
When HSBC Bank placed posters at airports such as Gatwick and Dubai in 2013 as part of its global campaign „The future is full of opportunity” – showing a fingerprint, a QR code, and the provocative slogan „Your DNA will be your data” – the message was unambiguous: the future will be personal, perhaps unsettlingly personal. The bank sought to position itself as a forward-looking company that addresses digitalization, artificial intelligence, sustainability, and genetics head-on.
The slogan seemed ambiguous – both a promising vision and an alarming prospect. Critics immediately sounded the alarm: your DNA becomes readable, analyzable, and digitally accessible – an open book about ancestry, health risks, perhaps even personality traits – comparable to biometric data such as fingerprints or facial scans. What was once considered the most intimate aspect of a person is now becoming a file: machine-decipherable, potentially storable, possibly tradable. The human being is reduced to information – „bits and bytes” that could be processed, sold, or misused.
Who owns the rights to your genetic data? And who has access to it? You yourself? The company that conducted the test? Do surveillance, data leaks, and misuse by state and private actors reach a new dimension as a result? The vision of designer babies, genetic selection, and personalized advertising based on biological markers raises serious ethical questions. Will progress ultimately be paid for at the price of individual freedom?
However, the biotech revolution has long since gained tremendous momentum, and more and more voices recognize the enormous potential of this development. The March 2024 e-book edition of HSBC Bank Frontiers – Biotech – Five Big Ideas Shaping the Biotech Revolution states:
„The idea of exploiting nature for goods and services is nothing new. But the practice entered its modern era in the 1970s, with the advent of genetic engineering and the establishment of the first biotech companies. This brought together science and business like never before, and the influx of capital galvanized research and development efforts.
Over the three decades that followed, the field was accelerated by breakthrough devices such as DNA sequencers and synthesizers, and biology itself slowly morphed into a computational, data-driven discipline. Following the turn of the millennium, as researchers successfully charted ever greater regions of the human genome, and the cost of sequencing that genome rapidly declined – from around $100 million in 2001 to less than $1,000 today – the potential of biotech to transform our lives has become electrifyingly apparent.
Over the past few years, the possibilities unlocked by biotech advances – from treating genetic diseases to de-extincting animal species – have been enlarged by progress in complementary fields such as artificial intelligence. This has underpinned a coming-of-age moment in this space.“
Nowhere else do benefits and risks collide as directly as in personalized – or precision-based – medicine. Its goal is to use individual genetic and biological information to refine diagnoses, personalize therapies, and detect diseases at an early stage. The vision is promising: fewer side effects, better chances of recovery, more effective prevention – medicine tailored to the individual.
But here, too, what is celebrated as medical progress brings societal, legal, and ethical challenges:
- Who decides which genetic data are relevant?
- What role does access to these technologies play – does it exacerbate social inequalities?
- And how can discrimination, for example through genetic scoring by insurance companies or employers, be prevented?
Between the hope for individual healing and the fear of collective disempowerment lie fundamental questions: Does access to one’s biological data empower the individual – or expose them? Is personalized medicine a blessing – or the beginning of an era of subtle dehumanization?
Hence the crucial question is: Is personalized medicine the promised ascent into heaven – or the plunge through the gates of hell?
This blog post invites readers to consider personalized medicine in all its complexity – beyond hype or alarmism. It is deliberately aimed as well at non-experts who wish to engage critically with the opportunities, risks, and ethical tensions of this medical revolution.
📑Table of Contents
1. Conceptual Classification and the Fundamental Paradigm
2. Driving Forces – Between Vision and Reality
3. The Future of Medicine Begins in the Cell
3.1. How Biology and Technology Come Together
3.2. Data, Responsibility, and the Bigger Picture
4. From DNA to Knowledge – A High-Speed Train of Innovation
4.1. DNA – The Blueprint of Life
a) DNA: The Molecular Information Archive of Life
b) From Code to Protein: How Instructions Become Reality
c) Epigenetics & Non-coding DNA: The Conducting Team of Our Genome
d) The Hidden Control Center: What Non-coding DNA Regulates
e) When the Operating System Makes You Sick
f) Open Mysteries in Research
g) The True Magic of Genes
4.2. Genetic Variation and Its Consequences
4.3. Genome Sequencing: Key to Individual Genetic Material
a) The Journey of DNA: From Umbilical Cord Blood to Genetic Information
b) Three Directives for Gene Diagnostics: WGS, WES, and Panel
4.4. Methods of Genome Sequencing
4.4.1. Method 1: Copying with Fluorescent Dye
a) Sanger Sequencing
b) Illumina Technology
c) Single-Molecule Real-Time (SMRT) Sequencing
4.4.2. Method 2: High-Tech Tunnel – Electrically Scanning DNA
a) Oxford Nanopore Sequencing
4.4.3. Genome Sequencing – Technologies Compared
a) Throughput vs. Usability – Quantity Does Not Always Equal Quality
b) Cost-Effectiveness – Cost per Base vs. Cost per Insight
c) From Sample to Result – How Quickly Does the Whole Genome Speak?
d) Clinical Applications – Which Technology Fits Which Purpose?
e) AI as Co-Pilot – Automation Without Relinquishing Responsibility
f) Comparison Table: Sanger, Illumina, PacBio, ONT
g) Key Takeaways: Between Maturity and Routine
4.5. Genome Sequencing: The Next Technological Leap
a) FENT: How a Microchip Is Revolutionizing DNA Analysis
b) SBX: When DNA Stretches to Be Understood
c) The G4X System: From the Genetic Recipe to the Spatial Atlas of the Cell
4.6. From Code to Cure – Bioinformatics as the Key to Tomorrow’s Medicine
4.6.1. From the Genetic Code to Computer-Aided Genome Analysis
4.6.2. Modern Bioinformatics – The Digital Toolbox of Biology
5. A Success Story
6. A Look into the Future: The Synthetic Human Genome Project
7. Personalized Medicine and Smart Governance
7.1. Power, Governance, and Smart Governance
7.2. Governmentality
7.3. Personalized Medicine as a Catalyst for Biomolecular Governmentality
7.4. The Global Trend: Worldwide Biomolecular Governmentality
7.5. Conclusion: The Global Ambivalence of Biomolecular Power
8. Epilogue

1. Conceptual Classification and the Fundamental Paradigm
The development of modern medicine toward tailored treatments is often described using terms such as „personalized medicine”, „individualized therapy” or „precision medicine”. But what exactly do these buzzwords mean?
Personalized medicine is currently considered one of the most popular terms – catchy like a marketing slogan. It carries the promise of tailoring therapies to each individual: to their genes, their lifestyle, their biology. Accordingly, the term is frequently used in the media and public debate.
Individualized therapy sounds less glamorous but conveys a similar idea: the adjustment of medications or dosages to the needs of individual patients. This term is used primarily in clinical practice.
Finally, precision medicine is the most sober of the three terms – a technical term that dominates research laboratories and scientific publications. It emphasizes the data-driven approach: algorithms, genetic analyses, and biomarkers determine which therapy works for which patient group. This is the language of science.
But regardless of which term is used, they all represent a paradigm shift: moving away from standardized procedures toward medicine that focuses on the biological uniqueness of each individual.
2. Driving Forces –Between Vision and Reality
The driving forces behind personalized medicine are diverse and closely interconnected. On one hand, there are scientific and technological developments that have enabled enormous progress in recent decades: new methods to read genetic material quickly and accurately, powerful data analysis through artificial intelligence, and the availability of large biomedical datasets. These technologies form the foundation for recognizing individual differences between patients and leveraging them medically.
On the other hand, social and political impulses act as accelerators of this transformation. Public health programs, targeted research funding, strategy papers from international health organizations, and specific legislative initiatives actively drive the integration of personalized medicine into healthcare practice. Increased health awareness among the population, as well as growing expectations for individualized treatment options, further reinforce this trend.
Personalized medicine is therefore not merely a product of technological feasibility, but emerges at the intersection of innovation, politics, and societal demand.
A current example of the practical implementation of this development comes from the United Kingdom: under a new 10-year plan, every newborn is to undergo a DNA test. The so-called whole-genome sequencing will screen for hundreds of genetically linked diseases – with the aim of detecting potentially life-threatening conditions early and preventing them in a targeted manner. According to British Health Secretary Wes Streeting, the goal is to fundamentally transform the National Health Service (NHS): moving away from mere disease treatment toward a system of prediction and prevention. Genomics – the comprehensive analysis of genetic information – will serve, in combination with artificial intelligence, as an early warning system.
„The revolution in medical science means that we can transform the NHS over the coming decade, from a service which diagnoses and treats ill health to one that predicts and prevents it“, said Streeting. The vision: personalized healthcare that identifies risks before symptoms even appear – thereby improving quality of life and alleviating pressure on the healthcare system.
But as convincing as this vision may sound, there are also cautionary voices coming from the scientific community. For example, geneticist Prof. Robin Lovell-Badge from the Francis Crick Institute warns against underestimating the complexity of genomic data. Not only is technical expertise required for data collection, but above all qualified personnel who can communicate the information obtained in a responsible and comprehensible manner. Data alone does not constitute a diagnosis – what is crucial is its meaningful and patient-oriented interpretation.
What does it concretely mean to rethink medicine?
The real revolution is taking place in laboratories and data centers, where the building blocks of personalized medicine are being developed. The following chapters take you behind the scenes, step by step, and reveal the key technologies needed to bring personalized medicine from concept to clinical practice.
3. The Future of Medicine Begins in the Cell
3.1. How Biology and Technology Come Together
Our bodies consist of billions of cells – tiny, specialised units that work together day after day to enable health and life. In every single cell, countless processes occur that are precisely coordinated with one another. These cellular mechanisms are crucial for understanding health, disease, and the development of new, targeted therapies.
One central mechanism is energy production: cells convert nutrients and oxygen into ATP (adenosine triphosphate) – the body’s „energy currency”. Without ATP, no cell could function, no muscle could move, no thought could arise.
At the same time, cells continuously produce proteins – based on genetic information. These complex molecules perform essential tasks in the body, serving as chemical catalysts, signaling molecules, structural components, or transporters.
Cellular cleaning and renewal are also vital: processes such as autophagy break down and recycle damaged cellular components – an internal waste disposal system that can help prevent diseases like Alzheimer’s or Parkinson’s.
For cells to work together in a coordinated way within the body, they need functioning signaling pathways – they „communicate” with one another through chemical messengers. This cell communication controls when cells should grow, rest, or respond to changes.
Another key process is cell division – essential for growth, wound healing, and tissue renewal. At the same time, DNA repair mechanisms ensure that errors in the genetic material are corrected. If this fails, apoptosis, the programmed cell death, takes over to limit potential damage.
All these mechanisms run in the background – constantly and with high precision. However, if one of these processes is disrupted, it can lead to serious diseases such as cancer, autoimmune diseases or metabolic disorders. This is exactly where personalized medicine comes in. It uses state-of-the-art technologies to better understand these cellular processes and influence them in a targeted manner.
Thanks to genome sequencing (for example through „next-generation sequencing”), it is possible to identify genetic alterations that influence protein production or repair mechanisms.
Biomarker analyses (the examination of specific molecules) in blood, tissue, or other bodily fluids reveal whether certain signaling pathways are over- or underactive – and help predict individual disease progressions or select appropriate therapies.
Single-cell analysis makes it possible to visualize differences even between individual cells – for example, in a tumor, where some cells respond to therapy and others do not. This enables more precise treatment.
Proteomics (the analysis of all proteins in a cell) and metabolomics (the analysis of metabolic products) also provide a current picture of how active specific cellular mechanisms really are – for example, whether a cell is stressed, has sufficient energy, or is in the process of dividing.
Artificial intelligence (AI) helps analyze these vast amounts of data and identify patterns – for example, which combination of cellular disorders is typical for a particular type of cancer. Digital health data (such as from wearables) complement this picture in daily life and enable precise long-term monitoring.
Even new therapeutic approaches are based on understanding cellular processes: gene therapies and CRISPR-based gene editing target the DNA to correct faulty mechanisms. In organoids – mini-organ models in the lab – drugs can be tested directly on patient-specific cell models, without any risk to humans.
Modern technologies now reveal what was previously hidden: how cells work, what causes imbalances in disease – and how to intervene in a targeted manner. This has given rise to individualized medicine that is no longer based on conjecture, but on measurable biology.
This new precision brings not only opportunities, but also new responsibilities.
3.2. Data, Responsibility, and the Bigger Picture
The complexity of these cellular processes is directly reflected in the complexity of personalized medicine: the more precisely we understand the intricate processes and interactions in our cells, the more targeted – but also more demanding – diagnosis and therapy become. This new precision requires new technologies, new ways of thinking, and a deep biological understanding. At the same time, it opens the door to more effective, sustainable, and tailored treatment concepts.
However, the more targeted and profound these interventions become, the greater the responsibility – and the challenge of identifying and controlling undesirable long-term effects or side effects at an early stage. Particularly in the case of innovative procedures such as gene therapy or immunomodulation, it is important to carefully weigh up the benefits against the potential risks. Personalized medicine therefore requires not only precision, but also foresight.
A central element of this foresight is the availability of personal health data – not only in individual cases, but on a large scale.
Only by combining individual data with population data is it possible to understand biological processes in detail, predict disease progression and develop personalised therapies. Genetic information, laboratory values, imaging data and digital everyday measurements provide clues as to how cellular mechanisms work in a specific person – and how they vary in others.
To interpret these individual patterns medically, large datasets are needed for comparison. Only in this way can AI systems detect complex relationships that escape the human eye – such as rare genetic variants, molecular risk constellations, or unexpected therapy effects. The rule here is: the larger and more diverse the data base, the more reliable the models – especially when they are continuously enriched with new information.
However, this progress can only be achieved if people are willing to share their health data – and can trust that it will be used securely and responsibly.
The future of medicine begins where all life originates: in the cell. But its success depends on the interplay of three forces: technological innovation, scientific depth – and ethical responsibility.

4. From DNA to Knowledge – A High-Speed Train of Innovation
How genome sequencing and bioinformatic analysis are revolutionizing medicine
4.1. DNA – The Blueprint of Life
a) DNA: The Molecular Information Archive of Life
b) From Code to Protein: How Instructions Become Reality
c) Epigenetics & Non-coding DNA: The Conducting Team of Our Genome
d) The Hidden Control Center: What Non-coding DNA Regulates
e) When the Operating System Makes You Sick
f) Open Mysteries in Research
g) The True Magic of Genes
4.2. Genetic Variation and Its Consequences
4.3. Genome Sequencing: Key to Individual Genetic Material
a) The Journey of DNA: From Umbilical Cord Blood to Genetic Information
b) Three Directives for Gene Diagnostics: WGS, WES, and Panel
4.4. Methods of Genome Sequencing
4.4.1. Method 1: Copying with Fluorescent Dye
4.4.2. Method 2: High-Tech Tunnel – Electrically Scanning DNA
4.4.3. Genome Sequencing – Technologies Compared
4.5. Genome Sequencing: The Next Technological Leap
4.6. From Code to Cure – Bioinformatics as the Key to Tomorrow’s Medicine
Imagine standing on a train platform and watching a train rush by at such speed that you can barely make out the image in the window. This is how many people perceive developments in personalized medicine: technologies are advancing at breakneck speed, terms seem abstract – and yet there is a common thread running through it all: the human genome.
As diverse as modern approaches may be – from organoids and CRISPR-based gene editing to RNA-based medicines – at the center lies the human genetic blueprint. Today, genome sequencing forms the foundation of many diagnoses and therapeutic decisions. It reveals which genetic alterations trigger diseases, which signaling pathways are affected – and where targeted interventions might be possible.
What once cost billions and took years can now be done in a matter of days: the entire human genome can be decoded. But the sequence alone does not constitute a diagnosis. The sequence of three billion base pairs only has medical value once it is understood – and this is where the real challenge begins.
Bioinformatics enters the stage: an interdisciplinary field that combines mathematics, computer science and biology to derive patterns, risks and treatment options from raw data. It is the real bottleneck of personalized medicine – because it determines whether genetic variants are considered harmless, risky or disease-causing.
Individual sequences are compared with international reference databases, algorithms analyze millions of genetic variants, and learning systems model the complex interplay of genetics, environment, and metabolism.
This process is highly complex – and at the same time essential. Only computer-assisted evaluation makes DNA medically usable. There is an inseparable symbiosis between genome sequencing and bioinformatic analysis: the sequence provides the data, the analysis provides the understanding. Without it, DNA remains a mere string of letters.
This is precisely why the following sections focus on the crucial question:
What does it mean to decode the genome – and to truly understand it?
What conclusions are possible – and where are their limits?
For anyone who wants to understand the future of medicine, it is essential to know how we read the human genome today – and how we are learning to interpret it.
The high-speed train of innovation has long since departed. Anyone who wants to ride along must understand where it is headed.
4.1. DNA – The Blueprint of Life
To embark on this journey, it is worth taking a brief look at the molecular foundations: how our DNA becomes a blueprint, how proteins are formed from it via the intermediate step of RNA, and how epigenetics fine-tunes these processes. Only this foundation makes it possible to understand what is actually happening when we decode the genome.
a) DNA: The Molecular Information Archive of Life
In every cell of our body lies a carefully packaged and astonishing molecule: DNA (deoxyribonucleic acid). It contains the complete blueprint and operating instructions of our body – a molecular archive of life’s information.
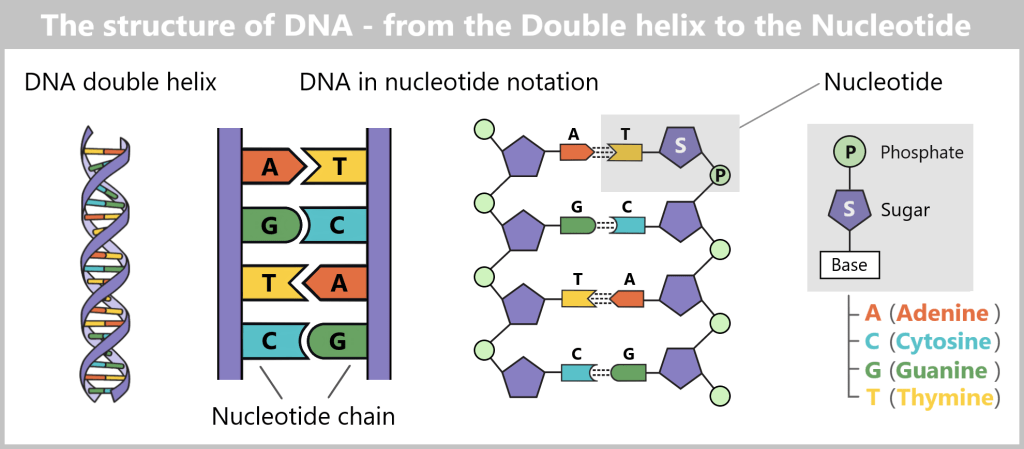
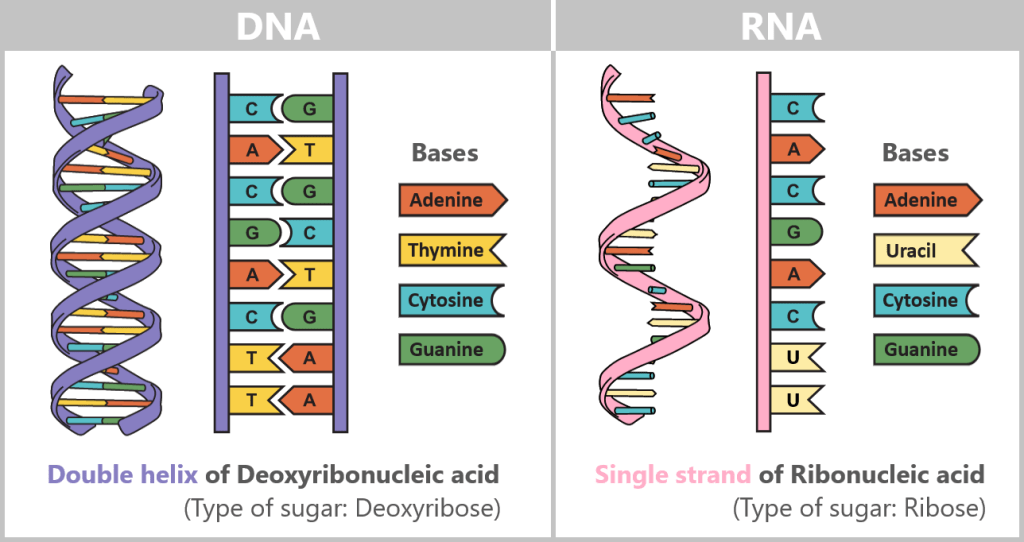
Structurally, it resembles a spiral-shaped rope ladder: two sugar-phosphate chains form the sides, with the „rungs“ – the bases adenine (A), cytosine (C), guanine (G) and thymine (T) – in between. They always pair up according to fixed rules: A with T, C with G. This complementary bond makes each strand a template for the other – a molecular backup system.
If you unravel the DNA from a single cell, it measures around two meters – with a diameter of just a few millionths of a millimeter. Life therefore literally hangs on a wafer-thin but astonishingly long thread.
The sequence of around three billion base pairs is as unique as a fingerprint. It forms the genetic code – the instructions for producing proteins, the molecules that control the structure, function and regulation of all biological processes.
As a central information archive, DNA is both extremely valuable and sensitive. That is why it is stored safely in the cell nucleus, comparable to a safe. In order for the body to access the information in this safe, it needs a clever transport system. After all, the blueprints for proteins – the molecular architects of all life processes – must reach the ribosomes, the so-called „protein factories“ in the cytoplasm. This is where proteins are produced – a process known as protein biosynthesis.
But how does a specific blueprint travel safely from the cell nucleus to these factories? In other words: How is the genetic information for a particular protein transferred from its protected archival location to the site of production?
b) From Code to Protein: How Instructions Become Reality
The journey begins with the genes – specific sections of DNA that contain the blueprints for proteins. There are around 20.000 such genes in the human genome.
DNA is not just a passive information archive in the cell nucleus – it contains an active repertoire of instructions. You can think of each gene as a chapter in a molecular cookbook: often with multiple versions of a recipe, adapted to the cell type, developmental stage, or external conditions.
First, a temporary „working copy” of a gene is created – in the form of RNA (ribonucleic acid). It resembles DNA but uses the base uracil (U) instead of thymine (T) and usually consists of a single strand. This transient structure makes RNA ideal for short-term use – it is degraded after fulfilling its function.
Alternative Splicing: The Flexible Kitchen of Genes
Cells must adapt flexibly to changing demands: responding to inflammation, defending against pathogens, building muscle, or regulating blood sugar levels. This requires not a rigid set of instructions, but a system capable of improvising depending on the situation.
Depending on the cell type and external signals, the RNA strand is dynamically modified by being cut at specific markers – transferred from the DNA. Unneeded sections (introns) are removed, and the remaining sections (exons) are reassembled. This process creates a multitude of possible messages from a single template. The result is messenger RNA (mRNA), which carries the genetic information from the nucleus to the ribosomes in the cytoplasm – the cell’s „protein factories”.
You can think of this process like following a recipe: depending on the need, the cell leaves out certain ingredients, adds spices, or adjusts the preparation steps. In this way, a single basic instruction can yield a wide variety of dishes.
Through this process – called alternative splicing – cells can produce a variety of different proteins from a single gene, each performing specific functions.
The entire process „from gene to protein” is often referred to as gene expression.
