
Als die HSBC Bank im Jahr 2013 im Rahmen ihrer globalen Kampagne „The future is full of opportunity“ an Flughäfen wie Gatwick und Dubai Plakate mit einem Fingerabdruck, einem QR-Code und dem provokanten Slogan „Your DNA will be your data“ platzierte, war die Botschaft unmissverständlich: Die Zukunft wird persönlich – vielleicht sogar beunruhigend persönlich. Die Bank wollte sich als zukunftsorientiertes Unternehmen positionieren, das Digitalisierung, Künstliche Intelligenz, Nachhaltigkeit und Genetik offensiv thematisiert.
Der Slogan wirkte doppeldeutig – als verheißungsvolle Vision ebenso wie als alarmierender Ausblick. Kritiker schlugen sofort Alarm: Deine DNA wird lesbar, analysierbar und digital verfügbar – ein offenes Buch über Herkunft, Gesundheitsrisiken, vielleicht sogar Persönlichkeitsmerkmale – vergleichbar mit biometrischen Daten wie Fingerabdrücken oder Gesichtsscans. Was einst als das Intimste am Menschen galt, wird nun zur Datei: maschinell entschlüsselbar, potenziell speicherbar, womöglich handelbar. Der Mensch wird reduziert auf Information – auf „Bits und Bytes“, die verarbeitet, verkauft oder missbraucht werden könnten.
Wer besitzt die Rechte an deinen genetischen Daten? Und wer hat Zugriff darauf? Du selbst? Die Firma, die den Test durchgeführt hat? Erreichen dadurch Überwachung, Datenlecks und Missbrauch durch staatliche und private Akteure eine neue Dimension? Die Vision von Designerbabys, genetischer Selektion und personalisierter Werbung auf Basis biologischer Marker wirft schwerwiegende ethische Fragen auf. Wird der Fortschritt am Ende mit dem Preis individueller Freiheit bezahlt?
Doch die Biotech-Revolution hat längst eine gewaltige Dynamik entfaltet, und immer mehr Stimmen erkennen in dieser Entwicklung ein enormes Potenzial. In der E-Book-Ausgabe der HSBC Bank Frontiers – Biotech – Five Big Ideas Shaping the Biotech Revolution von März 2024 heißt es:
„Die Idee, die Natur für Waren und Dienstleistungen zu nutzen, ist nicht neu. Doch in den 1970er Jahren, mit dem Aufkommen der Gentechnik und der Gründung der ersten Biotech-Unternehmen, begann die moderne Ära dieser Praxis. Dies brachte Wissenschaft und Wirtschaft zusammen wie nie zuvor, und der Kapitalzufluss beflügelte die Forschungs- und Entwicklungsbemühungen.
In den folgenden drei Jahrzehnten wurde das Feld durch bahnbrechende Geräte wie DNA-Sequenzierer und -Synthesizer beschleunigt, und die Biologie selbst wandelte sich langsam zu einer rechnergestützten, datengesteuerten Disziplin. Nach der Jahrtausendwende, als die Forscher immer größere Bereiche des menschlichen Genoms erfolgreich entschlüsseln konnten und die Kosten für die Sequenzierung dieses Genoms rapide sanken – von etwa 100 Millionen Dollar im Jahr 2001 auf weniger als 1.000 Dollar heute –, wurde das Potenzial der Biotechnologie, unser Leben zu verändern, auf elektrisierende Weise deutlich.
In den letzten Jahren wurden die Möglichkeiten der Biotechnologie – von der Behandlung genetisch bedingter Krankheiten bis zur Wiederbelebung ausgerotteter Tierarten – durch Fortschritte in ergänzenden Bereichen wie der künstlichen Intelligenz erweitert. Dies hat dazu beigetragen, dass in diesem Bereich ein neues Zeitalter angebrochen ist.“
Nirgendwo sonst prallen Nutzen und Risiko so direkt aufeinander wie in der personalisierten – oder präzisionsbasierten – Medizin. Ihr Anspruch: individuelle genetische und biologische Informationen zu nutzen, um Diagnosen zu präzisieren, Therapien zu individualisieren und Krankheiten frühzeitig zu erkennen. Die Vision ist vielversprechend: weniger Nebenwirkungen, bessere Heilungschancen, effizientere Prävention – Medizin, maßgeschneidert auf den Einzelnen.
Doch auch hier gilt: Was als medizinischer Fortschritt gefeiert wird, bringt gesellschaftliche, rechtliche und ethische Herausforderungen mit sich:
- Wer entscheidet, welche genetischen Daten relevant sind?
- Welche Rolle spielt der Zugang zu diesen Technologien – werden soziale Ungleichheiten dadurch vertieft?
- Und wie lassen sich Diskriminierung, etwa durch genetisches Scoring bei Versicherungen oder Arbeitgebern, verhindern?
Zwischen der Hoffnung auf individuelle Heilung und der Sorge vor kollektiver Entmündigung stehen grundlegende Fragen: Wird der Mensch durch den Zugriff auf seine biologischen Daten ermächtigt – oder entblößt? Ist personalisierte Medizin ein Segen – oder der Auftakt zu einer Ära subtiler Entmenschlichung?
Daher lautet die Gretchenfrage: Ist die personalisierte Medizin der verheißene Aufstieg in den Himmel – oder der Sturz durch die Tore der Hölle?
Dieser Blogbeitrag lädt dazu ein, die personalisierte Medizin in ihrer ganzen Komplexität zu betrachten – jenseits von Hype oder Alarmismus. Er richtet sich bewusst auch an fachfremde Leser, die sich kritisch mit den Chancen, Risiken und ethischen Spannungsfeldern dieser medizinischen Revolution auseinandersetzen möchten.
📑Inhaltsverzeichnis
1. Die begriffliche Einordnung und das grundlegende Paradigma
2. Treibende Kräfte – zwischen Vision und Wirklichkeit
3. Die Zukunft der Medizin beginnt in der Zelle
3.1. Wie Biologie und Technologie zusammenfinden
3.2. Daten, Verantwortung und der größere Rahmen
4. Von DNA zur Erkenntnis – ein Hochgeschwindigkeitszug der Innovation
4.1. Die DNA – Bauplan des Lebens
a) Die DNA: Das molekulare Informationsarchiv des Lebens
b) Vom Code zum Protein: Wie Anleitungen Wirklichkeit werden
c) Epigenetik & nicht-codierende DNA: Das Dirigenten-Team unseres Erbguts
d) Das verborgene Kontrollzentrum: Was die nicht-codierende DNA steuert
e) Wenn das Betriebssystem krank macht
f) Offene Rätsel der Forschung
g) Die wahre Magie der Gene
4.2. Genetische Variation und ihre Folgen
4.3. Genomsequenzierung: Schlüssel zum individuellen Erbgut
a) Die Reise der DNA: Vom Nabelschnurblut zur genetischen Information
b) Drei Regieanweisungen für die Gen-Diagnostik: WGS, WES und Panel
4.4. Methoden der Genomsequenzierung
4.4.1. Methode 1: Kopieren mit Leuchtfarbe
a) Sanger-Sequenzierung
b) Illumina-Technologie
c) Single-Molecule Real-Time(SMRT)-Sequenzierung
4.4.2. Methode 2: Hightech-Tunnel – DNA elektrisch abtasten
a) Oxford Nanopore Sequenzierung
4.4.3. Genomsequenzierung – Technologien im Vergleich
a) Durchsatz vs. Verwertbarkeit – Masse ist nicht immer gleich Klasse
b) Wirtschaftlichkeit – Kosten pro Base vs. Kosten pro Erkenntnis
c) Von der Probe zum Befund – Wie schnell spricht das ganze Genom?
d) Klinische Einsatzfelder – Welche Technologie passt wozu?
e) KI als Co-Pilot – Automatisierung ohne Verantwortung abzugeben
f) Vergleichstabelle: Sanger, Illumina, PacBio, ONT
g) Key Takeaways: Zwischen Reife und Routine
4.5. Genomsequenzierung: Der nächste Technologiesprung
a) FENT: Wie ein Mikrochip die DNA-Analyse revolutioniert
b) SBX: Wenn DNA sich streckt, um verstanden zu werden
c) Das G4X-System: Vom genetischen Rezept zum räumlichen Atlas der Zelle
4.6. Vom Code zur Heilung – Bioinformatik als Schlüssel der Medizin von morgen
4.6.1. Vom genetischen Code zur computergestützten Genomanalyse
4.6.2. Moderne Bioinformatik – Der digitale Werkzeugkasten der Biologie
5. Eine Erfolgsgeschichte
6. Ein Blick in die Zukunft: Das Synthetic Human Genome Project
7. Personalisierte Medizin und Smart Governance
7.1. Macht, Governance und Smart Governance
7.2. Gouvernementalität
7.3. Personalisierte Medizin als Katalysator biomolekularer Gouvernementalität
7.4. Der weltweite Trend: Globale biomolekulare Gouvernementalität
7.5. Schlussfolgerung: Globale Ambivalenz der biomolekularen Macht
8. Epilog

1. Die begriffliche Einordnung und das grundlegende Paradigma
Die Entwicklung der modernen Medizin hin zu maßgeschneiderten Behandlungen wird häufig mit Begriffen wie „personalisierte Medizin“, „individualisierte Therapie“ oder „Präzisionsmedizin“ beschrieben. Doch was genau verbirgt sich hinter diesen Schlagworten?
Personalisierte Medizin gilt derzeit als einer der populärsten Begriffe – eingängig wie ein Werbeslogan. Damit verbunden ist das Versprechen, Therapien auf jeden Menschen zuzuschneiden: auf seine Gene, seine Lebensgewohnheiten, seine Biologie. In den Medien und in der öffentlichen Debatte wird der Ausdruck entsprechend häufig verwendet.
Individualisierte Therapie klingt weniger glamourös, meint aber Ähnliches: die Anpassung von Medikamenten oder Dosierungen an die Bedürfnisse einzelner Patienten. Dieser Begriff findet vor allem im klinischen Alltag Verwendung.
Präzisionsmedizin schließlich ist der nüchternste der drei Begriffe – ein Fachwort, das in Forschungslaboren und wissenschaftlichen Publikationen dominiert. Es unterstreicht den datenbasierten Ansatz: Algorithmen, Genanalysen und Biomarker entscheiden darüber, welche Therapie bei welcher Patientengruppe wirkt. Das ist die Sprache der Wissenschaft.
Doch egal, welcher Begriff gewählt wird – sie alle stehen für einen Paradigmenwechsel: Weg von standardisierten Verfahren – hin zu einer Medizin, die den einzelnen Menschen in seiner biologischen Einzigartigkeit in den Mittelpunkt stellt.
2. Treibende Kräfte – zwischen Vision und Wirklichkeit
Die treibenden Kräfte hinter der personalisierten Medizin sind vielfältig und eng miteinander verflochten. Auf der einen Seite stehen wissenschaftlich-technologische Entwicklungen, die in den letzten Jahrzehnten enorme Fortschritte ermöglicht haben: neue Methoden, um Erbgut schnell und genau zu lesen, leistungsfähige Datenanalyse durch künstliche Intelligenz sowie die Verfügbarkeit großer biomedizinischer Datenmengen. Diese Technologien bilden die Grundlage dafür, individuelle Unterschiede zwischen Patienten überhaupt erst erkennen und medizinisch nutzen zu können.
Auf der anderen Seite wirken gesellschaftliche und politische Impulse als Beschleuniger dieses Wandels. Öffentliche Gesundheitsprogramme, gezielte Forschungsförderung, Strategiepapiere internationaler Gesundheitsorganisationen und gezielte Gesetzesinitiativen treiben die Integration der personalisierten Medizin in die Versorgungspraxis aktiv voran. Auch das gestiegene Gesundheitsbewusstsein in der Bevölkerung sowie die zunehmende Erwartung an individuelle Behandlungsangebote verstärken diesen Trend.
Personalisierte Medizin ist also kein bloßes Produkt technologischer Machbarkeit, sondern entsteht im Spannungsfeld von Innovation, Politik und gesellschaftlicher Nachfrage.
Ein aktuelles Beispiel für die praktische Umsetzung dieser Entwicklung liefert das Vereinigte Königreich: Im Rahmen eines neuen 10-Jahres-Plans soll künftig jedes Neugeborene einem DNA-Test unterzogen werden. Die sogenannte Ganzgenomsequenzierung soll nach hunderten genetisch bedingten Krankheiten suchen – mit dem Ziel, potenziell tödliche Erkrankungen frühzeitig zu erkennen und ihnen gezielt vorzubeugen. Nach Angaben des britischen Gesundheitsministers Wes Streeting geht es darum, den National Health Service (NHS) grundlegend zu transformieren: weg von der reinen Krankheitsbehandlung – hin zu einem System der Vorhersage und Prävention. Genomik, also die umfassende Analyse der Erbinformation, soll dabei in Kombination mit künstlicher Intelligenz als Frühwarnsystem dienen.
„Die Revolution in der medizinischen Wissenschaft bedeutet, dass wir den NHS von einem Dienst, der Krankheiten diagnostiziert und behandelt, in einen Dienst verwandeln können, der sie vorhersagt und verhindert“, so Streeting. Die Vision: eine personalisierte Gesundheitsversorgung, die Risiken erkennt, bevor Symptome überhaupt auftreten – und damit die Lebensqualität verbessert sowie das Gesundheitssystem entlastet.
Doch so überzeugend diese Vision auch klingt – aus der Wissenschaft kommen auch mahnende Stimmen. So warnt etwa der Genetiker Prof. Robin Lovell-Badge vom Francis Crick Institute davor, die Komplexität der genomischen Daten zu unterschätzen. Es brauche nicht nur technisches Know-how zur Datenerhebung, sondern vor allem qualifiziertes Personal, das die gewonnenen Informationen verantwortungsvoll und verständlich kommunizieren kann. Denn Daten allein sind noch keine Diagnose – entscheidend ist ihre sinnvolle und patientengerechte Interpretation.
Was bedeutet es konkret, Medizin neu zu denken?
Die wahre Revolution spielt sich in den Laboren und Rechenzentren ab, in denen die Bausteine der personalisierten Medizin entstehen. Die folgenden Kapitel führen Schritt für Schritt hinter die Kulissen und zeigen die Schlüsseltechnologien, die nötig sind, um die personalisierte Medizin vom Konzept in die Klinik zu bringen.
3. Die Zukunft der Medizin beginnt in der Zelle
3.1. Wie Biologie und Technologie zusammenfinden
Unser Körper besteht aus Milliarden Zellen – winzigen, spezialisierten Einheiten, die Tag für Tag zusammenarbeiten, um Gesundheit und Leben zu ermöglichen. In jeder einzelnen Zelle laufen unzählige Prozesse ab, die genau aufeinander abgestimmt sind. Diese zellulären Mechanismen sind entscheidend für das Verständnis von Gesundheit, Krankheit und der Entwicklung neuer, gezielter Therapien.
Ein zentraler Mechanismus ist die Energiegewinnung: Zellen verwandeln Nährstoffe und Sauerstoff in ATP (Adenosintriphosphat)– die „Energie-Währung“ des Körpers. Ohne ATP könnte keine Zelle funktionieren, kein Muskel sich bewegen, kein Gedanke entstehen.
Gleichzeitig produzieren Zellen fortwährend Proteine – auf Basis genetischer Informationen. Diese komplexen Moleküle übernehmen zentrale Aufgaben im Körper – als chemische Katalysatoren, Botenstoffe, Baustoffe oder Transportmoleküle.
Auch die Reinigung und Erneuerung innerhalb der Zelle ist lebenswichtig: Prozesse wie die Autophagie bauen beschädigte Zellbestandteile ab und recyceln sie – eine Art innere Müllabfuhr, die Krankheiten wie Alzheimer oder Parkinson vorbeugen kann.
Damit Zellen im Körper koordiniert zusammenarbeiten können, brauchen sie funktionierende Signalwege – sie „kommunizieren“ über chemische Botenstoffe miteinander. Diese Zellkommunikation steuert, wann Zellen wachsen, ruhen oder auf Veränderungen reagieren sollen.
Ein weiterer zentraler Vorgang ist die Zellteilung – notwendig für Wachstum, Wundheilung und Gewebeerneuerung. Gleichzeitig sorgen DNA-Reparaturmechanismen dafür, dass Fehler im Erbgut korrigiert werden. Gelingt das nicht, greift die Apoptose, der programmierte Zelltod, um Schäden zu begrenzen.
All diese Mechanismen laufen im Hintergrund ab – ständig und hochpräzise. Wenn jedoch einer dieser Abläufe gestört ist, kann das zu schweren Erkrankungen führen, etwa Krebs, Autoimmunerkrankungen oder Stoffwechselstörungen. Genau hier setzt die personalisierte Medizin an. Sie nutzt modernste Technologien, um diese zellulären Prozesse besser zu verstehen und gezielt zu beeinflussen.
Dank Genomsequenzierung (etwa durch „Next Generation Sequencing“) lassen sich genetische Veränderungen identifizieren, die z. B. die Proteinproduktion oder Reparaturmechanismen beeinflussen.
