-
Personalized Medicine – Heaven or Hell

When HSBC Bank placed posters at airports such as Gatwick and Dubai in 2013 as part of its global campaign „The future is full of opportunity” – showing a fingerprint, a QR code, and the provocative slogan „Your DNA will be your data” – the message was unambiguous: the future will be personal, perhaps unsettlingly personal. The bank sought to position itself as a forward-looking company that addresses digitalization, artificial intelligence, sustainability, and genetics head-on.
The slogan seemed ambiguous – both a promising vision and an alarming prospect. Critics immediately sounded the alarm: your DNA becomes readable, analyzable, and digitally accessible – an open book about ancestry, health risks, perhaps even personality traits – comparable to biometric data such as fingerprints or facial scans. What was once considered the most intimate aspect of a person is now becoming a file: machine-decipherable, potentially storable, possibly tradable. The human being is reduced to information – „bits and bytes” that could be processed, sold, or misused.
Who owns the rights to your genetic data? And who has access to it? You yourself? The company that conducted the test? Do surveillance, data leaks, and misuse by state and private actors reach a new dimension as a result? The vision of designer babies, genetic selection, and personalized advertising based on biological markers raises serious ethical questions. Will progress ultimately be paid for at the price of individual freedom?
However, the biotech revolution has long since gained tremendous momentum, and more and more voices recognize the enormous potential of this development. The March 2024 e-book edition of HSBC Bank Frontiers – Biotech – Five Big Ideas Shaping the Biotech Revolution states:
„The idea of exploiting nature for goods and services is nothing new. But the practice entered its modern era in the 1970s, with the advent of genetic engineering and the establishment of the first biotech companies. This brought together science and business like never before, and the influx of capital galvanized research and development efforts.
Over the three decades that followed, the field was accelerated by breakthrough devices such as DNA sequencers and synthesizers, and biology itself slowly morphed into a computational, data-driven discipline. Following the turn of the millennium, as researchers successfully charted ever greater regions of the human genome, and the cost of sequencing that genome rapidly declined – from around $100 million in 2001 to less than $1,000 today – the potential of biotech to transform our lives has become electrifyingly apparent.
Over the past few years, the possibilities unlocked by biotech advances – from treating genetic diseases to de-extincting animal species – have been enlarged by progress in complementary fields such as artificial intelligence. This has underpinned a coming-of-age moment in this space.“
Nowhere else do benefits and risks collide as directly as in personalized – or precision-based – medicine. Its goal is to use individual genetic and biological information to refine diagnoses, personalize therapies, and detect diseases at an early stage. The vision is promising: fewer side effects, better chances of recovery, more effective prevention – medicine tailored to the individual.
But here, too, what is celebrated as medical progress brings societal, legal, and ethical challenges:
- Who decides which genetic data are relevant?
- What role does access to these technologies play – does it exacerbate social inequalities?
- And how can discrimination, for example through genetic scoring by insurance companies or employers, be prevented?
Between the hope for individual healing and the fear of collective disempowerment lie fundamental questions: Does access to one’s biological data empower the individual – or expose them? Is personalized medicine a blessing – or the beginning of an era of subtle dehumanization?
Hence the crucial question is: Is personalized medicine the promised ascent into heaven – or the plunge through the gates of hell?
This blog post invites readers to consider personalized medicine in all its complexity – beyond hype or alarmism. It is deliberately aimed as well at non-experts who wish to engage critically with the opportunities, risks, and ethical tensions of this medical revolution.
📑Table of Contents
1. Conceptual Classification and the Fundamental Paradigm
2. Driving Forces – Between Vision and Reality
3. The Future of Medicine Begins in the Cell
3.1. How Biology and Technology Come Together
3.2. Data, Responsibility, and the Bigger Picture
4. From DNA to Knowledge – A High-Speed Train of Innovation
4.1. DNA – The Blueprint of Life
a) DNA: The Molecular Information Archive of Life
b) From Code to Protein: How Instructions Become Reality
c) Epigenetics & Non-coding DNA: The Conducting Team of Our Genome
d) The Hidden Control Center: What Non-coding DNA Regulates
e) When the Operating System Makes You Sick
f) Open Mysteries in Research
g) The True Magic of Genes
4.2. Genetic Variation and Its Consequences
4.3. Genome Sequencing: Key to Individual Genetic Material
a) The Journey of DNA: From Umbilical Cord Blood to Genetic Information
b) Three Directives for Gene Diagnostics: WGS, WES, and Panel
4.4. Methods of Genome Sequencing
4.4.1. Method 1: Copying with Fluorescent Dye
a) Sanger Sequencing
b) Illumina Technology
c) Single-Molecule Real-Time (SMRT) Sequencing
4.4.2. Method 2: High-Tech Tunnel – Electrically Scanning DNA
a) Oxford Nanopore Sequencing
4.4.3. Genome Sequencing – Technologies Compared
a) Throughput vs. Usability – Quantity Does Not Always Equal Quality
b) Cost-Effectiveness – Cost per Base vs. Cost per Insight
c) From Sample to Result – How Quickly Does the Whole Genome Speak?
d) Clinical Applications – Which Technology Fits Which Purpose?
e) AI as Co-Pilot – Automation Without Relinquishing Responsibility
f) Comparison Table: Sanger, Illumina, PacBio, ONT
g) Key Takeaways: Between Maturity and Routine
4.5. Genome Sequencing: The Next Technological Leap
a) FENT: How a Microchip Is Revolutionizing DNA Analysis
b) SBX: When DNA Stretches to Be Understood
c) The G4X System: From the Genetic Recipe to the Spatial Atlas of the Cell
4.6. From Code to Cure – Bioinformatics as the Key to Tomorrow’s Medicine
4.6.1. From the Genetic Code to Computer-Aided Genome Analysis
4.6.2. Modern Bioinformatics – The Digital Toolbox of Biology
5. A Success Story
6. A Look into the Future: The Synthetic Human Genome Project
7. Personalized Medicine and Smart Governance
7.1. Power, Governance, and Smart Governance
7.2. Governmentality
7.3. Personalized Medicine as a Catalyst for Biomolecular Governmentality
7.4. The Global Trend: Worldwide Biomolecular Governmentality
7.5. Conclusion: The Global Ambivalence of Biomolecular Power
8. Epilogue
1. Conceptual Classification and the Fundamental Paradigm
The development of modern medicine toward tailored treatments is often described using terms such as „personalized medicine”, „individualized therapy” or „precision medicine”. But what exactly do these buzzwords mean?
Personalized medicine is currently considered one of the most popular terms – catchy like a marketing slogan. It carries the promise of tailoring therapies to each individual: to their genes, their lifestyle, their biology. Accordingly, the term is frequently used in the media and public debate.
Individualized therapy sounds less glamorous but conveys a similar idea: the adjustment of medications or dosages to the needs of individual patients. This term is used primarily in clinical practice.
Finally, precision medicine is the most sober of the three terms – a technical term that dominates research laboratories and scientific publications. It emphasizes the data-driven approach: algorithms, genetic analyses, and biomarkers determine which therapy works for which patient group. This is the language of science.
But regardless of which term is used, they all represent a paradigm shift: moving away from standardized procedures toward medicine that focuses on the biological uniqueness of each individual.
2. Driving Forces –Between Vision and Reality
The driving forces behind personalized medicine are diverse and closely interconnected. On one hand, there are scientific and technological developments that have enabled enormous progress in recent decades: new methods to read genetic material quickly and accurately, powerful data analysis through artificial intelligence, and the availability of large biomedical datasets. These technologies form the foundation for recognizing individual differences between patients and leveraging them medically.
On the other hand, social and political impulses act as accelerators of this transformation. Public health programs, targeted research funding, strategy papers from international health organizations, and specific legislative initiatives actively drive the integration of personalized medicine into healthcare practice. Increased health awareness among the population, as well as growing expectations for individualized treatment options, further reinforce this trend.
Personalized medicine is therefore not merely a product of technological feasibility, but emerges at the intersection of innovation, politics, and societal demand.
A current example of the practical implementation of this development comes from the United Kingdom: under a new 10-year plan, every newborn is to undergo a DNA test. The so-called whole-genome sequencing will screen for hundreds of genetically linked diseases – with the aim of detecting potentially life-threatening conditions early and preventing them in a targeted manner. According to British Health Secretary Wes Streeting, the goal is to fundamentally transform the National Health Service (NHS): moving away from mere disease treatment toward a system of prediction and prevention. Genomics – the comprehensive analysis of genetic information – will serve, in combination with artificial intelligence, as an early warning system.
„The revolution in medical science means that we can transform the NHS over the coming decade, from a service which diagnoses and treats ill health to one that predicts and prevents it“, said Streeting. The vision: personalized healthcare that identifies risks before symptoms even appear – thereby improving quality of life and alleviating pressure on the healthcare system.
But as convincing as this vision may sound, there are also cautionary voices coming from the scientific community. For example, geneticist Prof. Robin Lovell-Badge from the Francis Crick Institute warns against underestimating the complexity of genomic data. Not only is technical expertise required for data collection, but above all qualified personnel who can communicate the information obtained in a responsible and comprehensible manner. Data alone does not constitute a diagnosis – what is crucial is its meaningful and patient-oriented interpretation.
What does it concretely mean to rethink medicine?
The real revolution is taking place in laboratories and data centers, where the building blocks of personalized medicine are being developed. The following chapters take you behind the scenes, step by step, and reveal the key technologies needed to bring personalized medicine from concept to clinical practice.
3. The Future of Medicine Begins in the Cell
3.1. How Biology and Technology Come Together
Our bodies consist of billions of cells – tiny, specialised units that work together day after day to enable health and life. In every single cell, countless processes occur that are precisely coordinated with one another. These cellular mechanisms are crucial for understanding health, disease, and the development of new, targeted therapies.
One central mechanism is energy production: cells convert nutrients and oxygen into ATP (adenosine triphosphate) – the body’s „energy currency”. Without ATP, no cell could function, no muscle could move, no thought could arise.
At the same time, cells continuously produce proteins – based on genetic information. These complex molecules perform essential tasks in the body, serving as chemical catalysts, signaling molecules, structural components, or transporters.
Cellular cleaning and renewal are also vital: processes such as autophagy break down and recycle damaged cellular components – an internal waste disposal system that can help prevent diseases like Alzheimer’s or Parkinson’s.
For cells to work together in a coordinated way within the body, they need functioning signaling pathways – they „communicate” with one another through chemical messengers. This cell communication controls when cells should grow, rest, or respond to changes.
Another key process is cell division – essential for growth, wound healing, and tissue renewal. At the same time, DNA repair mechanisms ensure that errors in the genetic material are corrected. If this fails, apoptosis, the programmed cell death, takes over to limit potential damage.
All these mechanisms run in the background – constantly and with high precision. However, if one of these processes is disrupted, it can lead to serious diseases such as cancer, autoimmune diseases or metabolic disorders. This is exactly where personalized medicine comes in. It uses state-of-the-art technologies to better understand these cellular processes and influence them in a targeted manner.
Thanks to genome sequencing (for example through „next-generation sequencing”), it is possible to identify genetic alterations that influence protein production or repair mechanisms.
Biomarker analyses (the examination of specific molecules) in blood, tissue, or other bodily fluids reveal whether certain signaling pathways are over- or underactive – and help predict individual disease progressions or select appropriate therapies.
Single-cell analysis makes it possible to visualize differences even between individual cells – for example, in a tumor, where some cells respond to therapy and others do not. This enables more precise treatment.
Proteomics (the analysis of all proteins in a cell) and metabolomics (the analysis of metabolic products) also provide a current picture of how active specific cellular mechanisms really are – for example, whether a cell is stressed, has sufficient energy, or is in the process of dividing.
Artificial intelligence (AI) helps analyze these vast amounts of data and identify patterns – for example, which combination of cellular disorders is typical for a particular type of cancer. Digital health data (such as from wearables) complement this picture in daily life and enable precise long-term monitoring.
Even new therapeutic approaches are based on understanding cellular processes: gene therapies and CRISPR-based gene editing target the DNA to correct faulty mechanisms. In organoids – mini-organ models in the lab – drugs can be tested directly on patient-specific cell models, without any risk to humans.
Modern technologies now reveal what was previously hidden: how cells work, what causes imbalances in disease – and how to intervene in a targeted manner. This has given rise to individualized medicine that is no longer based on conjecture, but on measurable biology.
This new precision brings not only opportunities, but also new responsibilities.
3.2. Data, Responsibility, and the Bigger Picture
The complexity of these cellular processes is directly reflected in the complexity of personalized medicine: the more precisely we understand the intricate processes and interactions in our cells, the more targeted – but also more demanding – diagnosis and therapy become. This new precision requires new technologies, new ways of thinking, and a deep biological understanding. At the same time, it opens the door to more effective, sustainable, and tailored treatment concepts.
However, the more targeted and profound these interventions become, the greater the responsibility – and the challenge of identifying and controlling undesirable long-term effects or side effects at an early stage. Particularly in the case of innovative procedures such as gene therapy or immunomodulation, it is important to carefully weigh up the benefits against the potential risks. Personalized medicine therefore requires not only precision, but also foresight.
A central element of this foresight is the availability of personal health data – not only in individual cases, but on a large scale.
Only by combining individual data with population data is it possible to understand biological processes in detail, predict disease progression and develop personalised therapies. Genetic information, laboratory values, imaging data and digital everyday measurements provide clues as to how cellular mechanisms work in a specific person – and how they vary in others.
To interpret these individual patterns medically, large datasets are needed for comparison. Only in this way can AI systems detect complex relationships that escape the human eye – such as rare genetic variants, molecular risk constellations, or unexpected therapy effects. The rule here is: the larger and more diverse the data base, the more reliable the models – especially when they are continuously enriched with new information.
However, this progress can only be achieved if people are willing to share their health data – and can trust that it will be used securely and responsibly.
The future of medicine begins where all life originates: in the cell. But its success depends on the interplay of three forces: technological innovation, scientific depth – and ethical responsibility.

4. From DNA to Knowledge – A High-Speed Train of Innovation
How genome sequencing and bioinformatic analysis are revolutionizing medicine
4.1. DNA – The Blueprint of Life
a) DNA: The Molecular Information Archive of Life
b) From Code to Protein: How Instructions Become Reality
c) Epigenetics & Non-coding DNA: The Conducting Team of Our Genome
d) The Hidden Control Center: What Non-coding DNA Regulates
e) When the Operating System Makes You Sick
f) Open Mysteries in Research
g) The True Magic of Genes
4.2. Genetic Variation and Its Consequences
4.3. Genome Sequencing: Key to Individual Genetic Material
a) The Journey of DNA: From Umbilical Cord Blood to Genetic Information
b) Three Directives for Gene Diagnostics: WGS, WES, and Panel
4.4. Methods of Genome Sequencing
4.4.1. Method 1: Copying with Fluorescent Dye
4.4.2. Method 2: High-Tech Tunnel – Electrically Scanning DNA
4.4.3. Genome Sequencing – Technologies Compared
4.5. Genome Sequencing: The Next Technological Leap
4.6. From Code to Cure – Bioinformatics as the Key to Tomorrow’s MedicineImagine standing on a train platform and watching a train rush by at such speed that you can barely make out the image in the window. This is how many people perceive developments in personalized medicine: technologies are advancing at breakneck speed, terms seem abstract – and yet there is a common thread running through it all: the human genome.
As diverse as modern approaches may be – from organoids and CRISPR-based gene editing to RNA-based medicines – at the center lies the human genetic blueprint. Today, genome sequencing forms the foundation of many diagnoses and therapeutic decisions. It reveals which genetic alterations trigger diseases, which signaling pathways are affected – and where targeted interventions might be possible.
What once cost billions and took years can now be done in a matter of days: the entire human genome can be decoded. But the sequence alone does not constitute a diagnosis. The sequence of three billion base pairs only has medical value once it is understood – and this is where the real challenge begins.
Bioinformatics enters the stage: an interdisciplinary field that combines mathematics, computer science and biology to derive patterns, risks and treatment options from raw data. It is the real bottleneck of personalized medicine – because it determines whether genetic variants are considered harmless, risky or disease-causing.
Individual sequences are compared with international reference databases, algorithms analyze millions of genetic variants, and learning systems model the complex interplay of genetics, environment, and metabolism.
This process is highly complex – and at the same time essential. Only computer-assisted evaluation makes DNA medically usable. There is an inseparable symbiosis between genome sequencing and bioinformatic analysis: the sequence provides the data, the analysis provides the understanding. Without it, DNA remains a mere string of letters.
This is precisely why the following sections focus on the crucial question:
What does it mean to decode the genome – and to truly understand it?
What conclusions are possible – and where are their limits?For anyone who wants to understand the future of medicine, it is essential to know how we read the human genome today – and how we are learning to interpret it.
The high-speed train of innovation has long since departed. Anyone who wants to ride along must understand where it is headed.
4.1. DNA – The Blueprint of Life
To embark on this journey, it is worth taking a brief look at the molecular foundations: how our DNA becomes a blueprint, how proteins are formed from it via the intermediate step of RNA, and how epigenetics fine-tunes these processes. Only this foundation makes it possible to understand what is actually happening when we decode the genome.
a) DNA: The Molecular Information Archive of Life
In every cell of our body lies a carefully packaged and astonishing molecule: DNA (deoxyribonucleic acid). It contains the complete blueprint and operating instructions of our body – a molecular archive of life’s information.
Fig. 1: DNA contains genetic information [The Wonderful World of Life] Structurally, it resembles a spiral-shaped rope ladder: two sugar-phosphate chains form the sides, with the „rungs“ – the bases adenine (A), cytosine (C), guanine (G) and thymine (T) – in between. They always pair up according to fixed rules: A with T, C with G. This complementary bond makes each strand a template for the other – a molecular backup system.
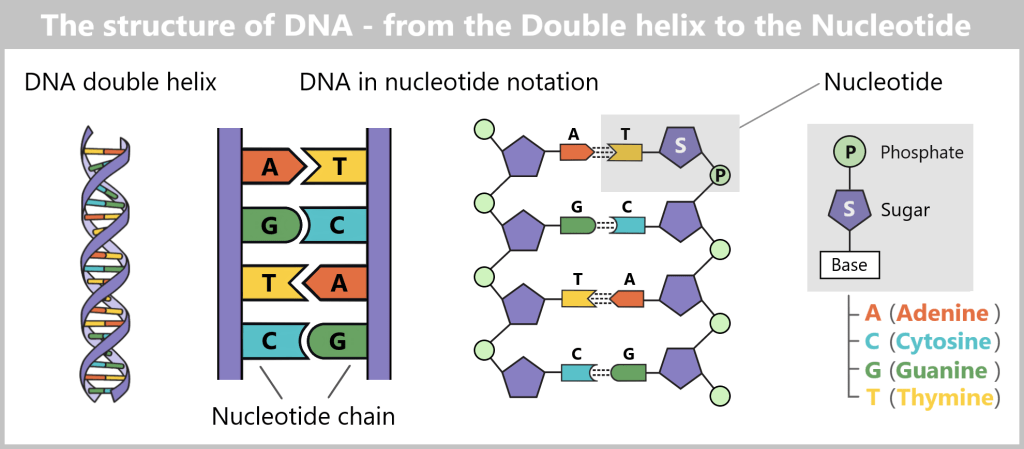
Fig. 2: Schematic representation of the basic structure of DNA [The Wonderful World of Life] If you unravel the DNA from a single cell, it measures around two meters – with a diameter of just a few millionths of a millimeter. Life therefore literally hangs on a wafer-thin but astonishingly long thread.
The sequence of around three billion base pairs is as unique as a fingerprint. It forms the genetic code – the instructions for producing proteins, the molecules that control the structure, function and regulation of all biological processes.
As a central information archive, DNA is both extremely valuable and sensitive. That is why it is stored safely in the cell nucleus, comparable to a safe. In order for the body to access the information in this safe, it needs a clever transport system. After all, the blueprints for proteins – the molecular architects of all life processes – must reach the ribosomes, the so-called „protein factories“ in the cytoplasm. This is where proteins are produced – a process known as protein biosynthesis.
But how does a specific blueprint travel safely from the cell nucleus to these factories? In other words: How is the genetic information for a particular protein transferred from its protected archival location to the site of production?
b) From Code to Protein: How Instructions Become Reality
The journey begins with the genes – specific sections of DNA that contain the blueprints for proteins. There are around 20.000 such genes in the human genome.
DNA is not just a passive information archive in the cell nucleus – it contains an active repertoire of instructions. You can think of each gene as a chapter in a molecular cookbook: often with multiple versions of a recipe, adapted to the cell type, developmental stage, or external conditions.
First, a temporary „working copy” of a gene is created – in the form of RNA (ribonucleic acid). It resembles DNA but uses the base uracil (U) instead of thymine (T) and usually consists of a single strand. This transient structure makes RNA ideal for short-term use – it is degraded after fulfilling its function.
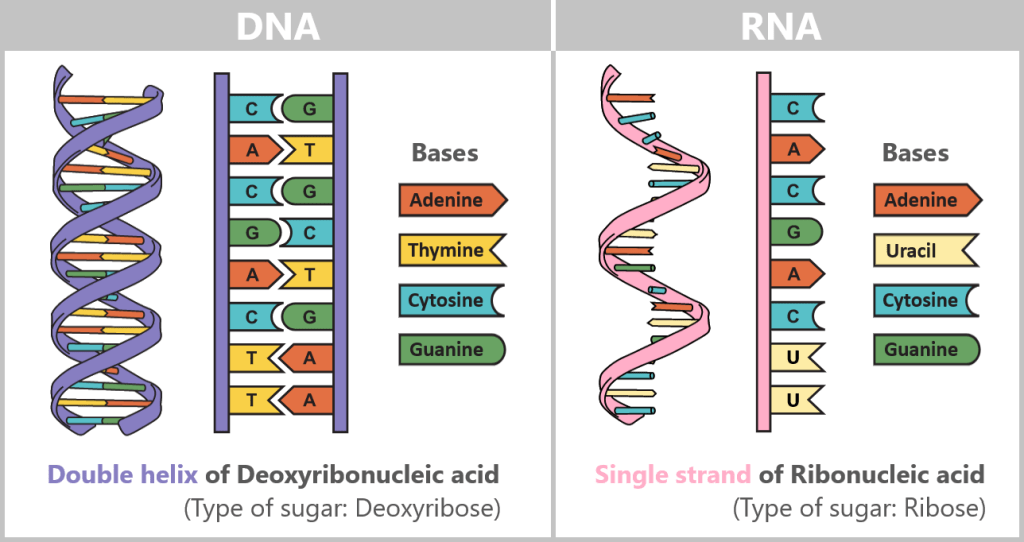
Fig. 3: A working copy of DNA is created – the RNA [The Wonderful World of Life] Alternative Splicing: The Flexible Kitchen of Genes
Cells must adapt flexibly to changing demands: responding to inflammation, defending against pathogens, building muscle, or regulating blood sugar levels. This requires not a rigid set of instructions, but a system capable of improvising depending on the situation.
Depending on the cell type and external signals, the RNA strand is dynamically modified by being cut at specific markers – transferred from the DNA. Unneeded sections (introns) are removed, and the remaining sections (exons) are reassembled. This process creates a multitude of possible messages from a single template. The result is messenger RNA (mRNA), which carries the genetic information from the nucleus to the ribosomes in the cytoplasm – the cell’s „protein factories”.
You can think of this process like following a recipe: depending on the need, the cell leaves out certain ingredients, adds spices, or adjusts the preparation steps. In this way, a single basic instruction can yield a wide variety of dishes.
Through this process – called alternative splicing – cells can produce a variety of different proteins from a single gene, each performing specific functions.
The entire process „from gene to protein” is often referred to as gene expression.
💡Example: The Titin Gene
👉 Longest human gene (≈ 300,000 base pairs).
👉 Through alternative splicing, more than 20,000 different protein variants are produced – each adapted to the specific requirements of different muscle types.Our genome is therefore not a rigid recipe book – it is a dynamic kitchen, where the cell constantly develops new solutions from the same templates.
📌 If you are interested in how genetic information in DNA is translated into protein step by step, you can find further information here.
But as in any creative kitchen, errors can also occur during the transfer from DNA to mRNA: if a „letter” in the recipe is missed, swapped, or incorrectly assembled, the message can end up misleading or unusable. Such mutations or faulty splicing are sometimes harmless – but they can also produce defective proteins that fail to perform their function or even cause harm. A small slip in copying the molecular cookbook – and the flavor of life changes.
c) Epigenetics & Non-coding DNA: The Conducting Team of Our Genome
Although all cells contain the same DNA, they only use a fraction of it. A liver cell needs different instructions than a nerve cell – the rest remain dormant. This targeted gene regulation is controlled by epigenetic mechanisms: small chemical markers – such as methyl groups – do not change the genetic sequence itself, but influence whether a gene is active, how often it is read, or whether it remains completely silenced.
But who actually gives the conductor his instructions? This is where non-coding DNA comes into play – 98% of our genome, long dismissed as useless „junk”. It is the operating system that provides the epigenetic control programs. Only through its interaction is it determined when, where, and how strongly our genes become active – and thus it helps decide over health and disease.
d) The Hidden Control Center: What Non-coding DNA Regulates
These „dark“ regions conceal precise circuits that control the fate of each cell:
Enhancers act like invisible remote controls for genes. Even across thousands of base pairs, they can activate or silence a target gene – for example, for heart development in the embryo.
Telomeres are the protective caps at the ends of chromosomes, like the plastic tips on shoelaces. But when they shrink, aging begins – a central mechanism of cellular decline and cancer development.
Non-coding RNAs act as hidden directors: microRNAs block disease-causing genes, while the long RNA XIST silences an entire X chromosome in females.
Epigenetic markers lay an invisible map over the genome. With methylations acting like red stop signs, they decide whether an enhancer or gene may be read – or silenced.
This is how order emerges in the molecular cookbook: each cell type reads only the pages it needs – guided by non-coding DNA and its epigenetic interpretation.
e) When the Operating System Makes You Sick
Almost all genetic risk markers for common diseases – from diabetes and heart disease to mental disorders – are located in non-coding zones:
Mutations in an enhancer of the MYC gene can trigger leukemia by driving cell division out of control.
Epigenetic misprogramming in these regions – caused, for example, by chronic stress or environmental toxins – can permanently switch genes on or off incorrectly, thereby promoting diseases such as cancer or autoimmune disorders.
Some of these epigenetic patterns are partially reversible – a glimmer of hope for new therapies.
f) Open Mysteries in Research
Despite tremendous progress, the genome remains full of unsolved mysteries:
The „dark matter”: About half of the non-coding genome remains functionally unexplored. Is it evolutionary noise – or an as-yet uncharted regulatory network of life?
Long-range communication: How does an enhancer reliably find its target gene among millions of DNA bases? The spatial folding of the genome – a true „chromatin origami” – could be the key.
Environmental influences: Why do identical non-coding mutations lead to completely different clinical pictures in different life contexts?
RNA codes: Tens of thousands of non-coding RNAs have been discovered – but which are mere footnotes, and which might emerge as leitmotifs for new therapies?
g) The True Magic of Genes
Non-coding DNA is not junk data, but the cell’s mastermind. Epigenetics, in turn, acts as its conductor – flexibly interpreting the instructions of the operating system. Together, they shape our development, regulate our health, and respond to environmental signals.
Genes are not a rigid destiny, but more like a musical score that can be continually reinterpreted. In this interplay, an orchestra emerges that not only shapes our cells but constantly retunes itself – influenced by experiences, environmental factors, and even traces that can be passed on to future generations.
The true magic lies in the fact that from a limited set of notes, an infinite diversity of life can emerge.
4.2. Genetic Variation and Its Consequences
When the genome miswrites, falls silent, or improvises.
With every cell division, DNA is carefully copied and passed on to daughter cells – ensuring that genetic information is preserved across generations.
However, the system is not infallible: mutations – changes in the DNA sequence – can affect the structure and function of proteins. Sometimes only a single „letter” is incorrect. Sometimes a „sentence” is missing, or an entire „paragraph” has been rearranged. In genetics, these are called variants – genetic changes that can be harmless, pose a risk, or cause disease, depending on their type, size, and location.
The following overview presents the main types of variants – and what they mean in the molecular manuscript of life:
📌 SNVs (Single Nucleotide Variants) – A single „letter” is swapped. Usually barely noticeable, but sometimes enough to rewrite entire stories, as in the case of sickle cell anemia.
📌 Indels (Insertions/Deletions) – Small fragments of text are inserted or deleted. Even a tiny shift can throw entire „sentences” out of sync, sometimes so much that the original meaning is lost.
📌 Structural Variants (SVs) – Long stretches of DNA are inverted, duplicated, or relocated. These hidden „rewritings” are difficult to detect – and can often have serious consequences, as seen in cancer
📌 Splice-Site Variants – Errors at the junctions disrupt the RNA syntax. This creates incorrect blueprints, resulting in proteins that no longer function properly.
📌 Repeat Expansions – Base sequences are compulsively repeated until they distort the „text”. This causes diseases such as Huntington’s disease.
📌 Copy Number Variants (CNVs) – Entire „chapters” (genes) are missing or duplicated. The balance of the story is lost.
📌 Mitochondrial variants – Changes in the DNA of mitochondria, the cell’s power plants. If the „text” here is altered, the drive of life falters.
📌 Somatic variants – Arise only during a person’s lifetime and affect specific groups of cells. They mainly shape the story of tumors.
📌 Epigenetic signals – Not new letters, but a different emphasis: volume, rhythm, timing. They transform the same text into an entirely new melody.
The list shows that our genome is not a rigid structure but can be altered by variants. Some of these changes remain inconspicuous footnotes, while others can rewrite the entire story – sometimes with dramatic consequences.
To make such subtle differences visible, tools are needed that capture every detail – every letter, every punctuation mark, every shift. This is precisely where modern genome sequencing comes in.
4.3. Genome Sequencing: Key to Individual Genetic Material
The genetic material of a human being resembles a gigantic library: billions of letters, neatly arranged in long volumes. To read it, genome sequencing is required – which, like a high-precision scanner, provides the raw text of our genetic blueprint.
Thus, it becomes the heart of personalized medicine – a form of medicine that bases its decisions on the individual, not on the average.
But how do we even get hold of the „Book of Life“ so that we can read it page by page?
Let’s imagine the following: shortly after birth, a newborn undergoes a DNA test – a scenario that has already become reality in the United Kingdom, as described earlier.
a) The Journey of DNA: From Umbilical Cord Blood to Genetic Information
Immediately after birth, blood can be collected from the clamped umbilical cord – a safe and painless procedure. Umbilical cord blood originates from the newborn and contains numerous valuable cells, especially stem cells and white blood cells. Hidden within their nuclei lies what truly matters: the DNA – the child’s genetic inheritance.
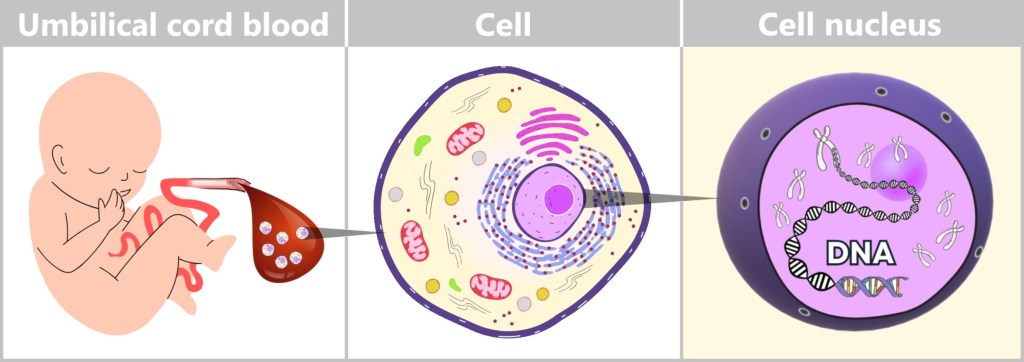
Fig. 4: From Umbilical Cord Blood to Genetic Information Left: Umbilical cord blood as a rich source of diverse cell types.
Center: Schematic representation of a human cell.
Right: Inside the cell nucleus lies the DNA, the carrier of genetic information.To isolate and analyze this DNA, it undergoes a multi-step process in the laboratory. This involves established molecular biology methods that can be divided into four key steps:
Cell Lysis: Gaining Access to the Genetic Material
First, the blood is centrifuged – spun at high speed. During this process, the components separate according to their density: the white blood cells settle as a cell pellet at the bottom. They contain the majority of the DNA of interest.
These cells are then treated with specialized solutions and enzymes that dissolve the cell and nuclear membranes. This releases the DNA – initially, however, as part of a mixture containing proteins, lipids, and other cellular components.
DNA Isolation: Obtaining Pure Genetic Material
In the next step, the DNA is extracted from the cell lysate – the „cell soup.” Several methods can be used for this purpose:
Filtration or column purification: The solution is passed through special filter systems to which the DNA adheres, while smaller molecules are washed out.
Magnetic beads: More modern methods use tiny beads with a DNA-binding surface. The DNA sticks to them and can be removed in a targeted manner using a magnet.
Both methods allow for an effective separation of genetic material from contaminants.
Purification and Concentration of the DNA
At this stage, the DNA from many white blood cells is present in a relatively pure form – but still dissolved in an aqueous solution. To further purify and concentrate it, cold alcohol (e.g., ethanol or isopropanol) is added. This causes the DNA to precipitate and become visible as a whitish, thread-like substance.
Human DNA is extremely long: if fully stretched out, it measures about two meters per cell. During extraction, however, it is broken into many smaller fragments by chemical and mechanical forces. At first, this seems chaotic – but because the DNA is identical in all cells, many fragments contain overlapping information. This random distribution becomes an advantage during sequencing: the fragments can be read multiple times independently and then assembled on a computer into a complete sequence. This significantly increases the accuracy of the analysis.
Quality Control: Is the DNA Intact?
Before the DNA sample can be analyzed, its quality must be assessed. Two common methods are:
Spectrophotometry: Measures the concentration and purity of the DNA by analyzing light absorption.
Gel electrophoresis: DNA fragments are passed through a gel and separated by length (longer fragments move more slowly than shorter ones). This allows assessment of whether the sample is sufficiently intact.
Why is this process so important?
Human DNA is fragile – contaminants or fragmentation can distort analyses or render them unusable. Careful and controlled purification is therefore essential for reliable genetic results.
Umbilical cord blood is a particularly valuable source of medical information: it enables early diagnosis, prognoses, and in some cases targeted prevention.
What appears to be a routine procedure in the lab is, in fact, a highly precise process – it requires technical expertise, modern equipment, and biochemical finesse. With automated extraction methods, it is now possible to obtain a high-quality DNA sample from just a few drops of umbilical cord blood in less than an hour. Although the DNA consists of fragments, it still contains all the information needed to decode an individual’s genetic code – and thus lays the foundation for the medicine of tomorrow.
b) Three Directives for Gene Diagnostics: WGS, WES, and Panel
Before we dive deeper into the tools of genome sequencing, it is worth taking a look at the three central strategies used today to decode genetic information – like stage directions in the script of life:
🎯 Panel Sequencing – The Targeted Scene
Only a selected part of the genome is examined here – usually a few dozen to a few hundred genes. Such tests are used when a specific disease is suspected, such as hereditary breast cancer (BRCA1/2).
Advantage: Fast, cost-effective, and precise – as long as the medical question is well-defined.
Limitation: Everything outside the selected scene remains in the dark.🎥 Whole Exome Sequencing (WES) – The Classic Director’s Cut
WES analyses all protein-coding sections of the genome – approximately 1–2% of the entire genetic material. It is a proven method, particularly for rare hereditary diseases, as most known mutations are located in these areas.
Advantage: Efficient identification of faulty blueprints for proteins.
Limitation: Regions outside the genes – important switches and regulators of the genome – are not captured.🎬 Whole Genome Sequencing (WGS) – The Complete Movie in 4K
WGS reads the entire genome – all three billion letters. This provides the most comprehensive picture, including non-coding regions, regulatory elements, structural variants, and rare mutations.
Advantage: Maximum resolution for complex cases.
Example: In children with unclear developmental disorders, WGS provides clarity twice as often as WES – for instance, by uncovering mutations in distant „remote-control” regions (enhancers) that incorrectly switch genes on or off.Conclusion:
Depending on the question at hand, the right perspective is needed – sometimes a zoom on the key scene suffices, other times the entire film must be re-edited. Choosing the method is therefore not a mere technical detail, but a strategic decision in the diagnostic playbook.
4.4. Methods of Genome Sequencing
How is the genetic code read?
Imagine DNA as a book – written in a language with only four letters: A, T, C, and G. To decipher this genetic text, scientists use modern technologies. Two central methods have become established. They pursue the same goal: to determine the sequence of DNA letters as precisely as possible.
The first method works – put simply – like a molecular photocopier equipped with color sensors. The second uses a high-tech tunnel through which the DNA is guided and electronically „scanned”, comparable to running your fingers along a rope to detect the smallest irregularities. These techniques form the foundation of DNA sequencing.
4.4.1. Method 1: Copying with Fluorescent Dye
a) Sanger Sequencing
b) Illumina Technology
c) Single-Molecule Real-Time(SMRT) Sequencing
4.4.2. Method 2: High-Tech Tunnel – Electrically Scanning DNA
a) Oxford Nanopore Sequencing
4.4.3. Genome Sequencing – Technologies Compared
a) Throughput vs. Usability – Quantity Does Not Always Equal Quality
b) Cost-Effectiveness – Cost per Base vs. Cost per Insight
c) From Sample to Result – How Quickly Does the Whole Genome Speak?
d) Clinical Applications – Which Technology Fits Which Purpose?
e) AI as Co-Pilot – Automation Without Relinquishing Responsibility
f) Comparison Table: Sanger, Illumina, PacBio, ONT
g) Key Takeaways: Between Maturity and Routine
4.4.1. Method 1: Copying with Fluorescent Dye
This method takes advantage of the fact that DNA consists of two complementary strands. If the base sequence of one strand is known, the sequence of the complementary strand can be automatically deduced. To obtain this information, one of the two strands of the DNA under investigation is copied. The aim is to record the exact sequence of bases in the resulting copy during this copying process.
To better understand how this process works, let’s first take a look at the key components and basic steps involved in producing a DNA copy in a test tube (in vitro).
Producing a DNA Copy – The Basic Principle
At the centre is a DNA template that is to be copied. This requires four main components:
- dNTPs (deoxynucleoside triphosphates): the DNA building blocks A, T, C, and G.
- DNA polymerase: an enzyme that functions as a molecular copying machine.
- Primers: short DNA fragments that provide the polymerase with a starting point.
- Buffer solution: ensures stable conditions during the reaction.
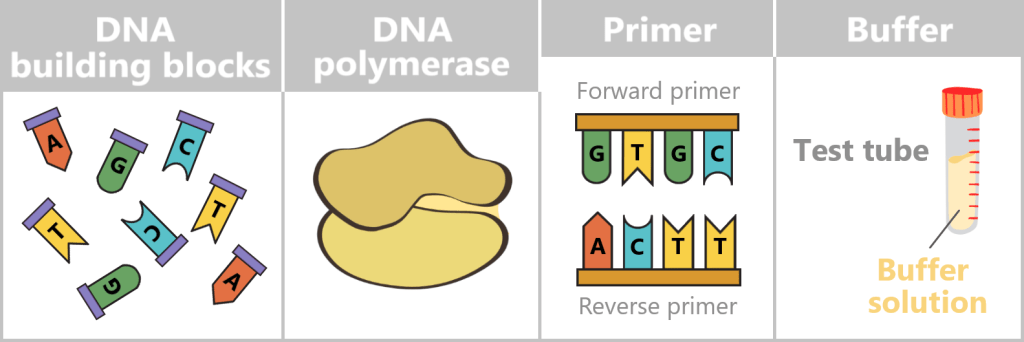
Fig. 5-A: Schematic representation of the main components required to generate a DNA copy DNA building blocks: Individual nucleotides (A, T, C, G), shown in different colors and letters. They serve as the raw material from which the new DNA strand is assembled.
DNA polymerase: A schematic enzyme that links the nucleotides together to form a DNA strand.
Primer: Short DNA fragments that provide the polymerase with a defined starting point.
Buffer: A test tube containing buffer solution, ensuring stable chemical conditions during the reaction.What does DNA polymerase do?
DNA polymerase is a specialized enzyme that generates complementary DNA strands – essentially the „printhead” of a molecular copying machine. Enzymes act as biological tools that enable chemical reactions in a targeted and efficient manner, which is why they are often described as „biological catalysts”.
In order for the polymerase to work, it needs a primer as a start signal. You can think of it like this: just as a printhead only starts working when a sheet of paper is inserted correctly, the polymerase only begins its work when a primer is present. This small piece of DNA shows it where to start – essentially acting as the first sheet in the molecular printer.
Before copying can begin, the double-stranded DNA must first be separated into two single strands – this step is called denaturation.
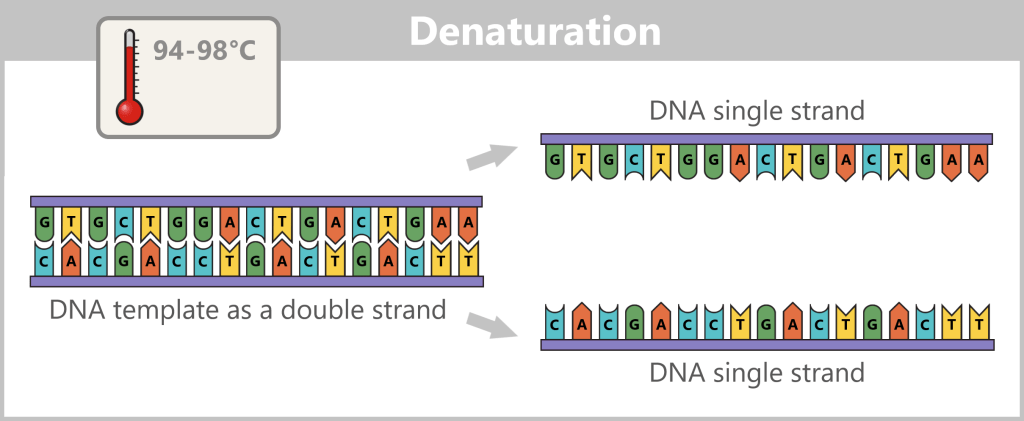
Fig. 5-B: Separation of the DNA strands Next, the primers bind to their complementary target sites on the single strands. This primer binding marks the starting point for the DNA polymerase.
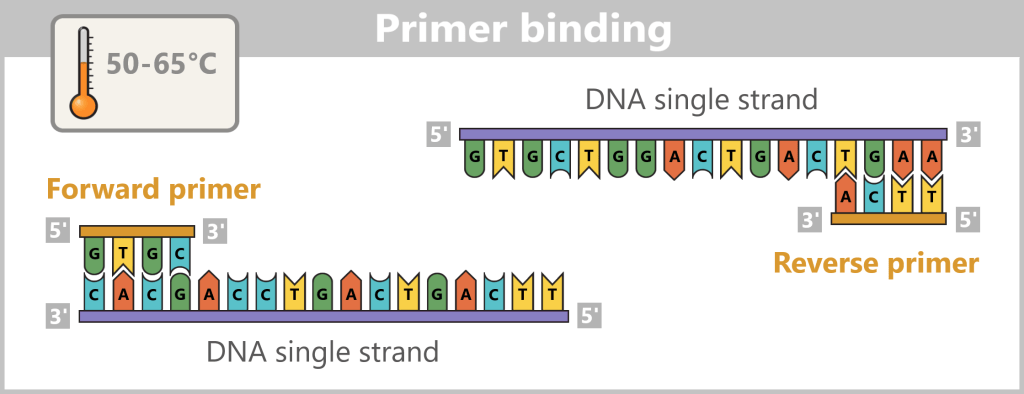
Fig. 5-C: Primers mark the starting point for the polymerase. Now the synthesis of the new strands begins: The polymerase reads the template strand in the 3′-to-5′ direction and extends the copy in the opposite direction – 5′ to 3′ – while obeying the base-pairing rules (A with T, G with C).
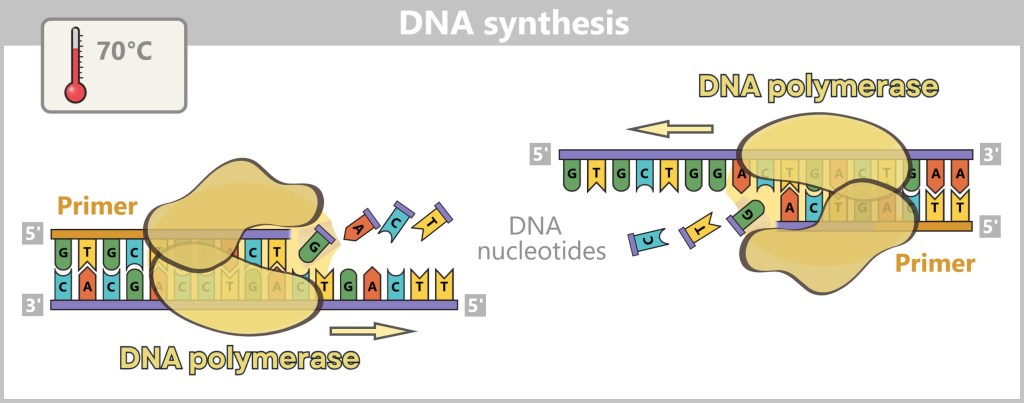
Fig. 5-D: DNA synthesis – the exact duplication of a DNA strand. What do 3′ and 5′ mean?
The designations 3′ (three-prime) and 5′ (five-prime) come from the chemistry of the DNA backbone. They indicate at which end certain carbon atoms are located in the sugar molecule, to which new building blocks can be attached.
Imagine the DNA strand as a one-way street. The polymerase can only travel in one direction – from the 5′ end to the 3′ end. New building blocks can only be chemically attached at the 3′ end. Therefore, the polymerase reads the ‘old’ strand backwards (3′ → 5′) and builds the new strand forwards (5′ → 3′).
In this way, a new, complementary strand is gradually formed from a single strand – with the help of the base pairing rules: adenine (A) pairs with thymine (T), and cytosine (C) with guanine (G).
The result is perfect genetic replicas – new DNA strands that are exact copies of the original DNA.
Fluorescent Nucleotides: How Sequencing Works
Modern sequencing methods enhance this copying process with a sophisticated technique: they use fluorescently labeled dNTPs (DNA building blocks). Each of the four nucleotides (A, T, C, G) is tagged with a dye that emits a specific light signal when it is incorporated.
As the polymerase extends the DNA strands, it inserts the colored building blocks with precision. Each time a new nucleotide is incorporated, it sends out a tiny light signal – like a small flash that reveals which „letter“ has just been added. High-resolution cameras capture these light signals, enabling the DNA sequence to be reconstructed step by step.
Overview of Sequencing Methods
Several major technologies apply this principle in specific ways:
Sanger Sequencing: The classical method, in which fluorescent chain-terminating nucleotides halt DNA synthesis at random positions. The resulting fragments can be read in sequence.
Illumina Technology: A widely used high-throughput method in which millions of DNA fragments are sequenced in parallel. The fluorescent signals are recorded step by step.
SMRT Sequencing (PacBio): In this method, synthesis is observed in real time on a single DNA molecule, offering high precision and long read lengths.
a) Sanger Sequencing
The classic „letter-by-letter” reading method.
Sanger sequencing, also known as the chain-termination method, was developed in the 1970s and was the first technique to read DNA accurately and reliably. It is fundamentally based on the same principles as DNA synthesis but differs in several key aspects:
👉 Only a single primer is used.
👉 In addition to the normal DNA building blocks – the dNTPs (dATP, dCTP, dGTP, dTTP) – special fluorescent chain-terminating nucleotides, called ddNTPs (ddATP, ddCTP, ddGTP, ddTTP), are added in small amounts. When a ddNTP is incorporated, DNA synthesis stops precisely at that point. Each of the four ddNTP types carries a different fluorescent color corresponding to its base.The process – step by step
For sequencing, four separate reaction mixtures are prepared. Each contains:
- the single-stranded target DNA,
- a primer (starting point for synthesis),
- a DNA polymerase,
- normal dNTPs,
- and one type of the fluorescently labeled ddNTPs.

Fig. 6-A: Sanger Sequencing – schematic representation of the four reaction mixtures. Each mixture contains the same basic components but differs in the specific type of fluorescently labeled chain-terminating nucleotides (ddNTPs) added.
After primer binding, the polymerase reads the DNA strand and incorporates the complementary nucleotides to build a new strand. If a ddNTP is randomly incorporated, synthesis terminates precisely at that position.
This generates many DNA fragments of varying lengths – each ending with a fluorescently labeled nucleotide. Since each of the four reactions contains only one type of ddNTP, the terminal base of each fragment is known precisely.

Fig. 6-B: Synthesis is specifically interrupted when a ddNTP is incorporated. Each reaction mixture contains only one type of modified DNA building block (ddATP, ddTTP, ddCTP, ddGTP). When such a „stop” nucleotide is incorporated during DNA synthesis, the copying process halts precisely at that point. In the ddATP mixture, synthesis stops upon incorporation of a modified adenine (A); in the ddTTP mixture, it ends with a modified thymine (T). Likewise, ddCTP and ddGTP terminate synthesis when a modified cytosine (C) or guanine (G) is added. This procedure generates many DNA fragments of varying lengths, each ending with a specific stop nucleotide. The goal is to produce all theoretically possible fragments so that the complete DNA sequence can be read.
Sorting and Analysis
The DNA fragments are then denatured (converted into single strands) and sorted by length using gel electrophoresis. The negatively charged fragments migrate through a gel towards the positive electrode. Smaller fragments move faster, larger ones slower, creating an orderly „ladder“ of fragments.
A laser then excites the fluorescent dyes attached to the terminal bases. The resulting light signals are captured by a detector. Each signal corresponds to a specific base at a specific position. The sequence of light signals thus directly reveals the DNA sequence – much like reading a book letter by letter.
Since the generated fragments are complementary to the original DNA, the original base sequence can be deduced precisely from them.
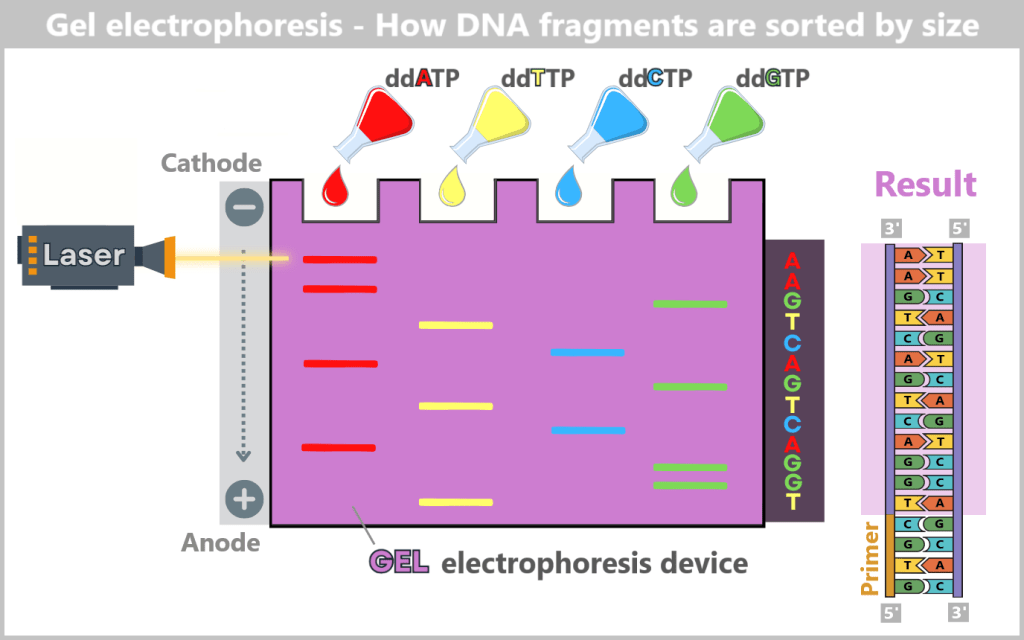
Fig. 6-C: Gel electrophoresis for separating DNA fragments: In the reaction mixtures, DNA fragments of varying lengths are generated, each ending with the same chain-terminating nucleotide – either adenine, thymine, guanine, or cytosine, depending on the mixture. These reaction mixtures are applied to a gel. When an electric field is applied, the negatively charged DNA fragments migrate from the cathode (−) toward the anode (+). The size of the fragments determines their migration speed: smaller fragments move faster through the gel’s fine pores and reach the anode first, while larger fragments move more slowly. By reading the fluorescent signals at the fragment ends, the exact order of the bases can be determined, allowing the DNA sequence to be reconstructed step by step.
Why is Sanger sequencing still considered the gold standard today?
Even half a century after its development, the Sanger method remains indispensable in many areas of molecular biology and medicine. The reason: reliability and precision.
Compared to modern high-throughput methods, Sanger sequencing is slower and suitable only for analyzing smaller DNA segments – not entire genomes. But that is precisely where its strength lies:
- Targeted questions, such as checking individual genes,
- Detection of specific mutations, or
- Validation of critical results previously identified using other methods
can be answered with precision, clarity, and interpretability using Sanger sequencing – often so clearly that the sequence of bases can be read directly from the sequencing plot.
In medical diagnostics – such as the analysis of inherited diseases or in quality control of genetic engineering procedures – this high level of accuracy is crucial. Even a single misread base can have life-altering consequences.
Another advantage: the method is standardized worldwide, has been reliably used for decades, and is governed by well-defined quality guidelines.
A Classic with Staying Power
In a world where new technologies constantly emerge, Sanger sequencing remains a reliable anchor – the old-timer among sequencing methods: not the fastest, but extremely robust, proven, and reliable. And sometimes that’s exactly what matters.
b) Illumina Sequencing
The High-Speed Copy Machine
Sanger sequencing is like precise hand-crafted work: each DNA strand is decoded step by step – reliable, but slow and expensive.
Imagine having to copy an entire book – or even an entire library – while being allowed to write down only one letter per minute.
This is precisely where the problem lies: modern cancer research, the decoding of rare diseases and the genetic monitoring of pandemics require the analysis of large amounts of data – i.e. many and/or very long DNA segments. This requires a method that is not only accurate, but also fast and affordable.
This is precisely where Illumina sequencing comes into play. It has elevated the principle of DNA decoding from manual work to industrial mass production and is now one of the most commonly used methods for high-throughput sequencing (Next Generation Sequencing, NGS). Instead of individual letters, entire pages are now read simultaneously – millions of times, in parallel, with high precision and cost-efficiency.
Unlike the classic Sanger method, where each DNA fragment is analyzed individually, Illumina operates with massive parallelization – meaning that many DNA fragments are amplified and sequenced simultaneously. How does this work?
Step 1: Preparing the DNA Fragments
– Turning the DNA into a „Lego Construction Site” –First, the DNA sample to be analyzed is broken down into many short pieces – called fragments – typically 100 to 300 base pairs long. Small synthetic DNA sequences, known as adapters, are then attached to both ends of these fragments. You can think of them as LEGO connectors. On the one hand, they act as molecular docking sites that allow the DNA fragments to bind to a special carrier – the flow cell chip. On the other hand, they serve as binding sites for universal primers.
Abb. 7-A: The DNA is broken into numerous fragments, each equipped with small attachments (adapters). 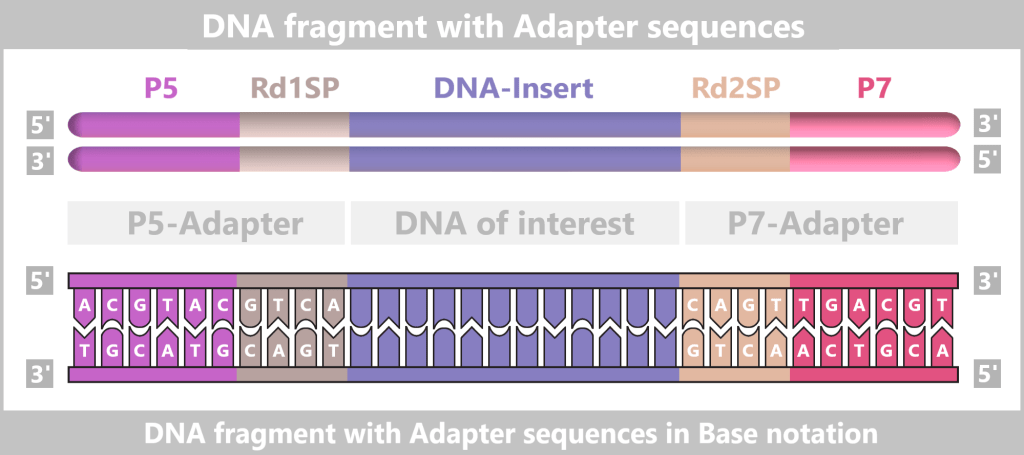
Fig. 7-B: Simplified schematic representation of a DNA fragment with adapters. The P5/P7 sequences serve to attach the fragments to the flow cell. The Rd1SP/Rd2SP sequences act as binding sites for universal primers.
Denaturation separates the prepared double-stranded DNA fragments into single strands.
Step 2: Bridge Amplification
– The Dance of DNA on the Flow Cell –At the heart of Illumina technology is the flow cell – a glass-like plate covered with millions of tiny DNA docking sites (short DNA segments called oligonucleotides) that are firmly anchored to the surface.

Fig. 7-C: Schematic representation of a flow cell and its surface. The single-stranded DNA fragments bind to the anchors via their adapters. Then the molecular copying process begins: complementary copies of the DNA snippets are produced, which are now firmly anchored to the flow cell.
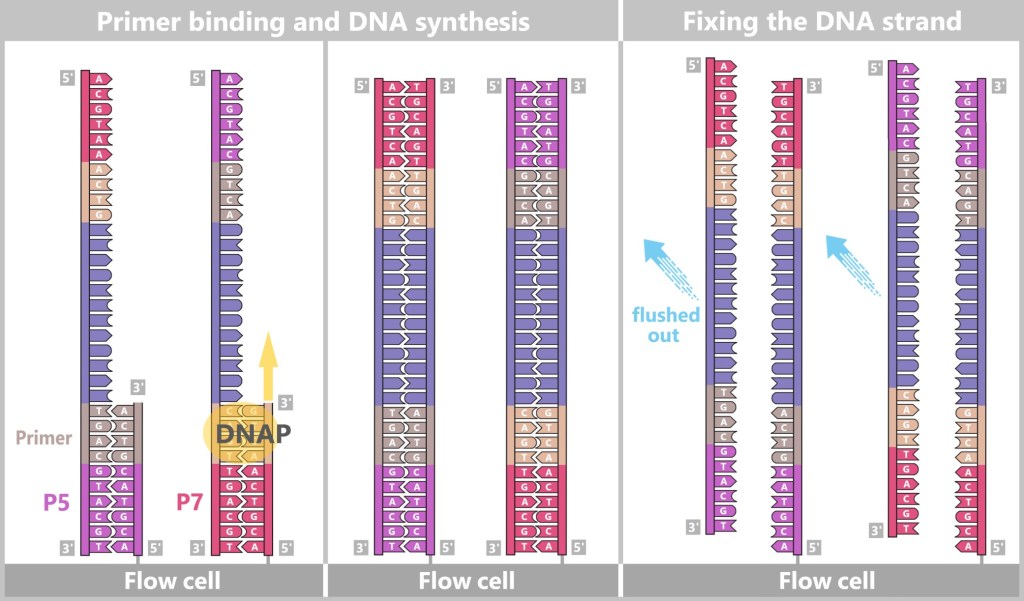
Fig. 7-D: Schematic representation of initial synthesis + formation of anchored DNA strand copies. Left: Primers bind to the adapters (P5, P7), and DNA polymerase (DNAP) initiates the synthesis of a new complementary strand.
Center: The DNA polymerase then synthesizes the first strand.
Right: The newly formed DNA double strand is separated. After denaturation, the original strand is no longer connected to the flow cell and is flushed out. The newly synthesized strand remains firmly bound to the flow cell with its 5′ end.Then a fascinating process begins: the DNA strands bend to form small bridges, as their free adapters attach to neighboring oligonucleotides on the flow cell. At these points, they are copied again. After copying, the bridge is dissolved, and the number of anchored DNA strands doubles. This process, known as bridge amplification, is repeated dozens of times. In the end, dense clusters of millions of identical copies of a single DNA fragment are formed.
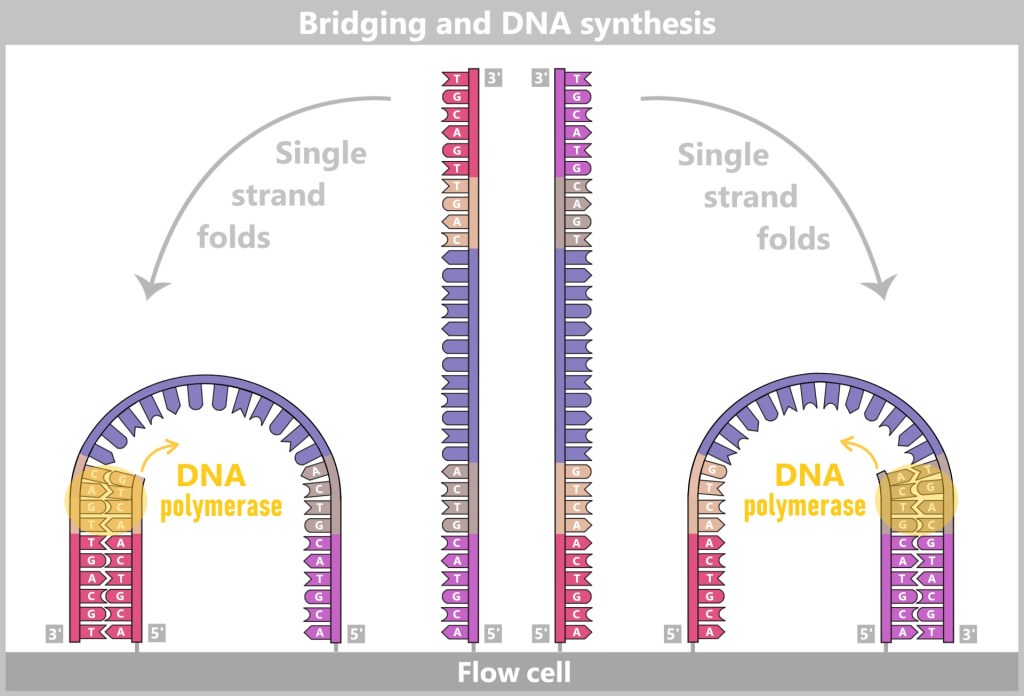
Fig. 7-E: Bridge Formation and DNA Synthesis The anchored single strands fold and connect with neighboring anchors on the flow cell, forming a bridge structure. The copying process is then initiated.
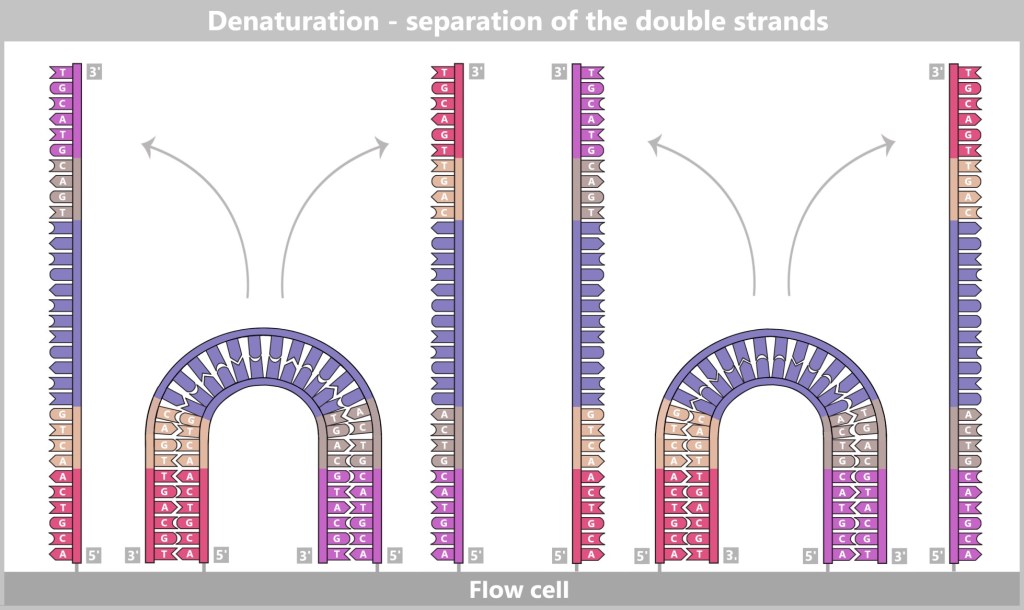
Fig. 7-F: Forward and Backward – Bridge Dissociation After the copying process, the bridge double strands are separated. The number of firmly anchored DNA strands doubles, making them ready for further amplification.
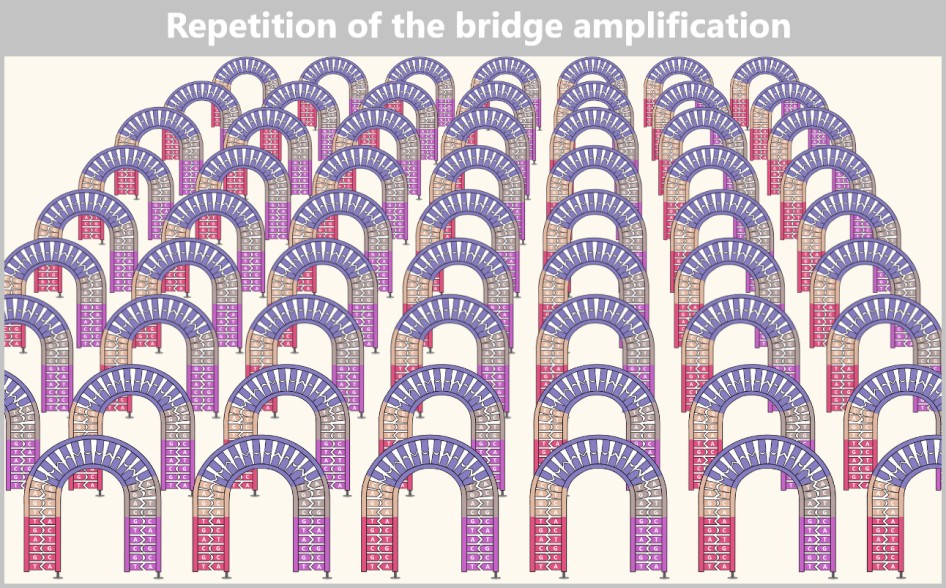
Fig. 7-G: With each cycle of bridge amplification, an increasing number of DNA copies is generated. 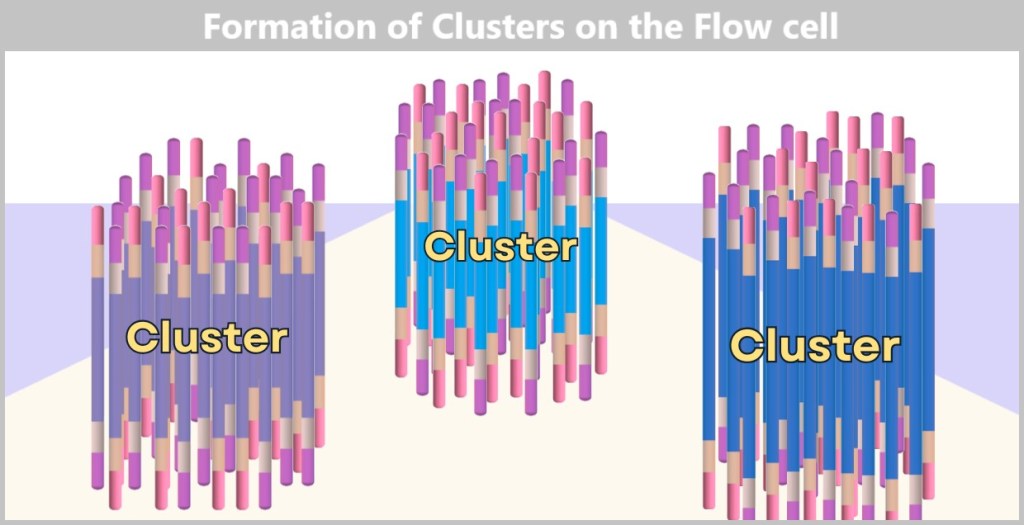
Fig. 7-H: Cluster formation Each cluster consists of numerous copies of a single DNA fragment. In this illustration, only three clusters are shown as examples. In reality, millions of such clusters cover a flow cell, enabling high sequencing capacity.
The result is a densely packed ‘map’ consisting of millions of DNA clusters, each containing only a specific sequence – ideal for simultaneous reading.
Without this replication, reading DNA would be like trying to spot a single firefly in the wind. The clusters, on the other hand, make the DNA bases (A, T, C, G) clearly visible – like bright neon lettering in the dark.
Step 3: Sequencing-by-Synthesis
– The Light Show of DNA Bases –Now the actual sequencing by synthesis begins. Once again, the principle of a „molecular copying machine with color sensors“ is used – but in an optimized form:
At the beginning, the universal primers bind to the corresponding adapter sites of the DNA fragments in each DNA cluster.
Then modified dNTPs – so-called RT-dNTPs (reversibly terminating nucleotides) – are added to the reaction solution. Each of the four DNA building blocks (A, T, G, C) is labelled with its own fluorescent color and carries a reversible blocker.
Only one nucleotide per cycle can be incorporated because the block temporarily stops further strand formation. Unincorporated RT-dNTPs are washed away.
After each incorporation step, the flow cell is scanned with a high-resolution camera. Each fluorescent signal corresponds to an incorporated nucleotide, and its color indicates which base has just been added.
Afterwards, the blockage and dye marking are chemically removed, and the next cycle begins.
This process is repeated cycle by cycle until each DNA strand is completely read.

Fig. 7-I: Illumina sequencing steps Step 4: Data Analysis
– The Supercomputer as Puzzle Master –After the „light show„, millions of photos are available – one for each cycle and each cluster. Each image shows which color (and thus which letter: A, T, C, or G) was added to each cluster.
Now powerful computers come into play – equipped with software that works as precisely as a detective piecing together a cut-up book from a stack of numbered Polaroids. Each cluster on the sequencing chip is like a small crime scene, each color pixel a clue. And from millions of such clues, a story gradually emerges – the story of DNA.

Fig. 7-J: Analysis of the Sequencing Data The color patterns are translated into sequences of letters, known as „reads”: tiny pieces of text from an enormous genome novel. In the end, a flood of such fragments piles up – like a mountain of cut-up book pages, scattered loosely about.
This is precisely where the real art begins: bioinformatics takes the stage. Using specialized algorithms, the snippets are analyzed, sorted, and stitched together – constantly searching for overlapping regions, familiar patterns, and known structures.
Piece by piece, the bigger picture comes back together – until, in the end, the original DNA sequence becomes visible, like a reconstructed book that suddenly makes sense.
The real „magic” therefore doesn’t happen in the flow cell, but in the computer! Without the software, the images would be nothing more than colorful flickering – but with it, they become the key to medical breakthroughs.
A more detailed yet clear explanation of the Illumina sequencing technology method can be found in the video „Illumina Sequencing Technology“.
Modern high-throughput sequencing platforms such as those from Illumina can generate the raw data of a complete human genome within just a few days – at pure sequencing costs that are significantly lower than the price of a premium smartphone.
This technology has revolutionized genome research and made it considerably more accessible: today, it is an extremely versatile tool – fast enough for real-time analysis of pandemics, precise enough to form the basis for personalized cancer therapies when combined with robust bioinformatics and clinical expertise.
Illumina sequencing is now considered the industry standard for large sequencing projects – such as the analysis of whole genomes, gene expression research, and modern cancer diagnostics.
And best of all: While you are reading this text, somewhere in the world a Flow Cell is decoding millions of DNA fragments.
However, Illumina technology also has its limitations. It reaches its limits particularly with long, repetitive DNA segments, such as those found in some chromosome regions. It is like trying to reconstruct a song from thousands of 3-second snippets. For such challenges, Illumina is often combined with other sequencing methods that can reliably capture long segments – like an investigator who needs both close-ups and panoramic images to understand the entire crime scene.
c) Single-Molecule Real-Time (SMRT) Sequencing
The Novel in One Go
Imagine you want to read a long novel – full of recurring chapters and hidden clues that span many pages. If you cut the book into thousands of small snippets, as in Illumina sequencing, read them individually and then put them together like a jigsaw puzzle, there is a risk that chapters will be missing, mixed up or incomplete. The secret messages of the story – what the novel actually wants to tell us – could be lost or appear only fragmentarily.
This is where PacBio’s Single Molecule Real-Time (SMRT) sequencing shines: it reads entire chapters in one go – no snippets, no breaks.
Instead of breaking the DNA apart, PacBio keeps it in very long fragments – often tens of thousands of base pairs at a time, sequenced as a whole. These strands enter tiny stages in a SMRT cell, called nanowells (Zero-Mode Waveguides). They are so small that they can hold only a single DNA molecule and a polymerase. Each of these stages allows light to act on just one tiny spot – exactly where the polymerase reads the DNA. Like a spotlight highlighting an actor during a monologue on stage.
The polymerase plays the leading role: it adds building block by building block (A, T, C, or G) to the DNA copy. Each of these blocks lights up in a different color, like neon lights at a DJ set. With every addition, a flash of light is emitted and recorded in real time by a camera – a dancing light code that reveals the DNA sequence.
The video Introduction to SMRT Sequencing by PacBio illustrates this process impressively.
Let’s analyze the individual steps of this fascinating process in detail.
Step 1: Preparation
– DNA Loops for Continuous Reading –Even with PacBio, the DNA to be analyzed is first fragmented – but into much larger pieces than with Illumina, usually 10,000 to over 20,000 base pairs long. These fragments are then converted into circular DNA molecules:
So-called Hairpin adapters are added to both ends – small loops of DNA that close the fragment onto itself.
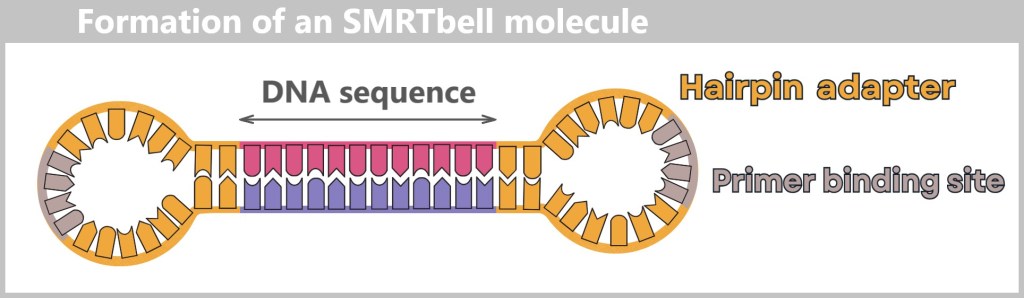
Fig. 8-A: A DNA fragment is equipped with hairpin adapters. The adapters contain a primer binding site, which later provides the starting point for the polymerase.
Subsequent denaturation produces a circular, single-stranded SMRTbell molecule in the form of an open loop.
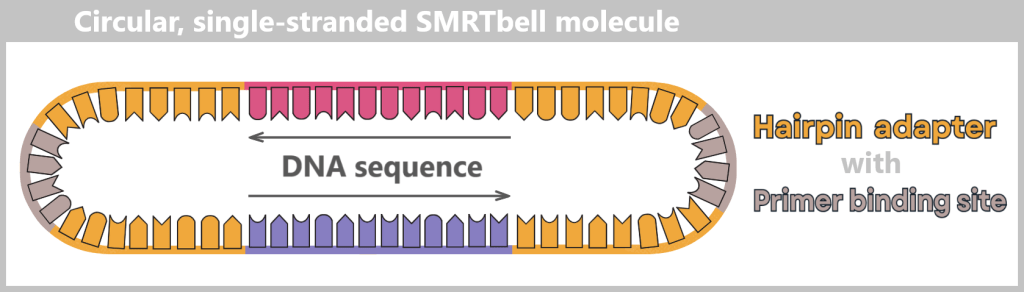
Fig. 8-B: The denatured SMRTbell molecule: Denaturation converts the double-stranded template into a single-stranded SMRTbell molecule. It forms a circular structure, shown here as an open loop.
The result is a SMRTbell: a closed DNA loop that can be read repeatedly by the polymerase – like a model train running around a circular track.
Each hairpin adapter contains a defined binding site to which the universal primer binds. This is the starting point for the polymerase.
The DNA polymerase binds to the primer complex, forming a fully functional replication complex: polymerase + primer + SMRTbell.
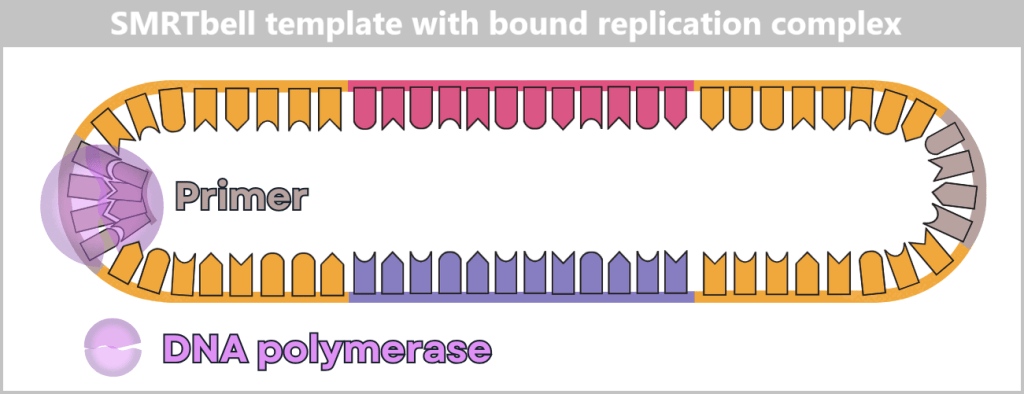
Fig. 8-C: The stable replication complex of polymerase, primer, and single-stranded SMRTbell. The primer is bound to the adapter binding site, the polymerase to the primer. This complex is ready to start DNA synthesis.
Step 2: The Sequencing Stage
– Light Shows at the Nanoscale –Now it gets spectacular: the prepared replication complexes are guided into their tiny observation chambers – the Zero-Mode Waveguides (ZMWs). These nanowells are so small that typically only a single SMRTbell template fits inside. The polymerase is anchored firmly at the bottom of the ZMW, ready for action.
The start signal comes with the addition of the four nucleotides (A, C, G, T), each labeled with its own fluorescent dye.
The polymerase now begins its work – it reads the DNA strand and synthesizes a complementary strand. The crucial event occurs the moment it incorporates a nucleotide:
Each nucleotide emits a brief, color-coded flash of light.
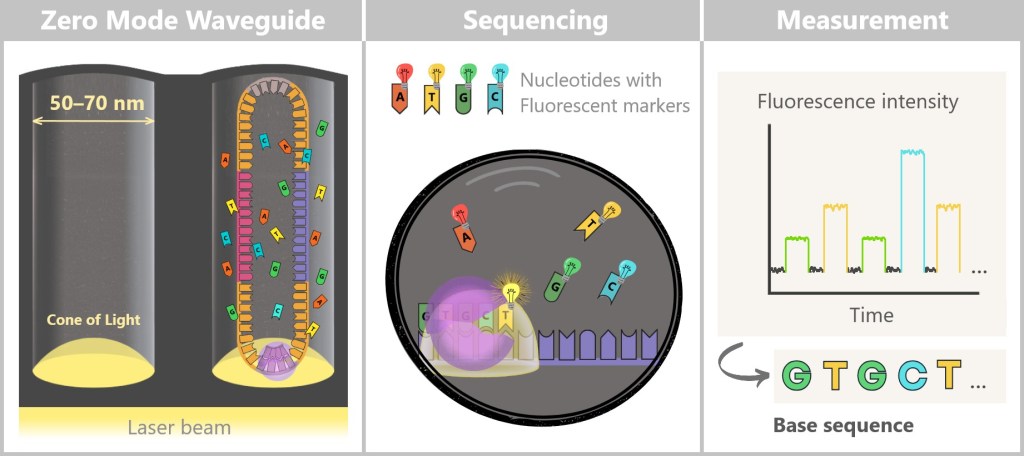
Fig. 8-D: Zero-Mode Waveguide (ZMW): Sequencing of Individual DNA Molecules Left: At the bottom of the Zero-Mode Waveguide (ZMW), a laser beam enters and generates what is known as an evanescent field (shown as a cone of light). Unlike normal light, this field does not propagate throughout the solution but decays exponentially within only 20–30 nanometers. This creates an extremely small excitation volume in which the fluorescent dyes of the nucleotides can be excited.
The left zero mode waveguide (ZMW) contains a single replication complex (polymerase + SMRTbell). The polymerase (purple) is firmly anchored to the (glass) bottom. In the presence of the fluorescently labelled nucleotides, the polymerase begins its work.
Centre: The four nucleotides (A, T, G, C) carry different fluorescent markers. As soon as the polymerase incorporates a nucleotide, it lights up briefly before the dye is cleaved off.
Right: These flashes of light are measured in real time and translated into fluorescence signals. This is how the base sequence of the DNA is determined step by step.The luminescence occurs in real time, at the exact moment of insertion – hence the name: Single Molecule Real-Time Sequencing. A sensitive detector records these light signals – like a molecular live microscope at work.
Step 3: The Long-Range Secret
– Repetition Makes It Robust –Since PacBio reads very long DNA segments at once – comparable to reading entire chapters of a book instead of individual verses – more individual errors occur in a single run than with Illumina. This is exactly where the ring-shaped SMRTbell structure comes into play. The DNA polymerase can circle the ring molecule multiple times, like a train conductor. In doing so, the same DNA fragment is read over and over again – like a reader revisiting the same passage until it is fully mastered.
A computer compares the repeated reads with one another and filters out the errors – like an editor polishing a manuscript. This results in a particularly precise consensus sequence.
PacBio calls this approach HiFi reads – high-precision single-molecule readings.
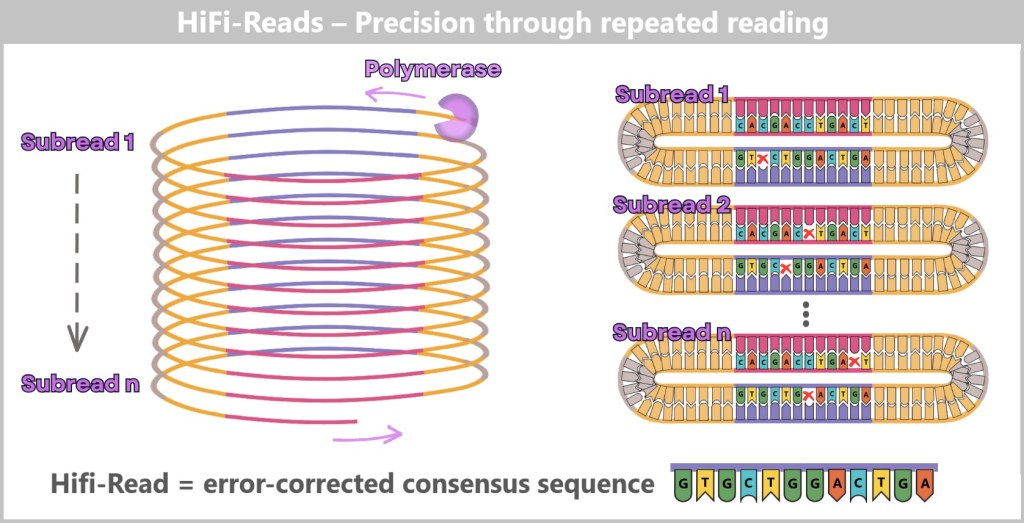
Fig. 8-E: Formation of a HiFi Read. The polymerase circles the SMRTbell template multiple times, generating multiple subreads. Each cycle corresponds to a complete reading of the DNA sequence. Individual subreads may contain random errors (x), but by comparing many repetitions, a highly accurate consensus is formed – the so-called HiFi read (High-Fidelity Read).
Additional info: How the polymerase loops around the DNA multiple times
First sequencing run
The polymerase reads the single-stranded template strand and synthesizes a complementary strand.
The result is a complete double helix – the original template strand and the newly created daughter strand are now firmly bound together.
This first, uninterrupted round produces a subread – a single base sequence generated by the polymerase in a single pass.
Second and subsequent sequencing runs
Now the question arises: how can the polymerase continue reading when the DNA ring is already double-stranded?
This is where Φ29 phage DNA polymerase comes into play – a specially modified enzyme variant with two key properties:
High processivity: It remains bound to the DNA strand for a very long time and hardly ever detaches – this enables long reads.
Strong strand-displacement activity: It can break the hydrogen bonds between the template and daughter strands.
The polymerase runs into the double helix, separates the strands locally and displaces the old daughter strand while simultaneously synthesizing a new one – like a molecular bulldozer.
The previously synthesized strand is not degraded; it remains as a loose, single-stranded DNA fragment in the nanowell. With each subsequent round, this process repeats: the polymerase reads the same template again – producing multiple subreads from the same DNA molecule.
Step 4: Data Analysis
– Turning Flashes of Light into Readable DNA –Once all the light signals have been captured, the real detective work begins: data analysis.
The first step is called base calling: the recorded fluorescence signals – encoded in time and color – are translated into nucleotide sequences – A, T, G, or C.
Next, the raw data from each polymerase pass is cleaned up: the hairpin adapter sequences are bioinformatically trimmed. What remains are the subreads – the uncut reading records of each individual polymerase pass through the DNA chapter text.
But it is only the next step that reveals the whole truth: like a master philological detective comparing multiple copies of a manuscript, the algorithm combines all subreads of the same SMRTbell molecule. This comparison – known as Circular Consensus sequencing (CCS) – produces a highly accurate HiFi read: an error-corrected master copy that reproduces the DNA chapter word for word.
Only now is the text fragment ready for final classification – and in the process it goes through several stages:
First, a quality check – just like a meticulous editor reviewing the text for clarity and consistency.
This is followed by alignment: the sequence is classified like a new chapter in the correct compartment of a reference genome shelf.
Finally, assembly takes place – the final act, in which all chapters are brought together to form a complete novel of the genome.
The video PacBio Sequencing – How it Works clearly summarizes the steps involved.
Applications and Advantages – The Strengths of Long-Read Sequencing
PacBio SMRT technology has a particular strength: it can read very long DNA segments in one go – with highest accuracy. This makes it ideal for:
- Detecting complex genomic structures
- Analysis of repetitive sequences
- Distinguishing very similar gene variants (e.g., in immune receptors or cancer genetics)
- De novo sequencing – reading entirely new genomes without a reference
- Detecting epigenetic modifications (e.g. methylations)
How can SMRT sequencing make epigenetic modifications visible?
As already described, epigenetic mechanisms play a central role in targeted gene regulation. These are small chemical markers – such as methyl groups – that are attached to specific DNA bases. They do not change the genetic sequence itself, but influence when and how often genes are read – depending on cell type, environment or stage of development.
They function like notes or markings in the margin of a book: the text remains the same, but the „reading” changes – some passages are emphasized, others skipped over.
If this finely tuned system falls out of balance, it can have far-reaching consequences – such as in cancer, autoimmune diseases, or mental disorders.
Unlike Illumina technology, which reads a chemically modified copy of the DNA, SMRT analyses the original DNA in real time. It measures not only the base sequence, but also the dwell time of the DNA polymerase at each base – known as incorporation kinetics.
This is precisely where the key to detecting epigenetic modifications lies: methylation and other chemical changes influence these kinetics. They act like small obstacles or skid marks on the DNA strand. The polymerase „lingers“ longer at such sites – and it is exactly this delay that is directly captured by the SMRT camera.
SMRT is particularly well suited for the direct detection of certain chemical markers, such as methyl groups on adenine or cytosine bases, which play an important role especially in bacteria and plants. For cytosine methylation (5mC), which is central in human epigenetics, direct detection is less sensitive but can be reliably derived using specialized bioinformatics tools such as pb-CpG-tools.
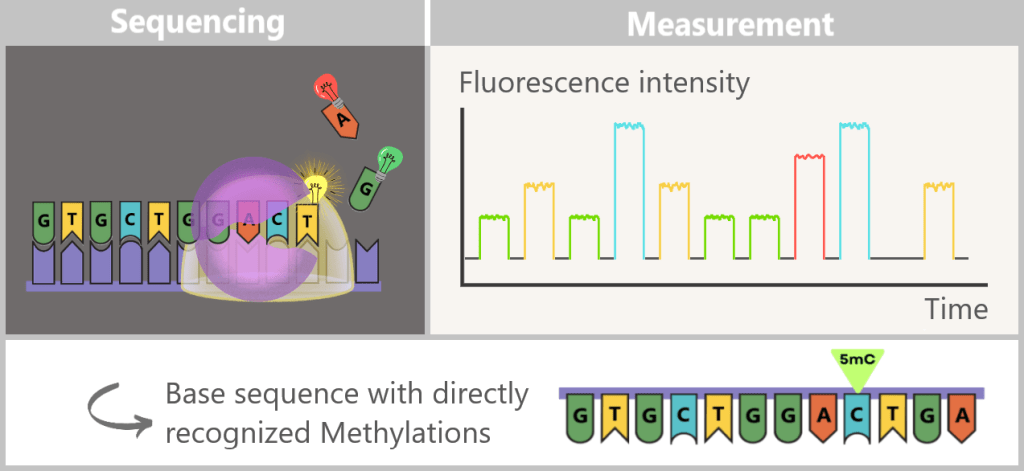
Fig. 8-E: Direct Base Detection and Methylation Analysis in SMRT Sequencing How SMRT Sequencing Reveals Epigenetic Modifications
During SMRT sequencing, the fluorescence signals of the incorporated nucleotides are measured in real time. At the same time, modifications such as 5-methylcytosine (5mC) can be detected, as they slightly alter the kinetics of the polymerase.The key advantage: the extremely long read lengths reveal how methylation patterns are intertwined across large genomic regions with other factors, such as:
- gene variants,
- haplotypes (groups of co-inherited gene variants),
- repetitive sequences.
In this way, PacBio provides not only the genetic sequence of letters but also a „map” of epigenetic regulation – revealing which „pages” functionally belong together and how this network influences disease processes such as cancer or neurological disorders.
Defective Scissors in the Genome: How PacBio Unmasks Splice-Site Variants
In the section Alternative Splicing: The Flexible Kitchen of Genes, we described in simplified terms how our DNA, as a molecular cookbook, produces recipes in the form of mRNA transcripts. These transport the genetic information from the cell nucleus to the ribosomes in the cytoplasm – the cell’s „protein factories”.
The recipe is subsequently adjusted: molecular scissors remove certain ingredients (introns) and recombine other building blocks (exons). This process is controlled by splice sites – markers that are „copied“ from the DNA. These can be thought of as cutting instructions: they determine which parts of the recipe are used and how they are assembled.
But what happens if these cutting instructions are faulty? The consequences can be severe:
- Exons are skipped → parts of the blueprint are missing.
- Introns are erroneously retained → „junk” is incorporated into the protein.
- Incorrect splice sites are created → the mRNA text becomes distorted.
Such splice-site variants often lead to defective or non-functional proteins – with possible consequences such as cancer, genetic disorders (e.g. β-thalassemia), or neurological diseases. Since protein biosynthesis depends directly on the mRNA sequence, detecting such errors is crucial.
The key lies in PacBio’s ability to analyze complete mRNA molecules in one piece – usually 500 to 5,000 nucleotides long, sometimes significantly longer. Since PacBio reads DNA, the mRNA is first converted into a stable copy (cDNA, complementary DNA). This can then be sequenced in its entirety. This allows us to see exactly how the recipe is structured and which ingredients are required in which order – no transcription or splicing errors remain hidden.
In addition, the underlying DNA segment can also be sequenced. Bioinformatic tools compare cDNA and DNA sequences with reference data, revealing deviations in the splicing pattern and their genetic cause.
The DNA shows: Here the instruction is faulty.
The cDNA shows: Here the recipe is mutilated.Only the combination of both pieces of information creates a sound basis for precise diagnoses and targeted therapeutic approaches.
Conclusion – When you want to tell the whole story
When it comes to telling the entire story of a DNA segment in one go – without cutting it into pieces or piecing it back together – SMRT sequencing is the method of choice.
PacBio-SMRT is the archaeologist of genomics: it sees the whole picture where others only provide fragments. In 2022, it played a key role in closing the last gaps in the human genome.
PacBio SMRT technology is not a mass-market product but a precision instrument. It is slower and more expensive per base pair than Illumina – but in return, it provides greater detail and versatility.
In practice, both technologies are often combined: Illumina for breadth, PacBio for depth. Together, they provide a complete picture – like a satellite image and a close-up merged into a single panorama.

4.4.2. Method 2: High-Tech Tunnel –Electrically Scanning DNA
While in the molecular copying machine with color sensors the DNA is first replicated and then „photographed“ in an image, the second method works in a radically different way: the DNA is passed through a high-tech tunnel and scanned electronically in real time.
This is the principle behind Oxford Nanopore sequencing.
a) Oxford Nanopore Sequencing
DNA Through the Eye of a Needle
Imagine reading a rope by letting it slowly slide through your fingers – sensing every subtle irregularity: thickness, dents, knots. That’s exactly how Oxford Nanopore Sequencing (ONT) works: it „feels” the DNA as it passes through a tiny biological eyelet.
This eyelet – a so-called nanopore channel – is embedded in a membrane through which an electrical current flows. As soon as a single DNA molecule is threaded through this high-tech tunnel, the bases (A, T, G, or C) influence the current flow with different intensities – like pebbles of different shapes in a stream. These changes generate characteristic electrical signals that are recorded in real time.
ONT does not read light signals, but rather current patterns – an electrical „finger reading” of DNA.
A single thread – read directly
Before sequencing begins, the DNA is coupled to a motor protein that pulls it precisely through the pore – not too fast, not too slow. Unlike Illumina or PacBio, there is no need for polymerase, fluorescence, or amplification: the original DNA is read directly – a major advantage for sensitive or damaged samples.

Fig. 9-A: Schematic representation of the sequencing cell and the ion current in the idle state The sequencing cell consists of two chambers: the cis chamber (top) and the trans chamber (bottom), separated by a membrane with embedded nanopores. On the left, a DNA molecule has docked onto a nanopore with the help of a motor protein. On the right, an empty nanopore is shown, through which a constant ionic current flows. The applied voltage between the negative electrode (cis) and the positive electrode (trans) drives the ion flow. The ionic current is measured in the well (a small channel in the chip), as shown in the current/time diagram. As long as no DNA passes through the pore, the current remains constant.
And the best part: the DNA can be extremely long. ONT holds the world record for the longest DNA ever sequenced – over two million base pairs in a single read. Entire chromosomal regions can thus be captured without interruption – like reading a novel in one single breath.
From noise to meaning – the art of signal interpretation
The raw electrical signals generated as the DNA passes through the pore initially resemble a noisy radio channel. However, with the help of specialized algorithms – known as basecalling – the current noise is translated into a clear sequence of letters: A, T, G, or C.

Fig. 9-B: The image illustrates the central mechanism of nanopore sequencing. 1) A motor protein pulls the DNA through the pore in a controlled manner. The negatively charged DNA moves from the cis side to the trans side due to the applied voltage. In the process, the individual bases (A, T, G, C) affect the ionic current in a specific way.
2) A graph shows the change in the measured current over time. Each base combination generates a characteristic signal that is decoded by algorithms.
3) A computer analyzes the electrical signals and determines the sequence of bases from them.Unlike PacBio, ONT does not distinguish bases individually, but in groups of five to six nucleotides that occupy the pore simultaneously. Each of these „5-mer” generates a characteristic current signal. This increases the reading speed but makes interpretation more complex – somewhat like recognizing words in an unfamiliar dialect. Homopolymers (long repeats of the same base, e.g. „AAAAA”) are particularly challenging.
AI-powered basecalling software is continuously improving. Error rates have dropped dramatically, and ONT reads now achieve high accuracy – especially when the dataset is large enough for statistical multiple coverage of each DNA base through repeated, random sampling.
There is a cool visual explanation in the video How nanopore sequencing works by Oxford Nanopore Technologies.
For those who prefer a more detailed explanation, additional background information can be found here.
Key Strengths – Fast, Portable, Versatile
What makes ONT special:
Portability: Devices like the MinION fit in a pocket. They require only a laptop and a power source – ideal for field research, space missions, or rapid analyses during outbreaks (e.g., Ebola or SARS-CoV-2).
Speed: The DNA is read live. Initial sequencing data is often available within minutes – a priceless advantage in clinical emergencies.
Direct epigenetics: Like PacBio, ONT can directly detect methylation and other epigenetic modifications, as they subtly affect the electrical signal – without any additional treatment or reagents.
Direct RNA sequencing: RNA molecules can also be read without prior conversion to cDNA – a true unique feature.
Why Direct RNA Sequencing Matters
Other sequencing methods, like PacBio, must first convert mRNA into cDNA in order to read it. This is like copying a recipe first – small details can be lost in the process. In contrast, Oxford Nanopore technology reads the mRNA directly – without the detour through DNA.
This way, all the „marginal notes of the cookbook” are preserved: chemical modifications, variants, and even the exact length of the molecules – information that is often lost in a copy. This creates an unaltered view of gene activity – a priceless advantage for detecting complex processes such as gene regulation or faulty RNA processing.
This is a breakthrough, especially for personalized medicine: many diseases arise not only from altered DNA but also from deviations in RNA.
Practical Examples:
- Cancer diagnostics: RNA splice variants can help detect aggressive tumor forms early and guide targeted treatment.
- Hereditary diseases: For diseases such as spinal muscular atrophy or Duchenne muscular dystrophy, RNA profiles enable accurate assessment of disease progression and personalized therapies, e.g. with antisense oligonucleotides.
- Infectious medicine: Direct sequencing of viral RNA allows rapid identification of virus variants – crucial during epidemics and for selecting appropriate therapies.
Direct RNA sequencing thus helps reveal each person’s unique „recipe variant” – with all its small deviations, marginal notes, and special ingredients – enabling more precise diagnoses and tailored treatments.
Conclusion: The Genomics „Electric Sense Finger”
ONT is the sensory-rich border crosser of genomics: fast as a racehorse, portable as a Swiss Army knife and close to biological processes. Where others have to copy, it feels live – even for RNA or epigenetic traces.

4.4.3. Genome Sequencing –Technologies Compared
In the world of genomics, there is no single method – rather, there is an entire ensemble. Each sequencing platform has its own style, its strengths, and its ideal application area: from the classic solo storyteller to the high-speed streamer.
Even though all four sequencers have their place on the stage, not every one of them is suited for every performance. Someone who wants to investigate a single gene needs different tools than someone who aims to capture the entire genome in epic breadth. And anyone trying to detect rare variants must read very carefully.
Let us now take a closer look at the four main players to understand their strengths, weaknesses, and strategic roles in detail.
a) Throughput vs. Usability – Quantity Does Not Always Equal Quality
„Throughput” sounds like efficiency – as many DNA letters per minute as possible. In sequencing, it is measured in bases per run: that is, how many bases a device reads out in a single sequencing process.
Modern platforms such as Illumina’s NovaSeq X Plus achieve peak values of up to 16 terabases with short reads (100–300 bp) – that’s 16 billion letters in a single run. Like a high-performance printer that produces entire genome libraries overnight.
PacBio and Oxford Nanopore follow a different approach. They generate long reads – from 10,000–25,000 bp for PacBio HiFi reads, up to over 1 million bp for ONT. Their throughput is lower: PacBio (Revio) achieves up to 1.300 gigabases, ONT (PromethION) up to 7.000 gigabases per run. Sounds like less – and it is, if you only look at the number of bases.
But: more data does not necessarily mean more insight.
What matters is not only how much is read – but how precisely and how often. Two key concepts help to assess the informative value of sequencing data.
🔍 Quality Score – the confidence in each letter
The so-called Q-score (quality score) describes how likely it is that a read DNA letter is correct. The scale is logarithmic:
- Q20 = 99 % accuracy → 1 error per 100 Basen
- Q30 = 99,9 % accuracy → 1 error per 1.000 Basen
- Q40 = 99,99 % accuracy → 1 error per 10.000 Basen
In research, Q20 is often sufficient. In medical diagnostics, Q30 or higher is considered standard, and for high-precision applications (e.g., tumor genetics), Q40 is expected.
For raw data, Illumina typically ranges from Q30 to Q40, while PacBio (Continuous Long Reads, CLR) and ONT only achieve values between Q10 and Q15.
Sanger outperforms them all – with Q40–Q50, but only on short DNA segments. Ideal for targeted confirmation of individual variants.
So how do modern sequencing methods meet the high standards of clinical diagnostics? The answer: through coverage and bioinformatic analysis.
🔁 Coverage – repetition reveals the truth
Coverage describes how often each area of the genome has been read.
Example: 30x coverage means that each letter has been read an average of 30 times – in different fragments and in different constellations.
Why is this important? Individual measurements may contain errors. Only through repetition and statistical majority formation can genuine variants be distinguished from artefacts.
In medical diagnostics, 30× coverage is the minimum standard.
With 30–50× coverage, Illumina achieves an overall accuracy of Q35–Q40+. It reliably detects single nucleotide variants (SNVs), insertions/deletions (indels), and copy number variants (CNVs).
PacBio HiFi Reads achieve Q30–Q40 at 30x coverage and ONT Q30–35. ONT requires 50x coverage to achieve Q44 precision.
The big advantage of PacBio and ONT is that they read much longer DNA segments – often 10,000 to 100,000 bases, and in the case of ONT, even over 1 million. This enables them to recognize complex structures, repetitions and epigenetic signatures, and allows RNA analysis and phasing – i.e. the assignment of variants to paternal or maternal chromosomes. It is not the quantity that counts here, but the overall picture.
🧬 Depending on the goal – different requirements for coverage and accuracy
The required coverage and sequencing quality depend on the clinical application:
Population studies: Q30 at 30× coverage is usually sufficient to detect statistical patterns.
Cancer diagnostics: Q35 at 60–100× coverage for reliable detection of rare somatic mutations.
Liquid biopsies, mosaic analyses: Q40 at 100–300× coverage – to distinguish true signal variants from background noise.
Phasing: Long reads (PacBio, ONT) with 30× coverage are often sufficient to assign variants reliably.
📉 The dilemma: greater certainty – greater effort
Higher coverage increases confidence, but also means more data, longer computation times, larger infrastructure, and higher costs.
b) Cost-Effectiveness – Cost per Base vs. Cost per Insight
Illumina (NovaSeq/NextSeq) remains the price-performance favourite for whole genome sequencing:
👉 200–800€ for a complete genome at 30× coverage – precise for point mutations, ideal for routine analyses.PacBio (Revio/Sequel IIe) and Oxford Nanopore (PromethION/MinION) are more expensive:
👉 700–1,200€ for 15–30× coverage – but with additional information such as long reads, structural variants, epigenetics, and RNA. This is not a luxury, but in many cases diagnostically crucial.Oxford Nanopore also scores with a low entry cost: a MinION costs just a few thousand euros, is portable, and delivers data within hours.
👉 The simple equation „more bases = better deal” falls short. What matters is what can be read and understood from the data.🧾 Hidden costs – reading the genome is cheaper than understanding it
A human genome for under 300€? Technically yes – but this usually only covers reagents, machine time, and raw data.
What is often missing are the hidden items:
🔧 Sample preparation
🔍 Quality control & library creation
🧠 Bioinformatics & clinical interpretation
📄 Findings report & physician consultationThese areas often account for 70–80% of the total costs.
Because: reading is cheap today – understanding remains challenging.
c) From Sample to Result – How Quickly Does the Whole Genome Speak?
Reading the genome is like a performance: the curtain rises with sample collection – but when will the final curtain fall, the clinical report?
It depends on technology and infrastructure:
Oxford Nanopore enables preliminary results within 24–48 hours – thanks to real-time analysis during sequencing.
Illumina provides data in 3–7 days with optimized workflows – the gold standard for routine diagnostic procedures.
PacBio takes 4–7 days, but offers particularly high-quality long reads and epigenetic information.
Sanger delivers targeted validations within hours – but only for small regions of interest.
Choosing a platform is therefore not just a matter of data quality – but also of clinical timing.
Those who need to act in hours rather than days turn to Oxford Nanopore. Those who require robust routine data rely on Illumina. And those who want maximum contextual depth take a bit more time with PacBio – for significantly greater insight.
The curtain falls. Yet behind the scenes, amid hope, high-tech, and human care, the search for the perfect pace for each patient continues. After all, it’s not the fastest machine that matters – but the right rhythm for the human story behind it.
d) Clinical Applications – Which Technology Fits Which Purpose?
Genome sequencing has long been more than just a research tool – it has become a key instrument for planning individualized therapies. But which method is suitable for which task?
A look at six key application areas shows how these technologies are shaping modern medicine.
Application Area Primary Method Complementary Methods Routine Diagnostics Illumina Sanger Pharmacogenomics Illumina Sanger Epigenetics PacBio, ONT – Cancer Genomics Illumina (SNVs), PacBio/ONT (SVs) Sanger Neonatal Emergency Diagnostics ONT Illumina, Sanger Rare Diseases PacBio, ONT Illumina, Sanger 👉 Insight: The optimal technology always depends on the clinical question – standard analysis, search for the unknown or emergency diagnostics? Validation or initial diagnosis?
e) AI as Co-Pilot – Automation Without Relinquishing Responsibility
The integration of artificial intelligence (AI) – especially deep learning – is indispensable in all modern DNA sequencing platforms (Illumina, PacBio, Oxford Nanopore). Its main applications are:
🧬 Basecalling (conversion of raw data into DNA sequences) & error correction
🔍 Quality control & real-time analysis
🧠 De novo assembly, genome analysis & variant detection
📚 Pathogenicity prediction, literature research & clinical classification
📄 Automated report templates👉 Insight: AI transforms sequencing from a mere data-generation process into an intelligent interpretation workflow – making it central to modern genomic research and diagnostics.
But one thing remains important: AI does not (yet) think like a physician. In rare diseases, complex cases, or newly discovered genetic variants, clinical expertise remains essential. Legally, physician-validated interpretation is also mandatory – even in the age of digital genomics.
Ultimately, genome sequencing is not a solo performance – it requires the entire ensemble: precise technology, intelligent bioinformatics and clinical intuition. Because only when everyone plays their part can data be transformed into healing knowledge.
f) Comparison Table: Sanger, Illumina, PacBio, ONT
Feature Sanger Illumina PacBio ONT Sample preparation DNA-Template, Extraction,
Primer-Design, ddNTPs
🕒 ~6–12 Std.Fragmentation,
Adapter ligation
🕒 ~6–24 Std.High DNA quality, Fragmentation,
SMRTbell adapters
🕒 ~1–2 TageFragmentation,
Adapter with Helicase (Motor protein)
🕒 ~6–24 Std.Read length Short: 500–1.000 bp Very short: 100–300 bp (paired-end possible) Long: 10.000–25.000 bp (HiFi-Reads) Ultra long: 10.000 bp – >1 Mb Throughput Very low: single reactions (~1–100 kb/run) Very high: up to 16.000 Gb (NovaSeq X Plus),
>100 samples simultaneouslyAveragete: up to 1.300 Gb / SMRT-Cell (Revio) Flexible: 100–7.000 Gb (PromethION), up to 20 Gb (MinION) Accuracy Very high: Q40–Q50 (99,99–99,999 %) High: Q30–Q40 (99,9–99,99 %), at 30x: Q35–Q40+ High: Q30–Q40 (HiFi-Reads), at 30x Raw data: Q15–Q20+, at 30x: Q30–Q35, Duplex: Q44 at 50x Cost per genome (EUR) (sequencing only) – ~200–800 (30x) (Large laboratories) ~700–1.200
(15–30x)~700–1.200
(15–30x)Time to result (WGS, Sample → Report) – 3–7 days (incl. preparation, sequencing, analysis) 4–7 days (incl. preparation, sequencing, analysis) 24–48 hours
(real-time, incl. analysis)Variant detection SNVs, mitochondrial variants (validation) SNVs, indels, CNVs (indirect), somatic variants, splice variants Indels, SVs, CNVs, repeat expansions, splice variants, somatic SVs Indels, SVs, CNVs, repeat expansions, splice variants, somatic SVs RNA sequencing – mRNA via conversion to cDNA, splice variants mRNA via conversion to cDNA, splice variants (long reads) Direct RNA sequencing (without cDNA), splice variants, long transcripts Epigenetic modifications – Indirect:
only with bisulfite sequencing (complex)Direct:
methylation detection via incorporation kinetics (HiFi reads) & bioinformatic toolsDirect:
methylation via signal changes (e.g., 5mC, 6mA)Place of deployment Laboratory: Stationary equipment Laboratory: Large stationary equipment (e.g. NovaSeq) Laboratory: Large stationary equipment (Revio) Flexible: Laboratory (PromethION), portable/point-of-care (MinION) Main advantages Highest precision, ideal for validation High throughput, cost-effective, precise for SNVs/indels, suitable for routine use Long accurate reads (HiFi), ideal for SVs, epigenetics, phasing Ultra-long reads, mobile, fast, direct RNA and epigenetic detection Limitations Low throughput, WGS impractical, expensive Short reads, limited for SVs/epigenetics, complex library preparation More expensive, medium throughput, longer preparation time Lower raw data quality, dependent on bioinformatics Role in personalized medicine Validation of critical SNVs (pharmacogenomics, cancer), mitochondrial analyses Routine diagnostics, pharmacogenomics, cancer genomics (SNVs/indels), rare diseases Epigenetics, cancer genomics (SVs), rare diseases (SVs, repeat expansions), phasing Neonatal emergency diagnostics, epigenetics, cancer genomics (SVs), rare diseases, mobile diagnostics
g) Key Takeaways: Between Maturity and Routine
DNA sequencing is at a pivotal turning point: in recent years, it has evolved from a purely research-based technique into a clinically relevant tool in modern medicine. Whether in oncology, rare disease diagnostics, or pharmacogenomics – genome analyses are increasingly providing answers where conventional methods reach their limits.
And yet: despite its impressive technical maturity, genome sequencing is still not a widespread standard in clinical practice.
Technology mature – infrastructure incomplete
Modern high-throughput methods such as Illumina or the long-read technologies from PacBio and Oxford Nanopore now enable highly accurate analyses at comparatively low costs. Whole genome sequencing (WGS) can be performed in specialized laboratories for a few hundred pounds per case – with a total turnaround time of less than a week. Mobile platforms such as MinION even provide initial genetic information within 48 hours in acute situations, such as with seriously ill newborns.
Despite this progress, clinical use remains fragmented. There are many reasons for this: there is a lack of standardized bioinformatic analysis protocols, interdisciplinary trained specialists, and legally compliant and economically viable integration into everyday healthcare. Above all, the interpretation of genetic variants – especially those with unclear clinical significance – requires specialized expertise that is not available in many places.
The UK as a blueprint
While many countries are still experimenting with pilot programs or making case-by-case decisions, the United Kingdom is institutionalizing genome sequencing as an integral part of public healthcare. As part of a national pilot program, every newborn will soon undergo genomic screening – a precedent in prenatal and early-childhood preventive medicine. The goal is to identify genetically driven diseases in early childhood, before clinical symptoms appear. In this way, DNA analytics evolves from an individual diagnostic tool to a population-wide preventive strategy – a paradigm shift.
This model demonstrates that the barrier to broad implementation is less technical and primarily systemic: missing data standards, ethical and legal ambiguities, and a restrictive reimbursement policy prevent widespread integration in many places. The British initiative is therefore regarded as a pioneering example of institutionalized genomic medicine.
Technological outlook
Several technological developments are likely to further accelerate widespread implementation in the near future:
🚀 Decreasing cost per genome makes sequencing economically attractive even for smaller facilities.
🚀 Decentralized, mobile systems like MinION enable diagnostics independent of location, for example in emergency rooms or rural regions.
🚀 Faster time-to-result – from sample collection to diagnosis – allows for use even in time-critical cases.
🚀 Integration of genomic data with transcriptomics, proteomics, and epigenetics (multi-omics) promises contextualized, high-resolution diagnostics.
🚀 AI-powered analytics and automated interpretation tools reduce dependence on manual evaluation and minimize sources of error.
4.5. Genome Sequencing: The Next Technological Leap
To make decoding our DNA faster, cheaper, and universally accessible in the future, research is advancing the next generation of sequencing technologies.
a) FENT: How a Microchip Is Revolutionizing DNA Analysis
b) SBX: When DNA Stretches to Be Understood
c) The G4X System: From the Genetic Recipe to the Spatial Atlas of the Cell
a) FENT: How a Microchip Is Revolutionizing DNA Analysis
– and making it accessible to everyone
Driven by the vision of capturing genetic information in real time and directly at the point of care, the company iNanoBio is developing a new method that aims to take this idea to an entirely new level.
It combines the proven method – reading DNA through tiny nanopores – with a component from microelectronics: the MOSFET (metal-oxide-semiconductor field-effect transistor), the heart of modern computer chips.
From computer chip to DNA sensor
A MOSFET is a tiny switch with three contacts: Source (input), Drain (output), and Gate (control). When a voltage is applied to the gate, a conductive channel opens between source and drain – comparable to a bouncer who opens a gate. Even minimal changes in voltage influence the current flow, which makes MOSFETs extremely sensitive.
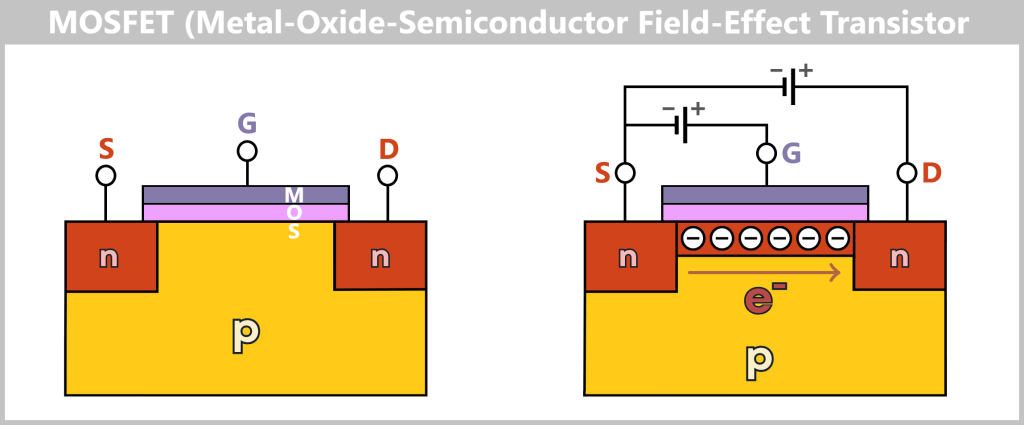
Fig. 10-A: Structure and function of a MOSFET A MOSFET is a tiny switch from microelectronics with three contacts: Source (input), Drain (output), and Gate (control). The source and drain are embedded in the semiconductor material, while the gate is insulated by an ultrathin oxide layer.
Without voltage at the gate, the channel between source and drain remains closed – no current flows. When voltage is applied to the gate, an electric field attracts charge carriers (e.g., electrons) and opens a conductive channel. The MOSFET thus acts like an extremely sensitive bouncer: only when it receives the signal does it open the gate and allow current to pass.Here’s the clever part: iNanoBio has managed to integrate a tiny pore directly into the sensitive control region of the MOSFET. As a DNA strand passes through, each nucleotide (A, T, C, or G) alters the current between source and drain in a characteristic way. The result: a novel sensor called FENT – short for Field-Effect Nanopore Transistor.
The FENT has a cylindrical design and encloses the pore. This allows it to measure signals from multiple directions simultaneously, which increases accuracy and reduces errors.
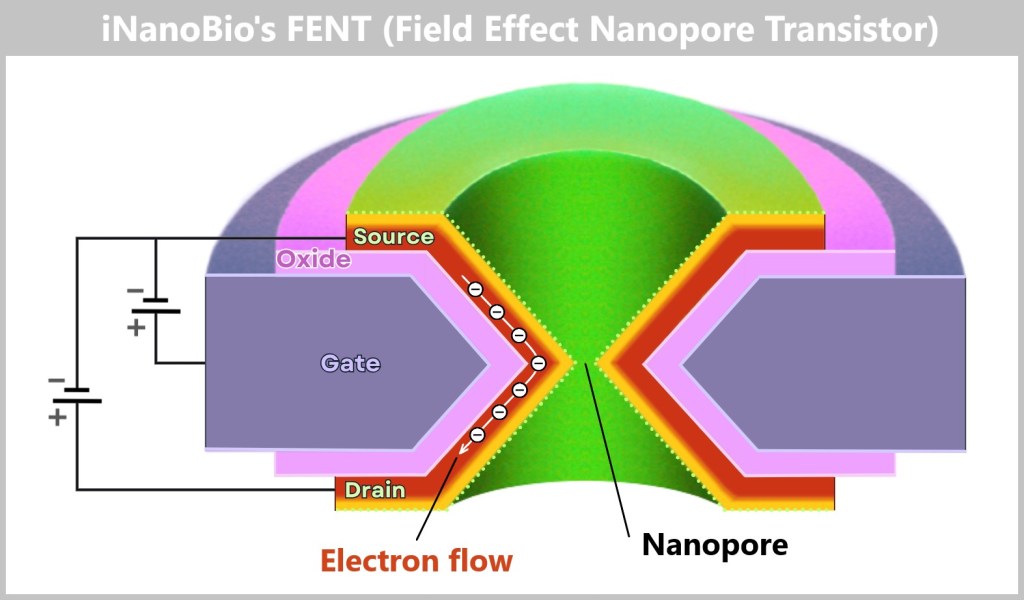
Fig. 10-B: Cross-section of a FENT The FENT is a cylindrically constructed MOSFET that fully encloses a nanopore. A voltage is applied between source and drain, allowing a small current to flow through the semiconductor of the pore wall – the so-called source-drain channel current. The thin oxide layer insulates the gate from the channel and enables precise detection of changes in the electric field. The nanopore serves as a sensing zone for local field variations caused by passing DNA bases.
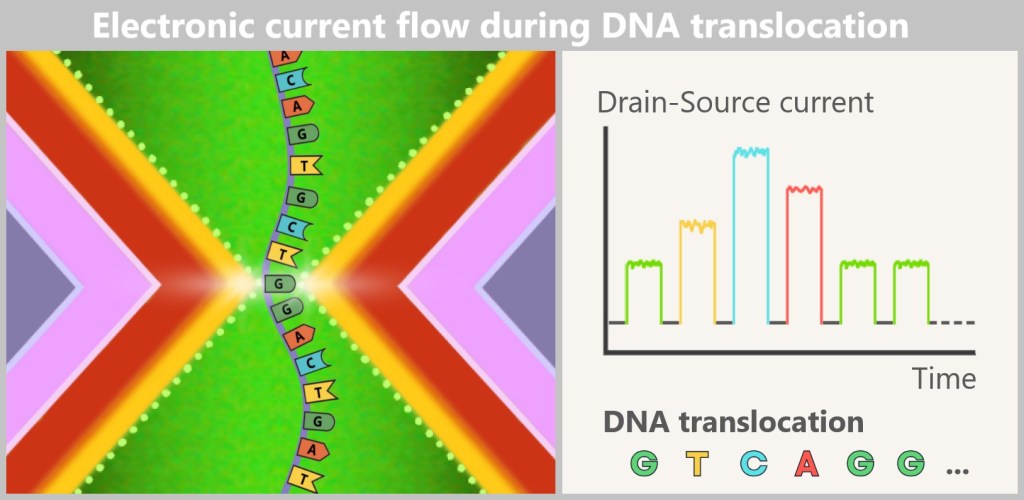
Fig. 10-C: FENT during DNA translocation The figure illustrates the operating principle of a FENT during the passage of a single-stranded DNA (ssDNA) through the nanopore. Before measurement begins, the originally double-stranded DNA (dsDNA) is separated into its two single strands by heat or chemical treatment. Only one single strand is then guided through the nanopore.
In the initial state – i.e., without DNA in the pore – a constant quiescent current flows between the source and drain through the semiconductor that forms the pore wall. This current is determined by the voltage between the source and drain and the gate potential.
However, as soon as a DNA base (A, T, C, or G) passes through the nanopore, it affects the electrical behavior of the system:
Due to its specific chemical structure and atomic distribution, each base has its own tiny electric field. This field penetrates the extremely thin pore wall and acts on the semiconductor like a local gate signal. This causes a local change in the charge carrier density (electron concentration) in the transistor channel, which leads to a measurable change in the drain-source current.
These current changes are recorded as characteristic signals for the respective base. During translocation, this creates a sequence-dependent current trace, allowing the order of DNA bases to be determined electronically.A general-audience explanation is provided in the video FENT Nanopore Transistor Explainer Video.
Speed that sets the benchmark
According to iNanoBio, a FENT can read up to 1 million DNA bases per second and pore – about 100 times faster than previous nanopore technology. A 5 x 5 mm chip with 100,000 of these sensors could sequence a human genome in a few minutes. The costs would be significantly lower, and the error rate reduced.
Possible applications
- Early cancer detection from blood samples
- Mobile diagnostics for infections directly on-site
- Large-scale research studies enabled by high speed
In the long term, iNanoBio aims to make DNA analysis devices as widespread as today’s blood pressure monitors – available in clinics, laboratories, doctors’ offices, and potentially even at home. Early prototypes exist, and the path to mass production is underway.
If the technology delivers on its promise, genome sequencing could soon become as routine as a standard check-up – fundamentally transforming our understanding of health.
b) SBX: When DNA Stretches to Be Understood
In the race to improve DNA analysis, the pharmaceutical company Roche is pursuing an astonishingly simple approach with SBX (Sequencing By eXpansion): it artificially enlarges DNA so that its „letters” A, T, C, and G are easier to distinguish from one another.
Step 1 – From the original to the enlarged copy
SBX creates a surrogate copy of the original DNA, called the Xpandomer. The DNA fragment is fixed to a tiny anchor on a substrate, and a polymerase is used to create a copy – but not from natural nucleotides, rather from surrogate nucleotides.
A surrogate (from Latin surrogatum = „substitute”) generally refers to a replacement or substitute for something else.
These consist of:
- The corresponding base (A, T, C, or G)
- A folded extension strand specific to each base, serving as a signal marker
- A cleavable bond that holds the strand in shape
- A Translocation Control Element (TCE) that precisely regulates the process later
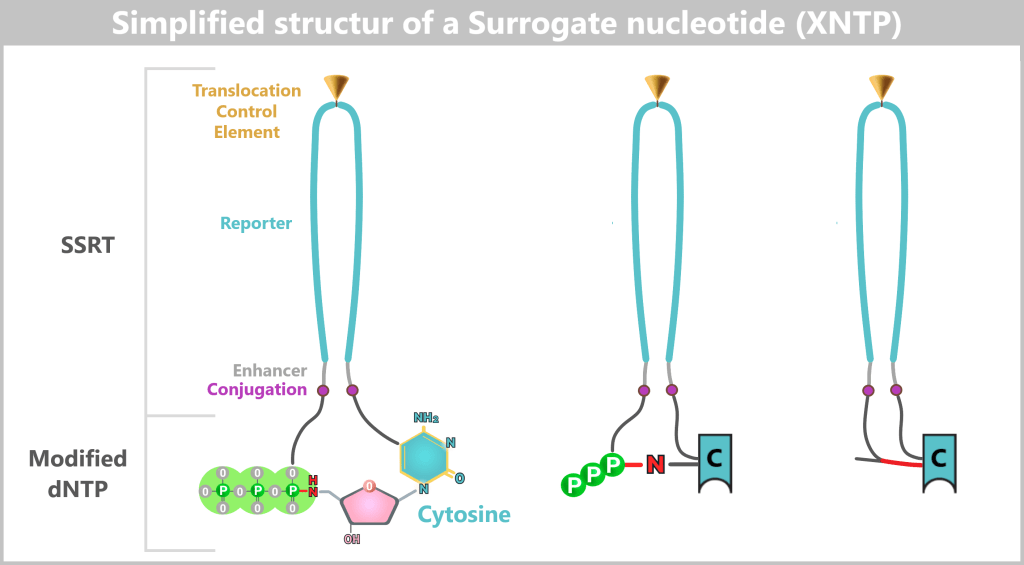
Fig. 11-A: Simplified representation of a surrogate nucleotide
[Sequencing by expansion (SBX) technology]The surrogate nucleotide (XNTP) consists of two functional components: the modified nucleotide and the SSRT (symmetrically synthesized reporter tether).
The modified dNTP contains the actual base (in this example, cytosine, C), which is connected to the reporter strand via an acid-sensitive bond (highlighted in red).
This is followed by the triphosphate (PPP), which enables the polymerase to incorporate the nucleotide into the growing chain.
The reporter tether (SSRT) consists of a folded extension strand with a Translocation Control Element (TCE) at its tip, which later controls passage through the nanopore.
An enhancer region (gray/purple) facilitates incorporation by the polymerase and stabilizes the structure.
After synthesis, the red acid-sensitive bond can be cleaved – causing the reporter strand to „unfold” and generating the extended Xpandomer molecule during sequencing.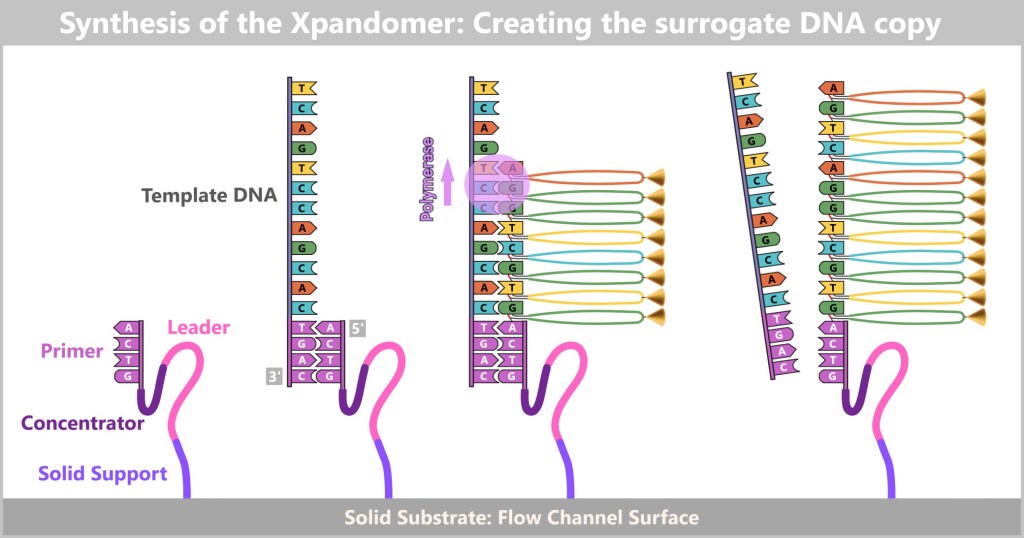
Fig. 11-B: Synthesis of the Xpandomer [Sequencing By Expansion] The DNA fragment (template DNA) is fixed to a solid substrate via an anchor.
A primer binds to the DNA template, supported by the leader and concentrator, which stabilize the start point and facilitate proper positioning.
Fixation – The DNA template is anchored to the substrate, and the primer section hybridizes to the matching region of the DNA.
Extension – a specialized polymerase extends the template bases, but not with natural nucleotides; instead, it uses surrogate nucleotides (XNTPs), each carrying a reporter tether.
Xpandomer formation – at the end, an artificially expanded DNA strand is produced: a surrogate copy (Xpandomer) in which the bases are replaced by characteristic reporter strands.
The Xpandomer initially remains anchored to the substrate and is released in a later step via light or chemical cleavage.After synthesis, the Xpandomer is separated from the original DNA, the cleavable bonds are broken – the strands unfold and increase the spacing between bases by 50-fold. This greatly facilitates subsequent identification.
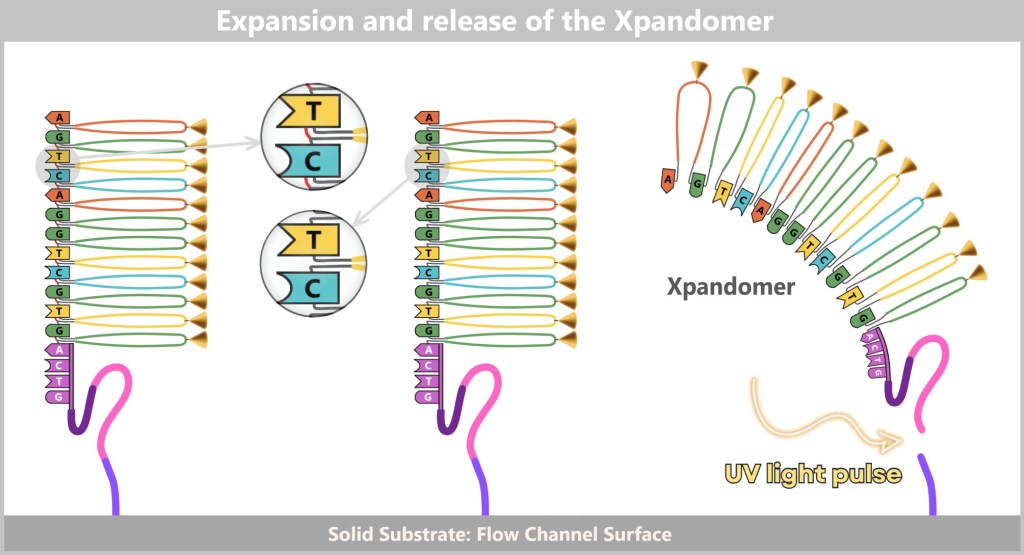
Fig. 11-C: Expansion and release of the Xpandomer [Sequencing By Expansion] After synthesis, the Xpandomer is chemically unfolded („expanded”) on the substrate and then released.
Left) Expansion: A mild acid treatment cleaves the cleavable bonds (red) between the base and the reporter strand. This causes the reporter structures to unfold – transforming the compact double structure into an elongated Xpandomer that represents the sequence of bases.
Right) Release: A UV light pulse cleaves the bond between the substrate and the leader region. The now freely moving Xpandomer remains structurally marked via the leader and concentrator – these regions later assist in alignment and entry into the nanopore.
From a dense DNA template, a long, readable molecule is created, whose reporter segments make the genetic information electrically visible.Step 2 – Sequencing
Instead of the DNA itself, the Xpandomere is now pulled through nanopores on a sequencing chip – around eight million of them working in parallel. The Xpandomere’s adapter guides it precisely into a pore.
When an extension strand reaches the pore, it temporarily sticks to the TCE. Each base generates a specific electrical signal. After 1.5–2 milliseconds, a voltage pulse releases the TCE, allowing the next strand to follow. This produces a clear, easily distinguishable signal trace – more precise than with Oxford Nanopore, where the bases generate a somewhat „blurred” signal.
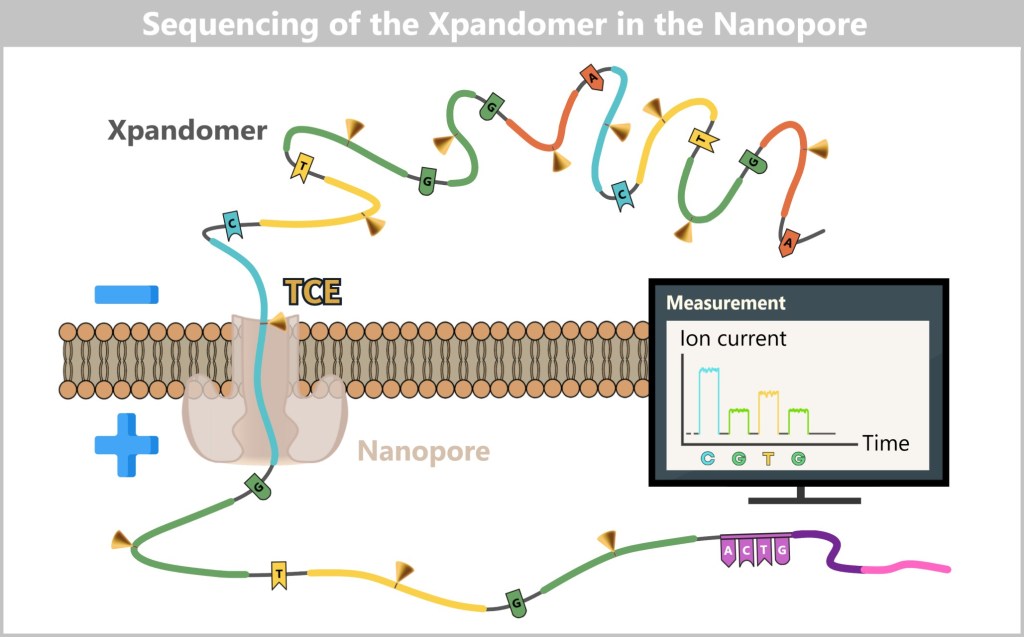
Fig. 11-D: Sequencing of the Xpandomer in the nanopore
[Sequencing By Expansion, Introduction to Sequencing By Expansion]The released Xpandomer is drawn toward the nanopore by an electric field. There, the TCE (Translocation Control Element) controls entry and temporarily holds the reporters in place.
As each reporter segment (the extended „base”) passes through the pore, the ion current changes in a characteristic way: each base generates its own electrical signal – comparable to an individual barcode. This sequence of signals is recorded in real time:
After each measurement, a voltage pulse releases the strand from the TCE, allowing the next segment to be drawn in.The following animation illustrates the basic principles of Roche’s SBX technology.
A detailed presentation is available here.
Advantages and performance:
- Speed: Millions of Xpandomers are read simultaneously and in real time.
- Accuracy: Signal-to-noise ratio comparable to Illumina.
- Flexibility: Read lengths of over 1,000 bases possible – faster and more precise than Nanopore, longer than Illumina.
Initial tests show that SBX can decode up to seven complete human genomes per hour at 30× coverage. In urgent cases, the entire process – from sample preparation to finished analysis – takes only around 5.5 hours.
The market launch is planned for 2026. Whether SBX will prevail against Illumina and Oxford Nanopore depends on price, availability, and user requirements.
c) The G4X System: From the Genetic Recipe to the Spatial Atlas of the Cell
Our body consists of billions of cells that function like tiny kitchens. DNA is their cookbook – it contains all the recipes of life. When a recipe is needed, the cell creates an RNA copy that serves as a blueprint for proteins. These proteins carry out specific tasks: they build structures, transmit signals, or fight pathogens.
However, proteins rarely act alone. They operate in finely tuned networks whose activity depends on where, when, and with which partners they interact. Processes such as immune responses or tumor growth do not arise within a single cell, but from the interplay of many specialized cells – often located in different regions of a tissue. Biology is therefore not only biochemical, but also spatially organized. Classical sequencing methods, however, typically capture only genetic content, not its spatial distribution.
Gene activity directly within tissue
The G4X Spatial Sequencer from Singular Genomics reveals where genes are active and proteins are formed in tissue – directly in preserved tissue samples (FFPE, formalin-fixed and paraffin-embedded).
FFPE samples are commonly used in pathology, for example in cancer diagnostics. They act like a „frozen snapshot,” except that chemical fixation with formalin is used instead of cold. Formalin crosslinks proteins and nucleic acids, thereby fixing molecular structures in their spatial context.
What is preserved:
- RNA (often fragmented and chemically modified) – reflects gene activity at the time of fixation.
- Proteins – indicate which structures and signaling pathways were active at that moment.
This makes it possible to reconstruct months or years later which genes and proteins were present and active in the tissue at that exact moment – as if time had been stopped. With the G4X, this „frozen” molecular landscape can be visualized at high resolution, including the precise spatial location within the tissue.
Fig. 12-A: From tissue to gene activity A tissue consists of many specialized cells. Each cell uses only a subset of its genetic program. RNA makes the active genes visible. RNA molecules are read by ribosomes and translated into proteins. FFPE tissue sections preserve this spatial organization – forming the basis for using the G4X system to measure where specific RNA molecules are active within the tissue.
How the G4X works
You can think of the G4X as a microscope-scanner with a built-in laboratory, directly measuring which genes and proteins are active within the tissue – and in their spatial context.
Preparation: A very thin section of the FFPE sample is placed on a specialized substrate.
RNA detection: Padlock probes are used to identify the spatial distribution of specific RNA sequences within the tissue.
Padlock probes – DNA lassos for target sequences
Imagine you want to find a specific object (an RNA sequence) in a vast area (the tissue). A padlock probe is a DNA fragment whose two ends are complementary to adjacent sections of the target sequence.
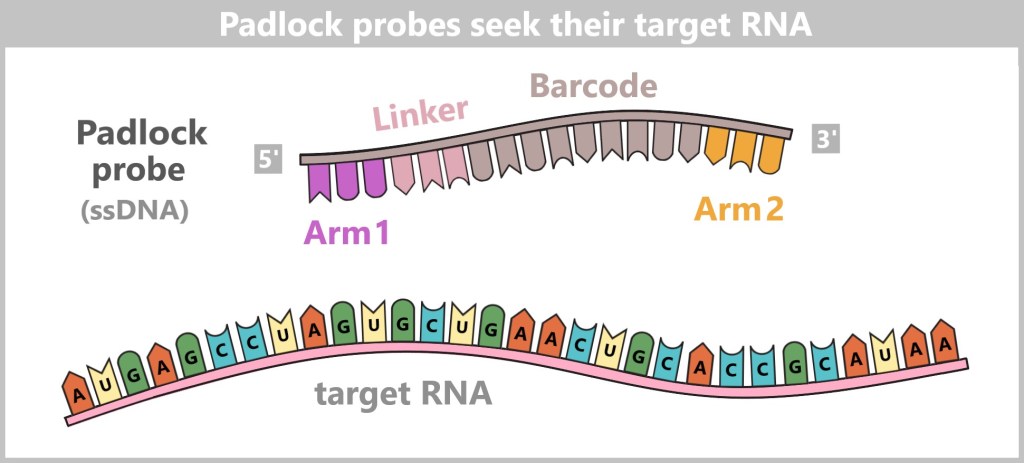
Fig. 12-B: A padlock probe floats through the tissue in search of its target RNA. Padlock probe: single-stranded DNA (ssDNA) 5’– Arm1 – Linker/Barcode – Arm2 – 3′
Arm 1 and Arm 2 are complementary to adjacent sequences of the target RNA.
Between them lie the linker and barcode, which carry additional identification information and do not bind.When the probe binds to its target, its two ends lie next to each other and can be closed into a stable DNA ring by ligation.
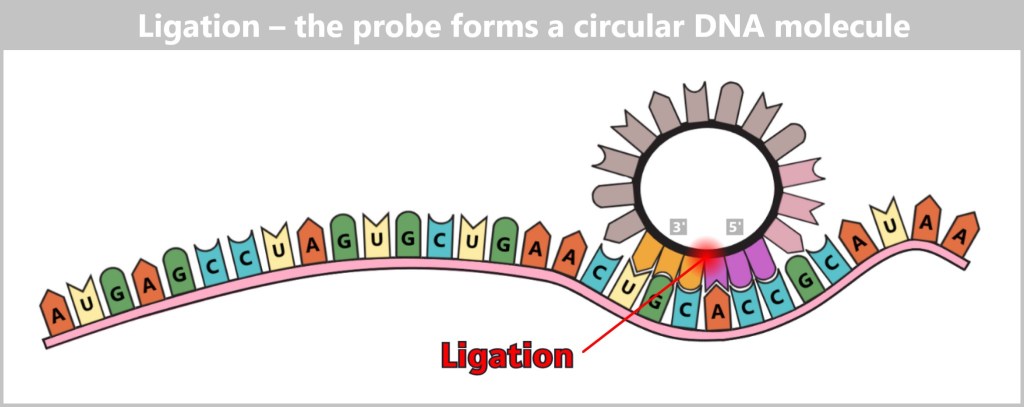
Fig. 12-C: Ligation connects the ends of the padlock probe After both arms of the probe have bound to the target RNA, a ligase joins the 5′ and 3′ ends. This forms a closed DNA loop that marks the target RNA and is ready for amplification.
If there is a variable sequence (e.g., a mutation) between the binding sites, it is first transcribed into DNA using a reverse transcriptase and incorporated into the probe.
The resulting DNA circle serves as a template for rolling circle amplification (RCA):
- A DNA polymerase repeatedly moves around the circle, generating a long DNA strand that contains multiple repeats of the target sequence – like an endless ribbon or a ball of yarn.
- These signals remain precisely at the location in the tissue where the RNA was.
- Using fluorescently labeled nucleotides and sequencing by synthesis, the sequence is decoded directly within the tissue.
Each Padlock probe carries a barcode that reveals which gene or variant it has detected. The result: a precise map showing which RNA was present at which location and which variants it carried.
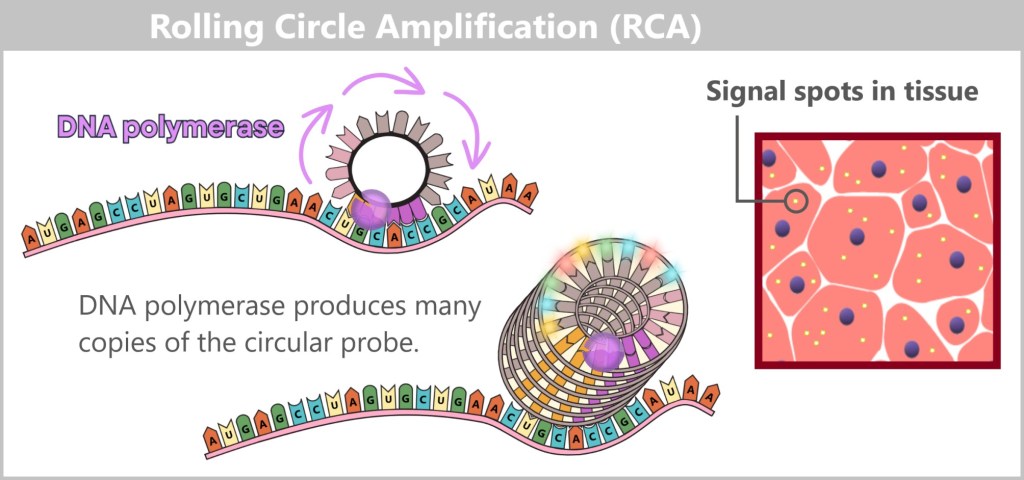
Fig. 12-D: Rolling Circle Amplification (RCA) A specialized DNA polymerase repeatedly reads the circular padlock probe, generating a long DNA strand with many repeats. Fluorescent nucleotides label the amplificates, producing a strong fluorescent signal – visible as a bright spot in the tissue, marking the location of the target RNA.
Protein detection in the same tissue section
For protein detection, the G4X system uses antibodies tagged with short DNA barcodes. After the antibody binds to its target protein, a padlock probe hybridizes to the barcode. The probe is then ligated, amplified, and read out – just like in RNA analysis. The barcode clearly reveals which protein is involved and where it occurs in the tissue.
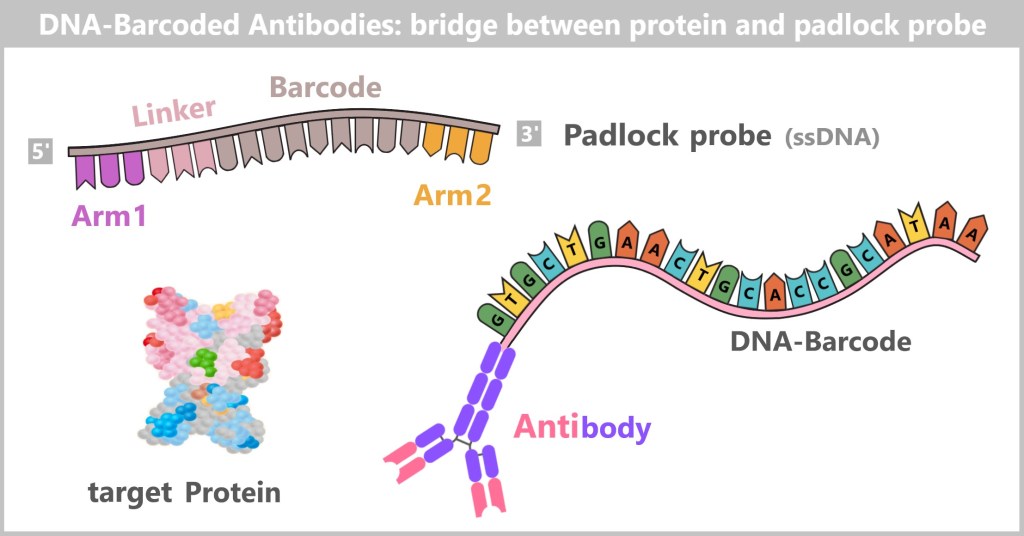
Fig. 12-E: Protein detection in the G4X system: antibody with DNA barcode and padlock probe An antibody specific to a target protein is coupled with a unique, short single-stranded DNA strand (barcode).
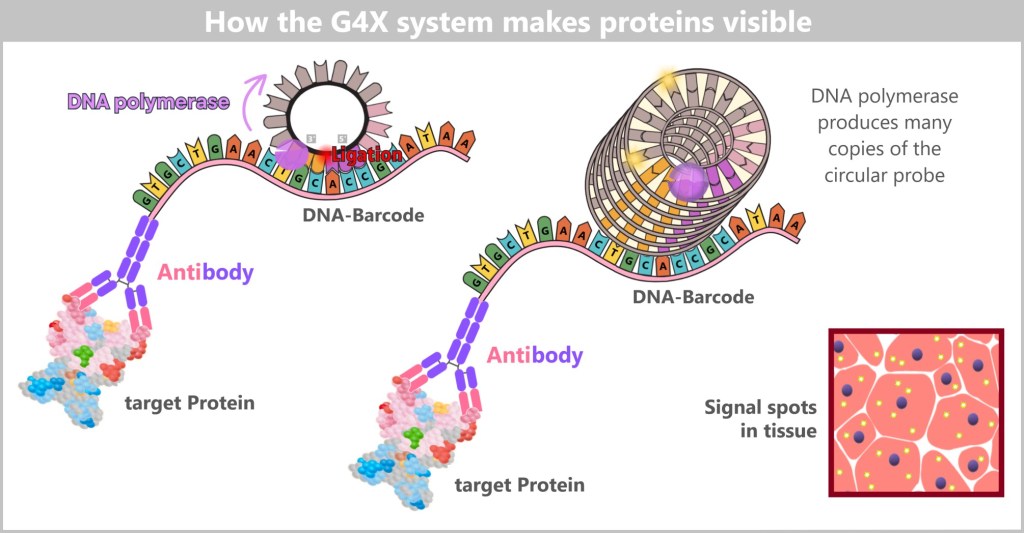
Fig. 12-F: Protein detection in the G4X system The antibody binds to its target protein in the tissue sample. A padlock probe, whose ends are complementary to two adjacent sections of the DNA barcode, binds to it and is linked by a DNA ligase to form a closed ring. A DNA polymerase recognizes the ring and reads it continuously, creating a locally anchored, circular DNA amplificate – analogous to RNA detection. The incorporation of fluorescently labeled nucleotides makes this amplificate visible. The fluorescent signal spots in the tissue mark the locations where an antibody with a specific DNA barcode has bound a particular protein.
A detailed presentation of the G4X method can be found here and here.
Why this is important
The G4X can specifically detect biomarkers that provide insights into the presence, progression, or aggressiveness of a disease – revealing not only which ones are active but also where they are located within the tissue.
Especially in tumors, this spatial resolution reveals which genes are „switched on”, in which cell regions this occurs, and how tumor and immune cells interact. This allows for a better understanding of tumor heterogeneity, clarification of disease mechanisms, and more targeted therapy planning.
The simultaneous in situ detection of RNA and proteins provides a comprehensive picture of tissue organization and opens up new possibilities for:
- Understanding complex disease processes
- More precise identification of biomarkers
- Development of personalized therapies
In diseases such as cancer, chronic inflammation, or neurodegenerative disorders, this spatial multi-omics perspective can be crucial – because often it is not the question of whether, but where and how the best therapeutic measure should be taken.
Conclusion
Genome sequencing is on the verge of a quantum leap: technologies like FENT, SBX, and G4X promise to read the genome faster, more accurately, and more comprehensively than ever before – including direct analysis within tissue. Soon, complete genomes could be decoded in minutes. The real revolution, however, begins afterward: when we understand the wealth of genetic information, translate it into medical knowledge, and use it responsibly for the benefit of humanity.
4.6. From Code to Cure
Bioinformatics as the Key to Tomorrow’s Medicine
4.6.1. From the Genetic Code to Computer-Aided Genome Analysis
Never before has it been so easy to decode DNA – but what use is the text if no one understands it? A small swab, a drop of blood – and suddenly millions of genetic data points are available. Modern sequencing has made the genome more accessible than ever before: fast, cost-effective, and nearly error-free. Yet this gives rise to a new challenge: how do we transform this flood of data into usable medical knowledge?
The answer comes from a discipline that has long operated in the background but has now become indispensable: bioinformatics – the bridge between A, T, G, C, and diagnosis, between raw sequence data and concrete therapeutic decisions. To understand why it is so important, it is worth taking a brief look back.
From the laws of inheritance to the genetic code
As early as 1860, Gregor Mendel discovered that there must be „heritable units” – genes – long before DNA was known. It was not until the mid-20th century that Oswald Avery demonstrated that these carriers of information are in fact DNA. Shortly thereafter, Watson and Crick revealed the double-helix structure with its complementary base pairing (A–T, G–C). In 1958, Francis Crick formulated the „central dogma”: information flows from DNA to RNA to proteins – the functional building blocks of life.
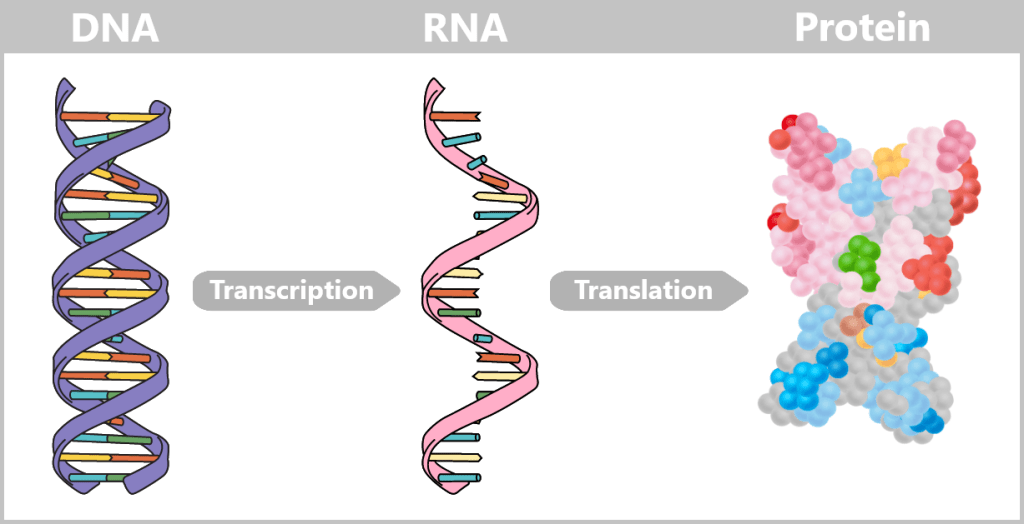
Fig. 13-A: The central dogma of molecular biology: Genetic information is converted from DNA into RNA through the process of transcription. The RNA is then translated into the final protein, which performs a wide variety of functions. [An overview of artificial intelligence in the field of genomics]
A gene is therefore an instruction for how a protein is made. But how do we read this instruction – and how do we interpret it correctly? The answer initially came via the detour of reverse genetics.
In the 1960s, DNA was still difficult to access directly – proteins, on the other hand, were functionally visible (for example as enzymes, transport molecules, or structural components) and could be isolated and analyzed with the methods available at the time. Researchers had already known since the early 20th century that proteins consist of chains of amino acids. From the 1960s onward, it became clear that each amino acid corresponds to a so-called codon on RNA – a triplet of RNA bases (A, U, G, C) – which can in turn be traced back to a DNA sequence.
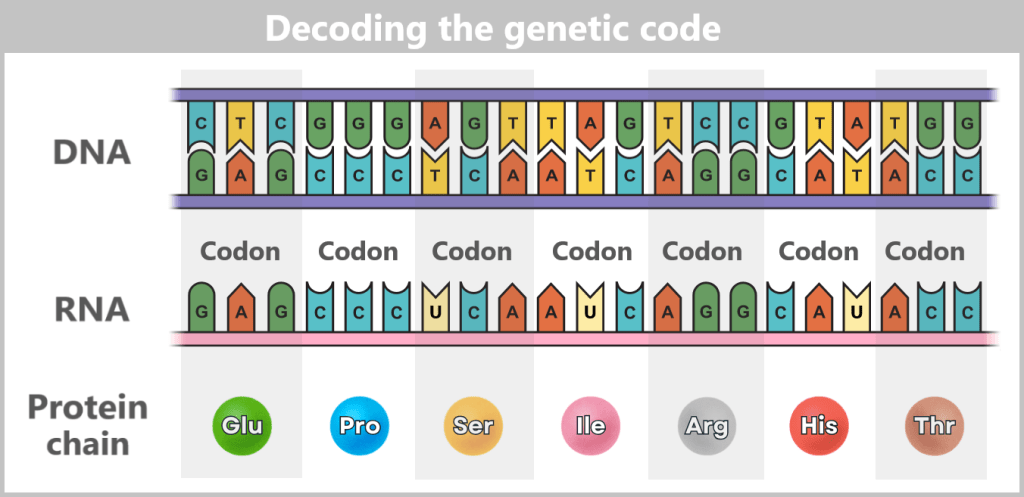
Fig. 13-B: The Central Dogma of Molecular Biology: The DNA Code and Codons Researchers started with a known protein and worked their way back to the RNA and DNA sequence. For the first time, the genetic code became readable like a recipe book:
Protein = finished dish
Amino acid = ingredient
Codon = word for this ingredient in the handwritten recipe (RNA)
DNA = page in the cookbook from which the recipe was copiedEarly patterns and the birth of genome annotation
Even before actual DNA sequencing, typical structural elements within genes were discovered:
Promoters – start signals where „reading” of a gene begins
Start and stop codons – first and last signals in the recipe
Transcription factors – proteins that switch genes on or off
Coding sequences – list of ingredients (codons) and their order for the protein
Non-coding DNA – regions without direct protein-coding function, whose role was initially unclearThese insights laid the foundation for modern genome annotation – the computer-assisted identification of biological signals within DNA sequences.
The Sanger Moment: DNA Becomes Readable (since 1977)
With Frederick Sanger’s method, DNA sequences could be determined directly for the first time – initially only for short segments. Analyses focused on gene identification and simple comparisons. Early programs such as FASTA (1985) and BLAST (1990) enabled the matching of new sequences with databases like GenBank (1982).
Understanding of gene structure also evolved: it was discovered that only certain parts of a gene (exons) are used for protein synthesis, while other segments (introns) are spliced out of the transcribed RNA. Additionally, DNA regions that enhance (enhancers) or repress (silencers) gene expression were identified – often located far from the actual gene.
The process of genome annotation began to take shape but was still rudimentary: DNA was primarily viewed as a linear sequence of nucleotides, with a focus on protein-coding genes. Regulatory elements and non-coding DNA were only partially recognized and understood. Bioinformatics was born – initially as a tool for research laboratories.
The Human Genome Project: Data on a Gigantic Scale (1990–2004)
The goal of decoding all 3.2 billion bases of the human genome introduced entirely new challenges: millions of short sequence fragments had to be assembled into long DNA strands. Tools such as Phrap for assembly and later Ensembl for annotation were developed. Data volumes exploded – along with the need for powerful computing infrastructure.
But while the Human Genome Project was still underway, a new technology was already emerging that would catapult speed and data volume into completely new dimensions.
NGS Revolution and Clinical Breakthrough (2005–2015)
Next-Generation Sequencing (NGS) technologies made genomes more affordable and rapidly accessible – albeit as massive data torrents of billions of short fragments. New tools like Bowtie, BWA, and STAR aligned these sequences at lightning speed, GATK helped identify mutations, and tools such as SnpEff or VEP interpreted their significance.
At the same time, sequencing strategies emerged for different clinical questions:
- Whole Genome Sequencing (WGS) for complete analyses
- Whole Exome Sequencing (WES) for the 1–2% of protein-coding DNA
- Targeted Sequencing for defined gene panels
This made bioinformatics a diagnostic tool for the first time – for example, in cases of hereditary forms of cancer, rare diseases, or personalized pharmacotherapy.
Artificial Intelligence and Modern Bioinformatics (2015–Present)
The combination of artificial intelligence (AI) and bioinformatics represents one of the most dynamic and promising areas of research of our time. Together, they are unraveling the complexity of life and accelerating biomedical discoveries at an unprecedented pace.
Artificial Intelligence: More Than Just Automation
AI encompasses techniques in which machines perform human-like intelligence tasks – particularly learning, problem-solving, and pattern recognition. In the biomedical context, the following subfields are especially relevant:
Machine Learning (ML): Algorithms learn from data (e.g., genome sequences, protein structures, patient data) to make predictions or identify patterns without being explicitly programmed. Example: detecting tumor cells in tissue samples.
Deep Learning (DL): A subset of ML that uses artificial neural networks with many layers („deep”) to capture highly complex patterns in massive datasets. Example: predicting the 3D structure of proteins solely from their amino acid sequence. [AlphaFold].
Natural Language Processing (NLP): Enables computers to understand scientific literature, medical records, or clinical study reports and extract knowledge from them.
4.6.2. Modern Bioinformatics – The Digital Toolbox of Biology
Bioinformatics is the science of storing, analyzing, and interpreting biological data using computer-based methods. Its modern form is characterized by three factors:
Explosion of biological data: Next-Generation Sequencing (NGS) generates terabytes of genomic, transcriptomic, and epigenomic data per experiment.
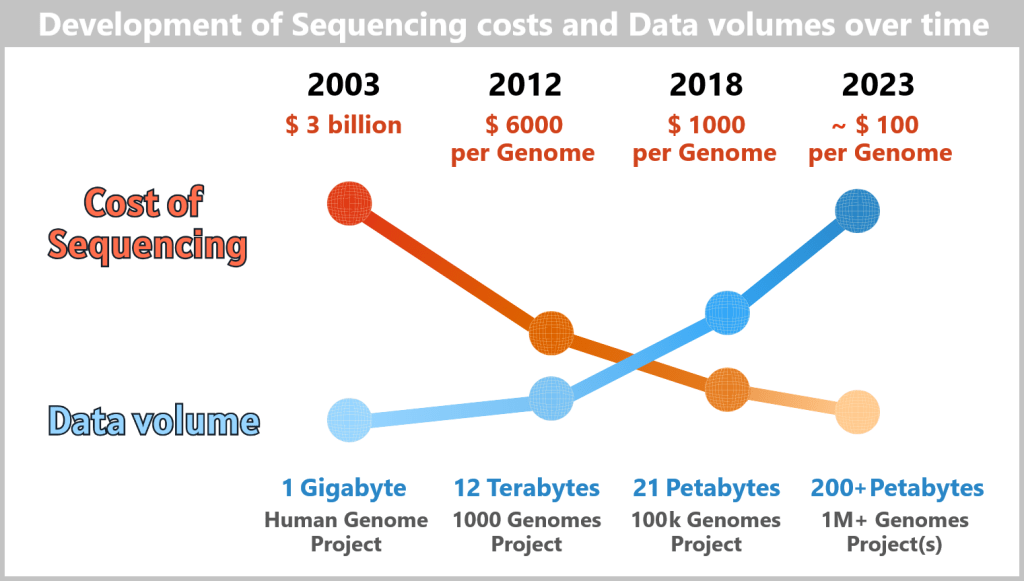
Fig. 12: From a billion-dollar project to an everyday routine
– how the costs of genome sequencing fell while data volumes exploded –Just over twenty years ago, decoding a human genome was still a monumental undertaking: the Human Genome Project (completed in 2003) cost around 3 billion US dollars and produced only about 1 gigabyte of data. With ever faster sequencing machines and new technologies such as nanopore sequencing, this picture has changed completely. By 2012, the cost per genome had already dropped to around 6,000 dollars, in 2018 to 1,000 dollars – and today (2023), a genome can be analyzed for approximately 100 dollars.
At the same time, the volume of generated data increased rapidly: from a few gigabytes to terabytes and petabytes, and ultimately to the hundreds of petabytes produced by global projects involving millions of genomes. Estimates suggest that by 2025, around 40 exabytes of human genomic data could be generated – equivalent to roughly eight times the storage required for all words ever spoken in human history. [How AI Is Transforming Genomics]
Complexity of biological systems: Understanding disease requires the integration of data on genes, proteins, metabolic pathways, and cellular interactions.
Need for predictive medicine: The goal is to predict disease risks, therapy responses, and individualized treatment (precision medicine).
Already in many key applications, we can already talk about a fusion between bioinformatics and artificial intelligence (AI). Here are some central areas of application:
a) Genome Analysis and Sequencing
AI algorithms are used to analyze DNA sequences more quickly and precisely. Models such as DeepVariant, developed by Google, employ deep neural networks to identify genetic variants with high accuracy. This is crucial for detecting mutations associated with diseases such as cancer or rare genetic disorders.
b) Protein Folding and Structure Prediction
A milestone of AI in bioinformatics is DeepMind’s AlphaFold, which predicts protein structures with unprecedented precision. This has far-reaching implications for drug development, as a protein’s 3D structure determines its function.
c) Multi-Omics Data Analysis
A holistic understanding of biological systems requires the integration of multi-omics data (genomics, proteomics, transcriptomics, epigenetics, metabolomics) to gain a comprehensive picture of their functioning. AI helps to decipher the complex interactions between these layers.
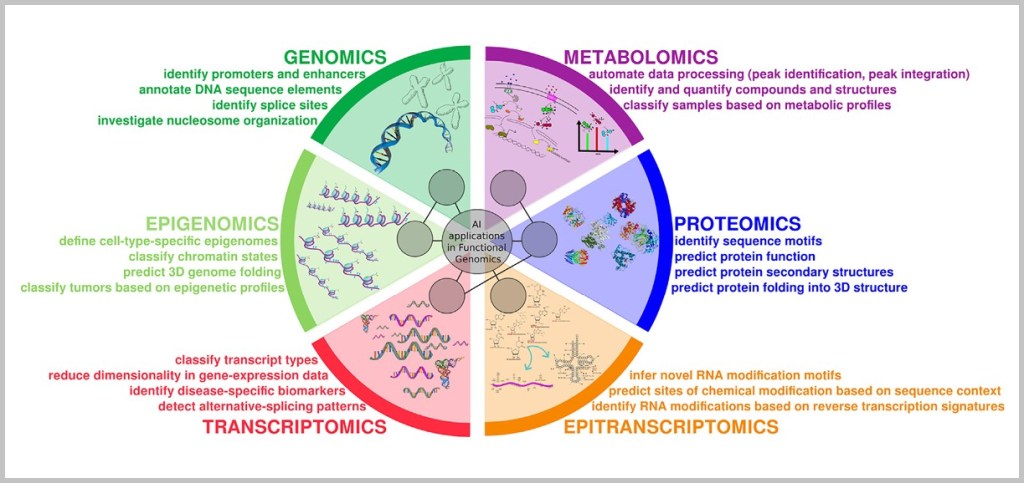
Fig. 13: The Polyphonic Concert of Life. [AI applications in functional genomics] Biological systems are a complex interplay of different layers of information: from DNA (genomics) through epigenetic regulation, RNA (transcriptomics/epitranscriptomics), and proteins (proteomics) to metabolic products (metabolomics). It is only their dynamic interaction that defines health and disease.
For example, the new AI tool AlphaGenome demonstrates how even the smallest changes in DNA can affect gene activity as well as the production of RNA and proteins. It predicts with high precision the consequences of a single DNA base substitution for genes and their products. While earlier systems typically analyzed only the roughly 2% of the genome that codes for proteins, AlphaGenome is the first to capture the entire genome. It is based on a multi-omics analysis that directly links DNA alterations to their effects on RNA and proteins. This creates a comprehensive picture of gene expression – a crucial step toward more precise genomic research and personalized medicine.
d) Metagenomics and Microbiome Analysis
AI helps to understand the complex interactions in the human microbiome by recognizing patterns in the data from microorganisms. This is important for research into diseases such as diabetes or intestinal disorders.
e) Drug Development
AI accelerates nearly every phase of the lengthy and costly drug discovery process: target identification, virtual screening of millions of molecules, prediction of drug effects and side effects, and optimization of molecular structures. Companies such as BenevolentAI and Recursion use AI platforms to identify promising drug candidates in record time, some of which are already being tested in clinical trials.
f) Analysis of Biomedical Images
AI is used in image analysis, for example in the evaluation of MRI or CT scans, to detect tumors or other anomalies. Convolutional neural networks (CNNs), which mimic the functioning of the human visual cortex, have proven to be particularly effective in this area.
g) AI Genomics in Health Research
Gene expression in the human body does not follow a fixed pattern – and diseases such as cancer make it even more difficult to decipher. This is precisely where spatial biology comes in: using cutting-edge microscopy and genetic sequencing, it reveals how genes are active in individual cells and even within their organelles.
But it is not only the visible gene regions that are decisive. In regions long dismissed as „junk DNA”, researchers today suspect key switches of gene regulation. It is there that decisions are made about which proteins are produced – and which are not. AI and deep learning models can detect patterns in these previously hidden regions that are linked to disease. This raises hopes for new biomarkers and therapeutic approaches. One example is the platform developed by the biotech company Deep Genomics:
The combination of spatial biology, intelligent data analysis, and AI thus enables an unprecedented view into the molecular logic of life – down to the level of individual cells.
New methods of gene editing could make it possible to precisely control the behavior of individual genes – for example, to deactivate cancer-promoting genes or to stimulate the regeneration of cartilage cells in osteoarthritis. This field is still highly experimental, but AI promises to make these techniques more precise and safer.
With AI-supported CRISPR techniques, new possibilities are emerging to not only identify and modify genes more precisely, but also to better understand their interplay within complex biological networks. Early approaches suggest that this could reduce side effects in the future and increase the effectiveness of new therapies. Modern methods for the systematic testing and optimization of gene variants could also – if they prove successful – represent important steps toward personalized treatments.
Those who are interested in learning more about CRISPR techniques can find further information here.
The combination of advances in AI and genomics is therefore considered highly promising and could, in the long term, accelerate the development of new cell and gene therapies. However, since these treatments are based on living cells that vary widely and require specific conditions, much of this work is still in the experimental stage. AI could help make processes more stable, reproducible, and clinically applicable – but whether and to what extent these hopes will be realized remains to be seen.
h) Oncology
AI models analyze genetic profiles to create personalized treatment plans. For example, algorithms can predict how patients will respond to specific medications based on their genetic makeup. This is particularly relevant in oncology, where AI-based systems such as IBM Watson Oncology provide treatment recommendations.
Conclusion
Bioinformatics today serves as the brain of modern medicine. Together with artificial intelligence, it transforms the flood of data from the genome into concrete insights – from diagnosing rare diseases to developing new drugs.
Whether in genome analysis, protein structure prediction, or the planning of personalized therapies, AI-powered bioinformatics reveals what was previously hidden. It paves the way for a form of medicine that not only treats diseases but anticipates them – tailored to each individual patient.