💡Example: The Titin Gene
👉 Longest human gene (≈ 300,000 base pairs).
👉 Through alternative splicing, more than 20,000 different protein variants are produced – each adapted to the specific requirements of different muscle types.
Our genome is therefore not a rigid recipe book – it is a dynamic kitchen, where the cell constantly develops new solutions from the same templates.
📌 If you are interested in how genetic information in DNA is translated into protein step by step, you can find further information here.
But as in any creative kitchen, errors can also occur during the transfer from DNA to mRNA: if a „letter” in the recipe is missed, swapped, or incorrectly assembled, the message can end up misleading or unusable. Such mutations or faulty splicing are sometimes harmless – but they can also produce defective proteins that fail to perform their function or even cause harm. A small slip in copying the molecular cookbook – and the flavor of life changes.
c) Epigenetics & Non-coding DNA: The Conducting Team of Our Genome
Although all cells contain the same DNA, they only use a fraction of it. A liver cell needs different instructions than a nerve cell – the rest remain dormant. This targeted gene regulation is controlled by epigenetic mechanisms: small chemical markers – such as methyl groups – do not change the genetic sequence itself, but influence whether a gene is active, how often it is read, or whether it remains completely silenced.
But who actually gives the conductor his instructions? This is where non-coding DNA comes into play – 98% of our genome, long dismissed as useless „junk”. It is the operating system that provides the epigenetic control programs. Only through its interaction is it determined when, where, and how strongly our genes become active – and thus it helps decide over health and disease.
d) The Hidden Control Center: What Non-coding DNA Regulates
These „dark“ regions conceal precise circuits that control the fate of each cell:
Enhancers act like invisible remote controls for genes. Even across thousands of base pairs, they can activate or silence a target gene – for example, for heart development in the embryo.
Telomeres are the protective caps at the ends of chromosomes, like the plastic tips on shoelaces. But when they shrink, aging begins – a central mechanism of cellular decline and cancer development.
Non-coding RNAs act as hidden directors: microRNAs block disease-causing genes, while the long RNA XIST silences an entire X chromosome in females.
Epigenetic markers lay an invisible map over the genome. With methylations acting like red stop signs, they decide whether an enhancer or gene may be read – or silenced.
This is how order emerges in the molecular cookbook: each cell type reads only the pages it needs – guided by non-coding DNA and its epigenetic interpretation.
e) When the Operating System Makes You Sick
Almost all genetic risk markers for common diseases – from diabetes and heart disease to mental disorders – are located in non-coding zones:
Mutations in an enhancer of the MYC gene can trigger leukemia by driving cell division out of control.
Epigenetic misprogramming in these regions – caused, for example, by chronic stress or environmental toxins – can permanently switch genes on or off incorrectly, thereby promoting diseases such as cancer or autoimmune disorders.
Some of these epigenetic patterns are partially reversible – a glimmer of hope for new therapies.
f) Open Mysteries in Research
Despite tremendous progress, the genome remains full of unsolved mysteries:
The „dark matter”: About half of the non-coding genome remains functionally unexplored. Is it evolutionary noise – or an as-yet uncharted regulatory network of life?
Long-range communication: How does an enhancer reliably find its target gene among millions of DNA bases? The spatial folding of the genome – a true „chromatin origami” – could be the key.
Environmental influences: Why do identical non-coding mutations lead to completely different clinical pictures in different life contexts?
RNA codes: Tens of thousands of non-coding RNAs have been discovered – but which are mere footnotes, and which might emerge as leitmotifs for new therapies?
g) The True Magic of Genes
Non-coding DNA is not junk data, but the cell’s mastermind. Epigenetics, in turn, acts as its conductor – flexibly interpreting the instructions of the operating system. Together, they shape our development, regulate our health, and respond to environmental signals.
Genes are not a rigid destiny, but more like a musical score that can be continually reinterpreted. In this interplay, an orchestra emerges that not only shapes our cells but constantly retunes itself – influenced by experiences, environmental factors, and even traces that can be passed on to future generations.
The true magic lies in the fact that from a limited set of notes, an infinite diversity of life can emerge.
4.2. Genetic Variation and Its Consequences
When the genome miswrites, falls silent, or improvises.
With every cell division, DNA is carefully copied and passed on to daughter cells – ensuring that genetic information is preserved across generations.
However, the system is not infallible: mutations – changes in the DNA sequence – can affect the structure and function of proteins. Sometimes only a single „letter” is incorrect. Sometimes a „sentence” is missing, or an entire „paragraph” has been rearranged. In genetics, these are called variants – genetic changes that can be harmless, pose a risk, or cause disease, depending on their type, size, and location.
The following overview presents the main types of variants – and what they mean in the molecular manuscript of life:
📌 SNVs (Single Nucleotide Variants) – A single „letter” is swapped. Usually barely noticeable, but sometimes enough to rewrite entire stories, as in the case of sickle cell anemia.
📌 Indels (Insertions/Deletions) – Small fragments of text are inserted or deleted. Even a tiny shift can throw entire „sentences” out of sync, sometimes so much that the original meaning is lost.
📌 Structural Variants (SVs) – Long stretches of DNA are inverted, duplicated, or relocated. These hidden „rewritings” are difficult to detect – and can often have serious consequences, as seen in cancer
📌 Splice-Site Variants – Errors at the junctions disrupt the RNA syntax. This creates incorrect blueprints, resulting in proteins that no longer function properly.
📌 Repeat Expansions – Base sequences are compulsively repeated until they distort the „text”. This causes diseases such as Huntington’s disease.
📌 Copy Number Variants (CNVs) – Entire „chapters” (genes) are missing or duplicated. The balance of the story is lost.
📌 Mitochondrial variants – Changes in the DNA of mitochondria, the cell’s power plants. If the „text” here is altered, the drive of life falters.
📌 Somatic variants – Arise only during a person’s lifetime and affect specific groups of cells. They mainly shape the story of tumors.
📌 Epigenetic signals – Not new letters, but a different emphasis: volume, rhythm, timing. They transform the same text into an entirely new melody.
The list shows that our genome is not a rigid structure but can be altered by variants. Some of these changes remain inconspicuous footnotes, while others can rewrite the entire story – sometimes with dramatic consequences.
To make such subtle differences visible, tools are needed that capture every detail – every letter, every punctuation mark, every shift. This is precisely where modern genome sequencing comes in.
4.3. Genome Sequencing: Key to Individual Genetic Material
The genetic material of a human being resembles a gigantic library: billions of letters, neatly arranged in long volumes. To read it, genome sequencing is required – which, like a high-precision scanner, provides the raw text of our genetic blueprint.
Thus, it becomes the heart of personalized medicine – a form of medicine that bases its decisions on the individual, not on the average.
But how do we even get hold of the „Book of Life“ so that we can read it page by page?
Let’s imagine the following: shortly after birth, a newborn undergoes a DNA test – a scenario that has already become reality in the United Kingdom, as described earlier.
a) The Journey of DNA: From Umbilical Cord Blood to Genetic Information
Immediately after birth, blood can be collected from the clamped umbilical cord – a safe and painless procedure. Umbilical cord blood originates from the newborn and contains numerous valuable cells, especially stem cells and white blood cells. Hidden within their nuclei lies what truly matters: the DNA – the child’s genetic inheritance.
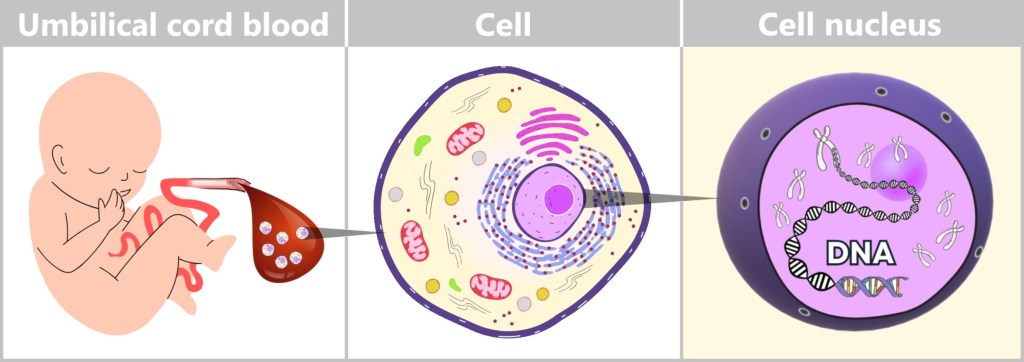
Left: Umbilical cord blood as a rich source of diverse cell types.
Center: Schematic representation of a human cell.
Right: Inside the cell nucleus lies the DNA, the carrier of genetic information.
To isolate and analyze this DNA, it undergoes a multi-step process in the laboratory. This involves established molecular biology methods that can be divided into four key steps:
Cell Lysis: Gaining Access to the Genetic Material
First, the blood is centrifuged – spun at high speed. During this process, the components separate according to their density: the white blood cells settle as a cell pellet at the bottom. They contain the majority of the DNA of interest.
These cells are then treated with specialized solutions and enzymes that dissolve the cell and nuclear membranes. This releases the DNA – initially, however, as part of a mixture containing proteins, lipids, and other cellular components.
DNA Isolation: Obtaining Pure Genetic Material
In the next step, the DNA is extracted from the cell lysate – the „cell soup.” Several methods can be used for this purpose:
Filtration or column purification: The solution is passed through special filter systems to which the DNA adheres, while smaller molecules are washed out.
Magnetic beads: More modern methods use tiny beads with a DNA-binding surface. The DNA sticks to them and can be removed in a targeted manner using a magnet.
Both methods allow for an effective separation of genetic material from contaminants.
Purification and Concentration of the DNA
At this stage, the DNA from many white blood cells is present in a relatively pure form – but still dissolved in an aqueous solution. To further purify and concentrate it, cold alcohol (e.g., ethanol or isopropanol) is added. This causes the DNA to precipitate and become visible as a whitish, thread-like substance.
Human DNA is extremely long: if fully stretched out, it measures about two meters per cell. During extraction, however, it is broken into many smaller fragments by chemical and mechanical forces. At first, this seems chaotic – but because the DNA is identical in all cells, many fragments contain overlapping information. This random distribution becomes an advantage during sequencing: the fragments can be read multiple times independently and then assembled on a computer into a complete sequence. This significantly increases the accuracy of the analysis.
Quality Control: Is the DNA Intact?
Before the DNA sample can be analyzed, its quality must be assessed. Two common methods are:
Spectrophotometry: Measures the concentration and purity of the DNA by analyzing light absorption.
Gel electrophoresis: DNA fragments are passed through a gel and separated by length (longer fragments move more slowly than shorter ones). This allows assessment of whether the sample is sufficiently intact.
Why is this process so important?
Human DNA is fragile – contaminants or fragmentation can distort analyses or render them unusable. Careful and controlled purification is therefore essential for reliable genetic results.
Umbilical cord blood is a particularly valuable source of medical information: it enables early diagnosis, prognoses, and in some cases targeted prevention.
What appears to be a routine procedure in the lab is, in fact, a highly precise process – it requires technical expertise, modern equipment, and biochemical finesse. With automated extraction methods, it is now possible to obtain a high-quality DNA sample from just a few drops of umbilical cord blood in less than an hour. Although the DNA consists of fragments, it still contains all the information needed to decode an individual’s genetic code – and thus lays the foundation for the medicine of tomorrow.
b) Three Directives for Gene Diagnostics: WGS, WES, and Panel
Before we dive deeper into the tools of genome sequencing, it is worth taking a look at the three central strategies used today to decode genetic information – like stage directions in the script of life:
🎯 Panel Sequencing – The Targeted Scene
Only a selected part of the genome is examined here – usually a few dozen to a few hundred genes. Such tests are used when a specific disease is suspected, such as hereditary breast cancer (BRCA1/2).
Advantage: Fast, cost-effective, and precise – as long as the medical question is well-defined.
Limitation: Everything outside the selected scene remains in the dark.
🎥 Whole Exome Sequencing (WES) – The Classic Director’s Cut
WES analyses all protein-coding sections of the genome – approximately 1–2% of the entire genetic material. It is a proven method, particularly for rare hereditary diseases, as most known mutations are located in these areas.
Advantage: Efficient identification of faulty blueprints for proteins.
Limitation: Regions outside the genes – important switches and regulators of the genome – are not captured.
🎬 Whole Genome Sequencing (WGS) – The Complete Movie in 4K
WGS reads the entire genome – all three billion letters. This provides the most comprehensive picture, including non-coding regions, regulatory elements, structural variants, and rare mutations.
Advantage: Maximum resolution for complex cases.
Example: In children with unclear developmental disorders, WGS provides clarity twice as often as WES – for instance, by uncovering mutations in distant „remote-control” regions (enhancers) that incorrectly switch genes on or off.
Conclusion:
Depending on the question at hand, the right perspective is needed – sometimes a zoom on the key scene suffices, other times the entire film must be re-edited. Choosing the method is therefore not a mere technical detail, but a strategic decision in the diagnostic playbook.
4.4. Methods of Genome Sequencing
How is the genetic code read?
Imagine DNA as a book – written in a language with only four letters: A, T, C, and G. To decipher this genetic text, scientists use modern technologies. Two central methods have become established. They pursue the same goal: to determine the sequence of DNA letters as precisely as possible.
The first method works – put simply – like a molecular photocopier equipped with color sensors. The second uses a high-tech tunnel through which the DNA is guided and electronically „scanned”, comparable to running your fingers along a rope to detect the smallest irregularities. These techniques form the foundation of DNA sequencing.
4.4.1. Method 1: Copying with Fluorescent Dye
a) Sanger Sequencing
b) Illumina Technology
c) Single-Molecule Real-Time(SMRT) Sequencing
4.4.2. Method 2: High-Tech Tunnel – Electrically Scanning DNA
a) Oxford Nanopore Sequencing
4.4.3. Genome Sequencing – Technologies Compared
a) Throughput vs. Usability – Quantity Does Not Always Equal Quality
b) Cost-Effectiveness – Cost per Base vs. Cost per Insight
c) From Sample to Result – How Quickly Does the Whole Genome Speak?
d) Clinical Applications – Which Technology Fits Which Purpose?
e) AI as Co-Pilot – Automation Without Relinquishing Responsibility
f) Comparison Table: Sanger, Illumina, PacBio, ONT
g) Key Takeaways: Between Maturity and Routine

4.4.1. Method 1: Copying with Fluorescent Dye
This method takes advantage of the fact that DNA consists of two complementary strands. If the base sequence of one strand is known, the sequence of the complementary strand can be automatically deduced. To obtain this information, one of the two strands of the DNA under investigation is copied. The aim is to record the exact sequence of bases in the resulting copy during this copying process.
To better understand how this process works, let’s first take a look at the key components and basic steps involved in producing a DNA copy in a test tube (in vitro).
Producing a DNA Copy – The Basic Principle
At the centre is a DNA template that is to be copied. This requires four main components:
- dNTPs (deoxynucleoside triphosphates): the DNA building blocks A, T, C, and G.
- DNA polymerase: an enzyme that functions as a molecular copying machine.
- Primers: short DNA fragments that provide the polymerase with a starting point.
- Buffer solution: ensures stable conditions during the reaction.
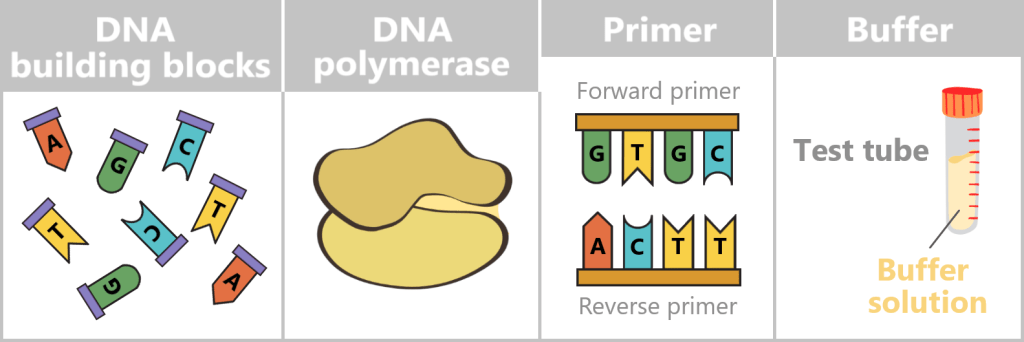
DNA building blocks: Individual nucleotides (A, T, C, G), shown in different colors and letters. They serve as the raw material from which the new DNA strand is assembled.
DNA polymerase: A schematic enzyme that links the nucleotides together to form a DNA strand.
Primer: Short DNA fragments that provide the polymerase with a defined starting point.
Buffer: A test tube containing buffer solution, ensuring stable chemical conditions during the reaction.
What does DNA polymerase do?
DNA polymerase is a specialized enzyme that generates complementary DNA strands – essentially the „printhead” of a molecular copying machine. Enzymes act as biological tools that enable chemical reactions in a targeted and efficient manner, which is why they are often described as „biological catalysts”.
In order for the polymerase to work, it needs a primer as a start signal. You can think of it like this: just as a printhead only starts working when a sheet of paper is inserted correctly, the polymerase only begins its work when a primer is present. This small piece of DNA shows it where to start – essentially acting as the first sheet in the molecular printer.
Before copying can begin, the double-stranded DNA must first be separated into two single strands – this step is called denaturation.
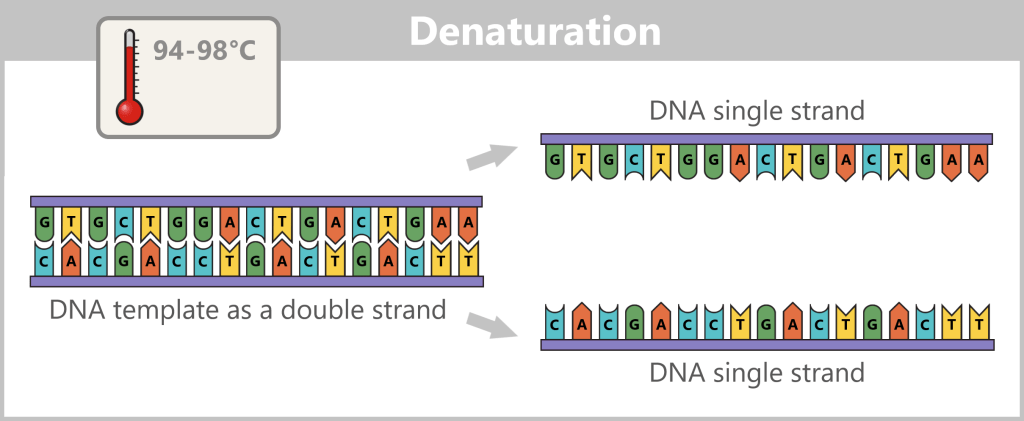
Next, the primers bind to their complementary target sites on the single strands. This primer binding marks the starting point for the DNA polymerase.
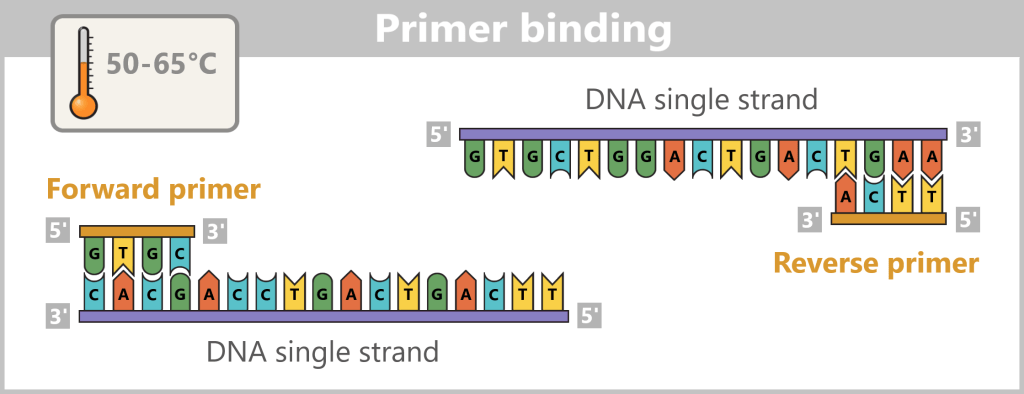
Now the synthesis of the new strands begins: The polymerase reads the template strand in the 3′-to-5′ direction and extends the copy in the opposite direction – 5′ to 3′ – while obeying the base-pairing rules (A with T, G with C).
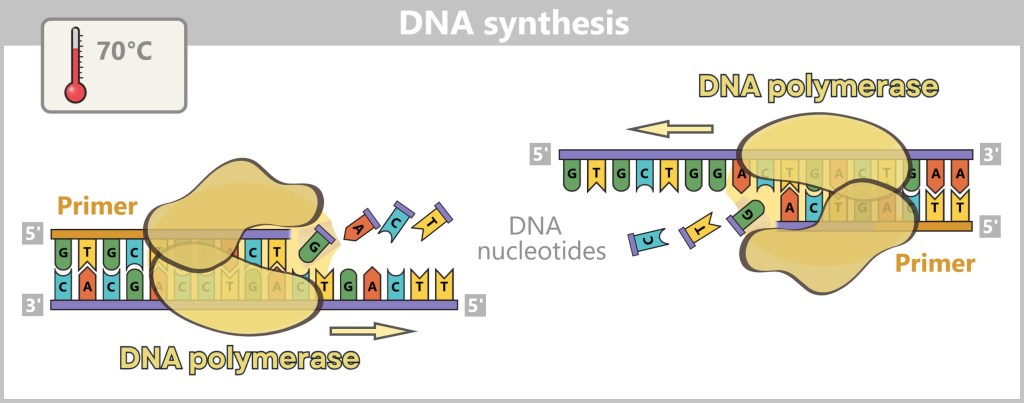
What do 3′ and 5′ mean?
The designations 3′ (three-prime) and 5′ (five-prime) come from the chemistry of the DNA backbone. They indicate at which end certain carbon atoms are located in the sugar molecule, to which new building blocks can be attached.
Imagine the DNA strand as a one-way street. The polymerase can only travel in one direction – from the 5′ end to the 3′ end. New building blocks can only be chemically attached at the 3′ end. Therefore, the polymerase reads the ‘old’ strand backwards (3′ → 5′) and builds the new strand forwards (5′ → 3′).
In this way, a new, complementary strand is gradually formed from a single strand – with the help of the base pairing rules: adenine (A) pairs with thymine (T), and cytosine (C) with guanine (G).
The result is perfect genetic replicas – new DNA strands that are exact copies of the original DNA.
Fluorescent Nucleotides: How Sequencing Works
Modern sequencing methods enhance this copying process with a sophisticated technique: they use fluorescently labeled dNTPs (DNA building blocks). Each of the four nucleotides (A, T, C, G) is tagged with a dye that emits a specific light signal when it is incorporated.
As the polymerase extends the DNA strands, it inserts the colored building blocks with precision. Each time a new nucleotide is incorporated, it sends out a tiny light signal – like a small flash that reveals which „letter“ has just been added. High-resolution cameras capture these light signals, enabling the DNA sequence to be reconstructed step by step.
Overview of Sequencing Methods
Several major technologies apply this principle in specific ways:
Sanger Sequencing: The classical method, in which fluorescent chain-terminating nucleotides halt DNA synthesis at random positions. The resulting fragments can be read in sequence.
Illumina Technology: A widely used high-throughput method in which millions of DNA fragments are sequenced in parallel. The fluorescent signals are recorded step by step.