Biomarker-Analysen (die Analyse bestimmter Moleküle)im Blut, Gewebe oder anderen Körperflüssigkeiten zeigen, ob bestimmte Signalwege über- oder unteraktiv sind – und helfen dabei, individuelle Krankheitsverläufe vorherzusagen oder passende Therapien auszuwählen.
Die Einzelzellanalytik ermöglicht es, Unterschiede selbst zwischen einzelnen Zellen sichtbar zu machen – etwa in einem Tumor, wo manche Zellen auf eine Therapie ansprechen und andere nicht. Dadurch wird eine präzisere Behandlung möglich.
Auch die Proteomik (Analyse aller Proteine in einer Zelle) und Metabolomik (Analyse von Stoffwechselprodukten) liefern ein aktuelles Bild davon, wie aktiv bestimmte zelluläre Mechanismen wirklich sind – z. B. ob eine Zelle gestresst ist, ausreichend Energie hat oder sich gerade teilt.
Künstliche Intelligenz (KI) hilft dabei, diese riesigen Datenmengen zu analysieren und Muster zu erkennen – zum Beispiel, welche Kombination von zellulären Störungen typisch für eine bestimmte Krebserkrankung ist. Digitale Gesundheitsdaten (etwa aus Wearables) ergänzen dieses Bild im Alltag und ermöglichen eine präzise Langzeitbeobachtung.
Sogar neue Therapieansätze beruhen auf dem Verständnis zellulärer Abläufe: Gentherapien und CRISPR-basierte Gen-Editierung greifen gezielt in die DNA ein, um fehlerhafte Mechanismen zu korrigieren. In Organoiden – Mini-Organmodellen im Labor – lassen sich Medikamente direkt an patientenspezifischen Zellmodellen testen, ganz ohne Risiko für den Menschen.
Moderne Technologien machen heute sichtbar, was früher im Verborgenen lag: Wie Zellen arbeiten, was bei Krankheit aus dem Gleichgewicht gerät – und wie man gezielt eingreifen kann. So entsteht eine individualisierte Medizin, die nicht mehr auf Vermutung basiert, sondern auf messbarer Biologie.
Diese neue Präzision bringt nicht nur Chancen, sondern auch neue Verantwortlichkeiten mit sich.
3.2. Daten, Verantwortung und der größere Rahmen
Die Komplexität dieser zellulären Prozesse spiegelt sich unmittelbar in der Komplexität der personalisierten Medizin wider: Je genauer wir die feinen Abläufe und Wechselwirkungen in unseren Zellen verstehen, desto gezielter – aber auch anspruchsvoller – werden Diagnose und Therapie. Diese neue Präzision erfordert neue Technologien, neue Denkweisen und ein tiefes biologisches Verständnis. Gleichzeitig eröffnet sie die Chance auf wirksamere, nachhaltigere und maßgeschneiderte Behandlungskonzepte.
Doch je gezielter und tiefer diese Eingriffe werden, desto größer ist auch die Verantwortung – und die Herausforderung, unerwünschte Langzeitwirkungen oder Nebenreaktionen frühzeitig zu erkennen und zu beherrschen. Gerade bei innovativen Verfahren wie Gentherapie oder Immunmodulation gilt es, den Nutzen sorgfältig gegen mögliche Risiken abzuwägen. Die personalisierte Medizin verlangt daher nicht nur Präzision, sondern auch Weitblick.
Ein zentrales Element dieses Weitblicks ist die Verfügbarkeit personenbezogener Gesundheitsdaten – und zwar nicht nur im Einzelfall, sondern im großen Maßstab.
Erst durch die Kombination individueller Daten mit Bevölkerungsdaten wird es möglich, biologische Prozesse im Detail zu verstehen, Krankheitsverläufe vorherzusagen und personalisierte Therapien zu entwickeln. Genetische Informationen, Laborwerte, Bildgebungsdaten oder digitale Alltagsmessungen liefern Hinweise darauf, wie zelluläre Mechanismen bei einem Menschen konkret funktionieren – und wie sie bei anderen variieren.
Um diese individuellen Muster medizinisch einzuordnen, braucht es den Vergleich mit großen Datensätzen. Nur so können KI-Systeme komplexe Zusammenhänge erkennen, die dem menschlichen Auge entgehen – etwa seltene Genvarianten, molekulare Risikokonstellationen oder unerwartete Therapieeffekte. Dabei gilt: Je größer und vielfältiger die Datenbasis, desto zuverlässiger die Modelle – besonders, wenn sie laufend mit neuen Informationen angereichert werden.
Doch diese Fortschritte gelingen nur, wenn Menschen bereit sind, ihre Gesundheitsdaten zu teilen – und darauf vertrauen können, dass sie sicher und verantwortungsvoll genutzt werden.
Die Zukunft der Medizin beginnt dort, wo alles Leben entsteht: in der Zelle. Doch ihr Erfolg hängt ab vom Zusammenspiel dreier Kräfte: technologischer Innovation, wissenschaftlicher Tiefe – und ethischer Verantwortung.

4. Von DNA zur Erkenntnis – ein Hochgeschwindigkeitszug der Innovation
Wie Genomsequenzierung und bioinformatische Analyse die Medizin revolutionieren
4.1. Die DNA – Bauplan des Lebens
a) Die DNA: Das molekulare Informationsarchiv des Lebens
b) Vom Code zum Protein: Wie Anleitungen Wirklichkeit werden
c) Epigenetik & nicht-codierende DNA: Das Dirigenten-Team unseres Erbguts
d) Das verborgene Kontrollzentrum: Was die nicht-codierende DNA steuert
e) Wenn das Betriebssystem krank macht
f) Offene Rätsel der Forschung
g) Die wahre Magie der Gene
4.2. Genetische Variation und ihre Folgen
4.3. Genomsequenzierung: Schlüssel zum individuellen Erbgut
a) Die Reise der DNA: Vom Nabelschnurblut zur genetischen Information
b) Drei Regieanweisungen für die Gen-Diagnostik: WGS, WES und Panel
4.4. Methoden der Genomsequenzierung
4.4.1. Methode 1: Kopieren mit Leuchtfarbe
4.4.2. Methode 2: Hightech-Tunnel – DNA elektrisch abtasten
4.4.3. Genomsequenzierung – Technologien im Vergleich
4.5. Genomsequenzierung: Der nächste Technologiesprung
4.6. Vom Code zur Heilung: Bioinformatik als Schlüssel der Medizin von morgen
Stellen Sie sich vor, Sie stehen am Bahnsteig, und ein Zug rauscht mit solch einer Geschwindigkeit vorbei, dass kaum ein Bild im Fenster zu erkennen ist. So ähnlich empfinden viele Menschen die Entwicklungen in der personalisierten Medizin: Technologien kommen in rasantem Tempo, Begriffe wirken abstrakt – und doch zieht sich durch alles ein roter Faden: das menschliche Genom.
So vielfältig die modernen Ansätze auch sind – von Organoiden über CRISPR-basierte Gen-Editierung bis zu RNA-Medikamenten – im Zentrum steht der genetische Bauplan des Menschen. Die Genomsequenzierung ist heute Grundlage vieler Diagnosen und Therapieentscheidungen. Sie zeigt, welche genetischen Veränderungen Krankheiten auslösen, welche Signalwege betroffen sind – und wo gezielte Eingriffe möglich sein könnten.
Was einst Milliarden kostete und Jahre dauerte, gelingt heute in wenigen Tagen: Das gesamte Erbgut eines Menschen kann entschlüsselt werden. Doch die reine Sequenz ist noch kein Befund. Die Abfolge von drei Milliarden Basenpaaren entfaltet erst dann medizinischen Wert, wenn sie auch verstanden wird – und genau hier beginnt die eigentliche Herausforderung.
Die Bioinformatik betritt die Bühne: ein interdisziplinäres Feld, das Mathematik, Informatik und Biologie vereint, um aus Rohdaten Muster, Risiken und Therapieoptionen abzuleiten. Sie ist das eigentliche Nadelöhr der personalisierten Medizin – denn sie entscheidet, ob genetische Varianten als harmlos, risikobehaftet oder krankheitsverursachend gelten.
Dazu werden individuelle Sequenzen mit internationalen Referenzdatenbanken abgeglichen, Algorithmen analysieren Millionen genetischer Varianten, und lernende Systeme modellieren das komplexe Zusammenspiel von Genetik, Umwelt und Stoffwechsel.
Dieser Prozess ist hochkomplex – und zugleich essenziell. Denn erst die rechnergestützte Auswertung macht die DNA medizinisch nutzbar. Zwischen Genomsequenzierung und bioinformatischer Analyse besteht eine untrennbare Symbiose: Die Sequenz liefert die Daten, die Analyse das Verständnis. Ohne sie bleibt die DNA eine bloße Aneinanderreihung von Buchstaben.
Genau deshalb richtet sich der Blick der folgenden Abschnitte auf die entscheidende Frage:
Was heißt es, das Genom zu entschlüsseln – und es wirklich zu verstehen?
Welche Aussagen sind möglich – und wo liegen ihre Grenzen?
Denn wer die Zukunft der Medizin begreifen will, muss wissen, wie wir heute das menschliche Erbgut lesen – und wie wir lernen, es zu deuten.
Der Hochgeschwindigkeitszug der Innovation ist längst abgefahren. Wer mitfahren will, muss verstehen, wohin er führt.
4.1. Die DNA – Bauplan des Lebens
Um diese Reise anzutreten, lohnt ein kurzer Blick auf die molekularen Grundlagen: Wie unsere DNA zur Bauanleitung wird, wie daraus über den Umweg der RNA Proteine entstehen – und wie die Epigenetik diese Prozesse feinjustiert. Erst dieses Fundament macht verständlich, was beim Entschlüsseln des Genoms eigentlich passiert.
a) Die DNA: Das molekulare Informationsarchiv des Lebens
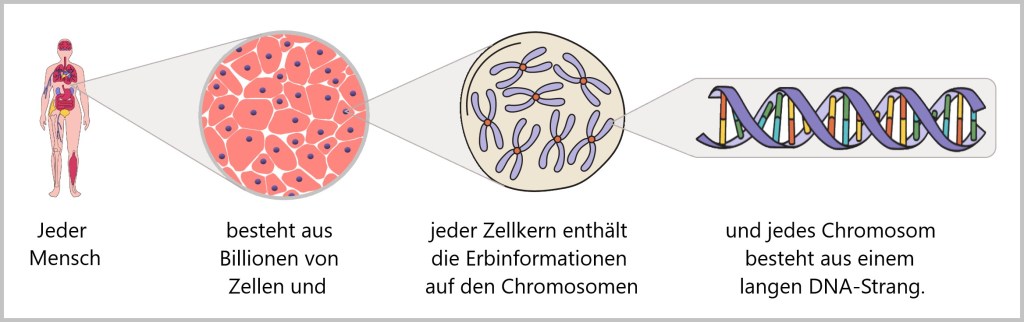
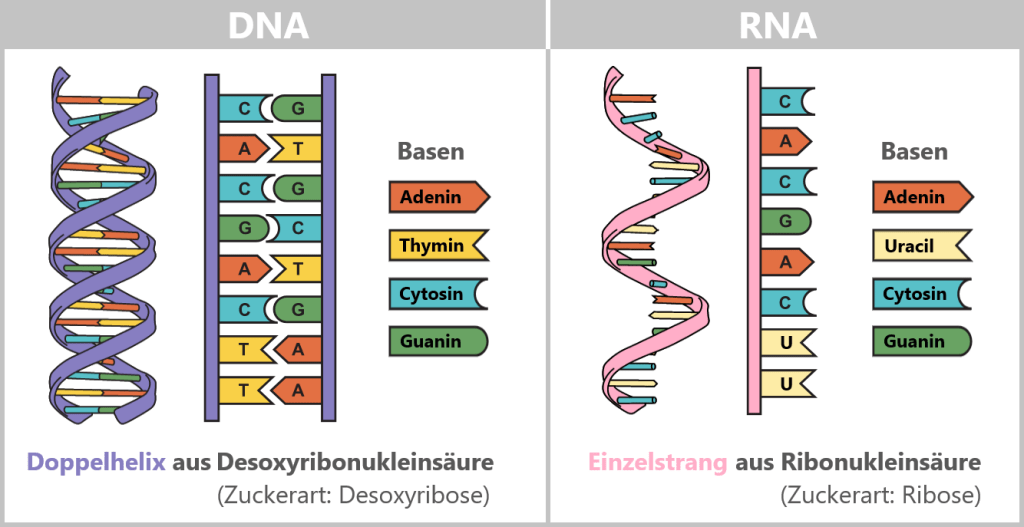
In jeder Zelle unseres Körpers liegt sorgfältig verpackt ein beeindruckendes Molekül: die DNA (Desoxyribonukleinsäure). Sie enthält die vollständige Bau- und Funktionsanleitung unseres Körpers – ein molekulares Informationsarchiv des Lebens.
Strukturell gleicht sie einer spiralförmig verdrehten Strickleiter: Zwei Zucker-Phosphat-Ketten bilden die Seiten, dazwischen liegen die „Sprossen“ – die Basen Adenin (A), Cytosin (C), Guanin (G) und Thymin (T). Sie paaren sich stets nach festen Regeln: A mit T, C mit G. Diese komplementäre Bindung macht jeden Strang zur Vorlage für den anderen – ein molekulares Backup-System.

Entrollt man die DNA aus nur einer einzigen Zelle, misst sie rund zwei Meter – bei einem Durchmesser von nur wenigen Millionstel Millimetern. Das Leben hängt also buchstäblich an einem hauchdünnen, aber erstaunlich langen Faden.
Die Abfolge der rund drei Milliarden Basenpaare ist so individuell wie ein Fingerabdruck. Sie bildet den genetischen Code – die Anweisungen zur Herstellung von Proteinen, jenen Molekülen, die Struktur, Funktion und Regulation aller biologischen Prozesse steuern.
Als zentrales Informationsarchiv ist die DNA zugleich äußerst wertvoll und empfindlich. Deshalb wird sie sicher im Zellkern aufbewahrt, vergleichbar mit einem Safe. Damit der Körper auf die Informationen in diesem Safe zugreifen kann, braucht es ein cleveres Transportsystem. Schließlich müssen die Baupläne für Proteine – jene molekularen Architekten aller Lebensprozesse – zu den Ribosomen gelangen, den sogenannten „Protein-Fabriken“ im Zellplasma. Dort werden die Proteine hergestellt – ein Prozess, den man als Proteinbiosynthese bezeichnet.
Doch wie kommt ein konkreter Bauplan sicher aus dem Zellkern zu diesen Fabriken? Mit anderen Worten: Wie wird die genetische Information für ein bestimmtes Protein vom geschützten Archivort an den Ort der Produktion übertragen?
b) Vom Code zum Protein: Wie Anleitungen Wirklichkeit werden
Die Reise beginnt bei den Genen – bestimmten Abschnitten der DNA, die die Baupläne für Proteine enthalten. Etwa 20.000 solcher Gene finden sich im menschlichen Genom.
Die DNA ist dabei nicht nur ein passives Informationsarchiv im Zellkern – sie enthält ein aktives Repertoire an Anleitungen. Man kann sich jedes Gen wie ein Kapitel in einem molekularen Kochbuch vorstellen: nicht selten mit mehreren Varianten eines Rezepts, angepasst an Zelltyp, Entwicklungsstand oder äußere Bedingungen.
Zunächst entsteht eine temporäre „Arbeitskopie“ eines Gens – in Form von RNA (Ribonukleinsäure). Sie ähnelt der DNA, verwendet jedoch die Base Uracil (U) anstelle von Thymin (T) und besteht meist nur aus einem einzelnen Strang. Diese flüchtige Struktur macht die RNA ideal für den kurzfristigen Gebrauch – sie wird nach Erfüllung ihrer Funktion wieder abgebaut.
Alternatives Splicing: Die flexible Küche der Gene
Zellen müssen sich flexibel an wechselnde Anforderungen anpassen: auf Entzündungen reagieren, Krankheitserreger abwehren, Muskeln aufbauen oder den Blutzuckerspiegel regulieren. Dafür braucht es keine starre Anleitung, sondern ein System, das je nach Situation improvisieren kann.
In Abhängigkeit vom Zelltyp und äußeren Signalen wird der RNA-Strang dynamisch umgebaut, indem er an bestimmten Markierungen – übertragen aus der DNA – geschnitten wird. Nicht benötigte Abschnitte (Introns) werden entfernt, die verbleibenden (Exons) neu zusammengesetzt. So entsteht aus einer Vorlage eine Vielzahl möglicher Botschaften. Das Ergebnis ist die messenger RNA (mRNA), die die genetische Information aus dem Zellkern zu den Ribosomen im Zytoplasma bringt – den „Protein-Fabriken“ der Zelle.
Man kann sich diesen Prozess vorstellen wie das Nachkochen eines Rezepts: je nach Bedarf lässt die Zelle Zutaten weg, fügt Gewürze hinzu oder passt die Zubereitungsschritte an. So wird aus einer Grundanleitung eine Vielfalt von Gerichten.
