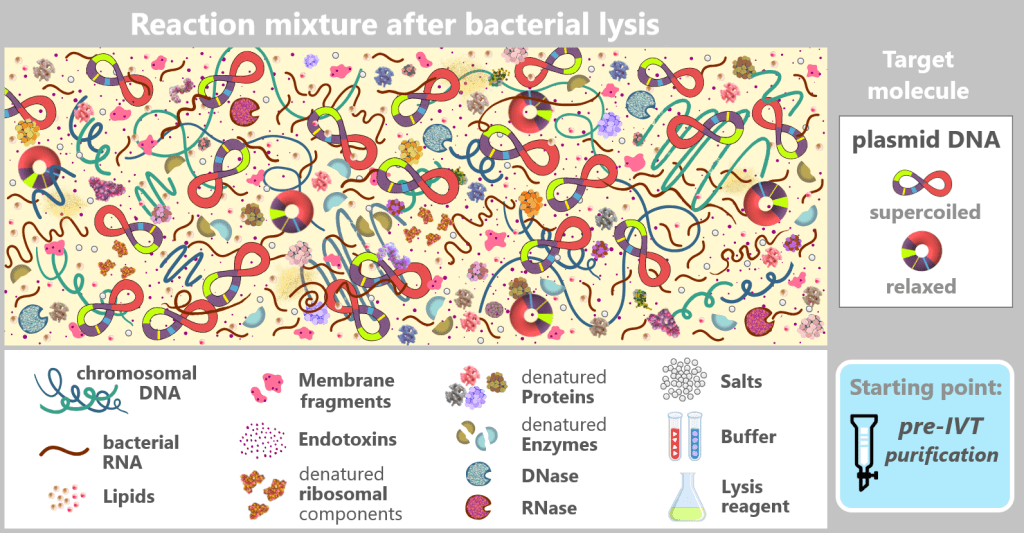
The figure schematically shows the composition of the solution after disruption of the E. coli cells in which the plasmid was amplified. In addition to the desired plasmid DNA, which contains the expression cassette for the mRNA, numerous bacterial components and process-related substances are present. The depicted components are embedded in a protein-rich cytosolic matrix derived from the bacterial cytoplasm (yellow background).
This complex and heterogeneous mixture illustrates that the plasmid DNA must first be isolated from a highly contaminated biological environment before it can be used as a template for in vitro transcription.
✧ ✧ ✧
1.2.2. Typical impurities and by-products after in vitro transcription
Although in vitro transcription (IVT) is an efficient enzymatic method for producing mRNA, the reaction does not yield a pure final product. After synthesis, a complex mixture is present that, in addition to the target mRNA, contains various process-related impurities and by-products.
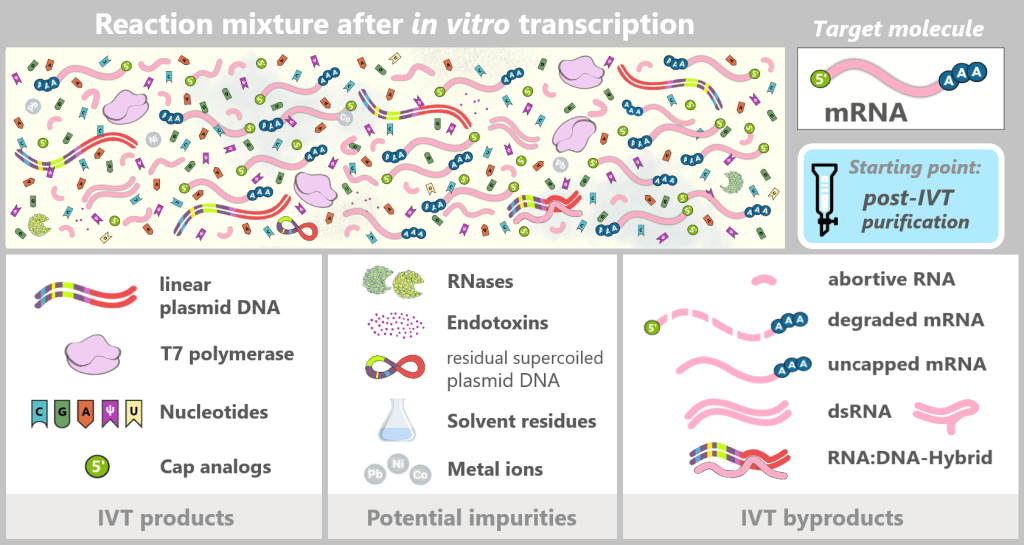
The figure schematically illustrates the complex composition of the IVT reaction solution. In addition to the desired, fully processed mRNA (target molecule), the mixture contains various RNA by-products such as uncapped, degraded, or abortive RNA, double-stranded RNA, and RNA:DNA hybrids. Additional components include:
IVT products
Linear plasmid DNA: DNA template containing the expression cassette for the spike protein.
T7 polymerase: Enzyme that recognizes the T7 promoter and synthesizes the mRNA.
Nucleotides: Building blocks of mRNA synthesis – cytosine (C), guanine (G), adenine (A), and N1-methylpseudouridine (m¹Ψ) instead of uridine (U). Uridine is listed because small amounts of U may remain from the production of modified nucleotides.
Cap analogs: Synthetic cap analogs used during IVT to form the Cap-1 structure and which may remain in excess after the reaction.
Potential impurities
RNases: Enzymes that can cleave and degrade RNA; they may be introduced unintentionally via raw materials or bacterial residues.
Endotoxins: Components of the outer membrane of Gram-negative bacteria such as E. coli.
Solvent residues: Traces from the production and purification of starting materials.
Metal ions: Trace impurities from raw materials, process water, or production equipment.
Overview: Typical RNA by-products after IVT (before purification)
a) Abortive mRNA
b) Degraded mRNA
c) Uncapped mRNA
d) Double-stranded RNA (dsRNA)
e) RNA:DNA hybrids
⚬ ⚬ ⚬
a) Abortive mRNA
At the start of transcription, T7 RNA polymerase binds tightly to the promoter and forms a stable initiation complex. However, the transition from this initiation phase into productive elongation is mechanically unstable.
The polymerase locally unwinds the DNA around the transcription start site and begins synthesizing a short RNA strand. In doing so, it pulls DNA into the enzyme without itself moving forward along the DNA. This so-called scrunching leads to the accumulation of mechanical stress within the transcription complex, as the enzyme remains anchored at the promoter while continuously drawing in more DNA.
If the polymerase is unable to relieve this tension by releasing from the promoter and transitioning into the elongation phase, the DNA snaps back into its original configuration. The short RNA fragment that has already been synthesized is released – resulting in an abortive transcript.
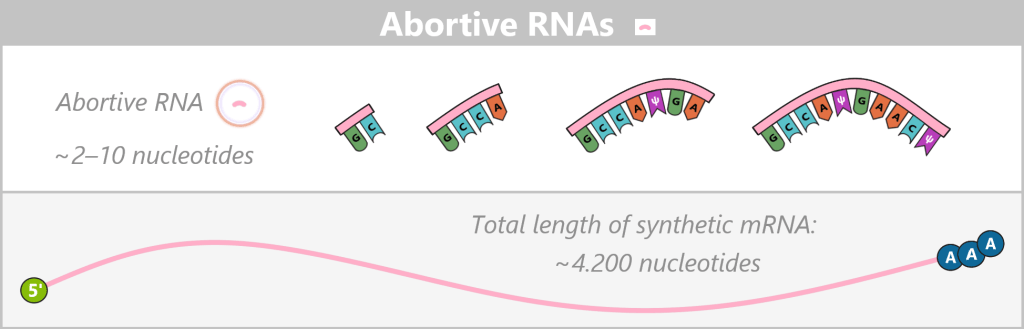
This process can repeat multiple times, meaning that a single polymerase can produce numerous abortive RNA fragments before successfully completing a full transcription event. Only once an RNA length of typically around 8–14 nucleotides is reached does the polymerase escape the promoter and form a stable elongation complex.
Appearance
Abortive RNAs are very short RNA fragments, typically only 2–10 nucleotides (nt) long, that arise during the unstable initiation phase of transcription. Despite the use of co-transcriptional capping strategies in which a cap analog serves as the initiating nucleotide, most abortive transcripts do not reach the length required for stable cap incorporation. Consequently, they predominantly exist as uncapped fragments carrying a 5′ triphosphate end. Only in rare cases – typically in longer abortive transcripts of approximately 10–12 nucleotides – can a cap analog be formally incorporated.
Biological impact
The biological effects of abortive RNA fragments are not yet fully understood. Individual isolated fragments of only a few nucleotides are generally too short to activate known innate immune RNA sensors and are not translated into proteins.
The potential risk therefore lies less in the individual 2–10-nt fragments themselves, but rather in possible secondary effects:
Double-strand formation: Short RNA fragments could act as primers or contribute to the formation of short double-stranded RNA molecules.
Complex formation: High amounts of such fragments could aggregate into more complex structures or potentially reduce the efficiency of downstream purification steps.
Non-specific stress effects: A high concentration of short RNA fragments could transiently increase cellular stress, even if they are rapidly degraded.
BioNTech notes that abortive by-products in the cytosol (the fluid interior of the cell) of transfected cells (cells into which the mRNA has been introduced) may potentially interact in unknown ways with endogenous RNAs or pattern recognition receptors (PRRs), which highlights the need for further research. [Understanding the impact of in vitro transcription byproducts and contaminants]
Approximate proportion (before purification)
BioNTech reports that approximately 44% of T7 RNA polymerases produce abortive transcripts before a full-length transcription is achieved. As a result, abortive RNAs can be the most frequent RNA species in terms of molecule number in the crude reaction mixture. However, under standard IVT conditions, they account for less than 1% of the total RNA mass. The exact proportion of abortive RNAs strongly depends on the template sequence and the reaction conditions.
b) Degraded mRNA
In contrast to abortive mRNA, degraded mRNA consists of formerly complete or long mRNA strands that have been partially or completely destroyed by external influences or enzymatic processes. Degradation can occur through several mechanisms:
Enzymatic degradation by RNases: This is the most common cause of degradation during and after IVT. RNases (ribonucleases) are extremely stable enzymes that are found almost everywhere (e.g. on skin, in dust, or in raw materials). They cleave RNA at specific or structurally preferred sites. Even minimal contamination of the reaction mixture can cause freshly synthesized mRNA to break down into fragments.
Chemical/physical degradation: Due to its chemical structure, RNA is significantly less stable than DNA. Elevated pH values, high temperatures, or certain metal ions promote hydrolytic cleavage of the RNA backbone and lead to strand breaks.
Mechanical degradation: If the solution is exposed to high shear forces – for example through vigorous stirring or pumping through narrow tubing – long mRNA strands can physically tear apart. This tends to generate relatively large fragments rather than the fine fragments typically caused by RNases.
Premature termination during elongation: Strictly speaking, this is not classical degradation, but rather incomplete mRNA generated during synthesis itself. If the polymerase prematurely dissociates from the DNA template during elongation – for example due to strong secondary structures or limited nucleotide availability – a shortened mRNA molecule is produced that lacks the 3′ end and therefore the poly-A tail.
Appearance
Degraded mRNA exists as a mixture of fragments of varying lengths. These fragments may contain individual strand breaks or may be extensively degraded, and they often lack a complete 5′ cap and/or poly-A tail.

… as a heterogeneous mixture of fragments that can arise during or after in vitro transcription. In contrast to intact, full-length mRNA (shown below as a reference), the degradation products vary in length and display characteristic damage: lost or damaged 5′ cap structures, exposed 5′ triphosphates (ppp) – which become exposed upon cap loss, shortened poly-A tails, and internal strand breaks. The fragments do not exist as a uniform species, but rather as a complex mixture.
Biological impact
Degraded mRNA is not a uniform substance but a heterogeneous mixture. Not all fragments are biologically problematic. The greatest risk arises from fragments that carry specific immunological danger signals, including:
- exposed 5′ triphosphates, which are recognized by cytosolic RNA sensors such as RIG-I,
- short double-stranded RNA structures that arise through secondary structure formation or hybridization and can activate receptors such as MDA5 or TLR3,
- unusual end structures (missing cap or poly-A tail), which may be recognized as „FOREIGN”.
Such fragments can trigger innate immune responses and impair the tolerability of the mRNA formulation.
Approximate proportion (before purification)
The proportion of degraded mRNA varies strongly depending on process conditions. Typically, it lies in the range of about 1–10% before purification. However, in well-optimized IVT processes, this fraction is deliberately reduced to a minimum.
c) Uncapped mRNA
In modern IVT processes, the 5′ cap structure is often introduced co-transcriptionally using so-called CleanCap analogs. These are synthetic initiator oligonucleotides that already contain the complete 5′ cap structure, including the first transcribed nucleotide.