SMRT Sequencing (PacBio): In this method, synthesis is observed in real time on a single DNA molecule, offering high precision and long read lengths.
a) Sanger Sequencing
The classic „letter-by-letter” reading method.
Sanger sequencing, also known as the chain-termination method, was developed in the 1970s and was the first technique to read DNA accurately and reliably. It is fundamentally based on the same principles as DNA synthesis but differs in several key aspects:
👉 Only a single primer is used.
👉 In addition to the normal DNA building blocks – the dNTPs (dATP, dCTP, dGTP, dTTP) – special fluorescent chain-terminating nucleotides, called ddNTPs (ddATP, ddCTP, ddGTP, ddTTP), are added in small amounts. When a ddNTP is incorporated, DNA synthesis stops precisely at that point. Each of the four ddNTP types carries a different fluorescent color corresponding to its base.
The process – step by step
For sequencing, four separate reaction mixtures are prepared. Each contains:
- the single-stranded target DNA,
- a primer (starting point for synthesis),
- a DNA polymerase,
- normal dNTPs,
- and one type of the fluorescently labeled ddNTPs.

Each mixture contains the same basic components but differs in the specific type of fluorescently labeled chain-terminating nucleotides (ddNTPs) added.
After primer binding, the polymerase reads the DNA strand and incorporates the complementary nucleotides to build a new strand. If a ddNTP is randomly incorporated, synthesis terminates precisely at that position.
This generates many DNA fragments of varying lengths – each ending with a fluorescently labeled nucleotide. Since each of the four reactions contains only one type of ddNTP, the terminal base of each fragment is known precisely.

Each reaction mixture contains only one type of modified DNA building block (ddATP, ddTTP, ddCTP, ddGTP). When such a „stop” nucleotide is incorporated during DNA synthesis, the copying process halts precisely at that point. In the ddATP mixture, synthesis stops upon incorporation of a modified adenine (A); in the ddTTP mixture, it ends with a modified thymine (T). Likewise, ddCTP and ddGTP terminate synthesis when a modified cytosine (C) or guanine (G) is added. This procedure generates many DNA fragments of varying lengths, each ending with a specific stop nucleotide. The goal is to produce all theoretically possible fragments so that the complete DNA sequence can be read.
Sorting and Analysis
The DNA fragments are then denatured (converted into single strands) and sorted by length using gel electrophoresis. The negatively charged fragments migrate through a gel towards the positive electrode. Smaller fragments move faster, larger ones slower, creating an orderly „ladder“ of fragments.
A laser then excites the fluorescent dyes attached to the terminal bases. The resulting light signals are captured by a detector. Each signal corresponds to a specific base at a specific position. The sequence of light signals thus directly reveals the DNA sequence – much like reading a book letter by letter.
Since the generated fragments are complementary to the original DNA, the original base sequence can be deduced precisely from them.
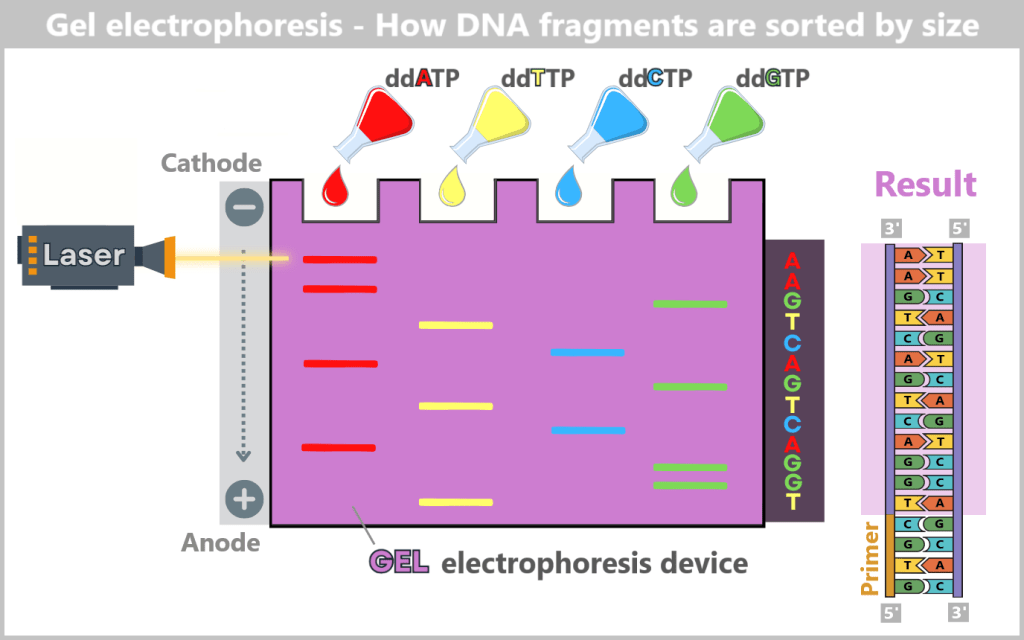
In the reaction mixtures, DNA fragments of varying lengths are generated, each ending with the same chain-terminating nucleotide – either adenine, thymine, guanine, or cytosine, depending on the mixture. These reaction mixtures are applied to a gel. When an electric field is applied, the negatively charged DNA fragments migrate from the cathode (−) toward the anode (+). The size of the fragments determines their migration speed: smaller fragments move faster through the gel’s fine pores and reach the anode first, while larger fragments move more slowly. By reading the fluorescent signals at the fragment ends, the exact order of the bases can be determined, allowing the DNA sequence to be reconstructed step by step.
Why is Sanger sequencing still considered the gold standard today?
Even half a century after its development, the Sanger method remains indispensable in many areas of molecular biology and medicine. The reason: reliability and precision.
Compared to modern high-throughput methods, Sanger sequencing is slower and suitable only for analyzing smaller DNA segments – not entire genomes. But that is precisely where its strength lies:
- Targeted questions, such as checking individual genes,
- Detection of specific mutations, or
- Validation of critical results previously identified using other methods
can be answered with precision, clarity, and interpretability using Sanger sequencing – often so clearly that the sequence of bases can be read directly from the sequencing plot.
In medical diagnostics – such as the analysis of inherited diseases or in quality control of genetic engineering procedures – this high level of accuracy is crucial. Even a single misread base can have life-altering consequences.
Another advantage: the method is standardized worldwide, has been reliably used for decades, and is governed by well-defined quality guidelines.
A Classic with Staying Power
In a world where new technologies constantly emerge, Sanger sequencing remains a reliable anchor – the old-timer among sequencing methods: not the fastest, but extremely robust, proven, and reliable. And sometimes that’s exactly what matters.
b) Illumina Sequencing
The High-Speed Copy Machine
Sanger sequencing is like precise hand-crafted work: each DNA strand is decoded step by step – reliable, but slow and expensive.
Imagine having to copy an entire book – or even an entire library – while being allowed to write down only one letter per minute.
This is precisely where the problem lies: modern cancer research, the decoding of rare diseases and the genetic monitoring of pandemics require the analysis of large amounts of data – i.e. many and/or very long DNA segments. This requires a method that is not only accurate, but also fast and affordable.
This is precisely where Illumina sequencing comes into play. It has elevated the principle of DNA decoding from manual work to industrial mass production and is now one of the most commonly used methods for high-throughput sequencing (Next Generation Sequencing, NGS). Instead of individual letters, entire pages are now read simultaneously – millions of times, in parallel, with high precision and cost-efficiency.
Unlike the classic Sanger method, where each DNA fragment is analyzed individually, Illumina operates with massive parallelization – meaning that many DNA fragments are amplified and sequenced simultaneously. How does this work?
Step 1: Preparing the DNA Fragments
– Turning the DNA into a „Lego Construction Site” –
First, the DNA sample to be analyzed is broken down into many short pieces – called fragments – typically 100 to 300 base pairs long. Small synthetic DNA sequences, known as adapters, are then attached to both ends of these fragments. You can think of them as LEGO connectors. On the one hand, they act as molecular docking sites that allow the DNA fragments to bind to a special carrier – the flow cell chip. On the other hand, they serve as binding sites for universal primers.
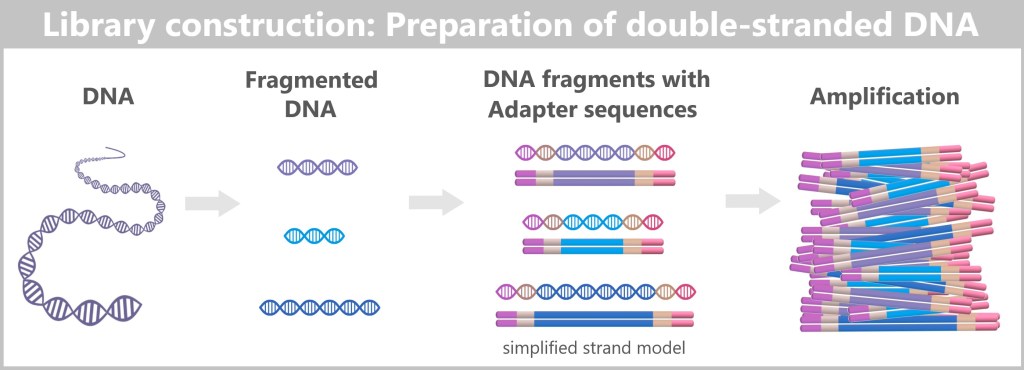
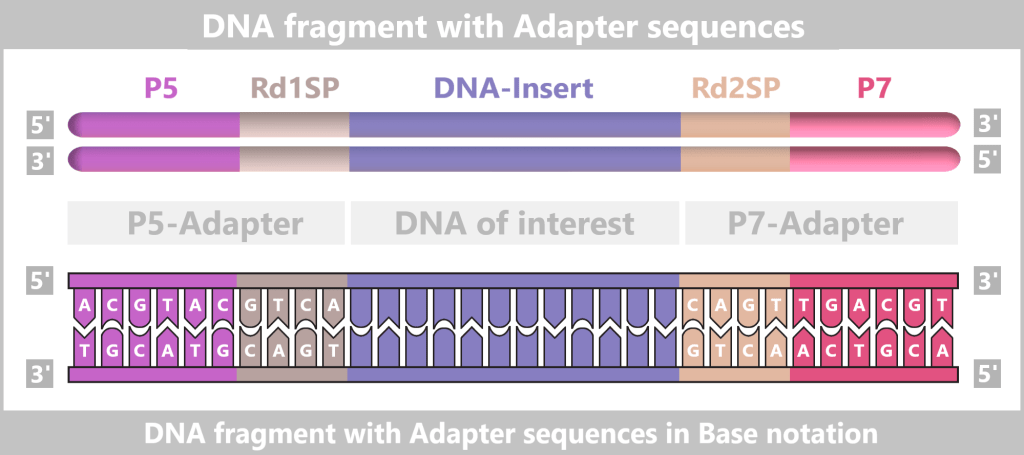
The P5/P7 sequences serve to attach the fragments to the flow cell. The Rd1SP/Rd2SP sequences act as binding sites for universal primers.
Denaturation separates the prepared double-stranded DNA fragments into single strands.
Step 2: Bridge Amplification
– The Dance of DNA on the Flow Cell –
At the heart of Illumina technology is the flow cell – a glass-like plate covered with millions of tiny DNA docking sites (short DNA segments called oligonucleotides) that are firmly anchored to the surface.

The single-stranded DNA fragments bind to the anchors via their adapters. Then the molecular copying process begins: complementary copies of the DNA snippets are produced, which are now firmly anchored to the flow cell.
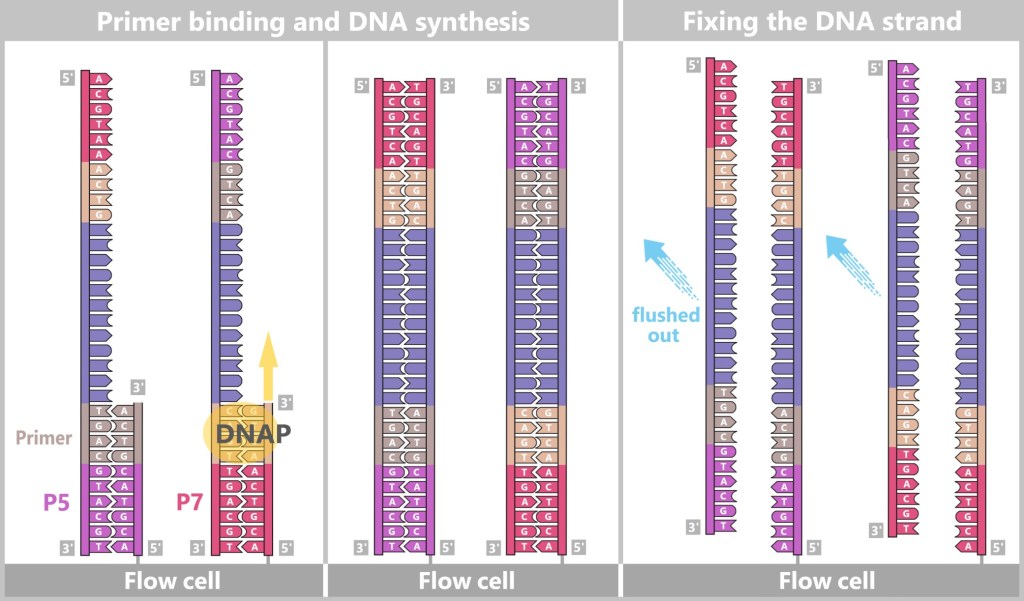
Left: Primers bind to the adapters (P5, P7), and DNA polymerase (DNAP) initiates the synthesis of a new complementary strand.
Center: The DNA polymerase then synthesizes the first strand.
Right: The newly formed DNA double strand is separated. After denaturation, the original strand is no longer connected to the flow cell and is flushed out. The newly synthesized strand remains firmly bound to the flow cell with its 5′ end.
Then a fascinating process begins: the DNA strands bend to form small bridges, as their free adapters attach to neighboring oligonucleotides on the flow cell. At these points, they are copied again. After copying, the bridge is dissolved, and the number of anchored DNA strands doubles. This process, known as bridge amplification, is repeated dozens of times. In the end, dense clusters of millions of identical copies of a single DNA fragment are formed.
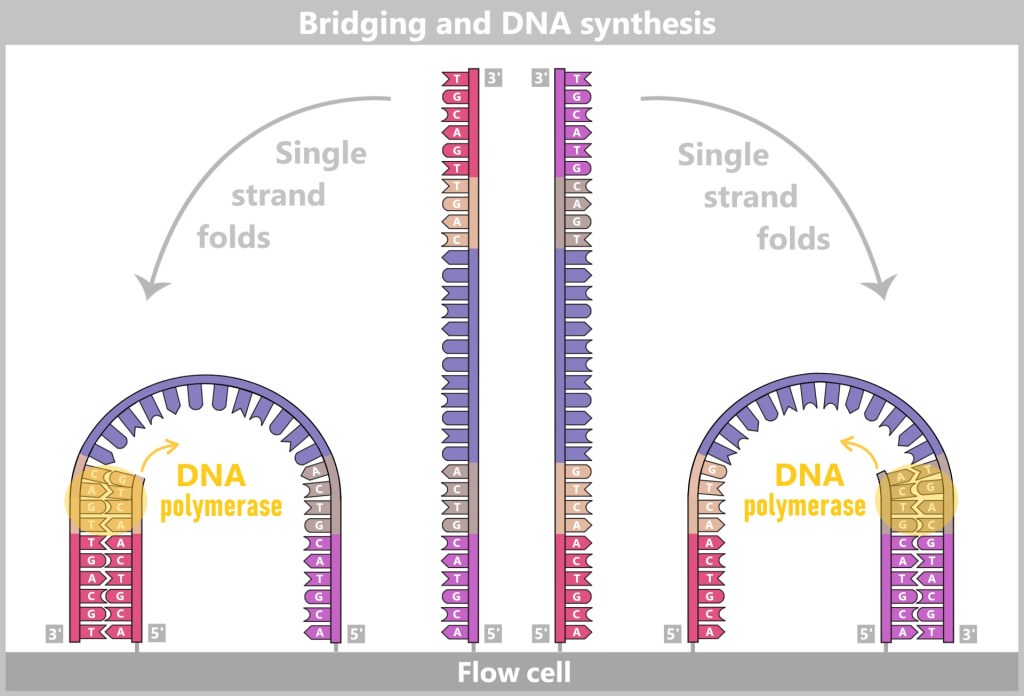
The anchored single strands fold and connect with neighboring anchors on the flow cell, forming a bridge structure. The copying process is then initiated.
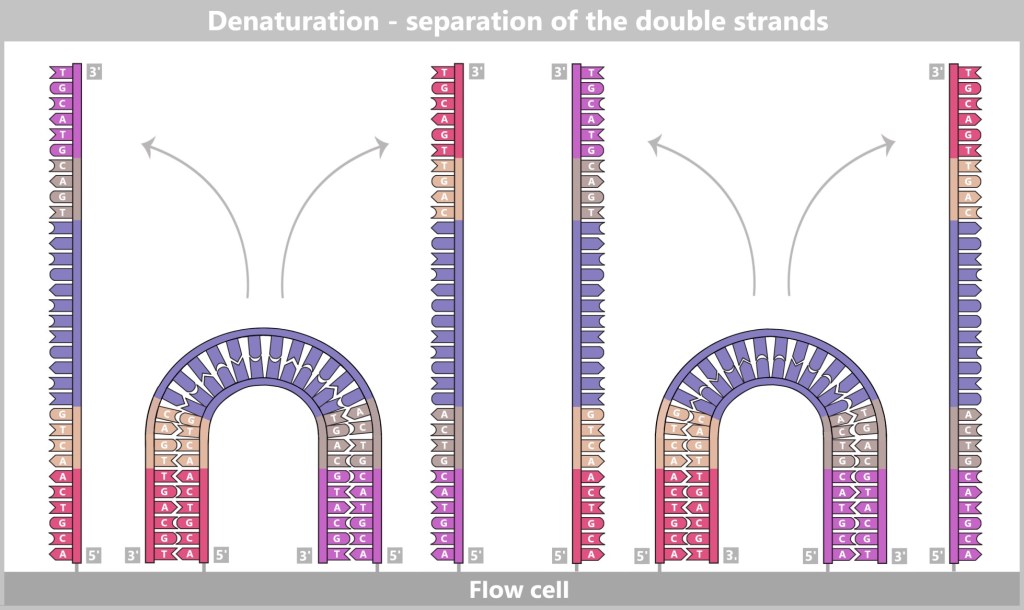
After the copying process, the bridge double strands are separated. The number of firmly anchored DNA strands doubles, making them ready for further amplification.
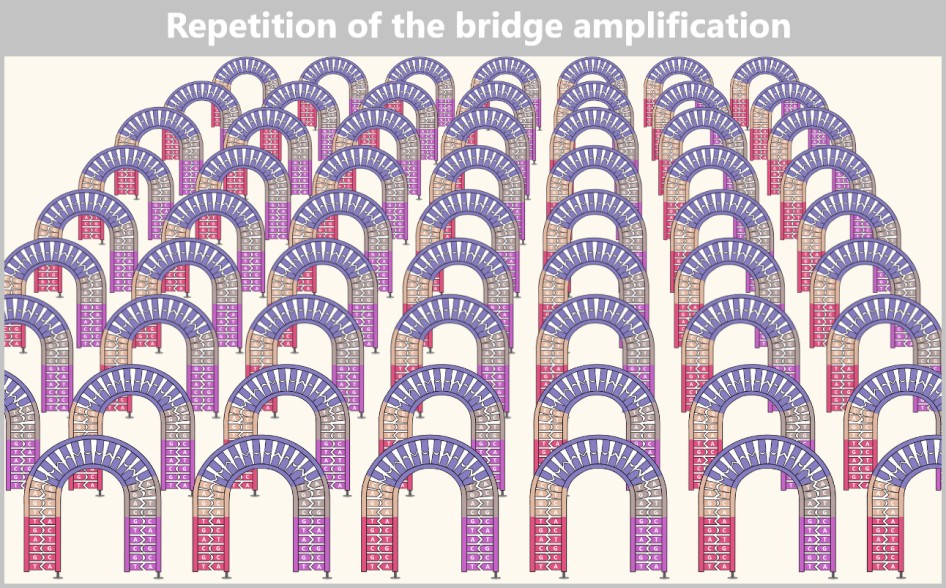
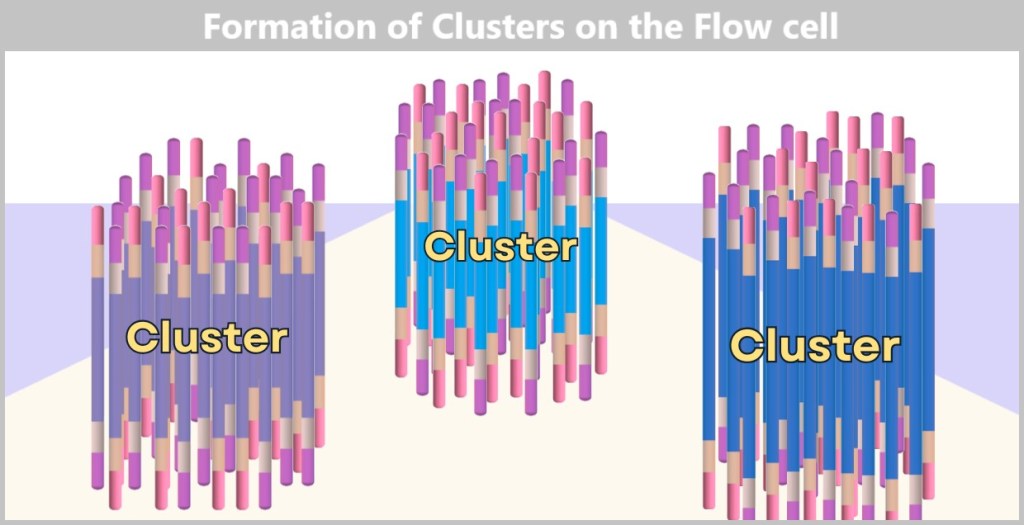
Each cluster consists of numerous copies of a single DNA fragment. In this illustration, only three clusters are shown as examples. In reality, millions of such clusters cover a flow cell, enabling high sequencing capacity.
The result is a densely packed ‘map’ consisting of millions of DNA clusters, each containing only a specific sequence – ideal for simultaneous reading.
Without this replication, reading DNA would be like trying to spot a single firefly in the wind. The clusters, on the other hand, make the DNA bases (A, T, C, G) clearly visible – like bright neon lettering in the dark.
Step 3: Sequencing-by-Synthesis
– The Light Show of DNA Bases –
Now the actual sequencing by synthesis begins. Once again, the principle of a „molecular copying machine with color sensors“ is used – but in an optimized form:
At the beginning, the universal primers bind to the corresponding adapter sites of the DNA fragments in each DNA cluster.
Then modified dNTPs – so-called RT-dNTPs (reversibly terminating nucleotides) – are added to the reaction solution. Each of the four DNA building blocks (A, T, G, C) is labelled with its own fluorescent color and carries a reversible blocker.
Only one nucleotide per cycle can be incorporated because the block temporarily stops further strand formation. Unincorporated RT-dNTPs are washed away.
After each incorporation step, the flow cell is scanned with a high-resolution camera. Each fluorescent signal corresponds to an incorporated nucleotide, and its color indicates which base has just been added.
Afterwards, the blockage and dye marking are chemically removed, and the next cycle begins.