5. A Success Story
The case of KJ Muldoon shows just how much these developments are already a reality today.
When the young boy received his diagnosis just a few hours after birth, his fate seemed sealed. He suffers from an extremely rare metabolic disorder caused by a single DNA point mutation (SNV), which prevents his body from breaking down toxic ammonia. Without a functioning enzyme, toxic metabolic products rapidly accumulate – posing a life-threatening risk even in infancy. Until now, only a risky liver transplant was considered capable of ensuring his survival.
But in KJ’s case, doctors and researchers took a new approach. Within just six months, they designed a therapy created exclusively for him: a personalized base-editing treatment.
The principle behind it is as ingenious as it is elegant: an RNA-guided system scans the genome, specifically locates KJ’s point mutation, and binds to that spot. Instead of cutting the DNA strand completely – as is the case with traditional CRISPR-Cas9 – an enzyme chemically modifies a single base: exchanging one DNA „letter” to correct the disease-causing mutation. To put it simply: it’s like fixing a single typo in a book without cutting out or rewriting the entire paragraph – extremely precise and gentle on the genome. The treatment was administered directly in the body („in vivo”) through multiple consecutive rounds, specifically targeting the liver.
The initial results are encouraging. KJ can now digest more proteins, infections are milder, and he is gaining weight. For his parents, this means a piece of everyday life without constant danger to his life – and, above all, new hope for their child’s future.
Behind this medical breakthrough lies a success story involving several disciplines. Without modern genome sequencing, the exact mutation would not have been visible. Without bioinformatics, it would not have been possible to design the appropriate base editing tool in such a short time and with such precision. And without the concept of personalized medicine, it would have been unthinkable to develop a cure exclusively for a single patient.
The therapy is still experimental, and no one knows whether its effects will be lasting or if long-term side effects might occur. Yet KJ’s case illustrates the direction medicine is heading: toward a form of healthcare that no longer relies on standard protocols, but on individualized solutions – driven by data, molecular biology, and tailored therapies. A medicine that not only treats diseases but, ideally, targets them precisely at their genetic root.
Further background information on this story can be found in this video.
6. A Look into the Future: The Synthetic Human Genome Project
Biology is on the verge of another quantum leap. After decoding and editing genomes, the vision now is to rewrite them from scratch. This is precisely the goal of the Synthetic Human Genome Project (SynHG) – an ambitious undertaking that could redefine the boundaries of biology and medicine.
With an initial funding of £10 million from the Wellcome Trust and under the leadership of Professor Jason Chin at the Generative Biology Institute in Oxford, leading universities such as Cambridge, Manchester, and Imperial College London are working on this project. The mission: to develop tools and technologies for creating synthetic human genomes – a step that could transform our understanding of life itself.
Recomposing a Genome
Unlike genome editing, which only modifies existing DNA, SynHG goes much further: it aims to reassemble the genome letter by letter from scratch. While CRISPR corrects individual typos in the text of life, this approach is about completely rewriting an entire chapter.
The first milestone: the synthesis of a complete human chromosome as proof of feasibility. It is the beginning of a journey that could take decades – and yet it builds on earlier pioneering achievements, such as the Human Genome Project (2003) and the Synthetic Yeast Project, in which synthetic yeast chromosomes were created for the first time in 2023.
Modern methods – generative AI, machine learning, and robot-assisted assembly – are intended to tackle this enormous task.
Potential Applications – Between Laboratory Vision and Clinical Practice
The opportunities are vast. At the level of somatic cells – i.e., cells whose changes are not passed on to offspring – new avenues for regenerative medicine could open up:
- tailor-made replacement tissues and organs grown in the lab,
- virus-resistant cells against HIV or SARS-CoV-2,
- gene therapies against cancer or hereditary diseases such as cystic fibrosis.
Such applications are not distant science fiction – they could become reality within the next 10 to 20 years, as technologies such as CRISPR and stem cell cultures are already well established.
The situation is quite different when it comes to germline editing – i.e., interventions in egg or sperm cells. In theory, this could permanently prevent serious hereditary diseases such as Huntington’s disease. However, the idea of „designer babies” with desired characteristics such as appearance or intelligence remains highly problematic from a scientific and ethical perspective, as such traits depend on hundreds of genes.
The project itself is limited to experiments in test tubes and cell cultures. It does not aim to create artificial life. Nevertheless, it opens up an unprecedented opportunity for researchers to intervene in fundamental human biological systems – with far-reaching consequences that must be critically examined.
Risks and Open Questions
The technical hurdles are enormous. In somatic cells, there is a risk of mosaicism (not all cells incorporate the new genome) or tumor formation due to unintended genetic alterations. In the germline, the risks are even greater: developmental disorders, epigenetic effects, or unpredictable consequences across generations.
Researchers are therefore discussing safety mechanisms such as genetic „kill switches”, which could deactivate synthetic cells in case of an emergency.
Ethics: Between Healing and Hubris
Somatic therapies are akin to organ transplants: controversial, but socially accepted when they cure. Germline interventions, however, touch taboo areas – fueled by concerns about social inequality or even eugenic scenarios. Many countries, including Germany, prohibit them through laws such as the Embryo Protection Act. International organizations like WHO and UNESCO call for strict regulation.
„To build trust, we need to actively involve society”, emphasizes sociologist Professor Joy Zhang from the University of Kent, who is investigating the ethical dimensions of the project. With her Care-full Synthesis initiative, she is attempting to initiate an open dialogue about the opportunities, but also the limitations, of this research.
Renowned geneticist Professor Bill Earnshaw from the University of Edinburgh, who has developed methods for producing artificial human chromosomes, puts it more bluntly: „The genie is out of the bottle. We could have a set of restrictions now, but if an organisation who has access to appropriate machinery decided to start synthesising anything, I don’t think we could stop them.“
In this context, the call by numerous professional organizations – including the International Society for Cell and Gene Therapy (ISCT) – for a ten-year moratorium on the use of CRISPR and related techniques in the human germline seems even more urgent. Such a postponement appears not only to be a precautionary measure, but also a necessary political response in order to enable social debates, ethical guidelines, and international control mechanisms in the first place.
Ethics expert Arthur Caplan from New York University describes the dilemma from a scientific perspective:
„… So if you ask me, will we see genetic engineering of children aimed at their improvement? I say yes, undoubtedly. Now when? I’m not sure what the answer to that is.“
„… It will come. There are traits that people will eagerly try to put into their kids in the future. They will try to design out genetic diseases, get rid of them. They will try to build in capacities and abilities that they agree are really wonderful. Will we hang up these interventions on ethical grounds? For the most part, no, would be my prediction, But not within the next 10 years. The tools are still too crude.“Outlook
SynHG opens a window into a future where we can not only read and edit the genome, but write it. Somatic applications – personalized therapies, regenerative tissues, and custom-designed cells – could soon shape the practice of medicine.
Interventions in the germline, however, remain a distant horizon – for technical, legal, and ethical reasons. The project represents not only a scientific challenge but also a delicate societal balancing act.
Given the potential for misuse of the technology, the question arises as to why Wellcome decided to provide funding at all. Dr. Tom Collins, who was responsible for this decision, explained to BBC News:
„We asked ourselves what was the cost of inaction. This technology is going to be developed one day, so by doing it now we are at least trying to do it in as responsible a way as possible and to confront the ethical and moral questions in as upfront way as possible.“
While Europe is still grappling with how to handle the topic carefully in public debate, across the Atlantic the term „new life forms” is approached much more openly. Eric Nguyen, a bioscientist at the renowned Stanford University, outlines a far more visionary perspective in his TED Talk, „How AI Could Generate New Life-Forms“.
In a striking way, he demonstrates how artificial intelligence (AI) could transform biology – not only by „reading” and „writing” DNA, but also by deliberately creating novel organisms. AI models such as machine learning and generative algorithms are already capable of recognizing complex patterns in biological data: in genomes, protein structures, or metabolic pathways. Nguyen goes further, arguing that in the future AI could also design entirely new DNA sequences – the starting point for previously unknown life forms.
This vision is undoubtedly forward-looking, but it raises fundamental questions: What priorities should be set? How can the use of such technologies be justified in a world of massive resource inequality? Who funds the research, who oversees the outcomes – and by what criteria is it decided which life forms are „useful” or „safe”?
Nguyen’s presentation makes it clear that AI-supported biotechnology could not only enable solutions to global challenges, but also forces us to think about the ethical, social, and philosophical dimensions of „creating life.”
The message: AI-powered biotechnology promises tremendous advances, yet simultaneously raises profound ethical and societal questions – challenging the very foundations of what it means to be human.
Klaus Schwab – Founder and former Chairman of the World Economic Forum
„We must address, individually and collectively, moral and ethical issues raised by cutting-edge research in artificial intelligence and biotechnology, which will enable significant life extension, designer babies, and memory extraction.“
[BrainyQuote]
7. Personalized Medicine and Smart Governance
The Biomolecular Revolution in Governance
7.1. Power, Governance, and Smart Governance
7.2. Governmentality
7.3. Personalized Medicine as a Catalyst for Biomolecular Governmentality
7.4. The Global Trend: Worldwide Biomolecular Governmentality
7.5. Conclusion: The Global Ambivalence of Biomolecular PowerThe decoding of the human genome marked not only a medical turning point, but also the beginning of a new era of political governance. What started as a large-scale scientific project in laboratories and data centers has long since reached the control apparatus of modern states: the precise measurement, categorization, and utilization of our biological foundations.
The vision of personalized medicine promises individualized therapies, precise predictions about disease risks, and more efficient use of scarce resources. But this narrative of progress falls short if we consider it solely from a medical, ethical, or data protection perspective. In fact, we are witnessing a fundamental transformation of power and governmentality – a biomolecular revolution in governance.
Before we delve deeper into the subject, it is important to clarify a few key terms.
7.1. Power, Governance, and Smart Governance
Governance refers to the art of steering societal processes, making decisions, and maintaining order. Traditionally, this occurs through the interaction of the state, markets, and civil society.
Smart governance transforms this understanding: it operates through digital technologies, data analytics, and algorithmic processes. Governance thus becomes continuous, procedural, and possible in real time. Power no longer manifests solely as political authority, but as a network of information flows, feedback loops, and predictions.
Power itself is not a possession, but a relationship: the ability to influence behavior, steer actions, or frame decisions. It operates both visibly – through institutions and laws – and subtly, by shaping expectations, norms, and bodies of knowledge.
7.2. Governmentality
Michel Foucault, a major thinker in the analysis of power structures, coined the term of governmentality to describe the totality of forms of knowledge, practices, and techniques through which modern societies are governed. In this sense, governing does not merely mean the exercise of coercion, but also the shaping of spaces for action that encourage individuals to regulate themselves in line with societal objectives.
Key dimensions of governmentality are:
The Interconnection of Knowledge and Power
Knowledge is never neutral. It emerges through the collection and interpretation of data, is embedded in power processes, and structures social orders. With digitalization, this connection becomes central: big data and artificial intelligence provide predictions, pattern recognition, and decision-making foundations that intervene deeply in societal processes. Smart governance operates precisely at this point: data become the foundation of political legitimacy and the exercise of power.
Biopolitics
Biopolitics refers to the regulation of life itself – health, reproduction, labor, education, migration, or security. The goal is to organize entire populations through laws, standards, and political programs. Today, biopolitics is linked to digital control: Smart Health, Smart Cities, and Smart Economies illustrate how knowledge is translated into concrete government practices through data.
Dispositives of Power
Biopolitics works through dispositifs: networks of institutions, laws, discourses, and practices. Hospitals, schools, media, and digital platforms are part of these structures. They standardize behavior not through overt coercion, but through subtle framing of what is considered „normal” or „desirable”. In this way, decisions are indirectly guided and behavior is embedded in everyday life.
Disciplinary Power
Alongside biopolitics, which regulates entire populations, disciplinary power targets the individual. It operates through surveillance, reward, punishment, and training. Its goal is not only obedience, but the productive shaping of bodies and behaviors. External directives are transformed, through sanctions and incentives, into internal motivations that drive individuals toward self-discipline.
Technologies of the Self
Technologies of the self refer to practices through which individuals internalize societal norms and autonomously adjust their behavior. Digital applications – such as fitness apps, learning platforms, or financial tools – translate societal directives into personal goals. This creates a form of power that operates not only from the outside, but is carried forward from within: people follow rules because they perceive them as meaningful and beneficial for themselves.
Those interested in exploring the art of governance in more detail can find further information here.
7.3. Personalized Medicine as a Catalyst for Biomolecular Governmentality
At first glance, personalized medicine, supported by modern genomics and bioinformatics, appears to be purely a medical and technical advance. But when viewed through the lens of governmentality, it emerges as a powerful catalyst for a profound transformation of power structures and governance practices. The precise analysis of individual genetic profiles and their linkage to comprehensive databases not only individualizes healthcare, but also changes the way societies are governed and individuals are regulated.
Knowledge & Power: From the Genome to the Governable Body
The decoding of the genome marked the initial explosion of knowledge, but it is personalized medicine that makes this knowledge governable. Bioinformatics and modern genomics do not merely generate data – they create predictive knowledge that forms the basis for interventions. This knowledge is by no means neutral: it categorizes individuals according to genetic risk profiles (e.g., „high risk for breast cancer,” „predisposed to cardiovascular diseases”). These categorizations become the foundation of new power structures. Both states and private actors use this data to predict disease risks, optimize healthcare budgets, and manage preventive measures. Social order is thereby reshaped – from access to insurance and labor markets to the prioritization of preventive resources.
Smart governance manifests here in algorithmic decision-making processes that appear objective but are, in fact, normative. Who gains access to which therapies, or which population groups are classified as „at risk”, is increasingly determined by data-driven models that invisibly reproduce existing power relations („We act based on the best available evidence”).
The CMS Health Tech Ecosystem Initiative, officially announced on July 30, 2025, during the „Make Health Tech Great Again” event at the White House, exemplifies the trend toward the emergence of new power structures. CMS (Centers for Medicare & Medicaid Services) is a U.S. federal agency within the Department of Health and Human Services (HHS) that manages healthcare programs such as Medicare and Medicaid and regulates the national healthcare market. The initiative aims to create smarter, safer, and more personalized healthcare, driven through partnerships with innovative private-sector companies. During the event, the U.S. government secured commitments from major health and IT firms – including Amazon, Anthropic, Apple, Google, and OpenAI – to lay the foundation for a next-generation digital health ecosystem.
According to U.S. President Donald Trump, this gives the U.S. healthcare systems a high-tech upgrade, ushering in „an era of convenience, profitability, speed, and – frankly – better health for the people”. (Note the order…)
Biopolitics 2.0: Optimizing the Population at the Molecular Level
Classical biopolitics regulated populations through statistics on birth rates or mortality. Personalized medicine elevates this to a new, molecular level. Genomics fragments the population into ever smaller, biomolecularly defined subgroups. People are no longer categorized merely by age or gender, but by genotype, risk alleles, and biomarkers („BRCA1 carriers”, „individuals with the LCT gene for lactose intolerance”). This enables hyper-precise and individualized regulation – biopolitics becomes granular.
The „healthy”, „normal” body is no longer defined solely as one without symptoms, but as one with a „normal”, „low-risk” genome. Deviation begins not with disease, but with genetic predisposition. This creates a new class of „presymptomatic patients” or „risk subjects”, who are managed biopolitically through more frequent screenings, prevention programs, vaccination campaigns, prophylactic surgeries, lifestyle recommendations, or reproductive decisions based on genetic forecasts.
While Foucault saw the nation state as the main actor in biopolitics, power is increasingly shifting to private actors today: big tech and biotech companies that control data, algorithms, and technologies. They effectively define what genetic „normality” and „risk” mean.
We now speak of the economization of biological life. Genetic data have become a valuable commodity – a bio-resource. Large technology and pharmaceutical companies accumulate bio-data to generate profits: in drug development, therapy recommendations, and personalized advertising. The human body and its biomolecular data thus become part of capitalist logic of exploitation. The individual themselves becomes „human capital”, seemingly obliged to optimize his genetic potential.
In an NBC News report on August 26, 2025, U.S. Health Secretary RFK Jr. stated that the 60 largest technology companies would soon allow Americans access to their personal health data – data that had been economically exploited for years without their consent: “You are going to be able to see, by next year, all your health records on your cellphone.”
What is presented here as a political achievement merely confirms the status quo.
Dispositifs of Power: The Network of Biomolecular Surveillance
The dispositive network that supports this new form of biopolitics is multi-layered: pharmaceutical companies develop targeted therapies; biobanks and genetic databases provide the raw material; algorithms analyze and interpret it; doctors communicate results and recommendations; digital health apps monitor compliance. Each link in this chain functions as a power dispositif: it defines what is considered „healthy” or „sick“ and subtly conveys what „responsible health care” means in the age of genetics. Genetic testing, for example, is marketed as „responsible” behavior – thus leading individuals to participate unknowingly in biopolitical goals such as prevention or cost efficiency.
This development is reflected in national health strategies that promote genetic screenings to minimize disease burdens. The goal is to maximize the „health dividend” while simultaneously reducing costs – a biomolecular rationalization strategy.
Initiatives such as the 100.000 Genomes Project or Genomics England’s Newborn Genomes Programme move in the same direction. By comparison, the European Health Data Space (EHDS) – an EU-wide initiative for the networking and secure use of health data – is far more comprehensive. The EHDS provides the data and regulatory infrastructure that makes large-scale personalized medicine possible in the first place.
For most citizens, this sounds like a new beginning: more participation, better therapies, more precise prevention. Yet this promising vision casts a shadow. In practice, unequal opponents face each other: on one side, the individual patient contributing their genetic profile, lab results, and medical history; on the other, global pharmaceutical companies, data platforms, and technology providers with multi-billion-dollar budgets that refine, patent, and commercialize this data. Cui bono? – who benefits? The answer is sobering: quick profits go to the big players, while the promises remain with the individual.
While the EHDS emphasizes citizens’ rights – more control, more transparency, more self-determination – anyone who has experienced the complexity of consent processes in health apps, insurance companies, or electronic patient records knows: there is a gap between formal options and actual capacity to act. While Goliath has lobbying channels, legal experts, and data centers at its disposal, David is left with informational brochures and trust.
The EHDS is thus both a government technology and a power project. It decides whether data is treated as a public good or as a commodity market. In any case, Goliath will benefit. The open question is whether David will be left with more than just the role of a fig leaf. The health data space will only become truly emancipatory if benefits and risks are distributed fairly – through binding public welfare requirements, transparent accountability, and real returns for patients. Otherwise, the great promise of „empowerment” will remain a narrative that merely conceals existing asymmetries.
Disciplinary Power: Internalized Biomolecular Surveillance
While biopolitics focuses on the population as a whole, personalized medicine directs its disciplinary effect at the individual. The genetic risk profile acts as a constant, internalized monitor. Those who know they have an increased risk of colon cancer adjust their behavior: dietary habits, preventive medical appointments, and exercise routines are adapted to the genetic forecast.
The electronic patient record (ePA) intensifies this dynamic. By centrally storing genetic and medical data and making them accessible at any time, it transforms medical recommendations into an ever-present digital authority. Personal notifications, reminders for medication intake, personalized reports, or interfaces with health apps reinforce self-monitoring: the external directive („You must get screened”) becomes an internal voice („I must take care of myself because my genome requires it”). The ePA structures and institutionally legitimizes this self-discipline, making it invisible and seemingly voluntary – governed simultaneously by digital infrastructure, social norms, and medical authority.
The result is a subtle yet pervasive form of power: it does not manifest through sanctions, but through the constant, digital concern for one’s own – genetically anticipated – future. The ePA turns the individual into a disciplined actor of their own risk profile, while the surveillance formally disappears but remains technically and institutionally omnipresent.
Technologies of the Self: The Genetic Self-Optimizer
Here, personalized medicine appears in its purest form as a technology of the self. At-home DNA tests, fitness trackers, and health apps are the tools through which individuals internalize societal norms of health and performance and actively work on themselves. They translate the abstract concept of genetic predisposition into concrete, everyday actions: an extra serving of vegetables, 10,000 steps, meditation to reduce stress. The individual governs themselves according to a biomolecular logic – not because the state commands it, but because they seek to optimize their genetic destiny. Power relations thus become invisible and present themselves as expressions of individual sovereignty and self-care.
7.4. The Global Trend: Worldwide Biomolecular Governmentality
Personalized medicine is no longer a national phenomenon, but part of a global transformation of power and governance. Worldwide, initiatives are emerging that pool genomic data, health information, and digital technologies in order to translate populations and individuals into ever more precise risk profiles. The logic of Foucault’s governmentality – the linkage of knowledge, power, and self-regulation – manifests itself here at a transnational level.
In the United States, programs such as the All of Us Research Program of the National Institutes of Health or the Million Veteran Program collect genetic and medical data from millions of citizens. The collection of diverse health data serves not only scientific research, but also forms the basis for algorithmic risk profiles, preventive interventions, and resource-optimized healthcare strategies.
Canada is pursuing a comparable goal with the Canadian Precision Health Initiative and the Pan-Canadian Genomics Network: the systematic collection and interconnection of biomolecular data, combined with an infrastructure that enables secure governance, international collaboration, and economic utilization.
This is where Biopolitics 2.0 becomes visible: populations are fragmented, governed, and optimized at the molecular level.
Europe constitutes an orchestrated network of transnational biopolitics. The European Health Data Space (EHDS), supported by initiatives such as ICPerMed and ERA PerMed, pools genetic, clinical, and lifestyle data across national borders.
National biobanks such as the UK Biobank or the French Health Data Hub act as dispositifs that generate both knowledge and power: they determine which data is used, how populations are classified, and which health policy interventions appear legitimate.
Through platforms such as SHIFT-HUB or the Helmholtz Initiative iMed, the international networking of science, industry, and society is institutionalized – constituting a transnational form of smart governance that scales Foucault’s concept of technologies of the self to the global level.
The global logic is also evident in Asia and the Pacific region: the GenomeAsia 100k Project, Singapore’s National Precision Medicine Strategy, and China’s National Health Data Platform link biomolecular data with AI-driven analyses to enable preventive, individualized, and population-based governance.
Australia and Israel rely on digital infrastructure that reinforces citizens’ self-discipline – electronic patient records, genetic databases, and personalized apps function as subtle yet omnipresent dispositifs of power.
Even global research projects such as the Human Cell Atlas illustrate the transnational dimension: by mapping the diversity of human cells, they create a universal network of knowledge and power that provides the foundation for personalized therapies worldwide. The boundaries between state governance, private-sector power, and scientific expertise blur: Foucault’s classical distinction between the nation-state and disciplinary power is replaced by global networks and international collaboration.
7.5. Conclusion: The Global Ambivalence of Biomolecular Power
The global expansion of personalized medicine clearly demonstrates that the biomolecular revolution is no longer confined to individual nation-states. It manifests as a transnational network of data, algorithms, and governance that steers both populations and individuals at the molecular level. Global initiatives – from the United States and Europe to Asia, Australia, and Israel – intertwine biopolitics, disciplinary power, and technologies of the self on an international scale.
The ambivalence remains central: on the one hand, these developments open up opportunities for more precise therapies, individualized prevention, and potentially greater health equity. On the other hand, power asymmetries arise in which states, international organizations, and private actors control access to data, resources, and technologies. Individuals are increasingly becoming disciplined subjects of their own genetic profiles, while global dispositifs unnoticedly enforce normative and economic goals.
This seamlessly combines the local experience of personalized medicine with a global governance logic: smart governance and biomolecular biopolitics have long been internationalized, and the challenge lies in distributing the benefits and risks fairly. The global dimension of this transformation makes it clear that the biomolecular revolution is irreversible not only medically, but also politically and socially – a global system of control that offers opportunities for health and risks for the concentration of power.

8. Epilogue
The desire to optimize health and longevity is a primal instinct of humankind. Even in ancient civilizations, health was not considered a matter of chance, but rather a desirable state that could be achieved through lifestyle and philosophy. Greek „dietetics” – a holistic practice combining nutrition, exercise, and mental hygiene – and the motto „mens sana in corpore sano” testify to a 2.500-year-old desire for optimization. This urge, fueled by the survival instinct, the fear of losing control, and the pursuit of quality of life, is not only ancient but eternal.
In modern times, personalized medicine embodies the latest expression of this dream. It promises to treat people not as averages, but as individuals, based on genetic code, biomarkers, and lifestyle data. But is it the fulfillment of this dream or a modern mirage? In its utopian vision, it suggests the complete controllability of biology, an overcoming of the „biological lottery”. The reality is more sobering: it enables more precise diagnoses and therapies, prevents treatment failures – but it does not abolish disease.
The idea of a disease-free existence remains an illusion. Evolutionary biology and biophysics set limits: pathogenic germs continue to evolve, cancer is a statistical consequence of cell division, and aging is an inevitable degenerative process. The realistic goal is therefore not the elimination of disease, but its transformation: converting fatal diseases into chronic conditions and maximizing the „healthspan” – the years spent in good health. This goal is radical, but achievable, as advances in the treatment of HIV or diabetes show.
But how far is humanity willing to go to achieve this goal? History shows that in the face of suffering and death, ethical boundaries often blur. Willingness ranges from everyday discipline and financial sacrifice to participation in experimental therapies. This raises an uncomfortable question: what compromises is society prepared to make? Personalized medicine requires data – genetic, medical, and personal. Collecting such data carries the risk of undermining privacy and autonomy. The COVID-19 pandemic was a stress test that showed how large parts of the population were willing to trade civil liberties for security. Models such as China’s social credit system illustrate one possible future scenario, in which transparency and control are rewarded in the name of health and safety. Yet such scenarios are not inevitable: they represent an extreme, not an unavoidable destiny.
Personalized medicine has two sides. Its pragmatic promise is more efficient, precise, and humane medicine that saves lives and alleviates suffering – for example, through tailored cancer therapies or early diagnosis. Its utopian but risky promise is the illusion of total control over life and death. It is thus the culmination of a journey that has lasted thousands of years. Whether this journey ends in emancipated self-optimization or a dystopian „biocracy” does not depend on technology, but on the ethical and political framework that society gives it.
The age-old dream of health seems more tangible today than ever before, yet it challenges us to find wise answers. Personalized medicine can be a tool of emancipation if we use it with care. The decisive question of modernity is therefore not only how much freedom we are willing to sacrifice for a longer life, but how we can reconcile health and freedom. Only in this way can the eternal pursuit of health become progress for all.
The world will not change all at once; it never does. Life will go on mostly the same in the short run, and people in 2025 will mostly spend their time in the same way they did in 2024. We will still fall in love, create families, get in fights online, hike in nature, etc.
But the future will be coming at us in a way that is impossible to ignore, and the long-term changes to our society and economy will be huge.
Sam Altman (CEO of OpenAI)
Sources (as of January 15, 2026)
AI, Artificial Intelligence, Bioinformatics, Biopolitics, Control, Crispr, Digital Health, DNA, DNA Sequencing, DNA Test, FENT, G4X Spatial Sequencer, Gene, Gene Defect, Gene Variation, Genetic Medicine, Genetics, Genome Sequencing, Genomics, Governance, Healthcare, Illumina, iNanoBio, Medicine, Michel Foucault, Next Generation Sequencing, NGS, Nvidia, ONT, Oxford Nanopore, PacBio, Personalized Medicine, Precision Medicine, Protein, RNA, Roche, Sanger sequencing, SBX Sequencing, Singular Genomics, Smart Governance, SMRT Sequencing, Surveillance, SynHG, Synthetic Human Genome Project, Transcriptomics, Xpandomer -
Personalisierte Medizin – Himmel oder Hölle

Als die HSBC Bank im Jahr 2013 im Rahmen ihrer globalen Kampagne „The future is full of opportunity“ an Flughäfen wie Gatwick und Dubai Plakate mit einem Fingerabdruck, einem QR-Code und dem provokanten Slogan „Your DNA will be your data“ platzierte, war die Botschaft unmissverständlich: Die Zukunft wird persönlich – vielleicht sogar beunruhigend persönlich. Die Bank wollte sich als zukunftsorientiertes Unternehmen positionieren, das Digitalisierung, Künstliche Intelligenz, Nachhaltigkeit und Genetik offensiv thematisiert.
Der Slogan wirkte doppeldeutig – als verheißungsvolle Vision ebenso wie als alarmierender Ausblick. Kritiker schlugen sofort Alarm: Deine DNA wird lesbar, analysierbar und digital verfügbar – ein offenes Buch über Herkunft, Gesundheitsrisiken, vielleicht sogar Persönlichkeitsmerkmale – vergleichbar mit biometrischen Daten wie Fingerabdrücken oder Gesichtsscans. Was einst als das Intimste am Menschen galt, wird nun zur Datei: maschinell entschlüsselbar, potenziell speicherbar, womöglich handelbar. Der Mensch wird reduziert auf Information – auf „Bits und Bytes“, die verarbeitet, verkauft oder missbraucht werden könnten.
Wer besitzt die Rechte an deinen genetischen Daten? Und wer hat Zugriff darauf? Du selbst? Die Firma, die den Test durchgeführt hat? Erreichen dadurch Überwachung, Datenlecks und Missbrauch durch staatliche und private Akteure eine neue Dimension? Die Vision von Designerbabys, genetischer Selektion und personalisierter Werbung auf Basis biologischer Marker wirft schwerwiegende ethische Fragen auf. Wird der Fortschritt am Ende mit dem Preis individueller Freiheit bezahlt?
Doch die Biotech-Revolution hat längst eine gewaltige Dynamik entfaltet, und immer mehr Stimmen erkennen in dieser Entwicklung ein enormes Potenzial. In der E-Book-Ausgabe der HSBC Bank Frontiers – Biotech – Five Big Ideas Shaping the Biotech Revolution von März 2024 heißt es:
„Die Idee, die Natur für Waren und Dienstleistungen zu nutzen, ist nicht neu. Doch in den 1970er Jahren, mit dem Aufkommen der Gentechnik und der Gründung der ersten Biotech-Unternehmen, begann die moderne Ära dieser Praxis. Dies brachte Wissenschaft und Wirtschaft zusammen wie nie zuvor, und der Kapitalzufluss beflügelte die Forschungs- und Entwicklungsbemühungen.
In den folgenden drei Jahrzehnten wurde das Feld durch bahnbrechende Geräte wie DNA-Sequenzierer und -Synthesizer beschleunigt, und die Biologie selbst wandelte sich langsam zu einer rechnergestützten, datengesteuerten Disziplin. Nach der Jahrtausendwende, als die Forscher immer größere Bereiche des menschlichen Genoms erfolgreich entschlüsseln konnten und die Kosten für die Sequenzierung dieses Genoms rapide sanken – von etwa 100 Millionen Dollar im Jahr 2001 auf weniger als 1.000 Dollar heute –, wurde das Potenzial der Biotechnologie, unser Leben zu verändern, auf elektrisierende Weise deutlich.
In den letzten Jahren wurden die Möglichkeiten der Biotechnologie – von der Behandlung genetisch bedingter Krankheiten bis zur Wiederbelebung ausgerotteter Tierarten – durch Fortschritte in ergänzenden Bereichen wie der künstlichen Intelligenz erweitert. Dies hat dazu beigetragen, dass in diesem Bereich ein neues Zeitalter angebrochen ist.“
Nirgendwo sonst prallen Nutzen und Risiko so direkt aufeinander wie in der personalisierten – oder präzisionsbasierten – Medizin. Ihr Anspruch: individuelle genetische und biologische Informationen zu nutzen, um Diagnosen zu präzisieren, Therapien zu individualisieren und Krankheiten frühzeitig zu erkennen. Die Vision ist vielversprechend: weniger Nebenwirkungen, bessere Heilungschancen, effizientere Prävention – Medizin, maßgeschneidert auf den Einzelnen.
Doch auch hier gilt: Was als medizinischer Fortschritt gefeiert wird, bringt gesellschaftliche, rechtliche und ethische Herausforderungen mit sich:
- Wer entscheidet, welche genetischen Daten relevant sind?
- Welche Rolle spielt der Zugang zu diesen Technologien – werden soziale Ungleichheiten dadurch vertieft?
- Und wie lassen sich Diskriminierung, etwa durch genetisches Scoring bei Versicherungen oder Arbeitgebern, verhindern?
Zwischen der Hoffnung auf individuelle Heilung und der Sorge vor kollektiver Entmündigung stehen grundlegende Fragen: Wird der Mensch durch den Zugriff auf seine biologischen Daten ermächtigt – oder entblößt? Ist personalisierte Medizin ein Segen – oder der Auftakt zu einer Ära subtiler Entmenschlichung?
Daher lautet die Gretchenfrage: Ist die personalisierte Medizin der verheißene Aufstieg in den Himmel – oder der Sturz durch die Tore der Hölle?
Dieser Blogbeitrag lädt dazu ein, die personalisierte Medizin in ihrer ganzen Komplexität zu betrachten – jenseits von Hype oder Alarmismus. Er richtet sich bewusst auch an fachfremde Leser, die sich kritisch mit den Chancen, Risiken und ethischen Spannungsfeldern dieser medizinischen Revolution auseinandersetzen möchten.
📑Inhaltsverzeichnis
1. Die begriffliche Einordnung und das grundlegende Paradigma
2. Treibende Kräfte – zwischen Vision und Wirklichkeit
3. Die Zukunft der Medizin beginnt in der Zelle
3.1. Wie Biologie und Technologie zusammenfinden
3.2. Daten, Verantwortung und der größere Rahmen
4. Von DNA zur Erkenntnis – ein Hochgeschwindigkeitszug der Innovation
4.1. Die DNA – Bauplan des Lebens
a) Die DNA: Das molekulare Informationsarchiv des Lebens
b) Vom Code zum Protein: Wie Anleitungen Wirklichkeit werden
c) Epigenetik & nicht-codierende DNA: Das Dirigenten-Team unseres Erbguts
d) Das verborgene Kontrollzentrum: Was die nicht-codierende DNA steuert
e) Wenn das Betriebssystem krank macht
f) Offene Rätsel der Forschung
g) Die wahre Magie der Gene
4.2. Genetische Variation und ihre Folgen
4.3. Genomsequenzierung: Schlüssel zum individuellen Erbgut
a) Die Reise der DNA: Vom Nabelschnurblut zur genetischen Information
b) Drei Regieanweisungen für die Gen-Diagnostik: WGS, WES und Panel
4.4. Methoden der Genomsequenzierung
4.4.1. Methode 1: Kopieren mit Leuchtfarbe
a) Sanger-Sequenzierung
b) Illumina-Technologie
c) Single-Molecule Real-Time(SMRT)-Sequenzierung
4.4.2. Methode 2: Hightech-Tunnel – DNA elektrisch abtasten
a) Oxford Nanopore Sequenzierung
4.4.3. Genomsequenzierung – Technologien im Vergleich
a) Durchsatz vs. Verwertbarkeit – Masse ist nicht immer gleich Klasse
b) Wirtschaftlichkeit – Kosten pro Base vs. Kosten pro Erkenntnis
c) Von der Probe zum Befund – Wie schnell spricht das ganze Genom?
d) Klinische Einsatzfelder – Welche Technologie passt wozu?
e) KI als Co-Pilot – Automatisierung ohne Verantwortung abzugeben
f) Vergleichstabelle: Sanger, Illumina, PacBio, ONT
g) Key Takeaways: Zwischen Reife und Routine
4.5. Genomsequenzierung: Der nächste Technologiesprung
a) FENT: Wie ein Mikrochip die DNA-Analyse revolutioniert
b) SBX: Wenn DNA sich streckt, um verstanden zu werden
c) Das G4X-System: Vom genetischen Rezept zum räumlichen Atlas der Zelle
4.6. Vom Code zur Heilung – Bioinformatik als Schlüssel der Medizin von morgen
4.6.1. Vom genetischen Code zur computergestützten Genomanalyse
4.6.2. Moderne Bioinformatik – Der digitale Werkzeugkasten der Biologie
5. Eine Erfolgsgeschichte
6. Ein Blick in die Zukunft: Das Synthetic Human Genome Project
7. Personalisierte Medizin und Smart Governance
7.1. Macht, Governance und Smart Governance
7.2. Gouvernementalität
7.3. Personalisierte Medizin als Katalysator biomolekularer Gouvernementalität
7.4. Der weltweite Trend: Globale biomolekulare Gouvernementalität
7.5. Schlussfolgerung: Globale Ambivalenz der biomolekularen Macht
8. Epilog1. Die begriffliche Einordnung und das grundlegende Paradigma
Die Entwicklung der modernen Medizin hin zu maßgeschneiderten Behandlungen wird häufig mit Begriffen wie „personalisierte Medizin“, „individualisierte Therapie“ oder „Präzisionsmedizin“ beschrieben. Doch was genau verbirgt sich hinter diesen Schlagworten?
Personalisierte Medizin gilt derzeit als einer der populärsten Begriffe – eingängig wie ein Werbeslogan. Damit verbunden ist das Versprechen, Therapien auf jeden Menschen zuzuschneiden: auf seine Gene, seine Lebensgewohnheiten, seine Biologie. In den Medien und in der öffentlichen Debatte wird der Ausdruck entsprechend häufig verwendet.
Individualisierte Therapie klingt weniger glamourös, meint aber Ähnliches: die Anpassung von Medikamenten oder Dosierungen an die Bedürfnisse einzelner Patienten. Dieser Begriff findet vor allem im klinischen Alltag Verwendung.
Präzisionsmedizin schließlich ist der nüchternste der drei Begriffe – ein Fachwort, das in Forschungslaboren und wissenschaftlichen Publikationen dominiert. Es unterstreicht den datenbasierten Ansatz: Algorithmen, Genanalysen und Biomarker entscheiden darüber, welche Therapie bei welcher Patientengruppe wirkt. Das ist die Sprache der Wissenschaft.
Doch egal, welcher Begriff gewählt wird – sie alle stehen für einen Paradigmenwechsel: Weg von standardisierten Verfahren – hin zu einer Medizin, die den einzelnen Menschen in seiner biologischen Einzigartigkeit in den Mittelpunkt stellt.
2. Treibende Kräfte – zwischen Vision und Wirklichkeit
Die treibenden Kräfte hinter der personalisierten Medizin sind vielfältig und eng miteinander verflochten. Auf der einen Seite stehen wissenschaftlich-technologische Entwicklungen, die in den letzten Jahrzehnten enorme Fortschritte ermöglicht haben: neue Methoden, um Erbgut schnell und genau zu lesen, leistungsfähige Datenanalyse durch künstliche Intelligenz sowie die Verfügbarkeit großer biomedizinischer Datenmengen. Diese Technologien bilden die Grundlage dafür, individuelle Unterschiede zwischen Patienten überhaupt erst erkennen und medizinisch nutzen zu können.
Auf der anderen Seite wirken gesellschaftliche und politische Impulse als Beschleuniger dieses Wandels. Öffentliche Gesundheitsprogramme, gezielte Forschungsförderung, Strategiepapiere internationaler Gesundheitsorganisationen und gezielte Gesetzesinitiativen treiben die Integration der personalisierten Medizin in die Versorgungspraxis aktiv voran. Auch das gestiegene Gesundheitsbewusstsein in der Bevölkerung sowie die zunehmende Erwartung an individuelle Behandlungsangebote verstärken diesen Trend.
Personalisierte Medizin ist also kein bloßes Produkt technologischer Machbarkeit, sondern entsteht im Spannungsfeld von Innovation, Politik und gesellschaftlicher Nachfrage.
Ein aktuelles Beispiel für die praktische Umsetzung dieser Entwicklung liefert das Vereinigte Königreich: Im Rahmen eines neuen 10-Jahres-Plans soll künftig jedes Neugeborene einem DNA-Test unterzogen werden. Die sogenannte Ganzgenomsequenzierung soll nach hunderten genetisch bedingten Krankheiten suchen – mit dem Ziel, potenziell tödliche Erkrankungen frühzeitig zu erkennen und ihnen gezielt vorzubeugen. Nach Angaben des britischen Gesundheitsministers Wes Streeting geht es darum, den National Health Service (NHS) grundlegend zu transformieren: weg von der reinen Krankheitsbehandlung – hin zu einem System der Vorhersage und Prävention. Genomik, also die umfassende Analyse der Erbinformation, soll dabei in Kombination mit künstlicher Intelligenz als Frühwarnsystem dienen.
„Die Revolution in der medizinischen Wissenschaft bedeutet, dass wir den NHS von einem Dienst, der Krankheiten diagnostiziert und behandelt, in einen Dienst verwandeln können, der sie vorhersagt und verhindert“, so Streeting. Die Vision: eine personalisierte Gesundheitsversorgung, die Risiken erkennt, bevor Symptome überhaupt auftreten – und damit die Lebensqualität verbessert sowie das Gesundheitssystem entlastet.
Doch so überzeugend diese Vision auch klingt – aus der Wissenschaft kommen auch mahnende Stimmen. So warnt etwa der Genetiker Prof. Robin Lovell-Badge vom Francis Crick Institute davor, die Komplexität der genomischen Daten zu unterschätzen. Es brauche nicht nur technisches Know-how zur Datenerhebung, sondern vor allem qualifiziertes Personal, das die gewonnenen Informationen verantwortungsvoll und verständlich kommunizieren kann. Denn Daten allein sind noch keine Diagnose – entscheidend ist ihre sinnvolle und patientengerechte Interpretation.
Was bedeutet es konkret, Medizin neu zu denken?
Die wahre Revolution spielt sich in den Laboren und Rechenzentren ab, in denen die Bausteine der personalisierten Medizin entstehen. Die folgenden Kapitel führen Schritt für Schritt hinter die Kulissen und zeigen die Schlüsseltechnologien, die nötig sind, um die personalisierte Medizin vom Konzept in die Klinik zu bringen.
3. Die Zukunft der Medizin beginnt in der Zelle
3.1. Wie Biologie und Technologie zusammenfinden
Unser Körper besteht aus Milliarden Zellen – winzigen, spezialisierten Einheiten, die Tag für Tag zusammenarbeiten, um Gesundheit und Leben zu ermöglichen. In jeder einzelnen Zelle laufen unzählige Prozesse ab, die genau aufeinander abgestimmt sind. Diese zellulären Mechanismen sind entscheidend für das Verständnis von Gesundheit, Krankheit und der Entwicklung neuer, gezielter Therapien.
Ein zentraler Mechanismus ist die Energiegewinnung: Zellen verwandeln Nährstoffe und Sauerstoff in ATP (Adenosintriphosphat)– die „Energie-Währung“ des Körpers. Ohne ATP könnte keine Zelle funktionieren, kein Muskel sich bewegen, kein Gedanke entstehen.
Gleichzeitig produzieren Zellen fortwährend Proteine – auf Basis genetischer Informationen. Diese komplexen Moleküle übernehmen zentrale Aufgaben im Körper – als chemische Katalysatoren, Botenstoffe, Baustoffe oder Transportmoleküle.
Auch die Reinigung und Erneuerung innerhalb der Zelle ist lebenswichtig: Prozesse wie die Autophagie bauen beschädigte Zellbestandteile ab und recyceln sie – eine Art innere Müllabfuhr, die Krankheiten wie Alzheimer oder Parkinson vorbeugen kann.
Damit Zellen im Körper koordiniert zusammenarbeiten können, brauchen sie funktionierende Signalwege – sie „kommunizieren“ über chemische Botenstoffe miteinander. Diese Zellkommunikation steuert, wann Zellen wachsen, ruhen oder auf Veränderungen reagieren sollen.
Ein weiterer zentraler Vorgang ist die Zellteilung – notwendig für Wachstum, Wundheilung und Gewebeerneuerung. Gleichzeitig sorgen DNA-Reparaturmechanismen dafür, dass Fehler im Erbgut korrigiert werden. Gelingt das nicht, greift die Apoptose, der programmierte Zelltod, um Schäden zu begrenzen.
All diese Mechanismen laufen im Hintergrund ab – ständig und hochpräzise. Wenn jedoch einer dieser Abläufe gestört ist, kann das zu schweren Erkrankungen führen, etwa Krebs, Autoimmunerkrankungen oder Stoffwechselstörungen. Genau hier setzt die personalisierte Medizin an. Sie nutzt modernste Technologien, um diese zellulären Prozesse besser zu verstehen und gezielt zu beeinflussen.
Dank Genomsequenzierung (etwa durch „Next Generation Sequencing“) lassen sich genetische Veränderungen identifizieren, die z. B. die Proteinproduktion oder Reparaturmechanismen beeinflussen.
Biomarker-Analysen (die Analyse bestimmter Moleküle)im Blut, Gewebe oder anderen Körperflüssigkeiten zeigen, ob bestimmte Signalwege über- oder unteraktiv sind – und helfen dabei, individuelle Krankheitsverläufe vorherzusagen oder passende Therapien auszuwählen.
Die Einzelzellanalytik ermöglicht es, Unterschiede selbst zwischen einzelnen Zellen sichtbar zu machen – etwa in einem Tumor, wo manche Zellen auf eine Therapie ansprechen und andere nicht. Dadurch wird eine präzisere Behandlung möglich.
Auch die Proteomik (Analyse aller Proteine in einer Zelle) und Metabolomik (Analyse von Stoffwechselprodukten) liefern ein aktuelles Bild davon, wie aktiv bestimmte zelluläre Mechanismen wirklich sind – z. B. ob eine Zelle gestresst ist, ausreichend Energie hat oder sich gerade teilt.
Künstliche Intelligenz (KI) hilft dabei, diese riesigen Datenmengen zu analysieren und Muster zu erkennen – zum Beispiel, welche Kombination von zellulären Störungen typisch für eine bestimmte Krebserkrankung ist. Digitale Gesundheitsdaten (etwa aus Wearables) ergänzen dieses Bild im Alltag und ermöglichen eine präzise Langzeitbeobachtung.
Sogar neue Therapieansätze beruhen auf dem Verständnis zellulärer Abläufe: Gentherapien und CRISPR-basierte Gen-Editierung greifen gezielt in die DNA ein, um fehlerhafte Mechanismen zu korrigieren. In Organoiden – Mini-Organmodellen im Labor – lassen sich Medikamente direkt an patientenspezifischen Zellmodellen testen, ganz ohne Risiko für den Menschen.
Moderne Technologien machen heute sichtbar, was früher im Verborgenen lag: Wie Zellen arbeiten, was bei Krankheit aus dem Gleichgewicht gerät – und wie man gezielt eingreifen kann. So entsteht eine individualisierte Medizin, die nicht mehr auf Vermutung basiert, sondern auf messbarer Biologie.
Diese neue Präzision bringt nicht nur Chancen, sondern auch neue Verantwortlichkeiten mit sich.
3.2. Daten, Verantwortung und der größere Rahmen
Die Komplexität dieser zellulären Prozesse spiegelt sich unmittelbar in der Komplexität der personalisierten Medizin wider: Je genauer wir die feinen Abläufe und Wechselwirkungen in unseren Zellen verstehen, desto gezielter – aber auch anspruchsvoller – werden Diagnose und Therapie. Diese neue Präzision erfordert neue Technologien, neue Denkweisen und ein tiefes biologisches Verständnis. Gleichzeitig eröffnet sie die Chance auf wirksamere, nachhaltigere und maßgeschneiderte Behandlungskonzepte.
Doch je gezielter und tiefer diese Eingriffe werden, desto größer ist auch die Verantwortung – und die Herausforderung, unerwünschte Langzeitwirkungen oder Nebenreaktionen frühzeitig zu erkennen und zu beherrschen. Gerade bei innovativen Verfahren wie Gentherapie oder Immunmodulation gilt es, den Nutzen sorgfältig gegen mögliche Risiken abzuwägen. Die personalisierte Medizin verlangt daher nicht nur Präzision, sondern auch Weitblick.
Ein zentrales Element dieses Weitblicks ist die Verfügbarkeit personenbezogener Gesundheitsdaten – und zwar nicht nur im Einzelfall, sondern im großen Maßstab.
Erst durch die Kombination individueller Daten mit Bevölkerungsdaten wird es möglich, biologische Prozesse im Detail zu verstehen, Krankheitsverläufe vorherzusagen und personalisierte Therapien zu entwickeln. Genetische Informationen, Laborwerte, Bildgebungsdaten oder digitale Alltagsmessungen liefern Hinweise darauf, wie zelluläre Mechanismen bei einem Menschen konkret funktionieren – und wie sie bei anderen variieren.
Um diese individuellen Muster medizinisch einzuordnen, braucht es den Vergleich mit großen Datensätzen. Nur so können KI-Systeme komplexe Zusammenhänge erkennen, die dem menschlichen Auge entgehen – etwa seltene Genvarianten, molekulare Risikokonstellationen oder unerwartete Therapieeffekte. Dabei gilt: Je größer und vielfältiger die Datenbasis, desto zuverlässiger die Modelle – besonders, wenn sie laufend mit neuen Informationen angereichert werden.
Doch diese Fortschritte gelingen nur, wenn Menschen bereit sind, ihre Gesundheitsdaten zu teilen – und darauf vertrauen können, dass sie sicher und verantwortungsvoll genutzt werden.
Die Zukunft der Medizin beginnt dort, wo alles Leben entsteht: in der Zelle. Doch ihr Erfolg hängt ab vom Zusammenspiel dreier Kräfte: technologischer Innovation, wissenschaftlicher Tiefe – und ethischer Verantwortung.
4. Von DNA zur Erkenntnis – ein Hochgeschwindigkeitszug der Innovation
Wie Genomsequenzierung und bioinformatische Analyse die Medizin revolutionieren
4.1. Die DNA – Bauplan des Lebens
a) Die DNA: Das molekulare Informationsarchiv des Lebens
b) Vom Code zum Protein: Wie Anleitungen Wirklichkeit werden
c) Epigenetik & nicht-codierende DNA: Das Dirigenten-Team unseres Erbguts
d) Das verborgene Kontrollzentrum: Was die nicht-codierende DNA steuert
e) Wenn das Betriebssystem krank macht
f) Offene Rätsel der Forschung
g) Die wahre Magie der Gene
4.2. Genetische Variation und ihre Folgen
4.3. Genomsequenzierung: Schlüssel zum individuellen Erbgut
a) Die Reise der DNA: Vom Nabelschnurblut zur genetischen Information
b) Drei Regieanweisungen für die Gen-Diagnostik: WGS, WES und Panel
4.4. Methoden der Genomsequenzierung
4.4.1. Methode 1: Kopieren mit Leuchtfarbe
4.4.2. Methode 2: Hightech-Tunnel – DNA elektrisch abtasten
4.4.3. Genomsequenzierung – Technologien im Vergleich
4.5. Genomsequenzierung: Der nächste Technologiesprung
4.6. Vom Code zur Heilung: Bioinformatik als Schlüssel der Medizin von morgenStellen Sie sich vor, Sie stehen am Bahnsteig, und ein Zug rauscht mit solch einer Geschwindigkeit vorbei, dass kaum ein Bild im Fenster zu erkennen ist. So ähnlich empfinden viele Menschen die Entwicklungen in der personalisierten Medizin: Technologien kommen in rasantem Tempo, Begriffe wirken abstrakt – und doch zieht sich durch alles ein roter Faden: das menschliche Genom.
So vielfältig die modernen Ansätze auch sind – von Organoiden über CRISPR-basierte Gen-Editierung bis zu RNA-Medikamenten – im Zentrum steht der genetische Bauplan des Menschen. Die Genomsequenzierung ist heute Grundlage vieler Diagnosen und Therapieentscheidungen. Sie zeigt, welche genetischen Veränderungen Krankheiten auslösen, welche Signalwege betroffen sind – und wo gezielte Eingriffe möglich sein könnten.
Was einst Milliarden kostete und Jahre dauerte, gelingt heute in wenigen Tagen: Das gesamte Erbgut eines Menschen kann entschlüsselt werden. Doch die reine Sequenz ist noch kein Befund. Die Abfolge von drei Milliarden Basenpaaren entfaltet erst dann medizinischen Wert, wenn sie auch verstanden wird – und genau hier beginnt die eigentliche Herausforderung.
Die Bioinformatik betritt die Bühne: ein interdisziplinäres Feld, das Mathematik, Informatik und Biologie vereint, um aus Rohdaten Muster, Risiken und Therapieoptionen abzuleiten. Sie ist das eigentliche Nadelöhr der personalisierten Medizin – denn sie entscheidet, ob genetische Varianten als harmlos, risikobehaftet oder krankheitsverursachend gelten.
Dazu werden individuelle Sequenzen mit internationalen Referenzdatenbanken abgeglichen, Algorithmen analysieren Millionen genetischer Varianten, und lernende Systeme modellieren das komplexe Zusammenspiel von Genetik, Umwelt und Stoffwechsel.
Dieser Prozess ist hochkomplex – und zugleich essenziell. Denn erst die rechnergestützte Auswertung macht die DNA medizinisch nutzbar. Zwischen Genomsequenzierung und bioinformatischer Analyse besteht eine untrennbare Symbiose: Die Sequenz liefert die Daten, die Analyse das Verständnis. Ohne sie bleibt die DNA eine bloße Aneinanderreihung von Buchstaben.
Genau deshalb richtet sich der Blick der folgenden Abschnitte auf die entscheidende Frage:
Was heißt es, das Genom zu entschlüsseln – und es wirklich zu verstehen?
Welche Aussagen sind möglich – und wo liegen ihre Grenzen?Denn wer die Zukunft der Medizin begreifen will, muss wissen, wie wir heute das menschliche Erbgut lesen – und wie wir lernen, es zu deuten.
Der Hochgeschwindigkeitszug der Innovation ist längst abgefahren. Wer mitfahren will, muss verstehen, wohin er führt.
4.1. Die DNA – Bauplan des Lebens
Um diese Reise anzutreten, lohnt ein kurzer Blick auf die molekularen Grundlagen: Wie unsere DNA zur Bauanleitung wird, wie daraus über den Umweg der RNA Proteine entstehen – und wie die Epigenetik diese Prozesse feinjustiert. Erst dieses Fundament macht verständlich, was beim Entschlüsseln des Genoms eigentlich passiert.
a) Die DNA: Das molekulare Informationsarchiv des Lebens
In jeder Zelle unseres Körpers liegt sorgfältig verpackt ein beeindruckendes Molekül: die DNA (Desoxyribonukleinsäure). Sie enthält die vollständige Bau- und Funktionsanleitung unseres Körpers – ein molekulares Informationsarchiv des Lebens.
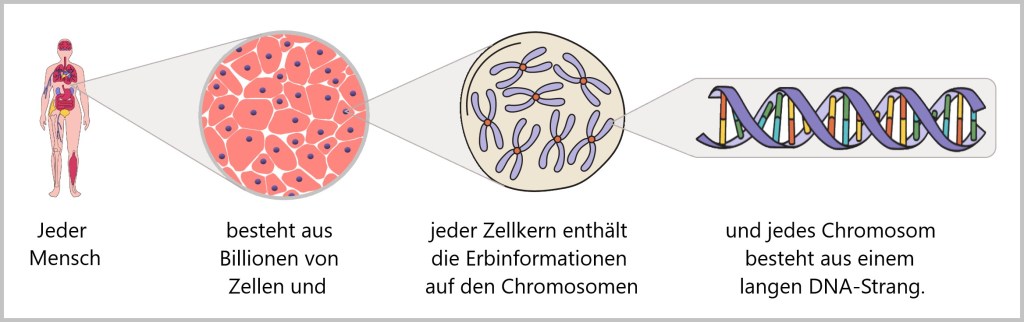
Abb. 1: Die DNA enthält die Erbinformation [Die Wunderwelt des Lebens] Strukturell gleicht sie einer spiralförmig verdrehten Strickleiter: Zwei Zucker-Phosphat-Ketten bilden die Seiten, dazwischen liegen die „Sprossen“ – die Basen Adenin (A), Cytosin (C), Guanin (G) und Thymin (T). Sie paaren sich stets nach festen Regeln: A mit T, C mit G. Diese komplementäre Bindung macht jeden Strang zur Vorlage für den anderen – ein molekulares Backup-System.

Abb. 2: Schematische Darstellung der Grundstruktur der DNA [Die Wunderwelt des Lebens] Entrollt man die DNA aus nur einer einzigen Zelle, misst sie rund zwei Meter – bei einem Durchmesser von nur wenigen Millionstel Millimetern. Das Leben hängt also buchstäblich an einem hauchdünnen, aber erstaunlich langen Faden.
Die Abfolge der rund drei Milliarden Basenpaare ist so individuell wie ein Fingerabdruck. Sie bildet den genetischen Code – die Anweisungen zur Herstellung von Proteinen, jenen Molekülen, die Struktur, Funktion und Regulation aller biologischen Prozesse steuern.
Als zentrales Informationsarchiv ist die DNA zugleich äußerst wertvoll und empfindlich. Deshalb wird sie sicher im Zellkern aufbewahrt, vergleichbar mit einem Safe. Damit der Körper auf die Informationen in diesem Safe zugreifen kann, braucht es ein cleveres Transportsystem. Schließlich müssen die Baupläne für Proteine – jene molekularen Architekten aller Lebensprozesse – zu den Ribosomen gelangen, den sogenannten „Protein-Fabriken“ im Zellplasma. Dort werden die Proteine hergestellt – ein Prozess, den man als Proteinbiosynthese bezeichnet.
Doch wie kommt ein konkreter Bauplan sicher aus dem Zellkern zu diesen Fabriken? Mit anderen Worten: Wie wird die genetische Information für ein bestimmtes Protein vom geschützten Archivort an den Ort der Produktion übertragen?
b) Vom Code zum Protein: Wie Anleitungen Wirklichkeit werden
Die Reise beginnt bei den Genen – bestimmten Abschnitten der DNA, die die Baupläne für Proteine enthalten. Etwa 20.000 solcher Gene finden sich im menschlichen Genom.
Die DNA ist dabei nicht nur ein passives Informationsarchiv im Zellkern – sie enthält ein aktives Repertoire an Anleitungen. Man kann sich jedes Gen wie ein Kapitel in einem molekularen Kochbuch vorstellen: nicht selten mit mehreren Varianten eines Rezepts, angepasst an Zelltyp, Entwicklungsstand oder äußere Bedingungen.
Zunächst entsteht eine temporäre „Arbeitskopie“ eines Gens – in Form von RNA (Ribonukleinsäure). Sie ähnelt der DNA, verwendet jedoch die Base Uracil (U) anstelle von Thymin (T) und besteht meist nur aus einem einzelnen Strang. Diese flüchtige Struktur macht die RNA ideal für den kurzfristigen Gebrauch – sie wird nach Erfüllung ihrer Funktion wieder abgebaut.
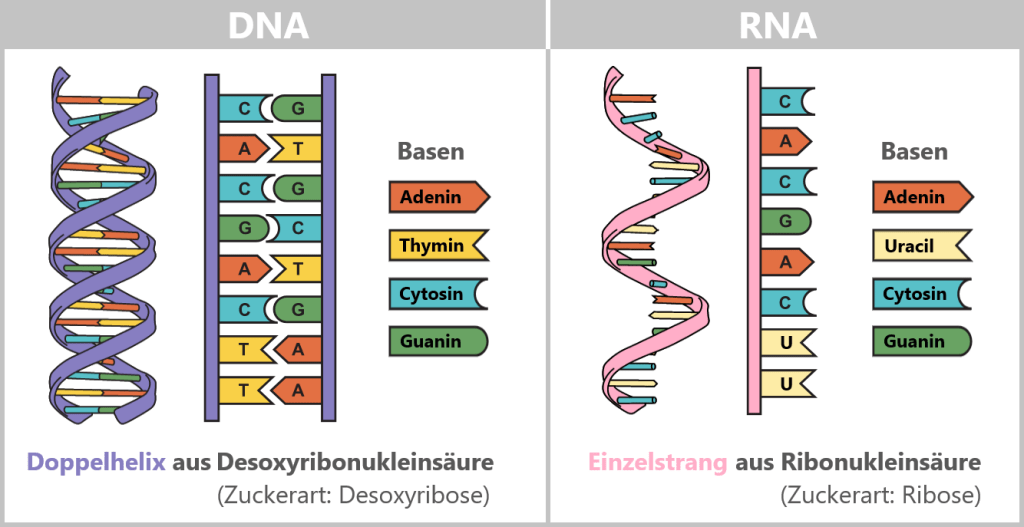
Abb. 3: Von der DNA wird eine Arbeitskopie erstellt, die RNA [Die Wunderwelt des Lebens] Alternatives Splicing: Die flexible Küche der Gene
Zellen müssen sich flexibel an wechselnde Anforderungen anpassen: auf Entzündungen reagieren, Krankheitserreger abwehren, Muskeln aufbauen oder den Blutzuckerspiegel regulieren. Dafür braucht es keine starre Anleitung, sondern ein System, das je nach Situation improvisieren kann.
In Abhängigkeit vom Zelltyp und äußeren Signalen wird der RNA-Strang dynamisch umgebaut, indem er an bestimmten Markierungen – übertragen aus der DNA – geschnitten wird. Nicht benötigte Abschnitte (Introns) werden entfernt, die verbleibenden (Exons) neu zusammengesetzt. So entsteht aus einer Vorlage eine Vielzahl möglicher Botschaften. Das Ergebnis ist die messenger RNA (mRNA), die die genetische Information aus dem Zellkern zu den Ribosomen im Zytoplasma bringt – den „Protein-Fabriken“ der Zelle.
Man kann sich diesen Prozess vorstellen wie das Nachkochen eines Rezepts: je nach Bedarf lässt die Zelle Zutaten weg, fügt Gewürze hinzu oder passt die Zubereitungsschritte an. So wird aus einer Grundanleitung eine Vielfalt von Gerichten.
Durch diesen Prozess – genannt alternatives Splicing – können Zellen aus einem Gen eine Vielzahl unterschiedlicher Proteine herstellen, die jeweils spezifische Funktionen übernehmen.
Der gesamte Vorgang „Vom Gen zum Protein“ wird häufig als Genexpression bezeichnet.
💡 Beispiel: Das Titin-Gen
👉 Längstes menschliches Gen (≈ 300.000 Basenpaare).
👉 Durch alternatives Splicing entstehen > 20.000 verschiedene Proteinvarianten – jeweils angepasst an die unterschiedlichen Anforderungen in verschiedenen Muskeltypen.Unser Erbgut ist also kein starres Rezeptbuch – es ist eine dynamische Küche, in der die Zelle aus denselben Vorlagen immer neue Lösungen entwickelt.
📌 Wer sich dafür interessiert, wie die Erbinformation in der DNA Schritt für Schritt in ein Protein übersetzt wird, findet hier weiterführende Informationen.
Doch wie in jeder kreativen Küche können auch hier beim Übergang der DNA zur mRNA Fehler passieren: Wird beim Abschreiben oder Montieren ein „Zutatenbuchstabe“ übersehen, vertauscht oder falsch zusammengesetzt, kann die Botschaft am Ende missverständlich oder unbrauchbar werden. Solche Mutationen oder fehlerhaftes Splicing sind manchmal harmlos – doch sie können auch defekte Proteine hervorbringen, die ihre Aufgabe nicht erfüllen oder sogar Schaden anrichten. Ein kleiner Patzer in der Abschrift des molekularen Kochbuchs – und der Geschmack des Lebens verändert sich.
c) Epigenetik & nicht-codierende DNA: Das Dirigenten-Team unseres Erbguts
Obwohl alle Zellen die gleiche DNA enthalten, nutzen sie jeweils nur einen Bruchteil davon. Eine Leberzelle benötigt andere Anleitungen als eine Nervenzelle – der Rest bleibt stillgelegt. Diese gezielte Genregulation wird durch epigenetische Mechanismen gesteuert: Kleine chemische Markierungen – etwa Methylgruppen – verändern nicht die genetische Sequenz selbst, sondern beeinflussen, ob ein Gen aktiv ist, wie oft es abgelesen wird oder ob es komplett stummgeschaltet bleibt.
Doch wer gibt dem Dirigenten eigentlich seine Anweisungen? Hier kommt die nicht-codierende DNA ins Spiel – 98 % unseres Erbguts, lange als nutzloser „Ballast“ abgetan. Sie ist das Betriebssystem, das die epigenetischen Schaltpläne bereitstellt. Erst ihr Zusammenspiel legt fest, wann, wo und wie stark unsere Gene aktiv werden – und entscheidet so über Gesundheit oder Krankheit.
d) Das verborgene Kontrollzentrum: Was die nicht-codierende DNA steuert
In diesen „dunklen“ Regionen verbergen sich präzise Schaltkreise, die das Schicksal jeder Zelle lenken:
Enhancer wirken wie unsichtbare Fernbedienungen der Gene. Selbst über Tausende Bausteine hinweg können sie ein Zielgen aktivieren oder zum Schweigen bringen – etwa für die Herzentwicklung im Embryo.
Telomere sind die Schutzkappen der Chromosomenenden, wie Plastikhüllen an Schnürsenkeln. Doch wenn sie schrumpfen, beginnt das Altern – ein zentraler Mechanismus von Zellverfall und Krebsentstehung.
Nicht-codierende RNAs agieren als heimliche Regisseure: Mikro-RNAs blockieren krankmachende Gene, während die lange RNA XIST bei Frauen ein gesamtes X-Chromosom zum Schweigen bringt.
Epigenetische Markierungen legen eine unsichtbare Landkarte über das Genom. Mit Methylierungen wie roten Stoppschildern entscheiden sie, ob ein Enhancer oder Gen gelesen werden darf – oder verstummt.
So entsteht Ordnung im molekularen Kochbuch: Jeder Zelltyp liest nur die Seiten, die er braucht – gesteuert durch nicht-codierende DNA und ihre epigenetische Interpretation.
e) Wenn das Betriebssystem krank macht
Fast alle genetischen Risikomarker für Volkskrankheiten – von Diabetes über Herzleiden bis zu psychischen Störungen – liegen in nicht-codierenden Zonen:
Mutationen in einem Enhancer für das MYC-Gen können Leukämie auslösen, indem sie die Zellteilung außer Kontrolle bringen.
Epigenetische Fehlprogrammierungen in diesen Bereichen – etwa durch chronischen Stress oder Umweltgifte – können Gene dauerhaft falsch an- oder abschalten und so Krankheiten wie Krebs oder Autoimmunstörungen begünstigen.
Solche epigenetischen Muster sind teils reversibel – ein Hoffnungsschimmer für neue Therapien.
f) Offene Rätsel der Forschung
Trotz gewaltiger Fortschritte bleibt das Genom voller ungelöster Rätsel:
Die „dunkle Materie“: Rund die Hälfte des nicht-codierenden Genoms ist funktional unergründet. Handelt es sich um evolutionäres Rauschen – oder um ein noch unkartiertes Steuernetz des Lebens?
Langstrecken-Kommunikation: Wie findet ein Enhancer zielsicher sein Gen inmitten von Millionen DNA-Bausteinen? Die räumliche Faltung des Genoms – ein wahres „Chromatin-Origami“ – könnte der Schlüssel sein.
Umwelt-Einflüsse: Weshalb führen identische nicht-codierende Mutationen in verschiedenen Lebenswelten zu völlig unterschiedlichen Krankheitsbildern?
RNA-Codes: Zehntausende nicht-codierende RNAs sind entdeckt – doch welche von ihnen sind nur Randnotizen, und welche erheben sich möglicherweise zu Leitmotiven neuer Therapien?
g) Die wahre Magie der Gene
Die nicht-codierende DNA ist kein Datenmüll, sondern das Mastermind der Zelle. Die Epigenetik wiederum ist ihr Dirigent – sie interpretiert flexibel, was das Betriebssystem vorgibt. Gemeinsam formen sie unsere Entwicklung, steuern unsere Gesundheit und reagieren auf Umweltsignale.
Gene sind kein starres Schicksal, mehr eine Partitur, die immer wieder neu interpretiert werden kann. Im Zusammenspiel entsteht ein Orchester, das nicht nur unsere Zellen formt, sondern sich ständig neu einstimmt – durch Erfahrungen, Umwelteinflüsse und sogar durch Spuren, die auch an kommende Generationen weitergegeben werden können.
Die wahre Magie liegt darin, dass aus begrenzten Noten eine unendliche Vielfalt von Leben erklingt.
4.2. Genetische Variation und ihre Folgen
Wenn sich das Erbgut verschreibt, verstummt oder improvisiert.
Bei jeder Zellteilung wird die DNA sorgfältig kopiert und an die Tochterzellen weitergegeben – so bleibt die genetische Information über Generationen hinweg erhalten.
Doch das System ist nicht unfehlbar: Mutationen – also Veränderungen in der DNA-Sequenz – können Struktur und Funktion von Proteinen beeinflussen. Manchmal stimmt nur ein Buchstabe nicht. Oder ein Satz fehlt. Oder ein ganzer Absatz wurde umgestellt. In der Genetik spricht man dann von Varianten – genetischen Veränderungen, die je nach Art, Größe und Position harmlos, risikobehaftet oder krankheitsauslösend sein können.
Die folgende Übersicht enthält die wichtigsten Variantentypen – und was sie im molekularen Manuskript des Lebens bedeuten:
📌 SNVs (Single Nucleotide Variants) – Ein einziger Buchstabe ist vertauscht. Meist kaum spürbar – doch manchmal genügt er, um ganze Geschichten umzuschreiben, wie bei der Sichelzellenanämie.
📌 Indels (Insertionen/Deletionen) – Kleine Textfragmente werden eingefügt oder gelöscht. Schon eine winzige Verschiebung kann ganze Sätze aus dem Takt bringen, manchmal so stark, dass der ursprüngliche Sinn verloren geht.
📌 Strukturelle Varianten (SVs) – Lange DNA-Passagen werden verdreht, verdoppelt oder verschoben. Diese verdeckten Umschreibungen sind schwer zu entdecken – und oft verhängnisvoll, etwa bei Krebs.
📌 Splice-Site-Varianten – Fehler an den Schnittstellen zerreißen die Syntax der RNA. Es entstehen falsche Baupläne – und Proteine, die nicht mehr stimmen.
📌 Repeat-Expansionen – Basenfolgen werden zwanghaft wiederholt, bis sie den Text entstellen. So entstehen Krankheiten wie Chorea Huntington.
📌 Copy Number Variants (CNVs) – Ganze Kapitel (Gene) fehlen oder erscheinen doppelt. Das Gleichgewicht der Erzählung geht verloren.
📌 Mitochondriale Varianten – Veränderungen in der DNA der Mitochondrien, der Kraftwerke der Zelle. Wenn hier der Text verfälscht wird, stockt der Antrieb des Lebens.
📌 Somatische Varianten – Treten erst im Laufe des Lebens auf und betreffen nur bestimmte Zellgruppen. Sie prägen vor allem die Geschichte von Tumoren.
📌 Epigenetische Signale – Keine neuen Buchstaben, sondern eine andere Betonung: Lautstärke, Rhythmus, Beginn. Sie verwandeln denselben Text in eine völlig neue Melodie.
Die Liste zeigt: Unser Erbgut ist kein starres Gebilde, sondern lässt sich durch Varianten verändern. Manche dieser Abweichungen bleiben unauffällige Randnotizen, andere aber können die gesamte Geschichte neu schreiben – manchmal mit dramatischen Folgen.
Um solche feinen Unterschiede sichtbar zu machen, braucht es Werkzeuge, die jedes Detail erfassen – jeden Buchstaben, jedes Satzzeichen, jede Verschiebung. Genau hier setzt die moderne Genomsequenzierung an.
4.3. Genomsequenzierung: Schlüssel zum individuellen Erbgut
Das Erbgut eines Menschen gleicht einer gigantischen Bibliothek: Milliarden von Buchstaben, fein säuberlich in langen Bänden angeordnet. Um darin zu lesen, bedarf es der Genomsequenzierung – die wie ein hochpräziser Scanner den Rohtext unseres genetischen Bauplans liefert.
So wird sie zum Herzstück der personalisierten Medizin: einer Medizin, die ihre Entscheidungen am Einzelnen ausrichtet, nicht am Durchschnitt.
Doch wie gelangen wir überhaupt an das „Buch des Lebens“, um es Seite für Seite lesen zu können?
Stellen wir uns vor: Bei einem Neugeborenen wird kurz nach der Geburt ein DNA-Test durchgeführt – ein Szenario, das im Vereinigten Königreich bereits Wirklichkeit geworden ist, wie zuvor beschrieben.
a) Die Reise der DNA: Vom Nabelschnurblut zur genetischen Information
Unmittelbar nach der Geburt kann Blut aus der abgetrennten Nabelschnur entnommen werden – ein sicherer und schmerzfreier Vorgang. Das Nabelschnurblut stammt vom Neugeborenen und enthält zahlreiche wertvolle Zellen, insbesondere Stammzellen und weiße Blutkörperchen. In ihren Zellkernen liegt das Entscheidende verborgen: die DNA – das genetische Erbe des Kindes.
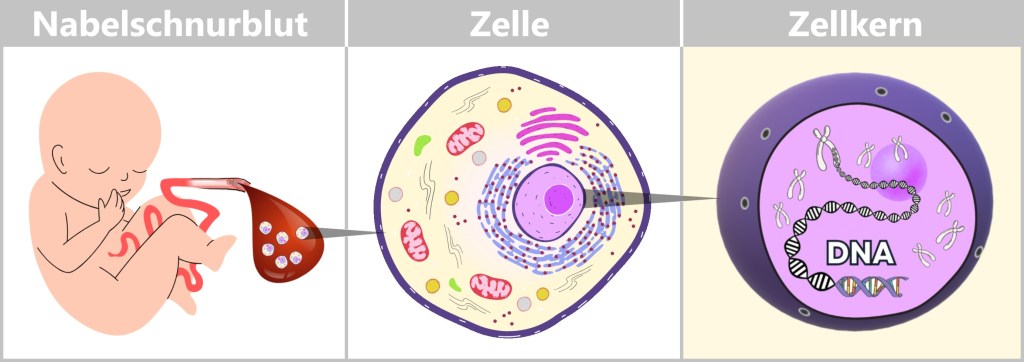
Abb. 4: Vom Nabelschnurblut zur genetischen Information Links: Nabelschnurblut als reiche Quelle unterschiedlicher Zellen.
Mitte: Schematische Darstellung einer menschlichen Zelle.
Rechts: Im Zellkern liegt die DNA, Trägerin der genetischen Information.Um diese DNA zu isolieren und analysieren zu können, durchläuft sie im Labor einen mehrstufigen Prozess. Dabei kommen bewährte molekularbiologische Methoden zum Einsatz, die sich in vier zentrale Schritte gliedern lassen:
Zellaufschluss: Zugang zum Erbgut schaffen
Zunächst wird das Blut zentrifugiert – also mit hoher Geschwindigkeit geschleudert. Dabei trennen sich die Bestandteile nach Dichte: Die weißen Blutkörperchen setzen sich als Zellpellet am Boden ab. Sie enthalten den größten Teil der interessierenden DNA.
Diese Zellen werden anschließend mit speziellen Lösungen und Enzymen behandelt, die die Zell- und Zellkernmembran auflösen. Dadurch wird die DNA freigesetzt – zunächst allerdings als Teil eines Gemischs aus Proteinen, Lipiden und anderen Zellbestandteilen.
DNA-Isolierung: Reines Erbgut gewinnen
Im nächsten Schritt wird die DNA aus dem Zelllysat – der „Zellsuppe“ – isoliert. Dazu gibt es verschiedene Verfahren:
Filtration oder Säulenreinigung: Die Lösung wird durch spezielle Filtersysteme geleitet, an denen die DNA haften bleibt, während kleinere Moleküle ausgespült werden.
Magnetkügelchen: Modernere Verfahren verwenden winzige Kügelchen mit DNA-bindenden Oberfläche. Die DNA haftet daran und kann per Magnet gezielt entnommen werden.
Beide Methoden ermöglichen eine effektive Trennung von Erbgut und Störstoffen.
Reinigung und Konzentration der DNA
Nun liegt die DNA vieler weißer Blutkörperchen in relativ reiner Form vor – allerdings noch in einer wässrigen Lösung. Um sie weiter zu reinigen und zu konzentrieren, wird kalter Alkohol (z. B. Ethanol oder Isopropanol) hinzugefügt. Dadurch fällt die DNA aus und wird sichtbar – als weißlicher, fadenartiger Niederschlag.
Die menschliche DNA ist extrem lang: Entrollt man sie, misst sie pro Zelle etwa zwei Meter. Bei der Extraktion wird sie jedoch durch chemische und mechanische Einflüsse in viele kleinere Abschnitte zerteilt. Das sieht zunächst nach Chaos aus – doch da die DNA in allen Zellen identisch ist, enthalten viele Fragmente überlappende Informationen. Diese zufällige Verteilung wird bei der Sequenzierung zum Vorteil: Die Fragmente können mehrfach unabhängig gelesen und anschließend am Computer zu einem Gesamtbild zusammengesetzt werden. Das erhöht die Genauigkeit der Analyse erheblich.
Qualitätskontrolle: Ist die DNA intakt?
Bevor die DNA-Probe analysiert werden kann, muss ihre Qualität überprüft werden. Zwei gängige Verfahren sind:
Spektrophotometrie: Misst die Konzentration und Reinheit der DNA über die Lichtabsorption.
Gelelektrophorese: DNA-Fragmente werden durch ein Gel geleitet und nach Länge getrennt. (Längere Fragmente wandern langsamer als kürzere.) So lässt sich beurteilen, ob die Probe ausreichend intakt ist.
Warum ist dieser Prozess so wichtig?
Die menschliche DNA ist empfindlich – Verunreinigungen oder Fragmentierungen können Analysen verfälschen oder unbrauchbar machen. Eine sorgfältige und kontrollierte Aufreinigung ist daher Voraussetzung für zuverlässige genetische Aussagen.
Gerade Nabelschnurblut stellt eine besonders wertvolle Quelle medizinischer Information dar: Es ermöglicht Frühdiagnostik, Prognosen und in manchen Fällen gezielte Prävention.
Was im Labor wie ein routinierter Ablauf wirkt, ist in Wahrheit ein hochpräziser Vorgang – erfordert technisches Know-how, moderne Geräte und biochemisches Feingefühl. Durch automatisierte Extraktionsmethoden lässt sich heute in weniger als einer Stunde aus wenigen Tropfen Nabelschnurblut eine hochwertige DNA-Probe gewinnen. Sie besteht zwar aus Fragmenten, enthält aber dennoch alle Informationen, um den individuellen genetischen Code zu entschlüsseln – und damit die Grundlage für eine Medizin von morgen zu legen.
b) Drei Regieanweisungen für die Gen-Diagnostik: WGS, WES und Panel
Bevor wir tiefer in die Werkzeuge der Genom-Sequenzierung eintauchen, lohnt sich ein Blick auf die drei zentralen Strategien, mit denen heute genetische Informationen entschlüsselt werden – gleichsam wie Regieanweisungen im Drehbuch des Lebens:
🎯 Panel-Sequenzierung – Die gezielte Szene
Hier wird nur ein ausgewählter Teil des Genoms untersucht – meist einige Dutzend bis wenige Hundert Gene. Solche Tests kommen zum Einsatz, wenn der Verdacht auf eine spezifische Erkrankung besteht, etwa bei erblich bedingtem Brustkrebs (BRCA1/2).
Vorteil: Schnell, kosteneffizient, präzise – solange die medizinische Fragestellung klar ist.
Limitierung: Alles außerhalb der gewählten Szene bleibt im Dunkeln.🎥 Whole Exome Sequencing (WES) – Der klassische Director’s Cut
WES analysiert alle protein-kodierenden Abschnitte des Genoms – also etwa 1–2 % des gesamten Erbguts. Gerade bei seltenen Erbkrankheiten ist es ein bewährtes Mittel, da die meisten bekannten Mutationen in diesen Bereichen liegen.
Vorteil: Effiziente Suche nach fehlerhaften Bauplänen für Proteine.
Limitierung: Bereiche außerhalb der Gene – wichtige Schalter und Regler des Erbguts – werden nicht erfasst.🎬 Whole Genome Sequencing (WGS) – Der vollständige Film in 4K
WGS liest das gesamte Genom – alle drei Milliarden Buchstaben. Damit liefert es das umfassendste Bild, inklusive nicht-kodierender Regionen, regulatorischer Elemente, struktureller Varianten und seltener Mutationen.
Vorteil: Maximale Auflösung für komplexe Fälle.
Beispiel: Bei Kindern mit unklaren Entwicklungsstörungen bringt WGS doppelt so häufig Klarheit wie WES – etwa durch das Aufdecken von Mutationen in entfernten „Fernsteuerungs“-Regionen (Enhancern), die Gene falsch an- oder abschalten.Fazit:
Je nach Fragestellung braucht es die passende Perspektive – manchmal reicht ein Zoom auf die Schlüsselszene, manchmal muss der ganze Film neu geschnitten werden. Die Wahl der Methode ist daher keine technische Nebensache, sondern eine strategische Entscheidung im Drehbuch der Diagnostik.
4.4. Methoden der Genomsequenzierung
Wie wird der genetische Code gelesen?
Stellen Sie sich vor, DNA ist ein Buch – geschrieben in einer Sprache mit nur vier Buchstaben: A, T, C und G. Um diesen genetischen Text zu entschlüsseln, nutzen Wissenschaftler moderne Technologien. Zwei zentrale Methoden haben sich etabliert. Sie verfolgen dasselbe Ziel: die Reihenfolge der DNA-Buchstaben möglichst genau zu bestimmen.
Die erste Methode funktioniert – vereinfacht gesagt – wie eine molekulare Kopiermaschine mit Farbsensoren. Die zweite nutzt einen Hightech-Tunnel, durch den die DNA geleitet und elektronisch „abgetastet“ wird – vergleichbar mit dem Abtasten eines Seils mit den Fingern, um kleinste Unebenheiten zu spüren. Diese Verfahren bilden die Grundlage der DNA-Sequenzierung.
4.4.1. Methode 1: Kopieren mit Leuchtfarbe
a) Sanger-Sequenzierung
b) Illumina-Technologie
c) Single-Molecule Real-Time(SMRT)-Sequenzierung
4.4.2. Methode 2: Hightech-Tunnel – DNA elektrisch abtasten
a) Oxford Nanopore Sequenzierung
4.4.3. Genomsequenzierung – Technologien im Vergleich
a) Durchsatz vs. Verwertbarkeit – Masse ist nicht immer gleich Klasse
b) Wirtschaftlichkeit – Kosten pro Base vs. Kosten pro Erkenntnis
c) Von der Probe zum Befund – Wie schnell spricht das ganze Genom?
d) Klinische Einsatzfelder – Welche Technologie passt wozu?
e) KI als Co-Pilot – Automatisierung ohne Verantwortung abzugeben
f) Vergleichstabelle: Sanger, Illumina, PacBio, ONT
g) Key Takeaways: Zwischen Reife und Routine4.4.1. Methode 1: Kopieren mit Leuchtfarbe
Diese Methode nutzt die Tatsache, dass DNA aus zwei komplementären Strängen besteht. Kennt man die Basenabfolge eines Strangs, lässt sich die Sequenz des Gegenstrangs automatisch ableiten. Um diese Information zu gewinnen, wird einer der beiden Stränge der zu untersuchenden DNA kopiert. Ziel ist es, während dieses Kopiervorgangs die exakte Reihenfolge der Basen in der entstehenden Kopie zu erfassen.
Um besser zu verstehen, wie dieses Verfahren funktioniert, werfen wir zunächst einen Blick auf die wichtigsten Bestandteile und die grundlegenden Schritte zur Herstellung einer DNA-Kopie im Reagenzglas (in vitro).
Herstellung einer DNA-Kopie – das Grundprinzip
Im Zentrum steht eine DNA-Vorlage, die kopiert werden soll. Dafür braucht es vier Hauptbestandteile:
- dNTPs (Desoxynukleosidtriphosphate): die DNA-Bausteine A, T, C und G.
- DNA-Polymerase: ein Enzym, das als molekularer Kopierer fungiert.
- Primer: kurze DNA-Stücke, die der Polymerase den Startpunkt liefern.
- Pufferlösung: sorgt für stabile Bedingungen während der Reaktion.
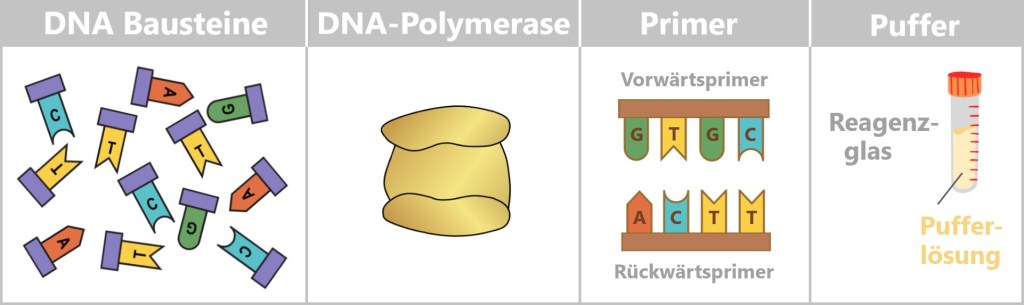
Abb. 5-A: Schematische Darstellung der Hauptbestandteile zur Herstellung einer DNA-Kopie DNA-Bausteine: Einzelne Nukleotide (A, T, C, G), dargestellt in verschiedenen Farben und Buchstaben. Sie bilden das Rohmaterial, aus dem der neue DNA-Strang zusammengesetzt wird.
DNA-Polymerase: Ein schematisch dargestelltes Enzym, das Nukleotide zu einem DNA-Strang verknüpft.
Primer: Kurze DNA-Stücke, die der Polymerase den Startpunkt vorgeben.
Puffer: Ein Reagenzglas mit Pufferlösung, die für stabile chemische Bedingungen während der Reaktion sorgt.Was macht die DNA-Polymerase?
Die DNA-Polymerase ist ein spezielles Enzym, das komplementäre DNA-Stränge erzeugt – gewissermaßen der „Druckkopf“ einer molekularen Kopiermaschine. Enzyme wirken als biologische Werkzeuge, die chemische Reaktionen gezielt und effizient ermöglichen – deshalb nennt man sie auch „biologische Katalysatoren“.
Damit die Polymerase arbeiten kann, benötigt sie einen Primer als Startsignal. Man kann sich das so vorstellen: Wie ein Druckkopf erst loslegt, wenn ein Blatt Papier richtig eingelegt ist, beginnt auch die Polymerase ihre Arbeit erst, wenn ein Primer vorhanden ist. Dieses kleine DNA-Stück zeigt ihr, wo sie starten soll – es ist sozusagen das erste Blatt im molekularen Drucker.
Vor Beginn der Kopie muss die doppelsträngige DNA zunächst in zwei Einzelstränge aufgetrennt werden – dieser Schritt heißt Denaturierung.
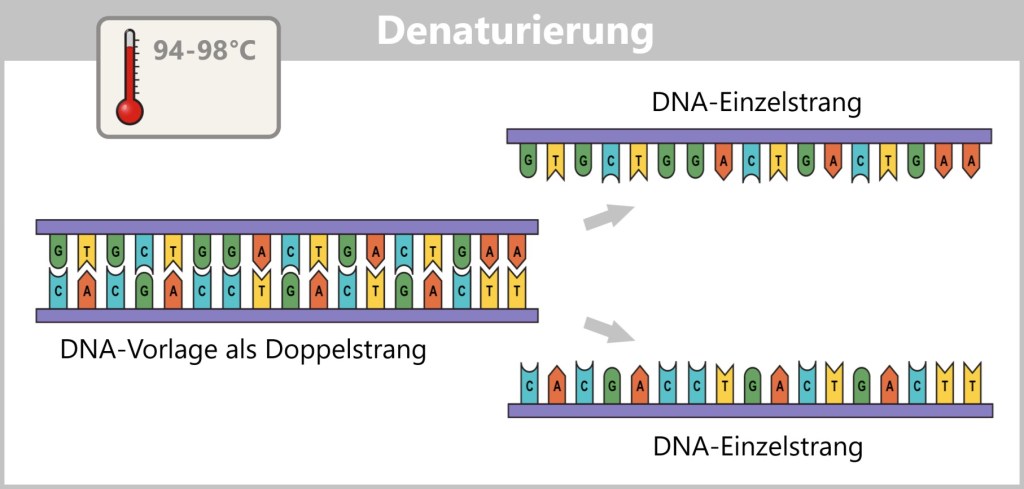
Abb. 5-B: Trennung der DNA-Stränge Anschließend lagern sich die Primer an ihre passenden Zielstellen der Einzelstränge. Diese Primerbindung markiert den Startpunkt für die DNA-Polymerase.
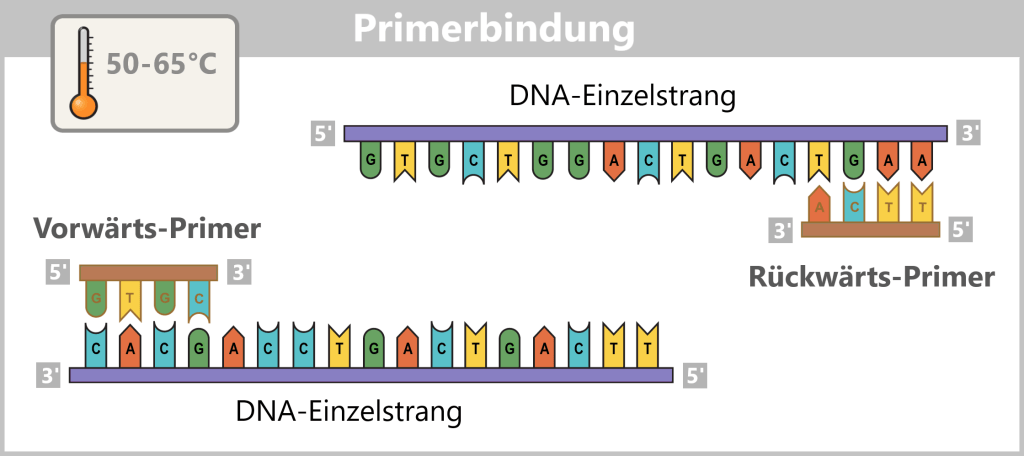
Abb. 5-C: Primer markieren den Startpunkt für die Polymerase. Nun beginnt die Synthese der neuen Stränge: Die Polymerase liest den Vorlage-Strang in 3′-zu-5′-Richtung und ergänzt die Kopie in Gegenrichtung – von 5′ nach 3′ – unter Beachtung der Basenpaarung (A mit T, G mit C).
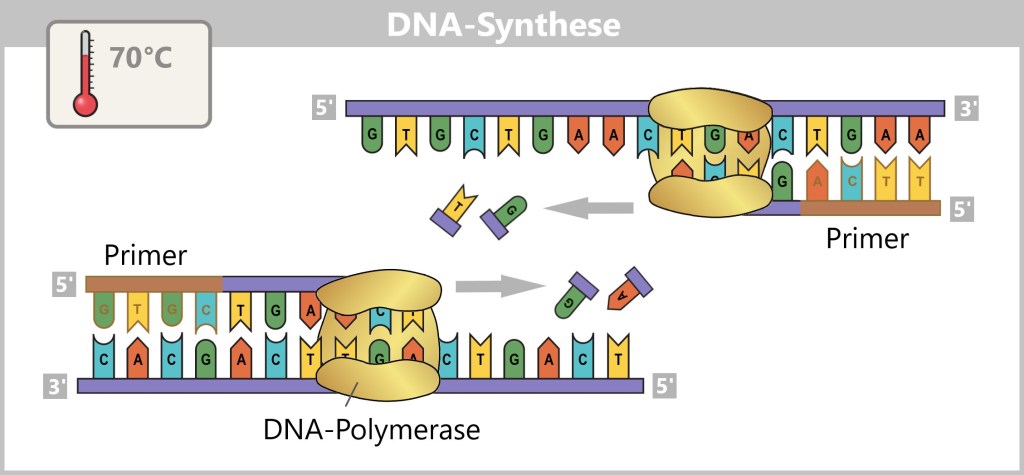
Abb. 5-D: DNA-Synthese – die exakte Verdopplung eines DNA-Strangs. Was bedeuten 3′ und 5′?
Die Bezeichnungen 3′ (sprich: drei-Strich) und 5′ (fünf-Strich) stammen aus der Chemie des DNA-Rückgrats. Sie geben an, an welchem Ende sich bestimmte Kohlenstoffatome im Zuckermolekül befinden, an die neue Bausteine angefügt werden können.
Stellen Sie sich den DNA-Strang wie eine Einbahnstraße vor. Die Polymerase kann nur in eine Richtung fahren – vom 5′-Ende zum 3′-Ende. Neue Bausteine lassen sich chemisch nur am 3′-Ende anfügen. Daher liest die Polymerase den „alten“ Strang rückwärts (3′ → 5′) und baut den neuen Strang vorwärts (5′ → 3′).
So entsteht aus einem Einzelstrang nach und nach ein neuer, passender Gegenstrang – mithilfe der Basenpaarungsregeln: Adenin (A) paart sich mit Thymin (T), und Cytosin (C) mit Guanin (G).
Das Ergebnis sind perfekte genetische Abbilder – neue DNA-Stränge, exakte Kopien der Ursprungs-DNA.
Leuchtende Buchstaben: Wie Sequenzierung funktioniert
Moderne Sequenziermethoden erweitern diesen Kopiervorgang um eine raffinierte Technik: Sie verwenden fluoreszierend markierte dNTPs (DNA-Bausteine). Jedes der vier Nukleotide (A, T, C, G) ist mit einem Farbstoff versehen, der beim Einbau ein spezifisches Lichtsignal aussendet.
Während die Polymerase die DNA-Stränge verlängert, fügt sie die farbigen Bausteine passgenau ein. Jedes Mal, wenn ein neues Nukleotid eingebaut wird, sendet es ein winziges Lichtsignal – wie ein kleiner Blitz, der verrät, welcher „Buchstabe“ gerade ergänzt wurde. Hochauflösende Kameras erfassen diese Lichtsignale und ermöglichen so die schrittweise Rekonstruktion der DNA-Sequenz.
Sequenziermethoden im Überblick
Mehrere bedeutende Technologien nutzen dieses Prinzip in spezifischer Form:
Sanger-Sequenzierung: Die klassische Methode, bei der fluoreszierende Kettenabbruch-Nukleotide die DNA-Synthese an zufälligen Stellen beenden. Die resultierenden Fragmente lassen sich der Reihenfolge nach auslesen.
Illumina-Technologie: Eine weit verbreitete Hochdurchsatzmethode, bei der Millionen DNA-Fragmente parallel sequenziert werden. Die fluoreszierenden Signale werden Schritt für Schritt erfasst.
SMRT-Sequenzierung (PacBio): Hier wird die Synthese in Echtzeit an einem einzigen DNA-Molekül beobachtet – mit hoher Präzision und langen Leselängen.
a) Sanger-Sequenzierung
Das klassische „Buchstaben-für-Buchstaben-Lesen“
Die Sanger-Sequenzierung, auch Kettenabbruchmethode genannt, wurde in den 1970er-Jahren entwickelt und war das erste Verfahren, mit dem sich DNA präzise und zuverlässig auslesen ließ. Sie beruht im Wesentlichen auf denselben Prinzipien wie die DNA-Synthese, unterscheidet sich jedoch in einigen entscheidenden Punkten:
- Es wird nur ein Primer verwendet.
- Neben den normalen DNA-Bausteinen – den dNTPs (dATP, dCTP, dGTP, dTTP) – kommen zusätzlich spezielle fluoreszierende Kettenabbruch-Bausteine, die sogenannten ddNTPs (ddATP, ddCTP, ddGTP, ddTTP) in geringer Konzentration zum Einsatz. Wenn ein solcher ddNTP eingebaut wird, stoppt die DNA-Synthese genau an dieser Stelle. Jeder der vier ddNTP-Typen trägt eine unterschiedliche Leuchtfarbe, je nach Base.
Der Ablauf – Schritt für Schritt
Für die Sequenzierung werden vier separate Reaktionsmischungen angesetzt. Jede enthält:
- die einzelsträngige Ziel-DNA,
- einen Primer (Startpunkt der Synthese),
- eine DNA-Polymerase,
- normale dNTPs,
- und jeweils eine Sorte der farblich markierten ddNTPs.
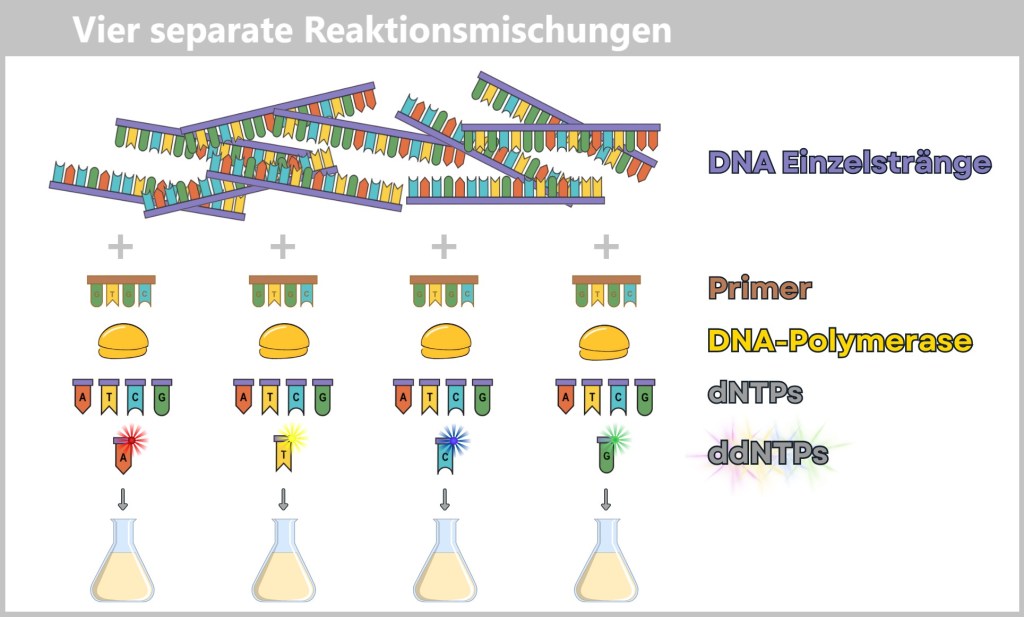
Abb. 6-A: Sanger-Sequenzierung – schematische Darstellung der vier Reaktionsmischungen. Jede Mischung enthält die gleichen Grundkomponenten, unterscheidet sich jedoch durch die jeweils zugegebene Variante der farblich markierten Stopp-Bausteine (ddNTPs).
Nach der Primerbindung liest die Polymerase den DNA-Strang aus und fügt passende Nukleotide ein, um einen komplementären Strang aufzubauen. Wird dabei zufällig ein ddNTP eingebaut, endet die Synthese an genau dieser Stelle.
So entstehen viele unterschiedlich lange DNA-Fragmente – jedes endet mit einem farbig markierten Basenbuchstaben. Da in jeder der vier Reaktionen nur ein ddNTP enthalten ist, weiß man genau, mit welchem Buchstaben die erzeugten DNA-Fragmente enden.
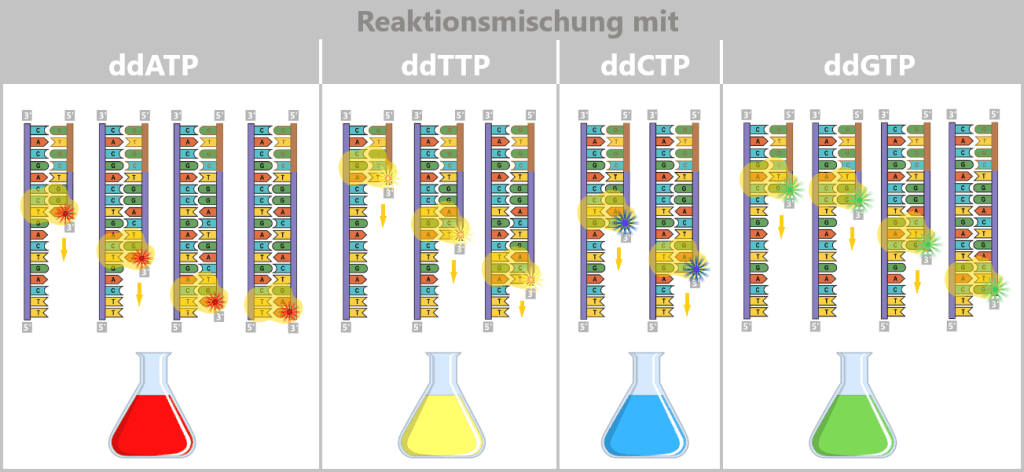
Abb. 6-B: Die Synthese wird gezielt unterbrochen – bei Einbau eines ddNTPs. In jeder Reaktionsmischung ist jeweils eine Sorte der modifizierten DNA-Bausteine (ddATP, ddTTP, ddCTP, ddGTP) enthalten. Wird einer dieser Stopp-Bausteine während der DNA-Synthese eingebaut, bricht der Kopiervorgang genau an dieser Stelle ab. In der ddATP-Mischung stoppt die Synthese, sobald ein modifiziertes Adenin (A) eingebaut wird. In der ddTTP-Mischung endet sie beim Einbau eines modifizierten Thymins (T). Genauso führen ddCTP und ddGTP zum Abbruch der Synthese, wenn ein modifiziertes Cytosin (C) bzw. Guanin (G) eingebaut wird. Durch dieses Verfahren entstehen viele DNA-Fragmente unterschiedlicher Länge, die jeweils mit einem spezifischen Stopp-Nukleotid enden. Ziel ist es, alle theoretisch möglichen Fragmente zu erzeugen, um daraus die vollständige DNA-Sequenz ablesen zu können.
Sortierung und Auswertung
Anschließend werden die DNA-Fragmente denaturiert (in Einzelstränge überführt) und durch Gelelektrophorese der Länge nach sortiert. Dabei wandern die negativ geladenen Fragmente durch ein Gel in Richtung der positiven Elektrode. Kleinere Fragmente bewegen sich schneller, größere langsamer – so entsteht eine geordnete „Leiter“ von Fragmenten.
Ein Laser regt nun die fluoreszierenden Farbstoffe der Endbasen an. Die dabei entstehenden Lichtsignale werden von einem Detektor erfasst. Jedes Signal steht für eine bestimmte Base an einer bestimmten Position. Die Reihenfolge der Lichtsignale ergibt damit direkt die DNA-Sequenz – wie bei einem Buchstaben-für-Buchstaben-Lesen.
Da die erzeugten Fragmente komplementär zur Ursprungs-DNA sind, lässt sich daraus die ursprüngliche Basenfolge exakt ableiten.
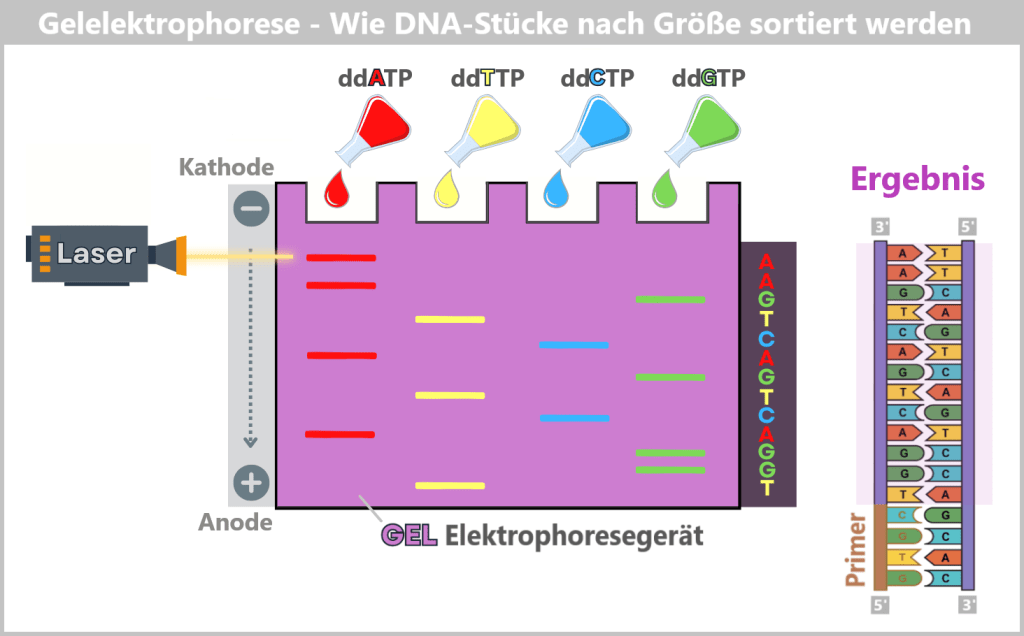
Abb. 6-C: Gelelektrophorese zur Trennung von DNA-Fragmenten: In den Reaktionsansätzen entstehen unterschiedlich lange DNA-Fragmente, die jeweils mit demselben Stopp-Nukleotid enden – je nach Ansatz entweder mit Adenin, Thymin, Guanin oder Cytosin. Diese Reaktionsmischungen werden auf ein Gel aufgetragen. Sobald ein elektrisches Feld angelegt wird, wandern die negativ geladenen DNA-Fragmente von der Kathode (−) zur Anode (+). Dabei bestimmt die Größe der Fragmente ihre Wandergeschwindigkeit: Kleinere Fragmente bewegen sich schneller durch die feinen Poren des Gels und gelangen früher zur Anode, während größere Fragmente langsamer sind. Durch das Auslesen der fluoreszierenden Farbsignale an den Fragment-Enden kann die genaue Reihenfolge der Basen bestimmt werden – und damit die DNA-Sequenz Schritt für Schritt rekonstruiert werden.
Warum gilt die Sanger-Sequenzierung bis heute als Goldstandard?
Auch ein halbes Jahrhundert nach ihrer Entwicklung bleibt die Sanger-Methode in vielen Bereichen der Molekularbiologie und Medizin unverzichtbar. Der Grund: Zuverlässigkeit und Präzision.
Im Vergleich zu modernen Hochdurchsatzverfahren ist die Sanger-Sequenzierung zwar langsamer und eignet sich nur für die Analyse kleinerer DNA-Abschnitte – nicht für ganze Genome. Aber genau darin liegt ihre Stärke:
- Gezielte Fragestellungen, wie das Überprüfen einzelner Gene,
- Nachweis spezifischer Mutationen, oder
- Validierung kritischer Ergebnisse, die zuvor mit anderen Methoden identifiziert wurden,
lassen sich mit der Sanger-Sequenzierung präzise, klar und interpretierbar beantworten – oft so deutlich, dass man die Abfolge der Basen direkt im Sequenzplot sehen kann.
In der medizinischen Diagnostik – etwa bei der Analyse vererbter Erkrankungen oder in der Qualitätskontrolle gentechnischer Verfahren – ist diese hohe Genauigkeit entscheidend. Denn ein falsch interpretierter Buchstabe kann hier lebenswichtige Folgen haben.
Ein weiterer Pluspunkt: Die Methode ist weltweit standardisiert, seit Jahrzehnten bewährt und unterliegt klaren Qualitätsrichtlinien.
Ein Klassiker mit Beständigkeit
In einer Welt, in der ständig neue Technologien auf den Markt drängen, bleibt die Sanger-Sequenzierung ein verlässlicher Anker – der Oldtimer unter den Sequenzierverfahren: nicht der Schnellste, aber extrem robust, bewährt und zuverlässig. Und manchmal ist genau das entscheidend.
b) Illumina-Sequenzierung
Die Hochgeschwindigkeits-Kopiermaschine
Die Sanger-Sequenzierung ist wie präzise Handarbeit: Jeder DNA-Strang wird Schritt für Schritt entziffert – zuverlässig, aber langsam und teuer.
Stellen Sie sich vor, Sie müssten ein ganzes Buch oder sogar eine Bibliothek abschreiben – und dürften dabei nur einen Buchstaben pro Minute notieren.
Genau hier liegt das Problem: Moderne Krebsforschung, die Entschlüsselung seltener Krankheiten oder die genetische Überwachung von Pandemien verlangen die Analyse großer Datenmengen – also vieler und/oder sehr langer DNA-Abschnitte. Dafür braucht es eine Methode, die nicht nur genau, sondern auch schnell und bezahlbar ist.
Und genau da kommt die Illumina-Sequenzierung ins Spiel. Sie hat das Prinzip der DNA-Entzifferung von der Handarbeit zur industriellen Massenproduktion erhoben und ist heute eine der am häufigsten eingesetzten Methoden für die Hochdurchsatz-Sequenzierung (Next Generation Sequencing, NGS). Statt einzelner Buchstaben werden nun ganze Seiten gleichzeitig gelesen – millionenfach, parallel, mit hoher Präzision und kosteneffizient.
Anders als bei der klassischen Sanger-Methode, bei der jedes DNA-Fragment einzeln analysiert wird, arbeitet Illumina mit einer massiven Parallelisierung – das heißt: Es werden viele DNA-Fragmente gleichzeitig vervielfältigt und sequenziert. Wie funktioniert das?
Schritt 1: Vorbereitung der DNA-Fragmente
– Die DNA wird zur „Lego-Baustelle“ –Zunächst wird die zu untersuchende DNA-Probe in viele kurze Stücke zerlegt – sogenannte Fragmente, die 100 bis 300 Basenpaaren lang sind. An beide Enden dieser Fragmente werden kleine künstliche DNA-Stücke – sogenannte Adapter – angehängt. Man kann sie sich wie LEGO-Steckverbinder vorstellen. Sie dienen einerseits als molekulare Andockstellen, um die DNA-Fragmente später auf einem speziellen Träger – dem Flowcell-Chip – zu befestigen. Andererseits dienen sie als Bindungsstellen für universelle Primer.
Abb. 7-A: Die DNA wird in zahlreiche DNA-Fragmente mit kleinen Anhängern (Adaptern) zerlegt. 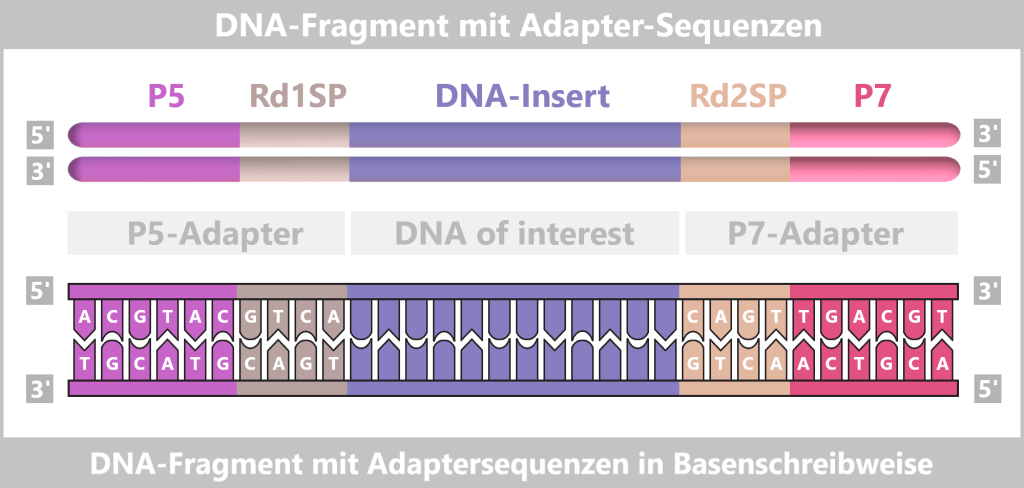
Abb. 7-B: Vereinfachte schematische Darstellung des DNA-Fragments mit Adaptern. Die P5/P7-Sequenzen dienen der Bindung an der Flow Cell. Die Rd1SP/Rd2SP-Sequenzen sind Bindungsstellen für universelle Primer.
Durch Denaturierung werden die so vorbereiteten doppelsträngigen DNA-Fragmente in Einzelstränge gespalten.
Schritt 2: Brückenamplifikation
– Der Tanz der DNA auf der Flow Cell –Das Herzstück der Illumina-Technologie ist die sogenannte Flow Cell – eine glasartige Platte, übersät mit Millionen winziger DNA-Andockstellen (kurzen DNA-Abschnitten, sogenannten Oligonukleotiden), die fest an der Oberfläche verankert sind.
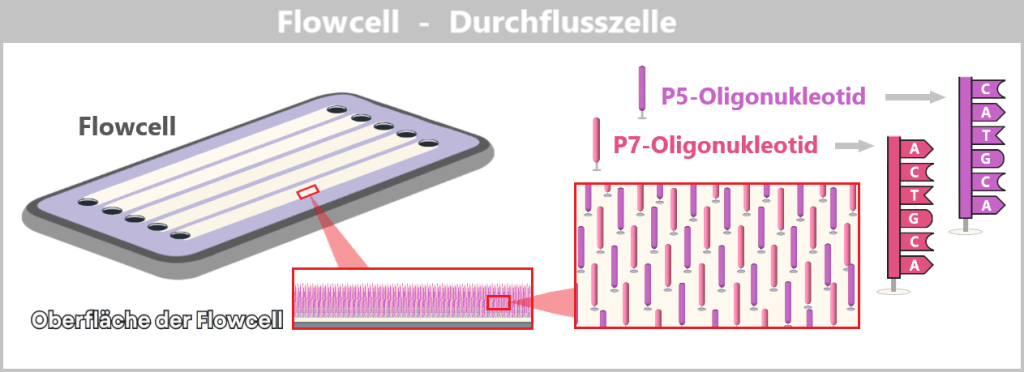
Abb. 7-C: Schematische Darstellung einer Flowcell und deren Oberfläche Über ihre Adapter binden sich die einzelsträngigen DNA-Fragmente an die Anker. Anschließend beginnt die molekulare Kopierarbeit: Es werden komplementäre Kopien der DNA-Schnipsel erzeugt, die nun fest mit der Flow Cell verankert sind.
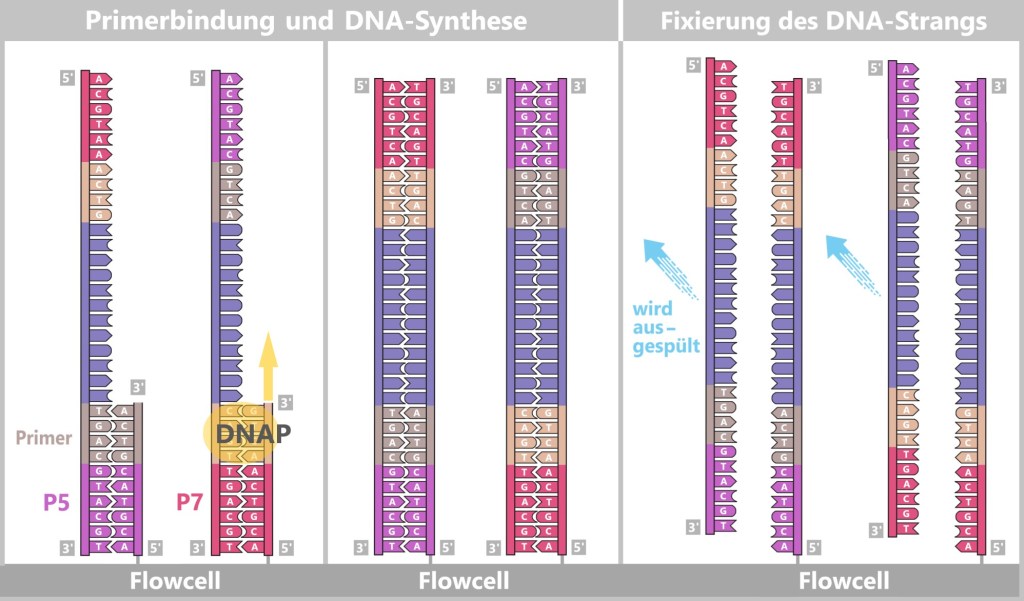
Abb. 7-D: Schematische Darstellung der ersten Synthese Links: Primer binden an die Adapter (P5, P7), und die DNA-Polymerase (DNAP) startet die Synthese eines neuen, komplementären Strangs.
Mitte: Die DNA-Polymerase synthetisiert daraufhin einen ersten Strang.
Rechts: Der neue entstandene DNA-Doppelstrang wird aufgetrennt. Der ursprüngliche Strang hat nach der Denaturierung keine Verbindung mehr zur Flowcell und wird ausgespült. Der neu synthetisierte Strang bleibt mit seinem 5′-Ende fest an der Flowcell gebunden.Dann beginnt ein faszinierender Prozess: Die DNA-Stränge biegen sich zu kleinen Brücken, indem ihre freien Adapter an benachbarte Oligonukleotide auf der Flow Cell binden. An diesen Stellen werden sie erneut kopiert. Nach dem Kopieren wird die Brücke wieder aufgelöst – und die Zahl der fest verankerten DNA-Stränge verdoppelt sich. Dieser Vorgang, bekannt als Brückenamplifikation, wiederholt sich dutzendfach. Am Ende entstehen dichte Cluster aus Millionen identischer Kopien eines einzelnen DNA-Fragments.
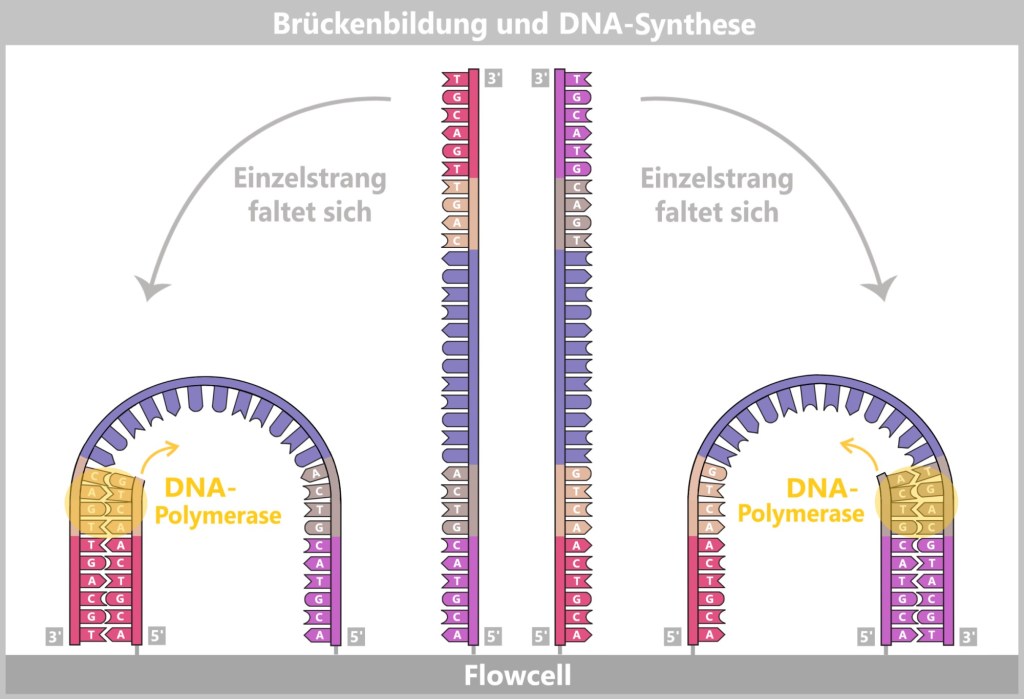
Abb. 7-E: Brückenbildung und DNA-Synthese Die verankerten Einzelstränge falten sich und verbinden sich mit den benachbarten Ankern auf der Flowcell, wodurch eine Brückenstruktur entsteht. Danach wird der Kopiervorgang initiiert.
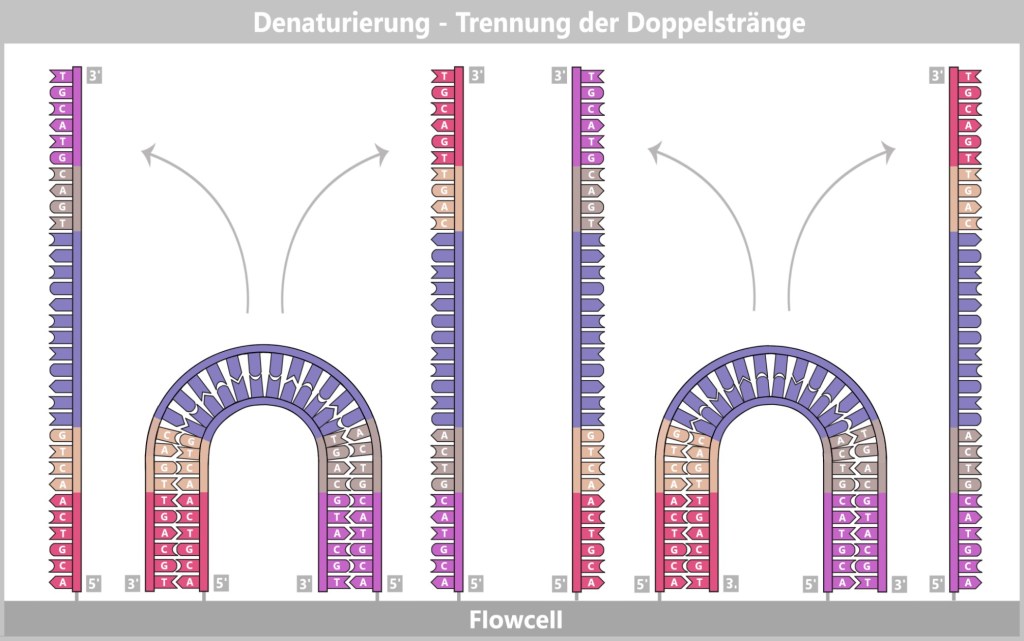
Abb. 7-F: Vorwärts und rückwärts: Die Brücken lösen sich. Nach dem Kopiervorgang werden die Brücken-Doppelstränge getrennt. Die Anzahl der fest verankerten DNA-Stränge verdoppelt sich. Sie sind bereit für die weitere Amplifikation.

Abb. 7-G: Mit jeder Wiederholung der Brückenamplifikation entstehen immer mehr DNA-Kopien. 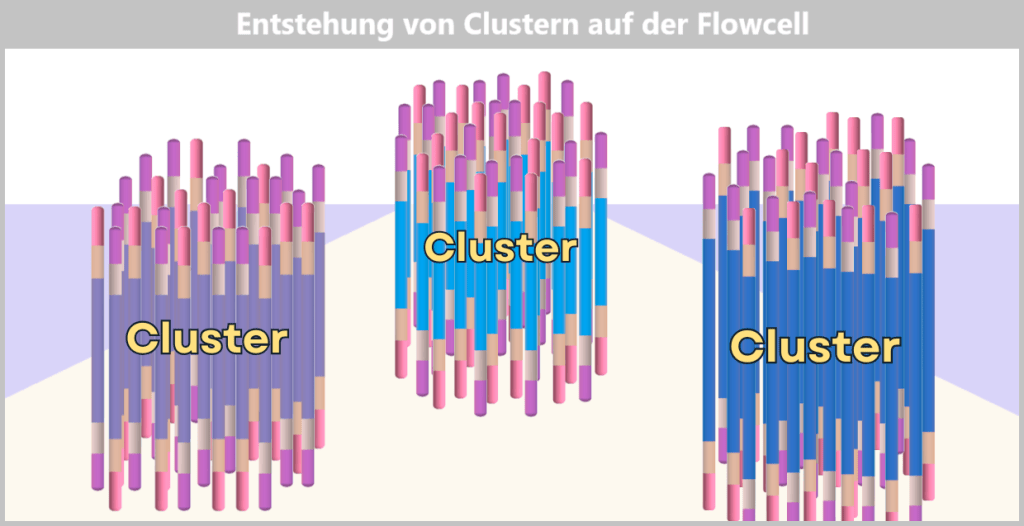
Abb. 7-H: Clusterbildung Das Ergebnis ist eine dicht gepackte „Landkarte“ aus Millionen DNA-Clustern, die jeweils nur eine bestimmte Sequenz enthalten – ideal für das gleichzeitige Auslesen.
Ohne diese Vervielfältigung wäre das Lesen der DNA, als würde man versuchen, ein einzelnes Glühwürmchen im Wind zu erkennen. Die Cluster hingegen machen die DNA-Basen (A, T, C, G) deutlich sichtbar – wie ein leuchtender Neon-Schriftzug in der Dunkelheit.
Schritt 3: Sequenzieren durch Synthese
– Die Lichtshow der DNA-Basen –Nun beginnt das eigentliche Sequenzieren durch Synthese. Auch hier kommt das Prinzip der „molekularen Kopiermaschine mit Farbsensoren“ zum Einsatz – aber in optimierter Form:
Zu Beginn binden die Universal-Primer an den entsprechenden Adapterstellen der DNA-Fragmente in jedem DNA-Clusters.
Dann werden modifizierte dNTPs – sogenannte RT-dNTPs (reversibel terminierende Nukleotide) – in die Reaktionslösung gegeben. Jeder der vier DNA-Bausteine (A, T, G, C) ist dabei mit einer eigenen Fluoreszenzfarbe markiert und trägt eine reversible Blockierung.
Nur ein Nukleotid pro Zyklus kann eingebaut werden, weil die Blockierung den weiteren Strangaufbau vorübergehend stoppt. Nicht eingebaute RT-dNTPs werden weggewaschen.
Nach jedem Einbau wird die Flowcell mit einer hochauflösenden Kamera gescannt. Jeder leuchtende Punkt steht für ein eingebautes Nukleotid und seine Farbe zeigt an, welcher Buchstabe gerade ergänzt wurde.
Danach wird die Blockierung und die Farbstoffmarkierung chemisch entfernt, und der nächste Zyklus beginnt.
Dieser Prozess wird Zyklus für Zyklus wiederholt – bis jeder DNA-Strang vollständig gelesen ist.
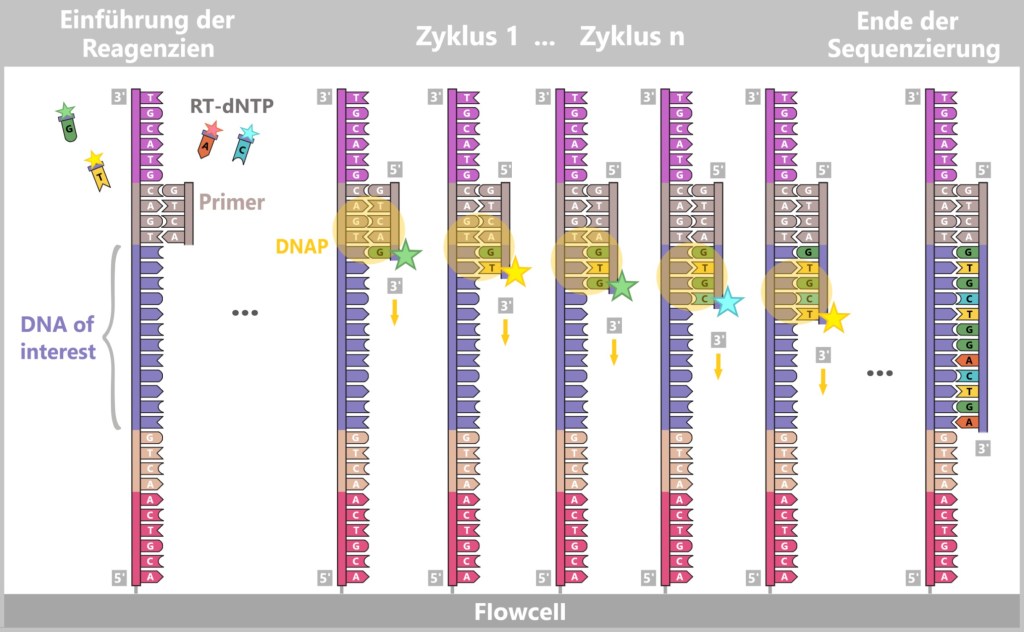
Abb. 7-I: Illumina Sequenzierungsschritte Schritt 4: Datenauswertung
– Der Supercomputer als Puzzlemeister –Nach der „Lichtshow“ liegen Millionen von Fotos vor – eines für jeden Zyklus und jedes Cluster. Jedes Bild zeigt, welche Farbe (und damit welcher Buchstabe: A, T, C oder G) in jedem Cluster hinzugefügt wurde.
Jetzt kommen leistungsfähige Computer ins Spiel – ausgestattet mit Software, die so präzise arbeitet wie ein Detektiv, der aus einem Stapel durchnummerierter Polaroids ein zerschnittenes Buch zusammensetzt. Jedes Cluster auf dem Sequenzierungschip ist wie ein kleiner Tatort, jeder Farbpixel eine Spur. Und aus Millionen solcher Spuren entsteht allmählich eine Geschichte – die Geschichte der DNA.
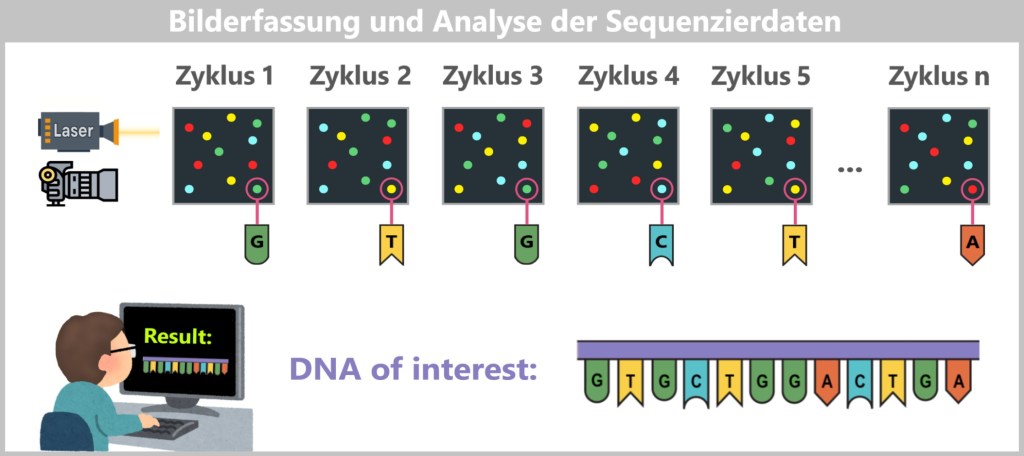
Abb. 7-J: Analyse der Sequenzierdaten Die Farbmuster werden in Buchstabenfolgen übersetzt, sogenannte „Reads“: winzige Textstücke eines gewaltigen Genom-Romans. Am Ende türmt sich eine Flut solcher Fragmente auf – wie ein Berg zerschnittener Buchseiten, lose verstreut.
Genau hier beginnt die eigentliche Kunst: Die Bioinformatik tritt auf den Plan. Mit Hilfe spezieller Algorithmen werden die Schnipsel analysiert, sortiert und aneinandergefügt – immer auf der Suche nach überlappenden Stellen, vertrauten Mustern, bekannten Strukturen.
Stück für Stück setzt sich das große Ganze wieder zusammen – bis am Ende die ursprüngliche DNA-Sequenz sichtbar wird, wie ein rekonstruiertes Buch, das plötzlich wieder Sinn ergibt.
Die eigentliche „Magie“ geschieht also nicht in der Flow Cell, sondern im Computer! Ohne die Software wären die Bilder nur buntes Flimmern – mit ihr aber werden sie zum Schlüssel für medizinische Durchbrüche.
Eine ausführlichere und dennoch anschauliche Erklärung der Illumina-Sequenzierungstechnologie Methode findet sich im Video „Illumina Sequencing Technology“.
Moderne Hochdurchsatz-Sequenzierplattformen wie die von Illumina können die Rohdaten eines vollständigen menschlichen Genoms innerhalb weniger Tage erzeugen – zu reinen Sequenzierkosten, die deutlich unter dem Preis eines Premium-Smartphones liegen.
Diese Technologie hat die Genomforschung revolutioniert und erheblich zugänglicher gemacht: Sie ist heute ein äußerst vielseitiges Werkzeug – schnell genug für die Echtzeit-Analyse von Pandemien, präzise genug, um in Kombination mit robuster Bioinformatik und klinischer Expertise die Grundlage für personalisierte Krebstherapien zu bilden.
Die Illumina-Sequenzierung gilt inzwischen als Industriestandard für große Sequenzierprojekte – etwa bei der Analyse ganzer Genome, in der Genexpressionsforschung oder in der modernen Krebsdiagnostik.
Und das Beste: Während man diesen Text liest, entschlüsselt irgendwo auf der Welt eine Flow Cell Millionen DNA-Fragmente.
Allerdings hat auch die Illumina-Technologie ihre Grenzen. Besonders bei langen, sich wiederholenden DNA-Abschnitten – wie sie in manchen Chromosomenbereichen vorkommen – gerät sie an ihre Grenzen. Das ist, als müsste man ein Lied aus tausenden 3-Sekunden-Schnipseln rekonstruieren. Für solche Herausforderungen kombiniert man Illumina häufig mit anderen Sequenzierverfahren, die auch lange Abschnitte zuverlässig erfassen können – wie ein Ermittler, der sowohl Nahaufnahmen als auch Panoramabilder braucht, um den ganzen Tatort zu verstehen.
c) Single-Molecule Real-Time(SMRT)-Sequenzierung
Der Roman in einem Zug
Stellen Sie sich vor, Sie wollen einen langen Roman lesen – voller wiederkehrender Kapitel und versteckter Hinweise, die sich über viele Seiten erstrecken. Wenn Sie das Buch wie bei der Illumina-Sequenzierung in tausende kleine Schnipsel zerschneiden, diese einzeln lesen und anschließend wie ein Puzzle zusammensetzen, besteht die Gefahr, dass Kapitel fehlen, vertauscht oder unvollständig bleiben. Die geheimen Botschaften der Geschichte – das, was der Roman uns eigentlich erzählen will – könnte dadurch verloren gehen oder nur bruchstückhaft erscheinen.
Hier glänzt die Single Molecule Real-Time (SMRT)-Sequenzierung von PacBio: Sie liest ganze Buchkapitel in einem Zug – ohne Schnipsel, ohne Pause.
Anstatt die DNA zu zerlegen, bleibt sie bei PacBio in sehr langen Stücken erhalten – oft Zehntausende Basenpaare am Stück, die als Ganzes sequenziert werden. Diese Fäden betreten winzige Bühnen in einer SMRT-Zelle, sogenannte Nanokammern (Zero-Mode Waveguides). Diese sind so klein, dass sie nur Platz für ein einziges DNA-Molekül und eine Polymerase bieten. Jede dieser Bühnen lässt Licht nur an einem winzigen Punkt wirken – genau dort, wo die Polymerase die DNA abliest. Wie ein Spotlicht, dass einen Schauspieler bei einem Monolog auf der Bühne ins Zentrum rückt.
Die Polymerase spielt die Hauptrolle: Sie fügt Baustein für Baustein (A, T, C oder G) an die DNA-Kopie an. Jeder dieser Bausteine leuchtet in einer anderen Farbe, ähnlich wie Neonlichter bei einem DJ. Bei jedem Einbau blitzt ein Lichtsignal auf, das von einer Kamera in Echtzeit aufgezeichnet wird – ein tanzender Lichtcode, der die DNA-Sequenz sichtbar macht.
Das Video Introduction to SMRT Sequencing von PacBio veranschaulicht diesen Vorgang eindrucksvoll.
Analysieren wir die einzelnen Schritte dieses faszinierenden Verfahrens im Detail.
Schritt 1: Die Vorbereitung
– DNA-Ringe für das Endloslesen –Auch bei PacBio wird die zu untersuchende DNA zunächst in Fragmente zerlegt – allerdings in deutlich größere Stücke als bei Illumina, meist 10.000 bis über 20.000 Basenpaare lang. Diese Fragmente werden zu ringförmigen DNA-Molekülen verarbeitet:
Man fügt an beide Enden sogenannte Hairpin-Adapter an – kleine Schlaufen aus DNA, die das Fragment in sich selbst schließen.
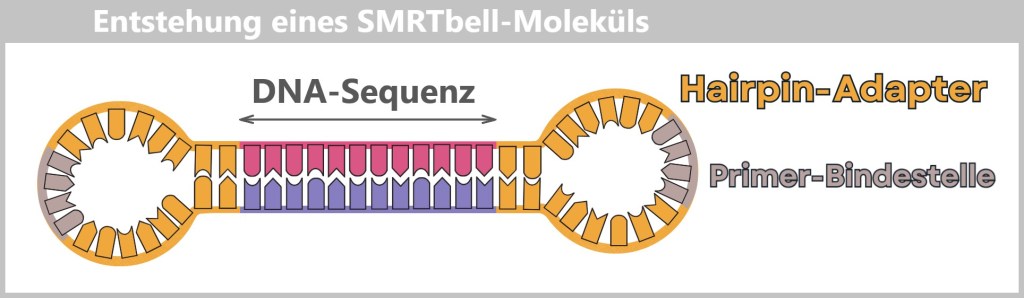
Abb. 8-A: Ein DNA-Fragment wird mit Hairpin-Adaptern versehen. Die Adapter enthalten eine Primer-Bindestelle, die später den Startpunkt für die Polymerase liefert.
Durch anschließende Denaturierung entsteht ein zirkuläres, einzelsträngiges SMRTbell-Molekül in Form einer offenen Schlaufe.
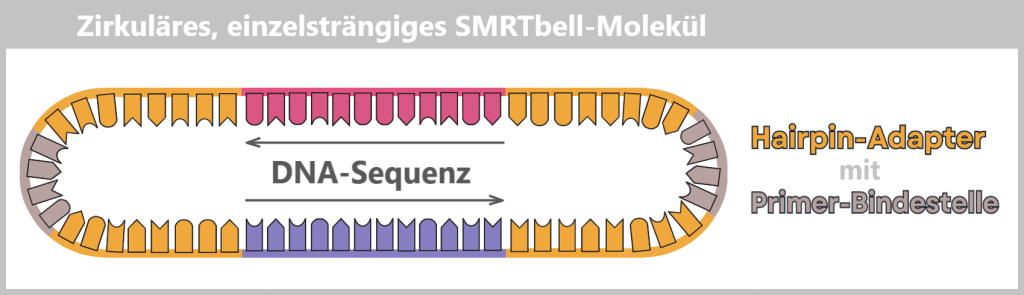
Abb. 8-B: Das denaturierte SMRTbell-Molekül: Durch Denaturierung entsteht aus der doppelsträngigen Vorlage ein einzelsträngiges SMRTbell-Molekül. Dieses bildet eine zirkuläre Struktur, die in der Darstellung als geöffnete Schlaufe erscheint.
Das Ergebnis ist ein SMRTbell: ein geschlossener DNA-Ring, der von der Polymerase beliebig oft gelesen werden kann – wie eine Modelleisenbahn, die im Kreis fährt.
Jeder Hairpin-Adapter enthält eine definierte Bindungsstelle, an der der universelle Primer bindet. Das ist der Startpunkt für die Polymerase.
Die DNA-Polymerase lagert sich an den Primer-Komplex an. Es entsteht ein voll funktionsfähiger Replikationskomplex: Polymerase + Primer + SMRTbell.
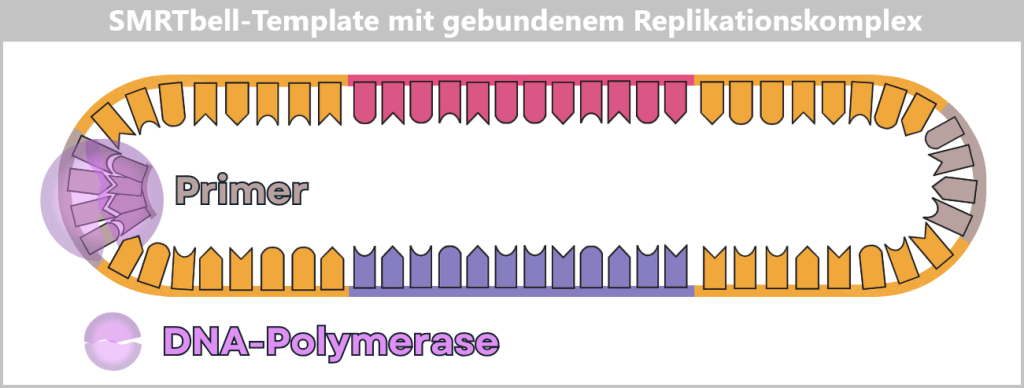
Abb. 8-C: Der stabile Replikationskomplex aus Polymerase, Primer und SMRTbell-Einzelstrang. Der Primer ist an die Adapter-Bindestelle gebunden, die Polymerase an den Primer. Dieser Komplex ist startbereit für die DNA-Synthese.
Schritt 2: Die Sequenzierbühne
– Lichtspiele im Nanomaßstab –Jetzt wird es spektakulär: Die vorbereiteten Replikationskomplexe werden in ihre winzigen Beobachtungskammern geleitet – die Zero Mode Waveguides (ZMWs). Diese Nanokammern sind so klein, dass in der Regel nur ein einziges SMRTbell-Template Platz findet. Die Polymerase ist dabei fest am Boden des ZMWs verankert und wartet auf ihren Einsatz.
Das Startsignal gibt die Zugabe der vier Nukleotide (A, C, G, T), die jeweils mit einem eigenen Fluoreszenzfarbstoff markiert sind.
Die Polymerase beginnt nun mit ihrer Arbeit – sie liest den DNA-Strang und baut einen komplementären Strang auf. Sobald sie ein Nukleotid einbaut, passiert das Entscheidende:
Jedes Nukleotid sendet einen kurzen, farbcodierten Lichtblitz aus.
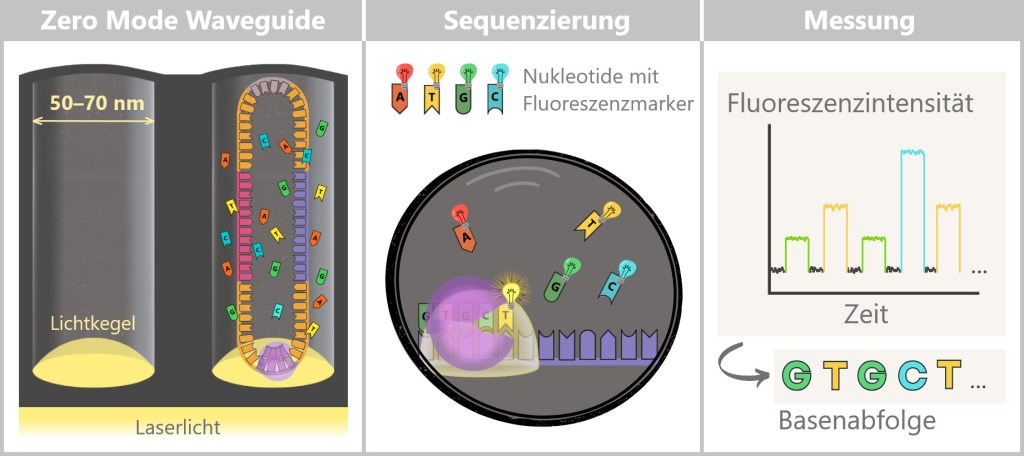
Abb. 8-D: Zero Mode Waveguide (ZMW): Sequenzierung einzelner DNA-Moleküle Links: Am Boden des Zero Mode Waveguides (ZMW) tritt ein Laserstrahl ein, der ein sogenanntes evaneszentes Feld (dargestellt als Lichtkegel) erzeugt. Dieses Feld breitet sich nicht wie normales Licht in die Lösung aus, sondern klingt innerhalb von nur 20–30 Nanometern exponentiell ab. Dadurch entsteht ein extrem kleines Anregungsvolumen, in dem die Fluoreszenzfarbstoffe der Nukleotide angeregt werden können.
Im linken Zero Mode Waveguide (ZMW) befindet sich ein einzelner Replikationskomplex (Polymerase + SMRTbell). Die Polymerase (lila) ist dabei fest am (Glas-)Boden verankert. Mit Anwesenheit der Fluoreszenz-markierten Nukleotide beginnt die Polymerase mit ihrer Arbeit.
Mitte: Die vier Nukleotide (A, T, G, C) tragen unterschiedliche Fluoreszenzmarker. Sobald die Polymerase ein Nukleotid einbaut, leuchtet es kurz auf, bevor der Farbstoff abgespalten wird.
Rechts: Diese Lichtblitze werden in Echtzeit gemessen und in Fluoreszenzsignale übersetzt. So entsteht Schritt für Schritt die Basenabfolge der DNA.Das Leuchten geschieht in Echtzeit, genau im Moment der Einfügung – daher der Name: Single Molecule Real-Time Sequencing. Ein empfindlicher Detektor zeichnet diese Lichtsignale auf –wie ein molekulares Live-Mikroskop bei der Arbeit.
Schritt 3: Das Geheimnis der Langstrecke
– Wiederholung macht’s robust –Da PacBio sehr lange DNA-Abschnitte auf einmal liest – vergleichbar mit dem Lesen ganzer Buchkapitel statt einzelner Verse, treten bei einem einzelnen Durchlauf mehr Einzelfehler auf als bei Illumina. Genau hier kommt die ringförmige SMRTbell-Struktur zum Einsatz. Die DNA-Polymerase kann wie ein Zugführer das Ringmolekül mehrfach umrunden. Dabei wird das gleiche DNA-Fragment immer wieder gelesen – wie ein Vorleser, der sich mehrfach an denselben Text wagt, bis er ihn sicher beherrscht.
Ein Computer gleicht die Wiederholungen miteinander ab und filtert die Fehler heraus – wie ein Lektor, der ein Manuskript glättet. So entsteht eine besonders präzise Konsens-Sequenz.
PacBio nennt dieses Verfahren HiFi-Reads – hochpräzise Einzelmolekül-Lesungen.
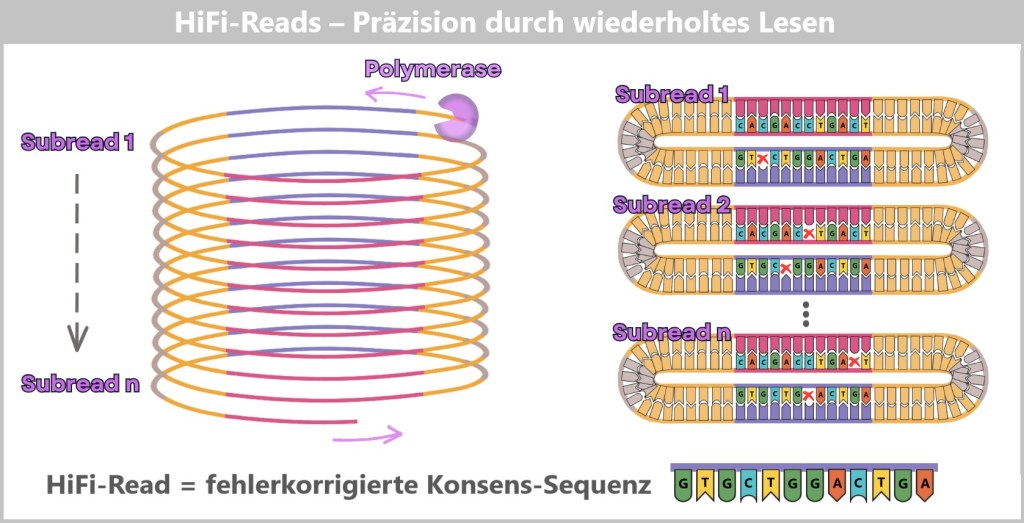
Abb. 8-E: Die Entstehung eines HiFi-Reads. Die Polymerase wandert mehrfach um das zirkuläre SMRTbell-Template und erzeugt dabei mehrere Subreads. Jeder Umlauf entspricht einer vollständigen Ablesung der DNA-Sequenz. Einzelne Subreads können zufällige Fehler (x) enthalten, doch durch den Vergleich vieler Wiederholungen wird ein hochpräziser Konsens gebildet – der sogenannte HiFi-Read (High-Fidelity Read).
Zusatzinfo: Wie die Polymerase mehrfach um die DNA läuft
Erster Sequenzierungsdurchlauf
Die Polymerase liest den einzelsträngigen Template-Strang ab und synthetisiert dabei einen komplementären Strang.
Das Ergebnis ist eine vollständige Doppelhelix – der ursprüngliche Template-Strang und der neu entstandene Tochterstrang sind nun fest miteinander verbunden.
Aus dieser ersten, ununterbrochenen Umrundung entsteht ein Subread – also eine einzelne Basenabfolge, die von der Polymerase in einem Durchlauf erzeugt wurde.
Zweite und weitere Sequenzierungsdurchläufe
Nun stellt sich die Frage: Wie kann die Polymerase weiterlesen, wenn doch der DNA-Ring bereits doppelsträngig ist?
Hier kommt die Φ29-Phagen-DNA-Polymerase ins Spiel – eine speziell modifizierte Enzymvariante mit zwei entscheidenden Eigenschaften:
Hohe Prozessivität: Sie bleibt sehr lange am DNA-Strang gebunden und löst sich kaum – das ermöglicht die langen Reads.
Starke Strangverdrängungsaktivität: Sie kann Wasserstoffbrückenbindungen zwischen Template- und Tochterstrang aufbrechen.
Die Polymerase läuft in die Doppelhelix hinein, trennt die Stränge lokal auf und verdrängt den alten Tochterstrang, während sie gleichzeitig einen neuen synthetisiert – wie ein molekularer Bulldozer.
Der zuvor gebildete Strang wird dabei nicht abgebaut, sondern bleibt als loser, einzelsträngiger DNA-Faden in der Nanokammer zurück. Bei jeder weiteren Runde wiederholt sich dieser Prozess: Die Polymerase liest dasselbe Template erneut ab – und erzeugt dadurch mehrere Subreads vom gleichen DNA-Molekül.
Schritt 4: Datenanalyse
– Aus Lichtblitzen wird lesbare DNA –Sind alle Lichtsignale eingefangen, beginnt die eigentliche Detektivarbeit: die Datenanalyse.
Der erste Schritt ist das sogenannte Base-Calling: Dabei werden die aufgezeichneten Fluoreszenzsignale – zeitlich und farblich codiert – in Nukleotidsequenzen übersetzt – A, T, G oder C.
Anschließend werden die Rohdaten jedes Polymerase-Umlaufs bereinigt: Die Hairpin-Adapter-Sequenzen werden bioinformatisch abgetrennt. Zurück bleiben die sogenannten Subreads – die ungekürzten Leseprotokolle jedes einzelnen Rundgangs der Polymerase durch den DNA-Kapiteltext.
Doch erst der nächste Schritt enthüllt die volle Wahrheit: Wie ein philologischer Meisterdetektiv, der mehrere Abschriften eines Manuskripts vergleicht, kombiniert der Algorithmus alle Subreads desselben SMRTbell-Moleküls. Aus diesem Vergleich – Circular Consensus Sequencing (CCS) genannt – entsteht ein hochpräziser HiFi-Read: eine fehlerbereinigte Masterkopie, die das DNA-Kapitel wortgetreu wiedergibt.
Erst jetzt ist das Textfragmentbereit für die finale Einordnung – und durchläuft dabei mehrere Stationen:
Zunächst eine Qualitätskontrolle, ganz wie ein sorgfältiges Lektorat, das den Text auf Klarheit und Stimmigkeit prüft.
Darauf folgt das Alignment: Die Sequenz wird wie ein neues Kapitel ins richtige Fach eines Referenzgenom-Regals eingeordnet.
Schließlich erfolgt die Assemblierung – der Schlussakt, bei dem alle Kapitel zu einem vollständigen Roman des Genoms zusammengefügt werden.
Das Video PacBio Sequencing – How it Works fasst die Schritte anschaulich zusammen.
Anwendung und Vorteile – Die Stärken der Langleser
Die PacBio-SMRT-Technologie hat eine besondere Stärke: Sie kann sehr lange DNA-Abschnitte am Stück lesen – mit höchster Genauigkeit. Das macht sie ideal für:
- Das Erkennen komplexer Genomstrukturen
- Die Analyse wiederholungsreicher Abschnitte
- Die Unterscheidung sehr ähnlicher Genvarianten (z. B. bei Immunrezeptoren oder in der Krebsgenetik)
- De-novo-Sequenzierung – also das vollständige Lesen neuer Genome ohne Referenz
- Das Aufspüren von epigenetischen Modifikationen (z.B. Methylierungen)
Wie kann SMRT epigenetische Modifikationen sichtbar machen?
Wie bereits beschrieben, spielen epigenetische Mechanismen eine zentrale Rolle bei der gezielten Genregulation. Es handelt sich um kleine chemische Markierungen – etwa Methylgruppen – die an bestimmte DNA-Basen angeheftet werden. Sie verändern nicht die genetische Sequenz selbst, sondern beeinflussen, wann und wie oft Gene abgelesen werden – abhängig von Zelltyp, Umgebung oder Entwicklungsstadium.
Sie funktionieren wie Notizen oder Markierungen am Seitenrand eines Buches: Der Text bleibt derselbe, aber die „Lesart“ ändert sich – manche Passagen werden betont, andere überblättert.
Gerät dieses fein abgestimmte System aus dem Gleichgewicht, kann das weitreichende Folgen haben – etwa bei Krebs, Autoimmunerkrankungen oder psychischen Störungen.
Im Gegensatz zur Illumina-Technologie, die eine chemisch veränderte Kopie der DNA liest, analysiert SMRT die Original-DNA in Echtzeit. Dabei wird nicht nur die Basenabfolge, sondern auch die Verweildauer der DNA-Polymerase an jeder Base – die sogenannte Einbaukinetik – gemessen.
Genau hier liegt der Schlüssel zum Nachweis epigenetischer Modifikationen: Methylierungen und andere chemische Veränderungen beeinflussen diese Kinetik. Sie wirken wie kleine Hindernisse oder Bremsspuren auf dem DNA-Strang. Die Polymerase „verharrt“ an solchen Stellen länger – und genau diese Verzögerung wird von der SMRT-Kamera direkt erfasst.
SMRT eignet sich besonders gut zur direkten Detektion bestimmter chemischer Markierungen, etwa Methylgruppen an Adenin- oder Cytosin-Basen, die vor allem in Bakterien und Pflanzen eine wichtige Rolle spielen. Für die in der humanen Epigenetik zentrale Cytosin-Methylierung (5mC) ist die direkte Detektion weniger sensitiv, kann aber mithilfe spezialisierter Bioinformatik-Tools wie pb-CpG-tools zuverlässig abgeleitet werden.
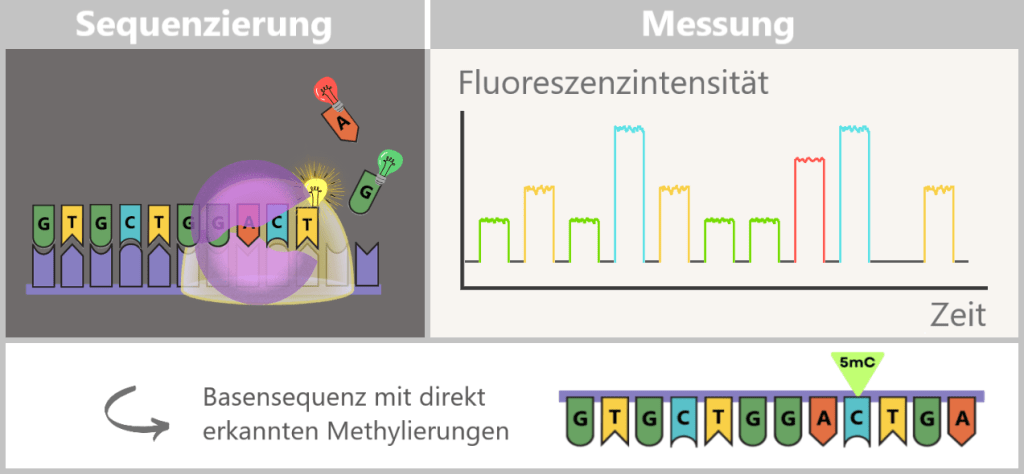
Abb. 8-E: Direkte Basendetektion und Methylierungsanalyse in der SMRT-Sequenzierung Wie SMRT epigenetische Modifikationen sichtbar macht
Während der SMRT-Sequenzierung werden die Fluoreszenzsignale der eingebauten Nukleotide in Echtzeit gemessen. Gleichzeitig können Modifikationen wie 5-Methylcytosin (5mC) erkannt werden, da sie die Kinetik der Polymerase leicht verändern.Der entscheidende Vorteil: Die extrem langen Leselängen enthüllen, wie sich Methylierungsmuster über große genomische Regionen mit anderen Faktoren verschränken – etwa mit:
- Genvarianten,
- Haplotypen (Gruppen von gemeinsam vererbten Genvarianten),
- repetitiven Sequenzen.
So liefert PacBio nicht nur die genetische Buchstabenfolge, sondern eine „Landkarte“ der epigenetischen Steuerung – und macht sichtbar, welche „Buchseiten“ funktionell zusammengehören und wie diese Vernetzung Krankheitsprozesse wie Krebs oder neurologische Störungen beeinflusst.
Defekte Scheren im Genom: Wie PacBio Splice-Site-Varianten enttarnt
Im Abschnitt Alternatives Splicing: Die flexible Küche der Gene wurde vereinfacht beschrieben, wie unsere DNA als molekulares Kochbuch Rezepte in Gestalt von mRNA-Abschriften erzeugt. Diese transportieren die genetische Information aus dem Zellkern zu den Ribosomen im Zytoplasma – den „Protein-Fabriken“ der Zelle.
Dabei wird das Rezept nachträglich angepasst: Eine molekulare Schere entfernt bestimmte Zutaten (Introns) und kombiniert andere Bausteine (Exons) neu. Gesteuert wird dieser Prozess durch Splice-Stellen – Markierungen, die aus der DNA „mitkopiert“ werden. Man kann sie sich wie eine Schnittanleitung vorstellen: Sie entscheidet, welche Teile des Rezepts verwendet und wie sie zusammengesetzt werden.
Doch was passiert, wenn diese Schnittanleitung fehlerhaft ist? Die Folgen können gravierend sein:
- Exons werden übersprungen → Teile der Bauanleitung fehlen.
- Introns bleiben fälschlich erhalten → „Müll“ wird ins Protein eingebaut.
- Falsche Spleißstellen entstehen → Der mRNA-Text wird sinnentstellt.
Solche Splice-Site-Varianten führen häufig zu fehlerhaften oder funktionslosen Proteinen – mit möglichen Folgen wie Krebs, genetischen Erkrankungen (z. B. β-Thalassämie) oder neurologischen Störungen. Da die Proteinbiosynthese direkt vom mRNA-Text abhängt, ist es entscheidend, solche Fehler zu erkennen.
Der Schlüssel liegt in PacBios Fähigkeit, vollständige mRNA-Moleküle in einem Stück zu analysieren – meist 500 bis 5.000 Nukleotide lang, manchmal deutlich länger. Da PacBio DNA liest, wird die mRNA zunächst in eine stabile Kopie (cDNA, komplementäre DNA) umgewandelt. Diese kann dann vollständig sequenziert werden. So lässt sich exakt erkennen, wie das Rezept aufgebaut ist und welche Zutaten in welcher Reihenfolge vorgesehen sind – kein Schreib- oder Schnittfehler bleibt verborgen.
Zusätzlich kann auch der zugrunde liegende DNA-Abschnitt sequenziert werden. Bioinformatische Tools vergleichen cDNA- und DNA-Sequenzen mit Referenzdaten – so werden Abweichungen im Spleißmuster und ihre genetische Ursache sichtbar.
Die DNA zeigt: Hier ist die Anleitung fehlerhaft.
Die cDNA zeigt: Hier ist das Rezept verstümmelt.Erst die Kombination beider Informationen schafft eine fundierte Basis für präzise Diagnosen und gezielte Therapieansätze.
Fazit – Wenn man die ganze Geschichte erzählen will
Wenn es darum geht, die ganze Geschichte eines DNA-Abschnitts am Stück zu erzählen – ohne zu zerschneiden oder zusammenzusetzen –, dann ist SMRT-Sequenzierung die Methode der Wahl.
PacBio-SMRT ist der Archäologe der Genomik: Es sieht das Ganze, wo andere nur Bruchstücke liefern. 2022 trug es maßgeblich dazu bei, die letzten Lücken im menschlichen Genom zu schließen.
Die PacBio-SMRT-Technologie ist kein Massenprodukt, sondern ein Präzisionsinstrument. Sie ist langsamer und teurer pro Basenpaar als Illumina – aber dafür auch detailreicher und vielseitiger.
In der Praxis werden beide Technologien oft kombiniert: Illumina für die Breite, PacBio für die Tiefe. Gemeinsam ergeben sie ein vollständiges Bild – wie Satellitenbild und Nahaufnahme in einem einzigen Panorama.
4.4.2. Methode 2: Hightech-Tunnel – DNA elektrisch abtasten
Während bei der molekularen Kopiermaschine mit Farbsensoren die DNA zuerst vervielfältigt und dann bildlich „fotografiert“ wird, funktioniert die zweite Methode radikal anders: Die DNA wird durch einen Hightech-Tunnel geschickt – und dabei live elektronisch abgetastet.
Nach diesem Prinzip funktioniert die Oxford Nanopore Sequenzierung.
a) Oxford Nanopore Sequenzierung
Die DNA geht durchs Nadelöhr
Stellen Sie sich vor, Sie lesen ein Seil, indem Sie es langsam durch Ihre Finger gleiten lassen – und dabei jede noch so feine Unebenheit ertasten: Dicke, Dellen, Knoten. Genau so funktioniert die Oxford Nanopore Sequenzierung (ONT): Sie „fühlt“ die DNA, während sie durch ein winziges biologisches Nadelöhr gleitet.
Dieses Nadelöhr – ein sogenannter Nanopore-Kanal – ist eingebettet in eine Membran, durch die ein elektrischer Strom fließt. Sobald ein einzelnes DNA-Molekül durch diesen Hightech-Tunnel gezogen wird, beeinflussen die Basen (A, T, G oder C) den Stromfluss unterschiedlich stark – wie verschieden geformte Kieselsteine in einem Bach. Diese Veränderungen erzeugen charakteristische elektrische Signale, die in Echtzeit aufgezeichnet werden.
ONT liest also keine Lichtsignale, sondern Strommuster – ein elektrisches Fingerlesen der DNA.
Ein einzelner Faden – direkt gelesen
Bevor die Lesung beginnt, wird die DNA mit einem Motorprotein gekoppelt, das sie präzise durch die Pore zieht – nicht zu schnell, nicht zu langsam. Anders als bei Illumina oder PacBio braucht es weder Polymerase noch Fluoreszenz oder Amplifikation: Die Original-DNA wird direkt gelesen – ein großer Vorteil bei empfindlichen oder beschädigten Proben.
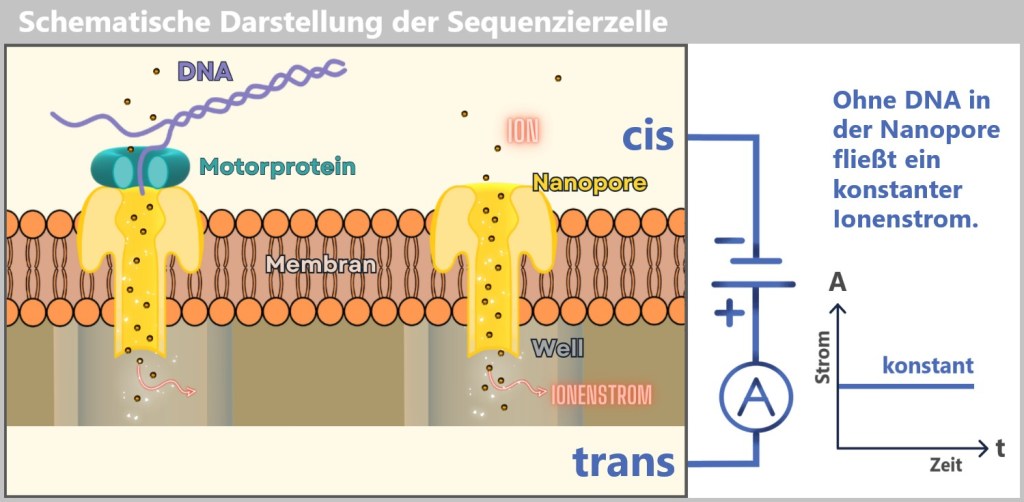
Abb. 9-A: Schematische Darstellung der Sequenzierzelle und des Ionenstroms im Ruhezustand Die Sequenzierzelle besteht aus zwei Kammern: der cis-Kammer (oben) und der trans-Kammer (unten), getrennt durch eine Membran mit eingebetteten Nanoporen. Links hat ein DNA-Molekül mithilfe eines Motorproteins an eine Nanopore angedockt. Rechts ist eine leere Nanopore dargestellt, durch die ein konstanter Ionenstrom fließt. Die angelegte Spannung zwischen der negativen Elektrode (cis) und der positiven Elektrode (trans) treibt den Ionenfluss an. Der Ionenstrom wird im Well (kleiner Kanal im Chip) gemessen, wie im Strom/Zeit-Diagramm dargestellt. Solange keine DNA durch die Pore wandert, bleibt der Strom konstant.
Und das Beste: Die DNA kann dabei extrem lang sein. ONT hält den Weltrekord für die längste je sequenzierte DNA – über zwei Millionen Basenpaare am Stück. Ganze Chromosomenabschnitte lassen sich so ohne Unterbrechung erfassen – wie ein Roman in einem einzigen Atemzug.
Aus Rauschen wird Sinn – Die Kunst der Signalinterpretation
Die elektrischen Rohsignale, die beim Durchgang der DNA durch die Pore entstehen, ähneln zunächst einem verrauschten Radiokanal. Doch mithilfe spezialisierter Algorithmen – dem sogenannten Basecalling – wird aus dem Stromrauschen eine klare Buchstabenfolge: A, T, G oder C.
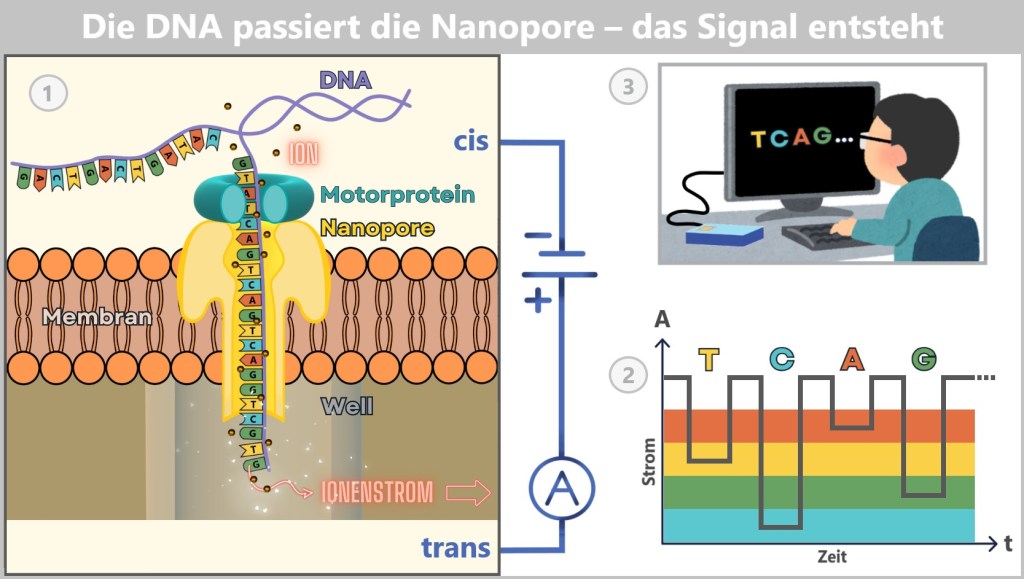
Abb. 9-B: Das Bild veranschaulicht den zentralen Mechanismus der Nanopore-Sequenzierung. 1) Hier sieht man eine schematische Darstellung der Nanopore in einer Membran. Ein Motorprotein zieht die DNA kontrolliert durch die Pore. Die negativ geladene DNA bewegt sich aufgrund der angelegten Spannung von der cis-Seite zur trans-Seite. Dabei beeinflussen die einzelnen Basen (A, T, G, C) den Ionenstrom auf spezifische Weise.
2) Ein Graph stellt die Veränderung des gemessenen Stroms über die Zeit dar. Jede Basen-Kombination erzeugt ein charakteristisches Signal, das durch Algorithmen entschlüsselt wird.
3) Ein Computer analysiert die elektrischen Signale und bestimmt daraus die Reihenfolge der Basen.Anders als bei PacBio werden bei ONT die Basen nicht einzeln unterschieden, sondern in Gruppen von fünf bis sechs Nukleotiden, die gleichzeitig in der Pore liegen. Jeder dieser „5-Mer“ erzeugt ein charakteristisches Stromsignal. Das erhöht die Lesegeschwindigkeit, macht die Interpretation aber komplexer – ein wenig wie das Erkennen von Wörtern in einem ungewohnten Dialekt. Besonders Homopolymeren (lange Wiederholungen derselben Base, z. B. „AAAAA“) stellen eine Herausforderung dar.
Die KI-gestützte Basecalling-Software verbessert sich kontinuierlich. Die Fehlerquoten sind heute drastisch gesunken, und ONT-Reads erreichen mittlerweile auch eine hohe Genauigkeit – besonders wenn die Datenmenge für eine statistische Mehrfachkontrolle jeder DNA-Base (durch wiederholte, zufällige Erfassung) hoch genug ist.
Eine coole visuelle Erklärung gibt’s im Video How nanopore sequencing works von Oxford Nanopore Technologies.
Wer es etwas ausführlicher mag, findet hier weitere Hintergrundinfos.
Besondere Stärken – Schnell, portabel, vielseitig
Was ONT besonders macht:
Mobilität: Geräte wie der MinION passen in eine Hosentasche. Sie benötigen nur einen Laptop und etwas Strom – ideal für Feldforschung, Raumfahrt oder schnelle Analysen bei Ausbrüchen (z. B. Ebola oder SARS-CoV-2).
Schnelligkeit: Die DNA wird live gelesen. Erste Sequenzdaten liegen oft schon nach wenigen Minuten vor – ein unschätzbarer Vorteil in klinischen Notfällen.
Direkte Epigenetik: Wie bei PacBio erkennt ONT Methylierungen und andere epigenetische Modifikationen direkt, da sie das elektrische Signal subtil beeinflussen – ganz ohne zusätzliche Behandlung oder Reagenzien.
Direkte RNA-Sequenzierung: Auch RNA-Moleküle können ohne vorherige Umwandlung in cDNA gelesen werden – ein echtes Alleinstellungsmerkmal.
Warum direkte RNA-Sequenzierung wichtig ist
Andere Sequenzierverfahren wie PacBio müssen mRNA erst in cDNA umwandeln, um sie lesen zu können. Das ist, als würde man ein Rezept zunächst abschreiben – dabei können kleine Details verloren gehen. Die Oxford-Nanopore-Technologie hingegen liest die mRNA direkt – ohne Umweg über DNA.
So bleiben alle „Randnotizen des Kochbuchs“ erhalten: chemische Modifikationen, Varianten und sogar die exakte Länge der Moleküle – Informationen, die bei einer Kopie oft verschwinden. Dadurch entsteht ein unverfälschtes Bild der Genaktivität – ein unschätzbarer Vorteil, um komplexe Abläufe wie Genregulation oder fehlerhafte RNA-Verarbeitung zu erkennen.
Gerade für die personalisierte Medizin ist das ein Durchbruch: Denn viele Krankheiten entstehen nicht allein durch veränderte DNA, sondern auch durch Abweichungen in der RNA.
Beispiele aus der Praxis:
- Krebsdiagnostik: RNA-Spleißvarianten können helfen, aggressive Tumorformen frühzeitig zu erkennen und gezielt zu behandeln.
- Erbkrankheiten: Bei Erkrankungen wie spinaler Muskelatrophie oder Duchenne-Muskeldystrophie ermöglichen RNA-Profile eine genaue Einschätzung des Verlaufs und personalisierte Therapien, z. B. mit Antisense-Oligonukleotiden.
- Infektionsmedizin: Die direkte Sequenzierung viraler RNA erlaubt eine schnelle Identifikation von Virusvarianten – entscheidend bei Epidemien und für die Wahl der Therapie.
Die direkte RNA-Sequenzierung hilft also, die persönliche Rezeptvariante jedes Menschen zu erkennen – mit all ihren kleinen Abweichungen, Randnotizen und besonderen Zutaten – und dadurch Diagnosen zu präzisieren sowie maßgeschneiderte Behandlungen zu ermöglichen.
Fazit: Der elektrische Sinnesfinger der Genomik
ONT ist der sinnesstarke Grenzgänger der Genomik: Schnell wie ein Rennpferd, portabel wie ein Schweizer Taschenmesser und nah dran am biologischen Geschehen. Wo andere kopieren müssen, fühlt es live – selbst bei RNA oder epigenetischen Spuren.
4.4.3. Genomsequenzierung – Technologien im Vergleich
In der Welt der Genomik gibt es nicht die eine Methode – sondern ein ganzes Ensemble. Jede Sequenzierplattform hat ihren eigenen Stil, ihre Stärken und ihr ideales Einsatzgebiet: vom klassischen Solo-Erzähler bis zum Hochgeschwindigkeitsstreamer.
Auch wenn alle vier Sequenzierer ihre Bühne haben – nicht jeder eignet sich für jede Inszenierung. Wer gezielt ein einzelnes Gen untersuchen will, braucht andere Mittel als jemand, der das gesamte Genom in epischer Breite erfassen möchte. Und wer seltene Varianten aufspüren will, muss genau hinlesen.
Betrachten wir die vier Hauptakteure im Detail, um ihre Stärken, Schwächen und strategischen Rollen zu verstehen.
a) Durchsatz vs. Verwertbarkeit – Masse ist nicht immer gleich Klasse
„Durchsatz“ klingt nach Effizienz – möglichst viele DNA-Buchstaben pro Minute. In der Sequenzierung misst man ihn in Basen pro Lauf: also wie viele Basen ein Gerät in einem einzigen Sequenzierprozess ausliest.
Moderne Plattformen wie Illuminas NovaSeq X Plus erreichen mit kurzen Reads (100–300 bp) Spitzenwerte von bis zu 16 Terabasen – das sind 16 Milliarden Buchstaben in einem einzigen Lauf. Wie ein Hochleistungsdrucker, der über Nacht ganze Genombibliotheken produziert.
PacBio und Oxford Nanopore verfolgen einen anderen Ansatz. Sie erzeugen lange Reads – von 10.000–25.000 bp bei PacBio HiFi-Reads, bis hin zu über 1 Million bp bei ONT. Ihr Durchsatz ist geringer: PacBio (Revio) erreicht bis zu 1.300 Gigabasen, ONT (PromethION) bis zu 7.000 Gigabasen pro Lauf. Klingt nach weniger – und das ist es auch, wenn man nur die Basenzahl betrachtet.
Doch: Mehr Daten bedeuten nicht zwangsläufig mehr Erkenntnis. Entscheidend ist nicht nur, wie viel gelesen wird – sondern wie präzise und wie oft. Zwei zentrale Konzepte helfen, die Aussagekraft von Sequenzdaten einzuordnen:
🔍 Quality Score – das Vertrauen in jeden Buchstaben
Der sogenannte Q-Score (Quality Score) beschreibt, wie wahrscheinlich ein gelesener DNA-Buchstabe korrekt ist. Die Skala ist logarithmisch:
- Q20 = 99 % Genauigkeit → 1 Fehler pro 100 Basen
- Q30 = 99,9 % Genauigkeit → 1 Fehler pro 1.000 Basen
- Q40 = 99,99 % Genauigkeit → 1 Fehler pro 10.000 Basen
In der Forschung ist häufig Q20 ausreichend. In der medizinischen Diagnostik gelten Q30 oder höher als Standard, bei Hochpräzisionsanwendungen (z. B. Tumorgenetik) wird Q40 erwartet.
Bei Rohdaten liegt Illumina typischerweise im Bereich von Q30–Q40, während PacBio (Continuous Long Reads, CLR) und ONT nur Werte zwischen Q10-Q15 aufweisen.
Sanger übertrifft alle – mit Q40–Q50, allerdings nur auf kurzen DNA-Abschnitten. Ideal zur gezielten Bestätigung einzelner Varianten.
Wie also erreichen moderne Sequenzierungsverfahren die hohen Anforderungen der klinischen Diagnostik? Die Antwort: durch Coverage und bioinformatische Analyse.
🔁 Coverage – die Wiederholung macht die Wahrheit
Coverage (Abdeckung) beschreibt, wie oft jeder Bereich des Genoms gelesen wurde.
Beispiel: 30x Coverage bedeutet, dass jeder Buchstabe im Durchschnitt 30-mal gelesen wurde – auf verschiedenen Fragmenten, in unterschiedlichen Konstellationen.
Warum ist das wichtig? Einzelmessungen können Fehler enthalten. Nur durch Wiederholung und statistische Mehrheitsbildung lassen sich echte Varianten von Artefakten unterscheiden.
In der medizinischen Diagnostik ist 30x Coverage der Mindeststandard.
Illumina erreicht bei 30–50x Coverage eine Gesamtgenauigkeit von Q35–Q40+. Es erkennt zuverlässigSingle Nucleotide Variants (SNVs), Insertions/Deletions (Indels) und Copy Number Variants (CNVs).
PacBio HiFi Reads erreichen bei 30x Coverage Q30–Q40 und ONT Q30-35. ONT benötigt 50x Coverage, um eine Präzision von Q44 zu erreichen.
Der große Vorteil von PacBio und ONT: Sie lesen wesentlich längere DNA-Abschnitte – oft 10.000 bis 100.000 Basen, bei ONT sogar über 1 Million. Dadurch erkennen sie komplexe Strukturen, Wiederholungen, epigenetische Signaturen und ermöglichen RNA-Untersuchung und die Phasierung – also die Zuordnung von Varianten zu väterlichen oder mütterlichen Chromosomen. Hier zählt nicht die Masse, sondern das Gesamtbild.
🧬 Je nach Ziel – unterschiedliche Anforderungen an Coverage und Genauigkeit
Die nötige Coverage und Sequenzierqualität hängen vom klinischen Einsatz ab:
Populationsstudien: Q30 bei 30x Coverage reicht meist aus, um statistische Muster zu erkennen.
Krebsdiagnostik: Q35 bei 60–100x zur sicheren Detektion seltener somatischer Mutationen.
Liquid Biopsies, Mosaik-Analysen: Q40 bei 100–300x – zur Unterscheidung von echten Signalvarianten vom Hintergrundrauschen.
Phasierung: Lange Reads (PacBio, ONT) mit 30x Coverage genügen oft, um Varianten zuverlässig zuzuordnen.
📉 Das Dilemma: Mehr Sicherheit – mehr Aufwand
Mehr Coverage erhöht die Sicherheit, bedeutet aber auch mehr Daten, längere Rechenzeiten, größere Infrastruktur – und höhere Kosten.
b) Wirtschaftlichkeit – Kosten pro Base vs. Kosten pro Erkenntnis
Illumina (NovaSeq/NextSeq) bleibt der Preis-Leistungs-Favorit für Whole-Genome-Sequencing:
👉 200–800 € für ein vollständiges Genom bei 30x-Abdeckung – präzise bei Punktmutationen, ideal für Routineanalysen.PacBio (Revio/Sequel IIe) und Oxford Nanopore (PromethION/MinION) sind teurer:
👉 700–1.200 € für 15–30x-Abdeckung – dafür mit zusätzlichen Informationen wie lange Reads, strukturelle Varianten, Epigenetik und RNA. Das ist kein Luxus, sondern in vielen Fällen diagnostisch entscheidend.Oxford Nanopore punktet zudem mit niedrigem Einstiegspreis: Ein MinION kostet wenige Tausend Euro, ist mobil und liefert Daten binnen Stunden.
👉 Die einfache Rechnung „mehr Basen = besserer Deal“ greift zu kurz. Entscheidend ist, was aus den Daten gelesen und verstanden werden kann.🧾 Versteckte Kosten – Das Genom ist günstiger zu lesen als zu verstehen
Ein menschliches Genom für unter 300 €? Technisch ja – aber meist sind nur Reagenzien, Maschinenzeit und Rohdaten abgedeckt.
Was oft fehlt, sind die unsichtbaren Posten:
🔧 Probenaufbereitung
🔍 Qualitätskontrolle & Library-Erstellung
🧠 Bioinformatik & klinische Interpretation
📄 Befundbericht & ärztliche RückspracheDiese Bereiche machen häufig 70–80 % der Gesamtkosten aus.
Denn: Lesen ist heute günstig – Verstehen bleibt anspruchsvoll.
c) Von der Probe zum Befund – Wie schnell spricht das ganze Genom?
Das Genomlesen ist wie eine Aufführung: Der Vorhang hebt sich mit der Probenentnahme – aber wann fällt der letzte Vorhang, der klinische Befund?
Es hängt von Technologie und Infrastruktur ab:
Oxford Nanopore ermöglicht vorläufige Ergebnisse binnen 24–48 Stunden – dank Echtzeit-Analyse während der Sequenzierung.
Illumina liefert mit optimierten Workflows Daten in 3–7 Tagen – der Goldstandard für routinierte Diagnostikabläufe.
PacBio benötigt 4–7 Tage, bietet dafür jedoch besonders hochwertige Langlesungen und epigenetische Informationen.
Sanger liefert Validierungen gezielt binnen Stunden – aber nur für kleine Zielregionen.
Die Plattformwahl ist also nicht nur eine Frage der Datenqualität – sondern auch des klinischen Timings.
Wer in Stunden statt Tagen handeln muss, greift zu Oxford Nanopore. Wer robuste Routinedaten braucht, setzt auf Illumina. Und wer maximale Kontexttiefe wünscht, nimmt sich mit PacBio etwas mehr Zeit – für deutlich mehr Einsicht.
Der Vorhang fällt. Doch hinter den Kulissen balanciert zwischen Hoffnung, Hightech und menschlicher Sorgfalt geht die Suche nach dem perfekten Tempo für jeden Patienten weiter. Denn am Ende zählt nicht die schnellste Maschine – sondern der richtige Takt für die menschliche Geschichte dahinter.
d) Klinische Einsatzfelder – Welche Technologie passt wozu?
Die Genomsequenzierung ist längst mehr als ein Forschungswerkzeug – sie ist zum zentralen Instrument geworden, um Therapien individuell zu planen. Doch welche Methode eignet sich für welche Aufgabe?
Ein Blick auf sechs zentrale Anwendungsfelder zeigt, wie diese Technologien die moderne Medizin prägen.
Anwendungsfeld Primäre Methode Ergänzende Methoden Routinediagnostik Illumina Sanger Pharmakogenomik Illumina Sanger Epigenetik PacBio, ONT – Krebsgenomik Illumina (SNVs), PacBio/ONT (SVs) Sanger Neonatale Notfalldiagnostik ONT Illumina, Sanger Seltene Erkrankungen PacBio, ONT Illumina, Sanger 👉 Erkenntnis: Die optimale Technologie hängt stets von der klinischen Fragestellung ab – Standardanalyse, Suche nach dem Unbekannten oder Notfalldiagnostik? Validierung oder Erstdiagnose?
e) KI als Co-Pilot – Automatisierung ohne Verantwortung abzugeben
Die Integration von Künstlicher Intelligenz (KI) – insbesondere Deep Learning – ist in allen modernen DNA-Sequenzierplattformen (Illumina, PacBio, Oxford Nanopore) unverzichtbar. Ihre Hauptanwendungsfelder sind:
🧬 Basecalling (Umwandlung von Rohdaten in DNA-Sequenzen) & Fehlerkorrektur
🔍 Qualitätskontrolle & Echtzeitanalyse
🧠 De-novo-Assembly, Genomanalyse & Variantenerkennung
📚 Pathogenitätsvorhersage, Literaturrecherche & Klinische Klassifikation
📄 Automatisierte Befundvorlagen👉 Erkenntnis: KI transformiert die Sequenzierung von einem reinen Datengenerierungs- zu einem intelligenten Interpretationsprozess – und ist damit Kern moderner genomischer Forschung und Diagnostik.
Doch wichtig bleibt: KI denkt (noch) nicht wie ein Arzt. Bei seltenen Erkrankungen, komplexen Verläufen oder neu entdeckten Genveränderungen bleibt die ärztliche Erfahrung zentral. Auch rechtlich ist eine ärztlich validierte Auswertung Pflicht – selbst im Zeitalter digitaler Genomik.
Am Ende ist Genomsequenzierung keine Solovorstellung – sie braucht das ganze Ensemble: Präzise Technik, kluge Bioinformatik und ärztliche Intuition. Denn nur wenn alle mitspielen, wird aus Daten heilsame Erkenntnis.
f) Vergleichstabelle: Sanger, Illumina, PacBio, ONT
Merkmal Sanger Illumina PacBio ONT Proben-vorbereitung DNA-Template, Extraktion,
Primer-Design, ddNTPs
🕒 ~6–12 Std.Fragmentierung,
Adapter-Ligation
🕒 ~6–24 Std.Hohe DNA-Qualität, Fragmentierung, SMRTbell-Adapter
🕒 ~1–2 TageFragmentierung,
Adapter mit Helikase (Motorprotein)
🕒 ~6–24 Std.Leselänge Kurz: 500–1.000 bp Sehr kurz: 100–300 bp (paired-end möglich) Lang: 10.000–25.000 bp (HiFi-Reads) Ultra-lang: 10.000 bp – >1 Mb Durchsatz Sehr niedrig: Einzelreaktionen (~1–100 kb/Lauf) Sehr hoch: bis 16.000 Gb (NovaSeq X Plus),
>100 Proben gleichzeitigMittel: bis 1.300 Gb / SMRT-Cell (Revio) Flexibel: 100–7.000 Gb (PromethION), bis 20 Gb (MinION) Genauigkeit Sehr hoch: Q40–Q50 (99,99–99,999 %) Hoch: Q30–Q40 (99,9–99,99 %), bei 30x: Q35–Q40+ Hoch: Q30–Q40 (HiFi-Reads), bei 30x Rohdaten: Q15–Q20+, bei 30x: Q30–Q35, Duplex: Q44 bei 50x Kosten/Genom (EUR) (nur Sequenzierung) – ~200–800 (30x) (Großlabore) ~700–1.200
(15–30x)~700–1.200
(15–30x)Zeit bis Ergebnis (WGS, Probe → Befund) – 3–7 Tage (inkl. Vorbereitung, Sequenzierung, Analyse) 4–7 Tage (inkl. Vorbereitung, Sequenzierung, Analyse) 24–48 Std.
(Echtzeit, inkl. Analyse)Varianten-erkennung SNVs, mitochondriale Varianten (Validierung) SNVs, Indels, CNVs (indirect), somatische Varianten, Splice-Varianten Indels, SVs, CNVs, Repeat-Expansionen, Splice-Varianten, somatische SVs Indels, SVs, CNVs, Repeat-Expansionen, Splice-Varianten, somatische SVs RNA-Sequenzierung – mRNA durch Umwandlung in cDNA, Splice-Varianten mRNA durch Umwandlung in cDNA, Splice-Varianten (lange Reads) Direkte RNA-Sequenzierung (ohne cDNA), Splice-Varianten, Langzeit-Transkripte Epigenetische Modifikationen – Indirekt: nur mit Bisulfit-Sequenzierung (aufwendig) Direkt: Methylierungserkennung durch Einbaukinetik (HiFi-Reads) & Bioinformatische Tools Direkt: Methylierung durch Signaländerungen (z. B. 5mC, 6mA) Einsatzort Labor: Stationäre Geräte Labor: Stationäre Großgeräte (z. B. NovaSeq) Labor: Stationäre Großgeräte (Revio) Flexibel: Labor (PromethION), mobil/Point-of-Care (MinION) Hauptvorteile Höchste Präzision, ideal zur Validierung Hoher Durchsatz, kostengünstig, präzise bei SNVs/Indels, routinetauglich Lange, präzise Reads (HiFi), ideal für SVs, Epigenetik, Phasierung Ultra-lange Reads, mobil, schnell, direkte RNA- und Epigenetik-Erkennung Limitationen Niedriger Durchsatz, WGS unpraktisch, teuer Kurze Reads, eingeschränkt bei SVs/Epigenetik, komplexe Library-Prep Teurer, mittlerer Durchsatz, längere Vorbereitungszeit Geringere Rohdatenqualität, abhängig von Bioinformatik Rolle in der personalisierten Medizin Validierung kritischer SNVs (Pharmakogenomik, Krebs), mitochondriale Analysen Routinediagnostik, Pharmakogenomik, Krebsgenomik (SNVs/Indels), seltene Erkrankungen Epigenetik, Krebsgenomik (SVs), seltene Erkrankungen (SVs, Repeat-Expansionen), Phasierung Neonatale Notfalldiagnostik, Epigenetik, Krebsgenomik (SVs), seltene Erkrankungen, mobile Diagnostik
g) Key Takeaways: Zwischen Reife und Routine
Die DNA-Sequenzierung befindet sich an einem entscheidenden Wendepunkt: In den vergangenen Jahren hat sie sich von einem rein forschungsbasierten Verfahren zu einem klinisch relevanten Werkzeug der modernen Medizin entwickelt. Ob in der Onkologie, der Diagnostik seltener Erkrankungen oder der Pharmakogenomik – Genomanalysen liefern zunehmend Antworten, wo konventionelle Methoden an ihre Grenzen stoßen.
Und doch: Trotz der beeindruckenden technischen Reife ist die Genomsequenzierung bislang kein flächendeckender Standard in der klinischen Praxis.
Technologie reif – Infrastruktur unvollständig
Moderne Hochdurchsatzverfahren wie Illumina oder die Langlesetechnologien von PacBio und Oxford Nanopore ermöglichen heute hochpräzise Analysen zu vergleichsweise geringen Kosten. Whole-Genome-Sequenzierungen (WGS) sind in spezialisierten Laboren für wenige Hundert Euro pro Fall durchführbar – mit einer Gesamtdauer von unter einer Woche. Mobile Plattformen wie MinION liefern in akuten Situationen – etwa bei schwer erkrankten Neugeborenen – erste genetische Hinweise sogar binnen 48 Stunden.
Trotz dieser Fortschritte bleibt der klinische Einsatz fragmentarisch. Gründe dafür sind vielfältig: Es mangelt an standardisierten bioinformatischen Auswertungsprotokollen, an interdisziplinär geschultem Fachpersonal sowie an einer rechtssicheren und wirtschaftlich tragfähigen Integration in den Versorgungsalltag. Vor allem die Interpretation genetischer Varianten – insbesondere solcher mit unklarer klinischer Bedeutung – erfordert spezialisiertes Know-how, das vielerorts nicht verfügbar ist.
Großbritannien als Blaupause
Während viele Länder noch mit Pilotprogrammen experimentieren oder Einzelfallentscheidungen treffen, institutionalisiert das Vereinigte Königreich die Genomsequenzierung als integralen Bestandteil der öffentlichen Gesundheitsvorsorge. Dort soll im Rahmen eines nationalen Pilotprogramms künftig jedes Neugeborene genomisch untersucht werden – ein Präzedenzfall für die pränatale und frühkindliche Vorsorgemedizin. Ziel ist es, genetisch bedingte Erkrankungen bereits im frühkindlichen Stadium zu erkennen – noch bevor klinische Symptome auftreten. Damit wird DNA-Analytik vom Einzelfall zur bevölkerungsweiten Präventionsstrategie – ein Paradigmenwechsel.
Dieses Modell demonstriert, dass die Hürde für eine breite Implementierung weniger technischer, sondern vor allem systemischer Natur ist: fehlende Datenstandards, ethische und rechtliche Unklarheiten sowie eine zurückhaltende Erstattungspolitik verhindern vielerorts eine flächendeckende Integration. Der britische Vorstoß gilt daher als wegweisendes Beispiel für die institutionalisierte Genommedizin.
Technologischer Ausblick
Mehrere technologische Entwicklungen dürften die Umsetzung in die Breite in naher Zukunft weiter beschleunigen:
🚀 Sinkende Kosten pro Genom machen Sequenzierung auch in kleineren Einrichtungen wirtschaftlich attraktiv.
🚀 Dezentrale, mobile Systeme wie MinION ermöglichen ortsunabhängige Diagnostik, etwa in Notaufnahmen oder ländlichen Regionen.
🚀 Schnellere Time-to-Result – von der Probenentnahme bis zur Befundung – erlaubt den Einsatz auch in zeitkritischen Fällen.
🚀 Die Integration von Genomdaten mit Transkriptomik, Proteomik und Epigenetik (Multi-Omics) verspricht eine kontextualisierte, hochauflösende Diagnostik.
🚀 KI-gestützte Analytik sowie automatisierte Interpretationstools senken die Abhängigkeit von manueller Auswertung und reduzieren Fehlerquellen.
4.5. Genomsequenzierung: Der nächste Technologiesprung
Damit die Entschlüsselung unserer DNA künftig schneller, günstiger und überall möglich wird, arbeitet die Forschung an der nächsten Generation von Sequenzierungstechnologien.
a) FENT: Wie ein Mikrochip die DNA-Analyse revolutioniert
b) SBX: Wenn DNA sich streckt, um verstanden zu werden
c) Das G4X-System: Vom genetischen Rezept zum räumlichen Atlas der Zelle
a) FENT: Wie ein Mikrochip die DNA-Analyse revolutioniert
– und für alle zugänglich macht
Angetrieben von der Vision, genetische Informationen in Echtzeit und patientennah erfassen zu können, entwickelt das Unternehmen iNanoBio ein neues Verfahren, das diese Idee auf ein völlig neues Niveau heben soll.
Es kombiniert die bewährte Methode – das Auslesen von DNA durch winzige Nanoporen – mit einem Bauteil aus der Mikroelektronik: dem MOSFET (Metal-Oxide-Semiconductor-Feldeffekttransistor), Herzstück moderner Computerchips.
Vom Computerchip zum DNA-Sensor
Ein MOSFET ist ein winziger Schalter mit drei Kontakten: Source (Eingang), Drain (Ausgang) und Gate (Steuerung). Liegt am Gate eine Spannung an, öffnet sich ein leitender Kanal zwischen Source und Drain – vergleichbar mit einem Türsteher, der ein Tor freigibt. Bereits minimale Spannungsänderungen beeinflussen den Stromfluss, was MOSFETs extrem empfindlich macht.
Abb. 10-A: Aufbau und Funktionsweise eines MOSFETs Ein MOSFET ist ein winziger Schalter aus der Mikroelektronik mit drei Kontakten: Source (Eingang), Drain (Ausgang) und Gate (Steuerung). Source und Drain sind im Halbleitermaterial verankert, das Gate ist durch eine hauchdünne Oxidschicht isoliert.
Ohne Spannung am Gate bleibt der Kanal zwischen Source und Drain geschlossen – kein Strom fließt. Legt man jedoch Spannung am Gate an, entsteht ein elektrisches Feld, das leitende Teilchen (z. B. Elektronen) anzieht und einen Stromkanal öffnet. Der MOSFET wirkt damit wie ein extrem sensibler Türsteher: Erst wenn er das Signal bekommt, öffnet er das Tor und lässt Strom passieren.Jetzt kommt der Clou: iNanoBio hat es geschafft, eine winzige Pore direkt in das empfindliche Steuerzentrum des MOSFETs zu integrieren. Wenn ein DNA-Strang hindurchgleitet, verändert jedes einzelne Nukleotid (A, T, C oder G) den Stromfluss zwischen Source und Drain charakteristisch. Das Ergebnis: ein neuartiger Sensor mit dem Namen FENT – kurz für Field-Effect Nanopore Transistor.
Der FENT ist zylindrisch aufgebaut und umschließt die Pore. Dadurch kann er Signale gleichzeitig aus mehreren Richtungen messen, was die Genauigkeit erhöht und Fehler reduziert.
Abb. 10-B: Querschnitt eines FENTs Der FENT ist ein zylindrisch gebauter MOSFET, der eine Nanopore vollständig umschließt. Zwischen Source und Drain wird eine Spannung angelegt, sodass ein kleiner Strom durch den Halbleiter der Porenwand fließt – der sogenannte Source-Drain-Kanalstrom. Die dünne Oxidschicht isoliert das Gate vom Kanal und ermöglicht die präzise Erfassung elektrischer Feldänderungen. Die Nanopore dient als Sensorzone für lokale Feldänderungen, verursacht durch vorbeigleitende DNA-Basen.
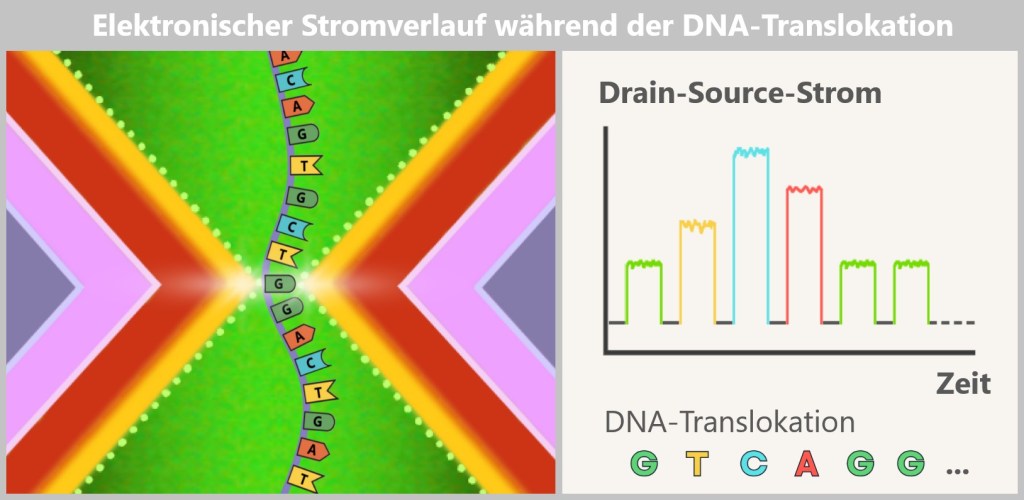
Abb. 10-C: FENT während der DNA-Translokation Die Abbildung zeigt das Funktionsprinzip eines FENT während der Passage einer DNA-Einzelstrangs (ssDNA) durch die Nanopore. Vor Beginn der Messung wird die ursprünglich doppelsträngige DNA (dsDNA) durch Hitze oder chemische Behandlung in ihre beiden Einzelstränge getrennt. Nur ein Einzelstrang wird anschließend durch die Nanopore geführt.
Im Ausgangszustand – also ohne DNA in der Pore – fließt zwischen Source und Drain ein konstanter Ruhestrom durch den Halbleiter, der die Porenwand bildet. Dieser Strom wird durch die Spannung zwischen Source und Drain sowie das Gate-Potenzial bestimmt.
Sobald jedoch eine DNA-Base (A, T, C oder G) die Nanopore passiert, beeinflusst sie das elektrische Verhalten des Systems:
Jede Base besitzt aufgrund ihrer spezifischen chemischen Struktur und Atomverteilung ein eigenes, winziges elektrisches Feld. Dieses Feld dringt durch die extrem dünne Porenwand und wirkt auf den Halbleiter wie ein lokales Gate-Signal. Dadurch verändert sich lokal die Ladungsträgerdichte (Elektronenkonzentration) im Transistorkanal, was zu einer messbaren Änderung des Drain-Source-Stroms führt.
Diese Stromänderungen werden als charakteristische Signale für die jeweilige Base aufgezeichnet. So entsteht während der Translokation eine sequenzabhängige Stromspur, die es ermöglicht, die Reihenfolge der DNA-Basen elektronisch zu bestimmen.Eine allgemeinverständliche Erklärung liefert das Video FENT Nanopore Transistor Explainer Video.
Geschwindigkeit, die Maßstäbe setzt
Laut iNanoBio kann ein FENT bis zu 1 Million DNA-Basen pro Sekunde und Pore auslesen – etwa 100-mal schneller als bisherige Nanopore-Technik. Ein 5 x 5 mm großer Chip mit 100.000 dieser Sensoren könnte ein menschliches Genom in wenigen Minuten sequenzieren. Die Kosten wären deutlich geringer, die Fehlerrate niedriger.
Mögliche Anwendungen
- Krebsfrüherkennung aus Blutproben
- Mobile Diagnostik für Infektionen direkt vor Ort
- Großstudien in der Forschung dank hoher Geschwindigkeit
Langfristig will iNanoBio DNA-Analysegeräte so verbreiten wie heutige Blutdruckmessgeräte – verfügbar in Kliniken, Laboren, Arztpraxen und möglicherweise auch zu Hause. Erste Prototypen existieren, der Weg zur Massenproduktion läuft.
Wenn die Technologie hält, was sie verspricht, könnte die Genomsequenzierung bald so selbstverständlich sein wie eine Routineuntersuchung – und unser Verständnis von Gesundheit grundlegend verändern.
b) SBX: Wenn DNA sich streckt, um verstanden zu werden
Im Wettlauf um die Verbesserung der DNA-Analyse verfolgt der Pharmakonzern Roche mit SBX (Sequencing By eXpansion) einen verblüffend einfachen Ansatz: Er macht DNA künstlich größer, damit ihre „Buchstaben“ A, T, C und G leichter auseinanderzuhalten sind.
Schritt 1 – Vom Original zur vergrößerten Kopie
SBX erstellt aus der Original-DNA eine Surrogat-Kopie, den Xpandomer. Dafür wird das DNA-Fragment an einem winzigen Anker auf einem Substrat fixiert, und mithilfe einer Polymerase wird eine Kopie erstellt – allerdings nicht aus natürlichen Bausteinen, sondern aus Surrogat-Nukleotiden.
Ein Surrogat (von lateinisch surrogatum = „Ersatz“) bezeichnet allgemein einen Ersatz oder Stellvertreter für etwas anderes.
Diese bestehen aus:
- der passenden Base (A, T, C oder G),
- einem für jede Base spezifischen gefalteten Verlängerungsfaden als Signalgeber,
- einer spaltbaren Bindung, die den Faden in Form hält,
- einem Translocation Control Element (TCE), das den Ablauf später präzise steuert.
Abb. 11-A: Vereinfachte Darstellung eines Surrogat-Nukleotids
[Sequencing by expansion (SBX) technology]Das Surrogat-Nukleotid (XNTP) besteht aus zwei funktionellen Teilen: dem modifizierten Nukleotid und dem SSRT (symmetrically synthesized reporter tether).
Das modifizierte dNTP enthält die eigentliche Base (hier beispielhaft Cytosin, C), die über eine säureempfindliche Bindung (rot markiert) mit dem Reporterfaden verbunden ist.
Daran schließt sich das Triphosphat (PPP) an, das die Einbindung des Nukleotids in die wachsende Kette durch die Polymerase ermöglicht.
Der Reporterfaden (SSRT) besteht aus einem gefalteten Verlängerungsfaden mit einem Translocation Control Element (TCE) an der Spitze, das den Durchtritt durch die Nanopore später steuert.
Ein Enhancer-Bereich (grau/lila) erleichtert die Einbindung durch die Polymerase und stabilisiert die Struktur.
Nach der Synthese kann die rote, säureempfindliche Bindung gelöst werden – dadurch „entfaltet“ sich der Reporterfaden und erzeugt beim Sequenzieren das ausgedehnte Xpandomer-Molekül.
Abb. 11-B: Synthese des Xpandomers [Sequencing By Expansion] Das DNA-Fragment (Template DNA) ist über einen Anker am festen Substrat fixiert. Ein Primer bindet an die DNA-Vorlage, unterstützt vom Leader und Concentrator, die den Startpunkt stabilisieren und die Positionierung erleichtern.
Fixierung – Die DNA-Vorlage ist am Substrat verankert, der Primer-Abschnitt hybridisiert an den passenden Bereich der DNA.
Verlängerung – Eine spezielle Polymerase ergänzt die Basen der Vorlage – aber nicht mit natürlichen Nukleotiden, sondern mit Surrogat-Nukleotiden (XNTPs), die jeweils einen Reporterfaden mitführen.
Xpandomer entsteht – Am Ende liegt ein künstlich erweiterter DNA-Strang vor: eine Surrogat-Kopie (Xpandomer), deren Basen durch charakteristische Reporterfäden ersetzt sind.
Der Xpandomer bleibt zunächst am Substrat fixiert und wird in einem späteren Schritt durch Licht oder chemische Spaltung freigesetzt.Nach der Synthese wird der Xpandomer von der Original-DNA gelöst, die spaltbaren Bindungen werden getrennt – die Fäden entfalten sich und vergrößern den Abstand zwischen den Basen um das 50-Fache. Das erleichtert die spätere Identifikation erheblich.