Durch diesen Prozess – genannt alternatives Splicing – können Zellen aus einem Gen eine Vielzahl unterschiedlicher Proteine herstellen, die jeweils spezifische Funktionen übernehmen.
Der gesamte Vorgang „Vom Gen zum Protein“ wird häufig als Genexpression bezeichnet.
💡 Beispiel: Das Titin-Gen
👉 Längstes menschliches Gen (≈ 300.000 Basenpaare).
👉 Durch alternatives Splicing entstehen > 20.000 verschiedene Proteinvarianten – jeweils angepasst an die unterschiedlichen Anforderungen in verschiedenen Muskeltypen.
Unser Erbgut ist also kein starres Rezeptbuch – es ist eine dynamische Küche, in der die Zelle aus denselben Vorlagen immer neue Lösungen entwickelt.
📌 Wer sich dafür interessiert, wie die Erbinformation in der DNA Schritt für Schritt in ein Protein übersetzt wird, findet hier weiterführende Informationen.
Doch wie in jeder kreativen Küche können auch hier beim Übergang der DNA zur mRNA Fehler passieren: Wird beim Abschreiben oder Montieren ein „Zutatenbuchstabe“ übersehen, vertauscht oder falsch zusammengesetzt, kann die Botschaft am Ende missverständlich oder unbrauchbar werden. Solche Mutationen oder fehlerhaftes Splicing sind manchmal harmlos – doch sie können auch defekte Proteine hervorbringen, die ihre Aufgabe nicht erfüllen oder sogar Schaden anrichten. Ein kleiner Patzer in der Abschrift des molekularen Kochbuchs – und der Geschmack des Lebens verändert sich.
c) Epigenetik & nicht-codierende DNA: Das Dirigenten-Team unseres Erbguts
Obwohl alle Zellen die gleiche DNA enthalten, nutzen sie jeweils nur einen Bruchteil davon. Eine Leberzelle benötigt andere Anleitungen als eine Nervenzelle – der Rest bleibt stillgelegt. Diese gezielte Genregulation wird durch epigenetische Mechanismen gesteuert: Kleine chemische Markierungen – etwa Methylgruppen – verändern nicht die genetische Sequenz selbst, sondern beeinflussen, ob ein Gen aktiv ist, wie oft es abgelesen wird oder ob es komplett stummgeschaltet bleibt.
Doch wer gibt dem Dirigenten eigentlich seine Anweisungen? Hier kommt die nicht-codierende DNA ins Spiel – 98 % unseres Erbguts, lange als nutzloser „Ballast“ abgetan. Sie ist das Betriebssystem, das die epigenetischen Schaltpläne bereitstellt. Erst ihr Zusammenspiel legt fest, wann, wo und wie stark unsere Gene aktiv werden – und entscheidet so über Gesundheit oder Krankheit.
d) Das verborgene Kontrollzentrum: Was die nicht-codierende DNA steuert
In diesen „dunklen“ Regionen verbergen sich präzise Schaltkreise, die das Schicksal jeder Zelle lenken:
Enhancer wirken wie unsichtbare Fernbedienungen der Gene. Selbst über Tausende Bausteine hinweg können sie ein Zielgen aktivieren oder zum Schweigen bringen – etwa für die Herzentwicklung im Embryo.
Telomere sind die Schutzkappen der Chromosomenenden, wie Plastikhüllen an Schnürsenkeln. Doch wenn sie schrumpfen, beginnt das Altern – ein zentraler Mechanismus von Zellverfall und Krebsentstehung.
Nicht-codierende RNAs agieren als heimliche Regisseure: Mikro-RNAs blockieren krankmachende Gene, während die lange RNA XIST bei Frauen ein gesamtes X-Chromosom zum Schweigen bringt.
Epigenetische Markierungen legen eine unsichtbare Landkarte über das Genom. Mit Methylierungen wie roten Stoppschildern entscheiden sie, ob ein Enhancer oder Gen gelesen werden darf – oder verstummt.
So entsteht Ordnung im molekularen Kochbuch: Jeder Zelltyp liest nur die Seiten, die er braucht – gesteuert durch nicht-codierende DNA und ihre epigenetische Interpretation.
e) Wenn das Betriebssystem krank macht
Fast alle genetischen Risikomarker für Volkskrankheiten – von Diabetes über Herzleiden bis zu psychischen Störungen – liegen in nicht-codierenden Zonen:
Mutationen in einem Enhancer für das MYC-Gen können Leukämie auslösen, indem sie die Zellteilung außer Kontrolle bringen.
Epigenetische Fehlprogrammierungen in diesen Bereichen – etwa durch chronischen Stress oder Umweltgifte – können Gene dauerhaft falsch an- oder abschalten und so Krankheiten wie Krebs oder Autoimmunstörungen begünstigen.
Solche epigenetischen Muster sind teils reversibel – ein Hoffnungsschimmer für neue Therapien.
f) Offene Rätsel der Forschung
Trotz gewaltiger Fortschritte bleibt das Genom voller ungelöster Rätsel:
Die „dunkle Materie“: Rund die Hälfte des nicht-codierenden Genoms ist funktional unergründet. Handelt es sich um evolutionäres Rauschen – oder um ein noch unkartiertes Steuernetz des Lebens?
Langstrecken-Kommunikation: Wie findet ein Enhancer zielsicher sein Gen inmitten von Millionen DNA-Bausteinen? Die räumliche Faltung des Genoms – ein wahres „Chromatin-Origami“ – könnte der Schlüssel sein.
Umwelt-Einflüsse: Weshalb führen identische nicht-codierende Mutationen in verschiedenen Lebenswelten zu völlig unterschiedlichen Krankheitsbildern?
RNA-Codes: Zehntausende nicht-codierende RNAs sind entdeckt – doch welche von ihnen sind nur Randnotizen, und welche erheben sich möglicherweise zu Leitmotiven neuer Therapien?
g) Die wahre Magie der Gene
Die nicht-codierende DNA ist kein Datenmüll, sondern das Mastermind der Zelle. Die Epigenetik wiederum ist ihr Dirigent – sie interpretiert flexibel, was das Betriebssystem vorgibt. Gemeinsam formen sie unsere Entwicklung, steuern unsere Gesundheit und reagieren auf Umweltsignale.
Gene sind kein starres Schicksal, mehr eine Partitur, die immer wieder neu interpretiert werden kann. Im Zusammenspiel entsteht ein Orchester, das nicht nur unsere Zellen formt, sondern sich ständig neu einstimmt – durch Erfahrungen, Umwelteinflüsse und sogar durch Spuren, die auch an kommende Generationen weitergegeben werden können.
Die wahre Magie liegt darin, dass aus begrenzten Noten eine unendliche Vielfalt von Leben erklingt.
4.2. Genetische Variation und ihre Folgen
Wenn sich das Erbgut verschreibt, verstummt oder improvisiert.
Bei jeder Zellteilung wird die DNA sorgfältig kopiert und an die Tochterzellen weitergegeben – so bleibt die genetische Information über Generationen hinweg erhalten.
Doch das System ist nicht unfehlbar: Mutationen – also Veränderungen in der DNA-Sequenz – können Struktur und Funktion von Proteinen beeinflussen. Manchmal stimmt nur ein Buchstabe nicht. Oder ein Satz fehlt. Oder ein ganzer Absatz wurde umgestellt. In der Genetik spricht man dann von Varianten – genetischen Veränderungen, die je nach Art, Größe und Position harmlos, risikobehaftet oder krankheitsauslösend sein können.
Die folgende Übersicht enthält die wichtigsten Variantentypen – und was sie im molekularen Manuskript des Lebens bedeuten:
📌 SNVs (Single Nucleotide Variants) – Ein einziger Buchstabe ist vertauscht. Meist kaum spürbar – doch manchmal genügt er, um ganze Geschichten umzuschreiben, wie bei der Sichelzellenanämie.
📌 Indels (Insertionen/Deletionen) – Kleine Textfragmente werden eingefügt oder gelöscht. Schon eine winzige Verschiebung kann ganze Sätze aus dem Takt bringen, manchmal so stark, dass der ursprüngliche Sinn verloren geht.
📌 Strukturelle Varianten (SVs) – Lange DNA-Passagen werden verdreht, verdoppelt oder verschoben. Diese verdeckten Umschreibungen sind schwer zu entdecken – und oft verhängnisvoll, etwa bei Krebs.
📌 Splice-Site-Varianten – Fehler an den Schnittstellen zerreißen die Syntax der RNA. Es entstehen falsche Baupläne – und Proteine, die nicht mehr stimmen.
📌 Repeat-Expansionen – Basenfolgen werden zwanghaft wiederholt, bis sie den Text entstellen. So entstehen Krankheiten wie Chorea Huntington.
📌 Copy Number Variants (CNVs) – Ganze Kapitel (Gene) fehlen oder erscheinen doppelt. Das Gleichgewicht der Erzählung geht verloren.
📌 Mitochondriale Varianten – Veränderungen in der DNA der Mitochondrien, der Kraftwerke der Zelle. Wenn hier der Text verfälscht wird, stockt der Antrieb des Lebens.
📌 Somatische Varianten – Treten erst im Laufe des Lebens auf und betreffen nur bestimmte Zellgruppen. Sie prägen vor allem die Geschichte von Tumoren.
📌 Epigenetische Signale – Keine neuen Buchstaben, sondern eine andere Betonung: Lautstärke, Rhythmus, Beginn. Sie verwandeln denselben Text in eine völlig neue Melodie.
Die Liste zeigt: Unser Erbgut ist kein starres Gebilde, sondern lässt sich durch Varianten verändern. Manche dieser Abweichungen bleiben unauffällige Randnotizen, andere aber können die gesamte Geschichte neu schreiben – manchmal mit dramatischen Folgen.
Um solche feinen Unterschiede sichtbar zu machen, braucht es Werkzeuge, die jedes Detail erfassen – jeden Buchstaben, jedes Satzzeichen, jede Verschiebung. Genau hier setzt die moderne Genomsequenzierung an.
4.3. Genomsequenzierung: Schlüssel zum individuellen Erbgut
Das Erbgut eines Menschen gleicht einer gigantischen Bibliothek: Milliarden von Buchstaben, fein säuberlich in langen Bänden angeordnet. Um darin zu lesen, bedarf es der Genomsequenzierung – die wie ein hochpräziser Scanner den Rohtext unseres genetischen Bauplans liefert.
So wird sie zum Herzstück der personalisierten Medizin: einer Medizin, die ihre Entscheidungen am Einzelnen ausrichtet, nicht am Durchschnitt.
Doch wie gelangen wir überhaupt an das „Buch des Lebens“, um es Seite für Seite lesen zu können?
Stellen wir uns vor: Bei einem Neugeborenen wird kurz nach der Geburt ein DNA-Test durchgeführt – ein Szenario, das im Vereinigten Königreich bereits Wirklichkeit geworden ist, wie zuvor beschrieben.
a) Die Reise der DNA: Vom Nabelschnurblut zur genetischen Information
Unmittelbar nach der Geburt kann Blut aus der abgetrennten Nabelschnur entnommen werden – ein sicherer und schmerzfreier Vorgang. Das Nabelschnurblut stammt vom Neugeborenen und enthält zahlreiche wertvolle Zellen, insbesondere Stammzellen und weiße Blutkörperchen. In ihren Zellkernen liegt das Entscheidende verborgen: die DNA – das genetische Erbe des Kindes.
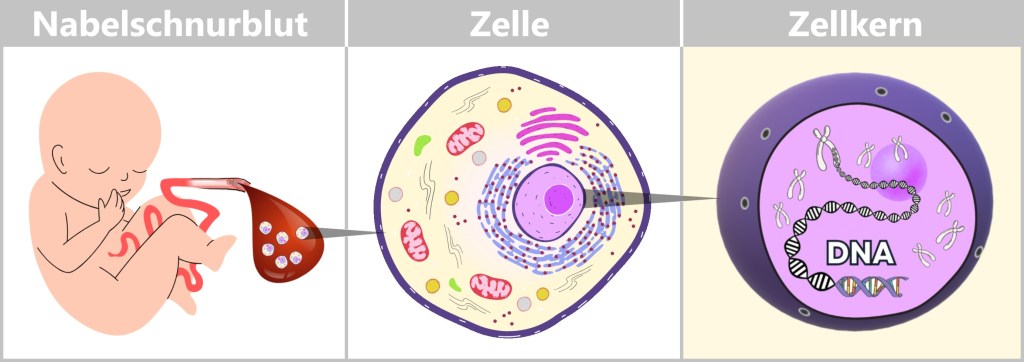
Links: Nabelschnurblut als reiche Quelle unterschiedlicher Zellen.
Mitte: Schematische Darstellung einer menschlichen Zelle.
Rechts: Im Zellkern liegt die DNA, Trägerin der genetischen Information.
Um diese DNA zu isolieren und analysieren zu können, durchläuft sie im Labor einen mehrstufigen Prozess. Dabei kommen bewährte molekularbiologische Methoden zum Einsatz, die sich in vier zentrale Schritte gliedern lassen:
Zellaufschluss: Zugang zum Erbgut schaffen
Zunächst wird das Blut zentrifugiert – also mit hoher Geschwindigkeit geschleudert. Dabei trennen sich die Bestandteile nach Dichte: Die weißen Blutkörperchen setzen sich als Zellpellet am Boden ab. Sie enthalten den größten Teil der interessierenden DNA.
Diese Zellen werden anschließend mit speziellen Lösungen und Enzymen behandelt, die die Zell- und Zellkernmembran auflösen. Dadurch wird die DNA freigesetzt – zunächst allerdings als Teil eines Gemischs aus Proteinen, Lipiden und anderen Zellbestandteilen.
DNA-Isolierung: Reines Erbgut gewinnen
Im nächsten Schritt wird die DNA aus dem Zelllysat – der „Zellsuppe“ – isoliert. Dazu gibt es verschiedene Verfahren:
Filtration oder Säulenreinigung: Die Lösung wird durch spezielle Filtersysteme geleitet, an denen die DNA haften bleibt, während kleinere Moleküle ausgespült werden.
Magnetkügelchen: Modernere Verfahren verwenden winzige Kügelchen mit DNA-bindenden Oberfläche. Die DNA haftet daran und kann per Magnet gezielt entnommen werden.
Beide Methoden ermöglichen eine effektive Trennung von Erbgut und Störstoffen.
Reinigung und Konzentration der DNA
Nun liegt die DNA vieler weißer Blutkörperchen in relativ reiner Form vor – allerdings noch in einer wässrigen Lösung. Um sie weiter zu reinigen und zu konzentrieren, wird kalter Alkohol (z. B. Ethanol oder Isopropanol) hinzugefügt. Dadurch fällt die DNA aus und wird sichtbar – als weißlicher, fadenartiger Niederschlag.
Die menschliche DNA ist extrem lang: Entrollt man sie, misst sie pro Zelle etwa zwei Meter. Bei der Extraktion wird sie jedoch durch chemische und mechanische Einflüsse in viele kleinere Abschnitte zerteilt. Das sieht zunächst nach Chaos aus – doch da die DNA in allen Zellen identisch ist, enthalten viele Fragmente überlappende Informationen. Diese zufällige Verteilung wird bei der Sequenzierung zum Vorteil: Die Fragmente können mehrfach unabhängig gelesen und anschließend am Computer zu einem Gesamtbild zusammengesetzt werden. Das erhöht die Genauigkeit der Analyse erheblich.
Qualitätskontrolle: Ist die DNA intakt?
Bevor die DNA-Probe analysiert werden kann, muss ihre Qualität überprüft werden. Zwei gängige Verfahren sind:
Spektrophotometrie: Misst die Konzentration und Reinheit der DNA über die Lichtabsorption.
Gelelektrophorese: DNA-Fragmente werden durch ein Gel geleitet und nach Länge getrennt. (Längere Fragmente wandern langsamer als kürzere.) So lässt sich beurteilen, ob die Probe ausreichend intakt ist.
Warum ist dieser Prozess so wichtig?
Die menschliche DNA ist empfindlich – Verunreinigungen oder Fragmentierungen können Analysen verfälschen oder unbrauchbar machen. Eine sorgfältige und kontrollierte Aufreinigung ist daher Voraussetzung für zuverlässige genetische Aussagen.
Gerade Nabelschnurblut stellt eine besonders wertvolle Quelle medizinischer Information dar: Es ermöglicht Frühdiagnostik, Prognosen und in manchen Fällen gezielte Prävention.