This process is repeated cycle by cycle until each DNA strand is completely read.

Step 4: Data Analysis
– The Supercomputer as Puzzle Master –
After the „light show„, millions of photos are available – one for each cycle and each cluster. Each image shows which color (and thus which letter: A, T, C, or G) was added to each cluster.
Now powerful computers come into play – equipped with software that works as precisely as a detective piecing together a cut-up book from a stack of numbered Polaroids. Each cluster on the sequencing chip is like a small crime scene, each color pixel a clue. And from millions of such clues, a story gradually emerges – the story of DNA.

The color patterns are translated into sequences of letters, known as „reads”: tiny pieces of text from an enormous genome novel. In the end, a flood of such fragments piles up – like a mountain of cut-up book pages, scattered loosely about.
This is precisely where the real art begins: bioinformatics takes the stage. Using specialized algorithms, the snippets are analyzed, sorted, and stitched together – constantly searching for overlapping regions, familiar patterns, and known structures.
Piece by piece, the bigger picture comes back together – until, in the end, the original DNA sequence becomes visible, like a reconstructed book that suddenly makes sense.
The real „magic” therefore doesn’t happen in the flow cell, but in the computer! Without the software, the images would be nothing more than colorful flickering – but with it, they become the key to medical breakthroughs.
A more detailed yet clear explanation of the Illumina sequencing technology method can be found in the video „Illumina Sequencing Technology“.
Modern high-throughput sequencing platforms such as those from Illumina can generate the raw data of a complete human genome within just a few days – at pure sequencing costs that are significantly lower than the price of a premium smartphone.
This technology has revolutionized genome research and made it considerably more accessible: today, it is an extremely versatile tool – fast enough for real-time analysis of pandemics, precise enough to form the basis for personalized cancer therapies when combined with robust bioinformatics and clinical expertise.
Illumina sequencing is now considered the industry standard for large sequencing projects – such as the analysis of whole genomes, gene expression research, and modern cancer diagnostics.
And best of all: While you are reading this text, somewhere in the world a Flow Cell is decoding millions of DNA fragments.
However, Illumina technology also has its limitations. It reaches its limits particularly with long, repetitive DNA segments, such as those found in some chromosome regions. It is like trying to reconstruct a song from thousands of 3-second snippets. For such challenges, Illumina is often combined with other sequencing methods that can reliably capture long segments – like an investigator who needs both close-ups and panoramic images to understand the entire crime scene.
c) Single-Molecule Real-Time (SMRT) Sequencing
The Novel in One Go
Imagine you want to read a long novel – full of recurring chapters and hidden clues that span many pages. If you cut the book into thousands of small snippets, as in Illumina sequencing, read them individually and then put them together like a jigsaw puzzle, there is a risk that chapters will be missing, mixed up or incomplete. The secret messages of the story – what the novel actually wants to tell us – could be lost or appear only fragmentarily.
This is where PacBio’s Single Molecule Real-Time (SMRT) sequencing shines: it reads entire chapters in one go – no snippets, no breaks.
Instead of breaking the DNA apart, PacBio keeps it in very long fragments – often tens of thousands of base pairs at a time, sequenced as a whole. These strands enter tiny stages in a SMRT cell, called nanowells (Zero-Mode Waveguides). They are so small that they can hold only a single DNA molecule and a polymerase. Each of these stages allows light to act on just one tiny spot – exactly where the polymerase reads the DNA. Like a spotlight highlighting an actor during a monologue on stage.
The polymerase plays the leading role: it adds building block by building block (A, T, C, or G) to the DNA copy. Each of these blocks lights up in a different color, like neon lights at a DJ set. With every addition, a flash of light is emitted and recorded in real time by a camera – a dancing light code that reveals the DNA sequence.
The video Introduction to SMRT Sequencing by PacBio illustrates this process impressively.
Let’s analyze the individual steps of this fascinating process in detail.
Step 1: Preparation
– DNA Loops for Continuous Reading –
Even with PacBio, the DNA to be analyzed is first fragmented – but into much larger pieces than with Illumina, usually 10,000 to over 20,000 base pairs long. These fragments are then converted into circular DNA molecules:
So-called Hairpin adapters are added to both ends – small loops of DNA that close the fragment onto itself.
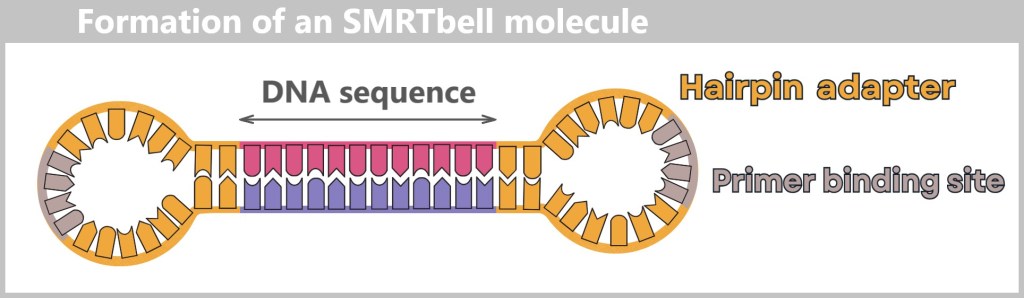
The adapters contain a primer binding site, which later provides the starting point for the polymerase.
Subsequent denaturation produces a circular, single-stranded SMRTbell molecule in the form of an open loop.
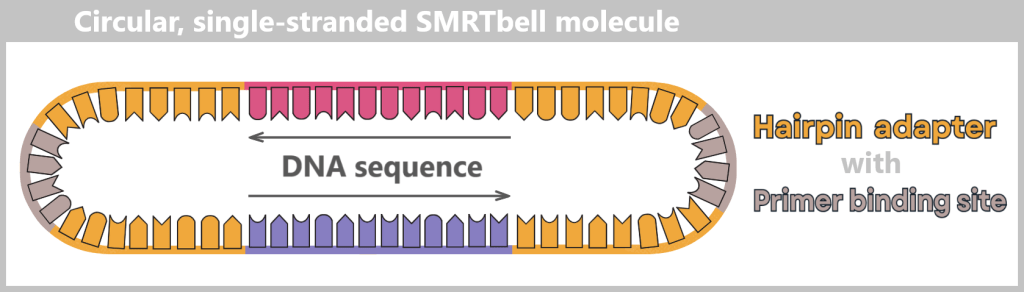
Denaturation converts the double-stranded template into a single-stranded SMRTbell molecule. It forms a circular structure, shown here as an open loop.
The result is a SMRTbell: a closed DNA loop that can be read repeatedly by the polymerase – like a model train running around a circular track.
Each hairpin adapter contains a defined binding site to which the universal primer binds. This is the starting point for the polymerase.
The DNA polymerase binds to the primer complex, forming a fully functional replication complex: polymerase + primer + SMRTbell.
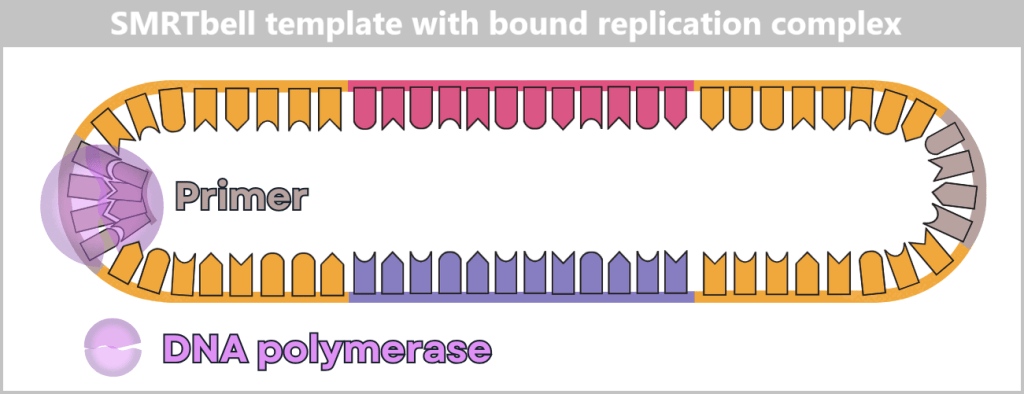
The primer is bound to the adapter binding site, the polymerase to the primer. This complex is ready to start DNA synthesis.
Step 2: The Sequencing Stage
– Light Shows at the Nanoscale –
Now it gets spectacular: the prepared replication complexes are guided into their tiny observation chambers – the Zero-Mode Waveguides (ZMWs). These nanowells are so small that typically only a single SMRTbell template fits inside. The polymerase is anchored firmly at the bottom of the ZMW, ready for action.
The start signal comes with the addition of the four nucleotides (A, C, G, T), each labeled with its own fluorescent dye.
The polymerase now begins its work – it reads the DNA strand and synthesizes a complementary strand. The crucial event occurs the moment it incorporates a nucleotide:
Each nucleotide emits a brief, color-coded flash of light.
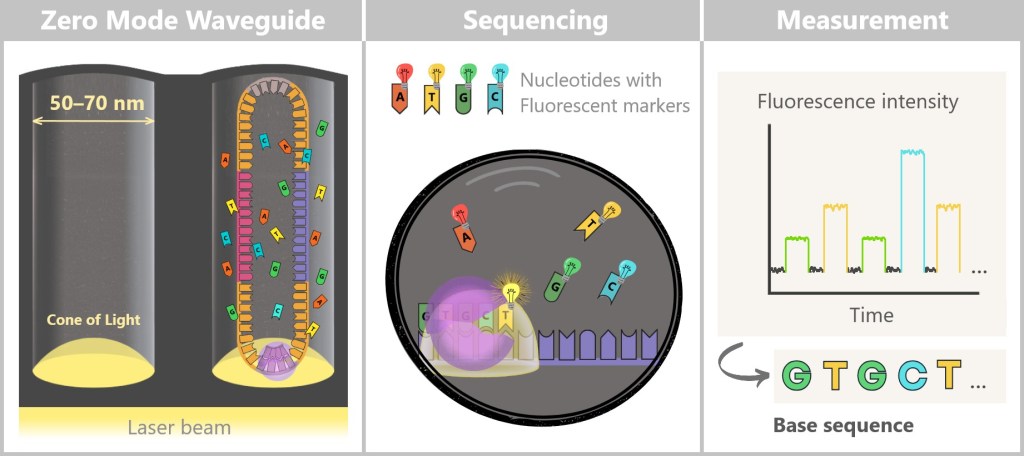
Left: At the bottom of the Zero-Mode Waveguide (ZMW), a laser beam enters and generates what is known as an evanescent field (shown as a cone of light). Unlike normal light, this field does not propagate throughout the solution but decays exponentially within only 20–30 nanometers. This creates an extremely small excitation volume in which the fluorescent dyes of the nucleotides can be excited.
The left zero mode waveguide (ZMW) contains a single replication complex (polymerase + SMRTbell). The polymerase (purple) is firmly anchored to the (glass) bottom. In the presence of the fluorescently labelled nucleotides, the polymerase begins its work.
Centre: The four nucleotides (A, T, G, C) carry different fluorescent markers. As soon as the polymerase incorporates a nucleotide, it lights up briefly before the dye is cleaved off.
Right: These flashes of light are measured in real time and translated into fluorescence signals. This is how the base sequence of the DNA is determined step by step.
The luminescence occurs in real time, at the exact moment of insertion – hence the name: Single Molecule Real-Time Sequencing. A sensitive detector records these light signals – like a molecular live microscope at work.
Step 3: The Long-Range Secret
– Repetition Makes It Robust –
Since PacBio reads very long DNA segments at once – comparable to reading entire chapters of a book instead of individual verses – more individual errors occur in a single run than with Illumina. This is exactly where the ring-shaped SMRTbell structure comes into play. The DNA polymerase can circle the ring molecule multiple times, like a train conductor. In doing so, the same DNA fragment is read over and over again – like a reader revisiting the same passage until it is fully mastered.
A computer compares the repeated reads with one another and filters out the errors – like an editor polishing a manuscript. This results in a particularly precise consensus sequence.
PacBio calls this approach HiFi reads – high-precision single-molecule readings.
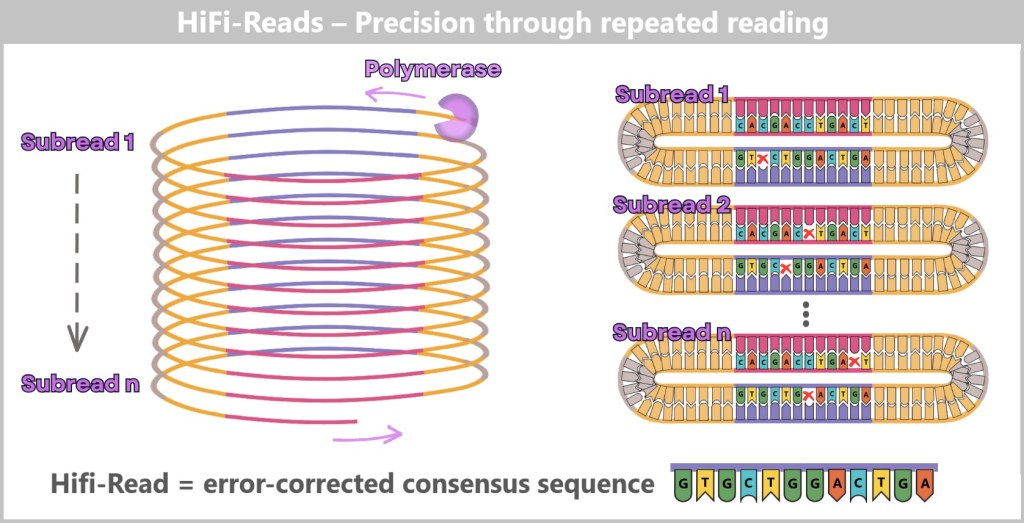
The polymerase circles the SMRTbell template multiple times, generating multiple subreads. Each cycle corresponds to a complete reading of the DNA sequence. Individual subreads may contain random errors (x), but by comparing many repetitions, a highly accurate consensus is formed – the so-called HiFi read (High-Fidelity Read).
Additional info: How the polymerase loops around the DNA multiple times
First sequencing run
The polymerase reads the single-stranded template strand and synthesizes a complementary strand.
The result is a complete double helix – the original template strand and the newly created daughter strand are now firmly bound together.
This first, uninterrupted round produces a subread – a single base sequence generated by the polymerase in a single pass.
Second and subsequent sequencing runs
Now the question arises: how can the polymerase continue reading when the DNA ring is already double-stranded?
This is where Φ29 phage DNA polymerase comes into play – a specially modified enzyme variant with two key properties:
High processivity: It remains bound to the DNA strand for a very long time and hardly ever detaches – this enables long reads.
Strong strand-displacement activity: It can break the hydrogen bonds between the template and daughter strands.
The polymerase runs into the double helix, separates the strands locally and displaces the old daughter strand while simultaneously synthesizing a new one – like a molecular bulldozer.
The previously synthesized strand is not degraded; it remains as a loose, single-stranded DNA fragment in the nanowell. With each subsequent round, this process repeats: the polymerase reads the same template again – producing multiple subreads from the same DNA molecule.
Step 4: Data Analysis
– Turning Flashes of Light into Readable DNA –
Once all the light signals have been captured, the real detective work begins: data analysis.
The first step is called base calling: the recorded fluorescence signals – encoded in time and color – are translated into nucleotide sequences – A, T, G, or C.
Next, the raw data from each polymerase pass is cleaned up: the hairpin adapter sequences are bioinformatically trimmed. What remains are the subreads – the uncut reading records of each individual polymerase pass through the DNA chapter text.
But it is only the next step that reveals the whole truth: like a master philological detective comparing multiple copies of a manuscript, the algorithm combines all subreads of the same SMRTbell molecule. This comparison – known as Circular Consensus sequencing (CCS) – produces a highly accurate HiFi read: an error-corrected master copy that reproduces the DNA chapter word for word.
Only now is the text fragment ready for final classification – and in the process it goes through several stages:
First, a quality check – just like a meticulous editor reviewing the text for clarity and consistency.
This is followed by alignment: the sequence is classified like a new chapter in the correct compartment of a reference genome shelf.
Finally, assembly takes place – the final act, in which all chapters are brought together to form a complete novel of the genome.
The video PacBio Sequencing – How it Works clearly summarizes the steps involved.
Applications and Advantages – The Strengths of Long-Read Sequencing
PacBio SMRT technology has a particular strength: it can read very long DNA segments in one go – with highest accuracy. This makes it ideal for:
- Detecting complex genomic structures
- Analysis of repetitive sequences
- Distinguishing very similar gene variants (e.g., in immune receptors or cancer genetics)
- De novo sequencing – reading entirely new genomes without a reference
- Detecting epigenetic modifications (e.g. methylations)
How can SMRT sequencing make epigenetic modifications visible?
As already described, epigenetic mechanisms play a central role in targeted gene regulation. These are small chemical markers – such as methyl groups – that are attached to specific DNA bases. They do not change the genetic sequence itself, but influence when and how often genes are read – depending on cell type, environment or stage of development.
They function like notes or markings in the margin of a book: the text remains the same, but the „reading” changes – some passages are emphasized, others skipped over.
If this finely tuned system falls out of balance, it can have far-reaching consequences – such as in cancer, autoimmune diseases, or mental disorders.
Unlike Illumina technology, which reads a chemically modified copy of the DNA, SMRT analyses the original DNA in real time. It measures not only the base sequence, but also the dwell time of the DNA polymerase at each base – known as incorporation kinetics.
This is precisely where the key to detecting epigenetic modifications lies: methylation and other chemical changes influence these kinetics. They act like small obstacles or skid marks on the DNA strand. The polymerase „lingers“ longer at such sites – and it is exactly this delay that is directly captured by the SMRT camera.
SMRT is particularly well suited for the direct detection of certain chemical markers, such as methyl groups on adenine or cytosine bases, which play an important role especially in bacteria and plants. For cytosine methylation (5mC), which is central in human epigenetics, direct detection is less sensitive but can be reliably derived using specialized bioinformatics tools such as pb-CpG-tools.
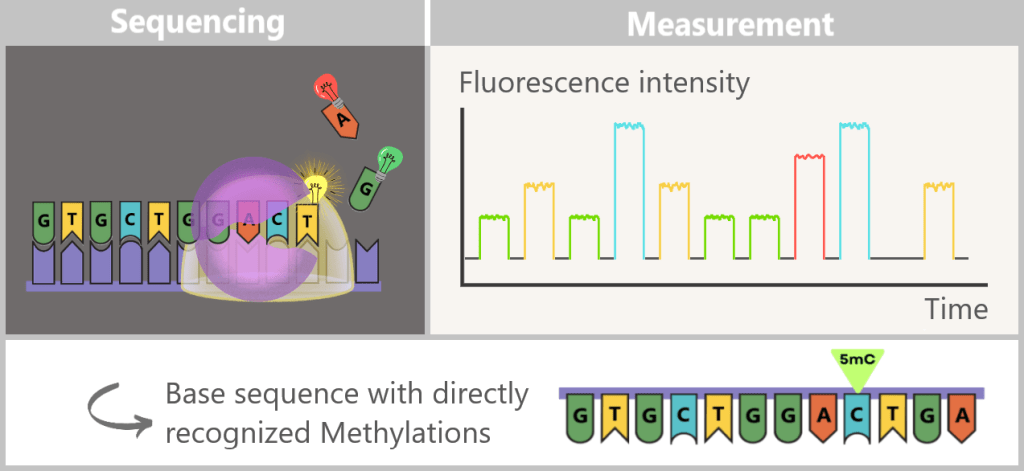
How SMRT Sequencing Reveals Epigenetic Modifications
During SMRT sequencing, the fluorescence signals of the incorporated nucleotides are measured in real time. At the same time, modifications such as 5-methylcytosine (5mC) can be detected, as they slightly alter the kinetics of the polymerase.
The key advantage: the extremely long read lengths reveal how methylation patterns are intertwined across large genomic regions with other factors, such as:
- gene variants,
- haplotypes (groups of co-inherited gene variants),
- repetitive sequences.
In this way, PacBio provides not only the genetic sequence of letters but also a „map” of epigenetic regulation – revealing which „pages” functionally belong together and how this network influences disease processes such as cancer or neurological disorders.
Defective Scissors in the Genome: How PacBio Unmasks Splice-Site Variants
In the section Alternative Splicing: The Flexible Kitchen of Genes, we described in simplified terms how our DNA, as a molecular cookbook, produces recipes in the form of mRNA transcripts. These transport the genetic information from the cell nucleus to the ribosomes in the cytoplasm – the cell’s „protein factories”.
The recipe is subsequently adjusted: molecular scissors remove certain ingredients (introns) and recombine other building blocks (exons). This process is controlled by splice sites – markers that are „copied“ from the DNA. These can be thought of as cutting instructions: they determine which parts of the recipe are used and how they are assembled.
But what happens if these cutting instructions are faulty? The consequences can be severe:
- Exons are skipped → parts of the blueprint are missing.
- Introns are erroneously retained → „junk” is incorporated into the protein.
- Incorrect splice sites are created → the mRNA text becomes distorted.
Such splice-site variants often lead to defective or non-functional proteins – with possible consequences such as cancer, genetic disorders (e.g. β-thalassemia), or neurological diseases. Since protein biosynthesis depends directly on the mRNA sequence, detecting such errors is crucial.
The key lies in PacBio’s ability to analyze complete mRNA molecules in one piece – usually 500 to 5,000 nucleotides long, sometimes significantly longer. Since PacBio reads DNA, the mRNA is first converted into a stable copy (cDNA, complementary DNA). This can then be sequenced in its entirety. This allows us to see exactly how the recipe is structured and which ingredients are required in which order – no transcription or splicing errors remain hidden.
In addition, the underlying DNA segment can also be sequenced. Bioinformatic tools compare cDNA and DNA sequences with reference data, revealing deviations in the splicing pattern and their genetic cause.
The DNA shows: Here the instruction is faulty.
The cDNA shows: Here the recipe is mutilated.
Only the combination of both pieces of information creates a sound basis for precise diagnoses and targeted therapeutic approaches.
Conclusion – When you want to tell the whole story
When it comes to telling the entire story of a DNA segment in one go – without cutting it into pieces or piecing it back together – SMRT sequencing is the method of choice.
PacBio-SMRT is the archaeologist of genomics: it sees the whole picture where others only provide fragments. In 2022, it played a key role in closing the last gaps in the human genome.
PacBio SMRT technology is not a mass-market product but a precision instrument. It is slower and more expensive per base pair than Illumina – but in return, it provides greater detail and versatility.
In practice, both technologies are often combined: Illumina for breadth, PacBio for depth. Together, they provide a complete picture – like a satellite image and a close-up merged into a single panorama.

4.4.2. Method 2: High-Tech Tunnel –Electrically Scanning DNA
While in the molecular copying machine with color sensors the DNA is first replicated and then „photographed“ in an image, the second method works in a radically different way: the DNA is passed through a high-tech tunnel and scanned electronically in real time.
This is the principle behind Oxford Nanopore sequencing.
a) Oxford Nanopore Sequencing
DNA Through the Eye of a Needle
Imagine reading a rope by letting it slowly slide through your fingers – sensing every subtle irregularity: thickness, dents, knots. That’s exactly how Oxford Nanopore Sequencing (ONT) works: it „feels” the DNA as it passes through a tiny biological eyelet.