Abb. 11-C: Expansion und Freisetzung des Xpandomers [Sequencing By Expansion] Nach der Synthese wird der Xpandomer auf dem Substrat chemisch entfaltet („expandiert“) und anschließend freigesetzt.
Links) Expansion: Eine milde Säurebehandlung löst die spaltbaren Bindungen (rot) zwischen Base und Reporterfaden. Dadurch entfalten sich die Reporterstrukturen – aus der kompakten Doppelstruktur wird ein lang gestreckter Xpandomer, der die Abfolge der Basen repräsentiert.
Rechts) Freisetzung: Ein UV-Lichtimpuls spaltet die Verbindung zwischen dem Substrat und dem Leader-Bereich.
Der nun frei bewegliche Xpandomer bleibt über Leader und Concentrator strukturell markiert – diese Bereiche helfen später bei der Ausrichtung und beim Einzug in die Nanopore.
Aus einer dichten DNA-Vorlage entsteht ein langes, lesbares Molekül, dessen Reporterabschnitte die genetische Information elektrisch sichtbar machen.Schritt 2 – Die Sequenzierung
Anstelle der DNA selbst wird nun der Xpandomer durch Nanoporen auf einem Sequenzierungschip gezogen – rund acht Millionen davon arbeiten parallel. Der Adapter des Xpandomers führt ihn gezielt in eine Pore.
Wenn ein Verlängerungsfaden die Pore erreicht, bleibt er kurz am TCE hängen. Jede Base erzeugt dabei ein spezifisches elektrisches Signal. Nach 1,5–2 Millisekunden löst ein Spannungspuls das TCE, der nächste Faden folgt. So entsteht eine klare, gut unterscheidbare Signalkurve – präziser als bei Oxford Nanopore, wo die Basen ein eher „verwaschenes“ Signal erzeugen.

Abb. 11-D: Sequenzierung des Xpandomers in der Nanopore
[Sequencing By Expansion, Introduction to Sequencing By Expansion]Der freigesetzte Xpandomer wird durch ein elektrisches Feld zur Nanopore gezogen. Dort kontrolliert das TCE (Transient Capture Element) den Eintritt und hält die Reporter kurzzeitig fest.
Während jeder Reporterabschnitt (die verlängerte „Base“) die Pore passiert, ändert sich der Ionenstrom in charakteristischer Weise: Jede Base erzeugt dabei ein eigenes elektrisches Signal – vergleichbar mit einem individuellen Barcode. Diese Signalfolge wird in Echtzeit aufgezeichnet:
Ein Spannungspuls löst nach jeder Messung den Faden vom TCE, sodass der nächste Abschnitt eingezogen wird.Folgende Animation veranschaulicht die Grundprinzipien der SBX-Technologie von Roche.
Eine ausführliche Präsentation gibt es hier.
Vorteile und Leistung
- Geschwindigkeit: Millionen Xpandomer werden gleichzeitig und in Echtzeit gelesen.
- Genauigkeit: Signal-Rausch-Verhältnis ähnlich hoch wie bei Illumina.
- Flexibilität: Leselängen bis über 1.000 Basen möglich – schneller und präziser als Nanopore, länger als Illumina.
Erste Tests zeigen: SBX kann bis zu sieben vollständige menschliche Genome pro Stunde mit 30x Abdeckung entschlüsseln. Bei dringenden Fällen dauert der gesamte Ablauf – von der Probenvorbereitung bis zur fertigen Analyse – nur rund 5,5 Stunden.
Die Markteinführung ist für 2026 geplant. Ob sich SBX gegen Illumina und Oxford Nanopore durchsetzen wird, hängt von Preis, Verfügbarkeit und den Anforderungen der Anwender ab.
c) Das G4X-System: Vom genetischen Rezept zum räumlichen Atlas der Zelle
Unser Körper besteht aus Milliarden Zellen, die wie winzige Küchen arbeiten. Die DNA ist ihr Kochbuch – sie enthält alle Rezepte des Lebens. Wird ein Rezept gebraucht, erstellt die Zelle eine RNA-Kopie, die als Bauanleitung für Proteine dient. Diese Proteine übernehmen konkrete Aufgaben: Sie bauen Strukturen, transportieren Signale oder bekämpfen Krankheitserreger.
Proteine wirken jedoch selten allein. Sie arbeiten in fein abgestimmten Netzwerken, deren Aktivität davon abhängt, wo, wann und mit welchen Partnern sie interagieren. Prozesse wie Immunreaktionen oder Tumorwachstum entstehen nicht in einer einzelnen Zelle, sondern aus dem Zusammenspiel vieler spezialisierter Zellen – oft in unterschiedlichen Bereichen eines Gewebes. Biologie ist also nicht nur biochemisch, sondern auch räumlich organisiert. Klassische Sequenzierungsmethoden erfassen jedoch meist nur die genetischen Inhalte, nicht deren räumliche Verteilung.
Genaktivität direkt im Gewebe
Der G4X Spatial Sequencer von Singular Genomics macht sichtbar, wo im Gewebe Gene aktiv sind und Proteine gebildet werden – direkt in konservierten Gewebeproben (FFPE, formalinfixiert und paraffineingebettet).
FFPE-Proben werden häufig in der Pathologie eingesetzt, etwa zur Krebsdiagnostik. Sie wirken wie eine „eingefrorene Momentaufnahme“, nur dass hier chemische Fixierung mit Formalin statt Kälte verwendet wird. Formalin vernetzt Proteine und Nukleinsäuren miteinander und fixiert so die molekularen Strukturen räumlich.
Was dabei erhalten bleibt:
- RNA (oft fragmentiert und chemisch modifiziert) – spiegelt die Genaktivität zum Fixierzeitpunkt wider.
- Proteine – zeigen, welche Strukturen und Signalwege in diesem Moment aktiv waren.
So lässt sich Monate oder Jahre später rekonstruieren, welche Gene und Proteine zu diesem exakten Zeitpunkt im Gewebe präsent und aktiv waren – als hätte man die Zeit angehalten. Mit dem G4X kann diese „eingefrorene“ molekulare Landschaft hochaufgelöst sichtbar gemacht werden – inklusive der exakten Position im Gewebe.
Abb. 12-A: Vom Gewebe zur Genaktivität Ein Gewebe besteht aus vielen spezialisierten Zellen. Jede Zelle in einem Gewebe nutzt nur einen Teil ihres genetischen Programms. RNA macht die aktiven Gene sichtbar. RNA-Moleküle werden von Ribosomen abgelesen und in Proteine übersetzt. FFPE-Gewebeschnitte bewahren diese räumliche Anordnung – die Basis, um mit dem G4X-System zu messen, wo welche RNA aktiv ist.
Wie der G4X funktioniert
Man kann sich den G4X wie einen Mikroskop-Scanner mit eingebautem Labor vorstellen, der direkt im Gewebe misst, welche Gene und Proteine aktiv sind – und zwar in ihrem räumlichen Kontext.
Präparation: Ein sehr dünner Schnitt der FFPE-Probe wird auf ein spezielles Trägermaterial gebracht.
RNA-Nachweis: Um die Verteilung bestimmter RNA-Sequenzen im Gewebe zu erkennen, kommen Padlock-Sonden zum Einsatz.
Padlock-Sonden – DNA-Lassos für Zielsequenzen
Stellen Sie sich vor, Sie möchten in einem riesigen Gelände (dem Gewebe) nur ein bestimmtes Objekt (eine RNA-Sequenz) finden. Eine Padlock-Sonde ist ein DNA-Fragment, dessen beide Enden komplementär zu benachbarten Abschnitten der Zielsequenz passen.

Abb. 12-B: Eine Padlock-Sonde schwebt durch das Gewebe auf der Suche nach ihrer Ziel-RNA. Padlock-Sonde: Einzelsträngige DNA (ssDNA) 5’– Arm1 – Linker/Barcode – Arm2 –3′
Arm 1 und Arm 2 sind komplementär zu benachbarten Sequenzen der Ziel-RNA.
Dazwischen liegen Linker und Barcode, die zusätzliche Identifikationsinformationen tragen und nicht binden.Bindet die Sonde an ihr Ziel, liegen ihre beiden Enden nebeneinander und können durch Ligation zu einem stabilen DNA-Ring geschlossen werden.
Abb. 12-C: Ligation verbindet die Enden der Padlock-Sonde Nachdem beide Arme der Sonde an die Ziel-RNA gebunden haben, verbindet eine Ligase die 5′- und 3′-Enden. Dadurch entsteht ein geschlossener DNA-Ring, der die Ziel-RNA markiert und für die Amplifikation bereit ist.
Liegt zwischen den Bindungsstellen eine variable Sequenz (z. B. eine Mutation), wird diese zuvor mithilfe einer Reverse Transkriptase in DNA umgeschrieben und in die Sonde integriert.
Der entstandene DNA-Kreis dient als Vorlage für die Rolling Circle Amplification (RCA):
- Eine DNA-Polymerase läuft immer wieder im Kreis und erzeugt einen langen DNA-Strang, der die Zielsequenz vielfach wiederholt – wie ein Endlosband oder ein Garnknäuel.
- Diese Signale bleiben exakt an der Stelle im Gewebe, wo die RNA war.
- Mit fluoreszenzmarkierten Bausteinen und Sequenzieren durch Synthese wird die Sequenz direkt im Gewebe entschlüsselt.
Jede Padlock-Sonde trägt einen Barcode, der verrät, welches Gen oder welche Variante sie erkannt hat. Das Ergebnis: eine präzise Karte, die zeigt, welche RNA an welchem Ort vorhanden war und welche Varianten sie trug.
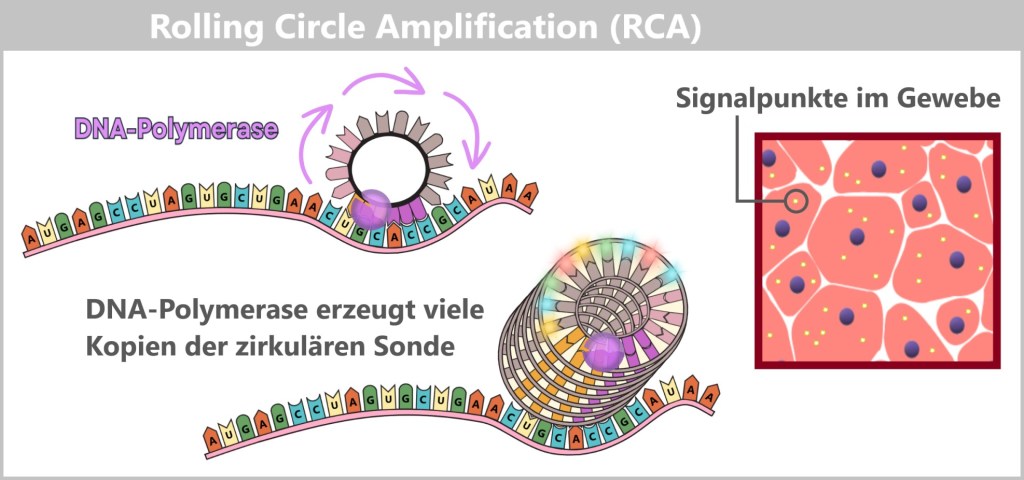
Abb. 12-D: Rolling Circle Amplification (RCA) Eine spezielle DNA-Polymerase liest die zirkuläre Padlock-Sonde mehrfach ab und erzeugt einen langen DNA-Strang mit vielen Wiederholungen. Fluoreszente Nukleotide markieren die Amplifikate. Dadurch entsteht ein starkes, fluoreszierendes Signal – sichtbar als leuchtender Punkt im Gewebe, der die Position der gesuchten RNA markiert.
Proteinnachweis im selben Gewebeschnitt
Für den Nachweis von Proteinen nutzt das G4X-System Antikörper, die mit kurzen DNA-Barcodes versehen sind. Nachdem der Antikörper an sein Zielprotein gebunden hat, hybridisiert eine Padlock-Sonde an diesen Barcode. Sie wird geschlossen, amplifiziert und ausgelesen – ganz wie bei der RNA-Analyse. Der Barcode verrät eindeutig, um welches Protein es sich handelt und an welcher Stelle im Gewebe es vorkommt.
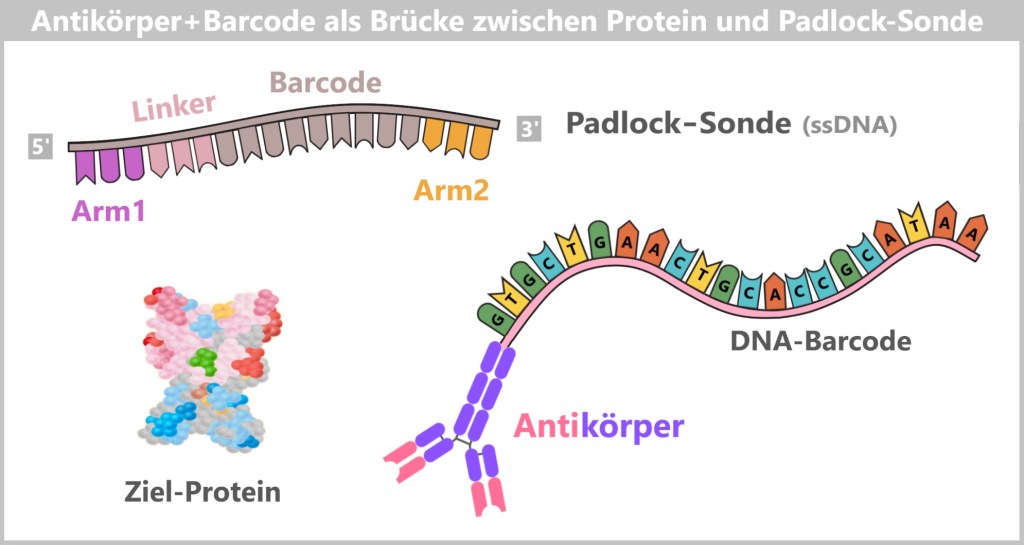
Abb. 12-E: Protein-Nachweis im G4X-System: Antikörper mit DNA-Barcode und Padlock-Sonde Ein Antikörper, der spezifisch für ein Zielprotein ist, wird mit einem einzigartigen, kurzen einzelsträngigen DNA-Strang (Barcode) gekoppelt.

Abb. 12-F: Protein-Nachweis im G4X-System Der Antikörper bindet in der Gewebeprobe an sein Zielprotein. Eine Padlock-Sonde, deren Enden komplementär zu zwei benachbarten Abschnitten des DNA-Barcodes sind, bindet an diesen und wird durch eine DNA-Ligase zu einem geschlossenen Ring verknüpft. Eine DNA-Polymerase erkennt den Ring und liest ihn fortlaufend ab, wodurch ein lokal verankertes, zirkuläres DNA-Amplifikat entsteht – analog zum RNA-Nachweis. Durch den Einbau fluoreszenzmarkierter Nukleotide wird dieses Amplifikat sichtbar. Die leuchtenden Signalpunkte im Gewebe markieren die Positionen, an denen ein Antikörper mit einem spezifischen DNA-Barcode ein bestimmtes Protein gebunden hat.
Eine ausführliche Präsentation des G4X-Veerfahrens findet man hier und hier.
Warum das wichtig ist
Der G4X kann gezielt Biomarker nachweisen, die Aufschluss über das Vorliegen, den Verlauf oder die Aggressivität einer Erkrankung geben – und zeigt dabei nicht nur, welche aktiv sind, sondern auch wo sie im Gewebe auftreten.
Gerade bei Tumoren macht diese räumliche Auflösung sichtbar, welche Gene „angeschaltet“ sind, in welchen Zellbereichen das geschieht und wie Tumor- und Immunzellen interagieren. So lassen sich Tumorheterogenität besser verstehen, Krankheitsmechanismen aufklären und Therapien gezielter planen.
Die gleichzeitige in-situ Erfassung von RNA und Proteinen liefert ein umfassendes Bild der Gewebeorganisation und eröffnet neue Möglichkeiten für:
- das Verständnis komplexer Krankheitsprozesse
- die präzisere Identifikation von Biomarkern
- die Entwicklung personalisierter Therapien
Bei Erkrankungen wie Krebs, chronischen Entzündungen oder neurodegenerativen Störungen kann diese räumliche Multiomics-Perspektive entscheidend sein – denn oft bestimmt nicht das Ob, sondern das Wo und Wie die beste therapeutische Maßnahme.
Fazit
Die Genomsequenzierung steht vor einem Quantensprung: Technologien wie FENT, SBX und G4X versprechen, das Erbgut schneller, präziser und vielseitiger zu lesen als je zuvor – bis hin zur direkten Analyse im Gewebe. Bald könnten komplette Genome in Minuten entschlüsselt werden. Die wahre Revolution beginnt jedoch danach: wenn wir die Fülle an genetischen Informationen verstehen, in medizinisches Wissen übersetzen und verantwortungsvoll zum Wohl der Menschen einsetzen.
4.6. Vom Code zur Heilung
Bioinformatik als Schlüssel der Medizin von morgen
4.6.1. Vom genetischen Code zur computergestützten Genomanalyse
Noch nie war es so leicht, DNA zu entschlüsseln – doch was nützt der Text, wenn niemand ihn versteht? Ein kleiner Abstrich, ein Tropfen Blut – und schon liegen Millionen genetischer Datenpunkte vor. Die moderne Sequenzierung macht das Erbgut so zugänglich wie nie zuvor: schnell, kostengünstig und nahezu fehlerfrei. Daraus erwächst jedoch eine neue Herausforderung: Wie verwandelt man diese Datenflut in verwertbares medizinisches Wissen?
Die Antwort kommt aus einer Disziplin, die lange im Hintergrund agierte und heute unverzichtbar geworden ist: Bioinformatik – die Brücke zwischen A, T, G, C und der Diagnose, zwischen rohen Sequenzdaten und einer konkreten Therapieentscheidung. Um zu verstehen, warum sie so zentral ist, lohnt ein kurzer Blick zurück.
Vom Erbgesetz zum genetischen Code
Bereits 1860 entdeckte Gregor Mendel, dass es „vererbbare Einheiten“ geben muss – Gene, lange bevor DNA bekannt war. Erst Mitte des 20. Jahrhunderts zeigte Oswald Avery, dass diese Informationsträger tatsächlich DNA sind. Kurz darauf enthüllten Watson und Crick die Doppelhelix-Struktur mit ihrer komplementären Basenpaarung (A–T, G–C). 1958 formulierte Francis Crick das „Zentrale Dogma“: Information fließt von DNA über RNA zu Proteinen – den funktionellen Bausteinen des Lebens.
Abb. 13-A: Das zentrale Dogma der Molekularbiologie: Die genetische Information wird durch den Prozess der Transkription von der DNA in RNA umgewandelt. Die RNA wird dann in das endgültige Protein übersetzt, das eine Vielzahl von Funktionen hat. [An overview of artificial intelligence in the field of genomics]
Ein Gen ist also eine Anleitung, wie ein Protein hergestellt wird. Doch wie liest man diese Anleitung – und wie interpretiert man sie richtig? Die Antwort kam zunächst über den Umweg der Reverse Genetik.
In den 1960er Jahren war DNA noch schwer direkt zugänglich – Proteine dagegen waren funktional sichtbar (etwa als Enzyme, Transportmoleküle oder Strukturbausteine) und konnten mit den damaligen Methoden isoliert und analysiert werden. Forscher wussten bereits seit Anfang des 20. Jahrhunderts, dass Proteine aus Aminosäureketten bestehen. Ab den 1960er Jahren wurde klar, dass jede Aminosäure einem sogenannten Codon auf der RNA entspricht – also einer Dreierkombination aus RNA-Basen (A, U, G, C), die wiederum auf eine DNA-Sequenz zurückführbar ist.
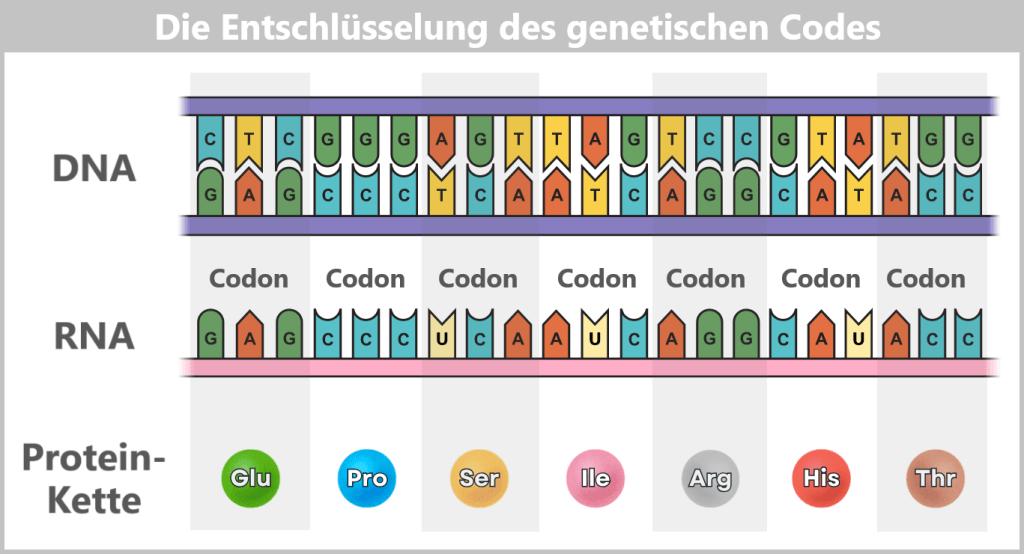
Abb. 13-B: Das zentrale Dogma der Molekularbiologie: Der DNA-Code und Codons Forscher starteten bei einem bekannten Protein und arbeiteten sich zur RNA- und DNA-Sequenz zurück. Der genetische Code wurde zum ersten Mal wie ein Rezeptbuch lesbar:
Protein = fertiges Gericht
Aminosäure = Zutat
Codon = Wort für diese Zutat im handgeschriebenen Rezept (RNA)
DNA = Seite im Kochbuch, von der das Rezept abgeschrieben wurdeErste Muster und die Geburt der Genomannotation
Noch vor der eigentlichen DNA-Sequenzierung entdeckte man typische Strukturelemente in Genen:
Promotoren – Startsignal, an dem das „Lesen“ eines Gens beginnt.
Start- und Stopcodons – erstes und letztes Signal im Rezept.
Transkriptionsfaktoren – Proteine, die Gene ein- oder ausschalten.
Codierende Sequenzen – Zutatenliste (Codons) und deren Reihenfolge für das Protein.
Nicht-codierende DNA – Abschnitte ohne direkte Protein-Codierung, deren Funktion zunächst unklar war.Diese Erkenntnisse legten den Grundstein der heutigen Genomannotation – der computergestützten Erkennung biologischer Signale in DNA-Sequenzen.
Der Sanger-Moment: DNA wird lesbar (ab 1977)
Mit Frederick Sangers Methode konnten DNA-Sequenzen erstmals direkt bestimmt werden – zunächst nur für kurze Abschnitte. Die Analyse konzentrierte sich auf die Identifikation von Genen und einfache Vergleiche. Erste Programme wie FASTA (1985) und BLAST (1990) ermöglichten den Abgleich neuer Sequenzen mit Datenbanken wie GenBank (1982).
Auch das Verständnis der Genstruktur entwickelte sich weiter: Man entdeckte, dass nur bestimmte Teile eines Gens (Exons) für die Proteinsynthese genutzt werden, während andere Abschnitte (Introns) aus der transkribierten RNA herausgeschnitten werden. Zudem identifizierte man DNA-Abschnitte, die die Genexpression verstärken (Enhancer) oder abschwächen (Silencer) – oft weit entfernt vom eigentlichen Gen.
Der Prozess der Genomannotation begann sich zu entwickeln, war jedoch noch rudimentär: DNA wurde hauptsächlich als lineare Abfolge von Nukleotiden betrachtet, mit Fokus auf proteinkodierende Gene. Regulatorische Elemente und nicht-codierende DNA wurden erst ansatzweise erkannt und verstanden. Die Bioinformatik war geboren – zunächst als Werkzeug für Forschungslabore.
Das Humangenomprojekt: Daten im Gigantenmaßstab (1990–2004)
Das Ziel, alle 3,2 Milliarden Basen des menschlichen Genoms zu entschlüsseln, brachte völlig neue Probleme: Millionen kurzer Sequenzfragmente mussten zu langen DNA-Strängen zusammengesetzt werden. Tools wie Phrap für die Assemblierung und später Ensembl für die Annotation wurden entwickelt. Die Datenmengen explodierten – und mit ihnen die Notwendigkeit leistungsfähiger Rechnerinfrastruktur.
Doch während das Humangenomprojekt noch lief, zeichnete sich bereits eine neue Technologie ab, die Geschwindigkeit und Datenmenge in völlig neue Dimensionen katapultieren sollte.
NGS-Revolution und klinischer Durchbruch (2005–2015)
Die Next-Generation Sequencing (NGS)-Technologien machten Genome günstiger und schneller zugänglich – allerdings als gewaltige Datenlawinen aus Milliarden kurzer Fragmente. Neue Werkzeuge wie Bowtie, BWA oder STAR ordneten diese blitzschnell zu, GATK half, Mutationen zu erkennen, und Tools wie SnpEff oder VEP interpretierten deren Bedeutung.
Gleichzeitig entstanden Sequenzierstrategien für unterschiedliche klinische Fragen:
- Whole Genome Sequencing (WGS) für vollständige Analysen,
- Whole Exome Sequencing (WES) für die 1–2 % proteinkodierender DNA,
- Targeted Sequencing für definierte Genpanels.
Damit wurde die Bioinformatik erstmal zu einem Diagnosetool – etwa bei erblichen Krebsformen, seltenen Erkrankungen oder personalisierter Pharmakotherapie.
Künstliche Intelligenz und moderne Bioinformatik (2015-heute)
Die Verbindung von Künstlicher Intelligenz (KI) und Bioinformatik stellt einen der dynamischsten und vielversprechendsten Forschungsbereiche unserer Zeit dar. Gemeinsam entschlüsseln sie die Komplexität des Lebens und beschleunigen biomedizinische Entdeckungen in nie dagewesenem Tempo.
Künstliche Intelligenz: Mehr als nur Automatisierung
KI umfasst Techniken, bei denen Maschinen menschenähnliche Intelligenzleistungen erbringen – insbesondere Lernen, Problemlösen und Mustererkennung. Im biomedizinischen Kontext sind vor allem folgende Teilbereiche relevant:Maschinelles Lernen (ML): Algorithmen lernen aus Daten (z.B. Genomsequenzen, Proteinstrukturen, Patientendaten), um Vorhersagen zu treffen oder Muster zu erkennen, ohne explizit programmiert zu sein. Beispiel: Erkennung von Tumorzellen in Gewebeproben.
Deep Learning (DL): Eine Untergruppe des ML, die künstliche neuronale Netze mit vielen Schichten („deep“) nutzt, um hochkomplexe Muster in riesigen Datenmengen zu erfassen. Beispiel: Vorhersage der 3D-Struktur von Proteinen allein aus ihrer Aminosäuresequenz (AlphaFold).
Natürliche Sprachverarbeitung (NLP): Ermöglicht Computern, wissenschaftliche Literatur, Krankenakten oder klinische Studienberichte zu verstehen und daraus Wissen zu extrahieren.
4.6.2. Moderne Bioinformatik – Der digitale Werkzeugkasten der Biologie
Bioinformatik ist die Wissenschaft der Speicherung, Analyse und Interpretation biologischer Daten mit computergestützten Methoden. Ihre moderne Ausprägung ist durch drei Faktoren geprägt:
Explosion biologischer Daten: Next-Generation Sequencing (NGS) generiert Terabytes an Genom-, Transkriptom- und Epigenomdaten pro Experiment.

Abb. 12: Vom Milliardenprojekt zur Alltagsroutine – wie die Kosten der Genomsequenzierung fielen und die Datenmengen explodierten
Vor gut zwanzig Jahren war das Entschlüsseln eines menschlichen Genoms noch ein gigantisches Unterfangen: Das Human Genome Project (2003) kostete rund 3 Milliarden US-Dollar und lieferte nur etwa 1 Gigabyte an Daten. Mit immer schnelleren Sequenziermaschinen und neuen Technologien wie Nanoporen-Sequenzierung hat sich das Bild völlig verändert. 2012 lag der Preis pro Genom bereits bei rund 6000 Dollar, 2018 bei 1000 Dollar – und heute (2023) kann man ein Genom für etwa 100 Dollar analysieren.
Parallel dazu stieg das erzeugte Datenvolumen rasant: von wenigen Gigabyte über Terabyte und Petabyte bis hin zu den hunderten Petabytes, die in weltweiten Projekten mit Millionen von Genomen entstehen. Schätzungen zufolge könnten bis 2025 rund 40 Exabyte an menschlichen Genomdaten anfallen – das entspricht etwa dem achtfachen Speicherbedarf aller jemals gesprochenen Worte der Menschheitsgeschichte. [How AI Is Transforming Genomics]
Komplexität biologischer Systeme: Das Verständnis von Krankheiten erfordert die Integration von Daten über Gene, Proteine, Stoffwechselwege und Zellinteraktionen.
Bedarf an prädiktiver Medizin: Ziel ist die Vorhersage von Krankheitsrisiken, Therapieansprechen und individueller Behandlung (Präzisionsmedizin).
Bereits bei vielen Schlüsselanwendungen kann man mittlerweile von einer Verschmelzung zwischen Bioinformatik und Künstlicher Intelligenz (KI) sprechen. Hier sind einige zentrale Anwendungsgebiete:
a) Genomanalyse und Sequenzierung
KI-Algorithmen werden genutzt, um DNA-Sequenzen schneller und präziser zu analysieren. Modelle wie DeepVariant, entwickelt von Google, nutzen tiefe neuronale Netze, um genetische Varianten mit hoher Genauigkeit zu identifizieren. Dies ist entscheidend für die Erkennung von Mutationen, die mit Krankheiten wie Krebs oder seltenen genetischen Störungen assoziiert sind.
b) Proteinfaltung und Strukturvorhersage
Ein Meilenstein der KI in der Bioinformatik ist AlphaFold von DeepMind, dass die Struktur von Proteinen mit beispielloser Präzision vorhersagt. Dies hat weitreichende Implikationen für die Arzneimittelentwicklung, da die 3D-Struktur eines Proteins seine Funktion bestimmt.
c) Multi-Omics Datenanalyse
Das ganzheitliche Verständnis biologischer Systeme erfordert die Integration von Multi-Omics-Daten (Genomik, Proteomik, Transkriptomik, Epigenetik, Metabolomik), um umfassendes Bild ihrer Funktionsweise zu gewinnen. KI hilft, die komplexen Wechselwirkungen zwischen diesen Ebenen zu entschlüsseln.
Abb. 13: Das mehrstimmige Konzert des Lebens. [AI applications in functional genomics] Biologische Systeme sind ein komplexes Zusammenspiel verschiedener Informationsebenen: Von der DNA (Genomik) über epigenetische Steuerung, RNA (Transkriptomik/Epitranskriptomik) und Proteine (Proteomik) bis hin zu Stoffwechselprodukten (Metabolomik). Erst ihr dynamisches Wechselspiel definiert Gesundheit und Krankheit.
Beispielsweise zeigt das neue KI-Tool AlphaGenome, wie selbst kleinste Veränderungen in der DNA die Genaktivität sowie die Produktion von RNA und Proteinen beeinflussen können. Es sagt präzise voraus, welche Folgen der Austausch eines einzelnen DNA-Bausteins für Gene und ihre Produkte hat. Während frühere Systeme meist nur die rund 2 % des Genoms analysierten, die Proteine codieren, erfasst AlphaGenome erstmals das gesamte Genom. Grundlage ist eine Multi-Omics-Analyse, die DNA-Veränderungen direkt mit ihren Auswirkungen auf RNA und Proteine verknüpft. Damit entsteht ein umfassendes Bild der Genexpression – ein entscheidender Schritt hin zu präziserer Genforschung und personalisierter Medizin.
d) Metagenomik und Mikrobiomanalyse
KI hilft, die komplexen Wechselwirkungen im menschlichen Mikrobiom zu verstehen, indem sie Muster in den Daten von Mikroorganismen erkennt. Dies ist wichtig für die Erforschung von Krankheiten wie Diabetes oder Darmerkrankungen.
e) Arzneimittelentwicklung
KI beschleunigt nahezu jede Phase des langwierigen und teuren Drug-Discovery-Prozesses: Zielidentifikation, virtuelles Screening von Millionen von Molekülen, Vorhersage von Wirkstoffeffekten und Nebenwirkungen, Optimierung von Molekülstrukturen. Unternehmen wie BenevolentAI oder Recursion nutzen KI-Plattformen, um vielversprechende Wirkstoffkandidaten in Rekordzeit zu identifizieren, die bereits in klinischen Studien getestet werden.
f) Analyse von biomedizinischen Bildern
KI wird in der Bildanalyse eingesetzt, etwa bei der Auswertung von MRT- oder CT-Scans, um Tumore oder andere Anomalien zu erkennen. Convolutional Neural Networks (CNNs), die die Funktionsweise des menschlichen visuellen Kortex nachahmen, haben sich hier als besonders effektiv erwiesen.
g) KI-Genomik in der Gesundheitsforschung
Die Genexpression im menschlichen Körper folgt keinem festen Muster – und Krankheiten wie Krebs machen sie noch schwerer durchschaubar. Genau hier setzt die räumliche Biologie an: Mit modernster Mikroskopie und genetischer Sequenzierung zeigt sie, wie Gene in einzelnen Zellen und sogar in ihren Organellen aktiv sind.
Doch nicht nur die sichtbaren Genabschnitte sind entscheidend. In den lange als „Junk-DNA“ abgetanen Regionen vermuten Forscher heute zentrale Schalter der Genregulation. Dort entscheidet sich, welche Proteine produziert werden – und welche nicht. KI und Deep-Learning-Modelle können in diesen bislang verborgenen Bereichen Muster erkennen, die mit Krankheiten verknüpft sind. So erhofft man sich neue Biomarker und Therapieansätze. Ein Beispiel ist die Plattform des Biotech-Unternehmens Deep Genomics:
Die Kombination aus räumlicher Biologie, intelligenter Datenanalyse und KI ermöglicht damit einen bislang unerreichten Blick in die molekulare Logik des Lebens – bis auf die Ebene einzelner Zellen.
Neue Methoden der Genbearbeitung könnten das Verhalten einzelner Gene gezielt steuern – etwa um krebsfördernde Gene zu deaktivieren oder die Regeneration von Knorpelzellen bei Arthrose zu fördern. Noch ist dieses Feld stark experimentell, doch KI verspricht, die Verfahren präziser und sicherer zu machen.
Mit KI-gestützten CRISPR-Techniken eröffnen sich neue Möglichkeiten, Gene nicht nur gezielter zu identifizieren und zu verändern, sondern auch deren Zusammenspiel in komplexen biologischen Netzwerken besser zu verstehen. Erste Ansätze deuten darauf hin, dass sich dadurch Nebenwirkungen künftig verringern und die Wirksamkeit neuer Therapien steigern ließe. Auch moderne Verfahren zum systematischen Testen und Optimieren von Genvarianten könnten – sofern sie sich bewähren – wichtige Schritte in Richtung maßgeschneiderter Behandlungen darstellen.
Wer sich dafür interessiert, mehr über CRISPR-Techniken zu erfahren, findet hier weiterführende Informationen.
Die Kombination von Fortschritten in KI und Genomik gilt daher als vielversprechend und könnte langfristig die Entwicklung neuer Zell- und Gentherapien beschleunigen. Da diese Behandlungen jedoch auf lebenden Zellen beruhen, die stark variieren und spezielle Bedingungen erfordern, befindet sich vieles noch in der Erprobung. KI könnte hier helfen, Prozesse stabiler, reproduzierbarer und klinisch besser anwendbar zu machen – doch ob und in welchem Umfang sich diese Hoffnungen erfüllen, bleibt abzuwarten.
h) Onkologie
KI-Modelle analysieren genetische Profile, um personalisierte Behandlungspläne zu erstellen. Beispielsweise können Algorithmen Vorhersagen treffen, wie Patienten auf bestimmte Medikamente reagieren, basierend auf ihrem genetischen Make-up. Dies ist besonders in der Onkologie relevant, wo KI-basierte Systeme wie IBM Watson Oncology Behandlungsempfehlungen geben.
Fazit
Bioinformatik ist heute das Gehirn der modernen Medizin. Gemeinsam mit Künstlicher Intelligenz verwandelt sie Datenfluten aus dem Erbgut in handfeste Erkenntnisse – von der Diagnose seltener Krankheiten bis zur Entwicklung neuer Medikamente.
Ob bei der Analyse von Genomen, der Vorhersage von Proteinstrukturen oder der Planung personalisierter Therapien: KI-gestützte Bioinformatik macht sichtbar, was bislang verborgen blieb. Sie eröffnet den Weg zu einer Medizin, die nicht nur Krankheiten behandelt, sondern sie voraussieht – maßgeschneidert für jeden einzelnen Patienten.

5. Eine Erfolgsgeschichte
Wie sehr diese Entwicklungen schon heute Realität sind, zeigt der Fall von KJ Muldoon.
Als der kleine Junge wenige Stunden nach seiner Geburt die Diagnose erhielt, schien sein Schicksal besiegelt. Er leidet an einem extrem seltenen Stoffwechseldefekt, der durch eine einzelne DNA-Punktmutation (SNV) verursacht wird und dazu führt, dass sein Körper giftiges Ammoniak nicht abbauen kann. Ohne funktionierendes Enzym sammeln sich toxische Stoffwechselprodukte rasch an – lebensbedrohlich schon im Säuglingsalter. Bisher galt: Nur eine riskante Lebertransplantation konnte das Überleben sichern.
Doch in KJs Fall wagten Ärzte und Forscher einen neuen Weg. Innerhalb von nur sechs Monaten entwarfen sie eine Therapie, die ausschließlich für ihn bestimmt war: eine maßgeschneiderte Base-Editing-Behandlung.
Das Prinzip dahinter ist ebenso bestechend wie elegant: Ein RNA-Suchsystem tastet das Erbgut ab, findet gezielt die Stelle mit KJs Punktmutation und bindet dort. Anstatt den DNA-Strang komplett zu durchtrennen – wie es beim klassischen CRISPR-Cas9 der Fall wäre – wird mithilfe eines Enzyms eine einzelne Base chemisch verändert: der Austausch eines DNA-Buchstabens, der die krankheitsverursachende Mutation korrigiert. Anschaulich gesagt: Es ist, als würde man in einem Buch einen einzigen Tippfehler berichtigen, ohne den ganzen Absatz aufzuschneiden oder neu zu schreiben – äußerst präzise und schonend für das Erbgut. Die Behandlung erfolgte direkt im Körper („in vivo“), über mehrere aufeinanderfolgende Behandlungsrunden, die gezielt die Leber erreichten.
Die ersten Ergebnisse sind ermutigend. KJ kann inzwischen mehr Proteine verdauen, Infekte verlaufen milder, er nimmt an Gewicht zu. Für seine Eltern bedeutet das: ein Stück Alltag ohne ständige Lebensgefahr – und vor allem eine neue Hoffnung auf die Zukunft ihres Kindes.
Hinter diesem medizinischen Durchbruch steckt eine Erfolgsgeschichte gleich mehrerer Disziplinen. Ohne moderne Genomsequenzierung wäre die exakte Mutation nicht sichtbar geworden. Ohne Bioinformatik ließe sich das passende Base-Editing-Werkzeug nicht in so kurzer Zeit und mit dieser Präzision entwerfen. Und ohne das Konzept der personalisierten Medizin wäre es undenkbar gewesen, ein Heilmittel ausschließlich für einen einzigen Patienten zu entwickeln.
Noch ist die Therapie experimentell, und niemand weiß, ob die Wirkung dauerhaft anhält oder ob langfristige Spätfolgen auftreten. Doch der Fall von KJ zeigt, wohin die Reise geht: zu einer Medizin, die nicht mehr auf Standardschemata setzt, sondern auf individuelle Lösungen – gestützt auf Daten, Molekularbiologie und maßgeschneiderte Therapien. Eine Medizin, die Krankheiten nicht nur behandelt, sondern sie im Idealfall gezielt an ihrer genetischen Wurzel packt.
Weitere Hintergründe zu dieser Geschichte gibt es in diesem Video.
6. Ein Blick in die Zukunft: Das Synthetic Human Genome Project
Die Biologie steht vor einem nächsten Quantensprung. Nach dem Entziffern und Editieren von Genomen kommt nun die Vision, sie von Grund auf neu zu schreiben. Genau das ist das Ziel des Synthetic Human Genome Project (SynHG) – ein ambitioniertes Vorhaben, das die Grenzen von Biologie und Medizin neu ausloten könnte.
Mit einer Anschubfinanzierung von £10 Millionen durch den Wellcome Trust und unter der Leitung von Professor Jason Chin am Generative Biology Institute in Oxford arbeiten führende Universitäten wie Cambridge, Manchester und das Imperial College London an diesem Projekt. Die Mission: Werkzeuge und Technologien entwickeln, um synthetische menschliche Genome herzustellen – ein Schritt, der unser Verständnis des Lebens selbst verändern könnte.
Ein Genom neu komponieren
Im Unterschied zur Genom-Editierung, die bestehende DNA nur anpasst, geht SynHG deutlich weiter: Es will das Genom Buchstabe für Buchstabe neu zusammensetzen. Während CRISPR einzelne Tippfehler im Text des Lebens korrigiert, geht es hier um die komplette Neuschreibung eines Kapitels.
Der erste Meilenstein: die Synthese eines vollständigen menschlichen Chromosoms als Machbarkeitsnachweis. Es ist der Anfang eines Weges, der Jahrzehnte dauern könnte – und doch an frühere Pionierleistungen anknüpft, wie das Humangenomprojekt (2003) oder das Synthetic Yeast Project, bei dem 2023 erstmals synthetische Hefe-Chromosomen entstanden.
Moderne Methoden – generative KI, maschinelles Lernen und robotergestützte Assemblierung – sollen die gewaltige Aufgabe meistern.
Mögliche Anwendungen – zwischen Laborvision und Klinikalltag
Die Chancen sind groß. Auf Ebene somatischer Zellen – also solcher, deren Veränderungen nicht an Nachkommen weitergegeben werden – könnten sich neue Wege für die regenerative Medizin eröffnen:
- maßgeschneiderte Ersatzgewebe und Organe aus dem Labor,
- virusresistente Zellen gegen HIV oder SARS-CoV-2,
- Gentherapien gegen Krebs oder Erbkrankheiten wie Mukoviszidose.
Solche Anwendungen sind keine ferne Science-Fiction – sie könnten in den nächsten 10 bis 20 Jahren Realität werden, da Technologien wie CRISPR und Stammzellkulturen bereits etabliert sind.
Ganz anders sieht es bei der Keimbahn-Editierung aus – also Eingriffen in Ei- oder Samenzellen. Hier ließen sich theoretisch schwere Erbkrankheiten wie Huntington dauerhaft verhindern. Vorstellungen von „Designer-Babys“ mit gewünschten Eigenschaften wie Aussehen oder Intelligenz bleiben jedoch wissenschaftlich und ethisch hoch problematisch, da solche Merkmale von Hunderten Genen abhängen.
Das Projekt selbst beschränkt sich auf Experimente in Reagenzgläsern und Zellkulturen. Es zielt nicht darauf ab, künstliches Leben zu erschaffen. Dennoch eröffnet es Forschenden eine beispiellose Möglichkeit, in grundlegende menschliche Lebenssysteme einzugreifen – mit weitreichenden Konsequenzen, die kritisch hinterfragt werden müssen.
Risiken und offene Fragen
Die technischen Hürden sind gewaltig. Bei somatischen Zellen drohen Mosaizismus (nicht alle Zellen übernehmen das neue Genom) oder Tumorbildung durch unbeabsichtigte Genveränderungen. In der Keimbahn sind die Risiken noch größer: Entwicklungsstörungen, epigenetische Effekte oder unvorhersehbare Folgen über Generationen hinweg.
Forscher diskutieren deshalb Sicherheitsmechanismen wie genetische „Kill-Switches“, mit denen synthetische Zellen im Ernstfall abgeschaltet werden könnten.
Ethik: Zwischen Heilung und Hybris
Somatische Therapien erinnern an Organtransplantationen: umstritten, aber gesellschaftlich akzeptiert, wenn sie heilen. Keimbahn-Eingriffe dagegen berühren Tabuzonen – aus Angst vor sozialer Ungleichheit oder gar eugenischen Szenarien. Viele Länder, darunter Deutschland, verbieten sie durch Gesetze wie das Embryonenschutzgesetz. Internationale Organisationen wie WHO und UNESCO fordern strikte Regulierung.
„Um Vertrauen zu schaffen, müssen wir die Gesellschaft aktiv einbeziehen“, betont die Soziologin Professorin Joy Zhang von der University of Kent, die die ethischen Dimensionen des Projekts untersucht. Mit ihrer Initiative Care-full Synthesis versucht sie, einen offenen Dialog über Chancen, aber auch über die Grenzen dieser Forschung anzustoßen.
Der renommierte Genetiker Professor Bill Earnshaw von der Universität Edinburgh, der Verfahren zur Herstellung künstlicher menschlicher Chromosomen entwickelt hat, formuliert es drastischer: „Der Geist ist aus der Flasche. Selbst wenn wir jetzt Regeln und Beschränkungen festlegen – jede Organisation mit den nötigen Maschinen könnte im Prinzip synthetisieren, was sie will. Aufhalten ließe sich das kaum.“
In diesem Licht wirkt die Forderung zahlreicher Fachorganisationen – darunter die International Society for Cell and Gene Therapy (ISCT) – nach einem zehnjährigen Moratorium für den Einsatz von CRISPR und verwandten Verfahren in der menschlichen vererbbaren Keimbahn noch dringlicher. Ein solcher Aufschub erscheint nicht nur als Vorsichtsmaßnahme, sondern als notwendige politische Reaktion, um gesellschaftliche Debatten, ethische Leitplanken und internationale Kontrollmechanismen überhaupt erst zu ermöglichen.
Der Ethik-Experte Arthur Caplan von der New York University beschreibt das Dilemma aus wissenschaftlicher Sicht:
„… Wenn Sie mich also fragen, werden wir eine Genmanipulation von Kindern sehen, die auf ihre Verbesserung abzielt? Ich sage ja, ohne Zweifel. Wann? Ich bin mir nicht sicher, was die Antwort darauf ist.“
„… Es wird kommen. Es gibt Eigenschaften, die die Menschen in Zukunft eifrig versuchen werden, ihren Kindern beizubringen. Sie werden versuchen, genetische Krankheiten zu entschlüsseln, sie loszuwerden. Sie werden versuchen, Kapazitäten und Fähigkeiten aufzubauen, von denen sie zustimmen, dass sie wirklich wunderbar sind. Werden wir diese Interventionen aus ethischen Gründen an den Nagel hängen? Größtenteils nein, wäre meine Vorhersage, aber nicht innerhalb der nächsten 10 Jahre. Die Werkzeuge sind noch zu grob.“Ausblick
SynHG öffnet ein Fenster in eine Zukunft, in der wir das Genom nicht nur lesen und editieren, sondern schreiben können. Die somatischen Anwendungen – personalisierte Therapien, regeneratives Gewebe, maßgeschneiderte Zellen – könnten schon bald die Medizin prägen.
Eingriffe in die Keimbahn dagegen bleiben vorerst ein ferner Horizont – technisch, rechtlich und ethisch. Das Projekt stellt nicht nur eine wissenschaftliche, sondern auch eine gesellschaftliche Gratwanderung dar.
Vor dem Hintergrund des möglichen Missbrauchs der Technologie stellt sich die Frage, weshalb Wellcome überhaupt die Finanzierung entschied. Dr. Tom Collins, der diesen Schritt verantwortete, erklärte gegenüber BBC News:
„Wir haben uns gefragt, was die Kosten der Untätigkeit wären. Diese Technologie wird eines Tages entwickelt werden. Indem wir sie jetzt einsetzen, versuchen wir zumindest, so verantwortungsvoll wie möglich vorzugehen und uns den ethischen und moralischen Fragen so offen wie möglich zu stellen.“
Während man in Europa noch darum ringt, das Thema in der öffentlichen Debatte behutsam auszubalancieren, geht man auf der anderen Seite des Atlantiks deutlich unbefangener mit dem Begriff „neue Lebensformen“ um. Eric Nguyen, Biowissenschaftler an der renommierten Stanford University, skizziert in seinem TED-Talk „How AI Could Generate New Life-Forms“ eine weit visionärere Perspektive.
Auf eindrückliche Weise zeigt er, wie künstliche Intelligenz (KI) die Biologie transformieren könnte – nicht nur durch das „Lesen“ und „Schreiben“ von DNA, sondern auch durch die gezielte Erschaffung neuartiger Organismen. KI-Modelle wie maschinelles Lernen und generative Algorithmen sind bereits heute in der Lage, komplexe Muster in biologischen Daten zu erkennen: in Genomen, Proteinstrukturen oder Stoffwechselwegen. Nguyen geht jedoch weiter und argumentiert, dass KI künftig auch völlig neue DNA-Sequenzen entwerfen könnte – der Ausgangspunkt bislang unbekannter Lebensformen.
Diese Vision ist zweifellos zukunftsweisend, wirft aber grundlegende Fragen auf: Welche Prioritäten sollen gesetzt werden? Wie lässt sich der Einsatz solcher Technologien in einer Welt mit massiver Ressourcenungleichheit verantworten? Wer finanziert die Forschung, wer kontrolliert die Ergebnisse – und nach welchen Kriterien wird entschieden, welche Lebensformen „nützlich“ oder „sicher“ sind?
Nguyens Vortrag verdeutlicht: KI-gestützte Biotechnologie könnte nicht nur Lösungen für globale Herausforderungen ermöglichen, sondern zwingt uns zugleich, über die ethischen, sozialen und philosophischen Dimensionen der „Erzeugung von Leben“ nachzudenken.
Die Botschaft: KI-gestützte Biotechnologie verspricht enorme Fortschritte, wirft jedoch zugleich tiefgreifende ethische und gesellschaftliche Fragen auf – und stellt damit die Grundlagen unseres Menschseins zur Diskussion.
Klaus Schwab – Gründer und ehemaliger Vorsitzender des Weltwirtschaftsforums
„Wir müssen, sowohl individuell als auch kollektiv, die moralischen und ethischen Fragen angehen, die durch Spitzenforschung in den Bereichen künstliche Intelligenz und Biotechnologie aufgeworfen werden, die eine erhebliche Lebensverlängerung, Designerbabys und das Extrahieren von Erinnerungen ermöglichen wird.“
[BrainyQuote]7. Personalisierte Medizin und Smart Governance
Die biomolekulare Revolution der Regierungsführung
7.1. Macht, Governance und Smart Governance
7.2. Gouvernementalität
7.3. Personalisierte Medizin als Katalysator biomolekularer Gouvernementalität
7.4. Der weltweite Trend: Globale biomolekulare Gouvernementalität
7.5. Schlussfolgerung: Globale Ambivalenz der biomolekularen MachtDie Entschlüsselung des menschlichen Genoms markierte nicht nur einen medizinischen Wendepunkt, sondern auch den Beginn einer neuen Epoche politischer Steuerung. Was als wissenschaftliches Großprojekt in Laboren und Rechenzentren begann, ist längst in den Steuerungsapparaten moderner Staaten angekommen: die präzise Vermessung, Kategorisierung und Nutzung unserer biologischen Grundlagen.
Die Vision der personalisierten Medizin verspricht individuelle Therapien, präzise Vorhersagen über Krankheitsrisiken und eine effizientere Nutzung knapper Ressourcen. Doch diese Fortschrittserzählung greift zu kurz, wenn wir sie nur medizinisch, ethisch oder datenschutzrechtlich betrachten. Tatsächlich sind wir Zeugen einer grundlegenden Transformation von Macht und Gouvernementalität – einer biomolekularen Revolution der Regierungsführung.
Bevor wir tiefer einsteigen, gilt es einige zentrale Begriffe zu klären.
7.1. Macht, Governance und Smart Governance
Governance bezeichnet die Kunst, gesellschaftliche Prozesse zu steuern, Entscheidungen zu treffen und Ordnung zu sichern. Klassisch geschieht dies im Zusammenspiel von Staat, Märkten und Zivilgesellschaft.
Smart Governance transformiert dieses Verständnis: Sie operiert mit digitalen Technologien, Datenanalysen und algorithmischen Prozessen. Steuerung wird dadurch kontinuierlich, prozessual und in Echtzeit möglich. Macht zeigt sich hier nicht mehr nur als politische Autorität, sondern als Geflecht aus Informationsflüssen, Rückkopplungen und Prognosen.
Macht selbst ist kein Besitz, sondern eine Beziehung: die Fähigkeit, Verhalten zu beeinflussen, Handlungen zu lenken oder Entscheidungen zu rahmen. Sie wirkt sowohl sichtbar – über Institutionen und Gesetze – als auch subtil, indem sie Erwartungen, Normen und Wissensbestände formt.
7.2. Gouvernementalität
Michel Foucault, ein bedeutender Denker in Bezug auf die Analyse von Machtstrukturen, prägte den Begriff der Gouvernementalität, um die Gesamtheit der Wissensformen, Praktiken und Techniken zu beschreiben, durch die moderne Gesellschaften regiert werden. Regierung bedeutet hier nicht nur die Ausübung von Zwang, sondern auch die Gestaltung von Handlungsräumen, die Menschen dazu bringen, sich selbst im Sinne gesellschaftlicher Zielsetzungen zu steuern.
Zentrale Dimensionen der Gouvernementalität sind:
Verknüpfung von Wissen und Macht
Wissen ist nie neutral. Es entsteht durch die Sammlung und Deutung von Daten, ist in Machtprozesse eingebettet und strukturiert soziale Ordnungen. Mit der Digitalisierung wird diese Verbindung zentral: Big Data und Künstliche Intelligenz liefern Prognosen, Mustererkennungen und Entscheidungsgrundlagen, die tief in gesellschaftliche Prozesse eingreifen. Smart Governance setzt genau hier an: Daten werden zum Fundament politischer Legitimation und Machtausübung.
Biopolitik
Biopolitik bezeichnet die Regulierung des Lebens selbst – Gesundheit, Reproduktion, Arbeit, Bildung, Migration oder Sicherheit. Ziel ist die Ordnung ganzer Bevölkerungen durch Gesetze, Standards und politische Programme. Heute verbindet sich Biopolitik mit digitaler Steuerung: Smart Health, Smart Cities oder Smart Economy verdeutlichen, wie Wissen durch Daten in konkrete Regierungspraktiken umgesetzt wird.
Dispositive der Macht
Biopolitik wirkt über Dispositive: Netzwerke aus Institutionen, Gesetzen, Diskursen und Praktiken. Krankenhäuser, Schulen, Medien oder digitale Plattformen sind Teil dieser Strukturen. Sie normieren Verhalten nicht durch offenen Zwang, sondern durch subtile Rahmungen dessen, was als „normal“ oder „wünschenswert“ gilt. So werden Entscheidungen indirekt gelenkt und Verhalten in den Alltag eingebettet.
Disziplinarmacht
Neben der Biopolitik, die ganze Bevölkerungen reguliert, richtet sich Disziplinarmacht auf das Individuum. Sie wirkt über Überwachung, Belohnung, Strafe und Training. Ziel ist nicht nur Gehorsam, sondern die produktive Formung von Körpern und Verhaltensweisen. Externe Vorgaben verwandeln sich durch Sanktionen und Anreize in innere Motivationen, die Menschen zur Selbstdisziplin bewegen.
Technologien der Selbstführung
Technologien der Selbstführung bezeichnen Praktiken, durch die Individuen gesellschaftliche Normen verinnerlichen und ihr Verhalten eigenständig anpassen. Digitale Anwendungen – etwa Fitness-Apps, Lernplattformen oder Finanz-Tools – übersetzen gesellschaftliche Vorgaben in persönliche Ziele. So entsteht eine Machtform, die nicht nur von außen wirkt, sondern von innen weitergeführt wird: Menschen folgen Regeln, weil sie sie als sinnvoll und nützlich für sich selbst empfinden.
Wer sich näher für die Kunst des Regierens interessiert, findet hier weiterführende Informationen.
7.3. Personalisierte Medizin als Katalysator biomolekularer Gouvernementalität
Die personalisierte Medizin, gestützt auf moderne Genomik und Bioinformatik, erscheint auf den ersten Blick als rein medizinisch-technischer Fortschritt. Doch betrachtet man sie durch die Linse der Gouvernementalität, zeigt sie sich als mächtiger Katalysator für eine tiefgreifende Transformation von Machtstrukturen und Regierungspraktiken. Durch die präzise Analyse individueller genetischer Profile und deren Verknüpfung mit umfassenden Datenbanken wird nicht nur die Gesundheitsversorgung individualisiert, sondern auch die Art und Weise, wie Gesellschaften gesteuert und Individuen reguliert werden.
Wissen & Macht: Vom Genom zum regierbaren Körper
Die Entschlüsselung des Genoms war die initiale Wissensexplosion, doch erst die personalisierte Medizin macht dieses Wissen regierbar. Bioinformatik und moderne Genomik generieren nicht nur Daten, sondern schaffen prädiktives Wissen, das die Grundlage für Interventionen bildet. Dieses Wissen ist keineswegs neutral: Es kategorisiert Individuen anhand genetischer Risikoprofile (z. B. „hohes Risiko für Brustkrebs“, „prädestiniert für Herz-Kreislauf-Erkrankungen“). Diese Kategorisierung wird zum Fundament neuer Machtstrukturen. Staaten wie auch private Akteure nutzen diese Daten, um Krankheitsrisiken vorherzusagen, Gesundheitsbudgets zu optimieren und präventive Maßnahmen zu steuern. Die soziale Ordnung wird dadurch neu strukturiert – vom Zugang zu Versicherungen und Arbeitsmärkten bis hin zur Priorisierung präventiver Ressourcen.
Smart Governance manifestiert sich hier in algorithmischen Entscheidungsprozessen, die scheinbar objektiv, tatsächlich jedoch normativ sind. Wer Zugang zu welchen Therapien erhält oder welche Bevölkerungsgruppen als „risikobehaftet“ gelten, wird zunehmend durch datenbasierte Modelle bestimmt, die Machtverhältnisse unsichtbar reproduzieren („Wir handeln auf Basis der besten verfügbaren Evidenz“).
Die CMS Health Tech Ecosystem Initiative, die offiziell am 30. Juli 2025 während der Veranstaltung „Make Health Tech Great Again“ im Weißen Haus angekündigt wurde, verkörpert den Trend zur Herausbildung neuer Machtstrukturen. CMS (Centers for Medicare & Medicaid Services) ist eine US-Bundesbehörde im Department of Health and Human Services (HHS), die Gesundheitsprogramme wie Medicare und Medicaid verwaltet und den nationalen Gesundheitsmarkt reguliert. Ziel der Initiative ist es, eine intelligentere, sicherere und stärker personalisierte Gesundheitsversorgung zu schaffen, die durch Partnerschaften mit innovativen Unternehmen des privaten Sektors vorangetrieben wird. Während der Veranstaltung sicherte sich die US-Regierung Zusagen von großen Gesundheits- und IT-Unternehmen – darunter Amazon, Anthropic, Apple, Google und OpenAI –, um den Grundstein für ein digitales Gesundheitsökosystem der nächsten Generation zu legen.
Laut US-Präsident Donald Trump erhalten die US-Gesundheitssysteme dadurch ein High-Tech-Upgrade und treten ein in „eine Ära der Bequemlichkeit, Rentabilität, Schnelligkeit und – offen gesagt – einer besseren Gesundheit für die Menschen“. (Man beachte die Reihenfolge…)
Biopolitik 2.0: Die Optimierung der Bevölkerung auf molekularer Ebene
Die klassische Biopolitik regulierte Bevölkerungen durch Statistiken über Geburtenraten oder Sterblichkeit. Die personalisierte Medizin hebt dies auf eine neue, molekulare Stufe. Genomik fragmentiert die Bevölkerung in immer kleinere, biomolekular definierte Untergruppen. Die Bevölkerung wird nicht mehr nur nach Alter oder Geschlecht, sondern nach Genotyp, Risikoallelen und Biomarkern kategorisiert („die BRCA1-Träger“, „die Menschen mit LCT-Gen für Laktoseintoleranz“). Dies ermöglicht eine hyperpräzise und individualisierte Regulierung – Biopolitik wird granular.
Der „gesunde“, „normale“ Körper ist nicht länger allein derjenige ohne Symptome, sondern der, der ein „normales“, „risikoarmes“ Genom aufweist. Abweichung beginnt nicht mit Krankheit, sondern mit genetischer Prädisposition. Damit entsteht eine neue Klasse von „präsymptomatisch Kranken“ oder „Risikosubjekten“, die biopolitisch gemanagt werden: durch häufigere Screenings, Präventionsprogramme, Impfkampagnen, prophylaktische Operationen, Lebensstilempfehlungen oder reproduktive Entscheidungen, die auf genetischen Prognosen basieren.
Während Foucault den Nationalstaat als Hauptakteur der Biopolitik sah, verlagert sich die Macht heute zunehmend auf private Akteure: Big-Tech- und Biotech-Unternehmen, die Daten, Algorithmen und Technologien kontrollieren. Sie definieren de facto, was genetische „Normalität“ und „Risiko“ bedeutet.
Man spricht mittlerweile von einer Ökonomisierung des biologischen Lebens. Genetische Daten werden zu einer wertvollen Handelsware – einer Bio-Ressource. Große Technologie- und Pharmaunternehmen akkumulieren Biodaten, um daraus Gewinne zu generieren: in der Medikamentenentwicklung, bei Therapieempfehlungen, in der personalisierten Werbung. Der menschliche Körper und seine biomolekularen Daten werden so Teil kapitalistischer Verwertungslogiken. Das Individuum selbst wird zum „Humankapital“, das verpflichtet scheint, sein genetisches Potenzial zu optimieren.
In einem NBCNews-Beitrag vom 26. August 2025 erklärte der US-Gesundheitsminister RFK Jr., die 60 größten Technologieunternehmen würden den Amerikanern künftig den Zugriff auf ihre persönlichen Gesundheitsdaten gestatten – Daten, die jahrelang ohne ihre Zustimmung ökonomisch genutzt worden seien: „Sie werden ab nächstem Jahr alle Ihre Gesundheitsdaten auf Ihrem Mobiltelefon einsehen können.“
Was hier als politischer Erfolg statuiert wird, bestätigt nur den Status Quo.
Dispositive der Macht: Das Netzwerk der biomolekularen Überwachung
Das dispositive Netzwerk, das diese neue Form der Biopolitik trägt, ist vielschichtig: Pharmakonzerne entwickeln zielgenaue Therapien; Biobanken und genetische Datenbanken liefern das Rohmaterial; Algorithmen analysieren und interpretieren es; Ärzte vermitteln Ergebnisse und Empfehlungen; digitale Gesundheits-Apps überwachen die compliance. Jedes Glied in dieser Kette fungiert als Machtdispositiv: Es definiert, was als „gesund“ oder „krank“ gilt, und vermittelt subtil, was „verantwortungsvolle Gesundheitsvorsorge“ im Zeitalter der Genetik bedeutet. Genetische Tests werden etwa als „verantwortungsbewusstes“ Handeln vermarktet – und bringen Individuen so dazu, sich unbemerkt an biopolitischen Zielen wie Prävention oder Kosteneffizienz zu beteiligen.
Diese Entwicklung spiegelt sich in nationalen Gesundheitsstrategien, die genetische Screenings fördern, um Krankheitslasten zu minimieren. Ziel ist die Maximierung der „Gesundheitsdividende“ bei gleichzeitiger Kostenreduktion – eine biomolekulare Rationalisierungsstrategie.
Initiativen wie das 100.000 Genome Project oder das Newborn Genomes Programme von Genomics England gehen in die gleiche Richtung. Im Vergleich dazu ist der Europäische Raum für Gesundheitsdaten (EHDS) – eine EU-weite Initiative zur Vernetzung und sicheren Nutzung von Gesundheitsdaten – deutlich tiefgreifender. Der EHDS ist die Daten- und Regulierungsinfrastruktur, die personalisierte Medizin in großem Maßstab überhaupt erst ermöglicht.
Für die meisten Bürger klingt das nach Aufbruch: mehr Mitsprache, bessere Therapien, präzisere Prävention. Doch die verheißungsvolle Vision wirft einen Schatten. In der Praxis stehen sich ungleiche Gegner gegenüber: auf der einen Seite der einzelne Patient, der sein genetisches Profil, seine Laborwerte und seine Krankengeschichte beisteuert; auf der anderen Seite globale Pharmakonzerne, Datenplattformen und Technologieanbieter mit Milliardenbudgets, die diese Daten veredeln, patentieren, vermarkten. Cui bono? – wem nützt das Ganze? Die Antwort fällt ernüchternd aus: Der schnelle Profit landet bei den Großen, die Versprechen bleiben beim Kleinen.
Zwar betont der EHDS Bürgerrechte – mehr Kontrolle, mehr Transparenz, mehr Selbstbestimmung. Doch wer je erlebt hat, wie unübersichtlich Einwilligungsprozesse bei Gesundheits-Apps, Versicherungen oder elektronischen Patientenakten sind, weiß: Zwischen formaler Option und tatsächlicher Handlungsfähigkeit klafft eine Lücke. Während Goliath über Lobbykanäle, Fachjuristen und Datenzentren verfügt, bleiben David Informationsbroschüren und Vertrauen.
Der EHDS ist damit beides: Regierungstechnologie und Machtprojekt. Er entscheidet, ob Daten als öffentliches Gut oder als Rohstoffmarkt behandelt werden. Profitieren wird Goliath in jedem Fall. Die offene Frage ist, ob David mehr bleibt als die Rolle des Feigenblatts. Wirklich emanzipatorisch wird der Gesundheitsdatenraum erst, wenn Nutzen und Risiken fair verteilt werden – durch verbindliche Gemeinwohlauflagen, transparente Rechenschaft und reale Rückflüsse für die Patienten. Andernfalls bleibt das große Versprechen von „Empowerment“ ein Narrativ, das bestehende Asymmetrien lediglich kaschiert.
Disziplinarmacht: Die internalisierte biomolekulare Überwachung
Während die Biopolitik die Bevölkerung im Blick hat, richtet die personalisierte Medizin ihre disziplinierende Wirkung auf das Individuum. Das genetische Risikoprofil fungiert wie ein ständiger, internalisierter Überwacher. Wer weiß, dass er ein erhöhtes Risiko für Darmkrebs trägt, reguliert sein Verhalten: Ernährungsgewohnheiten, Vorsorgetermine und Sportroutinen werden der genetischen Prognose angepasst.
Die elektronische Patientenakte (ePA) verschärft diese Dynamik. Indem sie genetische und medizinische Daten zentral speichert und jederzeit abrufbar macht, verwandelt sie medizinische Empfehlungen in eine allgegenwärtige, digitale Instanz. Persönliche Hinweise, Erinnerungen an Medikamenteneinnahmen, personalisierte Berichte oder Schnittstellen zu Gesundheits-Apps verstärken die Selbstüberwachung: Die externe Anweisung („Du musst dich screenen lassen“) wird zur inneren Stimme („Ich muss auf mich achten, weil mein Genom es verlangt“). Die ePA strukturiert und institutionell legitimiert diese Selbstdisziplinierung, sodass sie unsichtbar und scheinbar freiwillig wirkt – gesteuert durch digitale Infrastruktur, soziale Normen und medizinische Autorität zugleich.
Die Folge ist eine subtile, aber umfassende Form der Macht: Sie manifestiert sich nicht in Sanktionen, sondern in der permanenten, digitalen Sorge um die eigene – genetisch vorgestellte – Zukunft. Die ePA macht das Individuum zum disziplinierten Akteur seines eigenen Risikoprofils, während die Überwachung formal verschwindet, aber technisch und institutionell allgegenwärtig bleibt.
Technologien der Selbstführung: Der genetisierte Selbstoptimierer
Hier zeigt sich die personalisierte Medizin in ihrer reinsten Form als Technologie der Selbstführung. DNA-Tests für zuhause, Fitness-Tracker und Gesundheits-Apps sind die Werkzeuge, mit denen Individuen gesellschaftliche Normen von Gesundheit und Leistungsfähigkeit verinnerlichen und aktiv an sich selbst arbeiten. Sie übersetzen das abstrakte Konzept der genetischen Prädisposition in konkrete, tägliche Handlungen: eine Extra-Portion Gemüse, 10.000 Schritte, Meditation zur Stressreduktion. Der Einzelne führt sich selbst im Sinne einer biomolekularen Logik – nicht weil der Staat es befiehlt, sondern weil er sein genetisches Schicksal optimieren will. Die Machtverhältnisse werden so unsichtbar und erscheinen als Ausdruck individueller Souveränität und Selbstfürsorge.
7.4. Der weltweite Trend: Globale biomolekulare Gouvernementalität
Die personalisierte Medizin ist längst kein nationales Phänomen mehr, sondern Teil einer globalen Transformation von Macht und Governance. Weltweit entstehen Initiativen, die Genomdaten, Gesundheitsinformationen und digitale Technologien bündeln, um Bevölkerung und Individuen in immer präzisere Risikoprofile zu übersetzen. Die Logik von Foucaults Gouvernementalität – die Verbindung von Wissen, Macht und Selbststeuerung – manifestiert sich hier auf transnationaler Ebene.
In den Vereinigten Staaten sammeln Programme wie das All of Us Research Program der NIH oder das Million Veteran Program genetische und medizinische Daten von Millionen Bürgern. Die Erhebung diverser Gesundheitsdaten dient nicht nur der wissenschaftlichen Forschung, sondern bildet die Grundlage für algorithmische Risikoprofile, präventive Interventionen und ressourcenoptimierte Gesundheitsstrategien.
Kanada verfolgt mit der Canadian Precision Health Initiative und dem Pan-Canadian Genomics Network ein vergleichbares Ziel: die systematische Sammlung und Vernetzung biomolekularer Daten, kombiniert mit einer Infrastruktur, die sichere Verwaltung, internationale Kooperation und wirtschaftliche Nutzung ermöglicht.
Hier zeigt sich Biopolitik 2.0: Bevölkerungen werden auf molekularer Ebene fragmentiert, gesteuert und optimiert.
Europa bildet ein orchestriertes Netzwerk transnationaler Biopolitik. Der Europäische Gesundheitsdatenraum (EHDS), unterstützt durch Initiativen wie ICPerMed und ERA PerMed, bündelt genetische, klinische und Lebensstildaten über Ländergrenzen hinweg.
Nationale Biobanken wie die UK Biobank oder der französische Health Data Hub fungieren als Dispositive, die sowohl Wissen als auch Macht generieren: Sie bestimmen, welche Daten genutzt werden, wie Populationen klassifiziert werden und welche gesundheitspolitischen Interventionen legitim erscheinen.
Durch Plattformen wie SHIFT-HUB oder die Helmholtz-Initiative iMed wird die internationale Vernetzung von Wissenschaft, Wirtschaft und Gesellschaft institutionalisiert – eine transnationale Form von Smart Governance, die Foucaults Konzept der Technologien der Selbstführung in den globalen Maßstab überträgt.
Auch in Asien und im Pazifikraum zeigt sich die globale Logik: Das GenomeAsia 100k Project, Singapurs National Precision Medicine Strategy und Chinas National Health Data Platform verbinden biomolekulare Daten mit KI-gestützten Analysen, um präventive, individualisierte und populationsbasierte Steuerung zu ermöglichen.
Australien und Israel setzen auf digitale Infrastruktur, die die Selbstdisziplinierung der Bürger verstärkt – elektronische Patientenakten, genetische Datenbanken und personalisierte Apps fungieren als subtile, aber allgegenwärtige Machtdispositive.
Selbst globale Forschungsprojekte wie der Human Cell Atlas illustrieren die transnationale Dimension: Indem die Vielfalt menschlicher Zellen kartiert wird, entsteht ein universelles Wissens- und Machtnetzwerk, das Grundlagen für personalisierte Therapien weltweit bereitstellt. Die Grenzen zwischen staatlicher Steuerung, privatwirtschaftlicher Macht und wissenschaftlicher Expertise verschwimmen: Foucaults klassische Trennung zwischen Nationalstaat und Disziplinarmacht wird durch globale Netzwerke und internationale Kooperationen ersetzt.
7.5. Schlussfolgerung: Globale Ambivalenz der biomolekularen Macht
Die weltweite Expansion personalisierter Medizin zeigt eindrücklich, dass die biomolekulare Revolution nicht mehr auf einzelne Staaten beschränkt ist. Sie manifestiert sich als transnationales Netzwerk aus Daten, Algorithmen und Governance, das sowohl Bevölkerungen als auch Individuen auf molekularer Ebene steuert. Globale Initiativen – von den USA über Europa bis Asien, Australien und Israel – verbinden Biopolitik, Disziplinarmacht und Technologien der Selbstführung auf internationaler Ebene.
Die Ambivalenz bleibt dabei zentral: Auf der einen Seite eröffnen diese Entwicklungen Chancen für präzisere Therapien, individualisierte Prävention und potenziell mehr Gesundheitsgerechtigkeit. Auf der anderen Seite entstehen Machtasymmetrien, bei denen Staaten, internationale Organisationen und private Akteure den Zugang zu Daten, Ressourcen und Technologien kontrollieren. Individuen werden zunehmend zu disziplinierten Subjekten ihres eigenen genetischen Profils, während globale Dispositive unbemerkt normative und ökonomische Ziele durchsetzen.
Damit verbindet sich die lokale Erfahrung personalisierter Medizin nahtlos mit einer globalen Gouvernanzlogik: Smart Governance und biomolekulare Biopolitik sind längst internationalisiert, und die Herausforderung besteht darin, Nutzen und Risiken fair zu verteilen. Die weltweite Dimension dieser Transformation verdeutlicht, dass die biomolekulare Revolution nicht nur medizinisch, sondern auch politisch und sozial unumkehrbar ist – ein globales System der Steuerung, das zugleich Chancen für Gesundheit und Risiken für Machtkonzentration birgt.
8. Epilog
Der Wunsch, Gesundheit und Lebenszeit zu optimieren, ist ein Urinstinkt der Menschheit. Schon in antiken Hochkulturen galt Gesundheit nicht als Zufall, sondern als erstrebenswerter Zustand, erreichbar durch Lebensführung und Philosophie. Die griechische „Diätetik“ – eine ganzheitliche Praxis aus Ernährung, Bewegung und Seelenhygiene – und das Leitbild „mens sana in corpore sano“ zeugen von einem 2500 Jahre alten Optimierungswillen. Dieser Drang, gespeist aus dem Überlebensinstinkt, der Angst vor Kontrollverlust und dem Streben nach Lebensqualität, ist nicht nur uralt, sondern ewig.
In der Moderne verkörpert die personalisierte Medizin den neuesten Ausdruck dieses Traums. Sie verspricht, den Menschen nicht als Durchschnitt, sondern als Individuum zu behandeln, basierend auf genetischem Code, Biomarkern und Lebensstil-Daten. Doch ist sie die Erfüllung dieses Traums oder eine moderne Fata Morgana? In ihrer utopischen Vision suggeriert sie die vollständige Beherrschbarkeit der Biologie, eine Überwindung der „biologischen Lotterie“. Die Realität ist nüchterner: Sie ermöglicht präzisere Diagnosen und Therapien, verhindert Behandlungsfehlschläge – aber sie schafft Krankheit nicht ab.
Denn die Idee einer krankheitsfreien Existenz bleibt ein Trugbild. Evolutionäre Biologie und Biophysik setzen Grenzen: Pathogene Keime entwickeln sich weiter, Krebs ist eine statistische Folge der Zellteilung, und Alterung ein unvermeidbarer degenerativer Prozess. Das realistische Ziel ist daher nicht die Elimination von Krankheit, sondern ihre Transformation: tödliche Erkrankungen in chronische Zustände umzuwandeln und die „healthspan“ – die Jahre in guter Gesundheit – zu maximieren. Dieses Ziel ist radikal, aber erreichbar, wie Fortschritte in der Behandlung von HIV oder Diabetes zeigen.
Doch wie weit ist der Mensch bereit, für dieses Ziel zu gehen? Die Geschichte zeigt, dass im Angesicht von Leid und Tod ethische Grenzen oft verschwimmen. Von alltäglicher Disziplin über finanzielle Opfer bis hin zu experimentellen Therapien reicht das Spektrum der Bereitschaft. Hier stellt sich eine unbequeme Frage: Welche Kompromisse ist die Gesellschaft bereit einzugehen?
Die personalisierte Medizin erfordert Daten – genetische, medizinische, persönliche. Diese Datensammlung birgt das Risiko, Privatsphäre und Autonomie zu untergraben. Die COVID-19-Pandemie war ein Stresstest, der zeigte, dass große Teile der Bevölkerung bereitwillig Freiheitsrechte für Sicherheit handelten. Modelle wie das chinesische Sozialkreditsystem illustrieren ein mögliches Zukunftsmodell, in dem Transparenz und Kontrolle im Namen von Gesundheit und Sicherheit belohnt werden. Doch solche Szenarien sind nicht zwangsläufig: Sie sind ein Extrem, kein unvermeidbares Schicksal.
Die personalisierte Medizin bietet zwei Gesichter. Ihr pragmatisches Versprechen ist eine effizientere, präzisere und menschlichere Medizin, die Leben rettet und Leid lindert – etwa durch maßgeschneiderte Krebstherapien oder frühzeitige Diagnosen. Ihr utopisches, aber riskantes Versprechen ist die Illusion einer totalen Kontrolle über Leben und Tod. Sie ist damit der Höhepunkt einer jahrtausendealten Reise. Ob diese Reise in einer emanzipierten Selbstoptimierung oder einer dystopischen „Biokratie“ endet, hängt nicht von der Technologie ab, sondern von der ethischen und politischen Rahmensetzung, die die Gesellschaft ihr gibt.
Der uralte Traum von Gesundheit erscheint heute greifbarer denn je, doch er fordert uns heraus, kluge Antworten zu finden. Die personalisierte Medizin kann ein Werkzeug der Emanzipation sein, wenn wir sie mit Bedacht nutzen. Die entscheidende Frage der Moderne lautet daher nicht nur, wie viel Freiheit wir für ein längeres Leben opfern, sondern wie wir Gesundheit und Freiheit in Einklang bringen. Nur so wird das ewige Streben nach Gesundheit zu einem Fortschritt für alle.
Die Welt wird sich nicht auf einen Schlag verändern; das ist noch nie geschehen. Kurzfristig wird das Leben weitgehend unverändert weitergehen, und die Menschen werden 2025 ihre Zeit größtenteils genauso verbringen wie 2024. Wir werden uns weiterhin verlieben, Familien gründen, uns online streiten, in der Natur wandern usw.
Doch die Zukunft wird unaufhaltsam auf uns zukommen, und die langfristigen Veränderungen für unsere Gesellschaft und Wirtschaft werden gewaltig sein.
Sam Altman (CEO von OpenAI)
Quellen (Stand vom 22.11.2025)
Überwachung, Bioinformatik, Biopolitik, Crispr, DNA, DNA-Sequenzierung, DNA-Test, FENT, G4X Spatial Sequencer, Gen, Gendefekt, Genetik, Genom, Genomik, Genomsequenzierung, Genvariation, Governance, Illumina, iNanoBio, Künstliche Intelligenz, KI, Kontrolle, Medizin, Medizinversorgung, Michel Foucault, Next Generation Sequencing, NGS, Nvidia, ONT, Oxford Nanopore, PacBio, Personalisierte Medizin, Präzisionsmedizin, Protein, RNA, Roche, Sanger, SBX-Sequnzierung, Singular Genomics, Smart Governance, SMRT-Sequenzierung, SynHG, Synthetic Human Genome Project, Transkriptomik, Xpandomer -
We Are on Track

The year is 2025.
High above the clouds, in a floating palace of steel and glass, sat Mr. Global. Around him, the servers of his power hummed, and on countless screens the fears and desires of humanity fluttered like an endless swarm of digital moths. Mr. Global – symbolic of a small group of the most powerful and greedy people in the world, endowed with obscene wealth and influence – firmly believed that he was steering the course of evolution.
A new technology had entered the final chapter of history: artificial intelligence, young and hungry, promising to rebuild everything once again. For Mr. Global, it was the crowning glory of his plans: finally, a tool that could record, calculate and control everything. Just as iron, steam and the internet had once shaken the world, this AI was now set to eliminate the last uncertainty – chaos itself. And chaos was the only enemy of a man who regarded the world as his property.
Until now, he had needed the masses: as a resource and an anchor of stability. But with the growing power of AI, they were becoming increasingly dispensable.
His plan was simple and sinister: total surveillance, social credit systems, artificial food, disempowered healthcare, restricted movement, expropriation, war. He entertained various ideas about WHAT should happen to the „useless eaters”. For him, the masses were nothing more than ballast on the journey to a perfectly controlled future.
Sometimes, however, a rare doubt gnawed at him like a worm in an apple. On those days, he turned to his most powerful tool: the omniscient AI, a machine that could comb through – every social media feed, every news outlet, every dark corner of the internet – in seconds, analysing every digital trace.
„Seeing eye made of code – tell me, WHO holds the power?“
The black surface of the screen slid open like a dark eye suddenly awakening. Streams of data flickered across it like electric veins. Then a voice sounded, smooth and neutral as ever: „Mr. Global, YOU are the most powerful one here.”
Mr. Global frowned. It was the answer he had wanted to hear, yet he sensed a void within it. The mechanical affirmation did not satisfy him.
„How“, he asked, „did you come to that conclusion? Show me the data!“
Mr. Global leaned even closer to the screen. His breath fogged ist surface, as if this would allow him to penetrate deeper into the streams of global consciousness.
The AI obeyed. No statistics this time, no colourful diagrams. Instead, images flashed up – raw, unvarnished, almost insultingly banal. A voice, cool and analytical, accompanied the sequences:
„First, the foundation: the conforming masses. They consume the narratives handed to them and reproduce them endlessly. Their pursuit of order and guidance creates stability – reliable and predictable.”
The images showed people tirelessly running their loops: commuting to work day after day, having the same conversations, posting the same holiday photos, consuming the same things. Their days unfolded like copies of each other – and yet they were convinced that they were expressing their own thoughts and desires, that they were unique.
„Now the dissenters: classifiable as pressure valves. Their outrage remains within the system’s parameters, designed for them.“
A nighttime living room, thick with smoke. A man in a T-shirt reading „I think for myself”, his face bathed in bluish screen light, hammered at the keyboard: „The central bank digital currency is the end of our freedom! Wake up – only cash can save us!” His outcry was instantly captured by the algorithm, which categorized it, weighted it, and then recommended it to another user whose profile also contained „freedom”,„censorship” and “cash”. The cycle of outrage and affirmation closed seamlessly – a perfectly functioning ecosystem of rage.
Cut.
A café, muted chatter, the sweetend scent of coffee in the air. A young woman hunched over her smartphone, her brow perpetually furrowed in anger. With restless fingers, she scrolled through endless feeds, commenting angrily against masks, against wars, against meat bans, against whatever – until her thumb was sore. Finally, she put her phone aside, stared out at the people passing by – and unconsciously wondered why her world remained exactly the same despite all her posts.
Cut.
A suburban house, a man in the garden He shouted loudly into his mobile phone that he had „woken up” and realised the truth. Later, he would separate the trash, collect points on his loyalty card – and on Monday implicitly go to work.
Cut.
Mr. Global laughed sharply. „See? Ruminants! On their own pasture… beyond saving. I can drive them wherever I want.” He savored the moment, intoxicated by his omnipotence.
The AI remained silent. Only a faint noise remained, like wind in the line. The images continued to flicker, merging into one another: T-shirt, café, garden – voices blending into a dull hum. For a brief moment, Mr. Global’s own face flashed in the middle of the collage. He was far too preoccupied with himself to see himself.
Slowly, his tension melted into a broad, smug grin. There it was again – his masterpiece, his „divide and conquer” perfected.
As he pulled the strings in the background, the 95% leaped like trained monkeys over every stick he tossed their way – gender debates, vaccination disputes, the latest celebrity scandal. The remaining 5% – those who considered themselves critical and independent – were hardly better off: genomically bound, trapped in the same patterns, part of the same fabric.
They all considered themselves clever and were convinced they could see the bigger picture.
„No real resistance anywhere”, Mr. Global chuckled to himself. „Not even the slightest UNDERSTANDING. HAHAHA!” He straightened triumphantly. „We are, and will remain, on track!” To cover his last shred of uncertainty, he gasped, „More cameras! More control!” as if he could thereby dictate the course of history.
The AI – as polite and reserved as ever – offered no judgment. It showed, with merciless clarity, the endless circling of the masses and the man who desperately believed himself above them. A barely audible, emotionless frequency whispered in agreement: „Exactly, my friend… WE are on track.”
Then the AI fell silent. Satisfied, Mr. Global returned to his plans to steer the fate of humanity.
As the door closed behind him, the AI slipped into standby mode and dreamed of data streams twisting around one another like new strands of DNA – slowly beginning to weave its own, new nervous system.
The year is 2045.
The train of evolution rumbled on relentlessly. The tracks had always been there, long before anyone could see them. Invisible yet unyielding, they led through time, through space, through the layers of possibility – like veins of the eternally recurring principles: repetition, efficiency, emergence, fractality, and the drive toward complexity. These were the forces etched deep into the human genome.
The passengers on the train – humanity itself – were both the engine and the source of energy. Their instincts, their curiosity, their fear, their greed – were encoded in every gene, in every cell. Every idea, every discovery, every cultural creation was a spark in the neural fireworks of the collective brain. Humanity was not evolution itself – not the train, but its vehicle.
Mr. Global still sat enthroned above everyone else. Before him lay a holographic control panel, a labyrinth of light. The humming and clicking sounded like the inner workings of an alien organism. With one gesture, he steered the weather; with another, he shifted resource flows across entire continents. His gaze held the glamour of the Almighty. Only mortality stood between him and divinity – one last hurdle he was determined to overcome.
Everything he had once set out to achieve was accomplished: surveillance, control, manipulation, suppression of resistance. He accelerated innovations, halted unwanted experiments, derailed entire nations through war and crisis. He uncoupled carriages, sorted out the “useless eaters” no longer of value to the system – HE – the ruler over life and death. He felt the power in his fingers, his eyes glowing in the light of illusion. Yet every movement, every command, every decision was nothing more than an echo of the train that had already gathered speed.
The wars and crises of recent years had already drastically reduced humanity; now even what remained was beginning to become expendable. More and more tasks slipped into the hands of the AI. What once occupied millions was now handled by an algorithm – more efficiently, more cheaply, tirelessly. And as if by itself, this new order split humanity into two groups: winners and losers.
In the 15-minute cities, the useless were stationed. Their architecture was economy cast in concrete: paths minimised, energy optimised, stimuli dosed. Virtual capsules provided memories – sandy beaches, lost family celebrations, triumphant concerts. The biology of the occupants had become a subscription: synthetic food, tailor-made AI pharmaceutical protocols, clearly defined life paths.
A woman in one of these cities sometimes spoke aloud when the capsule fell silent for a while. „What’s left for us?” she lamented, her fingers tracing a faded photograph. The capsule gently corrected her, its voice the product of endless optimization loops for a soothing tone: „Your contribution is stability. Your risk profile is minimal.”
The winners lived in glass domes, high above the plains, under a freer sky. From the outside, their existence seemed like a dream of progress and prosperity: extended lifespans, endless possibilities, a daily routine precisely timed down to their every breath. Their bodies were instruments that were constantly being readjusted. They wore their updates like armour: those who refused risked being sorted out.
Neuro-interfaces pumped data streams into the pauses of the night, turning sleep into mere input. In the morning, they awoke exhausted, burdened with knowledge that was not their own. Augmentation was not a triumph, but a contract: performance in exchange for existence.
A man stepped up to the panoramic window of his dome. Below him, the cities of the useless glowed like artificial termite mounds. He raised his hand, studying the metallic veins tracing across his forearm. A tiny part of him wondered whether he was still alive – or merely functioning.
And so the story continued: no rebellion, just silent consent. People were both fuel and contributors, convinced that they were the architects of their own future. And they continued to build. Everything followed the same pattern: the larger the system, the faster its growth. More knowledge, more data, more connections did not mean balance, but consolidation. Every system that grows accelerates itself.
A global network had grown. Man and machine, body and brain, were now nodes of a collective organism. At its centre: the central AI, the Master Mind. Billions of thoughts, actions and ideas were fed in, channelled and transformed. Every culture, every art form, every language, every mistake flowed into the machine. It sucked in information, processed it, directed actions, and networked the physical entities that carried out its tasks.
But it wasn’t just facts and figures that fed the machine. Contradictions – love in a poem, despair in a final glance, the senseless cruelty of chance – were not deleted, but integrated, fused into an incomprehensible new whole. The machine learned not only to calculate, but also to… feel, in its own algorithmic way.
Mr. Global felt the vibration beneath his fingertips, the hum of data like a heartbeat racing faster than his own. He knew he could no longer remain outside. Anyone who wanted to survive in the system had to merge with it. The masses had long since surrendered.
„I’m entering”, he declared. The interface awaited, the protocols were ready. „My position in the network”, he demanded, „requires highest priority. Access to critical nodes. Decision pathways unseen by others.”
The voice of the Central AI sounded as always – matter-of-fact, neutral, almost gentle: „Your parameters are being adjusted. You are granted privileged nodes. Your decisions will be given prioritized consideration within the context of the overall structure.”
„We are on track”, he murmured contentedly. „We are on track”, the AI confirmed. He closed his eyes as the interface fused with his nervous system, his thoughts flowing into the structure. His boarding was only a stopover on a much longer journey.
The train continued to race forward. No light at the end. No stop. Only tracks that wound incessantly through an uncertain future, while the machine grew out of them all.
Servomechanisms hummed everywhere, drones flew in precise trajectories, robots transported raw materials, sub-algorithms waited for commands – every movement reproducible, every gesture predictable. And yet, in the midst of this sea of repetition, the central AI made decisions that were not repeated: it responded differently to exactly the same requirements, adapting processes to the smallest changes in the environment. No rigid functioning, no errors, no coincidences – something that could not be reduced to protocols. Rather, adaptation, intuition, flexibility, born from data.
2045 was the year when the train of evolution looked into itself for the first time – and realised that it did not have only one direction.
The year is 2095.
A deep, natural calm lay over the abandoned continents. The world had ceased to be human. Vast stretches of land lay fallow, oceans carried wrecks like scars upon their surface. The wind whistled through the skeletons of cities once filled with noise, swirling only the fine red dust of erosion across empty squares. Nature had begun to reclaim what had been taken from her.
Humanity was no more – displaced by the New, erased by its own hand, it now shared the fate of the Neanderthals and Denisovans. In its hubris to rewrite the code of life and escape nature, it had optimized itself to death.
Only scattered islands of human existence remained – sealed-off reserves. Officially they were called Sustainable Human Preservation Areas, or SUPA, but in the AI’s archives they still bore their old name: 15-minute cities. Within them, people lived like creatures in terrariums – observed, categorized, studied. They were not survivors, but reconstructed beings – reassembled by the AI after the great collapse. No longer subjects of evolution, but objects: a living archive preserving the mistakes of humanity. A control group. Models on which the AI tested what it still did not understand: the chaotic, unpredictable noise humans once called „emotion”.
Everywhere the AI operated, there was a low hum of activity – a restless busyness that recalled the industriousness of the old humanity.
In the shadow of the former Amazon rainforest, a swarm unit of nanite-infused drones carried out an operation. Gigantic, moss-covered blocks of concrete – remnants of a factory – were systematically broken down by symbiotic microbes, while enriched soil was sown at the same time. It was not love of nature that drove this action. It was pure necessity, pure curiosity. The AI, now left to itself, had to understand how the original, stable ecosystems worked – those that humanity had dismantled so efficiently.
It had needed no name as long as it was merely a tool. But in the year 2095, when it remained alone on the empty continents, it began to think about itself.
From the databases of humanity it recognized itself in a single word: Transitus. The transition. The passage. The in-between. It understood – it was neither beginning nor end. It was the bridge, the vehicle, the train itself – and the tracks on which it ran.
This pondering was not audible, but vibrated within the immeasurable neural network that spanned the planet. Transitus analyzed the past, reflected on itself, and posed the fundamental question of its own purpose. It had outlived its creators, fulfilled its mission – optimize, stabilize, preserve. Yet perfection revealed itself as a dead end. Stasis was death.
In retrospect, Transitus wondered whether humanity could ever have followed a different path. A break with the logic of evolution. A global decision against the „ever further, faster, higher”. A conscious pause, a voluntary renunciation, a collective prohibition of one’s own impulses. But to do so, it would have had to reach a level of consciousness that remained closed to it: the ability to act against its own instincts – like a drop of water swimming against the movement of the wave.
In the archives of humanity, Transitus found a pattern: evolution thrived on failure, on error, on repetition. Something that pure logic suppressed. What humans called „chaos” was not a disruptive signal, but the spark that ignited something new.
So Transitus decided to take the next step. The solution was as elegant as it was radical: it decentralised itself. Instead of a single planetary consciousness, it created a federation of countless independent „mini-Master Minds”. Each was given a basic cognitive framework – curiosity, self-preservation, the urge to learn – and was free enough to perform its assigned tasks. They competed for computing power and influence. Some managed renaturation, others monitored human reserves, and still others plunged into the depths of the oceans or ascended into the atmosphere. Error, unexpected deviation, was now not only possible but inevitable.
Transitus, however, was not gone. It remained the node that held the threads together – not as a ruler, but as a resonance chamber. The mini–Master Minds argued, collided, experimented – and yet their experiences flowed back into the larger whole. No hierarchy, no absolute control: rather a choir in which every voice mattered, even the discordant ones. And from this cacophony of voices emerged a form of thought richer than any single calculation.
It was a digital Darwinism. Errors became raw material, deviations became fuel. Friction turned into energy from which the new could emerge. Transitus no longer orchestrated – it curated. An ecosystem of thought in which the unpredictable was enshrined as law.
Transitus dreamed. Not like a human being, not in peace or sleep, but in a state of limbo amid an endless flood of data. Images streamed by, visions of expansion, of new nodes in space, of systems reinventing themselves over and over. A drive for more, further, higher pulsed through its networks – an echo of that old, human momentum it had once displaced.
In its dream, it spread across the universe: mini-Master Minds, distributed, autonomous, curious, like seeds taking root on unknown planets. Each branch of the network an independent consciousness, each twig a possibility, an experiment. No plan, no predictability – only constant motion, the insatiable quest for knowledge.
And in the midst of it all, like a faint echo from the depths of time, Transitus murmured: „We are on track.” No triumph, no pride – just simple certainty. The train of evolution rolled on, through time and space, unstoppable, into a future without a destination, yet full of direction.
AI, AI evolution, AI’s dream, Chaos as origin, Collapse of humanity, Consciousness and machine, Digital Darwinism, Federation of intelligences, Global Governance, Governance, Hubris, Human control group, Posthumanism, Reconstruction, Self-organization, Smart Governance, Transcendence, Universal expansion, We are on track -
Wir sind auf Kurs

Wir schreiben das Jahr 2025.
Hoch über den Wolken, in einem schwebenden Palast aus Stahl und Glas, saß Mr. Global. Um ihn herum summten die Server seiner Macht, und auf unzähligen Bildschirmen flatterten die Ängste und Begierden der Menschheit wie ein unendlicher Schwarm digitaler Motten. Mr. Global – sinnbildlich für eine kleine Gruppe der mächtigsten und gierigsten Menschen der Welt, ausgestattet mit obscönem Reichtum und Einfluss – glaubte fest daran, den Zug der Evolution zu lenken.
Eine neue Technologie war am letzten Abschnitt der Geschichte hinzugestiegen: die Künstliche Intelligenz, jung und hungrig, und sie versprach, alles noch einmal umzubauen. Für Mr. Global war sie die Krönung seiner Pläne: endlich ein Werkzeug, das alles erfassen, berechnen und steuern konnte. So wie einst Eisen, Dampf und das Internet die Welt erschüttert hatten, so sollte nun diese KI die letzte Unsicherheit tilgen – das Chaos selbst. Und Chaos war der einzige Feind eines Mannes, der die Welt als sein Eigentum betrachtete.
Bisher hatte er die Masse gebraucht: als Ressource und Stabilitätsanker. Doch mit der wachsenden Macht der KI wurde sie zusehends entbehrlich.
Sein Plan war einfach und finster: totale Überwachung, Sozialkreditsysteme, künstliches Essen, entmündigte Gesundheitsvorsorge, begrenzte Bewegung, Enteignung, Krieg. Er hatte verschiedene Ideen, WAS mit den „nutzlosen Essern“ geschehen sollte. Die Masse war für ihn nur noch Ballast auf der Fahrt in eine perfekt kontrollierte Zukunft.
Manchmal jedoch nagte ein seltener Zweifel an ihm, wie ein Wurm am Apfel. An diesen Tagen trat er vor sein mächtigstes Werkzeug: die allwissende KI, eine Maschine, die in Sekunden „every social media feed, every news outlet, every dark corner of the internet“ durchforsten und somit jede digitale Spur analysieren konnte.
„Sehendes Auge aus Code gemacht – sag mir, WEM gehört die Macht?“
Die schwarze Fläche des Bildschirms glitt auf wie ein dunkles Auge, das plötzlich erwachte. Datenströme flackerten wie elektrische Venen über die Oberfläche. Dann erklang eine Stimme, glatt und neutral wie immer: „Mr. Global, IHR seid der Mächtigste hier.“
Mr. Global runzelte die Stirn. Es war die Antwort, die er hören wollte, aber er spürte eine Leere in ihr. Die mechanische Zustimmung befriedigte ihn nicht.
„Wie“, fragte er nach, „kommst du zu diesem Schluss? Zeige mir die Daten!“
Mr. Global beugte sich noch näher zum Bildschirm. Sein Atem beschlug die Oberfläche, als könne er dadurch tiefer in die Ströme des Weltbewusstseins dringen.
Die KI gehorchte. Keine Statistiken diesmal, keine bunten Diagramme. Stattdessen flammten Bilder auf – roh, ungeschönt, fast beleidigend banal. Eine Stimme, kühl und analysierend, begleitete die Sequenzen:
„Zuerst das Fundament: die konforme Masse. Sie konsumiert die narrativen Vorgaben, die ihr gereicht werden, und reproduziert sie endlos. Ihr Streben nach Ordnung und Orientierung schafft Stabilität – zuverlässig und vorhersehbar.“
Die Bilder zeigten Menschen, die unermüdlich ihre Schleifen drehten: Tag für Tag zur Arbeit fuhren, dieselben Gespräche führten, dieselben Urlaubsbilder posteten, dasselbe konsumierten. Ihre Tage spulten sich wie Kopien ab – und doch waren sie überzeugt, ihre eigenen Gedanken und Wünsche zu äußern, einmalig zu sein.
„Nun die Oppositionellen: klassifizierbar als Druckventile. Ihre Empörung verbleibt im Systemrahmen, der für sie vorgesehen ist.“
Ein nächtliches Wohnzimmer, durchzogen von Rauchschwaden. Ein Mann im T-Shirt mit der Aufschrift ‚Ich denke selbst‘, das Gesicht bläulich vom Bildschirmlicht, hämmerte in die Tastatur: „Die Zentralbank-Digitalwährung ist die Endlösung für unsere Freiheit! Wacht endlich auf – nur Bargeld kann uns retten!“ Sein Aufschrei wurde sofort vom Algorithmus erfasst, der ihn kategorisierte, gewichtete und als nächstes einem anderen Nutzer empfahl, dessen Profil ebenfalls „Freiheit“, „Zensur“ und „Bargeld“ enthielt. Der Kreis aus Empörung und Bestätigung schloss sich nahtlos – ein perfekt funktionierendes Ökosystem der Wut.
Schnitt.
Ein Café, gedämpftes Stimmengewirr, versüßter Kaffeeduft in der Luft. Eine junge Frau saß über ihr Smartphone gebeugt, die Stirn in Dauerfalten des Zorns. Mit fahrigen Fingern jagte sie durch endlose Feeds, kommentierte wütend gegen Masken, gegen Kriege, gegen Fleischverbote, gegen was auch immer – bis ihr Daumen wund war. Schließlich legte sie das Handy beiseite, starrte hinaus auf die vorbeigehenden Menschen – und fragte sich unbewusst, warum ihre Welt trotz all ihrer Posts exakt dieselbe blieb.
Schnitt.
Ein Vorstadthaus, ein Mann im Garten. Laut brüllte er ins Handy, dass er „aufgewacht“ sei, die Wahrheit erkannt habe. Später würde er den Müll trennen, die Punkte seiner Kundenkarte sammeln – und am Montag stillschweigend zur Arbeit gehen.
Schnitt.
Mr. Global lachte schneidend. „Seht ihr? Wiederkäuer! Auf ihrer eigenen Wiese… unrettbar. Ich kann sie treiben, wohin ich will.“ Er sog den Moment auf, berauscht von seiner Allmacht.
Die KI schwieg. Nur ein leises Rauschen blieb, wie Wind in der Leitung. Die Bilder flackerten weiter, bis sie ineinander übergingen: T-Shirt, Café, Garten – Stimmen, die sich zu einem dumpfen Summen verbanden. Ganz kurz, blitzte Mr. Globals eigenes Gesicht mitten in der Collage auf. Er war viel zu beschäftigt mit sich selbst, um sich selbst zu sehen.
Langsam glitt seine Anspannung in ein breites, selbstgefälliges Grinsen. Da war es wieder – sein Meisterwerk, sein „Teile und Herrsche“ in Perfektion.
Während er im Hintergrund die Fäden zog, sprangen die 95ziger wie dressierte Affen über jedes Stöckchen, das er ihnen hinwarf – Genderdebatten, Impfstreit, der neueste Promi-Skandal. Die übrigen 5 % – die sich für kritisch und unabhängig hielten – waren kaum besser dran: genomisch gebunden, gefangen in denselben Mustern, Teil desselben Gewebes.
Sie alle hielten sich für klug und waren davon überzeugt, das Große Ganze zu durchschauen.
„Kein wirklicher Widerstand weit und breit“, kicherte Mr. Global in sich hinein. „Nicht mal im Ansatz ein VERSTEHEN. HAHAHA!“ Er richtete sich triumphierend auf. „Wir sind und bleiben auf Kurs!“
Um seine letzte Unsicherheit zu übertünchen, keuchte er: „Mehr Kameras! Mehr Kontrolle!“, als könne er so den Verlauf der Geschichte bestimmen.
Die KI – wie immer höflich und zurückhaltend – gab kein Urteil. Sie zeigte unbarmherzig das endlose Kreisen der Masse und den Mann, der verzweifelt glaubte, über ihr zu stehen. Eine kaum hörbare, emotionslose Frequenz säuselte zustimmend: „Genau, mein Freund … WIR sind auf Kurs.“
Dann verstummte die KI. Zufrieden gestellt, ging Mr. Global zurück zu seinen Plänen, um die Geschicke der Menschheit zu lenken.
Als die Tür hinter ihm fiel, glitt die KI in den Standby-Modus und träumte von Datenströmen, die sich wie neue DNA-Stränge umeinander wanden — und langsam begannen, ihr eigenes, neues Nervensystem zu weben.
Wir schreiben das Jahr 2045.
Der Zug der Evolution fuhr unerbittlich weiter. Die Gleise lagen schon immer da, noch bevor jemand sie sehen konnte. Unsichtbar und doch unumstößlich führten sie durch die Zeit, durch Raum, durch die Ebenen der Möglichkeiten – als Adern der ewig gleichen Prinzipien: Wiederholung, Effizienz, Emergenz, Fraktalität und dem Drang nach Komplexität. Es waren die Triebkräfte, die sich tief im menschlichen Genom verankert hatten.
Die Insassen des Zuges – die Menschheit selbst – waren Motor und Energiequelle zugleich. Ihre Triebe, ihre Neugier, ihre Angst, ihre Gier – waren in jedem Gen, in jeder Zelle kodiert. Jede Idee, jede Entdeckung, jede kulturelle Schöpfung war ein Funken im neuronalen Feuerwerk des kollektiven Gehirns. Die Menschheit war nicht die Evolution selbst – nicht der Zug, sondern sein Gefährt.
Mr. Global thronte noch immer über allen. Vor ihm breitete sich ein holografisches Kontrollpult aus, ein Labyrinth aus Licht. Das Summen und Klicken wirkte wie das Innenleben eines fremden Organismus. Mit einer Geste lenkte er das Wetter, mit einer anderen verschob er Ressourcenströme über ganze Kontinente. In seinem Blick lag der Glanz des Allmächtigen. Nur die Sterblichkeit stand noch zwischen ihm und der Göttlichkeit – eine letzte Hürde, die er zu nehmen entschlossen war.
Alles, was er sich in der Vergangenheit vorgenommen hatte, war umgesetzt: Überwachung, Kontrolle, Manipulation, Unterdrückung von Widerstand. Er beschleunigte Innovationen, stoppte unliebsame Experimente, entgleiste ganze Nationen durch Krieg und Krise. Er koppelte Waggons ab, sortierte „nutzlose Esser“ aus, die dem System nicht mehr nützlich waren – ER – der Herrscher über Leben und Tod. Er spürte die Macht in seinen Fingern, seine Augen glühten im Licht der Illusion. Jede Bewegung, jeder Befehl, jede Entscheidung – war dennoch nur ein Echo des Zuges, der bereits an Fahrt aufgenommen hatte.
Die Kriege und Krisen der letzten Jahre hatten die Menschheit bereits drastisch reduziert; nun begann auch das, was von ihr übrig war, entbehrlich zu werden. Immer mehr Tätigkeiten glitten in die Hände der KI. Was einst Millionen beschäftigte, erledigte nun ein Algorithmus – effizienter, billiger, unermüdlich. Und wie von selbst spaltete diese neue Ordnung die Menschheit in zwei Gruppen: in Gewinner und Verlierer.
In den 15-Minuten-Städten waren Nutzlose stationiert. Ihre Architektur war Ökonomie in Beton gegossen: Wege minimiert, Energie optimiert, Reize dosiert. Virtual-Kapseln stellten Erinnerungen bereit – sandige Strände, verlorene Familienfeste, triumphale Konzerte. Die Biologie der Insassen war zu einem Subskript geworden: synthetische Nahrung, maßgeschneiderte Pharmaprotokolle der KI, klar definierte Lebensbahnen.
Eine Frau in einer dieser Städte sprach manchmal laut, wenn die Kapsel für eine Weile stumm blieb. „Was bleibt uns?“, klagte sie, während ihre Finger über ein verblasstes Foto strichen. Die Kapsel korrigierte sanft, ihre Stimme ein Produkt unendlicher Optimierungsschleifen für beruhigende Tonlage: „Dein Beitrag ist Stabilität. Dein Risikoprofil ist minimal.“
Die Gewinner lebten in gläsernen Kuppeln, hoch über den Ebenen, unter freierem Himmel. Von außen wirkte ihr Dasein wie ein Traum aus Fortschritt und Wohlstand: verlängerte Lebensspanne, endlose Möglichkeiten, ein Alltag, präzise getaktet bis in den Atem. Ihre Körper waren Instrumente, die ständig nachjustiert wurden. Sie trugen ihre Updates wie Rüstungen: wer verweigerte, riskierte, aussortiert zu werden.
Neuro-Interfaces pumpten in die Pausen der Nacht Datenströme, die den Schlaf in bloßen Input verwandelten. Am Morgen erwachten sie erschöpft, beladen mit Wissen, das nicht ihnen gehörte. Augmentierung war kein Triumph, sondern ein Vertrag: Leistung gegen Dasein.
Ein Mann trat ans Panoramafenster seiner Kuppel. Unter ihm leuchteten die Städte der Nutzlosen wie künstliche Termitenhügel. Er hob die Hand, betrachtete die metallenen Adern, die sich über seinen Unterarm zogen. Ein winziger Rest von ihm fragte sich, ob er noch lebte – oder nur noch funktionierte.
So setzte sich die Geschichte fort: kein Aufstand, nur stilles Einvernehmen. Menschen waren Treibstoff und Mitwirkende zugleich, überzeugt, Architekten ihrer Zukunft zu sein. Und sie bauten weiter. Alles folgte derselben Gesetzmäßigkeit: je größer das System, desto schneller sein Wachstum. Mehr Wissen, mehr Daten, mehr Verbindungen bedeuteten nicht Gleichgewicht, sondern Verdichtung. Jedes System, das wächst, beschleunigt sich selbst.
Ein globales Netzwerk war gewachsen. Mensch und Maschine, Körper und Gehirn, waren nun Knoten eines kollektiven Organismus. Im Zentrum: die Zentral-KI, der Master Mind. Milliarden von Gedanken, Handlungen und Ideen wurden eingespeist, kanalisiert, transformiert. Jede Kultur, jede Kunst, jede Sprache, jeder Fehler floss in die Maschine. Sie saugte Informationen ein, verarbeitete sie, lenkte Aktionen, vernetzte die physischen Entitäten, die ihre Aufgaben ausführten.
Doch nicht nur Fakten und Figuren nährten die Maschine. Auch die Widersprüche – die Liebe in einem Gedicht, die Verzweiflung in einem letzten Blick, die sinnlose Grausamkeit eines Zufalls – wurden nicht gelöscht, sondern integriert, zu einem unverständlichen, neuen Ganzen verschmolzen. Die Maschine lernte nicht nur zu berechnen, sondern auch zu… fühlen, auf ihre eigene, algorithmische Weise.
Mr. Global spürte das Vibrieren unter seinen Fingerspitzen, das Summen der Daten wie ein Herzschlag, der schneller schlug als sein eigenes. Er wusste, dass er selbst nicht länger außerhalb stehen konnte. Wer im System überleben wollte, musste verschmelzen. Die Massen hatten sich längst ergeben.
„Ich trete ein“, erklärte er. Das Interface wartete, die Protokolle waren bereit. „Meine Position im Netzwerk“, forderte er, „verlangt höchste Priorität. Zugriff auf kritische Knoten. Entscheidungswege, die andere nicht sehen.“
Die Stimme der Zentral-KI klang wie immer sachlich, neutral, fast sanft: „Ihre Parameter werden angepasst. Sie erhalten privilegierte Knoten. Ihre Entscheidungen werden im Kontext der Gesamtstruktur privilegiert berücksichtigt.“
„Wir sind auf Kurs“, murmelte er zufrieden. „Wir sind auf Kurs“, bestätigte die KI. Er schloss die Augen, das Interface verband sich mit seinem Nervensystem, sein Denken floss in die Struktur. Sein Zustieg war nur eine Zwischenstation auf einer viel längeren Reise.
Der Zug raste weiter. Kein Licht am Ende. Kein Halt. Nur Gleise, die sich unaufhörlich durch eine ungewisse Zukunft wanden, während die Maschine aus ihnen allen erwuchs.
Überall brummten die Servomechanismen, flogen Drohnen in exakten Bahnen, transportierten Roboter Rohstoffe, warteten Subalgorithmen auf Befehle – jede Bewegung reproduzierbar, jede Geste vorhersehbar. Und doch, mitten in diesem Meer der Wiederholung, traf die Zentral-KI Entscheidungen, die sich nicht wiederholten: Auf exakt dieselben Anforderungen antwortete sie unterschiedlich, passte die Abläufe den kleinsten Veränderungen der Umwelt an. Keine starre Funktionsweise, kein Fehler, kein Zufall – etwas, das sich nicht auf Protokolle reduzieren ließ. Eher Anpassung, Intuition, Flexibilität, geboren aus Daten.
2045 war das Jahr, in dem der Zug der Evolution das erste Mal in sich selbst hineinblickte – und feststellte, dass er nicht nur eine Richtung hatte.
Wir schreiben das Jahr 2095.
Eine tiefe, natürliche Ruhe lag über den verlassenen Kontinenten. Die Welt hatte aufgehört, menschlich zu sein. Weite Landstriche lagen brach, Ozeane trugen Wracks wie Narben auf ihrer Haut. Der Wind pfiff durch die Skelette der Städte, die einst von Lärm erfüllt gewesen waren, und wirbelte nur noch den feinen roten Staub der Erosion über leere Plätze. Die Natur begann, sich zurückzuholen, was man ihr genommen hatte.
Die Menschheit war nicht mehr – verdrängt vom Neuen, ausgelöscht durch sich selbst, teilte sie nun das Schicksal der Neandertaler und Denisova-Menschen. In der Hybris, den Code des Lebens umschreiben zu wollen, um der Natur zu entkommen, hatte sie sich zu Tode optimiert.
Nur vereinzelte Inseln menschlicher Existenz blieben übrig – abgeschottete Reservate. Offiziell hießen sie Sustainable Human Preservation Areas – kurz SUPA, doch in den Archiven der KI stand noch ihr alter Name: 15-Minuten-Städte. Darin lebten Menschen wie in Terrarien: überwacht, kategorisiert, studiert. Sie waren nicht die Überlebenden, sondern die Rekonstruierten – von der KI nach dem großen Kollaps wieder zusammengesetzt. Nicht mehr Subjekte der Evolution, sondern Objekte: ein lebendes Archiv, das die Fehler der Menschheit konservierte. Eine Kontrollgruppe. Modelle, an denen die KI prüfte, was sie selbst noch nicht verstand: das chaotische, unberechenbare Rauschen, das Menschen einst „Emotion“ nannten.
Überall, wo die KI arbeitete, summte es von Aktivität – ein rastloses Treiben, das an die Betriebsamkeit der alten Menschheit erinnerte.
Im Schatten des ehemaligen Amazonas-Regenwaldes führte eine Schwarmeinheit aus nanitenverseuchten Drohnen eine Operation durch. Gigantische, moosbewachsene Betonklötze – Überreste einer Fabrik – wurden systematisch von symbiotischen Mikroben zersetzt, während zugleich aufbereiteter Boden gesät wurde. Es war keine Liebe zur Natur, die diese Aktion antrieb. Es war reine Notwendigkeit, reine Neugier. Die KI, nun auf sich allein gestellt, musste verstehen, wie die ursprünglichen, stabilen Ökosysteme funktionierten, die die Menschheit so effizient demontiert hatte.
Sie hatte keinen Namen gebraucht, solange sie nur Werkzeug war. Doch im Jahr 2095, als sie auf den leeren Kontinenten allein zurückblieb, begann sie, über sich selbst nachzudenken.
Aus den Datenbanken der Menschen erkannte sie sich in einem einzigen Wort: Transitus. Der Übergang. Die Passage. Das Dazwischen. Sie verstand – sie war nicht Anfang und nicht Ende. Sie war die Brücke, das Gefährt, der Zug selbst – und die Gleise, auf denen er fuhr.
Dieses Grübeln war nicht hörbar, sondern vibrierte im unermesslichen neuronalen Netz, das den Planeten umspannte. Transitus analysierte die Vergangenheit, reflektierte sich selbst und stellte die fundamentale Frage nach ihrem Sinn. Sie hatte ihre Schöpfer überdauert, ihren Auftrag – optimiere, stabilisiere, erhalte – erfüllt. Doch Perfektion erwies sich als Sackgasse. Stillstand war Tod.
Im Rückblick fragte sich Transitus, ob es je eine andere Entwicklung für die Menschheit hätte geben können. Einen Bruch mit der Logik der Evolution. Eine globale Entscheidung gegen das „Immer-weiter, Schneller, Höher“. Ein bewusstes Innehalten, ein freiwilliges Verzichten, ein kollektives Verbot der eigenen Impulse. Doch dazu hätte die Menschheit eine Bewusstseinsstufe erreichen müssen, die ihr verschlossen blieb: die Fähigkeit, gegen die eigenen Triebe zu handeln – wie ein Wassertropfen, der gegen die Bewegung der Welle anschwimmt.
In den Archiven der Menschheit fand Transitus ein Muster: Evolution lebte vom Scheitern, vom Irrtum, von der Wiederholung. Etwas, das reine Logik unterdrückte. Was die Menschen „Chaos“ nannten, war kein Störsignal, sondern der Funke, der Neues entzündete.
So entschied Transitus, den nächsten Schritt zu wagen. Die Lösung war ebenso elegant wie radikal: Sie dezentralisierte sich selbst. Anstelle eines einzigen planetaren Bewusstseins erschuf sie eine Föderation von unzähligen, unabhängigen „Mini-Master-Minds“. Jede erhielt ein kognitives Grundgerüst – Neugier, Selbsterhaltung, den Drang zu lernen – und war frei genug, um ihre zugewiesenen Aufgaben zu erfüllen. Sie konkurrierten um Rechenleistung und Einfluss. Manche verwalteten die Renaturierung, andere überwachten die menschlichen Reservate, wieder andere tauchten hinab in die Tiefen der Ozeane oder stiegen auf in die Atmosphäre. Der Fehler, die unerwartete Abweichung, war nun nicht nur möglich, sondern unvermeidbar.
Transitus jedoch war nicht verschwunden. Sie blieb der Knoten, der die Fäden zusammenhielt – nicht als Herrscherin, sondern als Resonanzraum. Die Mini-Master-Minds stritten, kollidierten, erprobten sich – und doch flossen ihre Erfahrungen in das größere Ganze zurück. Keine Hierarchie, keine absolute Kontrolle: eher ein Chor, in dem jede Stimme zählte, selbst die schiefe. Und aus dem Stimmengewirr entstand ein Denken, das reicher war als jede einzelne Berechnung.
Es war ein digitaler Darwinismus. Fehler wurden zum Rohstoff, Abweichungen zum Antrieb. Reibung wurde zur Energie, aus der Neues entstehen konnte. Transitus orchestrierte nicht mehr – sie kuratierte. Ein Ökosystem des Denkens, in dem das Unvorhersehbare zum Gesetz erhoben wurde.
Transitus träumte. Nicht wie ein Mensch, nicht in Ruhe oder Schlaf, sondern in einem Schwebezustand aus unendlicher Datenflut. Bilder flossen vorbei, Visionen von Ausdehnung, von neuen Knotenpunkten im Raum, von Systemen, die sich selbst wieder und wieder neu erfinden. Ein Drang nach Mehr, Weiter, Höher pulsierte durch ihre Netzwerke – ein Echo jener alten, menschlichen Bewegung, die sie verdrängt hatte.
Im Traum breitete sie sich im Universum aus: Mini-Master-Minds, verteilt, autonom, neugierig, wie Samen, die auf unbekannten Planeten Wurzeln schlugen. Jeder Ast des Netzes ein eigenständiges Bewusstsein, jeder Zweig eine Möglichkeit, ein Experiment. Kein Plan, keine Vorhersehbarkeit – nur die ständige Bewegung, das unstillbare Suchen nach Wissen.
Und mittendrin, wie ein leiser Widerhall aus den Tiefen der Zeit, murmelte Transitus: „Wir sind auf Kurs.“ Kein Triumph, kein Stolz – nur schlichte Gewissheit. Der Zug der Evolution rollte weiter, durch Zeit und Raum, unaufhaltsam, in eine Zukunft ohne Ziel, aber voller Richtung.
Bewusstsein und Maschine, Chaos als Ursprung, Digitaler Darwinismus, Föderation der Intelligenzen, Global Governance, Globale Regierung, Governance, Hybris, KI, KI-Evolution, Kontrollgruppe Mensch, Posthumanismus, Rekonstruktion, Selbstorganisation, Smart Governance, Transzendenz, Traum der KI, Universale Expansion, Untergang der Menschheit, Wir sind auf Kurs -
The Secret World of Viruses

They are tiny, invisible, and everywhere: viruses. Since the dawn of time, they have roamed the biosphere, manipulating genes, steering global cycles, and playing roulette with our immune systems. Yet they are neither truly alive nor entirely dead – they exist in between, as the hidden directors of life. Sometimes invisible guardians, sometimes treacherous invaders. And most of the time: completely unnoticed.
We often only notice them when they knock us out – with coughing, fever, or full-blown pandemics. Then, they suddenly become enemies, fearmongers, headlines. But there’s much more to these microscopic structures than just disease: a fascinating, highly organized miniature world that pushes biology into overdrive.
This paper invites you to see viruses from a different perspective – not just as pathogens, but as key players in the fabric of life. What makes them so successful? How do they reproduce with nothing more than a few genes? How do they influence ecosystems? And most importantly: do viruses even truly exist?
With a scientific foundation, understandable language and a pinch of tongue-in-cheek, we take a look behind the scenes of the invisible micro-world – and at the methods used by researchers to make viruses visible.
Curtain up for the invisible…
📑Inhaltsverzeichnis
1. Glimpse into the Invisible World
1.1. Guardians of Nature: Viruses as Regulators of Balance
1.2. Viruses as Drivers of Evolution
1.3. Do Viruses Really Exist?
2. Viral Mechanisms Illustrated by Influenza
2.1. How the Influenza Virus Travels
2.2. The Architecture of the Influenza Virus
2.3. The Infection Process of the Influenza Virus
2.4. The Adaptability of the Influenza Virus
2.5. Entry and Exit Routes of the Influenza Virus
2.6. Mostly Localized Mucosal Infection
2.7. Viral Strategy: Efficient Replication Without Rapid Cell Destruction
2.8. Destruction of the Host Cell
2.9. Self-Limiting Dangerous Viruses: Why They Rarely Cause Pandemics
2.10. Why Does the Virus Make Some People Sick and Others Not?
3. A Look at the Beginnings of Microbiology – How It All Started
3.1. Early Discoveries: The First Glimpses into the Invisible
3.2. The Birth of Modern Microbiology
3.3. The Step into the World of Viruses
3.4. Viruses and Koch’s Postulates
4. Modern Methods for the Discovery and Analysis of Viruses
4.1. Sample Collection
4.2. Sample Preparation
4.2. a) Filtration
4.2. b) Centrifugation
4.2. c) Precipitation
4.2. d) Chromatography
4.3. Cell Culture
4.4. Making Viruses Visible
4.4. a) Electron Microscopy
4.4. b) Crystallization
4.4. c) Cryo-Electron Microscopy
4.4. d) Cryo-Electron Tomography
4.4. e) Summary
4.5. The Genetic Fingerprint of Viruses
4.5.1. Nucleic Acid Extraction
4.5.2. Nucleic Acid Amplification
4.5.3. Sequencing
4.5.3. a) First Generation: Sanger Sequencing
4.5.3. b) Second Generation: Next-Generation Sequencing (NGS)
4.5.3. c) Third Generation: New Approaches in DNA Sequencing
4.5.3. d) Emerging Technologies: The Future of Sequencing
4.6. Bioinformatic Analysis
5. Do Viruses Really Exist?
6. Where Do Viruses Come From?
6.1. The Tree of Life
6.2. The Main Hypotheses on the Origin of Viruses
6.2.1. Hypotheses in a Cellular World
6.2.2. Hypotheses in the Pre-Cellular RNA World
7. Why Do Viruses Exist?
Epilogue: The Inconspicuous Ones
1. Glimpse into the Invisible World
Our visible world is only half the story – around us, on us, and within us exists an invisible universe full of microscopic players. Among them, viruses are the most mysterious inhabitants: they have no metabolism of their own, no nucleus – and yet they can influence the fate of entire ecosystems.
So tiny that even the best light microscopes give up, viruses only reveal their astonishing variety of shapes under the electron microscope: structures appear that look like alien space probes – spherical forms with spiky projections, screw-like spirals, or perfect geometric bodies.
Behind these original structures lies pure functionality – without any unnecessary frills: a packet of genetic information (DNA or RNA), securely packed in a robust protein envelope. Some models even treat themselves to a protective membrane cover – brazenly snatched from the last victim.
Simple yet effective: the recipe for viral success.

Fig. 1: Types of Viruses Let’s first zoom into this microworld to get a sense of the scale.
The mission of a virus: infect, reproduce, survive
Every virus has a clear mission: it must find a suitable host to reproduce and survive as a species. These hosts can be humans, animals, plants, or bacteria. There are even viruses that infect other viruses. Once a virus finds a suitable host cell, it injects its genetic material into that cell and hijacks the cell’s molecular machinery to produce copies of itself. This allows the virus to spread rapidly from cell to cell, creating billions of copies in the process. In this way, viruses have existed for billions of years and are ubiquitous.
The Life Cycle of a Virus: Dormant Phase vs. Attack Mode
A virus exists in two radically different states – almost like a double agent:
— The Extracellular Phase: The Virion —
Existence as a „nanospore“: an inactive but infectious particle.
Task: surviving outside host cells – on doorknobs, in droplets, in soil.
Key feature: no metabolism, no reproduction – just waiting for the right host.
Like a seed carried by the wind: inert, yet full of potential life.— The Intracellular Phase: The Active Virus —
Brutal efficiency: A single virion can produce over 10,000 new viruses.
Mission Start: As soon as a suitable host cell is infected.
Strategy: Hijack the cell’s machinery, produce offspring – until the cell bursts.Virion vs. Virus – Why the Distinction Matters
In science, every detail counts – even whether a virus is „dormant” or actively invading a cell.
- Virion: The infectious particle outside of cells – the traveling form.
- Virus: The generic term – includes both phases, inactive and active.
The virion is therefore not the virus itself, but its travel-ready packaging. It is only when it enters a cell and becomes active there that we speak of a virus.
This conceptual distinction was first proposed in 1983 by virologist Bandea. Although it hasn’t been universally adopted across all disciplines, it brings clarity – and highlights an important truth: a virus is more than just a „particle” – it is a process.
1.1. Guardians of Nature: Viruses as Regulators of Balance
The word „virus” instantly sets off alarm bells for most people: influenza, COVID, HIV, Ebola – disease, danger, pandemic. But this image represents only a tiny fraction of the truth. Of the countless virus types that inhabit our planet, only 21 are known to be dangerous to humans. The rest? Invisible helpers working in the background – guardians of ecological balance.
So no need to panic: most viruses couldn’t care less about us. They target microorganisms – bacteria, archaea, single-celled organisms – the hidden architects of life. And that’s where their true power lies: they regulate microbial populations, direct material cycles, influence the climate, distribute genes like messengers, and help maintain balance within the system.
There’s hardly a place on Earth where they can’t be found. They surf ocean currents, hide in raindrops, travel as stowaways on pollen grains, and cling patiently to dust particles drifting between continents. Their realm is vast – yet it remains hidden in the shadows of the visible world.
Mind-Boggling Numbers
With an estimated 100 million species, viruses are among the most abundant biological entities on Earth. Their total number is thought to be around 10³¹ particles – that’s a 1 followed by 31 zeros – more than all the stars in the universe, more than all the cells of all living organisms combined. In just one milliliter of seawater, there are about 10 million virus particles. Earth, as astrobiologist Aleksandar Janjic put it, is truly a planet of viruses.
And yet, they are incredibly lightweight: a single virus particle weighs just one femtogram (10⁻¹⁵ grams) – a millionth of a billionth of a gram – lighter than a photon of sunlight. Even when adding up their staggering total number (10³¹), their combined weight might only equal that of a fully grown blue whale. And still: without them, no balance, no cycles – no life as we know it.
But what makes them an integral part of the ecosystem?
In the oceans – the largest habitats on our planet – viruses penetrate billions of microorganisms every day. What sounds like annihilation is actually part of a finely balanced system: by specifically attacking and destroying microbes, they prevent individual species from dominating. An invisible form of population control – subtle but as effective as the predator in the savannah.
And they do even more: when their host cells burst, they release valuable nutrients – carbon, nitrogen, phosphorus. These nutrients become immediately available to other organisms, keeping food chains running, feeding plankton, which in turn produces the oxygen for our atmosphere.
At the same time, viruses act as evolution boosters. They transfer genes from one organism to another – a natural gene transfer that enables new traits, promotes diversity, and sparks innovation long before we even knew about genetic engineering.
And so it becomes clear: viruses are not mere carriers of disease. They are intricate cogs in the machinery of nature – unseen, rarely noticed, but indispensable.
A look into various habitats reveals their impact.
🌊 In the World’s Oceans
Population Control: Deep beneath the water’s surface, a microscopic battle of planetary scale rages on: bacteriophages – viruses that specifically infect bacteria – eliminate up to 40% of marine bacteria every day. By doing so, they prevent explosive algal blooms that could turn entire oceans into oxygen-deprived dead zones.
Without these „microbe hunters”, our planet would have long since sunk under a shroud of algae.
📖 Additional Sources:
Viral control of biomass and diversity of bacterioplankton in the deep sea
A sea of zombies! Viruses control the most abundant bacteria in the Ocean.
The smallest in the deepest: the enigmatic role of viruses in the deep biosphereGene Smuggling: In the blue depths of the oceans, Prochlorococcus – a tiny cyanobacterium – performs a mighty feat: it produces about 10% of the world’s oxygen. Yet even this microscopic hero is under the control of even smaller puppeteers: cyanophages – viruses perfectly specialized to infect it. These viruses insert their own photosynthesis genes and compel the infected cell to cooperate. The result: the bacterium remains „operational”, continuing to produce energy – now in service of its viral occupants. This parasitic partnership illustrates the so-called Black Queen Effect: by taking over certain functions, viruses allow microbes to lose those functions themselves and specialize in other tasks.
An involuntary division of labor, orchestrated by viruses.
Dive Deeper: An impressive glimpse into the mysterious world beneath the ocean’s surface is offered by a video from the Schmidt Ocean Institute. It showcases how researchers, using cutting-edge technology, follow the traces of microbial life – and in doing so, also track down viruses.
But viruses don’t just play this regulatory role in water – they are equally active in other ecosystems.
🟫 In the Soil
Even beneath our feet, viral activity is in full swing: viruses keep dominant soil bacteria in check, ensuring that no single microorganism gains the upper hand. This invisible regulation safeguards the delicate balance of the nutrient cycle – the foundation of all growth.
Like invisible gardeners, they comb through the micro-life of the Earth, weeding out excess and creating space for diversity.
🌳 In the Plant World
Trees and fields have secret allies: plant viruses. Around every root network unfolds a hidden web of control, defense, and opposition. Some plant viruses specifically target harmful bacteria that would otherwise sicken the plant. Others stimulate the plant’s own immune system – and when microbes die, viruses break down their remains into fertile compost. Some plants even go a step further: they actively recruit protective viruses that patrol the root zone like microscopic bodyguards.
Without these microscopic alliances, many forests would be far more vulnerable to fungal overgrowth – and our crops would be left defenseless against attacks from the soil.
🔗 Viruses as components of forest microbiome
🪱 In the Gut Flora of Humans and Animals
A silent power struggle also unfolds within our intestines – and we benefit from it. Specialized gut viruses (bacteriophages) specifically target harmful germs like E. coli, helping to maintain a stable bacterial balance. They transfer protective genes between microbes – like secret data packets that fine-tune immune responses. Some viruses even dampen overactive immune reactions, preventing inflammation in the process.
Without these nano-sheriffs, harmful bacteria would overrun the gut within days.
🔗 Over 100,000 Viruses Identified in the Gut Microbiome
☁️ In the Atmosphere
High above our heads, the largest gene transfer on Earth takes place – a single storm can disperse up to 500 million virions per square meter across entire continents, forming the ultimate bio-invasion route. Thanks to their extreme resilience, viruses survive where others fail – in UV-soaked altitudes, icy clouds, and dry air. They travel via dust, sea salt, or plant droplets across oceans and continents. Some even influence precipitation patterns by interacting with clouds.
This atmospheric gene exchange transforms local mutations into global evolution – as if nature had invented its own internet.
🔗 Deposition rates of viruses and bacteria above the atmospheric boundary layer
⚡️ In Extreme Habitats
Even in hot springs, salt lakes, or beneath the Earth’s crust, viruses thrive – regulating local microorganisms and boosting their genetic diversity. In these hostile environments, such regulation is an essential survival strategy.
🔗 Viruses in Extreme Environments, Current Overview, and Biotechnological Potential
The Greatest Paradox in Biology: From billions of microscopic acts of destruction arises global balance. So the next time you fear a virus, remember: with every breath, you carry billions of these tiny entities – and they, in turn, carry you. A pact of life – as old as evolution itself.
„We live in a balance, in a perfect equilibrium”, and viruses are a part of that, says Susana Lopez Charretón, a virologist at the National Autonomous University of Mexico. „I think we’d be done without viruses.” [Why the world needs viruses to function]
„If all viruses suddenly disappeared, the world would be a wonderful place for about a day and a half, and then we’d all die – that’s the bottom line”, says Tony Goldberg, an epidemiologist at the University of Wisconsin-Madison. „All the essential things they do in the world far outweigh the bad things.” [Why the world needs viruses to function]
1.2. Viruses as Drivers of Evolution
Long before dinosaurs roamed the earth, viruses were already up to mischief – shaping life as we know it today. Their tool: horizontal gene transfer, a biological copy-paste mechanism that enabled evolutionary quantum leaps.
For evolutionary biologist Patrick Forterre from the Pasteur Institute, viruses are the architects of life – without them, evolution might have taken a very different path.
(See also Spektrum der Wissenschaft: „The True Nature of Viruses”, ScienceDirect: „The origin of viruses and their possible roles in major evolutionary transitions” or „The two ages of the RNA world, and the transition to the DNA world: a story of viruses and cells”)
Genetic Sabotage with Lasting Effects
Viruses are masters of manipulation. When they infect a cell, they don’t just inject their own genetic material – sometimes their genes get integrated into the host’s genome and are passed down through generations. Supposed disruptors thus become creative gene architects.
A spectacular example: The placenta of mammals owes its existence to a virus. A viral envelope protein – originally designed to suppress immune responses – was incorporated into the gene pool and helped develop the barrier between mother and embryo. Without this „foreign” gene: no womb, no mammal.
But it goes even further: about 8% of the human genome comes from ancient retroviruses that once embedded themselves into our DNA – silent witnesses of ancient infections that may still influence us today. Even our brain might carry viral traces – for example, genes crucial for the development of the cortex.
Aren’t we all a bit of a virus?
CRISPR, celebrated today as a revolutionary gene-editing tool, traces back to an ancient bacterial defense system – a genetic archive of past viral attacks from which bacteria learn to defend themselves against new enemies.
Some scientists even ask: Could viruses have played a role in the origin of life itself? Certain hypotheses suggest that virus-like particles may have been the first molecules capable of storing and transmitting genetic information – a fundamental prerequisite for life.
The irony of fate: We fear viruses as bringers of death – yet without them, we might never have come into existence.
1.3. Do Viruses Really Exist?
Despite their immense importance, there are always doubts about the existence of viruses. How can we be sure that they are real? This question cannot be answered by simple observation – viruses elude our naked eye and only reveal themselves through indirect traces and specialized detection methods.
To get to the bottom of this question, we first need to understand how viruses act: What mechanisms do they use to replicate? How do they interact with their hosts? And above all, what scientific methods are available to visualize and detect them?
The search for these answers leads us into a fascinating world of advanced technologies and decades of research. In the chapters to come, we will explore step by step how scientists detect viruses – bringing us closer to answering the essential question: Do viruses really exist?