Was im Labor wie ein routinierter Ablauf wirkt, ist in Wahrheit ein hochpräziser Vorgang – erfordert technisches Know-how, moderne Geräte und biochemisches Feingefühl. Durch automatisierte Extraktionsmethoden lässt sich heute in weniger als einer Stunde aus wenigen Tropfen Nabelschnurblut eine hochwertige DNA-Probe gewinnen. Sie besteht zwar aus Fragmenten, enthält aber dennoch alle Informationen, um den individuellen genetischen Code zu entschlüsseln – und damit die Grundlage für eine Medizin von morgen zu legen.
b) Drei Regieanweisungen für die Gen-Diagnostik: WGS, WES und Panel
Bevor wir tiefer in die Werkzeuge der Genom-Sequenzierung eintauchen, lohnt sich ein Blick auf die drei zentralen Strategien, mit denen heute genetische Informationen entschlüsselt werden – gleichsam wie Regieanweisungen im Drehbuch des Lebens:
🎯 Panel-Sequenzierung – Die gezielte Szene
Hier wird nur ein ausgewählter Teil des Genoms untersucht – meist einige Dutzend bis wenige Hundert Gene. Solche Tests kommen zum Einsatz, wenn der Verdacht auf eine spezifische Erkrankung besteht, etwa bei erblich bedingtem Brustkrebs (BRCA1/2).
Vorteil: Schnell, kosteneffizient, präzise – solange die medizinische Fragestellung klar ist.
Limitierung: Alles außerhalb der gewählten Szene bleibt im Dunkeln.
🎥 Whole Exome Sequencing (WES) – Der klassische Director’s Cut
WES analysiert alle protein-kodierenden Abschnitte des Genoms – also etwa 1–2 % des gesamten Erbguts. Gerade bei seltenen Erbkrankheiten ist es ein bewährtes Mittel, da die meisten bekannten Mutationen in diesen Bereichen liegen.
Vorteil: Effiziente Suche nach fehlerhaften Bauplänen für Proteine.
Limitierung: Bereiche außerhalb der Gene – wichtige Schalter und Regler des Erbguts – werden nicht erfasst.
🎬 Whole Genome Sequencing (WGS) – Der vollständige Film in 4K
WGS liest das gesamte Genom – alle drei Milliarden Buchstaben. Damit liefert es das umfassendste Bild, inklusive nicht-kodierender Regionen, regulatorischer Elemente, struktureller Varianten und seltener Mutationen.
Vorteil: Maximale Auflösung für komplexe Fälle.
Beispiel: Bei Kindern mit unklaren Entwicklungsstörungen bringt WGS doppelt so häufig Klarheit wie WES – etwa durch das Aufdecken von Mutationen in entfernten „Fernsteuerungs“-Regionen (Enhancern), die Gene falsch an- oder abschalten.
Fazit:
Je nach Fragestellung braucht es die passende Perspektive – manchmal reicht ein Zoom auf die Schlüsselszene, manchmal muss der ganze Film neu geschnitten werden. Die Wahl der Methode ist daher keine technische Nebensache, sondern eine strategische Entscheidung im Drehbuch der Diagnostik.
4.4. Methoden der Genomsequenzierung
Wie wird der genetische Code gelesen?
Stellen Sie sich vor, DNA ist ein Buch – geschrieben in einer Sprache mit nur vier Buchstaben: A, T, C und G. Um diesen genetischen Text zu entschlüsseln, nutzen Wissenschaftler moderne Technologien. Zwei zentrale Methoden haben sich etabliert. Sie verfolgen dasselbe Ziel: die Reihenfolge der DNA-Buchstaben möglichst genau zu bestimmen.
Die erste Methode funktioniert – vereinfacht gesagt – wie eine molekulare Kopiermaschine mit Farbsensoren. Die zweite nutzt einen Hightech-Tunnel, durch den die DNA geleitet und elektronisch „abgetastet“ wird – vergleichbar mit dem Abtasten eines Seils mit den Fingern, um kleinste Unebenheiten zu spüren. Diese Verfahren bilden die Grundlage der DNA-Sequenzierung.
4.4.1. Methode 1: Kopieren mit Leuchtfarbe
a) Sanger-Sequenzierung
b) Illumina-Technologie
c) Single-Molecule Real-Time(SMRT)-Sequenzierung
4.4.2. Methode 2: Hightech-Tunnel – DNA elektrisch abtasten
a) Oxford Nanopore Sequenzierung
4.4.3. Genomsequenzierung – Technologien im Vergleich
a) Durchsatz vs. Verwertbarkeit – Masse ist nicht immer gleich Klasse
b) Wirtschaftlichkeit – Kosten pro Base vs. Kosten pro Erkenntnis
c) Von der Probe zum Befund – Wie schnell spricht das ganze Genom?
d) Klinische Einsatzfelder – Welche Technologie passt wozu?
e) KI als Co-Pilot – Automatisierung ohne Verantwortung abzugeben
f) Vergleichstabelle: Sanger, Illumina, PacBio, ONT
g) Key Takeaways: Zwischen Reife und Routine

4.4.1. Methode 1: Kopieren mit Leuchtfarbe
Diese Methode nutzt die Tatsache, dass DNA aus zwei komplementären Strängen besteht. Kennt man die Basenabfolge eines Strangs, lässt sich die Sequenz des Gegenstrangs automatisch ableiten. Um diese Information zu gewinnen, wird einer der beiden Stränge der zu untersuchenden DNA kopiert. Ziel ist es, während dieses Kopiervorgangs die exakte Reihenfolge der Basen in der entstehenden Kopie zu erfassen.
Um besser zu verstehen, wie dieses Verfahren funktioniert, werfen wir zunächst einen Blick auf die wichtigsten Bestandteile und die grundlegenden Schritte zur Herstellung einer DNA-Kopie im Reagenzglas (in vitro).
Herstellung einer DNA-Kopie – das Grundprinzip
Im Zentrum steht eine DNA-Vorlage, die kopiert werden soll. Dafür braucht es vier Hauptbestandteile:
- dNTPs (Desoxynukleosidtriphosphate): die DNA-Bausteine A, T, C und G.
- DNA-Polymerase: ein Enzym, das als molekularer Kopierer fungiert.
- Primer: kurze DNA-Stücke, die der Polymerase den Startpunkt liefern.
- Pufferlösung: sorgt für stabile Bedingungen während der Reaktion.
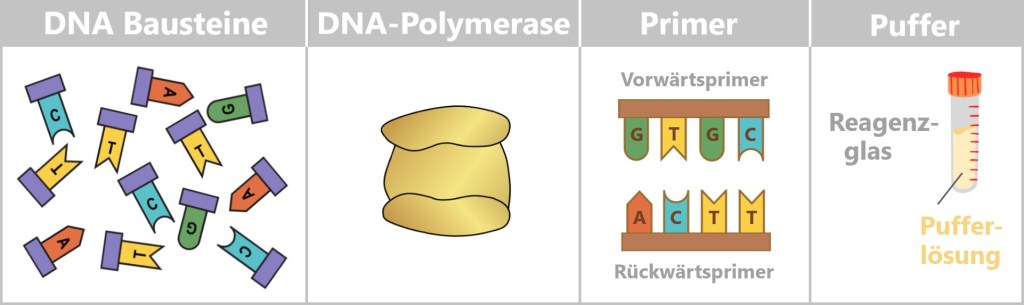
DNA-Bausteine: Einzelne Nukleotide (A, T, C, G), dargestellt in verschiedenen Farben und Buchstaben. Sie bilden das Rohmaterial, aus dem der neue DNA-Strang zusammengesetzt wird.
DNA-Polymerase: Ein schematisch dargestelltes Enzym, das Nukleotide zu einem DNA-Strang verknüpft.
Primer: Kurze DNA-Stücke, die der Polymerase den Startpunkt vorgeben.
Puffer: Ein Reagenzglas mit Pufferlösung, die für stabile chemische Bedingungen während der Reaktion sorgt.
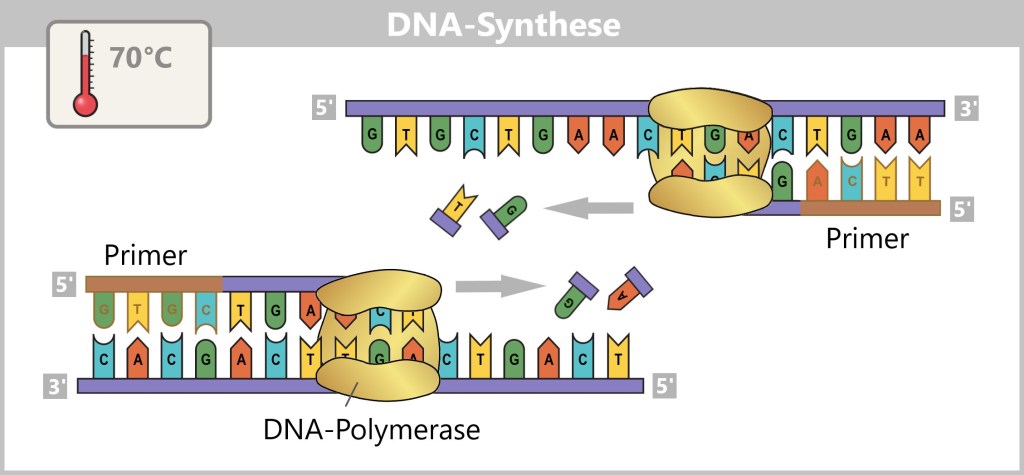
Was macht die DNA-Polymerase?
Die DNA-Polymerase ist ein spezielles Enzym, das komplementäre DNA-Stränge erzeugt – gewissermaßen der „Druckkopf“ einer molekularen Kopiermaschine. Enzyme wirken als biologische Werkzeuge, die chemische Reaktionen gezielt und effizient ermöglichen – deshalb nennt man sie auch „biologische Katalysatoren“.
Damit die Polymerase arbeiten kann, benötigt sie einen Primer als Startsignal. Man kann sich das so vorstellen: Wie ein Druckkopf erst loslegt, wenn ein Blatt Papier richtig eingelegt ist, beginnt auch die Polymerase ihre Arbeit erst, wenn ein Primer vorhanden ist. Dieses kleine DNA-Stück zeigt ihr, wo sie starten soll – es ist sozusagen das erste Blatt im molekularen Drucker.
Vor Beginn der Kopie muss die doppelsträngige DNA zunächst in zwei Einzelstränge aufgetrennt werden – dieser Schritt heißt Denaturierung.
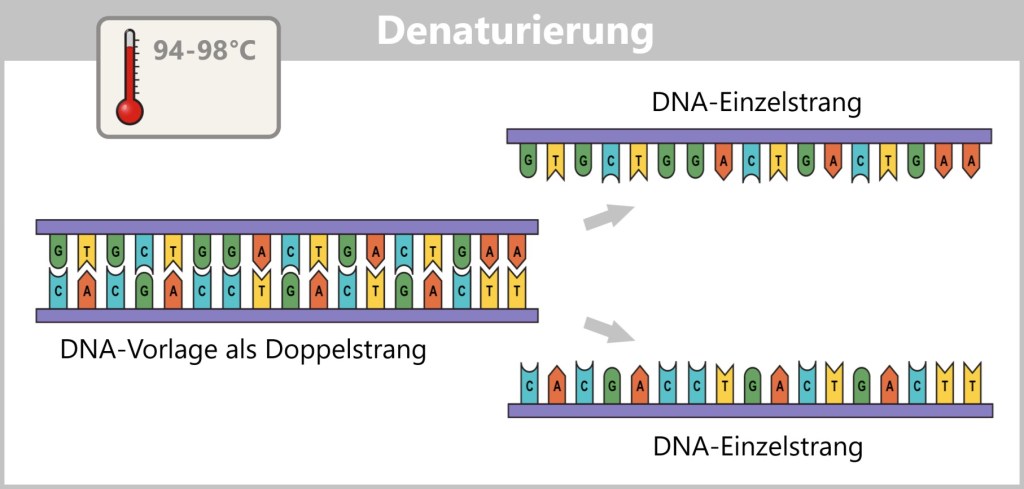
Anschließend lagern sich die Primer an ihre passenden Zielstellen der Einzelstränge. Diese Primerbindung markiert den Startpunkt für die DNA-Polymerase.
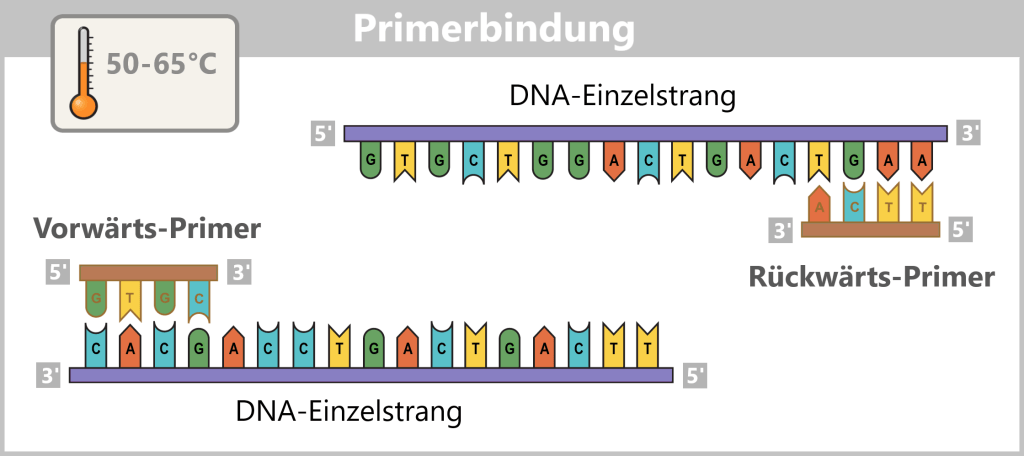
Nun beginnt die Synthese der neuen Stränge: Die Polymerase liest den Vorlage-Strang in 3′-zu-5′-Richtung und ergänzt die Kopie in Gegenrichtung – von 5′ nach 3′ – unter Beachtung der Basenpaarung (A mit T, G mit C).
Was bedeuten 3′ und 5′?
Die Bezeichnungen 3′ (sprich: drei-Strich) und 5′ (fünf-Strich) stammen aus der Chemie des DNA-Rückgrats. Sie geben an, an welchem Ende sich bestimmte Kohlenstoffatome im Zuckermolekül befinden, an die neue Bausteine angefügt werden können.
Stellen Sie sich den DNA-Strang wie eine Einbahnstraße vor. Die Polymerase kann nur in eine Richtung fahren – vom 5′-Ende zum 3′-Ende. Neue Bausteine lassen sich chemisch nur am 3′-Ende anfügen. Daher liest die Polymerase den „alten“ Strang rückwärts (3′ → 5′) und baut den neuen Strang vorwärts (5′ → 3′).
So entsteht aus einem Einzelstrang nach und nach ein neuer, passender Gegenstrang – mithilfe der Basenpaarungsregeln: Adenin (A) paart sich mit Thymin (T), und Cytosin (C) mit Guanin (G).
Das Ergebnis sind perfekte genetische Abbilder – neue DNA-Stränge, exakte Kopien der Ursprungs-DNA.
Leuchtende Buchstaben: Wie Sequenzierung funktioniert
Moderne Sequenziermethoden erweitern diesen Kopiervorgang um eine raffinierte Technik: Sie verwenden fluoreszierend markierte dNTPs (DNA-Bausteine). Jedes der vier Nukleotide (A, T, C, G) ist mit einem Farbstoff versehen, der beim Einbau ein spezifisches Lichtsignal aussendet.
Während die Polymerase die DNA-Stränge verlängert, fügt sie die farbigen Bausteine passgenau ein. Jedes Mal, wenn ein neues Nukleotid eingebaut wird, sendet es ein winziges Lichtsignal – wie ein kleiner Blitz, der verrät, welcher „Buchstabe“ gerade ergänzt wurde. Hochauflösende Kameras erfassen diese Lichtsignale und ermöglichen so die schrittweise Rekonstruktion der DNA-Sequenz.
Sequenziermethoden im Überblick
Mehrere bedeutende Technologien nutzen dieses Prinzip in spezifischer Form:
Sanger-Sequenzierung: Die klassische Methode, bei der fluoreszierende Kettenabbruch-Nukleotide die DNA-Synthese an zufälligen Stellen beenden. Die resultierenden Fragmente lassen sich der Reihenfolge nach auslesen.
Illumina-Technologie: Eine weit verbreitete Hochdurchsatzmethode, bei der Millionen DNA-Fragmente parallel sequenziert werden. Die fluoreszierenden Signale werden Schritt für Schritt erfasst.
SMRT-Sequenzierung (PacBio): Hier wird die Synthese in Echtzeit an einem einzigen DNA-Molekül beobachtet – mit hoher Präzision und langen Leselängen.
a) Sanger-Sequenzierung
Das klassische „Buchstaben-für-Buchstaben-Lesen“
Die Sanger-Sequenzierung, auch Kettenabbruchmethode genannt, wurde in den 1970er-Jahren entwickelt und war das erste Verfahren, mit dem sich DNA präzise und zuverlässig auslesen ließ. Sie beruht im Wesentlichen auf denselben Prinzipien wie die DNA-Synthese, unterscheidet sich jedoch in einigen entscheidenden Punkten:
- Es wird nur ein Primer verwendet.