This eyelet – a so-called nanopore channel – is embedded in a membrane through which an electrical current flows. As soon as a single DNA molecule is threaded through this high-tech tunnel, the bases (A, T, G, or C) influence the current flow with different intensities – like pebbles of different shapes in a stream. These changes generate characteristic electrical signals that are recorded in real time.
ONT does not read light signals, but rather current patterns – an electrical „finger reading” of DNA.
A single thread – read directly
Before sequencing begins, the DNA is coupled to a motor protein that pulls it precisely through the pore – not too fast, not too slow. Unlike Illumina or PacBio, there is no need for polymerase, fluorescence, or amplification: the original DNA is read directly – a major advantage for sensitive or damaged samples.

The sequencing cell consists of two chambers: the cis chamber (top) and the trans chamber (bottom), separated by a membrane with embedded nanopores. On the left, a DNA molecule has docked onto a nanopore with the help of a motor protein. On the right, an empty nanopore is shown, through which a constant ionic current flows. The applied voltage between the negative electrode (cis) and the positive electrode (trans) drives the ion flow. The ionic current is measured in the well (a small channel in the chip), as shown in the current/time diagram. As long as no DNA passes through the pore, the current remains constant.
And the best part: the DNA can be extremely long. ONT holds the world record for the longest DNA ever sequenced – over two million base pairs in a single read. Entire chromosomal regions can thus be captured without interruption – like reading a novel in one single breath.
From noise to meaning – the art of signal interpretation
The raw electrical signals generated as the DNA passes through the pore initially resemble a noisy radio channel. However, with the help of specialized algorithms – known as basecalling – the current noise is translated into a clear sequence of letters: A, T, G, or C.

1) A motor protein pulls the DNA through the pore in a controlled manner. The negatively charged DNA moves from the cis side to the trans side due to the applied voltage. In the process, the individual bases (A, T, G, C) affect the ionic current in a specific way.
2) A graph shows the change in the measured current over time. Each base combination generates a characteristic signal that is decoded by algorithms.
3) A computer analyzes the electrical signals and determines the sequence of bases from them.
Unlike PacBio, ONT does not distinguish bases individually, but in groups of five to six nucleotides that occupy the pore simultaneously. Each of these „5-mer” generates a characteristic current signal. This increases the reading speed but makes interpretation more complex – somewhat like recognizing words in an unfamiliar dialect. Homopolymers (long repeats of the same base, e.g. „AAAAA”) are particularly challenging.
AI-powered basecalling software is continuously improving. Error rates have dropped dramatically, and ONT reads now achieve high accuracy – especially when the dataset is large enough for statistical multiple coverage of each DNA base through repeated, random sampling.
There is a cool visual explanation in the video How nanopore sequencing works by Oxford Nanopore Technologies.
For those who prefer a more detailed explanation, additional background information can be found here.
Key Strengths – Fast, Portable, Versatile
What makes ONT special:
Portability: Devices like the MinION fit in a pocket. They require only a laptop and a power source – ideal for field research, space missions, or rapid analyses during outbreaks (e.g., Ebola or SARS-CoV-2).
Speed: The DNA is read live. Initial sequencing data is often available within minutes – a priceless advantage in clinical emergencies.
Direct epigenetics: Like PacBio, ONT can directly detect methylation and other epigenetic modifications, as they subtly affect the electrical signal – without any additional treatment or reagents.
Direct RNA sequencing: RNA molecules can also be read without prior conversion to cDNA – a true unique feature.
Why Direct RNA Sequencing Matters
Other sequencing methods, like PacBio, must first convert mRNA into cDNA in order to read it. This is like copying a recipe first – small details can be lost in the process. In contrast, Oxford Nanopore technology reads the mRNA directly – without the detour through DNA.
This way, all the „marginal notes of the cookbook” are preserved: chemical modifications, variants, and even the exact length of the molecules – information that is often lost in a copy. This creates an unaltered view of gene activity – a priceless advantage for detecting complex processes such as gene regulation or faulty RNA processing.
This is a breakthrough, especially for personalized medicine: many diseases arise not only from altered DNA but also from deviations in RNA.
Practical Examples:
- Cancer diagnostics: RNA splice variants can help detect aggressive tumor forms early and guide targeted treatment.
- Hereditary diseases: For diseases such as spinal muscular atrophy or Duchenne muscular dystrophy, RNA profiles enable accurate assessment of disease progression and personalized therapies, e.g. with antisense oligonucleotides.
- Infectious medicine: Direct sequencing of viral RNA allows rapid identification of virus variants – crucial during epidemics and for selecting appropriate therapies.
Direct RNA sequencing thus helps reveal each person’s unique „recipe variant” – with all its small deviations, marginal notes, and special ingredients – enabling more precise diagnoses and tailored treatments.
Conclusion: The Genomics „Electric Sense Finger”
ONT is the sensory-rich border crosser of genomics: fast as a racehorse, portable as a Swiss Army knife and close to biological processes. Where others have to copy, it feels live – even for RNA or epigenetic traces.

4.4.3. Genome Sequencing –Technologies Compared
In the world of genomics, there is no single method – rather, there is an entire ensemble. Each sequencing platform has its own style, its strengths, and its ideal application area: from the classic solo storyteller to the high-speed streamer.
Even though all four sequencers have their place on the stage, not every one of them is suited for every performance. Someone who wants to investigate a single gene needs different tools than someone who aims to capture the entire genome in epic breadth. And anyone trying to detect rare variants must read very carefully.
Let us now take a closer look at the four main players to understand their strengths, weaknesses, and strategic roles in detail.
a) Throughput vs. Usability – Quantity Does Not Always Equal Quality
„Throughput” sounds like efficiency – as many DNA letters per minute as possible. In sequencing, it is measured in bases per run: that is, how many bases a device reads out in a single sequencing process.
Modern platforms such as Illumina’s NovaSeq X Plus achieve peak values of up to 16 terabases with short reads (100–300 bp) – that’s 16 billion letters in a single run. Like a high-performance printer that produces entire genome libraries overnight.
PacBio and Oxford Nanopore follow a different approach. They generate long reads – from 10,000–25,000 bp for PacBio HiFi reads, up to over 1 million bp for ONT. Their throughput is lower: PacBio (Revio) achieves up to 1.300 gigabases, ONT (PromethION) up to 7.000 gigabases per run. Sounds like less – and it is, if you only look at the number of bases.
But: more data does not necessarily mean more insight.
What matters is not only how much is read – but how precisely and how often. Two key concepts help to assess the informative value of sequencing data.
🔍 Quality Score – the confidence in each letter
The so-called Q-score (quality score) describes how likely it is that a read DNA letter is correct. The scale is logarithmic:
- Q20 = 99 % accuracy → 1 error per 100 Basen
- Q30 = 99,9 % accuracy → 1 error per 1.000 Basen
- Q40 = 99,99 % accuracy → 1 error per 10.000 Basen
In research, Q20 is often sufficient. In medical diagnostics, Q30 or higher is considered standard, and for high-precision applications (e.g., tumor genetics), Q40 is expected.
For raw data, Illumina typically ranges from Q30 to Q40, while PacBio (Continuous Long Reads, CLR) and ONT only achieve values between Q10 and Q15.
Sanger outperforms them all – with Q40–Q50, but only on short DNA segments. Ideal for targeted confirmation of individual variants.
So how do modern sequencing methods meet the high standards of clinical diagnostics? The answer: through coverage and bioinformatic analysis.
🔁 Coverage – repetition reveals the truth
Coverage describes how often each area of the genome has been read.
Example: 30x coverage means that each letter has been read an average of 30 times – in different fragments and in different constellations.
Why is this important? Individual measurements may contain errors. Only through repetition and statistical majority formation can genuine variants be distinguished from artefacts.
In medical diagnostics, 30× coverage is the minimum standard.
With 30–50× coverage, Illumina achieves an overall accuracy of Q35–Q40+. It reliably detects single nucleotide variants (SNVs), insertions/deletions (indels), and copy number variants (CNVs).
PacBio HiFi Reads achieve Q30–Q40 at 30x coverage and ONT Q30–35. ONT requires 50x coverage to achieve Q44 precision.
The big advantage of PacBio and ONT is that they read much longer DNA segments – often 10,000 to 100,000 bases, and in the case of ONT, even over 1 million. This enables them to recognize complex structures, repetitions and epigenetic signatures, and allows RNA analysis and phasing – i.e. the assignment of variants to paternal or maternal chromosomes. It is not the quantity that counts here, but the overall picture.
🧬 Depending on the goal – different requirements for coverage and accuracy
The required coverage and sequencing quality depend on the clinical application:
Population studies: Q30 at 30× coverage is usually sufficient to detect statistical patterns.
Cancer diagnostics: Q35 at 60–100× coverage for reliable detection of rare somatic mutations.
Liquid biopsies, mosaic analyses: Q40 at 100–300× coverage – to distinguish true signal variants from background noise.
Phasing: Long reads (PacBio, ONT) with 30× coverage are often sufficient to assign variants reliably.
📉 The dilemma: greater certainty – greater effort
Higher coverage increases confidence, but also means more data, longer computation times, larger infrastructure, and higher costs.
b) Cost-Effectiveness – Cost per Base vs. Cost per Insight
Illumina (NovaSeq/NextSeq) remains the price-performance favourite for whole genome sequencing:
👉 200–800€ for a complete genome at 30× coverage – precise for point mutations, ideal for routine analyses.
PacBio (Revio/Sequel IIe) and Oxford Nanopore (PromethION/MinION) are more expensive:
👉 700–1,200€ for 15–30× coverage – but with additional information such as long reads, structural variants, epigenetics, and RNA. This is not a luxury, but in many cases diagnostically crucial.
Oxford Nanopore also scores with a low entry cost: a MinION costs just a few thousand euros, is portable, and delivers data within hours.
👉 The simple equation „more bases = better deal” falls short. What matters is what can be read and understood from the data.
🧾 Hidden costs – reading the genome is cheaper than understanding it
A human genome for under 300€? Technically yes – but this usually only covers reagents, machine time, and raw data.
What is often missing are the hidden items:
🔧 Sample preparation
🔍 Quality control & library creation
🧠 Bioinformatics & clinical interpretation
📄 Findings report & physician consultation
These areas often account for 70–80% of the total costs.
Because: reading is cheap today – understanding remains challenging.
c) From Sample to Result – How Quickly Does the Whole Genome Speak?
Reading the genome is like a performance: the curtain rises with sample collection – but when will the final curtain fall, the clinical report?
It depends on technology and infrastructure:
Oxford Nanopore enables preliminary results within 24–48 hours – thanks to real-time analysis during sequencing.
Illumina provides data in 3–7 days with optimized workflows – the gold standard for routine diagnostic procedures.
PacBio takes 4–7 days, but offers particularly high-quality long reads and epigenetic information.
Sanger delivers targeted validations within hours – but only for small regions of interest.
Choosing a platform is therefore not just a matter of data quality – but also of clinical timing.
Those who need to act in hours rather than days turn to Oxford Nanopore. Those who require robust routine data rely on Illumina. And those who want maximum contextual depth take a bit more time with PacBio – for significantly greater insight.
The curtain falls. Yet behind the scenes, amid hope, high-tech, and human care, the search for the perfect pace for each patient continues. After all, it’s not the fastest machine that matters – but the right rhythm for the human story behind it.
d) Clinical Applications – Which Technology Fits Which Purpose?
Genome sequencing has long been more than just a research tool – it has become a key instrument for planning individualized therapies. But which method is suitable for which task?
A look at six key application areas shows how these technologies are shaping modern medicine.
| Application Area | Primary Method | Complementary Methods |
|---|---|---|
| Routine Diagnostics | Illumina | Sanger |
| Pharmacogenomics | Illumina | Sanger |
| Epigenetics | PacBio, ONT | – |
| Cancer Genomics | Illumina (SNVs), PacBio/ONT (SVs) | Sanger |
| Neonatal Emergency Diagnostics | ONT | Illumina, Sanger |
| Rare Diseases | PacBio, ONT | Illumina, Sanger |
👉 Insight: The optimal technology always depends on the clinical question – standard analysis, search for the unknown or emergency diagnostics? Validation or initial diagnosis?
e) AI as Co-Pilot – Automation Without Relinquishing Responsibility
The integration of artificial intelligence (AI) – especially deep learning – is indispensable in all modern DNA sequencing platforms (Illumina, PacBio, Oxford Nanopore). Its main applications are:
🧬 Basecalling (conversion of raw data into DNA sequences) & error correction
🔍 Quality control & real-time analysis
🧠 De novo assembly, genome analysis & variant detection
📚 Pathogenicity prediction, literature research & clinical classification
📄 Automated report templates
👉 Insight: AI transforms sequencing from a mere data-generation process into an intelligent interpretation workflow – making it central to modern genomic research and diagnostics.
But one thing remains important: AI does not (yet) think like a physician. In rare diseases, complex cases, or newly discovered genetic variants, clinical expertise remains essential. Legally, physician-validated interpretation is also mandatory – even in the age of digital genomics.
Ultimately, genome sequencing is not a solo performance – it requires the entire ensemble: precise technology, intelligent bioinformatics and clinical intuition. Because only when everyone plays their part can data be transformed into healing knowledge.
f) Comparison Table: Sanger, Illumina, PacBio, ONT
| Feature | Sanger | Illumina | PacBio | ONT |
|---|---|---|---|---|
| Sample preparation | DNA-Template, Extraction, Primer-Design, ddNTPs 🕒 ~6–12 Std. | Fragmentation, Adapter ligation 🕒 ~6–24 Std. | High DNA quality, Fragmentation, SMRTbell adapters 🕒 ~1–2 Tage | Fragmentation, Adapter with Helicase (Motor protein) 🕒 ~6–24 Std. |
| Read length | Short: 500–1.000 bp | Very short: 100–300 bp (paired-end possible) | Long: 10.000–25.000 bp (HiFi-Reads) | Ultra long: 10.000 bp – >1 Mb |
| Throughput | Very low: single reactions (~1–100 kb/run) | Very high: up to 16.000 Gb (NovaSeq X Plus), >100 samples simultaneously | Averagete: up to 1.300 Gb / SMRT-Cell (Revio) | Flexible: 100–7.000 Gb (PromethION), up to 20 Gb (MinION) |
| Accuracy | Very high: Q40–Q50 (99,99–99,999 %) | High: Q30–Q40 (99,9–99,99 %), at 30x: Q35–Q40+ | High: Q30–Q40 (HiFi-Reads), at 30x | Raw data: Q15–Q20+, at 30x: Q30–Q35, Duplex: Q44 at 50x |
| Cost per genome (EUR) (sequencing only) | – | ~200–800 (30x) (Large laboratories) | ~700–1.200 (15–30x) | ~700–1.200 (15–30x) |
| Time to result (WGS, Sample → Report) | – | 3–7 days (incl. preparation, sequencing, analysis) | 4–7 days (incl. preparation, sequencing, analysis) | 24–48 hours (real-time, incl. analysis) |
| Variant detection | SNVs, mitochondrial variants (validation) | SNVs, indels, CNVs (indirect), somatic variants, splice variants | Indels, SVs, CNVs, repeat expansions, splice variants, somatic SVs | Indels, SVs, CNVs, repeat expansions, splice variants, somatic SVs |
| RNA sequencing | – | mRNA via conversion to cDNA, splice variants | mRNA via conversion to cDNA, splice variants (long reads) | Direct RNA sequencing (without cDNA), splice variants, long transcripts |
| Epigenetic modifications | – | Indirect: only with bisulfite sequencing (complex) | Direct: methylation detection via incorporation kinetics (HiFi reads) & bioinformatic tools | Direct: methylation via signal changes (e.g., 5mC, 6mA) |
| Place of deployment | Laboratory: Stationary equipment | Laboratory: Large stationary equipment (e.g. NovaSeq) | Laboratory: Large stationary equipment (Revio) | Flexible: Laboratory (PromethION), portable/point-of-care (MinION) |
| Main advantages | Highest precision, ideal for validation | High throughput, cost-effective, precise for SNVs/indels, suitable for routine use | Long accurate reads (HiFi), ideal for SVs, epigenetics, phasing | Ultra-long reads, mobile, fast, direct RNA and epigenetic detection |
| Limitations | Low throughput, WGS impractical, expensive | Short reads, limited for SVs/epigenetics, complex library preparation | More expensive, medium throughput, longer preparation time | Lower raw data quality, dependent on bioinformatics |
| Role in personalized medicine | Validation of critical SNVs (pharmacogenomics, cancer), mitochondrial analyses | Routine diagnostics, pharmacogenomics, cancer genomics (SNVs/indels), rare diseases | Epigenetics, cancer genomics (SVs), rare diseases (SVs, repeat expansions), phasing | Neonatal emergency diagnostics, epigenetics, cancer genomics (SVs), rare diseases, mobile diagnostics |
g) Key Takeaways: Between Maturity and Routine
DNA sequencing is at a pivotal turning point: in recent years, it has evolved from a purely research-based technique into a clinically relevant tool in modern medicine. Whether in oncology, rare disease diagnostics, or pharmacogenomics – genome analyses are increasingly providing answers where conventional methods reach their limits.
And yet: despite its impressive technical maturity, genome sequencing is still not a widespread standard in clinical practice.
Technology mature – infrastructure incomplete
Modern high-throughput methods such as Illumina or the long-read technologies from PacBio and Oxford Nanopore now enable highly accurate analyses at comparatively low costs. Whole genome sequencing (WGS) can be performed in specialized laboratories for a few hundred pounds per case – with a total turnaround time of less than a week. Mobile platforms such as MinION even provide initial genetic information within 48 hours in acute situations, such as with seriously ill newborns.
Despite this progress, clinical use remains fragmented. There are many reasons for this: there is a lack of standardized bioinformatic analysis protocols, interdisciplinary trained specialists, and legally compliant and economically viable integration into everyday healthcare. Above all, the interpretation of genetic variants – especially those with unclear clinical significance – requires specialized expertise that is not available in many places.
The UK as a blueprint
While many countries are still experimenting with pilot programs or making case-by-case decisions, the United Kingdom is institutionalizing genome sequencing as an integral part of public healthcare. As part of a national pilot program, every newborn will soon undergo genomic screening – a precedent in prenatal and early-childhood preventive medicine. The goal is to identify genetically driven diseases in early childhood, before clinical symptoms appear. In this way, DNA analytics evolves from an individual diagnostic tool to a population-wide preventive strategy – a paradigm shift.
This model demonstrates that the barrier to broad implementation is less technical and primarily systemic: missing data standards, ethical and legal ambiguities, and a restrictive reimbursement policy prevent widespread integration in many places. The British initiative is therefore regarded as a pioneering example of institutionalized genomic medicine.
Technological outlook
Several technological developments are likely to further accelerate widespread implementation in the near future:
🚀 Decreasing cost per genome makes sequencing economically attractive even for smaller facilities.
🚀 Decentralized, mobile systems like MinION enable diagnostics independent of location, for example in emergency rooms or rural regions.
🚀 Faster time-to-result – from sample collection to diagnosis – allows for use even in time-critical cases.
🚀 Integration of genomic data with transcriptomics, proteomics, and epigenetics (multi-omics) promises contextualized, high-resolution diagnostics.
🚀 AI-powered analytics and automated interpretation tools reduce dependence on manual evaluation and minimize sources of error.
4.5. Genome Sequencing: The Next Technological Leap
To make decoding our DNA faster, cheaper, and universally accessible in the future, research is advancing the next generation of sequencing technologies.
a) FENT: How a Microchip Is Revolutionizing DNA Analysis
b) SBX: When DNA Stretches to Be Understood
c) The G4X System: From the Genetic Recipe to the Spatial Atlas of the Cell
a) FENT: How a Microchip Is Revolutionizing DNA Analysis
– and making it accessible to everyone
Driven by the vision of capturing genetic information in real time and directly at the point of care, the company iNanoBio is developing a new method that aims to take this idea to an entirely new level.
It combines the proven method – reading DNA through tiny nanopores – with a component from microelectronics: the MOSFET (metal-oxide-semiconductor field-effect transistor), the heart of modern computer chips.
From computer chip to DNA sensor
A MOSFET is a tiny switch with three contacts: Source (input), Drain (output), and Gate (control). When a voltage is applied to the gate, a conductive channel opens between source and drain – comparable to a bouncer who opens a gate. Even minimal changes in voltage influence the current flow, which makes MOSFETs extremely sensitive.
A MOSFET is a tiny switch from microelectronics with three contacts: Source (input), Drain (output), and Gate (control). The source and drain are embedded in the semiconductor material, while the gate is insulated by an ultrathin oxide layer.
Without voltage at the gate, the channel between source and drain remains closed – no current flows. When voltage is applied to the gate, an electric field attracts charge carriers (e.g., electrons) and opens a conductive channel. The MOSFET thus acts like an extremely sensitive bouncer: only when it receives the signal does it open the gate and allow current to pass.
Here’s the clever part: iNanoBio has managed to integrate a tiny pore directly into the sensitive control region of the MOSFET. As a DNA strand passes through, each nucleotide (A, T, C, or G) alters the current between source and drain in a characteristic way. The result: a novel sensor called FENT – short for Field-Effect Nanopore Transistor.
The FENT has a cylindrical design and encloses the pore. This allows it to measure signals from multiple directions simultaneously, which increases accuracy and reduces errors.
The FENT is a cylindrically constructed MOSFET that fully encloses a nanopore. A voltage is applied between source and drain, allowing a small current to flow through the semiconductor of the pore wall – the so-called source-drain channel current. The thin oxide layer insulates the gate from the channel and enables precise detection of changes in the electric field. The nanopore serves as a sensing zone for local field variations caused by passing DNA bases.
The figure illustrates the operating principle of a FENT during the passage of a single-stranded DNA (ssDNA) through the nanopore. Before measurement begins, the originally double-stranded DNA (dsDNA) is separated into its two single strands by heat or chemical treatment. Only one single strand is then guided through the nanopore.
In the initial state – i.e., without DNA in the pore – a constant quiescent current flows between the source and drain through the semiconductor that forms the pore wall. This current is determined by the voltage between the source and drain and the gate potential.
However, as soon as a DNA base (A, T, C, or G) passes through the nanopore, it affects the electrical behavior of the system:
Due to its specific chemical structure and atomic distribution, each base has its own tiny electric field. This field penetrates the extremely thin pore wall and acts on the semiconductor like a local gate signal. This causes a local change in the charge carrier density (electron concentration) in the transistor channel, which leads to a measurable change in the drain-source current.
These current changes are recorded as characteristic signals for the respective base. During translocation, this creates a sequence-dependent current trace, allowing the order of DNA bases to be determined electronically.
A general-audience explanation is provided in the video FENT Nanopore Transistor Explainer Video.
Speed that sets the benchmark
According to iNanoBio, a FENT can read up to 1 million DNA bases per second and pore – about 100 times faster than previous nanopore technology. A 5 x 5 mm chip with 100,000 of these sensors could sequence a human genome in a few minutes. The costs would be significantly lower, and the error rate reduced.
Possible applications
- Early cancer detection from blood samples
- Mobile diagnostics for infections directly on-site
- Large-scale research studies enabled by high speed
In the long term, iNanoBio aims to make DNA analysis devices as widespread as today’s blood pressure monitors – available in clinics, laboratories, doctors’ offices, and potentially even at home. Early prototypes exist, and the path to mass production is underway.