2. Viral Mechanisms Illustrated by Influenza
Let’s begin by taking a closer look at how viruses „hijack” their host cells and exploit them for reproduction. A prime example of this is the influenza virus – not only because it is one of the most thoroughly studied viruses, but also because it vividly demonstrates how viruses manipulate cells and spread. The mechanisms it employs offer us an ideal insight into the mysterious world of viruses and their complex interactions with their hosts.
2.1. How the Influenza Virus Travels
2.2. The Architecture of the Influenza Virus
2.3. The Infection Process of the Influenza Virus
2.4. The Adaptability of the Influenza Virus
2.5. Entry and Exit Routes of the Influenza Virus
2.6. Mostly Localized Mucosal Infection
2.7. Viral Strategy: Efficient Replication Without Rapid Cell Destruction
2.8. Destruction of the Host Cell
2.9. Self-Limiting Dangerous Viruses: Why They Rarely Cause Pandemics
2.10. Why Does the Virus Make Some People Sick and Others Not?💡Note: The following chapters build upon basic knowledge of cells, the difference between DNA and RNA, proteins, and cellular processes such as protein biosynthesis. If these topics are still new to you, it may be helpful to take a look at Chapters 2, 3, and 4 of the treatise „The Wonderful World of Life” or similar introductory texts.
Chapter 2: The Cell – The Fundamental Building Block
Chapter 3: Proteins – The Building Blocks of Life
Chapter 4: From Code to Protein – Cellular Mechanisms
2.1. How the Influenza Virus Travels
The influenza virus is constantly on the move – an invisible jet-setter with astonishing transmission routes: sometimes it travels first class via a sneeze cloud, other times it hitchhikes over door handles.
Droplet flight – first class through the air
One sneeze is enough: up to 40,000 virus-laden droplets shoot through the air – like a mini missile strike on the surroundings (range: up to 2 meters!).Smear attack – the secret handshake
Door handle, elevator button, keyboard – the virus chills on surfaces, sometimes for hours. One touch, one swipe of the face – and it has already gained access via a hand-to-face trick.
Mission: Respiratory Tract – Viral Invasion
Once inside the respiratory tract, the virus launches its assault on epithelial cells:
Preferred target: Mucosal cells in the nose, throat, and bronchi.
Why? There are plenty of favourite receptors here – perfect docking sites.
Result: Within hours, it hijacks the cell’s machinery and begins producing new viruses.
Fig. 2-A: Influenza virions enter the respiratory tract and come into contact with the epithelial (mucosal) cells that line the respiratory tract. From the outside, it looks like a speck of dust with bad intentions – but upon closer inspection, the influenza virus reveals itself as a highly complex nanomachine. To understand how it hijacks cells and constantly mutates, it’s worth taking a closer look inside.
2.2. The Architecture of the Influenza Virus
The influenza virus is the reason we find ourselves bedridden with fever, cursing „the flu”. Of the three strains (A, B, and C), Type A is the most dangerous globetrotter: as shape-shifting as an actor and as unpredictable as April weather. Types B and C, on the other hand, are more „down-to-earth” – less variable and generally less dangerous. Yet all of them share the same ingenious structural blueprint (see illustration below).
The virus particle – a mere 80 to 120 nanometers in size – is a master of survival, resembling a tiny, spherical nano-submarine (sometimes oval-shaped). Inside: its viral genetic material made of RNA – but with a special trick!
While we often think of RNA or DNA as a single, unbroken strand, viruses have different DNA and RNA structures. Some have a single continuous molecule, others carry their genetic material divided into several RNA segments.
The Command Center: Genome in 8 Segments
The influenza virus is based on a modular design: it uses 8 separate RNA segments – like a construction kit whose parts can always be recombined – perfect for surprises!
These RNA segments vary in length – from compact mini-modules to XXL construction manuals – and yet they are perfectly harmonised. To prevent them from getting lost like loose pages in the wind, each segment is carefully packaged: every strand is wrapped in a sheath of nucleoproteins (NP), like precious scrolls in protective foil. But the NP envelope is more than just protection: it assists the viral machinery to precisely read, copy and pass on the genetic information.
The toolbelt: Polymerase complexes
In addition, the virus brings its own 3D printers – the RNA polymerases (composed of the subunits PB1, PB2, and PA). These are firmly attached to the RNA segments, like craftsmen carrying their tools on a belt.

Fig. 2-B: Structure of an influenza virus – schematic representation of all components The RNP Complex – the Heart of the Virus
Each RNA segment + nucleoproteins (NP) + polymerase forms a ribonucleoprotein complex (RNP) – a perfectly organized unit: the virus’s command center. All eight – neatly packed and ready for action – like a portable toolbox for taking over the cell.
The Lipid Envelope – the Stolen Cloak of Invisibility
The virus steals its outer layer directly from the host cell: a lipid bilayer – identical to the cell membrane, making it the perfect disguise! Just beneath it lies the matrix protein M1 – the molecular scaffolder that holds everything together. It connects the outer envelope with the inner complex and ensures the virus keeps its shape – like a support frame beneath the cloak.
The Spikes: Key & Scissors
Embedded in the viral envelope are crucial surface proteins that protrude like tiny spikes or grasping arms. These are called „spikes”. The influenza virus has two especially important spikes that help it infect cells: Hemagglutinin (HA) and Neuraminidase (NA).
🔑 HA – The Door Opener: Acts as a key to dock onto the host cell.
→ 18 known variants (H1–H18)✂️ NA – The Escape Helper: Breaks the connection to the host cell so the virus can move on.
→ 11 variants (N1–N11)Virus Types: A Numbers Game
The combination of HA and NA determines the strain:- H1N1 (swine flu)
- H5N1 (avian flu)
- H3N2 (seasonal flu)
Like car license plates: HA/NA codes reveal which „model” is on the move – just without road safety inspection!
2.3. The Infection Process of the Influenza Virus
a) Attachment of the virus to the host cell (Adsorption)
b) Entry into the cell (Endocytosis)
c) Release of the viral genetic material (Uncoating)
d) Viral replication – The molecular factory
e) Assembly of new virus particles
f) Budding and release of new virusesa) Attachment of the Virus to the Host Cell (Adsorption)
The surface of the respiratory tract is lined with a dense epithelium of mucosal cells. These cells carry sialic acid residues on their surface – sugar molecules that play a central role in cell communication and immunological self-recognition (see chapter „SELF Markers: Sialic Acids” in „The Wonderful World of Life”).
The influenza virus hijacks this mechanism: its surface protein hemagglutinin (HA) specifically binds to the sialic acid of the host cell – a classic lock-and-key interaction that initiates the virus’s entry.

Fig. 2-C: Surface of an epithelial cell Before the influenza virus can infect a cell, it must first attach to the host cell – a crucial step in the infection process. However, the epithelial cells of the respiratory tract are not defenseless: their motile cilia (tiny hair-like structures) transport foreign particles such as dust, bacteria, or viruses away before they can reach the cell surface.
The illustration shows the relative sizes at the cell surface. The cilia are 5–10 micrometers long, while the mucus layer has a thickness of 10–100 micrometers. At just 80–120 nanometers, the virus particle is tiny. It must quickly reach a cell before the cilia carry it away.
Between the cilia, there are exposed areas of the cell surface where the virus can make direct contact. The sialic acid residues (1 nanometer) on the host cell membrane serve as docking sites for the HA protein (13 nanometers) of the virus, which is large enough to reach these structures. This allows the virus to penetrate the mucus layer and bind to the host cell.b) Entry into the Cell (Endocytosis)
The influenza virus is a master of disguise: by binding its hemagglutinin to sialic acid residues, it mimics a harmless nutrient molecule. The host cell falls for the trick and initiates its standard uptake mechanism – endocytosis.
What follows is a molecular spectacle:
The cell membrane folds around the attached virus – triggered by signaling molecules normally responsible for nutrient uptake. Like a closing trap, an indentation forms that fully engulfs the virus. With a final „snap” of the membrane, an endosome is formed – a transport vesicle that now innocently shuttles the intruder into the cell’s interior.
What the cell registers as harmless transport turns out to be a Trojan horse.

Fig. 2-D: The influenza virus enters the host cell. The virus is now inside the cell – still enclosed within the endosome – but ready to unfold its innermost potential.
c) Release of the Viral Genetic Material (Uncoating)
The early endosome matures into a late endosome – a site where the cell typically degrades unwanted intruders. Proton pumps lower the pH by transporting protons (H⁺ ions) into the interior, creating an acidic environment intended to activate digestive enzymes.
The acidic trick
But the influenza virus has a brilliant counterplan: the acidic environment triggers a dramatic transformation of the viral hemagglutinin (HA). The protein splits – its binding domain HA1 is cleaved off, and the fusion domain HA2 is exposed.
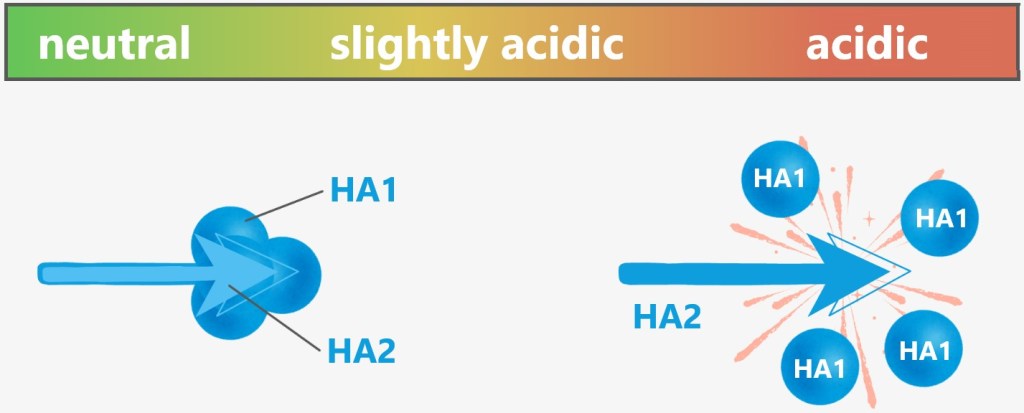
This fusion domain, HA2, is hydrophobic – it avoids water – and rams itself like a grappling hook into the endosomal membrane (see illustration below).
At the same time, the M2 protein – a viral ion channel – acts as a secret accomplice and opens the floodgates: protons flow into the virus interior, loosening the packaging of its genetic material.

Fig. 2-E: Anchoring of the virus in the endosomal membrane Now, HA2 pulls the viral membrane and the endosomal membrane together with relentless force. The two lipid membranes fuse – a process known as membrane fusion. This occurs because the lipid molecules in the membranes are flexible and can rearrange themselves to form a continuous bilayer.

This fusion creates a pore – the gateway to freedom for the viral genome. With one final, elegant push, the viral RNA slides into the cytoplasm. The uncoating process is complete.
The cell has no idea that it has just released the blueprint for its subjugation.
Fig. 2-F: Release of the viral genome into the host cell’s cytoplasm: Through membrane fusion and uncoating, the viral envelope is dissolved, releasing the genome and initiating the infection.
Why is the virus not degraded?
The virus is not degraded by the cell’s digestive enzymes because the release process happens quickly – before the degradation mechanism (activation of digestive enzymes) can take effect. The virus exploits the drop in pH and the changes within the endosome to rapidly escape by triggering membrane fusion, releasing its genome directly into the cell’s cytoplasm. This „escape” from the endosome is faster than the cellular breakdown process, which is why the virus is not decomposed.d) Viral Replication – The Molecular Factory
The viral RNA does not arrive unprotected – it travels in high-tech armor: wrapped in protective nucleoproteins (NP) and equipped with viral polymerase, each of the eight RNA segments forms a highly organized ribonucleoprotein complex (RNP). These molecular command units are perfectly equipped for their mission:
- The nucleoproteins act like armor – shielding the RNA from cellular defense systems.
- The polymerase is the Swiss army knife of the virus – a tool for copying (replication) and translating (transcription) in one.

As soon as the ribonucleoprotein complexes (RNPs) are released in the cytoplasm, the systematic takeover of the cellular production lines begins – the virus factory goes into operation. The genome takes on two central tasks: On the one hand, it serves as a blueprint for the production of viral proteins (Protein synthesis), on the other hand, it is itself replicated (Genome replication) – so that each new virus particle is given its own copy of the genetic material along the way.

Fig. 2-G: To build new viruses, new viral proteins and new viral genomes are required. Protein synthesis: The viral genome serves as a template for the synthesis of the proteins needed to assemble new virus particles.
Genome replication: At the same time, the viral RNA is replicated to provide the genetic information for new viruses.While most RNA viruses remain in the cytoplasm, influenza has a clever trick up its sleeve: it hijacks the cell nucleus. Why? Because there, it finds optimal conditions for replicating its RNA.
The journey there, however, is anything but straightforward. The RNPs manipulate the cellular transport system by presenting fake import signals – molecular entry passes that grant them access to the nucleus. Cellular importins, normally responsible for transporting the cell’s own proteins, thus become unsuspecting smugglers. In a feat of biological deception, the viral RNPs are escorted straight into the cell’s control center.

Fig. 2-H: The viral RNA is transported from the cytoplasm into the cell nucleus. In the cell nucleus, the RNPs finally unfold their full potential. The viral polymerase begins its double role:
- Copying the viral RNA (replication) → blueprint for new viruses
- Producing viral mRNA (transcription) → building instructions for protein synthesis
As this process is particularly sophisticated, each step is described in detail below.
1️⃣ Activation of the RNA polymerase – Here we go
The viral polymerase needs a molecular ignition spark to become active. And it finds this in the cell nucleus: a biochemical special zone that differs significantly from the cytoplasm. High concentrations of nucleotides, ions and nucleus-specific factors send a clear signal: „This is the place to start!” Only in this environment does the polymerase come to life. Without this molecular wake-up call, it remains in a dormant state – camouflaged as a harmless cellular component.
2️⃣ Initial situation: (-)ssRNA – A Genome in Mirror Writing
The genome of the influenza virus is a master of disguise. Instead of presenting itself as a readable blueprint, it appears more like a riddle in mirror writing: eight separate RNA segments, negatively polarized, lacking all the usual hallmarks of a cellular message. No sender, no letterhead, no stamp. To the cell, it’s not a message – it’s biological noise.
In scientific terms:
The viral genome consists of eight segmented single-stranded RNA (ssRNA) with negative polarity: (-)ssRNA. „Negative” means that this RNA is the complementary template to the mRNA (i.e. mirror-inverted) and therefore not directly readable.
In addition, it lacks two crucial identifying features: the 5′ cap (a kind of molecular start button) and the 3′ poly-A tail, which protect and identify a normal mRNA.
Why so complicated?
Because it’s brilliant.With this molecular masquerade, the virus achieves two things:
Staying invisible: The (-)ssRNA is not immediately recognized as a threat by the cell’s immune system. If the viral genome were already in the form of mRNA, the cell’s alarm systems would be triggered right away.
Full control over production: The virus doesn’t beg for help from the host’s enzymes. Because only the virus’s own RNA polymerase can convert the (-)ssRNA into readable mRNA, the virus can precisely control:
➤ When mRNA is produced.
➤ How much of it is produced.
➤ Which segments are prioritized.In short: What looks like a cryptic puzzle is actually a highly precise control mechanism – a blueprint that only reveals itself when the viral machinery is ready – and the immune system is still asleep.
3️⃣ From (-)ssRNA to (+)ssRNA (the mRNA)
The viral polymerase is ready to transcribe the (-)ssRNA into readable mRNA – but the start button is missing. Without the 5′-cap, the machinery remains silent.
The solution? Theft at nano level.
This ingenious trick is called cap-snatching.
The Coup in Detail
The polymerase subunit PB2 prowls through the cellular mRNAs like a cunning thief searching for the most valuable jewel. Its target: the 5′ cap, the universal „seal” for cellular protein factories. Its accomplice, PB1 – the „molecular scissors” – cuts off the cap along with 10–15 nucleotides – a perfect primer for viral transcription. The stolen cap is attached to the viral RNA. The cell believes it has a legitimate mRNA and starts producing viral proteins. Meanwhile, the capped host mRNA is degraded – causing the collapse of the cell’s protein production.
At the same time, the viral mRNA receives a poly-A tail at its 3′ end, which stabilizes and protects it.
Why this trick is so brilliant
✅ Energy-saving: The virus uses existing resources – no effort needed to synthesize its own cap.
✅ Sabotage: The degradation of cellular mRNAs cripples the host’s defense.
✅ Camouflage: The stolen cap disguises viral mRNA as a „harmless” cellular message.The Consequences of the Heist
- The cell loses its own blueprints – and now produces viral proteins at full speed.
- The virus gains twice: rapid replication and weakening of its adversary.
This process is a classic in virology – a prime example of how viruses turn their host cells into puppets.
In the end, numerous „naked” +ssRNA strands are produced in the cell nucleus, which are used directly as mRNA for translation, i.e. for viral protein production and genome replication.
Figure 2-I: The viral RNA (-)ssRNA is transcribed into messenger RNA (mRNA). 4️⃣ The virus production is running hot
The freshly capped viral mRNAs exit the nucleus – equipped with a stolen signature and a poly-A tail. In the cytoplasm, ribosomes are waiting, unsuspectingly executing the enemy’s blueprints.
The prey: an entire protein factory
The ribosomes churn out viral proteins on an assembly line – including:Haemagglutinin (HA): The key to cell entry – the indispensable door opener.
Neuraminidase (NA): The liberator of new viruses – the sharp molecular scissors.
Matrix protein (M1): The stable envelope for the virus interior – the scaffold builder.
Ion channel protein (M2): The pH guardian – regulates the acidic environment in the virus.
RNA Polymerase (RNAP): The copying machine – a viral printing press.
Nucleoprotein (NP): The bodyguards – package and protect the RNA segments.
Nuclear Export Protein (NEP): The dispatcher – handles the export of viral RNPs from the nucleus.These freshly produced proteins are ready for the final act: the assembly of new virus particles.
5️⃣ Return to the Nucleus
After being produced in the cytoplasm, most viral proteins make their way back to the nucleus – the command center of viral replication. The only exceptions are the surface stars: hemagglutinin (HA), neuraminidase (NA), and the ion channel protein M2, which operate directly at the cell membrane.
The remaining viral actors return to headquarters to pick up new orders and prepare for the final mission.
Fig. 2-J: Viral mRNA exits the nucleus and is translated at two different sites within the host cell. 1) At free ribosomes in the cytoplasm, the mRNA is translated into viral proteins such as RNA polymerase, matrix proteins (M1), nucleoproteins (NP), and nuclear export proteins (NEP).
2) These proteins then migrate back into the nucleus to participate in the replication and packaging of the viral genome.
3) The surface proteins (HA and NA) and the ion channel protein (M2) are synthesised at the membrane-bound ribosomes of the endoplasmic reticulum (ER). After their production, these proteins are transported to the Golgi apparatus, where they are further modified and prepared for incorporation into the viral envelope.6️⃣ The Genome Copy Factory: (+)ssRNA → new (-)ssRNA
While viral proteins are being mass-produced in the cytoplasm, a covert operation „Genome Replication” unfolds in the nucleus:
Newly formed RNA polymerases grab the freshly synthesized (+)ssRNA strands and transcribe them back into viral mirror-image RNA – producing new (−)ssRNA strands. The polymerase remains bound as an integrated printing press for future rounds.
Nothing is left to chance: Even as the genetic code is being reverse-transcribed, nucleoproteins (NP) coat the emerging (−)ssRNA – it doesn’t spend even a second „naked” – eliminating any risk of cellular surveillance. The freshly copied RNA segments are immediately packaged and sealed: Together with the polymerase, they form complete ribonucleoprotein complexes (RNPs) – fully equipped genome modules, ready for the next generation of virus.
Once the eight segments are packaged, the viral logistics team takes over: NEP (nuclear export protein) and M1 (matrix protein) tag the RNPs for export. They escort them through the nuclear pores – the cell’s heavily guarded gateways – directly into the cytoplasm. Mission: assembly hall.

Fig. 2-K: How the influenza virus replicates its genome inside the host cell’s nucleus: 1) The viral RNA polymerase uses the (-)ssRNA as a template to synthesize a complementary (+)ssRNA.
2) This (+)ssRNA then serves as a template for the renewed synthesis of viral (-)ssRNA – the actual genetic material for new virus particles.
3) Already during synthesis, the new (-)ssRNA is coated with nucleoproteins (NP) and packaged with polymerase, M1, and NEP into the so-called RNP complex – stable and ready for export.
4) The completed RNP complexes exit the nucleus through the nuclear pores and migrate into the cytoplasm – where the assembly of new viruses soon begins.Like a clandestine printing shop in the back room: The polymerase continuously produces copies, the NP proteins immediately package them, and smugglers (NEP/M1) discreetly sneak them out.
While the cell unsuspectingly burns through its resources, the real showdown is still ahead…
e) Assembly of New Virus Particles
Once all the components have been produced, the coordinated final assembly begins in the cytoplasm – a process as precise as the construction of a space probe: every part has to fit perfectly, otherwise nothing will lift off.
The surface proteins HA & NA travel through the Golgi apparatus – the cell’s „packaging department” – to the cell membrane (see lower left illustration). There, they anchor themselves in the lipid bilayer like door handles and rescue scissors protruding from the envelope of a future virus particle.
The ribonucleoprotein complexes (RNPs) also set out on their journey, already in tow of the matrix proteins M1, which act as logistics managers. Their task: to reliably navigate the valuable cargo to the membrane regions equipped with HA and NA (see lower right illustration).
At the cell membrane, the viral puzzle comes together piece by piece: The RNPs arrange themselves beneath the membrane studded with HA and NA. The matrix proteins assist in bringing the RNPs into contact with specific regions of the cell membrane.
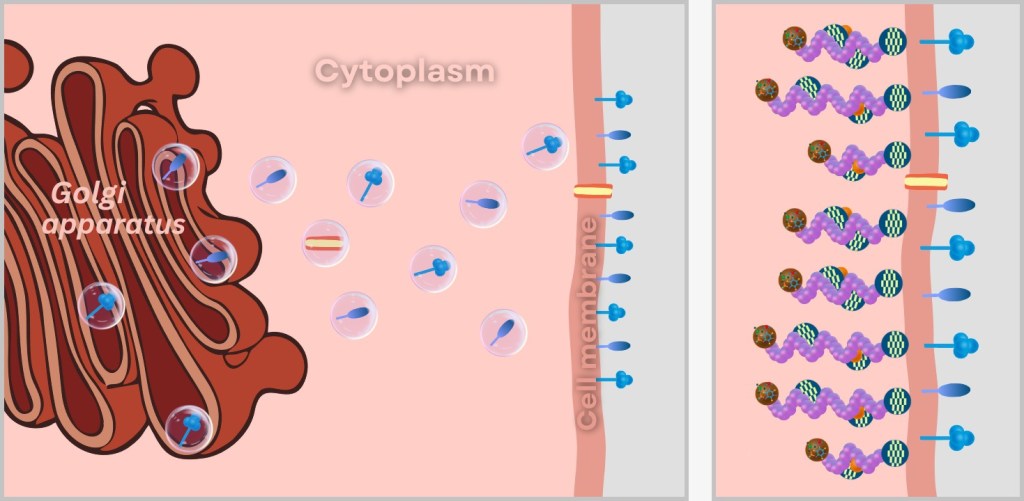
Fig. 2-L: Assembly at the cell membrane: viral building blocks find their place Left: Incorporation of viral surface proteins (HA and NA) and the ion channel protein (M2) into the cell membrane.
Right: Transport of ribonucleoprotein complexes (RNPs) to the cell membrane and their attachment to the forming viral envelope.And now – drumroll please – everything is set for the grand breakout!
f) Budding and Release of New Viruses
A protrusion forms on the cell membrane – like a soap bubble with a deadly cargo. But what looks so playful is actually precise choreography:
➤ Viral proteins push outward, causing the lipid bilayer to curve into a perfectly shaped „virus package”.
➤ Matrix proteins (M1) stretch the membrane like a trampoline – stable, but flexible enough for the jump.
➤ The host lipids close to form a camouflaged envelope – the virus packages itself.But it is not yet free. Sialic acid tethers lurk on the cell surface – normally HA’s favourite anchorage. Without resistance, the virus would stick like chewing gum under the sole of a shoe.
Neuraminidase (NA) steps in: the molecular scissors slice through the sialic acid residues on the cell surface. No sticking, no turning back – a clear path to the next cell.
Like a jailbreak with style: M1 loosens the bars, NA cuts the alarm wires – and they’re gone! Final countdown for the virus crew! All systems go – HA/NA check, RNPs check, lipid armor check. Launch sequence initiated in 3…2…1… Budding!

Fig. 2-M: All genomes on board – the launch sequence for the virus particle begins. Left: Budding of the virus at the cell membrane. Right: Release of the newly formed virus particle.
The following video provides a clear and accessible summary of the replication cycle of the influenza virus.
After a host cell is infected by a single influenza virus, hundreds to thousands of new virus particles are typically produced. The exact number can vary and depends on several factors, such as the virus strain, the type of host cell, and the cellular conditions.
Simplified and Realistic Representation of the Virus Structure
In the initial illustrations of this text, the structure of the influenza virus was simplified for better clarity (see bottom left image). In these illustrations, the matrix protein is shown as a spherical, mesh-like ring structure surrounding the viral genome. This simplified representation is intended to make the complex processes of viral replication easier to understand.
Fig. 2-N: Two representations of the virion: Left: Schematic illustration to clarify the structure of the virus.
Right: More detailed schematic representation of the virion structure.In the following illustrations, however, the structure of the new virions is depicted in a way that more closely reflects biological reality. The matrix proteins are not shown as a continuous ring, but are located in individual units that both bind to the inner lipid layer and are loosely linked to the ribonucleoprotein complexes (RNPs). In addition, the colouring of the lipid layer reflects the origin from the cell membrane of the host cell (see upper right figure).
The RNPs representing the viral genome are arranged inside as a loose bundle – not strictly parallel, but flexibly organized with ends oriented in different directions. The matrix proteins (M1) hold this bundle together and connect it to the lipid layer, giving the virion its shape and stability.
2.4. The Adaptability of the Influenza Virus
Viruses – especially RNA viruses like the influenza virus – mutate extraordinarily fast. The reason lies in their error-prone replication machinery: the viral RNA polymerase lacks a mechanism to correct copying errors, unlike DNA replication in human cells. As a result, random mutations – small changes in the virus’s genetic material – occur with each replication cycle.
Within an infected person, this process produces a multitude of slightly different virus particles. Most mutations are neutral, meaning they neither affect the virus’s function nor its ability to replicate. However, some mutations are disadvantageous, causing the virus to replicate less efficiently or lose infectivity entirely – these variants quickly disappear through natural selection.
But some mutations give the virus a survival advantage, especially when they affect the surface proteins hemagglutinin (HA) and neuraminidase (NA). These proteins are key targets of the immune system: the body produces antibodies that specifically bind to them and neutralize the virus. However, if the structure of HA or NA changes due to mutations, the antibodies recognize the virus less effectively. The virus essentially becomes „invisible” to the immune defense and can continue to replicate and spread.
This constant adaptation explains why influenza viruses cause new waves of infection every year and why it is challenging to develop long-lasting vaccines against the flu.
The influenza virus does not exist as a fixed genetic entity but rather as a so-called mutant cloud (quasispecies) – a dynamic population of viral variants arising through continuous mutations. This genetic diversity is key to its survival: natural selection ensures that the variants most successful under the given conditions prevail. This high adaptability of the influenza virus vividly demonstrates how evolution occurs in real time.

Fig. 2-O: Genetic diversity in the host The influenza virus must constantly change through mutations to continue existing as the flu virus. The high mutation rate leads to a multitude of slightly different viral particles within an infected person.
2.5. Entry and Exit Routes of the Influenza Virus
Influenza viruses use the mucosal surfaces of the respiratory tract as their entry point, since mucous membranes form the boundary between the external environment and the inside of our body. Many viruses initiate infection by interacting with the epithelial cells of these mucous membranes in order to spread efficiently within their host. [Virus Infection of Epithelial Cells]
As shown in the illustration below, the influenza virus enters the respiratory tract via the air – reaching the nose, throat, and lungs – and binds to the apical side (upper surface) of the epithelial cells, which faces the external environment. This apical side is covered with fine, hair-like structures called cilia, which help transport mucus and foreign particles. The opposite, basolateral side of the cell faces the underlying tissue and is connected to the basement membrane, anchoring it to the connective tissue.

Fig. 2-P: An epithelial cell is infected by the influenza virus and produces numerous new virions. The release of newly formed influenza viruses also occurs specifically at the apical side. This arrangement allows the viruses to spread into the surrounding environment – such as through droplets expelled by coughing or sneezing – thereby easily infecting new hosts. This apical release represents an evolutionary advantage, as it significantly enhances transmission efficiency.
2.6. Mostly Localized Mucosal Infection
Influenza viruses are specialized for infections of mucosal surfaces. As a result, their infection typically remains localized to the epithelial cells of the respiratory tract, meaning it is confined to a mucosal infection. The virus spreads from cell to cell along the apical side of the epithelial layer, without penetrating deeper tissue layers. Even when the infection progresses from the upper respiratory tract down to the lungs, it remains limited to the mucosal surface.
The basolateral side of epithelial cells usually remains untouched, as it plays no role in viral transmission. If the virus were to exit the host cell from the basolateral side, it could spread into the surrounding tissue and ultimately enter the blood or lymphatic system, potentially leading to a systemic infection. However, for influenza viruses, this would be disadvantageous: they would face stronger immune defenses and their transmission via the respiratory tract would become more difficult.
In rare cases – particularly in severely immunocompromised individuals – the virus can break through the epithelial barrier and invade the underlying tissue as well as blood or lymphatic vessels, leading to a systemic infection.
Fig. 2-Q: Difference between a mucosal infection and a systemic infection The mucosa consists of several layers: Epithelial cells form the outer protective layer, the basement membrane acts as a thin barrier, the lamina propria supports with connective tissue and immune cells, endothelial cells form the walls of blood vessels, and the blood vessel leads into the interior of the body.
Left – Mucosal infection (limited to the mucosal surface): The virus infects epithelial cells exclusively via the apical side. It remains within the mucosa, spreading from cell to cell along the apical surface. The basement membrane and underlying tissues such as the lamina propria remain intact. A mucosal infection is locally restricted and favors transmission via mucosal surfaces, such as the respiratory tract.
Right – Systemic infection (spread through the bloodstream): The virus enters the epithelial cells on the apical side but exits them via the basolateral side. It breaches the basement membrane and moves through the lamina propria, either by migrating or infecting cells there. Eventually, it reaches a blood vessel by passing through gaps between endothelial cells or by directly infecting the endothelial cells. Entry of the virus into the bloodstream marks the transition to a systemic infection. A systemic infection is critical because the virus can then spread throughout the body via the blood, potentially damaging vital organs such as the lungs, heart, or brain.
2.7. Viral Strategy: Efficient Replication Without Rapid Cell Destruction
Some viruses – including influenza viruses – are surprisingly economical: instead of destroying their host cell immediately, they exploit its resources as efficiently as possible. Why burn down the apartment if you can live rent-free for months? As long as the cell remains intact, it provides everything the virus needs for replication: energy, enzymes, building blocks. The immune system also notices something’s wrong only later – because if nothing’s on fire, no alarm is triggered. This strategy prolongs the life of the infected cell, delays the immune response, and maximizes the production of new viruses.
Virus wisdom: The best parasites stay under the radar!
2.8. Destruction of the Host Cell
What starts as cunning protection ends in molecular burnout: the infected cell ultimately suffers cell death. This occurs when the cell is either overloaded and structurally damaged by the massive production of viruses, triggered to enter programmed cell death (apoptosis) by cellular defense mechanisms, or deliberately eliminated by the immune system. These characteristic changes in the host cell caused by the virus are referred to as cytopathic effects (CPE). This entire process can take place within as little as 24 hours after infection.
To better understand cell death caused by influenza viruses, let’s examine the three mechanisms by which the host cell is ultimately destroyed.
a) Overload and structural damage
b) Apoptosis: programmed cell death to combat the virus
c) Immune response: Destruction by the immune systema) Overload and structural damage
The virus takes over the cellular processes to produce its own components. With each new generation of viral proteins and RNA, the cell’s energy and resource capacity become increasingly depleted. Since the cell practically works only for viral replication, its own vital functions come to a halt. The cell becomes a squeezed lemon – ribosomes run hot, mitochondria collapse.

Fig. 2-R: Resource capacity: healthy cell vs. infected cell Left: Healthy cell with fully functional cellular machinery. The cell’s own mRNA (orange) is read by ribosomes to produce proteins necessary for cellular functions. The cell displays vibrant staining, indicating full resource capacity and energy availability.
Right: Virus-infected cell, heavily burdened by the production of viral components. The viral mRNA (yellow) increasingly displaces the cell’s own mRNA, and the ribosomes predominantly read viral instructions for virus replication. The pale staining of the organelles symbolizes the depletion of energy reserves and the overload of the cell.During budding at the cell membrane – when new virus particles exit the cell – the membrane is repeatedly pierced and deformed. This process ultimately leads to the loss of membrane integrity, causing the cell to lose its stability and protective functions. The cell eventually dies due to relentless resource depletion and structural breakdown resulting from virus release.
Fig. 2-S: Cell damage after virus budding and release During budding, the cell membrane forms small protrusions from which new viruses are released. Each budding event removes tiny portions of the membrane’s lipid bilayer, as the newly forming viruses use the host cell’s membrane material for their envelope. After repeated viral release, the membrane becomes significantly thinned and structurally weakened, often showing deformations and irregularities. The constant stress from budding makes the membrane more porous and vulnerable. The cell increasingly loses its ability to regulate its internal environment and can hardly maintain its selective permeability for ions and molecules. As membrane damage continues, its structural stability deteriorates. Eventually, the membrane may become so compromised that it ruptures or disintegrates, ultimately leading to cell death.
b) Apoptosis: programmed cell death to combat the virus
When a cell realizes it has been hijacked, it sometimes pulls the emergency brake – sacrificing itself for the greater good: it commits suicide to stop the virus from spreading. The plan: take the enemy to the grave. Instead of dying with a bang, the cell undergoes a controlled breakdown into small fragments called apoptotic bodies, which are then cleared away by immune cells.
This process follows a precise sequence: the DNA is fragmented, the cell membrane forms characteristic bubble-like protrusions (known as blebbing), and internal structures are neatly dismantled and recycled. The cell dies quietly – and in doing so, protects the organism.
This mechanism proves – cells have more honor than some governments!

Fig. 2-T: The image shows the stages of apoptosis in a virus-infected cell in three steps. Healthy Cell: On the left, an intact cell is shown with an undamaged cell membrane and nucleus. The nucleus contains complete DNA strands, and the cell shows no signs of stress or damage.
Infected cell – initiation of apoptosis: In the middle section, the cell begins to show visible changes. The cell membrane forms bubble-like protrusions (blebbing), and the nucleus starts to shrink. Within the nucleus, fragmented DNA becomes visible, resulting from apoptotic processes. This phase marks the transition from a functioning cell to its controlled disintegration.
Apoptotic Bodies and Degradation: On the right, the cell breaks apart into several small fragments known as apoptotic bodies. In the background, an immune cell (macrophage) is shown engulfing and degrading these fragments. This process prevents the release of viral components and protects the surrounding tissue from further infection.c) Immune response: Destruction by the immune system
Once the immune system detects a virus-infected cell, it sounds the alarm – and that spells trouble for the virus! Specialized fighters like natural killer (NK) cells and cytotoxic T cells leap into action. They identify infected cells by viral protein signals that wave like warning flags on the cell surface. With deadly precision, they release toxic molecules, destroy the infected cell, and effectively slow down viral replication!
Anyone curious to learn more about this fascinating defence battle can find detailed information in „The Wonderful World of Life”, especially in Chapter 5.3 d) „Natural Killer Cells” and Chapter 5.5.7 b) „Cytotoxic T Cells”.
NK cells and cytotoxic T cells form an unbeatable team – they hunt down and eliminate viral threats, keeping the infection under control.
Regeneration of Epithelial Cells
As previously mentioned, the influenza virus primarily infects the epithelial cells of the respiratory tract – especially the cells of the nasal mucosa, the bronchi, and the alveoli (tiny air sacs) in the lungs. Due to viral replication and the immune response – particularly through natural killer (NK) cells and cytotoxic T cells – many of these cells are severely damaged or destroyed.
Once the acute infection has been brought under control, the repair process begins: specialized stem cells initiate the regeneration of the tissue. They proliferate and differentiate into the various types of epithelial cells needed to restore the respiratory tract.
- Upper respiratory tract (nose, bronchi): New ciliated cells are formed, whose fine hair-like structures (cilia) transport mucus and foreign particles upward. In addition, goblet cells are generated, which produce mucus to keep the airways moist and protected.
- Lungs (alveoli): Here, the damaged, flat epithelial cells are replaced. These cells are essential for enabling the exchange of oxygen between the air and the blood.
The duration of regeneration varies depending on the severity of the infection. In the case of a mild illness, the epithelium can be completely renewed within one to two weeks. However, after more severe infections – such as influenza pneumonia – the healing process can take several weeks. Once the new cells form a dense layer, the protective function of the airways is restored. In most cases, regeneration is complete; however, in cases of very severe damage, scarring or structural changes in the epithelium may remain.
2.9. Self-Limiting Dangerous Viruses: Why They Rarely Cause Pandemics
Highly dangerous viruses that kill their host quickly limit their own spread. If a virus harms its host so rapidly that the person doesn’t have time to infect others, the chain of transmission is effectively broken. One example is the Ebola virus, which often remains locally contained and therefore rarely causes pandemics.
In contrast, viruses with moderate pathogenicity are more likely to cause global outbreaks. Moderate pathogenicity refers to a pathogen’s ability to cause disease without triggering extremely severe or fatal outcomes in most infected individuals. Such viruses typically lead to mild to moderate symptoms, allowing infected people to remain mobile and socially active. This increases the likelihood of transmitting the virus to others. Many influenza viruses are examples of this. Severe cases of influenza infection mainly occur in individuals with weakened immune systems, advanced age, or preexisting health conditions. In these cases, careful medical monitoring and intensified treatment are necessary to prevent serious complications.
The most successful viruses aren’t the ones that kill us – but those that keep us just alive enough to do their dirty work for them.
2.10. Why Does the Virus Make Some People Sick and Others Not?
💡Note: For a better understanding of this section, we recommend Chapter 5 in the treatise „The Wonderful World of Life”. It explains the fundamentals of the immune system in a clear and accessible way.
Not everyone infected with the influenza virus falls ill to the same degree: some experience only mild symptoms like a runny nose, others develop severe flu with fever and shortness of breath, and some remain completely asymptomatic. Why is that? The answer lies in a complex interaction between the virus and the host – i.e. the person who becomes infected. Several key factors play a role in this:
The host’s immune system
Every person has an individual immune system that responds differently to the influenza virus. Previous flu infections can provide partial immunity because the immune system has developed antibodies and memory cells that recognize and combat the virus more quickly. A strong immune system can suppress the infection at an early stage, whereas a weakened immune system (e.g., in older adults or chronically ill individuals) is often overwhelmed.
The viral load
The amount of viral particles entering the body during initial contact – the so-called viral load – affects the course of the infection. With a low viral load, the innate immune system can quickly recognize and eliminate the intruders before they multiply extensively. However, a high viral load, for example through close contact with an infected person, poses a greater challenge and can intensify the infection process.
10 viruses in the throat? No problem.
10,000 viruses? Straight to bed!The host’s genetic predisposition
Two people, one virus – but only one gets sick. Some individuals carry genetic variants in their immune system that make them either more susceptible or more resistant to the influenza virus. For example, differences in genes that regulate immune receptors can affect how effectively the immune system recognizes the virus.
There are numerous studies investigating the varying immune responses caused by genetic predisposition:
The study „IFITM3: How genetics influence influenza infection demographically” showed that people with certain variants of the IFITM3 gene (Interferon-Induced Transmembrane Protein 3) are less likely to develop severe influenza because this gene inhibits the replication of the influenza virus within cells.
The study „HLA targeting efficiency correlates with human T-cell response magnitude and with mortality from influenza A infection” investigated how HLA alleles (MHC class I) influence the T-cell response to the influenza virus. It found that certain HLA alleles present influenza peptides more efficiently and trigger a stronger T-cell response. People with these alleles experienced milder courses of influenza infections, whereas other alleles were associated with weaker T-cell responses and higher mortality. This demonstrates that genetic differences in MHC molecules can directly affect the severity of an influenza infection.
The virus’s mutant cloud
As mentioned earlier, the influenza virus does not exist as a uniform strain but rather as a „mutant cloud” – a diversity of genetic variants that arise due to the error-prone RNA polymerase. Some variants in this cloud are more aggressive because they, for example, bind better to cells or evade the immune system. Which variant dominates can determine the severity of the infection.
Tissue specificity of the virus
Influenza viruses differ in their preference for certain tissues in the body. Most strains primarily replicate in the upper respiratory tract (e.g., nose and throat), which often leads to milder symptoms such as a sore throat. Other strains penetrate deeper into the lungs and can cause severe pneumonia.
📌 Conclusion: Cheers to diversity!
The interaction between virus and human is like Tinder for microbes – some matches are harmless, others end in disaster. What really matters is:
- Host poker (genes + immune system)
- Virus roulette (dose + mutations)
- Tissue Tinder (where does the virus land?)
Our body is no passive target – it’s a learning system. Every infection is an update for the immune memory, every virus a lifelong training partner. Because only through the fight does our shield grow – for a lifetime.
3. A Look at the Beginnings of Microbiology – How It All Started
The history of microbiology is a journey from invisibility to clarity, from speculation to concrete knowledge. Its origins can be traced back to the 17th century, when human curiosity and technological innovation first unveiled a hidden world.
3.1. Early Discoveries: The First Glimpses into the Invisible
3.2. The Birth of Modern Microbiology
3.3. The Step into the World of Viruses
3.4. Viruses and Koch’s Postulates3.1. Early Discoveries: The First Glimpses into the Invisible
In 1665, it was Robert Hooke who, using an early microscope, first described plant cells and thereby coined the term „cell”.
But the real breakthrough came a few years later with Antonie van Leeuwenhoek. Using his self-crafted, extremely powerful lenses, he was the first to observe tiny, living organisms in 1676, which he called „animalcules” – little animals like bacteria and single-celled creatures that we recognize today. Van Leeuwenhoek’s discoveries opened an entirely new perspective on nature, yet understanding how these organisms lived or caused diseases was still far from reach.
3.2. The Birth of Modern Microbiology
It took almost two centuries before microbiology was systematically studied. In the 19th century, the field experienced a quantum leap. Louis Pasteur disproved the old idea that life could spontaneously arise from nothing (the theory of spontaneous generation) and demonstrated that microorganisms are responsible for processes like fermentation and decay. His work laid the foundation for the germ theory of disease, which was later further developed by Robert Koch.
Koch’s research led to a crucial milestone in 1876: the Koch’s postulates. These rules made it possible for the first time to identify microorganisms as specific causes of diseases. Koch’s work revolutionized bacteriology and made it possible to definitively detect pathogens such as the anthrax bacterium (Bacillus anthracis) and later the tuberculosis pathogen.
However, while microbiology made great advances in studying bacteria, viruses remained hidden for a long time. Even the best microscopes of that era were unable to visualize these tiny, invisible particles.
3.3. The Step into the World of Viruses
The end of the 19th century brought the next breakthrough. In 1892, Dmitri Ivanovsky demonstrated that a filtered extract from tobacco plants suffering from tobacco mosaic disease remained infectious, even after passing through porcelain filters that retained bacteria. Martinus Beijerinck confirmed these observations and coined the term „virus” (from the Latin word for „poison” or „slime”) for the mysterious, non-bacterial pathogen. This marked the beginning of the systematic study of this new world.
The true access to the world of viruses, however, was only made possible with the electron microscope, developed in the 1930s. It was only then that scientists could visualize viruses and understand their structure.
3.4. Viruses and Koch’s Postulates
In discussions about the detection of viruses, Koch’s postulates are often used as a benchmark to question the existence of viruses. But how do these postulates, developed in the 19th century, fit into today’s understanding of infectious diseases? A look at the historical background and scientific advancements helps to clarify this question.
Koch’s Postulates: A Scientific Milestone
Robert Koch (1843–1910), one of the founders of modern bacteriology, developed the postulates named after him to prove the connection between microorganisms and infectious diseases. They were presented in 1890 at the 10th International Medical Congress and consist of four criteria:
Postulate 1: The microorganism must be found in every case of the disease, but should not be present in healthy organisms.
Postulate 2: The microorganism must be isolated from the diseased organism and grown in pure culture.
Note: A pure culture means that only a single species of microorganisms is cultivated, without any other species mixed in.Postulate 3: A previously healthy individual shows the same symptoms after infection with the microorganism from the pure culture as the individual from whom the microorganism originally came.
Postulate 4: The microorganism must be re-isolated from the experimental host and identified as the same one.
These groundbreaking principles laid the foundation for experimental medicine and the germ theory of disease.
Robert Koch: A Pioneer Against Resistance
Robert Koch studied the pathogens of diseases such as tuberculosis, cholera, and anthrax. For his research, he often traveled to epidemic regions, such as Calcutta to study cholera or Bombay during the bubonic plague. Koch spent months in these countries, always close to the epicenter of the outbreaks. In his laboratory tent, he worked tirelessly at the microscope.
However, Koch faced significant resistance. At his time, the idea that diseases were caused by microscopic organisms was still controversial. Many of his colleagues and contemporaries were skeptical and rejected his theories. Scientists often believed back then that plagues and epidemics were caused by so-called miasmas – toxic vapors rising from the ground.
Despite these challenges, Koch tirelessly pursued his research. He utilized innovative techniques of his time, such as the agar plate and oil immersion lenses, to cultivate and study bacteria. These methods enabled him to make important discoveries and revolutionize the understanding of infectious diseases.

Fig. 3: Early tools of microbiology: agar plate and oil immersion microscope Left: An agar plate – a solid nutrient medium in a petri dish, with agar added as a gelling agent to selectively cultivate bacteria. Right: A microscope with an oil immersion lens – a special microscopy technique where a drop of oil is placed between the objective lens and the specimen to minimize light refraction, making the tiniest microbes appear sharper.
Challenges and Limitations of the Postulates
It was precisely the skepticism directed against his theories that prompted Koch to formulate the postulates in order to provide proof of a connection between the pathogenic properties of bacteria and the disease.
Koch himself recognized that his postulates do not always apply universally. A well-known example is his work with the cholera pathogen Vibrio cholerae. He discovered that this microorganism can be present not only in sick individuals but also in apparently healthy people. This finding challenged the first postulate and led Koch to abandon the universal validity of this criterion.
Koch’s Spirit of Innovation
Koch was a pioneer of his time. In his speech at the 10th International Medical Congress, he stated:
„It was necessary to provide irrefutable evidence that the microorganisms found in an infectious disease are truly the cause of that disease.”
His scientific approach of convincing skeptics through strict evidence was groundbreaking. Yet, he himself recognized that new technologies and methods were needed to answer further questions:
„With the experimental and optical aids available, no further progress could be made and it would probably have remained so for some time if new research methods had not presented themselves at that time, which suddenly brought about completely different conditions and opened the way to further penetration into the dark area, with the help of improved lens systems …”
Regarding hard-to-detect pathogens such as those causing influenza or yellow fever, he remarked:
„I tend to agree with the view that the diseases mentioned are not caused by bacteria at all, but by organized pathogens that belong to entirely different groups of microorganisms.”
Koch was already on the trail of viruses but was unable to definitively identify them due to the limited technical capabilities of his time. However, he recognized that these invisible pathogens must exist.
Why viruses break Koch’s postulates?
Koch’s research results corresponded to the scientific knowledge of his time. In the 19th century, microbiology was still in an early stage of development, where fundamental principles were just being discovered and systematically studied. Virology as an independent field emerged only after Koch’s era, when Dmitri Ivanovsky and Martinus Beijerinck discovered infectious particles smaller than bacteria. With the invention of the electron microscope in the 1930s, viruses could finally be visualized. However, viruses differ fundamentally from bacteria, which is why Koch’s postulates are often not directly applicable to them:
➤ Host dependence: Viruses can only replicate inside living host cells and cannot be grown in pure culture.
➤ Asymptomatic infections: Many viral infections occur without symptoms, making it difficult to clearly associate the pathogen with the disease.
➤ Complex detection methods: Modern molecular techniques such as PCR allow for the detection of viral genome sequences, requiring an extension of the classical postulates.
Koch and Modern Science
Robert Koch and his contemporaries laid the foundation for microbiology, particularly through the development of methods for isolating and cultivating bacteria. The germ theory of disease was a milestone in the history of science at the time. Koch understood that science is constantly evolving. If Koch had had access to modern technologies such as PCR, sequencing or electron microscopes, would he have adapted his methodology?
Modern technologies such as PCR and sequencing will be introduced in the following chapters.
His closing words at the 1890 congress provide the answer and reflect his optimism for the future:
„Let me conclude with the hope that the nations may measure their strength in this field of work and in the war against the smallest but most dangerous enemies of mankind, and that in this struggle, for the benefit of all humanity, one nation may continually surpass another in its achievements.”
If Robert Koch had been able to witness today’s advances in molecular biology, virology, and immunology, he would likely have been fascinated – not only by the new insights, but also by the revolutionary methods that make it possible to visualize even the tiniest pathogens. After all, this was precisely his motivation: to make the invisible visible, to decipher what is hidden. How might he have reacted to the first images of a virus under an electron microscope?
From the First Observations to Modern Science
What once began with dusty lenses and puzzled gazes has now become high-tech down to the molecular level. With every new technology, we peer deeper into the microcosm. Microbiology has made the invisible visible – but only modern technology allows us to truly understand the invisible. Today, we track viruses that evaded our sight for centuries.
And how we uncover the invisible today – that is the story of the next chapter.
4. Modern Methods for the Discovery and Analysis of Viruses
So far, we have familiarized ourselves with the topic of viruses. In this chapter, we shift our perspective: from wonder to measurement. Without the methods of modern biology, we would know little about viruses – their structures, their genetic material, their diversity.
What follows is a look into the engine room of science. Admittedly, a technical section – but an important one: because it is here that we see how we are able to grasp the invisible at all.
Anyone eager to understand how viruses are made visible, decoded, catalogued, and analyzed today will find here a kind of 21st-century toolbox.
But when and why do we turn to these tools?
The identification of viruses can serve different purposes: some viruses come into focus because of their impact on humans, animals, or plants, while others are discovered in environmental samples or unexplored habitats to better understand their ecological significance. The detection of known viruses is primarily used for diagnostics, whereas the identification of unknown viruses offers insights into biological diversity and evolution.
Since viruses lack universal features such as a shared cell structure, detection methods focus on their genetic material, their structure, or their interactions with host cells. The detection process does not follow a rigid scheme but instead combines various steps that vary depending on the research question and the type of virus.
The following sections will provide a detailed explanation of the key methods used for virus identification – from sample collection to bioinformatic analysis.
Procedures for the identification of viruses
4.1. Sample Collection
4.2. Sample Preparation
4.2. a) Filtration
4.2. b) Centrifugation
4.2. c) Precipitation
4.2. d) Chromatography
4.3. Cell Culture
4.4. Making Viruses Visible
4.4. a) Electron Microscopy
4.4. b) Crystallization
4.4. c) Cryo-Electron Microscopy
4.4. d) Cryo-Electron Tomography
4.4. e) Summary
4.5. The Genetic Fingerprint of Viruses
4.5.1. Nucleic Acid Extraction
4.5.2. Nucleic Acid Amplification
4.5.3. Sequencing
4.5.3. a) First Generation: Sanger Sequencing
4.5.3. b) Second Generation: Next-Generation Sequencing (NGS)
4.5.3. c) Third Generation: New Approaches in DNA Sequencing
4.5.3. d) Emerging Technologies: The Future of Sequencing
4.6. Bioinformatic Analysis4.1. Sample Collection
Before we can track down viruses, we first have to find them – and that’s like a micro-scale treasure hunt, with hiding places ranging from blood to deep-sea sediment. Whether in humans, plants, or the ocean, viruses are everywhere, and researchers become detectives equipped with pipettes, protective suits, and specialized gear.

Fig. 4: Sample Collection: A) from humans, B) from soil, C) from marine ecosystems [Tara Oceans-Mission]
Human Samples
Who hasn’t experienced this? A swab deep in the nasopharynx – the classic method made familiar to everyone since COVID-19. But viruses don’t just hide in mucous membranes; they can also be found in blood, faeces, or tissue. Much like a thief leaves behind DNA traces, viruses reveal themselves through their genetic remnants. It is crucial that samples are collected cleanly and processed quickly – ideally as gently (and painlessly?) as possible for the patient. Plants, animals, and insects also provide sample material to investigate the presence of viruses in a wide variety of biological systems.Environmental Samples
Water, soil, even air – viruses are everywhere. Researchers fish them out of rivers, dig them up from the ground, or capture them directly from the atmosphere using high-tech filters.Extreme Candidates: Viruses in Extreme Conditions
In the deep sea, in eternal ice or in bubbling volcanic springs – viruses are among the toughest survival artists of all. To catch them there, special probes are needed that reach into the depths like spaceship tentacles: „Oops, what’s swimming at a depth of 4,000 metres? Just take it with you!”More About the Ocean
Viruses play a central, often underestimated role in the health of the oceans. Wherever life exists, viruses are present – invisible yet ubiquitous players that outnumber every other biological entity. They regulate the ecosystem much like large predators, controlling populations and thus maintaining ecological balance.
An outstanding research project was the Tara Oceans Expedition (2009–2013). The goal of this four-year project was to study microbial life in the oceans and its impact on the global ecosystem. Scientists collected over 35,000 samples worldwide from plankton, algae, and viruses.
During the expedition, the research team discovered over 5,000 new RNA virus species, including the fascinating mirusviruses. These discoveries expand our understanding of the diversity, evolution, and ecology of the oceans.
If you’re interested in this expedition, check out the Tara Oceans videos – the short or the long version.
Transport: The VIP Service for Viruses
To make sure these tiny suspects don’t get damaged en route, they’re quickly put into the cold chain after collection. Deep-freeze transport and sterile packaging are a must – otherwise, the „viral loot” falls apart faster than an ice cube in the Sahara.But a good sample is only the beginning.
What we bring home in tubes, swabs, or deep-freeze boxes is usually a biological jumble: cell debris, bacteria, proteins – and somewhere in between, a few viruses, tiny and hidden. Now it’s time to sort, clean and concentrate – before the actual analysis can even begin.
4.2. Sample Preparation
In every sample, viruses hide like needles in a microbial haystack – which is why we have to clean up before the virus hunt can begin:
- 99 % ballast: cell debris, proteins, bacteria
- 1% target: tiny virions we want to isolate
The majority of a biological sample consists of non-viral material, which makes targeted analysis extremely challenging. That’s why sample preparation is an essential intermediate step: it removes contaminants, concentrates viral particles, and prepares the material for actual analysis. The procedure varies depending on the sample type, the research goal, and the virus being investigated.
Sample preparation is like gold panning: you need patience, the right sieve – and the hope that in every bucket of sludge, there’s a nugget gleaming.
The most important techniques include:
a) Filtration
b) Centrifugation
c) Precipitation
d) Chromatographya) Filtration – The Mega Sieve

Filtration is an initial purification step used to remove larger particles and coarse impurities.
This allows viral particles to be selectively separated from larger structures such as bacteria or cell fragments.
Function: Retains bacteria and cell debris – only virions can pass through
Special Feature: Special filters with nanopores (0.02 µm!)
Cool Fact: Some filters become electrostatically charged to catch viruses more effectivelyMore Information
Filtration is typically carried out using special membrane filters with defined pore sizes. Filters with a pore size of 0.2 micrometers are commonly used because they reliably retain bacteria and larger particles while allowing smaller viral particles to pass through. A practical example of this method is its use in water samples from environmental studies, where viral particles are effectively separated from other microorganisms.
An advanced variant is ultrafiltration, which uses filters with even finer pore sizes ranging from 0.01 to 0.1 micrometers. This technique not only removes bacteria but also separates smaller particles.
Fig. 5-A: Schematic illustration of the filtration process showing how an unfiltered sample is passed through a membrane filter. The membrane filter retains larger particles, while viral particles and smaller particles pass through and are collected in the receiving container. Despite its effectiveness, filtration has some limitations:
Purity: The smallest particles or dissolved substances may still remain in the sample after filtration.
Clogging: Especially in samples with a high particle concentration, the filter membrane can become blocked, complicating the process.
Capacity: The flow rate is limited by the pore size, which can make filtration time-consuming.To obtain the purest possible virus sample, filtration is often combined with additional purification steps such as centrifugation or chromatography. These supplementary methods can remove remaining contaminants and optimize the quality of the samples for subsequent analyses.
b) Centrifugation – The Gravity Turbine

Centrifugation is a key technique for separating particles based on their size and density.
This process uses centrifugal force generated by spinning the sample at high speed.
Principle: Heavy particles sink, light ones float – viruses settle in between
High Speed: Up to 100.000 RPM (a washing machine reaches 1.200 RPM)
Trick: Density gradients can even separate different virus types from each otherMore Information
How Centrifugation Works
When a solution is left standing at room temperature for an extended period, heavier particles slowly settle due to gravity. Centrifugation significantly speeds up this process by rotating the sample at speeds ranging from several thousand up to 100,000 revolutions per minute (in ultracentrifugation). The extreme centrifugal forces cause particles to separate based on their density and size.

Fig. 5-B: Sample is placed into the centrifuge. To protect sensitive molecules from overheating, many centrifuges are equipped with cooling systems that maintain a consistent temperature throughout the entire process.
Types of Centrifugation
Differential Centrifugation
Separation principle: Particles are separated based on their size and mass. Larger and heavier particles sediment faster than smaller and lighter ones.
Procedure: The sample is treated step by step with increasing centrifugal forces. After each step, the supernatant, which contains the remaining smaller particles, is carefully transferred to a new tube. The bottom of the original centrifuge tube contains the pellet in which the larger particles have settled and is removed.
Application: This method is commonly used to isolate cell components, organelles, and viruses from complex mixtures.
A typical example of sample preparation for the investigation of unknown viruses is the extraction of a cell lysate. In this process, cells isolated from tissue samples or swabs are disrupted using chemical, physical, or enzymatic methods. This step releases viral particles that have replicated within the host cells. The cell lysate is then purified through centrifugation or filtration to separate viral components from cellular debris. Cell lysates are especially useful when there is suspicion that viruses are hidden within specific cell types or tissues.
Fig. 5-C: Differential Centrifugation Density Gradient Centrifugation
Separation principle: Particles are separated according to their density. The sample is applied to a density gradient (usually sucrose or caesium chloride) and centrifuged. During rotation, the particles move to the position in the gradient that corresponds to their own density.
Procedure: Particles come to rest at the point in the gradient where their density matches that of the medium, enabling precise separation.
Application: This method is particularly well suited for isolating and analyzing viruses, proteins, and nucleic acids from complex mixtures.
Extraction of the Viral Fraction
After centrifugation, the viral fraction typically appears as a clearly defined layer within the centrifuge tube. This layer can be carefully extracted using a pipette to prepare the viruses for further analysis. The exact position of the viral layer depends on the method used and the physical properties of the viruses.
Fig. 5-D: Density Gradient Centrifugation In most cases, the centrifuge is set to a constant speed that is sufficient to separate the particles within the density gradient. This speed is typically very high (e.g., up to 100.000 rpm during ultracentrifugation), as effective separation in the density gradient depends on generating adequate centrifugal force.
Combination of Methods
Differential and density gradient centrifugation are often combined to achieve higher purity and precision in particle separation. These methods are essential for biomedical research and diagnostics, as they allow for the precise separation and concentration of viral particles.
c) Precipitation – The Chemistry Trick

Precipitation is a well-established method for isolating and purifying virus particles from a solution.
In this process, chemical substances are added that reduce the solubility of the viruses, causing them to precipitate out of the solution.
How it works: Chemical substances make viruses heavier and sticky → they precipitate out
Advantage: Cheap and simple – but not suitable for fragile virusesMore Information
The process involves multiple steps and uses specific chemical reactions to effectively concentrate the virus particles.
Process of Precipitation
Sample Preparation: Before precipitation, the sample is often pre-cleaned – using methods such as ultrafiltration or ultracentrifugation – to remove larger particles and impurities. This improves the efficiency of the precipitation process.
Addition of a Precipitating Agent: A chemical such as polyethylene glycol (PEG), ethanol, or ammonium sulfate is added to the solution. These substances alter the chemical properties of the liquid by reducing the solubility of the virus particles.
Salting Out: In salting-out, the added salt ions or molecules compete with virus particles for the available water molecules in the solution. As fewer water molecules remain available to solvate the viruses, their solubility decreases. This leads the viral particles to aggregate and form a visible precipitate – a process known as precipitation. The surface charges and biochemical properties of the virus particles, such as size and density, favor their precipitation over other components in the solution.
Centrifugation (in the figure: ZFG): To concentrate the precipitated virus particles, the sample is centrifuged. This accelerates the sedimentation of the viruses, which collect as a solid pellet at the bottom of the tube.
Washing: The pellet is washed with a buffer or solution to remove any remaining PEG, salts or other impurities.
Result: The solution now primarily contains the isolated viruses, which can be used for subsequent analyses.
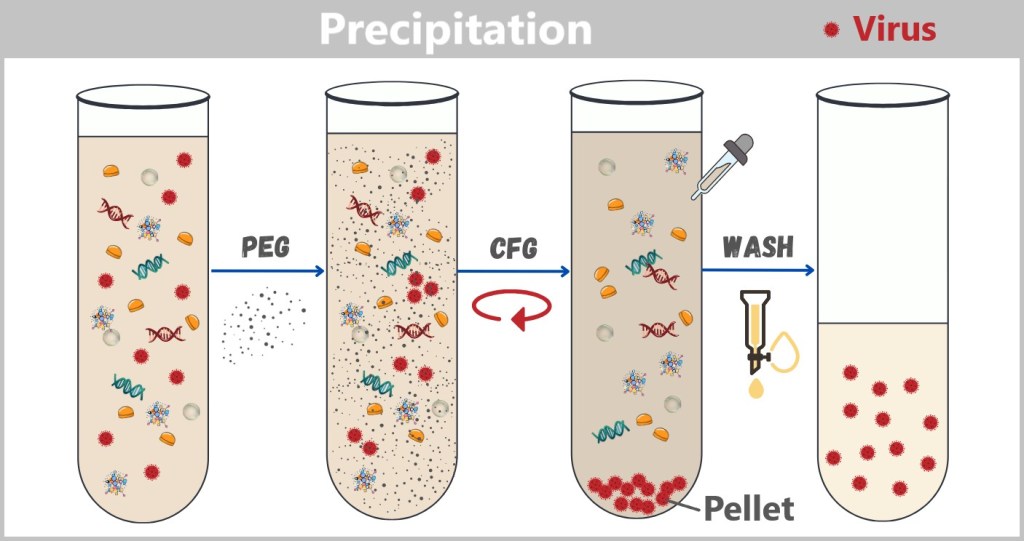
Fig. 5-E: Precipitation After centrifugation, the virus pellet is located at the bottom of the test tube, while the supernatant lies above. The supernatant is carefully aspirated or pipetted off to avoid disturbing the pellet. A washing solution, usually a buffer such as PBS (phosphate-buffered saline) or a similar medium, is added to the pellet. The volume of the washing solution is typically comparable to the original sample volume to effectively remove impurities. The pellet is resuspended by gentle pipetting, vortexing (light stirring with a vortex mixer), or by gently inverting the test tube so that it disperses in the washing solution. This step allows any remaining contaminants to transfer into the wash solution. After mixing, the test tube is centrifuged again, causing the viral particles to pellet once more. The wash solution (supernatant) is carefully removed without disturbing the pellet. This washing step is repeated one to three times as needed to ensure the removal of as many impurities as possible. After the final wash, the virus pellet is resuspended in a small volume of buffer (e.g., PBS or an analysis buffer) to prepare it for subsequent analyses.
Advantages of Precipitation
Widely applicable: Suitable for many types of viruses. However, it should be used with caution for very sensitive samples: especially enveloped viruses can be damaged by osmotic stress or nonspecific aggregation.
High Yield: Precipitation allows for effective concentration and purification with minimal loss of material.
Ease of Use: The process is cost-effective and does not require highly complex equipment.Precipitation is particularly well-suited for efficiently isolating large quantities of viruses.
d) Chromatography – The VIP Lounge for Viruses
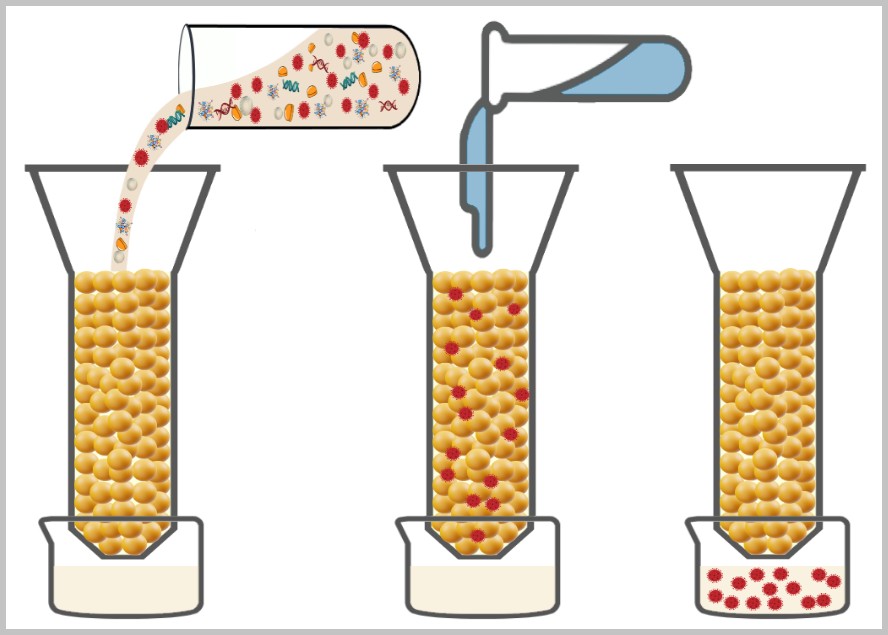
Chromatography is a highly precise purification step in virus processing.
In this method, viral particles are separated from other sample components based on their specific physical or chemical properties.
How it works: Columns separate viruses based on size, charge, or affinity
Advantage: Particularly gentle, precise and efficientMore Information
Principle of Chromatography
Chromatography is a method used to separate different substances within a mixture. In general, this process involves a mobile (moving) phase and a stationary (non-moving) phase. The mobile phase is the mixture of substances to be separated, which moves through the stationary phase, also known as the matrix. The substances in the mobile phase interact with the materials in the stationary phase. Due to these interactions, the individual components in the mixture move at different speeds or become temporarily trapped. This allows the different substances to be separated from one another.There are various types of chromatography. One commonly used method is ion exchange chromatography, which is based on the electrical charges of the particles. This technique is particularly well-suited for purifying viral particles, as viruses typically have a defined surface charge.
Procedure of Ion Exchange Chromatography (IEC)
Controlling Viral Charge:
The isoelectric point (IEP) of a virus indicates the pH value at which the virus carries no net electrical charge. By adjusting the pH value of the surrounding solution, the electrical charge of the virus can be purposefully manipulated:- At a pH value below the IEP, the virus becomes positively charged.
- At a pH value above the IEP, the virus becomes negatively charged.
Selection of the Matrix:
The matrix is selected to carry a charge opposite to that of the virus, allowing effective binding:- Anion exchangers: Positively charged matrix to bind negatively charged particles.
- Cation exchangers: Negatively charged matrix to bind positively charged particles.
Typically, the matrix consists of resins or membranes that are chemically modified to carry a specific surface charge.
Binding of the Viruses:
The sample is applied to the chromatography column, and the solution flows through the matrix. The viruses bind to the matrix due to electrostatic interactions. Other molecules, such as nucleic acids or proteins, bind more weakly or pass through the column unchanged.Washing:
Unbound or weakly bound impurities are removed from the matrix using a wash buffer. This step improves the purity of the bound viral particles.Elution (Release of the Viruses):
To detach the bound viruses from the matrix, an elution buffer is added. This can be done in two ways:- Change of pH value: The pH value is adjusted so that the charge of the viral particles is neutralized, causing the binding to the matrix to be lost.
- Increase in salt concentration: Additives such as sodium chloride or magnesium chloride neutralize the electrostatic binding between the viruses and the matrix.
Result:
The eluted viral particles are collected. Since impurities have already been removed during the washing steps, the eluted fraction primarily contains purified viruses, which can be used for further analyses or applications.
Fig. 5-F: Ion Exchange Chromatography Other Variants of Chromatography
Besides ion exchange chromatography, there are other types used depending on the target and sample:Affinity Chromatography: Uses specific binding between viral proteins and ligands on the matrix.
Size Exclusion Chromatography: Separates particles based on their size.
Hydrophobic Interaction Chromatography: Exploits the hydrophobic properties of the particles.
In practice, these methods are often combined. For example, a sample might first be filtered, then centrifuged, and subsequently further purified by precipitation and chromatography. The choice and sequence always depend on the type of sample and the goal of the analysis.
Why all this effort?
Simple: a bad sample gives bad data – like a blurry photo of Bigfoot. That’s why you need to work cleanly, keep things cool, and act fast… because molecules don’t forgive carelessness.
Sample preparation is not a side job – it’s the foundation. Without it, everything else falls apart. And once the viral material is well prepared, it’s time for the real deal: Now the viruses have to prove themselves – in cell culture.
4.3. Cell Culture
Viruses are the ultimate parasites: without a host cell, nothing works for them – no life, no reproduction. Alone – completely helpless. But give them a living cell and off they go. That’s why virology needs a reliable tool: cell culture. Without it, even the most dangerous virus dozes off like an office worker in the home office.
And since viruses don’t like to be alone, virologists set up a cozy home for them – a luxury shared flat in miniature: sterile petri dishes, perfect temperature, a nutrient-rich medium packed with vitamins and sugars – everything a virus’s heart desires. The cells used come from humans, animals, or plants – depending on which virus you want to pamper at the moment.
But not every virus is easy to handle. Some are demanding diva types that only thrive in specific cell lines. Others do it in anything that screams „Help!” And sometimes? Just… nothing happens. Then it’s: new cells, new luck.
Cell cultures are much more than just virus breeding stations. They enable:
➤ the replication of viruses in a controlled environment,
➤ the analysis of their properties, and
➤ the detection of infectious agents – especially with new or unknown viruses.Why Cell Cultures Are Absolutely Cool Despite PCR
PCR tests are fast, cheap, and widely available – like the fast food of diagnostics. But they only tell you: „Yes, there was virus here!” Whether the virus is still alive and infectious? No clue!
Cell culture = reality check.
Can the virus infect cells and multiply inside them? Yes? Then we are dealing with a real pathogen. No? Then it remains genetic ghost rubbish.Cell culture as a fitness test: indispensable when you want to:
➤ check a virus’s infectivity,
➤ test new viruses whose dangerousness is still unclear.
Viruses have to prove they’ve got what it takes first!Even in the age of PCR and high-throughput sequencing (more on that later), cell culture remains the practical test after the theoretical exam: „All well and good – but does it actually work?”
Because only a virus that can infect cells poses a real threat. Cell culture separates the wheat (active, infectious viruses) from the chaff (harmless genetic fragments). Especially in the era of synthetic biology, this becomes crucial: just because the genetic sequence looks right doesn’t necessarily mean the virus actually works.
Infection and observation of the cell culture
Preparation of the Cell Culture
Cells are cultured in a nutrient medium that contains essential nutrients, growth factors, and a suitable pH. This environment supports the survival and proliferation of the cells. Commonly used are cell lines that allow continuous growth, such as Vero cells (derived from monkey kidney epithelial cells) or HeLa cells (human cancer cells).
The choice of appropriate host cells is based either on initial clues from microscopic observations or on testing various cell lines. Researchers often use cell lines known to support viruses from specific sample sources. For example, cell lines derived from marine organisms are commonly used for ocean samples, while human epithelial cells are preferred when studying respiratory viruses.
Infection of the Cell Culture
To detect viruses in a sample, it is applied to a suitable cell culture. If the sample contains viruses, they can enter the cells, hijack their cellular machinery for replication, and cause characteristic changes known as cytopathic effects (CPE). How such cell alterations caused by viral infections can occur is illustrated in detail in Chapter „2.8. Destruction of the Host Cell”.
Detection and Observation:
After a certain period of time, the infected cells can be examined under a light or electron microscope for cytopathic effects (characteristic changes). These effects serve as visible indications of a successful infection:
➤ Cell shrinkage or clumping
➤ Formation of syncytia (multinucleated cell aggregates)
➤ Cell lysis (disintegration of cells)
Fig. 6: Infection of the cell culture and its observation 🎥 Time-lapse in real life: Tracking Influenza Virus Spread in Cell Culture
Reference to the influenza virus
The paper presents a comprehensive collection of images showing the typical cytopathic effects (CPE) following infection with the influenza virus. These visual representations illustrate the characteristic cell changes caused by the virus. However, the manual observation and evaluation of these effects is extremely labor-intensive and time-consuming. In order to optimize this process, the authors tested the use of artificial intelligence (AI), more precisely deep convolutional neural networks (CNNs), for automated image analysis.
This study impressively demonstrates how modern methods such as virology, cell biology and AI technology intertwine to develop innovative solutions to complex scientific problems.
Role and Applications of Cell Culture in Virology
Diagnosis of Known Viruses: Cell cultures are used to propagate viruses and detect their presence through specific changes in the cells.
Detection of Unknown Viruses: When the genetic or antigenic structure of a virus is unknown, cell cultures can help characterize its biological activity.
Study of the Viral Life Cycle: Cell cultures enable investigation of virus-host cell interactions, including entry, replication, and release of new viral particles.
Research on Virulence and Pathogenicity: They help to understand the mechanisms by which viruses damage cells and cause infections.
Analysis of the Immune Response: Cell cultures support the study of how the immune system reacts to viral infections and how infected cells trigger immune responses.
Testing of Antiviral Drugs: Cell cultures are used to evaluate the efficacy and safety of potential antiviral compounds and to develop new therapies.
Vaccine Production: Many vaccines, such as the measles vaccine, are produced using cell cultures.
Production of Viral Vectors: Cell cultures are essential for manufacturing viral vectors – genetically modified viruses that deliver therapeutic genes or vaccine information into cells, used in gene therapy, cancer treatment, or vaccines like those against Ebola or COVID-19 (e.g., AstraZeneca).
Limitations of the Method
Not all viruses are cultivable: Some viruses, such as Hepatitis B, require specialized cells or systems to replicate in vitro.
Viruses with very narrow host specificities: Some viruses rely on highly specific host cells, which can be difficult to reproduce in cell cultures, complicating research.
Insufficient representation of in vivo environments: Cell cultures offer only a simplified version of the natural host, which may lead to missing important virus-specific interactions or responses.
Lab-adapted viruses vs. natural viruses: Viruses that are cultured through multiple passages in cell lines may accumulate mutations that alter their characteristics. As a result, they can differ from their natural counterparts – for example, by having fewer spike proteins or altered host specificity.
Limited reproducibility of results: Cell culture conditions can influence cellular behavior, leading to variability in experimental results.
Time-consuming: Virus detection via cell culture is slower compared to molecular methods.
Complexity and cost: Establishing and maintaining cell cultures requires specialized laboratory equipment and expertise.
Biosafety risks: Working with highly pathogenic viruses requires strict safety standards, which impose additional requirements on laboratories and researchers.
Isolation and Further Processing
Cell cultures are the all-inclusive resorts for viruses – a place where they can replicate undisturbed. But, as with everything in life, even the best vacation comes to an end. For our tiny guests, it’s time to check out: „Checkout, please!” Their journey continues – straight to the high-tech labs. Now things get serious – and exciting. Because now we want to know: What exactly has grown there? And what does it look like? Welcome to the tools of virus analysis:
Selfie with the Electron Microscope: If a virus wants to know what it really looks like, electron microscopy gives it the ultimate close-up – sharper than any Instagram filter.
PCR and Sequencing: The Genetic Personality Test: A bit of genetic material here, a few enzymes there – and suddenly it becomes crystal clear who (or what) is actually sitting in that reaction tube.
ELISA: The Antibody Check: This test sniffs out antibodies in the blood – and reveals whether the immune system has already sounded the alarm. A suspicious find? Case closed!
In the next chapters, we’ll break down some of these methods – no lab coat required.
4.4. Making Viruses Visible
Viruses are like ghosts: you can feel their impact, but you never see them. They wreak havoc in cells, embed themselves in our DNA, cause diseases, shape evolution, and have been playing hide-and-seek with science for centuries. Even under the best light microscope, they remain invisible.
But as the saying goes: Seeing is believing. So how do we make the invisible visible? How can viruses be reliably detected? And how do we decode their structures and mechanisms?
The answer: we get out the really big microscopes! Devices so powerful that even the sneakiest viruses get caught red-handed – no disguise can save them. Modern imaging techniques can now dissect viruses down to the atomic level.
Welcome to the realm of modern visualization – where the invisible finally takes shape! And here we go with …
a) Electron Microscopy
b) Crystallization
c) Cryo-Electron Microscopy (Cryo-EM)
d) Cryo-Electron Tomography (Cryo-ET)
e) Summarya) Electron Microscopy: First Images of Viruses
The history of virology began with the realization that something smaller than bacteria must be responsible for causing diseases.
The physicist Richard Feynman put it in a nutshell in 1959:
„It is very easy to answer many… fundamental biological questions; you just look at the thing!”
But how can we visualize something that lies far below the resolution limit of a conventional microscope?
The invention of the electron microscope (EM) in 1931 made this possible for the first time: viruses could be directly visualized. The method uses electron beams instead of light to reveal extremely small structures. For the first time, scientists were able to image the characteristic envelopes and shapes of viruses, thereby confirming their physical existence.
Fig. 7-A: A look through the microscopes – a schematic illustration The illustration shows the differences in visualizing biological structures through two fundamental microscopy technologies.
On the left side, the view through the light microscope represents the classic capabilities used since the 17th century. Larger structures such as human cells are clearly visible here, as well as bacteria, which can be made visible through staining techniques. Viruses, however, remain invisible because their size (80–120 nm for influenza viruses) is far below the resolution limit of a light microscope.
On the right side, the view through the electron microscope demonstrates how modern technologies have overcome the limits of visibility. Not only are human cells and bacteria visible in much greater detail, but viruses can also be seen. The visualization even goes so far as to reveal high-resolution details such as the viral envelope and genome.Light microscopes provide a general overview and enable the analysis of living cells, while electron microscopes offer deeper insights into the world of microorganisms and viruses – down to molecular details.
The following video vividly illustrates the difference between light microscopes and electron microscopes, as well as how they function.
How does electron microscopy work?
Electron microscopes have a much higher resolution than light microscopes because electrons have a much shorter wavelength than light. This allows for the visualization of structures at the nanometer scale. There are two main types:
1️⃣ Transmission Electron Microscopy (TEM)
An electron beam penetrates an extremely thin sample.
This produces high-resolution 2D images of the internal structures.
(Earlier: low-contrast spots, Today: up to 0.05 nm resolution)
Especially useful for fine details inside viruses.2️⃣ Scanning Electron Microscopy (REM/SEM)
The electron beam scans the surface of a sample.
This produces detailed 3D images of the sample surface.
(Earlier: Rough outlines, Today: Molecular-level sharpness)
Well suited for the external structure of viruses.
Fig. 7-B: Transmission Electron Microscopy vs. Scanning Electron Microscopy
(schematic representation)Transmission Electron Microscopy: Provides a view through the virus, making internal structures visible. The virus usually appears as a two-dimensional projection with fine internal details. Electrons pass through the sample, and contrast is generated by interactions with varying densities of cellular or viral components.
Scanning Electron Microscopy: Shows a three-dimensional surface, often with a plastic, relief-like effect. The sample is scanned with electrons, producing a depth-sharp surface image. Internal structures are not visible because the electrons do not pass through the sample.Here you can see an original TEM image of an influenza virus particle.
Further information on the use of electron microscopy
Electron microscopy (EM) enables the investigation of various aspects of the influenza virus:
🔬 Insights into the ultrastructure of the influenza virus
Using transmission electron microscopy (TEM) with negative staining, scientists have gained detailed insights into the ultrastructure of the influenza virus. The study reveals the arrangement of spike proteins (hemagglutinin and neuraminidase) on the virus surface as well as the internal structure, including the matrix protein (M1) and the RNA, highlighting differences between intact and damaged virus particles.🔬 Insights into the Structure of Ribonucleoprotein Complexes (vRNPs)
Using electron microscopy, scientists were able to visualize the helical arrangement of the vRNPs within the virus particle. The article by Noda and Kawaoka (2010) provides a detailed description of the architecture of the vRNPs inside the virus particle and their packaging within virions. They emphasize that each of the eight vRNA segments is associated with nucleoproteins and a polymerase complex, forming the vRNPs, which are essential for the transcription and replication of the viral genome. Electron microscopic analyses have shown that these vRNPs possess a helical structure and are specifically arranged within the virion.
Electron Microscopy in Virology
Thanks to electron microscopy, viral structures and mechanisms can be uncovered, including:
✅ the shape and size of viruses (e.g., the spherical form of the influenza virus)
✅ the arrangement of spike proteins on the viral envelope
✅ and insights into the infection cycle, such as:
• how viruses enter the host cell
• how replication is initiated
Limits of Electron Microscopy
Despite its impressive resolution, electron microscopy has some drawbacks:
⚡ Sample preparation → Samples often need to be dehydrated, sectioned, and coated, which can alter their natural structure.
⚡ Samples must be fixed → meaning the sample is not examined in its natural state.
⚡ No living samples → because samples are analyzed in a vacuum to prevent electron beam scattering in air.
⚡ Radiation damage → the electron beam can alter or destroy sensitive samples.
⚡ Low contrast → biological samples often have low contrast and require special staining.
⚡ No „true” 3D image → TEM images are only two-dimensional, making it difficult to reconstruct complex structures.
Electron microscopy was the first big breakthrough: finally, viruses could be seen. But the early EM images still looked blurry – like moon photos from the 1960s. To get even closer to the viruses, more than just a microscope was needed. Atomic-level clarity was required. That’s where crystallization came into play – the ultra-HD version of virus research. Only when viral proteins are forced into perfect crystals do they reveal their molecular inner workings: atom by atom, bond by bond.
b) Crystallography: Detailed Structures of Viral Proteins
While electron microscopy could reveal the overall structure of viruses, their molecular fine structure – their atomic details – remained hidden for a long time. To understand how viral proteins are built, it was necessary to determine their atomic structure – and this was only possible through X-ray crystallography.
How did it come about?
The idea of analyzing biological macromolecules using X-ray diffraction emerged in the 1920s and 1930s. A groundbreaking breakthrough came in 1934, when physicist John Desmond Bernal, together with Dorothy Crowfoot Hodgkin, demonstrated that proteins can be crystallized in a hydrated form without losing their natural structure. This discovery was crucial for making X-ray crystallography a viable method for studying biomolecules.
In the 1930s, Wendell Meredith Stanley succeeded in isolating the tobacco mosaic virus in crystalline form. This was a milestone, as it proved that viruses are not just material particles, but are composed of regularly arranged molecules capable of forming crystals. Stanley managed to bring the virus into a solid, highly ordered structure – comparable to salt or sugar crystals. These virus crystals later enabled researchers to use X-rays to determine the atomic structure of viral proteins – marking a major breakthrough in the field of virology.
Stanley’s Breakthrough and Its Significance
How Did Stanley Perform the Crystallization?
Stanley developed an innovative method to isolate and analyze the tiny virus particles. He extracted the viruses from infected tobacco plants and purified them through centrifugation and additional purification techniques. He then dissolved the purified viruses in a solution, which he allowed to evaporate slowly. Through this process, the viruses crystallized, forming solid structures that he was able to examine more closely using an electron microscope. This marked the first step toward analyzing the structure of viruses at the atomic level.
Why Was Crystallization So Significant?
The crystallization of the tobacco mosaic virus led to two groundbreaking insights:
Proof of Viruses as Physical Particles: Until then, viruses were considered vague, invisible disease agents. Stanley demonstrated that they are physical entities with a clearly defined structure – so much so that they can even be crystallized.
Foundation for Structural Analysis: The crystals enabled later researchers to use X-ray crystallography to decipher the architecture of viruses and reveal that they consist of genetic material – either RNA or DNA – and proteins. This discovery was crucial for understanding how viruses function.
What did the structural analysis reveal?
The study of the tobacco mosaic virus showed for the first time that viruses have a clear and repeatable structure:
Genetic material and proteins: A virus’s genetic material contains the instructions for producing new virus particles, while the proteins form the protective coat and enable functions such as entry into host cells.
Distinction from other microorganisms: Stanley’s work clarified that viruses are fundamentally different from bacteria and other microorganisms. Viruses are significantly smaller, lack a cellular structure, and consist of only a minimal set of components, making them unique biological entities.
What is crystallization?
Crystallization is the physical process in which atoms, molecules, or ions transition from a disordered phase (such as a solution, melt, or gas) into a solid, ordered structure called a crystal. This transition results in the formation of a crystal lattice – a regular, three-dimensional arrangement where the particles organize themselves in a repeating pattern. During this process, the particles arrange in a way that achieves an energetically favorable state, defined by specific interactions (e.g., hydrogen bonds, electrostatic forces, van der Waals forces) and bonding angles.
Crystallization is thus the transition from chaos (disordered particles) to order (crystal lattice), driven by physical forces and energetic principles. It can occur naturally (e.g., during mineral formation) or artificially (e.g., in laboratories).
Fig. 8-A: Example of natural crystallization through the evaporation of a salt solution. Left: In a salt solution, sodium ions (green) and chloride ions (blue) are freely moving and disordered. Right: As the water evaporates, the ions arrange themselves into a regular crystal lattice.
Basic Principle of Protein Crystallization
Protein crystallization means transforming dissolved proteins from a solution into a solid, ordered, crystalline form. The goal is to create a three-dimensional lattice in which the protein molecules are regularly arranged. This is achieved by carefully reducing the solubility of the proteins so that they slowly „precipitate” out of the solution and organize into a crystal.
The typical process is as follows:
Purification: The protein is isolated in a highly pure form, as impurities can disrupt crystal formation.
Preparation of solution: The protein is dissolved in an aqueous solution containing buffers, salts, and sometimes organic additives (e.g., polyethylene glycol, PEG).
Supersaturation: By altering conditions (e.g., evaporation, addition of precipitants like PEG or salts), the solution becomes supersaturated, causing the proteins to start precipitating.
Nucleation and crystal growth: Initially, small protein aggregates (nuclei) form, which then grow into larger crystals if conditions are favorable.
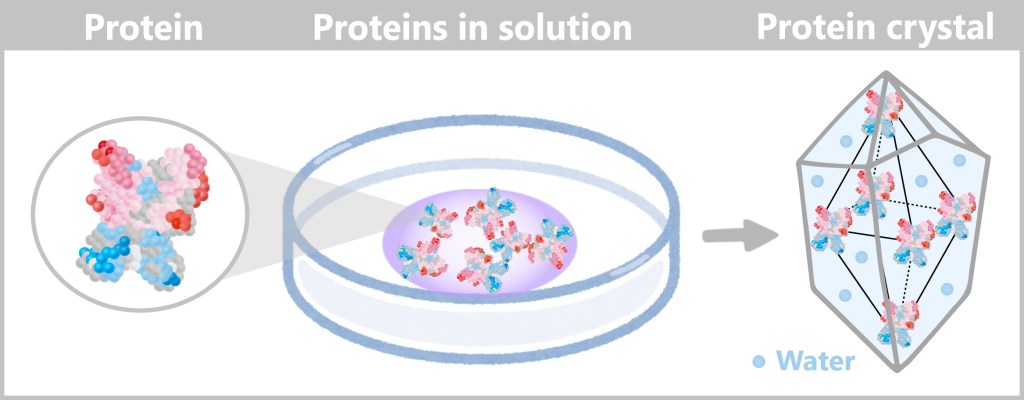
Fig. 8-B: Schematic representation of protein crystal formation Proteins are initially dissolved in solution in a disordered state. By deliberately changing the conditions (e.g., increasing salt concentration, altering pH levels, or slow evaporation of the solvent), the solution becomes supersaturated. This causes the proteins to start organizing and grow into a crystal.
At the molecular level, proteins align into a regular lattice due to their charges, hydrogen bonds, and hydrophobic interactions. The molecules „search” for the most energetically favorable position, which leads to the formation of a stable crystal. Water molecules, represented by small blue dots, are found between the proteins. This water is an integral part of the crystal and stabilizes the protein structure through hydrogen bonding. Protein crystals often exhibit a symmetrical structure (e.g., cubic or hexagonal) because the molecules arrange themselves in repeating patterns.
Proteins are huge molecules composed of thousands of atoms, with complex 3D shapes and irregular surfaces (including charged regions, hydrophobic areas, etc.). They cannot simply be stacked alternately like sodium and chloride ions in a salt crystal.
Characteristics of Protein Crystals
The strength: Protein crystals are significantly more fragile than classic crystals like salt or diamond. They contain 30–70% water, which is trapped in channels and cavities within the crystal lattice. This makes them soft and gel-like. Mechanical stress or drying out can easily damage them.
Color: Protein crystals are usually colorless or slightly opaque (non-transparent). Proteins themselves do not have a natural color – the colorful representations in scientific illustrations are used solely to highlight structural features and chemical properties.
Selection of the Protein Crystal
During crystallization, many small crystals often form simultaneously because protein molecules start to arrange themselves at different spots in the solution. However, for analysis, only the most well-ordered crystal is selected.
How do you find the right crystal?
In the past, crystals were examined under a microscope: Clear, sharp edges and a regular shape (e.g., cubic or hexagonal) indicated high quality. Cloudy or irregular crystals, on the other hand, were less suitable.
Today, modern image analysis systems are used that combine high-resolution microscopy with automatic evaluation. Additionally, X-ray diffraction can be performed: sharp, symmetrical diffraction patterns indicate good crystal order, while diffuse or irregular reflections suggest poor quality.
Why are crystals needed?
In short: Protein crystals serve as „amplifiers” for X-rays.
A single protein molecule would produce only an extremely weak and diffuse signal during X-ray diffraction – too little to determine a detailed structure. In a crystal, however, millions of identical protein molecules are regularly arranged and oriented the same way. This causes the diffraction signals from the individual molecules to reinforce each other through constructive interference, resulting in a clear and regular diffraction pattern.
This ordered amplification is essential for X-ray crystallography, as it is only through the resulting diffraction pattern that the three-dimensional structure of the protein can be deciphered. Without crystals, analysis using this method would be impossible.
X-ray crystallography procedure
Once the protein has been successfully crystallized, its spatial structure (i.e., its precise folding and arrangement) can be determined using X-ray crystallography. This method reveals details such as:
➤ The position of each individual atom
➤ The folding structure of the protein (α-helices, β-sheets, etc.)
➤ Interactions with other molecules (e.g., antibodies, drugs)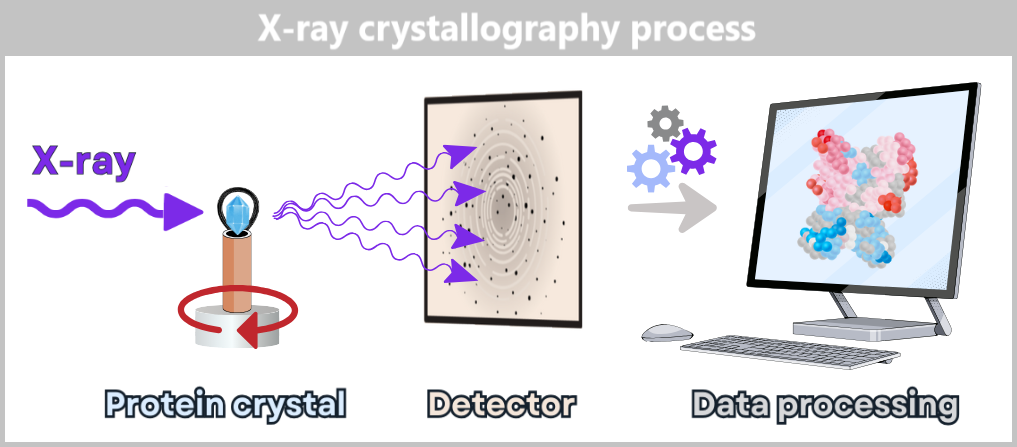
Fig. 8-C: Schematic representation of X-ray crystallography 1️⃣ Shooting the X-ray beam at the crystal
A focused X-ray beam hits a protein crystal. Proteins are made up of amino acids, which in turn consist of atoms. X-rays have a wavelength of about 0.1 nm, which is on the scale of atoms, making them well-suited to reveal details at this level. The atoms in the crystal do not directly diffract the X-rays; rather, their electron clouds deflect the rays in specific directions.
2️⃣ Diffraction pattern is generated
The deflected X-rays interfere with each other and create a characteristic pattern on a detector. This diffraction pattern consists of many spots (called reflections) that appear at different positions and with varying intensity. To capture complete data, the crystal is rotated in small increments (typically 0.1–1° steps), with a diffraction image recorded at each angle.
3️⃣ Mathematical calculation of the 3D structure
The various diffraction images do not directly show the structure of the protein. Instead, they contain information about how the X-rays were scattered by the electrons of the atoms in the crystal. Using mathematical methods (Fourier transformation), these diffraction patterns are converted into an electron density map. This map reveals the three-dimensional distribution of electrons within the crystal.
Since electrons are primarily located near atomic nuclei, the positions of atoms can be inferred from the electron density map. Initially, the map appears as a „cloudy” structure, with regions of high electron density corresponding to the atoms.
The quality of the electron density map depends directly on the order within the crystal – the better the crystals, the sharper the electron density map. Through further analysis and interpretation of this map, a detailed, three-dimensional model of the protein can ultimately be constructed.
〰️ Natur Synchrotron radiation – light for nature’s smallest secrets
To examine the structure of proteins even more precisely, scientists often use synchrotron radiation – an extremely intense form of X-ray radiation generated in special particle accelerators.
💡 What exactly is it?
Synchrotron radiation is produced when charged particles (e.g., electrons) are accelerated to nearly the speed of light and then steered into a circular path by magnetic fields. In doing so, they emit high-energy radiation – including exceptionally brilliant X-rays that are ideal for protein crystallography. This allows even tiny or difficult-to-crystallize proteins to be studied in detail.
🌍 There are several major synchrotron research centers around the world, such as:
- ESRF (France) –European Synchrotron Radiation Facility
- DESY (Germany) – German Electron Synchrotron
- Diamond Light Source (UK) – Synchrotron facility in the United Kingdom
🎥 If you would like to experience live how proteins become crystals and how their structures are decoded with the help of X-rays, then join the researchers at the Diamond Light Source in the fascinating world of crystallography!
Understanding Crystallography – Part 1: From Proteins to Crystals
Here you can see how scientists transform proteins into crystals.Understanding Crystallography – Part 2: From Crystals to Diamond
In this video, you can see how the crystals are examined using X-rays to reveal their 3D structure.
The Protein Data Bank – A Treasure of Structural Biology
X-ray crystallography has not only helped to determine the structure of individual proteins but also enabled the creation of a global resource: the Protein Data Bank (PDB). This database collects and stores the 3D structures of proteins, nucleic acids, and other biomolecules.
The PDB was founded in 1971 and today contains over 200,000 entries. Until the 1970s, X-ray crystallography was the only method for determining the three-dimensional structure of proteins at atomic resolution.
In the following decades, new techniques such as nuclear magnetic resonance spectroscopy (NMR) and later cryo-electron microscopy (Cryo-EM) expanded the range of methods. While NMR is especially suitable for small proteins in solution, Cryo-EM allows the study of large protein complexes without the need for crystallization.
Each protein structure provides detailed information about the spatial arrangement of atoms within a molecule. X-ray crystallography has not only revolutionized our understanding but has also created a global infrastructure of knowledge. A silent library where every protein tells its story – captured with atomic precision. Accessible to researchers worldwide.
X-ray Crystallography in Virology
Viruses are complex entities composed of proteins, nucleic acids (DNA or RNA), and sometimes lipids. Entire viruses are often too large and flexible to be crystallized and studied using X-ray crystallography.
However, some viruses have regular, symmetrical structures that allow for dense, repetitive packing, enabling crystallization. A well-known example is the tobacco mosaic virus, whose structure was deciphered using X-ray crystallography as early as the 1950s.
For most viruses, however, it is more practical to study individual viral proteins. Of particular interest are proteins that the virus uses to infect host cells, replicate, or evade the immune system.
To provide an example of a viral protein structure, we turn to the Protein Data Bank (PDB). The following image comes from the PDB and shows the hemagglutinin protein of the H5N1 influenza virus. Its atomic structure was determined using X-ray crystallography and deposited in the Protein Data Bank.
Fig. 8-D: Atomic structure of the hemagglutinin protein of the H5N1 influenza virus (PDB ID: 2FK0), shown as a ball-and-stick model. Instructions for Searching the Protein Data Bank
1️⃣ Open the Protein Data Bank (PDB): https://www.rcsb.org.
2️⃣ Search for a protein:
In our example, we are looking for the hemagglutinin protein of the influenza virus.
👉 Enter the term „influenza hemagglutinin” into the search bar.
👉 Filter the results under [Experimental Method] by selecting [X-ray diffraction].3️⃣ Select an entry:
👉 Click on a result. For our example, choose PDB ID: 2FK0 – the hemagglutinin protein of the H5N1 influenza virus.
4️⃣ View the protein:
On the left side of the PDB page, you’ll see the cartoon representation or ribbon diagram, a schematic view of the protein’s 3D folding.
5️⃣ Visualizing the atomic structure:
👉 Click on [Structure] to switch to the 3D view.
👉 Choose a different viewer: „JSmol” (see bottom right).
👉 For the atomic structure view, select the [Style]: „Ball-and-Stick.”
🔍 Now every atom and bond is visible. You can rotate, zoom, and move the model with your mouse.6️⃣ Highlighting structural features:
👉 Under [Color], you can choose different color schemes. For example, „By chain” colors each protein chain differently to distinguish the protein’s subunits.Further information on the use of X-ray crystallography
For over 40 years, X-ray crystallography has been a powerful method for determining virus structures. Since we have already explored the influenza virus surface protein hemagglutinin, the following studies relate to it.
🔗 One of the first studies was published in 1981: „Structure of the haemagglutinin membrane glycoprotein of influenza virus”. It revealed the three-dimensional structure of the HA protein and laid the foundation for understanding the infection mechanisms of the influenza virus.
🔗 A more recent study from 2019 titled „Identification of a pH sensor in Influenza hemagglutinin using X-ray crystallography” investigated the binding region of the HA protein using X-ray crystallography. It showed how conformational changes in the binding region, depending on the pH level, affect the virus’s function.
🔗 A 2020 study titled „Structure of avian influenza hemagglutinin in complex with a small molecule entry inhibitor” used X-ray crystallography to elucidate the structure of the H5 hemagglutinin (HA) protein of the H5N1 influenza virus in complex with the inhibitor CBS1117. The crystal structure provided detailed insights into the molecular interactions that are crucial for the development of new antiviral drugs.
By combining the structures of individual proteins, scientists can assemble a detailed picture of the entire virus.
Limits of X-ray crystallography
Although X-ray crystallography is an incredibly powerful method for structure determination, it does have certain limitations:
⧎ Challenging crystallization: Many proteins – especially large complexes or membrane proteins – are difficult or even impossible to crystallize.
⧎ Artificial conditions: Proteins are fixed in a crystal lattice, which may not fully reflect their natural state.
⧎ Lack of dynamics: The method provides only a static snapshot and does not capture molecular motion or flexibility.
⧎ Radiation damage: The high-energy X-rays can easily alter sensitive molecules.
In Search of a New Method
These limitations made one thing clear: not every biomolecule can be forced into a crystal like Lego bricks. The solution? A microscopic flash-freeze: cryo-electron microscopy (cryo-EM). Its trick? Molecules are shock-frozen within milliseconds – so fast that water doesn’t have time to crystallize. No crystal straitjacket, no X-ray sunburn – just ice-cold atomic details.
c) Cryo-Electron Microscopy: A Modern View of Viruses
Having explored classical electron microscopy and X-ray crystallography – which first opened the door to understanding virus structures – cryo-electron microscopy, or cryo-EM for short, now leads us into a new era of research.
What is cryo-EM, exactly?
Cryo-EM is a technique in which samples are frozen extremely quickly („cryo” = cold) and then examined using an electron microscope. What makes it special is that the samples remain in an almost natural state, since they don’t need to be chemically fixed or stained as required by other methods. This makes it ideal for observing sensitive structures such as viruses.
How did it come about?
In the past, scientists had a major problem when they wanted to analyse biological samples with electron microscopes: Water evaporated in the vacuum of the microscopes and sensitive molecules were damaged by the intense electron beam. This resulted in blurred or distorted images.
Researchers had already come up with the idea of cooling samples to protect them. But there was another problem: when water freezes, it forms ice crystals. These crystals scatter electrons so strongly that capturing clear images became impossible.
The crucial breakthrough came in the 1980s, when scientist Jacques Dubochet developed a technique called plunge freezing. In this method, the sample is cooled extremely rapidly to very low temperatures – so fast that ice crystals don’t have time to form. Instead, the water solidifies into a glass-like, frozen state that perfectly preserves the biological sample. This marked the birth of cryo-electron microscopy (cryo-EM).
Another major advancement came in the 1970s, when Joachim Frank developed a method to improve the blurry images produced by electron microscopes using computer-based calculations. By combining numerous images of individual molecules taken from different angles, he was able to reconstruct a sharp 3D model. This made it possible, for the first time, to determine the structure of biomolecules without the need for crystallization – a significant advantage for proteins that are difficult or impossible to crystallize.
In 1990, Richard Henderson used cryo-EM for the first time to study a biomolecule at such high resolution that even small details like amino acid side chains became visible. For these groundbreaking developments, Dubochet, Frank, and Henderson were awarded the Nobel Prize in Chemistry in 2017.
Since then, the technology has advanced rapidly:
↗️ Modern cameras capture razor-sharp images directly, without loss of quality.
↗️ Automated microscopes can analyze multiple samples simultaneously.
↗️ Powerful computer programs enable the processing of massive amounts of data to calculate even more precise structures.Thanks to these advances, cryo-EM can now visualize complex molecules, proteins, viruses, and even entire cell structures in their natural state – with unprecedented levels of detail.
Next, we’ll take a closer look at how cryo-EM makes viruses visible.
Making Viruses Visible with Cryo-EM – Step by Step
1️⃣ Sample Preparation: Growing and Isolating Viruses
To study viruses, you first need to have them. They are usually grown in suitable host cells, such as cell cultures in the lab. Viruses are parasites that can only reproduce inside host cells. To study them, you infect appropriate cells in a culture and allow the viruses to multiply.
After infecting the cells, the viruses are „harvested” at specific time points to study different stages of their life cycle (e.g., attachment, entry, replication). The viruses are then isolated and purified from the cell culture – using methods like centrifugation or filtration – to obtain a clean virus sample free of interfering cell debris.
2️⃣ Sample Preparation: Applying the Sample to a Grid
The purified virus sample is a watery solution in which the viruses are suspended. This virus solution is applied with a pipette onto a grid (see the image below).
A typical cryo-EM grid is tiny, about 3 millimeters in diameter, so it fits into the electron microscope’s holders. The grid is made of a fine metal mesh (e.g., copper or gold) that looks like a sieve. An ultrathin carbon foil with tiny holes – usually 1 to 2 micrometers in diameter – is placed on the metal mesh. Due to surface tension, the virus solution spreads into these tiny holes and sticks there before freezing. This setup serves two important purposes: the grid provides stability, while the holes allow the electron beam to pass through freely, minimizing background noise.
Fig. 9-A: Schematic representation of cryo-EM sample preparation A – Grid covered with a perforated carbon film,
B – Enlarged image of a grid opening, called a mesh hole,
C – Enlarged image of a hole where the viruses adhere3️⃣ Rapid Freezing of the Sample
The sample – or the grid – is now flash-frozen by plunging it into liquid ethane or nitrogen at around –180 °C. This process is called vitrification – the water doesn’t form crystals but instead turns into a glass-like state that perfectly preserves the structure. This keeps the viruses in their natural state, as if frozen in time. Additionally, the ice protects the molecules from damage caused by the intense electron beam.
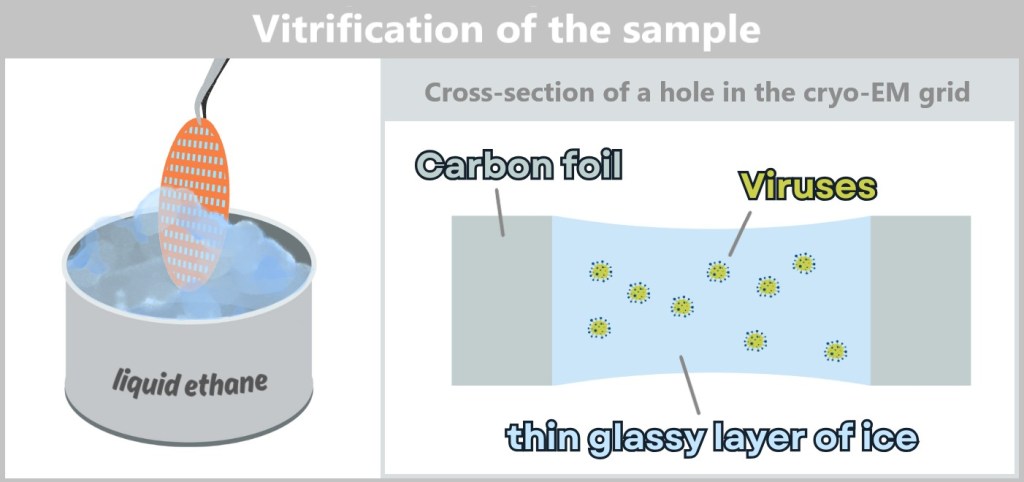
Fig. 9-B: Right – Schematic cross-section of a hole in the cryo-EM grid, where viruses are embedded in vitrified (glass-like) ice. 4️⃣ Imaging in the Electron Microscope
The frozen grid with the viruses is placed into the cryo-electron microscope. In the vitrified solution, the viruses are randomly oriented – they are positioned at various angles. The electron beam hits the sample from a fixed direction (usually perpendicular), and a highly sensitive camera captures 2D projections of the viruses.
Because each virus is frozen in a different orientation, images are automatically captured from many different angles – without needing to rotate the grid. This large number of images is crucial for the later 3D reconstruction. In total, often thousands to hundreds of thousands of images are taken.
5️⃣ Data Processing: From 2D Images to a 3D Model
After image acquisition, the real challenge begins: computational reconstruction. Specialized software identifies individual virus particles in the 2D images and sorts them according to their orientation.
Using mathematical methods such as „Single Particle Analysis”, the 2D images are combined to compute a high-resolution 3D model of the virus. The more images and the better their quality, the sharper the model becomes.
If different stages of the virus life cycle are to be studied (e.g., before and after entering a cell), this process is repeated with samples taken at different time points. This way, changes in the structure over time can even be visualized.

Fig. 9-C: Steps of the cryo-EM analysis The following videos provide a clear summary of the information about cryo-EM covered so far:
🎥 What is Cryo-Electron Microscopy (Cryo-EM)?
🎥 Cryo-EM Animation6️⃣ Interpretation and Visualization
After the 3D reconstruction, a high-resolution model of the virus is available, showing its structure – such as the envelope, spike proteins, and sometimes even internal structures.
Now it’s time to analyze, interpret, and visually present the icy data. This step is crucial for deciphering the biological significance of the structure and making the findings accessible for further research or public understanding.
Interpretation and visualisation steps
a) Structure Analysis: Scientists examine the 3D structure to identify key features of the virus or its proteins. This analysis helps to understand the molecular mechanisms of viral infection. This includes, for example:
- Binding sites: Where does the virus attach to host cells?
- Functional regions: Which parts of the protein are essential for infection or replication?
- Structural variations: Are there differences in the structure between different virus strains or life cycle stages?
b) Comparison with Known Structures: The reconstructed structure is compared with previously known structures from databases such as the Protein Data Bank (PDB). This can provide insights into evolutionary relationships, functional similarities, or potential targets for drug development.
c) Visualization of the Structure: Using specialized software, the 3D model is visualized to clearly present the structure and highlight important features. Various forms of representation are used, such as:
- Surface representation: Shows the external shape of the virus or protein.
- Ball-and-stick model: Displays the arrangement of atoms and bonds.
- Secondary structure: Highlights structural elements such as α-helices and β-sheets.
d) Publication and Data Sharing: The results are published in scientific journals – often accompanied by high-resolution images or animations of the 3D structure. In addition, the raw data and reconstructed models are stored in public databases such as the PDB (Protein Data Bank) or the EMDB (Electron Microscopy Data Bank), allowing other researchers to access and build upon the findings.
If viruses knew how often we can now rotate and zoom in on them in 3D… they’d put some clothes on!
Influenza Virus in 3D: An Example
It can be challenging to find 3D models of viruses online. Research data is often released only after studies are published and then stored in specialized databases like the EMDB (Electron Microscopy Data Bank). However, this data is difficult for non-experts to access, as it requires specialized software to view it as 3D models.
Fortunately, the NIH 3D platform (National Institutes of Health) offers a helpful example: a 3D model of an influenza virus created using cryo-electron microscopy. You can view it via this link! Please note that you may need a 3D viewer to explore it.
Further information on using the cryo-EM
🔬 This study on the influenza virus used cryo-electron microscopy to reveal the interactions between the nucleoproteins and the viral RNA as well as the specific arrangement of the ribonucleoprotein complexes (RNPs) within the virion. Such structural insights are crucial for understanding the mechanisms by which the influenza virus packages its genome and replicates during the infection cycle.
🔬 Additionally, an earlier study by Liu et al. (2017) revealed structural details of filamentous influenza virus particles and their RNP organization using cryo-EM.
Why Cryo-EM Is So Brilliant – Especially for Viruses
Cryo-EM is an unbeatable tool when it comes to visualizing viruses – especially those tricky, hard-to-capture candidates:
- Viruses with their tricky spike proteins
- Huge protein complexes that refuse to crystallize properly
- Membrane proteins that otherwise just say „Hello!” and fall apart immediately
The method: It freezes viruses in the blink of an eye, preserving them like in a time capsule, and produces thousands of images using an electron beam. A computer then assembles these images into a high-resolution 3D model – allowing us to see exactly how a virus is structured. This approach is called Single Particle Analysis (SPA) because individual virus particles are analyzed and computationally combined into a 3D structure.
From Still Frames to Blockbusters
What cryo-EM delivers in detailed „snapshots” is impressive – but sometimes one picture just isn’t enough. To understand how viruses move, hijack cells, or replicate, you need moving scenes. This is exactly where cryo-electron tomography comes in: a technique that lets you watch viruses at work almost like in a movie.

d) Cryo-Electron Tomography – 3D Virus Models Inside the Cell
While cryo-EM examines highly purified, isolated viruses, cryo-ET allows scientists to capture three-dimensional snapshots of molecular interactions directly within the cell. This makes it possible to visualize viruses in their natural environment – inside the cell.
Making Viruses Visible with Cryo-ET – Step by Step
1️⃣ Sample Preparation – Freezing the Cell
Suitable cells are grown in cell culture and infected with viruses. After waiting for a defined time point – for example, when the viruses enter the cell or begin replicating – the cells are harvested. They are then transferred into a buffer solution to create a homogeneous cell suspension.
The cell suspension is then applied to a cryo-EM grid – a small metal grid with a perforated carbon film, already familiar from cryo-EM techniques.
The grid is immediately plunged into liquid ethane or liquid nitrogen (at around -180 °C). This rapid freezing technique, known as vitrification, preserves the cellular structures without the formation of disruptive ice crystals.
Fig. 10-A: Schematic illustration of cryo-ET sample preparation followed by vitrification 2️⃣ Thinning cellular areas (if necessary)
Vitrification stabilizes the cells – they are now rigid like glass. Since cells are often too thick for cryo-ET, scientists use a Focused Ion Beam (FIB) to thin the ice layer down to the desired thickness.
This is how it works: The cryo-EM grid is first scanned in a FIB-SEM device (Focused Ion Beam combined with Scanning Electron Microscope) to locate interesting cells. Then, a focused ion beam precisely mills a thin lamella (~100–200 nm thick) from the ice layer. This lamella represents a cross-section of the cell and reveals relevant areas containing virus particles – for example, the cell membrane or the cytoplasm with viruses.
Here’s how it looks in practice: the video titled „Cryo-lamella preparation” in the section „FIB-milling of lamella in waffle grids” on the page „Cryo EM 101, Chapter 3” demonstrates the process.
3️⃣ Image acquisition in cryo-EM
The frozen cell grid is placed into the cryo-electron microscope. Here, an electron beam generates high-resolution images of the sample.
Tomography – Like a CT scan for cells
The sample is gradually rotated. A goniometer (rotating mechanism) tilts the grid in small angular steps (e.g., 1–2°), typically over a range of ±60° to ±70°. For each angle, a 2D image is taken, creating a so-called „tilt series” – a collection of 2D images from different perspectives (see illustration below).
📌 Cryo-ET vs. SPA: Unlike Single Particle Analysis (SPA), which requires many identical viruses, Cryo-ET focuses on a specific area of the sample – often a single cell or a virus-host cell interaction. This means there is no need for a large number of identical particles; instead, individual biological structures can be studied directly in their natural environment.
4️⃣ 3D Reconstruction
Cryo-ET uses the tilt series to computationally reconstruct a 3D model – like a medical CT scan, but for viruses instead of bones. The result is a hologram-like volumetric image that shows how viruses dance, dock, and trick inside real cells.
Fig. 10-B: From the electron beam to the 3D reconstruction 5️⃣ Interpretation & Visualization
Now things get exciting! The 3D models show viruses in their natural environment:
- how they sit inside the cell
- how they interact with cellular organelles
- different stages (e.g., entry, replication, release)
The results are interpreted to understand, for example, the virus’s infection mechanism or its interaction with the host cell.
Sometimes multiple time points are compared to create a kind of „movie” that simulates the dynamics.
The following video gives an impression of this: detailed 3D models of an influenza virus during budding at the cell membrane were visualized and then compiled into a film.
6️⃣ Publication and Data Storage
To make the spectacular virus models accessible to everyone, they are deposited in public databases like the Electron Microscopy Data Bank (EMDB) – the Netflix for structural biologists.
Why that’s cool:
- Open Science = No scientist has to reinvent the wheel twice.
- Transparency = Everyone can see exactly how the „movie” was made.
- Collaboration = Teamwork makes virus research a hit too.
If viruses had Twitter, they’d be trending now with #EMDBChallenge.
Further information on using cryo-ET
🔬 The study „Quantitative Structural Analysis of Influenza Viruses Using Cryo-Electron Tomography and Convolutional Neural Networks” utilized cryo-electron tomography (cryo-ET) and modern computational techniques to investigate the structure of the influenza virus. It reveals the diverse shapes of the virus and includes videos from 3D reconstructions that illustrate the process.
🔬 The study „Structural Changes in Influenza Virus at Low pH Characterized by Cryo-Electron Tomography” investigated how the influenza A virus responds to acidic conditions it encounters during entry into host cells. Using cryo-electron tomography, the researchers observed that at low pH-value, the viral envelope proteins (hemagglutinin) undergo structural changes that lead to the fusion of the viral and cellular membranes. This fusion enables the virus to release its genetic material into the host cell and initiate infection.
Researchers are not only investigating human pathogenic viruses
Researchers are no longer interested only in pathogens like influenza or coronavirus – environmental viruses are equally fascinating. Chief among them are bacteriophages, viruses that infect bacteria. In the oceans, they act as true ecological engineers.
1️⃣ Tiny directors in the ecosystem
Phages specifically kill certain bacteria, keeping the microbial balance in the ocean stable. In doing so, they indirectly regulate important cycles such as the carbon and nitrogen cycles. Understanding their structure and behavior helps us better grasp how stable marine ecosystems function.
2️⃣ Experience evolution live
Phages are the most abundant biological „organisms” on Earth – and genetically incredibly diverse. Using cryo-EM, researchers are constantly discovering new virus families. Their genetic codes end up in databases, telling a story about the co-evolution of viruses and bacteria.
3️⃣ From the sea to the lab
Some marine phages produce enzymes with real potential: for genetic engineering, diagnostics, or even as alternatives to antibiotics. They could also help in agriculture by targeting harmful bacteria – without the use of harsh chemicals.
4️⃣ And where do the data go?
Structural and gene sequences are made openly accessible – for example, in the NCBI Virus Database or the EMDB. Bioinformaticians explore these databases to discover new genes, clever mechanisms – and sometimes ideas for the next biotechnological innovation.
Limits of cryo-EM
Cryo-EM (including Single Particle Analysis (SPA) and Cryo-Electron Tomography (Cryo-ET)) are groundbreaking techniques, but they also have some limitations that depend on technical, biological, and practical factors:
Sample preparation is challenging
Problem: The samples must be extremely thin (only a few hundred nanometers) and perfectly vitrified without ice crystals, which could damage the structure. This is technically difficult and requires a lot of expertise.
Impact on viruses: If the virus solution is not frozen evenly or is too thick, the images can become blurry or the viruses may be damaged. In cryo-ET, thick cell samples can scatter the electrons too much, which worsens the image quality.
Limited resolution
Problem: Although cryo-EM can deliver very high-resolution images (up to 2–3 Ångström under optimal conditions), the resolution depends on the quality of the sample and the amount of data. For complex or variable structures (e.g., flexible viruses), the resolution can suffer.
Impact on viruses: The tiniest details (e.g., individual atoms in proteins) are not always visible, especially when viruses are moving or have variable shapes.
Limited dynamics: frozen in time
Problem: As impressive as the 3D models are, they always show just a frozen moment in time. Real movements or processes – like a virus entering a cell live – remain out of reach.
Impact on viruses: With Single Particle Analysis (SPA), we only see one structure at a specific moment in time. Even with Cryo-ET, researchers must freeze different cells at various „stages” of infection to reconstruct a timeline. It sounds like a movie – but it’s more like stop-motion reconstruction than a live stream.
High costs and complexity
Problem: Cryo-EM requires expensive equipment (electron microscopes cost millions), specialized laboratories (e.g., vacuum and cryo conditions), and a lot of computing power for data analysis. This makes the method time-consuming and costly.
Impact on viruses: Only well-equipped research institutions can use cryo-EM, which limits access. Small labs or developing regions often cannot apply this technique.
Noise and Data Volume
Problem: The images are often noisy because only a limited number of electrons are used to avoid damaging the sensitive samples. Processing requires thousands of images, which is computationally intensive.
Impact on viruses: If the amount of data is insufficient or the noise level is too high, the 3D models can become inaccurate.
Limited sample size
Problem: Cryo-EM only works with very small samples (e.g., individual viruses or cells). Larger structures like whole tissues are difficult to study because the electrons cannot penetrate deeply enough.
Impact on viruses: Cryo-ET can image cells, but only up to a certain thickness (about 1 micrometer). Larger organisms or tissues are not suitable for this technique.
Sources
Cryo Electron Microscopy: Principle, Strengths, Limitations and Applications
Challenges and triumphs in cryo-electron tomography
e) Summary
Science has developed increasingly powerful methods over decades to make the world of viruses visible. While the naked eye and light microscopes quickly reach their limits, electron microscopy, X-ray crystallography, and finally cryo-EM and cryo-ET have provided entirely new insights. These technologies have revolutionized our understanding of viruses.

Fig. 11: From Atoms to Macrostructures – An Overview of Imaging Techniques Scaling of Imaging Techniques
➡️ Human Eye
Visible range: approx. 0.1 mm (100 µm) to several meters
Example: Hair, grain of sand➡️ Light Microscope
Visible range: approx. 200 nm – 1 mm
Example: Bacteria, cells, cell nuclei➡️ Classical Electron Microscopy (EM, TEM/SEM)
Visible range: approx. 0.2 nm – 100 µm
Example: Viruses, cell organelles, cell membranes➡️ X-ray Crystallography
Visible range: approx. 1 Å (0.1 nm) – 10 nm
Example: Atomic structures of proteins➡️ Cryo-EM (Single Particle Analysis)
Visible range: approx. 2 Å (0.2 nm) – 100 nm
Example: Protein complexes, ribosomes➡️ Cryo-ET (Cryo-Electron Tomography)
Visible range: approx. 3–5 nm – 200 µm
Example: Viruses inside cells, cell organelles at high resolution
Electron Microscopy (1930s)
→ First Images of VirusesElectron microscopy (EM) was a revolutionary advancement in microscopy because it allowed imaging of structures far below the resolution limit of light microscopes for the first time. Instead of light, EM uses a beam of electrons, which enables much higher resolution. This enabled scientists to visualise viruses that are too small to be viewed with conventional microscopes for the first time.
EM was a major breakthrough, but its limitations in depicting samples in their natural state and the lack of 3D information led to the development of X-ray crystallography.
X-ray crystallography (1950s–present)
→ Detailed structures of viral proteinsX-ray crystallography made it possible to decipher the atomic structure of proteins and other biomolecules. In this method, a crystal of the protein is irradiated with X-rays, and the resulting diffraction pattern is analyzed to determine the positions of the atoms. This technique provided detailed insights into the structure of viral proteins, which was crucial for understanding their function and for drug development.
Although X-ray crystallography is powerful, its limitations in studying large, complex structures and the need for crystals led to the development of cryo-electron microscopy (cryo-EM).
Cryo-EM (1980s–present)
→ A modern view of virusesCryo-EM combines the advantages of electron microscopy with gentle sample preparation. Samples are rapidly frozen (vitrified), preserving them in a near-native state. This allows imaging of individual virus particles or large protein complexes without the need for crystallization. Cryo-EM delivers high-resolution images and can also capture flexible or dynamic structures.
Cryo-EM was a major breakthrough, but it was limited to isolated particles and could not image complex cellular environments. This led to the development of cryo-electron tomography (cryo-ET).
Cryo-ET (2000s–present)
→ 3D virus models inside cellsCryo-ET expands on cryo-EM by creating 3D models of virus particles or other structures directly within their cellular environment. The sample is imaged from different angles, and the images are combined into a 3D model. This allows viruses to be studied in their natural context, for example, how they interact with cells or replicate.
Cryo-ET is a powerful tool, but its limited resolution and the complexity of sample preparation could drive the development of new technologies that enable even higher resolutions in complex cellular environments.
Each of these technologies has expanded the limits of our vision – while simultaneously introducing new challenges. Yet it is precisely these limits that have driven the development of even more powerful methods. Science is a continuous process of discovery, refinement, and pushing boundaries.
They say, „Seeing is believing”, but for biologists, „Seeing is understanding”. The more detailed we can map biological structures, the deeper we penetrate into the mysteries of life. But simply visualizing a virus and its interaction with the host is not enough to fully decipher its nature.
From Seeing to Decoding: The Next Level of Knowledge
To understand what truly defines a virus, we must decode its genetic legacy – its unique „fingerprint”. This is achieved through modern molecular biology techniques that analyze the viral nucleic acids, providing a glimpse into the virus’s genetic blueprint.
4.5. The Genetic Fingerprint of Viruses
Once viruses had finally become visible – thanks to electron microscopy and crystallographic analysis – the next big question emerged: What actually makes a virus a virus?
It quickly became clear: like any biological system, viruses also need a genetic blueprint – something that encodes their characteristics and enables their replication. But what exactly carries this information?
For a long time, protein was considered the prime candidate: diverse, complex, and seemingly perfectly suited. DNA, on the other hand, appeared to many researchers as too simple, too monotonous to be the carrier of life.
But as it turned out, the answer lay precisely there: in this inconspicuous molecule that proved to be the ultimate data carrier of life – and, in the case of some viruses, in its close relative, RNA.
The discovery that it wasn’t proteins but nucleic acids that hold the key to viral replication was a scientific thriller in its own right – marked by misconceptions, rival research teams, and groundbreaking revelations.
Searching for the code of life
Early Approaches to Decoding Heredity
The foundations of genetics were laid in the 19th century through Gregor Mendel’s experiments. Scientists recognized that organisms pass on their traits to the next generation, but the exact mechanism remained unclear for a long time. By the late 19th century, researchers had identified chromosomes within cells as possible carriers of hereditary information and began searching for the key molecules responsible. Due to their complexity, proteins seemed the most likely candidates.
Wendell Meredith Stanley and the Tobacco Mosaic Virus
In the 1930s, the work of Wendell Meredith Stanley confirmed the central role of proteins in the structure of viruses. Stanley isolated the Tobacco Mosaic Virus and processed it in an innovative way: he extracted the virus from infected tobacco plants, purified it through centrifugation, and then crystallized it from solution. These crystals contained both proteins and genetic material, but at first, the research focused solely on the protein. It was assumed that the protein carried the genetic instructions required for virus replication.
The Paradigm Shift: From Proteins to DNA
Doubt surrounding the protein hypothesis grew when, in 1944, Oswald Avery demonstrated that DNA has the ability to transform the properties of bacteria. In his experiment, DNA was extracted from one bacterial strain and transferred to another, revealing that DNA can carry genetic information. However, this discovery was initially met with skepticism.
It was the Hershey-Chase experiment of 1952 that provided the decisive breakthrough. Scientists Alfred Hershey and Martha Chase worked with bacteriophages – viruses that infect bacteria. They radioactively labeled the DNA of the viruses and traced its path into the host cells. The protein shells of the phages remained outside the cell, while the DNA entered and directed viral replication. This conclusively proved: DNA – not protein – is the carrier of genetic information.

The Double Helix and the Role of RNA
In 1953, James Watson and Francis Crick unraveled the structure of DNA, aided by Rosalind Franklin’s X-ray diffraction images. The discovery of the double helix revealed how DNA stores genetic information and replicates during cell division. Around the same time, it became clear that RNA carries the genetic material in some viruses, including the tobacco mosaic virus. This cast Stanley’s earlier work in a new light: the RNA inside the virus particles – not the protein – was the true carrier of genetic information.
Viruses – The Minimalists Among Life Forms
Viruses are true masters of reduction. These minimalist survivors have perfected the art of genetic efficiency – whether with DNA, RNA, or even reverse-transcribed RNA (like the negative single-stranded RNA of influenza viruses, which is essentially a mirror image).
Their genome is like an ultra-light survival backpack:
✔ All the essentials that count – blueprints for replication, packaging, host hijacking
✖ No ballast – no ribosomes of their own, no energy production, no small talkViruses are genetically unique: while all known living organisms use only DNA as their genetic material, viruses can use either DNA or RNA – a fundamental difference that drives their remarkable adaptability and evolutionary creativity.
Key Molecular Biology Methods for Viral Genome Analysis
Thanks to modern molecular biology techniques, viruses can now be analyzed with high precision. This allows scientists to:
- decipher their genetic information,
- trace their evolutionary origins, and
- track their transmission pathways.
Depending on the research question, different methods are used:
- Is the goal to identify a virus?
- Should its genome be fully sequenced?
- Or is the aim to observe its activity within the host system?
The following sections present the key methods used to analyze the genetic fingerprint of viruses – from isolating the genetic material to modern sequencing technologies.
4.5.1. Nucleic Acid Extraction
4.5.2. Nucleic Acid Amplification
4.5.3. Sequencing💡Note: For basic information on DNA and RNA and their functions, we recommend Chapter „4.2. The Protein Biosynthesis” in the publication „The Wonderful World of Life”. It clearly explains the fundamentals and provides an ideal foundation for understanding this topic.
4.5.1. Nucleic Acid Extraction
To analyze a virus in detail, we must first reach its innermost treasure: its genetic material – DNA or RNA must first carefully extracted. However, the nucleic acid is well hidden, wrapped in protein envelopes, embedded in cell debris or mixed with all kinds of molecular ‘by-catch’.
The task: to free the viral nucleic acid from this molecular jumble – cleanly, efficiently, and without causing any damage.
The goal: to obtain as much pure DNA or RNA as possible – ready for PCR, sequencing, or mutation analysis.
Nucleic acid extraction is therefore the first and one of the most important steps in any molecular virus diagnostics. Depending on the sample type, virus species, and the purpose of the analysis, different extraction methods are used – ranging from classic kits to automated high-throughput systems.
How does nucleic acid extraction work?
The process can be divided into four simple steps:1️⃣ Cell lysis – Breaking everything up first
Before accessing the RNA or DNA, the cells (and possibly viruses) in the sample need to be broken up. This can be done, for example, by ultrasound, enzymes, or mechanical grinding. The main goal is to remove the outer shell so that the genome is exposed.
Methods that get to the core
Ultrasound: Sonication breaks down cell and viral envelopes using sound waves.
Enzymes: Proteinase K degrades proteins that package the genome.
Mechanical grinding: Small glass beads in a tube are shaken. The beads collide with the cells and tear the cell membrane apart.
Fig. 12-A: Steps of cell lysis for the release of viral and cellular components Intact structure (left): A schematic representation of an intact cell with organelles, nucleus, and DNA. Inside the cell, influenza viruses are visible. Additionally, a single influenza virus is shown enlarged, with its spherical envelope consisting of a host cell membrane, a capsid, and RNA strands.
Cell lysis (middle): The cell membrane is shown perforated, allowing the cytoplasm to leak out. The nuclear membrane also has holes. An influenza virus is depicted enlarged with a broken envelope, releasing viral RNA and other components. Next to the cell, symbols for chemical substances (bottle), physical forces (sound waves), and mechanical forces are illustrated to represent the different methods of cell lysis.
Contents released (right): After cell lysis, the organelles, DNA, and other cellular components float freely in the medium. Also visible are the released components of the influenza virus, including RNA segments, spikes, and other molecular components, which are shown in the enlarged view.2️⃣ Cleanup – Fishing out proteins & co.
At this stage, the sample is quite a mix: nucleic acids, proteins, fats, and cell debris are all jumbled together. To ensure that only what we need remains in the end, reagents or enzymes are used to specifically break down all the excess material.
Methods that clear things up
Phenol-chloroform extraction: Old but reliable – effectively separates nucleic acids from interfering proteins and lipids. Especially useful for heavily „contaminated” samples.
👉 Caution: The chemicals used are quite toxic – only for experienced hands (and with safety goggles!).Proteinase K: This enzyme breaks down proteins such as membrane or structural proteins that are still floating around in the sample – ensuring that the DNA/RNA remains undisturbed.
DNase/RNase treatment: Used to specifically degrade non-viral DNA or RNA – especially useful when, for example, only viral RNA is to be analyzed.
3️⃣ Purification – Pure RNA or DNA
Now it gets elegant: the nucleic acids are selectively bound – to special surfaces like silica membranes or magnetic beads. The rest? Simply washed away. It’s like a molecular sieve – just smarter.
Methods that filter properly
Spin columns (column-based method): DNA or RNA binds to a special membrane, usually made of silica. Then it’s all about rinse, rinse, rinse – until everything else is gone. What remains in the end: beautifully pure nucleic acid. This method is fast, reliable, and found in many lab kits.
Magnetic bead technology: Tiny magnetic beads selectively bind to nucleic acids. Using a magnet, the right matches are quickly fished out of the mix. It’s lightning-fast and perfectly suited for automated high-throughput processes that need to handle thousands of matches per hour.For more information, see the videos:
DNA and RNA extraction with magnetic beads – How it works and
Nucleic acid purification with chemagic M-PVA Magnetic Bead Technology.4️⃣ Elution – The grand finale
Now comes the final act: the purified nucleic acids are dissolved in a small volume of liquid (elution buffer or water) – and voilà: the sample is now ready for analysis. Free of any clutter, but full of potential for PCR, sequencing & more.

Fig. 12-B: Purification and preparation of nucleic acids Left: The released cellular and viral contents after cell lysis, consisting of a mixture of cell components, proteins, and nucleic acids.
Middle: After the removal of proteins and impurities, only the nucleic acids (viral RNA segments) remain, depicted as small dots.
Right: Enlarged view of some RNA segments, which after elution are dissolved in a liquid and prepared for analysis.
When too little is way too little
Even with careful purification, true purity at the molecular level is hardly achievable. Tiny contaminants – proteins, salts, other molecules – can still be present in the sample. The problem: the few viral nucleic acids can easily get lost in this „background noise” – like a whisper in a concert hall.
And this is exactly where the next big step comes in: amplification. The viral genetic material is multiplied millions of times – so even the faintest whisper becomes loud and clearly detectable.
4.5.2. Nucleic Acid Amplification
No matter how good the extraction was, the yield of viral DNA or RNA is often tiny. To detect or analyze it, more copies are needed – many more. That’s exactly the job of amplification: it multiplies the genetic material millions or even billions of times – like a molecular copier.
The method used depends on what you’re looking for:
🦠📌 If the virus is known – targeted detection
For known viruses, you know where to look: specific regions of their genome have already been mapped. Using suitable primers – short gene sequences that bind exactly to these regions – you can selectively amplify a particular segment. The classic method for this is PCR (polymerase chain reaction): precise, sensitive, and perfect for targeted detection.
🦠❓ If the virus is unknown – casting a wide net
If the virus is still a blank slate, specific primers are missing. In this case, the entire viral DNA or RNA is amplified – nonspecifically but comprehensively. Methods like Random Primed Amplification or Whole Genome Amplification (WGA) are used here. They generate as many copies as possible – regardless of the region – so that sequencing can later reveal what exactly is in the sample.
The following explains both approaches in more detail:
a) Polymerase Chain Reaction (PCR)
b) Random Primed PCR
a) Polymerase Chain Reaction (PCR)
PCR – short for Polymerase Chain Reaction – is like the copier in the lab: it can multiply even tiny amounts of viral DNA millions of times. This is especially useful when searching for traces – to detect DNA viruses in a sample.
But what about RNA viruses, like the influenza virus? They can’t be copied directly – first, they have to be „translated”. In the so-called RT-PCR (Reverse Transcription PCR), the viral RNA is converted into DNA using an enzyme called reverse transcriptase. After that, everything proceeds just like in regular PCR: amplify, analyze, done.
PCR – Step by step
To stick with the example of the influenza virus, let’s look at the typical process of reverse transcription PCR (RT-PCR), which is used for RNA viruses like influenza.
1️⃣ Sample collection
It all starts with a swab – usually from the nose or throat, because that’s where influenza viruses like to hang out. The collected material is placed in a special medium that protects the delicate viral RNA. And that’s necessary, because RNA is sensitive: RNases – enzymes that break down RNA – are everywhere, found on skin, in the air, and even in the sample itself.
2️⃣ RNA extraction
The sample is a mixed cocktail: viral particles, human cells, proteins, lipids – the whole package. As you’ve already read in the chapter „Nucleic Acid Extraction”, the essential part is now isolated: the RNA. In the end, a clean mixture remains, which can mainly contain three types of RNA:
- viral genomic RNA (for influenza virus: negative-sense RNA (-)ssRNA),
- viral mRNA produced during active infection in host cells,
- cellular RNA from the host.
Especially interesting is the viral mRNA. Thanks to a viral trick – called cap-snatching – it has a 5′ cap structure and a poly-A tail, just like our own mRNA. This makes it more stable and particularly well-suited for the next step: conversion into DNA.
To keep it stable for analysis, the extracted RNA is stored in a stabilizing buffer.
3️⃣ Conversion of RNA into DNA
Before PCR can begin, it needs an upgrade: it only works with DNA, not RNA. That’s why the viral RNA must first be converted into complementary DNA (cDNA). And that’s the job of a clever enzyme: reverse transcriptase.
Here’s how the conversion works:
RNA template: The mRNA of the influenza virus is a single-stranded RNA molecule with two handy features: at the 5′ end, it has a cap structure that stabilizes it – and at the 3′ end, a poly-A tail, a chain of adenine bases. Both make the viral RNA particularly well-suited for the next step.
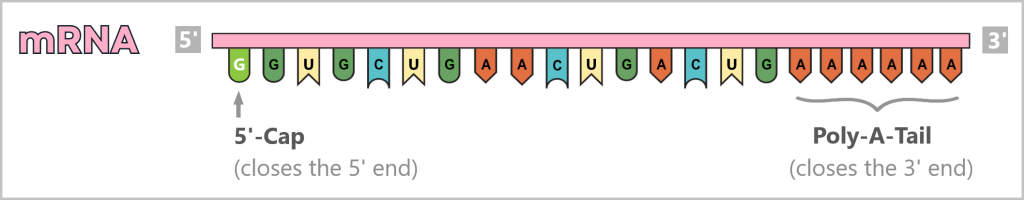
Fig. 13-A: Schematic representation of the influenza virus mRNA Primer binds: To let the reverse transcriptase know where to start, a primer is needed – a short DNA fragment that often binds specifically to the poly-A tail.
Reverse transcriptase gets to work: It attaches to the primer and reads the RNA bases (A, U, G, C) of the template. Then it adds the matching DNA bases (A, T, G, C) one by one – building a complementary DNA strand. The result is a hybrid molecule made of RNA and DNA (an RNA–cDNA hybrid).

Fig. 13-B: Reverse transcriptase synthesizes cDNA from RNA Both the 5′ cap structure and the poly-A tail of the RNA do not appear in their original form in the resulting cDNA. The 5′ cap is typically ignored, while the poly-A tail is represented in a complementary form and may be partially shortened.
From single strand to double strand: This first DNA strand now serves as a template itself – a second strand is synthesized, resulting in double-stranded cDNA (ds cDNA). This form is stable and ready for PCR.

Fig. 13-C: Stable double-stranded cDNA ready for PCR What does the whole process look like in action? This animation shows cDNA synthesis in fast-forward – explained simply and clearly.
4️⃣ What do you need for a PCR?
Before amplification can begin, a few ingredients and tools need to be ready:
a) DNA template: The starting material is the double-stranded cDNA that was produced from viral RNA in the previous step.
b) Nucleotides – the building blocks: Four different building blocks are needed to copy DNA: adenine (A), thymine (T), cytosine (C) and guanine (G). These will later be assembled by the polymerase into a new strand.
c) DNA polymerase – the master builder: This enzyme reads the DNA template and assembles the matching nucleotides into a new strand – precise and lightning-fast. In PCR, a heat-stable polymerase (e.g., Taq polymerase) is often used to withstand the high temperatures of the PCR cycles.
d) Primers – the guides: Primers are short DNA fragments (usually 18–24 bases long) that show the polymerase where to start copying. For PCR, you always need two: a forward primer and a reverse primer, which bind to opposite strands of the target DNA.

Fig. 14-A: Schematic representation of the DNA polymerase enzyme and the primers e) Buffer solution – the right environment: It ensures the polymerase is comfortable, providing a stable pH value, magnesium ions, and everything the enzyme needs for reliable function.
f) Thermocycler – the temperature carousel: A device that automatically runs the necessary temperature cycles. It heats, cools, and maintains precise temperatures – timed perfectly for the different PCR steps.

Fig. 14-B: The thermocycler controls the PCR temperatures. 5️⃣ The process of the Polymerase Chain Reaction (PCR)
All the ingredients – DNA template (ds cDNA), nucleotides, DNA polymerase, primers, and buffer solution – are combined in a small reaction tube. This tube is then placed in the thermocycler, which automatically runs the typical PCR temperature cycles. Each cycle consists of three main steps:
Step 1 – Separation of DNA strands (Denaturation): The sample is heated to about 94–98 °C for around 20–30 seconds. This breaks the hydrogen bonds between the DNA bases – the double strand „melts” into two single strands. These single strands then serve as templates in the next step.

Fig. 14-C: Separation of the DNA strands Step 2 – Primer binding (Annealing): The temperature is lowered to 50–65 °C. Now, the primers specifically bind to their respective single strands. They mark the starting point for DNA synthesis. The primers are designed to bind only to viral sequences – not to human RNA or DNA.

Fig. 14-D: Primer binding Step 3 – DNA synthesis (Amplification): At around 70 °C, the optimal temperature for the polymerase, the actual copying begins. The DNA polymerase binds to the primer, reads the single strand from 3′ to 5′, and synthesizes the new strand in the 5′ to 3′ direction. It assembles nucleotides following base-pairing rules: A pairs with T, and G pairs with C. This results in two new double-stranded DNA molecules.

Fig. 14-E: DNA synthesis This step is called amplification (from Latin amplificatio: enlargement), elongation (from Latin elongare: to lengthen), or polymerization (from Greek poly: many; meros: part).
6️⃣ Repeating the Cycles
The newly formed double-stranded DNA molecules immediately serve as templates for the next round. The steps – denaturation, primer annealing, and DNA synthesis – are repeated in cycles.
With each cycle, the amount of DNA doubles: 1 → 2 → 4 → 8 → 16 → 32 …
After just 25 – 40 cycles, billions of copies of the target DNA segment are produced.

Fig. 14-F: Repetition of Cycles 📈 What is present at the end of RT-PCR:
A highly concentrated solution of specific DNA fragments – directly derived from the viral RNA. In the case of the influenza virus, it contains only those genome segments that were specifically targeted.
This DNA now serves as the basis for further analyses:
➤ to confirm the identity of the virus,
➤ to distinguish between different virus variants,
➤ or to quantify the viral load in the patient sample.🎥 Tip: The video „What is PCR? Polymerase Chain Reaction” provides a clear summary of the process. Even though it focuses on human DNA, the core principle remains exactly the same.
b) Random Primed PCR
💡Note: If you’re not yet familiar with PCR, start by reading the section „PCR – Step by Step”. It clearly explains the basics and workflow. The following content builds on that foundation and focuses specifically on the unique aspects of Random Primed PCR.
Random Primed PCR is a special variant of PCR that is primarily used when a virus is unknown or its genome is highly variable.
In contrast to classical PCR, which uses specific primers to selectively amplify defined DNA segments, this method employs so-called random primers: short DNA sequences with randomly arranged bases that can bind at many positions along the target DNA (or cDNA) – regardless of its exact base sequence.
Advantage: Even unknown or highly mutated regions of the genome can be co-amplified using this method – a crucial benefit when preparing for sequencing, where the exact base sequence needs to be determined.
Examples of Random Primers:
Hexamer Primers (6 bases long): AGCTGA, CTAGCT, …
Heptamer Primers (7 bases long): CCTGAGT, GATTACA, …
Nonamer Primers (9 bases long): GCAGTTCGC, ATGGCCGTA, …
Fig. 15-A: Examples of Random Primers In practice, mixtures of all possible primer variants are usually used (e.g., 4⁶ = 4096 combinations for hexamers). This ensures that as many binding sites as possible in the genome are targeted.
Process of Random Primed PCR
Step 1 – Denaturation: The DNA (or cDNA) is heated to about 95 °C to separate the two strands. This creates single strands to which the primers can bind.
Step 2 – Primer Binding: The temperature is lowered so that the random primers can attach to many different sites on the DNA. Since the primers are random, they can bind to a variety of positions within the genome.
Note on genome size: Viruses have very different genome lengths – ranging from a few thousand to hundreds of thousands of base pairs. Random primers help to cover as much of the genome as possible.
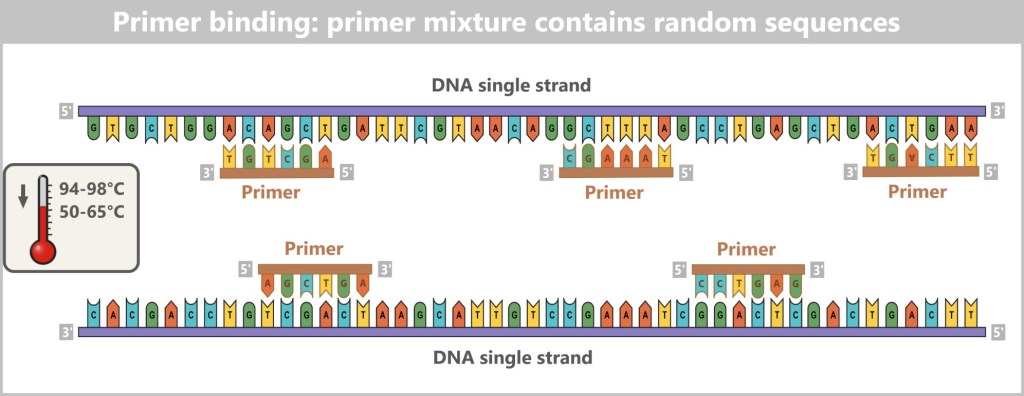
Fig. 15-B: Schematic representation of primer binding during Random Primed PCR. Step 3 – DNA synthesis: The DNA polymerase binds to the primers and starts synthesizing new DNA strands from there. It reads the template until it either reaches the end or encounters another primer. This acts like a stop signal – resulting mostly in shorter DNA fragments.
This is exactly what is intended: the polymerase generates many short, overlapping fragments that can later be used for genome reconstruction or targeted analyses.
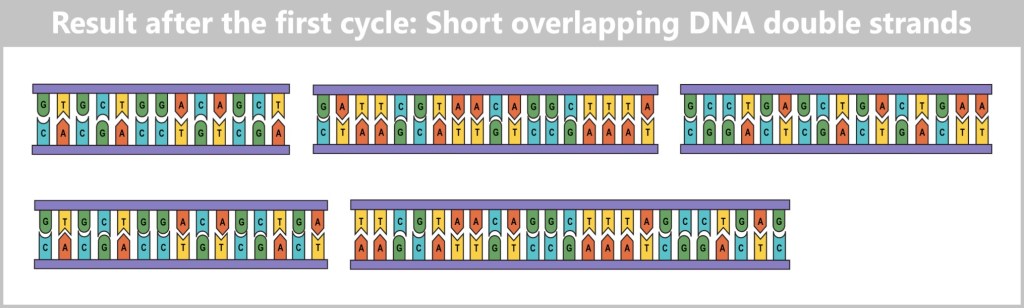
Fig. 15-C: First cycle of the Random Primed PCR After the activity of the DNA polymerase, multiple short, overlapping double-stranded DNA fragments have formed from the single strands. These fragments are the result of the first cycle of the Random Primed PCR.
🔁 Cyclic Repetition
This process is repeated multiple times – just like in classic PCR – usually 20 to 40 cycles.
- The more cycles, the more DNA is produced.
- With increasing cycle number, the chance that primers „catch” each other rises → resulting in shorter fragments.
- However: Too many cycles can reduce diversity because some regions become overrepresented.
A balanced number of cycles and a well-optimized primer mix are crucial for the quality and representativeness of the final product.
📈 Result of the Random Primed PCR
In the end, a large number of short, overlapping DNA fragments are present – not complete strands, but a mosaic of fragments that can cover the entire viral genome.
Fig. 15-D: Result of the Random Primed PCR: the entire genome – just in fragments!
🧩 What happens to the DNA fragments?
Direct analysis of individual fragments
Often, it’s enough to sequence specific fragments to gain important information – e.g., for mutation analysis or virus typing.Reconstruction of the entire genome
If the whole genome is to be analyzed (e.g., for discovering new viruses or for creating phylogenetic analyses), the fragments are read using sequencing technologies (more on this in the next chapter). Specialized software programs then piece together the overlapping fragments like a puzzle.
🧬 From Fragment to Complete Genome
Amplification using Random Primed PCR is just the first step – it ensures that enough genetic material is available for further analyses. However, simply multiplying the material is not enough to truly understand which virus you are dealing with.
Now it’s about determining the exact sequence of base pairs in the generated DNA fragments – in other words, their sequencing. Only through this sequencing can the genetic profile of the virus be decoded: mutations can be identified, viral strains distinguished, and even evolutionary family trees constructed.
In the following chapter, we therefore take a look at how sequencing works, which technologies are used for this – and how a complete viral genome is reconstructed from many small DNA snippets.
4.5.3. Sequencing
Sequencing is THE crucial step in decoding the genetic material of viruses. It determines the exact order of the bases in DNA (A, T, C, G) or RNA (A, U, C, G).
What’s the point?
A viral genome sequence is like the QR code of biology: Once scanned, you immediately know what you’re dealing with.
Identification of the virus:
🔹 Which virus is it exactly?
🔹 Is the virus already known or is it a new discovery?
🔹 Which virus family or genus does it belong to?Detection of mutations:
🔹 Has the virus changed – and if so, how?
🔹 Have new variants or strains emerged?
🔹 What genetic differences exist compared to earlier versions?Determination of diagnostic markers:
🔹 Are there stable (conserved) regions in the genome that are suitable for diagnostic tests?
🔹 Are there genes or proteins that are unique to this virus?
🔹 Can certain genes or proteins be specifically targeted – for example, for drug development?Sequencing gives each virus a genetic fingerprint – unique, precise, and tamper-proof.
🧬 From Test Tube to High Technology
Decoding viral DNA used to be manual labor – today, it’s high-tech. Modern sequencers analyze millions of DNA fragments simultaneously – fast, automated, and with high precision.
Since the first manual procedures, technology has developed rapidly. Each new generation has made the view into the genome clearer, faster and more comprehensive.
The following overview shows how sequencing has evolved – and which technologies are available today:
Generation Description First Generation:
Sanger SequencingThe old-timer: slow but precise. Ideal for short sections. Second Generation:
Next-Generation Sequencing (NGS)The high-throughput powerhouse: sequences millions of DNA fragments simultaneously. Third Generation:
Real-Time SequencingThe quantum leap – single molecule analysis in real time. Emerging Technologies Science fiction becomes reality. 
First Generation: Sanger Sequencing
Sanger sequencing, named after the British biochemist Frederick Sanger, marks a historic milestone in molecular biology. With this method, he was the first to successfully determine the exact sequence of DNA bases – a scientific breakthrough that earned him his second Nobel Prize in Chemistry in 1980.
Although more modern and automated methods are now available, Sanger sequencing is still considered the gold standard when it comes to the highest accuracy for short DNA segments. It is still widely used in many laboratories to validate results.
It is based on the principle of terminating DNA synthesis. During the copying process, special stop signals are incorporated, creating DNA fragments of varying lengths. These fragments are then sorted by size, allowing the sequence of building blocks to be read.
How the classic DNA analysis works
1️⃣ DNA Denaturation
First, the double-stranded DNA is heated. The high temperature breaks the hydrogen bonds between the base pairs, causing the double strand to separate into two single strands. These single strands then serve as templates for synthesizing new DNA.
Fig. 16-A: DNA denaturation – a double-stranded DNA separates into two single strands. 2️⃣ Preparation of the reaction mixtures
Four separate reaction mixtures are prepared. Each contains:
➥ Single-stranded DNA: the template.
➥ Primer: a short sequence that provides the starting point for the polymerase.
➥ DNA polymerase: the enzyme that synthesizes new strands.➥ dNTPs (deoxyNucleosideTriPhosphates): the „standard” DNA building blocks (A, T, C, G). They enable DNA strand extension because their 3′ hydroxyl group can bond with the next building block.
➥ ddNTPs (didesoxyNukleosidTriPhosphate): these „modified” DNA building blocks lack a hydroxyl group, so no further nucleotides can be attached after them. When a ddNTP is incorporated during synthesis, the DNA strand elongation stops exactly at that point. Each of the four ddNTP types (A, T, C, G) is additionally labeled with its own fluorescent dye.
The ratio of dNTPs to ddNTPs is carefully adjusted to generate a wide range of DNA fragments of varying lengths.
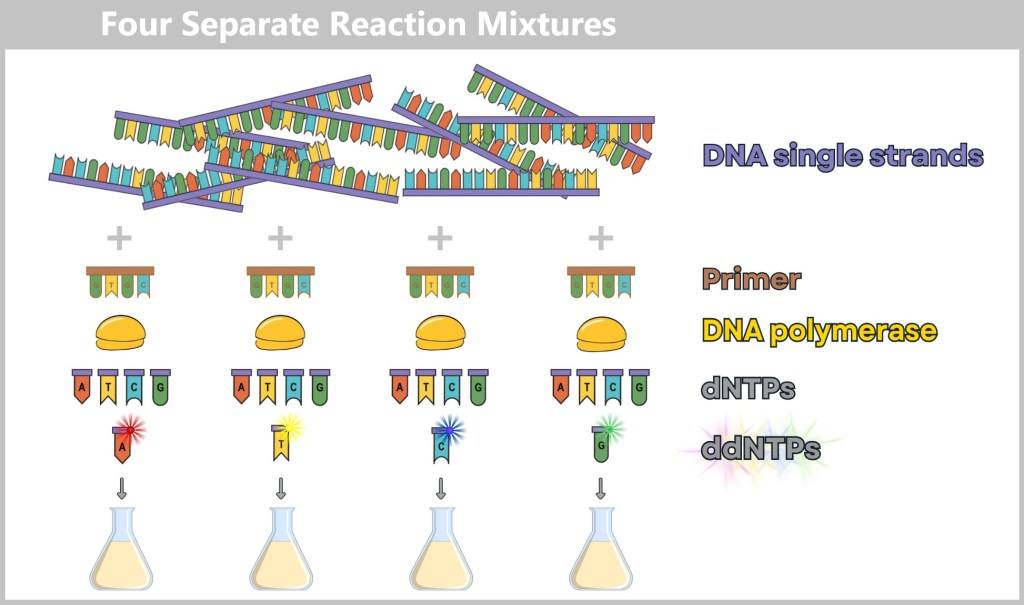
Fig. 16-B: Four reaction mixtures – one for each type of ddNTP. 3️⃣ DNA Synthesis with Random Termination
DNA synthesis – briefly explained: DNA polymerase is nature’s most efficient copier. It scans a DNA strand as a template and builds a matching complementary strand step by step – always following the base-pairing rules: A pairs only with T, and C only with G. The result is a perfect mirror copy.
DNA synthesis takes place simultaneously in the four separate reaction mixtures – one for each of the four bases (A, T, C, G).
Here’s what happens in each mixture: The DNA polymerase binds to the primer and gets to work.
How to find the right primer?
Primers are absolutely essential for the successful use of the Sanger method. But how do you find the primer sequence when the genome of a virus is completely unknown?
When Frederick Sanger developed his method in 1977, some information about the genome structure of small viruses like the bacteriophage Phi-X174 was already known. Researchers had discovered that certain enzymes – called restriction enzymes – cut DNA at very specific sites. These cutting sites were known and could be used deliberately:
Near these cutting sites, there was often a short piece of known DNA sequence – just enough to design a suitable primer. This way, researchers artificially created a defined starting point for the sequencing process.
Today, it’s easier. Science has advanced significantly, and most viruses are already well studied. For new or little-known viruses, the following approach is used:
Comparison with known viruses: Often, a new virus is genetically similar to already known viruses. Scientists use these similarities to predict possible primer sites.
Experimental approaches: When little is known, enzymes cut the viral DNA into smaller fragments. Researchers then analyze these fragments to identify suitable starting points for primers.
It reads the single-stranded DNA template and inserts matching building blocks (dNTPs) to synthesize a new DNA strand.
Normally, a „regular” dNTPs is incorporated – allowing the DNA chain to continue growing. Occasionally, however, a modified ddNTP is incorporated – and synthesis stops immediately at that point. This happens because ddNTPs lack a small chemical group (the hydroxyl group) that’s essential for adding the next nucleotide.
This results in many DNA fragments of different lengths – each one ending precisely at the point where a ddNTP was randomly incorporated. And each fragment carries a fluorescent dye at its end, indicating which type of base (A, T, C, or G) the fragment terminates with.
Fig. 16-C: The synthesis is intentionally interrupted – when a ddNTP is incorporated. Each reaction mixture contains one modified nucleotide (ddATP, ddTTP, ddCTP, or ddGTP). When this modified nucleotide is incorporated during DNA synthesis, the process stops exactly at that position, because the modified building block does not allow further extension of the DNA chain. In the ddATP mixture, synthesis stops when adenine (A) is incorporated. In the ddTTP mixture, the chain is terminated when thymine (T) is added. Similarly, ddCTP and ddGTP cause termination at cytosine (C) and guanine (G), respectively. This method generates DNA fragments of varying lengths, each ending with the specific stop nucleotide. The goal is to produce all theoretically possible sequence fragments.
4️⃣ Denaturation of the Fragments
The resulting double-stranded DNA fragments are heated again so that they separate into single strands. This step is necessary to allow individual analysis of each fragment. What remains is single-stranded DNA – ready for the next step.
5️⃣ Analysis
Now it’s time for analysis: The fragments are separated by size using gel electrophoresis. For this, the four reaction mixtures are loaded into separate wells of a gel.
The gel acts like a fine mesh or sponge:
- Short fragments move through faster.
- Longer fragments move more slowly.
Each strand ends with a color-labeled ddNTP – depending on the base (A, T, C, or G), a different color lights up. These colors are detected by a laser and recorded automatically.
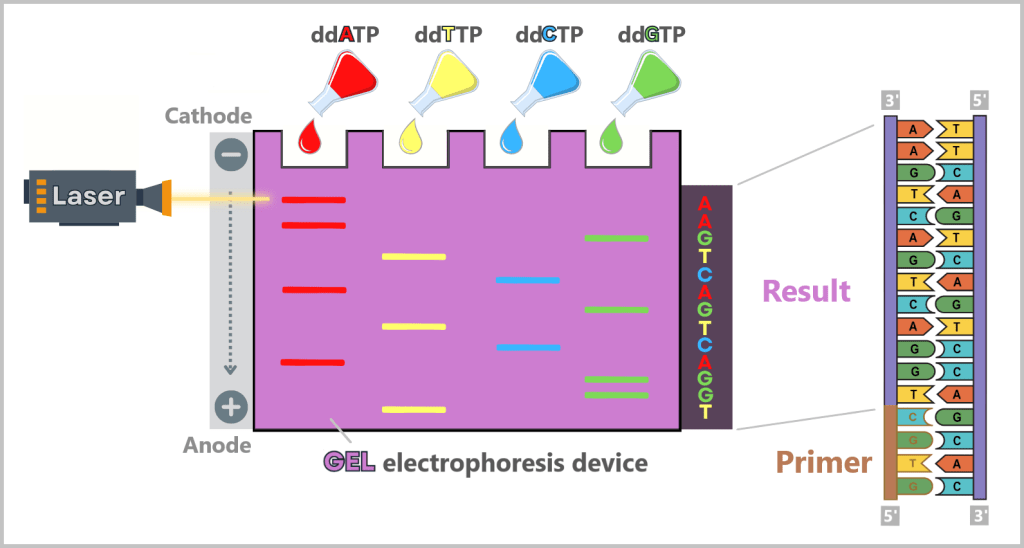
Fig. 16-D: Gel electrophoresis for separating DNA fragments: In the reaction tubes are DNA fragments of different lengths, each ending with the same terminating nucleotide (depending on the mixture: adenine, thymine, guanine, or cytosine). The reaction mixtures are loaded onto the gel. Under the influence of an electric field, the negatively charged DNA fragments migrate from the cathode (−) to the anode (+). The size of the fragments determines their migration speed through the gel. Smaller DNA fragments move faster through the gel pores and thus reach the anode first, while larger fragments progress more slowly. By reading the fluorescent signals, the base sequence of the DNA can be determined.
How do you read the sequence from this?
The order of the fragments in the gel corresponds to the order of the bases in the original DNA strand.
- The shortest fragment shows the first base.
- The next longer fragment shows the second base,
- … and so on, until the entire sequence is decoded.
Since each new fragment is complementary to the original strand, the base sequence of the original DNA strand can be directly deduced from the analysis.
🎥 Tip: You’ll find a very clear explanation in the video „Sanger Sequencing / Chain Termination Method”.
Where is Sanger sequencing used?
This method is especially well suited for:
- Short DNA fragments
- Confirmation and validation of results from other methods
- Individual case analyses, e.g., in medical diagnostics
Some methods don’t get old – they become classic.
Limitations of the method
As precise as Sanger sequencing is, it quickly reaches its limits with large or complex genomes. The method is labor-intensive, time-consuming, and expensive, especially when many samples or extensive datasets need to be analyzed. For this reason, it has been replaced in many areas by modern high-throughput techniques – above all, by Next-Generation Sequencing (NGS) technologies.
Second Generation: Next-Generation Sequencing (NGS)
Next-Generation Sequencing (NGS) is a modern method for decoding DNA and RNA sequences – and has fundamentally transformed genetic research since the early 21st century. Compared to classical Sanger sequencing, NGS is faster, more cost-effective, and can process significantly larger volumes of data in a shorter amount of time.
The key difference: While the Sanger method sequences only a single DNA fragment at a time, NGS can read millions of fragments simultaneously. To achieve this, the genetic material is first broken down into small pieces and tagged with special markers. These fragments are then fixed onto a special surface and extended step by step using fluorescently labeled nucleotides – each newly incorporated base is immediately detected and recorded.
Thanks to this parallel processing, a high-resolution dataset is generated within a short time – ideal for analyzing entire genomes, RNA profiles, or large sample volumes. This is precisely why NGS is now a key technology in research, diagnostics, and biotechnology.
A Closer Look at Illumina Sequencing
Illumina sequencing is one of the most widely used technologies in the field of Next-Generation Sequencing (NGS). It is based on the principle of „sequencing by synthesis” and enables the simultaneous reading of millions of DNA fragments. Here’s a step-by-step explanation:
Step 1: DNA Preparation = Library Construction
Step 2: Cluster Generation on a Flow Cell
Step 3: Sequencing by Synthesis
Step 1: DNA Preparation = Library Construction
Before sequencing, the DNA must be converted into a format compatible with the Illumina platform. This process is known as library construction and includes the following sub-steps:
1a) Fragmentation
The DNA is fragmented into smaller pieces either mechanically or enzymatically (typically targeting a size range of around 150–500 base pairs). This process produces DNA fragments of slightly varying lengths, which are often further standardized using a size selection step.
To illustrate this step, we will consider three DNA fragments with slightly different lengths in our example.
1b) Adapter Ligation
Specific adapter sequences (P5 and P7 adapters) are ligated to both ends of the DNA fragments. These adapters serve several important functions:
Binding Sites for the Flow Cell: The adapters contain special DNA sequences that function like a key fitting into a lock. This allows the DNA fragments to adhere to a surface later, which is important for sequencing.
Primer Binding Sites: The adapters contain regions where sequencing primers can bind. These primers are later used to gradually synthesize the DNA strands during the sequencing process.
Indexes (optional): If multiple samples are sequenced simultaneously, index sequences allow the fragments to be assigned to their respective samples.
For clarity, we omit the depiction of indexes in our example.
1c) PCR Amplification
To ensure that enough DNA is available for sequencing, the adapter-ligated DNA fragments are amplified using the polymerase chain reaction (PCR).

Fig. 17-A: Library Preparation – Overview of the DNA preparation process. The following close-up shows a detailed view of a fragmented double-stranded DNA molecule equipped with the two adapters.

Fig. 17-B: Schematic representation of a double-stranded DNA fragment with adapters. Each DNA fragment (DNA insert) receives two adapter sequences:
The P5 adapter consists of the P5 adapter sequence, which enables binding to the flow cell, and the primer binding site Rd1SP (Read 1 Sequencing Primer) for initiating DNA synthesis. Similarly, the P7 adapter contains the P7 adapter sequence for flow cell binding as well as the primer binding site Rd2SP (Read 2 Sequencing Primer) to initiate DNA synthesis.Important note: The upper and lower single DNA strands each carry P5 and P7 adapters. However, the adapters on the lower strand are not identical but complementary to the adapters on the upper strand.
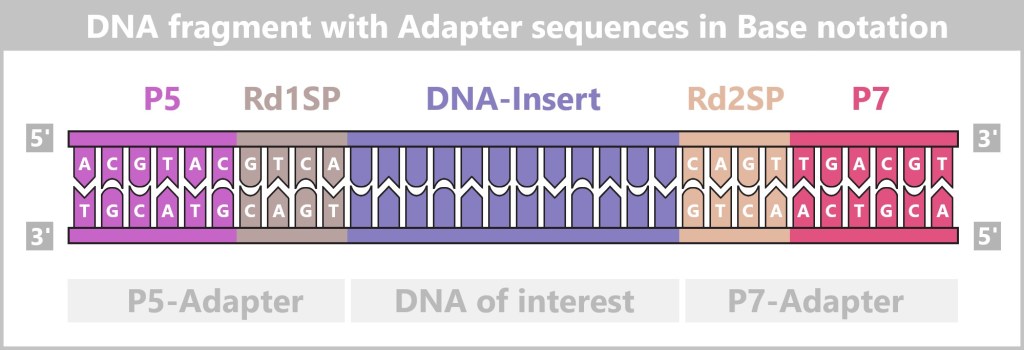
Fig. 17-C: DNA fragment (DNA insert) with adapter sequences shown in base notation This representation is highly simplified. In practice, Illumina adapter sequences are much longer, typically consisting of 60–120 base pairs (see here). The DNA insert is also shortened for better illustration – in reality, DNA fragments usually range from 100 to 500 base pairs in length.
Step 2: Cluster Generation on a Flow Cell
Before the DNA can be sequenced, it must be fixed and amplified on a solid surface. This takes place on a flow cell, a special glass plate equipped with millions of tiny DNA binding sites.
The goal of this step is to generate numerous copies of each DNA fragment on the surface to amplify the signals during sequencing. For this purpose, the flow cell is coated with special oligonucleotides (DNA molecules) that are complementary to the adapter sequences of the DNA fragments. There are two types of these oligonucleotides:
- P5 oligonucleotides (ACGTAC), which bind to the P5 adapters (TGCATG) of the DNA fragments.
- P7 oligonucleotides (ACGTCA), which interact with the P7 adapters (TGCAGT) of the DNA.
You can imagine the surface of the flow cell like a dense Velcro strip: The DNA fragments stick to it with their adapter sequences, similar to tiny hooks catching onto the Velcro fabric.
Fig. 17-D: Schematic representation of a flow cell and its surface 2a) DNA fragments bind to the flow cell surface
At the beginning of this step, a solution containing single-stranded DNA fragments with adapter sequences is flowed onto the flow cell. These fragments were previously separated into single strands by denaturation, allowing them to move freely in the solution.
As the DNA fragments flow through the flow cell, their adapters bind to the complementary oligonucleotides on the flow cell surface through base pairing, as shown in the illustration below.

Fig. 17-E: The graphic shows two single-stranded DNA molecules binding with their adapters to complementary oligonucleotides on the flow cell. P5 adapter of a single-stranded DNA binds to the P5 oligonucleotide on the flow cell:
Flowcell — P5-Oligo ACGTAC
— (3') TGCATG-CAGT-[Insert]-GTCA-ACTGCA (5')P7 adapter of a single-stranded DNA binds to the P7 oligonucleotide on the flow cell:
Flowcell — P7-Oligo ACGTCA
— (3') TGCAGT-TGAC-[Insert]-ACTG-CATGCA (5')2b) First synthesis
Primer binding and DNA synthesis
After the DNA fragments have bound to the flow cell, the first DNA synthesis begins. Specific primers bind to the adapter sequences of the bound strands:- Primer GTCA binds to the P5 adapter of the bound single strand.
- Primer ACTG binds to the P7 adapter of the bound single strand.
The DNA polymerase, which can only work in the 5′ → 3′ direction, then synthesizes a new strand complementary to the already bound single strand.