T7 RNA polymerase specifically uses CleanCap to initiate transcription; unlike classical cap analogs, there is no direct competition with GTP, the natural initiating nucleotide. As a result, predominantly correctly capped mRNA molecules are produced, while the proportion of uncapped mRNA is greatly reduced.
Appearance
Uncapped mRNA exists as a full-length RNA sequence but is characterized by the absence of the 5′ cap structure at its 5′ end.

Biological impact
Cells possess specialized enzymes that efficiently degrade RNA strands lacking a 5′ cap from the 5′ end. This mechanism serves to rapidly eliminate defective or aged endogenous RNAs.
Immune response: Uncapped RNA is recognized by cellular pattern recognition receptors such as RIG-I and can trigger a strong type I interferon response, placing the cell into an antiviral state.
In uncapped modRNA, the substitution of uridine with N1-methylpseudouridine (m¹Ψ) significantly reduces activation of these sensors. This creates a biological „tug-of-war” between the immunostimulatory uncapped 5′ end and the immunomodulatory nucleoside modification. Overall, uncapped modRNA would therefore act as a moderately immunogenic molecule but would still be degraded more rapidly than correctly capped modRNA.
Minimal protein production: Since the 5′ cap structure is also required for efficient ribosome binding, uncapped mRNA is translated into protein very poorly or not at all.
Approximate proportion (before purification)
The fraction of uncapped mRNA after IVT depends strongly on the capping strategy used. With modern co-transcriptional CleanCap approaches, the proportion of uncapped mRNA before purification typically lies in the range of approximately 1–6%.
d) Double-stranded RNA (dsRNA)
Another relevant by-product of in vitro transcription (IVT) is double-stranded RNA (dsRNA). While the desired vaccine mRNA is synthesized as a single-stranded molecule, RNA duplexes can also form during IVT in which two complementary RNA strands are linked via Watson–Crick base pairing.
The formation of such dsRNA species is not a random aggregation phenomenon but results from specific enzyme-mediated side reactions of T7 RNA polymerase.
Formation mechanisms
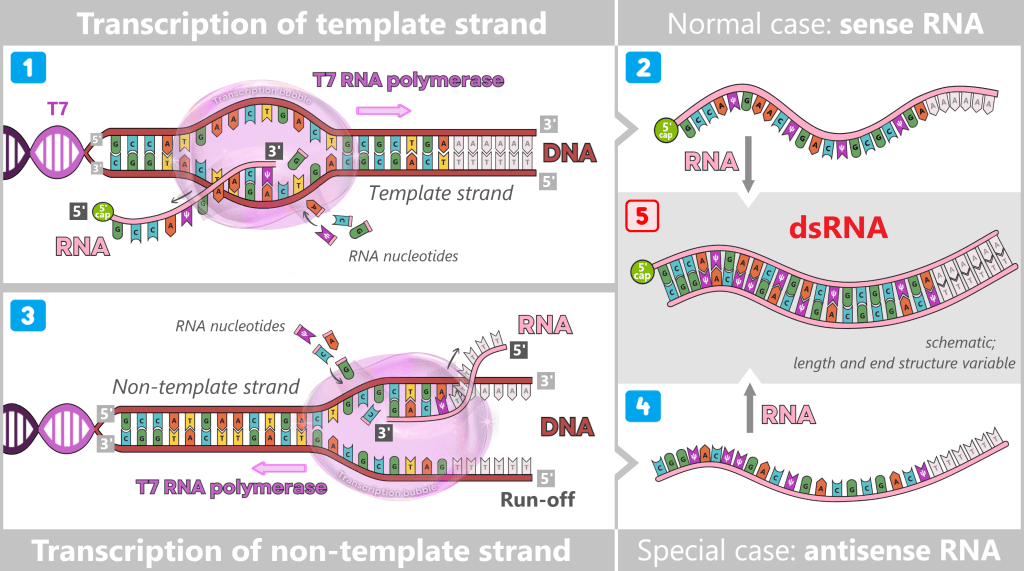
Promoter-independent transcription of the non-template strand
Under certain conditions, T7 RNA polymerase can synthesize RNA even without a canonical promoter initiation by reading the non-template strand of the DNA template. The resulting RNA molecules are largely complementary to the desired mRNA. When sense and antisense RNA strands encounter each other, they form extensive, nearly fully double-stranded RNA duplexes. This mechanism is particularly promoted when the DNA template is not fully linearized or when specific sequences are present at the 3′ end of the template.
(RNA is shown schematically; nucleoside modifications (e.g. m¹Ψ) are not explicitly depicted.)
1) The normal case: The T7 promoter has an exceptionally high affinity for T7 RNA polymerase. Therefore, the polymerase almost exclusively initiates transcription at this site. It reads DNA in the 3′ → 5′ direction and synthesizes a new RNA molecule exclusively in the 5′ → 3′ direction.
2) The result is the desired sense RNA, which corresponds exactly to the sequence of the non-template strand.
3) The special case: After run-off – the polymerase leaving the DNA template at the end of transcription – the enzyme dissociates from the DNA. In rare cases, it can bind nonspecifically to structurally accessible DNA ends. If it binds to the strand that was previously not used as a template, this strand becomes the new template.
Under certain reaction conditions, locally open or dynamically melting DNA regions may also form, allowing T7 RNA polymerase to bind in a promoter-independent manner. If this binding occurs on the previously unused strand, it is read as the template.
During promoter-independent transcription of the non-template strand, there is no defined initiation site, so no co-transcriptional capping occurs. The resulting antisense RNA therefore typically carries a free 5′ triphosphate (pppN).
4) The result is a complementary antisense RNA that corresponds to the sequence of the template strand.
5) Formation of long dsRNA duplexes: When sense RNA and antisense RNA encounter each other, double-stranded RNA (dsRNA) is formed. The resulting dsRNA by-products are not a homogeneous molecule but a structurally heterogeneous mixture of fully or partially complementary RNA duplexes. These may contain blunt ends or single-stranded overhangs and vary considerably in length and terminal structure.
RNA-dependent 3′ end extension (self-priming)
A second key mechanism is RNA-dependent extension of the 3′ end of the freshly synthesized mRNA. In this process, the 3′ end of the RNA folds back intramolecularly due to complementary sequences, forming a short double-stranded RNA hairpin. T7 RNA polymerase can bind to this structure again and extend the RNA strand, generating a sequence that is complementary to the original RNA. This process is independent of the DNA template and can even occur in already fully transcribed RNA molecules.
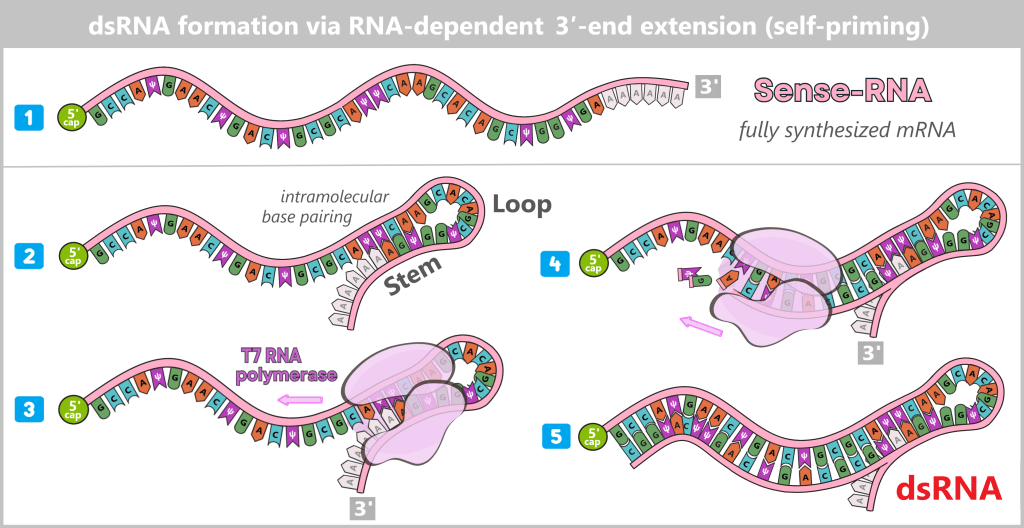
(RNA is shown schematically)
1) Sense RNA: A normal, fully synthesized mRNA molecule.
2) Hairpin formation: The 3′ region folds back on itself. A short dsRNA stem (e.g. 5–10 base pairs) forms with a single-stranded loop.
3) Polymerase binding: T7 RNA polymerase binds to this dsRNA stem as if it were a DNA template–primer complex.
4) Extension: The polymerase uses one strand of the hairpin as a template and extends the free 3′ end along the complementary RNA region, thereby synthesizing the antisense sequence directly into the extension of the sense strand.
5) Result: The polymerase extends the strand as far as possible. The newly synthesized antisense RNA immediately hybridizes with the sense RNA. This results in a partially or fully dsRNA-containing RNA molecule.
Distinction from RNA secondary structures
Single-stranded RNA naturally forms intramolecular secondary structures such as hairpins or internal loops (see upper figure – point 2). These are an integral part of functional mRNA and are typically short and thermodynamically flexible. Such structural elements are not considered dsRNA by-products in the strict sense and are not equivalent to the long dsRNA duplexes that are relevant as IVT impurities.
Both mechanisms lead to the formation of longer, more stable dsRNA regions, which are structurally clearly distinct from the short, dynamic secondary structures of correctly transcribed mRNA.
Appearance
dsRNA by-products typically exist as long, partially or fully complementary RNA duplexes. They may span nearly the entire length of the target mRNA or contain extended double-stranded regions. These duplexes often lack a 5′ cap structure and may feature single-stranded overhangs at their termini.
Biological impact
dsRNA as a structural danger signal
Double-stranded RNA (dsRNA) is not a typical structural motif of endogenous, translatable mRNA in the cytoplasm of eukaryotic cells. This means that while cellular mRNAs are normally single-stranded and serve as templates for protein synthesis via translation, dsRNA occurs physiologically only in tightly regulated, short-lived contexts. The presence of dsRNA in the cytoplasm is therefore not primarily interpreted by the cell as genetic information, but as a structural warning signal.
Cytosolic recognition of dsRNA
Cells possess specialized pattern recognition receptors (PRRs). These „danger detectors” patrol the cytoplasm and search for structural patterns such as duplex length, end structure, and chemical nucleotide modifications.
Cytosolic dsRNA sensors and their immunological consequences
| Feature / specificity | Cellular sensor | Biological consequence (for cell) |
| Short dsRNA (≈ 10–300 bp) with 5′ triphosphate and/or blunt ends | RIG-I (Retinoic acid-Inducible Gene I) short-ppp detector | Induction of inflammatory gene expression (type I interferons, cytokines). Activation of neighboring cells. |
| Long dsRNA (> ~500–1,000 bp), independent of end structures | MDA5 (Melanoma Differentiation-Associated protein 5) long-duplex scanner | Strong type I interferon response. Important antiviral defense mechanism. |
| Medium-length dsRNA (≈ ≥30–100 bp) | PKR (Protein Kinase R) intracellular effector | Global shutdown of protein synthesis via phosphorylation of translation factor eIF2α. Inhibition of viral replication. |
| Medium-length dsRNA (≈ ≥40–100 bp) | OAS (2′-5′-Oligoadenylate Synthetases) intracellular effector | Non-specific RNA degradation via activation of latent RNase L, which cleaves cellular and viral RNA. Can lead to cell death. |
| dsRNA in endosomes (extracellularly taken up or phagocytosed dsRNA) | TLR3 (Toll-like Receptor 3) gatekeeper receptor | Induction of type I interferons and pro-inflammatory cytokines. |
Effect of m¹Ψ
The use of nucleoside-modified mRNA (modRNA), for example through the incorporation of N¹-methylpseudouridine, can significantly reduce the activation of certain cytosolic RNA sensors. RIG-I and PKR are particularly affected, as they preferentially respond to single-stranded RNA or short-lived dsRNA structures. In contrast, length-dependent sensors such as MDA5 are only partially modulated by this modification and remain largely sensitive to extended dsRNA duplexes.
For this reason, dsRNA still possesses a high immunostimulatory potential even in the context of modRNA – meaning it can activate the immune system and trigger unwanted inflammatory responses. The efficient minimization of such dsRNA by-products through optimized transcription conditions, chemical modifications, and downstream purification processes is therefore a central aspect of mRNA manufacturing.
Approximate proportion (before purification)
The amount of dsRNA after IVT is highly process-dependent and is influenced by factors such as the quality of the DNA template, reaction conditions, and the polymerase used. Typically, the dsRNA fraction before purification is in the low single-digit percentage range, but under unfavorable conditions it can be significantly higher.
e) RNA:DNA hybrids
During in vitro transcription, under certain conditions, the newly synthesized RNA strand may not fully dissociate from the DNA template. It can remain partially bound to the template strand while displacing the complementary DNA strand. This leads to the formation of so-called RNA:DNA hybrids.