If the technology delivers on its promise, genome sequencing could soon become as routine as a standard check-up – fundamentally transforming our understanding of health.
b) SBX: When DNA Stretches to Be Understood
In the race to improve DNA analysis, the pharmaceutical company Roche is pursuing an astonishingly simple approach with SBX (Sequencing By eXpansion): it artificially enlarges DNA so that its „letters” A, T, C, and G are easier to distinguish from one another.
Step 1 – From the original to the enlarged copy
SBX creates a surrogate copy of the original DNA, called the Xpandomer. The DNA fragment is fixed to a tiny anchor on a substrate, and a polymerase is used to create a copy – but not from natural nucleotides, rather from surrogate nucleotides.
A surrogate (from Latin surrogatum = „substitute”) generally refers to a replacement or substitute for something else.
These consist of:
- The corresponding base (A, T, C, or G)
- A folded extension strand specific to each base, serving as a signal marker
- A cleavable bond that holds the strand in shape
- A Translocation Control Element (TCE) that precisely regulates the process later
[Sequencing by expansion (SBX) technology]
The surrogate nucleotide (XNTP) consists of two functional components: the modified nucleotide and the SSRT (symmetrically synthesized reporter tether).
The modified dNTP contains the actual base (in this example, cytosine, C), which is connected to the reporter strand via an acid-sensitive bond (highlighted in red).
This is followed by the triphosphate (PPP), which enables the polymerase to incorporate the nucleotide into the growing chain.
The reporter tether (SSRT) consists of a folded extension strand with a Translocation Control Element (TCE) at its tip, which later controls passage through the nanopore.
An enhancer region (gray/purple) facilitates incorporation by the polymerase and stabilizes the structure.
After synthesis, the red acid-sensitive bond can be cleaved – causing the reporter strand to „unfold” and generating the extended Xpandomer molecule during sequencing.
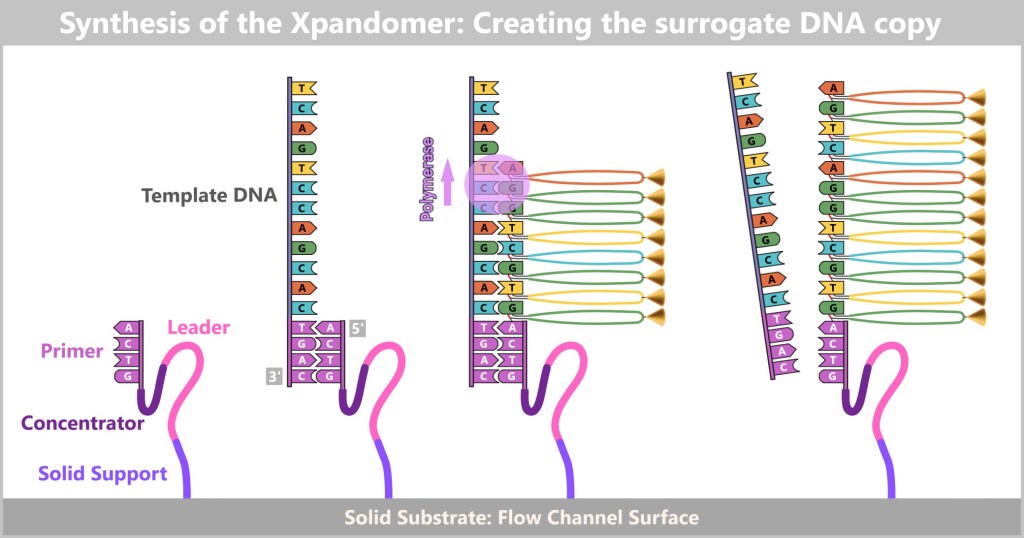
The DNA fragment (template DNA) is fixed to a solid substrate via an anchor.
A primer binds to the DNA template, supported by the leader and concentrator, which stabilize the start point and facilitate proper positioning.
Fixation – The DNA template is anchored to the substrate, and the primer section hybridizes to the matching region of the DNA.
Extension – a specialized polymerase extends the template bases, but not with natural nucleotides; instead, it uses surrogate nucleotides (XNTPs), each carrying a reporter tether.
Xpandomer formation – at the end, an artificially expanded DNA strand is produced: a surrogate copy (Xpandomer) in which the bases are replaced by characteristic reporter strands.
The Xpandomer initially remains anchored to the substrate and is released in a later step via light or chemical cleavage.
After synthesis, the Xpandomer is separated from the original DNA, the cleavable bonds are broken – the strands unfold and increase the spacing between bases by 50-fold. This greatly facilitates subsequent identification.
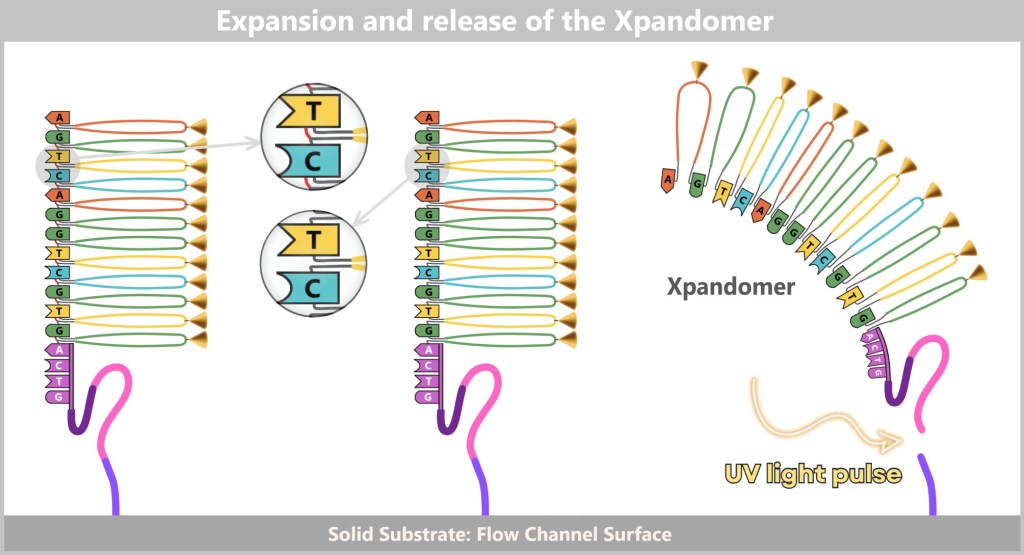
After synthesis, the Xpandomer is chemically unfolded („expanded”) on the substrate and then released.
Left) Expansion: A mild acid treatment cleaves the cleavable bonds (red) between the base and the reporter strand. This causes the reporter structures to unfold – transforming the compact double structure into an elongated Xpandomer that represents the sequence of bases.
Right) Release: A UV light pulse cleaves the bond between the substrate and the leader region. The now freely moving Xpandomer remains structurally marked via the leader and concentrator – these regions later assist in alignment and entry into the nanopore.
From a dense DNA template, a long, readable molecule is created, whose reporter segments make the genetic information electrically visible.
Step 2 – Sequencing
Instead of the DNA itself, the Xpandomere is now pulled through nanopores on a sequencing chip – around eight million of them working in parallel. The Xpandomere’s adapter guides it precisely into a pore.
When an extension strand reaches the pore, it temporarily sticks to the TCE. Each base generates a specific electrical signal. After 1.5–2 milliseconds, a voltage pulse releases the TCE, allowing the next strand to follow. This produces a clear, easily distinguishable signal trace – more precise than with Oxford Nanopore, where the bases generate a somewhat „blurred” signal.
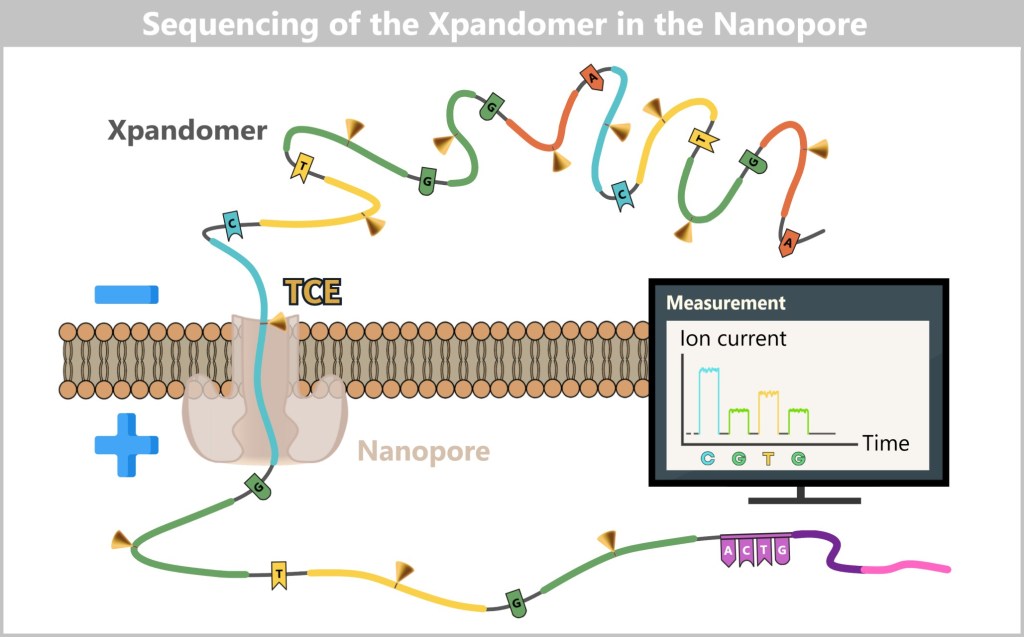
[Sequencing By Expansion, Introduction to Sequencing By Expansion]
The released Xpandomer is drawn toward the nanopore by an electric field. There, the TCE (Translocation Control Element) controls entry and temporarily holds the reporters in place.
As each reporter segment (the extended „base”) passes through the pore, the ion current changes in a characteristic way: each base generates its own electrical signal – comparable to an individual barcode. This sequence of signals is recorded in real time:
After each measurement, a voltage pulse releases the strand from the TCE, allowing the next segment to be drawn in.
The following animation illustrates the basic principles of Roche’s SBX technology.
A detailed presentation is available here.
Advantages and performance:
- Speed: Millions of Xpandomers are read simultaneously and in real time.
- Accuracy: Signal-to-noise ratio comparable to Illumina.
- Flexibility: Read lengths of over 1,000 bases possible – faster and more precise than Nanopore, longer than Illumina.
Initial tests show that SBX can decode up to seven complete human genomes per hour at 30× coverage. In urgent cases, the entire process – from sample preparation to finished analysis – takes only around 5.5 hours.
The market launch is planned for 2026. Whether SBX will prevail against Illumina and Oxford Nanopore depends on price, availability, and user requirements.
c) The G4X System: From the Genetic Recipe to the Spatial Atlas of the Cell
Our body consists of billions of cells that function like tiny kitchens. DNA is their cookbook – it contains all the recipes of life. When a recipe is needed, the cell creates an RNA copy that serves as a blueprint for proteins. These proteins carry out specific tasks: they build structures, transmit signals, or fight pathogens.
However, proteins rarely act alone. They operate in finely tuned networks whose activity depends on where, when, and with which partners they interact. Processes such as immune responses or tumor growth do not arise within a single cell, but from the interplay of many specialized cells – often located in different regions of a tissue. Biology is therefore not only biochemical, but also spatially organized. Classical sequencing methods, however, typically capture only genetic content, not its spatial distribution.
Gene activity directly within tissue
The G4X Spatial Sequencer from Singular Genomics reveals where genes are active and proteins are formed in tissue – directly in preserved tissue samples (FFPE, formalin-fixed and paraffin-embedded).
FFPE samples are commonly used in pathology, for example in cancer diagnostics. They act like a „frozen snapshot,” except that chemical fixation with formalin is used instead of cold. Formalin crosslinks proteins and nucleic acids, thereby fixing molecular structures in their spatial context.
What is preserved:
- RNA (often fragmented and chemically modified) – reflects gene activity at the time of fixation.
- Proteins – indicate which structures and signaling pathways were active at that moment.
This makes it possible to reconstruct months or years later which genes and proteins were present and active in the tissue at that exact moment – as if time had been stopped. With the G4X, this „frozen” molecular landscape can be visualized at high resolution, including the precise spatial location within the tissue.
A tissue consists of many specialized cells. Each cell uses only a subset of its genetic program. RNA makes the active genes visible. RNA molecules are read by ribosomes and translated into proteins. FFPE tissue sections preserve this spatial organization – forming the basis for using the G4X system to measure where specific RNA molecules are active within the tissue.
How the G4X works
You can think of the G4X as a microscope-scanner with a built-in laboratory, directly measuring which genes and proteins are active within the tissue – and in their spatial context.
Preparation: A very thin section of the FFPE sample is placed on a specialized substrate.
RNA detection: Padlock probes are used to identify the spatial distribution of specific RNA sequences within the tissue.
Padlock probes – DNA lassos for target sequences
Imagine you want to find a specific object (an RNA sequence) in a vast area (the tissue). A padlock probe is a DNA fragment whose two ends are complementary to adjacent sections of the target sequence.
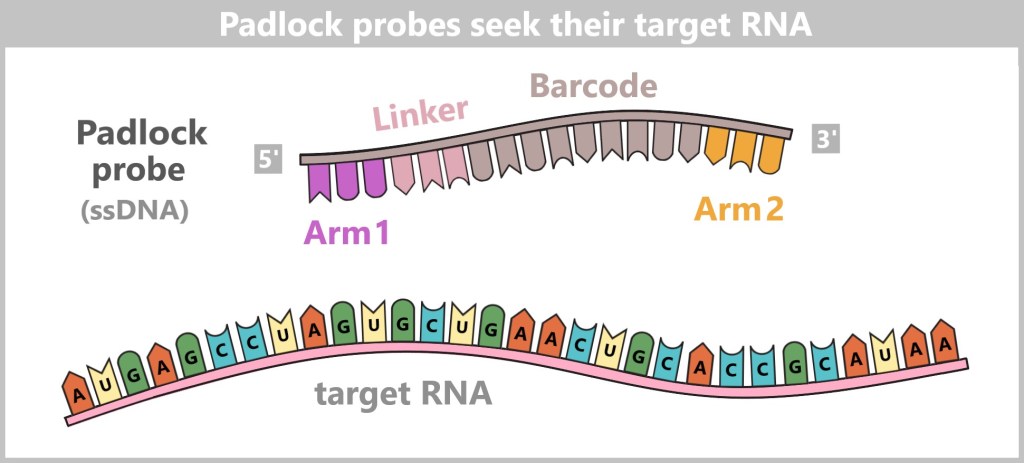
Padlock probe: single-stranded DNA (ssDNA) 5’– Arm1 – Linker/Barcode – Arm2 – 3′
Arm 1 and Arm 2 are complementary to adjacent sequences of the target RNA.
Between them lie the linker and barcode, which carry additional identification information and do not bind.
When the probe binds to its target, its two ends lie next to each other and can be closed into a stable DNA ring by ligation.
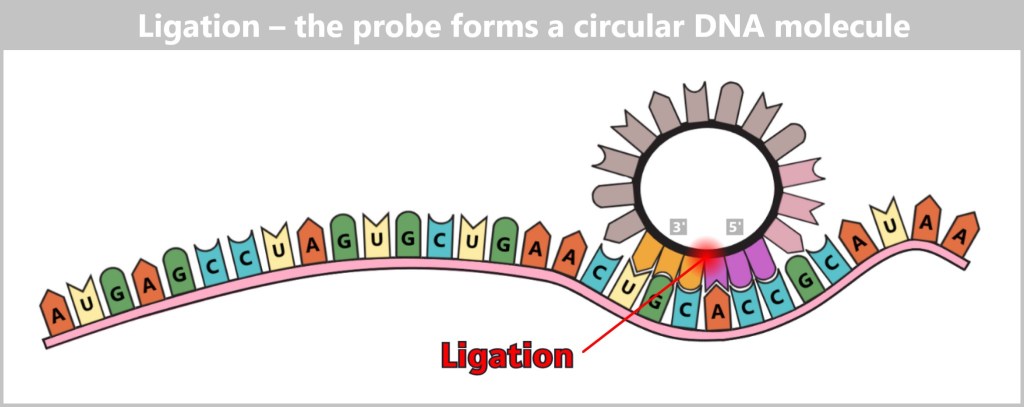
After both arms of the probe have bound to the target RNA, a ligase joins the 5′ and 3′ ends. This forms a closed DNA loop that marks the target RNA and is ready for amplification.
If there is a variable sequence (e.g., a mutation) between the binding sites, it is first transcribed into DNA using a reverse transcriptase and incorporated into the probe.
The resulting DNA circle serves as a template for rolling circle amplification (RCA):
- A DNA polymerase repeatedly moves around the circle, generating a long DNA strand that contains multiple repeats of the target sequence – like an endless ribbon or a ball of yarn.
- These signals remain precisely at the location in the tissue where the RNA was.
- Using fluorescently labeled nucleotides and sequencing by synthesis, the sequence is decoded directly within the tissue.
Each Padlock probe carries a barcode that reveals which gene or variant it has detected. The result: a precise map showing which RNA was present at which location and which variants it carried.
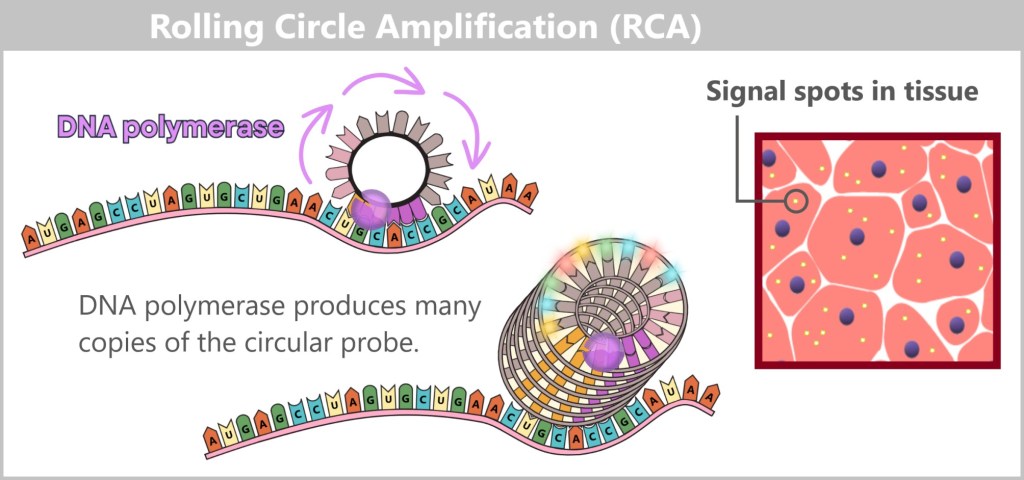
A specialized DNA polymerase repeatedly reads the circular padlock probe, generating a long DNA strand with many repeats. Fluorescent nucleotides label the amplificates, producing a strong fluorescent signal – visible as a bright spot in the tissue, marking the location of the target RNA.
Protein detection in the same tissue section
For protein detection, the G4X system uses antibodies tagged with short DNA barcodes. After the antibody binds to its target protein, a padlock probe hybridizes to the barcode. The probe is then ligated, amplified, and read out – just like in RNA analysis. The barcode clearly reveals which protein is involved and where it occurs in the tissue.
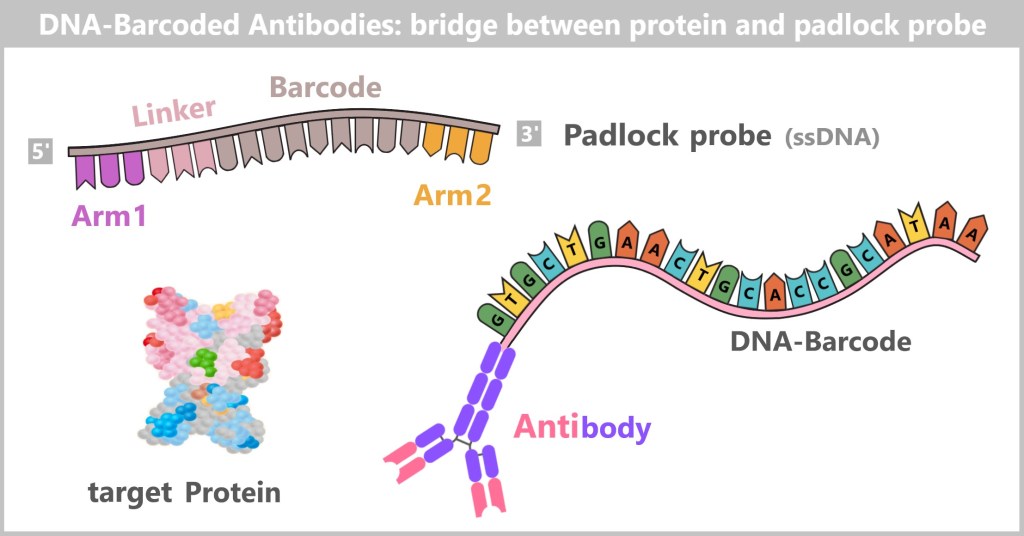
An antibody specific to a target protein is coupled with a unique, short single-stranded DNA strand (barcode).
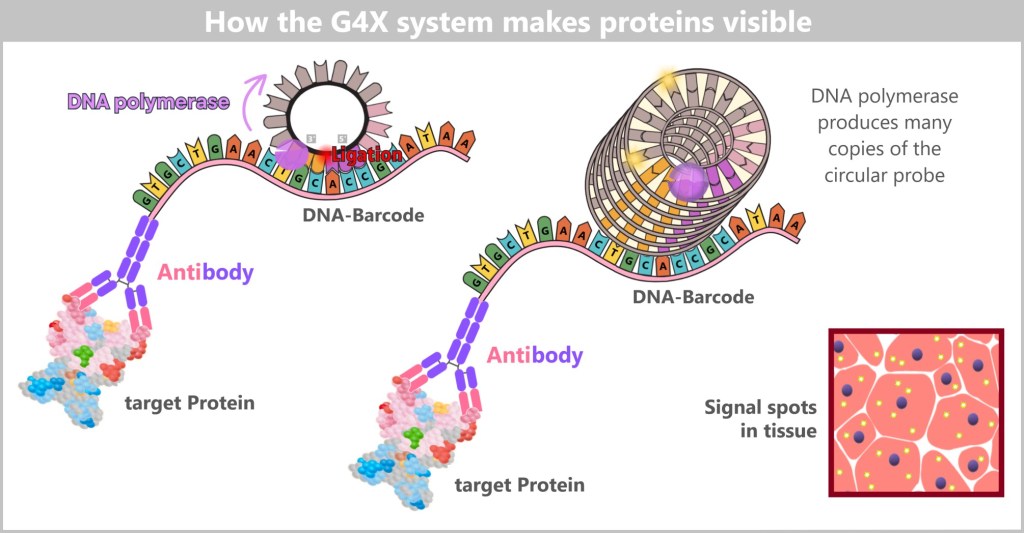
The antibody binds to its target protein in the tissue sample. A padlock probe, whose ends are complementary to two adjacent sections of the DNA barcode, binds to it and is linked by a DNA ligase to form a closed ring. A DNA polymerase recognizes the ring and reads it continuously, creating a locally anchored, circular DNA amplificate – analogous to RNA detection. The incorporation of fluorescently labeled nucleotides makes this amplificate visible. The fluorescent signal spots in the tissue mark the locations where an antibody with a specific DNA barcode has bound a particular protein.