- Neben den normalen DNA-Bausteinen – den dNTPs (dATP, dCTP, dGTP, dTTP) – kommen zusätzlich spezielle fluoreszierende Kettenabbruch-Bausteine, die sogenannten ddNTPs (ddATP, ddCTP, ddGTP, ddTTP) in geringer Konzentration zum Einsatz. Wenn ein solcher ddNTP eingebaut wird, stoppt die DNA-Synthese genau an dieser Stelle. Jeder der vier ddNTP-Typen trägt eine unterschiedliche Leuchtfarbe, je nach Base.
Der Ablauf – Schritt für Schritt
Für die Sequenzierung werden vier separate Reaktionsmischungen angesetzt. Jede enthält:
- die einzelsträngige Ziel-DNA,
- einen Primer (Startpunkt der Synthese),
- eine DNA-Polymerase,
- normale dNTPs,
- und jeweils eine Sorte der farblich markierten ddNTPs.
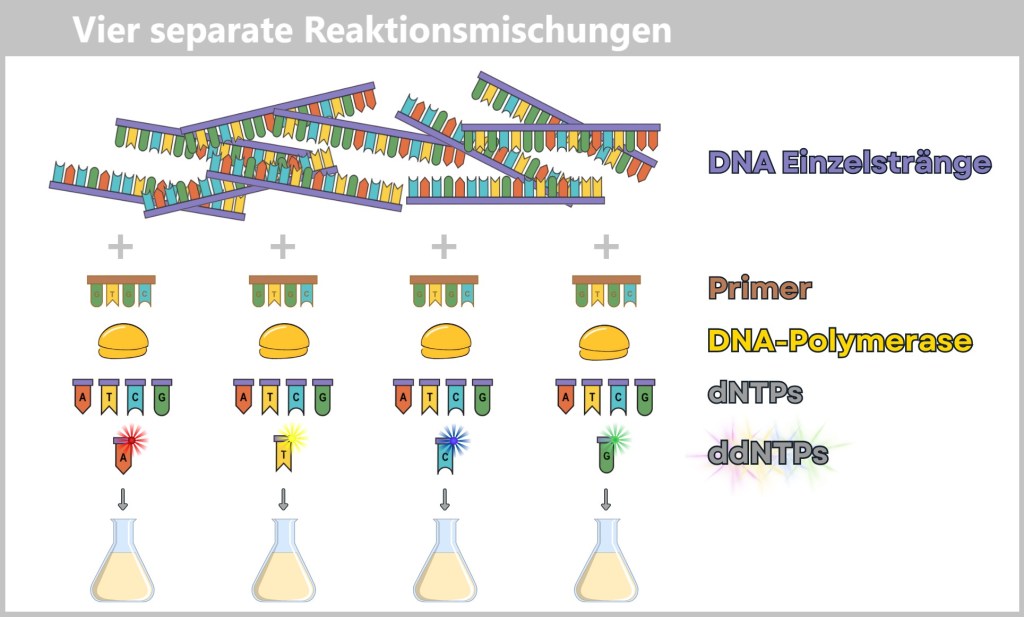
Jede Mischung enthält die gleichen Grundkomponenten, unterscheidet sich jedoch durch die jeweils zugegebene Variante der farblich markierten Stopp-Bausteine (ddNTPs).
Nach der Primerbindung liest die Polymerase den DNA-Strang aus und fügt passende Nukleotide ein, um einen komplementären Strang aufzubauen. Wird dabei zufällig ein ddNTP eingebaut, endet die Synthese an genau dieser Stelle.
So entstehen viele unterschiedlich lange DNA-Fragmente – jedes endet mit einem farbig markierten Basenbuchstaben. Da in jeder der vier Reaktionen nur ein ddNTP enthalten ist, weiß man genau, mit welchem Buchstaben die erzeugten DNA-Fragmente enden.
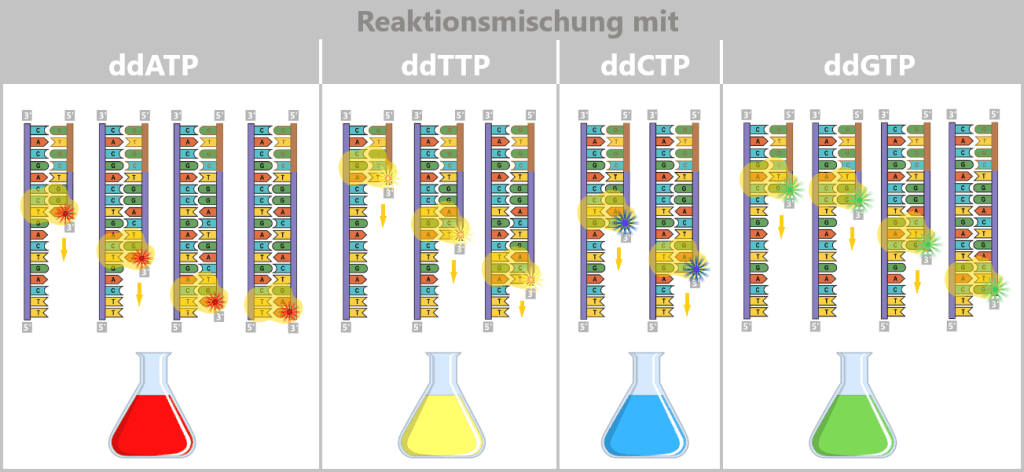
In jeder Reaktionsmischung ist jeweils eine Sorte der modifizierten DNA-Bausteine (ddATP, ddTTP, ddCTP, ddGTP) enthalten. Wird einer dieser Stopp-Bausteine während der DNA-Synthese eingebaut, bricht der Kopiervorgang genau an dieser Stelle ab. In der ddATP-Mischung stoppt die Synthese, sobald ein modifiziertes Adenin (A) eingebaut wird. In der ddTTP-Mischung endet sie beim Einbau eines modifizierten Thymins (T). Genauso führen ddCTP und ddGTP zum Abbruch der Synthese, wenn ein modifiziertes Cytosin (C) bzw. Guanin (G) eingebaut wird. Durch dieses Verfahren entstehen viele DNA-Fragmente unterschiedlicher Länge, die jeweils mit einem spezifischen Stopp-Nukleotid enden. Ziel ist es, alle theoretisch möglichen Fragmente zu erzeugen, um daraus die vollständige DNA-Sequenz ablesen zu können.
Sortierung und Auswertung
Anschließend werden die DNA-Fragmente denaturiert (in Einzelstränge überführt) und durch Gelelektrophorese der Länge nach sortiert. Dabei wandern die negativ geladenen Fragmente durch ein Gel in Richtung der positiven Elektrode. Kleinere Fragmente bewegen sich schneller, größere langsamer – so entsteht eine geordnete „Leiter“ von Fragmenten.
Ein Laser regt nun die fluoreszierenden Farbstoffe der Endbasen an. Die dabei entstehenden Lichtsignale werden von einem Detektor erfasst. Jedes Signal steht für eine bestimmte Base an einer bestimmten Position. Die Reihenfolge der Lichtsignale ergibt damit direkt die DNA-Sequenz – wie bei einem Buchstaben-für-Buchstaben-Lesen.
Da die erzeugten Fragmente komplementär zur Ursprungs-DNA sind, lässt sich daraus die ursprüngliche Basenfolge exakt ableiten.
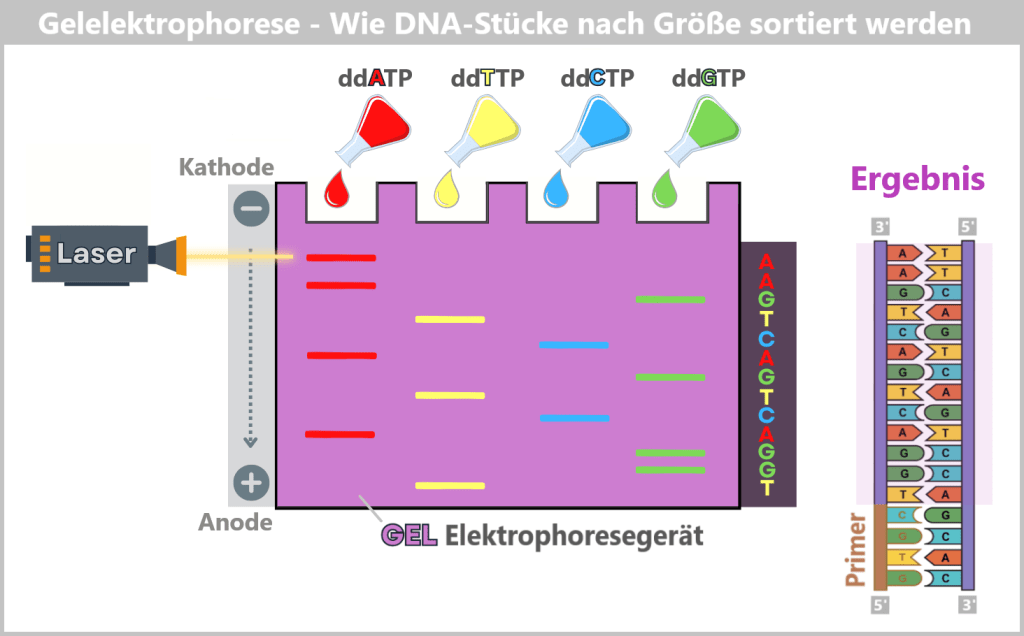
In den Reaktionsansätzen entstehen unterschiedlich lange DNA-Fragmente, die jeweils mit demselben Stopp-Nukleotid enden – je nach Ansatz entweder mit Adenin, Thymin, Guanin oder Cytosin. Diese Reaktionsmischungen werden auf ein Gel aufgetragen. Sobald ein elektrisches Feld angelegt wird, wandern die negativ geladenen DNA-Fragmente von der Kathode (−) zur Anode (+). Dabei bestimmt die Größe der Fragmente ihre Wandergeschwindigkeit: Kleinere Fragmente bewegen sich schneller durch die feinen Poren des Gels und gelangen früher zur Anode, während größere Fragmente langsamer sind. Durch das Auslesen der fluoreszierenden Farbsignale an den Fragment-Enden kann die genaue Reihenfolge der Basen bestimmt werden – und damit die DNA-Sequenz Schritt für Schritt rekonstruiert werden.
Warum gilt die Sanger-Sequenzierung bis heute als Goldstandard?
Auch ein halbes Jahrhundert nach ihrer Entwicklung bleibt die Sanger-Methode in vielen Bereichen der Molekularbiologie und Medizin unverzichtbar. Der Grund: Zuverlässigkeit und Präzision.
Im Vergleich zu modernen Hochdurchsatzverfahren ist die Sanger-Sequenzierung zwar langsamer und eignet sich nur für die Analyse kleinerer DNA-Abschnitte – nicht für ganze Genome. Aber genau darin liegt ihre Stärke:
- Gezielte Fragestellungen, wie das Überprüfen einzelner Gene,
- Nachweis spezifischer Mutationen, oder
- Validierung kritischer Ergebnisse, die zuvor mit anderen Methoden identifiziert wurden,
lassen sich mit der Sanger-Sequenzierung präzise, klar und interpretierbar beantworten – oft so deutlich, dass man die Abfolge der Basen direkt im Sequenzplot sehen kann.
In der medizinischen Diagnostik – etwa bei der Analyse vererbter Erkrankungen oder in der Qualitätskontrolle gentechnischer Verfahren – ist diese hohe Genauigkeit entscheidend. Denn ein falsch interpretierter Buchstabe kann hier lebenswichtige Folgen haben.
Ein weiterer Pluspunkt: Die Methode ist weltweit standardisiert, seit Jahrzehnten bewährt und unterliegt klaren Qualitätsrichtlinien.
Ein Klassiker mit Beständigkeit
In einer Welt, in der ständig neue Technologien auf den Markt drängen, bleibt die Sanger-Sequenzierung ein verlässlicher Anker – der Oldtimer unter den Sequenzierverfahren: nicht der Schnellste, aber extrem robust, bewährt und zuverlässig. Und manchmal ist genau das entscheidend.
b) Illumina-Sequenzierung
Die Hochgeschwindigkeits-Kopiermaschine
Die Sanger-Sequenzierung ist wie präzise Handarbeit: Jeder DNA-Strang wird Schritt für Schritt entziffert – zuverlässig, aber langsam und teuer.
Stellen Sie sich vor, Sie müssten ein ganzes Buch oder sogar eine Bibliothek abschreiben – und dürften dabei nur einen Buchstaben pro Minute notieren.
Genau hier liegt das Problem: Moderne Krebsforschung, die Entschlüsselung seltener Krankheiten oder die genetische Überwachung von Pandemien verlangen die Analyse großer Datenmengen – also vieler und/oder sehr langer DNA-Abschnitte. Dafür braucht es eine Methode, die nicht nur genau, sondern auch schnell und bezahlbar ist.
Und genau da kommt die Illumina-Sequenzierung ins Spiel. Sie hat das Prinzip der DNA-Entzifferung von der Handarbeit zur industriellen Massenproduktion erhoben und ist heute eine der am häufigsten eingesetzten Methoden für die Hochdurchsatz-Sequenzierung (Next Generation Sequencing, NGS). Statt einzelner Buchstaben werden nun ganze Seiten gleichzeitig gelesen – millionenfach, parallel, mit hoher Präzision und kosteneffizient.
Anders als bei der klassischen Sanger-Methode, bei der jedes DNA-Fragment einzeln analysiert wird, arbeitet Illumina mit einer massiven Parallelisierung – das heißt: Es werden viele DNA-Fragmente gleichzeitig vervielfältigt und sequenziert. Wie funktioniert das?
Schritt 1: Vorbereitung der DNA-Fragmente
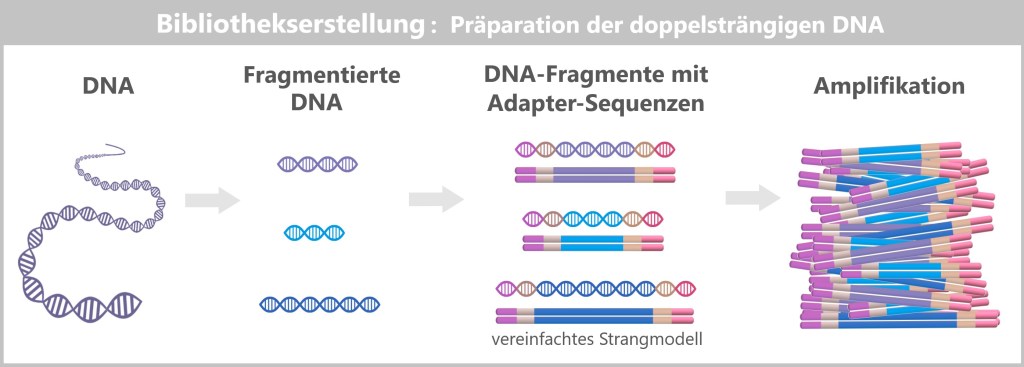
– Die DNA wird zur „Lego-Baustelle“ –
Zunächst wird die zu untersuchende DNA-Probe in viele kurze Stücke zerlegt – sogenannte Fragmente, die 100 bis 300 Basenpaaren lang sind. An beide Enden dieser Fragmente werden kleine künstliche DNA-Stücke – sogenannte Adapter – angehängt. Man kann sie sich wie LEGO-Steckverbinder vorstellen. Sie dienen einerseits als molekulare Andockstellen, um die DNA-Fragmente später auf einem speziellen Träger – dem Flowcell-Chip – zu befestigen. Andererseits dienen sie als Bindungsstellen für universelle Primer.
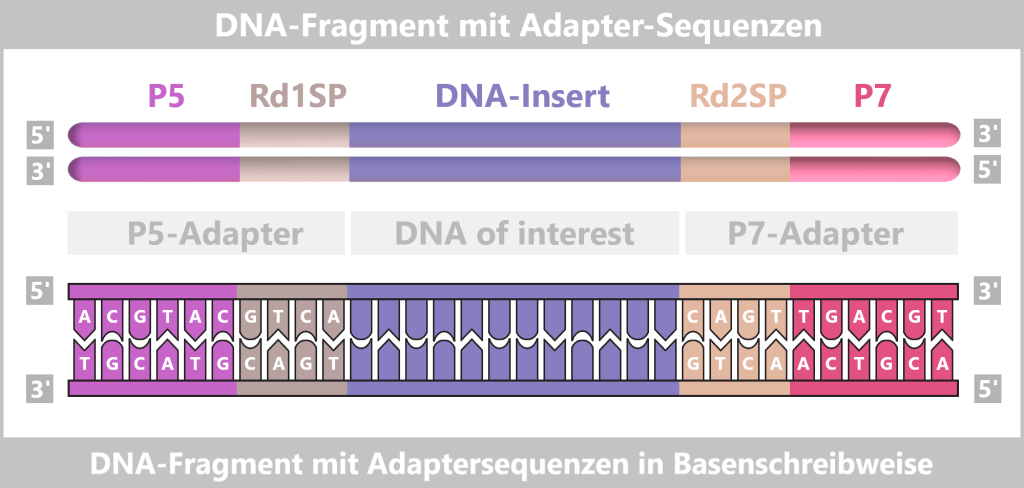
Die P5/P7-Sequenzen dienen der Bindung an der Flow Cell. Die Rd1SP/Rd2SP-Sequenzen sind Bindungsstellen für universelle Primer.