The formation of such hybrid structures is strongly sequence-dependent. In particular, purine-rich transcripts – RNA sequences containing high amounts of adenine and guanine – as well as DNA templates with repetitive GAA (guanine–adenine–adenine) motifs favor the formation of stable RNA:DNA duplexes. These by-products are often underestimated but can be experimentally detected using specific antibodies against RNA:DNA hybrids. [Understanding the impact of in vitro transcription byproducts and contaminants]
Appearance
RNA:DNA hybrids typically exist in the form of R-loop-like structures. These are locally restricted, three-stranded nucleic acid conformations consisting of:
- an RNA:DNA hybrid duplex,
- a displaced single-stranded DNA segment,
- and the adjacent double-stranded DNA outside the hybrid region.
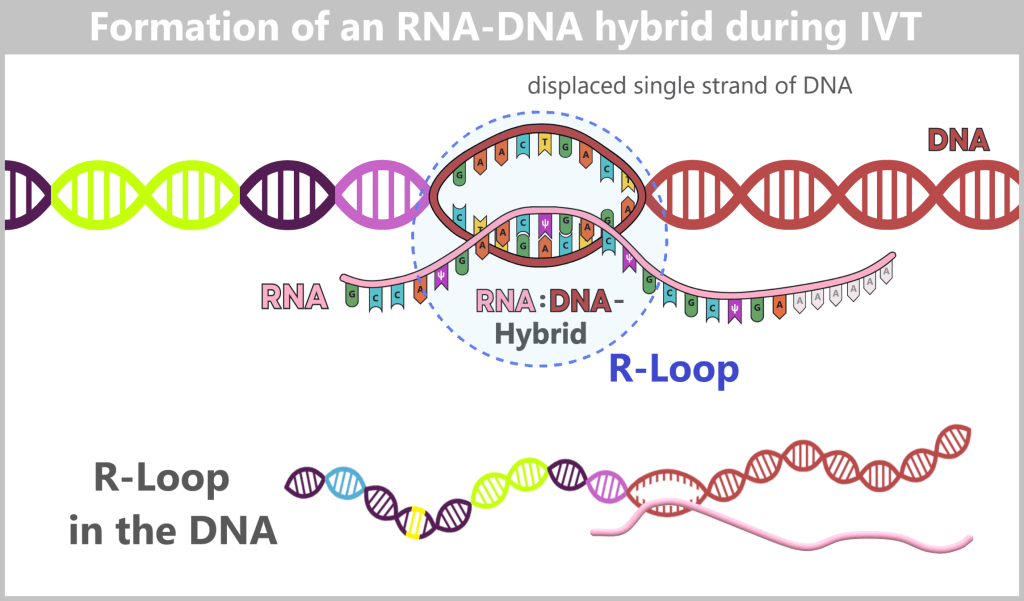
The newly synthesized RNA strand partially hybridizes with the DNA template strand, displacing the complementary non-template strand. This forms an R-loop consisting of an RNA:DNA hybrid duplex and a displaced single-stranded DNA region, embedded within an otherwise double-stranded DNA region. Outside the R-loop, the DNA remains in its regular double-helical structure.
Biological impact
Current research suggests that RNA:DNA hybrids may be recognized by cellular pattern recognition receptors (PRRs). These sensors of the innate immune system include cGAS, TLR9, and the inflammasome protein NLRP3, which can bind such hybrid structures. Their activation can trigger an immune response, leading to the production of pro-inflammatory cytokines and type I interferons.
However, whether and to what extent RNA:DNA hybrid contaminants actually contribute to unwanted immune reactions in therapeutically administered IVT mRNA has not yet been systematically investigated.
Against this background, the efficient removal of RNA:DNA hybrid impurities from IVT mRNA appears to be a relevant aspect of quality assurance. This is particularly important for therapeutic applications in which strong immune activation is not desired. [Understanding the impact of in vitro transcription byproducts and contaminants]
Approximate proportion (before purification)
The proportion of RNA:DNA hybrids after in vitro transcription is highly process-dependent. It is influenced, among other factors, by the sequence and quality of the DNA template as well as by the reaction conditions. Overall, their fraction prior to purification is considered low, but it can vary depending on the specific transcript and process parameters. Reliable publicly available quantitative data on the exact amount of RNA:DNA hybrids in therapeutic mRNA products are not currently available, as such measurements are typically part of proprietary manufacturing and quality control knowledge.
** Another possible mechanism for the formation of RNA:DNA duplexes during DNase treatment is described in section „1.3.1. a) Possible post-transcriptional RNA:DNA hybridization”. **
1.3. Purification Methods in the Manufacturing Process
Because mRNA vaccines must meet extremely high purity requirements, purification is not a single isolated process step. Rather, it runs like a common thread throughout the entire manufacturing process.
The exact industrial workflow used by manufacturers such as BioNTech/Pfizer or Moderna is proprietary (company-internal) and is not fully disclosed. However, patents and regulatory documents indicate that a combination of established biotechnological purification methods is employed.
A schematic, cross-industry overview of mRNA manufacturing, illustrating the continuous integration of purification activities throughout the overall process, can be found for example in the technical documentation of Merck / Sigma-Aldrich. (Manufacturing strategies for mRNA vaccines and therapies, Figure 1).
Within the manufacturing process, three particularly critical purification phases can be distinguished in simplified form:
- before in vitro transcription (pre-IVT): purification of the DNA template,
- after in vitro transcription (post-IVT): purification of the synthesized mRNA,
- after formulation: sterile filtration of the lipid nanoparticle formulation.
Rather than discussing these phases strictly in chronological order, the following sections present the most important purification principles and methods that are repeatedly applied at different stages of the manufacturing process.
1.3.1. DNase I digestion – enzymatic DNA degradation
1.3.2. Proteinase K – enzymatic degradation of residual proteins
1.3.3. Filtration
1.3.4. Chromatography – the molecular separation method
1.3.5. Magnetic bead purification
The following overview summarizes the key purification techniques. It serves as a framework for the subsequent detailed description of each method.
| Method | Removes | Principle | Advantages | Limitations |
| DNase I digestion | Template DNA | Specific enzymatic degradation | Highly selective, breaks down problematic nucleic acids | Enzymes must be completely removed; otherwise risk of residual activity |
| Proteinase K | Enzymes from transcription (e.g. polymerases, ligase, restriction enzymes) | Non-specific protein degradation | Broadly effective, removes many protein types | Additional purification to remove enzyme residues |
| Filtration & TFF (tangential flow filtration) | Salts, nucleotides, small molecules, buffer residues; also enables concentration | Size-based separation using membranes | Scalable, robust, versatile (concentration + buffer exchange in one step) | Does not distinguish similar nucleic acids; rather a „rough tool” |
| Chromatography (Poly(dT), AEX) | dsRNA, short RNA fragments, residual nucleotides, proteins | Separation by charge or specific binding to surfaces | Very precise, separates closely related nucleic acids; well established in industry | Technically demanding, cost-intensive, requires tight process control |
| Magnetic bead purification (Process 1) | Selects mature polyadenylated mRNA; removes short fragments & impurities | Poly(T)-coated beads bind specifically to the poly(A) tail of mRNA | High specificity, fast and efficient separation | Suitable for small to medium volumes; difficult to scale for large-scale production |
1.3.1. DNase I digestion – enzymatic DNA degradation
Deoxyribonuclease I (DNase I) is an enzyme that cleaves DNA molecules into smaller fragments. This process is often referred to as „digestion” of DNA – a figurative term describing the enzymatic breakdown of long DNA strands into shorter pieces. In biotechnology, DNase I is used to selectively fragment unwanted DNA contaminants.
DNase I is therefore not a complete purification method on its own, but rather a preparatory step that facilitates subsequent physical and chromatographic separation processes.
In mRNA vaccine production, DNase I is used after in vitro transcription (IVT) to fragment the DNA template (template DNA) as well as any DNA component of RNA:DNA hybrids that may have formed and could affect product quality or safety.
DNase I recognizes the three-dimensional structure of DNA – specifically the sugar-phosphate backbone. The enzyme binds to DNA like a saddle and inserts itself into the so-called minor groove. In doing so, it locally bends the DNA structure to gain access to the phosphodiester bond. Because the width of the minor groove varies slightly depending on the base sequence, DNase I exhibits mild sequence preference, cleaving somewhat more readily at certain regions (e.g. A/T-rich stretches).
The typical B-form DNA is a slender, right-handed double helix resembling a spiral staircase with two grooves of different sizes (a major and a minor groove) winding around it. The enzyme preferentially interacts with the minor groove of the DNA duplex and binds to the sugar-phosphate backbone. This binding induces a local distortion of the DNA, enabling cleavage of the phosphodiester bond. The major groove is shown for completeness but does not play a primary role in DNase I binding.
DNase I cleaves DNA non-specifically, meaning it cuts at many different positions along the strand. It acts on both single-stranded DNA (ssDNA) and double-stranded DNA (dsDNA). Its activity depends on several factors, in particular the presence of metal ions, which influence the nature of the cleavage sites [bioswisstec, YEASEN]:
In the presence of magnesium ions (Mg²⁺), DNase I primarily introduces single-strand cuts („nicks”) in double-stranded DNA (dsDNA). This results in the formation of short dsDNA fragments with overhangs, as well as a smaller proportion of single-stranded DNA (ssDNA) fragments.
In the presence of manganese ions (Mn²⁺), DNase I cleaves both strands of dsDNA at nearly the same position, producing predominantly blunt-ended dsDNA fragments.
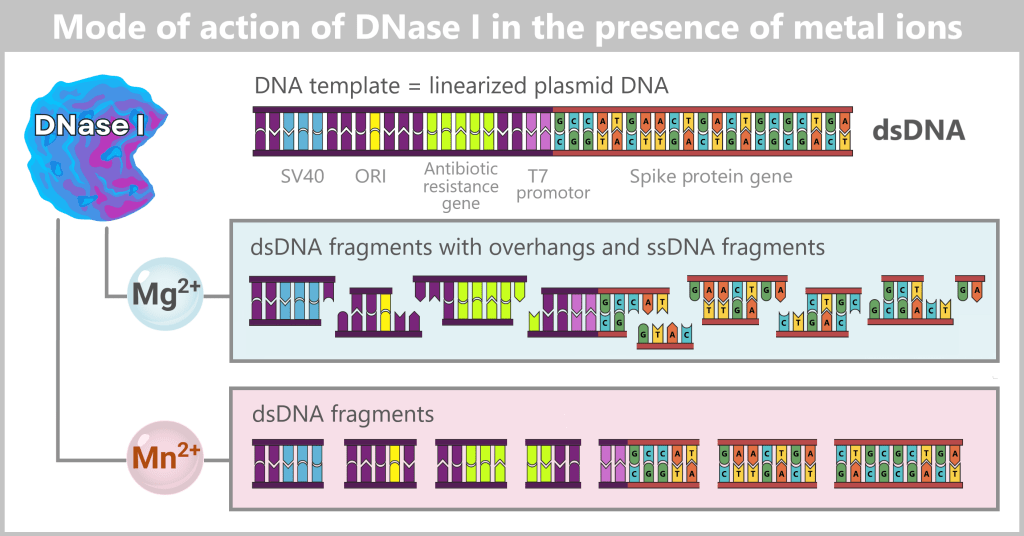
Effectiveness of DNase I
The manufacturer Thermo Fisher Scientific describes the mode of action and limitations of DNase I digestion in the online article „DNase I demystified”. The article highlights that the effectiveness of DNase I strongly depends on the reaction conditions and the type of DNA substrate present.
We are now in the production process after in vitro transcription (IVT). The reaction mixture is a complex mix consisting of:
- the target mRNA (main component),
- RNA by-products (e.g. truncated transcripts, RNA:DNA hybrids),
- the DNA template (in relevant but lower amounts than mRNA),
- rare residual forms of non-linear plasmid DNA (e.g. supercoiled),
- enzymes, free nucleotides, salts, and buffer components.
Like many enzymes, the activity of DNase I is also influenced by the composition of the reaction mixture.
Factors influencing the effectiveness of DNase I digestion
| Factor | Mechanism | Process relevance (mRNA production) |
| Substrate accessibility | DNase I preferentially cleaves accessible phosphodiester bonds; compact or topologically constrained DNA is less accessible. | Supercoiled or strongly constrained DNA structures (e.g. rare plasmid residual forms) are degraded more slowly or incompletely. |
| Substrate saturation | High DNA amounts can saturate the enzyme; high RNA concentrations can influence spatial accessibility of DNA. | In IVT mixtures with high nucleic acid load, an enzyme excess is required. |
| Helix geometry | The active site of DNase I is optimized for B-form DNA (deviations in helix structure reduce efficiency). | RNA:DNA hybrids adopt an A-form-like helix → cleavage activity < 2% compared to dsDNA |
| DNA structure (dsDNA vs ssDNA) | Highest activity on dsDNA; activity on ssDNA is ~500-fold lower | Short or partially single-stranded DNA fragments are degraded less efficiently. |
| Ions in buffer | Mg²⁺ is essential for catalytic activity; Ca²⁺ stabilizes enzyme structure | Therapeutic processes require precisely controlled ion concentrations. |
| Chelators | Chelators (e.g. EGTA, EDTA) bind Ca²⁺/Mg²⁺ and inactivate DNase I. | Residual buffer components from upstream steps can strongly reduce DNase activity. |
| Salt concentration | Increased ionic strength weakens electrostatic binding between enzyme and DNA. | IVT buffer conditions may reduce activity → compensated by higher enzyme amounts. |
The Thermo Fisher Scientific article notes that it is „probably impossible to remove every last DNA strand from an RNA preparation”.