A detailed presentation of the G4X method can be found here and here.
Why this is important
The G4X can specifically detect biomarkers that provide insights into the presence, progression, or aggressiveness of a disease – revealing not only which ones are active but also where they are located within the tissue.
Especially in tumors, this spatial resolution reveals which genes are „switched on”, in which cell regions this occurs, and how tumor and immune cells interact. This allows for a better understanding of tumor heterogeneity, clarification of disease mechanisms, and more targeted therapy planning.
The simultaneous in situ detection of RNA and proteins provides a comprehensive picture of tissue organization and opens up new possibilities for:
- Understanding complex disease processes
- More precise identification of biomarkers
- Development of personalized therapies
In diseases such as cancer, chronic inflammation, or neurodegenerative disorders, this spatial multi-omics perspective can be crucial – because often it is not the question of whether, but where and how the best therapeutic measure should be taken.
Conclusion
Genome sequencing is on the verge of a quantum leap: technologies like FENT, SBX, and G4X promise to read the genome faster, more accurately, and more comprehensively than ever before – including direct analysis within tissue. Soon, complete genomes could be decoded in minutes. The real revolution, however, begins afterward: when we understand the wealth of genetic information, translate it into medical knowledge, and use it responsibly for the benefit of humanity.
4.6. From Code to Cure
Bioinformatics as the Key to Tomorrow’s Medicine
4.6.1. From the Genetic Code to Computer-Aided Genome Analysis
Never before has it been so easy to decode DNA – but what use is the text if no one understands it? A small swab, a drop of blood – and suddenly millions of genetic data points are available. Modern sequencing has made the genome more accessible than ever before: fast, cost-effective, and nearly error-free. Yet this gives rise to a new challenge: how do we transform this flood of data into usable medical knowledge?
The answer comes from a discipline that has long operated in the background but has now become indispensable: bioinformatics – the bridge between A, T, G, C, and diagnosis, between raw sequence data and concrete therapeutic decisions. To understand why it is so important, it is worth taking a brief look back.
From the laws of inheritance to the genetic code
As early as 1860, Gregor Mendel discovered that there must be „heritable units” – genes – long before DNA was known. It was not until the mid-20th century that Oswald Avery demonstrated that these carriers of information are in fact DNA. Shortly thereafter, Watson and Crick revealed the double-helix structure with its complementary base pairing (A–T, G–C). In 1958, Francis Crick formulated the „central dogma”: information flows from DNA to RNA to proteins – the functional building blocks of life.
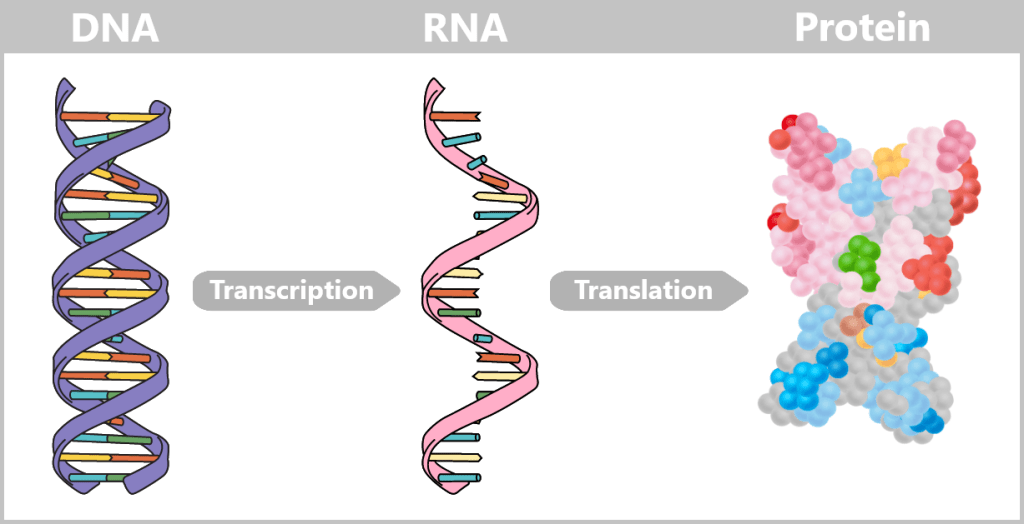
Genetic information is converted from DNA into RNA through the process of transcription. The RNA is then translated into the final protein, which performs a wide variety of functions. [An overview of artificial intelligence in the field of genomics]
A gene is therefore an instruction for how a protein is made. But how do we read this instruction – and how do we interpret it correctly? The answer initially came via the detour of reverse genetics.
In the 1960s, DNA was still difficult to access directly – proteins, on the other hand, were functionally visible (for example as enzymes, transport molecules, or structural components) and could be isolated and analyzed with the methods available at the time. Researchers had already known since the early 20th century that proteins consist of chains of amino acids. From the 1960s onward, it became clear that each amino acid corresponds to a so-called codon on RNA – a triplet of RNA bases (A, U, G, C) – which can in turn be traced back to a DNA sequence.
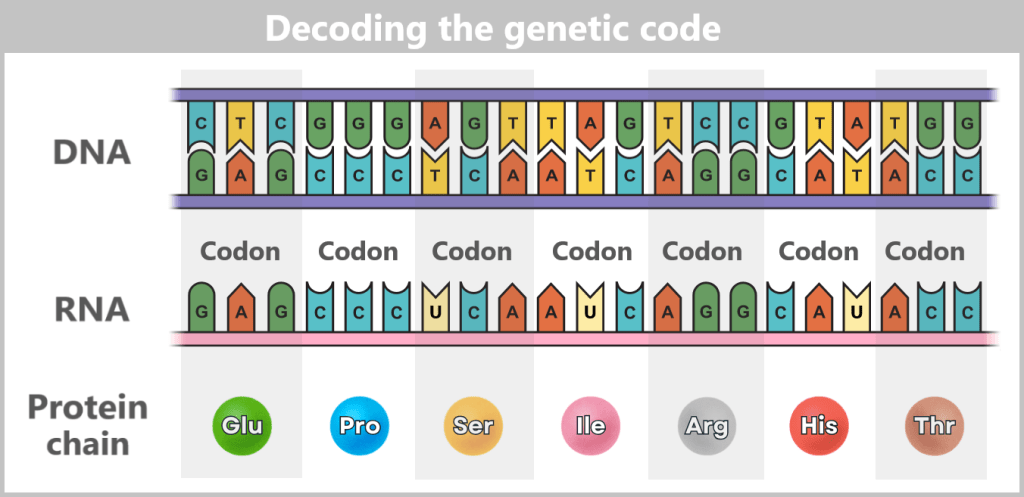
Researchers started with a known protein and worked their way back to the RNA and DNA sequence. For the first time, the genetic code became readable like a recipe book:
Protein = finished dish
Amino acid = ingredient
Codon = word for this ingredient in the handwritten recipe (RNA)
DNA = page in the cookbook from which the recipe was copied
Early patterns and the birth of genome annotation
Even before actual DNA sequencing, typical structural elements within genes were discovered:
Promoters – start signals where „reading” of a gene begins
Start and stop codons – first and last signals in the recipe
Transcription factors – proteins that switch genes on or off
Coding sequences – list of ingredients (codons) and their order for the protein
Non-coding DNA – regions without direct protein-coding function, whose role was initially unclear
These insights laid the foundation for modern genome annotation – the computer-assisted identification of biological signals within DNA sequences.
The Sanger Moment: DNA Becomes Readable (since 1977)
With Frederick Sanger’s method, DNA sequences could be determined directly for the first time – initially only for short segments. Analyses focused on gene identification and simple comparisons. Early programs such as FASTA (1985) and BLAST (1990) enabled the matching of new sequences with databases like GenBank (1982).
Understanding of gene structure also evolved: it was discovered that only certain parts of a gene (exons) are used for protein synthesis, while other segments (introns) are spliced out of the transcribed RNA. Additionally, DNA regions that enhance (enhancers) or repress (silencers) gene expression were identified – often located far from the actual gene.
The process of genome annotation began to take shape but was still rudimentary: DNA was primarily viewed as a linear sequence of nucleotides, with a focus on protein-coding genes. Regulatory elements and non-coding DNA were only partially recognized and understood. Bioinformatics was born – initially as a tool for research laboratories.
The Human Genome Project: Data on a Gigantic Scale (1990–2004)
The goal of decoding all 3.2 billion bases of the human genome introduced entirely new challenges: millions of short sequence fragments had to be assembled into long DNA strands. Tools such as Phrap for assembly and later Ensembl for annotation were developed. Data volumes exploded – along with the need for powerful computing infrastructure.
But while the Human Genome Project was still underway, a new technology was already emerging that would catapult speed and data volume into completely new dimensions.
NGS Revolution and Clinical Breakthrough (2005–2015)
Next-Generation Sequencing (NGS) technologies made genomes more affordable and rapidly accessible – albeit as massive data torrents of billions of short fragments. New tools like Bowtie, BWA, and STAR aligned these sequences at lightning speed, GATK helped identify mutations, and tools such as SnpEff or VEP interpreted their significance.
At the same time, sequencing strategies emerged for different clinical questions:
- Whole Genome Sequencing (WGS) for complete analyses
- Whole Exome Sequencing (WES) for the 1–2% of protein-coding DNA
- Targeted Sequencing for defined gene panels
This made bioinformatics a diagnostic tool for the first time – for example, in cases of hereditary forms of cancer, rare diseases, or personalized pharmacotherapy.
Artificial Intelligence and Modern Bioinformatics (2015–Present)
The combination of artificial intelligence (AI) and bioinformatics represents one of the most dynamic and promising areas of research of our time. Together, they are unraveling the complexity of life and accelerating biomedical discoveries at an unprecedented pace.
Artificial Intelligence: More Than Just Automation
AI encompasses techniques in which machines perform human-like intelligence tasks – particularly learning, problem-solving, and pattern recognition. In the biomedical context, the following subfields are especially relevant:
Machine Learning (ML): Algorithms learn from data (e.g., genome sequences, protein structures, patient data) to make predictions or identify patterns without being explicitly programmed. Example: detecting tumor cells in tissue samples.
Deep Learning (DL): A subset of ML that uses artificial neural networks with many layers („deep”) to capture highly complex patterns in massive datasets. Example: predicting the 3D structure of proteins solely from their amino acid sequence. [AlphaFold].
Natural Language Processing (NLP): Enables computers to understand scientific literature, medical records, or clinical study reports and extract knowledge from them.
4.6.2. Modern Bioinformatics – The Digital Toolbox of Biology
Bioinformatics is the science of storing, analyzing, and interpreting biological data using computer-based methods. Its modern form is characterized by three factors:
Explosion of biological data: Next-Generation Sequencing (NGS) generates terabytes of genomic, transcriptomic, and epigenomic data per experiment.
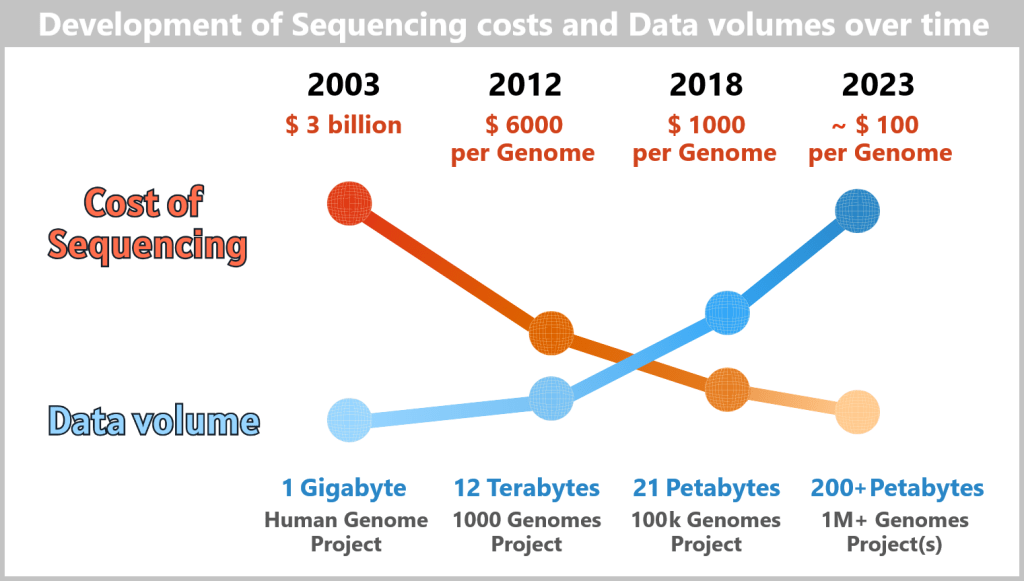
– how the costs of genome sequencing fell while data volumes exploded –
Just over twenty years ago, decoding a human genome was still a monumental undertaking: the Human Genome Project (completed in 2003) cost around 3 billion US dollars and produced only about 1 gigabyte of data. With ever faster sequencing machines and new technologies such as nanopore sequencing, this picture has changed completely. By 2012, the cost per genome had already dropped to around 6,000 dollars, in 2018 to 1,000 dollars – and today (2023), a genome can be analyzed for approximately 100 dollars.
At the same time, the volume of generated data increased rapidly: from a few gigabytes to terabytes and petabytes, and ultimately to the hundreds of petabytes produced by global projects involving millions of genomes. Estimates suggest that by 2025, around 40 exabytes of human genomic data could be generated – equivalent to roughly eight times the storage required for all words ever spoken in human history. [How AI Is Transforming Genomics]
Complexity of biological systems: Understanding disease requires the integration of data on genes, proteins, metabolic pathways, and cellular interactions.
Need for predictive medicine: The goal is to predict disease risks, therapy responses, and individualized treatment (precision medicine).
Already in many key applications, we can already talk about a fusion between bioinformatics and artificial intelligence (AI). Here are some central areas of application:
a) Genome Analysis and Sequencing
AI algorithms are used to analyze DNA sequences more quickly and precisely. Models such as DeepVariant, developed by Google, employ deep neural networks to identify genetic variants with high accuracy. This is crucial for detecting mutations associated with diseases such as cancer or rare genetic disorders.
b) Protein Folding and Structure Prediction
A milestone of AI in bioinformatics is DeepMind’s AlphaFold, which predicts protein structures with unprecedented precision. This has far-reaching implications for drug development, as a protein’s 3D structure determines its function.
c) Multi-Omics Data Analysis
A holistic understanding of biological systems requires the integration of multi-omics data (genomics, proteomics, transcriptomics, epigenetics, metabolomics) to gain a comprehensive picture of their functioning. AI helps to decipher the complex interactions between these layers.
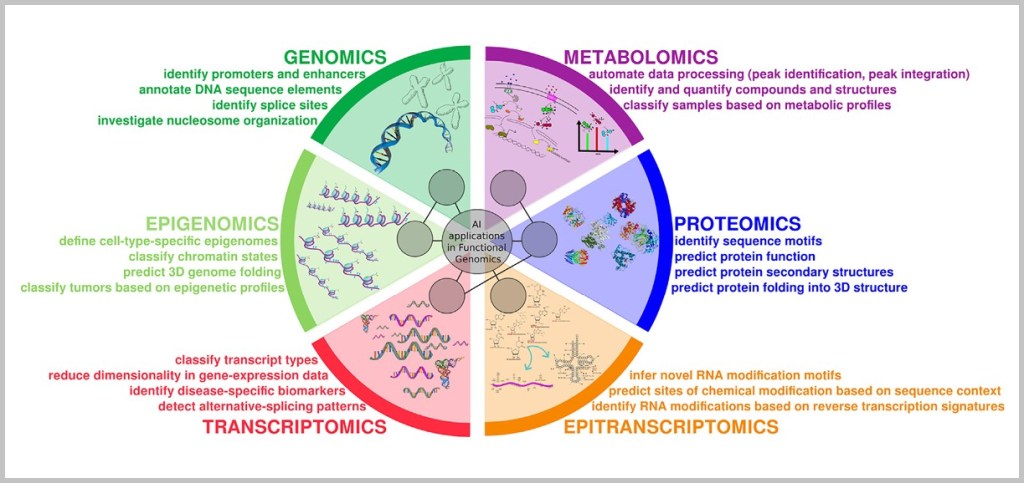
Biological systems are a complex interplay of different layers of information: from DNA (genomics) through epigenetic regulation, RNA (transcriptomics/epitranscriptomics), and proteins (proteomics) to metabolic products (metabolomics). It is only their dynamic interaction that defines health and disease.
For example, the new AI tool AlphaGenome demonstrates how even the smallest changes in DNA can affect gene activity as well as the production of RNA and proteins. It predicts with high precision the consequences of a single DNA base substitution for genes and their products. While earlier systems typically analyzed only the roughly 2% of the genome that codes for proteins, AlphaGenome is the first to capture the entire genome. It is based on a multi-omics analysis that directly links DNA alterations to their effects on RNA and proteins. This creates a comprehensive picture of gene expression – a crucial step toward more precise genomic research and personalized medicine.
d) Metagenomics and Microbiome Analysis
AI helps to understand the complex interactions in the human microbiome by recognizing patterns in the data from microorganisms. This is important for research into diseases such as diabetes or intestinal disorders.
e) Drug Development
AI accelerates nearly every phase of the lengthy and costly drug discovery process: target identification, virtual screening of millions of molecules, prediction of drug effects and side effects, and optimization of molecular structures. Companies such as BenevolentAI and Recursion use AI platforms to identify promising drug candidates in record time, some of which are already being tested in clinical trials.
f) Analysis of Biomedical Images
AI is used in image analysis, for example in the evaluation of MRI or CT scans, to detect tumors or other anomalies. Convolutional neural networks (CNNs), which mimic the functioning of the human visual cortex, have proven to be particularly effective in this area.
g) AI Genomics in Health Research
Gene expression in the human body does not follow a fixed pattern – and diseases such as cancer make it even more difficult to decipher. This is precisely where spatial biology comes in: using cutting-edge microscopy and genetic sequencing, it reveals how genes are active in individual cells and even within their organelles.
But it is not only the visible gene regions that are decisive. In regions long dismissed as „junk DNA”, researchers today suspect key switches of gene regulation. It is there that decisions are made about which proteins are produced – and which are not. AI and deep learning models can detect patterns in these previously hidden regions that are linked to disease. This raises hopes for new biomarkers and therapeutic approaches. One example is the platform developed by the biotech company Deep Genomics:
The combination of spatial biology, intelligent data analysis, and AI thus enables an unprecedented view into the molecular logic of life – down to the level of individual cells.
New methods of gene editing could make it possible to precisely control the behavior of individual genes – for example, to deactivate cancer-promoting genes or to stimulate the regeneration of cartilage cells in osteoarthritis. This field is still highly experimental, but AI promises to make these techniques more precise and safer.
With AI-supported CRISPR techniques, new possibilities are emerging to not only identify and modify genes more precisely, but also to better understand their interplay within complex biological networks. Early approaches suggest that this could reduce side effects in the future and increase the effectiveness of new therapies. Modern methods for the systematic testing and optimization of gene variants could also – if they prove successful – represent important steps toward personalized treatments.
Those who are interested in learning more about CRISPR techniques can find further information here.
The combination of advances in AI and genomics is therefore considered highly promising and could, in the long term, accelerate the development of new cell and gene therapies. However, since these treatments are based on living cells that vary widely and require specific conditions, much of this work is still in the experimental stage. AI could help make processes more stable, reproducible, and clinically applicable – but whether and to what extent these hopes will be realized remains to be seen.
h) Oncology
AI models analyze genetic profiles to create personalized treatment plans. For example, algorithms can predict how patients will respond to specific medications based on their genetic makeup. This is particularly relevant in oncology, where AI-based systems such as IBM Watson Oncology provide treatment recommendations.
Conclusion
Bioinformatics today serves as the brain of modern medicine. Together with artificial intelligence, it transforms the flood of data from the genome into concrete insights – from diagnosing rare diseases to developing new drugs.
Whether in genome analysis, protein structure prediction, or the planning of personalized therapies, AI-powered bioinformatics reveals what was previously hidden. It paves the way for a form of medicine that not only treats diseases but anticipates them – tailored to each individual patient.

5. A Success Story
The case of KJ Muldoon shows just how much these developments are already a reality today.
When the young boy received his diagnosis just a few hours after birth, his fate seemed sealed. He suffers from an extremely rare metabolic disorder caused by a single DNA point mutation (SNV), which prevents his body from breaking down toxic ammonia. Without a functioning enzyme, toxic metabolic products rapidly accumulate – posing a life-threatening risk even in infancy. Until now, only a risky liver transplant was considered capable of ensuring his survival.
But in KJ’s case, doctors and researchers took a new approach. Within just six months, they designed a therapy created exclusively for him: a personalized base-editing treatment.
The principle behind it is as ingenious as it is elegant: an RNA-guided system scans the genome, specifically locates KJ’s point mutation, and binds to that spot. Instead of cutting the DNA strand completely – as is the case with traditional CRISPR-Cas9 – an enzyme chemically modifies a single base: exchanging one DNA „letter” to correct the disease-causing mutation. To put it simply: it’s like fixing a single typo in a book without cutting out or rewriting the entire paragraph – extremely precise and gentle on the genome. The treatment was administered directly in the body („in vivo”) through multiple consecutive rounds, specifically targeting the liver.
The initial results are encouraging. KJ can now digest more proteins, infections are milder, and he is gaining weight. For his parents, this means a piece of everyday life without constant danger to his life – and, above all, new hope for their child’s future.
Behind this medical breakthrough lies a success story involving several disciplines. Without modern genome sequencing, the exact mutation would not have been visible. Without bioinformatics, it would not have been possible to design the appropriate base editing tool in such a short time and with such precision. And without the concept of personalized medicine, it would have been unthinkable to develop a cure exclusively for a single patient.
The therapy is still experimental, and no one knows whether its effects will be lasting or if long-term side effects might occur. Yet KJ’s case illustrates the direction medicine is heading: toward a form of healthcare that no longer relies on standard protocols, but on individualized solutions – driven by data, molecular biology, and tailored therapies. A medicine that not only treats diseases but, ideally, targets them precisely at their genetic root.
Further background information on this story can be found in this video.
6. A Look into the Future: The Synthetic Human Genome Project
Biology is on the verge of another quantum leap. After decoding and editing genomes, the vision now is to rewrite them from scratch. This is precisely the goal of the Synthetic Human Genome Project (SynHG) – an ambitious undertaking that could redefine the boundaries of biology and medicine.
With an initial funding of £10 million from the Wellcome Trust and under the leadership of Professor Jason Chin at the Generative Biology Institute in Oxford, leading universities such as Cambridge, Manchester, and Imperial College London are working on this project. The mission: to develop tools and technologies for creating synthetic human genomes – a step that could transform our understanding of life itself.
Recomposing a Genome
Unlike genome editing, which only modifies existing DNA, SynHG goes much further: it aims to reassemble the genome letter by letter from scratch. While CRISPR corrects individual typos in the text of life, this approach is about completely rewriting an entire chapter.
The first milestone: the synthesis of a complete human chromosome as proof of feasibility. It is the beginning of a journey that could take decades – and yet it builds on earlier pioneering achievements, such as the Human Genome Project (2003) and the Synthetic Yeast Project, in which synthetic yeast chromosomes were created for the first time in 2023.
Modern methods – generative AI, machine learning, and robot-assisted assembly – are intended to tackle this enormous task.