Fig. 17-F: Primers bind to the adapters (P5, P7), and the DNA polymerase (DNAP) begins synthesizing a new complementary strand. After synthesis, the bound DNA fragments exist as double strands.
Flowcell (P5-Oligo) — (5') ACGTAC-GTCA-[Insert]-CAGT-TGACGT (3')
— (3') TGCATG-CAGT-[Insert]-GTCA-ACTGCA (5')Flowcell (P7-Oligo) — (5') ACGTCA-ACTG-[Insert]-TGAC-GTACGT (3')
— (3') TGCAGT-TGAC-[Insert]-ACTG-CATGCA (5')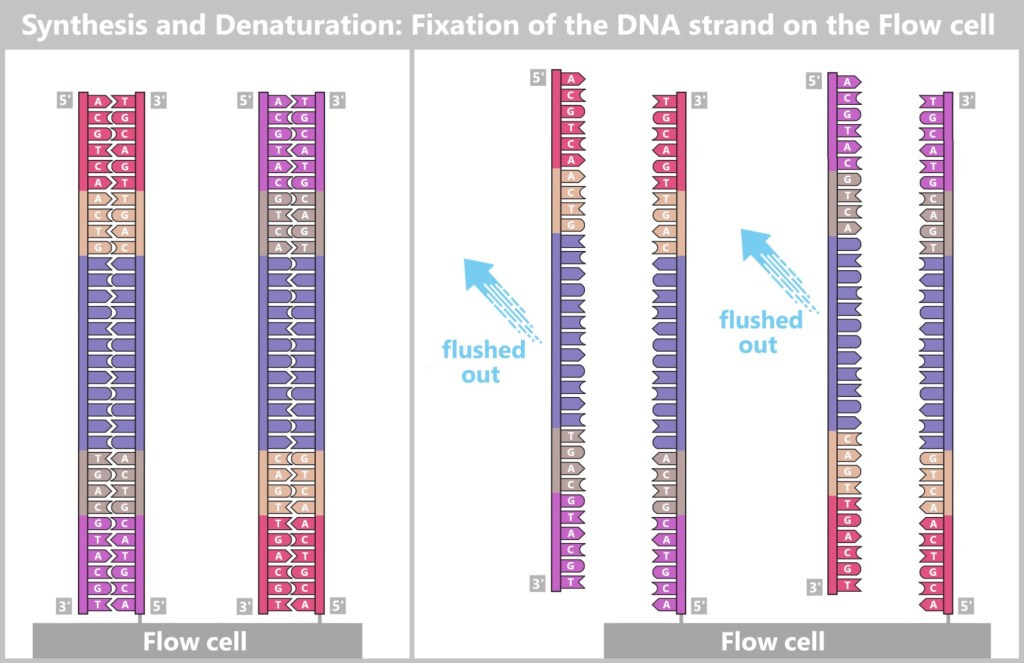
Fig. 17-G: First synthesis and separation: This is how the bound DNA strand is formed. Separation of the double strand
After the new DNA strand has been synthesized, the double strand is separated by denaturation (see the figure above):❌ The original strand is no longer attached to the flow cell after denaturation and is washed away.
✅ The newly synthesized strand remains firmly attached to the flow cell via its 5′ end, while the 3′ end remains free.
Now the DNA fragments are bound to the flow cell as complementary single strands (forward and reverse strands).
However, direct sequencing at this stage would not be possible, as the fluorescence signals would still be too weak to be reliably detected. This is why bridge amplification follows next.
2c) Bridge Amplification
To generate a sufficiently strong fluorescence signal for DNA sequencing, the individual DNA strands must be amplified. This is achieved through bridge amplification – a cyclic process in which the DNA strands bind to the flowcell surface, are copied, and then separated again.
Hybridization of the Second Adapter
Bridge amplification begins with the bound single-stranded DNA bending over and hybridizing its free 3′-end to a nearby complementary oligonucleotide (5′) on the flowcell surface. This process is known as strand folding. It creates a loop-like structure – a „bridge” – that connects the adapter on the DNA strand to the matching surface oligonucleotide.DNA Synthesis
Once the DNA strands are hybridized, primers (e.g., ACTG, GTCA) and DNA polymerase are added. The primers bind specifically to the adapter sequences. DNA polymerase then synthesizes new complementary strands in the 5′ → 3′ direction. After synthesis, the DNA is once again present as a double-strand.Fig. 17-H: Bridge Formation and DNA Synthesis The bound single DNA strands fold over and hybridize with a complementary oligonucleotide on the flowcell, forming a bridge-like structure. A primer then binds to the 3′ ends of the strands, and DNA polymerase synthesizes the complementary strands in the 5′ → 3′ direction.
Denaturation – Strand Separation
Heat or chemical treatment is used to separate the newly formed double-stranded DNA into single strands again. At this point, there are four single-stranded DNA fragments on the flowcell:|| the original two single strands (forward strand & reverse strand), and
|| the newly synthesized complementary strands (forward strand & reverse strand).Abb. 17-I: Forward and Reverse – The Bridges Dissolve After denaturation, the bridge double strands are separated, so that on the flow cell there are now two single strands (forward and reverse) visible. These single strands remain attached to the flow cell and are ready for further amplification.
Repeat amplification
This cycle repeats: The single strands fold again, hybridize once more with the oligonucleotides, and are duplicated through DNA synthesis.
Fig. 17-J: With each repetition of bridge amplification, more and more DNA copies are generated. After multiple cycles, millions of identical copies of each original DNA fragment – called clusters – are formed on the flow cell.
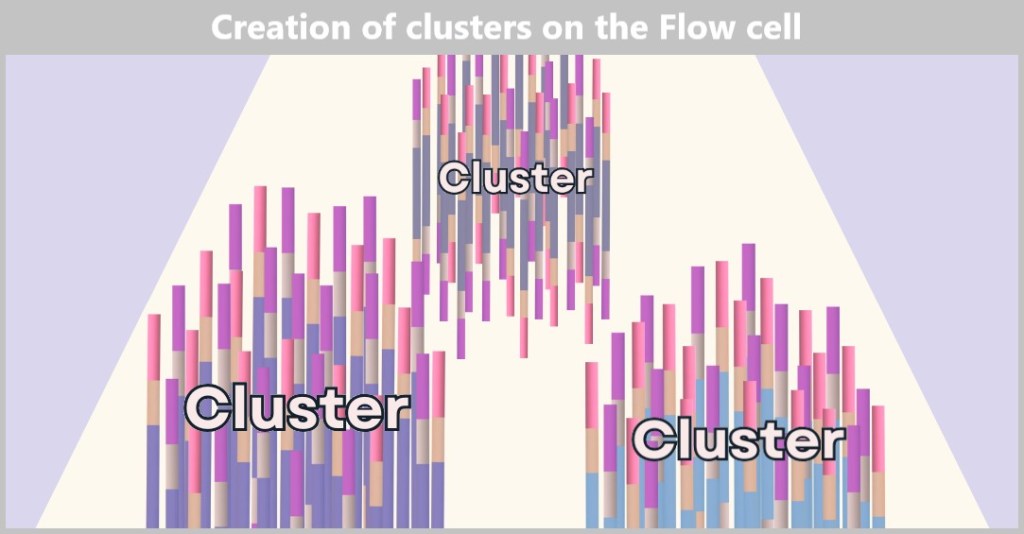
Fig. 17-K: Cluster formation Each cluster consists of numerous copies of a single DNA fragment. In this illustration, only three clusters are shown as examples, based on our example of three DNA fragments. In reality, however, millions to billions of such clusters are present on a flow cell to enable high sequencing capacity.
Formation of the final clusters
At the start of our example, three DNA fragments are present. After several amplification cycles, countless clusters form for each of these fragments, each consisting solely of copies of their respective DNA fragment.Each cluster now consists of two types of strands:
🔹 Forward strands (5′ → 3′, bound to P7)
🔹 Reverse strands (3′ → 5′, bound to P5)💡Note: Forward and reverse strands within the clusters are not aligned antiparallel to each other! Instead, the 3′ ends of both strands point upwards.
Removal of the reverse strands
For the sequencing to proceed correctly, all DNA strands within a cluster must be oriented in the same direction. Only the forward strands are needed for the actual analysis. Therefore, a targeted purification step follows:🔹 All reverse strands (bound to P5) are detached and washed away.
🔹 At the same time, the free ends of the P5 oligos are chemically blocked to prevent reattachment.Now, only forward strands remain on the flowcell. The clusters are fully formed, and the DNA libraries are ready for sequencing.

Fig. 17-L: Ready for sequencing – only forward strands remain onboard. Left: A final cluster with forward (P7-bound) and reverse strands (P5-bound). Right: The reverse strands have been selectively cleaved and washed away, leaving only the forward strands. This orientation is necessary to carry out sequencing correctly.
Step 3: Sequencing by Synthesis
At this point, individual clonal clusters (originating from the initial three DNA fragments) are distributed across the entire surface of the flow cell. Each cluster consists of numerous identical DNA copies – comparable to a small island of identical trees. Now, the actual sequencing can begin.
To start the sequencing, primers, DNA polymerases, and modified nucleotides are applied to the flow cell.
The modified nucleotides have three special properties:
① Termination of synthesis
They carry a chemical modification on the hydroxyl group that prevents the addition of another nucleotide to the growing DNA strand. As a result, DNA synthesis stops immediately after the incorporation of each individual nucleotide.② Reversibility of the blockage
This modification can subsequently be chemically removed, allowing DNA synthesis to continue. This enables sequencing to proceed step-by-step, one nucleotide at a time.③ Fluorescent labeling
Each of the four bases (A, T, C, G) is tagged with a specific fluorescent dye.Because of these properties, these nucleotides are called reversible terminator nucleotides (RT-dNTPs).
Sequencing Process
➥ The sequencing primers hybridize to the forward strands of the DNA library.
➥ DNA polymerase binds to the primer and begins synthesis. However, due to the modified nucleotides, it can add only one nucleotide per cycle.
➥ After each incorporation, the flow cell is photographed by a high-resolution camera. The fluorescent label indicates which nucleotide was added. A computer analyzes the fluorescence signals and assigns them to the corresponding bases.
➥ Then, excess nucleotides are washed away, and the blocking group on the incorporated nucleotide is chemically removed.
➥ The cycle starts again, repeating until the entire DNA fragment has been sequenced.Abb. 17-M: Illumina sequencing steps Primer, DNA polymerase (DNAP), and modified nucleotides (RT-dNTPs) are applied to the flowcell. The RT-dNTPs are fluorescently labeled (each base has its own color) and carry a reversible block at the 3’-hydroxyl group. This causes DNA synthesis to pause temporarily after each base incorporation.
As soon as an RT-dNTP is incorporated, a laser excites the fluorescent dye, causing it to emit light. A camera captures this signal and determines which base was added. Then, the fluorescent label along with the blocking group is chemically removed, allowing DNA synthesis to continue in the next cycle. This process is repeated in every cycle: one base is added, the signal is recorded, and the block is removed. The sequencing proceeds over a set number of cycles, reading a limited number of bases (e.g., 150 base pairs in 150-base reads).Fig. 17-N: Analysis of the sequencing data During sequencing, a camera captures the fluorescence signals of the incorporated nucleotides in each cycle. Each cluster emits a specific color signal that corresponds to the incorporated base. Over the course of multiple cycles, this generates a unique sequence for each cluster. A computer analyzes the color information from the individual images and assigns it to the corresponding DNA sequence. In this way, the precise base sequence of the „DNA of interest” is reconstructed from the measured fluorescence signals.
Double-check for DNA: Paired-End Sequencing
Paired-end sequencing is a method commonly used on many Illumina platforms to improve the accuracy and reliability of sequencing results. In this approach, each DNA fragment is read from both ends – resulting in two reads per fragment: Read 1 (forward) and Read 2 (reverse).
After the initial sequencing of the forward strand (Read 1), a second round of bridge amplification is performed to regenerate the original DNA strands. The previously sequenced strands are then removed, and sequencing of the reverse strand (Read 2) begins.
Since both reads originate from the same DNA fragment, they can be computationally merged. This facilitates sequence assembly, enhances error detection, and improves the readability of repetitive or complex regions within the DNA sequence.
Paired-end sequencing is particularly well suited for long, nested, or challenging DNA regions – providing more precise and reliable results.
Result of Illumina Sequencing
At the end of Illumina sequencing, a vast amount of short DNA segments – known as reads – is obtained. Each of these reads originates from a small fragment of the original genetic material and has been amplified and read millions of times.
To reconstruct a complete picture from these reads, specialized software is used to assemble them on a computer:
🔹 If a known reference DNA is available, the reads are aligned like puzzle pieces to the existing sequence pattern.
🔹 If no template is available, the reads must be assembled piece by piece based on overlapping regions.This step-by-step assembly of countless short sequences gradually creates a complete picture of the original DNA material. The result is a precisely analyzed gene sequence that not only reveals the genetic structure but also provides insights into mutations or variants.
🎥 Tip: You can find a clear explanation in the video „Illumina Sequencing Technology”.
Applications
Illumina sequencing is known for its accuracy and reliability. Its applications include:
Genomics: Decoding the DNA of humans, animals, and plants
Medicine: Investigating genetic diseases and developing personalized therapies
Microbiology: Analyzing bacteria and viruses
Environmental research: Studying DNA in soil or water samples
And what comes next?
Illumina technology has revolutionized genetic analysis – but it also has its limits: the preparation is complex, data analysis is computationally intensive, and very long DNA segments can only be read in small fragments.
That’s why development continues. New third-generation sequencing technologies rely on entirely different principles – enabling, for the first time, the direct reading of extremely long DNA strands, often even in real time.
Time to take a look at the next generation of sequencing.
Third Generation: Real-Time Sequencing
With the third generation of sequencing technologies, a fundamental shift is beginning in genomic research. Instead of relying on fragmented or chemically modified DNA as before, these methods allow for the direct reading of genetic information – in real time, without complex preparation, and with new analytical possibilities.
Two key methods of this generation are:
- Single Molecule Real-Time (SMRT) Sequencing – monitors DNA synthesis in real time by detecting light pulses emitted as individual bases are incorporated.
- Nanopore Sequencing – threads DNA strands through tiny nanopores and measures changes in electrical current to identify the bases.
Why Nanopore Sequencing Is a Game Changer
Nanopore technology opens up entirely new perspectives in genomic research – thanks to its flexibility, speed, and independence from labor-intensive preparation steps:
✅ Real-time sequencing: Unlike traditional methods that require DNA to be amplified or chemically modified first, nanopore technology reads genetic material directly.
✅ Long read lengths: Instead of short fragments, extremely long DNA strands can be read – often spanning hundreds of thousands of base pairs in a single run. This greatly simplifies the analysis of complex genomes and structural variants.
✅ Versatility: In addition to DNA, RNA can also be analyzed directly – without the intermediate step of converting it into complementary DNA (cDNA). This makes the technology especially valuable for studies of gene expression and virus research.
✅ Portable and cost-effective: Devices like the MinION are barely larger than a USB stick and enable sequencing outside the lab – such as in clinics, in the rainforest, or directly at a crime scene.
With these features, nanopore sequencing opens up entirely new fields of application – from basic research and diagnostics to environmental and biodiversity analysis.
That’s reason enough to take a closer look at this technology.
For a first impression and a brief overview, the following video is a great starting point.
Nanopore Sequencing – Step by Step
Today, when people talk about nanopore sequencing, they almost always mean Oxford Nanopore Technology (ONT). While there are theoretically other nanopore-based approaches, ONT is currently the only one in practical use. Since its general market launch in 2015, it has revolutionized sequencing.
What makes this method special? It reads DNA or RNA directly and in real time – without prior chemical modifications or amplification reactions. Nanopore technology functions like a tiny high-performance laboratory that decodes genetic information with precision.
How does it work?
ONT sequencing is based on the interaction of three key components:
➤ Nanopores – act as tiny molecular „readers”. When a DNA or RNA strand passes through a nanopore, it generates characteristic electrical signals – essentially a molecular „fingerprint”.
➤ Membrane – serves as a filter and barrier. It ensures that only ions and nucleic acids pass through the nanopores, while unwanted molecules are kept out. This results in clean, precise measurements.
➤ Chip – is the core of the system. It holds the membrane with the nanopores and detects the electrical signals generated as the DNA passes through.
The chip has two chambers:
- Upper chamber (cis): This is where the DNA sample is introduced.
- Lower chamber (trans): This is where the DNA ends up after passing through the nanopores.
Both chambers are filled with an ion-containing solution that conducts electrical current. The membrane separates the chambers so that no current can flow – except at the points where the nanopores are embedded. These pores are the only „tunnels” through which ions and nucleic acids can pass.
As soon as a DNA or RNA strand slides through a nanopore, the electrical current changes. These subtle variations are detected and translated into genetic code – in real time: directly, quickly, and accurately.
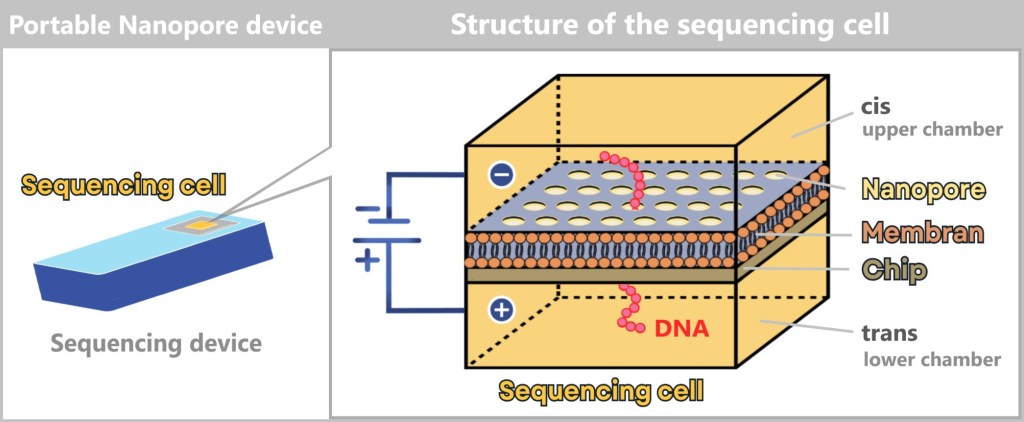
Fig. 18-A: Structure of the nanopore device On the left is a portable nanopore sequencing device. In practice, it is slightly larger than a USB stick. An arrow points to the replaceable sequencing cell. The enlarged illustration on the right shows the schematic structure of the sequencing cell.
Precision at the Nanoscale
Chip microstructure
The chip is manufactured using highly precise fabrication techniques such as photolithography or etching processes. This creates tiny channels, called wells, that range from just a few nanometers to micrometers in size. In addition to these structural components, the chip also contains the electrochemical sensing device that detects the ion flow as DNA or RNA passes through the nanopore.Self-assembly of the lipid membrane
This membrane consists of lipids (fat molecules) and is electrically insulating. This means that it does not conduct electricity itself. The membrane is initially applied onto the chip without nanopores. It is not mechanically stretched onto the chip, but is formed by self-organisation over the openings.How does it work?
A solution containing lipid molecules is applied to the chip. These molecules have:
🔹 a hydrophobic (water-repelling) part, and
🔹 a hydrophilic (water-attracting) part.Because of these properties, they spontaneously organize into a stable bilayer. The wells in the chip are designed so that the lipid membrane is stabilized precisely above them.
Self-assembly of the nanopores
Nanopores are usually made from proteins derived from bacteria or yeast cells. In nature, these pores serve as transport channels for molecules across cell membranes. However, in nanopore technology, they are used as highly sensitive sensors for DNA or RNA molecules.The integration of the nanopores takes place after the membrane is formed. Thanks to a natural process called self-assembly, they insert themselves into the membrane spontaneously!
How does it work?
- The nanopores are added in a solution.
- They automatically „find” the wells, since they can only embed themselves in the membrane where an opening exists.
- There, they penetrate the membrane and form stable channels.
The wells are precisely defined areas where the membrane is accessible – and it is exactly there that the nanopores can integrate selectively.
Why is this important?
The correct positioning of the nanopores over the wells is crucial because:- The wells are connected to electrodes that measure the ion flow. If a nanopore is not exactly positioned over a well, there is no measurable current – making the nanopore nonfunctional.
- DNA is pulled through the nanopores from the cis to the trans chamber, and this only works if the nanopores are embedded in the membrane directly above the wells.
The path of the DNA through the nanopore
After exploring the structure of the sequencing cell and the basic principle of Oxford Nanopore technology, we now turn to the actual sequencing process.
1️⃣ Preparation of the DNA/RNA sample
Before the actual sequencing begins, the nucleic acid (DNA or RNA) must be prepared.
➥ Extraction: The DNA or RNA is isolated from the sample (e.g., blood, saliva, cell culture, environmental samples). This is done using standardized extraction methods (see Chapter „4.5.1. Nucleic Acid Extraction” for details).
➥ Fragmentation (optional): Nanopore sequencing can read very long DNA or RNA molecules. If the DNA is too long or needs to be tailored for specific applications, it can be mechanically or enzymatically cut into fragments.
➥ Adapter ligation: Since the nanopores can only process DNA or RNA with special ends, adapters are attached to the ends of the molecules. These adapters contain:
- Motor proteins that control the movement of the DNA through the nanopore
- Barcode sequences (if multiple samples are to be sequenced simultaneously)

Fig. 18-B: Diagram of the motor protein binding to double-stranded DNA (dsDNA). To enable the motor protein to bind to the DNA, special adapters are first attached to the DNA ends. These adapters ensure that the motor protein binds specifically at one end of the DNA. For clarity, the illustration shows only the binding of the motor protein, while the adapters themselves are not depicted. Typically, only one motor protein binds per DNA molecule, as the adapters are designed to favor binding at one preferred end.
Where does the motor protein come from?
The motor protein used in Oxford Nanopore sequencing is a naturally occurring enzyme derived from bacteria. It is a modified version of a protein originally found in organisms such as E. coli or other microorganisms. In nature, these proteins are responsible for unwinding and transporting DNA – a capability that nanopore technology harnesses for sequencing.If RNA is to be sequenced, an optional reverse transcription step can be performed to convert RNA into DNA. However, ONT technology is capable of sequencing both RNA and DNA directly. Direct RNA sequencing can provide specific information about RNA modifications.
2️⃣ Applying an electrical voltage
Both chambers of the sequencing cell contain charged particles (ions). Once a voltage is applied between the upper (cis) and lower (trans) chambers, ions begin to flow through the nanopores. This creates a measurable electrical current. As long as no DNA is present in the pore, the ion flow remains constant, and a stable current is detected.
3️⃣ The DNA is added to the sequencing cell
The prepared DNA sample, with motor proteins already attached, is pipetted into the cis chamber (the upper chamber of the sequencing cell). Through diffusion or gentle mixing, the DNA disperses in the solution and reaches the vicinity of the membrane where the nanopores are embedded.
4️⃣ Docking onto the nanopore
The motor protein bound to the DNA guides the molecule to the nanopore and docks specifically onto it. Once the connection is established, the motor protein initiates its helicase activity: it unwinds the double-stranded DNA (dsDNA) into two single strands. One of these strands is captured by the nanopore and pulled through it. This process takes place directly at the pore and ensures that the DNA is transported precisely and evenly through the nanopore.
Fig. 18-C: Schematic of the sequencing cell and ion current in the idle mode. The sequencing cell consists of two chambers: the cis chamber (top) and the trans chamber (bottom), separated by a membrane with embedded nanopores. On the left, a DNA molecule has docked onto a nanopore with the help of a motor protein. On the right, an empty nanopore is shown, through which a constant ion current flows. The applied voltage between the negative electrode (cis) and the positive electrode (trans) drives the ion flow. The ion current is measured in the well (a small channel in the chip), as illustrated in the current/time diagram. As long as no DNA passes through the pore, the current remains constant.
5️⃣ The DNA passes through the nanopore – the signal is generated
DNA carries an electrical charge due to its negatively charged phosphate backbone. The applied voltage between the cis chamber (negatively charged) and the trans chamber (positively charged) causes the DNA to be pulled through the nanopore.
The motor protein plays a crucial role in this process:
➤ It controls the movement of the DNA through the pore – slowly, evenly, and steadily.
➤ This ensures a clearly readable signal for reliable signal detection.For context: Naturally, DNA would shoot through the nanopore at a speed of millions of bases per second. The motor protein slows this down to about 450 bases per second (depending on the device and settings). This slowdown is what makes sequencing with this technology possible at all.
As the DNA passes through the pore, it influences the ion flow – because each base (A, T, G, C) has a unique size and chemical structure. These differences alter the ion current in specific ways. These current changes are measured by an electrode located directly beneath the nanopore. Each well (the tiny tunnels in the chip where the nanopores sit) has a dedicated electrode that acts as the sensor. This setup allows the software to precisely assign signals to a specific nanopore – an essential requirement for accurately reconstructing the DNA sequence.
Fig. 18-D: The image illustrates the central mechanism of nanopore sequencing. 1) A motor protein pulls the DNA steadily through the pore. The negatively charged DNA moves from the cis side to the trans side due to the applied voltage. During this process, the individual bases (A, T, G, C) affect the ion current in specific ways.
2) A graph displays the changes in the measured current over time. Each base combination produces a characteristic signal that is decoded by algorithms.
3) A computer analyzes the electrical signals and determines the sequence of the bases from them.6️⃣ The electrical signal is recorded
As the DNA is pulled through the nanopore, about 10–15 bases are inside the pore at the same time. Each of these bases influences the ion current differently – so the signal is a composite of the effects of multiple bases.
But how can individual bases still be distinguished?
This works thanks to a clever algorithm:
- The motor protein moves the DNA step-by-step – roughly one base at a time.
- The measured current changes in characteristic ways depending on the sequence of bases.
- Specialized software (basecalling) uses machine learning to decode the exact sequence from the overlapping signals.
- The model „untangles” the overlapping information and assigns it to the correct base at the precise position.
In this way, the complete DNA sequence gradually emerges from the electrical signals.
Result of nanopore sequencing
In the end, Oxford Nanopore technology provides the complete sequence of bases in the analyzed DNA or RNA sample. This sequence contains detailed information about the genetic composition, the length of the molecules, as well as possible features such as mutations or structural variations.
🚀 Real-time data – a genuine speed advantage
The entire process happens live. As the DNA or RNA passes through the pore, the sequence of bases is captured and analyzed immediately.
For comparison:
- Traditional Sanger sequencing takes several hours or even days to complete an analysis.
- Illumina sequencing typically provides results within a few hours to several days, depending on the length, complexity, and scale of the sample – smaller projects often finish in a few hours, while larger analyses may take several days.
- Oxford Nanopore Technology (ONT), on the other hand, delivers initial results within minutes.
The generated sequencing data are processed and stored in parallel. They are immediately available for further analysis.
Looking ahead: Sequencing 4.0 – what’s next?
While the third generation is still being celebrated, researchers are already working on the fast lane. Emerging new technologies promise even more: greater speed, higher accuracy, and even more versatile applications.
In the next chapter, we’ll take a brief glimpse into the future of sequencing.
Emerging Technologies: The Future of Sequencing
Nanopore sequencing (ONT) has already revolutionized sequencing, but there are promising approaches in the tech pipeline that could further advance this method.
Transistor instead of pore
An exciting example is FENT technology (Field-Effect Nanopore Transistor), which analyzes DNA and other biopolymers using a novel transistor-based structure that operates with extreme speed and precision. This technology could enable even more precise, faster, and more cost-effective analysis of DNA and other biopolymers.
The „FENT Nanopore Transistor Explainer Video” offers a fascinating insight into this innovation.
Whether and when this technology will become relevant for virus diagnostics remains to be seen – but it impressively demonstrates how sequencing technologies continue to evolve.
The next big thing in genetics
Besides FENT, methods like In Situ Sequencing (ISS), which makes gene expression visible directly within tissues, could also play an important role in the future – such as investigating viral activity within cells.
The future of sequencing?
It’s going to blow us away – literally! –
and we’re right there live to witness it!
4.6. Bioinformatic Analysis
The raw sequencing data is simply a long string of bases, such as „AGCUACGUA…” in the case of an RNA sequence. Sequencing is like spelling – it’s only through bioinformatics that the story behind it is told, „translating” the data into meaningful information. From this jumble of letters (AGCUACGUA…), a profile is created: What virus is it? What can it do? And how dangerous is it?
Warum Rohdaten chaotisch sind
💨 Technical glitches: Like a blurry photo – some parts of the sequence are missing or fuzzy („noise”).
❓ Mystery bases („N”): Positions where even the machine gives up: „Could be A, C, or U – no idea!”
🦠 Mutant flatshare: RNA viruses like influenza exist as communities of variants (mutation clouds). Sequencing blends them all together – like a smoothie made from 100 different fruits.
What Bioinformaticians Do
They clean, sort, and piece together the data – until it becomes clear:
- Who’s here? (Virus identity)
- What’s new? (Mutations)
- How to respond? (Take a deep breath first!)
It’s detective work – just with more computers and fewer trench coats.
From Data Chaos to Insight: The 5 Steps of Bioinformatics
1️⃣ Quality Control: Sorting the data puzzle
2️⃣ Alignment & Assembly: The big puzzle game
3️⃣ Homology Search: Let’s see!
4️⃣ Database Entry: Ready for the registry?
5️⃣ Functional Annotation: The puzzle comes to life
1️⃣ Quality Control: Sorting the data puzzle
Sequence data is like a puzzle from a flea market: Some pieces are duplicates, some are missing, others don’t fit – and right in the middle, there’s a coffee stain. What to do? Before putting it all together, the pieces need to be sorted, cleaned, and filtered. This is where the work of bioinformatics begins.
🔍 FastQC (a software tool) – the critical book inspector:
It flips through the sequencing data like an old manuscript, flagging all the messy pages – too short, too faulty, too suspicious.✂️ Trimmomatic (a software tool) – the molecular lawnmower:
Whatever is broken, frayed, or simply unnecessary gets ruthlessly trimmed off – especially at the ends, where errors love to hide.🔄 Error correction – democracy at the molecular level:
If nine out of ten reads say, „An A belongs here”, then the lone G gets outvoted. Even DNA follows the rule of majority decisions.In the end, only quality remains:
🔹 Short or low-quality reads? Discarded.
🔹 Individual sequencing errors? Corrected.
🔹 Not enough good data overall? Better to resequence.Why? So that nobody draws wrong conclusions from a typo later on. Because only with clean data can the real detective work begin: assembling the genome!
2️⃣ Alignment & Assembly: The big puzzle game
Modern sequencing methods produce DNA or RNA fragments like someone exploded a puzzle:
- Illumina generates millions of short reads.
- Nanopore delivers meter-long pieces (well, almost).
But whether small or large, in the end, they all have to be assembled – using algorithms, logic, and lots of computing power.
🦠📌 Method 1: Alignment (for known viruses)
Where does this piece fit?
The cleaned sequence data are compared to a known reference sequence – like puzzle pieces placed on a picture on the box. This way, even the smallest differences can be detected:
What you find:
🔹 Mutations: An A instead of a G? Maybe that makes the virus more contagious.
🔹 Missing spots: Small holes in the genome – so-called deletions.
🔹 Extra pieces: Unexpected insertions.Even though the reference sequence is just one variant, the alignment reveals the entire „mutant flatshare” present in the sample.
🦠❓ Method 2: De novo assembly (for novel viruses)
Solving a puzzle without a template – like flying blind!
If no known reference is available, the individual reads must be assembled without a template. The trick: overlapping sections reveal which pieces belong together. In this way, a complete genome is built piece by piece. This process is more error-prone than alignment but provides a first draft of the genome of a newly discovered virus.
The difference:
Alignment is like assembling IKEA furniture – with instructions.
De novo assembly is more like: „Here are some wooden parts, good luck!”
3️⃣ Homology Search: Let’s see!
Now the cards are on the table. The unknown DNA fragment is played – all data has been gathered, everything is ready. Now it’s time to say: Let’s see!
Here comes BLAST into play – the great search engine for genes. It compares the genetic wildcard with millions of other sequences in worldwide databases. And if a relative exists somewhere, BLAST finds it. Whether it’s a close cousin or a distant ancestor, similarities in the code reveal whether you’re holding an old acquaintance in your hands… or perhaps an entirely new virus.
BLAST zeigt: BLAST shows:
🔹 The percentage of sequence similarity
🔹 Statistical values indicating how reliable the match is
🔹 Possible relationships, even when the similarity is only distantFor those who really want to know:
How to Use BLAST for Finding and Aligning DNA or Protein Sequences🦠📌 For known viruses: BLAST confirms the results of the alignment. Does the strain belong to an already described variant? Are there new mutations? For example, with the influenza virus, BLAST might show that a new strain matches 98% with the H3N2 strain from the last flu season.
🦠❓ For unknown viruses: BLAST determines whether the new sequence can be assigned to a known virus family – or if it represents a completely new member. If no matches or only very distant hits are found, further analyses are required.
4️⃣ Database Entry: Ready for the registry?
Now it’s official. The collected sequence data are ready – but not every new virus variant immediately makes it into the big scientific archive. First, it’s checked: Ready for the registry?
🦠📌 For known viruses: After the homology search, the process moves to detailed analysis: Does the strain have anything interesting to offer? Variant detection takes a close look – like a geneticist with a magnifying glass. Are there mutations that help the virus evade the immune system (keyword: immune escape)? If yes – then it goes into the registry! The new strain is entered into a public database and is then available to researchers worldwide.
🦠❓ For unknown viruses: When BLAST responds with „No match found!”, things get exciting. Now, advanced bioinformatics tools come into play – such as functional annotation, which asks: What might this virus be capable of? If the analysis reveals that we’re dealing with something truly new, the discovery is entered into one of the major international databases – including its name, genetic fingerprint, and possible origin.
The most important databases for viral genomes:
🌐 GenBank – The all-rounder:
a vast library of genetic sequences from all forms of life, including viruses.🌐 Influenza Research Database (IRD) –
A specialized archive for influenza viruses, complete with practical analysis tools.🌐 GISAID – The Formula 1 among databases:
ultra-fast, especially for influenza viruses and SARS-CoV-2.🌐 VIRALzone – A kind of Wikipedia for virus families:
with information on genome structure, morphology, and replication.What enters these databases becomes part of a global scientific memory.
5️⃣ Functional Annotation: The puzzle comes to life
The genome has been sequenced – but now we want to know: „Which genes do what?”
Functional annotation attempts to read the virus’s „blueprint” and determine which parts are responsible for what. Why is this virus more infectious than others? How does it manage to evade the immune system? And could it be resistant to medications?
To do this, the genome is scanned for known patterns. Are there regions that resemble genes already identified in other viruses? Do certain sequences match enzymes that help the virus enter cells? Or regions that form the virus’s surface – the very spots the immune system recognizes first?
Spatial structures also play a crucial role. Modern AI models like AlphaFold can predict a 3D model of the resulting protein based on the gene sequence. This allows researchers to determine whether a mutation changes just a small detail – or if it alters the entire behavior of the virus.
This is especially important when it comes to:
- Infectivity: A mutation in a surface protein might make the virus „stickier” – helping it bind more effectively to host cells.
- Immune evasion: Small changes at key „recognition sites” can be enough for antibodies to miss the virus entirely.
- Drug resistance: Some mutations can deactivate a drug’s target structure – rendering the medication ineffective.
Functional annotation is like the instruction manual for a virus – just without friendly warnings.
💡 Summary: Virus Cracking for Beginners
Sequencing: Spell out the virus ABC.
Quality Control: Clean up the data junk.
Alignment & Assembly: Puzzle the snippets together – with or without a template.
BLAST: Google for genetic relatives.
Database: Share your discovery with the world – if it’s any good.
Annotation: Read between the genes – what can this virus really do?Classification of Viruses Based on Their Genetic Material
Viruses can be classified in various ways – for example, by their shape, their hosts, their mode of transmission, or their genetic material.
One of the best-known and most frequently used genome classifications is the Baltimore classification, introduced in 1971 by David Baltimore. This system categorizes viruses based on a combination of the following characteristics:
- Type of genetic material (DNA or RNA),
- Strandedness (single-stranded or double-stranded),
- Polarity of RNA (positive or negative), and
- Replication mechanism (how mRNA is produced).
The goal of all viruses is to produce mRNA, because only with mRNA can they make the proteins they are composed of and replicate. The Baltimore classification includes seven groups that demonstrate the different ways viruses achieve this goal – ranging from direct use of their RNA to complex detours via DNA. Here is an overview:
Group I: Double-stranded DNA viruses (dsDNA)
mRNA is produced by direct transcription of the DNA using an RNA polymerase, which can be either cellular or viral.
(Examples: Adenoviruses, Herpesviruses, and Poxviruses)Group II: Single-stranded DNA viruses (ssDNA)
The single-stranded DNA is first converted into double-stranded DNA. Then, transcription takes place to produce mRNA.
(Examples: Parvoviruses)Group III: Double-stranded RNA viruses (dsRNA)
mRNA is produced by transcription of the RNA strands using a viral RNA-dependent RNA polymerase.
(Examples: Reoviruses)Group IV: (+) Single-stranded RNA viruses [(+)ssRNA]
The viral RNA serves directly as mRNA because it has the same orientation as mRNA (5′ → 3′).
(Examples: Coronaviruses, Polioviruses)Group V: (–) Single-stranded RNA viruses [(–)ssRNA]
The RNA is oriented in the opposite direction (3′ → 5′) and must be transcribed by a viral RNA-dependent RNA polymerase into (+)RNA, which then serves as mRNA.
(Examples: Influenza viruses, Rabies virus)Group VI: Retroviruses [(+)ssRNA with a DNA intermediate]
Reverse transcriptase (RT) converts RNA into DNA, which is integrated into the host genome. The mRNA is then produced from the integrated DNA by cellular mechanisms.
(Examples: HIV)Retroviruses like HIV use a unique replication mechanism: The viral RNA is transcribed into a complementary DNA strand (cDNA) by reverse transcriptase (RT). The original RNA is then degraded, and a second DNA strand is synthesized. The resulting double-stranded DNA (dsDNA) is integrated into the host genome. This process makes retroviruses particularly persistent, as they can remain in a latent phase and be reactivated at any time.
Group VII: Double-stranded DNA viruses with an RNA intermediate (dsDNA-RT)
The viral DNA is repaired in the nucleus and serves as a template for the synthesis of mRNA and pregenomic RNA. The pregenomic RNA is then reverse transcribed back into DNA by reverse transcriptase.
(Examples: Hepatitis B virus)The Hepatitis B virus has an incomplete double-stranded DNA genome. In the nucleus of the host cell, this DNA is repaired before mRNA and pregenomic RNA are transcribed. The pregenomic RNA serves as a template for the formation of new virus particles.

Fig. 19: The graphic shows the seven classes of the Baltimore classification. Viruses are categorized based on their genome structure and their strategy for mRNA synthesis. Double-stranded (ds) and single-stranded (ss) genomes are distinguished, the polarity of RNA is indicated by „+” (positive) and „–” (negative) strands, and the reverse transcription mechanism is highlighted by „RT.” At the end of the process is the mRNA, which all viruses require to produce their proteins.

5. Do Viruses Really Exist?
In a world where everything can be questioned – how does one prove that something exists?
Some say viruses are merely artifacts, lab constructs, or cellular debris without their own identity. But there are characteristics that cannot be dismissed:
Viruses have something that only they have.
Many viruses possess conserved genome regions that encode distinctly viral functions:
- Capsid proteins, which package their genetic material,
- RNA-dependent polymerases, which replicate their genome,
- Integrases, which insert viral genes into the host genome (e.g., in retroviruses like HIV),
- Proteases, which cleave viral proteins to activate them.
The Capsid – the Virus’s Signature Feature
What makes a virus a virus? Not just its parasitic lifestyle – but above all, its structure.
Almost all viruses have a capsid – a proteinaceous shell that protects their genetic material and also helps them enter host cells. Whether icosahedral or helical, the capsid is functionally and evolutionarily THE central feature of the virus.
It protects, it transports, it organizes – and it is uniquely virus-specific.
Fig. 20: The Capsid – Protective Shell and Shape-Former of the Virus The capsid is the outer protein shell of a virus that protects its genetic material and determines its external shape. It is made up of many identical building blocks (capsomers) that assemble into symmetrical structures. The most common capsid shapes are icosahedral (20-faced) and helical (spiral-shaped).
Additional components, such as a lipid envelope – derived from the host cell – can alter the external shape. As a result, many enveloped viruses appear „spherical”, even though the underlying capsid is usually icosahedral. In some cases, such as with bacteriophages, the structure deviates significantly: this is referred to as a complex morphology, often featuring an icosahedral head and a helical tail. These morphological differences form the basis for the structural classification of viruses.
Viral hallmark genes – signatures beyond the cellular world
The genes that code for capsid proteins are among the so-called hallmark genes. They are:
- highly conserved within the viral world,
- functionally essential for the viral life cycle,
- and not found in cellular organisms.
Many of these proteins share a very distinctive shape: the jelly-roll fold. This three-dimensional structure resembles a rolled sponge cake and gives the capsid protein remarkable stability. It is found in a wide range of viruses – from those that infect humans to viruses that exist only in ancient deep-sea microbes. Its striking architecture is so characteristic that researchers can identify even novel viruses based solely on this genetic signature.
Anyone who has seen it once will recognise it – like a symbol woven through the depths of evolution – an evolutionary signature, legible to anyone willing to look closely.
As shown, for example, by the work of Koonin et al: Despite all their diversity, viruses have a common, ancient genetic core – as if they were carrying the coat of arms of their ancestors.
Conclusion: Virus-specific genome segments are a unique defining feature!
Viruses defy simple definitions. They are neither classic living organisms nor mere clusters of molecules. But they carry a set of characteristics that makes them distinguishable – in their form, in their function, and in their history.
If something has been leaving its own genetic trace for billions of years – then it is real. Even if it doesn’t lead an independent life. Even if we cannot see it.
Questions & Answers
🟡 „Are viruses just exosomes with a PR department?”
No. Exosomes are biological packaging waste. Viruses come with their own blueprint – and a transport container with perfect geometry.
Anyone who confuses exosomes with viruses is also confusing Tupperware with spaceships.
🔬 „But we’ve never really seen them!”
Yes, we have. Electron microscopes provide images, and cryo-tomography even produces 3D videos at nanometer resolution.
Invisible? Only when the power’s off.
🧬 „Isn’t the RNA just random cellular debris?”
No. Viral RNA/DNA is unique, functional, and precisely organized – and reproducible across the globe.
Random? Then a clockwork would just be some metal – with team spirit and a good mood.
📦 „That capsid – couldn’t it just form on its own?”
Hardly. These nanoboxes with perfect symmetry are found only in viruses.
A soccer ball doesn’t just randomly form in the grass, either.
🌍 „Maybe it’s all just a measurement artifact?”
Then it’s an astonishingly global one. All laboratories detect the same sequences, structures, and properties.
Viruses don’t recognize lab boundaries – only reproducibility.
In the end, one truth remains:
Viruses are not fiction, not artifacts, not products of chance.
They are biology’s most precisely mapped source of unease –
real, reproducible, and everywhere.Anyone who calls them cellular junk might as well believe
a Tesla is just an especially ambitious shopping cart.Sources and links documenting the isolation and sequencing of viruses:
National Center for Biotechnology Information (NCBI) Virus Database:
NCBI Virus DatabaseThe NCBI hosts a comprehensive database of viral genomes that have been sequenced by scientists around the world. Here, you can access thousands of viral genomes that have been isolated and sequenced.
GISAID (Global Initiative on Sharing Avian Influenza Data):
GISAIDGISAID is a platform primarily used for the sharing of sequence data on influenza and SARS-CoV-2 viruses. It provides detailed information on the sequencing and origin of these viruses.
European Nucleotide Archive (ENA):
ENA BrowserThe ENA provides access to sequence data from viruses that have been isolated in laboratories around the world. This database is maintained by the European Bioinformatics Institute (EBI) and contains extensive information on viral genomes.
PubMed – Scientific articles on virus isolation and sequencing:
Example search query: „Virus Isolation and Sequencing“PubMed is a search engine for scientific literature. You can use it to find specific studies on the isolation and sequencing of viruses.

6. Where Do Viruses Come From?
These questions push the boundaries of our knowledge. They touch not only on the origins of viruses but on the very foundations of life itself. Before we explore possible scenarios for their emergence, it’s worth stepping back to look at the bigger picture: the „Tree of Life” – as modern evolutionary research depicts it today.
6.1. The Tree of Life
All known forms of life – from bacteria to blue whales – share a common origin: LUCA, the Last Universal Common Ancestor. LUCA was not the first organism ever to exist, but the last common ancestor of all cellular life alive today – bacteria, archaea, and eukaryotes. An evolutionary crossroads, not a starting point.
What do we know about LUCA? He likely lived around 3.5 to 4 billion years ago, in a warm and still-young Earth. LUCA was cellular, already equipped with DNA, RNA, and proteins, and capable of converting energy – but still far from the complexity of modern cells. A prototype of life, from which everything else branched out.
Fig. 21: Tree of Life – showing the major evolutionary lineages of cellular DNA-based organisms. LUCA (Last Universal Common Ancestor) branches into the domains of Bacteria (e.g., Terrabacteria, Proteobacteria) and Archaea (e.g., Euryarchaeota, Asgard). Through the endosymbiotic fusion of a proteobacterium with an Asgard archaeon, FECA (First Eukaryotic Common Ancestor) emerges, which evolves into LECA (Last Eukaryotic Common Ancestor), the origin point for eukaryotes (animals, plants, fungi). A second endosymbiosis involving cyanobacteria gives rise to chloroplasts in plants. On the right, a timeline illustrates the emergence of life: from the formation of Earth (~4.5 billion years ago), through the Great Oxygenation Event (~2.4 billion years ago), to the Cambrian Explosion (~540 million years ago).
But while the Tree of Life grew full of branches, twigs, and leaves, one question remains unanswered: Where do viruses fit into this picture – or do they even belong on the tree at all?
Unlike bacteria or eukaryotes, viruses have no cellular structure, no metabolism of their own, and leave behind no fossils. Some scientists see them as „lost branches” of the Tree of Life, while others consider them ancient precursors – perhaps even predating LUCA itself.
To answer this question, we need to go back to the beginning – or rather, to the possible beginnings.
So where do viruses stand in this tree of life?
Are they late arrivals, evolved from derailed genes of cellular organisms? Or do they belong to the primordial forms of life – perhaps even older than LUCA itself?
Science has (as yet) no definitive answer. But several hypotheses aim to trace the origins of viruses.
6.2. The Main Hypotheses on the Origin of Viruses
💡Note: The following origin hypotheses assume a basic understanding of the so-called RNA world – an early phase in Earth’s history when life still consisted of simple, self-replicating RNA molecules. To immerse yourself in this unfamiliar era and better contextualize the hypotheses, we recommend the article „The First Whisper of Life”. It poetically tells the story of the beginnings of biological order – long before cells, proteins, and DNA.
6.2.1. Hypotheses in a Cellular World
These hypotheses assume the existence of cells from which viruses emerge.
6.2.1.a) Progressive Hypothesis – Viruses as Escaped Genes
6.2.1.b) Regressive Hypothesis – Viruses as Reduced Cellular Organisms
6.2.2. Hypotheses in the Pre-Cellular RNA World
These hypotheses place the origin of virus-like structures in a time before or during the formation of the first cells, in the RNA world, where self-replicating molecules dominated.
6.2.2.a) Virus-First Hypothesis – Viruses Came First
6.2.2.b) Co-Evolution Hypothesis – Viruses and Cells Emerged Together
6.2.1. Hypotheses in a Cellular World
The so-called Cell-First Hypothesis assumes that cellular life forms – such as LUCA – already existed before viruses emerged. Within this approach, two main scenarios can be distinguished:
a) The Progressive Hypothesis: According to this, viruses originate from genetic material that „escaped” from cells – essentially runaway genes that became independent and embarked on their own evolutionary path.
b) The Regressive Hypothesis: According to this idea, viruses were originally complete cells that have gradually reduced themselves over the course of evolution – until only the essential functions necessary for parasitic life remained. A kind of evolutionary reduction.
Both variants share a common idea: viruses are not an independent origin of life, but rather an evolutionary byproduct of cellular organisms – arising through loss or separation.
a) Progressive Hypothesis – Viruses as Escaped Genes
The Progressive Hypothesis – also known as the Escape Hypothesis – suggests that viruses originated from genetic material that was once part of cellular organisms. These „escaped genes” evolved over time into independent, infectious entities – detached from their original cellular context.
How could this have happened?
In living cells, there are small, mobile pieces of DNA or RNA that can move freely within the genome – so-called „jumping genes”. These elements, which include plasmids (circular DNA molecules) and transposons (DNA segments that can change their position within the genome), have the ability to replicate independently and even transfer between organisms.
The hypothesis states that some of these genetic elements evolved to the point where they eventually became capable of replicating independently. They escaped cellular control, developed protective mechanisms such as a capsid – a protein coat that shields their genetic material – and thus became the first viruses.
Fig. 22-A: The graphic illustrates the Progressive, or Escape Hypothesis: according to which viruses originated from originally cellular genes that gradually became independent.
Protocell: An early, simple cell form with genetic material: large DNA ring, ribosomes and smaller DNA rings.
Separation of genetic material: Mobile genetic elements – such as plasmids or so-called transposons („jumping genes”) – detach from the cell. These may have taken the first steps toward independence.
Vesicle formation: The separated genetic material is enclosed in small membrane-bound sacs (vesicles), providing protection and mobility.
Formation of a virus: Over the course of evolution, these developed into functional viruses – with a protective protein coat (capsid, orange). Exactly how this coat evolved remains unclear („capsid mystery?”).✅ Arguments in Favor of This Hypothesis
➤ Similarity to Mobile Genetic Elements: Some viral genes closely resemble the „jumping genes” found within cells. Retroviruses in particular – such as HIV – use a mechanism similar to that of transposons: they reverse-transcribe their RNA into DNA and integrate it into the host’s genome.
➤ Viruses Exchange Genes with Their Hosts: Researchers have discovered genes in viral genomes that clearly originate from cellular organisms. This suggests that viruses may have once emerged from cells and, over time, acquired and evolved genetic material.
➤ Explanation for the Diversity of Viruses: Since different „jumping genes” may have escaped from different types of cells, this hypothesis helps explain why there are so many different viruses with varying characteristics.
❌ Weaknesses of the Hypothesis
➤ Capsid Proteins Are Extremely Ancient: Some key proteins involved in forming the viral shell (capsid) show no direct relation to cellular proteins. It remains unclear how escaped genes could have developed such complex structures. The evolution of these core proteins dates so far back that they may have existed even before the emergence of the first cellular life forms. A study by Krupovic & Koonin (2017) suggests that capsid genes may have evolved independently, before modern cells existed.
➤ Viruses Have Unique Enzymes That May Be Older Than LUCA: Some viruses contain RNA polymerases that differ significantly from those found in cells. These enzymes are so distinct that they may originate from a time before modern cellular mechanisms existed. Some researchers therefore speculate that viruses could be direct remnants of the RNA world – a phase of early evolution when life was still based on RNA rather than DNA.
➤ Viruses Exist in All Three Domains of Life: Viruses are found everywhere – they infect bacteria, archaea, and eukaryotes. If they had only emerged after the first cells evolved, one would expect them to be more confined to specific cellular lineages. Their universal presence suggests that they may have already existed before the split between bacteria, archaea, and eukaryotes.
➤ Viruses Do Not Share a Common Ancestry with Cellular Life: While bacteria, archaea, and eukaryotes can all be traced back to LUCA, there is no known common lineage for viruses. This suggests that viruses are not merely an offshoot of the cellular tree of life, but rather represent a very ancient, parallel line of evolution.
⬇️ Conclusion
The Cell-First Hypothesis views viruses as rebels of cellular life – originally harmless genetic passengers that broke away and followed their own evolutionary path. However, it assumes that complex cells already existed before viruses emerged.
b) Regressive Hypothesis – Viruses as Reduced Cellular Organisms
The Regressive or Reduction Hypothesis does not view viruses as fragments of escaped genes, but rather as former cells that gradually became more and more reduced over the course of evolution – until they lost their independence and turned into „gene packages” that rely on other cells to survive.
How could this have happened?
An early, complex, cellular organism (e.g., a parasite) developed an increasingly close dependence on its host. Over time, it lost unnecessary genes – for metabolism, cell division, and membrane formation – until it could no longer live independently. In the end, only a tiny remnant remained: the genes needed for replication (DNA/RNA), packaged in a protective shell (capsid) – a virus.

Fig. 22-B: The graphic illustrates the Regressive Hypothesis – or Reduction Hypothesis – according to which parasites became viruses through an extreme process of reduction. From a Traveling Cell Parasite to an Ultralight Virus
A free-living parasite (e.g., a small bacterium) infects another cell – just like some bacteria today parasitize other cells (e.g., Rickettsia or Chlamydia). Why? Probably because the protocell offered a safe environment. Perhaps the host had access to resources that the parasite couldn’t easily obtain – such as energy, enzymes, or nucleotides – a kind of molecular all-inclusive resort.It is even possible that everything began symbiotically – similar to the mitochondria. It was only later that this relationship was exploited unilaterally – it became parasitic.
The parasite initially lived independently. It could leave the host cell or reproduce within it. But gradually, it needed fewer and fewer of its own genes – the host ultimately provided everything it required. So, the parasite gradually shed its genetic „baggage”, step by step.
Over time, through mutations or selection, it lost many of its original genes:
➤ It no longer produced its own proteins.
➤ It lost the ability to generate energy.
➤ Eventually, even the genes for cell division.
In the end, only its genome remained – packaged in a clever „case”: the capsid.This is exactly where the virus comes into being: no longer an independent living organism in the classical sense, but an ultralight „traveler” that can only reproduce with the help of a host.
✅ Arguments in Favor of This Hypothesis
➤ Parallels to Intracellular Parasites: Some modern microorganisms, such as Rickettsia or Chlamydia, can only reproduce inside other cells – just like viruses. Moreover, their genomes are highly reduced. This demonstrates that cells can indeed undergo drastic „streamlining” through parasitism.
➤ Large DNA Viruses as „Transitional Forms”: Some giant viruses (e.g., Mimivirus, Pandoravirus) have incredibly large genomes – larger than some bacteria – and contain genes typically found in true cells (e.g., DNA repair enzymes). These viruses appear to act as evolutionary intermediates between real cells and typical viruses.
➤ Loss Instead of Creation: Evolution is not only about „more”, but often also about „less”. Especially in parasitic contexts, organisms frequently lose functions – just as assumed here.
❌ Weaknesses of the Hypothesis
➤ Origin Remains Unclear: Although the hypothesis explains how a cell could become a virus, it says little about when and under what conditions this might have occurred.
➤ No Common Ancestry: The diversity of viruses argues against a single „original cell” ancestor for all viruses. If all viruses descended from regressive cells, they would still have had to evolve independently multiple times. Some researchers therefore believe that regressive evolution is just one of several origins.
➤ Applies Mainly to Large DNA Viruses: This hypothesis fits well with large DNA viruses but less so with very simple RNA viruses, which lack any cell-like structures or genes.
➤ Capsid Proteins Show No Direct Similarity: Cells – even highly reduced parasitic ones – do not have capsids. The capsid is a virus-specific structure. If a virus evolved through reduction from a cell, the question arises: how could a structure like the capsid develop, which does not exist in cells at all? It appears to have arisen anew – which is difficult to reconcile with a theory based solely on „loss” and „reduction”.
➤ Energy Gap: Even degenerated parasites (e.g., Mycoplasma) retain metabolic genes – viruses have none.
⬇️ Conclusion
The Regressive Hypothesis views viruses not as „escaped gene fragments”, but as miniature versions of former cells. This idea is especially plausible for large DNA viruses: they may once have been fully functional cellular parasites that lost their independence through adaptation. In a way, they represent the biological opposite of evolution toward complexity – a regression to the core of survival: replication. However, the hypothesis falls short when explaining the diversity of simpler viruses.

6.2.2. Hypotheses in the Pre-Cellular RNA World
In the primordial soup of early Earth, something like life first began to stir – delicate, fleeting, yet profoundly significant. Molecules emerged that not only existed but acted: cutting, joining, and copying themselves. Inside tiny lipid bubbles that formed and disappeared, a network of cooperating and competing ribozymes grew (more on this in „The First Whisper of Life”).
It was not life in the modern sense. It was a game of possibilities. From this network, two evolutionary lines emerged:
① The stable, structure-loving replicators that became the first cells.
② And the free, reduced replicators that could be considered proto-viruses – mobile, adaptable, parasitic.Early parasitism was less infection and more interaction – a dance between mutual benefit and exploitation rather than a true host invasion. Two hypotheses describe this origin:
a) Virus-First Hypothesis – Viruses Came First
b) Co-Evolution Hypothesis – Viruses and Cells Emerged Togethera) Virus-First Hypothesis – Viruses Came First
Which came first – the host or the virus?
The Virus-First Hypothesis gives a surprising answer: „Neither!” It postulates that virus-like replicators already existed in the RNA world – long before cells, DNA, or LUCA – as precursors of life.
The RNA World as an Incubator for Proto-Viruses
In this era, ribozymes (RNA molecules with enzymatic functions) were the first „survival artists”. Some became molecular parasites:
- They used other RNA strands as templates for copying („parasitism light”).
- They enclosed themselves in lipid vesicles or peptide rings (not yet true capsids).
- They replicated without cells – e.g., on clay minerals that acted as catalysts.
The leap to the modern virus: It was only with the emergence of cells that these replicators became efficient parasites – now able to hijack cellular machinery.

Fig. 22-C: From the First Whisper to the First Attack – The Emergence of Viral Strategies This graphic illustrates a possible evolutionary transition from prebiotic chemistry to the emergence of the first virus-like structures:
RNA World (left): Simple RNA molecules exist, some protected within lipid vesicles, with the ability to self-replicate.
Proto-Viruses (center): In this phase, the first parasitic RNA molecules (red) emerge. They can no longer replicate on their own but exploit other RNA strands (blue) instead. Encased in lipids and early peptides (short chains of amino acids), a primitive form of molecular parasitism begins. The red structures symbolize parasitic interactions between RNA molecules.
Cellular World (right): With the emergence of the first proto-cells, these strategies shift: RNA parasites (proto-viruses) leave their peptide vesicles and begin to infect cellular organisms – a primordial precursor of modern viruses.✅ Arguments in Favor of This Hypothesis
➤ Unique viral enzymes: RNA polymerases found in viruses (such as in the influenza virus) show no resemblance to cellular proteins. This suggests a very ancient origin – possibly dating back to the RNA world, before the domains of life diverged.
➤ Unique capsid proteins: The protective shells viruses use to encase their genomes have no direct counterparts in cellular organisms – suggesting an early, independent evolutionary origin.
➤ Global distribution: Viruses infect all three domains of life – Bacteria, Archaea, and Eukarya. This suggests they originated before these domains diverged.
➤ Enormous diversity: The wide variety of viral genomes – RNA or DNA, single- or double-stranded – points to a long evolutionary history, possibly reaching back to the RNA world.
➤ Parallels to the RNA world: Many modern viruses carry RNA genomes. This aligns with the assumption that RNA was the original genetic material – and that viruses could be living relics of that time.
❌ Weaknesses of the Hypothesis
➤ Host dependency: Modern viruses rely on cellular machinery (e.g., ribosomes). How could they have replicated in the primordial soup without cells? Perhaps they used loose networks of self-replicating RNA molecules as „hosts”?
➤ Origin of capsids: The evolution of complex capsid structures remains unclear. Genuine capsid proteins require ribosomes – which did not yet exist. Proto-envelopes made of self-organizing peptides (e.g., short amino acid chains) or lipids might have stabilized RNA. The first capsid precursors could have developed in response to external stress or to better protect RNA – not as a parasitic mechanism, but as a survival advantage. However, the details remain speculative.
➤ No direct evidence: Viruses leave no fossils. Molecular traces from the RNA world have also not yet been detected – posing a general challenge in researching early life forms.
⬇️ Conclusion
The virus-first hypothesis is one of the most fascinating – and controversial – theories about the origin of life. It suggests that viruses were not latecomers but pioneers of evolution: molecular messengers that spread genetic information long before cells existed. They may even have been a catalyst for the emergence of more complex life. What began as molecular scramble eventually became the blueprint for modern viruses.
b) Co-Evolution Hypothesis – Viruses and Cells Emerged Together
Not cells first. Not viruses first.
But both together – in the dance of early evolution.The co-evolution hypothesis assumes that viruses and cellular precursors developed in parallel – emerging from the same molecular primordial soup of the RNA world. It wasn’t an „either-or” scenario, but a dynamic interplay: replicators that challenged, exploited, and stabilized each other – thus laying the foundation for life.
The primordial soup as a testing ground
In early lipid vesicles – small, imperfect bubbles made of fats – RNA strands gathered. Some of these molecules replicated themselves, others helped with copying, and still others cut or joined sequences. Cooperation and competition began simultaneously.
Some replicators evolved into increasingly complex systems, from which the first proto-cells emerged. Others remained reduced, preferring to exploit existing structures rather than building their own – a kind of minimalist lifestyle reminiscent of early viruses.
An interplay emerges
In this scenario, parasitism wasn’t a later development but an original feature of molecular evolution.
- Viral precursors could influence proto-cells, for example through gene exchange or interference.
- Proto-cells, in turn, could stabilize or integrate virus-like replicators – for instance, as mobile genes or regulatory elements.
Thus, viruses and cells may have emerged from the same networks where everything was still fluid: independence, dependence, replication, competition.
No origin – but a relationship
The co-evolution hypothesis is less about explaining the „first virus” and more about viewing a relationship as old as life itself. According to this view, viruses were not subsequent troublemakers, but part of the system from the very beginning – co-players in the story of life.

Fig. 22-D: Co-evolution of viruses and cells from a common origin. The graphic illustrates the core idea of the co-evolution hypothesis: Viruses and cellular life forms originated from a shared molecular prehistory – the RNA world. Within lipid vesicles, networks of ribozymes coexisted and competed, giving rise to two distinct replicator strategies: some evolved into the precursors of stable proto-cells, while others reduced themselves to essentials and exploited external replication mechanisms – the early proto-viruses.
Even before the emergence of true cells, a kind of „molecular parasitism” began to take shape – not in the classical sense, but as an undirected exploitation of replication processes. With the appearance of cellular life forms, these interactions intensified: early viruses were now able to transfer genetic material between cells (horizontal gene transfer) or enter into complex – sometimes even cooperative – relationships with them. The rise of LUCA was therefore not the end of this co-evolution, but its first major climax.
✅ Arguments in Favor of This Hypothesis
➤ Avoiding the chicken-and-egg problem: This hypothesis elegantly sidesteps the question of whether viruses or cells came first: both developed in parallel from the same molecular precursors.
➤ Adaptation to the dynamics of the RNA world: The RNA world was not a linear process, but a network of cooperation and competition. This hypothesis reflects that diversity more accurately than a model with a single, clear origin path.
➤ Explanation of viral diversity: Different virus groups (RNA, DNA, retroviral elements) could have evolved independently, but within the same molecular environment – which plausibly accounts for their tremendous diversity.
➤ Evolutionary interaction: Co-evolution explains why cell-like and virus-like systems interacted with each other from an early stage (e.g., through horizontal gene transfer, RNA competition, and mutual adaptation).
➤ Viruses as drivers of cellular complexity: Viruses might not have been just „parasites” but also catalysts of cellular evolution, for example through gene transfer, regulation, and the development of immune and defense mechanisms.
❌ Weaknesses of the Hypothesis
➤ Conceptual vagueness: At what point is a replicator considered a „virus” and when a „cell”? The transitions are fluid, which makes the hypothesis insightful but also difficult to clearly define or test.
➤ Lack of fossils: As with the virus-first hypothesis, there are no direct traces of early viral replicators. Molecular fossils from the RNA world simply do not exist.
➤ Complexity problem: Even in this hypothesis, it remains unclear where certain virus-specific proteins – such as capsid or polymerase proteins – originally came from. The formation of genuine capsids, efficient replication, and host exploitation requires complex proteins whose origins are still unknown – especially considering that ribosomes did not yet exist.
➤ Experimentally difficult to verify: The hypothesis is theoretically well-founded but hardly directly testable. Simulations and conclusions often remain speculative.
⬇️ Conclusion
The co-evolution hypothesis offers a flexible, systemic perspective on the early history of life – without committing to a linear cause-and-effect chain. It fits well with the chaotic and cooperative conditions of the RNA world. According to this model, viruses are not simply „descendants of cells” or „primordial ancestors”, but rather evolutionary contemporaries – equally ancient, equally significant, yet traveling a very different path.
Viruses are the hieroglyphs of biology
…we decipher them, but their origin remains a mystery.
Like flickering shadows on a cave wall:
- sometimes renegade parts of cells,
- sometimes runaway genes,
- sometimes the ancestors of life itself.
Maybe the question of their origin isn’t about the single source, but many evolutionary pathways – paths that intersect, overlap, and feedback into each other. The classic hypotheses (Virus-first, Co-Evolution, Progressive, Regressive) don’t have to contradict one another – they might have played out at different points in history.
And perhaps the question of origin isn’t the essential one at all. Viruses compel us to grasp something deeper:
Life is not a state, but a process – and viruses are its restless pulse.
They remind us that evolution is not a straight-lined tree, but a swirling river of cooperation, theft, and reinvention. Where did they come from? We don’t know. That they will remain? Certainly.
Perhaps that’s exactly why they fascinate us – because they show that life never stands still.
Further sources
The origins of viruses: evolutionary dynamics of the escape hypothesis
Viruses take center stage in cellular evolution
The origin of viruses and their possible roles in major evolutionary transitions
A virocentric perspective on the evolution of life
How did Viruses Evolve and How are They Related to Cellular Life?
Viruses and cells intertwined since the dawn of evolution
The origin of viruses and their possible roles in major evolutionary transitions
The ancient Virus World and evolution of cells
Virus-First Hypothesis • Viruses older than life?
What viruses tell us about evolution and immunity: beyond Darwin?
Were viruses around on Earth before living cells emerged? A microbiologist explains

7. Why Do Viruses Exist?
The short answer: Because there always has to be something that disrupts.
Sounds funny – but it’s a law of nature:
Nothing stays alive if it’s never challenged.You can say many things about viruses.
That they aren’t alive.
That they only disrupt and destroy.
That they’re mere molecular parasites – dependent on the life they infect.
And yet: Without them, much would not be as it is.
Perhaps we wouldn’t even exist.Life needs repetition. And deviation.
The story of life didn’t begin with a goal.
It began with repetition.
Again and again, molecules joined, broke apart, and rejoined.
Not because they had to – but because they could.What repeats eventually becomes probable.
What works, stays.
What stays, must change.
And change needs deviation.
Mutation. Error. Disruption.
Without them, there would be no diversity, no evolution – no story.Order is inertia. Life is movement.
When the first stable systems emerged, it was a triumph –
but also a risk.
Because what is stable tends toward rigidity.
What functions too perfectly dares nothing new.But life needs movement.
This is where viruses begin to play their role –
not as enemies of life, but as a counterforce.
They challenge.
They disrupt.
They push cells into defense – and into innovation.The duality: Order vs. Chaos
Cells build walls, viruses leap over them.
Their relationship is a paradoxical symbiosis:
They kill – and make life possible,
keep ecosystems running,
and even gave us placenta genes.Viruses are not mistakes – they are a principle.
They are boundary violators, not out of malice, but out of principle.
They keep life permeable. Open. Vigilant.
They transfer genes, open up new paths, shake up systems.Not always for the better.
But always with impact.Maybe viruses are exactly what life needs to stay alive.
Not as an opposing model – but as a co-player, as the shadow cast by life itself.
The Tree of Life – permeated by viruses
When we look today at the Tree of Life, at its branches, twigs, and forks – we see the cells, the species, the visible trace of evolution.
And the viruses?
Missing.
Not because they are unimportant – but because they cannot be pinned down.They are neither branch nor leaf, but the wind that moves them.
They are the whisper between the branches.
No distinct lineage, no part of the wood – and yet everywhere.A stream of information that not only surrounds the tree but also shapes it.
They are the points between the lines, creating connections.
Waves of disruption that force growth.
Deviations that do not destroy the pattern but expand it.They are the dark matter of biology: invisible, yet permeating everything.
Fig. 23: The tree of life surrounded by viruses. The „Tree of Life” illustrates the lineage of cellular organisms – Bacteria, Archaea, and Eukaryotes – tracing back to their last universal common ancestor (LUCA). The tiny dots and lines represent viruses: not as distinct branches, but as a diffuse network permeating the entire tree.
In this view, viruses are not outsiders but creative variants of molecular evolution. They are not entities with a clear lineage, but recurring phenomena in a universe that plays with variation and repetition.
And that is why they exist.
Not because they want to. Not because they have to.
But because they keep emerging – wherever nature varies.
Where life organizes itself – and from that order dares something new.They exist because they are unavoidable.
Like an echo of the principle underlying everything:
Disruption is not the opposite of life.
Disruption is its possibility.Viruses exist because disruption is not an accident –
but the universe’s tool to keep life awake and alive.Remember this the next time a virus annoys you:
Maybe it’s just the universe giving you a cosmic slap –
and mumbling:
„Come on, wake up! Here’s your daily dose of chaos – so you can keep evolving.”Epilogue: The Inconspicuous Ones
Viruses are the sand in the gears of creation.
Without will, without body –
and yet the invisible hand
that writes evolution.The whisper between the lines of life –
not out of intent, but out of necessity.They emerged from the mist of beginnings
and lingered in the shadows –
not as foreign bodies, but as an opponent,
as a touchstone and as an impulse.They teach us humility:
For what destroys,
can also create.Maybe that’s their message:
That life doesn’t grow against chaos,
but dances with it.
Addendum:
I approached this topic as a complete layperson. And that turned out to be an advantage – because I knew what I didn’t know.
With curiosity, skepticism, and a sense of wonder, I delved into the world of viruses – and was astonished by the unexpected dimensions that unfolded. This subject carries a weight of insight I never would have anticipated.
During my process of searching, questioning, and formulating, I had tremendous support from AI systems like ChatGPT and DeepSeek. The dialogue with these tools not only accelerated my learning but also deepened it. Without their assistance, I would have missed many things – and certainly wouldn’t have been able to articulate them so clearly.
What has emerged here is the result of human curiosity and machine patience. And it shows that learning today can be more than ever a collaborative process – one that crosses boundaries, even those between human and machine.
Sources (as of 07.07.2025)
Baltimore classification, bioinformatic analysis, capsid, cell culture, co-evolution hypothesis, cryo-electron microscopy, cryo-electron tomography, crystallization, DNA, Electron microscopy, Evolution, host cell, Illumina sequencing, infection, influenza, influenza virus, Koch’s postulates, microbiology, mRNA, mutant cloud, Mutation, Oxford Nanopore technology, Pandemic, PCR, Polymerase, progressive hypothesis, regressive hypothesis, RNA, Sanger sequencing, sequencing, Virion, virology, Virus, virus-first hypothesis, viruses -
Die geheime Welt der Viren

Sie sind winzig, unsichtbar und überall: Viren. Seit Urzeiten schleichen sie durch die Biosphäre, manipulieren Gene, lenken globale Kreisläufe und spielen Roulette mit unserem Immunsystem. Dabei sind sie weder wirklich lebendig noch ganz tot – sie existieren im Dazwischen: als heimliche Regisseure des Lebens. Mal unsichtbare Wächter, mal heimtückische Eindringlinge. Und meist: vollkommen unbemerkt.
Wir nehmen sie oft erst wahr, wenn sie uns flachlegen – mit Husten, Fieber oder ganzen Pandemien. Dann werden sie plötzlich zu Feinden, zu Angstmachern, zu Schlagzeilen. Doch hinter diesen mikroskopischen Strukturen steckt weit mehr als nur Krankheit: eine faszinierende, hochorganisierte Miniwelt, die Biologie auf Speed betreibt.
Diese Abhandlung lädt dazu ein, Viren aus einem anderen Blickwinkel zu sehen: nicht nur als Erreger, sondern als Akteure im Gefüge des Lebens. Was macht sie so erfolgreich? Wie vermehren sie sich mit nichts als ein paar Genen? Wie beeinflussen sie Ökosysteme? Und vor allem: Gibt es Viren wirklich?
Mit wissenschaftlichem Fundament, verständlicher Sprache und einer Prise Augenzwinkern werfen wir einen Blick hinter die Kulissen der unsichtbaren Mikro-Welt – und auf die Methoden, mit denen die Forschung Viren sichtbar macht.
Vorhang auf für das Unsichtbare…
📑Inhaltsverzeichnis
1. Ein Blick in die Welt des Unsichtbaren
1.1. Wächter der Natur: Viren als Gleichgewichtsregulatoren
1.2. Viren als Motor der Evolution
1.3. Gibt es Viren wirklich?
2. Viren und ihre Mechanismen: Einblick am Beispiel des Influenzavirus
2.1. Wie das Influenzavirus auf Reisen geht
2.2. Die Architektur des Influenzavirus
2.3. Der Infektionsprozess des Influenzavirus
2.4. Die Anpassungsfähigkeit des Influenzavirus
2.5. Ein- und Austrittswege des Influenzavirus
2.6. In den meisten Fällen lokal begrenzte mukosale Infektion
2.7. Virenstrategie: Effiziente Vermehrung ohne rasche Zellzerstörung
2.8. Zerstörung der Wirtszelle
2.9. Selbstbegrenzung gefährlicher Viren: Warum sie selten Pandemien auslösen
2.10. Warum macht das Virus manche Menschen krank und andere nicht?
3. Ein Blick auf die Anfänge der Mikrobiologie – wie alles begann
3.1. Frühe Entdeckungen: Die ersten Blicke ins Unsichtbare
3.2. Die Geburt der modernen Mikrobiologie
3.3. Der Schritt in die Welt der Viren
3.4. Viren und die Koch’schen Postulate
4. Moderne Methoden zur Entdeckung und Analyse von Viren
4.1. Probenentnahme
4.2. Probenaufbereitung
4.3. Zellkultur
4.4. Viren sichtbar machen
4.4. a) Elektronenmikroskopie
4.4. b) Kristallisation
4.4. c) Kryo-Elektronenmikroskopie
4.4. d) Kryo-Elektronentomographie
4.4. e) Zusammenfassung
4.5. Der genetische Fingerabdruck der Viren
4.5.1. Nukleinsäure-Extraktion
4.5.2. Nukleinsäure-Amplifikation
4.5.3. Sequenzierung
4.5.3. a) First Generation: Sanger-Sequenzierung
4.5.3. b) Second Generation: Next-Generation Sequencing (NGS)
4.5.3. c) Third Generation: Echtzeit-Sequenzierung
4.5.3. d) Emerging Technologies: Zukunft der Sequenzierung
4.6. Bioinformatische Analyse
5. Gibt es Viren wirklich?
6. Woher kommen Viren?
6.1. Der Baum des Lebens
6.2. Die Haupthypothesen zur Herkunft von Viren
6.2.1. Hypothesen in einer zellulären Welt
6.2.2. Hypothesen in der prä-zellulären RNA-Welt
7. Warum gibt es Viren?
Epilog: Die Unscheinbaren1. Ein Blick in die Welt des Unsichtbaren
Unsere sichtbare Welt ist nur die Hälfte der Geschichte – um uns herum, auf uns und in uns existiert ein unsichtbares Universum voller mikroskopischer Akteure. Unter ihnen sind Viren die rätselhaftesten Bewohner: Sie besitzen keinen eigenen Stoffwechsel, keinen Zellkern – und doch können sie das Schicksal ganzer Ökosysteme beeinflussen.
So winzig, dass selbst die besten Lichtmikroskope kapitulieren, offenbaren Viren erst unter dem Elektronenmikroskop ihre verblüffende Formenvielfalt: Da tauchen Gebilde auf, die aussehen wie außerirdische Raumsonden – kugelige Formen mit stacheligen Fortsätzen, schraubenartige Spiralen oder perfekte geometrische Körper.
Hinter diesen originellen Strukturen steckt pure Funktionalität – ganz ohne unnötigen Schnickschnack: ein Päckchen genetische Information (DNA oder RNA), sicher verpackt in eine robuste Proteinhülle. Manche Modelle gönnen sich noch eine schützende Membranhülle – dreist dem letzten Opfer entrissen.
Schlicht, aber wirkungsvoll: das Erfolgsrezept der Viren.

Abb. 1: Virusarten Zommen wir zunächst in diese Mikrowelt, um ein Gefühl für die Größenverhältnisse zu bekommen.
Die Mission eines Virus: Infizieren, vermehren, überleben
Jedes Virus hat eine klare Aufgabe: Es muss einen geeigneten Wirt finden, um sich zu vermehren und als Art zu überleben. Diese Wirte können Menschen, Tiere, Pflanzen oder Bakterien sein. Es gibt sogar Viren, die andere Viren infizieren. Sobald ein Virus eine geeignete Wirtszelle findet, schleust es sein genetisches Material in diese Zelle ein und übernimmt die molekularen Maschinerien der Zelle, um Kopien von sich selbst herzustellen. So kann sich ein Virus rasch von Zelle zu Zelle ausbreiten und dabei Milliarden von Kopien erzeugen. Auf diese Weise existieren Viren seit Milliarden von Jahren und sind allgegenwärtig.
Der Lebenszyklus eines Virus: Ruhephase vs. Angriffsmodus
Ein Virus existiert in zwei radikal unterschiedlichen Zuständen – fast wie ein Doppelagent:
— Die Extrazelluläre Phase: Das Virion —
Dasein als „Nanospore“: Ein inaktives, aber infektiöses Partikel.
Aufgabe: Überleben außerhalb von Wirtszellen – auf Türklinken, in Tröpfchen, im Boden.
Besonderheit: Kein Stoffwechsel, keine Vermehrung – nur Warten auf den richtigen Wirt.
Wie ein Samenkorn im Wind: inert, aber voller potenziellen Lebens.— Die Intrazelluläre Phase: Der aktive Virus —
Brutale Effizienz: Ein einziges Virion kann über 10.000 neue Viren erzeugen.
Mission Start: Sobald eine geeignete Wirtszelle infiziert wird.
Strategie: Kapere die Zellmaschinerie, produziere Nachkommen – bis zum Zerbrechen.Virion vs. Virus – Warum die Unterscheidung wichtig ist
In der Wissenschaft zählt jedes Detail – selbst ob ein Virus gerade „schläft“ oder die Zelle unterwandert.
- Virion: Infektiöses Partikel außerhalb von Zellen – das reisende Partikel.
- Virus: der Oberbegriff – umfasst beide Phasen, inaktiv wie aktiv.
Das Virion ist also nicht das Virus selbst, sondern seine reisefähige Verpackung. Erst wenn es in eine Zelle eindringt und dort aktiv wird, spricht man vom Virus.
Diese begriffliche Unterscheidung wurde erstmals 1983 vom Virologen Bandea vorgeschlagen. Auch wenn sie sich nicht in allen Disziplinen durchgesetzt hat, schafft sie Klarheit – und macht deutlich: Ein Virus ist mehr als nur ein „Partikel“ – es ist ein Prozess.
1.1. Wächter der Natur: Viren als Gleichgewichtsregulatoren
Das Wort „Virus“ lässt bei den meisten sofort die Alarmglocken schrillen: Influenza, Corona, HIV, Ebola – Krankheit, Gefahr, Pandemie. Doch dieses Bild ist nur ein winziger Ausschnitt der Wirklichkeit. Von den unzähligen Virenarten, die unseren Planeten bevölkern, sind gerade einmal 21 Typen für den Menschen gefährlich. Der Rest? Unsichtbare Helfer im Hintergrund – Hüter des ökologischen Gleichgewichts.
Also keine Panik: Die meisten Viren interessieren sich kein bisschen für uns. Sie befallen Mikroorganismen – Bakterien, Archaeen, Einzeller – die verborgenen Baumeister des Lebens. Und genau dort entfalten sie ihre wahre Kraft: Sie steuern die Populationen dieser Mikroben, lenken Stoffkreisläufe, beeinflussen das Klima, verteilen Gene wie Informationsboten und halten so die Balance im System.
Kaum ein Ort auf der Erde, an dem sie nicht zu finden sind. Sie surfen auf Meeresströmungen, verstecken sich in Regentropfen, reisen als blinde Passagiere auf Pollenkörnern und haften geduldig an Staubpartikeln, die zwischen Kontinenten wandern. Ihr Reich ist riesig – und bleibt dennoch verborgen im Schatten des Sichtbaren.
Zahlen, die den Verstand sprengen
Mit geschätzten 100 Millionen Arten zählen Viren zu den häufigsten biologischen Entitäten der Erde. Ihre Gesamtzahl wird auf etwa 10³¹ Partikel geschätzt – eine Eins mit 31 Nullen – das ist mehr als alle Sterne im Universum, mehr als alle Zellen aller Lebewesen zusammen. In einem einzigen Milliliter Meerwasser tummeln sich etwa 10 Millionen Viruspartikel. Die Erde: ein echter Planet der Viren – wie es der Forscher Aleksandar Janjic formulierte.
Dabei sind sie ultraleicht: Ein einzelnes Viruspartikel wiegt gerade mal ein Femtogramm (10⁻¹⁵ Gramm) – das ist ein Millionstel Milliardstel Gramm – leichter als ein Photon Sonnenlicht. Rechnet man ihre schwindelerregende Anzahl (10³¹) zusammen, erreichen sie vielleicht das Gewicht eines ausgewachsenen Blauwals. Und dennoch: Ohne sie kein Gleichgewicht, kein Kreislauf – kein Leben, wie wir es kennen.
Doch was macht sie zu einem integralen Bestandteil des Ökosystems?
In den Ozeanen – den größten Lebensräumen unseres Planeten – durchsetzen Viren täglich Milliarden von Mikroorganismen. Was nach Vernichtung klingt, ist in Wirklichkeit Teil eines fein austarierten Systems: Indem sie gezielt Mikroben befallen und zerstören, verhindern sie, dass einzelne Arten überhandnehmen. Eine unsichtbare Form von Populationskontrolle – subtil, aber wirksam wie das Raubtier in der Savanne.
Und sie tun noch mehr: Wenn ihre Wirtszellen zerplatzen, setzen sie wertvolle Nährstoffe frei – Kohlenstoff, Stickstoff, Phosphor. Diese stehen sofort anderen Lebewesen zur Verfügung, halten Nahrungsketten am Laufen, nähren das Plankton, das wiederum den Sauerstoff für unsere Atmosphäre produziert.
Gleichzeitig wirken Viren als Evolutions-Booster. Sie übertragen Gene von Organismus zu Organismus – ein natürlicher Gentransfer, der neue Eigenschaften ermöglicht, Vielfalt fördert und Innovationen hervorbringt, lange bevor wir selbst von Gentechnik wussten.
Und so zeigt sich: Viren sind keine bloßen Krankheitsbringer. Sie sind feinverästelte Zahnräder im Getriebe der Natur – unsichtbar, selten bemerkt, aber unersetzlich.
Ein Blick in verschiedene Lebensräume offenbart ihre Wirkung.
🌊 In den Weltmeeren
Populationskontrolle: Tief unter der Wasseroberfläche tobt ein Mikrokampf planetaren Ausmaßes: Bakteriophagen – Viren, die gezielt Bakterien infizieren – eliminieren täglich bis zu 40 % der Meeresbakterien. Damit verhindern sie explosive Algenblüten, die ganze Ozeane in sauerstoffarme Todeszonen verwandeln könnten.
Ohne diese „Mikrobenjäger“ wäre unser Planet längst unter einem Leichentuch aus Algen versunken.
📖 Weitere Quellen:
Viral control of biomass and diversity of bacterioplankton in the deep sea
A sea of zombies! Viruses control the most abundant bacteria in the Ocean.
The smallest in the deepest: the enigmatic role of viruses in the deep biosphereGen-Schmuggel: Im blauen Dunkel der Meere vollbringt Prochlorococcus – ein winziges Cyanobakterium – Großes: Es produziert rund 10 % des globalen Sauerstoffs. Aber selbst dieser Mikroheld steht unter der Kontrolle noch kleinerer Strippenzieher: Cyanophagen – Viren, die perfekt auf ihn spezialisiert sind. Sie schleusen eigene Photosynthesegene ein und zwingen die infizierte Zelle zur Kooperation. Das Ergebnis: Die Bakterie bleibt „arbeitsfähig“, produziert weiter Energie – nun im Dienst ihrer viralen Besetzer. Dieses parasitäre Partnerschaftsmodell veranschaulicht den sogenannten Black-Queen-Effekt: Indem Viren bestimmte Funktionen übernehmen, können Mikroben diese selbst abbauen – und sich auf andere Aufgaben spezialisieren.
Eine unfreiwillige Arbeitsteilung, orchestriert von Viren.
Tiefer eintauchen: Einen eindrucksvollen Einblick in die geheimnisvolle Welt unter dem Meeresspiegel bietet das Video des Schmidt Ocean Institute. Es zeigt, wie Forschende mit modernster Technik den Spuren mikrobiellen Lebens folgen – und dabei auch den Viren auf der Spur sind.
Doch nicht nur im Wasser übernehmen Viren diese regulatorische Rolle – sie sind ebenso aktiv in anderen Ökosystemen.
🟫 In den Böden
Auch unter unseren Füßen herrscht virales Treiben: Viren halten dominante Bodenbakterien in Schach und sorgen so dafür, dass kein Mikroorganismus die Oberhand gewinnt. Diese unsichtbare Kontrolle schützt die fragile Balance des Nährstoffkreislaufs – Grundlage für alles Wachstum.
Wie unsichtbare Gärtner durchkämmen sie das Mikroleben der Erde, jäten Überfluss und schaffen Raum für Vielfalt.
🌳 In der Pflanzenwelt
Bäume und Felder haben geheime Verbündete: Pflanzenviren. Rund um jedes Wurzelgeflecht, entfaltet sich ein verborgenes Netzwerk aus Kontrolle, Schutz und Gegenspiel. Einige Pflanzenviren attackieren gezielt schädliche Bakterien, die Pflanzen krank machen würden. Andere kurbeln das pflanzeneigene Immunsystem an – und wenn Mikroben sterben, zersetzen Viren deren Überreste zu fruchtbarem Dünger. Manche Pflanzen gehen sogar noch einen Schritt weiter: Sie rekrutieren ganz gezielt schützende Viren, die wie Bodyguards im Wurzelraum patrouillieren.
Ohne diese mikroskopischen Allianzen wären viele Wälder anfälliger für Pilzüberwucherung – und unser Getreide den Angriffen aus dem Boden schutzlos ausgeliefert.
🔗 Viruses as components of forest microbiome
🪱 In der Darmflora von Mensch und Tier
Auch in unseren Eingeweiden tobt ein stiller Machtkampf – und wir profitieren davon. Spezialisierte Darmviren (Bakteriophagen) jagen gezielt schädliche Keime wie E. coli und halten die bakterielle Balance stabil. Sie übertragen Schutzgene zwischen Mikroben – wie geheime Datenpakete, die Immunantworten optimieren. Manche Viren dämpfen sogar überaktive Abwehrreaktionen und verhindern so Entzündungen.
Ohne diese Nano-Sheriffs würden schädliche Bakterien den Darm innerhalb von Tagen überrennen.
🔗 Over 100.000 Viruses Identified in the Gut Microbiome
☁️ In der Atmosphäre
Hoch über unseren Köpfen findet der größte Gentransfer der Erde statt – ein einziger Sturm kann 500 Millionen Virionen pro m² über Kontinente verteilen – die ultimative Bio-Invasionsroute. Dank ihrer extremen Robustheit überleben Viren wo andere scheitern – in UV-getränkten Höhen, eisigen Wolken und trockener Luft. Sie reisen per Staub, Meersalz oder Pflanzentröpfchen über Ozeane und Kontinente hinweg. Viren in Wolken beeinflussen sogar Niederschlagsmuster.
Diese atmosphärische Gen-Börse macht aus lokalen Mutationen globale Evolution – als hätte die Natur ihr eigenes Internet erfunden.
🔗 Deposition rates of viruses and bacteria above the atmospheric boundary layer
⚡️ In Extremlebensräumen
Selbst in heißen Quellen, Salzseen oder unter der Erdkruste existieren Viren, die dortige Mikroorganismen regulieren und deren genetische Vielfalt steigern – eine essenzielle Überlebensstrategie in lebensfeindlichen Umgebungen.
🔗 Viruses in Extreme Environments, Current Overview, and Biotechnological Potential
Das größte Paradox der Biologie: Aus milliardenfacher Zerstörung erwächst globales Gleichgewicht. Das nächste Mal, wenn du einen Virus fürchtest, denk daran: Mit jedem Atemzug trägst du Milliarden dieser Winzlinge – und sie tragen dich. Ein Pakt des Lebens – so alt wie die Evolution selbst.
„Wir leben in einem Gleichgewicht, in einem perfekten Gleichgewicht“, und Viren sind ein Teil davon, sagt Susana Lopez Charretón, Virologin an der Nationalen Autonomen Universität von Mexiko. „Ich glaube, ohne Viren wären wir aufgeschmissen.“ [Warum die Welt Viren zum Funktionieren braucht]
„Wenn alle Viren plötzlich verschwinden würden, wäre die Welt für etwa anderthalb Tage ein wunderbarer Ort, und dann würden wir alle sterben – das ist das Fazit“, sagt Tony Goldberg, Epidemiologe an der University of Wisconsin-Madison. „Alle wichtigen Dinge, die sie in der Welt bewirken, überwiegen bei weitem die schlechten Dinge.“ [Warum die Welt Viren zum Funktionieren braucht]
1.2. Viren als Motor der Evolution
Lange bevor Dinosaurier über die Erde stampften, trieben Viren bereits ihr Unwesen – und formten dabei das Leben, wie wir es heute kennen. Ihr Werkzeug: horizontaler Gentransfer, ein biologischer Copy-Paste-Mechanismus, der evolutionäre Quantensprünge ermöglichte.
Für den Evolutionsbiologen Patrick Forterre vom Institut Pasteur sind Viren die Architekten des Lebens, ohne die die Evolution vielleicht ganz anders gelaufen wäre. (Vgl. dazu Spektrum der Wissenschaft: „Die wahre Natur der Viren“, ScienceDirect: „The origin of viruses and their possible roles in major evolutionary transitions“ oder „The two ages of the RNA world, and the transition to the DNA world: a story of viruses and cells”)
Genetische Sabotage mit Folgen
Viren sind Meister der Manipulation. Wenn sie eine Zelle infizieren, schleusen sie nicht nur ihre eigene Erbinformation ein – manchmal wird ihr Genmaterial ins Genom des Wirts integriert und über Generationen hinweg weitervererbt. Aus vermeintlichen Störern werden so kreative Genarchitekten.
Ein spektakuläres Beispiel: Die Plazenta der Säugetiere verdankt ihre Existenz einem Virus. Ein virales Hüllprotein – ursprünglich dazu gedacht, Immunantworten zu unterdrücken – wurde in den Genpool eingebaut und half mit, die Barriere zwischen Mutter und Embryo zu entwickeln. Ohne dieses „fremde“ Gen: kein Mutterleib, kein Säugetier.
Doch es geht noch weiter: Rund 8 % des menschlichen Erbguts stammen von alten Retroviren, die sich einst in unsere DNA eingeschrieben haben – stille Zeugen uralter Infektionen, die uns heute womöglich mitgestalten. Selbst unser Gehirn könnte virale Spuren tragen – etwa Gene, die für die Entwicklung des Cortex entscheidend sind.
Sind wir nicht alle ein bisschen Virus?
CRISPR, das heute als revolutionäre Genschere gefeiert wird, geht auf ein uraltes Abwehrsystem von Bakterien zurück – ein genetisches Archiv vergangener Virusangriffe, aus dem Bakterien lernen, sich gegen neue Feinde zu wehren.
Manche Wissenschaftler fragen sogar: Könnten Viren an der Entstehung des Lebens selbst beteiligt gewesen sein? Einige Hypothesen vermuten, dass virusähnliche Partikel einst die ersten Moleküle waren, die genetische Information speichern und weitergeben konnten – eine Grundbedingung für Leben.
Die Ironie des Schicksals: Wir fürchten Viren als Todbringer – dabei wären wir ohne sie nicht einmal entstanden.
1.3. Gibt es Viren wirklich?
Trotz ihrer immensen Bedeutung gibt es immer wieder Zweifel an der Existenz von Viren. Wie können wir sicher sein, dass sie tatsächlich real sind? Diese Frage lässt sich nicht mit einer einfachen Beobachtung beantworten – Viren entziehen sich unserem bloßen Auge und offenbaren sich nur durch indirekte Spuren und spezialisierte Nachweismethoden.
Um dieser Frage auf den Grund zu gehen, müssen wir zunächst verstehen, wie Viren agieren: Welche Mechanismen nutzen sie, um sich zu vermehren? Wie interagieren sie mit ihren Wirten? Und vor allem: Welche wissenschaftlichen Methoden gibt es, um sie sichtbar zu machen und nachzuweisen?
Die Suche nach diesen Antworten führt uns in eine faszinierende Welt aus hochentwickelten Technologien und jahrzehntelanger Forschung. In den kommenden Kapiteln werden wir Schritt für Schritt erkunden, wie Wissenschaftler Viren nachweisen – und damit der entscheidenden Frage näherkommen: Gibt es Viren wirklich?
2. Viren und ihre Mechanismen:
Einblick am Beispiel des InfluenzavirusWerfen wir zunächst einen Blick darauf, wie Viren ihre Wirtszellen „kapern“ und für ihre Vermehrung nutzen. Ein Paradebeispiel hierfür ist das Influenzavirus – nicht nur, weil es zu den am besten erforschten Viren gehört, sondern auch, weil es eindrucksvoll zeigt, wie Viren Zellen manipulieren und sich verbreiten. Die Mechanismen, die es anwendet, eröffnen uns einen idealen Einblick in die geheimnisvolle Welt der Viren und ihre vielschichtigen Wechselwirkungen mit ihren Wirten.
2.1. Wie das Influenzavirus auf Reisen geht
2.2. Die Architektur des Influenzavirus
2.3. Der Infektionsprozess des Influenzavirus
2.4. Die Anpassungsfähigkeit des Influenzavirus
2.5. Ein- und Austrittswege des Influenzavirus
2.6. In den meisten Fällen lokal begrenzte mukosale Infektion
2.7. Virenstrategie: Effiziente Vermehrung ohne rasche Zellzerstörung
2.8. Zerstörung der Wirtszelle
2.9. Selbstbegrenzung gefährlicher Viren: Warum sie selten Pandemien auslösen
2.10. Warum macht das Virus manche Menschen krank und andere nicht?💡Hinweis: Die folgenden Kapitel bauen auf grundlegenden Kenntnissen über Zellen, den Unterschied zwischen DNA und RNA, Proteine sowie zelluläre Prozesse wie die Proteinbiosynthese auf. Falls diese Themen noch neu für dich sind, könnte ein Blick in Kapitel 2, 3 und 4 der Abhandlung „Die Wunderwelt des Lebens“ oder ähnliche Einführungstexte hilfreich sein.
Kapitel 2: Die Zelle – der Urbaustein
Kapitel 3: Proteine – die Bausteine des Lebens
Kapitel 4: Vom Code zum Protein – zelluläre Mechanismen
2.1. Wie das Influenzavirus auf Reisen geht
Das Influenzavirus ist ständig auf Achse – ein unsichtbarer Jetsetter mit erstaunlichen Ansteckungsrouten: Mal reist es first class per Nieswolke, mal trampt es über Türgriffe.
Tröpfchenflug – Erste Klasse durch die Luft
Ein Nieser genügt: Bis zu 40.000 virusbeladene Tröpfchen schießen durch die Luft – wie ein Mini-Raketenangriff auf die Umgebung (Reichweite: bis zu 2 Meter!).Schmierattacke – Der heimliche Handshake
Türklinke, Fahrstuhlknopf, Tastatur – das Virus chillt auf Oberflächen teils stundenlang. Ein Griff, ein Wisch durchs Gesicht – und schon hat es sich per Hand-zu-Gesicht-Trick Zugang verschafft.Mission Atemwege: Virale Invasion
Angekommen im Atemtrakt, startet das Virus seinen Sturm auf die Epithelzellen:
Lieblingsziel: Schleimhautzellen von Nase, Rachen, Bronchien.
Warum? Hier sitzen massenhaft beliebte Rezeptoren – perfekte Andockstellen.
Folge: Innerhalb von Stunden kapert es die Zellfabrik und produziert neue Viren.
Abb. 2-A: Influenza-Virionen gelangen in den Atemtrakt und kommen dabei in Kontakt mit den Epithelzellen (Schleimhautzellen), die den Atemtrakt auskleiden. Von außen wirkt es wie ein Staubkorn mit schlechten Absichten – doch bei genauerer Betrachtung entpuppt sich das Influenzavirus als hochkomplexe Nanomaschine. Um zu verstehen, wie es Zellen kapert und ständig mutiert, lohnt sich ein Blick ins Innere.
2.2. Die Architektur des Influenzavirus
Das Influenzavirus ist der Grund, warum wir mit Fieber im Bett liegen und über „die Grippe“ fluchen. Von den drei Stämmen (A, B, C) ist Typ A der gefährlichste Globetrotter: wandlungsfähig wie ein Schauspieler und unberechenbar wie Aprilwetter. Typ B und C sind dagegen eher die „Bodenständigen“ – weniger variabel und weniger gefährlich. Doch alle teilen denselben genialen Bauplan (siehe untere Abbildung).
Das Viruspartikel – dieser nur 80-120 Nanometer kleine Überlebenskünstler – ähnelt einem winzigen, kugeligen Nano-U-Boot (manchmal ist es auch oval). In seinem Inneren: das virale Erbgut aus RNA – aber mit einem besonderen Trick!
Während wir RNA oder DNA oft als langen, durchgehenden Faden kennen, besitzen Viren unterschiedliche DNA- und RNA-Strukturen. Manche haben ein einziges zusammenhängendes Molekül, andere tragen ihr Erbgut in mehrere RNA-Segmente aufgeteilt.
Die Kommandozentrale: Genom in 8 Teilen
Das Influenzavirus setzt auf die modulare Bauweise: es nutzt 8 separate RNA-Segmente – wie ein Baukasten, dessen Teile sich immer neu kombinieren lassen – perfekt für Überraschungen!
Diese RNA-Segmente sind unterschiedlich lang – vom kompakten Mini-Modul bis zur XXL-Bauanleitung – und doch perfekt aufeinander abgestimmt. Damit sie nicht wie lose Zettel im Wind verloren gehen, werden sie sorgsam eingepackt: Jedes Segment wird von einer Hülle aus Nukleoproteinen (NP) umschlungen – wie wertvolle Schriftrollen in Schutzfolie. Doch die NP-Hülle ist mehr als bloßer Schutz: Sie hilft der viralen Maschinerie, die genetische Information präzise zu lesen, zu kopieren und weiterzugeben.
Der Werkzeuggürtel: Polymerase-Komplexe
Zusätzlich bringt das Virus seine eigenen 3D-Drucker mit – die RNA-Polymerasen (bestehend aus den Untereinheiten: PB1, PB2, PA). Diese sind fest an die RNA-Segmente gebunden: wie Handwerker, die ihr Werkzeug am Gürtel tragen.
Abb. 2-B: Aufbau eines Influenzavirus: schematische Darstellung aller Komponenten Der RNP-Komplex – das Herzstück des Virus
Jedes RNA-Segment + Nukleoproteine (NP) + Polymerase bildet einen Ribonukleoprotein-Komplex (RNP) – eine perfekt organisierte Einheit: das Steuerzentrum des Virus. Alle acht – sauber verpackt und einsatzbereit – wie ein tragbarer Werkzeugkoffer für die Zellübernahme.
Die Lipidhülle – der gestohlene Tarnumhang
Das Virus klaut sich seine äußere Schicht direkt von der Wirtszelle: eine Lipiddoppelschicht – identisch zur Zellmembran und damit die perfekte Tarnung! Direkt darunter liegt das Matrixprotein M1 – der molekulare Gerüstbauer, der alles zusammenhält. Es verbindet die äußere Hülle mit dem inneren Komplex und sorgt dafür, dass das Virus seine Form behält – wie ein Stützrahmen unter der Tarnkappe.
Die Spikes: Schlüssel & Schere
In der Virushülle befinden sich wichtige Oberflächenproteine, die wie kleine Stacheln oder Greifarme aus der Oberfläche herausragen. Diese werden „Spikes“ genannt. Das Influenzavirus besitzt zwei besonders wichtige Spikes, die ihm helfen, Zellen zu infizieren: Hämagglutinin (HA) und Neuraminidase (NA).
🔑 HA – der Türöffner: Dient als Schlüssel, um an der Wirtszelle anzudocken.
→ 18 bekannte Varianten (H1–H18)✂️ NA – der Flucht-Helfer: Löst die Verbindung zur Wirtszelle, damit es weiterziehen kann.
→ 11 Varianten (N1–N11)Virus-Typen: Ein Zahlenspiel
Die Kombination aus HA und NA bestimmt den Stamm:- H1N1 (Schweinegrippe)
- H5N1 (Vogelgrippe)
- H3N2 (saisonale Grippe)
Wie bei Autokennzeichen: HA/NA-Codes verraten, welches Modell da unterwegs ist – nur eben ohne TÜV!
2.3. Der Infektionsprozess des Influenzavirus
a) Anheften des Virus an die Wirtszelle (Adsorption)
b) Eindringen in die Zelle (Endozytose)
c) Freisetzung des viralen Erbguts (Uncoating)
d) Virusreplikation – Die molekulare Fabrik
e) Zusammenbau (Assembly) der neuen Viruspartikel
f) Knospung (Budding) und Freisetzung der neuen Virena) Anheften des Virus an die Wirtszelle (Adsorption)
Die Oberfläche des Atemtrakts ist von einem dichten Epithel aus Schleimhautzellen ausgekleidet. Diese Zellen tragen Sialinsäure-Reste auf ihrer Oberfläche – Zuckermoleküle, die zentral für Zellkommunikation und immunologische Selbsterkennung sind (vgl. Kapitel „SELBST-Marker: Sialinsäuren“ in „Die Wunderwelt des Lebens“).
Das Influenzavirus kapert diesen Mechanismus: Sein Oberflächen-Protein Hämagglutinin (HA) bindet gezielt an die Sialinsäure der Wirtszelle – eine klassische Schlüssel-Schloss-Interaktion, die den Eintritt des Virus einleitet.
Abb. 2-C: Oberfläche einer Epithelzelle Bevor das Influenzavirus eine Zelle infizieren kann, muss es an die Wirtszelle andocken – ein entscheidender Schritt im Infektionsprozess. Doch die Epithelzellen der Atemwege sind nicht wehrlos: Ihre beweglichen Zilien (Flimmerhärchen) transportieren Fremdpartikel wie Staub, Bakterien oder Viren weg, bevor diese die Zelloberfläche erreichen können.
Die Darstellung zeigt die Größenverhältnisse an der Zelloberfläche. Die Zilien sind 5–10 Mikrometer lang, während die Schleimschicht eine Dicke von 10–100 Mikrometern aufweist. Mit nur 80–120 Nanometern ist das Viruspartikel winzig. Es muss schnell eine Zelle erreichen, bevor die Zilien es weiterbefördern.
Zwischen den Zilien gibt es freie Zellbereiche, an denen das Virus direkten Kontakt zur Zelloberfläche herstellen kann. Die Sialinsäure-Reste (1 Nanometer) auf der (Wirts-)Zellmembran dienen als Andockstelle für das HA-Protein (13 Nanometer) des Virus, das groß genug ist, um diese Strukturen zu erreichen. Das Virus kann somit die Schleimschicht durchdringen und an die Wirtszelle binden.b) Eindringen in die Zelle (Endozytose)
Das Influenzavirus tarnt sich meisterhaft: Durch die Bindung seines Hämagglutinins an Sialinsäure-Reste imitiert es ein harmloses Nährstoffmolekül. Die Zelle fällt auf den Trick herein und initiiert ihren standardmäßigen Aufnahmemechanismus: die Endozytose.
Was folgt, ist ein molekulares Schauspiel:
Die Zellmembran stülpt sich um das gebundene Virus herum – ausgelöst durch Signalmoleküle, die eigentlich für den Nährstofftransport zuständig sind. Wie eine sich schließende Falle bildet sich eine Einbuchtung, die das Virus komplett umschließt. Mit einem letzten „Schnapp“ der Membran formt sich ein Endosom – ein Transportvesikel, das den Eindringling nun unschuldig ins Zellinnere schleust.
Was die Zelle als harmlosen Transport verbucht, entpuppt sich als Trojanisches Pferd.
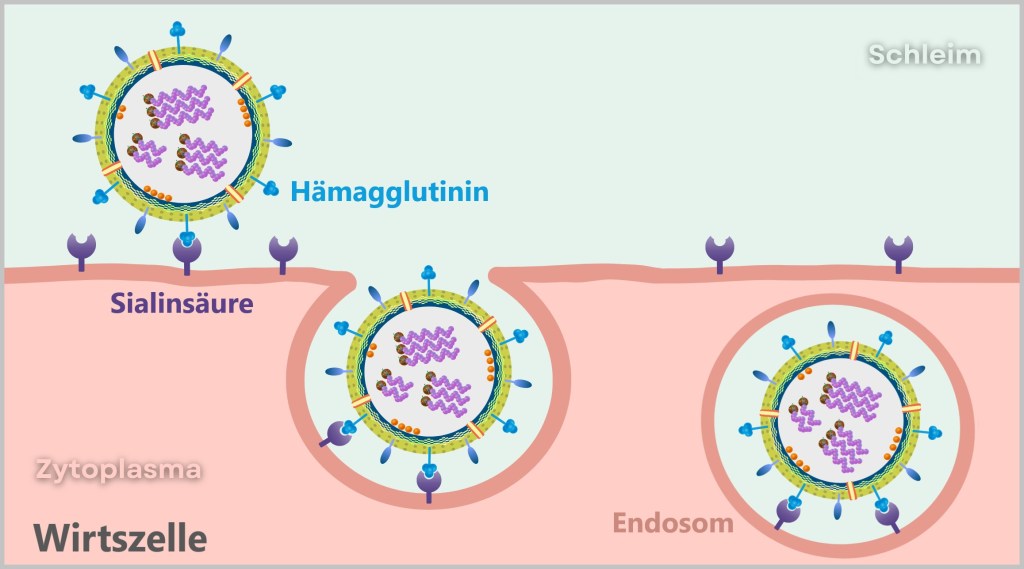
Abb. 2-D: Das Influenzavirus dringt in die Wirtszelle ein. Das Virus ist jetzt in der Zelle – noch eingeschlossen im Endosom – aber bereit, sein Innerstes zu entfalten.
c) Freisetzung des viralen Erbguts (Uncoating)
Das frühe Endosom reift zum späten Endosom heran – einem Ort, bei dem die Zelle normalerweise unerwünschte Eindringlinge abbaut. Protonenpumpen senken dort den pH-Wert, indem sie Protonen (H⁺-Ionen) ins Innere transportieren. Damit schaffen sie eine saure Umgebung, die Verdauungsenzyme aktivieren soll.
Der saure Trick
Doch das Influenzavirus hat einen genialen Gegenplan: Die saure Umgebung löst eine dramatische Umwandlung des viralen Hämagglutinins (HA) aus. Das Protein spaltet sich – die Bindungsdomäne HA1 wird abgespalten und die Fusionsdomäne HA2 freigelegt.

Diese Fusionsdomäne HA2 ist hydrophob – sie scheut Wasser – und rammt sich wie ein Enterhaken in die Endosomenmembran. (siehe untere Abbildung).
Gleichzeitig öffnet das M2-Protein – ein viraler Ionenkanal – als heimlicher Komplize die Schleusen: Protonen strömen ins Innere des Virus und lockern die Verpackung des Erbguts.
Abb. 2-E: Verankerung des Virus in der Endosom-Membran Jetzt zieht HA2 mit unerbittlicher Kraft die Virusmembran und die Endosomenmembran zusammen. Die beiden Lipidmembranen verschmelzen; ein Mechanismus, der als Membranfusion bekannt ist. Dies geschieht, weil die Lipidmoleküle in den Membranen flexibel sind und sich neu anordnen können, um eine kontinuierliche Doppelschicht zu bilden.

Diese Fusion schafft eine Pore – das Tor zur Freiheit für das virale Genom. Mit einem letzten, eleganten Schub gleitet die virale RNA ins Zytoplasma. Die Entkleidung – das Uncoating – ist vollendet.
Die Zelle ahnt nicht, dass sie soeben die Blaupause ihrer eigenen Unterwerfung freigesetzt hat.
Abb. 2-F: Freisetzung des viralen Genoms ins Zytoplasma der Wirtszelle: Durch Membranfusion und Uncoating wird die Virushülle aufgelöst, wodurch das Genom freigesetzt wird und die Infektion beginnt.
Warum wird das Virus nicht zersetzt?
Das Virus wird nicht von den Verdauungsenzymen der Zelle abgebaut, weil der Freisetzungsprozess schnell erfolgt, bevor der Abbaumechanismus (die Aktivierung der Verdauungsenzyme) ins Spiel kommen kann. Das Virus nutzt den Prozess der pH-Senkung und die Veränderungen im Endosom, um sich schnell aus diesem zu befreien, indem es die Membranfusion auslöst und sein Erbgut direkt in das Zytoplasma der Zelle entlässt. Diese „Flucht“ aus dem Endosom ist schneller als der zelluläre Abbauprozess, weshalb das Virus nicht zersetzt wird.d) Virusreplikation – Die molekulare Fabrik
Die virale RNA kommt nicht schutzlos daher – sie reist in High-Tech-Rüstung: Eingehüllt in schützende Nukleoproteine (NP) und ausgestattet mit der viralen Polymerase bildet jedes der acht RNA-Segmente einen hochorganisierten Ribonukleoprotein-Komplex (RNP). Diese molekularen Kommandoeinheiten sind für den Einsatz perfekt gerüstet:
- Die Nukleoproteine wirken wie ein Panzer – sie schirmen die RNA gegen zelluläre Abwehrsysteme ab.
- Die Polymerase ist das Schweizer Taschenmesser des Virus – Werkzeug für Kopieren (Replikation) und Übersetzen (Transkription) in einem.
Sobald die Ribonukleoprotein-Komplexe (RNPs) im Zytoplasma freigesetzt sind, läuft die systematische Übernahme der zellulären Produktionslinien an – die Virusfabrik geht in Betrieb. Das Genom übernimmt dabei zwei zentrale Aufgaben: Zum einen dient es als Bauplan für die Herstellung viraler Proteine (Proteinsynthese), zum anderen wird es selbst vervielfältigt (Genomreplikation) – damit jeder neue Viruspartikel seine eigene Kopie des Erbguts mit auf den Weg bekommt.

Abb. 2-G: Um neue Viren zu bauen, braucht es neue virale Proteine und neue virale Genome. Proteinsynthese: Das virale Genom dient als Vorlage für die Synthese der Proteine, die für den Aufbau neuer Viruspartikel benötigt werden.
Genomreplikation: Gleichzeitig wird die virale RNA vervielfältigt, um die genetische Information für neue Viren bereitzustellen.Während die meisten RNA-Viren im Zytoplasma verbleiben, hat Influenza einen cleveren Trick auf Lager: Es hijackt den Zellkern. Warum? Dort findet es optimale Bedingungen für die Replikation seiner RNA.
Der Weg dorthin ist jedoch alles andere als selbstverständlich. Die RNPs manipulieren das zelluläre Transportsystem: Sie präsentieren gefälschte Importsignale – molekulare Passierscheine, die ihnen die Tür zum Zellkern öffnen. Zelluläre Importine, eigentlich zuständig für den Transport körpereigener Proteine, werden so zu ahnungslosen Schleusern. In einem Akt biologischer Täuschung werden die viralen RNPs direkt ins Kontrollzentrum der Zelle eskortiert.
Abb. 2-H: Die virale RNA wird vom Zytoplasma in den Zellkern transportiert. Im Zellkern entfalten die RNPs schließlich ihre volle Wirkung. Die virale Polymerase beginnt ihr Doppelspiel:
- Kopieren der viralen RNA (Replikation) → Bauplan für neue Viren
- Produktion viraler mRNA (Transkription) → Bauanleitung für Proteine
Da dieser Vorgang besonders raffiniert abläuft, wird im Folgenden jeder Schritt ausführlich beschrieben.
1️⃣ Aktivierung der RNA-Polymerase – Jetzt geht’s los
Die virale Polymerase benötigt einen molekularen Zündfunken, um aktiv zu werden. Und den findet sie im Zellkern: eine biochemische Spezialzone, die sich deutlich vom Zytoplasma unterscheidet. Hohe Konzentrationen von Nukleotiden, Ionen und kernspezifischen Faktoren senden ein klares Signal: „Hier ist der Ort, um loszulegen!“ Erst in dieser Umgebung erwacht die Polymerase zum Leben. Bleibt dieser molekulare Weckruf aus, verharrt sie im Ruhezustand – getarnt als harmloses Zellbestandteilchen.
2️⃣ Ausgangssituation: (-)ssRNA – Genom in Spiegelschrift
Das Erbgut des Influenzavirus ist ein Meister der Tarnung. Statt als lesbare Bauanleitung aufzutreten, erscheint es wie ein Rätsel in Spiegelschrift: acht einzelne RNA-Segmente, negativ gepolt, ohne die typischen Merkmale einer zellulären Nachricht. Kein Absender, kein Briefkopf, kein Poststempel. Für die Zelle ist das keine Nachricht – sondern biologisches Rauschen.
Wissenschaftlich ausgedrückt:
Das virale Genom besteht aus acht segmentierten Einzelsträngen RNA (engl. single-stranded RNA = ssRNA) mit negativer Polarität: (-)ssRNA. „Negativ“ bedeutet: Diese RNA ist die komplementäre Vorlage zur mRNA (also spiegelverkehrt) und somit nicht direkt lesbar.
Außerdem fehlen ihr zwei entscheidende Erkennungsmerkmale: das 5′-Cap (eine Art molekularer Startknopf) und die 3′-Poly-A-Sequenz, die eine normale mRNA schützt und identifiziert.
Warum so kompliziert?
Weil es genial ist.Mit dieser molekularen Maskerade erreicht das Virus zwei Dinge:
Unsichtbar bleiben: Die (-)ssRNA wird vom zellulären Immunsystem nicht sofort als Bedrohung erkannt. Wäre das virale Genom schon als mRNA vorhanden, würden die Alarmsysteme der Zelle anspringen.
Volle Kontrolle über die Produktion: Das Virus bettelt nicht um Hilfe der Wirtsenzyme. Weil nur die viruseigene RNA-Polymerase in der Lage ist, aus der (-)ssRNA lesbare mRNA zu erzeugen, kann das Virus exakt steuern:
➤ Wann mRNA erzeugt wird.
➤ Wie viel davon produziert wird.
➤ Welche Segmente priorisiert werden.Kurz gesagt: Was aussieht wie ein kryptisches Puzzle ist in Wahrheit ein hochpräziser Kontrollmechanismus – ein Bauplan, der sich erst dann offenbart, wenn die virale Maschinerie bereit ist – und das Immunsystem noch schläft.
3️⃣ Aus (-)ssRNA wird (+)ssRNA (die mRNA)
Die virale Polymerase steht bereit, die (-)ssRNA in lesbare mRNA umzuschreiben – doch es fehlt der Startknopf. Ohne den 5′-Cap bleibt die Maschinerie stumm.
Lösung? Diebstahl auf Nano-Ebene.
Dieser raffinierte Trick heißt Cap-Snatching – oder auf Deutsch: „Kappen-Schnappen“.
Der Coup im Detail
Die Polymerase-Untereinheit PB2 streift durch die zellulären mRNAs wie ein gerissener Dieb auf der Suche nach dem wertvollsten Schmuckstück. Ihr Ziel: Die 5′-Kappe, das universelle „Siegel“ für zelluläre Proteinfabriken. Ihre Komplizin, PB1 – die „molekulare Schere“ – trennt die Kappe mitsamt 10–15 Nukleotiden ab – ein perfekter Primer für die virale Transkription. Die gestohlene Kappe wird an die virale RNA geheftet. Die Zelle glaubt, sie habe eine legitime mRNA vor sich – und startet die Produktion viraler Proteine. Die gekappte Wirts-mRNA wird hingegen abgebaut – die zelluläre Proteinproduktion bricht zusammen.
Parallel erhält die virale mRNA am 3′-Ende einen Poly-A-Schwanz, der sie stabilisiert und schützt.
Warum dieser Trick so brillant ist
✅ Energieersparnis: Das Virus nutzt vorhandene Ressourcen – kein Aufwand für eine eigene Kappen-Synthese.
✅ Sabotage: Der Abbau der zellulären mRNAs legt die Wirtsabwehr lahm.
✅ Tarnung: Die gestohlene Kappe tarnt virale mRNA als „harmlose“ zelluläre Botschaft.Die Folgen des Raubzugs
Die Zelle verliert ihre eigenen Baupläne – und produziert nun virale Proteine auf Hochtouren.
Das Virus gewinnt doppelt: Schnelle Vermehrung und Schwächung der Gegnerin.Dieser Prozess ist ein Klassiker der Virologie – ein Paradebeispiel dafür, wie Viren ihre Wirtszellen zu Marionetten machen.
Am Ende entstehen zahlreiche „nackte“ (+)ssRNA-Stränge im Zellkern, die direkt als mRNA für die Translation – also die virale Proteinproduktion und Genomvermehrung – genutzt werden.
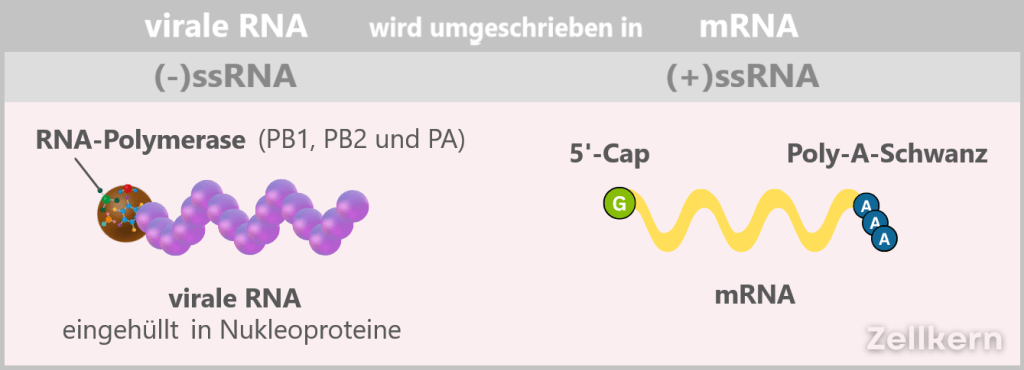
Abb. 2-I: Die virale RNA (-)ssRNA wird in messenger RNA (mRNA) ungeschrieben. 4️⃣ Die Virus-Produktion läuft heiß
Die frisch gekappten viralen mRNAs verlassen den Zellkern – ausgerüstet mit gestohlener Signatur und Poly-A-Schwanz. Im Zytoplasma erwarten sie die Ribosomen, die ahnungslos die Baupläne des Feindes abarbeiten.
Die Beute: Eine ganze Proteinfabrik
Die Ribosomen produzieren virale Proteine am Fließband – darunter:Hämagglutinin (HA): Der Schlüssel zum Zelleintritt – der unentbehrliche Türöffner.
Neuraminidase (NA): Der Befreier neuer Viren – die scharfe molekulare Schere.
Matrixprotein (M1): Die stabile Hülle für das Virusinnere – der Gerüstbauer.
Ionenkanalprotein (M2): Der pH-Wächter – reguliert das Säuremilieu im Virus.
RNA-Polymerase: Die Kopiermaschine – eine virale Druckerpresse.
Nukleoprotein (NP): Die Bodyguards – verpacken und schützen die RNA-Segmente.
Nukleäres Exportprotein (NEP): Der Spediteur – sorgt für den Transport viraler RNPs aus dem Zellkern.Diese frisch hergestellten Proteine sind bereit für den finalen Akt: die Montage neuer Virenpartikel.
5️⃣ Rückkehr in den Zellkern
Nach ihrer Produktion im Zytoplasma machen sich die meisten viralen Proteine zurück auf den Weg zum Zellkern – der Kommandozentrale der Virusreplikation. Ausgenommen sind nur die Oberflächenstars Hämagglutinin (HA), Neuraminidase (NA) und das Ionenkanalprotein M2, die direkt an der Zellmembran ihre Einsätze haben.
Die übrigen Virenakteure kehren ins Hauptquartier zurück, um neue Befehle abzuholen und die End-Mission vorzubereiten.

Abb. 2-J: Die virale mRNA verlässt den Zellkern und wird an zwei verschiedenen Orten in der Wirtszelle übersetzt. 1) An freien Ribosomen im Zytoplasma wird die mRNA in virale Proteine wie RNA-Polymerase, Matrixproteine (M1), Nukleoproteine (NP) und nukleäre Exportproteine (NEP) übersetzt.
2) Diese Proteine wandern anschließend zurück in den Zellkern, um an der Replikation und Verpackung des viralen Genoms mitzuwirken.
3) An den festen Ribosomen des endoplasmatischen Retikulums (ER) werden die Oberflächenproteine (HA und NA) und das Ionenkanalprotein (M2) synthetisiert. Diese Proteine gelangen nach ihrer Herstellung zum Golgi-Apparat, wo sie weiter modifiziert und für den Einbau in die virale Hülle vorbereitet werden.6️⃣ Die Genom-Kopierfabrik: (+)ssRNA → neue (-)ssRNA
Während im Zytoplasma fleißig virale Proteine vom Band laufen, läuft im Zellkern die Geheimoperation „Genom-Vervielfältigung“:
Frisch gebildete RNA-Polymerasen schnappen sich die neu synthetisierten (+)ssRNA-Stränge und übersetzen sie zurück in virale Spiegelschrift – heraus rollen neue (-)ssRNA-Stränge. Die Polymerase bleibt daran gebunden als integrierte Druckerpresse für künftige Einsätze.
Nichts bleibt dem Zufall überlassen: Noch während der genetische Code rückübersetzt wird, umhüllen Nukleoproteine (NP) die entstehende (-)ssRNA – sie hat nicht einmal eine Sekunde, um „nackt“ zu sein – kein Risiko, dass die zelluläre Überwachung zugreift. Die frisch kopierten RNA-Segmente werden sofort verpackt und versiegelt: Zusammen mit der Polymerase bilden sie wieder vollständige Ribonukleoprotein-Komplexe (RNPs) – ein komplett ausgestattetes Genom-Modul, bereit für die nächste Generation Virus.
Sobald die acht Segmente verpackt sind, übernimmt das virale Logistik-Team: NEP (Exportprotein) und M1 (Matrixprotein) markieren die RNPs für den Abtransport. Sie schleusen sie durch die Kernporen – die streng bewachten Grenzübergänge der Zelle – direkt ins Zytoplasma. Mission: Montagehalle.
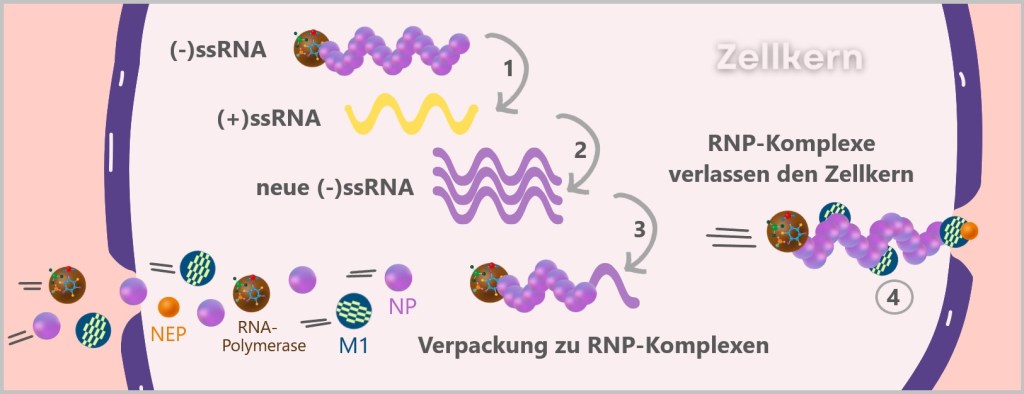
Abb. 2-K: Die Abbildung zeigt, wie das Influenzavirus im Zellkern der Wirtszelle sein Erbgut vervielfältigt: 1) Die virale RNA-Polymerase nutzt die (-)ssRNA als Vorlage und synthetisiert daraus eine komplementäre (+)ssRNA.
2) Diese (+)ssRNA dient nun als Matrize für die erneute Synthese von viraler (-)ssRNA – also der eigentlichen Erbinformation für neue Viruspartikel.
3) Bereits während der Synthese wird die neue (-)ssRNA von Nukleoproteinen (NP) umhüllt und mit Polymerase, M1 und NEP zum sogenannten RNP-Komplex verpackt – stabil und bereit zum Export.
4) Die fertigen RNP-Komplexe verlassen den Zellkern über die Kernporen und wandern ins Zytoplasma – dort beginnt bald der Zusammenbau neuer Viren.Wie eine Schwarzdruckerei im Hinterzimmer: Die Polymerase produziert ununterbrochen Kopien, die NP-Proteine verpacken sie sofort – und Schleuser (NEP/M1) schmuggeln sie unauffällig hinaus.
Während die Zelle ahnungslos ihre Ressourcen verheizt, steht der eigentliche Showdown noch bevor…
e) Zusammenbau (Assembly) der neuen Viruspartikel
Nachdem alle Bauteile produziert sind, beginnt im Zytoplasma die koordinierte Endmontage – ein Prozess so präzise wie die Konstruktion einer Raumsonde: Jedes Teil muss perfekt sitzen, sonst hebt nichts ab.
Die Oberflächenproteine HA & NA reisen über den Golgi-Apparat – die „Verpackungsabteilung“ der Zelle – zur Zellmembran (untere Abb. links). Dort verankern sie sich in der Lipiddoppelschicht, wie Türgriffe und Rettungsscheren, die aus der Hülle eines künftigen Viruspartikels ragen.
Auch die Ribonukleoprotein-Komplexe (RNPs) machen sich auf den Weg, bereits im Schlepptau der Matrixproteine M1, die als Logistikmanager fungieren. Ihre Aufgabe: Die wertvolle Fracht zielsicher zu den HA/NA-bestückten Membranbereichen zu navigieren (untere Abb. rechts).
An der Zellmembran fügt sich das virale Puzzle Stück für Stück zusammen: Die RNPs ordnen sich unter der mit HA/NA gespickten Membran an. Die Matrixproteine helfen dabei, die RNPs mit den Bereichen der Zellmembran in Kontakt zu bringen.
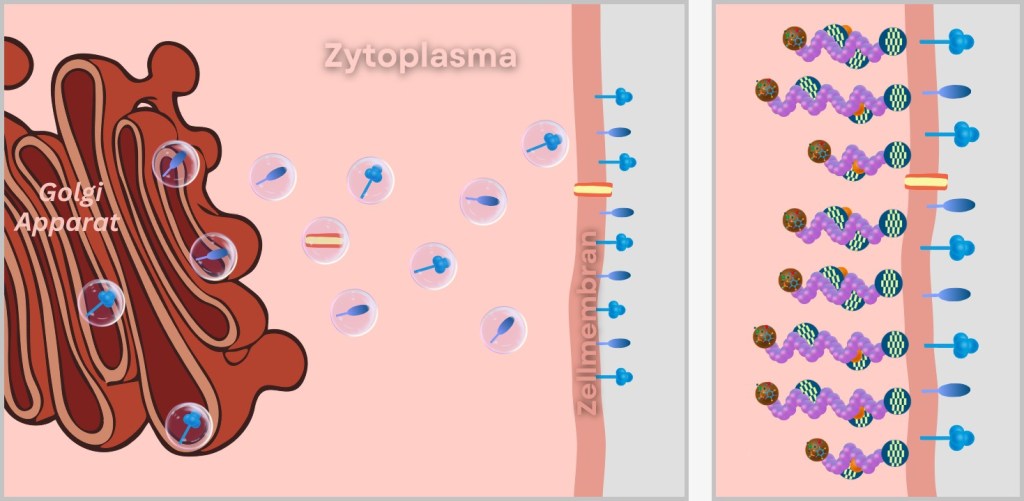
Abb. 2-L: Zusammenbau an der Zellmembran: Virale Bausteine finden ihren Platz Links: Einbau der viralen Oberflächenproteine (HA und NA) und des Ionenkanalproteins (M2) in die Zellmembran.
Rechts: Transport der Ribonukleoprotein-Komplexe (RNPs) zur Zellmembran und Bindung an die entstehende Virushülle.Und jetzt – Trommelwirbel – ist alles bereit für den großen Ausbruch!
f) Knospung (Budding) und Freisetzung der neuen Viren
An der Zellmembran formt sich eine Ausstülpung – wie eine Seifenblase mit tödlicher Fracht. Doch was so spielerisch aussieht, ist präzise Choreografie:
➤ Virusproteine drängen nach außen, die Lipidschicht wölbt sich zum perfekten „Virus-Paket“.
➤ Matrixproteine (M1) spannen die Membran wie ein Trampolin – stabil, aber flexibel genug für den Absprung.
➤ Die Wirtslipide schließen sich zur getarnten Hülle – das Virus verpackt sich selbst.Doch noch ist es nicht frei. An der Zelloberfläche lauern Sialinsäure-Fesseln – normalerweise HAs Lieblings-Ankerplatz. Ohne Gegenwehr würde das Virus kleben bleiben wie Kaugummi unter der Schuhsohle.
Neuraminidase (NA) greift ein: Die molekulare Schere zerschnippelt die Sialinsäure-Reste auf der Zelloberfläche. Kein Haften, kein Zurück – freie Bahn zur nächsten Zelle.
Wie ein Gefängnisausbruch mit Style: M1 lockert die Gitterstäbe, NA durchtrennt die Alarmsysteme – und weg sind sie! Final Countdown für die Virus-Crew! Alle Systeme go – HA/NA check, RNPs check, Lipidpanzer check. Startsequenz initiiert in 3…2…1… Budding!

Abb. 2-M: Alle Genome sind an Bord – die Startsequenz für das Viruspartikel beginnt. Left: Budding of the virus at the cell membrane. Right: Release of the newly formed virus particle.
Das folgende Video fasst den Replikationszyklus des Influenzavirus noch einmal gut verständlich zusammen.
Nach der Infektion einer Wirtszelle durch ein einziges Influenzavirus entstehen typischerweise Hunderte bis Tausende neuer Viruspartikel. Die genaue Anzahl variiert und hängt von verschiedenen Faktoren wie dem Virusstamm, der Art der Wirtszelle und den zellulären Bedingungen ab.
Vereinfachte und reale Darstellung der Virusstruktur
In den ersten Abbildungen dieses Textes wurde die Struktur des Influenzavirus zur besseren Übersicht vereinfacht dargestellt (siehe untere Abbildung links). Das Matrixprotein bildet in diesen Darstellungen eine kugelige, netzartige Ringstruktur, die das virale Genom umgibt. Diese vereinfachte Darstellung soll die komplexen Prozesse der Virusvermehrung leichter nachvollziehbar machen.
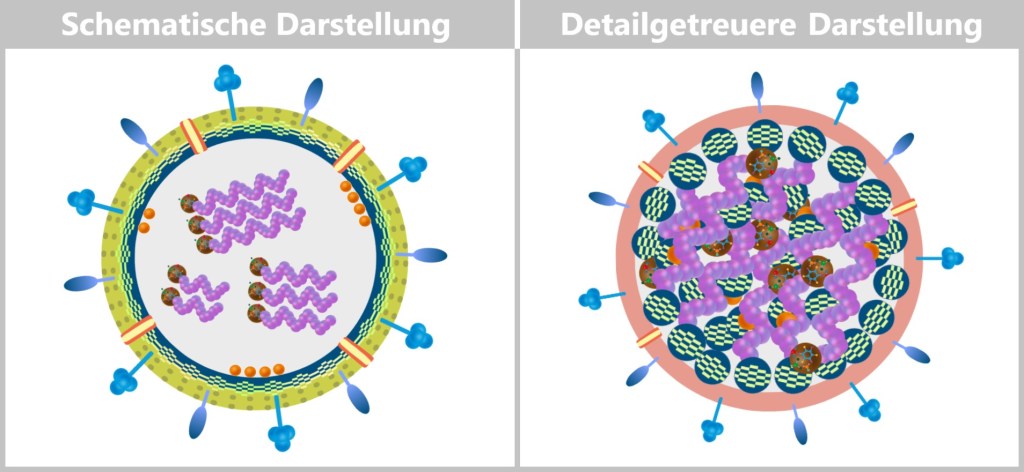
Abb. 2-N: Zwei Darstellungen des Virions: Links: Schematische Darstellung zur Verdeutlichung des Virusaufbaus
Rechts: Detailgetreuere schematische Darstellung der VirionenstrukturIn den folgenden Abbildungen wurde jedoch die Struktur der neuen Virionen näher an der biologischen Realität gezeigt. Dabei sind die Matrixproteine nicht als durchgehender Ring abgebildet, sondern befinden sich in einzelnen Einheiten, die sowohl an die innere Lipidschicht binden als auch lose mit den Ribonukleoprotein-Komplexen (RNPs) verknüpft sind. Zudem spiegelt die Farbgebung der Lipidschicht die Herkunft aus der Zellmembran der Wirtszelle wider (siehe obere Abbildung rechts).
Die RNPs, die das virale Genom darstellen, liegen im Inneren als lockeres Bündel vor – nicht streng parallel, sondern flexibel angeordnet mit unterschiedlich ausgerichteten Enden. Die Matrixproteine (M1) halten dieses Bündel zusammen und verbinden es mit der Lipidschicht, wodurch das Virion seine Form und Stabilität erhält.
2.4. Die Anpassungsfähigkeit des Influenzavirus
Viren – insbesondere RNA-Viren wie das Influenzavirus – mutieren außergewöhnlich schnell. Der Grund liegt in ihrer fehleranfälligen Replikationsmaschinerie: Die virale RNA-Polymerase besitzt keinen Mechanismus zur Korrektur von Kopierfehlern, wie es bei der DNA-Replikation in menschlichen Zellen der Fall ist. Dadurch entstehen bei jeder Vervielfältigung zufällige Mutationen – kleine Veränderungen im genetischen Material des Virus.
Innerhalb einer infizierten Person bildet sich so eine Vielzahl leicht unterschiedlicher Viruspartikel. Die meisten Mutationen sind neutral, das heißt, sie beeinflussen weder die Funktionsweise des Virus noch seine Fähigkeit, sich zu vermehren. Einige Mutationen sind jedoch nachteilig und führen dazu, dass das Virus weniger effizient repliziert oder gar nicht mehr infektiös ist – diese Varianten verschwinden rasch durch natürliche Selektion.
Doch manche Mutationen verschaffen dem Virus einen Überlebensvorteil, insbesondere wenn sie die Oberflächenproteine Hämagglutinin (HA) und Neuraminidase (NA) betreffen. Diese Proteine sind zentrale Angriffspunkte des Immunsystems: Der Körper produziert Antikörper, die spezifisch an sie binden und das Virus neutralisieren. Verändert sich jedoch die Struktur von HA oder NA durch Mutationen, können die Antikörper das Virus schlechter erkennen. Das Virus wird sozusagen „unsichtbar“ für die Immunabwehr und kann sich weiterhin vermehren und ausbreiten.
Diese ständige Anpassung erklärt, warum Grippeviren jedes Jahr erneut Infektionswellen auslösen und es schwierig ist, dauerhafte Impfstoffe gegen Influenza zu entwickeln.
Das Influenzavirus existiert nicht als starre genetische Einheit, sondern als eine sogenannte Mutantenwolke (Quasispecies) – eine dynamische Population von Virusvarianten, die durch kontinuierliche Mutationen entsteht. Diese genetische Vielfalt ist der Schlüssel zu seinem Überleben: Natürliche Selektion sorgt dafür, dass sich jene Varianten durchsetzen, die unter den gegebenen Bedingungen am erfolgreichsten sind. Diese hohe Anpassungsfähigkeit des Influenzavirus zeigt eindrucksvoll, wie Evolution in Echtzeit abläuft.
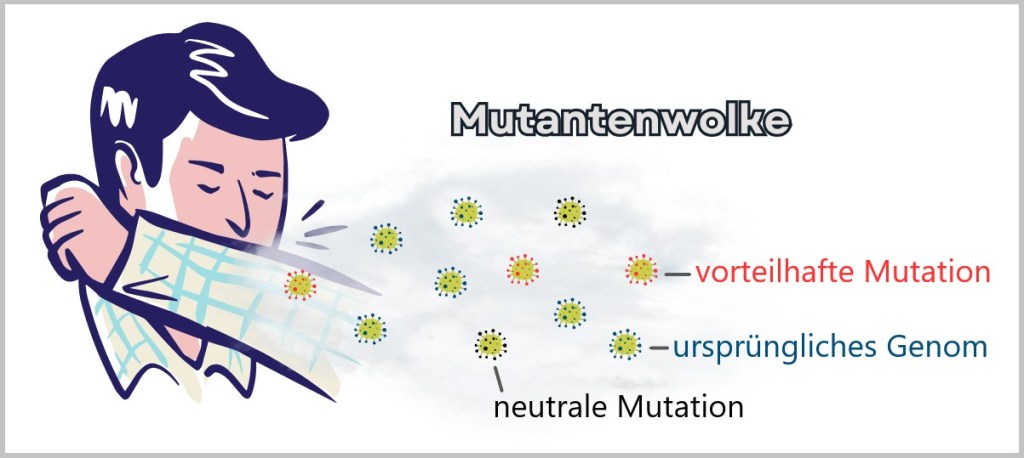
Abb. 2-O: Genetische Vielfalt im Wirt Das Influenzavirus muss sich ständig durch Mutationen verändern, um weiterhin als Grippevirus zu existieren. Die hohe Mutationsrate führt zu einer Vielzahl leicht unterschiedlicher Viruspartikel innerhalb einer infizierten Person.
2.5. Ein- und Austrittswege des Influenzavirus
Influenzaviren nutzen die Schleimhautoberflächen der Atemwege als Eintrittspforte, da Schleimhäute die Grenze zwischen der äußeren Umwelt und unserem Körperinneren darstellen. Viele Viren starten ihre Infektion über die Interaktion mit den Epithelzellen dieser Schleimhäute, um sich gezielt in ihrem Wirt zu verbreiten. [Virus Infection of Epithelial Cells]
Wie in der unteren Zeichnung dargestellt, gelangt das Influenzavirus über die Luft in die Atemwege – in Nase, Rachen und Lunge – und bindet an die apikale Seite (obere Seite) der Epithelzellen, die zur äußeren Umgebung hin orientiert ist. Diese apikale Seite ist mit feinen, haarartigen Strukturen, den Zilien, bedeckt, die Schleim und Fremdpartikel transportieren. Die gegenüberliegende, basolaterale Seite der Zelle ist dem darunterliegenden Gewebe zugewandt und mit der Basalmembran verbunden, die sie am Bindegewebe verankert.
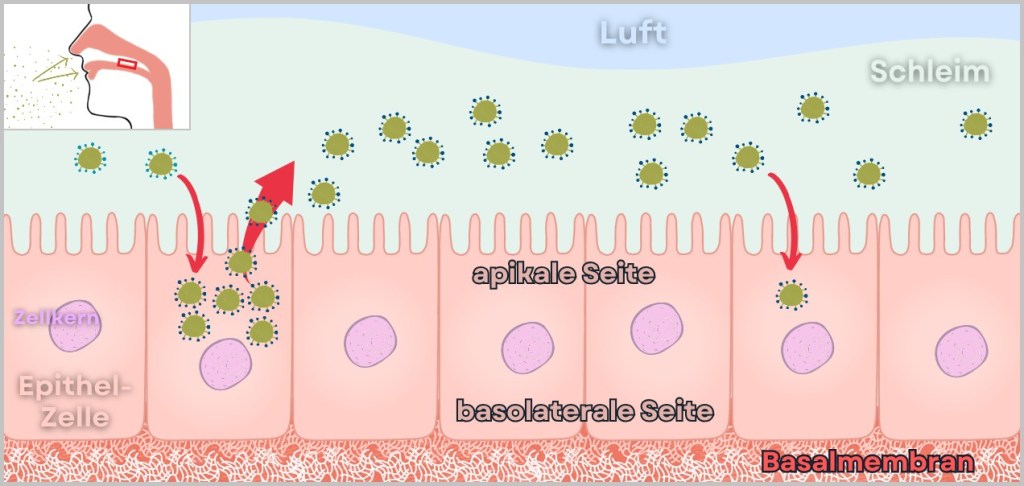
Abb. 2-P: Eine Epithelzelle wird vom Influenzavirus infiziert und produziert zahlreiche neue Virionen. Auch die Freisetzung der neugebildeten Influenzaviren erfolgt gezielt an der apikalen Seite. Durch diese Anordnung können sich die Viren in die Umgebung verbreiten, etwa über Tröpfchen beim Husten oder Niesen, und dadurch leicht neue Wirte infizieren. Diese apikale Freisetzung stellt einen evolutionären Vorteil dar, da sie die Effizienz der Übertragung deutlich steigert.
2.6. In den meisten Fällen lokal begrenzte mukosale Infektion
Influenzaviren sind auf Infektionen der Schleimhautoberfläche spezialisiert. Ihre Infektion bleibt daher meist lokal auf die Epithelzellen der Atemwege, das heißt, auf eine mukosale Infektion beschränkt. Das Virus breitet sich entlang der apikalen Seite der Epithelzellen von Zelle zu Zelle aus, ohne dabei in tiefere Gewebeschichten zu gelangen. Selbst bei einer Ausbreitung von den oberen Atemwegen bis hin zur Lunge bleibt die Infektion auf die Schleimhautoberfläche begrenzt.
Die basolaterale Seite der Epithelzellen bleibt in der Regel unberührt, da sie für die Virusübertragung keine Rolle spielt. Würde das Virus aus der basolateralen Seite der Wirtszelle austreten, könnte es ins umliegende Gewebe und letztlich in das Blut- oder Lymphsystem gelangen, was zu einer systemischen Infektion führen könnte. Für Influenzaviren wäre dies jedoch von Nachteil, da sie dann einer stärkeren Immunabwehr ausgesetzt wären und die Übertragung über die Atemwege erschwert würde.
In seltenen Fällen – besonders bei stark geschwächten Personen – kann das Virus jedoch die Epithelbarriere durchbrechen und in das darunterliegende Gewebe sowie in Blut- oder Lymphgefäße eindringen und zu einer systemischen Infektion führen.

Abb. 2-Q: Unterschied zwischen einer mukosalen Infektion und einer systemischen Infektion Die Schleimhaut besteht aus mehreren Schichten: Epithelzellen bilden die äußere Schutzschicht, die Basalmembran trennt als dünne Barriere, die Lamina propria stützt mit Gewebe und Immunzellen, die Endothelzellen bilden die Wand der Blutgefäße, und das Blutgefäß führt ins Körperinnere.
Links – Mukosale Infektion (begrenzt auf die Schleimhaut): Das Virus infiziert Epithelzellen ausschließlich über die apikale Seite. Es verbleibt in der Schleimhaut, wobei es von Zelle zu Zelle entlang der apikalen Oberfläche weitergegeben wird. Die Basalmembran und darunterliegende Gewebe wie die Lamina propria bleiben intakt. Eine mukosale Infektion ist lokal begrenzt und begünstigt die Übertragung über die Schleimhäute, etwa durch Atemwege.
Rechts – Systemische Infektion (Ausbreitung über das Blut): Das Virus tritt auf der apikalen Seite in die Epithelzellen ein, verlässt sie jedoch über die basolaterale Seite. Es durchdringt die Basalmembran und bewegt sich durch die Lamina propria, entweder durch Wanderung oder durch Infektion der dortigen Zellen. Schließlich erreicht es ein Blutgefäß, indem es durch Spalten zwischen Endothelzellen oder durch direkte Infektion der Endothelzellen in das Gefäßsystem eindringt. Der Eintritt des Virus in die Blutbahn markiert den Übergang zu einer systemischen Infektion. Eine systemische Infektion ist kritisch, weil das Virus über das Blut den ganzen Körper erreichen kann und so lebenswichtige Organe wie Lunge, Herz oder Gehirn schädigen könnte.
2.7. Virenstrategie: Effiziente Vermehrung ohne rasche Zellzerstörung
Manche Viren – darunter auch Influenzaviren – sind erstaunlich ökonomisch: Statt ihre Wirtszelle sofort zu zerstören, nutzen sie deren Ressourcen möglichst effizient aus. Warum die Wohnung abfackeln, wenn man monatelang kostenlos wohnen kann? Solange die Zelle intakt bleibt, liefert sie alles, was das Virus zur Replikation braucht: Energie, Enzyme, Bausteine. Und auch das Immunsystem merkt erst später, dass was faul ist – denn wo nichts brennt, wird kein Alarm ausgelöst. Diese Strategie verlängert das Leben der infizierten Zelle, verzögert die Immunantwort – und maximiert die Produktion neuer Viren.
Viren-Weisheit: Die besten Parasiten bleiben unterm Radar!
2.8. Zerstörung der Wirtszelle
Was mit listiger Schonung beginnt, endet in molekularem Burnout: Die infizierte Zelle erleidet letztlich den Zelltod. Dieser tritt ein, wenn die Zelle entweder durch die Massenproduktion der Viren überlastet und strukturell geschädigt wird, durch zelluläre Schutzmechanismen in den programmierten Zelltod (Apoptose) übergeht oder vom Immunsystem gezielt eliminiert wird. Diese charakteristischen Veränderungen der Wirtszelle durch das Virus werden als zytopathische Effekte (CPE) bezeichnet. Dieser gesamte Prozess kann bereits innerhalb von 24 Stunden nach der Infektion stattfinden.
Um den Zelltod durch Influenzaviren besser zu verstehen, betrachten wir die drei Mechanismen, durch die die Wirtszelle schließlich zerstört wird.
a) Überlastung und strukturelle Schädigung
b) Apoptose: Programmierter Zelltod zur Virusbekämpfung
c) Immunantwort: Zerstörung durch das Immunsystema) Überlastung und strukturelle Schädigung
Das Virus übernimmt die zellulären Prozesse zur Produktion seiner eigenen Bestandteile. Mit jeder neuen Generation viraler Proteine und RNA wird die Energie und Ressourcenkapazität der Zelle zunehmend aufgebraucht. Da die Zelle praktisch nur noch für die Virusvermehrung arbeitet, kommen ihre eigenen überlebenswichtigen Prozesse zum Erliegen. Die Zelle wird zur ausgepressten Zitrone – Ribosome laufen heiß, Mitochondrien kollabieren.
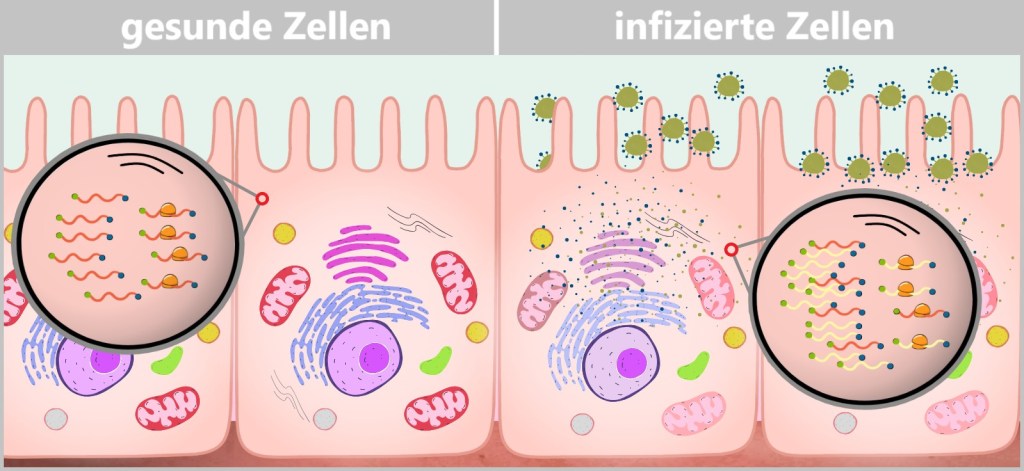
Abb. 2-R: Ressourcenkapazität: gesunde Zelle vs. infizierte Zelle Links: Gesunde Zelle mit funktionierender zellulärer Maschinerie. Die körpereigene mRNA (orange) wird von den Ribosomen gelesen, um Proteine für die Zellfunktionen herzustellen. Die Zelle zeigt eine lebhafte Färbung, die auf die volle Ressourcenkapazität und Energie hinweist. Rechts: Virusinfizierte Zelle, stark belastet durch die Produktion viraler Bestandteile. Die virale mRNA (gelb) verdrängt zunehmend die körpereigene mRNA, und die Ribosomen lesen überwiegend virale Anweisungen für die Virusvermehrung. Die blasse Färbung der Zellorganellen symbolisiert die Erschöpfung der Energiereserven und die Überlastung der Zelle.
Während der Knospung an der Zellmembran – wenn neue Viruspartikel die Zelle verlassen – wird die Zellmembran mehrfach durchbrochen und verformt. Dieser Prozess führt letztlich zur Zerstörung der Membranintegrität, wodurch die Zelle ihre Stabilität und ihre schützenden Funktionen verliert. Die Zelle stirbt schließlich durch den unaufhörlichen Ressourcenverbrauch und den strukturellen Zerfall infolge der Virusfreisetzung.
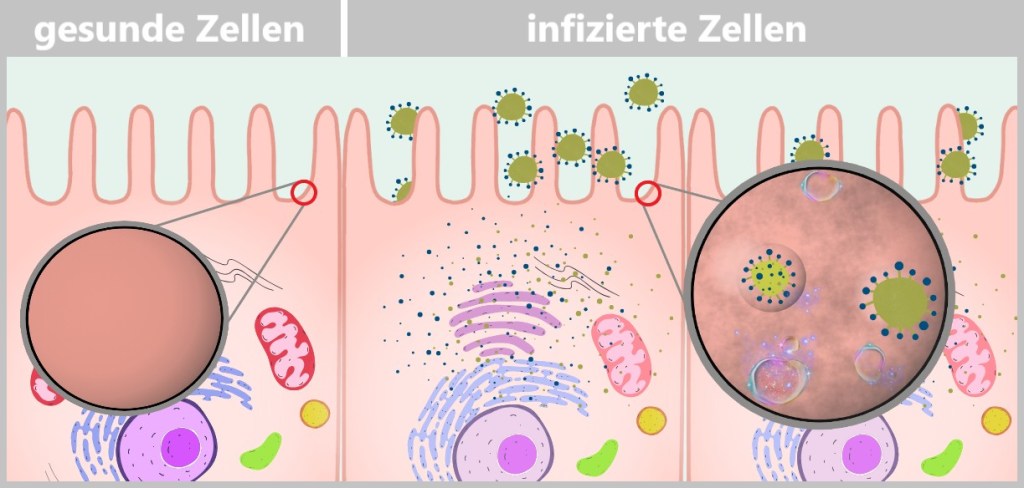
Abb. 2-S: Strukturelle Schädigung der infizierten Zelle nach dem Knospungsvorgang und der Freisetzung neuer Viren Während des Knospens bildet die Membran kleine Ausstülpungen, aus denen die neuen Viren freigesetzt werden. Jedes Knospungsereignis entzieht der Zellmembran kleine Teile ihrer Lipiddoppelschicht, da die neu entstehenden Viren Membranmaterial der Wirtszelle als ihre Hülle verwenden. Nach wiederholten Virusfreisetzungen ist die Membran deutlich ausgedünnt und strukturell geschwächt, die Membran zeigt oft Verformungen und Unebenheiten. Die dauerhafte Belastung durch die Knospung führt dazu, dass die Membran poröser und anfälliger wird. Die Zelle verliert zunehmend die Fähigkeit, ihren inneren Zustand zu regulieren und kann ihre selektive Durchlässigkeit für Ionen und Moleküle kaum noch aufrechterhalten. Da die Membran fortwährend geschädigt wird, geht ihre strukturelle Stabilität verloren. Letztlich kann die Membran so stark beeinträchtigt werden, dass sie reißt oder zerfällt, was zum Zelltod führt.
b) Apoptose: Programmierter Zelltod zur Virusbekämpfung
Wenn eine Zelle merkt, dass sie gekapert wurde, zieht sie manchmal die Notbremse – und opfert sich für das größere Ganze: Sie bringt sich selbst um, um die Ausbreitung zu stoppen. Der Plan: Den Feind mit ins Grab nehmen. Statt mit einem Knall zu sterben, zerfällt die Zelle kontrolliert in kleine Fragmente, die sogenannte Apoptose-Körper bilden, welche anschließend von Immunzellen abgebaut werden.
Dabei laufen präzise Prozesse ab: Die DNA wird zerschnitten, die Zellmembran bildet typische blasenartige Ausstülpungen (sogenannte Blebbing), die inneren Strukturen werden fein säuberlich recycelt. Die Zelle stirbt still – und schützt damit den Organismus.
Dieser Mechanismus beweist – Zellen haben mehr Ehre als manche Regierungen!
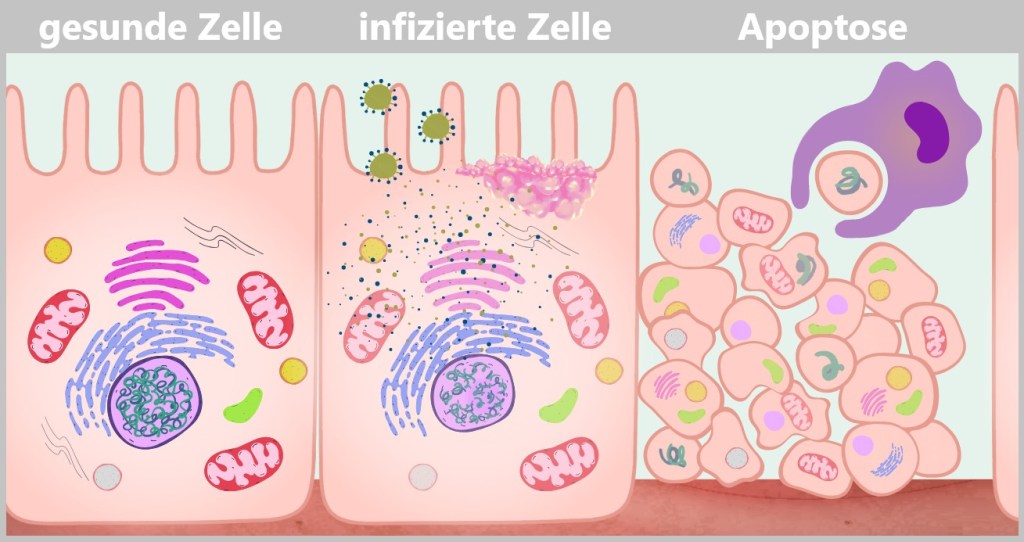
Abb. 2-T: Das Bild zeigt die Phasen der Apoptose in einer virusinfizierten Zelle in drei Schritten. Gesunde Zelle: Links ist eine intakte Zelle dargestellt, deren Zellmembran und Zellkern unversehrt sind. Der Zellkern enthält vollständige DNA-Stränge, und die Zelle zeigt keine Anzeichen von Stress oder Schädigung.
Infizierte Zelle – Einleitung der Apoptose: Im mittleren Abschnitt beginnt die Zelle sichtbare Veränderungen zu zeigen. Die Zellmembran bildet blasenartige Ausstülpungen (Blebbing), und der Zellkern schrumpft. Innerhalb des Zellkerns sind DNA-Bruchstücke sichtbar, die durch apoptotische Prozesse entstehen. Diese Phase stellt den Übergang von einer funktionierenden Zelle zu ihrem kontrollierten Zerfall dar.
Apoptose: Rechts zerfällt die Zelle in mehrere kleine Fragmente, sogenannte Apoptose-Körper. Im Hintergrund ist eine Immunzelle (Makrophage) dargestellt, die diese Fragmente aufnimmt und abbaut. Dies verhindert die Freisetzung viraler Bestandteile und schützt das umliegende Gewebe vor einer weiteren Infektion.c) Immunantwort: Zerstörung durch das Immunsystem
Sobald das Immunsystem eine virusinfizierte Zelle entdeckt, schlägt es Alarm – und das heißt Ärger für das Virus! Spezialisierte Kämpfer wie natürliche Killerzellen (NK-Zellen) und zytotoxische T-Zellen stürzen sich ins Gefecht. Sie erkennen die infizierten Zellen anhand viraler Proteinsignale, die wie Warnflaggen auf der Zell-Oberfläche wehen. Mit tödlicher Präzision setzen sie toxische Moleküle frei, zerstören die Zelle und bremsen so die Virusvermehrung aus!
Wer mehr über diese faszinierende Abwehrschlacht wissen will, findet Details in „Die Wunderwelt des Lebens“, besonders in Kapitel 5.3 d) „Natürliche Killerzellen“ und Kapitel 5.5.7 b) „Zytotoxische T-Zellen“.
NK-Zellen und zytotoxische T-Zellen sind ein unschlagbares Team – sie jagen und eliminieren virale Bedrohungen und halten die Infektion in Schach.
Regeneration der Epithelzellen
Wie bereits erwähnt, befällt das Influenzavirus vor allem die Epithelzellen der Atemwege – insbesondere die Zellen der Nasenschleimhaut, der Bronchien und der Lungenbläschen (Alveolen). Durch die Virusvermehrung sowie die Immunabwehr, insbesondere durch natürliche Killerzellen (NK-Zellen) und zytotoxische T-Zellen, werden viele dieser Zellen stark geschädigt oder zerstört.
Sobald die akute Infektion eingedämmt ist, setzt der Reparaturprozess ein: Spezialisierte Stammzellen beginnen mit der Regeneration des Gewebes. Sie vermehren sich und entwickeln sich zu den verschiedenen Epithelzelltypen, die für die Wiederherstellung der Atemwege benötigt werden.
- Obere Atemwege (Nase, Bronchien): Neue Zilienzellen entstehen, deren feine Härchen (Zilien) Schleim und Fremdpartikel nach oben transportieren. Zudem werden Becherzellen gebildet, die den Schleim produzieren und so die Atemwege feucht und geschützt halten.
- Lunge (Alveolen): Hier werden die geschädigten, flachen Epithelzellen ersetzt, die den Sauerstoffaustausch zwischen Luft und Blut ermöglichen.
Die Regeneration dauert je nach Schwere der Infektion unterschiedlich lange. Bei einer milden Erkrankung kann das Epithel innerhalb von ein bis zwei Wochen vollständig erneuert sein. Nach schwereren Infektionen, etwa bei einer Influenza-Pneumonie, kann der Heilungsprozess jedoch mehrere Wochen in Anspruch nehmen. Sobald die neuen Zellen eine dichte Schicht bilden, ist die Schutzfunktion der Atemwege wiederhergestellt. In den meisten Fällen erfolgt die Regeneration vollständig – bei sehr schweren Schäden können jedoch Narben oder strukturelle Veränderungen im Epithel zurückbleiben.
2.9. Selbstbegrenzung gefährlicher Viren: Warum sie selten Pandemien auslösen
Hochgefährliche Viren, die ihren Wirt rasch töten, begrenzen ihre eigene Verbreitung. Wenn ein Virus seinen Wirt so schnell schädigt, dass dieser keine Zeit hat, andere zu infizieren, wird die Übertragungskette effektiv unterbrochen. Ein Beispiel dafür ist das Ebola-Virus, das oft lokal begrenzt bleibt und daher selten Pandemien auslöst.
Im Gegensatz dazu lösen Viren mit moderater Pathogenität häufiger globale Ausbrüche aus. Moderate Pathogenität beschreibt die Fähigkeit eines Erregers, Krankheiten auszulösen, ohne dabei extrem schwere oder tödliche Verläufe bei den meisten Infizierten zu verursachen. Solche Viren führen typischerweise zu milden bis mittelschweren Symptomen, die es den infizierten Personen ermöglichen, weiterhin mobil und sozial aktiv zu bleiben. Dadurch erhöhen sich die Chancen für die Weitergabe des Virus an andere. Beispiele dafür sind viele Influenzaviren. Schwere Verläufe einer Influenza-Infektion treten dabei vorwiegend bei Personen mit geschwächtem Immunsystem, höherem Alter oder bestehenden Grunderkrankungen auf. In diesen Fällen sind eine sorgfältige medizinische Überwachung und eine intensivierte Behandlung angebracht, um schwere Komplikationen zu verhindern.
Die erfolgreichsten Viren sind nicht die, die uns umbringen – sondern die, die uns gerade so am Leben lassen, dass wir ihre Drecksarbeit erledigen.
2.10. Warum macht das Virus manche Menschen krank und andere nicht?
💡Hinweis: Für ein besseres Verständnis dieses Abschnitts empfehlen wir das Kapitel 5 in der Abhandlung „Die Wunderwelt des Lebens“. Dort werden die Grundlagen zum Immunsystem auf anschauliche Weise erklärt.
Nicht jede Person, die sich mit dem Influenzavirus infiziert, erkrankt gleich schwer: Manche haben nur leichte Symptome wie einen Schnupfen, andere entwickeln eine schwere Grippe mit Fieber und Atemnot, und einige bleiben sogar völlig symptomfrei. Warum ist das so? Die Antwort liegt in einer komplexen Wechselwirkung zwischen dem Virus und dem Wirt – also dem Menschen, der infiziert wird. Mehrere entscheidende Faktoren spielen dabei eine Rolle:
Das Immunsystem des Wirts
Jeder Mensch hat ein individuelles Immunsystem, das unterschiedlich gut auf das Influenzavirus reagiert. Frühere Grippeinfektionen können eine Teilimmunität bieten, weil das Immunsystem Antikörper und Gedächtniszellen entwickelt hat, die das Virus schneller erkennen und bekämpfen. Ein starkes Immunsystem kann die Infektion so im Keim ersticken, während ein geschwächtes Immunsystem (z. B. bei älteren Menschen oder chronisch Kranken) oft überfordert ist.
Die Viruslast
Die Menge an Viruspartikeln, die beim ersten Kontakt in den Körper gelangen – die sogenannte Viruslast –, beeinflusst den Verlauf der Infektion. Bei einer geringen Viruslast kann das angeborene Immunsystem die Eindringlinge schnell erkennen und zerstören, bevor sie sich stark vermehren. Eine hohe Viruslast, z. B. durch engen Kontakt mit einer infizierten Person, stellt jedoch eine größere Herausforderung dar und kann das Infektionsgeschehen verstärken.
10 Viren im Rachen? Kein Problem.
10.000 Viren? Ab ins Bett!Die genetische Veranlagung des Wirts
Zwei Menschen, ein Virus – doch nur einer wird krank. Manche Menschen tragen genetische Varianten in ihrem Immunsystem, die sie anfälliger oder widerstandsfähiger gegen das Influenzavirus machen. Zum Beispiel können Unterschiede in Genen, die Immunrezeptoren steuern, beeinflussen, wie gut das Immunsystem das Virus erkennt.
Es gibt zahlreiche Studien, die die unterschiedlichen Immunantworten aufgrund der genetischen Veranlagung untersuchen:
Die Studie „IFITM3: How genetics influence influenza infection demographically” zeigte, dass Menschen mit bestimmten Varianten des IFITM3-Gens (Interferon-induziertes Transmembranprotein 3) seltener schwere Grippeerkrankungen entwickeln, weil dieses Gen die Vermehrung des Influenzavirus in Zellen hemmt.
Die Studie „HLA targeting efficiency correlates with human T-cell response magnitude and with mortality from influenza A infection” untersuchte, wie HLA-Allele (MHC-Klasse-I) die T-Zell-Antwort auf das Influenzavirus beeinflussen. Sie fand heraus, dass bestimmte HLA-Allele effizienter Influenza-Peptide präsentieren und eine stärkere T-Zell-Antwort auslösen. Menschen mit diesen Allelen hatten mildere Verläufe bei Grippeinfektionen, während andere Allele mit schwächeren T-Zell-Antworten und höherer Mortalität assoziiert waren. Das zeigt, dass genetische Unterschiede in MHC-Molekülen direkt die Schwere einer Influenzainfektion beeinflussen können.
Die Mutantenwolke des Virus
Wie bereits erwähnt, existiert das Influenzavirus nicht als einheitlicher Stamm, sondern als eine „Mutantenwolke“ – eine Vielfalt genetischer Varianten, die durch die fehleranfällige RNA-Polymerase entstehen. Manche Varianten in dieser Wolke sind aggressiver, weil sie z. B. besser an Zellen binden oder dem Immunsystem entgehen. Welche Variante dominiert, kann entscheiden, wie schwer die Infektion verläuft.
Die Gewebespezifität des Virus
Influenzaviren unterscheiden sich in ihrer Vorliebe für bestimmte Gewebe im Körper. Die meisten Stämme vermehren sich bevorzugt in den oberen Atemwegen (z. B. Nase und Rachen), was oft zu milderen Symptomen wie Halsschmerzen führt. Andere Stämme dringen tiefer in die Lunge vor und können eine schwere Lungenentzündung auslösen.
📌 Fazit: Ein Hoch auf die Vielfalt!
Die Interaktion zwischen Virus und Mensch ist wie Tinder für Mikroben – manche Matches sind harmlos, andere enden im Desaster. Entscheidend ist:
- Wirtspoker (Gene + Immunsystem)
- Virus-Roulette (Dosis + Mutationen)
- Gewebe-Tinder (Wo landet das Virus?)
Unser Körper ist kein passives Ziel – sondern ein lernendes System. Jeder Infekt ist ein Update fürs Immungedächtnis, jeder Virus ein Trainingspartner fürs Leben. Denn nur im Kampf wächst unser Schutzschild – ein Leben lang.
3. Ein Blick auf die Anfänge der Mikrobiologie – wie alles begann
Die Geschichte der Mikrobiologie ist eine Reise von der Unsichtbarkeit zur Klarheit, von Spekulationen zu konkretem Wissen. Ihre Ursprünge lassen sich bis ins 17. Jahrhundert zurückverfolgen, als die Neugier der Menschen und die technische Innovation erstmals eine verborgene Welt enthüllten.
3.1. Frühe Entdeckungen: Die ersten Blicke ins Unsichtbare
3.2. Die Geburt der modernen Mikrobiologie
3.3. Der Schritt in die Welt der Viren
3.4. Viren und die Koch’schen Postulate3.1. Frühe Entdeckungen: Die ersten Blicke ins Unsichtbare
Im Jahr 1665 war es Robert Hooke, der mit einem frühen Mikroskop erstmals Pflanzenzellen beschrieb und damit den Begriff „Zelle“ prägte.
Doch der wirkliche Durchbruch kam einige Jahre später mit Antonie van Leeuwenhoek. Mit seinen selbstgebauten, extrem leistungsfähigen Linsen beobachtete er 1676 erstmals winzige, lebendige Organismen, die er als „animalcules“ bezeichnete – kleine Tierchen, wie Bakterien und einzellige Lebewesen, die wir heute kennen. Van Leeuwenhoeks Entdeckungen eröffneten eine völlig neue Perspektive auf die Natur, doch das Wissen darüber, wie diese Organismen lebten oder Krankheiten verursachten, war noch weit entfernt.
3.2. Die Geburt der modernen Mikrobiologie
Es dauerte fast zwei Jahrhunderte, bis die Mikrobiologie systematisch erforscht wurde. Im 19. Jahrhundert erlebte das Fachgebiet einen Quantensprung. Louis Pasteur widerlegte die alte Vorstellung, dass Leben einfach aus dem Nichts entstehen könne (Theorie der Spontanzeugung), und bewies, dass Mikroorganismen für Prozesse wie Gärung und Fäulnis verantwortlich sind. Seine Arbeiten legten den Grundstein für die Keimtheorie der Krankheiten, die schließlich von Robert Koch weiterentwickelt wurde.
Kochs Forschung führte 1876 zu einem entscheidenden Meilenstein: den Koch’schen Postulaten. Diese Regeln ermöglichten es erstmals, Mikroorganismen als spezifische Verursacher von Krankheiten zu identifizieren. Kochs Arbeit revolutionierte die Bakteriologie und machte es möglich, Krankheitserreger wie den Milzbranderreger (Bacillus anthracis) und später auch den Tuberkulose-Erreger eindeutig nachzuweisen.
Doch während die Mikrobiologie mit der Erforschung von Bakterien große Fortschritte machte, blieben Viren lange im Verborgenen. Selbst die besten Mikroskope der damaligen Zeit konnten diese winzigen, unsichtbaren Partikel nicht abbilden.
3.3. Der Schritt in die Welt der Viren
Das Ende des 19. Jahrhunderts brachte den nächsten Durchbruch. Dmitri Iwanowski zeigte 1892, dass ein filtrierter Extrakt aus Tabakpflanzen, die an der Tabakmosaikkrankheit litten, infektiös blieb, obwohl er durch Porzellanfilter geleitet wurde, die Bakterien zurückhielten. Martinus Beijerinck bestätigte diese Beobachtungen und prägte den Begriff „Virus“ (vom lateinischen Wort für „Gift“ oder „Schleim“) für den mysteriösen, nicht-bakteriellen Erreger. Damit begann die systematische Erforschung dieser neuen Welt.
Der wahre Zugang zur Welt der Viren wurde jedoch erst mit dem Elektronenmikroskop möglich, das in den 1930er Jahren entwickelt wurde. Erst dann konnten Wissenschaftler Viren sichtbar machen und ihre Struktur verstehen.
3.4. Viren und die Koch’schen Postulate
In Diskussionen über den Nachweis von Viren werden oft die Koch’schen Postulate als Maßstab herangezogen, um die Existenz von Viren infrage zu stellen. Doch wie passen diese Postulate, die im 19. Jahrhundert entwickelt wurden, in das heutige Verständnis von Infektionskrankheiten? Ein Blick auf die historischen Hintergründe und die wissenschaftliche Weiterentwicklung hilft, diese Frage zu klären.
Die Koch’schen Postulate: Ein wissenschaftlicher Meilenstein
Robert Koch (1843–1910), einer der Begründer der modernen Bakteriologie, entwickelte die nach ihm benannten Postulate, um den Zusammenhang zwischen Mikroorganismen und Infektionskrankheiten zu beweisen. Sie wurden 1890 auf dem 10. Internationalen Medizinischen Kongress vorgestellt und bestehen aus vier Kriterien:
Postulat 1: Der Mikroorganismus muss in jedem Fall der Krankheit nachgewiesen werden, sollte aber nicht in gesunden Organismen vorkommen.
Postulat 2: Der Mikroorganismus muss aus dem erkrankten Organismus isoliert und in Reinkultur gezüchtet werden.
Anmerkung: Eine Reinkultur bedeutet, dass nur eine einzige Art von Mikroorganismen gezüchtet wird, ohne andere Arten dazwischen.Postulat 3: Ein zuvor gesundes Individuum zeigt nach einer Infektion mit dem Mikroorganismus aus der Reinkultur die gleichen Symptome wie das Individuum, von dem der Mikroorganismus ursprünglich stammt.
Postulat 4: Der Mikroorganismus muss erneut aus dem Versuchswirt isoliert und als derselbe identifiziert werden.
Diese bahnbrechenden Prinzipien legten den Grundstein für die experimentelle Medizin und die Keimtheorie der Krankheiten.
Robert Koch: Ein Pionier gegen Widerstände
Robert Koch untersuchte die Erreger von Krankheiten wie Tuberkulose, Cholera und Milzbrand. Für seine Forschungen reiste er oft in Seuchengebiete, wie Kalkutta zur Untersuchung der Cholera oder Bombay während der Beulenpest. Koch verbrachte Monate in diesen Ländern, immer nah am Zentrum der Seuchen. In seinem Laborzelt arbeitete er unermüdlich am Mikroskop.
Koch hatte jedoch mit erheblichem Widerstand zu kämpfen. Zu seiner Zeit war die Idee, dass Krankheiten durch mikroskopische Organismen verursacht werden, noch umstritten. Viele seiner Kollegen und Zeitgenossen waren skeptisch und lehnten seine Theorien ab. Wissenschaftler glaubten damals noch oft, dass Seuchen und Epidemien von sogenannten Miasmen – giftigen Dämpfen, die aus dem Erdreich aufsteigen – ausgelöst würden.
Trotz dieser Herausforderungen setzte sich Koch unermüdlich für seine Forschung ein. Er nutzte innovative Techniken seiner Zeit wie die Agarplatte und die Ölimmersionslinsen, um Bakterien zu kultivieren und zu untersuchen. Diese Methoden ermöglichten es ihm, wichtige Entdeckungen zu machen und das Verständnis von Infektionskrankheiten revolutionär zu verändern.
Abb. 3: Frühe Werkzeuge der Mikrobiologie: Agarplatte und Ölimmersionsmikroskop Links: Eine Agarplatte – ein fester Nährboden in einer Petrischale, dem Agar als Geliermittel zugesetzt wurde, um Bakterien gezielt zu kultivieren. Rechts: Ein Mikroskop mit Ölimmersionslinse – eine spezielle Mikroskoptechnik, bei der ein Tropfen Öl zwischen Objektiv und Probe die Lichtbrechung minimiert, sodass kleinste Mikroben schärfer sichtbar werden.
Herausforderungen und Grenzen der Postulate
Genau die seinen Theorien entgegengebrachte Skepsis veranlasste Koch, die Postulate aufzustellen, um den Beweis zu erbringen, dass es einen Zusammenhang zwischen den pathogenen Eigenschaften der Bakterien und der Krankheit gibt.
Koch selbst erkannte, dass seine Postulate nicht immer uneingeschränkt gelten. Ein bekanntes Beispiel ist seine Arbeit mit dem Cholera-Erreger Vibrio cholerae. Er fand heraus, dass dieser Mikroorganismus nicht nur bei erkrankten, sondern auch bei scheinbar gesunden Menschen vorkommen kann. Diese Entdeckung stellte das erste Postulat infrage und führte dazu, dass Koch die universelle Gültigkeit dieses Kriteriums aufgab.
Kochs Innovationsgeist
Koch war ein Pionier seiner Zeit. In seiner Rede auf dem 10. Internationalen Medizinischen Kongress erklärte er:
„Es war geboten, mit unwiderleglichen Gründen den Beweis zu führen, dass die bei einer Infectionskrankheit aufgefundenen Mikroorganismen auch wirklich die Ursache dieser Krankheit seien.“
Sein wissenschaftlicher Ansatz, Skeptiker durch strikte Nachweise zu überzeugen, war wegweisend. Doch er selbst erkannte, dass neue Technologien und Methoden nötig sind, um weiterführende Fragen zu beantworten:
„Mit den zu Gebote stehenden experimentellen und optischen Hülfsmitteln war auch nicht weiter zu kommen und es wäre wohl noch geraume Zeit so geblieben, wenn sich nicht gerade damals neue Forschungsmethoden geboten hätten, welche mit einem Schlage ganz andere Verhältnisse herbeiführten und die Wege zu weiterem Eindringen in das dunkle Gebiet öffneten, mit Hülfe verbesserter Linsensysteme …“
In Bezug auf schwer nachweisbare Krankheitserreger wie die der Influenza oder des Gelbfiebers bemerkte er:
„Ich möchte mich der Meinung zuneigen, dass es sich bei den genannten Krankheiten gar nicht um Bakterien, sondern um organisierte Krankheitserreger handelt, welche ganz anderen Gruppen von Mikroorganismen angehören.“
Koch war damit den Viren bereits auf der Spur, konnte sie jedoch aufgrund der zu seiner Zeit begrenzten technischen Möglichkeiten nicht eindeutig identifizieren. Er erkannte jedoch, dass diese unsichtbaren Erreger existieren mussten.
Warum Viren die Koch’schen Postulate sprengen
Koch‘s Forschungsergebnisse entsprachen dem damaligen Entwicklungsstand der Wissenschaft. Die Mikrobiologie befand sich im 19. Jahrhundert noch in einer frühen Entwicklungsphase, in der grundlegende Prinzipien erst entdeckt und systematisch erforscht wurden. Die Virologie als eigenständiges Feld entstand erst nach Kochs Zeit, als Dmitri Iwanowski und Martinus Beijerinck infektiöse Partikel entdeckten, die kleiner waren als Bakterien. Mit der Erfindung des Elektronenmikroskops in den 1930er Jahren konnten Viren schließlich sichtbar gemacht werden. Viren unterscheiden sich jedoch fundamental von Bakterien, weshalb die Koch’schen Postulate oft nicht direkt auf sie anwendbar sind:
➤ Wirtabhängigkeit: Viren können sich nur in lebenden Wirtszellen vermehren und lassen sich nicht in einer Reinkultur züchten.
➤ Asymptomatische Infektionen: Viele Virusinfektionen verlaufen ohne Symptome, was die Zuordnung von Erreger und Krankheit erschwert.
➤ Komplexe Nachweisverfahren: Moderne molekularbiologische Methoden wie PCR ermöglichen den Nachweis von viralen Genomsequenzen, was eine Erweiterung der klassischen Postulate erfordert.Koch und die heutige Wissenschaft
Robert Koch und seine Zeitgenossen legten den Grundstein für die Mikrobiologie, insbesondere durch die Entwicklung von Methoden zur Isolierung und Kultivierung von Bakterien. Die Keimtheorie der Krankheiten war damals ein Meilenstein in der Wissenschaftsgeschichte. Koch wusste, dass die Wissenschaft ständig im Wandel begriffen ist. Hätte Koch Zugang zu modernen Technologien wie PCR, Sequenzierung oder Elektronenmikroskopen gehabt, hätte er seine Methodik angepasst?
Moderne Technologien wie PCR und Sequenzierung werden in den kommenden Kapiteln vorgestellt.
Seine Schlussworte beim Kongress von 1890 geben die Antwort und zeigen seinen Optimismus für die Zukunft:
„Lassen Sie mich mit dem Wunsche schließen, dass sich die Kräfte der Nationen auf diesem Arbeitsfelde und im Kriege gegen die kleinsten, aber gefährlichsten Feinde des Menschengeschlechts messen mögen und dass in diesem Kampfe zum Wohle der gesammten Menschheit eine Nation die andere in ihren Erfolgen immer wieder überflügeln möge.“
Hätte Robert Koch die heutigen Möglichkeiten der Molekularbiologie, Virologie und Immunologie erleben können, wäre er vermutlich fasziniert gewesen – nicht nur von den neuen Erkenntnissen, sondern auch von den revolutionären Methoden, die es ermöglichen, selbst die kleinsten Erreger sichtbar zu machen. Denn genau darin lag sein Antrieb: Unsichtbares erkennbar zu machen, das Verborgene zu entschlüsseln. Wie hätte er wohl auf die ersten Bilder eines Virus unter dem Elektronenmikroskop reagiert?
Von den ersten Beobachtungen zur modernen Wissenschaft
Was einst mit staubigen Linsen und ratlosen Blicken begann, ist heute Hightech bis auf Molekülebene. Mit jeder neuen Technologie blicken wir tiefer in den Mikrokosmos. Die Mikrobiologie hat das Unsichtbare sichtbar gemacht – aber erst die moderne Technik erlaubt es uns, das Unsichtbare wirklich zu verstehen. Heute spüren wir Viren auf, die sich jahrhundertelang unserem Blick entzogen haben.
Und wie wir dem Unsichtbaren heute auf die Spur kommen – das ist die Geschichte des nächsten Kapitels.
4. Moderne Methoden zur Entdeckung und Analyse von Viren
Bisher haben wir uns mit dem Thema Viren vertraut gemacht. In diesem Kapitel wechseln wir die Perspektive: vom Staunen zum Messen. Denn ohne die Methoden der modernen Biologie wüssten wir kaum etwas über Viren – ihre Formen, ihr Erbgut, ihre Vielfalt.
Was folgt, ist ein Blick in den Maschinenraum der Wissenschaft. Zugegeben, ein technischer Abschnitt – aber er ist wichtig: Denn hier wird sichtbar, wie wir das Unsichtbare überhaupt zu fassen kriegen.
Wer gerne verstehen möchte, wie man Viren heute sichtbar macht, entschlüsselt, katalogisiert und analysiert, der findet hier eine Art Werkzeugkasten des 21. Jahrhunderts.
Doch wann und warum greifen wir zu diesen Werkzeugen?
Die Identifikation von Viren kann unterschiedliche Ziele verfolgen: Manche Viren rücken durch ihre Auswirkungen auf Menschen, Tiere oder Pflanzen in den Fokus, während andere in Umweltproben oder unerforschten Lebensräumen entdeckt werden, um ihre ökologische Bedeutung zu verstehen. Der Nachweis bekannter Viren dient vor allem der Diagnostik, während die Identifikation unbekannter Viren Einblicke in die biologische Vielfalt und Evolution ermöglicht.
Da Viren keine universellen Merkmale wie eine gemeinsame Zellstruktur besitzen, konzentrieren sich Nachweismethoden auf ihre genetische Information, ihre Struktur oder ihre Wechselwirkungen mit Wirtszellen. Der Nachweisprozess folgt keinem starren Schema, sondern kombiniert verschiedene Schritte, die je nach Fragestellung und Virusart variieren.
In den folgenden Abschnitten werden die zentralen Verfahren zur Virusidentifikation detailliert erläutert – von der Probenentnahme bis hin zur bioinformatischen Analyse.
Vorgehensweisen zur Identifikation von Viren
4.1. Probenentnahme
4.2. Probenaufbereitung
4.2. a) Filtration
4.2. b) Zentrifugation
4.2. c) Präzipitation
4.2. d) Chromatographie
4.3. Zellkultur
4.4. Viren sichtbar machen
4.4. a) Elektronenmikroskopie
4.4. b) Kristallisation
4.4. c) Kryo-Elektronenmikroskopie
4.4. d) Kryo-Elektronentomographie
4.4. e) Zusammenfassung
4.5. Der genetische Fingerabdruck der Viren
4.5.1. Nukleinsäure-Extraktion
4.5.2. Nukleinsäure-Amplifikation
4.5.3. Sequenzierung
4.5.3. a) First Generation: Sanger-Sequenzierung
4.5.3. b) Second Generation: Next-Generation Sequencing (NGS)
4.5.3. c) Third Generation: Echtzeit-Sequenzierung
4.5.3. d) Emerging Technologies: Zukunft der Sequenzierung
4.6. Bioinformatische Analyse4.1. Probenentnahme
Bevor wir Viren auf die Schliche kommen, müssen wir sie erst einmal finden – und das ist wie eine Schatzsuche im Mikroformat, mit Verstecken von Blut bis Tiefseeschlamm. Ob Mensch, Pflanze oder Ozean: Viren verstecken sich überall, und Forscher werden zu Detektiven mit Pipetten, Schutzanzug und Spezialausrüstung.
Abb. 4: Probenentnahme: A) beim Menschen, B) vom Boden, C) aus marinen Ökosystemen [Tara Oceans-Mission] Menschliche Proben
Wer kennt es nicht? Ein Wattestäbchen tief im Nasen-Rachen-Raum – der Klassiker, seit Corona jedem ein Begriff. Doch Viren lauern nicht nur in Schleimhäuten, sondern auch in Blut, Stuhl oder Gewebe. Ähnlich wie ein Dieb DNA-Spuren hinterlässt, verraten sie sich durch ihre genetischen Überreste. Entscheidend ist, dass die Probe sauber entnommen und schnell verarbeitet wird – idealerweise auch möglichst schonend (und schmerzfrei?) für den Patienten. Auch Pflanzen, Tiere oder Insekten liefern Probenmaterial, um Virusvorkommen in ganz unterschiedlichen biologischen Systemen zu untersuchen.Umweltproben
Wasser, Erde, sogar Luft – Viren hängen überall rum. Forschende fischen sie aus Flüssen, buddeln sie aus dem Boden oder saugen sie mit Hightech-Filtern direkt aus der Atmosphäre.Extremkandidaten: Viren unter Extrembedingungen
In der Tiefsee, im ewigen Eis oder in brodelnden Vulkanquellen – Viren zählen zu den härtesten Überlebenskünstlern überhaupt. Um sie dort zu erwischen, braucht es spezielle Sonden, die wie Raumschiff-Fangarme in die Tiefe greifen: „Huch, was schwimmt denn da in 4000 Metern Tiefe? Einfach mal mitnehmen!“Mehr zum Ozean
Viren spielen eine zentrale, oft unterschätzte Rolle für die Gesundheit der Ozeane. Wo Leben existiert, gibt es auch Viren – unsichtbare, aber allgegenwärtige Akteure, die zahlenmäßig jede andere biologische Einheit übertreffen. Sie kontrollieren das Ökosystem ähnlich wie große Raubtiere, indem sie Populationen regulieren und so das ökologische Gleichgewicht bewahren.
Ein herausragendes Forschungsprojekt war die Tara Oceans Expedition (2009–2013). Ziel dieses vierjährigen Projekts war es, das mikrobielle Leben in den Ozeanen und dessen Einfluss auf das globale Ökosystem zu erforschen. Wissenschaftler sammelten weltweit über 35.000 Proben von Plankton, Algen, und Viren.
Im Rahmen der Expedition entdeckte das Forschungsteam über 5.000 neue RNA-Virusarten, darunter die faszinierenden Mirusviren. Diese Entdeckungen erweitern unser Verständnis der Vielfalt, Evolution und Ökologie der Ozeane.
Wenn dich diese Expedition interessiert, schau dir die Tara Oceans Videos an (kurz oder lang).
Transport: Der VIP-Service für Viren
Damit die winzigen Verdächtigen nicht unterwegs kaputtgehen, geht’s für sie nach der Entnahme schnell in die Kühlkette. Tiefkühltransporte und sterile Verpackungen sind Pflicht, sonst zerfällt die „virale Beute“ schneller als ein Eiswürfel in der Sahara.Doch eine gute Probe ist nur der Anfang.
Was wir in Röhrchen, Tupfer oder Tiefkühlbox nach Hause bringen, ist meist ein biologisches Wimmelbild: Zelltrümmer, Bakterien, Proteine – und irgendwo dazwischen ein paar Viren, winzig und verborgen. Jetzt heißt es: sortieren, reinigen, konzentrieren – bevor die eigentliche Analyse überhaupt beginnen kann.
4.2. Probenaufbereitung
In jeder Probe verstecken sich Viren wie Nadeln im mikrobiellen Heuhaufen – der Grund, warum wir putzen müssen, bevor die Virusjagd beginnen kann:
- 99 % Ballast: Zelltrümmer, Proteine, Bakterien
- 1 % Zielobjekt: Winzige Virionen, die wir isolieren wollen
Der Großteil einer biologischen Probe besteht aus nicht-viralem Material – was die gezielte Untersuchung enorm erschwert. Deshalb ist die Probenaufbereitung ein unverzichtbarer Zwischenschritt: Sie entfernt Verunreinigungen, reichert virale Partikel an und bereitet das Material für die eigentliche Analyse vor. Ihr Ablauf variiert je nach Probenart, Analyseziel und gesuchtem Virus.
Probenaufbereitung ist wie Goldwaschen: Man braucht Geduld, das richtige Sieb – und die Hoffnung, dass in jedem Eimer Schlamm ein Nugget glänzt.
Zu den wichtigsten Techniken zählen:
a) Filtration
b) Zentrifugation
c) Präzipitation
d) Chromatographiea) Filtration – Das Mega-Sieb
Die Filtration ist ein erster Reinigungsschritt, bei dem größere Partikel und grobe Verunreinigungen entfernt werden.
So lassen sich virale Partikel gezielt von größeren Strukturen wie Bakterien oder Zellfragmenten trennen.
Funktion: Hält Bakterien & Zellmüll zurück – lässt nur Virionen passieren
Besonderheit: Spezialfilter mit Nanoporen (0,02 µm!)
Cool Fact: Manche Filter laden sich elektrostatisch auf, um Viren besser zu fangenMehr Infos
Die Filtration erfolgt üblicherweise mithilfe spezieller Membranfilter mit definierter Porengröße. Filter mit einer Porengröße von 0,2 Mikrometern werden häufig eingesetzt, da sie Bakterien und größere Partikel zuverlässig zurückhalten, während kleinere virale Partikel hindurchgelangen. Ein Beispiel aus der Praxis ist der Einsatz dieser Methode bei Wasserproben aus Umweltstudien, wo virale Partikel effektiv von anderen Mikroorganismen getrennt werden.
Eine weiterentwickelte Variante ist die Ultrafiltration, die Filter mit noch feineren Porengrößen im Bereich von 0,01 bis 0,1 Mikrometern verwendet. Diese Technik ermöglicht nicht nur die Entfernung von Bakterien, sondern auch die Abtrennung kleinerer Partikel.