This highlights that the complete elimination of residual DNA is technically challenging. Even after careful DNase I digestion, short DNA fragments or RNA:DNA hybrids may remain.
These small DNA fragments are subsequently removed in downstream process steps – for example through magnetic bead purification (in process 1) or chromatography (in process 2) (cf. EPAR, section 2.2).
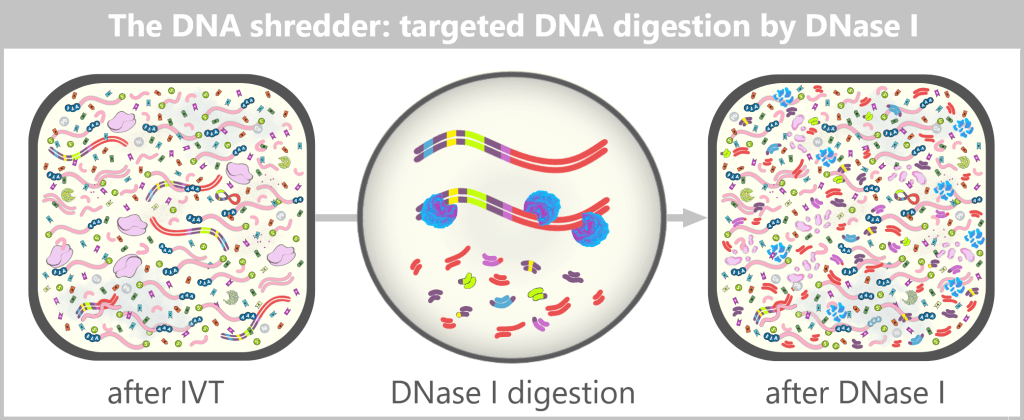
In industrial mRNA production, DNase I digestion is performed immediately after in vitro transcription (IVT), as the reaction mixture already contains a Mg²⁺-based buffer suitable for enzyme activity. The Mg²⁺-dependent activity of DNase I predominantly leads to fragmentation of DNA into short double-stranded DNA fragments with nicks (strand breaks) and short overhangs. (Mn²⁺ is not used in industrial processes due to its less specific and less controllable cleavage activity.)
After completion of the reaction, DNase I is typically inactivated or removed, for example by heat treatment, addition of EDTA, or Proteinase K treatment, to ensure that no enzymatic activity affects the mRNA.
1.3.1. a) Possible post-transcriptional RNA:DNA hybridization
In addition to R-loop structures formed during in vitro transcription, a second potential pathway for the formation of RNA:DNA hybrids is discussed in the recent literature.
DNase I digestion fragments the remaining DNA templates into pieces of varying length – predominantly double-stranded, but in some cases also with single-stranded overhangs or as short single-stranded fragments.
Fragments whose sequence is complementary to the mRNA – in particular those derived from the template strand of the original DNA template – may, under suitable conditions, hybridize with the mRNA and form RNA:DNA duplexes.
For thermodynamically stable hybridization, contiguous complementary sequences of approximately 8–12 nucleotides or more are generally required. Very short overhangs of only a few nucleotides (e.g. 2–4 bases), as typically generated in a near-complete DNase digestion, are not sufficient to form stable binding interactions. This mechanism therefore requires the presence of sufficiently long complementary DNA fragments, such as those arising from incomplete digestion or specific fragmentation patterns.
Regarding enzymatic degradability, it should be considered that DNase I preferentially cleaves double-stranded DNA. RNA:DNA hybrids represent a structurally distinct substrate and are more efficiently degraded by specialized enzymes such as RNase H or DNase I-XT. In practice, this may mean that once formed, RNA:DNA hybrids are relatively more resistant to subsequent DNase I digestion than free DNA fragments. Additional stabilization may occur when such structures are encapsulated within lipid nanoparticles.
This proposed mechanism of post-IVT hybridization has not yet been comprehensively characterized experimentally. It is based on theoretical considerations and isolated experimental indications and should therefore be regarded as a plausible but not yet conclusively demonstrated possibility.
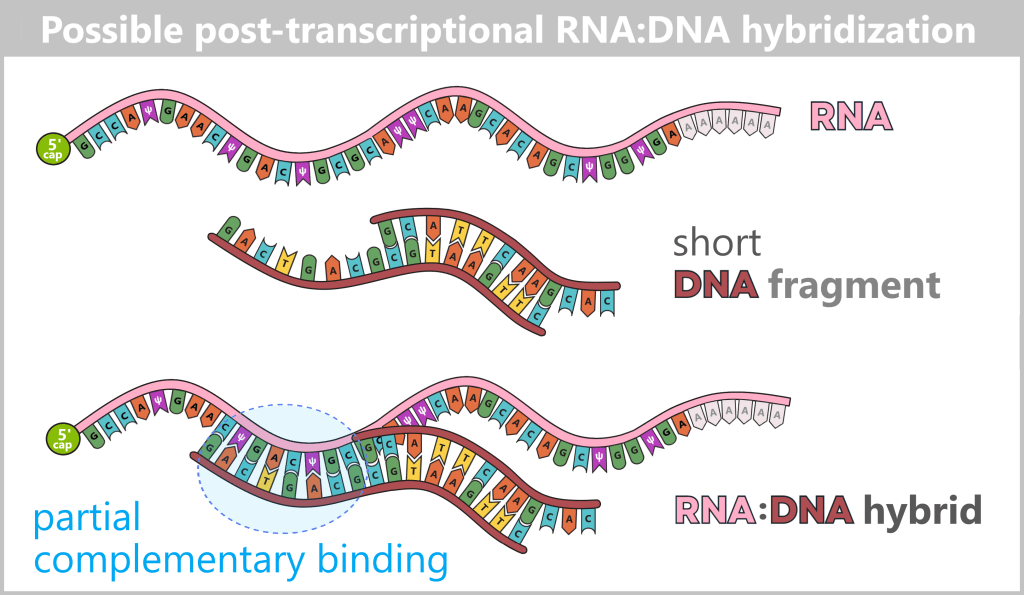
Short DNA fragments may, under suitable conditions, partially bind to complementary regions of the mRNA. In contrast to an R-loop, no three-stranded structure is formed; instead, a locally restricted RNA:DNA duplex arises.
1.3.2. Proteinase K – enzymatic degradation of residual proteins
Proteinase K is a broad-spectrum serine protease used in biotechnology to non-specifically hydrolyze proteins, breaking them down into smaller peptide fragments. It belongs to the class of proteases – enzymes that cleave peptide bonds between amino acids and thereby irreversibly disrupt the three-dimensional structure of proteins.
In mRNA vaccine production, Proteinase K can be applied at different stages of the process, in particular:
After cell lysis in plasmid production: Here, it serves to degrade bacterial proteins, nucleases, and other cellular components that could compromise the purity of the plasmid DNA.
After in vitro transcription (IVT): Following transcription, during which mRNA is synthesized from the DNA template, several enzymes remain in the reaction mixture, including T7 RNA polymerase as well as DNase I from the preceding process step. Proteinase K can be used to selectively hydrolyze these enzymatic residues, thereby further increasing the purity of the mRNA.
Mechanism of action
Proteinase K initially binds to accessible regions of a target protein and begins cleaving peptide bonds at those sites. These initial cuts progressively disrupt the protein’s compact three-dimensional structure. As destabilization increases, previously shielded internal regions become exposed, allowing the protein to gradually break down into increasingly smaller peptide fragments – effectively “falling apart.” At the end of the process, only short, biologically inactive peptide chains remain, which no longer retain enzymatic activity.
Proteinase K attacks DNase I and cleaves its peptide bonds. Initially, outer protein regions are hydrolyzed, leading to structural destabilization. Step by step, the protein breaks down into smaller fragments until only short, biologically inactive peptide fragments remain.
Proteinase K is itself a protein and is not retained in the final product. Its activity can be reduced by process conditions, for example through the removal of stabilizing calcium ions. In practice, however, Proteinase K is primarily removed physically: both the enzyme itself and the resulting peptide fragments are reliably eliminated in subsequent purification steps, particularly through ultrafiltration (e.g. tangential flow filtration) and chromatographic methods.
Relevance in the process
Through the targeted use of Proteinase K, residual process enzymes, bacterial proteins, and other protein contaminants can be effectively reduced. This creates well-defined starting conditions for subsequent filtration and chromatographic purification steps. The use of Proteinase K is not an obligatory component of every manufacturing process, but it increases the robustness and reproducibility of downstream processing, particularly in large-scale industrial production.
1.3.3. Filtration
Filtration is a physical separation process that separates particles or molecules primarily based on their size.
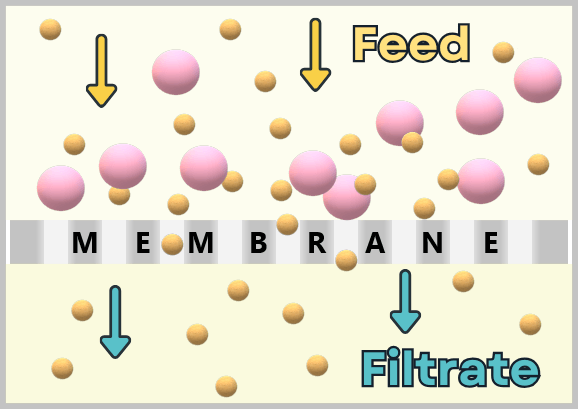
In this process, a mixture is passed through a porous filter material. In its simplest form, the pores act like a mechanical sieve: particles larger than the pore size are retained, while smaller molecules pass through.
a) Filtration – separation by size
b) Tangential flow filtration (TFF)
c) Sterile filtration
a) Filtration – separation by size
In plasmid purification, two fundamental principles are used:
- separation of insoluble particles (clarification), in order to clear a turbid suspension,
- separation of dissolved molecules by size (membrane filtration), for example to exchange buffers or concentrate molecules.
Clarification: Filtration after lysis of E. coli bacterial cells
Starting point: cell lysis
The addition of sodium hydroxide (NaOH) and the detergent SDS disrupts the lipid-rich cell envelope of the bacteria. Alkaline lysis affects not only membranes but also nucleic acids and proteins. The DNA double helix is denatured, and the long, fibrous chromosomal DNA is additionally mechanically fragmented. Under strongly basic conditions, proteins lose their three-dimensional structure and thus their biological function.
Neutralization: system reorganization
Upon addition of an acidic solution, the alkaline reaction mixture is neutralized. Under these conditions, the small circular plasmid DNA molecules can correctly renature and remain in solution. In contrast, fragmented and mispaired chromosomal DNA as well as denatured proteins aggregate together with lipids and membrane components and precipitate as a whitish precipitate.
Intermediate state
After alkaline lysis and neutralization, the sample exists as a two-phase system: a soluble phase containing plasmid DNA and an insoluble precipitate of aggregated components. These aggregates consist of fragmented genomic DNA, denatured proteins, ribosomal components, and membrane debris.
Clarification and depth filtration
At industrial scale, these solids are efficiently removed by depth filtration. In this process, the suspension passes through a multilayer porous filter material. Particles are retained not only on the surface but also within the filter matrix, providing high loading capacity and effective clarification.
Result
The clear filtrate contains the dissolved plasmid DNA, largely free of cell debris and precipitated aggregates. This crudely purified solution forms the basis for subsequent high-resolution chromatographic purification steps.
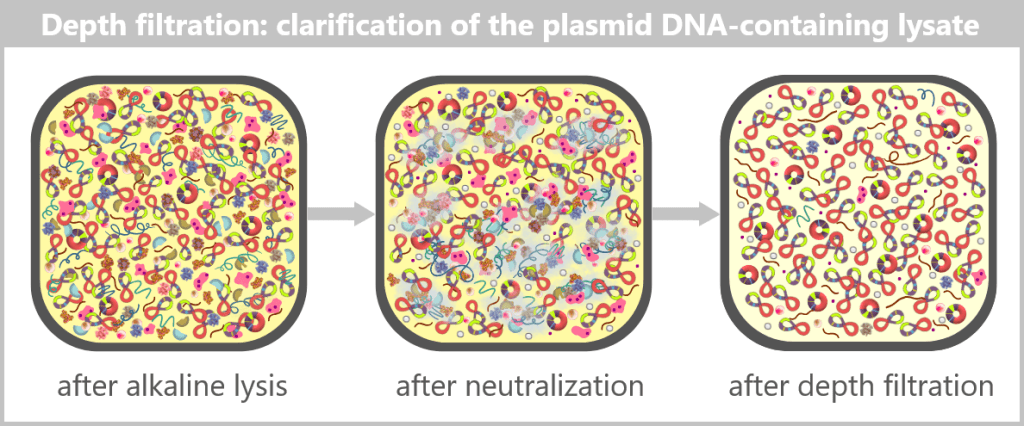
After alkaline lysis, the lysate contains, in addition to the desired plasmid DNA, numerous bacterial components and process-related substances.