Potential Applications – Between Laboratory Vision and Clinical Practice
The opportunities are vast. At the level of somatic cells – i.e., cells whose changes are not passed on to offspring – new avenues for regenerative medicine could open up:
- tailor-made replacement tissues and organs grown in the lab,
- virus-resistant cells against HIV or SARS-CoV-2,
- gene therapies against cancer or hereditary diseases such as cystic fibrosis.
Such applications are not distant science fiction – they could become reality within the next 10 to 20 years, as technologies such as CRISPR and stem cell cultures are already well established.
The situation is quite different when it comes to germline editing – i.e., interventions in egg or sperm cells. In theory, this could permanently prevent serious hereditary diseases such as Huntington’s disease. However, the idea of „designer babies” with desired characteristics such as appearance or intelligence remains highly problematic from a scientific and ethical perspective, as such traits depend on hundreds of genes.
The project itself is limited to experiments in test tubes and cell cultures. It does not aim to create artificial life. Nevertheless, it opens up an unprecedented opportunity for researchers to intervene in fundamental human biological systems – with far-reaching consequences that must be critically examined.
Risks and Open Questions
The technical hurdles are enormous. In somatic cells, there is a risk of mosaicism (not all cells incorporate the new genome) or tumor formation due to unintended genetic alterations. In the germline, the risks are even greater: developmental disorders, epigenetic effects, or unpredictable consequences across generations.
Researchers are therefore discussing safety mechanisms such as genetic „kill switches”, which could deactivate synthetic cells in case of an emergency.
Ethics: Between Healing and Hubris
Somatic therapies are akin to organ transplants: controversial, but socially accepted when they cure. Germline interventions, however, touch taboo areas – fueled by concerns about social inequality or even eugenic scenarios. Many countries, including Germany, prohibit them through laws such as the Embryo Protection Act. International organizations like WHO and UNESCO call for strict regulation.
„To build trust, we need to actively involve society”, emphasizes sociologist Professor Joy Zhang from the University of Kent, who is investigating the ethical dimensions of the project. With her Care-full Synthesis initiative, she is attempting to initiate an open dialogue about the opportunities, but also the limitations, of this research.
Renowned geneticist Professor Bill Earnshaw from the University of Edinburgh, who has developed methods for producing artificial human chromosomes, puts it more bluntly: „The genie is out of the bottle. We could have a set of restrictions now, but if an organisation who has access to appropriate machinery decided to start synthesising anything, I don’t think we could stop them.“
In this context, the call by numerous professional organizations – including the International Society for Cell and Gene Therapy (ISCT) – for a ten-year moratorium on the use of CRISPR and related techniques in the human germline seems even more urgent. Such a postponement appears not only to be a precautionary measure, but also a necessary political response in order to enable social debates, ethical guidelines, and international control mechanisms in the first place.
Ethics expert Arthur Caplan from New York University describes the dilemma from a scientific perspective:
„… So if you ask me, will we see genetic engineering of children aimed at their improvement? I say yes, undoubtedly. Now when? I’m not sure what the answer to that is.“
„… It will come. There are traits that people will eagerly try to put into their kids in the future. They will try to design out genetic diseases, get rid of them. They will try to build in capacities and abilities that they agree are really wonderful. Will we hang up these interventions on ethical grounds? For the most part, no, would be my prediction, But not within the next 10 years. The tools are still too crude.“
Outlook
SynHG opens a window into a future where we can not only read and edit the genome, but write it. Somatic applications – personalized therapies, regenerative tissues, and custom-designed cells – could soon shape the practice of medicine.
Interventions in the germline, however, remain a distant horizon – for technical, legal, and ethical reasons. The project represents not only a scientific challenge but also a delicate societal balancing act.
Given the potential for misuse of the technology, the question arises as to why Wellcome decided to provide funding at all. Dr. Tom Collins, who was responsible for this decision, explained to BBC News:
„We asked ourselves what was the cost of inaction. This technology is going to be developed one day, so by doing it now we are at least trying to do it in as responsible a way as possible and to confront the ethical and moral questions in as upfront way as possible.“
While Europe is still grappling with how to handle the topic carefully in public debate, across the Atlantic the term „new life forms” is approached much more openly. Eric Nguyen, a bioscientist at the renowned Stanford University, outlines a far more visionary perspective in his TED Talk, „How AI Could Generate New Life-Forms“.
In a striking way, he demonstrates how artificial intelligence (AI) could transform biology – not only by „reading” and „writing” DNA, but also by deliberately creating novel organisms. AI models such as machine learning and generative algorithms are already capable of recognizing complex patterns in biological data: in genomes, protein structures, or metabolic pathways. Nguyen goes further, arguing that in the future AI could also design entirely new DNA sequences – the starting point for previously unknown life forms.
This vision is undoubtedly forward-looking, but it raises fundamental questions: What priorities should be set? How can the use of such technologies be justified in a world of massive resource inequality? Who funds the research, who oversees the outcomes – and by what criteria is it decided which life forms are „useful” or „safe”?
Nguyen’s presentation makes it clear that AI-supported biotechnology could not only enable solutions to global challenges, but also forces us to think about the ethical, social, and philosophical dimensions of „creating life.”
The message: AI-powered biotechnology promises tremendous advances, yet simultaneously raises profound ethical and societal questions – challenging the very foundations of what it means to be human.
Klaus Schwab – Founder and former Chairman of the World Economic Forum
„We must address, individually and collectively, moral and ethical issues raised by cutting-edge research in artificial intelligence and biotechnology, which will enable significant life extension, designer babies, and memory extraction.“
[BrainyQuote]
7. Personalized Medicine and Smart Governance
The Biomolecular Revolution in Governance
7.1. Power, Governance, and Smart Governance
7.2. Governmentality
7.3. Personalized Medicine as a Catalyst for Biomolecular Governmentality
7.4. The Global Trend: Worldwide Biomolecular Governmentality
7.5. Conclusion: The Global Ambivalence of Biomolecular Power
The decoding of the human genome marked not only a medical turning point, but also the beginning of a new era of political governance. What started as a large-scale scientific project in laboratories and data centers has long since reached the control apparatus of modern states: the precise measurement, categorization, and utilization of our biological foundations.
The vision of personalized medicine promises individualized therapies, precise predictions about disease risks, and more efficient use of scarce resources. But this narrative of progress falls short if we consider it solely from a medical, ethical, or data protection perspective. In fact, we are witnessing a fundamental transformation of power and governmentality – a biomolecular revolution in governance.
Before we delve deeper into the subject, it is important to clarify a few key terms.
7.1. Power, Governance, and Smart Governance
Governance refers to the art of steering societal processes, making decisions, and maintaining order. Traditionally, this occurs through the interaction of the state, markets, and civil society.
Smart governance transforms this understanding: it operates through digital technologies, data analytics, and algorithmic processes. Governance thus becomes continuous, procedural, and possible in real time. Power no longer manifests solely as political authority, but as a network of information flows, feedback loops, and predictions.
Power itself is not a possession, but a relationship: the ability to influence behavior, steer actions, or frame decisions. It operates both visibly – through institutions and laws – and subtly, by shaping expectations, norms, and bodies of knowledge.
7.2. Governmentality
Michel Foucault, a major thinker in the analysis of power structures, coined the term of governmentality to describe the totality of forms of knowledge, practices, and techniques through which modern societies are governed. In this sense, governing does not merely mean the exercise of coercion, but also the shaping of spaces for action that encourage individuals to regulate themselves in line with societal objectives.
Key dimensions of governmentality are:
The Interconnection of Knowledge and Power
Knowledge is never neutral. It emerges through the collection and interpretation of data, is embedded in power processes, and structures social orders. With digitalization, this connection becomes central: big data and artificial intelligence provide predictions, pattern recognition, and decision-making foundations that intervene deeply in societal processes. Smart governance operates precisely at this point: data become the foundation of political legitimacy and the exercise of power.
Biopolitics
Biopolitics refers to the regulation of life itself – health, reproduction, labor, education, migration, or security. The goal is to organize entire populations through laws, standards, and political programs. Today, biopolitics is linked to digital control: Smart Health, Smart Cities, and Smart Economies illustrate how knowledge is translated into concrete government practices through data.
Dispositives of Power
Biopolitics works through dispositifs: networks of institutions, laws, discourses, and practices. Hospitals, schools, media, and digital platforms are part of these structures. They standardize behavior not through overt coercion, but through subtle framing of what is considered „normal” or „desirable”. In this way, decisions are indirectly guided and behavior is embedded in everyday life.
Disciplinary Power
Alongside biopolitics, which regulates entire populations, disciplinary power targets the individual. It operates through surveillance, reward, punishment, and training. Its goal is not only obedience, but the productive shaping of bodies and behaviors. External directives are transformed, through sanctions and incentives, into internal motivations that drive individuals toward self-discipline.
Technologies of the Self
Technologies of the self refer to practices through which individuals internalize societal norms and autonomously adjust their behavior. Digital applications – such as fitness apps, learning platforms, or financial tools – translate societal directives into personal goals. This creates a form of power that operates not only from the outside, but is carried forward from within: people follow rules because they perceive them as meaningful and beneficial for themselves.
Those interested in exploring the art of governance in more detail can find further information here.
7.3. Personalized Medicine as a Catalyst for Biomolecular Governmentality
At first glance, personalized medicine, supported by modern genomics and bioinformatics, appears to be purely a medical and technical advance. But when viewed through the lens of governmentality, it emerges as a powerful catalyst for a profound transformation of power structures and governance practices. The precise analysis of individual genetic profiles and their linkage to comprehensive databases not only individualizes healthcare, but also changes the way societies are governed and individuals are regulated.
Knowledge & Power: From the Genome to the Governable Body
The decoding of the genome marked the initial explosion of knowledge, but it is personalized medicine that makes this knowledge governable. Bioinformatics and modern genomics do not merely generate data – they create predictive knowledge that forms the basis for interventions. This knowledge is by no means neutral: it categorizes individuals according to genetic risk profiles (e.g., „high risk for breast cancer,” „predisposed to cardiovascular diseases”). These categorizations become the foundation of new power structures. Both states and private actors use this data to predict disease risks, optimize healthcare budgets, and manage preventive measures. Social order is thereby reshaped – from access to insurance and labor markets to the prioritization of preventive resources.
Smart governance manifests here in algorithmic decision-making processes that appear objective but are, in fact, normative. Who gains access to which therapies, or which population groups are classified as „at risk”, is increasingly determined by data-driven models that invisibly reproduce existing power relations („We act based on the best available evidence”).
The CMS Health Tech Ecosystem Initiative, officially announced on July 30, 2025, during the „Make Health Tech Great Again” event at the White House, exemplifies the trend toward the emergence of new power structures. CMS (Centers for Medicare & Medicaid Services) is a U.S. federal agency within the Department of Health and Human Services (HHS) that manages healthcare programs such as Medicare and Medicaid and regulates the national healthcare market. The initiative aims to create smarter, safer, and more personalized healthcare, driven through partnerships with innovative private-sector companies. During the event, the U.S. government secured commitments from major health and IT firms – including Amazon, Anthropic, Apple, Google, and OpenAI – to lay the foundation for a next-generation digital health ecosystem.
According to U.S. President Donald Trump, this gives the U.S. healthcare systems a high-tech upgrade, ushering in „an era of convenience, profitability, speed, and – frankly – better health for the people”. (Note the order…)
Biopolitics 2.0: Optimizing the Population at the Molecular Level
Classical biopolitics regulated populations through statistics on birth rates or mortality. Personalized medicine elevates this to a new, molecular level. Genomics fragments the population into ever smaller, biomolecularly defined subgroups. People are no longer categorized merely by age or gender, but by genotype, risk alleles, and biomarkers („BRCA1 carriers”, „individuals with the LCT gene for lactose intolerance”). This enables hyper-precise and individualized regulation – biopolitics becomes granular.
The „healthy”, „normal” body is no longer defined solely as one without symptoms, but as one with a „normal”, „low-risk” genome. Deviation begins not with disease, but with genetic predisposition. This creates a new class of „presymptomatic patients” or „risk subjects”, who are managed biopolitically through more frequent screenings, prevention programs, vaccination campaigns, prophylactic surgeries, lifestyle recommendations, or reproductive decisions based on genetic forecasts.
While Foucault saw the nation state as the main actor in biopolitics, power is increasingly shifting to private actors today: big tech and biotech companies that control data, algorithms, and technologies. They effectively define what genetic „normality” and „risk” mean.
We now speak of the economization of biological life. Genetic data have become a valuable commodity – a bio-resource. Large technology and pharmaceutical companies accumulate bio-data to generate profits: in drug development, therapy recommendations, and personalized advertising. The human body and its biomolecular data thus become part of capitalist logic of exploitation. The individual themselves becomes „human capital”, seemingly obliged to optimize his genetic potential.
In an NBC News report on August 26, 2025, U.S. Health Secretary RFK Jr. stated that the 60 largest technology companies would soon allow Americans access to their personal health data – data that had been economically exploited for years without their consent: “You are going to be able to see, by next year, all your health records on your cellphone.”
What is presented here as a political achievement merely confirms the status quo.
Dispositifs of Power: The Network of Biomolecular Surveillance
The dispositive network that supports this new form of biopolitics is multi-layered: pharmaceutical companies develop targeted therapies; biobanks and genetic databases provide the raw material; algorithms analyze and interpret it; doctors communicate results and recommendations; digital health apps monitor compliance. Each link in this chain functions as a power dispositif: it defines what is considered „healthy” or „sick“ and subtly conveys what „responsible health care” means in the age of genetics. Genetic testing, for example, is marketed as „responsible” behavior – thus leading individuals to participate unknowingly in biopolitical goals such as prevention or cost efficiency.
This development is reflected in national health strategies that promote genetic screenings to minimize disease burdens. The goal is to maximize the „health dividend” while simultaneously reducing costs – a biomolecular rationalization strategy.
Initiatives such as the 100.000 Genomes Project or Genomics England’s Newborn Genomes Programme move in the same direction. By comparison, the European Health Data Space (EHDS) – an EU-wide initiative for the networking and secure use of health data – is far more comprehensive. The EHDS provides the data and regulatory infrastructure that makes large-scale personalized medicine possible in the first place.
For most citizens, this sounds like a new beginning: more participation, better therapies, more precise prevention. Yet this promising vision casts a shadow. In practice, unequal opponents face each other: on one side, the individual patient contributing their genetic profile, lab results, and medical history; on the other, global pharmaceutical companies, data platforms, and technology providers with multi-billion-dollar budgets that refine, patent, and commercialize this data. Cui bono? – who benefits? The answer is sobering: quick profits go to the big players, while the promises remain with the individual.
While the EHDS emphasizes citizens’ rights – more control, more transparency, more self-determination – anyone who has experienced the complexity of consent processes in health apps, insurance companies, or electronic patient records knows: there is a gap between formal options and actual capacity to act. While Goliath has lobbying channels, legal experts, and data centers at its disposal, David is left with informational brochures and trust.
The EHDS is thus both a government technology and a power project. It decides whether data is treated as a public good or as a commodity market. In any case, Goliath will benefit. The open question is whether David will be left with more than just the role of a fig leaf. The health data space will only become truly emancipatory if benefits and risks are distributed fairly – through binding public welfare requirements, transparent accountability, and real returns for patients. Otherwise, the great promise of „empowerment” will remain a narrative that merely conceals existing asymmetries.
Disciplinary Power: Internalized Biomolecular Surveillance
While biopolitics focuses on the population as a whole, personalized medicine directs its disciplinary effect at the individual. The genetic risk profile acts as a constant, internalized monitor. Those who know they have an increased risk of colon cancer adjust their behavior: dietary habits, preventive medical appointments, and exercise routines are adapted to the genetic forecast.
The electronic patient record (ePA) intensifies this dynamic. By centrally storing genetic and medical data and making them accessible at any time, it transforms medical recommendations into an ever-present digital authority. Personal notifications, reminders for medication intake, personalized reports, or interfaces with health apps reinforce self-monitoring: the external directive („You must get screened”) becomes an internal voice („I must take care of myself because my genome requires it”). The ePA structures and institutionally legitimizes this self-discipline, making it invisible and seemingly voluntary – governed simultaneously by digital infrastructure, social norms, and medical authority.
The result is a subtle yet pervasive form of power: it does not manifest through sanctions, but through the constant, digital concern for one’s own – genetically anticipated – future. The ePA turns the individual into a disciplined actor of their own risk profile, while the surveillance formally disappears but remains technically and institutionally omnipresent.
Technologies of the Self: The Genetic Self-Optimizer
Here, personalized medicine appears in its purest form as a technology of the self. At-home DNA tests, fitness trackers, and health apps are the tools through which individuals internalize societal norms of health and performance and actively work on themselves. They translate the abstract concept of genetic predisposition into concrete, everyday actions: an extra serving of vegetables, 10,000 steps, meditation to reduce stress. The individual governs themselves according to a biomolecular logic – not because the state commands it, but because they seek to optimize their genetic destiny. Power relations thus become invisible and present themselves as expressions of individual sovereignty and self-care.
7.4. The Global Trend: Worldwide Biomolecular Governmentality
Personalized medicine is no longer a national phenomenon, but part of a global transformation of power and governance. Worldwide, initiatives are emerging that pool genomic data, health information, and digital technologies in order to translate populations and individuals into ever more precise risk profiles. The logic of Foucault’s governmentality – the linkage of knowledge, power, and self-regulation – manifests itself here at a transnational level.
In the United States, programs such as the All of Us Research Program of the National Institutes of Health or the Million Veteran Program collect genetic and medical data from millions of citizens. The collection of diverse health data serves not only scientific research, but also forms the basis for algorithmic risk profiles, preventive interventions, and resource-optimized healthcare strategies.
Canada is pursuing a comparable goal with the Canadian Precision Health Initiative and the Pan-Canadian Genomics Network: the systematic collection and interconnection of biomolecular data, combined with an infrastructure that enables secure governance, international collaboration, and economic utilization.
This is where Biopolitics 2.0 becomes visible: populations are fragmented, governed, and optimized at the molecular level.
Europe constitutes an orchestrated network of transnational biopolitics. The European Health Data Space (EHDS), supported by initiatives such as ICPerMed and ERA PerMed, pools genetic, clinical, and lifestyle data across national borders.
National biobanks such as the UK Biobank or the French Health Data Hub act as dispositifs that generate both knowledge and power: they determine which data is used, how populations are classified, and which health policy interventions appear legitimate.
Through platforms such as SHIFT-HUB or the Helmholtz Initiative iMed, the international networking of science, industry, and society is institutionalized – constituting a transnational form of smart governance that scales Foucault’s concept of technologies of the self to the global level.
The global logic is also evident in Asia and the Pacific region: the GenomeAsia 100k Project, Singapore’s National Precision Medicine Strategy, and China’s National Health Data Platform link biomolecular data with AI-driven analyses to enable preventive, individualized, and population-based governance.
Australia and Israel rely on digital infrastructure that reinforces citizens’ self-discipline – electronic patient records, genetic databases, and personalized apps function as subtle yet omnipresent dispositifs of power.
Even global research projects such as the Human Cell Atlas illustrate the transnational dimension: by mapping the diversity of human cells, they create a universal network of knowledge and power that provides the foundation for personalized therapies worldwide. The boundaries between state governance, private-sector power, and scientific expertise blur: Foucault’s classical distinction between the nation-state and disciplinary power is replaced by global networks and international collaboration.
7.5. Conclusion: The Global Ambivalence of Biomolecular Power
The global expansion of personalized medicine clearly demonstrates that the biomolecular revolution is no longer confined to individual nation-states. It manifests as a transnational network of data, algorithms, and governance that steers both populations and individuals at the molecular level. Global initiatives – from the United States and Europe to Asia, Australia, and Israel – intertwine biopolitics, disciplinary power, and technologies of the self on an international scale.
The ambivalence remains central: on the one hand, these developments open up opportunities for more precise therapies, individualized prevention, and potentially greater health equity. On the other hand, power asymmetries arise in which states, international organizations, and private actors control access to data, resources, and technologies. Individuals are increasingly becoming disciplined subjects of their own genetic profiles, while global dispositifs unnoticedly enforce normative and economic goals.
This seamlessly combines the local experience of personalized medicine with a global governance logic: smart governance and biomolecular biopolitics have long been internationalized, and the challenge lies in distributing the benefits and risks fairly. The global dimension of this transformation makes it clear that the biomolecular revolution is irreversible not only medically, but also politically and socially – a global system of control that offers opportunities for health and risks for the concentration of power.

8. Epilogue
The desire to optimize health and longevity is a primal instinct of humankind. Even in ancient civilizations, health was not considered a matter of chance, but rather a desirable state that could be achieved through lifestyle and philosophy. Greek „dietetics” – a holistic practice combining nutrition, exercise, and mental hygiene – and the motto „mens sana in corpore sano” testify to a 2.500-year-old desire for optimization. This urge, fueled by the survival instinct, the fear of losing control, and the pursuit of quality of life, is not only ancient but eternal.
In modern times, personalized medicine embodies the latest expression of this dream. It promises to treat people not as averages, but as individuals, based on genetic code, biomarkers, and lifestyle data. But is it the fulfillment of this dream or a modern mirage? In its utopian vision, it suggests the complete controllability of biology, an overcoming of the „biological lottery”. The reality is more sobering: it enables more precise diagnoses and therapies, prevents treatment failures – but it does not abolish disease.
The idea of a disease-free existence remains an illusion. Evolutionary biology and biophysics set limits: pathogenic germs continue to evolve, cancer is a statistical consequence of cell division, and aging is an inevitable degenerative process. The realistic goal is therefore not the elimination of disease, but its transformation: converting fatal diseases into chronic conditions and maximizing the „healthspan” – the years spent in good health. This goal is radical, but achievable, as advances in the treatment of HIV or diabetes show.
But how far is humanity willing to go to achieve this goal? History shows that in the face of suffering and death, ethical boundaries often blur. Willingness ranges from everyday discipline and financial sacrifice to participation in experimental therapies. This raises an uncomfortable question: what compromises is society prepared to make? Personalized medicine requires data – genetic, medical, and personal. Collecting such data carries the risk of undermining privacy and autonomy. The COVID-19 pandemic was a stress test that showed how large parts of the population were willing to trade civil liberties for security. Models such as China’s social credit system illustrate one possible future scenario, in which transparency and control are rewarded in the name of health and safety. Yet such scenarios are not inevitable: they represent an extreme, not an unavoidable destiny.
Personalized medicine has two sides. Its pragmatic promise is more efficient, precise, and humane medicine that saves lives and alleviates suffering – for example, through tailored cancer therapies or early diagnosis. Its utopian but risky promise is the illusion of total control over life and death. It is thus the culmination of a journey that has lasted thousands of years. Whether this journey ends in emancipated self-optimization or a dystopian „biocracy” does not depend on technology, but on the ethical and political framework that society gives it.
The age-old dream of health seems more tangible today than ever before, yet it challenges us to find wise answers. Personalized medicine can be a tool of emancipation if we use it with care. The decisive question of modernity is therefore not only how much freedom we are willing to sacrifice for a longer life, but how we can reconcile health and freedom. Only in this way can the eternal pursuit of health become progress for all.
The world will not change all at once; it never does. Life will go on mostly the same in the short run, and people in 2025 will mostly spend their time in the same way they did in 2024. We will still fall in love, create families, get in fights online, hike in nature, etc.
But the future will be coming at us in a way that is impossible to ignore, and the long-term changes to our society and economy will be huge.
Sam Altman (CEO of OpenAI)
Sources (as of January 15, 2026)