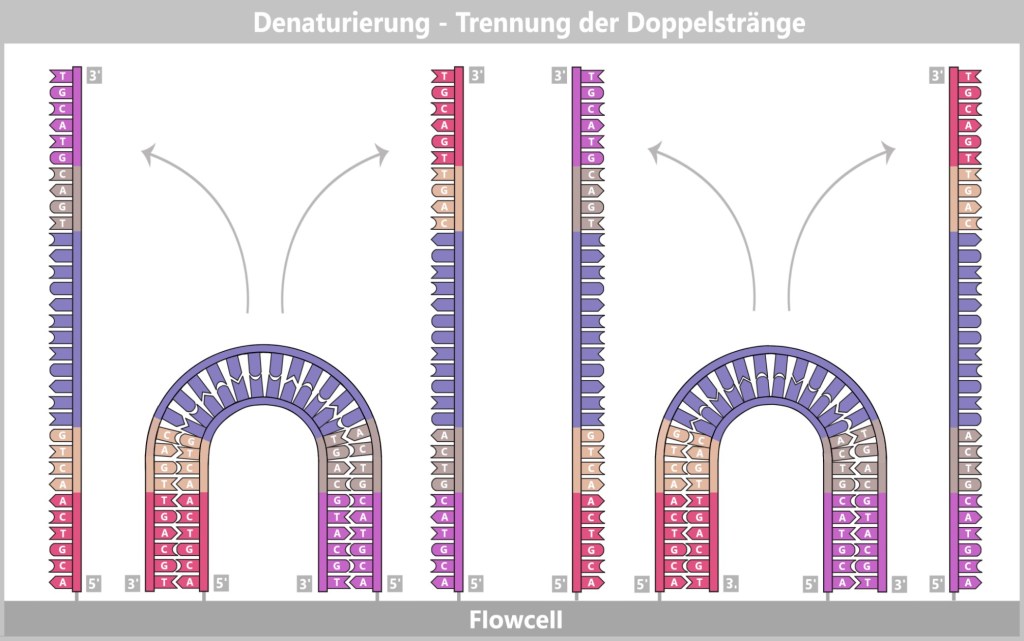
Durch Denaturierung werden die so vorbereiteten doppelsträngigen DNA-Fragmente in Einzelstränge gespalten.
Schritt 2: Brückenamplifikation
– Der Tanz der DNA auf der Flow Cell –
Das Herzstück der Illumina-Technologie ist die sogenannte Flow Cell – eine glasartige Platte, übersät mit Millionen winziger DNA-Andockstellen (kurzen DNA-Abschnitten, sogenannten Oligonukleotiden), die fest an der Oberfläche verankert sind.
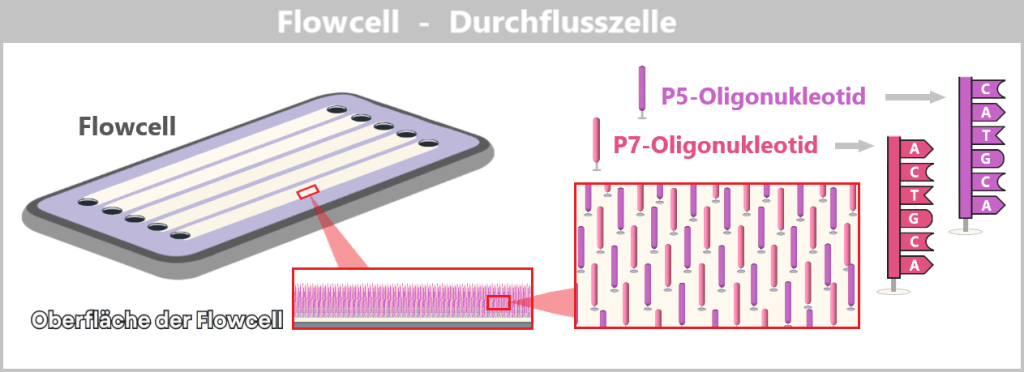
Über ihre Adapter binden sich die einzelsträngigen DNA-Fragmente an die Anker. Anschließend beginnt die molekulare Kopierarbeit: Es werden komplementäre Kopien der DNA-Schnipsel erzeugt, die nun fest mit der Flow Cell verankert sind.
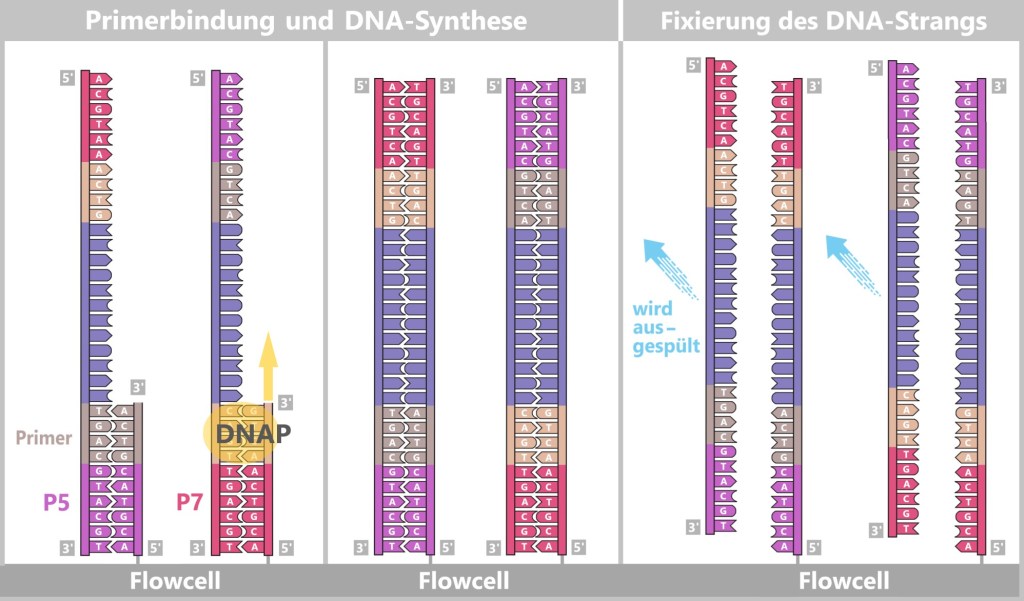
Links: Primer binden an die Adapter (P5, P7), und die DNA-Polymerase (DNAP) startet die Synthese eines neuen, komplementären Strangs.
Mitte: Die DNA-Polymerase synthetisiert daraufhin einen ersten Strang.
Rechts: Der neue entstandene DNA-Doppelstrang wird aufgetrennt. Der ursprüngliche Strang hat nach der Denaturierung keine Verbindung mehr zur Flowcell und wird ausgespült. Der neu synthetisierte Strang bleibt mit seinem 5′-Ende fest an der Flowcell gebunden.
Dann beginnt ein faszinierender Prozess: Die DNA-Stränge biegen sich zu kleinen Brücken, indem ihre freien Adapter an benachbarte Oligonukleotide auf der Flow Cell binden. An diesen Stellen werden sie erneut kopiert. Nach dem Kopieren wird die Brücke wieder aufgelöst – und die Zahl der fest verankerten DNA-Stränge verdoppelt sich. Dieser Vorgang, bekannt als Brückenamplifikation, wiederholt sich dutzendfach. Am Ende entstehen dichte Cluster aus Millionen identischer Kopien eines einzelnen DNA-Fragments.
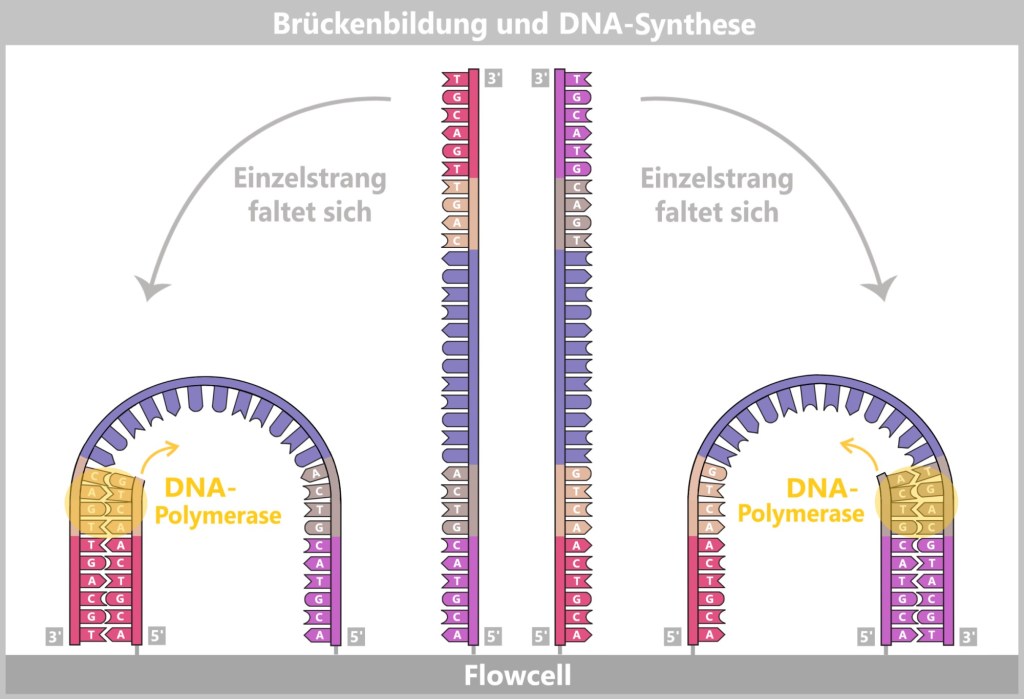
Die verankerten Einzelstränge falten sich und verbinden sich mit den benachbarten Ankern auf der Flowcell, wodurch eine Brückenstruktur entsteht. Danach wird der Kopiervorgang initiiert.
Nach dem Kopiervorgang werden die Brücken-Doppelstränge getrennt. Die Anzahl der fest verankerten DNA-Stränge verdoppelt sich. Sie sind bereit für die weitere Amplifikation.

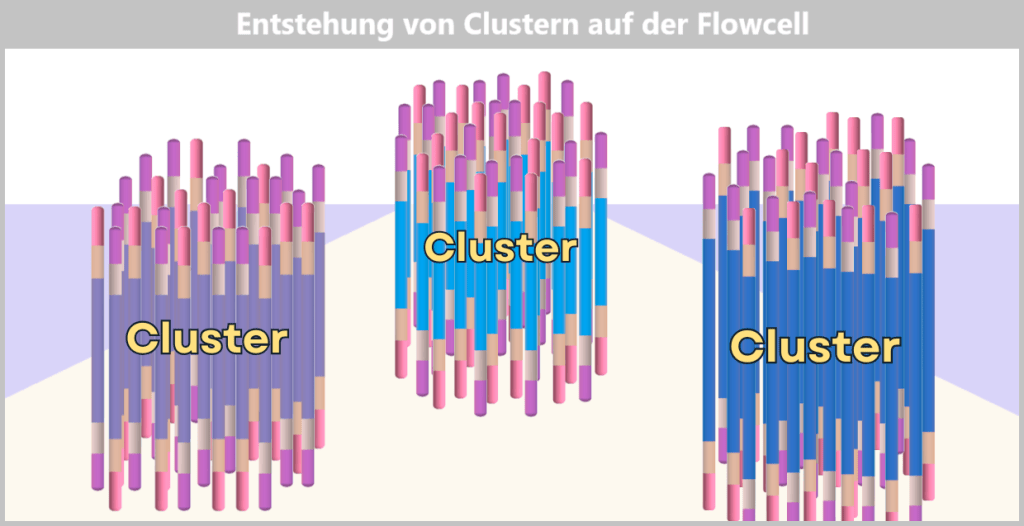
Das Ergebnis ist eine dicht gepackte „Landkarte“ aus Millionen DNA-Clustern, die jeweils nur eine bestimmte Sequenz enthalten – ideal für das gleichzeitige Auslesen.
Ohne diese Vervielfältigung wäre das Lesen der DNA, als würde man versuchen, ein einzelnes Glühwürmchen im Wind zu erkennen. Die Cluster hingegen machen die DNA-Basen (A, T, C, G) deutlich sichtbar – wie ein leuchtender Neon-Schriftzug in der Dunkelheit.
Schritt 3: Sequenzieren durch Synthese
– Die Lichtshow der DNA-Basen –
Nun beginnt das eigentliche Sequenzieren durch Synthese. Auch hier kommt das Prinzip der „molekularen Kopiermaschine mit Farbsensoren“ zum Einsatz – aber in optimierter Form:
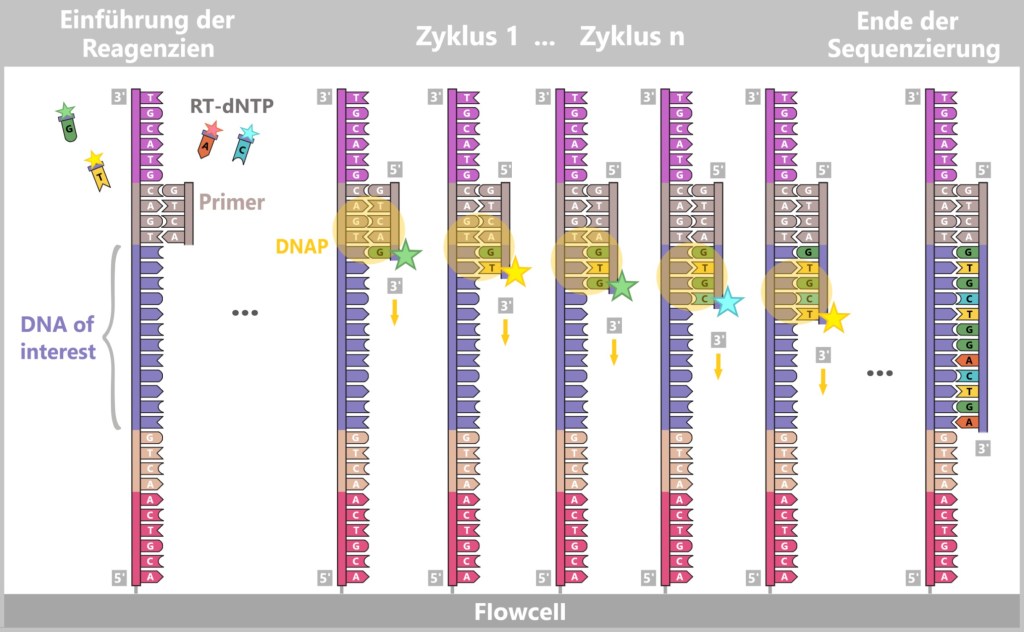
Zu Beginn binden die Universal-Primer an den entsprechenden Adapterstellen der DNA-Fragmente in jedem DNA-Clusters.
Dann werden modifizierte dNTPs – sogenannte RT-dNTPs (reversibel terminierende Nukleotide) – in die Reaktionslösung gegeben. Jeder der vier DNA-Bausteine (A, T, G, C) ist dabei mit einer eigenen Fluoreszenzfarbe markiert und trägt eine reversible Blockierung.
Nur ein Nukleotid pro Zyklus kann eingebaut werden, weil die Blockierung den weiteren Strangaufbau vorübergehend stoppt. Nicht eingebaute RT-dNTPs werden weggewaschen.
Nach jedem Einbau wird die Flowcell mit einer hochauflösenden Kamera gescannt. Jeder leuchtende Punkt steht für ein eingebautes Nukleotid und seine Farbe zeigt an, welcher Buchstabe gerade ergänzt wurde.
Danach wird die Blockierung und die Farbstoffmarkierung chemisch entfernt, und der nächste Zyklus beginnt.
Dieser Prozess wird Zyklus für Zyklus wiederholt – bis jeder DNA-Strang vollständig gelesen ist.
Schritt 4: Datenauswertung
– Der Supercomputer als Puzzlemeister –
Nach der „Lichtshow“ liegen Millionen von Fotos vor – eines für jeden Zyklus und jedes Cluster. Jedes Bild zeigt, welche Farbe (und damit welcher Buchstabe: A, T, C oder G) in jedem Cluster hinzugefügt wurde.
Jetzt kommen leistungsfähige Computer ins Spiel – ausgestattet mit Software, die so präzise arbeitet wie ein Detektiv, der aus einem Stapel durchnummerierter Polaroids ein zerschnittenes Buch zusammensetzt. Jedes Cluster auf dem Sequenzierungschip ist wie ein kleiner Tatort, jeder Farbpixel eine Spur. Und aus Millionen solcher Spuren entsteht allmählich eine Geschichte – die Geschichte der DNA.