During subsequent neutralization, fragmented genomic DNA, denatured proteins, as well as lipids and membrane remnants form irregular, voluminous aggregates (depicted as clouds).
Through depth filtration, these precipitated aggregates are effectively removed. The resulting filtrate is a clear solution containing the dissolved plasmid DNA. In addition, soluble RNA residues, endotoxins (LPS), salts, buffer components, and small amounts of dissolved protein residues may still be present.
Membrane filtration: ultrafiltration and diafiltration
A particularly important variant of membrane filtration in biotechnology is ultrafiltration (UF). It is used to separate dissolved macromolecules (e.g. nucleic acids or proteins) from smaller molecules such as salts, buffers, or nucleotides.
When fresh buffer is continuously added during ultrafiltration, the process is referred to as diafiltration (DF). This allows molecules not only to be concentrated but also to be transferred into a new solution in a controlled manner. UF and DF are often combined and referred to as the UF/DF process.
The predominant technique used to perform UF/DF is tangential flow filtration (TFF). [ROCKER]
✧ ✧ ✧
b) Tangential flow filtration (TFF)
In classical filtration methods (dead-end filtration or direct-flow filtration), the solution flows perpendicular to the membrane, causing large molecules to accumulate on the surface and form a „filter cake” layer. Over time, this reduces flow rate and membrane lifetime.
Tangential flow filtration (TFF) overcomes this limitation: here, the fluid moves tangentially along the membrane surface. A portion of the flow (the permeate) passes through the membrane, while the remaining stream (the retentate) continues to flow parallel to the surface and helps remove deposited material. As a result, the membrane remains permeable for longer periods, and concentration and diafiltration can be performed simultaneously.
Definitions:
Retentate: The fraction retained by the membrane (usually the desired macromolecule).
Permeate: The fraction that passes through the membrane (usually small molecules and impurities).
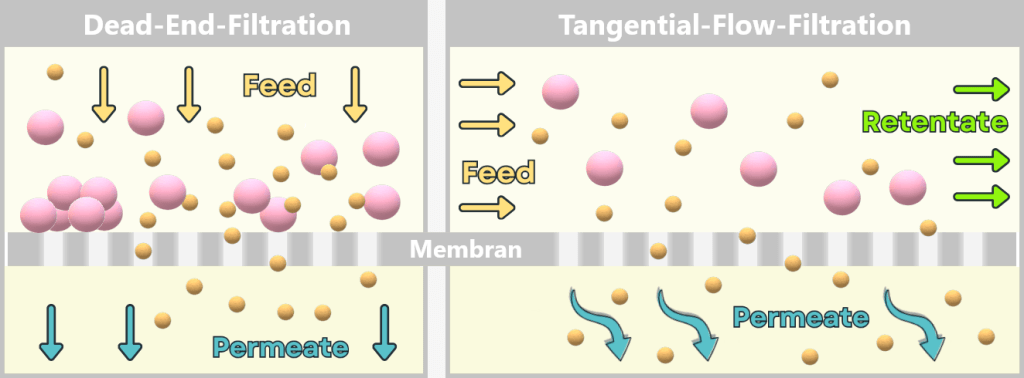
In dead-end filtration, the entire feed stream flows perpendicular to the membrane. Small molecules (permeate) pass through the pores, while larger molecules are retained. Over time, this leads to the formation of a „filter cake”, which reduces efficiency.
In tangential flow filtration, the solution flows parallel to the membrane. While small molecules pass through into the permeate, larger molecules are carried along in the retentate stream. The tangential flow continuously washes the membrane surface, preventing fouling and maintaining stable, efficient filtration over extended periods.
TFF is now a standard technique in biopharmaceutical production because it is robust, scalable, and versatile.
TFF in vaccine manufacturing
Tangential flow filtration (TFF) is used at multiple points during mRNA production: for example after transcription, after enzymatic processing steps, or before and after chromatographic purification steps. Its functions range from removing small molecules to buffer exchange and product concentration.
In this chapter, we focus on a central but not exhaustive purification step:
TFF directly after in vitro transcription (IVT)
Here, it is used for the initial („rough”) purification of freshly synthesized mRNA. It separates the mRNA from enzymes, reagents, and by-products, thereby preparing it for subsequent high-resolution purification steps.
Starting point: the „crude solution” after IVT, DNase I, and Proteinase K treatment
At this stage, the mRNA is present in a reaction-containing solution that still includes a variety of unwanted components:
- enzymes (e.g. T7 polymerase, DNase I),
- unconsumed nucleotides,
- residual DNA from the template,
- buffer salts,
- reaction by-products (e.g. short RNA fragments),
- any residual components from upstream biological processes.
To process the mRNA solution, it is passed through a filtration unit with membranes of defined pore size. Molecules below this cutoff – such as salts, nucleotides, or small protein remnants – pass through the membrane (permeate), while the large mRNA molecules are retained in the retentate.
During the process, fresh buffer is continuously added (diafiltration). This gradually washes out low-molecular-weight impurities while simultaneously transferring the mRNA into the desired buffer and concentrating it (ultrafiltration).
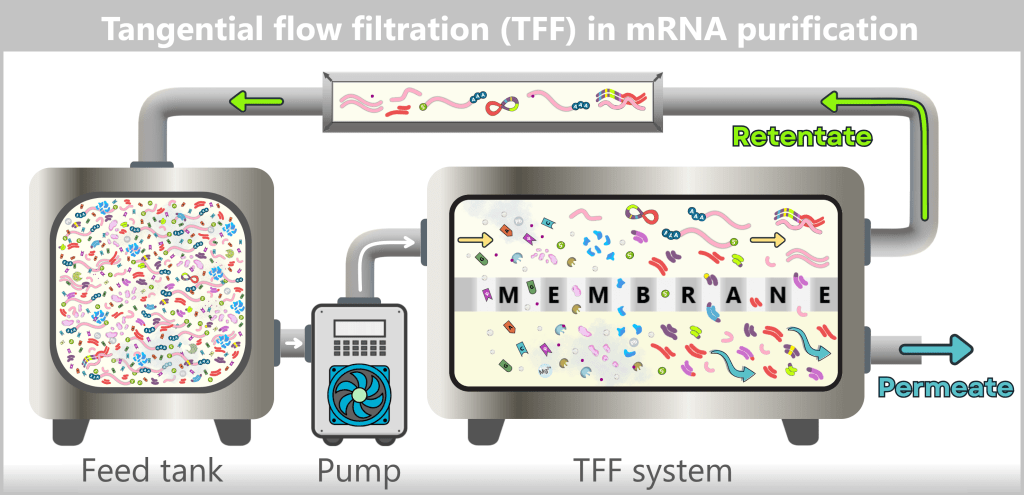
The mRNA solution is continuously circulated through the UF/DF system. Small molecules pass through the membrane and are removed as permeate. A pump ensures that the retentate – the mRNA-containing fraction – is repeatedly recirculated across the membrane. Through repeated cycles and buffer addition, impurities are removed while the mRNA fraction is simultaneously concentrated and buffer-exchanged.
Important to note:
TFF is a coarse separation based on size. Impurities with a size or shape similar to mRNA – such as double-stranded RNA, RNA fragments of comparable length, or RNA:DNA hybrids – cannot be removed in this step. These critical by-products remain in the retentate and must be separated in subsequent high-resolution chromatographic purification steps.
✧ ✧ ✧
c) Sterile filtration
Sterile filtration is a key final step prior to filling an mRNA vaccine. In this process, the finished formulation (lipid nanoparticles containing the mRNA) is passed through a very fine filter. The pore size is sufficiently small to reliably retain all bacteria, fungi, and larger particles.
Principle of sterile filtration
Sterile filtration is a purely size-based sieving process:
- The membrane has precisely defined pores of 0,22 µm.
- Microorganisms are significantly larger and are retained.
- The active substance particles (LNPs) are much smaller (60–100 nm = 0,06–0,1 µm) and pass through the filter without difficulty.
Since no heat or chemical treatment is required, this step is particularly suitable for sensitive biological preparations such as mRNA-LNPs, which cannot be sterilized by autoclaving (steam, pressure, and heat).
Process in detail
The LNP-mRNA solution is present in formulation buffer. Sterile filtration is carried out in a cleanroom environment (Grade A/B) under strictly aseptic conditions. Only sterile single-use components are used: filters, tubing, bags, pump heads, and collection containers.
The formulation is gently driven through the 0,22 µm filter using low pressure (peristaltic pump or pressurized gas).
| Retained by the filter: ▪️Bacteria (~0,5–5 µm) ▪️Yeasts / Fungal spores (~1–10 µm) ▪️Particles or aggregates > 0,22 µm | Pass through the filter: ▪️LNPs (60–100 nm) ▪️Buffer components ▪️Dissolved molecules |
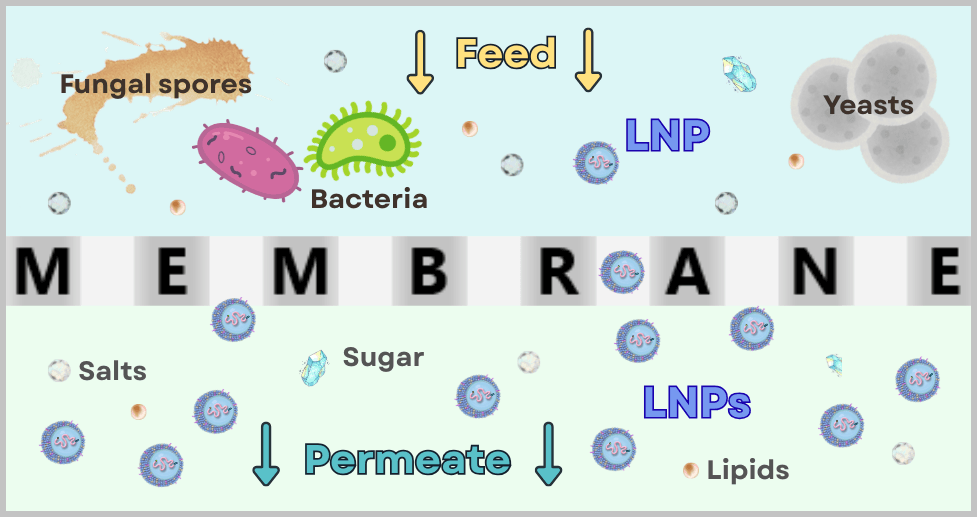
This ensures that the formulation is free of microorganisms and visible particles.
Why sterile filtration is necessary
According to GMP guidelines, sterile medicinal products must be manufactured using validated sterile filtration processes. Sterile filtration is therefore mandatory and ensures that:
- the final vaccine solution is microbiologically sound,
- no large particles, aggregates, or contaminants enter the final product,
- the subsequent sterile filling („aseptic fill & finish”) can be performed correctly.
Sterile filtration is not a chemical purification step, but an essential safety and quality control process. It forms the bridge between the biotechnological purification of the mRNA and the final sterile medicinal product in the vial.
1.3.4. Chromatography – the molecular separation method
Chromatography is one of the most important methods for purifying biological molecules such as DNA, RNA, or proteins. The principle is based on the differing interactions of substances with two phases: a stationary phase (a solid support material within a column) and a mobile phase (a liquid solution that transports the sample mixture).
As the mobile phase – that is, the mixture of molecules to be separated – flows through the stationary phase, the individual molecules are retained to different extents depending on properties such as size, charge, or other chemical characteristics. This leads to temporal separation, in which the components of the mixture elute (are washed out) sequentially.
Put simply, chromatography can be thought of as a „molecular obstacle course”: some molecules interact only weakly with the stationary phase and pass through quickly, while others „stick” more frequently and are therefore delayed. As a result, each component reaches the end of the column at a different time and can be collected separately.
Chromatographic methods in mRNA purification
In industrial processes, such as those described by Merck („Manufacturing strategies for mRNA vaccines”), two chromatographic methods are commonly used in combination:
| Method | Separation principle | Typical application |
| Affinity chromatography (Poly(dT)) | Specific binding | Primary purification: isolation of intact mRNA from the IVT reaction mixture |
| Ion-exchange chromatography | Electrical charge | Fine purification: removal of dsRNA, RNA:DNA hybrids, DNA fragments, and other product-related impurities |
✧ ✧ ✧
a) Poly(dT) affinity chromatography
Poly(dT)-AC = Poly(dT) Affinity Chromatography
Affinity chromatography utilizes specific interactions between biomolecules and a functionalized surface. In the production of mRNA vaccines, poly(dT) affinity chromatography is specifically used to isolate mature mRNA from the complex reaction mixture after in vitro transcription (IVT).