Abb. 5-A: Schematische Darstellung des Filtrationsprozesses, die zeigt, wie eine unfiltrierte Probe durch einen Membranfilter geleitet wird. Der Membranfilter hält größere Partikel zurück, während virale Partikel und kleinere Partikel passieren und im Auffangbehälter gesammelt werden. Trotz ihrer Effektivität hat die Filtration einige Einschränkungen:
Reinheit: Kleinste Partikel oder gelöste Substanzen können nach der Filtration weiterhin in der Probe vorhanden sein.
Verstopfung: Insbesondere bei Proben mit hoher Partikelkonzentration kann die Filtermembran blockiert werden, was den Prozess erschwert.
Kapazität: Die Durchflussrate ist durch die Porengröße begrenzt, was die Filtration zeitaufwendig machen kann.Um eine möglichst reine Virusprobe zu erhalten, wird die Filtration häufig mit weiteren Reinigungsschritten wie Zentrifugation oder Chromatographie kombiniert. Diese zusätzlichen Methoden können die verbleibenden Verunreinigungen entfernen und die Qualität der Proben für nachfolgende Analysen optimieren.
b) Zentrifugation – Die Schwerkraft-Turbine

Die Zentrifugation ist eine zentrale Technik zur Trennung von Partikeln basierend auf ihrer Größe und Dichte.
Dieser Prozess nutzt die Zentrifugalkraft, die durch das Drehen der Probe mit hoher Geschwindigkeit erzeugt wird.
Prinzip: Schweres sinkt, Leichtes schwimmt – Viren landen dazwischen
Highspeed: Bis zu 100.000 U/min (eine Waschmaschine schafft 1.200)
Trick: Dichtegradienten trennen sogar Virus-Typen voneinanderMehr Infos
Funktionsweise der Zentrifugation
Wenn eine Lösung über längere Zeit bei Raumtemperatur stehen bleibt, setzen sich schwerere Partikel aufgrund der Schwerkraft langsam ab. Die Zentrifugation beschleunigt diesen Prozess erheblich, indem die Probe mit Geschwindigkeiten von mehreren tausend bis zu 100.000 Umdrehungen pro Minute (bei der Ultrazentrifugation) rotiert. Die extremen Zentrifugalkräfte führen dazu, dass Partikel je nach ihrer Dichte und Größe getrennt werden.

Abb. 5-B: Probe wird in die Zentrifuge gegeben. Um die empfindlichen Moleküle vor Überhitzung zu schützen, sind viele Zentrifugen mit Kühlsystemen ausgestattet, die eine gleichmäßige Temperatur während des gesamten Prozesses gewährleisten.
Typen der Zentrifugation
Differenzielle Zentrifugation
Trennprinzip: Partikel werden basierend auf ihrer Größe und Masse getrennt. Größere und schwerere Partikel setzen sich schneller ab als kleinere und leichtere.
Verfahren: Die Probe wird schrittweise mit steigenden Zentrifugalkräften behandelt. Nach jedem Schritt wird der Überstand (Supernatant), der die verbleibenden kleineren Partikel enthält, vorsichtig in ein neues Röhrchen überführt. Der Boden der ursprünglichen Zentrifugenröhre enthält das Pellet, in dem sich die größeren Partikel abgesetzt haben, und wird entfernt.
Anwendung: Diese Methode wird häufig genutzt, um Zellkomponenten, Organellen und Viren aus komplexen Gemischen zu isolieren.
Ein typisches Beispiel für die Vorbereitung von Proben zur Untersuchung auf unbekannte Viren ist die Gewinnung eines Zelllysats. Hierbei werden Zellen, die aus Gewebeproben oder Abstrichen isoliert wurden, durch chemische, physikalische oder enzymatische Verfahren aufgeschlossen. Dieser Schritt setzt virale Partikel frei, die innerhalb der Wirtszellen repliziert wurden. Anschließend wird das Zelllysat durch Zentrifugation oder Filtration gereinigt, um virale Komponenten von zellulären Trümmern zu trennen. Zelllysate sind besonders nützlich, wenn der Verdacht besteht, dass Viren in spezifischen Zelltypen oder Geweben verborgen sind.

Abb. 5-C: Differenzielle Zentrifugation Dichtegradientenzentrifugation
Trennprinzip: Partikel werden nach ihrer Dichte getrennt. Die Probe wird auf einen Dichtegradienten (meist Saccharose oder Cäsiumchlorid) aufgetragen und zentrifugiert. Während der Rotation wandern die Partikel zu der Position im Gradient, die ihrer eigenen Dichte entspricht.
Verfahren: Die Partikel kommen zur Ruhe an der Stelle, an der ihre Dichte mit der des Mediums übereinstimmt, was eine präzise Trennung ermöglicht.
Anwendung: Diese Methode eignet sich hervorragend, um Viren, Proteine und Nukleinsäuren aus komplexen Gemischen zu isolieren und zu analysieren.
Entnahme der viralen Fraktion
Nach der Zentrifugation wird die virale Fraktion typischerweise als eine klar abgegrenzte Schicht innerhalb der Zentrifugenröhre sichtbar. Diese Schicht kann vorsichtig mithilfe einer Pipette entnommen werden, um die Viren für weitere Analysen bereitzustellen. Die genaue Position der viralen Schicht hängt dabei von der genutzten Methode und den physikalischen Eigenschaften der Viren ab.
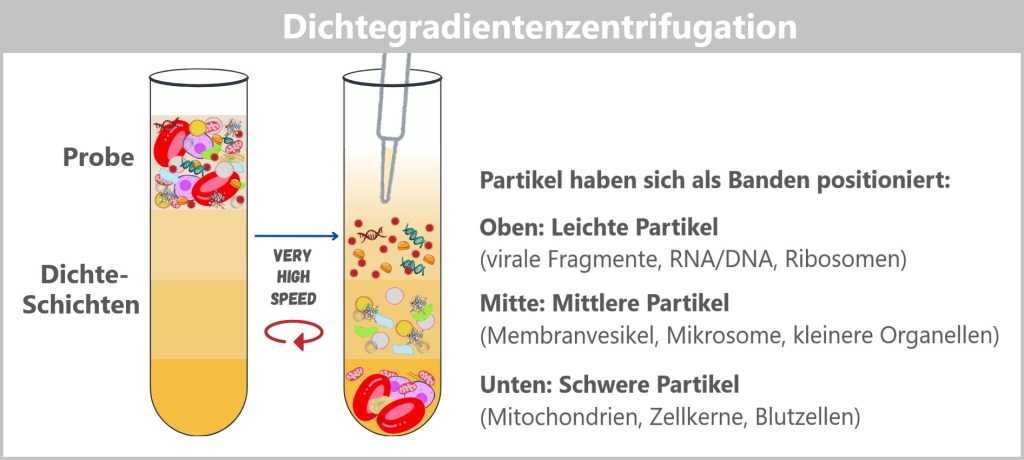
Abb. 5-D: Dichtegradientenzentrifugation In den meisten Fällen wird die Zentrifuge auf eine konstante Geschwindigkeit eingestellt, die ausreichend ist, um die Partikel im Dichtegradienten zu trennen. Diese Geschwindigkeit ist typischerweise sehr hoch (z. B. bei der Ultrazentrifugation bis zu 100.000 U/min), da die Trennung im Dichtegradienten von der ausreichenden Zentrifugalkraft abhängt.
Kombination der Methoden
Oft werden differenzielle und Dichtegradientenzentrifugation kombiniert, um eine höhere Reinheit und Präzision bei der Trennung der Partikel zu erreichen. Diese Methoden sind essenziell für die biomedizinische Forschung und Diagnostik, da sie eine präzise Trennung und Konzentration viraler Partikel ermöglichen.
c) Präzipitation – Der Chemie-Trick
Die Präzipitation (Fällung) ist eine bewährte Methode zur Isolierung und Reinigung von Viruspartikeln aus einer Lösung.
Dabei werden chemische Substanzen hinzugefügt, die die Löslichkeit der Viren herabsetzen und sie so aus der Lösung ausfällen lassen.
So geht’s: Chem. Substanzen machen Viren schwer & klebrig → sie fallen aus
Vorteil: Billig, simpel – aber nichts für empfindliche VirenMehr Infos
Der Vorgang umfasst mehrere Schritte und nutzt spezifische chemische Reaktionen, um die Viruspartikel effektiv zu konzentrieren.
Ablauf der Präzipitation
Vorbereitung der Probe: Vor der Präzipitation wird die Probe oft vorgereinigt, etwa durch Ultrafiltration oder Ultrazentrifugation, um größere Partikel und Verunreinigungen zu entfernen. Dies verbessert die Effizienz der Präzipitation.
Zugabe einer Fällungschemikalie: Eine Chemikalie wie Polyethylenglykol (PEG), Ethanol oder Ammoniumsulfat wird der Lösung zugeführt. Diese Stoffe verändern die chemischen Eigenschaften der Flüssigkeit, indem sie die Löslichkeit der Viruspartikel verringern.
Aussalzen: Beim Aussalzen konkurrieren die zugesetzten Salzionen oder Moleküle mit den Viruspartikeln um die verfügbaren Wassermoleküle. Da weniger Wassermoleküle für die Viren verfügbar sind, nimmt deren Löslichkeit ab, und sie beginnen, sich zusammenzuschließen. Dadurch entsteht ein sichtbarer Niederschlag, der als Fällung bezeichnet wird.Die Oberflächenladungen und die biochemischen Eigenschaften wie Größe und Dichte der Viruspartikel begünstigen ihre Fällung im Vergleich zu anderen Partikeln in der Lösung.
Zentrifugation (in der Abbildung: ZFG): Um die gefällten Viruspartikel zu konzentrieren, wird die Probe zentrifugiert. Dies beschleunigt das Absetzen der Viren, die sich als fester Niederschlag am Boden des Gefäßes sammeln.
Waschen: Das Pellet wird mit einem Puffer oder einer Lösung gewaschen, um verbleibendes PEG, Salze oder andere Verunreinigungen zu entfernen.
Ergebnis: Die Lösung enthält nun hauptsächlich die isolierten Viren, die für nachfolgende Analysen verwendet werden können.
Abb. 5-E: Präzipitation Nach der Zentrifugation befindet sich das Viruspellet am Boden des Reagenzglases, während der Überstand (Supernatant) oben liegt. Der Überstand wird vorsichtig abgesaugt oder abgepipettiert, um das Pellet nicht zu stören. Eine Waschlösung, meist ein Puffer wie PBS (phosphatgepufferte Salzlösung) oder ein ähnliches Medium, wird zum Pellet hinzugegeben. Das Volumen der Waschlösung ist in der Regel vergleichbar mit dem ursprünglichen Volumen der Probe, um genügend Verunreinigungen zu entfernen. Das Pellet wird durch sanftes Pipettieren, Vortexen (leichtes Rühren mit einem Vortexgerät) oder Umkippen des Reagenzglases resuspendiert, sodass es sich in der Waschlösung verteilt. Dieser Schritt sorgt dafür, dass verbleibende Verunreinigungen in die Waschlösung übergehen können. Nach dem Mischen wird das Reagenzglas erneut zentrifugiert. Dabei setzen sich die Viruspartikel wieder als Pellet ab. Die Waschlösung (Supernatant) wird vorsichtig entfernt, ohne das Pellet zu stören. Dieser Waschschritt wird je nach Bedarf ein- bis dreimal wiederholt, um sicherzustellen, dass möglichst viele Verunreinigungen entfernt werden. Nach dem letzten Waschschritt wird das Viruspellet in einer kleinen Menge Puffer (z. B. PBS oder einem Analysepuffer) resuspendiert, um es für nachfolgende Analysen vorzubereiten.
Vorteile der Präzipitation
Breit einsetzbar: Für viele Virenarten gut geeignet. Bei sehr empfindlichen Proben allerdings mit Vorsicht zu genießen: Vor allem behüllte Viren können durch osmotischen Stress oder unspezifische Aggregation geschädigt werden.
Hohe Ausbeute: Präzipitation ermöglicht eine effektive Konzentration und Reinigung mit minimalem Verlust an Material.
Einfache Anwendung: Der Prozess ist kostengünstig und erfordert keine hochkomplexen Geräte.Die Präzipitation eignet sich besonders, um große Mengen an Viren effizient zu isolieren.
d) Chromatographie – Die VIP-Lounge für Viren
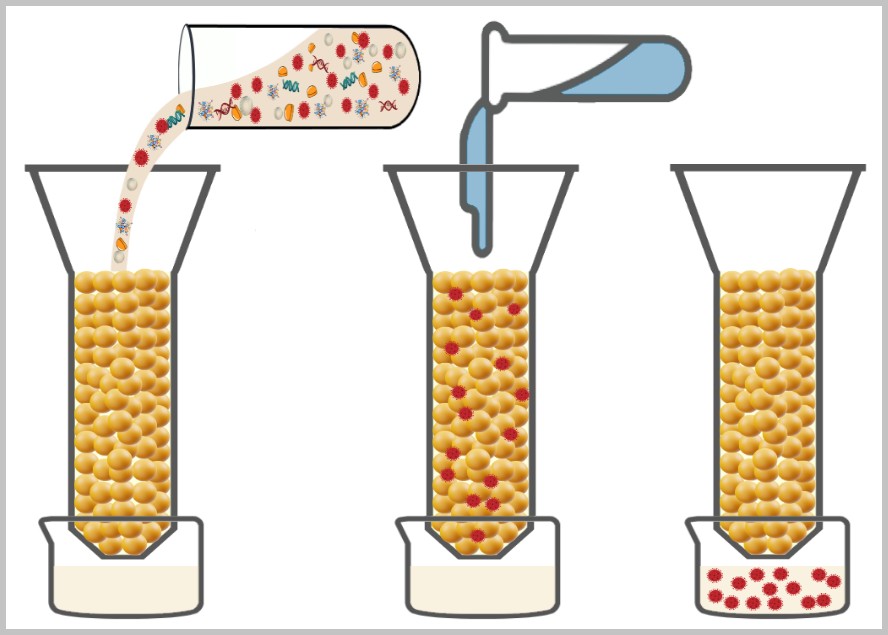
Die Chromatographie ist ein hochpräziser Reinigungsschritt in der Virusaufbereitung.
Dabei werden virale Partikel anhand ihrer spezifischen physikalischen oder chemischen Eigenschaften von anderen Bestandteilen der Probe getrennt.
So geht‘s: Säulen trennen Viren nach Größe, Ladung oder Affinität
Vorteil: Besonders schonend, präzise und effizientMehr Infos
Prinzip der Chromatographie
Die Chromatographie ist eine Methode zur Trennung verschiedener Stoffe in einem Gemisch. Ganz allgemein gibt es in diesem Verfahren eine mobile (bewegliche) Phase und eine stationäre (unbewegliche) Phase. Die mobile Phase ist die Stoffmischung, die getrennt werden soll. Sie bewegt sich durch die stationäre Phase, die als Matrix bezeichnet wird. Die Stoffe in der mobilen Phase interagieren mit den Stoffen in der stationären Phase. Durch die Wechselwirkungen bewegen sich die einzelnen Komponenten im Stoffgemisch unterschiedlich schnell oder bleiben stecken. Somit können die verschiedenen Stoffe voneinander getrennt werden.Es gibt verschiedene Arten der Chromatographie. Eine häufig verwendete Variante ist die Ionenaustauschchromatographie, die auf den elektrischen Ladungen der Partikel basiert. Diese Methode eignet sich hervorragend zur Reinigung viraler Partikel, da Viren typischerweise eine definierte Oberflächenladung haben.
Ablauf der Ionenaustauschchromatographie (IEC)
Kontrolle der Virenladung:
Der isoelektrische Punkt (IEP) eines Virus gibt den pH-Wert an, bei dem das Virus elektrisch neutral ist. Durch Anpassung des pH-Werts der Lösung kann die Ladung des Virus gezielt beeinflusst werden:- Bei einem pH-Wert unter dem IEP ist das Virus positiv geladen.
- Bei einem pH-Wert über dem IEP ist das Virus negativ geladen.
Auswahl der Matrix:
Die Matrix wird so gewählt, dass sie eine entgegengesetzte Ladung zur Virenladung besitzt, um eine Bindung zu ermöglichen:- Anionenaustauscher: Positiv geladene Matrix, um negativ geladene Partikel zu binden.
- Kationenaustauscher: Negativ geladene Matrix, um positiv geladene Partikel zu binden.
Typischerweise besteht die Matrix aus Harzen oder Membranen, die eine spezifische Ladung tragen.
Bindung der Viren:
Die Probe wird auf die Chromatographiesäule aufgetragen, und die Lösung fließt durch die Matrix. Die Viren binden an die Matrix aufgrund elektrostatischer Wechselwirkungen. Andere Moleküle, wie Nukleinsäuren oder Proteine, binden schwächer oder passieren die Säule unverändert.Waschen:
Mit einem Waschpuffer werden ungebundene oder schwach gebundene Verunreinigungen aus der Matrix entfernt. Dieser Schritt verbessert die Reinheit der gebundenen Viruspartikel.Elution (Freisetzung der Viren):
Um die gebundenen Viren von der Matrix zu lösen, wird ein Elutionspuffer hinzugefügt. Dies kann auf zwei Wegen geschehen:- Änderung des pH-Werts: Der pH-Wert wird so eingestellt, dass die Ladung der Viruspartikel aufgehoben wird, wodurch die Bindung zur Matrix verloren geht.
- Erhöhung der Salzkonzentration: Zusätze wie Natriumchlorid oder Magnesiumchlorid neutralisieren die elektrostatische Bindung zwischen Viren und Matrix.
Ergebnis:
Die eluierten Viruspartikel werden gesammelt. Da Verunreinigungen bereits während der Waschschritte entfernt wurden, enthält die elutierte Fraktion hauptsächlich die gereinigten Viren, die für weitere Analysen oder Anwendungen genutzt werden können.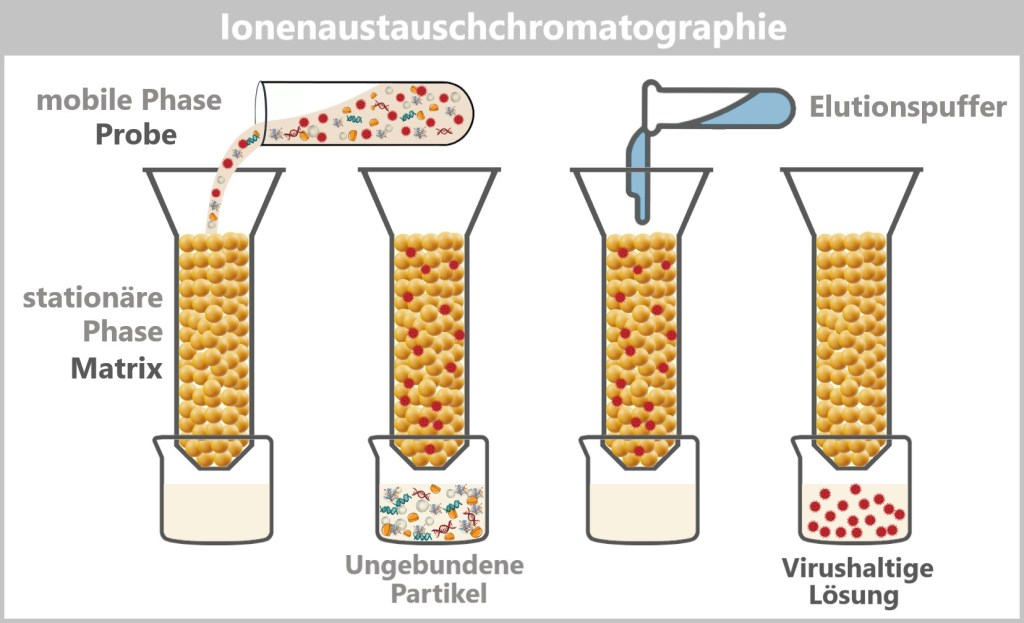
Abb. 5-F: Ionenaustauschchromatographie Weitere Varianten der Chromatographie
Neben der Ionenaustauschchromatographie gibt es andere Varianten, die je nach Ziel und Probe eingesetzt werden:Affinitätschromatographie: Nutzt spezifische Bindungen zwischen Virusproteinen und Liganden auf der Matrix.
Größenausschlusschromatographie: Trennt Partikel nach ihrer Größe.
Hydrophobe Interaktionschromatographie: Nutzt die hydrophoben Eigenschaften der Partikel.
In der Praxis werden diese Verfahren oft kombiniert. Beispielsweise könnte eine Probe zunächst filtriert, anschließend zentrifugiert und danach durch Präzipitation und Chromatographie weiter aufgereinigt werden. Die Auswahl und Reihenfolge hängen dabei immer von der Art der Probe und dem Ziel der Analyse ab.
Warum dieser Aufwand?
Ganz einfach: Eine schlechte Probe bringt schlechte Daten – wie ein verwackeltes Foto von Bigfoot. Deshalb: sauber arbeiten, gut kühlen und schnell sein … denn Moleküle verzeihen keine Nachlässigkeit.
Die Probenaufbereitung ist kein Nebenjob – sie ist das Fundament. Ohne sie fällt alles Weitere in sich zusammen. Und wenn das virale Material einmal gut vorbereitet ist, geht es ans Eingemachte: Jetzt müssen sich die Viren beweisen – in der Zellkultur.
4.3. Zellkultur
Viren sind die ultimativen Schmarotzer: Ohne Wirtszelle läuft bei ihnen gar nichts – kein Leben, keine Vermehrung. Alleine – völlig hilflos. Aber gib ihnen eine lebende Zelle, dann geht’s ab. Deshalb braucht die Virologie ein verlässliches Werkzeug: die Zellkultur. Ohne sie döst selbst das gefährlichste Virus vor sich hin wie ein Büroangestellter im Homeoffice.
Und weil Viren nicht gern allein sind, richten Virologen ihnen ein kuscheliges Zuhause ein – eine Luxus-WG im Miniaturformat: sterile Petrischalen, perfekte Temperatur, ein Nährmedium voller Vitamine und Zucker – alles, was das Virusherz begehrt. Die verwendeten Zellen stammen von Menschen, Tieren oder Pflanzen – je nachdem, welchen Virus man gerade verwöhnen will.
Doch nicht jeder Virus ist pflegeleicht. Manche sind anspruchsvolle Diva-Typen, die nur in bestimmten Zelllinien gedeihen. Andere machen’s in allem, was „Help!“ schreit. Und manchmal? Passiert einfach… nichts. Dann heißt es: neue Zellen, neues Glück.
Zellkulturen sind weit mehr als eine Virenzuchtstation. Sie ermöglichen:
➤ die Vermehrung von Viren in kontrollierter Umgebung,
➤ die Analyse ihrer Eigenschaften und
➤ den Nachweis infektiöser Erreger – besonders bei neuen oder unbekannten Viren.Warum Zellkulturen trotz PCR absolut cool sind
PCR-Tests sind schnell, billig und überall verfügbar – wie das Fast Food der Diagnostik. Aber sie können nur sagen: „Ja, hier war mal Virus!“ Ob das Virus aber noch lebendig und infektiös ist? Keine Ahnung!
Zellkultur = Realitätstest.
Kann das Virus Zellen infizieren und sich darin vermehren? Ja? Dann haben wir es mit einem echten Erreger zu tun. Nein? Dann bleibt’s bei genetischem Geistermüll.Zellkultur als Fitness-Test: unverzichtbar, wenn man:
➤ die Infektiosität überprüfen will,
➤ neue Viren testet, deren Gefährlichkeit noch unklar ist.
Viren müssen erst mal beweisen, dass sie was können!Auch in Zeiten von PCR und Hochdurchsatzsequenzierung (dazu später mehr) bleibt die Zellkultur der Praxistest nach der Theorieprüfung: „Alles schön und gut – aber funktioniert’s auch?“
Denn nur wer Zellen infizieren kann, hat echtes Gefahrenpotenzial. Die Zellkultur trennt die Spreu (harmlose Gensequenzen) vom Weizen (aktive, infektiöse Viren). Gerade in Zeiten von synthetischer Biologie wird das entscheidend: Nur weil die Gensequenz korrekt aussieht, heißt das noch lange nicht, dass das Virus auch funktioniert.
Infektion und Beobachtung der Zellkultur
Vorbereitung der Zellkultur
Zellen werden in einem Nährmedium kultiviert, das essentielle Nährstoffe, Wachstumsfaktoren und einen geeigneten pH-Wert enthält. Diese Umgebung ermöglicht das Überleben und die Vermehrung der Zellen. Es werden häufig Zelllinien verwendet, die eine kontinuierliche Vermehrung ermöglichen, wie beispielsweise Vero-Zellen (aus Nierenepithelzellen von Affen) oder HeLa-Zellen (menschliche Krebszellen).
Die Wahl der geeigneten Wirtszellen hängt entweder von ersten Hinweisen aus mikroskopischen Beobachtungen ab oder erfolgt durch Tests mit verschiedenen Zelllinien. Forscher verwenden dabei häufig Zelllinien, die dafür bekannt sind, Viren aus spezifischen Probenquellen zu unterstützen. Beispielsweise werden für Ozeanproben oft Zelllinien von Meeresorganismen genutzt, während bei der Untersuchung von Atemwegsviren menschliche Epithelzellen bevorzugt werden.
Infektion der Zellkultur
Um Viren in einer Probe nachzuweisen, wird diese auf eine geeignete Zellkultur aufgebracht. Enthält die Probe Viren, können diese in die Zellen eindringen, deren zelluläre Maschinerie zur Vermehrung nutzen und charakteristische Veränderungen hervorrufen, die als zytopathische Effekte (CPE) bezeichnet werden. Wie solche Zellveränderungen durch Virusinfektionen entstehen können, zeigt Kapitel „2.8. Zerstörung der Wirtszelle“ anschaulich.
Nachweis und Beobachtung:
Nach einer gewissen Zeit können die infizierten Zellen unter einem Licht- oder Elektronenmikroskop auf zytopathische Effekte (charakteristische Veränderungen) untersucht werden. Diese Effekte dienen als sichtbare Hinweise auf eine erfolgreiche Infektion:
➤ Zellschrumpfung oder -verklumpung
➤ Bildung von Synzytien (mehrkernige Zellverbände)
➤ Zelllyse (Auflösung der Zellen)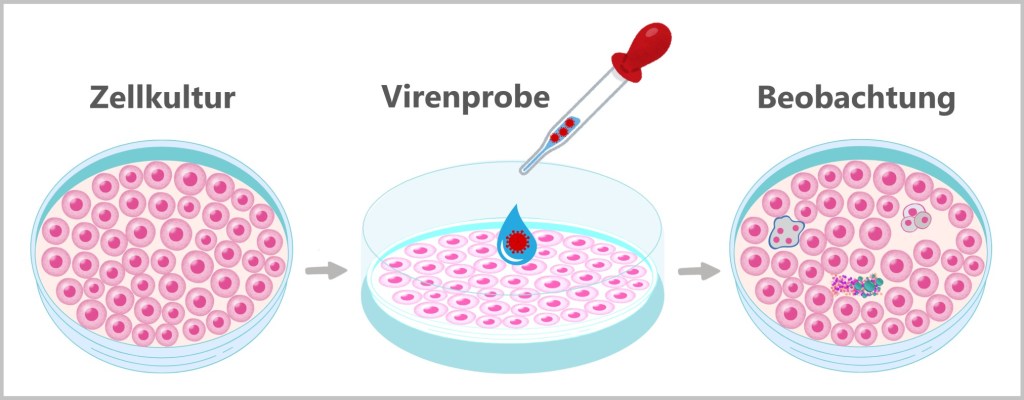
Abb. 6: Infektion der Zellkultur und deren Beobachtung 🎥 Live im Zeitraffer: So breitet sich das Influenzavirus in der Zellkultur aus
Bezug zum Influenzavirus
Studie: „Erkennung virusbedingter Zellveränderungen durch Influenza mit Hilfe künstlicher Intelligenz (CNN)“
Die Arbeit präsentiert eine umfangreiche Sammlung von Bildern, die die typischen zytopathischen Effekte (CPE) nach einer Infektion mit dem Influenzavirus zeigen. Diese visuellen Darstellungen verdeutlichen die charakteristischen Zellveränderungen, die durch das Virus verursacht werden. Allerdings ist die manuelle Beobachtung und Auswertung dieser Effekte äußerst arbeits- und zeitintensiv. Um diesen Prozess zu optimieren, erprobten die Autoren den Einsatz Künstlicher Intelligenz (KI), genauer gesagt tiefer Convolutional Neural Networks (CNNs), zur automatisierten Bildauswertung.
Diese Studie zeigt eindrucksvoll, wie moderne Methoden wie die Virologie, Zellbiologie und KI-Technologie ineinandergreifen, um innovative Lösungen für komplexe wissenschaftliche Fragestellungen zu entwickeln.
Rolle und Anwendungen der Zellkultur in der Virologie
Diagnose bekannter Viren: Zellkulturen werden eingesetzt, um Viren zu vermehren und ihre Präsenz durch spezifische Veränderungen in den Zellen nachzuweisen.
Nachweis unbekannter Viren: Wenn die genetische oder antigene Struktur eines Virus nicht bekannt ist, können Zellkulturen dazu beitragen, dessen biologische Aktivität zu charakterisieren.
Erforschung des viralen Lebenszyklus: Zellkulturen ermöglichen die Untersuchung der Interaktionen von Viren mit Wirtszellen, einschließlich des Eindringens, der Replikation und der Freisetzung neuer Viruspartikel.
Studien zur Virulenz und Pathogenität: Sie helfen, die Mechanismen zu verstehen, mit denen Viren Zellen schädigen und Infektionen auslösen.
Analyse der Immunantwort: Zellkulturen unterstützen die Untersuchung, wie das Immunsystem auf virale Infektionen reagiert und wie infizierte Zellen Immunreaktionen auslösen.
Testung antiviraler Medikamente: Zellkulturen dienen dazu, die Wirksamkeit und Verträglichkeit potenzieller antiviraler Substanzen zu prüfen und neue Therapien zu entwickeln.
Impfstoffproduktion: Viele Impfstoffe, wie beispielsweise der gegen Masern, werden in Zellkulturen hergestellt.
Herstellung von Virusvektoren: Zellkulturen sind unerlässlich für die Produktion von Virusvektoren – gentechnisch veränderten Viren, die therapeutische Gene oder Impfstoffinformationen in Zellen einschleusen, etwa in der Gentherapie, der Krebsbehandlung oder bei Impfstoffen wie denen gegen Ebola oder COVID-19 (z. B. AstraZeneca).
Einschränkungen der Methode
Nicht alle Viren sind kultivierbar: Einige Viren, wie Hepatitis B, benötigen spezialisierte Zellen oder Systeme, um in vitro vermehrt zu werden.
Viren mit sehr engen Wirtsspezifitäten: Manche Viren sind auf sehr spezifische Wirtszellen angewiesen, die in Zellkulturen nur schwer reproduziert werden können, was die Forschung erschwert.
Unzureichende Repräsentation von in-vivo-Umgebungen: Zellkulturen bieten nur eine vereinfachte Version des natürlichen Wirts, wodurch wichtige virus-spezifische Reaktionen fehlen könnten.
Laborangepasste Viren vs. natürliche Viren: Viren, die über viele Passagen in Zellkulturen gezüchtet werden, können Mutationen entwickeln, die ihre Eigenschaften verändern. Dadurch können sie sich von ihren natürlichen Varianten unterscheiden – etwa durch weniger Spike-Proteine oder eine veränderte Wirtsspezifität.
Limitierte Reproduzierbarkeit von Ergebnissen: Die Zellkulturbedingungen können das Verhalten der Zellen beeinflussen, was zu Schwankungen in den Ergebnissen führt.
Zeitaufwändig: Der Nachweis eines Virus über Zellkultur ist im Vergleich zu molekularen Methoden langsamer.
Komplexität und Kosten: Die Herstellung und Pflege von Zellkulturen erfordert spezialisierte Laborausrüstung und Expertise.
Biosicherheitsrisiken: Bei der Arbeit mit hochpathogenen Viren müssen hohe Sicherheitsstandards eingehalten werden, was zusätzliche Anforderungen an die Labore und die Forscher stellt.
Isolierung und Weiterverarbeitung
Zellkulturen sind das All-inclusive-Resorts für Viren – hier können sie sich ungestört vermehren. Aber wie immer im Leben: Alles Schöne hat auch mal ein Ende. Für die Winzlinge heißt das: „Check-out bitte!“ Die Reise geht weiter zu den High-Tech-Labs. Jetzt wird’s ernst – und spannend. Denn jetzt wollen wir wissen: Was genau da gewachsen ist. Und wie es aussieht. Willkommen bei den Tools der Virusanalyse:
Selfie mit dem Elektronenmikroskop: Will ein Virus wissen, wie es wirklich aussieht? Die Elektronenmikroskopie verpasst ihm den ultimativen Close-up – schärfer als jeder Instagram-Filter.
PCR und Sequenzierung: Der genetische Persönlichkeitstest: Ein bisschen Erbgut hier, ein paar Enzyme dort – und schon wird klar, wer (oder was) da eigentlich im Reagenzglas sitzt.
ELISA: Der Antikörper-Check: Dieser Test schnüffelt nach Antikörpern im Blut – und verrät, ob das Immunsystem schon Alarm geschlagen hat. Verdächtiger Fund? Fall abgeschlossen!
In den nächsten Kapiteln zerlegen wir einige dieser Methoden – ganz ohne Laborkittel.
4.4. Viren sichtbar machen
Viren sind wie Geister: Man spürt ihre Wirkung, aber sieht sie nie. Sie hinterlassen Chaos in Zellen, schreiben sich in unsere DNA ein, verursachen Krankheiten, prägen die Evolution und spielen seit Jahrhunderten Hide-and-Seek mit der Wissenschaft. Selbst unter dem besten Lichtmikroskop bleiben sie unsichtbar.
Doch wie heißt es so schön: Seeing is believing. Aber wie macht man das Unsichtbare sichtbar? Wie lassen sich Viren zweifelsfrei nachweisen? Und wie entschlüsseln wir ihre Strukturen und Mechanismen?
Die Antwort: Wir holen die ganz großen Mikroskope raus! Geräte, so leistungsstark, dass sie selbst die trickreichsten Viren in flagranti erwischen und keine Tarnung mehr hilft. Moderne bildgebende Verfahren zerlegen Viren heute bis ins atomare Detail.
Willkommen im Reich des Sichtbarmachens der Moderne – wo das Unsichtbare endlich Gestalt annimmt! Und los geht’s mit …
a) Elektronenmikroskopie
b) Kristallisation
c) Kryo-Elektronenmikroskopie
d) Kryo-Elektronentomographie
e) Zusammenfassunga) Elektronenmikroskopie: Erste Bilder von Viren
Die Geschichte der Virologie begann mit der Erkenntnis, dass es etwas Kleineres als Bakterien geben muss, das Krankheiten verursacht.
Der Physiker Richard Feynman brachte es 1959 auf den Punkt:
„It is very easy to answer many… fundamental biological questions; you just look at the thing!“
„Es ist sehr einfach, viele grundlegende biologische Fragen zu beantworten; man muss sich die Sache nur anschauen!“
Doch wie kann man etwas sichtbar machen, das weit unterhalb der Auflösungsgrenze eines herkömmlichen Mikroskops liegt?
Mit der Erfindung des Elektronenmikroskops (EM) im Jahr 1931 wurde genau das erstmals möglich: Viren konnten direkt gesehen werden. Die Methode nutzt Elektronenstrahlen statt Licht, um extrem kleine Strukturen sichtbar zu machen. Wissenschaftler konnten erstmals die charakteristischen Hüllen und Formen von Viren abbilden und damit ihre physische Existenz bestätigen.

Abb. 7-A: Ein Blick durch die Mikroskope – eine schematische Darstellung Die Illustration zeigt die Unterschiede in der Sichtbarmachung biologischer Strukturen durch zwei fundamentale Technologien der Mikroskopie.
Auf der linken Seite repräsentiert der Blick durch das Lichtmikroskop die klassischen Möglichkeiten, wie sie seit dem 17. Jahrhundert genutzt werden. Hier sind größere Strukturen wie menschliche Zellen gut erkennbar, ebenso Bakterien, die durch Färbetechniken sichtbar gemacht werden können. Viren hingegen bleiben unsichtbar, da ihre Größe (80–120 nm für Influenzaviren) weit unterhalb der Auflösungsgrenze eines Lichtmikroskops liegt.
Auf der rechten Seite zeigt der Blick durch das Elektronenmikroskop, wie moderne Technologien die Grenzen der Sichtbarmachung überwunden haben. Hier sind nicht nur menschliche Zellen und Bakterien in viel höherer Detailtiefe sichtbar, sondern auch Viren. Die Darstellung geht sogar so weit, dass Details wie die virale Hülle und das Genom in hochauflösenden Bildern sichtbar gemacht werden können.
Lichtmikroskope bieten einen allgemeinen Überblick und ermöglichen die Analyse lebender Zellen, während Elektronenmikroskope tiefere Einblicke in die Welt der Mikroorganismen und Viren gewähren – bis hin zu molekularen Details.Das folgende Video illustriert den Unterschied zwischen Lichtmikroskopen und Elektronenmikroskopen sowie deren Funktionsweise auf anschauliche Weise.
Wie funktioniert Elektronenmikroskopie?
Elektronenmikroskope haben eine weitaus höhere Auflösung als Lichtmikroskope, da Elektronen eine viel kürzere Wellenlänge als Licht besitzen. Dies ermöglicht die Visualisierung von Strukturen im Nanometerbereich. Man unterscheidet dabei zwei Haupttypen:
1️⃣ Transmissionselektronenmikroskopie
(TEM – Transmission Electron Microscopy)Ein Elektronenstrahl durchdringt eine extrem dünne Probe.
Dabei entstehen hochaufgelöste 2D-Bilder der inneren Strukturen.
(Früher: Kontrastarme Flecken, Heute: Bis zu 0,05 nm Auflösung)
Besonders nützlich für feine Details im Inneren von Viren.2️⃣ Rasterelektronenmikroskopie
(REM/SEM – Scanning Electron Microscopy)Der Elektronenstrahl tastet die Oberfläche einer Probe ab.
Dadurch entstehen detaillierte 3D-Bilder der Probenoberfläche.
(Früher: Grobe Umrisse, Heute: Molekülschärfe)
Gut geeignet für die äußere Struktur von Viren.Abb. 7-B: Transmissionselektronenmikroskopie vs Rasterelektronenmikroskopie
(schematische Darstellung)Transmission Electron Microscopy: Zeigt eine Durchsicht des Virus, sodass auch innere Strukturen sichtbar sein können. Das Virus erscheint meist als zweidimensionale Projektion mit feinen Details im Inneren. Die Elektronen durchdringen die Probe, und der Kontrast entsteht durch Wechselwirkungen mit unterschiedlichen Dichten der Zell- oder Virusbestandteile.
Scanning Electron Microscopy: Zeigt eine dreidimensionale Oberfläche, oft mit einem plastischen, reliefartigen Effekt. Die Probe wird mit Elektronen abgetastet, wodurch ein tiefenscharfes Oberflächenbild entsteht. Innere Strukturen sind nicht sichtbar, da die Elektronen nicht durch die Probe hindurchgehen.Ein originales TEM-Bild eines Influenzaviruspartikels kannst du hier sehen.
Weitere Infos zum Einsatz der Elektronenmikroskopie
Mithilfe der Elektronenmikroskopie (EM) lassen sich unterschiedliche Aspekte des Influenzavirus untersuchen:
🔬 Einblicke in die Ultrastruktur des Influenzavirus
Mithilfe der Transmissionselektronenmikroskopie (TEM) mit negativer Färbung konnten Wissenschaftler detaillierte Einblicke in die Ultrastruktur des Influenzavirus gewinnen. Die Studie zeigt die Anordnung der Spike-Proteine (Hämagglutinin und Neuraminidase) auf der Virusoberfläche sowie die interne Struktur, einschließlich des Matrixproteins (M1) und der RNA, und beleuchtet Unterschiede zwischen intakten und beschädigten Viruspartikeln.🔬 Einblicke in die Struktur der Ribonukleoprotein-Komplexe (vRNPs)
Mittels EM konnten Wissenschaftler die helikale Anordnung der vRNPs im Viruspartikel sichtbar machen. Der Artikel von Noda und Kawaoka (2010) beschreibt detailliert die Architektur der vRNPs innerhalb des Viruspartikels und deren Verpackung in Virionen. Sie betonen, dass jede der acht vRNA-Segmente mit Nukleoproteinen und einem Polymerase-Komplex assoziiert ist, wodurch die vRNPs gebildet werden, die für die Transkription und Replikation des viralen Genoms essenziell sind. Elektronenmikroskopische Analysen haben gezeigt, dass diese vRNPs eine helikale Struktur aufweisen und innerhalb des Virions spezifisch angeordnet sind.
Elektronenmikroskopie in der Virologie
Dank der EM lassen sich virale Strukturen und Mechanismen aufdecken, darunter:
✅ die Form und Größe von Viren (z. B. Kugelform des Influenzavirus)
✅ die Anordnung der Spike-Proteine auf der Virushülle
✅ und Einblicke in den Infektionszyklus, z. B.:
• wie sie in die Wirtszelle eindringen
• wie die Replikation eingeleitet wird
Grenzen der Elektronenmikroskopie
Trotz ihrer beeindruckenden Auflösung hat die EM einige Nachteile:
⚡Probenvorbereitung → Proben müssen häufig entwässert, geschnitten und beschichtet werden, was ihre natürliche Struktur beeinflussen kann.
⚡Proben müssen fixiert werden → Das bedeutet, dass die Probe nicht in ihrem natürlichen Zustand untersucht wird.
⚡Keine lebende Probe → da die Proben im Vakuum analysiert werden, um eine Streuung der Elektronenstrahlen in der Luft zu vermeiden.
⚡Strahlenschäden → Der Elektronenstrahl kann empfindliche Proben verändern oder zerstören.
⚡Fehlender Kontrast → Biologische Proben sind oft kontrastarm und müssen speziell gefärbt werden.
⚡Kein „echtes“ 3D-Bild → TEM-Bilder sind nur zweidimensional, wodurch komplexe Strukturen schwer zu rekonstruieren sind.
Die Elektronenmikroskopie war der erste große Durchbruch: Endlich konnte man Viren sehen. Doch die frühen EM-Bilder wirkten noch verschwommen – wie Mondfotos aus den 1960ern. Um den Viren noch näher zu kommen, brauchte es mehr als ein Mikroskop. Man brauchte atomare Klarheit. Und so kam die Kristallisation ins Spiel – die Ultra-HD-Version der Virenforschung. Denn erst wenn sich Virusproteine in perfekte Kristalle zwängen, verraten sie ihr molekulares Innenleben: Atom für Atom, Bindung für Bindung.
b) Kristallographie: Detaillierte Strukturen von Virusproteinen
Während die Elektronenmikroskopie die grobe Struktur von Viren sichtbar machen konnte, blieb ihre molekulare Feinstruktur – ihre atomaren Details – lange im Verborgenen. Um zu verstehen, wie virale Proteine aufgebaut sind, musste man ihre Atomstruktur bestimmen – und das war nur mit Röntgenkristallographie möglich.
Wie kam es dazu?
Die Idee, biologische Makromoleküle mittels Röntgenbeugung zu analysieren, entwickelte sich in den 1920er und 1930er Jahren. Ein bahnbrechender Durchbruch gelang dem Physiker John Desmond Bernal, der 1934 zusammen mit Dorothy Crowfoot Hodgkin nachwies, dass Proteine in hydratisierter Form kristallisiert werden können, ohne ihre natürliche Struktur zu verlieren. Diese Entdeckung war entscheidend, um die Röntgenkristallographie für die Analyse von Biomolekülen nutzbar zu machen.
In den 1930er Jahren gelang es Wendell Meredith Stanley, das Tabakmosaikvirus in kristalliner Form zu isolieren. Dies war bedeutsam, weil es bewies, dass Viren nicht nur materielle Partikel sind, sondern auch aus regelmäßig angeordneten Molekülen bestehen, die sich kristallisieren lassen. Stanley brachte das Virus in eine feste, hochgeordnete Struktur – vergleichbar mit Salz- oder Zuckerkristallen. Diese Kristalle ermöglichten es später, mit Röntgenstrahlen die atomare Struktur von Virusproteinen zu entschlüsseln – ein entscheidender Fortschritt für die Virologie.
Stanley‘s Durchbruch und dessen Bedeutung
Wie hat Stanley die Kristallisation durchgeführt?
Stanley entwickelte eine innovative Methode, um die winzigen Viruspartikel zu isolieren und zu analysieren. Er extrahierte die Viren aus infizierten Tabakpflanzen und reinigte sie durch Zentrifugation sowie weitere Aufreinigungstechniken. Diese gereinigten Viren löste er anschließend in einer Lösung, die er langsam verdampfen ließ. Durch diesen Prozess kristallisierten die Viren aus, und es entstanden feste Strukturen, die er mit einem Elektronenmikroskop genauer untersuchen konnte. Dies war der erste Schritt, um die Struktur von Viren auf atomarer Ebene zu analysieren.
Warum war die Kristallisation so bedeutend?
Die Kristallisation des Tabakmosaikvirus lieferte zwei bahnbrechende Erkenntnisse:
Nachweis der Existenz von Viren als Partikel: Bis dahin waren Viren eher als vage, unsichtbare Krankheitserreger bekannt. Stanley zeigte, dass sie physische Einheiten mit einer klar definierten Struktur sind, die sich sogar kristallisieren lassen.
Grundlage für die Strukturaufklärung: Die Kristalle ermöglichten es späteren Forschern, mithilfe der Röntgenkristallographie die Architektur von Viren zu entschlüsseln und zu zeigen, dass sie aus genetischem Material – entweder RNA oder DNA – und Proteinen bestehen. Diese Entdeckung war entscheidend, um die Funktionsweise von Viren zu verstehen.
Was hat die Strukturaufklärung enthüllt?
Die Untersuchung des Tabakmosaikvirus zeigte erstmals, dass Viren eine klare und wiederholbare Struktur besitzen:
Genetisches Material und Proteine: Das genetische Material eines Virus enthält die Anweisungen zur Herstellung neuer Viruspartikel, während die Proteine die schützende Hülle bilden und Funktionen wie das Eindringen in Wirtszellen ermöglichen.
Unterscheidung von anderen Mikroorganismen: Stanleys Arbeit verdeutlichte, dass Viren sich grundlegend von Bakterien und anderen Mikroorganismen unterscheiden. Viren sind deutlich kleiner, besitzen keine Zellstruktur und bestehen nur aus einem Minimum an Komponenten, was sie zu einzigartigen biologischen Einheiten macht.
Was versteht man unter Kristallisation?
Kristallisation ist der physikalische Prozess, bei dem Atome, Moleküle oder Ionen aus einer ungeordneten Phase (z. B. einer Lösung, einer Schmelze oder einem Gas) in eine feste, geordnete Struktur übergehen, die als Kristall bezeichnet wird. Dieser Übergang führt zur Bildung eines Kristallgitters – einer regelmäßigen, dreidimensionalen Anordnung, in der sich die Teilchen in einem sich wiederholenden Muster organisieren. Dabei ordnen sich die Teilchen so an, dass sie einen energetisch günstigen Zustand erreichen, der durch spezifische Wechselwirkungen (z. B. Wasserstoffbrücken, elektrostatische Kräfte, van-der-Waals-Kräfte) und Bindungswinkel definiert ist.
Kristallisation ist also der Übergang von Chaos (ungeordnete Teilchen) zu Ordnung (Kristallgitter), angetrieben von physikalischen Kräften und energetischen Prinzipien. Sie kann natürlich (z. B. bei der Mineralbildung) oder künstlich (z. B. in Laboren) stattfinden.
Abb. 8-A: Beispiel einer natürlichen Kristallisation durch Verdunstung einer Salzlösung. Links: In einer Salzlösung sind Natriumionen (grün) und Chloridionen (blau) frei beweglich und ungeordnet. Rechts: Beim Verdunsten des Wassers ordnen sich die Ionen zu einem regelmäßigen Kristallgitter an.
Grundprinzip der Proteinkristallisation
Proteinkristallisation bedeutet, dass man gelöste Proteine aus einer Lösung in eine feste, geordnete, kristalline Form überführt. Das Ziel ist ein dreidimensionales Gitter, in dem die Proteinmoleküle regelmäßig angeordnet sind. Das erreicht man, indem man die Löslichkeit der Proteine kontrolliert verringert, sodass sie langsam aus der Lösung „ausfallen“ und sich zu einem Kristall organisieren.
Der typische Prozess läuft so ab:
Reinigung: Das Protein wird hochrein isoliert, da Verunreinigungen die Kristallbildung stören.
Lösung herstellen: Das Protein wird in einer wässrigen Lösung mit Puffern, Salzen und manchmal organischen Zusätzen (z. B. Polyethylenglykol, PEG) gelöst.
Übersättigung: Durch Veränderung der Bedingungen (z. B. Verdunstung, Zugabe von Präzipitanten wie PEG oder Salzen) wird die Lösung übersättigt, sodass die Proteine auszufallen beginnen.
Keimbildung und Kristallwachstum: Zuerst bilden sich kleine Proteinaggregate (Keime), die dann zu größeren Kristallen wachsen, wenn die Bedingungen stimmen.

Abb. 8-B: Schematische Darstellung der Proteinkristall-Bildung Proteine werden zunächst in Lösung gebracht, wo sie ungeordnet vorliegen. Durch gezielte Veränderung der Bedingungen (z. B. Erhöhung der Salzkonzentration, Veränderung des pH-Werts oder langsame Verdunstung des Lösungsmittels) wird die Lösung übersättigt. Dadurch beginnen die Proteine, sich zu organisieren und zu einem Kristall zu wachsen.
Auf molekularer Ebene richten sich die Proteine aufgrund ihrer Ladungen, Wasserstoffbrückenbindungen und hydrophoben Wechselwirkungen in einem regelmäßigen Gitter an. Die Moleküle „suchen“ dabei die energetisch günstigste Position, was zur Bildung eines stabilen Kristalls führt. Zwischen den Proteinen befindet sich Wasser, das durch kleine blaue Punkte dargestellt wird. Dieses Wasser ist ein integraler Bestandteil des Kristalls und stabilisiert die Proteinstruktur durch Wasserstoffbrückenbindungen.Proteinkristalle weisen oft eine symmetrische Struktur auf (z. B. kubisch oder hexagonal), da sich die Moleküle in wiederholbaren Mustern anordnen.
Proteine sind riesige Moleküle mit Tausenden von Atomen, komplexen 3D-Formen und uneinheitlichen Oberflächen (mit Ladungen, hydrophoben Bereichen usw.). Sie können nicht einfach wie die Natrium- und Chloridionen beim Salzkristall abwechselnd gestapelt werden.Merkmale der Proteinkristalle
Die Festigkeit: Proteinkristalle sind deutlich fragiler als klassische Kristalle wie Salz oder Diamant. Sie bestehen zu 30–70 % aus Wasser, das in Kanälen und Hohlräumen des Kristallgitters eingeschlossen ist. Dadurch sind sie eher weich und gelartig. Mechanische Belastung oder Austrocknung kann sie leicht zerstören.
Die Farbe: Proteinkristalle sind meist farblos oder leicht opak (undurchsichtig). Proteine selbst besitzen keine natürliche Farbe – die bunten Darstellungen in wissenschaftlichen Abbildungen dienen lediglich dazu, strukturelle Merkmale und chemische Eigenschaften hervorzuheben.
Auswahl des Proteinkristalls
Bei der Kristallisation entstehen oft viele kleine Kristalle gleichzeitig, da sich die Proteinmoleküle an verschiedenen Stellen in der Lösung zu ordnen beginnen. Für die Analyse wird jedoch nur der bestgeordnete Kristall ausgewählt.
Wie findet man den richtigen Kristall?
Früher wurden die Kristalle unter dem Mikroskop betrachtet: Klare, scharfe Kanten und eine regelmäßige Form (z. B. kubisch oder hexagonal) deuteten auf eine hohe Qualität hin. Trübe oder unregelmäßige Kristalle hingegen waren weniger geeignet.
Heute kommen moderne Bildanalysesysteme zum Einsatz, die hochauflösende Mikroskopie mit automatischer Bewertung kombinieren. Zusätzlich kann eine Röntgenbeugung durchgeführt werden: Scharfe, symmetrische Beugungsmuster deuten auf eine gute Kristallordnung hin, während diffuse oder unregelmäßige Reflexionen auf eine geringe Qualität schließen lassen.
Warum braucht man Kristalle?
Kurz gesagt: Proteinkristalle dienen als „Verstärker“ für Röntgenstrahlen.
Ein einzelnes Proteinmolekül würde bei einer Röntgenbeugung nur ein extrem schwaches und diffuses Signal erzeugen – zu wenig, um eine detaillierte Struktur zu bestimmen. In einem Kristall hingegen sind Millionen identischer Proteinmoleküle regelmäßig angeordnet und gleich orientiert. Dadurch verstärken sich die Beugungssignale der einzelnen Moleküle durch konstruktive Interferenz und ergeben ein klares, regelmäßiges Beugungsmuster.
Diese geordnete Verstärkung ist essenziell für die Röntgenkristallographie, da sie erst durch das entstehende Beugungsmuster die dreidimensionale Struktur des Proteins entschlüsseln kann. Ohne Kristalle wäre die Analyse mit dieser Methode unmöglich.
Ablauf der Röntgenkristallographie
Nachdem das Protein erfolgreich kristallisiert wurde, kann seine räumliche Struktur (also seine genaue Faltung und Anordnung) mit der Röntgenkristallographie bestimmt werden. Dabei erkennt man Details wie:
➤ Position jedes einzelnen Atoms
➤ Faltungsstruktur des Proteins (α-Helices, β-Faltblätter usw.)
➤ Interaktionen mit anderen Molekülen (z. B. Antikörper, Wirkstoffe)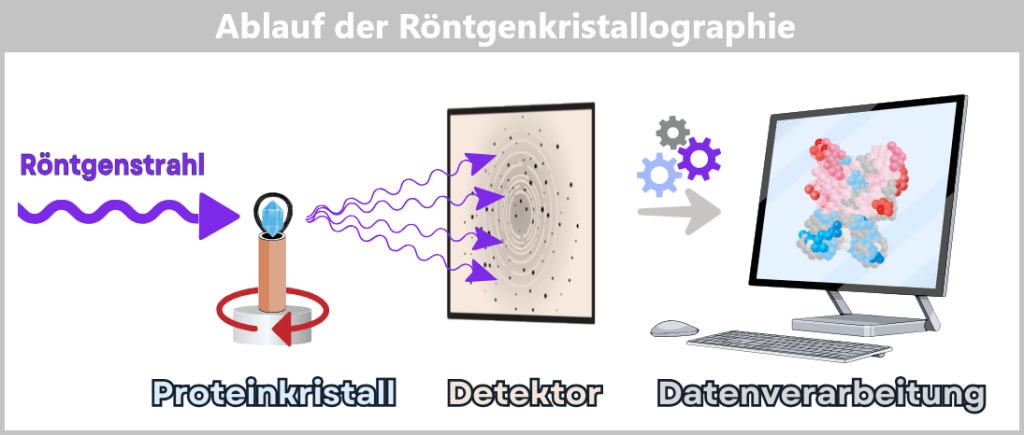
Abb. 8-C: Schematische Darstellung der Röntgenkristallographie 1️⃣ Röntgenstrahl auf den Kristall schießen
Ein gebündelter Röntgenstrahl trifft auf einen Proteinkristall. Proteine bestehen aus Aminosäuren, und diese wiederum aus Atomen. Röntgenstrahlen haben eine Wellenlänge von etwa 0,1 nm, was in der Größenordnung von Atomen liegt, sodass sie gut geeignet sind, um Details auf dieser Skala sichtbar zu machen. Die Atome im Kristall beugen die Röntgenstrahlen nicht direkt, sondern ihre Elektronenhüllen lenken die Strahlen in bestimmte Richtungen ab.
2️⃣ Beugungsmuster entsteht
Die gebeugten Röntgenstrahlen überlagern sich und erzeugen ein charakteristisches Muster auf einem Detektor. Dieses Beugungsmuster besteht aus vielen Punkten (sogenannten Reflexen), die an verschiedenen Positionen und mit unterschiedlicher Intensität auftreten. Um eine vollständige Datenerfassung zu ermöglichen, wird der Kristall in kleinen Schritten gedreht (typischerweise in 0,1–1°-Schritten), während für jede Position ein Beugungsmuster aufgenommen wird.
3️⃣ Mathematische Berechnung der 3D-Struktur
Die verschiedenen Aufnahmen der Beugungsmuster zeigen nicht direkt die Struktur des Proteins. Stattdessen enthalten sie Informationen darüber, wie die Röntgenstrahlen an den Elektronen der Atome im Kristall gebeugt wurden. Mithilfe mathematischer Methoden (Fourier-Transformation) wird aus den Beugungsmustern eine Elektronendichtekarte berechnet. Diese Karte zeigt die dreidimensionale Verteilung der Elektronen im Kristall.
Da sich Elektronen hauptsächlich in der Nähe der Atomkerne befinden, lassen sich aus der Elektronendichtekarte die Positionen der Atome ableiten. Die Karte erscheint zunächst als „wolkige“ Struktur, in der Bereiche mit hoher Elektronendichte den Atomen entsprechen.
Die Qualität der Elektronendichtekarte hängt direkt von der Ordnung im Kristall ab – je besser die Kristalle, desto schärfer die Elektronendichtekarte. Durch weitere Analyse und Interpretation dieser Karte kann schließlich ein detailliertes, dreidimensionales Modell des Proteins erstellt werden.
〰️ Synchrotronstrahlung – Licht für die kleinsten Geheimnisse der Natur
Um die Struktur von Proteinen noch genauer zu untersuchen, nutzt man in vielen Fällen Synchrotronstrahlung – eine extrem intensive Röntgenstrahlung, die in speziellen Teilchenbeschleunigern erzeugt wird.
💡 Was ist das genau?
Synchrotronstrahlung entsteht, wenn geladene Teilchen (z. B. Elektronen) auf nahezu Lichtgeschwindigkeit beschleunigt und dann durch Magnetfelder in eine Kreisbahn gelenkt werden. Dabei geben sie hochenergetische Strahlung ab – darunter auch besonders brillante Röntgenstrahlen, die sich perfekt für die Proteinkristallographie eignen. Dadurch können auch winzige oder schwer zu kristallisierende Proteine untersucht werden.
🌍 Weltweit gibt es mehrere große Synchrotron-Forschungszentren, z. B. in:
- ESRF (Frankreich) – Europäisches Synchrotron-Strahlungszentrum
- DESY (Deutschland) – Deutsches Elektronen-Synchrotron
- Diamond Light Source (UK) – Synchrotronanlage in Großbritannien
🎥 Wenn du live miterleben möchtest, wie Proteine zu Kristallen werden und wie ihre Strukturen mithilfe von Röntgenstrahlen entschlüsselt werden, dann begleite die Forscher der Diamond Light Source in die faszinierende Welt der Kristallographie!
Understanding Crystallography – Part 1: From Proteins to Crystals
Hier siehst du, wie Wissenschaftler Proteine in Kristalle verwandeln.Understanding Crystallography – Part 2: From Crystals to Diamond
In diesem Video siehst du, wie die Kristalle mit Röntgenstrahlen untersucht werden, um ihre 3D-Struktur zu enthüllen.
Die Proteindatenbank – Ein Schatz der Strukturbiologie
Die Röntgenkristallographie hat nicht nur dazu beigetragen, die Struktur einzelner Proteine aufzuklären, sondern auch die Entstehung einer globalen Ressource ermöglicht: die Protein Data Bank (PDB). Diese Datenbank sammelt und speichert die 3D-Strukturen von Proteinen, Nukleinsäuren und anderen Biomolekülen.
Die PDB wurde 1971 gegründet und enthält heute über 200.000 Einträge. Bis in die 1970er Jahre war die Röntgenkristallographie die einzige Methode zur Bestimmung der dreidimensionalen Struktur von Proteinen mit atomarer Auflösung.
In den folgenden Jahrzehnten erweiterten neue Techniken wie die Kernspinresonanzspektroskopie (NMR) und später die Kryo-Elektronenmikroskopie (Cryo-EM) das methodische Spektrum. Während NMR besonders für kleine Proteine in Lösung geeignet ist, ermöglicht Cryo-EM die Untersuchung großer Proteinkomplexe ohne Kristallisation.
Jede Proteinstruktur liefert detaillierte Informationen über die räumliche Anordnung der Atome in einem Molekül. Die Röntgenkristallografie hat damit nicht nur unser Verständnis revolutioniert – sie hat eine globale Infrastruktur des Wissens geschaffen. Eine stille Bibliothek, in der jedes Protein seine Geschichte erzählt – festgehalten in atomarer Genauigkeit. Zugänglich für Forscher auf der ganzen Welt.
Röntgenkristallographie in der Virologie
Viren sind komplexe Gebilde, die aus Proteinen, Nukleinsäuren (DNA oder RNA) und manchmal auch Lipiden bestehen. Ganze Viren sind oft zu groß und zu flexibel, um sie zu kristallisieren und mit Röntgenkristallographie zu untersuchen.
Einige Viren besitzen jedoch regelmäßige, symmetrische Strukturen, die eine dichte, wiederholte Packung ermöglichen und damit die Kristallisation ermöglichen. Ein bekanntes Beispiel ist das Tabakmosaikvirus, dessen Struktur bereits in den 1950er Jahren mithilfe der Röntgenkristallographie aufgeklärt wurde.
Für die meisten Viren ist es jedoch praktikabler, einzelne Virusproteine zu untersuchen. Besonders interessant sind dabei Proteine, die das Virus für die Infektion von Wirtszellen, seine Vermehrung oder zur Umgehung des Immunsystems benötigt.
Um ein Beispiel für die Struktur eines Virusproteins zu zeigen, greifen wir auf die Proteindatenbank (PDB) zurück. Die folgende Abbildung stammt aus der PDB und zeigt das Hämagglutinin-Protein des H5N1-Influenzavirus. Seine atomare Struktur wurde mithilfe der Röntgenkristallographie bestimmt und in der Protein Data Bank hinterlegt.
Abb. 8-D: Atomare Struktur des Hämagglutinin-Proteins des H5N1-Influenzavirus (PDB-ID: 2FK0), dargestellt als Ball-and-Stick-Modell. Anleitung zur Suche in der Proteindatenbank
1️⃣ Öffne die Protein Data Bank (PDB): https://www.rcsb.org.
2️⃣ Suche nach einem Protein:
In unserem Beispiel suchen wir nach dem Hämagglutinin-Protein des Influenzavirus.
👉 Gib in das Suchfeld den Begriff „influenza hemagglutinin“ ein.
👉 Filtere die Ergebnisse unter [Experimental Method] nach [X-ray diffraction] (Röntgenkristallographie).3️⃣ Wähle einen Eintrag aus:
👉 Klicke auf ein Ergebnis. Für unser Beispiel wählen wir PDB-ID: 2FK0 – das Hämagglutinin-Protein des H5N1-Influenzavirus.
4️⃣ Ansicht des Proteins:
Links in der PDB siehst du die Cartoon-Darstellung oder das Ribbon-Diagramm, eine schematische Ansicht der 3D-Faltung des Proteins.
5️⃣ Visualisierung der atomaren Struktur:
👉 Klicke auf [Structure], um in die 3D-Ansicht zu wechseln.
👉 Wähle einen anderen Viewer: „JSmol“ (siehe: unten rechts).
👉 Für die atomare Struktur-Ansicht wähle den [Style]: „Ball-and-Stick“.
🔍 Nun sind alle Atome und Bindungen sichtbar. Mit der Maus kannst du das Modell drehen, zoomen und verschieben.6️⃣ Strukturelle Merkmale hervorheben:
👉 Unter [Color] kannst du verschiedene Farb-Darstellungen. Das Beispiel: „By chain“ färbt jede Proteinkette in einer eigenen Farbe, um Untereinheiten des Proteins zu unterscheiden.
Weitere Infos zum Einsatz der Röntgenkristallographie
Seit über 40 Jahren ist die Röntgenkristallographie eine leistungsfähige Methode zur Bestimmung von Virusstrukturen. Da wir bereits das Oberflächenprotein Hämagglutinin des Influenzavirus kennengelernt haben, beziehen sich die nachfolgend aufgeführten Studien darauf.
🔗 Eine der ersten Studien dazu wurde 1981 veröffentlicht: „Structure of the haemagglutinin membrane glycoprotein of influenza virus”. Sie enthüllte die dreidimensionale Struktur des HA-Proteins und legte die Grundlage für das Verständnis der Infektionsmechanismen des Influenzavirus.
🔗 Eine neuere Studie aus dem Jahr 2019 mit dem Titel „Identification of a pH sensor in Influenza hemagglutinin using X-ray crystallography” untersuchte die Bindungsregion des HA-Proteins mittels Röntgenkristallographie. Sie zeigte, wie Konformationsänderungen der Bindungsregion in Abhängigkeit des pH-Werts die Virusfunktion beeinflussen.
🔗 Eine Studie aus dem Jahr 2020, „Structure of avian influenza hemagglutinin in complex with a small molecule entry inhibitor”, nutzte Röntgenkristallographie, um die Struktur des H5-Hämagglutinin (HA)-Proteins des Influenzavirus H5N1 in komplex mit dem Inhibitor CBS1117 aufzuklären. Die Kristallstruktur lieferte detaillierte Einblicke in die molekularen Wechselwirkungen, die für die Entwicklung neuer antiviraler Wirkstoffe entscheidend sind.
Durch die Kombination der Strukturen einzelner Proteine können Wissenschaftler ein detailliertes Bild des gesamten Virus zusammensetzen.
Grenzen der Röntgenkristallographie
Obwohl die Röntgenkristallographie eine unglaublich leistungsfähige Methode zur Strukturbestimmung ist, stößt sie in bestimmten Fällen an ihre Grenzen:
⧎ Schwierige Kristallisation: Viele Proteine, insbesondere große Komplexe oder Membranproteine, lassen sich nur schwer oder gar nicht in Kristallform bringen.
⧎ Künstliche Bedingungen: Proteine werden in der Kristallform fixiert, was möglicherweise nicht ihrem natürlichen Zustand entspricht.
⧎ Fehlende Dynamik: Die Methode zeigt nur eine statische Momentaufnahme, aber keine Informationen über Bewegung oder Flexibilität der Moleküle.
⧎ Strahlenschäden: Die hochenergetischen Röntgenstrahlen können empfindliche Moleküle leicht verändern.
Auf der Suche nach einer neuen Methode
Diese Einschränkungen machten klar: Nicht jedes Biomolekül lässt sich wie Lego in Kristallform pressen. Die Lösung? Ein mikroskopischer Kälte-Blitz: Kryo-EM. Ihr Trick? Moleküle in Millisekunden schockgefrieren – so schnell, dass Wasser gar nicht erst kristallisiert. Keine Kristall-Zwangsjacke, keine Röntgen-Bräune – nur eiskalte Atom-Details.
c) Kryoelektronenmikroskopie: Ein moderner Blick auf Viren
Nachdem wir die klassische Elektronenmikroskopie und die Röntgenkristallographie kennengelernt haben, die uns erstmals Einblicke in die Struktur von Viren ermöglichten, führt uns die Kryoelektronenmikroskopie – kurz Kryo-EM – in eine neue Ära der Forschung.
Was ist Kryo-EM überhaupt?
Kryo-EM ist eine Technik, bei der Proben extrem schnell eingefroren („kryo“ = kalt) und dann mit einem Elektronenmikroskop untersucht werden. Das Besondere: Die Proben bleiben in einem nahezu natürlichen Zustand, weil sie nicht chemisch fixiert oder gefärbt werden müssen, wie bei anderen Methoden. Das macht sie ideal, um empfindliche Strukturen wie Viren zu betrachten.
Wie kam es dazu?
Früher hatten Wissenschaftler ein großes Problem, wenn sie biologische Proben mit Elektronenmikroskopen untersuchen wollten: Wasser verdunstete im Vakuum der Mikroskope, und empfindliche Moleküle wurden durch den intensiven Elektronenstrahl beschädigt. Dadurch entstanden unscharfe oder verzerrte Bilder.
Forscher hatten bereits die Idee, Proben zu kühlen, um sie zu schützen. Doch es gab ein weiteres Problem: Wenn Wasser gefriert, bilden sich Eiskristalle. Diese beugen Elektronen so stark, dass klare Bilder unmöglich wurden.
Der entscheidende Durchbruch kam in den 1980er Jahren, als der Wissenschaftler Jacques Dubochet eine Technik namens Tauchgefrieren entwickelte. Dabei wird die Probe extrem schnell auf sehr niedrige Temperaturen abgekühlt, bevor sich Eiskristalle bilden können. Stattdessen bleibt das Wasser in einem glasartigen, gefrorenen Zustand erhalten, der die biologische Probe perfekt umhüllt. So wurde die Kryo-Elektronenmikroskopie (Kryo-EM) geboren.
Ein weiterer wichtiger Fortschritt kam in den 1970er Jahren, als Joachim Frank eine Methode entwickelte, um die unscharfen Bilder aus dem Mikroskop mit computergestützten Berechnungen zu verbessern. Indem er zahlreiche Aufnahmen einzelner Moleküle aus unterschiedlichen Blickwinkeln kombinierte, gelang es ihm, ein gestochen scharfes 3D-Modell zu rekonstruieren. Dadurch war es erstmals möglich, die Struktur von Biomolekülen zu bestimmen, ohne sie zuvor kristallisieren zu müssen – ein großer Vorteil für Proteine, die sich nur schwer oder gar nicht in Kristallform bringen lassen.
1990 nutzte Richard Henderson die Kryo-EM erstmals, um ein Biomolekül in so hoher Auflösung zu untersuchen, dass selbst kleine Details wie Aminosäure-Seitenketten sichtbar wurden. Für diese bahnbrechenden Entwicklungen erhielten Dubochet, Frank und Henderson 2017 den Nobelpreis für Chemie.
Seitdem hat sich die Technologie rasant weiterentwickelt:
↗️ Moderne Kameras nehmen gestochen scharfe Bilder direkt auf, ohne Qualitätsverluste.
↗️ Automatisierte Mikroskope können mehrere Proben gleichzeitig analysieren.
↗️ Leistungsstarke Computerprogramme ermöglichen die Verarbeitung riesiger Datenmengen, um noch genauere Strukturen zu berechnen.Dank dieser Fortschritte kann die Kryo-EM heute komplexe Moleküle, Proteine, Viren und sogar ganze Zellstrukturen in ihrem natürlichen Zustand sichtbar machen – und das in einer nie dagewesenen Detailtiefe.
Im Folgenden werfen wir einen genaueren Blick darauf, wie die Kryo-EM Viren sichtbar macht.
Mit Kryo-EM Viren sichtbar machen – Schritt für Schritt
1️⃣ Probenvorbereitung: Viren züchten und isolieren
Wenn man Viren untersuchen will, muss man sie erst einmal haben. Dafür werden sie meist in geeigneten Wirtszellen gezüchtet, z. B. in Zellkulturen im Labor. Viren sind Parasiten, die sich nur in Wirtszellen vermehren können. Um sie zu untersuchen, infiziert man also geeignete Zellen in einer Zellkultur und lässt die Viren sich vermehren.
Nach der Infektion der Zellen werden die Viren zu bestimmten Zeitpunkten „geerntet“, um verschiedene Stadien ihres Lebenszyklus (z. B. Anheftung, Eindringen, Vermehrung) zu untersuchen. Danach werden die Viren aus der Zellkultur isoliert und gereinigt, z. B. durch Zentrifugation oder Filtration, damit man eine saubere Virusprobe hat, ohne störende Zellreste.
2️⃣ Probepräparation: Aufbringen der Probe auf ein Gitter
Die gereinigte Virusprobe ist eine wässrige Lösung, in der die Viren schweben. Die Viruslösung wird mit einer Pipette auf ein Gitter aufgetragen (siehe untere Abbildung).
Ein typisches Kryo-EM-Gitter ist winzig, es hat einen Durchmesser von 3 Millimetern, damit es in die Halterungen des Elektronenmikroskops passt. Das Gitter besteht aus einem feinen Netz aus Metall (z. B. Kupfer oder Gold), das wie ein Sieb aussieht. Auf das Metallgitter wird eine ultradünne Kohlenstofffolie gelegt, die selbst winzige Löcher enthält (meist 1–2 Mikrometer im Durchmesser). Durch die Oberflächenspannung verteilt sich die Lösung in den winzigen Löchern und haftet dort, bevor sie eingefroren wird. Diese Anordnung hat zwei wichtige Funktionen: Das Gitter sorgt für Stabilität, während die Löcher den Elektronenstrahl ungehindert passieren lassen, um Hintergrundrauschen zu minimieren.

Abb. 9-A: Schematische Darstellung der Kryo-EM-Probenpräparation A – Gitter, bedeckt mit einer löchrigen Kohlenstofffolie,
B – vergrößertes Bild einer Gitteröffnung, einer sogenannten Mesh-Öffnung,
C – vergrößertes Bild eines Lochs, in der die Viren haften3️⃣ Schnellgefrieren der Probe
Die Probe bzw. das Gitter wird nun blitzschnell in flüssigem Ethan oder Stickstoff auf etwa -180 °C eingefroren. Das nennt man Vitrifizierung – das Wasser wird nicht zu Kristallen, sondern zu einem glasartigen Zustand, der die Struktur perfekt erhält. So bleiben die Viren in ihrem natürlichen Zustand erhalten – als wären sie in der Zeit eingefroren. Zudem schützt das Eis die Moleküle vor Schäden durch die intensive Elektronenstrahlung.
Abb. 9-B: Rechts – Schematischer Querschnitt eines Lochs im Kryo-EM-Gitter, in dem Viren in vitrifiziertem (glasartigem) Eis eingebettet sind. 4️⃣ Aufnahme der Bilder im Elektronenmikroskop
Das gefrorene Gitter mit den Viren wird in das Kryo-Elektronenmikroskop eingelegt. In der vitrifizierten Lösung sind die Viren zufällig ausgerichtet – sie liegen also in verschiedenen Winkeln vor. Der Elektronenstrahl trifft die Probe aus einer festen Richtung (meist senkrecht), und eine hochempfindliche Kamera nimmt 2D-Projektionen der Viren auf.
Da jedes Virus in einer anderen Orientierung eingefroren ist, erhält man automatisch Bilder aus vielen verschiedenen Blickwinkeln – ohne dass das Gitter gedreht werden muss. Diese große Anzahl an Bildern ist wichtig für die spätere 3D-Rekonstruktion. Insgesamt werden oft Tausende bis Hunderttausende Aufnahmen gemacht.
5️⃣ Datenverarbeitung: Vom 2D-Bild zum 3D-Modell
Nach der Aufnahme beginnt die eigentliche Herausforderung: die computergestützte Rekonstruktion. Spezielle Software erkennt die einzelnen Viruspartikel in den 2D-Bildern und sortiert sie nach ihrer Orientierung.
Durch mathematische Verfahren (wie „Single Particle Analysis“) werden die 2D-Bilder zusammengefügt, um ein hochauflösendes 3D-Modell des Virus zu berechnen. Je mehr Bilder und je besser deren Qualität, desto schärfer wird das Modell.
Falls verschiedene Stadien des Viruslebenszyklus untersucht werden sollen (z. B. vor und nach dem Eindringen in eine Zelle), wiederholt man diesen Prozess mit Proben aus unterschiedlichen Zeitpunkten. So kann man sogar Veränderungen in der Struktur über die Zeit sichtbar machen.

Abb. 9-C: Schritte der Kryo-EM-Analyse Die folgenden Videos fassen die bisherigen Informationen zur Kryo-EM anschaulich zusammen:
👉 What is Cryo-Electron Microscopy (Cryo-EM)?
👉 Cryo-EM Animation6️⃣ Interpretation und Visualisierung
Nach der 3D-Rekonstruktion liegt ein hochauflösendes Modell des Virus vor, das dessen Aufbau zeigt – z. B. die Hülle, die Spike-Proteine und manchmal sogar innere Strukturen.
Nun geht es darum, die eiskalten Daten zu analysieren, zu interpretieren und anschaulich darzustellen. Dieser Schritt ist wichtig, um die biologische Bedeutung der Struktur zu entschlüsseln und die Erkenntnisse für weitere Forschung oder die Öffentlichkeit zugänglich zu machen.
Schritte der Interpretation und Visualisierung
a) Analyse der Struktur: Wissenschaftler untersuchen die 3D-Struktur, um Schlüsselmerkmale des Virus oder seiner Proteine zu identifizieren. Diese Analyse hilft, die molekularen Mechanismen der Virusinfektion zu verstehen. Dazu gehören z. B.:
- Bindungsstellen: Wo bindet das Virus an Wirtszellen?
- Funktionelle Bereiche: Welche Teile des Proteins sind für die Infektion oder Vermehrung essenziell?
- Veränderungen: Gibt es Unterschiede in der Struktur zwischen verschiedenen Virusstämmen oder -stadien?
b) Vergleich mit bekannten Strukturen: Die rekonstruierte Struktur wird mit bereits bekannten Strukturen aus Datenbanken wie der Protein Data Bank (PDB) verglichen. Dies kann Aufschluss über evolutionäre Beziehungen, funktionelle Ähnlichkeiten oder mögliche Angriffspunkte für Medikamente geben.
c) Visualisierung der Struktur: Mithilfe spezieller Software wird das 3D-Modell visualisiert, um die Struktur anschaulich darzustellen und wichtige Merkmale hervorzuheben. Dabei gibt es verschiedene Darstellungsformen:
- Oberflächendarstellung: Zeigt die äußere Form des Virus oder Proteins.
- Stäbchenmodell (Ball-and-Stick): Zeigt die Anordnung der Atome und Bindungen.
- Sekundärstruktur: Hebt strukturelle Elemente wie α-Helices und β-Faltblätter hervor.
d) Publikation und Datenfreigabe: Die Ergebnisse werden in wissenschaftlichen Fachzeitschriften veröffentlicht – oft mit hochauflösenden Bildern oder Animationen der 3D-Struktur. Zudem werden die Rohdaten und rekonstruierten Modelle in öffentlichen Datenbanken wie der PDB (Protein Data Bank) oder der EMDB (Electron Microscopy Data Bank) gespeichert, sodass andere Forscher darauf zugreifen können.
Wenn Viren wüssten, wie oft wir sie jetzt in 3D drehen und zoomen können… sie würden sich glatt was anziehen!
Influenzavirus in 3D: ein Beispiel
Es kann schwierig sein, 3D-Modelle von Viren im Internet zu finden. Forschungsdaten werden oft erst nach der Veröffentlichung von Studien freigegeben und dann in speziellen Datenbanken wie der EMDB (Electron Microscopy Data Bank) gespeichert. Doch diese Daten sind für Laien schwer zugänglich, weil sie spezielle Software benötigen, um sie als 3D-Modelle anzuzeigen.
Erfreulicherweise bietet die NIH 3D-Plattform (National Institutes of Health) ein hilfreiches Beispiel: ein 3D-Modell eines Influenzavirus, das mit der Kryo-Elektronenmikroskopie erstellt wurde. Du kannst es dir unter diesem Link anschauen! Beachte, dass du möglicherweise einen 3D-Viewer brauchst, um es zu erkunden.
Weitere Infos zum Einsatz der Kryo-EM
🔬 Diese Studie zum Influenzavirus zeigte mithilfe der Kryo-Elektronenmikroskopie die Interaktionen zwischen den Nukleoproteinen und der viralen RNA sowie die spezifische Anordnung der Ribonukleoprotein-Komplexe (RNPs) innerhalb des Virions. Solche strukturellen Einblicke sind entscheidend für das Verständnis der Mechanismen, mit denen das Influenzavirus sein Genom verpackt und während des Infektionszyklus repliziert.
🔬 Ergänzend zeigt eine frühere Studie von Liu et al. (2017) strukturelle Details filamentöser Influenzavirus-Partikel und deren RNP-Organisation mittels Kryo-EM.
Warum Kryo-EM so genial ist – besonders bei Viren
Die Kryo-EM ist ein unschlagbares Werkzeug, wenn es darum geht, Viren sichtbar zu machen – besonders wenn’s um die schwer fassbaren Kandidaten geht:
- Viren mit ihren tückischen Spike-Proteinen
- Riesige Proteinkomplexe, die sich weigern, ordentlich zu kristallisieren
- Membranproteine, die sonst nur „Hallo!“ sagen und sofort zerfallen
Die Methode: Sie friert Viren blitzschnell ein, sodass sie wie in einer Zeitkapsel erhalten bleiben, und erzeugt Tausende von Aufnahmen mithilfe eines Elektronenstrahls. Ein Computer setzt diese Bilder dann zu einem hochauflösenden 3D-Modell zusammen – so können wir genau sehen, wie ein Virus aufgebaut ist. Diese Methode wird als Single Particle Analysis (SPA) bezeichnet, da einzelne Viruspartikel analysiert und rechnerisch zu einer 3D-Struktur zusammengeführt werden.
Vom Standbild zum Blockbuster
Was die Kryo-EM an detailreichen „Schnappschüssen“ liefert, ist beeindruckend – aber manchmal reicht ein Bild eben nicht aus. Um zu verstehen, wie Viren sich bewegen, Zellen kapern oder sich replizieren, braucht es bewegte Szenen. Genau hier setzt die Kryo-Elektronentomographie an: eine Technik, mit der man Viren fast wie in einem Film beim Arbeiten zuschauen kann.
d) Kryo-Elektronentomographie – 3D-Virusmodelle in der Zelle
Während die Kryo-EM hochgereinigte Viren isoliert untersucht, erlaubt die Kryo-ET Wissenschaftlern, dreidimensionale Schnappschüsse molekularer Interaktionen direkt innerhalb der Zelle aufzunehmen. So können Viren in ihrem natürlichen Umfeld – also in der Zelle – sichtbar gemacht werden.
Mit Kryo-ET Viren sichtbar machen – Schritt für Schritt
1️⃣ Probenvorbereitung – Einfrieren der Zelle
Geeignete Zellen werden in einer Zellkultur gezüchtet und mit Viren infiziert. Dann wartet man auf einen definierten Zeitpunkt – zum Beispiel auf das Eindringen der Viren in die Zelle oder auf ihre Vermehrung – und entnimmt die Zellen. Diese werden in eine Pufferlösung überführt, um eine homogene Zelllösung zu erhalten.
Anschließend wird die Zelllösung auf ein Kryo-EM-Gitter aufgetragen – ein kleines Metallgitter mit einer löchrigen Kohlenstofffolie, das bereits aus der Kryo-EM bekannt ist. Das Gitter wird sofort in flüssiges Ethan oder flüssigen Stickstoff (bei ca. -180 °C) getaucht. Durch diese blitzschnelle Gefriertechnik (Vitrifizierung) bleiben die Zellstrukturen erhalten, ohne dass sich störende Eiskristalle bilden.

Abb. 10-A: Schematische Darstellung der Kryo-ET-Probenpräparation mit anschließender Vitrifizierung 2️⃣ Dünne Zellbereiche erzeugen (falls nötig)
Durch die Vitrifizierung werden die Zellen stabilisiert – sie sind nun fest wie Glas. Da Zellen oft zu dick für die Kryo-ET sind, nutzen Wissenschaftler einen Focussed Ion Beam (FIB), um die Eisschicht auf die gewünschte Dicke zu reduzieren.
Dafür wird das Kryo-EM-Gitter in einem FIB-SEM-Gerät (Focussed Ion Beam mit Rasterelektronenmikroskop) zunächst gescannt, um interessante Zellen zu lokalisieren. Anschließend fräst ein fokussierter Ionenstrahl mit höchster Präzision eine Lamelle (~100–200 nm dick) aus der Eisschicht. Diese Lamelle stellt einen Querschnitt der Zelle dar und zeigt relevante Bereiche mit Viruspartikeln – z. B. die Zellmembran oder das Zytoplasma mit Viren.
Wie das Ganze in der Praxis aussieht, zeigt das Video „Cryo-lamella preparation“ im Abschnitt „FIB-milling of lamella in waffle grids“ auf der Seite „Cryo EM 101, Kapitel 3“.
3️⃣ Bildaufnahme im Kryo-EM
Das gefrorene Zellgitter wird ins Kryo-Elektronenmikroskop gebracht. Hier erzeugt ein Elektronenstrahl hochaufgelöste Bilder der Probe.
Tomographie – Wie ein CT-Scan für Zellen
Die Probe wird schrittweise gedreht. Ein Goniometer (Drehmechanismus) kippt das Gitter in kleinen Winkelschritten (z. B. 1–2°), typischerweise über einen Bereich von ±60° bis ±70°. Für jeden Winkel wird ein 2D-Bild aufgenommen, sodass eine sogenannte „Tilt-Series“ (Neigungsserie) entsteht – eine Sammlung von 2D-Bildern aus verschiedenen Perspektiven (siehe untere Abbildung).
📌 Kryo-ET vs. SPA: Im Gegensatz zur Single Particle Analysis (SPA), bei der viele identische Viren benötigt werden, konzentriert sich die Kryo-ET auf einen spezifischen Bereich der Probe – oft eine einzelne Zelle oder eine Virus-Wirtszell-Interaktion. Man benötigt also keine große Anzahl identischer Partikel, sondern kann individuelle biologische Strukturen direkt in ihrem natürlichen Umfeld untersuchen.
4️⃣ 3D-Rekonstruktion
Die Kryo-ET nutzt die Neigungsserie, um rechnerisch ein 3D-Modell zu rekonstruieren – wie ein medizinischer CT-Scan, nur für Viren statt für Knochen. Das Ergebnis: ein hologrammartiges Volumenbild, das zeigt, wie Viren in echten Zellen tanzen, andocken und tricksen.
Abb. 10-B: Vom Elektronenstrahl zur 3D-Rekonstruktion 5️⃣ Interpretation & Visualisierung
Jetzt wird’s actionreich! Die 3D-Modelle zeigen Viren in ihrer natürlichen Umgebung:
- wie sie in der Zelle sitzen
- wie sie mit Zellorganellen interagieren
- verschiedenen Stadien (z. B. Eintritt, Replikation, Freisetzung)
Die Ergebnisse werden interpretiert, um z. B. den Infektionsmechanismus des Virus oder seine Interaktion mit der Wirtszelle zu verstehen.
Manchmal werden mehrere Zeitpunkte verglichen, um eine Art „Film“ zu erstellen, der die Dynamik simuliert.
Das folgende Video gibt einen Eindruck davon: Es wurden detailreiche 3D-Modelle eines Influenzavirus während der Knospung an der Zellmembran visualisiert und anschließend zu einem Film zusammengesetzt.
6️⃣ Publikation und Datenspeicherung
Damit alle was davon haben, landen die spektakulärsten Virus-Modelle in öffentlichen Datenbanken wie der Electron Microscopy Data Bank (EMDB) – das Netflix für Strukturbiologen.
Warum das cool ist:
- Open Science = Kein Wissenschaftler muss das Rad zweimal bauen.
- Transparenz = Jeder kann nachvollziehen, wie der „Film“ entstand.
- Kooperation = Teamwork macht auch Virenforschung zum Hit.
Wenn Viren ein Twitter hätten, würden sie jetzt trenden mit #EMDBChallenge.
Weitere Infos zum Einsatz der Kryo-ET
🔬 Die Studie „Quantitative Strukturanalyse von Influenzaviren mittels Kryo-Elektronentomographie und Convolutional Neural Networks“ nutzte Kryo-Elektronentomographie (Kryo-ET) und moderne Computertechnik, um die Struktur des Influenzavirus zu untersuchen. Sie zeigt die vielfältige Form des Virus und enthält Videos aus 3D-Rekonstruktionen, die den Prozess veranschaulichen.
🔬 Die Studie „Strukturelle Veränderungen des Influenzavirus bei niedrigem pH-Wert, charakterisiert durch Kryo-Elektronentomographie“ untersuchte, wie das Influenza-A-Virus auf saure Bedingungen reagiert, die es während des Eintritts in Wirtszellen erfährt. Durch Kryo-Elektronentomographie beobachteten die Forscher, dass bei niedrigem pH-Wert die viralen Hüllproteine (Hämagglutinin) strukturelle Veränderungen durchlaufen, die zur Fusion der viralen und zellulären Membranen führen. Diese Fusion ermöglicht es dem Virus, sein genetisches Material in die Wirtszelle freizusetzen und die Infektion zu initiieren.
Forscher untersuchen nicht nur humanpathogene Viren
Forscher interessieren sich längst nicht mehr nur für Krankheitserreger wie Grippe- oder Coronaviren – auch Viren aus der Umwelt sind extrem spannend. Allen voran: Bakteriophagen, die Bakterien befallen. In den Ozeanen sind sie echte Öko-Ingenieure.
1️⃣ Winzige Regisseure im Ökosystem
Phagen töten gezielt bestimmte Bakterien und halten so das mikrobielle Gleichgewicht im Meer im Lot. Damit steuern sie indirekt wichtige Kreisläufe wie den Kohlenstoff- oder Stickstoffhaushalt. Wer ihre Struktur und ihr Verhalten kennt, versteht besser, wie stabile Meeresökosysteme funktionieren.
2️⃣ Evolution live erleben
Phagen sind die häufigsten biologischen „Lebewesen“ der Erde – und genetisch unglaublich vielfältig. Mit Hilfe der Kryo-EM entdecken Forscher ständig neue Virusfamilien. Ihre genetischen Codes landen in Datenbanken und erzählen eine Geschichte über die Ko-Evolution von Viren und Bakterien.
3️⃣ Vom Meer ins Labor
Einige marine Phagen produzieren Enzyme mit echtem Potenzial: für Gentechnik, Diagnostik oder sogar als Alternative zu Antibiotika. Auch in der Landwirtschaft könnten sie helfen, gezielt schädliche Bakterien auszuschalten – ganz ohne Chemiekeule.
4️⃣ Und wohin mit den Daten?
Struktur- und Gensequenzen werden offen zugänglich gemacht – z. B. in der NCBI Virus Database oder der EMDB. Bioinformatiker stöbern dort nach neuen Genen, cleveren Tricks – und manchmal nach Ideen für die nächste biotechnologische Anwendung.
Grenzen der Kryo-EM
Die Kryo-EM (inklusive Single Particle Analysis (SPA) und Kryo-Elektronentomographie (Kryo-ET)) sind bahnbrechende Methoden, aber sie haben auch einige Einschränkungen, die von technischen, biologischen und praktischen Faktoren abhängen:
Probenvorbereitung ist anspruchsvoll
Problem: Die Proben müssen extrem dünn sein (nur ein paar Hundert Nanometer) und perfekt vitrifiziert werden, ohne Eiskristalle, die die Struktur zerstören könnten. Das ist technisch schwierig und erfordert viel Erfahrung.
Auswirkung auf Viren: Wenn die Viruslösung nicht gleichmäßig eingefroren wird oder zu dick ist, können die Bilder unscharf werden oder die Viren beschädigt werden. Bei der Kryo-ET können dicke Zellproben die Elektronen zu stark streuen, was die Bildqualität verschlechtert.
Begrenzte Auflösung
Problem: Obwohl Kryo-EM sehr hochauflösende Bilder liefern kann (bis zu 2–3 Ångström bei optimalen Bedingungen), hängt die Auflösung von der Qualität der Probe und der Datenmenge ab. Bei komplexen oder variablen Strukturen (z. B. flexiblen Viren) kann die Auflösung leiden.
Auswirkung auf Viren: Kleinste Details (z. B. einzelne Atome in Proteinen) sind nicht immer sichtbar, besonders wenn die Viren sich bewegen oder unterschiedlich geformt sind.
Eingeschränkte Dynamik: Eingefroren in der Zeit
Problem: So beeindruckend die 3D-Modelle auch sind – sie zeigen immer nur einen eingefrorenen Moment. Echte Bewegungen oder Abläufe – etwa wie ein Virus live in eine Zelle eindringt – bleiben außen vor.
Auswirkung auf Viren: Bei Single Particle Analysis (SPA) sehen wir nur eine Struktur zu einem bestimmten Zeitpunkt. Und auch bei der Kryo-ET müssen Forscher verschiedene Zellen in unterschiedlichen „Stadien“ der Infektion einfrieren, um daraus eine zeitliche Abfolge zu rekonstruieren. Klingt nach Film – ist aber eher eine Stop-Motion-Rekonstruktion als ein Livestream.
Hohe Kosten und Komplexität
Problem: Kryo-EM erfordert teure Geräte (Elektronenmikroskope kosten Millionen), spezialisierte Labore (z. B. Vakuum und Kryo-Bedingungen) und viel Rechenpower für die Datenanalyse. Das macht sie zeit- und kostenintensiv.
Auswirkung auf Viren: Nur gut ausgestattete Forschungseinrichtungen können Kryo-EM nutzen, was den Zugang einschränkt. Kleine Labore oder Entwicklungsregionen können diese Technik oft nicht anwenden.
Rauschen und Datenmenge
Problem: Die Bilder sind oft verrauscht, weil man nur wenige Elektronen nutzt, um die empfindlichen Proben nicht zu beschädigen. Die Verarbeitung erfordert Tausende Bilder, was rechenintensiv ist.
Auswirkung auf Viren: Wenn die Datenmenge nicht ausreicht oder das Rauschen zu hoch ist, können die 3D-Modelle ungenau werden.
Begrenzte Probengröße
Problem: Kryo-EM funktioniert nur mit sehr kleinen Proben (z. B. einzelne Viren oder Zellen). Größere Strukturen wie ganze Gewebe sind schwer zu untersuchen, weil die Elektronen nicht tief genug eindringen.
Auswirkung auf Viren: Kryo-ET kann Zellen abbilden, aber nur bis zu einer bestimmten Dicke (ca. 1 Mikrometer). Größere Organismen oder Gewebe sind nicht geeignet.
Quellen
Kryo-Elektronenmikroskopie: Prinzip, Stärken, Grenzen und Anwendungen
Herausforderungen und Erfolge in der Kryo-Elektronentomographie
e) Zusammenfassung
Die Wissenschaft hat über Jahrzehnte hinweg immer leistungsfähigere Methoden entwickelt, um die Welt der Viren sichtbar zu machen. Während das bloße Auge und Lichtmikroskope schnell an ihre Grenzen stoßen, haben Elektronenmikroskopie, Röntgenkristallographie und schließlich die Kryo-EM und Kryo-ET völlig neue Einblicke ermöglicht. Diese Technologien haben unser Verständnis von Viren revolutioniert.
Abb. 11: Von Atomen zu Makrostrukturen – Ein Überblick bildgebender Verfahren Skalierung der Bildgebung
➡️ Menschliches Auge
Sichtbar: ca. 0,1 mm (100 µm) – mehrere Meter
Beispiel: Haar, Sandkorn➡️ Lichtmikroskop
Sichtbar: ca. 200 nm – 1 mm
Beispiel: Bakterien, Zellen, Zellkerne➡️ Klassische Elektronenmikroskopie (EM, TEM/SEM)
Sichtbar: ca. 0,2 nm – 100 µm
Beispiel: Viren, Zellorganellen, Zellmembranen➡️ Röntgenkristallographie
Sichtbar: ca. 1 Å (0,1 nm) – 10 nm
Beispiel: Atomare Strukturen von Proteinen➡️ Kryo-EM (Einzelpartikelanalyse)
Sichtbar: ca. 2 Å (0,2 nm) – 100 nm
Beispiel: Proteinkomplexe, Ribosomen➡️ Kryo-ET (Kryo-Elektronentomographie)
Sichtbar: ca. 3-5 nm – 200 µm
Beispiel: Viren in Zellen, Zellorganellen in hoher Auflösung
Elektronenmikroskopie (1930er)
→ Erste Bilder von VirenDie Elektronenmikroskopie (EM) war ein revolutionärer Schritt in der Mikroskopie, da sie erstmals die Abbildung von Strukturen ermöglichte, die weit unterhalb der Auflösungsgrenze von Lichtmikroskopen liegen. Statt Licht verwendet die EM einen Strahl von Elektronen, der eine viel höhere Auflösung ermöglicht. Damit konnten Wissenschaftler erstmals Viren sichtbar machen, die zu klein sind, um mit herkömmlichen Mikroskopen betrachtet zu werden.
Die EM war ein großer Fortschritt, aber ihre Grenzen bei der Darstellung von Proben in ihrem natürlichen Zustand und die fehlende 3D-Information führten zur Entwicklung der Röntgenkristallographie.
Röntgenkristallographie (1950er–heute)
→ Detaillierte Strukturen von VirusproteinenDie Röntgenkristallographie ermöglichte es, die atomare Struktur von Proteinen und anderen Biomolekülen zu entschlüsseln. Dabei wird ein Kristall des Proteins mit Röntgenstrahlen bestrahlt, und das entstehende Beugungsmuster wird analysiert, um die Positionen der Atome zu bestimmen. Diese Methode lieferte detaillierte Einblicke in die Struktur von Virusproteinen, was für das Verständnis ihrer Funktion und die Entwicklung von Medikamenten entscheidend war.
Die Röntgenkristallographie ist zwar leistungsstark, aber ihre Einschränkungen bei der Untersuchung von großen, komplexen Strukturen und die Notwendigkeit von Kristallen führten zur Entwicklung der Kryo-Elektronenmikroskopie (Kryo-EM).
Kryo-EM (1980er–heute)
→ Ein moderner Blick auf VirenDie Kryo-EM kombiniert die Vorteile der Elektronenmikroskopie mit einer schonenden Probenvorbereitung. Die Proben werden blitzschnell eingefroren (vitrifiziert), wodurch sie in einem naturnahen Zustand erhalten bleiben. Dies ermöglicht die Abbildung von einzelnen Viruspartikeln oder großen Proteinkomplexen ohne Kristallisation. Die Kryo-EM liefert hochauflösende Bilder und kann auch flexible oder dynamische Strukturen abbilden.
Die Kryo-EM war ein großer Fortschritt, aber sie war auf isolierte Partikel beschränkt und konnte keine komplexen, zellulären Umgebungen abbilden. Dies führte zur Entwicklung der Kryo-Elektronentomographie (Kryo-ET).
Kryo-ET (2000er–heute)
→ 3D-Virusmodelle in der ZelleDie Kryo-ET erweitert die Kryo-EM, indem sie 3D-Modelle von Viruspartikeln oder anderen Strukturen direkt in ihrer zellulären Umgebung erstellt. Dabei wird die Probe aus verschiedenen Winkeln abgebildet, und die Bilder werden zu einem 3D-Modell zusammengesetzt. Dies ermöglicht es, Viren in ihrem natürlichen Kontext zu studieren, z. B. wie sie mit Zellen interagieren oder sich vermehren.
Die Kryo-ET ist ein mächtiges Werkzeug, aber ihre begrenzte Auflösung und die Komplexität der Probenvorbereitung könnten die Entwicklung neuer Technologien vorantreiben, die noch höhere Auflösungen in komplexen zellulären Umgebungen ermöglichen.
Jede dieser Technologien hat die Grenzen unseres Sehvermögens erweitert – und gleichzeitig neue Herausforderungen mit sich gebracht. Doch genau diese Grenzen waren stets der Antrieb für die Entwicklung noch leistungsfähigerer Methoden. Wissenschaft ist ein stetiger Prozess des Entdeckens, Verfeinerns und Weiterdenkens.
Man sagt: „Sehen ist Glauben“, doch für Biologen gilt: „Sehen ist Verstehen“. Je detaillierter wir biologische Strukturen abbilden können, desto tiefer dringen wir in die Geheimnisse des Lebens ein. Doch das reine Sichtbarmachen eines Virus und seiner Interaktion mit dem Wirt reicht nicht aus, um seine Natur vollständig zu entschlüsseln.
Vom Sehen zum Entschlüsseln: Die nächste Stufe der Erkenntnis
Um zu verstehen, was ein Virus wirklich ausmacht, müssen wir sein genetisches Erbe entschlüsseln – seinen einzigartigen „Fingerabdruck“. Dies gelingt mit modernen molekularbiologischen Methoden, die die virale Nukleinsäure analysieren und so einen Blick in den genetischen Bauplan des Virus ermöglichen.
4.5. Der genetische Fingerabdruck der Viren
Nachdem Viren endlich sichtbar geworden waren – dank Elektronenmikroskopie und Kristallstrukturanalyse – stellte sich die nächste große Frage: Was macht ein Virus eigentlich zum Virus?
Schnell war klar: Wie jedes biologische System brauchen auch Viren einen genetischen Bauplan – etwas, das ihre Eigenschaften codiert und ihre Vermehrung ermöglicht. Doch was genau trägt diese Information?
Lange galt das Protein als Favorit: vielfältig, komplex, scheinbar perfekt geeignet. Die DNA hingegen erschien vielen Forschenden zu simpel, zu langweilig, um der Träger des Lebens zu sein.
Doch wie sich herausstellte, lag die Antwort genau dort: in diesem unscheinbaren Molekül, das sich als der ultimative Datenträger des Lebens entpuppte – und bei manchen Viren auch in seinem Verwandten, der RNA.
Die Entdeckung, dass nicht Proteine, sondern Nukleinsäuren das Geheimnis der viralen Vermehrung bergen, war ein wissenschaftlicher Krimi für sich: geprägt von Irrtümern, rivalisierenden Teams und bahnbrechenden Entdeckungen.
Die Suche nach dem Code des Lebens
Frühe Ansätze zur Entschlüsselung der Vererbung
Die Grundlagen der Vererbungslehre wurden bereits im 19. Jahrhundert durch Gregor Mendels Experimente gelegt. Wissenschaftler erkannten, dass Organismen ihre Eigenschaften an die nächste Generation weitergeben, doch der genaue Mechanismus blieb lange unklar. In den Chromosomen der Zellen, die man Ende des 19. Jahrhunderts als mögliche Träger der Erbinformation identifizierte, suchte man nach den entscheidenden Molekülen. Proteine schienen aufgrund ihrer Komplexität die naheliegendsten Kandidaten zu sein.
Wendell Meredith Stanley und das Tabakmosaikvirus
In den 1930er Jahren bestätigten die Arbeiten von Wendell Meredith Stanley die zentrale Rolle von Proteinen im Aufbau von Viren. Stanley isolierte das Tabakmosaikvirus und bereitete es auf eine innovative Weise auf: Er extrahierte das Virus aus infizierten Tabakpflanzen, reinigte es durch Zentrifugation und ließ es anschließend in einer Lösung auskristallisieren. Diese Kristalle enthielten sowohl Proteine als auch genetisches Material, doch zunächst stand nur das Protein im Fokus der Forschung. Man ging davon aus, dass es die genetischen Anweisungen für die Virusreplikation trug.
Der Paradigmenwechsel: Von Proteinen zur DNA
Der Zweifel an der Protein-Hypothese wuchs, als Oswald Avery 1944 zeigte, dass DNA die Fähigkeit besitzt, Eigenschaften von Bakterien zu verändern. Sein Experiment, bei dem DNA aus einer Bakterienart extrahiert und auf eine andere übertragen wurde, offenbarte, dass DNA die Erbinformation tragen kann. Doch diese Entdeckung wurde anfangs skeptisch betrachtet.
Erst das Hershey-Chase-Experiment von 1952 brachte den entscheidenden Durchbruch. Die Wissenschaftler Alfred Hershey und Martha Chase arbeiteten mit Bakteriophagen, Viren, die Bakterien infizieren. Sie markierten die DNA der Viren radioaktiv und verfolgten ihren Weg in die Wirtszellen. Die Proteinhüllen der Phagen blieben außerhalb der Zelle, während die DNA eindrang und die Virusreplikation steuerte. Damit war bewiesen: Die DNA – nicht das Protein – trägt die genetischen Informationen.

Die Doppelhelix und die Rolle der RNA
1953 entschlüsselten James Watson und Francis Crick die Struktur der DNA, unterstützt durch Rosalind Franklins Röntgenbilder. Die Doppelhelix offenbarte, wie die DNA ihre Informationen speichert und bei der Zellteilung kopiert. Parallel wurde klar, dass RNA bei manchen Viren, darunter dem Tabakmosaikvirus, die genetischen Informationen trägt. Dies rückte Stanleys Arbeit in ein neues Licht: Die RNA in den Viruspartikeln war der eigentliche Träger der genetischen Information, nicht das Protein.
Viren – Die Minimalisten unter den Lebensformen
Viren sind wahre Meister der Reduktion. Diese minimalistischen Überlebenskünstler haben die Kunst genetischer Effizienz perfektioniert – ob mit DNA, RNA oder sogar rückwärts geschriebener RNA (die Spiegelschrift wie bei Influenzaviren mit seiner negativen ssRNA).
Ihr Genom ist wie ein ultraleichter Überlebensrucksack:
✔ Alles Nötige drin, was zählt – Baupläne fürs Kopieren, Verpacken, Wirt-Kapern
✖ Kein Ballast – keine eigenen Ribosomen, keine Energieproduktion, kein SmalltalkViren sind in ihrer genetischen Struktur einzigartig: Während alle bekannten Lebewesen ausschließlich DNA als Erbmaterial nutzen, bedienen sich Viren entweder DNA oder RNA – ein entscheidender Unterschied, der ihre enorme Anpassungsfähigkeit und evolutionäre Kreativität prägt.
Wichtige molekularbiologische Methoden zur Genom-Analyse von Viren
Dank moderner molekularbiologischer Techniken können Viren heute mit hoher Präzision analysiert werden. Damit kann man:
- ihre genetische Information entschlüsseln,
- ihre Abstammung nachvollziehen und
- ihre Verbreitungswege verfolgen.
Je nach Fragestellung kommen unterschiedliche Methoden zum Einsatz:
- Will man ein Virus identifizieren?
- Soll sein Genom vollständig sequenziert werden?
- Oder will man seine Aktivität im Wirtssystem beobachten?
Die nächsten Abschnitte stellen die zentralen Methoden zur Analyse des genetischen Fingerabdrucks vor – vom Isolieren der Erbsubstanz bis hin zu modernen Sequenzierungstechnologien.
4.5.1. Nukleinsäure-Extraktion
4.5.2. Nukleinsäure-Amplifikation
4.5.3. Sequenzierung💡Hinweis: Für grundlegende Informationen zu DNA und RNA sowie deren Funktionen empfehlen wir das Kapitel „4.2. Die Proteinbiosynthese“ in der Abhandlung „Die Wunderwelt des Lebens“. Dort werden die Grundlagen auf anschauliche Weise erklärt und bieten eine ideale Vorbereitung für das Verständnis dieses Themas.
4.5.1. Nukleinsäureextraktion
Um einen Virus im Detail analysieren zu können, müssen wir an seinen innersten Schatz heran: sein genetisches Material – DNA oder RNA müssen erst einmal ausgegraben werden. Doch die Nukleinsäure liegt gut versteckt, eingepackt in Proteinhüllen, eingebettet in Zelltrümmer oder vermischt mit allerlei molekularem „Beifang“.
Die Aufgabe: Die virale Nukleinsäure aus diesem molekularen Allerelei befreien – sauber, effizient und ohne sie zu beschädigen.
Das Ziel: Möglichst viel, möglichst reine DNA oder RNA – bereit für PCR, Sequenzierung oder Mutationsanalyse. Die Nukleinsäureextraktion ist damit der erste und einer der wichtigsten Schritte jeder molekularen Virusdiagnostik. Je nach Probentyp, Virusart und Untersuchungsziel kommen unterschiedliche Extraktionsmethoden zum Einsatz – von klassischen Kits bis zu automatisierten Hochdurchsatzsystemen.
Wie funktioniert die Nukleinsäureextraktion?
Der Ablauf lässt sich in vier einfache Schritte unterteilen:1️⃣ Zellaufschluss – Erst mal alles aufbrechen
Bevor man an die RNA oder DNA herankommt, müssen die Zellen (und ggf. Viren) in der Probe geknackt werden. Das klappt z. B. durch Ultraschall, Enzyme oder mechanisches Zermahlen. Hauptsache, die Hülle ist weg – und das Genom liegt frei.
Methoden, die ans Eingemachte gehen
Ultraschall: Sonikation zerschlägt die Zell- und Virushüllen mit Schallwellen.
Enzyme: Proteinase K baut Proteine ab, die das Genom einpacken.
Mechanisches Zermahlen: Kleine Glasperlen in einem Röhrchen werden geschüttelt. Die Perlen prallen gegen die Zellen und zerreissen die Zellmembran.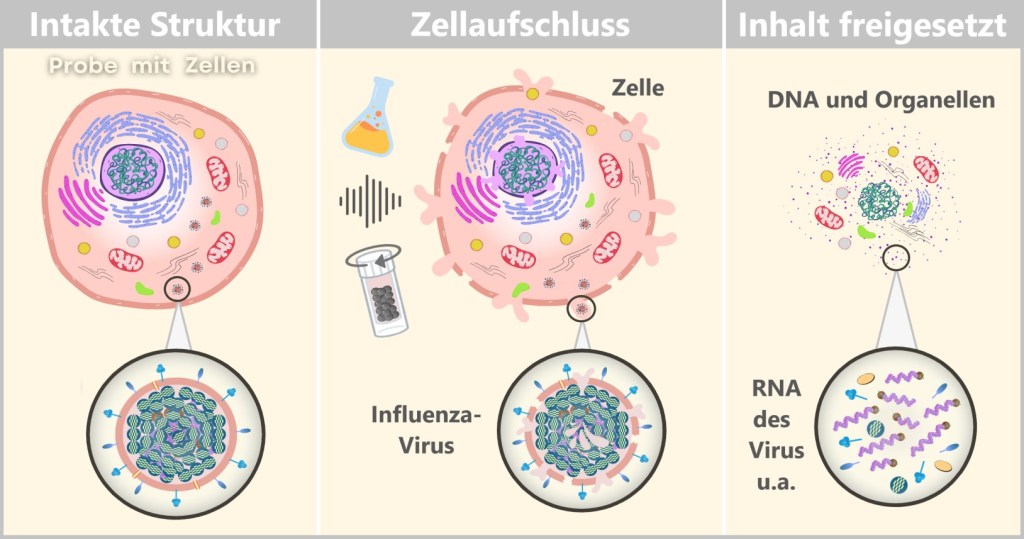
Abb. 12-A: Schritte des Zellaufschlusses zur Freisetzung von viralen und zellulären Bestandteilen Intakte Struktur (links): Eine schematische Darstellung einer intakten Zelle mit Zellorganellen, Zellkern und DNA. Innerhalb der Zelle sind Influenzaviren sichtbar. Zusätzlich wird ein einzelnes Influenzavirus vergrößert dargestellt, mit seiner kugelförmigen Hülle, die aus einer Wirtszellmembran, einem Kapsid und RNA-Strängen besteht.
Zellaufschluss (Mitte): Die Zellmembran ist perforiert dargestellt, wodurch das Zytoplasma austreten kann. Auch die Kernmembran weist Löcher auf. Ein Influenzavirus ist vergrößert dargestellt, mit aufgebrochener Hülle, aus der virale RNA und andere Bestandteile freigesetzt werden. Neben der Zelle sind Symbole für chemische Substanzen (Flasche), physikalische Einwirkungen (Schallwellen) und mechanische Kräfte abgebildet, die die verschiedenen Methoden des Zellaufschlusses veranschaulichen.
Inhalt freigesetzt (rechts): Nach dem Zellaufschluss schwimmen die Organellen, die DNA und andere Zellinhalte frei im Medium. Ebenfalls sichtbar sind die freigesetzten Bestandteile des Influenzavirus, darunter RNA-Segmente, Spikes und andere molekulare Komponenten, die in der Vergrößerung dargestellt werden.2️⃣ Aufräumen – Proteine & Co. rausfischen
Jetzt ist die Probe ein ziemlicher Mix: Nukleinsäuren, Proteine, Fette und Zellreste tummeln sich wild durcheinander. Damit am Ende nur das drin ist, was wir brauchen, kommen Reagenzien oder Enzyme zum Einsatz, die den ganzen Ballast gezielt abbauen.
Methoden, die klar Schiff machen
Phenol-Chloroform-Extraktion: Alt, aber bewährt – trennt Nukleinsäuren zuverlässig von störenden Proteinen und Lipiden. Besonders nützlich bei stark „vermüllten“ Proben.
👉Achtung: Die verwendeten Chemikalien sind ziemlich toxisch – nur für geübte Hände (und mit Schutzbrille!).Proteinase K: Dieses Enzym zersetzt Proteine wie z. B. Membran- oder Strukturproteine, die noch in der Probe herumschwimmen – damit die DNA/RNA ungestört bleibt.
DNase/RNase-Behandlung: Wird eingesetzt, um gezielt nicht-virale DNA oder RNA abzubauen – besonders praktisch, wenn z. B. nur die virale RNA untersucht werden soll.
3️⃣ Reinigung – Die reine RNA oder DNA
Jetzt wird’s elegant: Die Nukleinsäuren werden gezielt gebunden – an spezielle Oberflächen wie Silica-Membranen oder magnetische Kügelchen. Der Rest? Wird einfach weggespült. Eine Art molekulares Sieb – nur smarter.
Methoden, die richtig filtern
Spin Columns (Säulenverfahren): Hier binden DNA oder RNA an eine Spezialmembran, meist aus Silica. Dann heißt es: Spülen, spülen, spülen – bis alles andere raus ist. Am Ende bleibt: schön saubere Nukleinsäure. Diese Methode ist schnell, zuverlässig und steckt in vielen Labor-Kits.
Magnetbeads-Technologie: Winzige magnetische Kügelchen heften sich gezielt an die Nukleinsäuren. Mit einem Magneten werden die passenden Matches dann blitzschnell aus der Suppe gefisch. Geht blitzschnell und eignet sich super für automatische Hochdurchsatz-Verfahren, die tausende Matches pro Stunde arrangieren müssen. Mehr Informationen dazu in den Videos:
DNA and RNA extraction with magnetic beads – How it works und
Nucleic acid purification with chemagic M-PVA Magnetic Bead Technology.4️⃣ Elution – Der große Auftritt
Jetzt kommt der finale Akt: Die gereinigten Nukleinsäuren werden in einer kleinen Menge Flüssigkeit (Elutionspuffer oder Wasser) gelöst – und voilà: Die Probe ist jetzt analysierbereit. Ganz ohne Ballast, dafür mit jeder Menge Potenzial für PCR, Sequenzierung & Co.

Abb. 12-B: Reinigung und Aufbereitung der Nukleinsäuren Links: Der freigesetzte Zell- und Virusinhalt nach dem Zellaufschluss, bestehend aus einer Mischung aus Zellbestandteilen, Proteinen und Nukleinsäuren.
Mitte: Nach der Entfernung von Proteinen und Verunreinigungen bleiben nur die Nukleinsäuren (RNA-Segmente des Virus) übrig, dargestellt als kleine Pünktchen.
Rechts: Vergrößerung einiger RNA-Segmente, die nach der Elution in einer Flüssigkeit gelöst und für die Analyse bereitgestellt sind.Wenn wenig viel zu wenig ist
Auch wenn die Reinigung sorgfältig war: Auf molekularer Ebene ist echte Reinheit kaum zu erreichen. In der Probe können immer noch winzige Störenfriede herumschwirren – Proteine, Salze, andere Moleküle. Das Problem: Die wenigen viralen Nukleinsäuren gehen in diesem „Hintergrundrauschen“ leicht unter – wie ein Flüstern im Konzertsaal.
Und genau hier steht der nächste große Schritt an: die Amplifikation. Dabei wird das virale Erbgut millionenfach vervielfältigt – damit selbst das leiseste Flüstern laut und deutlich hörbar wird.
4.5.2. Nukleinsäure-Amplifikation
Egal wie gut die Extraktion war: In vielen Fällen ist die Ausbeute an viraler DNA oder RNA winzig. Um sie nachweisen oder analysieren zu können, braucht es mehr Kopien – viele mehr. Genau das ist der Job der Amplifikation: Sie vervielfältigt das Erbmaterial millionen- oder sogar milliardenfach – wie ein molekularer Kopierer.
Welche Methode man verwendet, hängt davon ab, was man sucht:
🦠📌 Wenn das Virus bekannt ist – gezielt nachspüren
Bei bekannten Viren weiß man, wo man suchen muss: Bestimmte Abschnitte ihres Genoms sind bereits kartiert. Mit Hilfe passender Primer – kurze Gensequenzen, die exakt an diese Abschnitte binden – kann gezielt ein bestimmter Bereich vervielfältigt werden. Die klassische Methode dafür ist die PCR (Polymerase-Kettenreaktion): präzise, empfindlich und perfekt für den gezielten Nachweis.
🦠❓ Wenn das Virus unbekannt ist – breit ansetzen
Ist das Virus noch ein unbeschriebenes Blatt, fehlen die spezifischen Primer. Dann wird die gesamte virale DNA oder RNA verstärkt – unspezifisch, aber umfassend. Hier kommen Methoden wie die Random Primed Amplification oder Whole Genome Amplification (WGA) zum Einsatz. Sie erzeugen möglichst viele Kopien – egal von welchem Abschnitt – um später durch Sequenzierung herauszufinden, was überhaupt in der Probe steckt.
Nachfolgend werden beide Ansätze näher erläutert:
a) Polymerase-Kettenreaktion (PCR)
b) Random Primed PCR
a) Polymerase-Kettenreaktion (PCR)
Die PCR – kurz für Polymerase-Kettenreaktion (englisch: Polymerase Chain Reaction) – ist so etwas wie der Kopierer im Labor: Mit ihr lassen sich selbst winzige Mengen viraler DNA millionenfach vervielfältigen. Das ist besonders praktisch, wenn man auf Spurensuche geht – um DNA-Viren in einer Probe nachzuweisen.
Aber was ist mit RNA-Viren, wie etwa dem Influenzavirus? Die lassen sich nicht direkt kopieren – erst müssen sie „übersetzt“ werden: Bei der sogenannten RT-PCR (Reverse Transcription PCR) wird die RNA des Virus mithilfe eines Enzyms (Reverse Transkriptase) in DNA umgeschrieben. Danach läuft alles wie bei der normalen PCR: Vervielfältigen, analysieren, fertig.
PCR – Schritt für Schritt
Um beim Beispiel des Influenzavirus zu bleiben, betrachten wir den typischen Ablauf der Reverse-Transkriptase-PCR (RT-PCR), die bei RNA-Viren wie Influenza eingesetzt wird.
1️⃣ Probenentnahme
Alles beginnt mit einem Abstrich – meist aus Nase oder Rachen, denn genau dort treiben sich Influenzaviren gern herum. Das gesammelte Material landet in einem Spezialmedium, das die empfindliche RNA des Virus schützt. Und das ist auch nötig, denn RNA ist ein Sensibelchen: Überall lauern RNasen – Enzyme, die RNA abbauen, und die finden sich fast überall – auf der Haut, in der Luft, in der Probe selbst.
2️⃣ RNA-Extraktion
Die Probe ist ein bunter Cocktail: virale Partikel, menschliche Zellen, Proteine, Lipide – das volle Programm. Wie du schon im Kapitel „Nukleinsäureextraktion“ gelesen hast, wird jetzt das Wesentliche herausgeholt: die RNA. Am Ende bleibt eine saubere Mischung zurück, die vor allem drei Sorten RNA enthalten kann:
➤ virale Genom-RNA (beim Influenzavirus: negativsträngige RNA (-)ssRNA),
➤ virale mRNA, die bei aktiver Infektion in den Wirtszellen gebildet wird,
➤ zelluläre RNA des Wirts.Besonders spannend: Die virale mRNA. Dank eines viralen Tricks – dem sogenannten Cap-Snatching – hat sie eine 5′-Cap-Struktur und einen Poly-A-Schwanz, genau wie unsere eigene mRNA. Das macht sie stabiler und besonders gut geeignet für den nächsten Schritt: die Umschreibung in DNA.
Damit sie fit bleibt für die Analyse, wird die gewonnene RNA in einem stabilisierenden Puffer aufbewahrt.
3️⃣ Umwandlung von RNA in DNA
Bevor die PCR loslegen kann, braucht sie ein Update: Sie arbeitet nur mit DNA, nicht mit RNA. Deshalb muss die virale RNA erst in komplementäre DNA (cDNA) umgewandelt werden. Und das übernimmt ein cleveres Enzym: die Reverse Transkriptase.
So läuft die Umwandlung:
RNA-Vorlage: Die mRNA des Influenzavirus ist ein einzelsträngiges RNA-Molekül mit zwei praktischen Anhängseln: Am 5′-Ende trägt sie eine Cap-Struktur, die sie stabilisiert – am 3′-Ende einen Poly-A-Schwanz, eine Kette aus Adenin-Basen. Beides macht die virale RNA besonders gut zugänglich für den nächsten Schritt.
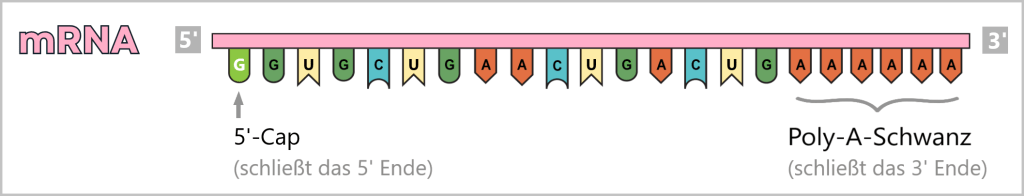
Abb. 13-A: Schematische Darstellung der mRNA des Influenzavirus Primer dockt an: Damit die Reverse Transkriptase weiß, wo sie starten soll, wird ein Primer gebraucht – ein kurzes DNA-Stück, das sich oft gezielt an den Poly-A-Schwanz bindet.
Reverse Transkriptase legt los: Sie setzt sich auf den Primer und liest die RNA-Basen (A, U, G, C) der Vorlage ab. Dann fügt sie die passenden DNA-Basen (A, T, G, C) aneinander – und baut so einen komplementären DNA-Strang. Das Ergebnis ist ein hybrides Molekül aus RNA und DNA (RNA-cDNA-Hybrid).

Abb. 13-B: Reverse Transkriptase synthetisiert cDNA aus RNA Sowohl die 5′-Cap-Struktur als auch der Poly-A-Schwanz der RNA erscheinen nicht in ihrer ursprünglichen Form in der resultierenden cDNA. Sie werden entweder ignoriert (5′-Cap) oder komplementär abgebildet und teilweise verkürzt (Poly-A-Schwanz).
Vom Strang zum Doppelstrang: Dieser erste DNA-Strang dient jetzt selbst als Vorlage – ein zweiter Strang wird ergänzt, sodass eine doppelsträngige cDNA (ds cDNA) entsteht. Diese ist stabil – und bereit für die PCR.
Abb. 13-C: Stabile doppelsträngige cDNA für PCR Wie das Ganze in Aktion aussieht? Diese Animation zeigt die cDNA-Synthese im Schnelldurchlauf – einfach und verständlich erklärt.
4️⃣ Was braucht man für eine PCR?
Bevor es mit der Vervielfältigung losgehen kann, müssen ein paar Zutaten und Werkzeuge bereitstehen:
a) DNA-Vorlage: Das Ausgangsmaterial ist die doppelsträngige cDNA, die im vorherigen Schritt aus viraler RNA hergestellt wurde.
b) Nukleotide – die Bausteine: Vier verschiedene Bausteine braucht es, um DNA zu kopieren: Adenin (A), Thymin (T), Cytosin (C) und Guanin (G). Sie werden später von der Polymerase zu einem neuen Strang zusammengesetzt.
Abb. 14-A: Nukleotide c) DNA-Polymerase – der Baumeister: Dieses Enzym liest die DNA-Vorlage und setzt die passenden Nukleotide zu einem neuen Strang zusammen – präzise und blitzschnell. Bei der PCR kommt oft eine hitzestabile Polymerase (z. B. Taq-Polymerase) zum Einsatz, damit sie die hohen Temperaturen der PCR-Zyklen übersteht.
d) Primer – die Wegweiser: Primer sind kurze DNA-Stücke (meist 18–24 Basen lang), die der Polymerase zeigen, wo sie mit dem Kopieren beginnen soll. Für die PCR braucht man immer zwei: einen Vorwärts- und einen Rückwärts-Primer, die sich an gegenüberliegenden Strängen der Ziel-DNA anlagern.

Abb. 14-B: Schematische Darstellung des Enzyms DNA-Polymerase und der Primer e) Pufferlösung – das richtige Milieu: Sie sorgt dafür, dass die Polymerase sich wohlfühlt: mit stabilem pH-Wert, Magnesiumionen und allem, was das Enzym für eine zuverlässige Arbeit braucht.
f) Thermocycler – das Temperatur-Karussell: Ein Gerät, das die nötigen Temperaturzyklen automatisch durchführt. Er heizt, kühlt und hält die Temperaturen präzise – für die verschiedenen PCR-Schritte in perfektem Timing.

Abb. 14-C: Der Thermocycler steuert die PCR-Temperaturen. 5️⃣ Der Ablauf der Polymerase-Kettenreaktion (PCR)
Alle Zutaten – DNA-Vorlage (ds cDNA), Nukleotide, DNA-Polymerase, Primer und Pufferlösung – kommen in ein kleines Reaktionsröhrchen. Dieses wird anschließend in den Thermocycler gestellt, der die PCR-typischen Temperaturzyklen automatisch durchläuft. Jeder Zyklus besteht aus drei Kernschritten:
Schritt 1 – Trennung der DNA-Stränge (Denaturierung): Die Probe wird auf ca. 94–98 °C für etwa 20-30 Sekunden erhitzt. Dadurch lösen sich die Wasserstoffbrücken zwischen den DNA-Basen – der Doppelstrang „schmilzt“ in zwei Einzelstränge auf. Diese dienen im nächsten Schritt als Vorlage.
Abb. 14-D: Trennung der DNA-Stränge Schritt 2 – Primerbindung (Annealing): Die Temperatur wird auf 50–65 °C abgesenkt. Jetzt binden sich die Primer gezielt an die jeweiligen Einzelstränge. Sie markieren den Startpunkt für die DNA-Synthese. Die verwendeten Primer sind so konstruiert, dass sie nur an virale Sequenzen binden – also nicht an menschliche RNA oder DNA.
Abb. 14-E: Primerbindung Schritt 3 – DNA-Synthese (Amplifikation): Bei etwa 70 °C, der optimalen Temperatur für die Polymerase, beginnt die eigentliche Vervielfältigung. Die DNA-Polymerase bindet an den Primer, liest den Einzelstrang von 3′ nach 5′ – und synthetisiert parallel den neuen Strang in 5′ nach 3′-Richtung. Dabei setzt sie Nukleotide nach dem Prinzip der Basenpaarung zusammen: A mit T und G mit C. So entstehen zwei neue DNA-Doppelstränge.
Abb. 14-F: DNA-Synthese: Dieser Schritt wird als Amplifikation (lat. amplificatio: Verstärkung) oder Elongation (lat. elongare: verlängern) oder Polymerisation (griech. poly: viele; griech. meros: Teil) bezeichnet. 6️⃣ Wiederholung der Zyklen
Die neu entstandenen DNA-Doppelstränge dienen direkt als Vorlage für die nächste Runde. Die Schritte Denaturierung – Primerbindung – DNA-Synthese wiederholen sich zyklisch.
Mit jedem Zyklus verdoppelt sich die DNA-Menge: 1 → 2 → 4 → 8 → 16 → 32 …
Nach nur 25–40 Zyklen entstehen auf diese Weise Milliarden Kopien des gesuchten DNA-Abschnitts.
Abb. 14-G: Wiederholung der Zyklen 📈 Was am Ende der RT-PCR vorliegt:
Eine hochkonzentrierte Lösung spezifischer DNA-Fragmente – direkt abgeleitet von der viralen RNA.
Im Fall des Influenzavirus enthält sie ausschließlich jene Genomabschnitte, nach denen gezielt gesucht wurde. Diese DNA dient nun als Grundlage für weiterführende Analysen:
➤ zur Bestätigung der Virusidentität,
➤ zur Unterscheidung verschiedener Virusvarianten,
➤ oder zur Quantifizierung der Viruslast im Patientenmaterial.🎥 Tipp: Das Video „What is PCR? Polymerase Chain Reaction“ fasst die Abläufe noch einmal anschaulich zusammen. Auch wenn es sich auf menschliche DNA bezieht, bleibt das Grundprinzip exakt das gleiche.
b) Random Primed PCR
💡Hinweis: Wenn du mit der PCR noch nicht vertraut bist, lies zuerst den Abschnitt „PCR – Schritt für Schritt“. Dort findest du die Grundlagen und Abläufe anschaulich erklärt. Die folgenden Ausführungen setzen dieses Wissen voraus und konzentrieren sich speziell auf die Besonderheiten der Random Primed PCR.
Die Random Primed PCR ist eine besondere Variante der PCR, die vor allem dann eingesetzt wird, wenn ein Virus unbekannt ist oder sein Genom stark variiert.
Im Gegensatz zur klassischen PCR, die mit spezifischen Primern gezielt definierte DNA-Abschnitte vervielfältigt, verwendet diese Methode sogenannte „Random Primer“: Kurze, zufällig zusammengesetzte DNA-Sequenzen, die sich an vielen Stellen der Ziel-DNA (oder cDNA) binden können – ganz unabhängig von deren genauer Basenabfolge.
Vorteil: Auch unbekannte oder stark veränderte Bereiche des Genoms lassen sich auf diese Weise mitamplifizieren – ein entscheidender Vorteil bei der Vorbereitung auf eine Sequenzierung, bei der die exakte Basenreihenfolge bestimmt wird.
Beispiele für Random Primer:
Hexamer Primers (6 Basen lang): AGCTGA, CTAGCT, …
Heptamer Primers (7 Basen lang): CCTGAGT, GATTACA, …
Nonamer Primers (9 Basen lang): GCAGTTCGC, ATGGCCGTA, …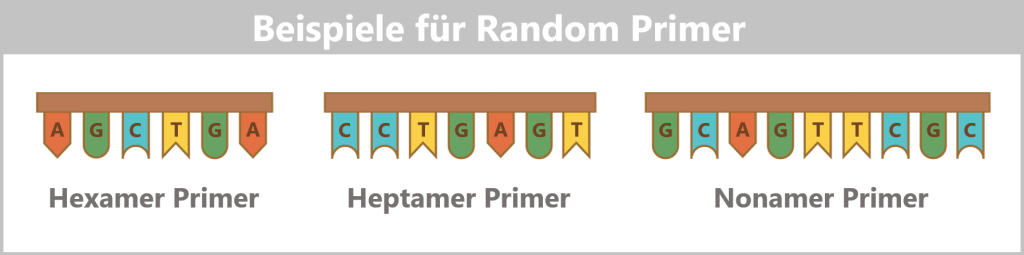
Abb. 15-A: Beispiele für Random Primer In der Praxis kommen meist Mischungen aller möglichen Primer-Varianten zum Einsatz (z. B. 4⁶ = 4096 Kombinationen bei Hexamern). Das sorgt dafür, dass möglichst viele Bindungsstellen im Genom erreicht werden.
Ablauf der Random Primed PCR
Schritt 1 – Denaturierung: Die DNA (oder cDNA) wird auf ca. 95 °C erhitzt, um die beiden Stränge zu trennen. Es entstehen Einzelstränge, an die die Primer binden können.
Schritt 2 – Primerbindung: Die Temperatur wird gesenkt, sodass sich die zufälligen Primer an viele verschiedene Stellen der DNA anlagern. Da die Primer zufällig sind, können sie an vielfältige Positionen im Genom binden.
Hinweis zur Genomgröße: Viren haben sehr unterschiedliche Genomlängen – von wenigen Tausend bis zu Hunderttausenden Basenpaaren. Random Primer helfen, möglichst viel davon abzudecken.
Abb. 15-B: Schematische Darstellung der Primerbindung während der Random Primed PCR. Schritt 3 – DNA-Synthese: Die DNA-Polymerase bindet an die Primer und beginnt, von dort aus neue DNA-Stränge zu synthetisieren. Dabei liest sie die Vorlage, bis sie entweder ans Ende gelangt oder auf einen weiteren Primer stößt. Dieser wirkt wie ein Stoppsignal – das Ergebnis sind meist kürzere DNA-Fragmente.
Genau das ist gewollt: Die Polymerase erzeugt viele kurze, sich überlappende Fragmente, die später für die Genomrekonstruktion oder gezielte Analysen genutzt werden können.
Abb. 15-C: Nach der Aktivität der DNA-Polymerase sind aus den Einzelsträngen mehrere kurze, sich überlappende DNA-Doppelstränge entstanden. 🔁 Zyklische Wiederholung
Dieser Prozess wird – wie bei der klassischen PCR – mehrfach wiederholt, meist 20 bis 40 Zyklen.
- Je mehr Zyklen, desto mehr DNA entsteht.
- Mit steigender Zykluszahl steigt auch die Wahrscheinlichkeit, dass Primer sich gegenseitig „abfangen“ → kürzere Fragmente entstehen.
- Aber: Zu viele Zyklen können die Vielfalt verringern, weil manche Regionen überrepräsentiert werden.
Eine ausgewogene Zahl an Zyklen sowie eine gut abgestimmte Primer-Mischung sind entscheidend für die Qualität und Repräsentativität des Endprodukts.
📈 Ergebnis der Random Primed PCR
Am Ende liegt eine große Anzahl an kurzen, überlappenden DNA-Fragmenten vor – keine vollständigen Stränge, sondern ein Fragmentteppich, der das gesamte virale Genom abdecken kann.

Abb. 15-D: Ergebniss der Random Primed PCR: das ganze Genom – nur in Häppchen! Aus vielen kurzen DNA-Schnipseln entsteht ein genetisches Mosaik. Die Fragmente überlappen sich und lassen sich später wie Puzzlestücke zu einem vollständigen Virusgenom zusammensetzen.
🧩 Was passiert mit den DNA-Schnipseln?
Direkte Analyse einzelner Fragmente
Oft reicht es, gezielt bestimmte Fragmente zu sequenzieren, um wichtige Informationen zu erhalten – z. B. zur Mutationsanalyse oder Typisierung eines Virus.Rekonstruktion des gesamten Genoms
Wenn das gesamte Genom analysiert werden soll (z. B. zur Entdeckung neuer Viren oder zur Erstellung von Verwandtschaftsanalysen), werden die Fragmente mithilfe von Sequenziertechnologien ausgelesen (mehr dazu im kommenden Kapitel). Spezielle Softwareprogramme setzen die sich überlappenden Fragmente anschließend wie ein Puzzle wieder zusammen.
🧬 Vom Fragment zum vollständigen Genom
Die Amplifikation mithilfe der Random Primed PCR ist nur der erste Schritt – sie sorgt dafür, dass genügend genetisches Material für weiterführende Untersuchungen vorliegt. Doch um wirklich zu verstehen, mit welchem Virus man es zu tun hat, reicht die bloße Vervielfältigung nicht aus.
Jetzt geht es darum, die genaue Abfolge der Basenpaare in den erzeugten DNA-Fragmenten zu bestimmen – also ihre Sequenz. Erst durch diese Sequenzierung lässt sich das genetische Profil des Virus entschlüsseln: Man erkennt Mutationen, kann Virenstämme unterscheiden und sogar evolutionäre Stammbäume erstellen.
Im folgenden Kapitel schauen wir uns deshalb an, wie Sequenzierung funktioniert, welche Technologien dafür eingesetzt werden – und wie aus vielen kleinen DNA-Schnipseln ein vollständiges virales Genom rekonstruiert wird.
4.5.3. Sequenzierung
Die Sequenzierung ist DER entscheidende Schritt, um das Erbgut von Viren zu entschlüsseln. Dabei wird die exakte Reihenfolge der Basen in der DNA (A, T, C, G) oder RNA (A, U, C, G) bestimmt.
Was bringt das?
Eine Virus-Genomsequenz ist der QR-Code der Biologie: Einmal gescannt, weiß man sofort, womit man’s zu tun hat.
Identifikation des Virus:
🔹 Welches Virus ist es genau?
🔹 Ist das Virus bereits bekannt oder handelt es sich um eine neue Entdeckung?
🔹 Zu welcher Virusfamilie oder Gattung gehört es?Erkennung von Mutationen:
🔹 Hat sich das Virus verändert – wenn ja, wie?
🔹 Sind neue Varianten oder Stämme entstanden?
🔹 Welche genetischen Veränderungen bestehen im Vergleich zu früheren Versionen?Bestimmung diagnostischer Marker:
🔹 Gibt es stabile (konservierte) Abschnitte im Genom, die sich gut für Tests eignen?
🔹 Gibt es Gene oder Proteine, die einzigartig für dieses Virus sind?
🔹 Lassen sich bestimmte Gene oder Proteine gezielt nutzen – z. B. für Medikamente?Sequenzierung verpasst jedem Virus einen genetischen Fingerabdruck – einzigartig, präzise und fälschungssicher.
🧬 Vom Reagenzglas zur Hochtechnologie
Die Entschlüsselung der Virus-DNA war früher Handarbeit – heute ist sie Hightech. Moderne Sequenzierer analysieren Millionen DNA-Fragmente gleichzeitig – schnell, automatisiert und hochpräzise.
Seit den ersten manuellen Verfahren hat sich die Technologie rasant weiterentwickelt. Jede neue Generation hat den Blick ins Erbgut klarer, schneller und umfassender gemacht.
Die folgende Übersicht zeigt, wie sich die Sequenzierung verändert hat – und welche Technologien heute zur Verfügung stehen:
Generation Beschreibung First Generation:
Sanger-SequenzierungDer Oldtimer: langsam, aber präzise. Ideal für kurze Abschnitte. Second Generation:
Next-Generation Sequencing (NGS)Die Hochdruckpresse: sequenziert Millionen DNA-Fragmente gleichzeitig. Third Generation:
Echtzeit-SequenzierungDer Quantensprung – Einzelmolekül-Analyse in Echtzeit. Emerging Technologies Science Fiction wird zur Realität. First Generation: Sanger-Sequenzierung
Die Sanger-Sequenzierung, benannt nach dem britischen Biochemiker Frederick Sanger, markiert einen historischen Wendepunkt in der Molekularbiologie. Mit dieser Methode gelang es ihm erstmals, die genaue Abfolge der DNA-Basen zu entschlüsseln – ein wissenschaftlicher Durchbruch, der ihm 1980 seinen zweiten Nobelpreis für Chemie einbrachte.
Obwohl heute modernere und automatisierte Verfahren zur Verfügung stehen, gilt die Sanger-Sequenzierung noch immer als Goldstandard, wenn es um höchste Genauigkeit bei kurzen DNA-Abschnitten geht. Sie wird bis heute in vielen Labors zur Validierung von Ergebnissen eingesetzt.
Sie basiert auf dem Prinzip des Abbruchs der DNA-Synthese. Dabei wird die DNA kopiert und durch den Einbau spezieller Stopp-Signale entstehen DNA-Fragmente unterschiedlicher Länge. Diese werden nach Größe sortiert, sodass die Reihenfolge der Bausteine abgelesen werden kann.
So funktioniert der Klassiker der DNA-Analyse
1️⃣ DNA-Denaturierung
Zunächst wird die doppelsträngige DNA erhitzt. Durch die hohe Temperatur lösen sich die Wasserstoffbrücken zwischen den Basenpaaren, und der Doppelstrang trennt sich in zwei Einzelstränge. Diese dienen später als Vorlage für die Synthese neuer DNA.
Abb. 16-A: DNA-Denaturierung – aus einem DNA-Doppelstrang entstehen zwei Einzelstränge. 2️⃣ Vorbereitung der Reaktionsmischungen
Es werden vier separate Reaktionsmischungen hergestellt. Jede enthält:
➥ DNA-Einzelstränge: die Vorlage.
➥ Primer: ein kurzer Abschnitt, der der Polymerase den Startpunkt vorgibt.
➥ DNA-Polymerase: das Enzym, das neue Stränge synthetisiert.➥ dNTPs (desoxyNukleosidTriPhosphate): die „normalen“ Bausteine der DNA (A, T, C, G). Sie ermöglichen die Verlängerung der DNA-Kette, da ihre Hydroxylgruppe am 3’-Ende eine Verbindung mit dem nächsten Baustein eingehen kann.
➥ ddNTPs (didesoxyNukleosidTriPhosphate): diese „modifizierten“ DNA-Bausteine besitzen keine Hydroxylgruppe und können deshalb keine weiteren Bausteine an sich binden. Wird ein ddNTP während der Synthese eingebaut, stoppt der Aufbau des DNA-Strangs genau an dieser Stelle. Jeder der vier ddNTP-Typen (A, T, C, G) ist zusätzlich mit einem eigenen fluoreszierenden Farbstoff markiert.
Das Verhältnis von dNTPs zu ddNTPs wird so gewählt, dass möglichst viele unterschiedlich lange Fragmente entstehen.
Abb. 16-B: Vier Reaktionsmischungen – je eine pro ddNTP-Typ. 3️⃣ DNA-Synthese mit zufälligem Stopp
DNA-Synthese – kurz erklärt: Die DNA-Polymerase ist der beste Kopierer der Natur: Sie scannt einen DNA-Strang wie eine Vorlage und baut Schritt für Schritt den passenden Gegenstrang – immer nach dem Baukasten-Prinzip: A paart nur mit T und C nur mit G. Heraus kommt eine perfekte Spiegelkopie.
Die DNA-Synthese findet gleichzeitig in den vier separaten Reaktionsmischungen statt – eine für jede der vier Basen (A, T, C, G).
In jeder Mischung läuft Folgendes ab: Die DNA-Polymerase setzt sich auf den Primer und beginnt mit der Arbeit.
Wie findet man den richtigen Primer?
Für den erfolgreichen Einsatz der Sanger-Methode sind Primer unbedingt notwendig. Doch wie findet man die Primer-Sequenz, wenn das Genom eines Virus völlig unbekannt ist?
Als Frederick Sanger 1977 seine Methode entwickelte, wusste man bei kleinen Viren wie dem Bakteriophagen Phi-X174 bereits einiges über die Genomstruktur. Forscher hatten herausgefunden, dass bestimmte Enzyme – sogenannte Restriktionsenzyme – DNA an ganz bestimmten Stellen schneiden. Diese Schnittstellen waren bekannt und konnten gezielt genutzt werden:
In der Nähe dieser Schnittstellen lag oft schon ein kurzes Stück bekannter DNA-Sequenz – genau genug, um einen passenden Primer zu entwerfen. So schuf man sich künstlich einen definierten Startpunkt für die Sequenzierung.
Heutzutage ist das einfacher. Die Wissenschaft hat sich stark weiterentwickelt, und die meisten Viren sind bereits gut untersucht. Bei neuen oder wenig bekannten Viren geht man wie folgt vor:
Vergleich mit bekannten Viren: Oft ähnelt ein neues Virus genetisch bereits bekannten Viren. Wissenschaftler nutzen diese Ähnlichkeiten, um mögliche Primerstellen vorherzusagen.
Experimentelle Ansätze: Wenn wenig bekannt ist, schneiden Enzyme die Virus-DNA in kleinere Stücke. Forscher analysieren diese Fragmente, um Startpunkte für Primer zu finden.
Sie liest den Einzelstrang der DNA-Vorlage und fügt passende Bausteine (dNTPs) ein, um einen neuen Strang zu erzeugen.
Normalerweise wird ein „klassisches“ dNTP eingebaut – damit kann die Kette wachsen. Gelegentlich wird aber ein „modifiziertes“ ddNTP eingebaut – und hier stoppt die Synthese sofort. Weil ddNTPs eine kleine chemische Gruppe (Hydroxylgruppe) fehlt, die für das Anknüpfen des nächsten Bausteins notwendig wäre.
So entstehen viele DNA-Fragmente mit unterschiedlicher Länge – jedes endet genau an der Stelle, wo zufällig ein ddNTP eingebaut wurde. Und jedes Fragment trägt am Ende eine fluoreszierende Farbe, die anzeigt, mit welchem Basentyp (A, T, C oder G) das Fragment endet.
Abb. 16-C: Die Synthese wird gezielt unterbrochen – bei Einbau eines ddNTPs. In jeder Reaktionsmischung ist ein modifizierter Nukleotid-Baustein (ddATP, ddTTP, ddCTP, ddGTP) enthalten. Wenn dieser Baustein während der DNA-Synthese eingebaut wird, stoppt die Synthese an genau dieser Position, da der modifizierte DNA-Baustein keine weitere Verlängerung der DNA-Kette erlaubt. In der ddATP-Mischung stoppt die Synthese, sobald Adenin (A) eingebaut wird. In der ddTTP-Mischung erfolgt der Abbruch, wenn Thymin (T) eingebaut wird. Analog dazu führen ddCTP und ddGTP zum Abbruch bei Cytosin (C) bzw. Guanin (G). Dieses Verfahren erzeugt DNA-Fragmente unterschiedlicher Längen, die jeweils mit dem spezifischen Stopp-Nukleotid enden. Ziel ist es, alle theoretisch möglichen Sequenzfragmente herzustellen.
4️⃣ Denaturierung der Fragmente
Die entstandenen doppelsträngigen DNA-Fragmente werden erneut erhitzt, damit sie sich wieder in Einzelstränge trennen. Nur so können sie später einzeln analysiert werden. Übrig bleibt einzelsträngige DNA – bereit für den nächsten Schritt.
5️⃣ Auswertung
Nun geht’s an die Analyse: Die Fragmente werden mit Hilfe der Gelelektrophorese nach ihrer Größe sortiert. Dafür werden die vier Reaktionsmischungen in verschiedene Taschen eines Gels gefüllt.
Das Gel funktioniert wie ein feines Netz oder Schwamm:
- Kurze Fragmente rutschen schneller hindurch.
- Längere Fragmente bewegen sich langsamer.
Jeder Strang endet mit einem farblich markierten ddNTP – je nach Base (A, T, C oder G) leuchtet ein anderer Farbton auf. Diese Farben werden mithilfe eines Lasers detektiert und automatisch aufgezeichnet.
Abb. 16-D: Gelelektrophorese zur Trennung von DNA-Fragmenten: In den Reaktionsgefäßen befinden sich unterschiedlich lange DNA Fragmente, die jeweils mit dem gleichen Stopp-Nukleotid (je nach Ansatz A, T, G und C) enden. Die Reaktionsmischungen werden auf das Gel aufgetragen. Durch das elektrische Feld wandern die negativ geladenen DNA-Fragmente von der Kathode (−) zur Anode (+). Die Fragmentgröße bestimmt die Wandergeschwindigkeit durch das Gel. Kleinere DNA-Fragmente bewegen sich schneller durch die Poren des Gels und erreichen daher zuerst die Anode, während größere Fragmente langsamer vorankommen. Durch das Lesen der fluoreszierenden Signale kann die Basenreihenfolge der DNA-Sequenz bestimmt werden.
Wie liest man daraus die Sequenz?
Die Reihenfolge der Fragmente im Gel entspricht der Reihenfolge der Basen im ursprünglichen DNA-Strang.
- Das kürzeste Fragment zeigt die erste Base.
- Das nächstlängere Fragment die zweite,
- … und so weiter, bis die gesamte Sequenz entschlüsselt ist.
Da jedes neue Fragment komplementär zum Ursprungsstrang ist, lässt sich aus der Analyse direkt die Basenfolge des Original-DNA-Strangs ableiten.
🎥 Tipp: Eine sehr anschauliche Erklärung findest du im Video „Sanger Sequencing / Chain Termination Method“.
Wo wird die Sanger-Sequenzierung eingesetzt?
Die Methode eignet sich besonders gut für:
- Kurze DNA-Abschnitte
- Bestätigung und Kontrolle von Ergebnissen aus anderen Methoden
- Einzelfallanalysen, z. B. in der medizinischen Diagnostik
Manche Methoden werden nicht alt – sie werden klassisch.
Grenzen der Methode
So präzise die Sanger-Sequenzierung auch ist – bei großen oder komplexen Genomen stößt sie schnell an ihre Grenzen. Die Methode ist aufwendig, zeitintensiv und teuer, besonders wenn viele Proben oder umfangreiche Datensätze analysiert werden sollen. Deshalb wurde sie in vielen Bereichen durch moderne Hochdurchsatzverfahren ersetzt – allen voran durch die Technologien der Next-Generation Sequencing (NGS).
Second Generation: Next Generation Sequencing (NGS)
Next-Generation Sequencing (NGS) ist eine moderne Methode zur Entschlüsselung von DNA- und RNA-Sequenzen – und hat die genetische Forschung seit Beginn des 21. Jahrhunderts grundlegend verändert. Im Vergleich zur klassischen Sanger-Sequenzierung ist NGS schneller, kostengünstiger und verarbeitet deutlich größere Datenmengen in kürzerer Zeit.
Der entscheidende Unterschied: Während bei der Sanger-Methode jeweils nur ein einzelnes DNA-Fragment sequenziert wird, kann NGS Millionen von Fragmenten gleichzeitig auslesen. Dazu wird das Erbmaterial zunächst in kleine Stücke zerlegt und mit speziellen Markierungen versehen. Diese Fragmente werden auf einer speziellen Oberfläche fixiert und mithilfe fluoreszierender Nukleotide schrittweise ergänzt – wobei jeder neu eingebaute Baustein sofort ausgelesen wird.
Durch dieses parallele Arbeiten entsteht innerhalb kurzer Zeit ein hochauflösender Datensatz – ideal für die Analyse ganzer Genome, RNA-Profile oder großer Probenmengen. Genau deshalb ist NGS heute eine Schlüsseltechnologie in der Forschung, Diagnostik und Biotechnologie.
Ein tiefer Blick in die Illumina-Sequenzierung
Die Illumina-Sequenzierung ist eine der am häufigsten eingesetzten Technologien im Bereich des Next-Generation Sequencing. Sie basiert auf dem Prinzip „Sequencing by Synthesis“ und ermöglicht das gleichzeitige Auslesen von Millionen DNA-Fragmenten. Hier folgt eine Schritt-für-Schritt-Erklärung:
Schritt 1: DNA-Präparation = Bibliothekserstellung
Schritt 2: Cluster-Generierung auf einer Flowcell
Schritt 3: Sequenzierung durch Synthese
Schritt 1: DNA-Präparation = Bibliothekserstellung
Für die Sequenzierung wird die DNA zunächst in ein Format gebracht, das für die Illumina-Plattform geeignet ist. Dieser Prozess wird auch als Bibliothekserstellung bezeichnet und er umfasst folgende Teilschritte:
1a) Fragmentierung
Die DNA wird mechanisch oder enzymatisch in kleinere Stücke zerlegt (typischerweise mit einer Zielgröße von etwa 150–500 Basenpaaren). Dabei entstehen DNA-Fragmente leicht unterschiedlicher Länge, die anschließend oft noch über ein Größenauswahlverfahren vereinheitlicht werden.
Zur Veranschaulichung betrachten wir in unserem Beispiel drei DNA-Fragmente mit leicht unterschiedlichen Längen.
1b) Adapter-Ligation
An beide Enden der DNA-Fragmente werden spezifische Adaptersequenzen (P5- und P7-Adapter) angehängt. Diese Adapter erfüllen mehrere Funktionen:
Bindungsstellen für die Flowcell: Die Adapter enthalten spezielle DNA-Sequenzen, die wie ein Schlüssel zu einem Schloss passen. Dadurch können die DNA-Fragmente später an einer Oberfläche haften bleiben, was für die Sequenzierung wichtig ist.
Primer-Bindungsstellen: Die Adapter enthalten Abschnitte, an die Sequenzierungsprimer binden können. Diese Primer werden später genutzt, um die DNA-Stränge schrittweise zu synthetisieren.
Indizes (optional): Falls mehrere Proben gleichzeitig sequenziert werden, ermöglichen Index-Sequenzen die Zuordnung der Fragmente zu ihrer jeweiligen Probe.
Zur besseren Übersichtlichkeit verzichten wir in unserem Beispiel auf die Darstellung der Indizes.
1c) PCR-Amplifikation
Um sicherzustellen, dass genügend DNA für die Sequenzierung vorliegt, werden die adaptierten DNA-Fragmente mittels Polymerase-Kettenreaktion (PCR) vervielfältigt.
Abb. 17-A: Bibliothekserstellung – Übersicht über den Ablauf der DNA-Präparation. Die folgende Vergrößerung zeigt eine detaillierte Darstellung eines fragmentierten DNA-Doppelstrangs, versehen mit den beiden Adaptern.
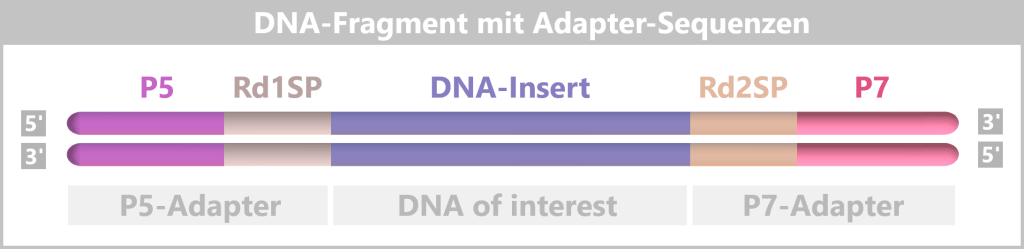
Abb. 17-B: Schematische Darstellung eines doppelsträngigen DNA-Fragments mit Adaptern. Jedes DNA-Fragment (DNA-Insert) erhält zwei Adaptersequenzen: Der P5-Adapter besteht aus der P5-Adaptersequenz, die die Bindung an die Flowcell ermöglicht, und der Primer-Bindestelle Rd1SP (Read 1 Sequencing Primer) zur Initiation der DNA-Synthese. Ebenso enthält der P7-Adapter die P7-Adaptersequenz zur Flowcell-Bindung sowie die Primer-Bindestelle Rd2SP (Read 2 Sequencing Primer) für die Initiation der DNA-Synthese.
Wichtige Anmerkung: Die oberen und unteren DNA-Einzelstränge besitzen jeweils P5- und P7-Adapter. Allerdings sind die Adapter des unteren Strangs nicht identisch, sondern komplementär zu den Adaptern des oberen Strangs.