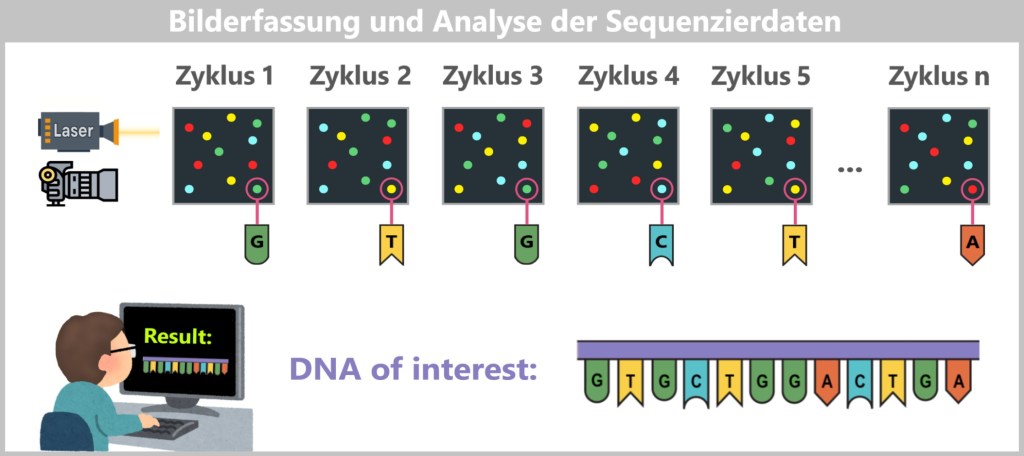
Die Farbmuster werden in Buchstabenfolgen übersetzt, sogenannte „Reads“: winzige Textstücke eines gewaltigen Genom-Romans. Am Ende türmt sich eine Flut solcher Fragmente auf – wie ein Berg zerschnittener Buchseiten, lose verstreut.
Genau hier beginnt die eigentliche Kunst: Die Bioinformatik tritt auf den Plan. Mit Hilfe spezieller Algorithmen werden die Schnipsel analysiert, sortiert und aneinandergefügt – immer auf der Suche nach überlappenden Stellen, vertrauten Mustern, bekannten Strukturen.
Stück für Stück setzt sich das große Ganze wieder zusammen – bis am Ende die ursprüngliche DNA-Sequenz sichtbar wird, wie ein rekonstruiertes Buch, das plötzlich wieder Sinn ergibt.
Die eigentliche „Magie“ geschieht also nicht in der Flow Cell, sondern im Computer! Ohne die Software wären die Bilder nur buntes Flimmern – mit ihr aber werden sie zum Schlüssel für medizinische Durchbrüche.
Eine ausführlichere und dennoch anschauliche Erklärung der Illumina-Sequenzierungstechnologie Methode findet sich im Video „Illumina Sequencing Technology“.
Moderne Hochdurchsatz-Sequenzierplattformen wie die von Illumina können die Rohdaten eines vollständigen menschlichen Genoms innerhalb weniger Tage erzeugen – zu reinen Sequenzierkosten, die deutlich unter dem Preis eines Premium-Smartphones liegen.
Diese Technologie hat die Genomforschung revolutioniert und erheblich zugänglicher gemacht: Sie ist heute ein äußerst vielseitiges Werkzeug – schnell genug für die Echtzeit-Analyse von Pandemien, präzise genug, um in Kombination mit robuster Bioinformatik und klinischer Expertise die Grundlage für personalisierte Krebstherapien zu bilden.
Die Illumina-Sequenzierung gilt inzwischen als Industriestandard für große Sequenzierprojekte – etwa bei der Analyse ganzer Genome, in der Genexpressionsforschung oder in der modernen Krebsdiagnostik.
Und das Beste: Während man diesen Text liest, entschlüsselt irgendwo auf der Welt eine Flow Cell Millionen DNA-Fragmente.
Allerdings hat auch die Illumina-Technologie ihre Grenzen. Besonders bei langen, sich wiederholenden DNA-Abschnitten – wie sie in manchen Chromosomenbereichen vorkommen – gerät sie an ihre Grenzen. Das ist, als müsste man ein Lied aus tausenden 3-Sekunden-Schnipseln rekonstruieren. Für solche Herausforderungen kombiniert man Illumina häufig mit anderen Sequenzierverfahren, die auch lange Abschnitte zuverlässig erfassen können – wie ein Ermittler, der sowohl Nahaufnahmen als auch Panoramabilder braucht, um den ganzen Tatort zu verstehen.
c) Single-Molecule Real-Time(SMRT)-Sequenzierung
Der Roman in einem Zug
Stellen Sie sich vor, Sie wollen einen langen Roman lesen – voller wiederkehrender Kapitel und versteckter Hinweise, die sich über viele Seiten erstrecken. Wenn Sie das Buch wie bei der Illumina-Sequenzierung in tausende kleine Schnipsel zerschneiden, diese einzeln lesen und anschließend wie ein Puzzle zusammensetzen, besteht die Gefahr, dass Kapitel fehlen, vertauscht oder unvollständig bleiben. Die geheimen Botschaften der Geschichte – das, was der Roman uns eigentlich erzählen will – könnte dadurch verloren gehen oder nur bruchstückhaft erscheinen.
Hier glänzt die Single Molecule Real-Time (SMRT)-Sequenzierung von PacBio: Sie liest ganze Buchkapitel in einem Zug – ohne Schnipsel, ohne Pause.
Anstatt die DNA zu zerlegen, bleibt sie bei PacBio in sehr langen Stücken erhalten – oft Zehntausende Basenpaare am Stück, die als Ganzes sequenziert werden. Diese Fäden betreten winzige Bühnen in einer SMRT-Zelle, sogenannte Nanokammern (Zero-Mode Waveguides). Diese sind so klein, dass sie nur Platz für ein einziges DNA-Molekül und eine Polymerase bieten. Jede dieser Bühnen lässt Licht nur an einem winzigen Punkt wirken – genau dort, wo die Polymerase die DNA abliest. Wie ein Spotlicht, dass einen Schauspieler bei einem Monolog auf der Bühne ins Zentrum rückt.
Die Polymerase spielt die Hauptrolle: Sie fügt Baustein für Baustein (A, T, C oder G) an die DNA-Kopie an. Jeder dieser Bausteine leuchtet in einer anderen Farbe, ähnlich wie Neonlichter bei einem DJ. Bei jedem Einbau blitzt ein Lichtsignal auf, das von einer Kamera in Echtzeit aufgezeichnet wird – ein tanzender Lichtcode, der die DNA-Sequenz sichtbar macht.
Das Video Introduction to SMRT Sequencing von PacBio veranschaulicht diesen Vorgang eindrucksvoll.
Analysieren wir die einzelnen Schritte dieses faszinierenden Verfahrens im Detail.
Schritt 1: Die Vorbereitung
– DNA-Ringe für das Endloslesen –
Auch bei PacBio wird die zu untersuchende DNA zunächst in Fragmente zerlegt – allerdings in deutlich größere Stücke als bei Illumina, meist 10.000 bis über 20.000 Basenpaare lang. Diese Fragmente werden zu ringförmigen DNA-Molekülen verarbeitet:
Man fügt an beide Enden sogenannte Hairpin-Adapter an – kleine Schlaufen aus DNA, die das Fragment in sich selbst schließen.
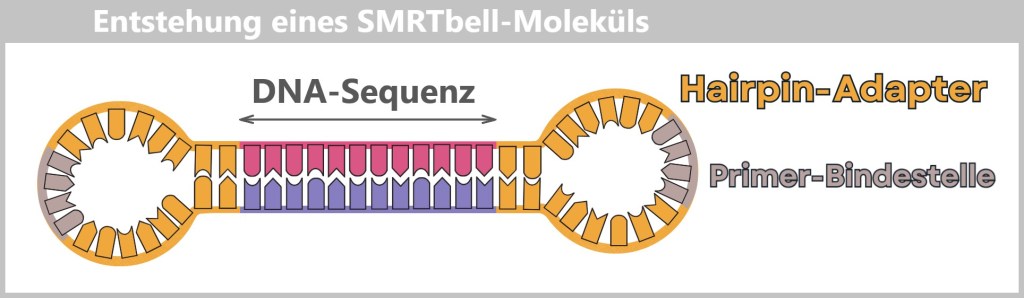
Die Adapter enthalten eine Primer-Bindestelle, die später den Startpunkt für die Polymerase liefert.
Durch anschließende Denaturierung entsteht ein zirkuläres, einzelsträngiges SMRTbell-Molekül in Form einer offenen Schlaufe.
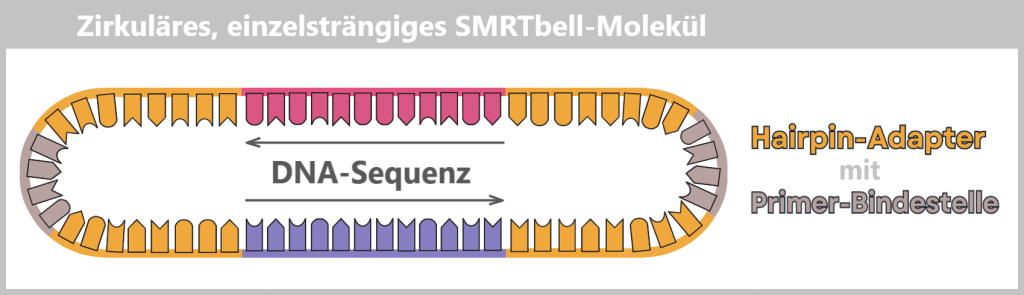
Durch Denaturierung entsteht aus der doppelsträngigen Vorlage ein einzelsträngiges SMRTbell-Molekül. Dieses bildet eine zirkuläre Struktur, die in der Darstellung als geöffnete Schlaufe erscheint.
Das Ergebnis ist ein SMRTbell: ein geschlossener DNA-Ring, der von der Polymerase beliebig oft gelesen werden kann – wie eine Modelleisenbahn, die im Kreis fährt.
Jeder Hairpin-Adapter enthält eine definierte Bindungsstelle, an der der universelle Primer bindet. Das ist der Startpunkt für die Polymerase.
Die DNA-Polymerase lagert sich an den Primer-Komplex an. Es entsteht ein voll funktionsfähiger Replikationskomplex: Polymerase + Primer + SMRTbell.
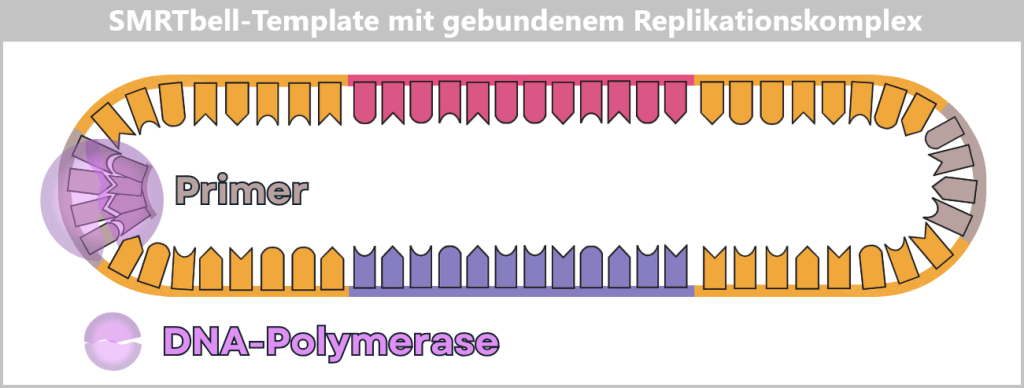
Der Primer ist an die Adapter-Bindestelle gebunden, die Polymerase an den Primer. Dieser Komplex ist startbereit für die DNA-Synthese.
Schritt 2: Die Sequenzierbühne
– Lichtspiele im Nanomaßstab –
Jetzt wird es spektakulär: Die vorbereiteten Replikationskomplexe werden in ihre winzigen Beobachtungskammern geleitet – die Zero Mode Waveguides (ZMWs). Diese Nanokammern sind so klein, dass in der Regel nur ein einziges SMRTbell-Template Platz findet. Die Polymerase ist dabei fest am Boden des ZMWs verankert und wartet auf ihren Einsatz.
Das Startsignal gibt die Zugabe der vier Nukleotide (A, C, G, T), die jeweils mit einem eigenen Fluoreszenzfarbstoff markiert sind.
Die Polymerase beginnt nun mit ihrer Arbeit – sie liest den DNA-Strang und baut einen komplementären Strang auf. Sobald sie ein Nukleotid einbaut, passiert das Entscheidende:
Jedes Nukleotid sendet einen kurzen, farbcodierten Lichtblitz aus.
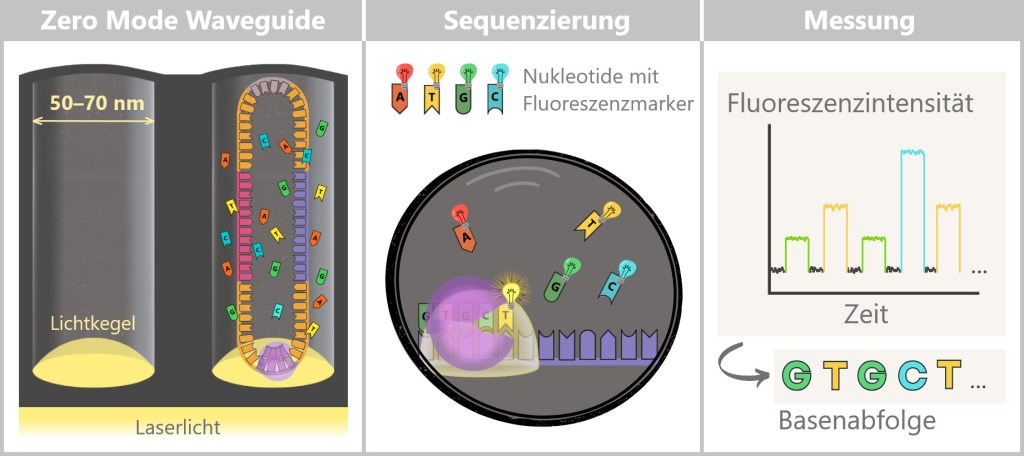
Links: Am Boden des Zero Mode Waveguides (ZMW) tritt ein Laserstrahl ein, der ein sogenanntes evaneszentes Feld (dargestellt als Lichtkegel) erzeugt. Dieses Feld breitet sich nicht wie normales Licht in die Lösung aus, sondern klingt innerhalb von nur 20–30 Nanometern exponentiell ab. Dadurch entsteht ein extrem kleines Anregungsvolumen, in dem die Fluoreszenzfarbstoffe der Nukleotide angeregt werden können.
Im linken Zero Mode Waveguide (ZMW) befindet sich ein einzelner Replikationskomplex (Polymerase + SMRTbell). Die Polymerase (lila) ist dabei fest am (Glas-)Boden verankert. Mit Anwesenheit der Fluoreszenz-markierten Nukleotide beginnt die Polymerase mit ihrer Arbeit.
Mitte: Die vier Nukleotide (A, T, G, C) tragen unterschiedliche Fluoreszenzmarker. Sobald die Polymerase ein Nukleotid einbaut, leuchtet es kurz auf, bevor der Farbstoff abgespalten wird.
Rechts: Diese Lichtblitze werden in Echtzeit gemessen und in Fluoreszenzsignale übersetzt. So entsteht Schritt für Schritt die Basenabfolge der DNA.
Das Leuchten geschieht in Echtzeit, genau im Moment der Einfügung – daher der Name: Single Molecule Real-Time Sequencing. Ein empfindlicher Detektor zeichnet diese Lichtsignale auf –wie ein molekulares Live-Mikroskop bei der Arbeit.
Schritt 3: Das Geheimnis der Langstrecke
– Wiederholung macht’s robust –
Da PacBio sehr lange DNA-Abschnitte auf einmal liest – vergleichbar mit dem Lesen ganzer Buchkapitel statt einzelner Verse, treten bei einem einzelnen Durchlauf mehr Einzelfehler auf als bei Illumina. Genau hier kommt die ringförmige SMRTbell-Struktur zum Einsatz. Die DNA-Polymerase kann wie ein Zugführer das Ringmolekül mehrfach umrunden. Dabei wird das gleiche DNA-Fragment immer wieder gelesen – wie ein Vorleser, der sich mehrfach an denselben Text wagt, bis er ihn sicher beherrscht.
Ein Computer gleicht die Wiederholungen miteinander ab und filtert die Fehler heraus – wie ein Lektor, der ein Manuskript glättet. So entsteht eine besonders präzise Konsens-Sequenz.
PacBio nennt dieses Verfahren HiFi-Reads – hochpräzise Einzelmolekül-Lesungen.