Starting point: After in vitro transcription (IVT), enzymatic post-treatment, and tangential flow filtration (TFF)
Principle and process
Stationary phase: The resin in the column is coated with long single-stranded stretches of thymine (T) bases, known as a poly(dT) matrix.
Mobile phase: The pre-purified IVT solution – consisting of mRNA, RNA by-products (e.g. dsRNA, truncated transcripts), residual DNA fragments, and defined buffer salts – is applied to the column.
The target molecule is mature mRNA, i.e. „polyadenylated mRNA”. It carries a stretch of adenine (A) bases at its 3′ end, known as the poly(A) tail.
Specific binding via base pairing
Adenine (A) and thymine (T) form specific hydrogen bonds, as in DNA. Figuratively speaking, the poly(A) tail of the mRNA binds strongly to the poly(dT) „hooks” on the resin.
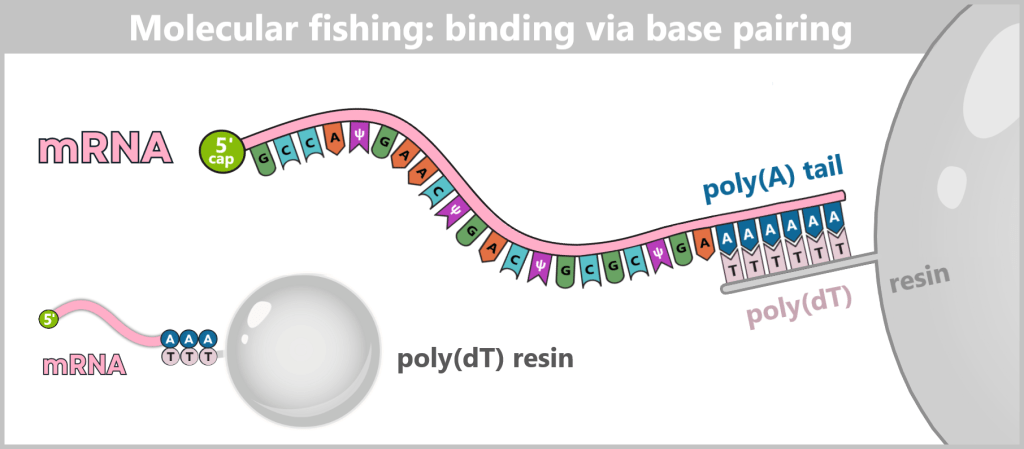
The poly(A) tail of therapeutic mRNA typically consists of about 100–120 adenine bases; in the figure, it is shown in a shortened form for illustrative purposes.
Only molecules with an intact poly(A) tail bind strongly to the resin; all other components of the IVT reaction flow through. These include:
(a) residual free nucleotides (only in trace amounts)
(b) protein fragments from enzymatic degradation
(c) short RNA fragments without a poly(A) tail
(d) DNA fragments and DNA-containing by-products
(e) residual supercoiled plasmid DNA
(f) free dsRNA
(g) buffer salts

Washing
Wash buffers remove weakly bound, non-specifically adhering molecules, while the mRNA remains firmly attached to the poly(dT) matrix.
Elution (release)
To release the purified mRNA from the „bait”, the specific interactions between A and T are deliberately disrupted. This is typically achieved by reducing ionic strength and/or moderately increasing the temperature. The mRNA detaches from the matrix and is eluted.
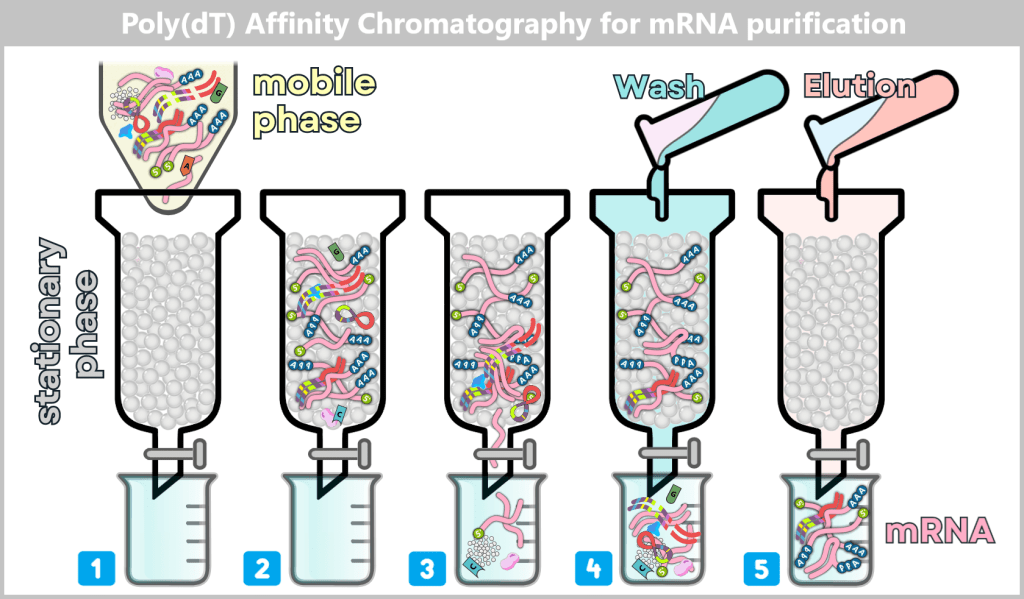
1) The IVT reaction mixture is applied to a column functionalized with poly(dT).
2) Polyadenylated mRNA binds specifically to the resin via base pairing,
3) while non-binding components flow through.
4) A wash buffer removes weakly or non-specifically bound molecules.
5) Addition of an elution buffer releases the mRNA from the poly(dT) matrix, yielding it in purified form.
Advantages
- very high selectivity due to poly(A)/poly(dT) hybridization
- efficient removal of most non-specific RNA and protein impurities
- enrichment of full-length, correctly polyadenylated mRNA
- comparatively simple and robust method
Limitations – why further purification steps are necessary
Despite its high specificity, poly(dT) affinity chromatography cannot remove all critical impurities:
- dsRNA: may bind to the matrix via polyadenylated ends or through non-specific interactions
- RNA:DNA hybrids: if DNA is bound to mRNA, it is co-eluted together with it
- fragmented mRNA with residual poly(A) sequences also binds to the resin
- intramolecular dsRNA structures within mRNA can lead to co-elution
- larger RNA complexes associated with mRNA are co-purified
- endotoxins are not specifically removed and may co-elute
Manufacturers such as MERCK, Lonza, or Cytiva therefore recommend anion-exchange chromatography (AEX) as a subsequent step for fine purification, particularly for the removal of dsRNA and residual DNA contaminants.
✧ ✧ ✧
b) Ion-exchange chromatography
Ion-exchange chromatography (IEX) is a well-established method for purifying nucleic acids and proteins. It separates molecules based on their electrical charge. In nucleic acids, this charge arises from the high density of negatively charged phosphate groups along the molecule. In contrast, the charge of proteins depends on their specific amino acid sequence and folding and can include both positively and negatively charged regions.
Depending on the type of charged target molecules, two main types are distinguished:
▪️Anion exchangers (AEX, Anion Exchange Chromatography):
bind negatively charged molecules such as DNA or RNA.
▪️Cation exchangers (CEX, Cation Exchange Chromatography):
bind positively charged molecules, for example certain proteins.
Due to their continuous phosphate backbone, nucleic acids carry a high and uniform negative charge density.
The sugar-phosphate backbone as the source of the negative charge in DNA/RNA
Schematic representation of the structural basis of the negative charge of nucleic acids
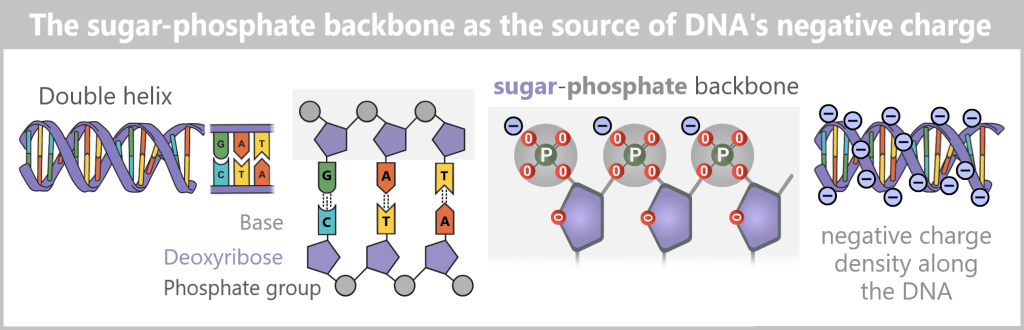
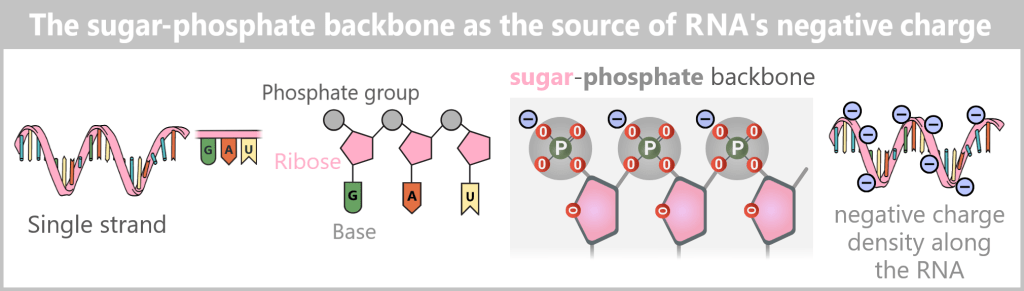
DNA (and analogously RNA) consists of a sugar-phosphate backbone, in which each phosphate group carries a negative charge. These regularly repeating phosphate groups give the molecule a high and uniformly distributed negative charge density along its length. The bases do not contribute to the overall charge.
This property makes anion-exchange chromatography (AEX) the method of choice for their purification. In the overall process, AEX is therefore used twice: first for the purification of the plasmid DNA template from the bacterial lysate. Then, after in vitro transcription (IVT) and initial crude purification, it is used for high-resolution separation of the desired mRNA from structurally similar impurities, in particular double-stranded RNA (dsRNA).
AEX in plasmid DNA purification
Starting point
After lysis of E. coli cells, neutralization, and subsequent depth filtration, a clarified lysate is obtained containing the plasmid DNA. In addition, residual bacterial RNA, endotoxins, salts, buffer components, and small amounts of dissolved protein residues may still be present.

Principle and process
Stationary phase: The support material (resin) consists of small, positively charged polymer beads. These serve as a binding surface for negatively charged molecules.
Mobile phase: The clarified lysate is applied to the column. Initially, negatively charged biomolecules – including plasmid DNA, RNA, and endotoxins – bind to the positively charged resin. Unbound or weakly charged components are removed with wash buffer.
The target molecule is plasmid DNA, which carries a uniformly distributed negative charge along its backbone.
Selective elution: Subsequently, a buffer with increasing salt concentration is passed through the column. The positively charged ions (e.g. Na⁺) in the buffer compete with the biomolecules for binding sites on the resin. As a result, molecules are released in a characteristic order depending on their charge density and binding strength:
- Endotoxins and small RNA fragments elute early, as they have a lower charge density.
- Plasmid DNA – predominantly in supercoiled, but also in relaxed circular form – binds most strongly due to its high negative charge density and therefore elutes only at higher salt concentrations.
Result
This selective elution yields a solution enriched in highly pure plasmid DNA, predominantly in the supercoiled form, with a smaller proportion of relaxed circular forms. Most other cellular components and impurities have already been removed. This DNA fraction subsequently serves as the template for the following in vitro transcription (IVT).