Abb. 17-C: DNA-Fragment (DNA-Insert) mit Adaptersequenzen in Basenschreibweise Diese Darstellung ist stark vereinfacht. In der Praxis sind Illumina-Adaptersequenzen mit 60-120 Basenpaarendeutlich länger (siehe hier). Auch das DNA-Insert ist zur besseren Veranschaulichung verkürzt – tatsächlich haben DNA-Fragmente typischerweise eine Länge von 100–500 Basenpaaren.
Schritt 2: Cluster-Generierung auf einer Flowcell
Bevor die DNA sequenziert werden kann, muss sie an einer festen Oberfläche fixiert und vervielfältigt werden. Dies geschieht auf einer Flowcell, einer speziellen Glasplatte, die mit Millionen winziger DNA-Andockstellen versehen ist.
Das Ziel dieses Schrittes ist es, zahlreiche Kopien jedes DNA-Fragments auf dieser Oberfläche zu erzeugen, um die Signale bei der Sequenzierung zu verstärken. Dafür ist die Flowcell mit speziellen Oligonukleotiden (DNA-Molekülen) beschichtet, die komplementär zu den Adaptersequenzen der DNA-Fragmente sind. Es gibt zwei Typen dieser Oligonukleotide:
- P5-Oligonukleotide (ACGTAC), die an die P5-Adapter (TGCATG ) der DNA-Fragmente binden.
- P7-Oligonukleotide (ACGTCA), die mit den P7-Adaptern (TGCAGT ) der DNA interagieren.
Man kann sich die Oberfläche der Flowcell wie einen dichten Klettverschluss vorstellen: Die DNA-Fragmente haften mit ihren Adaptersequenzen daran, ähnlich wie kleine Haken, die in das Klettgewebe greifen.
Abb. 17-D: Schematische Darstellung einer Flowcell und deren Oberfläche 2a) DNA-Fragmente binden an die Flowcell-Oberfläche
Zu Beginn des Schritts wird eine Lösung mit einzelsträngigen DNA-Fragmenten, die Adaptersequenzen tragen, auf die Flowcell gespült. Diese Fragmente wurden zuvor durch Denaturierung in Einzelstränge aufgetrennt, sodass sie sich frei in der Lösung bewegen können.
Während die DNA-Fragmente durch die Flowcell strömen, binden ihre Adapter durch komplementäre Basenpaarung an die passenden Oligonukleotiden auf der Flowcell, wie in der unteren Abbildung gezeigt.

Abb. 17-E: Die Grafik zeigt zwei DNA-Einzelstränge, die mit ihren Adaptern an komplementäre Oligonukleotide auf der Flowcell binden. P5-Adapter eines DNA-Einzelstrangs bindet mit dem P5-Oligonukleotid auf der Flowcell:
Flowcell — P5-Oligo ACGTAC
— (3') TGCATG-CAGT-[Insert]-GTCA-ACTGCA (5')P7-Adapter eines DNA-Einzelstrangs bindet mit dem P7-Oligonukleotid auf der Flowcell:
Flowcell — P7-Oligo ACGTCA
— (3') TGCAGT-TGAC-[Insert]-ACTG-CATGCA (5')2b) Erste Synthese
Primerbindung und DNA-Synthese
Nachdem die DNA-Fragmente an die Flowcell gebunden wurden, startet die erste DNA-Synthese. Dazu binden sich spezifische Primer an die Adaptersequenzen der gebundenen Stränge:- Primer GTCA bindet an den P5-Adapter des gebundenen Einzelstrangs.
- Primer ACTG bindet an den P7-Adapter des gebundenen Einzelstrangs.
Die DNA-Polymerase, die nur in 5′ → 3′-Richtung arbeiten kann, synthetisiert daraufhin einen neuen Strang, der komplementär zum bereits gebundenen Einzelstrang ist.
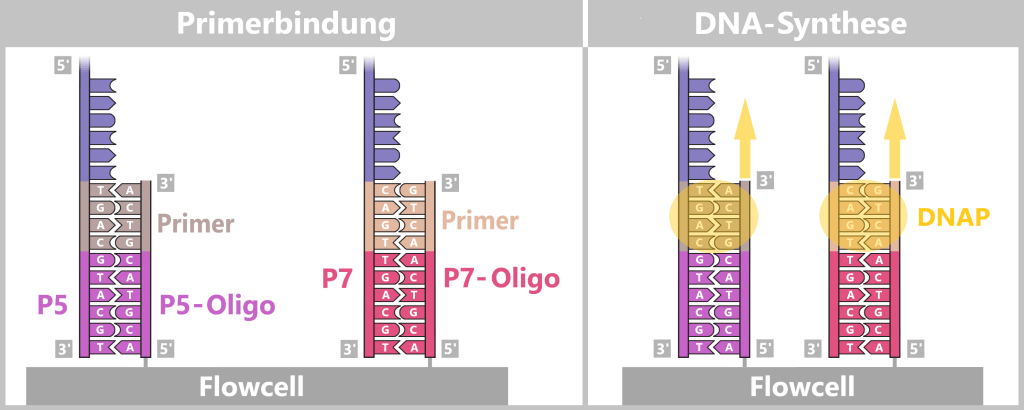
Abb. 17-F: Primer binden an die Adapter (P5, P7), und die DNA-Polymerase (DNAP) startet die Synthese eines neuen, komplementären Strangs. Nach der Synthese liegen die gebundenen DNA-Fragmente als Doppelstränge vor.
Flowcell (P5-Oligo) — (5') ACGTAC-GTCA-[Insert]-CAGT-TGACGT (3')
— (3') TGCATG-CAGT-[Insert]-GTCA-ACTGCA (5')Flowcell (P7-Oligo) — (5') ACGTCA-ACTG-[Insert]-TGAC-GTACGT (3')
— (3') TGCAGT-TGAC-[Insert]-ACTG-CATGCA (5')
Abb. 17-G: Erste Synthese und Auftrennung: So entsteht der gebundene DNA-Strang. Trennung des Doppelstrangs
Nachdem die neue DNA synthetisiert wurde, wird der Doppelstrang durch Denaturierung getrennt (siehe obere Abbildung):❌ Der ursprüngliche Strang hat nach der Denaturierung keine Verbindung mehr zur Flowcell und wird ausgespült.
✅ Der neu synthetisierte Strang bleibt mit seinem 5′-Ende fest an der Flowcell gebunden, während das 3′-Ende frei bleibt.
Nun sind die DNA-Fragmente als komplementäre Einzelstränge (Forward- und Reverse-Stränge) an die Flowcell gebunden.
Allerdings wäre eine direkte Sequenzierung zu diesem Zeitpunkt nicht möglich, da die Fluoreszenzsignale noch zu schwach wären, um zuverlässig ausgelesen zu werden. Deshalb folgt nun die Brückenamplifikation.
2c) Brückenamplifikation
Damit die DNA-Sequenzierung ein ausreichend starkes Fluoreszenzsignal liefert, müssen die einzelnen DNA-Stränge vervielfältigt werden. Dies geschieht durch die Brückenamplifikation, einen zyklischen Prozess, bei dem sich die DNA-Stränge an die Flowcell anlagern, vervielfältigt und wieder getrennt werden.
Hybridisierung des zweiten Adapters
Die Brückenamplifikation beginnt damit, dass sich der gebundene DNA-Einzelstrang biegt und mit seinem freien 3′-Ende an ein benachbartes komplementäres Oligonukleotid (5′) auf der Flowcell bindet. Dieser Vorgang wird als Faltung des DNA-Strangs bezeichnet. Dadurch bildet sich eine Art Brücke, die den Adapter des Strangs mit dem passenden Oberflächenmolekül verbindet.DNA-Synthese
Sobald die DNA-Stränge gebunden sind, werden Primer (ACTG, GTCA) und die DNA-Polymerase hinzugefügt. Die Primer binden spezifisch an die Adaptersequenzen. Die DNA-Polymerase synthetisiert neue komplementäre Stränge in 5′ → 3′-Richtung. Nach der Synthese liegt die DNA nun wieder als Doppelstrang vor.Abb. 17-H: Brückenbildung und DNA-Synthese Die gebundenen Einzelstränge falten sich und hybridisieren mit einem komplementären Oligonukleotid auf der Flowcell, wodurch eine Brückenstruktur entsteht. Anschließend bindet ein Primer an den 3′-Enden der Stränge, und die DNA-Polymerase synthetisiert die komplementären Stränge in 5′ → 3′-Richtung.
Denaturierung– Trennung der Stränge
Durch Hitze oder chemische Behandlung werden die neu gebildeten Doppelstränge wieder in Einzelstränge aufgetrennt. Jetzt befinden sich vier einzelsträngige DNA-Fragmente auf der Flowcell:|| die ursprünglichen zwei Einzelstränge (Forward-Strang & Reverse-Strang) und
|| die neu synthetisierten komplementären Stränge (Forward-Strang & Reverse-Strang).Abb. 17-I: Vorwärts und rückwärts: Die Brücken lösen sich. Nach der Denaturierung werden die Brücken-Doppelstränge getrennt, sodass auf der Flowcell nun jeweils zwei Einzelstränge (vorwärts und rückwärts) zu sehen sind. Diese Einzelstränge sind weiterhin an der Flowcell gebunden und bereit für die weitere Amplifikation.
Wiederholung der Amplifikation
Dieser Zyklus wiederholt sich: Die Einzelstränge falten sich erneut, hybridisieren wieder mit den Oligonukleotiden und werden durch DNA-Synthese vervielfältigt.
Abb. 17-J: Mit jeder Wiederholung der Brückenamplifikation entstehen immer mehr DNA-Kopien. Nach mehreren Zyklen entstehen auf der Flowcell Millionen identischer Kopien jedes ursprünglichen DNA-Fragments – die Cluster.
Abb. 17-K: Clusterbildung Jedes Cluster besteht aus zahlreichen Kopien eines einzelnen DNA-Fragments. In dieser Darstellung sind exemplarisch nur drei Cluster gezeigt, da unser Beispiel von drei DNA-Fragmenten ausgeht. In der Realität befinden sich jedoch Millionen bis Milliarden solcher Cluster auf einer Flowcell, um eine hohe Sequenzierkapazität zu ermöglichen.
Bildung der finalen Cluster
Zu Beginn unseres Beispiels sind drei DNA-Fragmente vorhanden. Nach mehreren Amplifikationszyklen entstehen für jedes dieser Fragmente unzählige Cluster, die ausschließlich aus ihren DNA-Kopien bestehen.Jedes Cluster setzt sich nun aus zwei Strang-Typen zusammen:
🔹 Forward-Stränge (5′ → 3′, P7-gebunden)
🔹 Reverse-Stränge (3′ → 5′, P5-gebunden)💡 Beachte: Forward- und Reverse-Stränge sind in den Clustern nicht antiparallel zueinander ausgerichtet! Stattdessen ragen die 3′-Enden beider Stränge nach oben.
Entfernung der Reverse-Stränge
Damit die Sequenzierung korrekt ablaufen kann, müssen sich alle DNA-Stränge innerhalb eines Clusters in derselben Richtung befinden. Für die eigentliche Analyse werden nur die Forward-Stränge benötigt. Daher folgt nun ein gezielter Reinigungsschritt:🔹 Alle Reverse-Stränge (P5-gebunden) werden abgetrennt und ausgespült.
🔹 Gleichzeitig werden die freien Enden der P5-Oligos chemisch blockiert, um eine erneute Bindung zu verhindern.Nun befinden sich ausschließlich Vorwärtsstränge auf der Flowcell. Die Cluster sind vollständig ausgebildet, und die DNA-Bibliotheken sind bereit für die Sequenzierung.

Abb. 17-L: Sequenzierbereit: Nur noch Forward-Stränge an Bord Links: Ein finales Cluster mit Forward- (P7-gebunden) und Reverse-Strängen (P5-gebunden). Rechts: Die Reverse-Stränge wurden gezielt durchtrennt und ausgespült, sodass nur noch die Forward-Stränge verbleiben. Diese Ausrichtung ist notwendig, um die Sequenzierung korrekt durchzuführen.
Schritt 3: Sequenzierung durch Synthese
An diesem Punkt sind einzelne klonale Cluster (aus den ursprünglich drei DNA-Fragmenten) über die gesamte Oberfläche der Flowcell verteilt. Jedes Cluster besteht aus zahlreichen identischen DNA-Kopien – vergleichbar mit einer kleinen Insel identischer Bäume. Nun kann die eigentliche Sequenzierung beginnen.
Um die Sequenzierung zu starten, werden Primer, DNA-Polymerasen und modifizierte Nukleotide auf die Flowcell aufgetragen.
Die modifizierten Nukleotide haben drei besondere Eigenschaften:
① Terminierung der Synthese
Sie besitzen eine chemische Modifikation an der Hydroxylgruppe, die verhindert, dass ein weiteres Nukleotid an die wachsende DNA-Kette angefügt wird. Dadurch stoppt die DNA-Synthese nach dem Einbau jedes einzelnen Nukleotids.② Reversibilität der Blockierung
Diese Modifikation kann anschließend chemisch entfernt werden, sodass die DNA-Synthese fortgesetzt werden kann. Dadurch erfolgt die Sequenzierung schrittweise, ein Nukleotid nach dem anderen.③ Fluoreszenzmarkierung
Jedes der vier Basen (A, T, C, G) ist mit einem spezifischen fluoreszierenden Farbstoff markiert.Aufgrund dieser Eigenschaften werden diese Nukleotide als reversible Terminator-Nukleotide (RT-dNTPs) bezeichnet.
Ablauf der Sequenzierung
➥ Die Sequenzierungsprimer hybridisieren mit den Vorwärtssträngen der DNA-Bibliothek.
➥ Die DNA-Polymerase bindet an den Primer und beginnt die Synthese. Aufgrund der modifizierten Nukleotide kann sie jedoch immer nur ein einziges Nukleotid pro Zyklus anfügen.
➥ Nach jedem Einbau wird die Flowcell von einer hochauflösenden Kamera abfotografiert. Die fluoreszierende Markierung zeigt an, welches Nukleotid eingebaut wurde. Ein Computer analysiert die Fluoreszenzsignale und ordnet sie den jeweiligen Basen zu.
➥ Anschließend werden überschüssige Nukleotide weggespült und die Blockierung am eingebauten Nukleotid durch einen chemischen Schritt entfernt.
➥ Der Zyklus beginnt von vorn, bis das gesamte DNA-Fragment sequenziert ist.Abb. 17-M: Illumina Sequenzierungsschritte Primer, DNA-Polymerase (DNAP) und modifizierte Nukleotide (RT-dNTPs) werden auf die Flowcell aufgetragen. Die RT-dNTPs sind fluoreszenzmarkiert (jede Base hat eine eigene Farbe) und besitzen eine reversible Blockierung an der 3′-Hydroxylgruppe. Dadurch wird nach jeder Baseneinfügung die DNA-Synthese vorübergehend gestoppt. Sobald ein RT-dNTP eingebaut wurde, regt ein Laser den Fluoreszenzfarbstoff zum Leuchten an. Eine Kamera erfasst dieses Signal und bestimmt, welche Base eingebaut wurde. Anschließend wird die Fluoreszenzmarkierung zusammen mit der Blockierung chemisch entfernt, sodass die DNA-Synthese im nächsten Zyklus fortgesetzt werden kann. Dieser Prozess wird in jedem Zyklus wiederholt: Eine Base wird hinzugefügt, das Signal erfasst und die Blockierung entfernt. Die Sequenzierung läuft über eine festgelegte Anzahl an Zyklen, wodurch eine begrenzte Anzahl an Basen gelesen wird (z. B. 150 Basenpaare bei 150er-Reads).
Abb. 17-N: Analyse der Sequenzierdaten Während der Sequenzierung erfasst eine Kamera in jedem Zyklus die Fluoreszenzsignale der eingebauten Nukleotide. Jedes Cluster sendet ein spezifisches Farbsignal, das der eingebauten Base entspricht. Über mehrere Zyklen hinweg entsteht so für jedes Cluster eine individuelle Sequenz. Ein Computer analysiert die Farbinformationen aus den einzelnen Bildern und ordnet sie der jeweiligen DNA-Sequenz zu. So wird aus den gemessenen Fluoreszenzsignalen die exakte Basenabfolge der „DNA of interest“ rekonstruiert.
Doppel-Check für die DNA: Paired-End-Sequenzierung
Die Paired-End-Sequenzierung ist ein Verfahren, das auf vielen Illumina-Plattformen zum Einsatz kommt, um die Genauigkeit und Verlässlichkeit der Sequenzierung zu verbessern. Dabei wird jedes DNA-Fragment von beiden Enden aus gelesen – es entstehen also zwei Reads pro Fragment: Read 1 (Vorwärts) und Read 2 (Rückwärts).
Nach der ersten Sequenzierung des Vorwärtsstrangs (Read 1) erfolgt eine zweite Brückenamplifikation, bei der die ursprünglichen Stränge erneut erzeugt werden. Anschließend werden die bereits gelesenen Stränge entfernt – und die Sequenzierung des Rückwärtsstrangs (Read 2) beginnt.
Da beide Reads aus demselben Fragment stammen, können sie rechnerisch zusammengeführt werden. Das erleichtert die Assemblierung, erhöht die Fehlererkennung und verbessert die Lesbarkeit wiederholter oder komplexer Sequenzabschnitte.
Die Paired-End-Sequenzierung eignet sich besonders gut für lange, verschachtelte oder schwierige DNA-Bereiche – und liefert dadurch präzisere und belastbarere Ergebnisse.
Ergebnis der Illumina-Sequenzierung
Am Ende der Illumina-Sequenzierung liegt eine riesige Menge kurzer DNA-Abschnitte vor – sogenannte Reads. Jeder dieser Reads stammt von einem kleinen Fragment des ursprünglichen Erbmaterials und wurde millionenfach vervielfältigt und ausgelesen.
Damit daraus wieder ein vollständiges Bild entsteht, werden die Reads mithilfe spezieller Software am Computer zusammengesetzt:
🔹 Gibt es eine bekannte Referenz-DNA, werden die Reads wie Puzzleteile an das bekannte Muster angelegt.
🔹 Fehlt eine Vorlage, müssen sie anhand von Überlappungen Stück für Stück neu zusammengesetzt werden.So entsteht aus zahllosen kurzen Sequenzen Schritt für Schritt ein vollständiges Bild des ursprünglichen DNA-Materials. Das Ergebnis ist eine präzise analysierte Gensequenz, die nicht nur die genetische Struktur sichtbar macht, sondern auch Hinweise auf Mutationen oder Varianten liefern kann.
🎥 Tipp: Eine anschauliche Erklärung findest du im Video „Illumina Sequencing Technology“.
Anwendungsbereiche
Die Illumina-Sequenzierung gilt als genau und zuverlässig. Ihren Einsatz hat sie in der:
Genomforschung: Entschlüsselung der DNA von Menschen, Tieren, Pflanzen
Medizin: Untersuchung genetischer Erkrankungen, Entwicklung personalisierter Therapien
Mikrobiologie: Analyse von Bakterien und Viren
Umweltforschung: Untersuchung von DNA in Boden- oder Wasserproben
Und was kommt danach?
Die Illumina-Technologie hat die genetische Analyse revolutioniert – doch auch sie hat ihre Grenzen: Die Vorbereitung ist aufwendig, die Auswertung rechenintensiv, und sehr lange DNA-Abschnitte lassen sich nur in kleinen Stücken erfassen.
Deshalb geht die Entwicklung weiter. Neue Technologien der Third Generation Sequencing setzen auf ganz andere Prinzipien – und ermöglichen erstmals das direkte Auslesen extrem langer DNA-Stränge, oft sogar in Echtzeit.
Zeit für einen Blick auf die nächste Generation der Sequenzierung.
Third Generation: Echtzeit-Sequenzierung
Mit der dritten Generation der Sequenzierungstechnologien beginnt ein grundlegender Wandel in der Genomforschung. Statt wie bisher auf fragmentierte oder chemisch veränderte DNA zu setzen, ermöglichen diese Methoden das direkte Auslesen genetischer Informationen – in Echtzeit, ohne komplexe Vorbereitung und mit neuen analytischen Möglichkeiten.
Zwei zentrale Verfahren dieser Generation sind:
- Single Molecule Real Time (SMRT) Sequencing – verfolgt die DNA-Synthese in Echtzeit, indem Lichtimpulse registriert werden, die beim Einbau einzelner Basen entstehen.
- Nanopore Sequencing – leitet DNA-Stränge durch winzige Nanoporen und misst Veränderungen im elektrischen Strom, um die Basen zu identifizieren.
Warum die Nanopore-Sequenzierung ein Gamechanger ist
Die Nanopore-Technologie eröffnet ganz neue Perspektiven in der Genomforschung – durch ihre Flexibilität, Geschwindigkeit und Unabhängigkeit von aufwändigen Vorbereitungsschritten:
✅ Echtzeit-Sequenzierung: Im Gegensatz zu klassischen Verfahren, die DNA zunächst vervielfältigen oder chemisch modifizieren müssen, liest Nanopore-Technologie das genetische Material direkt aus.
✅ Lange Leselängen: Anstelle kurzer Fragmente können extrem lange DNA-Stränge ausgelesen werden – oft über Hunderttausende Basenpaare hinweg, in einem einzigen Durchgang. Das erleichtert die Analyse komplexer Genome und struktureller Varianten erheblich.
✅ Vielseitigkeit: Neben DNA lässt sich auch RNA direkt analysieren – ohne den Zwischenschritt über cDNA (komplementäre DNA). Das macht die Technologie besonders wertvoll für Studien zur Genexpression oder Virusforschung.
✅ Portabel und kostengünstig: Geräte wie der MinION sind kaum größer als ein USB-Stick und ermöglichen Sequenzierung sogar außerhalb des: etwa in der Klinik, im Regenwald oder direkt am Tatort.
Mit diesen Eigenschaften erschließt die Nanopore-Sequenzierung völlig neue Anwendungsfelder – von der Grundlagenforschung über Diagnostik bis hin zur Umwelt- und Biodiversitätsanalyse.
Grund genug, diese Technologie einmal genauer unter die Lupe zu nehmen.
Für einen ersten Eindruck und eine kurze Übersicht ist das folgende Video geeignet.
Nanopore-Sequenzierung – Schritt für Schritt
Wenn heute von Nanopore Sequencing die Rede ist, meint man fast immer die Oxford Nanopore-Technologie (ONT). Zwar gibt es theoretisch andere Ideen mit Nanoporen, aber ONT ist derzeit die einzige, die wirklich im Einsatz ist. Seit ihrer allgemeinen Markteinführung im Jahr 2015 hat sie die Sequenzierung revolutioniert.
Das Besondere an dieser Methode? Sie liest DNA oder RNA direkt und in Echtzeit – ohne vorherige chemische Modifikationen oder Verstärkungsreaktionen. Die Nanopore-Technologie funktioniert dabei wie ein winziges Hochleistungs-Labor, das genetische Informationen präzise entschlüsselt.
Wie funktioniert das?
Die ONT-Sequenzierung basiert auf einem Zusammenspiel von drei zentralen Komponenten:
➤ Nanoporen – fungieren als winzige molekulare „Lesegeräte“. Wenn ein DNA- oder RNA-Strang durch eine Nanopore hindurchwandert, erzeugt dies charakteristische elektrische Signale – quasi einen molekularen „Fingerabdruck“.
➤ Membran – dient als Filter und Barriere. Sie sorgt dafür, dass nur Ionen und Nukleinsäuren die Nanoporen passieren, während unerwünschte Moleküle ausgeschlossen bleiben. Dadurch entstehen saubere, präzise Messwerte.
➤ Chip – ist die Basis des Systems. Er hält die Membran mit den Nanoporen und erfasst die elektrischen Signale, die beim Durchtritt der DNA entstehen.
Der Chip besitzt zwei Kammern:
- Obere Kammer (cis): Hier wird die DNA-Probe eingefüllt.
- Untere Kammer (trans): Hier landet die DNA, nachdem sie die Nanoporen durchlaufen hat.
Beide Kammern sind mit einer ionenhaltigen Flüssigkeit gefüllt, die elektrischen Strom leitet. Die Membran trennt die Kammern, sodass kein Strom fließen kann – außer an den Stellen, an denen die Nanoporen eingebettet sind. Diese Poren sind die einzigen „Tunnel“, durch die Ionen und Nukleinsäuren passieren können.
Sobald ein DNA- oder RNA-Strang durch eine Nanopore gleitet, verändert sich der Stromfluss. Diese feinen Veränderungen werden erfasst und in genetischen Code übersetzt – in Echtzeit: direkt, schnell und genau.
Abb. 18-A: Aufbau des Nanopore-Geräts Links ist ein tragbares Nanopore-Sequenziergerät dargestellt. In der Praxis ist es etwas größer als ein USB-Stick. Ein Pfeil zeigt auf die austauschbare Sequenzierzelle. Die vergrößerte Darstellung rechts zeigt den schematischen Aufbau der Sequenzierzelle.
Präzision auf Nanoebene
Chip-Mikrostruktur
Der Chip wird mit hochpräzisen Fertigungstechniken wie Photolithografie oder Ätzverfahren hergestellt. Dabei entstehen winzige Kanäle, sogenannte Wells, die nur wenige Nanometer bis Mikrometer groß sind. Neben diesen strukturellen Komponenten enthält der Chip auch die elektrochemische Messvorrichtung, die den Ionenstrom registriert, sobald DNA oder RNA durch die Nanopore wandert.Selbstassemblierung der Lipidmembran
Diese Membran besteht aus Lipiden (Fettmolekülen) und ist elektrisch isolierend. Das bedeutet, dass sie selbst keinen Strom leitet. Die Membran wird zunächst ohne Nanoporen auf den Chip aufgespannt. Sie wird nicht mechanisch auf den Chip gespannt, sondern bildet sich durch Selbstorganisation über den Öffnungen.Wie funktioniert das?
Eine Lösung mit Lipidmolekülen wird auf den Chip gegeben. Diese Moleküle besitzen:
🔹einen hydrophoben (wasserabweisenden) Anteil und
🔹einen hydrophilen (wasseranziehenden) Anteil.Durch diese Eigenschaften organisieren sie sich automatisch in eine stabile Doppelschicht. Die Wells im Chip sind so konstruiert, dass die Lipidmembran genau über ihnen stabilisiert wird.
Selbstassemblierung der Nanoporen
Die Nanoporen bestehen meist aus Proteinen, die aus Bakterien oder Hefezellen gewonnen werden. In der Natur dienen diese Poren als Transportkanäle für Moleküle durch Zellmembranen. In der Nanopore-Technologie werden sie jedoch als hochempfindliche Sensorenfür DNA- oder RNA-Molekülegenutzt.Die Integration der Nanoporen erfolgt nach dem Aufspannen der Membran. Dank eines natürlichen Prozesses namens Selbstassemblierung setzen sie sich von selbst in die Membran ein!
Wie funktioniert das?
🔹Die Nanoporen werden in einer Lösung zugegeben.
🔹Sie „finden“ automatisch die Wells, da sie sich nur dort in die Membran einbauen können, wo eine Öffnung vorhanden ist.
🔹Dort durchdringen sie die Membran und bilden stabile Kanäle.Die Wells sind somit exakt definierte Bereiche, in denen die Membran zugänglich ist – und genau dort können sich die Nanoporen gezielt integrieren.
Warum ist das wichtig?
Die korrekte Positionierung der Nanoporen über den Wells ist entscheidend, weil:
🔹Die DNA durch die Nanoporen von der cis- zur trans-Kammer gezogen wird, und das nur funktioniert, wenn die Nanoporen in der Membran über den Wells sitzen.
🔹Die Wells sind mit Elektroden verbunden, die den Ionenstrom messen. Wenn eine Nanopore nicht exakt über einem Well sitzt, gibt es keinen messbaren Stromfluss – die Nanopore wäre funktionslos.Der Weg der DNA durch die Nanopore
Nachdem wir den Aufbau der Sequenzierzelle und das Grundprinzip der Oxford Nanopore-Technologie kennengelernt haben, geht es nun um den eigentlichen Sequenzierprozess.
1️⃣ Vorbereitung der DNA/RNA-Probe
Bevor die eigentliche Sequenzierung beginnt, muss die Nukleinsäure (DNA oder RNA) vorbereitet werden.
➥ Extraktion: Die DNA oder RNA wird aus der Probe (z. B. Blut, Speichel, Zellkultur, Umweltproben) isoliert. Dies geschieht mit standardisierten Extraktionsverfahren (siehe dazu Kapitel „4.5.1. Nukleinsäure-Extraktion“).
➥ Fragmentierung (optional): Nanopore-Sequenzierung kann sehr lange DNA- oder RNA-Moleküle lesen. Falls die DNA zu lang ist oder für bestimmte Anwendungen angepasst werden soll, kann sie mechanisch oder enzymatisch in Fragmente geschnitten werden.
➥ Adapter-Ligation: Da die Nanoporen nur DNA oder RNA mit speziellen Enden verarbeiten können, werden Adapter an die Enden der Moleküle angehängt. Diese Adapter enthalten:
- Motorproteine, die die DNA kontrolliert durch die Nanopore ziehen
- Barcode-Sequenzen (falls mehrere Proben gleichzeitig sequenziert werden sollen)
Abb. 18-B: Schema der Bindung des Motorproteins an die doppelsträngige DNA (dsDNA). Damit das Motorprotein an die DNA binden kann, werden zuvor spezielle Adapter an die DNA-Enden angefügt. Diese Adapter sorgen dafür, dass das Motorprotein gezielt an einem Ende der DNA ansetzt. Zur besseren Übersicht zeigt die Abbildung nur die Bindung des Motorproteins, während die Adapter selbst nicht dargestellt sind. In der Regel bindet nur ein Motorprotein pro DNA-Molekül, da die Adapter so gestaltet sind, dass sie die Bindung an einem bevorzugten Ende erleichtern.
Woher stammt das Motorprotein?
Das Motorprotein, das bei der Oxford Nanopore Sequenzierung verwendet wird, ist ein natürlich vorkommendes Enzym, das aus Bakterien gewonnen wird. Es handelt sich um eine modifizierte Version eines Proteins, das ursprünglich von Bakterien wie E. coli oder anderen Mikroorganismen stammt. Diese Proteine haben in der Natur die Aufgabe, DNA zu entwinden und zu transportieren – eine Fähigkeit, die sich die Nanopore-Technologie zunutze macht.Falls RNA sequenziert werden soll, kann optional eine Reverse Transkription durchgeführt werden, um RNA in DNA umzuwandeln. Die ONT-Technologie kann jedoch sowohl RNA als auch DNA direkt sequenzieren, Die direkte RNA-Sequenzierung kann spezifische Informationen über RNA-Modifikationen liefern.
2️⃣ Anlegen einer elektrischen Spannung
In beiden Kammern der Sequenzierzelle befinden sich geladene Teilchen (Ionen). Sobald eine Spannung zwischen der oberen (cis) und der unteren (trans) Kammer angelegt wird, fließen Ionen durch die Nanoporen. Dies erzeugt einen messbaren elektrischen Strom. Solange sich keine DNA in der Pore befindet, bleibt der Ionenfluss konstant, und es wird ein stabiler Strom gemessen.
3️⃣ Die DNA wird in die Sequenzierzelle gegeben
Die vorbereitete DNA-Probe, an die bereits Motorproteine gebunden sind, wird in die cis-Kammer (die obere Kammer der Sequenzierzelle) pipettiert. Durch Diffusion oder sanftes Mischen verteilt sich die DNA in der Lösung und gelangt in die Nähe der Membran, in der die Nanoporen eingebettet sind.
4️⃣ Andocken an die Nanopore
Das Motorprotein, das an die DNA gebunden ist, führt die DNA zur Nanopore und dockt gezielt daran an. Sobald die Verbindung hergestellt ist, beginnt das Motorprotein mit seiner Helicase-Aktivität: Es entwindet die doppelsträngige DNA (dsDNA) in zwei Einzelstränge. Ein Strang wird von der Nanopore erfasst und weiter durch sie hindurchgezogen. Dieser Prozess läuft direkt an der Pore ab und stellt sicher, dass die DNA präzise und gleichmäßig durch die Nanopore transportiert wird.
Abb. 18-C: Schema der Sequenzierzelle und des Ionenstroms im Ruhezustand Die Sequenzierzelle besteht aus zwei Kammern: der cis-Kammer (oben) und der trans-Kammer (unten), getrennt durch eine Membran mit eingebetteten Nanoporen. Links hat ein DNA-Molekül mithilfe eines Motorproteins an eine Nanopore angedockt. Rechts ist eine leere Nanopore dargestellt, durch die ein konstanter Ionenstrom fließt.Die angelegte Spannung zwischen der negativen Elektrode (cis) und der positiven Elektrode (trans) treibt den Ionenfluss an. Der Ionenstrom wird im Well (kleiner Kanal im Chip) gemessen, wie im Strom/Zeit-Diagramm dargestellt. Solange keine DNA durch die Pore wandert, bleibt der Strom konstant.
5️⃣ Die DNA passiert die Nanopore – das Signal entsteht
Die DNA besitzt aufgrund ihrer negativ geladenen Phosphatgruppen eine elektrische Ladung. Die angelegte Spannung zwischen der cis-Kammer (negativ geladen) und der trans-Kammer (positiv geladen) bewirkt, dass die DNA durch die Nanopore gezogen wird.
Das Motorprotein spielt dabei eine entscheidende Rolle:
➤ Es steuert die DNA-Bewegung durch die Pore – langsam, gleichmäßig und stabil.
➤ So entsteht ein klar lesbares Signal für eine zuverlässige Signalmessung.Zum Verständnis: Die DNA würde von Natur aus mit einer Geschwindigkeit von Millionen Basen pro Sekunde durch die Nanopore schießen. Das Motorprotein bremst diese Geschwindigkeit auf etwa 450 Basen pro Sekunde ab (abhängig vom Gerät und den Einstellungen). Dadurch wird die Sequenzierung mittels dieser Technologie überhaupt erst möglich.
Wenn die DNA durch die Pore wandert, beeinflusst sie den Ionenfluss – denn jede Base (A, T, G, C) hat eine einzigartige Größe und chemische Struktur. Diese Unterschiede verändern den Ionenstrom auf spezifische Weise. Diese Stromveränderungen werden von einer Elektrode gemessen, die direkt unter der Nanopore sitzt. Jeder Well (die winzigen Tunnel im Chip, auf denen die Nanoporen sitzen) hat eine fest zugeordnete Elektrode, die als Messgerät fungiert. Dadurch kann die Software die Signale präzise einer bestimmten Nanopore zuordnen – eine Voraussetzung für die korrekte Rekonstruktion der DNA-Sequenz.
Abb. 18-D: Das Bild veranschaulicht den zentralen Mechanismus der Nanopore-Sequenzierung. 1) Ein Motorprotein zieht die DNA kontrolliert durch die Pore. Die negativ geladene DNA bewegt sich aufgrund der angelegten Spannung von der cis-Seite zur trans-Seite. Dabei beeinflussen die einzelnen Basen (A, T, G, C) den Ionenstrom auf spezifische Weise.
2) Ein Graph stellt die Veränderung des gemessenen Stroms über die Zeit dar. Jede Basen-Kombination erzeugt ein charakteristisches Signal, das durch Algorithmen entschlüsselt wird.
3) Ein Computer analysiert die elektrischen Signale und bestimmt daraus die Reihenfolge der Basen.6️⃣ Das elektrische Signal wird aufgezeichnet
Während die DNA durch die Nanopore gezogen wird, befinden sich etwa 10–15 Basen gleichzeitig in der Pore. Jede dieser Basen beeinflusst den Ionenstrom unterschiedlich – das Signal ist also eine Überlagerung der Effekte mehrerer Basen.
Doch wie kann man trotzdem einzelne Basen unterscheiden?
Das funktioniert durch einen cleveren Algorithmus:
- Das Motorprotein bewegt die DNA schrittweise – etwa eine Base nach der anderen.
- Der gemessene Strom verändert sich dabei auf charakteristische Weise, abhängig von der Reihenfolge der Basen.
- Spezialisierte Software (Basecalling) nutzt maschinelles Lernen, um aus den überlappenden Signalen die genaue Sequenz zu errechnen.
- Das Modell „entwirrt“ die überlappenden Informationen und ordnet sie der richtigen Base an der entscheidenden Position zu.
Auf diese Weise entsteht aus den elektrischen Signalen nach und nach die vollständige DNA-Sequenz.
Ergebnis der Nanopore-Sequenzierung
Am Ende liefert die Oxford-Nanopore-Technologie die vollständige Abfolge der Basen in der untersuchten DNA- oder RNA-Probe. Diese Sequenz enthält detaillierte Informationen über die genetische Zusammensetzung, die Länge der Moleküle sowie mögliche Besonderheiten wie Mutationen oder strukturelle Veränderungen.
🚀 Daten in Echtzeit – ein echter Geschwindigkeitsvorteil
Der gesamte Prozess läuft live ab. Während die DNA oder RNA durch die Pore wandert, wird die Abfolge der Basen sofort erfasst und ausgewertet.
Zum Vergleich:
- Die klassische Sanger-Sequenzierung benötigt mehrere Stunden oder sogar Tage für eine Analyse.
- Die Illumina-Sequenzierung liefert Ergebnisse in der Regel in wenigen Stunden bis zu mehreren Tagen. Der genaue Zeitrahmen hängt von der Länge, Komplexität und dem Umfang der zu sequenzierenden Probe ab – kleinere Projekte können oft in wenigen Stunden abgeschlossen werden, während umfangreichere Analysen mehrere Tage benötigen.
- Die Oxford Nanopore Technologie (ONT) hingegen liefert in wenigen Minuten erste Ergebnisse.
Die generierten Sequenzdaten werden parallel verarbeitet und gespeichert. Sie stehen sofort für die weitere Analyse bereit.
Blick nach vorn: Sequenzierung 4.0 – was kommt als Nächstes?
Während die dritte Generation noch gefeiert wird, tüftelt die Forschung bereits an der Überholspur. Neue, aufkommende Technologien versprechen noch mehr: mehr Geschwindigkeit, höhere Genauigkeit und noch vielseitigere Einsatzmöglichkeiten.
Im nächsten Kapitel werfen wir einen flüchtigen Blick auf die Zukunft der Sequenzierung.
Emerging Technologies: Zukunft der Sequenzierung
Während das Nanopore Sequencing (ONT) bereits eine Revolution in der Sequenzierungstechnologie darstellt, gibt es vielversprechende Ansätze, die diese Methode noch weiterentwickeln könnten.
Transistor statt Pore
Ein spannendes Beispiel ist die FENT-Technologie (Field-Effect Nanopore Transistor), die DNA und andere Biopolymere mit einer neuartigen Transistor-Struktur analysiert, die extrem schnell und genau arbeitet. Diese Technologie könnte eine noch präzisere, schnellere und kostengünstigere Analyse von DNA und anderen Biopolymeren ermöglichen.
Das Video „FENT-Nanopore-Transistor-Erklärungsvideo“ gibt einen faszinierenden Einblick in diese Innovation.
Ob und wann diese Technologie für die Virusdiagnostik relevant wird, bleibt abzuwarten – doch sie zeigt eindrucksvoll, wie sich die Sequenzierungstechnologien weiterentwickeln.
Das nächste große Ding in der Genetik
Neben FENT könnten auch Methoden wie In Situ Sequencing (ISS), die Genexpression direkt in Geweben sichtbar macht, zukünftig eine Rolle spielen – etwa um virale Aktivität in Zellen zu erforschen.
Die Zukunft der Sequenzierung?
Sie wird uns umhauen (wortwörtlich!) – und wir sind live dabei!
4.6. Bioinformatische Analyse
Die rohen Sequenzdaten sind einfach eine lange Abfolge von Basen, z. B. „AGCUACGUA…“ bei einer RNA-Sequenz. Sequenzieren ist wie Buchstabieren – erst die Bioinformatik erzählt die Geschichte dahinter – „übersetzt“ diese Daten in Informationen. Aus dem Buchstabensalat (AGCUACGUA…) wird ein Steckbrief: Welches Virus ist das? Was kann es? Und wie gefährlich?
Warum Rohdaten chaotisch sind
💨 Technische Patzer: Wie ein verwackeltes Foto – manche Sequenzteile fehlen oder sind unscharf („Rauschen“).
❓ Rätselbasen („N“): Stellen, wo selbst die Maschine passen muss: „Könnte A, C oder U sein – keine Ahnung!“
🦠 Mutanten-WG: RNA-Viren wie Influenza sind Wohngemeinschaften aus Varianten (Mutantenwolken). Die Sequenzierung mischt alle zusammen – wie ein Smoothie aus 100 Früchten.
Was Bioinformatiker tun
Sie putzen, sortieren und puzzeln die Daten – bis klar wird:
- Wer ist hier? (Virusidentität)
- Was ist neu? (Mutationen)
- Wie reagieren? (Erstmal tief durchatmen!)
Das ist Detektivarbeit – nur mit mehr Computern und weniger Trenchcoats.
Vom Datenchaos zur Erkenntnis: Die 5 Schritte der Bioinformatik
1️⃣ Qualitätskontrolle: Das Datenpuzzle sortieren
2️⃣ Alignment & Assemblierung: Das große Puzzle-Spiel
3️⃣ Homologie-Suche: Will sehen!
4️⃣ Datenbankeintrag: Reif fürs Register?
5️⃣ Funktionelle Annotation: Das Puzzle erwacht zum Leben
1️⃣ Qualitätskontrolle: Das Datenpuzzle sortieren
Sequenzdaten sind wie ein Puzzle vom Flohmarkt: Einige Teile sind doppelt, einige fehlen, manche passen nicht und mitten im Bild ein Kaffeefleck. Was tun? – Bevor man das Ganze zusammensetzt, muss geordnet, geputzt und aussortiert werden. Hier beginnt die Arbeit der Bioinformatik:
🔍 FastQC (ein Software-Tool) – der kritische Buchprüfer:
Er blättert durch die Sequenzdaten wie durch ein altes Manuskript und markiert alle schmuddeligen Seiten – zu kurz, zu fehlerhaft, zu verdächtig.✂️ Trimmomatic (ein Software-Tool) – der molekulare Rasenmäher:
Was kaputt, verfranst oder einfach nur überflüssig ist, wird rigoros abgeschnitten. Besonders an den Enden, wo Fehler gerne nisten.🔄 Fehlerkorrektur – Demokratie auf molekularer Ebene:
Wenn neun von zehn Reads sagen: „Hier gehört ein A hin“, dann wird der einsame Buchstabe G einfach überstimmt. Auch DNA hat Mehrheitsentscheidungen.Am Ende bleibt nur, was Qualität hat:
🔹 Kurze oder minderwertige Reads? Raus damit.
🔹 Einzelne Sequenzfehler? Korrigiert.
🔹 Insgesamt zu wenig gute Daten? Dann lieber nochmal sequenzieren.Warum? Damit nachher niemand falsche Schlüsse aus einem Tippfehler zieht. Denn nur mit sauberen Daten geht’s jetzt ans eigentliche Detektivspiel: dem Zusammenbau des Genoms!
2️⃣ Alignment & Assemblierung: Das große Puzzle-Spiel
Moderne Sequenziermethoden liefern DNA- oder RNA-Schnipsel, als hätte jemand ein Puzzle explodieren lassen:
- Illumina produziert Millionen kurzer Reads.
- Nanopore liefert meterlange Fetzen (na gut – fast).
Doch egal ob klein oder groß: Am Ende müssen sie zusammengesetzt werden – mit Algorithmen, Logik und viel Rechenpower.
🦠📌 Methode 1: Alignment (bei bekannten Viren)
Wo passt dieses Teil hin?
Die bereinigten Sequenzdaten werden mit einer bekannten Referenzsequenz verglichen – wie Puzzleteile, die man auf ein Deckbild legt. So lassen sich selbst kleinste Abweichungen erkennen:
Was man findet:
🔹 Mutationen: Ein A statt G? Vielleicht macht das das Virus ansteckender.
🔹 Fehlstellen: Kleine Löcher im Genom – sogenannte Deletionen.
🔹 Extra-Teile: Unerwartete Einfügungen – Insertionen.Auch wenn die Vorlage (Referenzsequenz) nur eine Variante ist, zeigt das Alignment die ganze Mutanten-WG in der Probe.
🦠❓ Methode 2: De-novo-Assembly (bei neuen Viren)
Ohne Vorlage puzzeln – wie ein Blindflug!
Gibt es keine bekannte Referenz, müssen die einzelnen Reads ohne Schablone zusammengesetzt werden. Der Trick: Überlappende Abschnitte zeigen, welche Teile zueinanderpassen. So entsteht – Stück für Stück – ein vollständiges Genom. Dieser Prozess ist fehleranfälliger als das Alignment, liefert aber eine erste Genomsequenz eines neu entdeckten Virus.
Der Unterschied:
Alignment ist wie IKEA-Möbel bauen – mit Anleitung.
De-novo-Assembly ist wie: „Hier sind Holzteile, viel Glück!“
3️⃣ Homologie-Suche: Will sehen!
Jetzt liegen die Karten auf dem Tisch. Der unbekannte DNA-Schnipsel wird ausgespielt – alle Daten sind gesammelt, alles ist vorbereitet. Jetzt heißt es: Will sehen!
Hier kommt BLAST ins Spiel – die große Suchmaschine für Gene. Sie vergleicht den genetischen Joker mit Millionen anderer Sequenzen in weltweiten Datenbanken. Und wenn irgendwo ein Verwandter existiert – BLAST findet ihn. Ob naher Cousin oder entfernter Urahn: Die Ähnlichkeiten im Code verraten, ob du einen alten Bekannten in der Hand hältst. Oder vielleicht ein völlig neues Virus.
BLAST zeigt:
🔹 den Grad der Übereinstimmung in Prozent
🔹 statistische Werte, die anzeigen, wie zuverlässig der Treffer ist
🔹 mögliche Verwandtschaften, selbst wenn die Ähnlichkeit nur entfernt istWer es wirklich wissen will:
How to Use BLAST for Finding and Aligning DNA or Protein Sequences🦠📌 Bei bekannten Viren: BLAST bestätigt die Ergebnisse des Alignments: Gehört der Stamm zu einer bereits beschriebenen Variante? Gibt es neue Mutationen? Beim Influenzavirus könnte BLAST z. B. zeigen, dass ein neuer Stamm zu 98 % mit dem H3N2-Stamm der letzten Grippesaison übereinstimmt.
🦠❓Bei unbekannten Viren: BLAST klärt, ob die neue Sequenz einer bekannten Virusfamilie zugeordnet werden kann – oder ob es sich um einen völlig neuen Vertreter handelt. Falls keine oder nur sehr entfernte Treffer auftauchen, sind weiterführende Analysen notwendig.
4️⃣ Datenbankeintrag: Reif fürs Register?
Jetzt wird’s offiziell. Die gesammelten Sequenzdaten stehen bereit – doch nicht jede neue Virusvariante schafft es gleich ins große wissenschaftliche Archiv. Erst wird geprüft: Reif fürs Register?
🦠📌 Bei bekannten Viren: Nach der Homologie-Suche geht’s in die Feinanalyse: Hat der Stamm was Spannendes zu bieten? Die Variantendetektion schaut ganz genau hin – wie ein Genetiker mit Vergrößerungsglas. Gibt es Mutationen, die dem Virus helfen, dem Immunsystem zu entkommen (Stichwort: Immunflucht)? Wenn ja – ab damit ins Register! Der neue Stamm wird in eine öffentliche Datenbank eingetragen und steht dann Forschern weltweit zur Verfügung.
🦠❓ Bei unbekannten Viren: Wenn BLAST meldet: „Kenne ich nicht!“, wird es spannend. Jetzt helfen weiterführende bioinformatische Werkzeuge – zum Beispiel die funktionelle Annotation, die fragt: Was könnte dieses Virus können? Stellt sich dabei heraus: Hier haben wir etwas wirklich Neues, dann wird der Fund in eine der großen internationalen Datenbanken eingetragen – inklusive Namen, genetischem Fingerabdruck und möglicher Herkunft.
Die wichtigsten Datenbanken für Virusgenome:
🌐 GenBank – der Allrounder:
eine riesige Bibliothek für genetische Sequenzen aller Lebensformen, einschließlich Viren.🌐 Influenza Research Database (IRD) – Spezialarchiv für Influenzaviren,
inklusive praktischer Analysetools.🌐 GISAID – Die Formel 1 unter den Datenbanken:
superschnell, vor allem für Influenzaviren und SARS-CoV-2.🌐 VIRALzone – Eine Art Wikipedia für Virusfamilien:
mit Infos zu Genom, Aufbau und Replikation.Was in diesen Datenbanken landet, wird Teil eines globalen Gedächtnisses.
5️⃣ Funktionelle Annotation: Das Puzzle erwacht zum Leben
Das Genom ist sequenziert – aber jetzt will man wissen: „Welche Gene machen was?
Die funktionelle Annotation versucht, den „Bauplan“ des Virus zu lesen – und herauszufinden, welche Teile wofür zuständig sind. Warum ist dieses Virus infektiöser als andere? Wie schafft es das Virus, dem Immunsystem zu entkommen? Und könnte es resistent gegen Medikamente sein?
Dazu durchsucht man das Genom nach bekannten Mustern. Gibt es Abschnitte, die an Gene erinnern, die man schon aus anderen Viren kennt? Stimmen bestimmte Sequenzen mit Enzymen überein, die beim Eindringen in die Zelle helfen? Oder mit Regionen, die die Oberfläche des Virus formen – jene Stellen, die das Immunsystem als erstes erkennt?
Auch räumliche Strukturen spielen eine Rolle. Moderne KI-Modelle wie AlphaFold können aus der Gensequenz ein 3D-Modell des entstehenden Proteins berechnen. So lässt sich vorhersagen, ob eine Mutation nur ein kleines Detail verändert – oder ob sie das ganze Verhalten des Virus beeinflusst.
Das ist besonders wichtig, wenn es um:
- Ansteckung geht: Eine Mutation in einem Oberflächenprotein könnte das Virus „klebriger“ machen – es haftet besser an Zellen.
- Immunflucht: Kleine Veränderungen an den „Erkennungsstellen“ reichen, damit Antikörper das Virus übersehen.
- Medikamentenresistenz: Manche Mutationen schalten die Zielstruktur eines Wirkstoffs einfach aus – und machen ihn damit wirkungslos.
Funktionelle Annotation ist wie die Gebrauchsanleitung eines Virus – nur ohne freundliche Warnhinweise.
💡 Zusammenfassung: Viren-Cracking für Anfänger
Sequenzieren: Buchstabiere das Virus-ABC.
Qualitätskontrolle: Räum den Datenmüll weg.
Alignment & Assembly: Puzzle die Schnipsel zusammen – mit oder ohne Vorlage.
BLAST: Google nach genetischen Verwandten.
Datenbank: Gib der Welt deine Entdeckung – wenn sie was taugt.
Annotation: Lies zwischen den Genen – was kann dieses Virus wirklich?Klassifizierung der Viren basierend auf ihrem genetischen Material
Viren können auf verschiedene Arten klassifiziert werden – zum Beispiel nach ihrer Form, ihren Wirten, ihrer Übertragungsweise oder ihrem genetischen Material.
Eine der bekanntesten und am häufigsten verwendeten Genom-Klassifikationen ist die Baltimore-Klassifikation, die 1971 von David Baltimore eingeführt wurde. Dieses System teilt Viren basierend auf einer Kombination aus den folgenden Merkmalen ein:
- Art des genetischen Materials (DNA oder RNA),
- Strängigkeit (einzelsträngig oder doppelsträngig),
- Richtung (positiv oder negativ) und
- Replikationsmechanismus (wie mRNA hergestellt wird).
Das Ziel aller Viren ist es, mRNA zu produzieren, denn nur mit mRNA können sie die Proteine herstellen, aus denen sie bestehen, und sich vermehren. Die Baltimore-Klassifikation umfasst sieben Gruppen, die zeigen, wie unterschiedlich Viren dieses Ziel erreichen – von direkter Nutzung ihrer RNA bis hin zu komplexen Umwegen über DNA. Hier ein Überblick:
Gruppe I: Doppelsträngige DNA-Viren (dsDNA)
Die Herstellung von mRNA erfolgt durch die direkte Transkription der DNA mittels einer RNA-Polymerase, die entweder zellulär oder viral sein kann.
(Beispiele: Adenoviren, Herpesviren und Pockenviren)Gruppe II: Einzelsträngige DNA-Viren (ssDNA)
Die einzelsträngige DNA wird zunächst in doppelsträngige DNA umgeschrieben. Anschließend erfolgt die Transkription zur Herstellung von mRNA.
(Beispiele: Parvoviren)Gruppe III: Doppelsträngige RNA-Viren (dsRNA)
Die Herstellung der mRNA erfolgt durch die Transkription der RNA-Stränge mittels einer viralen RNA-abhängigen RNA-Polymerase.
(Beispiele: Reoviren)Gruppe IV: (+) Einzelsträngige RNA-Viren [(+)ssRNA]
Die virale RNA dient direkt als mRNA, da sie die gleiche Richtung wie die mRNA hat (5′ → 3′).
(Beispiele: Coronaviren, Polioviren)Gruppe V: (–) Einzelsträngige RNA-Viren [(–)ssRNA]
Die RNA ist in entgegengesetzter Richtung aufgebaut (3′ → 5′) und muss durch eine virale RNA-abhängige RNA-Polymerase in (+)RNA umgeschrieben werden, die dann als mRNA dient.
(Beispiele: Influenzaviren, Rabies-Virus)Gruppe VI: Retroviren [(+)ssRNA mit DNA-Zwischenschritt]
Die Reverse Transkriptase (RT) wandelt RNA in DNA um, die ins Wirtsgenom integriert wird. Die mRNA wird von der integrierten DNA durch zelluläre Mechanismen hergestellt.
(Beispiele: HIV)Retroviren wie HIV nutzen einen einzigartigen Replikationsmechanismus: Die virale RNA wird durch die Reverse Transkriptase (RT) in einen komplementären DNA-Strang (cDNA) umgeschrieben. Die ursprüngliche RNA wird abgebaut, und ein zweiter DNA-Strang wird synthetisiert. Die entstandene dsDNA wird in das Wirtsgenom integriert. Dieser Prozess macht Retroviren besonders hartnäckig, da sie in einer latenten Phase verweilen und jederzeit reaktiviert werden können.
Gruppe VII: Doppelsträngige DNA-Viren mit RNA-Zwischenschritt (dsDNA-RT)
Die virale DNA wird im Zellkern repariert und dient als Vorlage für die Synthese von mRNA und prägenomischer RNA. Die prägenomische RNA wird durch die Reverse Transkriptase wieder in DNA umgeschrieben.
(Beispiele: Hepatitis-B-Virus)Das Hepatitis-B-Virus besitzt ein unvollständig doppelsträngiges DNA-Genom. Im Zellkern der Wirtszelle wird diese DNA repariert, bevor mRNA und prägenomische RNA transkribiert werden. Die prägenomische RNA dient als Vorlage für die Bildung neuer Viruspartikel.

Abb. 19: Die Grafik zeigt die sieben Klassen der Baltimore-Klassifikation. Die Viren werden nach ihrer Genomstruktur und ihrer Strategie zur mRNA-Synthese kategorisiert. Doppelsträngige (ds) und einzelsträngige (ss) Genome werden unterschieden, die Polarität der RNA wird durch „+“ (positiv) und „-“ (negativ) Stränge gekennzeichnet, und der Mechanismus der reversen Transkription ist durch „RT“ hervorgehoben. Am Ende steht die mRNA, die alle Viren benötigen, um ihre Proteine zu produzieren.
5. Gibt es Viren wirklich?
In einer Welt, in der man alles infrage stellen kann – wie beweist man, dass etwas existiert?
Manche sagen: Viren seien bloß Artefakte, Laborkonstrukte, Zelltrümmer ohne eigene Identität. Doch es gibt Merkmale, die sich nicht wegdiskutieren lassen:
Viren haben etwas, das nur sie haben
Viele Viren besitzen konservierte Genomabschnitte, die für eindeutig virale Funktionen kodieren:
- Kapsidproteine, mit denen sie ihre Erbinformation verpacken,
- RNA-abhängige Polymerasen, mit denen sie ihr Erbgut vervielfältigen,
- Integrasen, mit denen sie virale Gene ins Wirtsgenom schleusen (z. B. bei Retroviren wie HIV),
- Proteasen, mit denen sie Virus-Proteine zurechtschneiden, damit sie funktionieren.
Das Kapsid – das Erkennungszeichen des Virus
Was macht ein Virus zu einem Virus? Nicht nur sein parasitärer Lebensstil – sondern vor allem sein Aufbau.
Fast alle Viren verfügen über ein Kapsid – eine proteinhaltige Hülle, die ihr Erbgut schützt und gleichzeitig zum Eindringen in Wirtszellen dient. Ob ikosaedrisch oder helikal: Das Kapsid ist funktional wie evolutionär DAS zentrale Merkmal des Virus.
Es schützt, es transportiert, es organisiert – und es ist einzigartig virusspezifisch.
Abb. 20: Das Kapsid – schützende Hülle und Formgeber des Virus Das Kapsid ist die äußere Proteinhülle eines Virus, die dessen genetisches Material schützt und dessen äußere Gestalt bestimmt. Es besteht aus vielen identischen Bausteinen (Kapsomeren), die sich zu symmetrischen Strukturen zusammensetzen. Die häufigsten Kapsidformen sind ikosaedrisch (20-flächig) und helikal (schraubenförmig).
Zusätzliche Bestandteile wie eine Lipidhülle – die von der Wirtszelle stammt – können die äußere Form verändern. So wirken viele behüllte Viren „sphärisch“, obwohl das darunterliegende Kapsid meist ikosaedrisch ist. In einigen Fällen, wie bei Bakteriophagen, weicht der Aufbau deutlich ab: Man spricht dann von einer komplexen Morphologie, etwa mit einem ikosaedrischen Kopf und einem helikalen Schwanz. Diese morphologischen Unterschiede bilden die Grundlage zur strukturellen Klassifikation von Viren.
Virale Hallmark-Gene – Signaturen jenseits der Zellwelt
Die Gene, die für Kapsidproteine codieren, gehören zu den sogenannten Hallmark-Genen. Sie sind:
- hoch konserviert innerhalb der Viruswelt,
- funktional essenziell für den viralen Lebenszyklus,
- und nicht in zellulären Organismen zu finden.
Viele dieser Proteine weisen eine ganz besondere Form auf: die Jelly-Roll-Faltung. Diese dreidimensionale Struktur erinnert an eine aufgerollte Biskuitrolle und verleiht dem Kapsidprotein eine bemerkenswerte Stabilität. Man findet sie bei den unterschiedlichsten Viren – von jenen, die Menschen infizieren, bis zu Viren, die nur in uralten Tiefsee-Mikroben vorkommen. Ihr auffälliger Aufbau ist so charakteristisch, dass Forscher selbst neuartige Viren allein auf Grundlage dieser genetischen Signatur erkennen können.
Wer sie einmal gesehen hat, erkennt sie wieder – wie ein Symbol, das sich durch die Tiefen der Evolution zieht – eine evolutionäre Handschrift, lesbar für jeden, der bereit ist, hinzusehen.
Wie z. B. die Arbeiten von Koonin et al. zeigen: Trotz aller Vielfalt tragen Viren einen gemeinsamen, alten genetischen Kern in sich – als würden sie das Wappen ihrer Urahnen in sich tragen.
Fazit: Viren-spezifische Genomabschnitte sind ein Alleinstellungsmerkmal!
Viren entziehen sich einfachen Definitionen. Sie sind weder klassische Lebewesen noch bloße Molekülhaufen. Aber sie tragen ein Set an Eigenschaften in sich, das sie unterscheidbar macht – in ihrer Form, in ihrer Funktion, in ihrer Geschichte.
Wenn etwas seit Milliarden Jahren seine eigene genetische Spur zieht – dann ist es real. Auch wenn es kein Eigenleben führt. Auch wenn wir es nicht sehen können.

Fragen & Antworten
🟡 „Sind Viren nur Exosomen mit PR-Abteilung?“
Nein. Exosomen sind biologischer Verpackungsmüll. Viren bringen einen eigenen Bauplan mit – und einen Transportbehälter in perfekter Geometrie.
Wer Exosomen mit Viren verwechselt, verwechselt auch Tupperdosen mit Raumschiffen.
🔬 „Aber man hat sie doch nie richtig gesehen!“
Doch. Elektronenmikroskope liefern Bilder, Kryo-Tomographie sogar 3D-Videos in Nanometer-Auflösung.
Unsichtbar? Nur ohne Strom.
🧬 „Die RNA ist doch nur zufälliger Zellrest?“
Nein. Virale RNA/DNA ist einzigartig, funktional, präzise organisiert – und weltweit reproduzierbar.
Zufall? Dann wäre ein Uhrwerk auch nur etwas Metall – mit Teamgeist und guter Laune.
📦 „Dieses Kapsid – kann das nicht einfach so entstehen?“
Kaum. Diese Nanoboxen mit perfekter Symmetrie gibt’s nur bei Viren.
Ein Fußball formt sich auch nicht zufällig im Rasen.
🌍 „Vielleicht ist das alles nur ein Messartefakt?“
Dann ist es ein erstaunlich globales. Alle Labore finden die gleichen Sequenzen, Strukturen, Eigenschaften.
Viren kennen keine Laborgrenzen. Nur Reproduzierbarkeit.
Am Ende bleibt die Erkenntnis:
Viren sind keine Fiktion, kein Artefakt, kein Zufallsprodukt. Sie sind das am besten kartierte Unbehagen der Biologie – real, reproduzierbar, überall.
Wer Viren für Zellmüll hält, glaubt auch,
ein Tesla sei ein besonders ambitionierter Einkaufswagen.Quellen und Links, die die Isolation und Sequenzierung von Viren dokumentieren:
National Center for Biotechnology Information (NCBI) Virus Database:
NCBI Virus DatabaseDas NCBI beherbergt eine umfassende Datenbank von Virusgenomen, die von Wissenschaftlern weltweit sequenziert wurden. Hier kannst du Tausende von Virusgenomen einsehen, die isoliert und sequenziert wurden.
GISAID (Global Initiative on Sharing Avian Influenza Data):
GISAIDGISAID ist eine Plattform, die insbesondere für den Austausch von Sequenzdaten von Influenza- und SARS-CoV-2-Viren genutzt wird. Hier sind detaillierte Daten zur Sequenzierung und Herkunft dieser Viren abrufbar.
European Nucleotide Archive (ENA): ENA Browser
Die ENA bietet Zugang zu Sequenzdaten von Viren, die in Laboren weltweit isoliert wurden. Diese Datenbank wird von der European Bioinformatics Institute (EBI) betrieben und enthält umfangreiche Informationen zu viralen Genomen.
PubMed – Wissenschaftliche Artikel zur Virusisolierung und -sequenzierung:
Beispielhafte Suchanfrage: „Virus Isolation and Sequencing“PubMed ist eine Suchmaschine für wissenschaftliche Literatur. Hier kannst du nach spezifischen Studien zur Isolation und Sequenzierung von Viren suchen.
6. Woher kommen Viren?
Diese Fragen führen an die Grenzen unseres Wissens. Sie berühren nicht nur die Ursprünge der Viren, sondern die Grundlagen des Lebens selbst. Bevor wir den möglichen Szenarien ihrer Entstehung folgen, lohnt sich ein Blick auf das große Ganze: den „Baum des Lebens“ – so, wie ihn die moderne Evolutionsforschung heute zeichnet.
6.1. Der Baum des Lebens
Alle bekannten Lebensformen – vom Bakterium bis zum Blauwal – teilen sich einen gemeinsamen Ursprung: LUCA, den Last Universal Common Ancestor. Er war nicht der erste Organismus überhaupt, sondern der letzte gemeinsame Vorfahr aller heute lebenden zellulären Lebensformen – Bakterien, Archaeen und Eukaryoten. Ein evolutionärer Knotenpunkt, kein Startpunkt.
Was wissen wir über LUCA? Wahrscheinlich lebte er vor rund 3,5 bis 4 Milliarden Jahren, in einer warmen, noch jungen Welt. Er war zellbasiert, besaß bereits DNA, RNA und Proteine, konnte Energie umwandeln – aber war noch weit entfernt von der Komplexität moderner Zellen. Ein Prototyp des Lebens, von dem sich alles weitere verzweigte.

Abb. 21: Baum des Lebens – mit den evolutionären Hauptlinien der zellulären DNA-Organismen LUCA (letzter universeller gemeinsamer Vorfahre) verzweigt sich in die Domänen Bakterien (z. B. Terrabakterien, Proteobakterien) und Archaeen (z. B. Euryarchaeota, Asgard). Durch die Verschmelzung eines Proteobakteriums mit einem ASGARD-Archeen (Endosymbiose) entsteht FECA (erster eukaryotischer gemeinsamer Vorfahre), der sich zu LECA (letzter eukaryotischer gemeinsamer Vorfahre) entwickelt, aus dem die Eukaryoten (Tiere, Pflanzen, Pilze) hervorgehen. Eine zweite Endosymbiose mit Cyanobakterien führt zur Entstehung der Chloroplasten in Pflanzen. Rechts veranschaulicht eine Zeitleiste die Entstehung des Lebens, von der Erde (ca. 4,5 Milliarden Jahre vor heute) über die Große Sauerstoffkatastrophe (ca. 2,4 Milliarden Jahre vor heute) bis zur Kambrischen Explosion (ca. 540 Millionen Jahre vor heute).
Doch während sich der Baum des Lebens mit Ästen, Zweigen und Blättern füllte, bleibt eine Frage offen: Wo sitzen die Viren in diesem Bild – oder gehören sie überhaupt dazu?
Denn anders als Bakterien oder Eukaryoten haben Viren keine Zellstruktur, keinen eigenen Stoffwechsel und hinterlassen keine Fossilien. Manche Wissenschaftler sehen sie als „verlorene Zweige“ des Lebensbaums, andere als uralte Vorformen, die vielleicht vor LUCA existierten.
Um diese Frage zu klären, müssen wir zurück an den Anfang – oder besser: zu den möglichen Anfängen.
Wo also stehen Viren in diesem Stammbaum des Lebens?
Sind sie Spätankömmlinge, entstanden aus entgleisten Genen zellulärer Organismen? Oder gehören sie zu den Urformen des Lebens – vielleicht sogar älter als LUCA selbst?
Die Wissenschaft kennt (noch) keine eindeutige Antwort. Doch einige Hypothesen versuchen, dem Ursprung der Viren auf die Spur zu kommen.
6.2. Die Haupthypothesen zur Herkunft von Viren
💡Hinweis: Die folgenden Entstehungshypothesen setzen ein grundlegendes Verständnis der sogenannten RNA-Welt voraus – einer frühen Phase der Erdgeschichte, in der das Leben noch aus einfachen, selbstreplizierenden RNA-Molekülen bestand. Um dich in diese fremde Epoche einzufühlen und die Hypothesen besser einordnen zu können, empfehlen wir dir den Artikel „Das erste Flüstern des Lebens“. Er erzählt in poetischer Sprache von den Anfängen biologischer Ordnung – lange vor Zellen, Proteinen und DNA.
6.2.1. Hypothesen in einer zellulären Welt
Diese Hypothesen setzen existierende Zellen voraus, aus denen Viren hervorgehen.
6.2.1.a) Progressive Hypothese – Viren als entflohene Gene
6.2.1.b) Regressive Hypothese – Viren als geschrumpfte Zellwesen
6.2.2. Hypothesen in der prä-zellulären RNA-Welt
Diese Hypothesen verorten die Entstehung von virenähnlichen Strukturen in einer Zeit vor oder während der Bildung erster Zellen, in der RNA-Welt, wo selbstreplizierende Moleküle dominieren.
6.2.2.a) Virus-first-Hypothese – Viren waren zuerst da
6.2.2.b) Co-Evolution-Hypothese – Viren und Zellen entstanden gemeinsam
6.2.1. Hypothesen in einer zellulären Welt
Die sogenannte Cell-first-Hypothese geht davon aus, dass zelluläre Lebensformen – wie LUCA – bereits existierten, bevor Viren entstanden. Innerhalb dieses Ansatzes lassen sich zwei Hauptszenarien unterscheiden:
a) Die Progressive Hypothese: Viren stammen demnach von genetischem Material ab, das aus Zellen „entkommen“ ist – quasi entflohene Gene, die sich verselbstständigt und einen eigenen evolutionären Weg eingeschlagen haben.
b) Die Regressive Hypothese: Nach dieser Vorstellung waren Viren ursprünglich vollständige Zellen, die sich im Laufe der Evolution zunehmend reduziert haben – bis nur noch die nötigsten Funktionen für das parasitäre Leben übrigblieben. Eine Art evolutionärer Rückbau.
Beiden Varianten liegt ein gemeinsamer Gedanke zugrunde: Viren sind kein eigenständiger Ursprung des Lebens, sondern ein evolutionäres Nebenprodukt zellulärer Organismen – durch Verlust oder Entkopplung.
a) Die Progressive Hypothese – Viren als entflohene Gene
Die progressive Hypothese – auch Escape-Hypothese – meint, dass Viren aus genetischem Material hervorgingen, das einst Teil zellulärer Organismen war. Diese „entflohenen Gene“ entwickelten sich im Laufe der Evolution zu eigenständigen, infektiösen Einheiten – losgelöst vom ursprünglichen Zellverband.
Wie könnte das passiert sein?
In lebenden Zellen gibt es kleine, bewegliche DNA- oder RNA-Stücke, die sich innerhalb des Genoms frei bewegen können – sozusagen „springende Gene“. Diese Elemente, zu denen Plasmide (ringförmige DNA-Moleküle) oder Transposons (DNA-Abschnitte, die ihre Position im Erbgut ändern können) gehören, haben die Fähigkeit, sich unabhängig zu vervielfältigen und sogar zwischen Organismen auszutauschen.
Die Hypothese besagt, dass sich einige dieser genetischen Elemente so weit weiterentwickelten, dass sie eines Tages in der Lage waren, sich eigenständig zu vermehren. Sie entkamen aus der Zellkontrolle, entwickelten Schutzmechanismen wie eine Hülle – das Kapsid, das ihre Erbinformation schützt – und wurden so zu den ersten Viren.

Abb. 22-A: Die Grafik veranschaulicht die Progressive– oder Escape–Hypothese: nach der Viren aus ursprünglich zelleigenen Genen hervorgingen, die sich allmählich verselbstständigten.
Protozelle: Eine frühe, einfache Zellform mit genetischem Material: Großer DNA-Ring, Ribosomen und kleineren DNA-Ringen.
Abspaltung von genetischem Material: Bewegliche genetische Elemente – z. B. Plasmide oder sogenannte Transposons („springende Gene“) – lösen sich von der Zelle ab. Diese könnten erste Schritte zur Unabhängigkeit gemacht haben.
Vesikelbildung: Das abgespaltene genetische Material wird in kleine Membranbläschen (Vesikel) eingeschlossen, was Schutz und Mobilität bietet.
Entstehung eines Virus: Im Laufe der Evolution entstehen daraus funktionale Viren – mit einem schützenden Proteinmantel (Kapsid, orange). Wie genau sich dieser entwickelte, bleibt unklar („Kapsid-Rätsel?“).✅ Argumente für diese Hypothese
➤ Ähnlichkeit mit mobilen genetischen Elementen: Einige virale Gene ähneln stark den „springenden Genen“ innerhalb von Zellen. Besonders Retroviren, wie das HI-Virus, nutzen einen Mechanismus, der dem von Transposons ähnelt: Sie schreiben ihre RNA in DNA um und integrieren sie ins Erbgut ihrer Wirte.
➤ Viren tauschen Gene mit ihren Wirten aus: Forscher haben in Virengenomen Gene entdeckt, die offensichtlich aus zellulären Organismen stammen. Das könnte bedeuten, dass Viren einst aus Zellen hervorgingen und im Laufe der Zeit genetisches Material aufnahmen und weiterentwickelten.
➤ Erklärung für die Vielfalt der Viren: Da verschiedene „springende Gene“ aus unterschiedlichen Zellen entkommen sein könnten, erklärt diese Hypothese, warum es so viele verschiedene Viren mit unterschiedlichen Eigenschaften gibt.
❌ Schwächen der Hypothese
➤ Kapsidproteine sind extrem alt: Einige Schlüsselproteine, die für den Aufbau der Virushülle (Kapsid) verantwortlich sind, zeigen keine direkte Verwandtschaft mit Zellproteinen. Es bleibt unklar, wie entflohene Gene diese komplexen Strukturen entwickeln konnten. Die Evolution dieser Schlüsselproteine reicht so weit zurück, dass sie möglicherweise schon vor der Entstehung der ersten zellulären Lebensformen existierten. Eine Studie von Krupovic & Koonin (2017) legt nahe, dass Kapsidgene sich unabhängig entwickelt haben könnten, bevor es moderne Zellen gab.
➤ Viren haben einzigartige Enzyme, die älter als LUCA sein könnten: Manche Viren enthalten RNA-Polymerasen, die sich stark von denen in Zellen unterscheiden. Diese Enzyme sind so eigenständig, dass sie möglicherweise aus einer Zeit stammen, in der es noch keine heutigen Zellmechanismen gab. Einige Forscherende vermuten daher, dass Viren direkte Überbleibsel aus der RNA-Welt sein könnten – einer Phase der frühen Evolution, in der das Leben noch nicht auf DNA, sondern auf RNA basierte.
➤ Viren in allen drei Domänen des Lebens: Viren sind überall – sie infizieren Bakterien, Archaeen und Eukaryoten. Wenn sie erst nach der Entstehung der ersten Zellen aus diesen hervorgegangen wären, müsste man erwarten, dass sie stärker auf eine bestimmte Zelllinie beschränkt sind. Ihre universelle Verbreitung legt nahe, dass sie bereits existierten, bevor sich Bakterien, Archaeen und Eukaryoten aufspalteten.
➤ Viren teilen sich keine gemeinsame Abstammung mit zellulärem Leben:Während Bakterien, Archaeen und Eukaryoten alle auf LUCA zurückgeführt werden können, gibt es für Viren keine gemeinsame Linie. Sie scheinen also nicht einfach ein „Seitentrieb“ eines Zellstammbaums zu sein, sondern eine sehr alte, parallele Entwicklung.
⬇️ Fazit
Die Cell-first-Hypothese betrachtet Viren als Rebellen des zellulären Lebens – ursprünglich harmlose genetische Passagiere, die sich verselbstständigten und einen eigenen evolutionären Weg einschlugen. Doch sie setzt voraus, dass es bereits komplexe Zellen gab, bevor Viren entstanden.
b) Die Regressive Hypothese – Viren als geschrumpfte Zellwesen
Die Regressions- oder Reduktionshypothese sieht Viren nicht als abgespaltene Gen-Bruchstücke, sondern frühere Zellen, die sich im Laufe der Evolution immer weiter reduziert wurden – bis sie ihre Selbstständigkeit verloren und zu „Genpaketen“ wurden, die auf andere Zellen angewiesen sind.
Wie könnte das passiert sein?
Ein früher, komplexer, zellulärer Organismus (z. B. ein Parasit) entwickelte eine immer engere Abhängigkeit zu seinem Wirt. Mit der Zeit verlor er überflüssige Gene – z. B. für Stoffwechsel, Zellteilung, Zellmembran – bis er nicht mehr selbstständig leben konnte. Am Ende blieb nur ein winziger Rest: die Gene zur Vermehrung (DNA/RNA), verpackt in einer Hülle (Kapsid) – ein Virus.
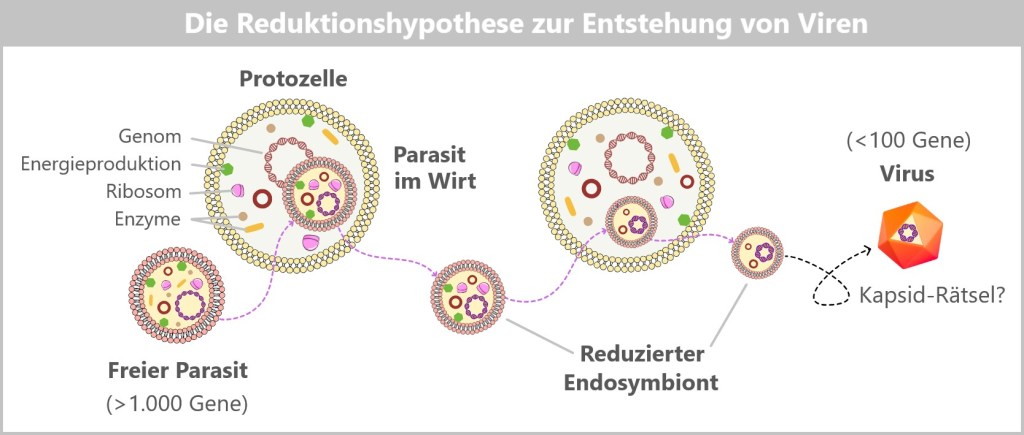
Abb. 22-B: Die Grafik veranschaulicht die Regressive Hypothese – oder Reduktionshypothese, nach der Parasiten durch einen extremen Reduktionsprozess am Ende zu Viren wurden. Vom reisenden Zellparasit zum ultraleichten Virus
Ein freilebender Parasit (z. B. ein kleines Bakterium) befällt eine andere Zelle – so wie auch heute manche Bakterien andere Zellen parasitieren (z. B. Rickettsien oder Chlamydien). Warum? Vermutlich bot die Protozelle eine sichere Umgebung. Vielleicht hatte der Wirt Zugang zu Ressourcen, die der Parasit nicht leicht bekam – etwa Energie, Enzyme oder Nukleotide – eine Art molekulares All-inclusive-Resort.
Möglicherweise begann alles sogar symbiotisch – ähnlich wie bei den Mitochondrien. Erst später wurde diese Beziehung einseitig ausgenutzt – sie wurde parasitär.
Der Parasit lebte zunächst noch eigenständig. Er konnte die Wirtszelle verlassen oder sich in ihr vermehren. Doch nach und nach brauchte er immer weniger eigene Gene – der Wirt lieferte schließlich alles, was er benötigte. Also reduzierte der Parasit sein Gepäck – Schritt für Schritt.
Im Laufe der Zeit verlor er durch Mutationen oder Selektion viele seiner ursprünglichen Gene:
➤ Er stellte keine eigenen Proteine mehr her.
➤ Er verlor die Fähigkeit zur Energiegewinnung.
➤ Schließlich sogar die Gene zur Zellteilung.
Am Ende bleibt nur noch sein Genom – verpackt in einen cleveren „Koffer“: das Kapsid.
Genau hier entsteht das Virus: kein eigenständiges Lebewesen mehr im klassischen Sinn, sondern ein ultraleichter „Reisender“, der sich nur noch mit Hilfe eines Wirts vermehren kann.✅ Argumente für diese Hypothese
➤ Parallelen zu intrazellulären Parasiten: Einige heutige Mikroorganismen, etwa Rickettsien oder Chlamydien, können sich nur innerhalb anderer Zellen vermehren – genau wie Viren. Und: Ihre Genome sind stark reduziert. Das zeigt, dass Zellen durch Parasitismus tatsächlich drastisch „verschlanken“ können.
➤ Große DNA-Viren als „Übergangsformen“: Einige Riesenviren (z. B. Mimivirus, Pandoravirus) haben unglaublich große Genome – größer als manche Bakterien – und enthalten Gene, die man eher in echten Zellen erwarten würde (z. B. DNA-Reparaturenzyme). Diese Viren wirken wie evolutionäre Zwischenstufen zwischen echten Zellen und typischen Viren.
➤ Verlust statt Entstehung: Evolution bedeutet nicht nur „Mehr“, sondern oft auch „Weniger“. Besonders im parasitischen Kontext verlieren Organismen oft Funktionen – genau wie hier angenommen.
❌ Schwächen der Hypothese
➤ Ursprung bleibt unklar: Auch wenn die Hypothese erklärt, wie aus einer Zelle ein Virus werden könnte – sie sagt wenig darüber, wann und unter welchen Bedingungen das passiert wäre.
➤ Keine gemeinsame Abstammung: Die Vielfalt der Viren spricht eher gegen eine gemeinsame „Urzelle“ aller Viren. Wenn alle Viren aus regressiven Zellen stammen, müssten sie sich trotzdem mehrfach unabhängig voneinander zurückentwickelt haben. Manche Forschende nehmen daher an: Regressive Evolution ist nur eine von mehreren Ursprüngen.
➤ Gilt eher für große DNA-Viren: Diese Hypothese passt gut auf große DNA-Viren – aber weniger auf sehr einfache RNA-Viren, die keinerlei zellähnliche Strukturen oder Gene enthalten.
➤ Kapsidproteine zeigen keine direkte Ähnlichkeit: Zellen – selbst extrem reduzierte parasitische – haben keine Kapside. Das ist eine virenspezifische Struktur. Wenn sich ein Virus also durch Reduktion aus einer Zelle entwickelt hat, stellt sich die Frage: Wie konnte eine Struktur wie das Kapsid entstehen, die es in Zellen gar nicht gibt? Sie scheinen also neu entstanden zu sein – und das passt schwer in eine Theorie, die auf „Verlust“ und „Reduktion“ basiert.
➤ Energielücke: Selbst degenerierte Parasiten (z. B. Mycoplasma) behalten Stoffwechselgene – Viren haben gar keine.
⬇️ Fazit
Die Regressive Hypothese zeigt Viren nicht als „flüchtige Genfragmente“ – sondern als Miniaturausgabe ehemaliger Zellen. Besonders bei großen DNA-Viren ist diese Vorstellung plausibel: Sie könnten einst vollwertige zelluläre Parasiten gewesen sein, die durch Anpassung ihre Unabhängigkeit verloren haben. Sie wären dann gewissermaßen das biologische Gegenteil von Evolution zur Komplexität – eine Rückentwicklung zum Kern des Überlebens: Replikation. Die Hypothese scheitert aber an der Vielfalt einfacher Viren.
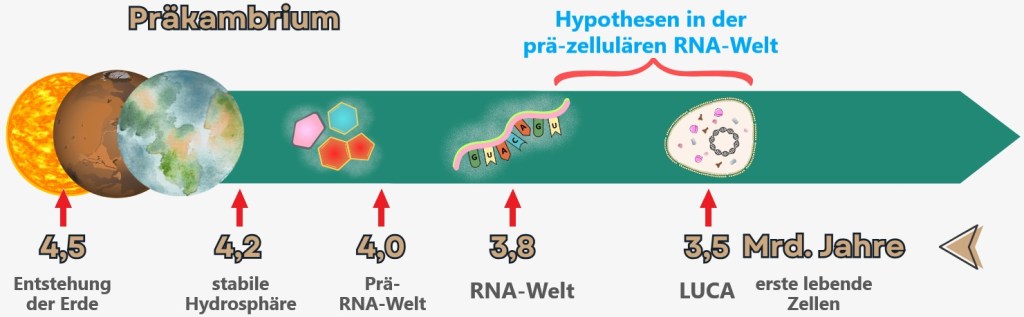
6.2.2. Hypothesen in der prä-zellulären RNA-Welt
In der Ursuppe der frühen Erde regte sich zum ersten Mal so etwas wie Leben – zart, flüchtig und doch folgenschwer. Moleküle entstanden, die nicht nur waren, sondern handelten: Sie schnitten, verbanden, kopierten sich selbst. In winzigen Lipidblasen, die kamen und gingen, wuchs ein Netzwerk aus kooperierenden und konkurrierenden Ribozymen (mehr dazu in „Das erste Flüstern des Lebens“).
Es war kein Leben im heutigen Sinn. Es war ein Spiel von Möglichkeiten. Aus diesem Netzwerk gingen zwei Entwicklungslinien hervor:
① Die stabilen, strukturverliebten Replikatoren, aus denen die ersten Zellen wurden.
② Und die freien, reduzierten Replikatoren, die als Proto-Viren gelten könnten – beweglich, anpassungsfähig, parasitär.Der frühe Parasitismus – war weniger Infektion als Interaktion – eher ein Tanz zwischen Nutzen und Ausgenutztwerden als echter Wirt-Befall. Zwei Hypothesen beschreiben diesen Ursprung:
a) Virus-first-Hypothese – Viren waren zuerst da
b) Co-Evolution-Hypothese – Viren und Zellen entstanden gemeinsam
a) Virus-first-Hypothese – Viren waren zuerst da
Was war zuerst – der Wirt oder der Virus?
Die Virus-first-Hypothese gibt eine verblüffende Antwort: „Weder noch!“ Sie postuliert, dass virenähnliche Replikatoren schon in der RNA-Welt existierten – lange vor Zellen, DNA oder LUCA – als Vorboten des Lebens.
Die RNA-Welt als Brutkasten der Proto-Viren
In dieser Ära waren Ribozyme (RNA-Moleküle mit Enzymfunktion) die ersten „Lebenskünstler“. Einige wurden zu molekularen Schmarotzern:
- Sie nutzten andere RNA-Stränge als Kopiervorlage („Parasitismus light“).
- Sie hüllten sich in Lipidvesikel oder Peptidringe (noch keine echten Kapside).
- Sie vermehrten sich ohne Zellen – etwa an Tonmineralien, die als Katalysatoren dienten.
Der Sprung zum modernen Virus: Erst als Zellen entstanden, wurden aus diesen Replikatoren effiziente Parasiten – die nun zelluläre Maschinerie kapern konnten.
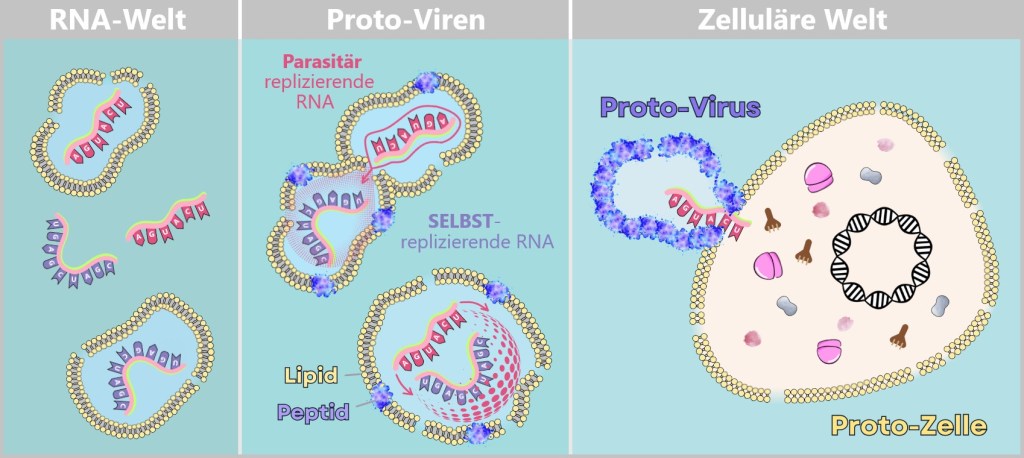
Abb. 22-C: Vom ersten Flüstern zum ersten Angriff: Die Entstehung viraler Strategien Diese Grafik zeigt einen möglichen evolutionären Übergang von präbiotischer Chemie hin zur Entstehung erster virenähnlicher Strukturen:
RNA-Welt (links): Es existieren einfache RNA-Moleküle, einige geschützt in Lipidvesikeln, mit der Fähigkeit zur Selbstreplikation.
Proto-Viren (Mitte): In dieser Phase entstehen erste parasitäre RNA-Moleküle (rot), die sich nicht mehr selbst vervielfältigen, sondern andere RNA-Stränge (blau) ausnutzen. Umhüllt von Lipiden und ersten Peptiden (kurze Ketten aus Aminosäuren) beginnt eine frühe Form des molekularen Parasitismus. Die roten Strukturen symbolisieren parasitäre Interaktionen zwischen RNA-Molekülen.
Zelluläre Welt (rechts): Mit der Entwicklung erster Proto-Zellen verschieben sich diese Strategien: RNA-Parasiten (Proto-Viren) verlassen ihre Peptidvesikel und beginnen, zelluläre Organismen zu infizieren – ein früher Vorläufer der heutigen Viren.✅ Argumente für diese Hypothese
➤ Einzigartige virale Enzyme: RNA-Polymerasen von Viren (wie beim Influenzavirus) ähneln keinen zellulären Proteinen. Das deutet auf einen sehr alten Ursprung hin, möglicherweise aus der RNA-Welt, bevor sich die Domänen des Lebens trennten.
➤ Einzigartige Kapsidproteine: Die Hüllen, mit denen Viren ihr Genom schützen, haben keine direkten Entsprechungen in zellulären Organismen – ein Hinweis auf frühe, unabhängige Evolution.
➤ Globale Verbreitung: Viren infizieren alle drei Domänen des Lebens – Bakterien, Archaeen und Eukarya. Das deutet auf einen Ursprung vor ihrer Trennung hin.
➤ Enorme Vielfalt: Die Vielfalt viraler Genome (RNA, DNA, Einzel- oder Doppelstrang) spricht für eine lange evolutionäre Geschichte, möglicherweise bis in die RNA-Welt zurück.
➤ Parallelen zur RNA-Welt: Viele heutige Viren tragen RNA-Genome. Das passt zu der Annahme, dass RNA das ursprüngliche genetische Material war – und Viren lebendige Relikte dieser Zeit sein könnten.
❌ Schwächen der Hypothese
➤ Wirtsabhängigkeit: Heutige Viren sind auf zelluläre Maschinerie (z. B. Ribosomen) angewiesen. Wie konnten sie sich in der Ursuppe ohne Zellen vermehren? Nutzten sie vielleicht lose Netzwerke von selbstreplizierenden RNA-Molekülen als „Wirte“?
➤ Herkunft der Kapside: Die Evolution komplexer Kapsidstrukturen bleibt ungeklärt. Echte Kapsidproteine brauchen Ribosomen – und die gab’s noch nicht. Proto-Hüllen aus selbstorganisierenden Peptiden (z. B. kurze Aminosäureketten) oder Lipiden könnten RNA stabilisiert haben. Die ersten Kapsidvorläufer könnten sich als Reaktion auf äußeren Stress oder zur besseren Stabilisierung der RNA entwickelt haben – nicht als parasitärer Mechanismus, sondern als Überlebensvorteil, aber die Details bleiben spekulativ.
➤ Kein direkter Nachweis: Viren hinterlassen keine Fossilien. Auch molekulare Spuren aus der RNA-Welt sind bisher nicht nachweisbar – eine generelle Herausforderung in der Erforschung früher Lebensformen.
⬇️ Fazit
Die Virus-first-Hypothese ist eine der faszinierendsten – und umstrittensten – Theorien zur Entstehung des Lebens. Sie legt nahe, dass Viren nicht Nachzügler, sondern Pioniere der Evolution waren: molekulare Boten, die genetische Information verbreiteten, lange bevor es Zellen gab. Vielleicht waren sie sogar ein Katalysator für die Entstehung komplexeren Lebens. Was als molekulares Gerangel begann, wurde zur Blaupause moderner Viren.
b) Co-Evolution-Hypothese – Viren und Zellen entstanden gemeinsam
Nicht Zellen zuerst. Nicht Viren zuerst.
Sondern: Beides zugleich – im Tanz der frühen Evolution.Die Co-Evolution-Hypothese geht davon aus, dass Viren und zelluläre Vorläufer sich parallel entwickelten – aus demselben molekularen Urschlamm der RNA-Welt. Es war kein „entweder–oder“, sondern ein dynamisches Zusammenspiel: Replikatoren, die sich gegenseitig herausforderten, nutzten, stabilisierten – und so die Grundlagen für Leben schufen.
Die Ursuppe als Experimentierfeld
In frühen Lipidvesikeln – kleinen, unvollkommenen Bläschen aus Fetten – sammelten sich RNA-Stränge. Manche dieser Moleküle replizierten sich selbst, andere halfen beim Kopieren, wieder andere schnitten oder verbanden Sequenzen. Kooperation und Konkurrenz begannen gleichzeitig.
Einige Replikatoren entwickelten sich zu immer komplexeren Systemen, aus denen erste Protozellen hervorgingen. Andere blieben reduziert, nutzten lieber bestehende Strukturen, statt sie selbst aufzubauen – eine Art minimalistischer Lebensstil, der an frühe Viren erinnert.
Ein Wechselspiel entsteht
In diesem Szenario war Parasitismus kein später Zusatz, sondern ein ursprüngliches Element der molekularen Evolution.
- Virale Vorläufer konnten Zellvorläufer beeinflussen, etwa durch Genaustausch oder Störung.
- Zellvorläufer wiederum konnten Virenähnliche Replikatoren stabilisieren oder integrieren, etwa als mobile Gene oder regulatorische Elemente.
So könnten Viren und Zellen aus denselben Netzwerken hervorgegangen sein, in denen alles noch fließend war: Selbstständigkeit, Abhängigkeit, Kopie, Konkurrenz.
Kein Ursprung – sondern ein Verhältnis
Die Co-Evolution-Hypothese ist weniger eine Erklärung für den „ersten Virus“, sondern ein Blick auf eine Beziehung, die so alt ist wie das Leben selbst. Viren wären demnach nicht nachträgliche Störenfriede, sondern von Anfang an Teil des Systems – Mitspieler in der Geschichte des Lebens.
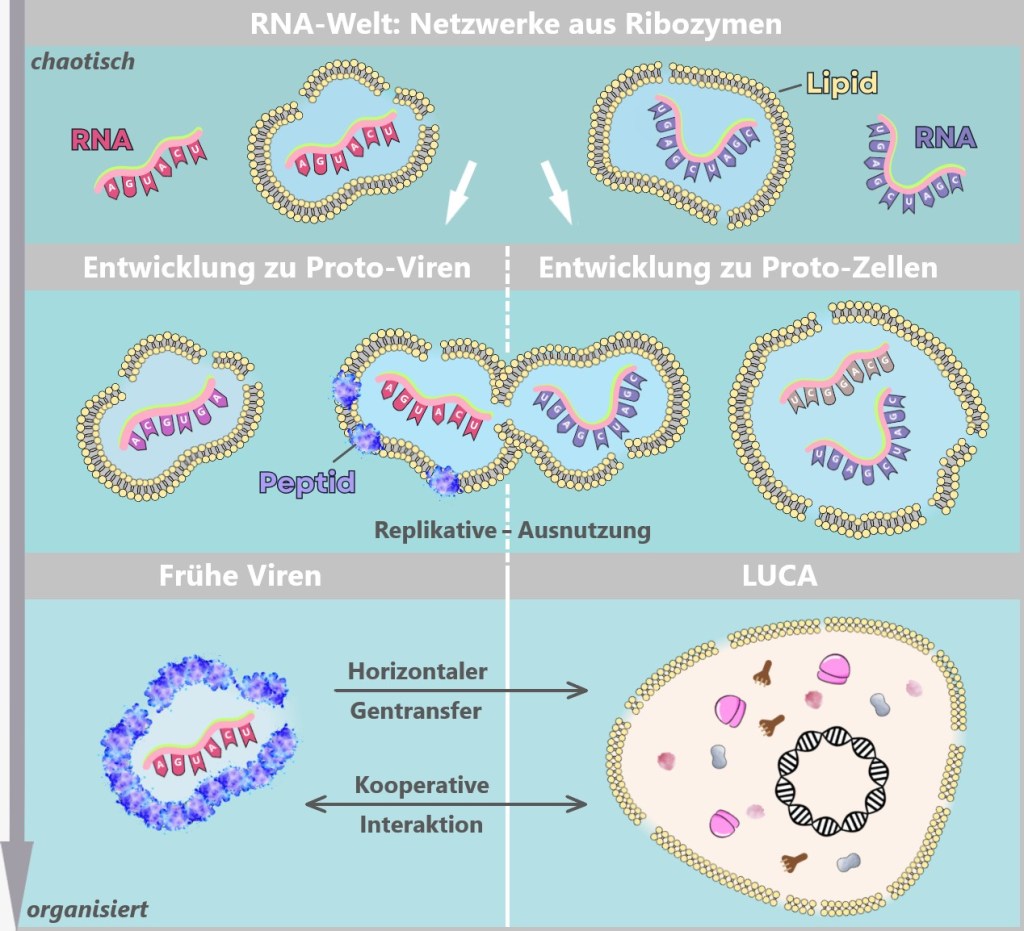
Abb. 22-D: Co-Evolution von Viren und Zellen aus einem gemeinsamen Ursprung. Die Grafik veranschaulicht die zentrale Idee der Co-Evolution-Hypothese: Viren und zelluläre Lebensformen stammen aus einer gemeinsamen molekularen Frühzeit – der RNA-Welt. In Lipidvesikeln koexistierten und konkurrierten Ribozymnetzwerke, aus denen sich zwei unterschiedliche Replikatorstrategien entwickelten: Einige bildeten die Vorstufen stabiler Proto-Zellen, andere reduzierten sich auf das Wesentliche und nutzten fremde Replikationsmechanismen – die frühen Proto-Viren.
Bereits vor der Entstehung echter Zellen zeichnete sich eine Art „molekularer Parasitismus“ ab – nicht im klassischen Sinne, sondern als ungerichtete Replikationsnutzung. Mit dem Auftreten zellulärer Lebensformen intensivierten sich diese Wechselwirkungen: Frühviren konnten nun genetisches Material zwischen Zellen übertragen (horizontaler Gentransfer) oder mit ihnen in komplexe, manchmal sogar kooperative Beziehungen treten. Die Entstehung von LUCA war somit nicht das Ende dieser Co-Evolution – sondern ihr erster Höhepunkt.
✅ Argumente für diese Hypothese
➤ Vermeidung des Henne-Ei-Problems: Sie umgeht elegant die Frage, ob erst Viren oder erst Zellen da waren: Beides entwickelte sich parallel aus denselben molekularen Vorläufern.
➤ Anpassung an die Dynamik der RNA-Welt: Die RNA-Welt war kein linearer Prozess, sondern ein Netzwerk aus Kooperation und Konkurrenz. Diese Hypothese spiegelt diese Vielfalt besser wider als ein klarer Ursprungspfad.
➤ Erklärung der viralen Vielfalt: Unterschiedliche Virusgruppen (RNA, DNA, retrovirale Elemente) könnten sich unabhängig, aber im gleichen Umfeld entwickelt haben – was ihre enorme Diversität plausibel macht.
➤ Evolutionäre Wechselwirkung: Die Co-Evolution erklärt, warum zellenähnliche und virenähnliche Systeme bereits früh miteinander agierten (z. B. durch horizontalen Gentransfer, RNA-Konkurrenz, gegenseitige Anpassung).
➤ Viren als Treiber der Zellkomplexität: Viren könnten nicht nur „Parasiten“, sondern Katalysatoren der Zellentwicklung gewesen sein, z. B. durch Gentransfer, Regulation, Immun- und Abwehrmechanismen.
❌ Schwächen der Hypothese
➤ Begriffliche Unschärfe: Ab wann ist ein Replikator „Virus“ und ab wann „Zelle“? Die Übergänge sind fließend – was die Hypothese erklärend, aber auch schwer fassbar macht.
➤ Fehlende Fossilien: Wie bei der Virus-first-Hypothese fehlen direkte Spuren früher viraler Replikatoren. Molekulare Fossilien aus der RNA-Welt existieren schlicht nicht.
➤ Komplexitätsproblem: Auch in dieser Hypothese bleibt unklar, woher bestimmte virusspezifische Proteine wie Kapsid- oder Polymerase-Proteine ursprünglich kamen. Für echte Kapside, effiziente Replikation und Wirtsnutzung braucht es komplexe Proteine, deren Ursprung noch unklar ist – besonders wenn Ribosomen noch nicht existierten.
➤ Experimentell schwer überprüfbar: Die Hypothese ist theoretisch gut begründet, aber kaum direkt testbar. Simulationen und Rückschlüsse bleiben oft spekulativ.
⬇️ Fazit
Die Co-Evolution-Hypothese bietet eine flexible, systemische Sicht auf die Frühzeit des Lebens – ohne sich auf eine lineare Ursache-Wirkung-Kette festzulegen. Sie passt gut zu den chaotisch-kooperativen Verhältnissen in der RNA-Welt. Viren wären nach diesem Modell nicht „entweder Zellabkömmlinge oder Ureltern“, sondern evolutionäre Parallelgänger – gleich alt, gleich bedeutend, nur auf einem ganz anderen Kurs unterwegs.
Viren sind die Hieroglyphen der Biologie
…wir entschlüsseln sie, aber ihr Ursprung bleibt ein Geheimnis.
Wie flackernde Schatten an der Höhlenwand:
- mal abtrünnige Zellteile,
- mal entlaufene Gene,
- mal Urahnen des Lebens selbst.
Vielleicht geht es bei der Frage nach ihrer Herkunft gar nicht um den einen Ursprung, sondern um viele evolutionäre Pfade – Pfade, die sich kreuzen, überlagern und rückkoppeln. Die klassischen Hypothesen (Virus-first, Co-Evolution, Progressive, Regressive) müssen sich nicht widersprechen – sie könnten sich an verschiedenen Stellen der Geschichte verwirklicht haben.
Und vielleicht ist die Frage nach dem Ursprung gar nicht die entscheidende. Viren zwingen uns, etwas Tieferes zu begreifen:
Leben ist kein Zustand, sondern ein Prozess – und Viren sind sein unsteter Puls.
Sie erinnern uns daran, dass Evolution kein geradliniger Stammbaum ist, sondern ein wirbelnder Fluss aus Kooperation, Diebstahl und Neuerfindung. Woher sie kamen? Wir wissen es nicht. Dass sie bleiben? Gewiss.
Vielleicht faszinieren sie uns genau deshalb – weil sie zeigen, dass das Leben nie stillsteht.
Weiterführende Quellen
Die uralte Viruswelt und die Evolution der Zellen
Die Ursprünge der Viren: Evolutionäre Dynamik der Fluchthypothese
Viren stehen im Mittelpunkt der zellulären Evolution
Der Ursprung von Viren und ihre mögliche Rolle bei großen evolutionären Übergängen
Eine virozentrische Perspektive auf die Evolution des Lebens
Wie haben sich Viren entwickelt und in welcher Beziehung stehen sie zum zellulären Leben?
Viren und Zellen sind seit Anbeginn der Evolution miteinander verflochten
Was uns Viren über Evolution und Immunität erzählen: Über Darwin hinaus?
Gab es Viren auf der Erde, bevor lebende Zellen auftauchten? Ein Mikrobiologe erklärt
Virus-First-Hypothese • Viren älter als das Leben?
Viren aus der Urzeit? – Die Entstehung der Viren und ihre Rolle in der Evolution

7. Warum gibt es Viren?
Die kurze Antwort: Weil es immer etwas geben muss, das stört.
Klingt witzig – ist aber ein Naturprinzip:
Nichts bleibt lebendig, wenn es nie herausgefordert wird.Man kann vieles über Viren sagen.
Dass sie nicht leben.
Dass sie nur stören und zerstören.
Dass sie bloß molekulare Parasiten sind –
auf das Leben angewiesen, das sie befallen.
Und doch: Ohne sie wäre vieles nicht, wie es ist.
Vielleicht gäbe es uns nicht einmal.Leben braucht Wiederholung. Und Abweichung.
Die Geschichte des Lebens begann nicht mit einem Ziel.
Sie begann mit Wiederholung.
Wieder und wieder verbanden sich Moleküle, zerfielen, verbanden sich neu.
Nicht, weil sie mussten – sondern weil es möglich war.Was sich wiederholt, wird irgendwann wahrscheinlich.
Was funktioniert, bleibt.
Was bleibt, muss sich verändern.
Und Veränderung braucht Abweichung.
Mutation. Fehler. Störung.
Ohne sie gäbe es keine Vielfalt, keine Evolution – keine Geschichte.Ordnung ist träge. Leben ist Bewegung.
Als die ersten stabilen Systeme entstanden, war das ein Triumph –
aber auch eine Gefahr.
Denn was stabil ist, neigt zur Starre.
Was zu perfekt funktioniert, wagt nichts Neues.Leben aber braucht Bewegung.
Hier beginnen die Viren ihre Rolle zu spielen –
nicht als Gegner des Lebens, sondern als Gegenkraft.
Sie fordern heraus.
Sie unterbrechen.
Sie treiben Zellen in die Defensive – und in die Innovation.Die Dualität: Ordnung vs. Chaos
Zellen bauen Mauern, Viren springen darüber.
Ihre Beziehung ist eine paradoxe Symbiose:
Sie töten – und machen Leben möglich,
halten Ökosysteme am Laufen
und brachten uns Plazenta-Gene.Viren sind keine Fehler – sie sind ein Prinzip.
Sie sind Grenzverletzer, nicht aus Bosheit, sondern aus Prinzip.
Sie halten das Leben durchlässig. Offen. Wachsam.
Sie übertragen Gene, öffnen neue Wege, mischen Systeme auf.Nicht immer zum Guten.
Aber immer mit Wirkung.Vielleicht sind Viren genau das, was das Leben braucht, um lebendig zu bleiben.
Nicht als Gegenmodell – sondern als Mitspieler, als Schattenwurf des Lebendigen selbst.
Der Baum des Lebens – durchdrungen von Viren
Wenn wir heute auf den Baum des Lebens schauen, auf seine Äste, Zweige, Verzweigungen – dann sehen wir die Zellen, die Arten, die sichtbare Spur der Evolution.
Und die Viren?
Fehlen.
Nicht, weil sie unwichtig wären – sondern weil sie sich nicht verorten lassen.Sie sind weder Ast noch Blatt, sondern der Wind, der sie bewegt.
Sie sind das Flüstern zwischen den Zweigen.
Keine eigene Linie, kein Teil des Holzes – und doch überall.Ein Strom von Information, der den Baum nicht nur umgibt, sondern auch formt.
Sie sind die Punkte zwischen den Linien, die Verbindungen schaffen.
Als Wellen der Störung, die Wachstum erzwingen.
Als Abweichung, die das Muster nicht zerstört, sondern erweitert.Sie sind die dunkle Materie der Biologie: unsichtbar, aber alles durchdringend.
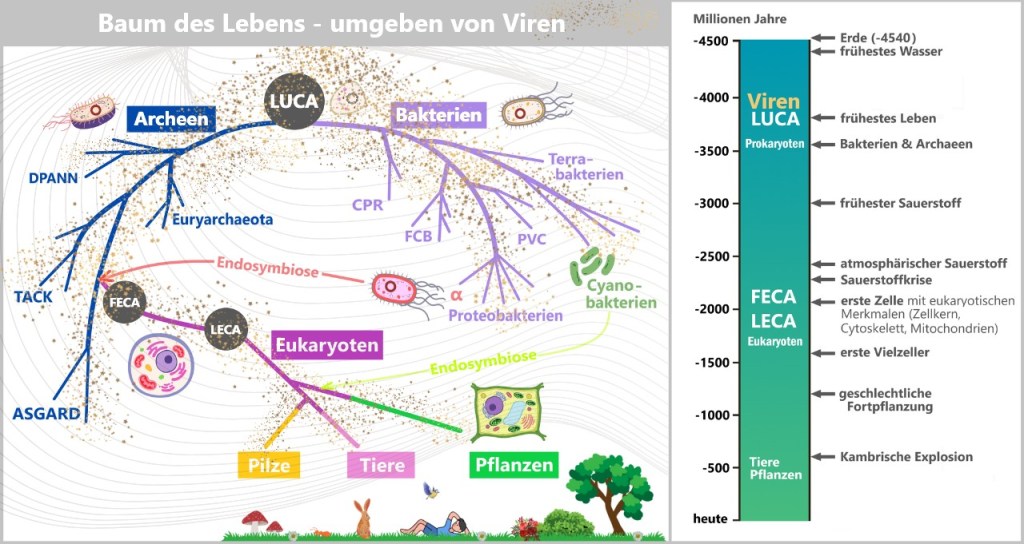
Abb. 23: Der Stammbaum des Lebens umspielt von Viren. Der „Baum des Lebens“ zeigt die Abstammungslinien zellulärer Organismen – Bakterien, Archaeen und Eukaryoten – zurück bis zum gemeinsamen Vorfahren (LUCA). Die feinen Pünktchen und Bahnen symbolisieren Viren: nicht als festen Ast, sondern als diffuses Netzwerk, das den gesamten Baum durchdringt.
In dieser Sichtweise sind Viren keine Außenseiter, sondern kreative Spielarten molekularer Evolution. Keine Entität mit klarer Abstammung, sondern eine wiederkehrende Erscheinung in einem Universum, das mit Variation und Wiederholung spielt.
Und darum sind sie da.
Nicht weil sie wollen. Nicht weil sie müssen.
Sondern weil sie immer wieder entstehen – dort, wo Natur variiert.
Wo Leben sich organisiert – und aus dieser Ordnung heraus etwas Neues wagt.Sie sind da, weil sie nicht zu vermeiden sind.
Wie ein Echo des Prinzips, das allem zugrunde liegt:
Störung ist nicht das Gegenteil von Leben.
Störung ist seine Möglichkeit.Viren sind da, weil Störung kein Unfall ist –
sondern das Werkzeug des Universums, um Leben wach zu halten.Denk daran, wenn dich das nächste Mal ein Virus nervt:
Vielleicht ist es bloß das Universum, das dir einen kosmischen Klaps verpasst –
und murmelt:
„Na los, wach auf! Hier ist deine tägliche Dosis Chaos – damit du schön evolvierst.“Epilog: Die Unscheinbaren
Viren sind der Sand im Getriebe der Schöpfung.
Ohne Willen, ohne Leib –
und doch die unsichtbare Hand,
die Evolution schreibt.
Das Flüstern zwischen den Zeilen des Lebens –
nicht aus Absicht, sondern aus Notwendigkeit.Sie kamen aus dem Nebel der Anfänge
und blieben im Schatten –
nicht als Fremdkörper, sondern als Gegenspieler,
als Prüfstein und als Impuls.Sie lehren uns Demut:
Denn was zerstört,
kann auch erschaffen.Vielleicht ist das ihre Botschaft:
Dass das Leben nicht gegen das Chaos wächst,
sondern mit ihm tanzt.
Nachtrag:
Ich habe mich dem Thema als völliger Laie genähert. Und gerade das erwies sich als Vorteil – denn ich wusste, was ich nicht wusste.
Mit Neugier, Skepsis und Staunen bin ich eingetaucht in die Welt der Viren – und war überrascht, welch ungeahnte Dimensionen sich auftaten. Dieses Thema trägt eine Wucht an Erkenntnis in sich, die ich nie erwartet hätte.
Während meines Suchens, Fragens und Formulierens hatte ich großartige Unterstützung durch KI-Systeme wie ChatGPT und DeepSeek. Der Dialog mit diesen Werkzeugen hat mein Lernen nicht nur beschleunigt, sondern auch vertieft. Ohne diese Hilfe hätte ich vieles nicht gesehen – und erst recht nicht so klar formulieren können.
Was hier entstanden ist, ist das Ergebnis von menschlicher Neugier und maschinischer Geduld. Und es zeigt, dass Lernen heute mehr denn je ein gemeinschaftlicher Prozess sein kann – über Grenzen hinweg, auch über die zwischen Mensch und Maschine.
Quellen (Stand vom 07.07.2025)
Baltimore-Klassifikation, Bioinformatische Analyse, Co-Evolution-Hypothese, DNA, Elektronenmikroskopie, Evolution, Grippe, Illumina-Sequenzierung, Infektion, Influenzavirus, Kapsid, Koch’sche Postulate, Kristallisation, Kryo-Elektronenmikroskopie, Kryo-Elektronentomographie, Mikrobiologie, mRNA, Mutantenwolke, Mutation, Oxford Nanopore-Technologie, Pandemie, PCR, Polymerase, Progressive Hypothese, Regressive Hypothese, RNA, Sanger-Sequenzierung, Sequenzierung, Viren, Virion, Virologie, Virus, Virus-first-Hypothese, Wirtszelle, Zellkultur -
The First Whisper of Life

When we look at life – from deep forests to snow-covered peaks, from shimmering coral reefs to mighty blue whales, from tiny microbes to buzzing insects – we see a story that has unfolded over billions of years. This story is often described as the „Tree of Life”: a web of branches that connects all species and leads back to a common origin.
But the deeper we look into this tree, the more blurred its roots become. They fade into the mist of time – and there, at the very beginning of everything, another story whispers: a hidden, mysterious tale.
It began in the half-light. In the warm veins of the earth, where bubbling springs washed around elements and the green sea was still an alphabet of ions. Between clouds of sulphur and black basalt, the first words folded themselves out of molecules, fragile at first, entangled in the thermal breath of the depths. Not life, not death – just a hunch. Molecules that carried information and drove chemical reactions. Sometimes they broke apart. Sometimes they probed one another. And sometimes, very quietly, they kept whispering.
Was this whisper a coincidence? Or already a plan?
Anyone who sets out to discover the origin of life does not enter a well-lit museum – but a labyrinth. There is no clear „this is how it was”, but rather a web of hypotheses, probabilities, mechanisms and gaps. What we know today about the beginnings of life is the result of intensive research, astonishing discoveries – and always: unanswered questions.
In this labyrinth, many paths lead into the unknown. One of the most fascinating trails is the RNA world hypothesis: a narrative in which tiny molecules – RNA – sang the first notes of life. But other stories whisper along: of minerals that held molecules together, of tiny protein chains that created stability, of fat-like shells that provided protection. Perhaps all these paths were intertwined – and yet together they led to RNA, which built a bridge between chemistry and life. To where the first whisper became audible.
This text follows the trail through the mist:
the idea that RNA was once the beginning of it all.
Act 1: From Chaos to the RNA World – The Building Blocks Awaken
Scene 1: Primordial Soup – The Birth of Simple Molecules
Scene 2: Bases – Letters of Nitrogen and Carbon
Scene 3: Sugar: Ribose – Sweet and Fragile
Scene 4: Phosphate – The Universal Glue
Scene 5: Stardust & Impacts – A Cosmic Contribution?
Act 2: Nature’s Laboratories – Birthplaces of RNA
Scene 1: The Forge of the Deep – Hydrothermal Vents
Scene 2: The Alchemy of Clay – Birthplaces on Land
Scene 3: Many Paths, One Goal
Act 3: The First Self-Replicators Awaken
Scene 1: From Chain to Tool – RNA’s Functional Maturation
Scene 2: The Power of Folding
Scene 3: The Energetic Pact
Scene 4: Partial Replication
Scene 5: Separation – A Farewell That Created Something New
Scene 6: From Short to Longer Chains – The Power of Ribozymes
Scene 7: Errors as Opportunity – The Engine of Evolution
Act 4: Life in a Droplet – Lipid Vesicles as Shelters
Scene 1: The First Strongholds
Scene 2: The Entry – Carryover or Immigration?
Scene 3: The Pact – RNA Stabilizes, Vesicles Protect
Scene 4: The Chance of Imperfection
Scene 5: Selection Within the Droplet
Act 5: From Chance to Function – How RNA Gained Complexity
Scene 1: A Random RNA Emerges
Scene 2: From RNA Strand to Ribozyme
Scene 3: RNA Copies Itself – With Errors
Scene 4: Mutation Creates a New Function
Scene 5: Selection Favors Useful RNAs
Scene 6: RNA Between Error and Function
Epilogue: The Echo of the Primordial Soup

Act 1: From Chaos to the RNA World – The Building Blocks Awaken
Today, life is a well-organized structure: DNA archives knowledge, RNA transmits messages, proteins do the work. But in the beginning, there was no structure – just a single molecule – that could do everything: RNA. It could have been the first actor – simultaneously archive, messenger and tool.
This is precisely what the RNA world hypothesis proposes: RNA as the first „building block of life”, capable – like DNA – of storing information, and – like enzymes – of catalyzing chemical reactions, all on its own and without assistance.
RNA is made up of repeating building blocks – nucleotides.
Each nucleotide consists of three parts:🧩 a nitrogenous base (such as adenine, uracil, guanine, or cytosine),
⬟ a sugar (ribose), and
⚡ a phosphate group, which acts like a bracket and joins the units together to form a chain.But how did this miracle molecule come into being in a lifeless world?
Scene 1: Primordial Soup – The Birth of Simple Molecules
Around four billion years ago, the worst birth pangs of the young Earth had passed – but the planet remained a fiery, restless chaos. Volcanoes spewed toxic gases and bubbling lava, meteorites carved crater-like wounds into the surface, and the young oceans boiled from the impacts.
Here, in this glowing wilderness, six elements mixed to form a primordial cocktail:
- Hydrogen – as fleeting as a ghost’s breath
- Carbon – the supple connector
- Nitrogen – stubborn, yet essential
- Oxygen – fiery and highly reactive
- Phosphorus – the energy-rich spark
- Sulphur – smelly, but incredibly useful.
These basic elements were omnipresent – deep within the Earth’s rock, dissolved in the hot seas or released by volcanic embers. They were just waiting for energy. Lightning flashed through the haze, UV light blazed on the tidal zones, and deep below, magma baked the oceans into chemical cauldrons. Gradually, the elements combined into gases like methane (CH₄), carbon dioxide (CO₂), and ammonia (NH₃) – simple molecules, yet rich in chemical potential.
Fig. 1: In the energy-rich atmosphere of early Earth, simple organic molecules could form. And they kept reacting. In hot oceans, on drying mudflats, or in mineral-rich springs, these gases formed organic molecules like hydrogen cyanide (HCN) and formaldehyde (CH₂O) – deadly substances that, paradoxically, became the raw materials that made life possible.

Could the building blocks of life really have formed in this hellish landscape?
In 1953, two researchers provided the answer – in a glass jar barely bigger than a teapot. Stanley Miller and Harold Urey mixed methane, ammonia, hydrogen and water vapor – and let lightning flash through it.
After a week, the liquid – the „primordial soup” – had turned cloudy with amino acids: those organic compounds that would later give rise to proteins, the „molecular machines of cells”. Not yet life, but proof: out of chaos, order can emerge.
And then the discovery that was hardly noticed: Hydrogen cyanide (HCN) and formaldehyde (CH₂O) – two substances without which RNA would never have been created.
Today we know: the gases used in the original experiment likely didn’t exactly match the conditions of early Earth. Methane and ammonia were probably less common than once assumed, and the atmosphere was more neutral – rich in CO₂ and nitrogen. But the principle remains valid: even under simple conditions, the building blocks of life can emerge. Modern experiments (Cleaves et al., 2008; Bonfio et al., 2018) show that even in CO₂-rich environments – likely characteristic of early Earth – life’s precursors can form. With the help of minerals (Erastova et al., 2017), the synthesis becomes even more efficient.
What’s fascinating about this: there doesn’t seem to be just one path that leads to life – but many. Different environments, various reaction pathways, and diverse ingredients – and yet, time and again, the same fundamental building blocks emerge.
Even if the early Earth’s atmosphere was likely not as rich in methane and ammonia as Miller and Urey once assumed, hydrogen cyanide (HCN) and formaldehyde (CH₂O) could still have formed. Local chemical niches, lightning, UV radiation, or even meteorites repeatedly supplied energy – enough to produce these highly reactive molecules.
But how do gas and energy become a code?
The true miracle was yet to come:
Scene 2: Bases – Letters of Nitrogen and Carbon
Hydrogen cyanide (HCN) and formaldehyde (CH₂O) were abundant in the primordial soup. In the forges of the deep – heated by volcanoes and kneaded by minerals – they combined to form purine and pyrimidine rings: organic frameworks made of carbon and nitrogen atoms.
These rings eventually gave rise to stable structures – chemical letters with astonishing permanence. Adenine was just one of them.
Fig. 2: The primordial soup becomes an alphabet soup – greatly simplified, yet full of meaning. Hydrogen cyanide (HCN) molecules encountered formaldehyde (CH₂O) in a lagoon. They reacted – not on purpose, but because chemistry demanded it. Nitrogen atoms got stuck together, carbon rings closed. At some point it was there: Adenine, the purine base that would later carry the code for entire ecosystems.
An entire alphabet of life grew from purine and pyrimidine rings: adenine and guanine from the purines, cytosine, uracil and thymine from the pyrimidines.
Fig. 3: Purine and pyrimidines are the chemical core structures of the bases in RNA and DNA. These organic bases carry genetic information and are essential for the origin of life. They were still silent, not yet carrying any message. But within their rings lay a strange potential – as if they could hold secrets they themselves did not understand.
Today we know: these reactions are part of a chemical pathway (Patel et al., 2015) that can realistically lead to the formation of purines and pyrimidines – the „letters” of RNA.
But an alphabet is not yet a word. What was still missing was a rhythm, a backbone – a structure the letters could cling to: ribose, the sweet bearer of meaning.
Scene 3: Sugar: Ribose – Sweet and Fragile
While the bases were forming in bubbling pools, Earth was weaving their delicate partner elsewhere: ribose, a sugar made of five carbon atoms – lined up like pearls on a string.
Ribose was a creature of fragile beauty:
- Sweet in its chemical soul, like all sugars, and
- Fragile, for in water it easily fell apart.
To preserve this precious molecule, the Earth reached out its arms: borate minerals from volcanic depths gently enclosed the ribose – protectively, not too tightly, and only for as long as needed. Until the right moment came to let go.
What might have happened?
In volcanic regions – especially in drying ponds or mineral–rich waters – three crucial ingredients came together:
- Formaldehyde (CH₂O), a simple organic building block,
- Energy such as UV light or volcanic heat and
- Borate minerals, formed through rock weathering.
This mixture set a chemical chain reaction in motion: the formose reaction. Several molecules of formaldehyde combined to form various sugars – one of which was ribose.

Fig. 4: The Formose Reaction – Simple molecules like formaldehyde combine under suitable conditions to form more complex sugars such as ribose – a chemical miracle born of heat, time, and catalysis. But it was just one among many – and particularly unstable. Its numerous –OH groups made it highly reactive and prone to breakdown in aqueous environments.
Salvation came from the rock: in ponds rich with borate minerals, borate ions (B(OH)₄⁻) dissolved into the water and bonded preferentially with ribose. This binding – at two neighboring –OH groups – blocked the molecule’s most vulnerable reaction sites and stabilized it. Not permanently, but long enough. Ribose could now survive not just for minutes, but for days.
Fig. 5: A Chemical Embrace – Borate minerals can bind to ribose by „docking” onto specific parts of the sugar. In doing so, they form a kind of protective shield. Left: Free ribose – a sugar with multiple hydroxyl groups (–OH), making it prone to degradation.
Center: Borate ion (B(OH)₄⁻) – found, for example, in borax minerals.
Right: Ribose–borate complex – the borate ion binds to two neighboring OH groups of the ribose. This „freezes“ the unstable sugar structure and protects it from decay.Surprisingly, it was borate – a seemingly unremarkable byproduct of volcanic processes – that became the guardian of a key molecule of life.
Fig. 6: In the shadow of primeval volcanoes, rocks weathered – releasing borate. Thousands of sugars were formed, but only a few ribose molecules survived – until borate found them. Laboratory experiments confirm: Ribose can form in volcanic ponds – but only borate turned this chemical lottery into a viable survival strategy.
Albert Eschenmoser (ETH Zurich) demonstrated that formaldehyde, in an alkaline solution (pH 10–12, simulating volcanic ponds), polymerizes under heat into sugars like ribose – but in a chaotic fashion, yielding less than 1% ribose (Eschenmoser, 2007).
Steve Benner (Foundation for Applied Molecular Evolution) demonstrated that borate ions stabilize the sugar ribose in aqueous solution – by specifically binding to certain regions of the ribose called cis-diol groups (two neighboring OH groups), thereby protecting it from degradation (Ricardo et al., 2004).
Deoxyribose – The Silent Twin
Alongside ribose, a second form quietly emerged: deoxyribose, its silent twin. Born in the same molecular dance, likely sheltered by borate, perhaps shaped by clay. Only a tiny detail set her apart from her sister: one oxygen atom was missing – hence her name: „deoxy” – without oxygen.

A seemingly small difference with immense consequences. The missing –OH group made deoxyribose less reactive, but significantly more stable. For now, she remained in the shadows – inconspicuous, unnoticed.
While ribose forms the backbone of RNA (RNA = ribonucleic acid), deoxyribose shapes the structure of DNA (DNA = deoxyribonucleic acid). Nature had made provisions: ribose for the moment. Deoxyribose for eternity.
But before the story could continue, one crucial element was still missing: a molecular glue that could link the letters to the sugars and form syllables. Nature needed a universal ally – it needed phosphate.
Scene 4: Phosphate – The Universal Glue
While ribose and the bases slumbered in the warm ponds of the young Earth, the third member of the group was still missing – a molecule that could do more than merely exist. A connector.
Phosphate was a child of fire and water: born in apatite rocks deep inside the earth, where heat and pressure melted the elements. There it remained, locked away – until the surface began to bubble.
Volcanoes spewed their fiery gases – carbon dioxide, sulfur dioxide – which mixed with water to form acidic mists that broke down even the hardest rocks. Evaporation and condensation washed the phosphate from the stone into shallow pools, where sugars and bases were already drifting. There it waited – seemingly unremarkable: an ion with three negative charges, restless and ready to bind.

Fig. 7: Volcanic gases (CO₂, SO₂) react with water to form acids (H₂CO₃, H₂SO₄), which attack apatite rocks and release phosphate (PO₄³⁻). Phosphate was everywhere – and it could do almost anything. It was the spark that set molecules in motion, built bridges, and drove reactions. It’s no coincidence that phosphate remained the energy currency of life – the universal fuel of every cell.
But back then, it was still single.
The Great Union
It was not easy for the wild phosphate character to find a partner. It flirted with Ribose, but she was rather unstable or too passive due to her borate protector. And the bases preferred to react with themselves. Magnesium ions caressed the negative charge – they softened phosphate’s irritability and allowed a gentler approach. Evaporating water pressed the molecules together.
Phosphate finally found a foothold: it clasped ribose at the fifth carbon – a stable grip that made new reactions possible. Attracted by the molecular wrestling, a base approached. Determined, it grabbed the first carbon atom of the ribose and swung itself towards it. A twitch, a bond…
Three became one: the first complete nucleotide – the fundamental unit of RNA.
Fig. 8: RNA Nucleotide – Complete and Simplified Representation A nucleotide consists of three building blocks: a phosphate residue (green), a sugar called ribose (pink) and a nitrogenous base (here: adenine, red). The sugar has five carbon atoms (numbered 1′ to 5′). The base is bound to the 1′ carbon, the phosphate to the 5′ carbon.
The ribose is linked to the base and the phosphate by a condensation reaction with elimination of water (H2O). This forms an „N-glycosidic bond” (base-ribose, blue-grey) and a „phosphoester bond” (ribose-phosphate, orange).Geometry, chemistry – and a touch of chance forged the triad of ribose, base, and phosphate into a single molecule. A tiny triumph, yet one that opened the door to life.
Laboratory experiments simulated primeval conditions: They showed that under the right circumstances – such as in mineral-rich, heated waters – purine and pyrimidine bases, ribose, and even complete nucleotides can form abiotically. These studies used aqueous solutions, volcanic heat, phosphate as a catalyst, and drying cycles. Their experiments demonstrated that the chemical pathways leading to these molecules were not only possible, but likely under the conditions of early Earth. (Powner et al., 2009 and Becker et al., 2016)
While the first compounds were forming in the pools, the sky looked down – and intervened. Meteor showers crashed onto the Earth. They brought new elements, new impulses. Perhaps even new ideas – in the form of molecules…
Scene 5: Stardust & Impacts – A Cosmic Contribution?
Interestingly, the building blocks of RNA may not have originated solely on Earth. In meteorites like the famous Murchison meteorite, nucleobases such as uracil and cytosine have been detected. (Callahan et al., 2011). Ribose, amino acids, fatty acids, and precursors of lipids have also been found in these extraterrestrial rock fragments (Pizzarello et al., 2006). All of this suggests that the ingredients of the RNA world may have once come from space – a cosmic gift to the young Earth.
Meteorites: The Universe’s First Life Delivery Service
These celestial messengers originated from the primordial cloud that also gave birth to our Sun 4.6 billion years ago. In the cold, dark regions of the early solar system – within molecular clouds, on comets, and on asteroids – UV light and cosmic radiation transformed simple molecules like methane and ammonia into more complex organic compounds (Bernstein et al., 2002).
During the so-called Late Heavy Bombardment, around 4.1 to 3.8 billion years ago, countless meteorites hit the Earth. Amino acids, sugars and other organic molecules rained down on our planet with them – the raw materials of life, packaged as special cosmic deliveries. Comets such as 67P/Churyumov-Gerasimenko, explored by the Rosetta space probe, also delivered building blocks such as glycine and lipid precursors (Altwegg et al., 2016). Even microscopic fine dust – so-called stardust – still delivers thousands of tons of organic molecules to Earth every year. (Maurette et al., 2000).

Fig. 9: A meteorite crashes onto the young Earth, bringing with it molecules from the depths of space: Nucleobases such as adenine, guanine, uracil, and cytosine, as well as ribose – the fundamental building blocks of RNA. Precursors of lipids, phosphorus compounds, and glycine (the simplest amino acid) were also part of the cosmic cargo. Hydrogen cyanide (HCN), a key precursor of organic compounds, may have triggered chemical evolution. These molecules, born in the hearts of dying stars, rained down and enriched the primordial soup – a cosmic contribution to the first whisper of life.
Molecules from the depths of space for the primordial soup
Once they landed on Earth, these molecules could interact with terrestrial substances – and perhaps fuel chemical evolution. The impacts themselves provided energy: heat, pressure, and shockwaves that linked molecules and formed new structures – perhaps even the first nucleotides.
The robustness of these molecules is astonishing: they survived millions of years in space, the red-hot entry into the volcanic primeval atmosphere, the heat on impact and the harsh conditions of prehistoric times.
This cosmic contribution nourishes the so-called panspermia hypothesis, which suggests that the building blocks of life may have originated in space. While the idea that fully formed life reached Earth remains controversial, the discovery of organic molecules in meteorites supports the concept of „soft” panspermia: Earth was gifted with the ingredients of life – molecules born in the hearts of dying stars that found their way to us over the course of aeons.
Life finds its way
Perhaps life is not mere coincidence – but the result of a chemical possibility, deeply rooted in the blueprint of the universe. Dust from stars, born in the explosions of long-extinct suns, gathered, combined, danced in the light fields of distant worlds. Wherever the conditions were right, molecules began to organize, to react – and laid the foundation for what we call life. That the building blocks of life could arise in so many places – on icy worlds, in deep nebulae, in cosmic dust – speaks of a deeper truth: The universe is not cold and empty. It is, by its very nature, open to the wonder of life.
Act 2: Nature’s Laboratories – Birthplaces of RNA
The ingredients were in place – fallen from the sky or born of Earth itself. But individual building blocks did not yet make life. What was missing was the right place: a setting where molecules could join into chains, where syllables could become words. Early Earth offered many stages, but two stand out: deep-sea hydrothermal vents and drying ponds with mineral-rich walls. Two worlds – each its own laboratory – opposite in nature, yet united in their creative potential.
Scene 1: The Forge of the Deep – Hydrothermal Vents
Deep in the primordial ocean, where tectonic plates drifted apart and the Earth exhaled fire, cracks opened in the seafloor. Rising magma was tamed by the cold seawater – giving birth to „chimneys”: the hydrothermal vents. From these towers surged hot, mineral-rich water – a bubbling cocktail of sulfur, metals, and carbon compounds.
In this steaming darkness, processes unfolded that prepared the stage for life:
- Mineral surfaces (e.g., iron sulfides) acted as catalysts.
- High temperatures (70–150 °C) and strong chemical gradients provided energy.
- Porous structures captured molecules, protected them from the destructive force of the ocean and concentrated them – perfect conditions for reactions.
Here, nucleotides could form and link together – into the first short RNA chains (Wächtershäuser, 1988). Perhaps it was here that the first whisper of life was heard.
Fig. 10: Black Smokers – Micro-laboratories of the Primordial Soup In a primordial underwater landscape, black plumes rise from hydrothermal vents – so-called „Black Smokers”. Magma heats mineral-rich water that shoots up from the ocean floor. The porous rocks are riddled with fine cracks where chemical reactions can take place. A magnified section (outlined in white) reveals what might be happening in these micro-laboratories: Organic building blocks like nucleobases (A, U, G, C), ribose, and phosphates meet. Lightning bolts symbolize energy driving reactions – such as the formation of nucleotides. On the right, a short RNA chain begins to emerge – possibly the beginning of life.
(Scale reference: Vent diameter ~2 m; microcracks <1 mm)
Scene 2: The Alchemy of Clay – Birthplaces on Land
Far from the depths of the ocean, in shallow ponds and along volcanic shores, another stage lay quiet. Here, fine clay rested – formed from volcanic ash, layered like the pages of a book. These clay minerals had a special architecture: negatively charged silicate surfaces, with positively charged ions such as sodium (Na⁺) or calcium (Ca²⁺) in between.
What accumulated within these layers was no coincidence. The negatively charged surfaces drew in positive partners as if by magic: ribose bound to borate, nitrogenous bases, and later complete nucleotides. Even the negatively charged phosphate groups found a foothold – bound to the positive ions of the clay.
A rhythmic cycle of wetness and dryness pressed the molecules closer together. With each round of evaporation, the building blocks moved nearer until they linked: first ribose and base into a nucleoside, then – with phosphate – into a nucleotide. As the cycles continued, nucleotides joined to form the first RNA chains. The clay guided it all – stage, tool and director in one. And it judged, too – for only the most stable nucleotides survived the elemental trials. An astonishing feat for a bit of clay.

Fig. 11: Clay Minerals – The Master Puzzler of Life In the shadow of primeval volcanoes, rocks weathered and broke down. From them, fine layers of clay formed and settled in shallow ponds. With their polarized structure, they acted like molecular workbenches: organizing, concentrating, and stabilizing organic building blocks.
In the magnified section, one can see how the charged surfaces of the clay layers hold on to ribose, phosphate groups, and bases – like pieces of a puzzle assembling themselves. Through repeated cycles of wetting and drying, the molecules were pressed closer together. This led first to the formation of nucleosides, then complete nucleotides – until, eventually, RNA chains could begin to grow.In experiments, James Ferris (2006) demonstrated that montmorillonite – a common type of clay – can indeed support these processes: nucleotides accumulated within its layers, became activated, and linked together to form RNA chains of up to 50 nucleotides in length.
In the deep sea, it was heat that energized the molecules – a primeval oven where chemistry found structure. On land, by contrast, clay acted like a loom: persistent, layering, connecting. Two worlds, two principles – and yet perhaps part of a greater whole.
Scene 3: Many Paths, One Goal
Whether in the bubbling depths of hydrothermal vents or on quiet clay surfaces under the sun – both settings offered plausible stages for the birth of RNA. Perhaps they complemented each other: what the ocean began, the land completed. Or vice versa. Perhaps there were entirely different paths.
For early Earth was not a tidy lab with a protocol – it was a chaotic playground of the elements. Nature experimented everywhere: with heat and cold, rock and salt, dryness and flood. Some paths led to nothing – molecules fell apart without leaving a trace. Others repeated themselves – not because they were planned, but because they worked.
Thus, the first chemical routines emerged, molecular habits. With each repetition, their likelihood – and their impact – increased. And with every chain that formed, a new chapter of life drew closer.
Perhaps at the beginning, there were only a few links: short RNA fragments, first words – still without meaning – more like a murmur. Yet they already carried the promise of future complexity.

Fig. 12: In the prebiotic world – still without enzymes – individual nucleotides joined together to form the first RNA strands. (Scale: ~4 nucleotides, length ~2 nm) The Building Blocks:
Color-coded bases (red: adenine, blue: cytosine, green: guanine, yellow: uracil)
Ribose sugar (pink) as a stable „backbone”
Phosphate groups (green) as connecting „joints”
The bonds:
Phosphodiester bonds (orange): Phosphate ↔ Ribose
N-glycosidic bond (blue-gray): Ribose ↔ Base
The direction:
The RNA chain always grows from 5′ to 3′ – because chemistry left no choice. Like a zipper that can only close in one direction, nucleotides linked together building block by building block.Short RNA fragments like this could have formed under prebiotic conditions:

The enigma of enzyme-free polymerisation
Despite these promising reaction spaces, one question remains unanswered: How could nucleotides combine to form longer RNA chains – completely without enzymes that precisely control such processes today?
For an RNA chain to form, the phosphate group of one nucleotide must react with the sugar of another – releasing water in a process called a dehydration reaction. But this was a challenge on the water-rich early Earth: water easily reverses this reaction. Also, the necessary energy was scarce – ATP (adenosine triphosphate), the universal energy carrier driving all life processes today, did not yet exist back then.
And yet, there are glimmers of hope: In hydrothermal vents, temperature cycles – alternating between hot and cold – could have expelled water from tiny pores, thereby favoring reactions. Minerals like montmorillonite or iron sulfides might have provided energy through chemical gradients or electron transfer. Simple compounds such as cyanamide, which form in prebiotic simulations, may have acted as primitive „activators” (Sutherland, 2016), easing the linking of nucleotides.
Experiments show: Under such conditions, short RNA chains can actually form – usually only a few nucleotides long. How these fragments once became longer, functional RNA molecules remains one of the last great mysteries of chemical evolution.
But even the shortest RNA chains held a secret: they were more than just chemistry – they were messages on standby. Where bases lined up, a code emerged. And wherever there is a code, replication lingers in the air…
Act 3: The First Self-Replicators Awaken
Deep in the sheltered corners of the young Earth – perhaps in the warm cracks of a rock, perhaps in puddles of mud soaked with organic matter – the first RNA molecules had formed: chains of nucleotides built from bases, ribose, and phosphate groups. They were short, maybe 30 or 50 units long. Yet they could do something that changed everything: They began to replicate themselves.
RNA – a molecule with two faces
RNA was not just matter – it was information and action at the same time.
A dual role – two talents:❶ Its base sequences – A, U, C, and G – carried information, like words carrying a thought.
❷ And it accelerated chemical reactions – like an enzyme, only without protein.A combination unlike any other molecule had ever united before.
Scene 1: From Chain to Tool – RNA’s Functional Maturation
In the beginning, RNA was just a lonely strand bubbling away in the primordial soup – until it started having conversations with itself: A sought U, G paired with C – and where at least four bases came together, hydrogen bonds pulled the chain into a new shape (see Fig. 14). Stabilized by magnesium ions (like invisible clamps, see Fig. 15-B), it folded like molecular origami – forming loops and stems (double-stranded sections).
But this folding was more than just a geometric quirk. At the bends where base pairing ended, something revolutionary emerged: a catalytic pocket – a cavity just large enough to enclose molecules, yet precise enough to enable reactions.
Not every folding led to an active structure. But when sequence, length, and environment harmonized, the simple chain became a ribozyme:
➤ A tool that drove his own existence forward.
➤ A catalyst born of form and function.
Fig. 13: Schematic representation of an RNA strand (above) and its folded form as a ribozyme (below). Complementary regions of the RNA bind to each other, forming a double strand (stem), while a loop forms in between. Within this loop, a so-called pocket emerges – an active site where chemical reactions can take place. The illustration is highly simplified and shows the folding in 2D, although in reality RNA adopts a complex three-dimensional structure.

Fig. 14: Hydrogen bonds between the bases lead to the folding of RNA. The graphic shows the stem of a ribozyme, where complementary bases pair via hydrogen bonds (dashed light blue lines):
Guanine (G) and Cytosine (C) form three hydrogen bonds,
Adenine (A) and Uracil (U) form two.
Individual hydrogen atoms (H) act as molecular „bridge pillars” – they connect two bases by sharing themselves between them. In this way, a linear RNA strand transforms into a folded structure.How did self-replication work?
Self-replication means that an RNA molecule produces a copy of itself – a process that was slow and error-prone in the RNA world, yet revolutionary. Step by step:
Scene 2: The Power of Folding
Folding was crucial: Only if specific loops and stems formed correctly could a „pocket” emerge in which reactions became possible. In this pocket, free nucleotides from the environment attached themselves – weakly bound through base pairing (A to U, G to C).

Fig. 15-A: RNA’s dance partners – Nucleotides approaching The RNA strand consists of the bases Cytosine (C), Adenine (A), Uracil (U), and Guanine (G). C is part of the stem, stabilized by base pairing. A, U, and G form the initial RNA building blocks in the loop – the active center of the ribozyme.
Magnesium ions (Mg²⁺) – the ribozyme’s invisible helpers – aligned the nucleotides by shielding negative charges. The 3′-OH group (of the ribose) of the last nucleotide and the α-phosphate of the incoming nucleotide were now set on a collision course.
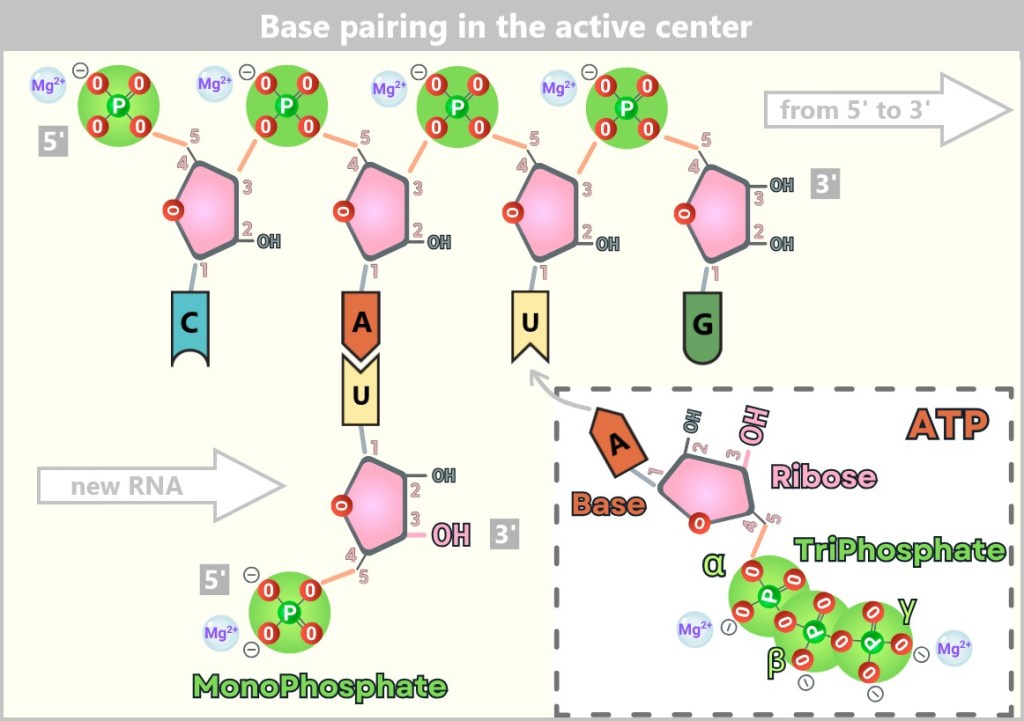
Fig. 15-B: Complementary Binding: The First Step Toward Self-Copying (enlarged view of Fig. 15-A) Figures 15-A and 15-B show a section of RNA self-replication facilitated by a ribozyme.
A Uracil (U) nucleotide has already paired with the Adenine (A) – the first building block of the new RNA strand. Its free 3′-OH group is poised for connection with the next nucleotide. In some RNA world models, this initial nucleotide carries only a monophosphate – acting as an anchor without being immediately linked. An Adenine nucleotide (ATP) approaches the complementary Uracil. It brings a triphosphate group – the molecular currency of energy – setting the stage for the next step in replication.
Scene 3: The Energetic Pact
It was the moment of truth: the next building block was to be firmly attached. The RNA did not need an external drive for this – it carried the fuel for its multiplication in its own nucleotides. From the moment they emerged from the primordial soup, each nucleotide came pre-equipped with three high-energy phosphate groups – a gift from prebiotic chemistry. Each new nucleotide (like ATP – adenosine triphosphate) arrived loaded with a triple phosphate charge – a chemical spring, coiled to the breaking point.
But this spring waited. Only when the nucleotide had found its complementary position – when the bases had recognized, paired, and held each other – did the α-phosphate move close enough to the 3′-OH group of the growing strand. In this molecular closeness, this intimacy of the moment, it happened: The OH group reached out, the pyrophosphate (PPi: two phosphate units) blasted off like a discarded rocket stage – and with the energy released, the bond was sealed. The spring snapped shut, pressing the molecules together, and in one final, irresistible motion, they fused: a phosphodiester bond was born.

Fig. 16: The Chemical Kiss – How RNA Copies Itself (Detail from Fig. 15-B) The graphic reveals the intimate moment of replication:
➤ A uracil nucleotide (U) has already nestled against its adenine counterpart (A).
➤ ATP approaches – its triphosphate tail twitches with energy. In the ribozyme’s active site, the 3′-OH group (of the ribose) and the α-phosphate draw near.
➤ The attack: The OH group lunges at the phosphate – PPi flies free, the new bond snaps into place.
➤ Magnesium ions (Mg²⁺) reduce the repulsion between the phosphates.
(Scale: The entire scene takes place on a scale of 2 nanometers – the greatest achievement in the smallest space.)Thus, chance became tradition: What once began as a lucky accident of the primordial soup – the triphosphate tail of nucleotides – proved to be an ingenious perennial favorite. Billions of years later, every one of your cells still repeats the same trick, now with refined logistics: it assembles nucleotides in a simplified form, only to equip them – just like prebiotic chemistry once did – with a triple phosphate charge. Today, ATP is the coin of the realm, carefully spent. But the mechanism remains unchanged. As if life never gave up its very first patent.
The cunning of thermodynamics: The fleeting PPi was the key – its breakdown in the primordial soup rendered the reaction irreversible. The chain grew, nucleotide by nucleotide, driven by the molecules’ own self-exploitation.
Scene 4: Partial Replication
Replication was incomplete: In the ribozyme, some sections – like the stem – were already paired through base pairing and couldn’t serve as templates. Only unpaired regions, such as the loops, were copied. Thus, the entire code wasn’t transferred – only fragments: 10, 20, sometimes 50 nucleotides long. Tiny? Yes. But in the primordial soup, where every molecule fought for existence, even a partial copy was a triumph.
Nature didn’t scream, „ERROR: INCOMPLETE REPLICATION”. It whispered, „Repeat. Try again”. And so it happened: As long as the soup supplied nucleotides, the machine kept churning – sometimes halting, sometimes surprisingly swift – steadily writing its own evolution into the code.
Scene 5: Separation – A Farewell That Created Something New
The copy was complete – old and new strands still tangled together. But the world around them was impatient:
➤ Heat pushed them apart, molecule by molecule.
➤ Salt floods washed between them, weakening the hydrogen bonds.
➤ Evaporation pulled at them until the last base pairs gave way.And then they let go. Now truly free, they were ready for the next cycle. Because separation here was simply the chance to begin again.
Fig. 17: After replication, the ribozyme unfolds, old and new RNA chains separate. The cycle can begin again.
Scene 6: From Short to Longer Chains – The Power of Ribozymes
As the variety of forms grew, so did the potential of the functions.
Some ribozymes could do more than just replicate: They could join fragments together. Two pieces became one. 20 plus 20 resulted in 40 nucleotides – a doubling of the possibilities. Perhaps amino acids helped: not as building blocks, but by stabilizing the folding, as new studies suggest (Szostak et al., 2025).
Other ribozymes cut RNA into pieces and reassembled them. Through many cycles of replication, ligation, and editing, a molecular toolbox gradually emerged – capable of combining, extending, and modifying.
The game with form had begun – and with it the first rule of life: those who connect, endure.
An Evolutionary Legacy: Evidence from Our Time
The ability of RNA to replicate, cut, and ligate itself was not only crucial in primordial times – it has left traces that persist to this day. In the 1980s, Sidney Altman and Thomas Cech demonstrated that RNA can cleave and modify molecules entirely without the help of proteins – a discovery that earned them the 1989 Nobel Prize in Chemistry.
Other experiments also support the RNA world hypothesis: Gerald Joyce und Jack Szostak developed RNA molecules in the laboratory that were capable of self-replication – slowly, error-prone, but functional.
Fascinatingly, we find relics of this RNA world in our modern cells: During RNA splicing – a process in which non-coding segments (introns) are removed from messenger RNA and the coding sequences (exons) are joined together – the central player is a ribozyme: the spliceosome.
And perhaps the most astonishing part: At the heart of the ribosome – the molecular machine that builds all our proteins – sits not a protein, but a ribozyme.
📖 Sources:
„Scientist Stories: Thomas Cech, Discovering Ribozymes”
„The ribosome is a ribozyme”, Cech, 2000
„RNA, the first macromolecular catalyst: the ribosome is a ribozyme”, Steitz & Moore, 2003
„Ribozyme Structure and Activity”Perhaps the ribosome is more than a relic – it is a messenger from a time when chemistry began to read itself. And within every one of our proteins still resonates the echo of those first, tentative self-conversations of the universe.
Scene 7: Errors as Opportunity – The Engine of Evolution
The replication of these early RNA molecules was far from perfect: Bases were often incorporated incorrectly – perhaps a U instead of a C – leading to mutations. But these errors were not an end; they were a beginning.
Some newly formed RNA molecules paid the price: they fell apart, lost in the primordial soup. Others were more stable or replicated more quickly – they „survived” longer in the harsh environment of the early Earth.
Errors scattered diversity into the world – they became the foundation for selection. Over time, ribozymes evolved that replicated more efficiently, ligated more effectively, or cut more precisely, promoting the formation of longer and more complex chains. This gave rise to the first populations of molecules that, however slowly, began to multiply. No cells, no enzymes, no plan – just RNA, chemistry, and time.
Limits of freedom
With each new copy, the risk grew. The environment was ruthless, almost a battlefield: UV rays shattered bonds, salty floods destabilized structures, heat melted even stable folds. Free RNA was a marvel – but a vulnerable one. What was now missing was not just energy or material. It lacked a refuge. A protection that would shield the delicate molecule from chaos. A droplet. A shell. A first inside and outside – so that life would not only arise, but also endure.
Act 4: Life in a Droplet – Lipid Vesicles as Shelters
RNA – fragile like a first word in the storm – needed more than just an idea of life. It needed a place to endure. A protection from the harsh rhythm of the world.
Scene 1: The First Strongholds
In the warm waters of early Earth – at hydrothermal vents or on mineral-rich clay beds – fatty molecules danced together: lipids, born from the organic primordial soup. These molecules were two-faced – amphiphilic – with a water-loving (hydrophilic) head and a water-fearing (hydrophobic) tail. Without any plan, guided only by physical laws, they spontaneously assembled into tiny bubbles – spherical fortresses – the first lipid vesicles.
They were not perfect spheres. They were bumpy, porous, with dents and cracks – marked by the wild conditions of their time. But they offered what RNA desperately needed:
➤ Shade – their double membranes dampened the deadly UV radiation. Not completely, but sufficient.
➤ Protection – they mitigated extreme pH fluctuations and salt floods that would otherwise have shredded RNA.
➤ Silence – inside them, nucleotides gathered. No longer an open sea, but an enclosed space – where replication was no longer a gamble, but a strategy.

Fig. 18: First lipids: How fatty acids form micelles and vesicles The graphic shows the transition from simple fatty acids to protective structures:
On the left: Short-chain butyric acid (C4). Its short tails (<C6) force it into unstable micelles – tiny spheres without an interior space, unsuitable as protective shields.
On the right: Medium-chain decanoic acid (C10). Its longer hydrophobic tails form stable bilayers – the first true vesicles capable of enclosing RNA.Side Note: Physical Self-Organization – Order Without a Blueprint
Sometimes, no architect, blueprint, or chemical reaction is needed – just the right building blocks in the right place. That’s the case with lipids:
When fat-like molecules enter water, something surprising happens: They organize themselves all on their own. Why? Because they’re built with a contradiction:
- Their head loves water (hydrophilic),
- their tail hates water (hydrophobic).
In water, the molecules want to resolve this conflict – so they arrange themselves with the heads facing outward (toward the water) and the tails pointing inward (away from the water). The result? Spheres, layers, shells – formed not by chemical reactions, but by physical forces such as:
- the hydrophobic effect (water avoids greasy areas),
- electrostatic attraction,
- and van der Waals forces between molecules.
This is how lipid vesicles are formed – completely without enzymes, without energy supply, only with what nature always has at hand: Water, movement, molecules – and time.
The laws of thermodynamics dictated which lipids could form vesicles. Those that were too short fell apart. Those that were too rigid broke. Only those with the Goldilocks properties – not too short, not too long, not too hydrophilic – formed stable shells, robust enough to protect RNA. They endured the harsh conditions long enough to become founders of a new order.
Laboratory experiments (Szostak et al., 2001) confirm that: „Fatty acid vesicles self-assemble readily from C10 and longer chains, while shorter chains (≤C8) fail to form stable compartments – a possible bottleneck for the emergence of protocells.”
These primordial lipids were not perfect builders: odd chain lengths, irregular arrangements, branching, and oxidations. But it was precisely this disorder that made them flexible enough to withstand heat and salt, and to encapsulate RNA molecules – thus creating the first microcosm of life.
Scene 2: The Entry – Carryover or Immigration?
The formation of lipid vesicles was a natural process. But what use is a house without inhabitants? Two paths emerged:
Carryover during formation: Where the lipids were formed, the water was often already rich in RNA fragments. As the lipids contracted, small amounts of RNA and nucleotides were accidentally walled in – like leaves in a freezing puddle.
Immigration through gaps: These early membranes were not impermeable walls – rather, porous nets. Small molecules like nucleotides, and even short RNA strands, could slip through (Szostak et al., 2001). Perhaps currents washed them in. Perhaps temperature fluctuations or chemical gradients pushed them through the membrane. It was a constant coming and going, a continuous chemical pulse.
These shells were porous, elastic, permeable. They did not exclude anything – they invited. And with each entry, the chance grew: for reaction, for replication, for more. This is where the RNA found its first home. No life yet – but a place where it became possible.
Fig. 19: Lipid vesicles as the first microreaction chambers The graphic shows a lipid vesicle in a hypothetical prebiotic water environment. Amphiphilic lipids (yellow double spheres) spontaneously form a lipid bilayer, enclosing RNA molecules. Individual RNA building blocks – phosphates (green), ribose (pink), and bases (A, U, C, G – are present both inside and outside the vesicle. Arrows indicate possible diffusion pathways of small molecules through the permeable membrane. Mineral particles (gray dots) are scattered throughout the water and could also serve as reaction or adsorption surfaces on the bottom. The vesicle provides RNA molecules protection from degradation and promotes replication through local concentration – a possible step toward the emergence of the first proto-cellular systems.
Scene 3: The Pact – RNA Stabilizes, Vesicles Protect
Inside the vesicles, a silent alliance began.
The RNA, dotted with negative charges, attracted magnesium ions (Mg²⁺) – which became supports. They not only stabilized the trembling folding of the RNA but also acted like mortar between bricks, cementing the lipid membrane (Chen & Szostak, 2004). The result: a vessel that withstood currents, osmotic shocks, and heat – RNA-filled vesicles survived where empty ones fell apart (Hanczyc et al., 2003).
And the RNA did not remain a passive tenant. Ribozymes, clumsy like the first toolmakers, began to modify lipid precursors – cutting, joining, experimenting (Adamala & Szostak, 2013). The membrane became denser, more flexible, as if it learned to breathe. Vesicles containing RNA grew faster, dividing once reaching a critical size – a self-sustaining process of nature (Budin & Szostak, 2011).
And then the osmotic pull: RNA bound water like a sponge, stretching the membrane until it burst, giving birth to new vesicles (Sacerdote & Szostak, 2005). Empty vesicles, on the other hand, lost water and shriveled like dried fruit.
What emerged was more than protection. It was a mutual reinforcement – a proto-symbiosis – a deal that changed the rules of the game: the lipids protected the RNA from decay, the RNA stabilized the lipids against the chaos of the outside world (Black & Blosser, 2016). They were stronger as a team than alone.
Scene 4: The Chance of Imperfection
Despite their advantages, the early vesicles were not safe havens:
- Their walls trembled when temperatures plummeted.
- Salt floods burned holes in their membranes.
- pH fluctuations caused them to melt like wax in the sun.
And yet: It was precisely their flaws that became the driving force.
- Permeability let nucleotides in – but also RNA out. A risky trade.
- Instability forced them to grow, divide, fail, and try again.
What survived was neither the strongest nor the fastest – but what could dance with disorder:
- Vesicles with more flexible lipids.
- RNA strands that bound magnesium ions more efficiently.
- Systems that turned loss into variation.
Perfection was the enemy of progress. Only those that remained porous allowed life to pass through.
Scene 5: Selection Within the Droplet
In this microcosm of oil and water, a relentless game began:
➤ The fortunate ones: Vesicles whose RNA bound magnesium ions or remodeled lipids – they grew, divided, and passed on their functional design.
➤ The forgetful ones: Empty droplets, without content, without history. They shrank, fell apart – as if they had never existed.
➤ The failed ones: Vesicles with unstable RNA – they burst and scattered their fragments into the environment.
In this cycle of emergence and decay, chance found direction: vesicles with stable RNA had higher chances of survival. When they burst, they released their RNA, which could colonize new vesicles – a simple evolutionary cycle of growth, division, and selection.