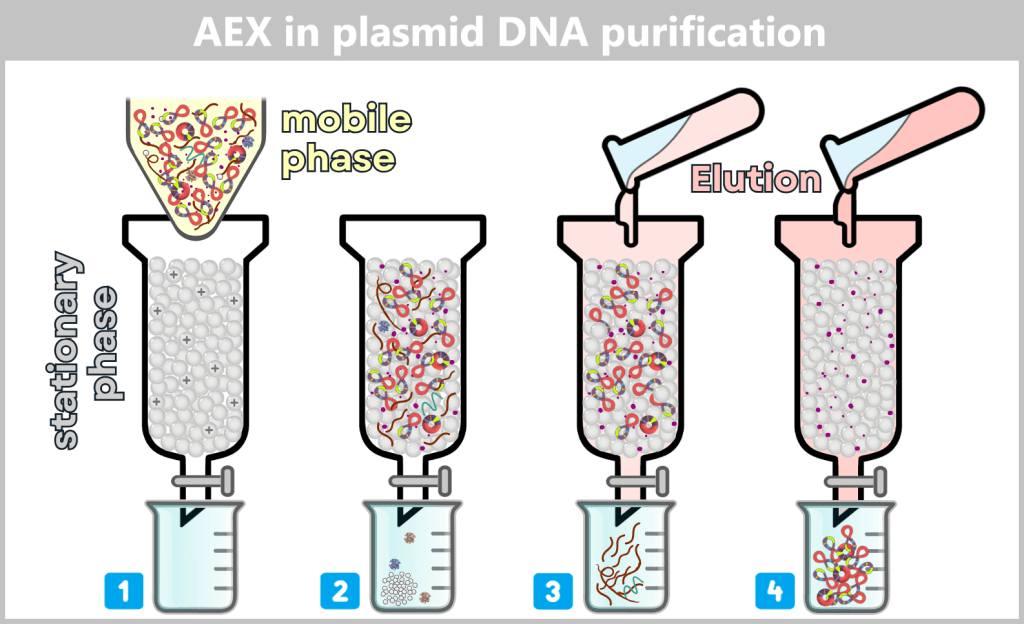
1) The clarified cell lysate (mobile phase) is applied to a column with a positively charged stationary phase (anion-exchange resin). 2) Negatively charged molecules in the sample bind to the resin; non-bound or weakly bound components (salts, protein residues) elute early. 3) Elution begins: a buffer with increasing salt concentration is applied. Dissolved ions (e.g. Na⁺ and Cl⁻) compete with bound molecules for binding sites. At low to moderate salt concentrations, mainly short and/or single-stranded RNA fragments elute. 4) Plasmid DNA elutes only at higher salt concentrations due to its high, uniformly distributed negative charge density. Endotoxins (LPS) bind very strongly to the matrix, which is generally advantageous from a purification perspective. The only problematic fraction consists of components that may co-elute with plasmid DNA due to non-specific interactions or association with DNA.
After anion-exchange chromatography, the plasmid DNA is obtained in high purity. The product consists predominantly of supercoiled plasmid DNA and may contain small proportions of relaxed circular forms. In low concentrations, residual short RNA fragments or endotoxins may also be detectable, while cellular macromolecules and process-related impurities have largely been removed.
AEX in mRNA purification: separation of mRNA and dsRNA
Starting point: After in vitro transcription (IVT), enzymatic post-treatment, tangential flow filtration (TFF), and poly(dT) affinity chromatography
After these upstream process steps, a highly enriched mRNA fraction is obtained. However, this mRNA crude solution typically still contains certain by-products and impurities that cannot be fully removed by poly(dT) affinity chromatography, including:
(a) double-stranded RNA by-products (dsRNA)
(b) RNA:DNA hybrids (in low, process-dependent amounts)
(c) fragmented mRNA with residual poly(A) sequences
(d) incorrectly capped mRNA transcripts
(e) endotoxins (LPS), particularly when associated with nucleic acids

The subsequent anion-exchange chromatography therefore does not serve further enrichment of mRNA, but rather high-resolution removal of structurally similar RNA by-products.
Principle and process
Stationary phase: The stationary phase consists of positively charged anion-exchange resins, typically polymer beads functionalized with quaternary ammonium groups. These provide binding sites for negatively charged nucleic acids.
Mobile phase: The mRNA crude solution is applied to the column as the mobile phase. Under low salt conditions, negatively charged nucleic acids in the sample bind to the resin.
The target molecule is single-stranded mRNA. It carries a negative charge along its backbone but, due to its single-stranded nature, secondary structure, and flexibility, it has a lower effective charge density than double-stranded RNA.
Selective elution
In the next step, an elution buffer with increasing salt concentration is passed through the column. The ions in the buffer (e.g. Na⁺) compete with the bound RNA molecules for binding sites on the resin. As a result, the bound components are eluted stepwise according to their binding strength:
Single-stranded mRNA elutes at low to moderate salt concentrations, as its effective charge density is lower.
Double-stranded RNA (dsRNA) binds significantly more strongly to the anion-exchange resin and therefore elutes only at higher salt concentrations or may remain on the column.
RNA:DNA hybrids represent a particular challenge for chromatographic purification. Their binding properties lie between those of single-stranded RNA (ssRNA) and double-stranded RNA (dsRNA). This hybrid character results not only from the RNA:DNA duplex itself, but is also influenced by three factors: the length of the hybrid region, the proportion of free single-stranded RNA, and the three-dimensional conformation of the molecule.
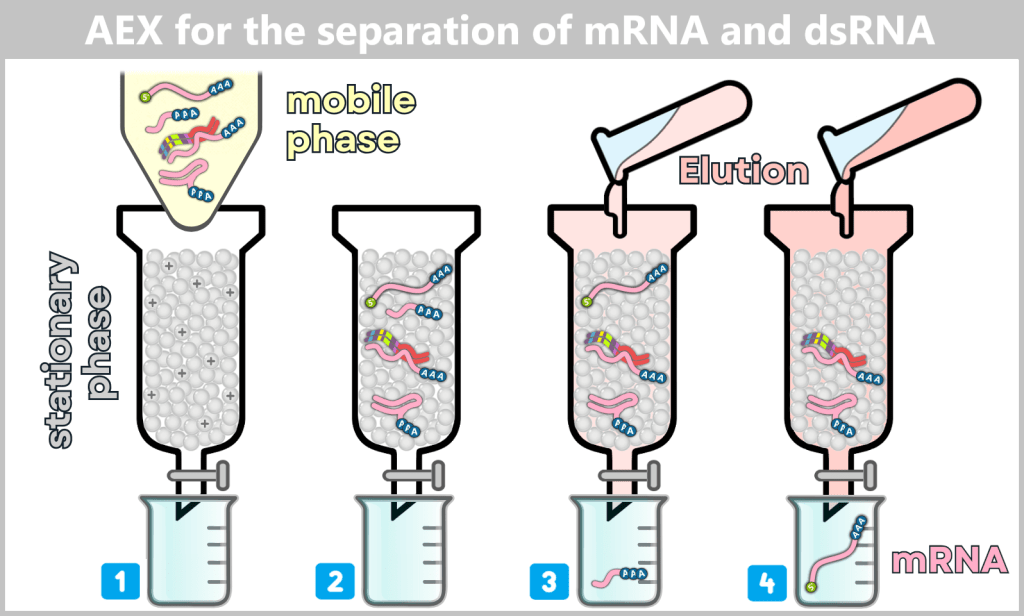
The representation is simplified and shows each component only once as an example.
1) The mRNA-containing solution (mobile phase) – consisting of the desired mRNA, dsRNA, and other by-products – is applied to the column. The stationary phase consists of a positively charged anion-exchange resin. 2) Under low salt conditions, all negatively charged RNA molecules bind to the resin. Due to its higher charge density and more rigid helical structure, dsRNA binds more strongly than flexible single-stranded mRNA. 3) With the addition of a buffer of increasing salt concentration, competing anions (e.g. Cl⁻) are introduced. These gradually displace the bound RNA molecules from the column. First, weakly bound impurities such as short RNA fragments are released. 4) At low to moderate salt concentrations, the desired single-stranded mRNA elutes. Double-stranded RNA remains bound longer due to its stronger interaction and elutes only at higher salt concentrations or may partially remain on the matrix.
Result
Anion-exchange chromatography (AEX) enables the selective separation of mRNA from more strongly or more weakly binding nucleic acid structures such as dsRNA. This yields an mRNA fraction that is largely free of dsRNA contaminants and hybrid structures.
The purified mRNA obtained in this way serves as the starting material for subsequent formulation into lipid nanoparticles (LNPs).
✧ ✧ ✧
Interplay of chromatographic steps in IVT mRNA purification
The purification of IVT mRNA does not rely on a single separation technique, but rather on the deliberate combination of multiple chromatographic principles with complementary strengths and limitations.
Poly(dT) affinity chromatography is used for the highly selective enrichment of polyadenylated mRNA. It efficiently removes non-polyadenylated by-products, enzymes, and low-molecular-weight process components. However, due to its purely sequence-based binding mechanism, it cannot fully discriminate between structurally similar RNA impurities. In particular, dsRNA, fragmented mRNA with residual poly(A) stretches, and RNA:DNA hybrids may co-elute with the target mRNA.
The subsequent anion-exchange chromatography (AEX) addresses this limitation by separating the remaining nucleic acids based on their effective charge density and conformation. Single-stranded mRNA, double-stranded RNA by-products, and complex RNA structures exhibit characteristic, but partially overlapping, binding and elution profiles.
RNA:DNA hybrids occupy a special position in this context. Due to their hybrid structural nature, they cannot be clearly assigned to either single- or double-stranded RNA. Their chromatographic separability strongly depends on the extent of the hybrid region, the proportion of free single-stranded RNA, and the overall spatial organization of the molecule. Accordingly, RNA:DNA hybrids may, depending on process conditions, either co-elute with mRNA or remain bound to the column.
Only the stepwise combination of affinity and ion-exchange chromatography therefore enables the substantial removal of relevant RNA by-products without compromising the structural integrity of the sensitive mRNA. At the same time, this approach highlights that complete elimination of all potential hybrid and structural variants is technically challenging and strongly dependent on process conditions.
1.3.5. Magnetic bead purification
dT-MBP = Poly(dT)-magnetic bead purification
A method described in regulatory documents (EMA EPAR, section 2.2, p. 32) for the purification of PCR products in process 1 is the use of magnetic beads.
Starting point: After in vitro transcription (IVT), DNase I digestion, and filtration
After in vitro transcription (IVT), enzymatic post-treatment with DNase I, and an initial coarse purification step by filtration (UF/DF or TFF), an mRNA-containing solution is obtained. In addition to the desired mRNA, this still contains various transcription by-products, including:
(a) residual free nucleotides (only in trace amounts)
(b) protein residues (e.g. enzymes or protein fragments)
(c) RNA fragments with and without a poly(A) tail
(d) incompletely capped RNA
(e) double-stranded RNA (dsRNA), partially with polyadenylated strands
(f) RNA:DNA hybrids
(g) residual DNA template fragments (especially short fragments)
(h) buffer salts from previous process steps
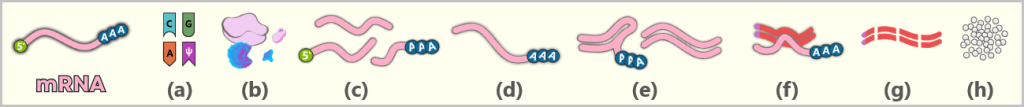
Principle
Magnetic bead purification is based on sequence-specific affinity binding. Magnetic polymer beads are functionalized with oligo(dT) sequences that are complementary to the poly(A) tail of mRNA.
mRNA molecules with a poly(A) tail bind specifically to these oligo(dT) ligands, while molecules without a poly(A) sequence remain in solution.
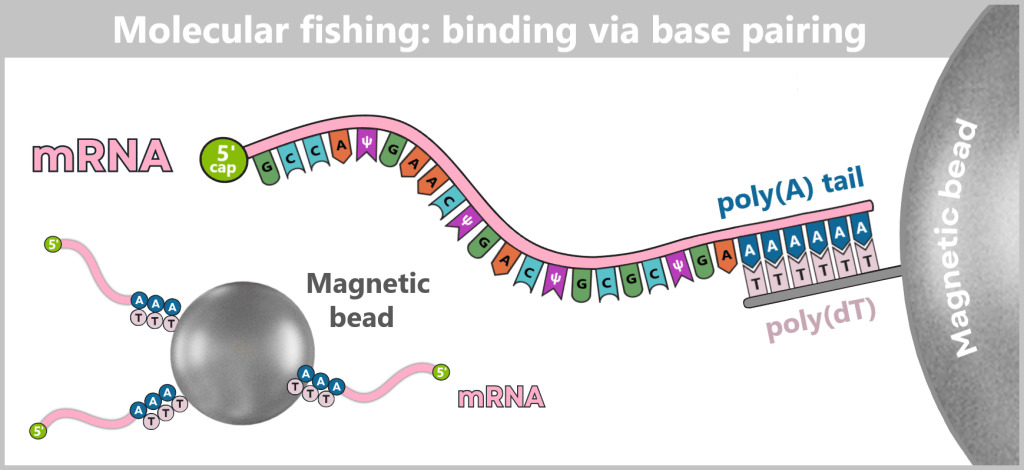
The process consists of several steps:
Binding: Poly(dT)-functionalized magnetic beads are added to the solution. Polyadenylated RNA binds to the bead surface via specific A–T base pairing.
Magnetic separation: The vessel is placed on a magnetic rack. The beads with bound RNA are drawn to the wall of the container, allowing unbound components to be removed.