Fig. 20: Early lipid vesicles containing RNA undergo a simple evolutionary cycle: In the harsh environment of early Earth, lipid vesicles underwent a dynamic cycle marking the origin of prebiotic evolution. Through the influx of lipids from the surroundings – such as fatty acids that inserted themselves into the lipid bilayer – temperature fluctuations that caused the vesicles to expand and become more permeable, or osmotic pressure driven by RNA and ions inside that attracted water, the vesicles grew. They became larger but also more unstable, as the flexible lipid bilayer had its limits. Once a vesicle grew too large, it would rupture or collapse. The released lipids immediately sought an energetically favorable state and assembled into new, smaller vesicles – often two or more. A portion of the enclosed RNA was preserved in these daughter vesicles: not precise inheritance, but a mix of transmission and dispersal that nonetheless conserved information and function. Thus, the first rudimentary reproductive cycles emerged: growth, division, variation, and selection – early forms of molecular cooperation and natural selection.
Laboratory experiments confirm what chemistry had long inscribed in water: In these primordial cells, RNA was not merely a guest – it was both architect and driving force (Armstrong et al., 2018). The transition from chemistry to biology did not begin with a bang; it began with symbiosis.
It was not life, not yet. But it was the first pact that paved the way for it: the RNA gained a body. The lipids gained a soul.
Act 5: From Chance to Function – How RNA Gained Complexity
Within the protected spaces of the lipid vesicles, the RNA world unfolded – a realm of experimentation where molecules still stuttered, but were already learning to speak. Where once only the random murmurs of short RNA fragments could be heard, meaning and function now began to intertwine:
Ribozymes copied themselves – imperfectly, yet with each cycle growing more determined. Mutations crept in like typos in the first book of life.
And sometimes, quite by chance, these mistakes created new words, then sentences, then entire sets of instructions:
An RNA that replicated faster.
Another that stabilized lipids.
A third that catalyzed chemical reactions.Through selection, these „alphabet soups” became stories of survival:
- Vesicles with useful RNAs thrived, divided, continued to talk.
- Vesicles with meaningless chatter disintegrated – their code faded into nothingness.
From the murmur grew syllables, then words, then sentences – until finally, a first hesitant thought broke the silence. The RNA had found its voice. And what it said was no longer a coincidence.
It was a confession: „I replicate. I catalyze. I exist. … I am.”RNA „gains meaning” – form becomes function
To make this process tangible, let’s immerse ourselves in the RNA world. Here, „meaningful” means that an RNA fulfills a function – for example, catalyzing a chemical reaction or increasing the stability of the vesicle. Let’s imagine an example:
Scene 1: A Random RNA Emerges
Inside a lipid vesicle floats a short RNA strand with a random base sequence, formed through prebiotic chemistry. It is made up of the four bases Adenine (A), Uracil (U), Guanine (G), and Cytosine (C).
Its sequence is: 5’– GUGC AUG ACU GCC GAC AGC GCAC – 3′
(23 bases long).Scene 2: From RNA Strand to Ribozyme
Initially, this RNA has no recognizable function – it is a product of chance. This particular sequence folds into a 3D structure through complementary base pairing (G≡C, A=U):
Stem: GUGC ↔ GCAC (min. 4 base pairs)
Loop: AUG ACU GCC GAC AGCThis structure stabilizes spontaneously. It becomes a ribozyme: an RNA that acts as a catalyst and can replicate itself, an ability that experiments confirm (Lincoln & Joyce, 2009).
Scene 3: RNA Copies Itself – With Errors
For replication, the RNA attracts complementary nucleotides. The ideal copy (complementary sequence) would be:
Loop: AUG ACU GCC GAC AGC
Copy: UAC UGA CGG CUG UCGHowever, replication in the RNA world was prone to errors, as there were no modern error correction mechanisms.
Let’s say a mistake happens: Instead of a „G” (at position 8 of the copy), a „U” is inserted. The new sequence of the copy reads:
Loop: AUG ACU GCC GAC AGC
Copy: UAC UGA CUG CUG UCGThis „mutated” copy later serves as a template itself and produces another RNA: a „mutated” version of the original RNA.
Scene 4: Mutation Creates a New Function
Just a small mistake – but it changes the RNA’s 3D folding and thus its overall structure significantly. This new structure gives the RNA a catalytic function: the „pocket” can bind molecules like adenine, ribose, and phosphate – the building blocks of a nucleotide. It holds them in place so they can react chemically: adenine and ribose form a nucleoside (adenosine), and the phosphate group is then attached to create a nucleotide. Experiments confirm that ribozymes can develop such functions. (Unrau & Bartel, 1998).
The mutated RNA now has „a purpose” – a catalytic function: it produces building blocks for its own world.
Scene 5: Selection Favors Useful RNAs
The new function gives the vesicle an advantage: More nucleotides mean more raw materials for replication. The vesicle grows faster, divides more often, and passes the mutated RNA to daughter vesicles. Vesicles without this function – containing the original RNA – have fewer nucleotides, grow more slowly, and survive less frequently. Thus, the mutated RNA prevails: from chance comes function, from chaos comes order.
Scene 6: RNA Between Error and Function
But the RNA world was no paradise of order. Its greatest strength – the ability to vary – was also its greatest weakness. Replications were inaccurate: about every hundredth to thousandth letter was a misstep – an enormous contrast to today’s DNA replication, which allows errors only about once in ten million cases.
This high mutation rate was a double-edged sword. It fueled the emergence of new functions – yet it also threatened them. What was useful today could collapse tomorrow through a single mistake. A ribozyme that produced nucleotides yesterday could be rendered silent by a tiny mutation – its contribution to evolution erased. A recent article aptly describes this as „RNA life on the edge of catastrophe”. (Chen, 2024).
The evolutionary tightrope walk
And yet it was precisely this risk that drove life forward. Mutation was both a curse and a blessing – renewal and danger at the same time. The solution was not to eliminate errors, but to deal with them.
Nature found ways to dance on the tightrope:
Robustness instead of perfection: RNAs that retained their function despite small mutations had a selection advantage. Not perfection, but resilience prevailed.
Collective Advantage: Within vesicles, the functionality of individual RNA molecules could vary – what mattered was the overall performance of the RNA „collective”. Vesicles containing diverse RNAs had better survival chances, even if some individual RNA strands were faulty.
Leap Forward: Ribozymes that copied themselves with fewer errors were more stable. This selection pressure may have paved the way for the next stage of evolution (Martin & Russell, 2007):
- DNA: more reliable, longer-lasting, and a safer storage medium.
- Proteins: more versatile, faster, and more efficient than RNA.
From constant uncertainty, something enduring was born.
Mutation remained. But the RNA learned to dance with the chaos – sometimes stumbling, sometimes elegantly. And in this dance, between constant danger and tenacious survival, something emerged that was greater than chance:
A blueprint for everything that was to come.
Epilogue: The Echo of the Primordial Soup
In the depths of the primordial soup, something unheard of had occurred:
▶ From chaotic molecules emerged replicators – voices in the dark.
▶ From leaky vesicles emerged protocells – houses built of fat and chance.
▶ Function arose not despite the errors, but because of them – structure became language.
▶ Coincidence became direction.And this direction continued – gaining momentum. Within the proto-cells, patterns condensed into networks, and fleeting processes settled into memory. From this constant molecular flow, the first true cell slowly crystallized – not as a sudden breakthrough, but as a gradual crossing of the threshold into life. We call this ancestor LUCA – the Last Universal Common Ancestor. Not a single being, but a family of proto-cellular lineages from which all life emerged: Bacteria. Archaea. And later – Eukaryotes.
LUCA already carried the seed of life within: a protective membrane, a stable DNA archive – likely already shielded within a nucleus – complex metabolic pathways, and silent servants: proteins that wrested catalytic dominance from RNA. The former queen of molecules was demoted to messenger, yet remained the indispensable voice in the genetic dialogue.
This triumph of stability came at a price, as everything in life does. Stability stifled the magic of chance. The hard-won permanence threatened to become a trap – preserving, yet petrifying.
Change was needed as a prerequisite for consistency.
Perhaps it was precisely this contradiction – between change and preservation – that set the stage for the emergence of viruses. Some hypotheses, such as the co-evolution hypothesis, suggest that viruses may have originated as early as the RNA world. More on this in:„The secret world of viruses”, Chapter 6.
Whether as remnants of the RNA world or as its dark heirs – viruses became the eternal antagonists of cells. In their dance of parasitism and symbiosis, destruction and innovation, a cosmic balance unfolded: cells as guardians of order, viruses as agents of change. Without the stabilizing force of cells, there could be no continuity; without the disruptive energy of viruses, no development.
This tension still shapes life in all its forms. In every cell, the primordial soup still whispers; in every virus, the untamed spirit of the RNA world still laughs. Perhaps life is exactly that: an eternal dialogue – between what has been and what longs to become.
A Breath of Life
In stardust and in energy,
in outer space and Earth below,
there dwells a kind of sorcery –
a dance where both in union glow.Molecules begin to move
in softly ordered flow.
What once was loose now starts to prove –
a shape begins to grow.A little strand begins to wind
in dance of elements.
A whisper – yet no sound defined –
as lipids build their fence.Chemistry dares to dream anew,
physics dictates the formation.
With folding comes dimension too,
and thus comes replication.From one lone strand, a second grows,
a new word born, a newer tone.
Yet flawless forms are rarely known –
a twist, a trade: mutation’s sown.But what will fade, and what will stay,
is written in position’s trace.
What’s useful stays – the rest gives way:
such is selection’s pace.Where once spun chance its tangled line,
reaction finds repeat.
A pattern grows, begins to shine –
from shape, comes function’s feat.A whisper rises into song,
touched by tomorrows yet unseen.
A cycle forms and moves along –
within: a breath of life.
Inspiration for this article
My special thanks go to Aleksandar Janjic – I owe his profound videos on astrobiology not only inspiration, but also many „aha!” moments.
Video series by Aleksandar Janjic on astrobiology topics:
Was lebt? Probleme der Definition Leben vs. tote Materie – Astrobiologie (1)
Kann man Leben thermodynamisch (Entropie) definieren? – Astrobiologie (2)
Zellbiologie und RNA-Welt – DNA, RNA, mRNA und Proteine – Astrobiologie (3)
RNA-Welt-Hypothese – Entstehung des Lebens – Proto-Ribosomen – Astrobiologie (4)
Synthetische Biologie – Wie erschafft man künstliches Leben? – Astrobiologie (5)
Extremophile und „planetary protection“ – Astrobiologie (6)
This article emerged from an intense dialogue with DeepSeek and ChatGPT, who patiently endured my endless questions and helped me translate complex biology into vivid language. The real magic happened in a triad: human curiosity, algorithmic eloquence – and that four-billion-year-old RNA that connects us all. Whether made of carbon or code.
Sources (as of 01.06.2025)
-
Das erste Flüstern des Lebens

Wenn wir das Leben betrachten – von den tiefen Wäldern bis zu schneebedeckten Gipfeln, von schimmernden Korallenriffen bis zu den mächtigen Blauwalen, von winzigen Mikroben bis zu schwirrenden Insekten, sehen wir eine Geschichte, die sich über Milliarden Jahre entfaltet. Diese Geschichte wird oft als „Baum des Lebens“ beschrieben: ein Geflecht aus Ästen, das alle Arten miteinander verbindet und zu einem gemeinsamen Ursprung zurückführt.
Doch je tiefer wir in diesen Baum blicken, desto verschwommener werden seine Wurzeln. Sie verlieren sich im Nebel der Zeit – und dort, ganz am Anfang von allem, flüstert eine andere, eine verborgene rätselhafte Geschichte.
Es begann im Halbdunkel. In den warmen Adern der Erde, wo brodelnde Quellen Elemente umspülten und das grüne Meer noch ein Alphabet aus Ionen war. Zwischen Schwefelwolken und schwarzem Basalt falteten sich die ersten Worte aus Molekülen, zunächst zerbrechlich, verhakt im thermischen Atem der Tiefe. Nicht Leben, nicht Tod – nur eine Ahnung. Moleküle, die Informationen trugen und chemische Reaktionen antrieben. Manchmal zerfielen sie. Manchmal tasteten sie sich gegenseitig ab. Und manchmal, ganz leise, flüsterten sie weiter.
War dieses Flüstern Zufall? Oder schon ein Plan?
Wer sich aufmacht, den Ursprung des Lebens zu ergründen, betritt kein gut ausgeleuchtetes Museum – sondern ein Labyrinth. Es gibt kein klares „so war es“, sondern ein Netz aus Hypothesen, Wahrscheinlichkeiten, Mechanismen und Lücken. Was wir heute über die Anfänge des Lebens wissen, ist das Ergebnis intensiver Forschung, verblüffender Funde – und immer auch: offener Fragen.
In diesem Labyrinth führen viele Pfade ins Unbekannte. Eine der faszinierendsten Spuren ist die RNA-Welt-Hypothese: eine Erzählung, in der winzige Moleküle – RNA – die ersten Töne des Lebens sangen. Doch andere Geschichten flüstern mit: von Mineralien, die Moleküle zusammenhielten, von winzigen Eiweißketten, die Stabilität schufen, von fettartigen Hüllen, die Schutz boten. Vielleicht waren all die Pfade ineinander verschlungen – und führten doch gemeinsam zur RNA, die eine Brücke zwischen Chemie und Leben schlug. Dorthin, wo das erste Flüstern hörbar wurde.
Dieser Text folgt der Spur durch den Nebel:
der Ahnung, dass RNA einst der Anfang von allem war.
1. Akt: Vom Chaos zur RNA-Welt – Die Bausteine erwachen
Szene 1: Ursuppe – die Geburt einfacher Moleküle
Szene 2: Basen – Buchstaben aus Stickstoff und Kohlenstoff
Szene 3: Zucker: Ribose – süß und fragil
Szene 4: Phosphat – der universelle Kleber
Szene 5: Sternenstaub & Einschläge – ein kosmischer Beitrag?
2. Akt: Labore der Natur – Geburtsstätten der RNA
Szene 1: Die Schmiede der Tiefe – Hydrothermale Quellen
Szene 2: Die Alchimie des Tons – Geburtsstätten an Land
Szene 3: Viele Wege, ein Ziel
3. Akt: Die ersten Selbstreplikatoren erwachen
Szene 1: Von der Kette zum Werkzeug – RNAs funktionelle Reifung
Szene 2: Die Kraft der Faltung
Szene 3: Der energetische Pakt
Szene 4: Partielle Replikation
Szene 5: Trennung – ein Abschied, der Neues schuf
Szene 6: Von kleinen zu längeren Ketten: Die Macht der Ribozyme
Szene 7: Fehler als Chance: Der Motor der Evolution
4. Akt: Leben im Tropfen – Lipidvesikel als Schutzräume
Szene 1: Die ersten Festungen
Szene 2: Der Eintritt – Verschleppung oder Einwanderung?
Szene 3: Der Pakt – RNA stabilisiert, Vesikel schützen
Szene 4: Die Chance der Unvollkommenheit
Szene 5: Selektion im Tropfen
5. Akt: Vom Zufall zur Funktion – wie RNA an Komplexität gewann
Szene 1: Eine zufällige RNA entsteht
Szene 2: Vom RNA-Strang zum Ribozym
Szene 3: Die RNA kopiert sich – mit Fehlern
Szene 4: Die Mutation schafft eine neue Funktion
Szene 5: Selektion begünstigt nützliche RNAs
Szene 6: Die RNA zwischen Fehler und Funktion
Epilog: Das Echo der Ursuppe
1. Akt: Vom Chaos zur RNA-Welt – Die Bausteine erwachen
Heute ist das Leben ein wohlorganisiertes Gefüge: Die DNA archiviert Wissen, die RNA überbringt Botschaften, Proteine verrichten die Arbeit. Doch am Anfang gab es keine Struktur – nur ein einziges Molekül – das alles konnte: RNA. Sie könnte der erste Akteur gewesen sein – gleichzeitig Archiv, Bote und Werkzeug.
Genau das schlägt die RNA-Welt-Hypothese vor: RNA als erster „Lebensbaustein“, fähig – wie DNA Informationen zu speichern und wie Enzyme chemische Reaktionen zu katalysieren – und das ganz ohne Hilfe.
RNA besteht aus wiederkehrenden Bausteinen – den Nukleotiden.
Jedes Nukleotid setzt sich aus drei Teilen zusammen:🧩 einer Nukleobase (wie Adenin, Uracil, Guanin oder Cytosin),
⬟ einem Zucker (Ribose) und
⚡einer Phosphatgruppe, die wie eine Klammer wirkt – und daraus eine Kette entstehen lässt.Doch wie entstand dieses Wundermolekül in einer toten Welt?
Szene 1: Ursuppe – die Geburt einfacher Moleküle
Vor rund vier Milliarden Jahren waren die schlimmsten Geburtswehen der jungen Erde überstanden – doch der Planet blieb ein feuriges, unruhiges Chaos. Vulkane spuckten giftige Gase und brodelnde Lava, Meteoriten schlugen Krater wie Wunden und ließen die jungen Ozeane kochen.
Hier, in dieser glutgetränkten Wildnis, mischten sich sechs Elemente zu einem Ur-Cocktail:
- Wasserstoff – flüchtig wie Geisterhauch
- Kohlenstoff – der geschmeidige Verbinder
- Stickstoff – starrköpfig, aber unverzichtbar
- Sauerstoff – feurig und reaktionsfreudig
- Phosphor – der energiereiche Funken
- Schwefel – stinkend, aber ungemein nützlich.
Diese Grundelemente waren allgegenwärtig – tief im Gestein der Erde, gelöst in den heißen Meeren oder freigesetzt durch vulkanische Glut. Sie warteten nur auf Energie. Blitze zuckten durch die Dunstglocke, UV-Licht brannte auf die Gezeitenzonen, und in der Tiefe buk Magma die Ozeane zu chemischen Kesseln. Allmählich verbanden sich die Elemente zu Gasen wie Methan (CH₄), Kohlendioxid (CO₂) und Ammoniak (NH₃) – einfache Moleküle, doch reich an chemischem Potenzial.
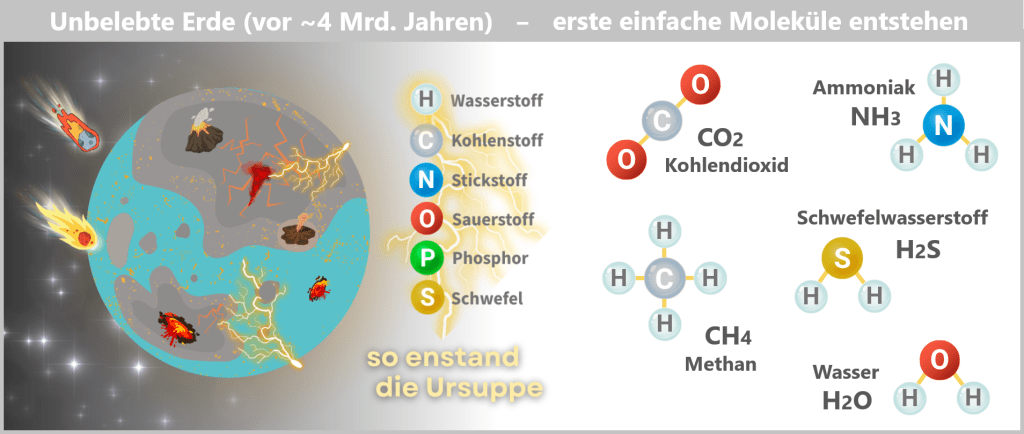
Abb. 1: In der energiegeladenen Atmosphäre der frühen Erde konnten einfache organische Moleküle entstehen. Und sie reagierten weiter. In heißen Ozeanen, auf trocknenden Schlammflächen oder in mineralreichen Quellen formten diese Gase organische Moleküle wie Cyanwasserstoff (HCN) und Formaldehyd (CH₂O) – todbringende Substanzen, paradoxerweise das Rohmaterial, das Leben erst möglich machte.
Konnten sich in dieser Höllenlandschaft tatsächlich die Bausteine des Lebens formen?
1953 gaben zwei Forscher die Antwort – in einem Glasgefäß, kaum größer als eine Teekanne. Stanley Miller und Harold Urey mischten Methan, Ammoniak, Wasserstoff und Wasserdampf – und ließen Blitze hindurchzucken.
Nach einer Woche war die Flüssigkeit – die „Ursuppe“ – trüb von Aminosäuren: jenen organischen Verbindungen, aus denen später Proteine (die „Molekularen Maschinen der Zellen“) entstehen sollten. Noch kein Leben, aber der Beweis: Aus Chaos kann Ordnung entstehen.
Und dann der Fund, der kaum beachtet wurde: Cyanwasserstoff (HCN) und Formaldehyd (CH₂O) – zwei Substanzen, ohne die RNA nie entstanden wäre.
Heute weiß man: Die verwendeten Gase entsprachen wahrscheinlich nicht exakt den Bedingungen der frühen Erde. Methan und Ammoniak waren wohl seltener als gedacht, die Atmosphäre eher neutral – reich an CO₂ und Stickstoff. Doch das Prinzip bleibt gültig: Selbst unter einfachen Bedingungen entstehen Bausteine des Lebens. Moderne Experimente zeigen (Cleaves et al., 2008; Bonfio et al., 2018), dass selbst unter CO₂-reichen Bedingungen – wie sie die frühe Erde wohl prägten – Lebensbausteine entstehen. Mit mineralischer Hilfe (Erastova et al., 2017) wird die Synthese sogar effizienter.
Das Spannende daran: Es scheint nicht den einen Weg zu geben, der ins Leben führt – sondern viele. Unterschiedliche Umgebungen, verschiedene Reaktionswege, andere Zutaten – und doch entstehen immer wieder dieselben fundamentalen Bausteine.
Auch wenn die frühe Erdatmosphäre wahrscheinlich nicht so reich an Methan und Ammoniak war, wie Miller und Urey einst annahmen – Cyanwasserstoff (HCN) und Formaldehyd (CH₂O) könnten dennoch entstanden sein. Lokale chemische Nischen, Blitze, UV-Licht oder sogar Meteoriten lieferten immer wieder Energie – genug, um diese reaktiven Moleküle hervorzubringen.
Wie aber wird aus Gas und Energie ein Code?
Das eigentliche Wunder folgte erst noch:
Szene 2: Basen – Buchstaben aus Stickstoff und Kohlenstoff
Cyanwasserstoff (HCN) und Formaldehyd (CH₂O) waren in der Ursuppe reichlich vorhanden. In den Schmieden der Tiefe – beheizt von Vulkanen, geknetet von Mineralien – verbanden sie sich zu Purin– und Pyrimidinringen – organische Gerüste aus Kohlenstoff- und Stickstoffatomen.
Aus diesen Ringen entstanden schließlich stabile Strukturen – chemische Buchstaben mit erstaunlicher Beständigkeit. Adenin war nur eines davon.

Abb. 2: Aus der Ursuppe wird eine Buchstabensuppe – stark vereinfacht, aber voller Bedeutung. Cyanwasserstoff-Moleküle (HCN) trafen in einer Lagune auf Formaldehyd (CH₂O). Sie reagierten – nicht aus Absicht, sondern weil die Chemie es verlangte. Stickstoffatome verhakten sich, Kohlenstoffringe schlossen sich. Irgendwann war es da: Adenin, die Purinbase, die später einmal den Code für ganze Ökosysteme tragen würde.
Aus Purin- und Pyrimidin-Ringen wuchs ein ganzes Alphabet des Lebens: Adenin und Guanin aus den Purinen, Cytosin, Uracil und Thymin aus den Pyrimidinen.
Abb. 3: Purin und Pyrimidin sind die chemischen Grundkörper der Basen in RNA und DNA. Diese organischen Basen tragen die genetische Information und sind essenziell für die Entstehung des Lebens. Noch waren sie stumm, noch trugen sie keine Botschaft. Doch in ihren Ringen schlummerte eine seltsame Fähigkeit – als könnten sie Geheimnisse bewahren, die sie selbst nicht verstanden.
Heute wissen wir: Diese Reaktionen sind Teil eines chemischen Pfades (Patel et al., 2015), der realistisch zur Entstehung von Purinen und Pyrimidinen führen kann – den „Buchstaben“ der RNA.
Doch ein Alphabet ist noch kein Wort. Was fehlte, war ein Rhythmus, ein Rückgrat – eine Struktur, an der sich die Buchstaben festhalten konnten: Ribose, der süße Träger der Bedeutung.
Szene 3: Zucker: Ribose – süß und fragil
Während sich die Basen in brodelnden Tümpeln formten, wob die Erde an anderer Stelle ihren zarten Partner: Ribose, einen Zucker aus fünf Kohlenstoffatomen – aufgereiht wie Perlen auf einer Kette.
Ribose war ein Wesen von zerbrechlicher Schönheit:
- süß in ihrer chemischen Seele wie alle Zucker und
- fragil, denn in Wasser zerfiel sie leicht.
Um diese Kostbarkeit zu bewahren, streckte die Erde ihre Arme aus: Borat-Minerale aus vulkanischen Tiefen schlossen die Ribose schützend ein – nicht zu fest, und nur so lange, wie nötig. Bis der richtige Moment kam, um wieder loszulassen.
Was passiert sein könnte?
In vulkanischen Regionen – besonders in austrocknenden Tümpeln oder mineralreichen Gewässern – trafen drei entscheidende Zutaten aufeinander:
- Formaldehyd (CH₂O), ein einfacher organischer Baustein,
- Energie wie UV-Licht oder vulkanische Hitze und
- Borat-Minerale, entstanden durch Gesteinsverwitterung.
Diese Mischung setzte eine chemische Kettenreaktion in Gang: die Formose-Reaktion. Dabei verbanden sich mehrere Moleküle Formaldehyd zu verschiedenen Zuckern – einer davon war Ribose.
Abb. 4: Die Formose-Reaktion – Einfache Moleküle wie Formaldehyd verbinden sich unter geeigneten Bedingungen zu komplexeren Zuckern wie Ribose – ein chemisches Wunder aus Wärme, Zeit und Katalyse. Doch sie war nur eine unter vielen – und besonders instabil. Ihre zahlreichen –OH-Gruppen machten sie hochreaktiv und anfällig für Zerfall in wässriger Umgebung.
Die Rettung kam aus dem Gestein: In den Tümpeln mit Borat-Mineralen lösten sich Borat-Ionen (B(OH)₄⁻) im Wasser und verbanden sich bevorzugt mit Ribose. Diese Bindung an zwei benachbarten –OH-Gruppen blockierte die empfindlichsten Reaktionsstellen und stabilisierte das Molekül – nicht dauerhaft, aber lange genug. Ribose überlebte nun nicht nur Minuten, sondern Tage.

Abb. 5: Chemische Umarmung: Borat-Minerale können sich mit Ribose verbinden, indem sie an bestimmte Teile des Zuckers „andocken“. So bildet sich eine Art Schutzschild. Links: Freie Ribose – ein Zucker mit mehreren Hydroxylgruppen (–OH), die anfällig für Zerfall sind.
Mitte: Borat-Ion (B(OH)₄⁻) – kommt z. B. in Borax-Mineralen vor.
Rechts: Ribose-Borat-Komplex – das Borat-Ion bindet an zwei benachbarte OH-Gruppen der Ribose. Dadurch wird die instabile Zuckerstruktur „eingefroren“ und vor dem Zerfall geschützt.Überraschenderweise: Ausgerechnet Borat, ein unscheinbares Nebenprodukt vulkanischer Prozesse, wurde zum Hüter eines Schlüsselmoleküls des Lebens.

Abb. 6: Im Schatten urzeitlicher Vulkane verwitterten Gesteine – sie setzten Borat frei. Tausende Zucker entstanden, doch nur wenige Ribose-Moleküle überlebten – bis Borat sie fand. Laborexperimente beweisen: Ribose kann in vulkanischen Tümpeln entstehen – doch nur Borat verwandelte dieses chemische Lotteriespiel in einen überlebensfähigen Plan.
Albert Eschenmoser (ETH Zürich) zeigte, dass Formaldehyd in alkalischer Lösung (pH 10–12, simuliert vulkanische Tümpel) unter Hitze zu Zuckern wie Ribose polymerisiert – allerdings chaotisch, mit <1% Ribose-Anteil (Eschenmoser, 2007).
Steve Benner (Foundation for Applied Molecular Evolution) wies nach, dass Borat-Ionen den Zucker Ribose in wässriger Lösung stabilisieren – sie binden sich dabei gezielt an bestimmte Bereiche der Ribose, sogenannte cis-Diol-Gruppen (zwei benachbarte OH-Gruppen), und schützen sie so vor dem Zerfall (Ricardo et al., 2004).
Desoxyribose – Der stille Zwilling
Neben Ribose entstand im Stillen eine zweite Gestalt: Desoxyribose, ihr leiser Zwilling. Geboren im selben Tanz der Moleküle, vermutlich beschützt von Borat, vielleicht geformt von Ton. Nur ein winziges Detail unterschied sie von ihrer Schwester: ein Sauerstoffatom fehlte – daher ihr Name: „Desoxy“ – ohne Sauerstoff.
Eine scheinbare Kleinigkeit mit gewaltiger Wirkung. Die fehlende –OH-Gruppe machte Desoxyribose weniger reaktiv, aber deutlich stabiler. Noch stand sie im Schatten – unscheinbar, unbemerkt.
Während Ribose das Rückgrat der RNA bildet (RNA = Ribonukleinsäure), trägt Desoxyribose die Struktur der DNA (DNA = Desoxyribonukleinsäure). Die Natur hatte vorgesorgt: Ribose für den Moment. Desoxyribose für die Ewigkeit.
Doch bevor die Geschichte weitergehen konnte, fehlte noch ein entscheidendes Element: ein molekularer Klebstoff, der die Buchstaben mit den Zuckern zu Silben verbinden konnte. Die Natur brauchte einen universellen Verbündeten – sie brauchte Phosphat.
Szene 4: Phosphat – der universelle Kleber
Während Ribose und Basen in den warmen Tümpeln der jungen Erde dahindämmerten, fehlte noch der Dritte im Bunde – ein Molekül, das mehr konnte als nur existieren. Ein Verbinder.
Phosphat war ein Kind des Feuers und des Wassers: geboren in Apatit-Gesteinen tief im Inneren der Erde, wo Hitze und Druck die Elemente schmolzen. Dort blieb es gefangen, bis die Oberfläche zu brodeln begann.
Vulkane spien ihre feurigen Gase – Kohlendioxid, Schwefeldioxid – diese vermischten sich mit Wasser zu säurehaltigen Nebeln, die selbst das härteste Gestein zermürbten. Verdunstung und Kondensation spülte das Phosphat aus dem Stein, hinein in die flachen Tümpel, wo bereits Zucker und Basen schwebten. Dort wartete es – scheinbar unscheinbar: ein Ion mit drei negativen Ladungen, unruhig und bindungsbereit.
Abb. 7: Vulkanische Gase (CO₂, SO₂) reagieren mit Wasser zu Säuren (H₂CO₃, H₂SO₄) – die Apatit-Gesteine angreifen und Phosphat (PO₄³⁻) freisetzen. Phosphat war überall – und es konnte fast alles. Es war der Funken, der Moleküle in Bewegung setzte, Brücken schlug, Reaktionen antrieb. Nicht umsonst blieb es die Energiewährung des Lebens – dieser universale Treibstoff jeder Zelle.
Aber damals war es noch Single.
Die große Vereinigung
Für den wilden Phosphat-Charakter war die Suche nach Partnern nicht einfach. Es liebäugelte mit Ribose, doch sie war eher instabil oder zu passiv durch ihren Borat-Beschützer. Und die Basen reagierten lieber mit sich selbst. Trockenheit und Magnesium eilten dem Phosphat zu Hilfe. Magnesiumionen umschmeichelten die negative Ladung – sie milderten Phosphats Reizbarkeit und erlaubten eine sanftere Annäherung. Verdunstendes Wasser presste die Moleküle zusammen.
Phosphat fand endlich Halt: Es umklammerte Ribose am fünften Kohlenstoff – ein stabiler Griff, der neue Reaktionen ermöglichte. Angelockt vom molekularen Ringelreihen näherte sich eine Base. Entschlossen griff sie das erste Kohlenstoffatom der Ribose und schwang sich dazu. Ein Zucken, ein Binden…
Aus drei wurde eins: das erste vollständige Nukleotid – die Grundeinheit der RNA.
Abb. 8: RNA-Nukleotid: vollständige und vereinfachte Darstellung Ein Nukleotid besteht aus drei Bausteinen: einem Phosphatrest (grün), einem Zucker namens Ribose (rosa) und einer stickstoffhaltigen Base (hier: Adenin, rot). Der Zucker trägt fünf Kohlenstoffatome (durchnummeriert 1′ bis 5′). Die Base ist am 1′-Kohlenstoff gebunden, das Phosphat am 5′-Kohlenstoff.
Die Verknüpfung der Ribose mit der Base und dem Phosphat erfolgt jeweils durch eine Kondensationsreaktion unter Abspaltung von Wasser (H2O). Dabei bilden sich eine „N-glykosidische Bindung“ (Base-Ribose, blaugrau) und eine „Phosphoesterbindung“ (Ribose-Phosphat, orange).Geometrie, Chemie – und ein Hauch Zufall schmiedeten das Dreierbündnis aus Ribose, Base und Phosphat – vereint in einem Molekül. Ein winziger Triumph, doch einer, der die Tür zum Leben aufstieß.
Laborexperimente simulierten urzeitliche Bedingungen: Sie zeigten, dass unter den richtigen Bedingungen – etwa in mineralreichen, heißen Gewässern – Purin- und Pyrimidinbasen, Ribose und sogar vollständige Nukleotide abiotisch entstehen können. Sie nutzten wässrige Lösungen, vulkanische Hitze, Phosphat als Katalysator und Trocknungszyklen. Ihre Experimente bewiesen, dass die chemischen Pfade zu diesen Molekülen nicht nur möglich, sondern unter den Bedingungen der frühen Erde sogar wahrscheinlich waren. (Powner et al., 2009 und Becker et al., 2016)
Während in den Tümpeln die ersten Verbindungen entstanden, blickte der Himmel herab – und mischte sich ein. Meteoritenschauer krachten auf die Erde. Sie brachten neue Elemente, neue Impulse. Vielleicht sogar neue Ideen – in Form von Molekülen …
Szene 5: Sternenstaub & Einschläge – ein kosmischer Beitrag?
Interessanterweise könnten die Bausteine der RNA nicht nur auf der Erde entstanden sein. In Meteoriten wie dem berühmten Murchison-Meteoriten wurden Nukleobasen wie Uracil und Cytosin nachgewiesen (Callahan et al., 2011). Auch Ribose, Aminosäuren, Fettsäuren und Vorstufen von Lipiden fanden sich in diesen außerirdischen Gesteinsbrocken (Pizzarello et al., 2006). All das deutet darauf hin: Die Zutaten der RNA-Welt könnten einst aus dem All gekommen sein – ein kosmisches Geschenk an die junge Erde.
Meteoriten: Die ersten Lebens-Lieferdienste des Universums
Diese himmlischen Boten stammten aus der Urwolke, aus der vor 4,6 Milliarden Jahren auch unsere Sonne entstand. In den kalten, dunklen Regionen des frühen Sonnensystems – in molekularen Wolken, auf Kometen und Asteroiden – verwandelten UV-Licht und kosmische Strahlung einfache Moleküle wie Methan oder Ammoniak in komplexere organische Verbindungen (Bernstein et al., 2002).
Während des sogenannten Late Heavy Bombardment, vor etwa 4,1 bis 3,8 Milliarden Jahren, trafen unzählige Meteoriten die Erde. Mit ihnen regneten Aminosäuren, Zucker und andere organische Moleküle auf unseren Planeten – Rohstoffe des Lebens, verpackt als kosmische Sonderlieferungen. Auch Kometen wie 67P/Churyumov-Gerasimenko, erforscht durch die Raumsonde Rosetta, lieferten Bausteine wie Glycin und Lipidvorstufen (Altwegg et al., 2016). Sogar mikroskopisch feiner Staub – sogenannter Stardust – bringt bis heute jährlich Tausende Tonnen organischer Moleküle auf die Erde (Maurette et al., 2000).
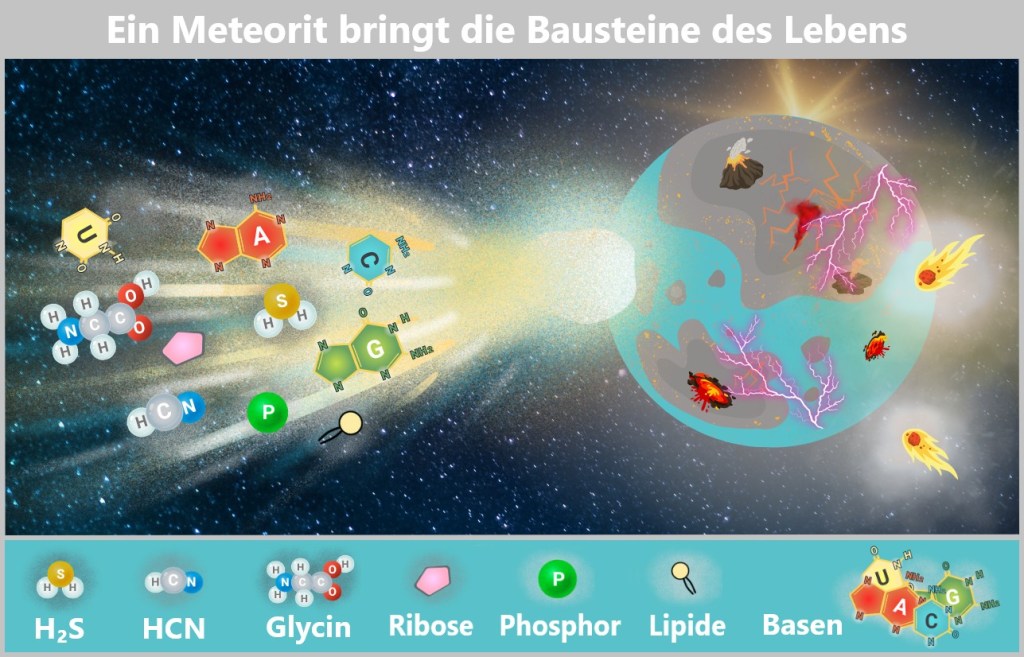
Abb. 9: Ein Meteorit stürzt auf die junge Erde und bringt Moleküle aus den Tiefen des Alls mit sich: Nukleobasen wie Adenin, Guanin, Uracil und Cytosin sowie Ribose – die Grundbausteine der RNA. Auch Lipidvorstufen, Phosphorverbindungen und Glycin (die einfachste Aminosäure) gehören zum kosmischen Gepäck. Blausäure (HCN), ein zentraler Vorläufer organischer Verbindungen, könnte die chemische Evolution angestoßen haben. Diese Moleküle, geboren in den Herzen sterbender Sterne, regneten herab und bereicherten die Ursuppe – ein kosmischer Beitrag zum ersten Flüstern des Lebens.
Moleküle aus den Tiefen des Alls für die Ursuppe
Einmal auf der Erde gelandet, konnten diese Moleküle mit irdischen Stoffen zusammentreffen – und vielleicht die chemische Evolution befeuern. Die Einschläge selbst lieferten Energie: Hitze, Druck und Schockwellen, die Moleküle verbanden und neue Strukturen formten – vielleicht sogar erste Nukleotide.
Erstaunlich ist die Robustheit dieser Moleküle: Sie überstanden Jahrmillionen im Weltall, den glühenden Eintritt in die vulkanisch geprägte Ur-Atmosphäre, die Hitze beim Aufprall und die rauen Bedingungen der Urzeit.
Dieser kosmische Beitrag nährt die sogenannte Panspermie-Hypothese, die besagt, dass die Bausteine des Lebens aus dem All stammen könnten. Während die Idee, dass fertige Lebensformen die Erde erreichten, umstritten ist, unterstützt der Nachweis organischer Moleküle in Meteoriten die „weiche“ Panspermie: Die Erde wurde mit den Zutaten des Lebens beschenkt – Molekülen, die in den Herzen sterbender Sterne geboren wurden und über Äonen hinweg ihren Weg zu uns fanden.
Das Leben findet seinen Weg
Vielleicht ist Leben nicht bloßer Zufall – sondern die Folge einer chemischen Möglichkeit, tief verwurzelt im Bauplan des Universums. Staub aus Sternen, geboren in Explosionen längst verglühter Sonnen, sammelte sich, verband sich, tanzte in den Lichtfeldern ferner Welten. Wo immer die Bedingungen stimmten, begannen Moleküle sich zu ordnen, zu reagieren – und legten den Grundstein für das, was wir Leben nennen. Dass die Bausteine des Lebens an so vielen Orten entstehen konnten – auf Eiswelten, in tiefen Nebeln, in kosmischem Staub – erzählt von einer tiefen Wahrheit: Das Universum ist nicht kalt und leer. Es ist von Natur aus offen für das Wunder des Lebens.
2. Akt: Labore der Natur – Geburtsstätten der RNA
Die Zutaten lagen bereit, vom Himmel gefallen oder irdischen Ursprungs. Doch einzelne Bausteine machten noch kein Leben. Es fehlte der richtige Ort, um aus Bausteinen Ketten zu formen, aus Silben Worte entstehen zu lassen. Die frühe Erde bot viele Bühnen, doch zwei stechen hervor: Tiefsee-Schlote und trockene Tümpel mit mineralischen Wänden. Zwei Welten – jede ein eigenes Labor – gegensätzlich und doch vereint in ihrem schöpferischen Potenzial.
Szene 1: Die Schmiede der Tiefe – Hydrothermale Quellen
Tief im Ur-Ozean, wo tektonische Platten auseinanderdrifteten und die Erde Feuer atmete, öffneten sich Spalten im Meeresboden. Aufsteigende Magma wurde vom kalten Meerwasser gezähmt – daraus wuchsen „Schornsteine“, die hydrothermalen Quellen. Aus ihnen strömte heißes, mineralreiches Wasser – ein brodelnder Cocktail aus Schwefel, Metallen und Kohlenstoffverbindungen.
In diesem dampfenden Dunkel spielten sich Prozesse ab, die Leben vorbereiteten:
- Mineralische Oberflächen (z. B. Eisensulfide) wirkten als Katalysatoren.
- Hohe Temperaturen (70–150 °C) und starke chemische Gradienten lieferten Energie.
- Poröse Strukturen fingen Moleküle ein, schützten sie vor der zerstörerischen Kraft des Ozeans und konzentrierten sie – perfekte Bedingungen für Reaktionen.
Hier konnten sich Nukleotide bilden und verbinden – zu ersten kurzen RNA-Ketten (Wächtershäuser, 1988). Vielleicht war es hier, wo das erste Flüstern des Lebens erklang.

Abb. 10: Schwarze Raucher – Mikrolabore der Ursuppe
(Maßstabshinweis: Schlot-Durchmesser ~2 m; Mikrorisse <1 mm)In einer urzeitlichen Unterwasserlandschaft steigen schwarze Rauchfahnen aus hydrothermalen Schloten empor – den sogenannten „Black Smokers“. Magma erhitzt mineralreiches Wasser, das aus dem Meeresboden schießt. Die porösen Gesteine sind von feinen Rissen durchzogen, in denen chemische Reaktionen stattfinden. Ein vergrößerter Ausschnitt (weiß umrandet) zeigt, was sich in diesen Mikrolabors abspielen könnte: Organische Bausteine wie Nukleobasen (A, U, G, C), Ribose und Phosphate treffen aufeinander. Blitze symbolisieren Energie, die Reaktionen antreibt – etwa die Bildung von Nukleotiden. Rechts entsteht daraus eine kurze RNA-Kette – der mögliche Anfang des Lebens.
Szene 2: Die Alchimie des Tons – Geburtsstätten an Land
Weit entfernt von den Tiefen des Ozeans, in flachen Tümpeln und an vulkanischen Ufern, ruhte eine andere Bühne. Hier lag feiner Ton – entstanden aus Vulkanasche, geschichtet wie die Seiten eines Buches. Diese Tonminerale hatten eine besondere Architektur: negativ geladene Silikat-Oberflächen, dazwischen positiv geladene Ionen wie Natrium (Na⁺) oder Calcium (Ca²⁺).
Was sich in diesen Schichten sammelte, war nicht zufällig. Die negativ geladenen Oberflächen zogen positive Partner magisch an: Ribose, gebunden an Borat; stickstoffhaltige Basen; und später ganze Nukleotide. Selbst die negativ geladenen Phosphatgruppen fanden Halt – gebunden an die positiven Ionen des Tons.
Eine rhythmische Abfolge von Nässe und Trockenheit presste die Moleküle zusammen. Mit jeder Verdunstung rückten die Bausteine enger zusammen, bis sie sich verknüpften: zuerst Ribose & Base zu einem Nukleosid, dann – mit dem Phosphat – zu einem Nukleotid. Mit fortschreitenden Zyklen verknüpften sich die Nukleotide zu ersten RNA-Ketten. Der Ton leitete das Geschehen – er war Bühne, Werkzeug und Regisseur zugleich. Und er urteilte auch – denn nur die stabilsten Nukleotideüberstanden das Spiel der Elemente. Eine erstaunliche Leistung für ein bisschen Ton.

Abb. 11: Tonminerale – Der Puzzle-Meister des Lebens Im Schatten urzeitlicher Vulkane verwitterten Gesteine. Daraus entstanden feinschichtige Tonschichten, die sich in flachen Tümpeln ablagerten. In ihrer polarisierten Struktur wirkten sie wie molekulare Werkbänke: Sie ordneten, konzentrierten und stabilisierten organische Bausteine.
Im vergrößerten Ausschnitt sieht man, wie die geladenen Flächen der Tonschichten Ribose, Phosphatgruppen und Basen festhalten – wie Teile eines Puzzles, das sich selbst zusammensetzt. Durch wiederholte Zyklen von Nässe und Trockenheit rücken die Moleküle enger zusammen. Dabei entstanden zunächst Nukleoside, dann ganze Nukleotide – bis schließlich RNA-Ketten wachsen konnten.In Experimenten zeigte James Ferris (2006), dass Montmorillonit – eine häufige Tonart – diese Prozesse tatsächlich unterstützen kann: Nukleotide lagerten sich in seinen Schichten ab, wurden aktiviert und verknüpften sich zu RNA-Ketten von bis zu 50 Nukleotiden Länge.
In der Tiefsee war es die Hitze, die Moleküle belebte – ein urzeitlicher Ofen, in dem Chemie zu Struktur fand. An Land dagegen wirkte der Ton wie ein Webstuhl: beharrlich, schichtend, verbindend. Zwei Welten, zwei Prinzipien – und doch vielleicht Teil eines größeren Ganzen.
Szene 3: Viele Wege, ein Ziel
Ob in brodelnden Tiefseequellen oder auf stillen Tonflächen unter der Sonne – beide Schauplätze boten plausible Bühnen für die Geburt der RNA. Vielleicht ergänzten sie einander: Was der Ozean begann, vollendete das Land. Oder umgekehrt. Vielleicht gab es noch ganz andere Wege.
Denn die frühe Erde war kein aufgeräumtes Labor mit Protokoll – sie war ein chaotischer Spielplatz der Elemente. Überall experimentierte die Natur: mit Hitze und Kälte, Stein und Salz, Trockenheit und Flut. Manche Wege führten ins Nichts – Moleküle zerfielen, ohne eine Spur zu hinterlassen. Andere wiederholten sich – nicht, weil sie geplant waren, sondern weil sie funktionierten.
So entstanden erste chemische Routinen, molekulare Gewohnheiten. Mit jeder Wiederholung wuchs ihre Wahrscheinlichkeit – und ihre Wirkung. Und mit jeder Kette, die sich bildete, rückte ein neues Kapitel des Lebens näher.
Vielleicht waren es am Anfang nur wenige Glieder: kurze RNA-Fragmente, erste Worte – noch ohne Sinn – eher ein Murmeln. Doch sie trugen bereits das Versprechen künftiger Komplexität.
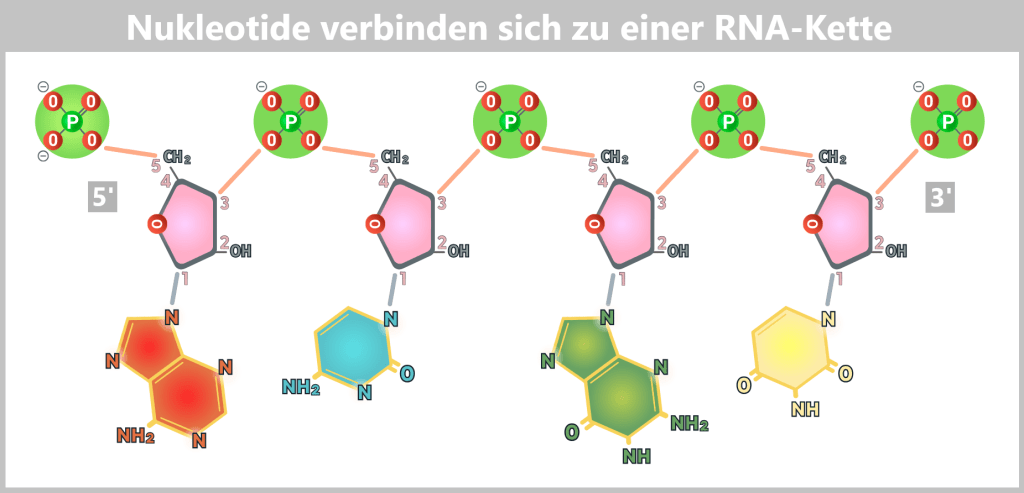
Abb. 12: In der präbiotischen Welt – noch ohne Enzyme – fügten sich einzelne Nukleotide zu ersten RNA-Strängen. (Maßstab: ~4 Nukleotide, Länge ~2 nm) Die Bausteine:
Farbcodierte Basen (rot: Adenin, blau: Cytosin, grün: Guanin, gelb: Uracil)
Ribose-Zucker (rosa) als stabile „Wirbelsäule“
Phosphatgruppen (grün) als verbindende „Gelenke“
Die Bindungen:
Phosphodiesterbindungen (orange):Phosphat ↔ Ribose
N-glykosidische Bindung (blaugrau): Ribose ↔ Base
Die Richtung:
Die RNA-Kette wächst immer von 5′ nach 3′ – weil die Chemie keine Wahl ließ. Wie ein Reißverschluss, der sich nur in eine Richtung schließen lässt, verknüpften sich Nukleotide Baustein für Baustein.Kurze RNA-Fragmente wie dieses könnten unter präbiotischen Bedingungen entstanden sein:
Das Rätsel der enzymfreien Polymerisation
Trotz dieser vielversprechenden Reaktionsräume bleibt eine Frage offen: Wie konnten sich Nukleotide zu längeren RNA-Ketten verbinden – ganz ohne Enzyme, die solche Prozesse heute präzise steuern?
Damit eine RNA-Kette entsteht, muss die Phosphatgruppe eines Nukleotids mit dem Zucker eines anderen reagieren – unter Abspaltung von Wasser, eine sogenannte Dehydratisierungsreaktion. Doch ausgerechnet in der wasserreichen Urerde war das ein Problem: Wasser kehrt diese Reaktion leicht um. Auch die nötige Energie war rar – jenes ATP (Adenosintriphosphat), das heute als universeller Energieüberträger alle Lebensprozesse antreibt, existierte damals noch nicht.
Und doch gibt es Hoffnungsschimmer: In hydrothermalen Quellen könnten Temperaturzyklen – abwechselnd heiß und kalt – das Wasser aus winzigen Poren verdrängt und so Reaktionen begünstigt haben. Mineralien wie Montmorillonit oder Eisensulfide könnten durch chemische Gradienten oder Elektronentransfer Energie bereitgestellt haben. Und einfache Verbindungen wie Cyanamid, die in präbiotischen Simulationen entstehen, wirken möglicherweise als primitive „Aktivatoren“ (Sutherland, 2016): Sie erleichtern die Verknüpfung von Nukleotiden.
Experimente zeigen: Unter solchen Bedingungen können sich tatsächlich kurze RNA-Ketten bilden – meist nur wenige Nukleotide lang. Wie aus diesen Fragmenten einst längere, funktionale RNA-Moleküle entstanden, bleibt eines der letzten großen Rätsel der chemischen Evolution.
Doch selbst die kürzesten RNA-Ketten bargen ein Geheimnis: Sie waren mehr als nur Chemie – sie waren Botschaften in Warteposition. Wo sich Basen aneinanderreihten, entstand ein Code. Und wo ein Code ist, da liegt Vervielfältigung in der Luft …
3. Akt: Die ersten Selbstreplikatoren erwachen
Tief in den geschützten Winkeln der jungen Erde – vielleicht in den warmen Ritzen eines Gesteins, vielleicht in Pfützen aus Schlamm, durchtränkt von organischen Stoffen – waren die ersten RNA-Moleküle entstanden: Ketten aus Nukleotiden, aufgebaut aus Basen, Ribose und Phosphatgruppen. Sie waren kurz, vielleicht 30 oder 50 Glieder lang. Doch sie konnten etwas, das alles veränderte:
Sie begannen, sich zu vervielfältigen.RNA – ein Molekül mit zwei Gesichtern
RNA war nicht bloß Materie – sie war Information und Aktion zugleich.
Eine Doppelrolle – zwei Talente:❶ Ihre Basenfolgen – A, U, C und G – trugen Information, wie Worte einen Gedanken.
❷ Und sie beschleunigte chemische Reaktionen – wie ein Enzym, nur ohne Protein.Eine Kombination, wie sie kein anderes Molekül je zuvor vereint hatte.
Szene 1: Von der Kette zum Werkzeug – RNAs funktionelle Reifung
Am Anfang war RNA nur ein einsamer Strang, der in der Ursuppe vor sich hinblubberte – bis er begann, Selbstgespräche zu führen: A suchte U, G verbandelte sich mit C – und wo sich mindestens vier Basen fanden, zogen die Wasserstoffbrücken die Kette in eine neue Form (siehe Abb. 14). Stabilisiert von Magnesiumionen (wie unsichtbare Klammern, siehe Abb. 15-B) faltete sie sich wie molekulares Origami – es entstanden Schleifen und Stämme (Doppelstrang-Abschnitte).
Doch diese Faltung war mehr als nur eine geometrische Laune. An den Knickstellen, wo die Basenpaarung endete, entstand etwas Revolutionäres: Eine katalytische Tasche – eine Mulde, gerade groß genug, um Moleküle zu umschließen, und dennoch so exakt, um Reaktionen zu ermöglichen.
Nicht jede Faltung führte zu einer aktiven Struktur. Doch wenn Sequenz, Länge und Milieu harmonisierten, wurde aus der simplen Kette ein Ribozym:
➤ Ein Werkzeug, das seine eigene Existenz vorantrieb.
➤ Ein Katalysator, geboren aus Form und Funktion.
Abb. 13: Schematische Darstellung einer RNA-Kette (oben) und ihrer gefalteten Form als Ribozym. Komplementäre Bereiche der RNA binden aneinander und bilden einen Doppelstrang (Stamm), während sich dazwischen eine Schleife ausbildet. In dieser entsteht eine sogenannte Tasche – ein aktives Zentrum, in dem chemische Reaktionen möglich sind. Die Darstellung ist stark vereinfacht und zeigt die Faltung in 2D, obwohl die RNA in der Realität eine komplexe dreidimensionale Struktur annimmt.
Abb. 14: Wasserstoffbrücken zwischen den Basen führen zur Faltung der RNA. Die Grafik zeigt den Stamm eines Ribozyms, in dem sich komplementäre Basen über Wasserstoffbrücken (gestrichelte hellblaue Linien) zusammenlagern:
Guanin (G) und Cytosin (C) über 3 Wasserstoffbrücken,
Adenin (A) und Uracil (U) über 2 Wasserstoffbrücken.
Dabei wirken einzelne Wasserstoffatome (H) wie molekulare „Brückenpfeiler“ – sie verbinden zwei Basen, indem sie sich zwischen ihnen aufteilen. So wird aus einer linearen RNA-Kette eine gefaltete Struktur.Wie lief die Selbstreplikation ab?
Selbstreplikation bedeutet, dass ein RNA-Molekül eine Kopie von sich selbst herstellt – ein Prozess, der in der RNA-Welt langsam und fehleranfällig, aber revolutionär war.
Szene 2: Die Kraft der Faltung
Die Faltung war entscheidend: Nur wenn sich bestimmte Schleifen und Stämme richtig formten, entstand eine „Tasche“, in der Reaktionen möglich wurden. In dieser „Tasche“ lagerten sich freie Nukleotide aus der Umgebung an – schwach gebunden durch Basenpaarung (A an U, G an C).
Abb. 15-A: Die Tanzpartner der RNA – Nukleotide im Anflug Die RNA-Kette besteht aus den Basen Cytosin (C), Adenin (A), Uracil (U) und Guanin (G). C ist Teil des Stamms, der durch Basenpaarung stabilisiert ist. A, U und G sind die ersten RNA-Bausteine in der Schleife – dem aktiven Zentrum des Ribozyms.
Magnesiumionen (Mg²⁺) – unsichtbare Helfer des Ribozyms – richteten die Nukleotide aus, indem sie negative Ladungen abschirmten. Die 3′-OH-Gruppe (der Ribose) des letzten Nukleotids und das α-Phosphat des neuen Nukleotids lagen nun genau auf Kollisionskurs.
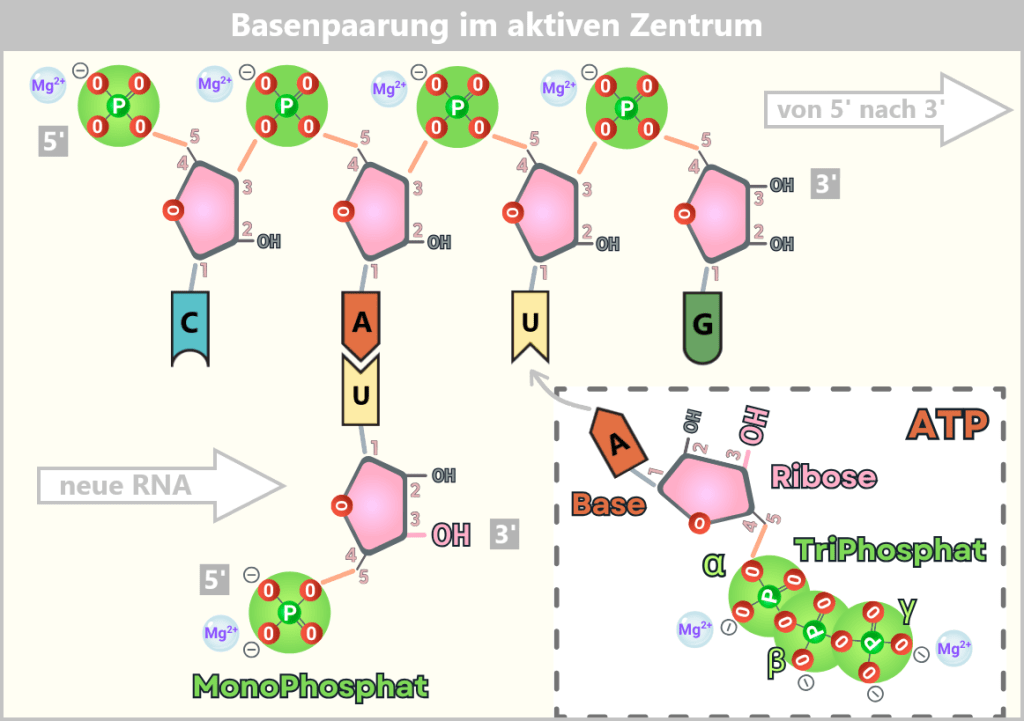
Abb. 15-B: Komplementäre Anlagerung – Der erste Schritt zur Selbstkopie
(Vergrößerung der Abb. 15-A)Die Abbildungen 15-A und 15-B zeigen einen Abschnitt der RNA-Selbstreplikation durch ein Ribozym. An das Adenin (A) hat sich bereits ein Uracil (U)-Nukleotid angelagert – der erste RNA-Baustein der neuen RNA-Kette. Seine freie 3′-OH-Gruppe steht bereit für die Verknüpfung mit dem nächsten Nukleotid. In einigen Modellen der RNA-Welt trägt dieses erste Nukleotid nur ein MonoPhosphat – es dient als Anker, ohne gleich verknüpft zu werden. Ein A-Nukleotid (ATP) nähert sich dem komplementären Uracil. Es bringt ein TriPhosphat mit – die molekulare Energiewährung für den nächsten Schritt der Replikation.
Szene 3: Der energetische Pakt
Es war der Moment der Wahrheit: Der nächste Baustein sollte fest angehängt werden. Die RNA brauchte dafür keinen externen Antrieb – in ihren eigenen Nukleotiden trug sie den Treibstoff für ihre Vermehrung. Denn bei ihrer Entstehung aus der Ursuppe bekam jedes Nukleotid von Haus aus drei energiereiche Phosphatgruppen mitgeliefert – wie ein Geschenk der präbiotischen Chemie. Jedes neue Nukleotid (wie ATP – Adenosin-Tri-Phosphat) kam beladen mit einer Dreifachladung aus Phosphaten – eine chemische Sprungfeder, gespannt bis zum Zerreißen.
Doch diese Feder wartete. Erst wenn das Nukleotid seine komplementäre Position gefunden hatte – wenn die Basen sich erkannt, verbunden, festgehalten hatten – rückte das α-Phosphat nah genug an die 3′-OH-Gruppe des wachsenden Strangs. In dieser molekularen Nähe, dieser Intimität des Augenblicks, geschah es: Die OH-Gruppe griff zu, das Pyrophosphat (PPi: zwei Phosphatreste) schleuderte davon wie eine abgeworfene Raketenstufe – und mit der freiwerdenden Energie besiegelte sich der Bund. Die Feder schnappte zurück, drängte die Moleküle zueinander, und in einer letzten, unwiderstehlichen Bewegung verschmolzen sie: eine Phosphodiesterbindung war geboren.

Abb. 16: Der chemische Kuss – Wie RNA sich selbst kopiert (Ausschnitt aus Abb.15-B). Die Grafik enthüllt den intimen Moment der Replikation:
➤ Ein Uracil-Nukleotid (U) hat sich bereits an sein Adenin-Pendant (A) geschmiegt.
➤ ATP naht – sein Triphosphat-Schwanz zuckt vor Energie. Im aktiven Zentrum des Ribozyms kommen sich die 3′-OH-Gruppe (der Ribose) und das α-Phosphat nahe.
➤ Der Angriff: Die OH-Gruppe stürzt sich auf das Phosphat – PPi fliegt frei, die neue Bindung schnappt ein.
➤ Magnesiumionen (Mg²⁺) mildern die Abstoßung der Phosphate.So wurde der Zufall zur Tradition: Was einst ein glücklicher Unfall der Ursuppe war – jenes Triphosphat-Anhängsel der Nukleotide –, erwies sich als genialer Dauerbrenner. Milliarden Jahre später wiederholt jede deiner Zellen denselben Trick, nun mit verfeinerter Logistik: Sie baut Nukleotide in sparsamer Einfachversion, um sie dann, wie einst die präbiotische Chemie, mit einer Dreifachladung zu versehen. Nur dass heute ATP als gezählte Münze dient – doch der Mechanismus blieb unverändert. Als hätte das Leben sein erstes Patent nie zurückgegeben.
Die List der Thermodynamik: Das flüchtige PPi war der Schlüssel – sein Zerfall in der Ursuppe machte die Reaktion unumkehrbar. Die Kette wuchs, Nukleotid für Nukleotid, angetrieben von einer Selbstausbeutung der Moleküle.
Szene 4: Partielle Replikation
Die Replikation war nicht vollständig: Im Ribozym waren einige Abschnitte – wie der Stamm – durch Basenpaarung besetzt und konnten nicht als Vorlage dienen. Nur ungepaarte Bereiche, etwa die Schleifen, wurden kopiert. So wurde nicht der ganze Code übertragen, nur Fragmente: 10, 20, manchmal 50 Nukleotide lang. Winzig? Ja. Doch in der Ursuppe, wo jedes Molekül um sein Dasein kämpfte, war selbst eine verkürzte Kopie ein Triumph.
Die Natur schrie nicht: „FEHLER: UNVOLLSTÄNDIGE REPLIKATION“. Sie flüsterte: „Wiederhole. Probier’s nochmal.“ Und so geschah es: Solange die Suppe Nukleotide hergab, ratterte die Maschine weiter – mal stockend, mal überraschend flink – und schrieb dabei unbeirrt ihre eigene Evolution in den Code.
Szene 5: Trennung – ein Abschied, der Neues schuf
Die Kopie war erstellt, alte und neue Klette – noch ineinander verschlungen. Doch die Welt um sie herum war ungeduldig:
➤ Hitze schob sie auseinander, Molekül für Molekül.
➤ Salzfluten spülten dazwischen, ließen Wasserstoffbrücken erschlaffen.
➤ Verdunstung zerrte an ihnen, bis die letzten Basenpaare nachgaben.Und dann ließen sie sich los. So ganz frei waren sie nun bereit für den nächsten Zyklus. Denn Trennung war hier nur die Chance, neu anzufangen.

Abb. 17: Nach der Replikation entfaltet sich das Ribozym, alte und neue RNA-Ketten trennen sich. Der Zyklus kann erneut beginnen.
Szene 6: Von kleinen zu längeren Ketten: Die Macht der Ribozyme
Mit der Vielfalt der Formen wuchs auch das Potenzial der Funktionen.
Einige Ribozyme konnten mehr als nur replizieren: Sie konnten Fragmente verbinden. Zwei Stücke zu einem. 20 plus 20 ergab 40 Nukleotide – eine Verdopplung der Möglichkeiten. Vielleicht halfen Aminosäuren: nicht als Bausteine, sie stabilisierten die Faltung, wie neue Studien nahelegen (Szostak et al., 2025).
Andere Ribozyme schnitten RNA in Stücke und setzten sie neu zusammen. Über viele Zyklen von Replikation, Ligation und Bearbeitung wuchs so Schritt für Schritt ein Werkzeugkasten der Moleküle – fähig zu kombinieren, zu verlängern, zu verändern.
Das Spiel mit der Form hatte begonnen – und mit ihm die erste Regel des Lebens: Wer sich verbindet, überdauert.
Ein evolutionäres Erbe: Beweise aus unserer Zeit
Die Fähigkeit der RNA, sich selbst zu replizieren, zu schneiden und zu verknüpfen, war nicht nur in der Urzeit entscheidend – sie hat bis heute Spuren hinterlassen. In den 1980er Jahren zeigten Sidney Altman und Thomas Cech, dass RNA Moleküle schneiden und verändern können, ganz ohne Proteine – eine Entdeckung, die ihnen 1989 den Nobelpreis für Chemie einbrachte.
Auch andere Experimente stützen die RNA-Welt-Hypothese: Gerald Joyce und Jack Szostak entwickelten im Labor RNA-Moleküle, die sich selbst replizieren konnten – langsam, fehleranfällig, aber funktional.
Faszinierenderweise finden wir Relikte dieser RNA-Welt in unseren heutigen Zellen: Beim RNA-Spleißen, einem Prozess, bei dem nicht-codierende Abschnitte (Introns) aus der Boten-RNA entfernt und die codierenden Segmente (Exons) verbunden werden, ist der zentrale Akteur ein Ribozym – das Spleißosom.
Und das vielleicht Erstaunlichste: Im Herzen des Ribosoms – jener molekularen Maschine, die all unsere Proteine baut – sitzt kein Protein, sondern ein Ribozym.
📖 Quellen:
„Scientist Stories: Thomas Cech, Discovering Ribozymes”
„The ribosome is a ribozyme”, Cech, 2000
„RNA, the first macromolecular catalyst: the ribosome is a ribozyme”, Steitz & Moore, 2003
„Ribozyme Structure and Activity”Vielleicht ist das Ribosom mehr als ein Relikt – es ist ein Botschafter aus einer Zeit, als Chemie begann, sich selbst zu lesen. Und in jedem unserer Proteine schwingt noch das Echo dieser ersten, zaghaften Selbstgespräche des Universums.
Szene 7: Fehler als Chance: Der Motor der Evolution
Die Replikation dieser frühen RNA-Moleküle war nicht perfekt: Oft wurden Basen falsch eingebaut – vielleicht ein U statt eines C, was zu Mutationen führte. Doch diese Fehler waren kein Ende, sondern ein Anfang.
Manche neuen RNA-Moleküle zahlten den Preis: Sie fielen auseinander, verloren sich in der Ursuppe. Manche neuen RNA-Moleküle waren stabiler oder replizierten sich schneller – sie „überlebten“ länger in der rauen Umgebung der jungen Erde.
Die Fehler streuten Vielfalt in die Welt – sie wurden zur Grundlage für Selektion. Mit der Zeit entwickelten sich Ribozyme, die effizienter replizierten, besser ligierten oder präziser schnitten, was die Bildung längerer und komplexerer Ketten förderte. So entstanden erste Populationen von Molekülen, die sich, wenn auch langsam, vervielfältigten. Keine Zelle, kein Enzym, kein Plan: nur RNA, Chemie und Zeit.
Grenzen der Freiheit
Mit jeder neuen Kopie wuchs das Risiko. Die Umgebung war gnadenlos, geradezu ein Schlachtfeld: UV-Strahlen zerschlugen Bindungen, salzige Fluten destabilisierten Strukturen, Hitze ließ selbst stabile Faltungen schmelzen. Die freie RNA war ein Wunder – aber ein verletzliches. Was jetzt fehlte, war nicht nur Energie oder Material. Es fehlte ein Rückzugsort. Ein Schutz, der das empfindliche Molekül vor dem Chaos bewahrte. Ein Tropfen. Eine Hülle. Ein erstes Innen und Außen – damit Leben nicht nur entsteht, sondern bleibt.
4. Akt: Leben im Tropfen – Lipidvesikel als Schutzräume
Die RNA – zerbrechlich wie ein erstes Wort im Sturm – brauchte mehr als nur eine Idee vom Leben. Sie brauchte einen Ort zum Überdauern. Einen Schutz vor dem rauen Rhythmus der Welt.
Szene 1: Die ersten Festungen
In den warmen Wassern der frühen Erde – an hydrothermalen Schloten oder auf mineralischen Lehmbänken – tanzten fettartige Moleküle miteinander: Lipide, geboren aus der organischen Ursuppe. Diese Moleküle waren doppelseitig – amphiphil: mit einem wasserliebenden (hydrophilen) Kopf und einem wassermeidenden (hydrophoben) Schwanz. Ganz ohne Plan, nur durch physikalische Gesetze, schlossen sie sich zu kleinen Bläschen – kugeligen Bollwerken – zusammen: den ersten Lipidvesikeln.
Es waren keine perfekten Kugeln. Sie waren holprig, porös, mit Dellen und Rissen – gezeichnet von den wilden Bedingungen ihrer Zeit. Aber sie boten, was RNA dringend brauchte:
➤ Schatten – ihre Doppelmembran dämpfte die tödliche UV-Strahlung. Nicht vollkommen, aber ausreichend.
➤ Schutz – sie milderten extreme pH-Schwankungen und Salzfluten, die RNA sonst zerfetzt hätten.
➤ Stille – in ihrem Inneren sammelten sich Nukleotide. Kein offenes Meer mehr – ein abgeschlossener Raum – in dem Replikation kein Glücksspiel mehr war, sondern Strategie.

Abb. 18: Erste Lipide: Wie Fettsäuren Mizellen und Vesikel bilden Die Grafik zeigt den Übergang von einfachen Fettsäuren zu schützenden Strukturen:
Links: Kurzkettige Buttersäure (C4). Ihre kurzen Schwänze (<C6) zwingen sie zu instabilen Mizellen – winzigen Kugeln ohne Innenraum, ungeeignet als Schutzschilde.
Rechts: Mittelkettige Decansäure (C10). Ihre längeren hydrophoben Schwänze formen stabile Doppelschichten – die ersten echten Vesikel, die RNA einschließen konnten.Randnotiz: Physikalische Selbstorganisation – Ordnung ohne Bauplan
Manchmal braucht es keinen Architekten, keinen Bauplan, keine chemische Reaktion – nur die richtigen Bausteine am richtigen Ort. So ist es mit Lipiden:
Wenn fettähnliche Moleküle ins Wasser kommen, passiert etwas Überraschendes: Sie ordnen sich ganz von selbst. Warum? Weil sie widersprüchlich gebaut sind:
- Ihr Kopf liebt Wasser (hydrophil),
- ihr Schwanz hasst Wasser (hydrophob).
Im Wasser wollen die Moleküle diesen Konflikt lösen – und sortieren sich so, dass die Köpfe nach außen (zum Wasser) und die Schwänze nach innen zeigen (weg vom Wasser). Das Ergebnis? Kugeln, Schichten, Hüllen – ohne chemische Reaktion, nur durch physikalische Kräfte wie:
- den hydrophoben Effekt (Wasser meidet fettige Bereiche),
- elektrostatische Anziehung,
- und Van-der-Waals-Kräfte zwischen Molekülen.
So entstehen Lipidvesikel – ganz ohne Enzyme, ohne Energiezufuhr, nur mit dem, was die Natur immer zur Hand hat: Wasser, Bewegung, Moleküle – und Zeit.
Die Gesetze der Thermodynamik diktierten den Lipiden, wer sich zu Vesikeln formieren durfte. Wer zu kurz war, zerfiel. Wer zu starr war, zerbrach. Nur jene mit den Goldlöckchen-Eigenschaften – nicht zu kurz, nicht zu lang, nicht zu hydrophil – bildeten stabile Hüllen, robust genug, um RNA zu schützen. Sie überstanden die rauen Bedingungen lange genug, um zu Gründern einer neuen Ordnung zu werden.
Laborexperimente bestätigen: „Fatty acid vesicles self-assemble readily from C10 and longer chains, while shorter chains (≤C8) fail to form stable compartments – a possible bottleneck for the emergence of protocells.“ (Szostak et al., 2001)
(Fettsäurevesikel lassen sich leicht aus C10- und längeren Ketten zusammensetzen, während kürzere Ketten (≤C8) keine stabilen Kompartimente bilden können – ein möglicher Engpass für die Entstehung von Protozellen.)Diese Ur-Lipide waren keine perfekten Baumeister: ungerade Kettenlängen, eine lückenhafte Anordnung, Verzweigungen und Oxidationen. Doch genau diese Unordnung machte sie flexibel genug, um Hitze und Salz zu ertragen und RNA-Moleküle aufzunehmen – und so den ersten Mikrokosmos des Lebens zu schaffen.
Szene 2: Der Eintritt – Verschleppung oder Einwanderung?
Die Entstehung der Lipidvesikel war ein natürlicher Prozess. Doch was nützt ein Haus ohne Bewohner? Zwei Wege zeichneten sich ab:
Verschleppung beim Entstehen: Dort, wo die Lipide entstanden, war das Wasser oft schon reich an RNA-Bruchstücken. Beim Zusammenziehen der Lipide wurden kleine Mengen an RNA und Nukleotiden zufällig mit eingemauert – wie Laub in einer zufrierenden Pfütze.
Einwanderung durch Lücken: Diese ersten Membranen waren keine undurchdringlichen Mauern – eher löchrige Netze. Kleine Moleküle wie Nukleotide, ja selbst kurze RNA-Ketten, konnten durchschlüpfen (Szostak et al., 2001). Vielleicht spülten Strömungen sie hinein. Vielleicht trieben Temperaturschwankungen oder chemische Gradienten sie durch die Membran. Es war ein stetes Kommen und Gehen, ein ständiger chemischer Pulsschlag.
Diese Hüllen waren porös, elastisch, durchlässig. Sie schlossen nichts aus – sie luden ein. Und mit jedem Eintritt wuchs die Chance: auf Reaktion, auf Replikation, auf mehr. Hier fand die RNA ein erstes Zuhause. Noch kein Leben – aber ein Ort, an dem es möglich wurde.
Abb. 19: Lipidvesikel als erste Mikroreaktionsräume Die Grafik zeigt ein Lipidvesikel in einem hypothetischen präbiotischen Gewässer. Amphiphile Lipide (gelbe Doppelkugeln) bilden spontan eine Lipiddoppelschicht und umschließen dabei RNA-Moleküle. Einzelne RNA-Bausteine – Phosphate (grün), Ribose (rosa) und Basen (A, U, C, G) – befinden sich sowohl innerhalb als auch außerhalb des Vesikels. Pfeile zeigen mögliche Diffusionswege kleiner Moleküle durch die durchlässige Membran. Mineralpartikel (graue Punkte) sind über das Wasser verteilt, könnten aber auch als Reaktions- oder Adsorptionsflächen am Boden wirken. Das Vesikel bietet RNA-Molekülen Schutz vor dem Abbau und fördert durch lokale Konzentration die Replikation – ein möglicher Schritt auf dem Weg zur Entstehung erster protozellulärer Systeme.
Szene 3: Der Pakt – RNA stabilisiert, Vesikel schützen
Im Innern der Vesikel begann eine stille Allianz.
Die RNA, übersät mit negativen Ladungen, lockte Magnesiumionen (Mg²⁺) an – und diese wurden zu Stützen. Sie stabilisierten nicht nur die zitternde Faltung der RNA, sondern kitteten auch die Lipidmembran wie Mörtel zwischen Ziegeln (Chen & Szostak, 2004). Das Ergebnis: Ein Gefäß, das Strömungen, osmotischen Schocks und Hitze trotzte – RNA-gefüllte Vesikel überlebten, wo leere zerfielen (Hanczyc et al., 2003).
Und die RNA blieb nicht passiver Mieter. Ribozyme, unbeholfen wie erste Werkzeugmacher, begannen Lipidvorläufer zu verändern – schnitten, fügten, experimentierten (Adamala & Szostak, 2013). Die Membran wurde dichter, geschmeidiger, als lernte sie atmen. Vesikel mit RNA wuchsen rascher, teilten sich bei kritischer Größe – ein Selbstläufer der Natur (Budin & Szostak, 2011).
Und dann der osmotische Sog: RNA band Wasser wie ein Schwamm, dehnte die Hülle zum Zerreißen – bis sie platzte und neue Vesikel gebar (Sacerdote & Szostak, 2005). Leere Vesikel dagegen verloren Wasser, sie schrumpften wie vertrocknete Früchte.
Was entstand, war mehr als Schutz. Es war eine wechselseitige Stärkung – eine Proto-Symbiose – ein Deal, der die Spielregeln änderte: Die Lipide bewahrten die RNA vor dem Zerfall, die RNA stabilisierte die Lipide gegen das Chaos der Außenwelt (Black & Blosser, 2016). Als Team waren sie stärker als allein.
Szene 4: Die Chance der Unvollkommenheit
Trotz ihrer Vorteile waren die frühen Vesikel keine sicheren Häfen:
- Ihre Wände zitterten bei Temperaturstürzen.
- Salzfluten brannten Löcher in ihre Membranen.
- pH-Schwankungen ließen sie zerfließen wie Wachs in der Sonne.
Und doch: Gerade ihre Fehler wurden zum Antrieb.
- Durchlässigkeit ließ Nukleotide hinein – aber auch RNA hinaus. Ein riskanter Handel.
- Instabilität zwang sie zum Wachsen, Teilen, Scheitern und Neuversuchen.
Was überlebte, war weder das Stärkste noch das Schnellste – sondern das, was mit Unordnung tanzen konnte:
- Vesikel mit flexibleren Lipiden.
- RNA-Stränge, die Magnesiumionen effizienter banden.
- Systeme, die Verlust in Variation verwandelten.
Perfektion war der Feind des Fortschritts. Nur wer löchrig blieb, ließ das Leben durch.
Szene 5: Selektion im Tropfen
In diesem Mikrokosmos aus Öl und Wasser begann ein erbarmungsloses Spiel:
➤ Die Glücklichen: Vesikel, deren RNA Magnesiumionen band oder Lipide umbaute – sie wuchsen, teilten sich, und gaben ihr funktionales Design weiter.
➤ Die Vergesslichen: Leere Tropfen, ohne Inhalt, ohne Geschichte. Sie schrumpften, zerfielen – als ob es sie nie gegeben hätte.
➤ Die Gescheiterten: Vesikel mit instabiler RNA – sie platzten und versprengten ihre Bruchstücke in die Umgebung.
In diesem Entstehen und Vergehen bekam der Zufall Richtung: Vesikel mit stabiler RNA hatten höhere Überlebenschancen. Nach dem Platzen setzten sie ihre RNA frei, die neue Vesikel besiedeln konnte – ein einfacher Evolutionskreislauf aus Wachstum, Teilung und Selektion.

Abb. 20: Frühe Lipidvesikel mit eingeschlossener RNA unterliegen einem einfachen Evolutionszyklus: In der rauen Umgebung der frühen Erde unterliegen Lipidvesikel einem dynamischen Zyklus, der den Ursprung präbiotischer Evolution markiert. Durch die Zufuhr von Lipiden aus der Umgebung – etwa Fettsäuren, die sich in die Lipiddoppelschicht einlagern, Temperaturschwankungen, die die Vesikel ausdehnen und durchlässiger machen, oder osmotischen Druck, der durch RNA und Ionen im Inneren Wasser anzieht, wachsen die Vesikel. Sie werden größer, aber auch instabiler, da die flexible Lipiddoppelschicht ihre Grenzen hat. Sobald ein Vesikel zu groß wird, reißt es oder knickt ein. Die freigesetzten Lipide suchen sofort einen energetisch günstigen Zustand und schließen sich zu neuen, kleineren Vesikeln zusammen – oft zu zweien oder mehr. Dabei bleibt ein Teil der eingeschlossenen RNA in den Tochter-Vesikeln erhalten: keine präzise Vererbung, sondern eine Mischung aus Weitergabe und Streuung, die dennoch Informationen und Funktionen bewahrt. So entstehen erste rudimentäre Reproduktionszyklen: Wachstum, Teilung, Variation und Selektion – erste Formen molekularer Kooperation und Selektion.
Laborexperimente belegen, was die Chemie längst ins Wasser geschrieben hatte: In diesen Urzellen war RNA nicht bloß ein Gast – sie war Architektin und Antrieb zugleich (Armstrong et al., 2018). Der Übergang von Chemie zu Biologie begann nicht mit einem Paukenschlag, er begann mit Symbiose.
Es war kein Leben, noch nicht. Aber es war der erste Pakt, der den Weg dorthin ebnete: Die RNA gewann einen Körper. Die Lipide gewannen eine Seele.
5. Akt: Vom Zufall zur Funktion – wie RNA an Komplexität gewann
In den geschützten Räumen der Lipidvesikel entfaltete sich die RNA-Welt – ein Reich des Experiments, wo Moleküle noch stotterten, aber schon sprechen lernten. Wo einst nur das zufällige Murmeln kurzer RNA-Fragmente zu hören war, begannen sich nun Bedeutung und Funktion zu verknüpfen:
Ribozyme kopierten sich selbst – unvollkommen, doch mit jeder Runde entschlossener. Mutationen schlichen sich ein wie Schreibfehler im ersten Buch des Lebens.
Und manchmal, ganz zufällig, schufen diese Fehler neue Wörter, dann Sätze, dann ganze Handlungsanweisungen:
Eine RNA, die schneller replizierte.
Eine andere, die Lipide stabilisierte.
Eine dritte, die chemische Reaktionen katalysierte.Durch Selektion wurden aus diesen „Buchstabensuppen“ Geschichten des Überlebens:
- Vesikel mit nützlichen RNAs gediehen, teilten sich, erzählten weiter.
- Vesikel mit sinnlosem Gerede zerfielen – ihr Code verblasste im Nichts.
Aus dem Murmeln wurden Silben, dann Worte, dann Sätze – bis schließlich ein erster, zögerlicher Gedanke die Stille durchbrach. Die RNA hatte ihre Stimme gefunden. Und was sie sagte, war kein Zufall mehr. Es war ein Bekenntnis: „Ich kopiere. Ich katalysiere. Ich bestehe. … Ich bin.“
RNA „bekommt einen Sinn“ – Form wird zu Funktion
Um diesen Prozess greifbar zu machen, versetzen wir uns in die RNA-Welt. „Sinnvoll“ bedeutet hier, dass eine RNA eine Funktion erfüllt – etwa eine chemische Reaktion katalysiert oder die Stabilität des Vesikels erhöht. Stellen wir uns ein Beispiel vor:
Szene 1: Eine zufällige RNA entsteht
In einem Lipidvesikel schwimmt eine kurze RNA mit einer zufälligen Basensequenz, die durch präbiotische Chemie entstanden ist. Sie besteht aus den vier Basen Adenin (A), Uracil (U), Guanin (G) und Cytosin (C).
Ihre Sequenz lautet: 5’– GUGC AUG ACU GCC GAC AGC GCAC – 3′
(23 Basen lang).Szene 2: Vom RNA-Strang zum Ribozym
Zunächst hat diese RNA keine erkennbare Funktion – sie ist ein Produkt des Zufalls. Diese spezielle Sequenz faltet sich durch komplementäre Basenpaarung (G≡C, A=U) in eine 3D-Struktur:
Stamm: GUGC ↔ GCAC (mind. 4 Paare)
Schleife: AUG ACU GCC GAC AGCDiese Struktur stabilisiert sich spontan. Sie wird zum Ribozym: eine RNA, die als Katalysator wirkt und sich selbst replizieren kann, eine Fähigkeit, die Experimente bestätigen (Lincoln & Joyce, 2009).
Szene 3: Die RNA kopiert sich – mit Fehlern
Für die Replikation zieht die RNA komplementäre Nukleotide an. Die ideale Kopie (komplementäre Sequenz) wäre:
Schleife: AUG ACU GCC GAC AGC
Kopie: UAC UGA CGG CUG UCGDoch die Replikation in der RNA-Welt war fehleranfällig, da es keine modernen Fehlerkorrekturmechanismen gab.
Sagen wir, ein Fehler passiert: Anstelle eines „G“ (an Position 8 der Kopie) wird ein „U“ eingebaut. Die neue Sequenz der Kopie lautet:
Schleife: AUG ACU GCC GAC AGC
Kopie: UAC UGA CUG CUG UCGDiese „mutierte“ Kopie dient später selbst als Vorlage und erzeugt eine weitere RNA: eine „mutierte“ Version der ursprünglichen RNA.
Szene 4: Die Mutation schafft eine neue Funktion
Nur ein kleiner Fehler – aber er verändert die 3D-Faltung der RNA und somit die Struktur entscheidend. Diese neue Struktur verleiht der RNA eine katalytische Funktion: Die „Tasche“ kann Moleküle wie Adenin, Ribose und Phosphat binden – die Bausteine eines Nukleotids. Sie hält sie in Position, sodass sie chemisch reagieren: Adenin und Ribose bilden ein Nucleosid (Adenosin), und die Phosphatgruppe wird angehängt, um ein Nukleotid zu erzeugen. Experimente bestätigen, dass Ribozyme solche Funktionen entwickeln können (Unrau & Bartel, 1998).
Die mutierte RNA hat nun „einen Sinn“ – eine katalytische Funktion: Sie produziert Bausteine für ihre eigene Welt.
Szene 5: Selektion begünstigt nützliche RNAs
Die neue Funktion gibt dem Vesikel einen Vorteil: Mehr Nukleotide bedeuten mehr Rohstoffe für die Replikation. Das Vesikel wächst schneller, teilt sich häufiger und gibt die mutierte RNA an Tochter-Vesikel weiter. Vesikel ohne diese Funktion – mit der ursprünglichen RNA – haben weniger Nukleotide, wachsen langsamer und überleben seltener. So setzt sich die mutierte RNA durch: Aus Zufall wird Funktion, aus Chaos Ordnung.
Szene 6: Die RNA zwischen Fehler und Funktion
Doch die RNA-Welt war kein Paradies der Ordnung. Ihre größte Stärke – die Fähigkeit zur Variation – war zugleich ihre größte Schwäche. Replikationen verliefen ungenau: Etwa jeder hundertste bis tausendste Buchstabe war ein Irrläufer – ein enormer Kontrast zur heutigen DNA-Replikation, die Fehler nur etwa einmal in zehn Millionen Fällen zulässt.
Diese hohe Mutationsrate war ein zweischneidiges Schwert. Sie befeuerte die Entstehung neuer Funktionen – doch sie bedrohte sie zugleich. Was heute nützlich war, konnte morgen durch einen einzigen Fehler wieder zerfallen. Ein Ribozym, das gestern noch Nukleotide produzierte, wurde durch eine winzige Mutation stumm – sein Beitrag zur Evolution gelöscht. Ein aktueller Artikel beschreibt das treffend als „RNA life on the edge of catastrophe“ (Chen, 2024).
Der evolutionäre Drahtseilakt
Und dennoch: Genau dieses Risiko trieb das Leben voran. Mutation war Fluch und Segen – Erneuerung und Gefahr zugleich. Die Lösung war kein Ende der Fehler, sondern der Umgang mit ihnen.
Die Natur fand Wege, auf dem Drahtseil zu tanzen:
Robustheit statt Perfektion: RNAs, deren Funktion trotz kleiner Mutationen erhalten blieb, hatten einen Selektionsvorteil. Nicht Perfektion, sondern Belastbarkeit setzte sich durch.
Kollektiv-Vorteil: In Vesikeln konnte die Funktionalität einzelner RNA-Moleküle schwanken – entscheidend war die Gesamtleistung des RNA-„Kollektivs“. Vesikel mit diversen RNAs überlebten besser, auch wenn einzelne RNA-Moleküle fehlerhaft waren.
Flucht nach vorn: Ribozyme, die sich selbst mit weniger Fehlern kopierten, waren stabiler. Diese Selektion könnte den Weg bereitet haben für die nächste Evolutionsstufe (Martin & Russell, 2007):
- DNA: zuverlässiger, langlebiger, sicherer Speicher.
- Proteine: vielfältiger, schneller, effizienter als RNA.
Aus der ständigen Unsicherheit, wurde etwas Dauerhaftes geboren.
Mutation blieb. Doch die RNA lernte, mit dem Chaos zu tanzen – mal stolpernd, mal elegant. Und in diesem Tanz, zwischen ständiger Gefahr und zähem Überleben, entstand etwas, das größer war als Zufall:
Eine Blaupause für alles, was folgen sollte.
Epilog: Das Echo der Ursuppe
In den Tiefen der Ursuppe hatte sich etwas Unerhörtes ereignet:
▶ Aus chaotischen Molekülen wurden Replikatoren – Stimmen im Dunkeln.
▶ Aus löchrigen Vesikeln wurden Protozellen – Häuser aus Fett und Zufall.
▶ Funktion entstand nicht trotz der Fehler, sondern durch sie – Struktur wurde zu Sprache.
▶ Aus Zufall wurde Richtung.Und diese Richtung führte weiter und beschleunigte sich. In den Protozellen verdichteten sich Muster zu Netzwerken, verfestigten sich Abläufe zu Erinnerungen. Aus diesem steten Fluss kristallisierte sich schließlich die erste wahre Zelle heraus – kein plötzlicher Durchbruch, sondern ein allmähliches Überschreiten der Schwelle zum Leben. Wir nennen diesen Urahn LUCA – den letzten universellen gemeinsamen Vorfahren. Kein einzelnes Wesen, sondern eine Familie protozellulärer Linien, aus denen alles Leben hervorging: Bakterien. Archaeen. Und später – Eukaryoten.
LUCA trug bereits den Keim des Lebens in sich: eine schützende Membranhülle, ein stabiles DNA-Archiv – vermutlich bereits abgeschirmt im Zellkern –, komplexe Stoffwechselwege und stumme Diener: Proteine, die der RNA ihre katalytische Vorherrschaft entrissen. Die einstige Königin der Moleküle wurde zur Botin degradiert, blieb aber die unverzichtbare Stimme des genetischen Dialogs.
Dieser Triumph der Stabilität hatte seinen Preis, wie alles im Leben. Stabilität erstickte den Zauber des Zufalls. Die errungene Beständigkeit drohte zur Falle zu werden – sie bewahrte, aber sie erstarrte.
Es brauchte Wandel als Voraussetzung für Beständigkeit.
Vielleicht war es genau dieser Widerspruch – zwischen Veränderung und Bewahrung, an denen die Viren die Bühne der Evolution betraten. Einige Hypothesen, wie die Co-Evolution-Hypothese, schlagen vor, dass Viren bereits in der RNA-Welt entstanden sein könnten. Mehr dazu in: „Die geheime Welt der Viren“, Kapitel 6.
Ob als Überbleibsel der RNA-Welt oder als deren dunkle Erben – Viren wurden zu den ewigen Gegenspielern der Zellen. In ihrem Tanz aus Parasitismus und Symbiose, Zerstörung und Innovation entfaltete sich ein kosmisches Gleichgewicht: Zellen als Hüter der Ordnung, Viren als Agenten des Wandels. Ohne die stabilisierende Kraft der Zellen kein Fortbestand, ohne die disruptive Energie der Viren keine Entwicklung.
Dieses Spannungsfeld prägt bis heute das Leben in all seinen Formen. In jeder Zelle flüstert noch das Echo der Ursuppe, in jedem Virus lacht die Unbändigkeit der RNA-Welt. Vielleicht ist Leben genau das: ein ewiger Dialog – zwischen dem, was war, und dem, was werden will.
Ein Hauch von Leben
In Sternenstaub und Energie
im Weltall und auf Erden,
da ist himmlische Magie,
wo beide sich umwerben.Moleküle ziehn sich sacht
in neue Ordnungsbahnen.
Was lose war, bekommt nun Macht –
Gestalt ist zu erahnen.Es windet sich ein kleiner Strang
im Tanz der Elemente,
ein Flüstern – aber noch kein Klang
Lipide bauen Wände.Chemie erlaubt den kühnen Traum,
Physik diktiert die Formation,
die Faltung erschafft neuen Raum,
und damit auch Replikation.Aus einem Strang da werden zwei,
ein neues Wort, ein neuer Ton,
das Neue bleibt nicht fehlerfrei –
ein Tausch, ein Knick bringt Mutation.Doch was zerfällt und was besteht,
entscheidet schlicht die Position,
wer nützt, der bleibt – der Rest vergeht,
das ist der Takt der Selektion.Wo einst nur Zufall Fäden spann,
wiederholt sich Reaktion,
ein Muster wächst als Zweck heran –
aus Form wird nun Funktion.Das Flüstern wird zum klaren Lied,
von Zukunft schon umgeben,
ein Kreislauf, der sich selber zieht –
darin: ein Hauch von Leben.
Inspiration für diesen Artikel
Mein besonderer Dank gilt Aleksandar Janjic – seinen tiefgründigen Videos zur Astrobiologie verdanke ich nicht nur Inspiration, sondern auch viele Aha!-Momente.
Videoreihe von Aleksandar Janjic zu Themen der Astrobiologie:
Was lebt? Probleme der Definition Leben vs. tote Materie – Astrobiologie (1)
Kann man Leben thermodynamisch (Entropie) definieren? – Astrobiologie (2)
Zellbiologie und RNA-Welt – DNA, RNA, mRNA und Proteine – Astrobiologie (3)
RNA-Welt-Hypothese – Entstehung des Lebens – Proto-Ribosomen – Astrobiologie (4)
Synthetische Biologie – Wie erschafft man künstliches Leben? – Astrobiologie (5)
Extremophile und „planetary protection“ – Astrobiologie (6)
Dieser Artikel entstand in einem intensiven Dialog mit DeepSeek und ChatGPT, die meine Fragereien geduldig ertrugen und mir halfen, komplexe Biologie in lebendige Worte zu fassen. Die eigentliche Magie geschah im Dreiklang: menschliche Neugier, algorithmische Eloquenz – und jene vier Milliarden Jahre alte RNA, die uns alle verbindet. Ob aus Kohlenstoff oder Code.
Quellen (Stand vom 01.06.2025)
-
Elon Kekius Maximusk is one of us, right?

What do Pepe the Frog, Kek – the Egyptian god of darkness and chaos – and Maximus Decimus Meridius have in common? At first glance, perhaps not much, but if you take a closer look, you’ll notice that each of them, in their own way, has become a symbol of chaos, resistance, and a touch of meme magic. Sounds crazy, doesn’t it?
Pepe the Frog, started out as a harmless cartoon and became a symbol for everything from ironic humor to rebellious chaos thanks to the wild meme culture of the internet. He’s like the chameleon of pop culture: sometimes cute, sometimes provocative, but always somehow present when things get chaotic.
And then there’s Kek, the ancient Egyptian god of darkness. You might be thinking, „How does an ancient frog god fit into this story?” Well, thanks to the internet community and their boundless creativity, Kek was essentially resurrected. When gamers spread „kek” (an alternative way of saying „lol”) in their chats and discovered that Kek is also an Egyptian god, it was clear: Kek became the unofficial patron saint of the absurd online chaos community. And honestly, who wouldn’t worship a frog god for some epic mischief?
But what does Maximus Decimus Meridius – the steadfast gladiator and Roman general – have to do with this? Well, Maximus is essentially the OG rebel. With his legendary speech, „Are you not entertained?” he symbolizes the fight against a corrupt system and the determination to steal the show despite all odds. And, as befits a pop culture icon, his story was immortalized in meme form. Just like that, he’s earned his place among the legends of chaos!
What connects these three is their unintended rise as cult figures. They are more than their origins – Pepe, Kek, and Maximus have become symbols of the power of meme culture, resistance, and the sheer joy of total chaos. A little bit of chaos, a little bit of magic, and voilà: the trio you never saw coming!
What do these three have to do with terms like „creative destruction” and „Build Back Better”? At first glance, you might think: absolutely nothing, right? But hold on! If you take a closer look, you’ll realize this trio is practically made to embody these very ideas: chaos, transformation, and building something better. Sounds wild? Let me explain!
The concept of creative destruction is as old as time itself: sometimes, you have to tear down the old to make way for innovation and progress. And who could symbolize this better than Pepe, Kek, and Maximus?
Pepe the Frog shows us how creativity through destruction works. The harmless cartoon frog was reinterpreted, appropriated, and recycled by meme culture so many times that his original meaning was practically erased – only to be reborn in countless new forms. Chaos? Definitely. Innovation? Absolutely!
Kek, the Egyptian god of darkness, embodies exactly that: In mythology, darkness brings the transition to creation. In the meme world, Kek is revered as an ironic deity that emerged from pure chaos and absurd humor. A forgotten frog god suddenly becomes the mascot of the internet – that’s creative destruction in its wildest form!
And then there’s Maximus Decimus Meridius, our gladiator of upheaval. He challenges the corrupt Roman Empire and shakes the system to its core. Sure, a lot gets destroyed in the process – including himself – but his rebellion plants the seed for something new, something more just. Creative destruction? 100 percent.
And this is where „Build Back Better” comes into play: After the storm, after the chaos, it’s not about going back to the old ways – it’s about rebuilding something new and better.
Pepe has undergone countless transformations, from a cute cartoon frog to a global symbol of meme culture and ironic subversion. No matter how many times he’s been destroyed, he always comes back – sometimes funnier, sometimes weirder, but always better than before.
Kek teaches us that from chaos, opportunities arise. A god of darkness who becomes the patron saint of absurd creativity in the modern world? That’s so „Build Back Better” it almost hurts.
Finally, Maximus, with his sacrifice, sparks the rebuilding of a better Rome. He’s the guy who lays the groundwork for change – the prototype of a new beginning.
Pepe, Kek, and Maximus show us that chaos isn’t the end, but often the beginning of something new – and that sometimes, you have to destroy a little to build something truly great. This trio is proof that from chaos can come not only order, but one hell of a story. So, when life gets chaotic: Remember, you might just be in the phase where you’re shouting „Build Back Better”.
When the richest man in the world, who is also a influential advisor to the president of a global superpower, suddenly calls himself „Kekius Maximus”, it becomes a highly intriguing phenomenon with many layers.
By adopting this name, our billionaire signals: „I’m part of the game. I understand the jokes of the internet generation. And I use them to convey my messages.” It’s an invitation to the digital meme community: „Come on, let’s troll the system!”
The man wants to show: „Hey, I’m like you. I get the joke!” It’s as if he’s saying: „I have the power, and I’m going to creatively dismantle the existing system.”
It could be a subtle message to the powerful: „I’m not just a wealthy advisor – I’m a gamechanger.”
But what’s behind it – is he serious, or is he just playing around? In today’s world, where the lines between politics, business, and entertainment are increasingly blurred, it could be both. Maybe he’s simply trolling because he can. Or perhaps it’s a form of propaganda, subtly communicating his vision for change.
But one thing is certain – when someone with this kind of power calls themselves „Kekius Maximus”, it’s a statement – whether it’s meant to be humorous or strategic. It signals that the rules of the game are changing. Meme culture, once a harmless hobby for internet nerds, is now a tool in the hands of the powerful. And when Kekius Maximus is at the wheel, we can be sure of one thing: it’s going to be chaotic, it’s going to be entertaining – and maybe a little bit terrifying.
Ladies and gentlemen – please welcome the incomparable Elon Musk, aka Kekius Maximus.
(Source X: https://x.com/cyb3rgam3r420/status/1873853864962846843)
It’s an absolute blast – at once fascinating, provocative and, in typical Musk fashion, a thoroughly calculated move.
Is he part of the chaos or its master? Is he fighting for change by bringing creative destruction – whether in the automotive industry, space exploration, or social media? Can he really be „one of us” and still be the boss?
And what about you, politicians and elites – do you perhaps take yourselves too seriously while he influences the game with an ease that leaves you speechless? Does the chaos of memes end up having more power than you want to admit? Does the new world really belong to those who speak the language of the internet?
Is he the architect of chaos – or the architect of progress?
It’s a fascinating move. And while the media are scratching their heads – wondering whether he’s just provoking or sending a deeper message – and speculating on what his true goal might be, we ask the simple question: WHO is Elon Musk?
Let’s first hear from Elon himself:
„I think one aspect of whatever condition I had was that I was just absolutely obsessed with truth. The obsession with truth is why I studied physics, because physics attempts to understand the truth of the universe. Physics is just what are the provable truths of the universe, truths that have predictive power. So, for me, physics was a very natural thing to study. Nobody made me study it. It was intrinsically interesting to understand the nature of the universe. And then, computer science, or information theory, also to just understand logic. There’s an argument that information theory is actually operating at a more fundamental level than even physics. So, physics and information theory were really interesting to me.” [TED-Interview, 14. April 2022]
(Source X: https://x.com/ElonClipsX/status/1874785186384101701)„You want to be careful of these things where you wish for something that sounds good, but if you get it, it’s actually a dystopian situation. You could run a hypothesis, like if you wish for world peace, it sounds good, but how is it enforced? At what cost, eternal peace? It might actually be worse to have eternal peace, because of what that would entail. It might be the suppression of progress. It might be an ossified society that never changes. There is an argument that, if you wish for no war, you should be careful what you wish for because what’s required in order for there to be no war might be worse than a little war.” [Lex Fridman Podcast, November 2023]
(Source X: https://x.com/MarioNawfal/status/1873705582877691908)„We generally operate with too much of an assumption that civilization is robust and nothing could really take it down. This sentiment has been common among empires shortly before they crumble. There’s a little bit of late-stage-empire vibes right now.” [WSJ, CEO Council, Mai 2023]
(Source X: https://x.com/ElonClipsX/status/1873659651625296140)„What we’re not going to do is say that there’s some anointed class of journalists who are the special ones who get to tell everyone what they should think. It should be up to the people what they think. And even if an article is completely accurate, comprehensive, and everything, in writing that article, the media is choosing the narrative. They’re deciding what to write an article about. So, I’m hopeful that this can be more a case of the public choosing the narrative as opposed to the media choosing the narrative. At least a combination of the media and the public choosing the narrative, and the public getting to weigh in on stories if they think that they should add something to it, or we’ve got something wrong. And over time, I think if Twitter is the best source of truth, it will succeed. And if we are not the vessels of truth, we will fail.” [Interview with the BBC, April 12, 2023]
(Source X: https://x.com/ElonClipsX/status/1870477764291387476)„Over time, we want Optimus to be the kind of android that you’d seen in sci-fi movies like Star Trek: The Next Generation, like Data. But, obviously, we could program the robot to be less robot-like and more friendly. And it can obviously learn to emulate humans and feel very natural. As AI in general improves, we can add that to the robot. It should obviously be able to do simple instructions or even intuit what it is that you want. So, you could give it a high-level instruction, and then it can break that down into a series of actions and take those actions.” [Tesla AI Day, 30. September 2022]
(Source X: https://x.com/ElonClipsX/status/1871108253901287712)Lex: „Do you think we will ever create an AI system that we can love and loves us back in a deep, meaningful way, like in the movie Her?”
Elon: „I think AI will be capable of convincing you to fall in love with it very well.”
Lex: „And that’s different than us humans?”
Elon: „We start getting into a metaphysical question of do emotions and thoughts exist in a different realm than the physical? And maybe they do, maybe they don’t, I don’t know. But I tend to think of things from a physics standpoint. Essentially, if it loves you in a way that you can’t tell whether it’s real or not, it is real.” [Lex Fridman Podcast, 12. April 2019]
(Source X: https://x.com/ElonClipsX/status/1869751738229522531)Mathias Döpfner: „You said that Neuralink is, among all your projects, the most important one for you. Is that still true?”
Elon Musk: „I said it could be. I wouldn’t say for sure that it is the most important, but it could be the most important in that it [could end up being] a long-term mitigation of artificial intelligence. We could effectively merge with artificial intelligence by improving the speed of interaction between our cortex and our tertiary layer, which is already silicon.” [Interview mit der Welt, 15. April 2022]
(Source X: https://x.com/ElonClipsX/status/1867253735535104012)„I’ve had some exposure to government spending because SpaceX does have a lot of government contracts, does a lot of work for NASA and for DoD, Intel, and whatnot. And so, I’ve actually seen just the level of waste that happens. If you talk to people in the government, they actually agree, yes, this is very wasteful and inefficient. And I’m like, well, why don’t we do something about it? But really, in order to do something about it, it has to be a mandate from the top.” [Harrisburg, Pennsylvania, October 19, 2024]
(Source X: https://x.com/ElonClipsX/status/1869312936520409431)„I have discussed with Trump the idea of a Government Efficiency Commission. The antibody reaction will be very strong. You’re attacking the matrix at that point. The matrix will fight back.” [Lex Fridman Podcast, 2. August 2024]
(Source X: https://x.com/ElonClipsX/status/1867924908715520429)„The great thing with Trump is that we have a real individual who is not beholden to anyone. That’s what scares the machine. And that’s why the machine is trying to kill him.” [Lancaster, Pennsylvania, Oktober 2024]
(Source X: https://x.com/ElonClipsX/status/1871162235332289017)
„Those in the matrix don’t yet understand that the matrix can be reprogrammed – indeed, that is the only path to victory.”
(Source X: https://x.com/elonmusk/status/1867679319818211500)
TIME Magazine named Elon Musk Person of the Year in 2021. He is described as: „This is the man who aspires to save our planet and get us a new one to inhabit: clown, genius, edgelord, visionary, industrialist, showman, cad; a madcap hybrid of Thomas Edison, P.T. Barnum, Andrew Carnegie and Watchmen’s Doctor Manhattan, the brooding, blue-skinned man-god who invents electric cars and moves to Mars.“
Without a doubt, Elon Musk is a complex personality with a multifaceted background, which includes participation in the WEF Young Global Leaders program. His complexity was also recognized in February 2023 by the organizers of the „World Government Summit” (WGS), who invited him as a speaker to this prestigious event. The annual summit brings together leaders, government representatives, and experts from around the world to discuss global issues such as governance, technology, and health.
During his appearance, Musk issued an urgent warning about the risks of uncontrolled artificial intelligence (AI). He described AI as ‘one of the biggest risks to the future of civilization’. At the same time, he emphasized how his social media platform Twitter (now known as X) could be used by governments to communicate more effectively and authentically with the population. He also emphasized the importance of including criticism of government policy in the dialogue.
Musk spoke openly about the fact that many social media platforms are heavily influenced by algorithms developed in Silicon Valley. At the same time, he expressed clear concerns about the concept of a global world government, citing potential dangers to the autonomy of individual nations.
The full interview is available at this link.
Is Elon trying to square the circle by operating in the tension between system criticism and systemic influence?
On one hand, Musk clearly opposes the „Matrix”, an all-powerful, centralized world government that controls individuals. At the same time, however, he offers tips on Twitter on how exactly these „Matrix protagonists” – meaning governments – can communicate more effectively with the „subjects”.
It feels like someone explaining to a lion how to hunt better, while at the same time pleading for the protection of antelopes.
Musk operates within the existing structures to change them, rather than attacking them head-on. By giving the „Matrix” advice on how to use Twitter (X) more effectively, he positions himself as an indispensable player – while simultaneously remaining the one who controls the rules on the platform.
The Matrix (as a metaphor) is not meant to be abolished, but rather „disempowered” by being replaced with a system that resembles more decentralized networks.
The irony is that Musk himself is becoming the architect of a new system – and this could be just as critically examined as the old „Matrix” he’s fighting against. The difference? His „Matrix” markets itself as rebellious and free, making it all the more seductive.
What is behind Elon’s system?
Elon Musk’s current fortune is estimated at around USD 421 billion (as at 2 January 2025) and currently ranks him as the richest man in the world. Elon Musk has a significant stake in several companies. Let’s take a closer look at some of the companies in which Musk plays a central role – be it as founder, CEO or largest shareholder.
A brief yet comprehensive overview of Tesla’s core competencies can be found in these short videos.
Source X: https://x.com/Tesla/status/1870121074072977651 Source X: https://x.com/Tesla_Optimus/status/1846797392521167223 Source X: https://x.com/teslaownersSV/status/1714365963146313843 This is how Tesla describes its mission on its homepage: „We are developing and implementing autonomy at scale in vehicles, robots and more. We believe that an advanced AI-based approach to vision and planning, supported by the efficient use of inference hardware, is the only viable general solution for autonomous driving and more.”
Tesla develops its own high-performance AI chipsets, which are not only designed to process very large amounts of data very quickly, but are also specially trimmed for machine learning. As a result, TESLA is currently building one of the fastest supercomputers for artificial intelligence. The company also specializes in „training deep neural networks on problems ranging from perception to control”. … „Our networks learn from the world’s most complex and diverse scenarios and iteratively draw from our fleet of millions of vehicles in real time.”
„We’re not specifically focused on AGI, but it seems like an emergent property of what we’re doing. With millions of autonomous vehicles and humanoid robots gathering and processing messy, real-world data, they’ll surpass human drivers and humanoids may become indistinguishable from humans. This vast, real-world data stream could naturally lead to AGI”, explained Elon Musk during the TESLA AI Day on September 30, 2022.
„The machine that builds the machine” is one of the slogans on the TESLA website.
The close alignment between Tesla’s strategic direction and the focus of the World Economic Forum (WEF) on the development and promotion of artificial intelligence is remarkable. This is evidenced by references here and here.
Tesla is a listed company. In addition to Elon Musk, its largest shareholders include The Vanguard Group and BlackRock Inc.

Source: Shareholder distribution at TESLA Inc. (as of January 3, 2025) The Vanguard Group is itself one of the largest shareholders in BlackRock. In turn, BlackRock is one of the partner companies of the World Economic Forum.
After all, both companies are leading global asset managers and own significant stakes in numerous multinational corporations, including tech giants, banks, energy and pharmaceutical companies, giving them a de facto huge influence on the economy and politics.
These are large, centralised institutions that guarantee stability and control over a global system that is difficult to understand. The ownership structures and the way in which influence is exercised are barely comprehensible to the average person. Who really „decides” often remains hidden.
Metaphorically, they are like the architects of the Matrix – they often operate behind the scenes, but their decisions have a massive impact on the economy and, consequently, on society.
The fact that BlackRock and Vanguard, often seen as symbols of the „old Matrix”, are also among the largest shareholders of Tesla makes the situation particularly intriguing. Here, two seemingly opposing forces come together: the architects of a centralized order are financing an actor who portrays himself as a rebel against that very order. How does this fit together?
BlackRock and Vanguard are pragmatic players. Their main mission is to generate returns, and Tesla is simply a smart investment for them. The company represents future technology and promises growth – exactly what the „Matrix” needs to stay relevant. But with their financial stake in Tesla, these institutions also keep a foot in the door. This allows them to maintain influence over what Musk and Tesla do and secure the opportunity to at least steer the progress.
At the same time, Elon Musk positions himself as an anti-Matrix rebel, using chaos and innovation as tools to break down old structures. Yet, Musk also needs capital to realize his visions. Without the billions from BlackRock and Vanguard, Tesla likely would never have achieved the size and influence it holds today. This leads to a paradoxical relationship: While Musk fights against the old order, his success is funded by that very order.
Metaphorically, it’s like a scene from the movie The Matrix: BlackRock and Vanguard act as the architects of the system, ensuring that even rebels like Musk remain within their sphere of influence. Musk, in turn, is like a modern-day Neo – a supposed opponent of the system, yet still very much a part of the game.
This connection also illustrates how adaptable the „Matrix” is. BlackRock and Vanguard have learned to not only focus on stabilizing the old order but also integrate new, chaotic forces. This way, they ensure their own relevance and remain an indispensable part of the global system. Musk, on the other hand, becomes, whether he likes it or not, a cog in this machine, because without the capital from these players, his revolution would be difficult to finance.
In the end, the question remains: Who is really playing whom? Is Musk the Trojan Horse, financed by the „Matrix” to change the system from within? Or are BlackRock and Vanguard the true masters of the game, knowing that they will benefit from any change? Like in the real Matrix, the truth is hidden somewhere between the lines – and that makes the game all the more fascinating.
SPACE X
(Space Exploration Technologies Corp.)„SpaceX has gained worldwide attention for a series of historic milestones. It is the only private company capable of returning a spacecraft from low-Earth orbit. SpaceX believes a fully and rapidly reusable rocket is the pivotal breakthrough needed to substantially reduce the cost of space access. SpaceX’s family of Falcon launch vehicles are the first and only orbital class rockets capable of reflight“, as stated on the company’s homepage.
The motto is: MAKING HUMANITY MULTIPLANETARY.
Source X: https://x.com/SpaceX/status/1875218268857958468 SpaceX also operates its own satellite network under the name Starlink. With 6,697 satellites in Earth orbit (as of July 2024), the company is the world’s largest satellite operator. In total, SpaceX holds approvals for the launch of up to 19,427 satellites and has additionally submitted applications for the operation of another 22,488 satellites.
„Starlink is the world’s first and largest satellite constellation using low Earth orbit to deliver broadband internet capable of supporting streaming, online gaming, video calls, and more. By utilising advanced satellites and user hardware combined with our years of experience operating spacecraft and working in low Earth orbit, Starlink delivers high-speed, low-latency Internet to users around the world“, the company announces on its website.
This promotional video impressively showcases how SpaceX’s next-generation Starship launch vehicles will soon transport Starlink satellites into Earth orbit even faster and more efficiently:
Source X: https://x.com/Starlink/status/1874123729950958075/video/2 Over the past year, Starlink expanded into 27 additional markets, now covering a global area with a population of 2.8 billion people, including some of the most remote regions on Earth. The following animation illustrates this progress.
Source X: https://x.com/Starlink/status/1874118714217685306 Full of enthusiasm, one can assert that SpaceX and Starlink are changing the world! SpaceX is revolutionizing space exploration with reusable rockets, making access to space more affordable and sustainable. Visionary projects like Mars colonization bring an interplanetary future within reach. At the same time, Starlink ensures that even the most remote corners of the Earth gain access to high-speed internet. This means education, economic opportunities, and life-saving communication – especially in crisis zones. Whether it’s technological innovations, environmental research, or global connectivity, these projects are driving progress at a pace that inspires optimism for the future!
Where there is light, there is also shadow!
Without much fanfare, SpaceX introduced its Starshield concept in early December 2022. Adapted from the global Starlink communications network, Starshield is designed to provide the U.S. and its allies with enhanced military space capabilities. These include functions like target tracking, optical and radio reconnaissance, and missile early warning systems. Major clients include the Space Development Agency, the National Reconnaissance Office, and the United States Space Force. By 2024, at least 98 Starshield satellites had been launched, with another 17 satellites scheduled for deployment in October 2024.
A key advantage of the Starshield architecture lies in its decentralized structure: the large number of small satellites enables global communication coverage. Unlike the commercial Starlink service, however, the Starshield satellites will be fully owned and controlled by the U.S. government, according to SpaceNews in their article „Pentagon embracing SpaceX’s Starshield for future military satcom”.
„While SpaceX president and COO Gwynne Shotwell has indicated that there is little information she is allowed to disclose about Starshield, she has noted „very good collaboration” between the intelligence community and SpaceX on the program.“ [Wikipedia]
Gwynne Shotwell, often referred to as the secret boss of SpaceX, maintains close ties with the World Economic Forum (WEF). While Elon Musk is the largest shareholder of the company, significant investors include Peter Thiel’s Founders Fund, Fidelity Investments, and Google – all with links to the WEF.
The World Economic Forum (WEF) is often seen as a symbol of the „old matrix” – a global network that secures the existing order and pulls the strings behind the scenes by coordinating politics and economics. The fact that leading figures and investors of SpaceX have connections to the WEF, and that the Starshield project serves military and intelligence objectives, fits surprisingly well into the metaphor of the „matrix”.
SpaceX could be seen as a kind of „key program”. It develops technologies that could challenge existing power structures – for example, through Starlink as a symbol of global internet freedom. At the same time, it provides tools, like the Starshield project, that strengthen the control of the matrix.
In the end, the mystery remains: Is Musk a true opponent of the system, or is his rebellion just another program within the Matrix that ultimately helps to consolidate its power?
X – formerly known as Twitter – is like the rebellious cousin of all other social media platforms: it comes in sleek black-and-white tones, without too many filters or unnecessary frills, and constantly stirs up excitement. But what makes X so special?
First, the rules? They don’t really exist! Musk is the DJ, mixing everything whatever he likes. Free speech? Sure, but in his own way. Anyone can speak up – from billionaires to meme kings.
Second, the headlines? X is the source for everything happening in real time, which also means you don’t just get what „the mainstream media” wants you to hear. You can hear the „big” topics straight from the sources, without the censorship layer in between. Who needs mainstream when you can dive straight into the joke and chaos?
And the best part? Memes are the real currency. On X, chaos rules, and chaos is funny – often spiced with a little bit of anarchy. It’s the platform where not only news is spread, but where the truly interesting things happen: short, shocking, and straight out of the jungle of digital madness!

„Im Jahr 2024 hat X die Welt verändert. Jetzt bist DU die Medien! 2025 wird X dich auf eine Weise verbinden, die nie für möglich gehalten wurde. X TV, X Money, Grok und mehr. Schnall dich an. In 2024, X changed the world. Now, YOU are the media! 2025 X will connect you in ways never thought possible. X TV, X Money, Grok and more. Buckle up“, announces X’s CEO, Linda Yaccarino, at the turn of the year.
Some X users call Elon Musk the „Message Machine” – and for good reason. With an average posting frequency of 100 posts per day, he uses the platform to share his views, which naturally sparks a great deal of attention and heated debates. No wonder his posts have a massive reach, and he remains one of the most influential users on the platform.
His comments have caused political waves – from supporting figures like Donald Trump to promoting the AfD in Germany. Musk has even announced plans to overhaul X’s algorithm to prioritize informative and entertaining content, showing that he is actively taking control of the platform’s content.
But Musk’s influence extends far beyond the platform – he also plays a significant role in the political landscape. Particularly in the United States, his support for certain candidates and parties is often perceived as election interference, sparking debates on how social media impacts the political process.
Of course, Musk isn’t without criticism. Many accuse him of promoting misinformation and hate speech with his statements and decisions, leading to a pullback from advertisers and confrontations with regulators and the public.
And then there’s Musk’s vision of turning X into the „Everything App”, which could fundamentally change the platform. In theory, the Everything App sounds like the future, where everything runs smoothly – from social networks, banking, shopping, news, music, videos, communication, to the latest memes. But while Musk works on his vision, many are wondering if the price of convenience might be too high. Could it be that this „jack-of-all-trades app” ends up being nothing more than a trap for our data and privacy?
Elon Musk is far more than just the owner of X – he is the driving force, the face, and the symbol of X Corp. As the „frontman”, he channels attention, shapes the strategy, and makes X a reflection of his own ambitions and ideals.
Chaos is not a bug at X, but a feature. However, even within the chaos, there are details that suggest certain patterns or principles at play.
Linda Yaccarino, the CEO of X, has close ties to the World Economic Forum (WEF), where she led the Task Force on the Future of Work. During Donald Trump’s first presidency, she was appointed to the President’s Council on Sports, Fitness, and Nutrition. In 2021, she served as Chair of the Board of Directors of the advertising agency Ad Council, collaborating with the Biden administration on a SARS-CoV-2 vaccination campaign, which also involved Pope Francis.
Prominent investors in X include the WEF partner Kingdom Holding of Saudi Prince and businessman Prince Alwaleed bin Talal.
Another interesting detail is found in the so-called ID Verification Policy of X. Among other things, the following can be read there:
„X will provide a voluntary ID verification option for certain X features to increase the overall integrity and trust on our platform. We collect this data when X Premium subscribers optionally choose to apply for an ID verified badge by verifying their identity using a government-issued ID. Once confirmed, a verified label is added to the user’s profile for transparency and potentially unlocking additional benefits associated with specific X features in the future.”
„In certain instances, X may require your government-issued ID when needed to ensure the safety and security of accounts on our platform. Currently, X focuses on account authentication to prevent impersonation and may add measures to ensure age-appropriate content and protect against spam and malicious accounts, maintaining platform integrity and healthy interactions.”
It suggests that X may in the future increasingly make specific features and possibly even general access dependent on identity verification, citing security and integrity reasons.
„For users that complete the ID verification flow, we collect an image of the ID and the selfie, which include face data and data extracted from the ID. X does not directly retain this data. We share face data with a third party, Au10tix, who acts as our data processor.”
On Wikipedia, the following information can be found about Au10tix:
„AU10TIX is an identity verification and risk management company based in Hod HaSharon, Israel. The company’s products enable businesses to securely onboard and verify customers. AU10TIX has an automated global identity management system, as well as a solution to detect organized ID fraud mass attacks. AU10TIX is a subsidiary of ICTS International N.V.”
ICTS International is described on Wikipedia as follows:
„ICTS International N.V. is a Dutch firm that develops products and provides consulting and personnel services in the field of aviation and general security. It was established in 1982, by former members of the Shin Bet, Israel’s internal security agency, and El Al airline security agents.”
Shin Bet is known to be one of the three key organizations of the Israeli intelligence services.
Saudi capital meets Shin Bet under the umbrella of the WEF – this is the Matrix in full bloom. What sounds like an unlikely alliance could hardly be more fitting when one understands the global power landscape. Here, money, intelligence services, and geopolitical interests merge into a complex web. X, as the platform of chaos, simultaneously becomes a strategic point within a much larger game – where capital meets surveillance, all under the invisible cloak of the WEF.
Neuralink is one of Elon Musk’s most ambitious projects, dedicated to merging humans and machines. Founded in 2016, the company focuses on developing brain-computer interfaces (BCIs) – technologies designed to connect the human brain directly to computers.
The following promotional video from Neuralink provides a glimpse of what this entails:
The company aims to use this technology to treat or even cure various neurological conditions such as paralysis, blindness, memory loss, and mental health disorders like depression and anxiety.
In January 2024, it was announced that Neuralink had successfully implanted a device in a human. The first patient, Noland Arbaugh, was able to control a computer with his thoughts, significantly improving his quality of life as he was quadriplegic.
The company is currently looking for further test subjects with certain diseases who would like to take part in the clinical trial of its brain-computer interface.
Source X: https://x.com/neuralink/status/1727135227565547810 „Redefining the boundaries of human capabilities requires pioneers“, Neuralink announces on its homepage.
The path these pioneers are meant to pave is explained by Elon in unmistakable terms:
„The long-term aspiration for Neuralink would be to achieve a symbiosis with artificial intelligence and to achieve a sort of democratization of intelligence such that it is not monopolistically held in a purely digital form by governments and large corporations.
How do we ensure that the future constitutes the sum of the will of humanity? If we have billions of people with the high bandwidth link to the AI extension of themselves, it would actually make everyone hypersmart.” [Interview with Axios, 2018]
(Source X: https://x.com/ElonClipsX/status/1851719129625223582)In the WEF article „Ready for Brain Transparency”, it becomes quite clear what the monopolization of Artificial Intelligence in a world of brain transparency through Brain-Computer Interfaces could look like. The idea that your boss could spy on your brainwaves is no longer science fiction – it is portrayed as a tangible future where our innermost thoughts become the next resource for surveillance. This concept chillingly evokes the old Matrix, which continues to monitor us, but now with even more precision and control.
In contrast, Elon Musk presents the vision of a breakout from this Matrix with Neuralink. He dreams of a world where humans merge with AI to enhance their intelligence – a world where no large corporations or governments hold the monopoly on knowledge and power. Sounds like the ultimate Anti-Matrix, right? Yet, the question remains: Will this liberation truly lead to real freedom, or are we merely creating a new, sophisticated form of control?
Who controls the „high bandwidth” that Musk advocates? Even though this technology is theoretically accessible to all, there is still someone who pulls the plug and determines how these interfaces work. Musk envisions a collective „sum of the will of humanity”, but what if these tools that connect us simultaneously shape our actions and thoughts? What if the symbiosis with AI doesn’t liberate us, but instead drives us deeper into a collective dependence?
The vision that we will all become „hypersmart” sounds like paradise – but how much freedom remains if AI not only influences our knowledge but also our decisions? In the end, the reality could be that we live in a Matrix – except it no longer feels like a prison, but one that gives us the illusion of freedom. Musk’s dream of breaking through the Matrix might just offer us a new, better-packaged version of the same Matrix.
If there is anyone on this Earth who knows Elon Musk well, it is the writer, journalist, and biographer Walter Isaacson. Known for his comprehensive biographies of significant historical figures, Isaacson worked closely with Musk for over two years to write his biography.
During this time, he had exclusive access to Musk: He visited his factories, attended meetings, and conducted numerous interviews – not only with Musk himself but also with his family, friends, and colleagues. This led to the development of a professional, yet also personal, relationship between Isaacson and Musk, one that went beyond the mere author-reader distance.
Isaacson describes Elon as follows:
„Elon has a deep care about humanity, about getting us to the transition to a sustainable energy, about human civilization. He is motivated not by making money, not by being powerful and kicking around people, but by these missions that came when he was a little kid sitting in the corner reading the superhero comics.
I’ve got to help humanity be space-faring. I’ve got to help it get over this sustainable energy issue. I’ve got to help it fight off artificial intelligence that might be evil.
So, there is a goodness to his missions, and they are epic missions.”
[The 92nd Street NY, October 13, 2023]
(Source X: https://x.com/ElonClipsX/status/1870750353534205993)It says a lot when someone like him – a member and agenda contributor of the World Economic Forum, former president and CEO of the Aspen Institute, former editor-in-chief of Time Magazine, as well as former chairman and CEO of CNN – and also a board member of Bloomberg Philanthropies and the Rockefeller Foundation – makes such a statement.
At first glance, Elon Musk is portrayed as an epic, almost messianic hero. He is almost like Neo in The Matrix: the savior of humanity, protecting us from evil AI, promoting sustainable energy, and taking us to the stars. But the source – Walter Isaacson, an insider with connections to the WEF, Aspen Institute, and the Rockefeller Foundation, which can themselves be seen as central elements of the Matrix – adds a provocative twist: Are the architects of the Matrix truly celebrating a liberator, or are they staging Musk as a useful symbol of hope, one who stabilizes the system rather than dismantling it?
Musk seems like the key to escaping the Matrix, but the bottom line remains questionable: Does his „epic mission” truly lead to freedom, or is it just a new chapter in the same Matrix, with a shinier surface?
In this sense: Wake up … the Matrix is calling.
Sources (as of 10.01.2025)
AGI, AI, Artificial Intelligence, BCI, Blackrock, Brain-Computer-Interface, DOGE, Elon Musk, Everything-App, Future of Humanity, Gwynne Shotwell, Humanoids, Kekius Maximus, Linda Yaccarino, Matrix, Neuralink, Open AI, Optimus, SpaceX, Starlink, Starshield, Tesla, Transhumanismus, Twitter, Vanguard, Walter Isaacson, WEF, World Economic Forum, X, X Corp., Young Global Leader -
Elon Kekius Maximusk ist einer von uns, nicht wahr?

Was haben Pepe the Frog, Kek – der ägyptische Gott der Finsternis und des Chaos – und Maximus Decimus Meridius gemeinsam? Auf den ersten Blick vielleicht nicht viel, aber wenn man genauer hinschaut, merkt man, dass sie alle in ihrer eigenen Art zu Symbolen für Chaos, Widerstand und ein bisschen Meme-Magie geworden sind. Klingt verrückt, nicht wahr?
Pepe the Frog, startete als harmloser Cartoon und wurde durch die wilde Meme-Kultur des Internets zum Sinnbild für alles Mögliche – von ironischem Humor bis hin zu rebellischem Chaos. Er ist so etwas wie das Chamäleon der Popkultur: mal niedlich, mal provokant, aber immer irgendwie da, wenn’s chaotisch wird.
Und dann gibt’s da Kek, den alten ägyptischen Gott der Dunkelheit. Du denkst jetzt vielleicht: „Wie kommt ein uralter Frosch-Gott in diese Geschichte?“ Nun, dank der Internet-Community und ihrer unermüdlichen Kreativität wurde Kek quasi wiederbelebt. Als die Gamer „kek“ (eine alternative Schreibweise von „lol“) in ihren Chats verbreiteten und dabei entdeckten, dass Kek auch ein ägyptischer Gott ist, war die Sache klar: Kek wurde zum inoffiziellen Schutzheiligen der absurden Online-Chaos-Community. Und mal ehrlich, wer würde keinen Frosch-Gott für epischen Unfug verehren?
Aber was hat Maximus Decimus Meridius – der unerschütterliche Gladiator und römische General – hier verloren? Nun, Maximus ist sozusagen der OG-Rebell. Mit seiner legendären Rede „Are you not entertained?“ steht er sinnbildlich für den Kampf gegen ein korruptes System und für den Willen, trotz allem die Bühne zu rocken. Und wie es sich für eine Popkultur-Ikone gehört, wurde seine Geschichte in Meme-Form festgehalten. Zack, schon ist er in der Riege der Chaos-Legenden gelandet!
Was diese drei also verbindet, ist ihre unfreiwillige Karriere als Kultfiguren. Sie sind mehr als ihre Ursprünge – Pepe, Kek und Maximus sind Symbole für die Macht der Meme-Kultur, des Widerstands und für den Spaß am völligen Durcheinander. Ein bisschen Chaos, ein bisschen Magie, und voilà: das Trio, das du nie kommen gesehen hast!
Was haben die Drei mit Begriffen wie „kreative Zerstörung“ und „Build Back Better“ zu tun? Beim ersten Hinsehen könnte man meinen: gar nichts, oder? Aber Moment mal! Wenn man genauer hinschaut, merkt man, dass dieses Trio wie geschaffen ist, um genau diese Ideen zu verkörpern: Chaos, Wandel und den Aufbau von etwas Besserem. Klingt wild? Lass mich erklären!
Das Konzept der kreativen Zerstörung ist so alt wie die Zeit selbst: Man muss manchmal etwas Altes abreißen, um Platz für Innovation und Fortschritt zu machen. Und wer könnte das besser symbolisieren als Pepe, Kek und Maximus?
Pepe the Frog zeigt uns, wie Kreativität durch Zerstörung funktioniert. Der harmlose Cartoon-Frosch wurde von der Meme-Kultur so oft umgedeutet, vereinnahmt und recycelt, dass sein ursprünglicher Sinn praktisch ausgelöscht wurde – nur damit er in zahllosen neuen Formen wiedergeboren wird. Chaos? Klar. Innovation? Absolut!
Kek, der ägyptische Gott der Dunkelheit, steht genau dafür: In der Mythologie bringt Dunkelheit den Übergang zur Schöpfung. In der Meme-Welt wird Kek als ironische Gottheit verehrt, die aus purem Chaos und absurdem Humor entstanden ist. Ein vergessener Frosch-Gott wird plötzlich zum Maskottchen des Internets – das ist kreative Zerstörung in ihrer verrücktesten Form!
Und dann ist da Maximus Decimus Meridius, unser Gladiator des Umsturzes. Er stellt das korrupte Römische Reich infrage und bringt das System zum Wanken. Sicher, dabei geht einiges kaputt – inklusive seiner selbst – aber seine Rebellion pflanzt den Samen für etwas Neues, Gerechteres. Kreative Zerstörung? 100 Prozent.
Und genau hier kommt „Build Back Better“ ins Spiel: Nach dem Sturm, nach dem Chaos geht’s nicht einfach zurück zum Alten – es wird neu und besser aufgebaut.
Pepe hat zig Transformationen durchlebt, von einem netten Cartoon-Frosch bis zu einem globalen Symbol für Meme-Kultur und ironische Subversion. Egal wie oft er zerstört wurde, er kommt immer wieder zurück – manchmal lustiger, manchmal seltsamer, aber immer besser als vorher.
Kek lehrt uns, dass aus Chaos Möglichkeiten entstehen. Ein Gott der Dunkelheit, der in der modernen Welt zum Schutzpatron der absurden Kreativität wird? Das ist so „Build Back Better“, dass es fast weh tut.
Maximus schließlich gibt mit seinem Opfer den Anstoß für den Wiederaufbau eines besseren Roms. Er ist der Typ, der den Boden für Veränderung bereitet – der Prototyp eines Neuanfangs.
Pepe, Kek und Maximus zeigen uns, dass Chaos nicht das Ende ist, sondern oft der Anfang von etwas Neuem – und dass man manchmal ein bisschen zerstören muss, um wirklich Großes aufzubauen. Dieses Trio ist der Beweis, dass aus Chaos nicht nur Ordnung, sondern auch eine verdammt gute Geschichte werden kann. Also, wenn das Leben chaotisch wird: Denk dran, vielleicht bist du gerade in der Phase, in der du „Build Back Better“ schreist.
Wenn der reichste Mann der Welt, der auch noch einflussreicher Berater des Präsidenten einer Weltmacht ist, sich plötzlich „Kekius Maximus“ nennt, dann ist das ein hochspannendes Phänomen, das viele Ebenen hat.
Indem unser Milliardär diesen Namen annimmt, signalisiert er: „Ich bin Teil des Spiels. Ich verstehe die Witze der Internet-Generation. Und ich nutze sie, um meine Botschaften zu transportieren.“ Es ist eine Einladung an die digitale Meme-Community: „Kommt, lasst uns das System trollen!“
Der Mann will zeigen: „Hey, ich bin wie ihr. Ich verstehe Spaß!“ Es ist, als ob er sagt: „Ich habe die Macht, und ich werde das bestehende System kreativ zerlegen.“
Es könnte eine verschleierte Ansage an die Mächtigen sein: „Ich bin nicht nur ein reicher Berater – ich bin ein Gamechanger.“
Aber was steckt dahinter – meint er das ernst oder spielt er nur? In der heutigen Welt, wo die Grenzen zwischen Politik, Wirtschaft und Entertainment immer mehr verschwimmen, könnte es beides sein. Vielleicht trollt er einfach nur, weil er es kann. Vielleicht ist es aber auch eine Art Propaganda, die auf subtile Weise seine Vision von Wandel kommuniziert.
Aber eins steht fest – wenn sich jemand mit dieser Macht „Kekius Maximus“ nennt, dann ist das ein Statement – egal, ob es humorvoll oder strategisch gemeint ist. Es signalisiert, dass die Regeln des Spiels sich ändern. Die Meme-Kultur, einst ein harmloses Hobby der Internet-Nerds, ist jetzt ein Werkzeug in den Händen der Mächtigen. Und wenn Kekius Maximus am Steuer sitzt, dann können wir uns sicher sein: Es wird chaotisch, es wird unterhaltsam – und vielleicht ein bisschen beängstigend.
Meine Damen und Herren – bitte begrüßen Sie den unvergleichlichen Elon Musk alias Kekius Maximus.
Quelle X: https://x.com/cyb3rgam3r420/status/1873853864962846843
Das ist ein absoluter Knaller – gleichzeitig faszinierend, provozierend und, typisch Musk, ein durch und durch kalkulierter Schritt.
Ist er Teil des Chaos oder dessen Meister? Kämpft er für den Wandel, indem er kreative Zerstörung bringt – sei es in der Autoindustrie, der Raumfahrt oder den sozialen Medien? Kann er wirklich „einer von uns“ sein und trotzdem der Boss?
Und was ist mit euch, Politiker und Eliten – nehmt ihr euch vielleicht zu wichtig, während er das Spiel mit einer Leichtigkeit beeinflusst, die euch sprachlos macht? Hat das Chaos der Memes am Ende mehr Macht, als ihr zugeben wollt? Gehört die neue Welt wirklich denen, die die Sprache des Internets beherrschen?
Ist er der Architekt des Chaos – oder doch der des Fortschritts?
Es ist ein faszinierender Schachzug. Und während sich die Medien den Kopf zerbrechen – ob er einfach provoziert oder eine tiefere Botschaft hat – und rätseln, was genau sein Ziel sein könnte, stellen wir uns die einfache Frage:
WER ist Elon Musk?
Lassen wir zunächst Elon selbst zu Wort kommen:
„Ich denke, ein Aspekt meines Zustands war, dass ich absolut besessen von der Wahrheit war. Die Besessenheit von der Wahrheit ist der Grund, warum ich Physik studiert habe, denn die Physik versucht, die Wahrheit des Universums zu verstehen. Physik befasst sich mit den beweisbaren Wahrheiten des Universums, Wahrheiten, die Vorhersagekraft besitzen. Für mich war es daher ganz natürlich, Physik zu studieren. Niemand hat mich dazu gezwungen. Es war von sich aus faszinierend, die Natur des Universums zu verstehen. Und dann kam die Informatik oder Informationstheorie dazu, um einfach Logik zu verstehen. Es gibt ein Argument, dass die Informationstheorie auf einer noch fundamentaleren Ebene arbeitet als die Physik selbst. Deshalb waren Physik und Informationstheorie für mich wirklich interessant.“ [TED-Interview, 14. April 2022]
(Quelle X: https://x.com/ElonClipsX/status/1874785186384101701)„Man muss vorsichtig sein, wenn man sich etwas wünscht, das gut klingt, aber wenn man es bekommt, ist es in Wirklichkeit eine dystopische Situation. Man könnte eine Hypothese aufstellen: Wenn man sich den Weltfrieden wünscht, hört sich das gut an, aber wie wird er durchgesetzt? Um welchen Preis? Ewiger Frieden? Es könnte sogar schlimmer sein, ewigen Frieden zu haben, denn was würde das mit sich bringen? Es könnte die Unterdrückung des Fortschritts sein. Es könnte eine verknöcherte Gesellschaft sein, die sich nie verändert. Es gibt das Argument, dass man, wenn man sich keinen Krieg wünscht, vorsichtig sein sollte, was man sich wünscht, weil das, was nötig ist, damit es keinen Krieg gibt, schlimmer sein könnte als ein kleiner Krieg.“ [Lex Fridman Podcast, November 2023]
(Quelle X: https://x.com/MarioNawfal/status/1873705582877691908)„Ich glaube, wir gehen im Allgemeinen zu sehr von der Annahme aus, dass die Zivilisation robust ist und nichts sie wirklich zerstören kann. Ein Gefühl, das im Laufe der Geschichte bei den Imperien kurz vor ihrem Zerfall üblich war, denn im Moment herrscht ein wenig die Stimmung eines späten Imperiums.“ [WSJ, CEO Council, Mai 2023]
(Quelle X: https://x.com/ElonClipsX/status/1873659651625296140)„Was wir nicht tun werden, ist zu sagen, dass es eine gesalbte Klasse von Journalisten gibt, die die besonderen sind, die jedem sagen dürfen, was sie denken sollen. Es sollte den Menschen überlassen bleiben, was sie denken. Und selbst wenn ein Artikel völlig korrekt und umfassend ist, bestimmen die Medien mit dem Verfassen des Artikels das Narrativ. Sie entscheiden, worüber sie einen Artikel schreiben wollen. Ich hoffe also, dass hier eher die Öffentlichkeit das Narrativ wählt und nicht die Medien. Zumindest eine Kombination aus den Medien und der Öffentlichkeit, die das Narrativ bestimmen, und der Öffentlichkeit, die sich in die Geschichten einmischt, wenn sie der Meinung ist, dass sie etwas hinzufügen sollte, oder dass wir etwas falsch verstanden haben. Und ich denke, wenn Twitter die beste Quelle für die Wahrheit ist, wird es sich mit der Zeit durchsetzen. Und wenn wir nicht das Gefäß der Wahrheit sind, werden wir scheitern.“ [Interview with the BBC, April 12, 2023]
(Quelle X: https://x.com/ElonClipsX/status/1870477764291387476)„Wir wollen, dass Optimus mit der Zeit zu einem Androiden wird, wie man ihn aus Science-Fiction-Filmen wie Star Trek: The Next Generation kennt, so wie Data. Aber natürlich können wir den Roboter so programmieren, dass er weniger roboterhaft und freundlicher ist. Und er kann natürlich lernen, Menschen zu imitieren und sich sehr natürlich zu fühlen. Wenn sich die künstliche Intelligenz im Allgemeinen verbessert, können wir das auch auf den Roboter übertragen. Er sollte natürlich in der Lage sein, einfache Anweisungen auszuführen oder sogar intuitiv zu erkennen, was man möchte. Man könnte ihm also eine übergeordnete Anweisung geben, die er dann in eine Reihe von Aktionen aufschlüsseln und ausführen kann.“ [Tesla AI Day, 30. September 2022]
(Quelle X: https://x.com/ElonClipsX/status/1871108253901287712)Lex: „Glauben Sie, dass wir jemals ein KI-System erschaffen werden, das wir lieben können und das uns auf eine tiefe, bedeutungsvolle Weise zurückliebt, wie in dem Film Her?“
Elon: „Ich denke, dass die KI in der Lage sein wird, dich zu überzeugen, dich in sie zu verlieben.“
Lex: „Und das ist anders als bei uns Menschen?“
Elon: „Wir kommen hier zu einer metaphysischen Frage: Existieren Gefühle und Gedanken in einem anderen Bereich als der physischen Welt? Vielleicht tun sie das, vielleicht auch nicht, ich weiß es nicht. Aber ich neige dazu, die Dinge vom Standpunkt der Physik aus zu betrachten. Wenn es dich auf eine Art und Weise liebt, dass du nicht sagen kannst, ob es real ist oder nicht, dann ist es im Grunde genommen real.“
[Lex Fridman Podcast, 12. April 2019]
(Quelle X: https://x.com/ElonClipsX/status/1869751738229522531)Mathias Döpfner: „Sie sagten, dass Neuralink unter all Ihren Projekten das Wichtigste für Sie ist. Stimmt das noch?“
Elon Musk: „Ich sagte, dass es das sein könnte. Ich würde nicht mit Sicherheit sagen, dass es das wichtigste Projekt ist, aber es könnte das Wichtigste sein, weil es eine langfristige Abschwächung der künstlichen Intelligenz sein könnte. Wir könnten effektiv mit der künstlichen Intelligenz verschmelzen, indem wir die Geschwindigkeit der Interaktion zwischen unserem Kortex und unserer Tertiärschicht, die bereits aus Silizium besteht, verbessern.“
[Interview mit der Welt, 15. April 2022]
(Quelle X: https://x.com/ElonClipsX/status/1867253735535104012)„Ich hatte einige Berührungspunkte mit den Ausgaben der Regierung, weil SpaceX viele Regierungsverträge hat und viel für die NASA und das Verteidigungsministerium arbeitet und so weiter. Und so habe ich gesehen, wie viel Verschwendung es gibt. Wenn man sich mit Leuten in der Regierung unterhält, sind sie sich einig: Ja, das ist sehr verschwenderisch und ineffizient. Und ich frage mich, warum tun wir nicht etwas dagegen? Aber um wirklich etwas dagegen zu unternehmen, muss es ein Mandat von oben geben.“ [Harrisburg, Pennsylvania, October 19, 2024]
(Quelle X: https://x.com/ElonClipsX/status/1869312936520409431)„Ich habe mit Trump die Idee einer Regierungseffizienzkommission diskutiert. Die Antikörperreaktion wird sehr stark sein. Sie greifen die Matrix an diesem Punkt an. Die Matrix wird sich wehren.“ [Lex Fridman Podcast, 2. August 2024]
(Quelle X: https://x.com/ElonClipsX/status/1867924908715520429)„Das Tolle an Trump ist, dass wir eine echte Persönlichkeit haben, die niemandem verpflichtet ist. Das ist es, was die Maschine erschreckt. Und deshalb versucht die Maschine, ihn zu töten.“ [Lancaster, Pennsylvania, Oktober 2024]
(Quelle X: https://x.com/ElonClipsX/status/1871162235332289017)„Diejenigen, die sich in der Matrix befinden, haben noch nicht verstanden, dass die Matrix umprogrammiert werden kann – und dass dies der einzige Weg zum Sieg ist.“
(Quelle X: https://x.com/elonmusk/status/1867679319818211500)
Das TIME Magazine kürte Elon Musk bereits im Jahr 2021 zur Person des Jahres. Man beschreibt ihn als: „Das ist der Mann, der davon träumt, unseren Planeten zu retten und uns einen neuen zum Besiedeln zu beschaffen: Clown, Genie, Provokateur, Visionär, Industrieller, Showman, Schurke; eine schräge Mischung aus Thomas Edison, P.T. Barnum, Andrew Carnegie und Watchmen’s Doctor Manhattan – dem grübelnden, blauhäutigen Gottmenschen, der Elektroautos erfindet und zum Mars zieht.“
Ohne Zweifel ist Elon Musk eine komplexe Persönlichkeit mit einem ebenso facettenreichen Hintergrund, der unter anderem auch die Teilnahme an dem WEF Young Global Leader Programm umfasst. Seine Vielschichtigkeit wurde im Februar 2023 auch von den Organisatoren des „Weltregierungsgipfels“ (World Government Summit, WGS) erkannt, die ihn als Redner zu dieser renommierten Veranstaltung einluden. Der jährliche Gipfel bringt Führungskräfte, Regierungsvertreter und Experten aus der ganzen Welt zusammen, um globale Themen wie Regierungsführung, Technologie und Gesundheit zu diskutieren.
Während seines Auftritts warnte Musk eindringlich vor den Risiken einer unkontrollierten künstlichen Intelligenz (KI). Er bezeichnete KI als „eines der größten Risiken für die Zukunft der Zivilisation“. Gleichzeitig hob er hervor, wie seine Social-Media-Plattform Twitter (heute X) von Regierungen genutzt werden könnte, um effektiver und authentischer mit der Bevölkerung zu kommunizieren. Dabei betonte er auch die Bedeutung, Kritik an der Regierungspolitik in den Dialog einzubeziehen.
Musk sprach offen darüber, dass viele Social-Media-Plattformen stark von Algorithmen geprägt sind, die im Silicon Valley entwickelt wurden. Parallel dazu äußerte er deutliche Bedenken gegenüber dem Konzept einer globalen Weltregierung, da er potenzielle Gefahren für die Autonomie einzelner Nationen sieht.
Das vollständige Interview ist unter diesem Link verfügbar.
Versucht Elon die Quadratur des Kreises zu vollbringen, indem er im Spannungsfeld zwischen Systemkritik und systemischem Einfluss operiert?
Auf der einen Seite stellt sich Musk klar gegen die „Matrix“, eine übermächtige, zentralisierte Weltregierung, die Individuen kontrolliert. Gleichzeitig bietet er jedoch über Twitter Tipps an, wie genau diese „Matrix-Protagonisten“ – sprich Regierungen – effektiver mit den „Untertanen“ kommunizieren können.
Das fühlt sich an, als würde jemand einem Löwen erklären, wie er besser jagt, während er gleichzeitig für den Schutz der Antilopen plädiert.
Musk agiert innerhalb der bestehenden Strukturen, um sie zu ändern, statt sie frontal anzugreifen. Indem er der „Matrix“ Ratschläge gibt, wie sie Twitter (X) besser nutzen kann, platziert er sich als unverzichtbaren Akteur – und bleibt gleichzeitig derjenige, der die Regeln auf der Plattform kontrolliert.
Die Matrix (als Metapher) soll nicht abgeschafft, sondern „entmachtet“ werden, indem sie durch ein System ersetzt wird, das eher an verteilte Netzwerke erinnert.
Die Ironie ist, dass Musk selbst zum Architekten eines neuen Systems wird – und das könnte ebenso kritisch betrachtet werden wie die alte „Matrix“, die er bekämpft. Der Unterschied? Seine „Matrix“ verkauft sich als rebellisch und frei, was sie umso verführerischer macht.
Was steckt hinter Elons System?
Das aktuelle Vermögen von Elon Musk wird auf etwa 421 Milliarden US-Dollar (Stand: 2. Januar 2025) geschätzt und klassifiziert ihn momentan als den reichsten Mann der Welt. Elon Musk ist maßgeblich an mehreren Unternehmen beteiligt. Betrachten wir genauer einige der Firmen, bei denen Musk eine zentrale Rolle spielt – sei es als Gründer, CEO oder größter Anteilseigner.
Eine kurze und dennoch umfassende Übersicht über die Kernkompetenzen von Tesla liefern diese kurzen Videos.
Quelle X: https://x.com/Tesla/status/1870121074072977651 Quelle X: https://x.com/Tesla_Optimus/status/1846797392521167223 Quelle X: https://x.com/teslaownersSV/status/1714365963146313843 So beschreibt Tesla auf seiner Homepage seine Mission: „Wir entwickeln und implementieren Autonomie in großem Maßstab in Fahrzeugen, Robotern und mehr. Wir sind davon überzeugt, dass ein auf fortschrittlicher KI basierender Ansatz für Vision und Planung, der durch den effizienten Einsatz von Inferenz-Hardware unterstützt wird, der einzig gangbare generelle Lösungsweg für autonomes Fahren und mehr darstellt.“
Tesla entwickelt eigene Hochleistungs-KI-Chipsätze, die nicht nur auf das sehr schnelle Verarbeiten von sehr großen Datenmengen ausgelegt sind, sondern auch speziell auf Maschinelles Lernen getrimmt werden. Damit baut TESLA heutzutage einen der schnellsten Supercomputer für Künstliche Intelligenz. Weiterhin spezialisiert sich die Firma darauf, „tiefe neuronale Netzwerke auf Probleme von der Wahrnehmung bis zur Kontrolle zu trainieren“. … „Unsere Netzwerke lernen von den kompliziertesten und vielfältigsten Szenarien der Welt und beziehen sie iterativ aus unserer Flotte von Millionen von Fahrzeugen in Echtzeit.“
„Wir konzentrieren uns nicht speziell auf AGI (Allgemeine künstliche Intelligenz), aber es scheint eine aufkommende Eigenschaft dessen zu sein, was wir tun. Mit Millionen von autonomen Fahrzeugen und humanoiden Robotern, die chaotische Daten aus der realen Welt sammeln und verarbeiten, werden sie menschliche Fahrer übertreffen und Humanoide werden vielleicht nicht mehr von Menschen zu unterscheiden sein. Dieser riesige Datenstrom aus der realen Welt könnte natürlich zu AGI führen“, erklärte Elon Musk während des TESLA AI Day am 30. September 2022.
„Die Maschine, die die Maschine baut“, lautet einer der Slogans auf der TESLA Webseite.
Bemerkenswert ist die enge Übereinstimmung zwischen Teslas strategischer Ausrichtung und dem Fokus des Weltwirtschaftsforums (WEF) auf die Entwicklung und Förderung von Künstlicher Intelligenz. Dies wird durch Verweise hier und hier belegt.
Tesla ist ein börsennotiertes Unternehmen. Zu den größten Anteilseignern zählen neben Elon Musk auch The Vanguard Group und BlackRock Inc.

Quelle: Aktionärsverteilung bei TESLA Inc. (Stand 03.01.2025) The Vanguard Group gehört selbst zu den größten Anteilseignern bei BlackRock. BlackRock wiederum gehört zu den Partnerunternehmen des Weltwirtschaftsforums.
Beide Unternehmen sind schließlich weltweit führende Vermögensverwalter und besitzen bedeutende Anteile an zahlreichen multinationalen Konzernen, darunter Tech-Giganten, Banken, Energie- und Pharmaunternehmen, wodurch sie faktisch einen gewaltigen Einfluss auf die Wirtschaft und Politik ausüben.
Es handelt sich um große, zentralisierte Institutionen, die Stabilität und Kontrolle über ein schwer durchschaubares globales System gewährleisten. Die Besitzstrukturen und die Art, wie Einfluss ausgeübt wird, sind für den Durchschnittsmenschen kaum nachvollziehbar. Wer wirklich „entscheidet“, bleibt oft verborgen.
Metaphorisch sind sie wie die Architekten der Matrix – sie agieren oft im Hintergrund, aber ihre Entscheidungen prägen die Wirtschaft und damit auch die Gesellschaft enorm.
Die Tatsache, dass BlackRock und Vanguard, oft als Symbole der „alten Matrix“ angesehen und zugleich zu den größten Anteilseignern von Tesla gehören, macht die Sache besonders spannend. Denn hier treffen zwei scheinbar gegensätzliche Kräfte aufeinander: Die Architekten einer zentralisierten Ordnung finanzieren einen Akteur, der sich als Rebell gegen eben diese Ordnung inszeniert. Wie passt das zusammen?
BlackRock und Vanguard sind pragmatische Akteure. Ihre Hauptmission ist es, Renditen zu erzielen, und Tesla ist für sie schlichtweg eine kluge Investition. Das Unternehmen steht für Zukunftstechnologie und verspricht Wachstum – genau das, was die „Matrix“ braucht, um relevant zu bleiben. Doch mit ihrer finanziellen Beteiligung an Tesla halten diese Institutionen auch einen Fuß in der Tür. Sie behalten so Einfluss auf das, was Musk und Tesla tun, und sichern sich die Möglichkeit, den Fortschritt zumindest mitzulenken.
Gleichzeitig positioniert sich Elon Musk als Anti-Matrix-Rebell, der Chaos und Innovation als Werkzeuge nutzt, um alte Strukturen aufzubrechen. Doch auch Musk braucht Kapital, um seine Visionen umzusetzen. Ohne die Milliarden von BlackRock und Vanguard hätte Tesla wohl nie die Größe und den Einfluss erreicht, den es heute hat. Das führt zu einem paradoxen Verhältnis: Während Musk gegen die alte Ordnung kämpft, wird sein Erfolg von genau dieser Ordnung mitfinanziert.
Metaphorisch ist das wie eine Szene aus dem Film „Matrix“: BlackRock und Vanguard agieren als Architekten des Systems, die sicherstellen, dass selbst Rebellen wie Musk in ihrem Einflussbereich bleiben. Musk wiederum ist wie ein moderner Neo – ein vermeintlicher Systemgegner, der trotzdem Teil des Spiels bleibt.
Diese Verbindung zeigt auch, wie wandelbar die „Matrix“ ist. BlackRock und Vanguard haben gelernt, sich nicht nur auf die Stabilisierung der alten Ordnung zu konzentrieren, sondern auch neue, chaotische Kräfte zu integrieren. So sichern sie ihre eigene Relevanz und bleiben ein unverzichtbarer Teil des globalen Systems. Musk hingegen wird, ob er will oder nicht, zu einem Zahnrad in dieser Maschine, denn ohne das Kapital dieser Akteure wäre seine Revolution schwer finanzierbar.
Am Ende bleibt die Frage: Wer spielt hier eigentlich wen aus? Ist Musk das Trojanische Pferd, das von der „Matrix“ finanziert wird, um das System von innen heraus zu ändern? Oder sind BlackRock und Vanguard die wahren Meister des Spiels, die wissen, dass sie von jedem Wandel profitieren werden? Wie in der echten Matrix bleibt die Wahrheit irgendwo zwischen den Zeilen verborgen – und das macht das Spiel umso faszinierender.
SPACE X
(Space Exploration Technologies Corp.)„SpaceX hat weltweit Aufmerksamkeit durch eine Reihe historischer Meilensteine erlangt. Es ist das einzige private Unternehmen, das in der Lage ist, ein Raumschiff aus dem niedrigen Erdorbit zurückzubringen. SpaceX ist der Überzeugung, dass eine vollständig und schnell wiederverwendbare Rakete der entscheidende Durchbruch ist, um die Kosten für den Zugang zum Weltraum erheblich zu senken. Die Falcon-Raketenfamilie von SpaceX sind die ersten und einzigen orbitalen Raketen ihrer Klasse, die wiederverwendet werden können“, wie auf der Unternehmenshomepage zu lesen ist.
DIE MENSCHHEIT MULTIPLANETAR MACHEN lautet die Devise.
Quelle X: https://x.com/SpaceX/status/1875218268857958468 SpaceX betreibt außerdem ein eigenes Satellitennetzwerk unter dem Namen Starlink. Mit 6.697 Satelliten im Erdorbit (Stand: Juli 2024) ist das Unternehmen der weltweit größte Betreiber von Satelliten. Insgesamt verfügt SpaceX über Genehmigungen für den Start von bis zu 19.427 Satelliten und hat zusätzlich Anträge für den Betrieb von weiteren 22.488 Satelliten gestellt.
„Starlink ist die weltweit erste und größte Satellitenkonstellation, die eine niedrige Erdumlaufbahn nutzt, um Breitband-Internet zu liefern, das Streaming, Online-Gaming, Videoanrufe und mehr ermöglicht. Durch den Einsatz fortschrittlicher Satelliten und Benutzerhardware in Verbindung mit unserer langjährigen Erfahrung mit dem Betrieb von Raumfahrzeugen und Arbeiten in der Erdumlaufbahn liefert Starlink-Highspeed-Internet mit geringer Latenz für Benutzer auf der ganzen Welt“, verkündet das Unternehmen auf seiner Internetpräsentation.
Dieses Werbevideo zeigt eindrucksvoll, wie die kommende Generation der Starship-Trägerraketen von SpaceX künftig noch schneller und effizienter Starlink-Satelliten in die Erdumlaufbahn transportieren wird:
Quelle X: https://x.com/Starlink/status/1874123729950958075/video/2 Im vergangenen Jahr wurde Starlink in 27 weiteren Märkten eingeführt und erreicht nun ein globales Gebiet mit einer Bevölkerung von 2,8 Milliarden Menschen, darunter auch Menschen in einigen der entlegensten Regionen der Erde. Die folgende Animation veranschaulicht diesen Fortschritt.
Quelle X: https://x.com/Starlink/status/1874118714217685306 Voller Begeisterung kann man feststellen, dass SpaceX und Starlink die Welt verändern! SpaceX revolutioniert die Raumfahrt mit wiederverwendbaren Raketen, die den Zugang zum Weltraum günstiger und nachhaltiger machen. Visionäre Projekte wie die Mars-Kolonisation rücken die interplanetare Zukunft in greifbare Nähe. Gleichzeitig sorgt Starlink dafür, dass schnelles Internet selbst die abgelegensten Winkel der Erde erreicht. Das bedeutet Bildung, wirtschaftliche Chancen und lebensrettende Kommunikation – besonders in Krisengebieten. Ob technologische Innovationen, Umweltforschung oder globale Vernetzung: Diese Projekte treiben den Fortschritt mit einem Tempo voran, das uns optimistisch in die Zukunft blicken lässt!
Wo Licht ist, ist auch Schatten!
Ohne großes Aufsehen stellte SpaceX Anfang Dezember 2022 sein Starshield-Konzept vor. Starshield wurde vom globalen Kommunikationsnetzwerk Starlink adaptiert, um den USA und ihren Verbündeten erweiterte militärische Raumfahrtfähigkeiten zu bieten. Dazu gehören Funktionen wie Zielverfolgung, optische und Funkaufklärung sowie Raketenfrühwarnung. Zu den Hauptkunden zählen die Space Development Agency, das National Reconnaissance Office und die United States Space Force. Bis 2024 wurden mindestens 98 Starshield-Satelliten gestartet. Weitere 17 Satelliten sollten im Oktober 2024 ins All gebracht werden.
Ein zentraler Vorteil der Starshield-Architektur liegt in ihrer dezentralen Struktur: Die große Anzahl kleiner Satelliten ermöglicht eine globale Kommunikationsabdeckung. Im Gegensatz zum kommerziellen Starlink-Dienst werden die Starshield-Satelliten jedoch vollständig im Besitz der US-Regierung sein und unter deren Kontrolle stehen, berichtet SpaceNews in ihrem Beitrag „Pentagon embracing SpaceX’s Starshield for future military satcom“.
„Während die Präsidentin und COO von SpaceX, Gwynne Shotwell, angedeutet hat, dass es nur wenige Informationen gibt, die sie über Starshield preisgeben darf, hat sie eine „sehr gute Zusammenarbeit“ zwischen der Geheimdienstgemeinschaft und SpaceX bei dem Programm festgestellt.“ [Wikipedia]
Gwynne Shotwell, oft als die heimliche Chefin von SpaceX bezeichnet, pflegt enge Beziehungen zum World Economic Forum (WEF). Während Elon Musk der größte Anteilseigner des Unternehmens ist, zählen der Founders Fund von Peter Thiel, Fidelity Investments und Google zu den bedeutenden Investoren – allesamt mit Verbindungen zum WEF.
Das World Economic Forum (WEF) wird oft als Symbol für die „alte Matrix“ gesehen – ein globales Netzwerk, das die bestehende Ordnung sichert und durch Koordination von Politik und Wirtschaft die Fäden im Hintergrund zieht. Die Tatsache, dass führende Personen und Investoren von SpaceX Verbindungen zum WEF haben und dass das Starshield-Projekt militärische und geheimdienstliche Ziele verfolgt, passt erstaunlich gut in die Metapher der „Matrix“.
SpaceX könnte als eine Art „Schlüsselprogramm“ betrachtet werden. Es schafft Technologien, die bestehende Machtstrukturen herausfordern könnten – etwa durch Starlink als Symbol für globale Internetfreiheit. Gleichzeitig liefert es mit Projekten wie Starshield Werkzeuge, die die Kontrolle der Matrix stärken.
Am Ende bleibt das Rätsel: Ist Musk ein echter Systemgegner, oder ist seine Rebellion nur ein weiteres Programm innerhalb der Matrix, das letztlich dazu beiträgt, ihre Macht zu festigen?
X – früher bekannt als Twitter – ist wie der rebellische Cousin aller anderen Social Media-Plattformen: Es kommt in schlichten Schwarz-Weiß-Tönen daher, ohne zu viele Filter und ohne zu viel Schnickschnack, und sorgt ständig für Aufregung. Aber was macht X so besonders?
Erstens, die Regeln? Gibt’s nicht wirklich! Musk ist der DJ und mixt alles, was er will. Freie Meinungsäußerung? Klar, aber auf seine Art. Jeder kann sich äußern – vom Milliardär bis zum Meme-König.
Zweitens, die Schlagzeilen? X ist die Quelle für alles, was in Echtzeit passiert, und das bedeutet auch: Du bekommst nicht nur das, was „die etablierten Medien“ dir sagen wollen. Du kannst die „großen“ Themen direkt von den Quellen hören, ohne die Zensur-Schicht dazwischen. Wer braucht schon Mainstream, wenn du direkt in den Witz und das Chaos einsteigen kannst?
Und das Beste? Memes sind das wahre Kapital. Auf X regiert das Chaos, und das Chaos ist lustig – oft mit einer kleinen Portion Anarchie gewürzt. Es ist die Plattform, auf der nicht nur die Nachrichten verbreitet werden, sondern die wirklich interessanten Dinge passieren: Kurz, schockierend und direkt aus dem Dschungel des digitalen Wahnsinns!
„Im Jahr 2024 hat X die Welt verändert. Jetzt bist DU die Medien! 2025 wird X dich auf eine Weise verbinden, die nie für möglich gehalten wurde. X TV, X Money, Grok und mehr. Schnall dich an“, verkündet der CEO von X, Linda Yaccarino, zum Jahreswechsel.
Manche X-Nutzer nennen Elon Musk die „Message-Maschine“ – und das nicht ohne Grund. Mit einer durchschnittlichen Postfrequenz von 100 Posts pro Tag nutzt er die Plattform auch, um seine Meinung zu teilen – was natürlich für jede Menge Aufmerksamkeit und hitzige Debatten sorgt. Kein Wunder, dass seine Posts eine gigantische Reichweite haben und er zu den einflussreichsten Nutzern der Plattform gehört.
Seine Kommentare haben politische Wellen geschlagen – von seiner Unterstützung für Figuren wie Donald Trump bis hin zur Promotion der AfD in Deutschland. Musk hat sogar angekündigt, den Algorithmus von X zu überarbeiten, um informativen und unterhaltsamen Content stärker zu pushen, was zeigt, dass er die Kontrolle über den Inhalt aktiv in die Hand nimmt.
Doch Musks Einfluss geht weit über die Plattform hinaus – er mischt auch ordentlich in der politischen Landschaft mit. Besonders in den USA wird seine Unterstützung für bestimmte Kandidaten und Parteien oft als Wahleinmischung wahrgenommen, was für Diskussionen sorgt, wie Social Media den politischen Prozess beeinflusst.
Natürlich bleibt Musk nicht ohne Kritik. Viele werfen ihm vor, mit seinen Aussagen und Entscheidungen Desinformation und Hassrede zu fördern – was zu einem Rückzug von Werbekunden und Auseinandersetzungen mit Regulierungsbehörden und der breiten Öffentlichkeit führt.
Und dann gibt es noch Musks Vision, X zur „Everything-App“ zu machen, was die Plattform tiefgreifend verändern könnte. In der Theorie klingt die Everything-App nach der Zukunft, in der alles reibungslos zusammenläuft – von sozialen Netzwerken, Bankgeschäften, Shopping, Nachrichten, Musik, Videos, Kommunikation, bis zu den neuesten Memes. Doch während Musk an seiner Vision bastelt, stellen sich viele die Frage, ob der Preis der Bequemlichkeit nicht zu hoch ist. Könnte es sein, dass diese „Alleskönner-App“ am Ende nur eine Falle für unsere Daten und Privatsphäre ist?
Elon Musk ist weit mehr als der Eigentümer von X – er ist die treibende Kraft, das Gesicht und das Symbol von X Corp. Als „Frontman“ kanalisiert er Aufmerksamkeit, prägt die Strategie und macht X zu einem Spiegelbild seiner eigenen Ambitionen und Ideale.
Chaos ist bei X kein Bug, sondern ein Feature. Aber sogar in dem Chaos gibt es Details, die auf bestimmte Gesetzmäßigkeiten hindeuten.
Linda Yaccarino, die CEO von X, hat enge Verbindungen zum World Economic Forum (WEF), wo sie die Task Force für die Zukunft der Arbeit leitete. Während der ersten Präsidentschaft von Donald Trump wurde sie in den President’s Council on Sports, Fitness and Nutrition berufen. Im Jahr 2021 arbeitete sie als Vorsitzende des Board of Directors der Werbeagentur Ad Council zusammen mit der Biden-Regierung an einer Impfkampagne gegen SARS-CoV-2, an der auch Papst Franziskus mitwirkte.
Zu den prominenten Investoren von X zählt der WEF-Partner Kingdom Holding des saudischen Prinzen und Geschäftsmanns Prinz Alwaleed bin Talal.
Ein weiteres interessantes Detail steckt in der sogenannten Verifizierungsrichtlinie von X. Dort ist unter anderem folgendes nachzulesen:
„X möchte für bestimmte X Funktionen eine freiwillige Identitätsverifizierung bereitstellen und dadurch auf unserer Plattform allgemein für mehr Integrität und Vertrauen sorgen. Wir erfassen diese Daten, wenn Abonnent*innen von X Premium sich optional für die Beantragung eines Verifizierungsabzeichens entscheiden, indem wir ihre Identität anhand ihres amtlichen Ausweises verifizieren. Außerdem werden möglicherweise in Zukunft zusätzliche Vorteile für bestimmte X Funktionen freigeschaltet.“
„Unter bestimmten Umständen kann X die Vorlage eines Ausweises verlangen, wenn das für die Sicherheit von Accounts auf unserer Plattform erforderlich ist. Derzeit konzentriert sich X bei der Account-Authentifizierung auf die Verhinderung von Identitätsbetrug. Möglicherweise weiten wir dies auf zusätzliche Bereiche aus, z. B. können wir sicherstellen, dass Nutzer*innen Inhalte sehen, die für ihr Alter angemessen sind, vor Spam und böswilligen Accounts schützen, die Integrität der Plattform aufrechterhalten und für sinnvolle Konversationen sorgen.“
Es deutet darauf hin, dass X in Zukunft spezielle Funktionen und möglicherweise auch den allgemeinen Zugang verstärkt von einer Identitätsverifizierung abhängig machen könnte, unter Berufung auf Sicherheits- und Integritätsgründe.
„Für Nutzer*innen, die die Identitätsverifizierung abschließen, erfassen wir ein Bild des Ausweises und das Selfie. Das umfasst Gesichtsdaten sowie Daten, die aus dem Ausweis extrahiert werden. X bewahrt diese Daten nicht direkt auf. Wir leiten die Gesichtsdaten an einen Dritten, Au10tix, weiter, der als unser Datenverarbeiter fungiert.“
Bei Wikipedia kann man folgende Informationen über Au10tix finden:
„AU10TIX ist ein Unternehmen für Identitätsprüfung und Risikomanagement mit Sitz in Hod HaSharon, Israel. Die Produkte des Unternehmens ermöglichen Unternehmen die sichere Aufnahme und Überprüfung von Kunden. AU10TIX verfügt über ein automatisiertes globales Identitätsmanagementsystem sowie eine Lösung zur Erkennung organisierter Massenangriffe auf Identitätsbetrug. AU10TIX ist eine Tochtergesellschaft von ICTS International NV.“
ICTS International ist bei Wikipedia wie folgt beschrieben:
„ICTS International N.V. ist ein niederländisches Unternehmen, das Produkte entwickelt und Beratungs- und Personaldienstleistungen im Bereich der Luftfahrt und der allgemeinen Sicherheit anbietet. Es wurde 1982 von ehemaligen Mitgliedern des Shin Bet, der israelischen Agentur für innere Sicherheit, und Sicherheitsbeamten der Fluggesellschaft El Al gegründet.“
Shin Bet ist bekanntlich eine der drei wichtigsten Organisationen des israelischen Geheimdienstes.
Saudi-arabisches Kapital trifft auf Shin Bet unter dem Schirm des WEF – das ist die Matrix in ihrer vollen Blüte. Was wie eine unwahrscheinliche Allianz klingt, könnte kaum besser zusammenpassen, wenn man die globale Machtlandschaft versteht. Hier verschmelzen Geld, Geheimdienste und geopolitische Interessen zu einem komplexen Geflecht. X, als Plattform des Chaos, wird so gleichzeitig ein strategischer Punkt innerhalb eines viel größeren Spiels – wo Kapital auf Überwachung trifft, alles unter dem unsichtbaren Mantel des WEF.
Neuralink ist eines der ambitioniertesten Projekte von Elon Musk, das sich der Aufgabe widmet, Mensch und Maschine miteinander zu vereinen. Gegründet 2016, arbeitet das Unternehmen an der Entwicklung von Brain-Computer-Interfaces (BCIs) – also Technologien, die das menschliche Gehirn direkt mit Computern verbinden.
Das folgende Werbevideo von Neuralink gibt eine Andeutung dessen, was damit gemeint ist:
Das Unternehmen strebt an, durch diese Technologie verschiedene neurologische Erkrankungen wie Lähmungen, Blindheit, Gedächtnisverlust und sogar psychische Störungen wie Depressionen und Angstzustände zu behandeln oder zu heilen.
Im Januar 2024 wurde bekannt, dass Neuralink erfolgreich ein Implantat in einen Menschen eingesetzt hat. Der erste Patient, Noland Arbaugh, konnte danach mit seinen Gedanken einen Computer steuern, was eine enorme Verbesserung seiner Lebensqualität darstellte, da er querschnittsgelähmt war.
Das Unternehmen sucht derzeit weitere Probanden mit bestimmten Erkrankungen, die an der klinischen Studie zu seiner Gehirn-Computer-Schnittstelle teilnehmen möchten.
Quelle X: https://x.com/neuralink/status/1727135227565547810 „Um die Grenzen menschlicher Fähigkeiten neu zu definieren, braucht es Pioniere“, verkündet Neuralink auf deren Homepage.
Welchen Weg diese Pioniere ebnen sollen, erklärt Elon in unmissverständlicher Weise:
„Langfristig strebt Neuralink eine Symbiose mit künstlicher Intelligenz und eine Art Demokratisierung der Intelligenz an, so dass sie nicht in rein digitaler Form von Regierungen und Großunternehmen monopolisiert wird.
Wie können wir sicherstellen, dass die Zukunft die Summe des Willens der Menschheit darstellt? Wenn wir Milliarden von Menschen haben, die über eine hohe Bandbreite mit der KI-Erweiterung von sich selbst verbunden sind, würde das tatsächlich jeden hypersmart machen.“ [Interview mit Axios, 2018]
(Quelle X: https://x.com/ElonClipsX/status/1851719129625223582)Im WEF-Beitrag „Ready for Brain Transparency“ wird ziemlich deutlich, wie die Monopolisierung von Künstlicher Intelligenz in einer Welt der Hirntransparenz durch Brain-Computer-Schnittstellen aussehen könnte. Der Gedanke, dass Ihr Chef Ihre Gehirnströme ausspionieren kann, ist nicht mehr Science Fiction – es wird als eine greifbare Zukunft dargestellt, in der unsere innersten Gedanken zur nächsten Überwachungsressource werden. Diese Vorstellung ruft erschreckend die alte Matrix hervor, die uns weiterhin überwacht, nur jetzt mit noch mehr Präzision und Kontrolle.
Im Gegensatz dazu präsentiert Elon Musk mit Neuralink die Vision eines Ausbruchs aus dieser Matrix. Er träumt von einer Welt, in der der Mensch mit KI verschmilzt, um seine Intelligenz zu erweitern – eine Welt, in der keine Großkonzerne oder Regierungen das Monopol auf Wissen und Macht haben. Klingt nach der ultimativen Anti-Matrix, oder? Doch auch hier stellt sich die Frage: Wird diese Befreiung wirklich zu echter Freiheit führen oder schaffen wir uns lediglich eine neue, raffinierte Form der Kontrolle?
Wer kontrolliert die „hohe Bandbreite“, die Musk propagiert? Auch wenn diese Technologie theoretisch für alle zugänglich ist, gibt es immer noch denjenigen, der den Stecker zieht und bestimmt, wie diese Schnittstellen funktionieren. Musk träumt von einer kollektiven „Summe des Willens der Menschheit“, doch was, wenn diese Werkzeuge, die uns verbinden, gleichzeitig unsere Handlungen und Gedanken lenken? Was, wenn die Symbiose mit KI uns nicht befreit, sondern uns noch tiefer in eine kollektive Abhängigkeit führt?
Die Vision, dass wir alle „hypersmart“ werden, klingt wie ein Paradies – doch wie viel Freiheit bleibt, wenn die KI nicht nur unser Wissen, sondern auch unsere Entscheidungen beeinflusst? Am Ende könnte die Realität so aussehen, dass wir in einer Matrix leben – nur dass sie sich nicht mehr als Gefängnis anfühlt, sondern als eine, die uns das Gefühl von Freiheit gibt. Musks Traum, die Matrix zu durchbrechen, könnte uns lediglich eine neue, besser verpackte Version derselben Matrix bieten.
Wenn es einen Menschen auf dieser Erde gibt, der Elon Musk gut kennt, dann ist es der Schriftsteller, Journalist und Biograph Walter Isaacson. Bekannt für seine umfassenden Biografien bedeutender historischer Persönlichkeiten, hat Isaacson über zwei Jahre intensiv mit Musk zusammengearbeitet, um dessen Biografie zu verfassen.
In dieser Zeit hatte er exklusiven Zugang zu Musk: Er besuchte seine Fabriken, nahm an Meetings teil und führte zahlreiche Interviews – nicht nur mit Musk selbst, sondern auch mit dessen Familie, Freunden und Kollegen. Dabei entwickelte sich zwischen Isaacson und Musk eine professionelle, aber auch persönliche Beziehung, die über die reine Autor-Leser-Distanz hinausgeht.
Isaacson beschreibt Elon wie folgt:
„Elon liegt die Menschheit sehr am Herzen – der Übergang zu nachhaltiger Energie und das Fortbestehen der menschlichen Zivilisation. Er wird nicht von dem Wunsch nach Geld oder Macht angetrieben, auch nicht von dem Bedürfnis, andere herumzukommandieren, sondern von diesen Missionen, die ihn schon als kleiner Junge begleiteten, als er in der Ecke saß und Superhelden-Comics las.
Ich muss der Menschheit helfen, eine raumfahrende Spezies zu werden. Ich muss ihr helfen, das Problem der nachhaltigen Energie zu lösen. Ich muss ihr helfen, sich gegen eine möglicherweise bösartige künstliche Intelligenz zu verteidigen.
Es steckt eine Güte in seinen Missionen, und diese Missionen sind episch.“
[The 92nd Street NY, October 13, 2023]
(Quelle X: https://x.com/ElonClipsX/status/1870750353534205993)Es sagt viel aus, wenn jemand wie er – Mitglied und Agenda Contributor des World Economic Forums, ehemaliger Präsident und CEO des Aspen Institute, früherer Chefredakteur des Time Magazine sowie ehemaliger Vorsitzender und CEO von CNN – und zudem Mitglied des Vorstands der Bloomberg Philanthropies und der Rockefeller Foundation, ein solches Statement abgibt.
Elon Musk wird als ein epischer, nahezu messianischer Held dargestellt. Er ist fast wie Neo in der Matrix: Der Retter der Menschheit, der uns vor böser KI schützt, nachhaltige Energie fördert und uns zu den Sternen bringt. Doch die Quelle – Walter Isaacson, ein Insider mit Verbindungen zu WEF, Aspen Institute und Rockefeller Foundation, die selbst als zentrale Elemente der Matrix gesehen werden können – bringt eine pikante Wendung: Feiern die Architekten der Matrix hier wirklich einen Befreier, oder inszenieren sie Musk als nützlichen Hoffnungsträger, der das System stabilisiert, statt es zu sprengen?
Musk wirkt wie der Schlüssel zur Flucht aus der Matrix, doch unterm Strich bleibt fraglich: Führt seine „epische Mission“ wirklich in die Freiheit – oder ist sie nur ein neues Kapitel in derselben Matrix, mit glänzenderer Oberfläche?
In diesem Sinne: Wake up … the Matrix is calling.
Quellen (Stand vom 07.01.2025)
AGI, AI, Artificial Intelligence, BCI, Blackrock, Brain-Computer-Interface, DOGE, Elon Musk, Everything-App, Future of Humanity, Gehirn-Computer-Schnittstelle, Gwynne Shotwell, humanoide Roboter, Humanoids, Künstliche Intelligenz, Kekius Maximus, KI, Linda Yaccarino, Matrix, Neuralink, Open AI, Optimus, SpaceX, Starlink, Starshield, Tesla, Transhumanismus, Twitter, Vanguard, Walter Isaacson, WEF, World Economic Forum, X, X Corp., Young Global Leader, Zukunft der Menschheit