Washing: Several washing steps reduce non-specifically bound molecules. The washing conditions influence binding stringency but do not enable complete removal of dsRNA or RNA:DNA hybrids if they contain polyadenylated RNA.
Elution: Under suitable elution conditions (e.g. altered salt concentration or temperature), the bound RNA is released from the beads and transferred back into solution.
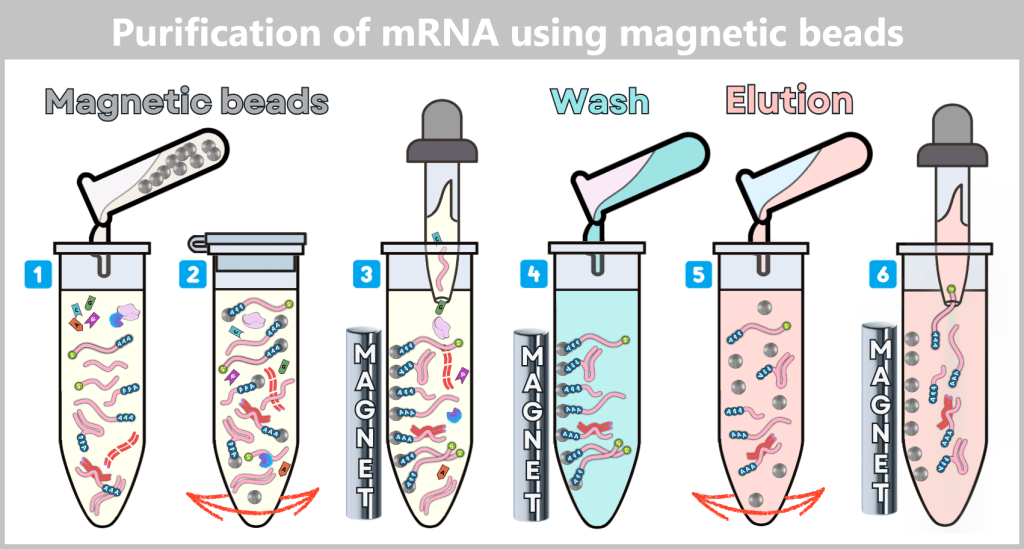
The representation is simplified and shows each component only once as an example.
1) Addition of magnetic beads: Poly(dT)-functionalized magnetic beads are added to the solution containing polyadenylated mRNA.
2) Mixing: The sample is gently mixed by rocking or brief rotation. Polyadenylated RNA hybridizes to the poly(dT) sequences on the magnetic beads via A–T base pairing.
3) Magnetic separation and removal of supernatant: The vessel is placed on a magnetic rack. The beads with bound RNA are pulled to the side of the tube, forming a compact bead aggregate. The liquid supernatant is carefully removed. It contains non-bound molecules such as DNA fragments, short RNA fragments, dsRNA without a poly(A) tail, buffer components, etc.
4) Washing steps: The magnet remains in place. Wash buffer is added to remove loosely bound or non-specifically adsorbed impurities. The sample is briefly mixed (gentle rocking), after which the beads are again attracted to the tube wall by the magnet. This step is repeated 2–3 times until loosely bound or non-specifically associated components are largely removed.
5) mRNA elution: After removal of the final wash buffer, the magnet is briefly removed. An elution buffer (e.g. low-salt water) or slightly elevated temperature disrupts the hybridization between polyadenylated RNA and poly(dT) oligos. The RNA is released back into solution.
6) Return to magnetic separation: The tube is placed on the magnet again, immobilizing the beads at the wall. The supernatant now contains the enriched polyadenylated RNA in elution buffer and is carefully transferred into a new sterile vessel.
Note: dsRNA, RNA:DNA hybrids, or fragmented RNA containing poly(A) regions may co-elute during magnetic bead purification.
Separation performance and limitations
Magnetic bead purification is highly selective for polyadenylated RNA but has inherent limitations:
- dsRNA by-products may co-elute, especially if they contain poly(A) sequences or are structurally associated with mRNA
- RNA:DNA hybrids are co-purified if the RNA component is polyadenylated
- fragmented mRNA retaining a poly(A) tail also binds to the beads
- intramolecular double-stranded structures within mRNA are not selectively removed
For these reasons, magnetic bead purification does not represent a complete purification step but rather an enrichment and pre-purification stage.
Downstream purification
Since dsRNA and RNA:DNA hybrids with poly(A) regions may co-elute during magnetic bead purification, an additional purification step is generally required. This is often achieved using anion-exchange chromatography (AEX) or a comparable high-resolution separation method.
Position in the manufacturing process
Poly(dT)-based magnetic bead purification is mainly used at laboratory scale, in process development, and in smaller production volumes. For large-scale industrial manufacturing processes (process 2), however, it is less practical, as magnetic separation becomes less efficient with increasing volume and handling becomes more complex. Therefore, scalable chromatographic methods are predominantly used in industrial production.
Further details on its application can be found in the manufacturer documentation of Thermo Fisher Scientific.
1.4. Comparison: Process 1 vs. Process 2
The following overview compares the two manufacturing processes, showing which starting materials and by-products arise at each stage and which methods are typically used to remove them.
Typical steps – depending on the manufacturer:
Feature
Process 1 – PCR-based
Process 2 – plasmid-based
Type of process
Laboratory process
(small-scale, flexible)
Industrial process
(large-scale production)
DNA template
Minimalistic.
Contains only the essential elements required for IVT:
• T7 promotor
• 5′ UTR
• Spike gene
• 3′ UTR
• Poly(A) sequence
Complex.
Includes additional elements required for propagation in E. coli:
• Origin of Replication (ORI)
• Antibiotic resistance gene
• bacterial REgulatory sequences
• Multiple Cloning Site (MCS)
• SV40 sequence elements
(for Pfizer/BNT)
Template structure
[T7] – [5′ UTR] – [Spike gene] – [3′ UTR] – [AAAA…]
[ORI] – [Resistance gene] – [bREs] – [MCS] – [SV40] – [T7] – [5′ UTR] – [Spike gene] – [3′ UTR] – [AAAA…]
DNA amplification
PCR-based amplification
Plasmid propagation in E. coli (bioreactor)
Pre-IVT preparation
/
Cell lysis: release of plasmid DNA + bacterial debris
Linearization: restriction enzyme cuts plasmid at a defined site
Impurities before IVT
PCR fragments: enzymes, primers, nucleotides, salts
Bacterial components: host DNA, RNAs, proteins, endotoxins, lipids, cell wall fragments, …
Purification
of PCR DNA
of plasmid DNA (pDNA)
Methods
Silica binding, UF/DF
TFF + multistep chromatography
Effort
moderate
high
IVT (in vitro transcription)
Template: PCR DNA
Expression cassette:
[T7] – [5′ UTR] – [Spike gene] – [3′ UTR] – [AAAA…]
→ mRNA is synthesized
Template: linearized pDNA
Expression cassette:
[T7] – [5′ UTR] – [Spike gene] – [3′ UTR] – [AAAA…]
→ mRNA is synthesized
IVT processing
5′ cap and poly(A) tail are generated either during or immediately after IVT depending on the process
→ functional mRNA obtained
5′ cap and poly(A) tail are generated either during or immediately after IVT depending on the process
→ functional mRNA obtained
Impurities after IVT (*)
Removal of IVT by-products:
template DNA, RNA fragments, RNA:DNA hybrids, double-stranded RNA (dsRNA), enzymes, salts, free nucleotides
Removal of IVT by-products + removal of additional plasmid- and bacterial-derived impurities (more challenging)
Purification
of mRNA
of mRNA
Upstream burden
low – moderate
high
Methods
▪️DNase I digestion
▪️Precipitation or centrifugation
▪️UF/DF or TFF
▪️Magnetic bead purification (**)
▪️DNase I digestion
▪️Proteinase K Treatment (**)
▪️UF/DF and/or TFF (**)
▪️Multistep chromatography
Effort
moderate
high
LNP formulation
mRNA is packaged into lipid nanoparticles
mRNA is packaged into lipid nanoparticles
Sterile filtration
removes microbial contamination risk
removes microbial contamination risk
Filling
aseptic sealing and labeling
aseptic sealing and labeling
Advantages
very fast, highly flexible
highly scalable, cost-efficient mass production
Disadvantages
PCR-based methods are only limitedly suitable for large-scale industrial production volumes
template contains many bacterial elements → significantly higher purification effort
(*) Although the post-IVT reaction mixtures for process 1 and process 2 may appear similar at first glance, their „upstream burden” differs qualitatively. Upstream burden refers to the amount and complexity of accompanying substances present prior to purification. Plasmid-based processes introduce more structurally complex DNA forms, a higher tendency for hybrid formation, and trace bacterial-derived impurities that, even after DNase digestion, make subsequent mRNA purification more demanding.
(**) Information on the manufacturing process according to the EMA „EPAR for the BioNTech/Pfizer COVID-19 vaccine” (Section 2.2, p. 32).
Terminology:
T7 promoter – start signal for transcription
5′ UTR – stabilizes the mRNA
Spike gene – blueprint for the protein
3′ UTR – stop signal and stabilizing element
Poly(A) sequence – protects against premature degradation
UTR stands for untranslated region – a segment of mRNA that is not translated into protein. These regions are located before the coding sequence (5′ UTR) and after it (3′ UTR). They perform important regulatory functions (see step 4).
Origin of replication (ORI) – starting point for plasmid replication
Antibiotic resistance gene – enables selection in E. coli
Regulatory bacterial sequences – control expression of the resistance gene
Multiple cloning site (MCS) – short region containing restriction sites for inserting the target sequence
SV40 sequence elements – regulatory viral sequences used in certain applications in eukaryotic cell culture (in Pfizer/BNT)
1.5. Summary: Production and Purification of modRNA
The production of modified mRNA for therapeutic and prophylactic applications is a multistep process that extends well beyond the actual in vitro transcription. The goal is to obtain from a complex reaction mixture a highly homogeneous, functionally active RNA molecule that is efficiently translated in cells while inducing only a controlled and desired immune response.
From DNA template to mRNA crude solution
The starting point of production is a defined DNA template, which serves as the template for RNA synthesis during in vitro transcription. Even at this early stage, various by-products are inevitably generated alongside the desired mRNA, including truncated transcripts, double-stranded RNA structures, RNA:DNA hybrids, and improperly processed RNA molecules. These by-products are not exceptions but a well-known consequence of enzymatic transcription under technical conditions.
After completion of transcription, the DNA template is typically enzymatically degraded. This is followed by an initial coarse purification step, such as filtration or tangential flow filtration, which removes small molecules, excess reagents, and enzymes. At this stage, a concentrated mRNA-containing solution is obtained; however, it remains structurally heterogeneous.
Enrichment of the target mRNA
To selectively enrich the desired mRNA, poly(A)-based affinity methods are employed. These include both classical poly(dT) chromatography and poly(dT)-functionalized magnetic beads. These approaches use the poly(A) sequence of mRNA as a selection feature and enable efficient separation of non-polyadenylated RNA fragments and many accompanying impurities.
At the same time, a key limitation of these methods becomes apparent: anything carrying a poly(A) sequence or structurally associated with polyadenylated RNA may be co-purified. This includes fragmented mRNA molecules, certain dsRNA by-products, and RNA:DNA hybrids, provided that the RNA component is polyadenylated. Poly(A)-based methods are therefore highly selective enrichment techniques, but not complete purification methods.
High-resolution separation by chromatography
To further reduce remaining structural heterogeneity, high-resolution separation techniques are applied, in particular anion-exchange chromatography (AEX). This method exploits differences in effective charge density and conformation of nucleic acids to separate single-stranded mRNA from more strongly bound by-products such as dsRNA.
AEX represents a critical step for the targeted depletion of immunologically active impurities. At the same time, it also highlights that certain molecular classes – especially RNA:DNA hybrids – do not exhibit uniform chromatographic behavior and may elute differently depending on their structure. Their complete removal therefore requires careful process design and, where necessary, the combination of multiple purification steps.
Classification and outlook
Overall, the production of modified mRNA is not a linear „input–output” process, but rather a finely tuned interplay of enzymatic, physical, and chromatographic methods. Each purification step reduces sample complexity, but none achieves absolute separation of all theoretically possible by-products.
Against this background, it is scientifically more appropriate to consider not only the final product itself, but also the logic of the manufacturing and purification process when discussing potential residual components such as DNA fragments or RNA-associated impurities. The key question is therefore less whether such residual components can arise in principle, but rather in which forms, at what levels, and with what biological relevance they may remain in the final product.
In view of the described manufacturing and purification processes, a central question emerges: how remaining nucleic acid components – particularly potential residual DNA – can be reliably detected and quantified.
The second part therefore addresses analytical detection methods, regulatory thresholds, and published experimental data on residual DNA in mRNA-based vaccines.
Further part of the article series
Part 2: Detection Methods and the Current State of Research
Current as of June 2026.

{kind=link}