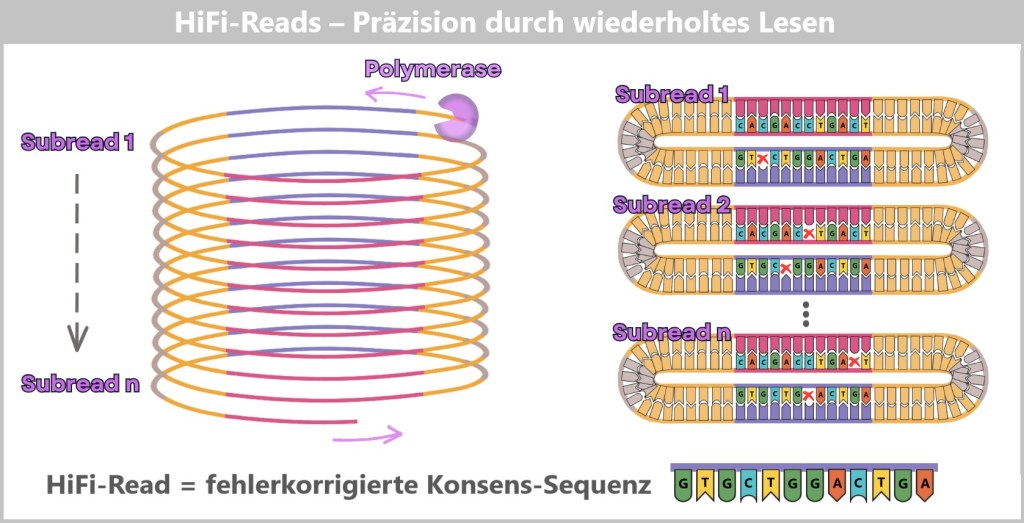
Die Polymerase wandert mehrfach um das zirkuläre SMRTbell-Template und erzeugt dabei mehrere Subreads. Jeder Umlauf entspricht einer vollständigen Ablesung der DNA-Sequenz. Einzelne Subreads können zufällige Fehler (x) enthalten, doch durch den Vergleich vieler Wiederholungen wird ein hochpräziser Konsens gebildet – der sogenannte HiFi-Read (High-Fidelity Read).
Zusatzinfo: Wie die Polymerase mehrfach um die DNA läuft
Erster Sequenzierungsdurchlauf
Die Polymerase liest den einzelsträngigen Template-Strang ab und synthetisiert dabei einen komplementären Strang.
Das Ergebnis ist eine vollständige Doppelhelix – der ursprüngliche Template-Strang und der neu entstandene Tochterstrang sind nun fest miteinander verbunden.
Aus dieser ersten, ununterbrochenen Umrundung entsteht ein Subread – also eine einzelne Basenabfolge, die von der Polymerase in einem Durchlauf erzeugt wurde.
Zweite und weitere Sequenzierungsdurchläufe
Nun stellt sich die Frage: Wie kann die Polymerase weiterlesen, wenn doch der DNA-Ring bereits doppelsträngig ist?
Hier kommt die Φ29-Phagen-DNA-Polymerase ins Spiel – eine speziell modifizierte Enzymvariante mit zwei entscheidenden Eigenschaften:
Hohe Prozessivität: Sie bleibt sehr lange am DNA-Strang gebunden und löst sich kaum – das ermöglicht die langen Reads.
Starke Strangverdrängungsaktivität: Sie kann Wasserstoffbrückenbindungen zwischen Template- und Tochterstrang aufbrechen.
Die Polymerase läuft in die Doppelhelix hinein, trennt die Stränge lokal auf und verdrängt den alten Tochterstrang, während sie gleichzeitig einen neuen synthetisiert – wie ein molekularer Bulldozer.
Der zuvor gebildete Strang wird dabei nicht abgebaut, sondern bleibt als loser, einzelsträngiger DNA-Faden in der Nanokammer zurück. Bei jeder weiteren Runde wiederholt sich dieser Prozess: Die Polymerase liest dasselbe Template erneut ab – und erzeugt dadurch mehrere Subreads vom gleichen DNA-Molekül.
Schritt 4: Datenanalyse
– Aus Lichtblitzen wird lesbare DNA –
Sind alle Lichtsignale eingefangen, beginnt die eigentliche Detektivarbeit: die Datenanalyse.
Der erste Schritt ist das sogenannte Base-Calling: Dabei werden die aufgezeichneten Fluoreszenzsignale – zeitlich und farblich codiert – in Nukleotidsequenzen übersetzt – A, T, G oder C.
Anschließend werden die Rohdaten jedes Polymerase-Umlaufs bereinigt: Die Hairpin-Adapter-Sequenzen werden bioinformatisch abgetrennt. Zurück bleiben die sogenannten Subreads – die ungekürzten Leseprotokolle jedes einzelnen Rundgangs der Polymerase durch den DNA-Kapiteltext.
Doch erst der nächste Schritt enthüllt die volle Wahrheit: Wie ein philologischer Meisterdetektiv, der mehrere Abschriften eines Manuskripts vergleicht, kombiniert der Algorithmus alle Subreads desselben SMRTbell-Moleküls. Aus diesem Vergleich – Circular Consensus Sequencing (CCS) genannt – entsteht ein hochpräziser HiFi-Read: eine fehlerbereinigte Masterkopie, die das DNA-Kapitel wortgetreu wiedergibt.
Erst jetzt ist das Textfragmentbereit für die finale Einordnung – und durchläuft dabei mehrere Stationen:
Zunächst eine Qualitätskontrolle, ganz wie ein sorgfältiges Lektorat, das den Text auf Klarheit und Stimmigkeit prüft.
Darauf folgt das Alignment: Die Sequenz wird wie ein neues Kapitel ins richtige Fach eines Referenzgenom-Regals eingeordnet.
Schließlich erfolgt die Assemblierung – der Schlussakt, bei dem alle Kapitel zu einem vollständigen Roman des Genoms zusammengefügt werden.
Das Video PacBio Sequencing – How it Works fasst die Schritte anschaulich zusammen.
Anwendung und Vorteile – Die Stärken der Langleser
Die PacBio-SMRT-Technologie hat eine besondere Stärke: Sie kann sehr lange DNA-Abschnitte am Stück lesen – mit höchster Genauigkeit. Das macht sie ideal für:
- Das Erkennen komplexer Genomstrukturen
- Die Analyse wiederholungsreicher Abschnitte
- Die Unterscheidung sehr ähnlicher Genvarianten (z. B. bei Immunrezeptoren oder in der Krebsgenetik)
- De-novo-Sequenzierung – also das vollständige Lesen neuer Genome ohne Referenz
- Das Aufspüren von epigenetischen Modifikationen (z.B. Methylierungen)
Wie kann SMRT epigenetische Modifikationen sichtbar machen?
Wie bereits beschrieben, spielen epigenetische Mechanismen eine zentrale Rolle bei der gezielten Genregulation. Es handelt sich um kleine chemische Markierungen – etwa Methylgruppen – die an bestimmte DNA-Basen angeheftet werden. Sie verändern nicht die genetische Sequenz selbst, sondern beeinflussen, wann und wie oft Gene abgelesen werden – abhängig von Zelltyp, Umgebung oder Entwicklungsstadium.
Sie funktionieren wie Notizen oder Markierungen am Seitenrand eines Buches: Der Text bleibt derselbe, aber die „Lesart“ ändert sich – manche Passagen werden betont, andere überblättert.
Gerät dieses fein abgestimmte System aus dem Gleichgewicht, kann das weitreichende Folgen haben – etwa bei Krebs, Autoimmunerkrankungen oder psychischen Störungen.
Im Gegensatz zur Illumina-Technologie, die eine chemisch veränderte Kopie der DNA liest, analysiert SMRT die Original-DNA in Echtzeit. Dabei wird nicht nur die Basenabfolge, sondern auch die Verweildauer der DNA-Polymerase an jeder Base – die sogenannte Einbaukinetik – gemessen.
Genau hier liegt der Schlüssel zum Nachweis epigenetischer Modifikationen: Methylierungen und andere chemische Veränderungen beeinflussen diese Kinetik. Sie wirken wie kleine Hindernisse oder Bremsspuren auf dem DNA-Strang. Die Polymerase „verharrt“ an solchen Stellen länger – und genau diese Verzögerung wird von der SMRT-Kamera direkt erfasst.
SMRT eignet sich besonders gut zur direkten Detektion bestimmter chemischer Markierungen, etwa Methylgruppen an Adenin- oder Cytosin-Basen, die vor allem in Bakterien und Pflanzen eine wichtige Rolle spielen. Für die in der humanen Epigenetik zentrale Cytosin-Methylierung (5mC) ist die direkte Detektion weniger sensitiv, kann aber mithilfe spezialisierter Bioinformatik-Tools wie pb-CpG-tools zuverlässig abgeleitet werden.
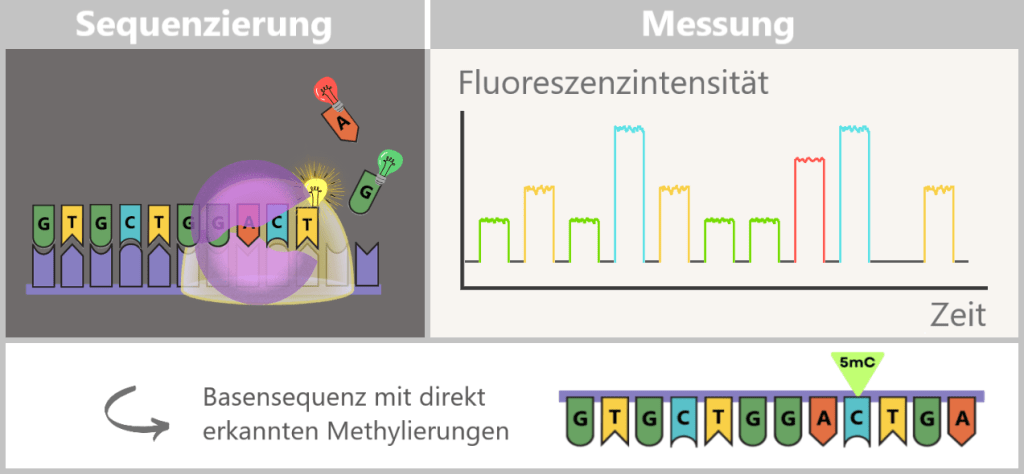
Wie SMRT epigenetische Modifikationen sichtbar macht
Während der SMRT-Sequenzierung werden die Fluoreszenzsignale der eingebauten Nukleotide in Echtzeit gemessen. Gleichzeitig können Modifikationen wie 5-Methylcytosin (5mC) erkannt werden, da sie die Kinetik der Polymerase leicht verändern.
Der entscheidende Vorteil: Die extrem langen Leselängen enthüllen, wie sich Methylierungsmuster über große genomische Regionen mit anderen Faktoren verschränken – etwa mit:
- Genvarianten,
- Haplotypen (Gruppen von gemeinsam vererbten Genvarianten),
- repetitiven Sequenzen.
So liefert PacBio nicht nur die genetische Buchstabenfolge, sondern eine „Landkarte“ der epigenetischen Steuerung – und macht sichtbar, welche „Buchseiten“ funktionell zusammengehören und wie diese Vernetzung Krankheitsprozesse wie Krebs oder neurologische Störungen beeinflusst.
Defekte Scheren im Genom: Wie PacBio Splice-Site-Varianten enttarnt
Im Abschnitt Alternatives Splicing: Die flexible Küche der Gene wurde vereinfacht beschrieben, wie unsere DNA als molekulares Kochbuch Rezepte in Gestalt von mRNA-Abschriften erzeugt. Diese transportieren die genetische Information aus dem Zellkern zu den Ribosomen im Zytoplasma – den „Protein-Fabriken“ der Zelle.
Dabei wird das Rezept nachträglich angepasst: Eine molekulare Schere entfernt bestimmte Zutaten (Introns) und kombiniert andere Bausteine (Exons) neu. Gesteuert wird dieser Prozess durch Splice-Stellen – Markierungen, die aus der DNA „mitkopiert“ werden. Man kann sie sich wie eine Schnittanleitung vorstellen: Sie entscheidet, welche Teile des Rezepts verwendet und wie sie zusammengesetzt werden.
Doch was passiert, wenn diese Schnittanleitung fehlerhaft ist? Die Folgen können gravierend sein:
- Exons werden übersprungen → Teile der Bauanleitung fehlen.
- Introns bleiben fälschlich erhalten → „Müll“ wird ins Protein eingebaut.
- Falsche Spleißstellen entstehen → Der mRNA-Text wird sinnentstellt.
Solche Splice-Site-Varianten führen häufig zu fehlerhaften oder funktionslosen Proteinen – mit möglichen Folgen wie Krebs, genetischen Erkrankungen (z. B. β-Thalassämie) oder neurologischen Störungen. Da die Proteinbiosynthese direkt vom mRNA-Text abhängt, ist es entscheidend, solche Fehler zu erkennen.
Der Schlüssel liegt in PacBios Fähigkeit, vollständige mRNA-Moleküle in einem Stück zu analysieren – meist 500 bis 5.000 Nukleotide lang, manchmal deutlich länger. Da PacBio DNA liest, wird die mRNA zunächst in eine stabile Kopie (cDNA, komplementäre DNA) umgewandelt. Diese kann dann vollständig sequenziert werden. So lässt sich exakt erkennen, wie das Rezept aufgebaut ist und welche Zutaten in welcher Reihenfolge vorgesehen sind – kein Schreib- oder Schnittfehler bleibt verborgen.
Zusätzlich kann auch der zugrunde liegende DNA-Abschnitt sequenziert werden. Bioinformatische Tools vergleichen cDNA- und DNA-Sequenzen mit Referenzdaten – so werden Abweichungen im Spleißmuster und ihre genetische Ursache sichtbar.
Die DNA zeigt: Hier ist die Anleitung fehlerhaft.
Die cDNA zeigt: Hier ist das Rezept verstümmelt.
Erst die Kombination beider Informationen schafft eine fundierte Basis für präzise Diagnosen und gezielte Therapieansätze.
Fazit – Wenn man die ganze Geschichte erzählen will
Wenn es darum geht, die ganze Geschichte eines DNA-Abschnitts am Stück zu erzählen – ohne zu zerschneiden oder zusammenzusetzen –, dann ist SMRT-Sequenzierung die Methode der Wahl.
PacBio-SMRT ist der Archäologe der Genomik: Es sieht das Ganze, wo andere nur Bruchstücke liefern. 2022 trug es maßgeblich dazu bei, die letzten Lücken im menschlichen Genom zu schließen.
Die PacBio-SMRT-Technologie ist kein Massenprodukt, sondern ein Präzisionsinstrument. Sie ist langsamer und teurer pro Basenpaar als Illumina – aber dafür auch detailreicher und vielseitiger.
In der Praxis werden beide Technologien oft kombiniert: Illumina für die Breite, PacBio für die Tiefe. Gemeinsam ergeben sie ein vollständiges Bild – wie Satellitenbild und Nahaufnahme in einem einzigen Panorama.

4.4.2. Methode 2: Hightech-Tunnel – DNA elektrisch abtasten
Während bei der molekularen Kopiermaschine mit Farbsensoren die DNA zuerst vervielfältigt und dann bildlich „fotografiert“ wird, funktioniert die zweite Methode radikal anders: Die DNA wird durch einen Hightech-Tunnel geschickt – und dabei live elektronisch abgetastet.
Nach diesem Prinzip funktioniert die Oxford Nanopore Sequenzierung.
a) Oxford Nanopore Sequenzierung
Die DNA geht durchs Nadelöhr
Stellen Sie sich vor, Sie lesen ein Seil, indem Sie es langsam durch Ihre Finger gleiten lassen – und dabei jede noch so feine Unebenheit ertasten: Dicke, Dellen, Knoten. Genau so funktioniert die Oxford Nanopore Sequenzierung (ONT): Sie „fühlt“ die DNA, während sie durch ein winziges biologisches Nadelöhr gleitet.
Dieses Nadelöhr – ein sogenannter Nanopore-Kanal – ist eingebettet in eine Membran, durch die ein elektrischer Strom fließt. Sobald ein einzelnes DNA-Molekül durch diesen Hightech-Tunnel gezogen wird, beeinflussen die Basen (A, T, G oder C) den Stromfluss unterschiedlich stark – wie verschieden geformte Kieselsteine in einem Bach. Diese Veränderungen erzeugen charakteristische elektrische Signale, die in Echtzeit aufgezeichnet werden.
ONT liest also keine Lichtsignale, sondern Strommuster – ein elektrisches Fingerlesen der DNA.
Ein einzelner Faden – direkt gelesen
Bevor die Lesung beginnt, wird die DNA mit einem Motorprotein gekoppelt, das sie präzise durch die Pore zieht – nicht zu schnell, nicht zu langsam. Anders als bei Illumina oder PacBio braucht es weder Polymerase noch Fluoreszenz oder Amplifikation: Die Original-DNA wird direkt gelesen – ein großer Vorteil bei empfindlichen oder beschädigten Proben.
Die Sequenzierzelle besteht aus zwei Kammern: der cis-Kammer (oben) und der trans-Kammer (unten), getrennt durch eine Membran mit eingebetteten Nanoporen. Links hat ein DNA-Molekül mithilfe eines Motorproteins an eine Nanopore angedockt. Rechts ist eine leere Nanopore dargestellt, durch die ein konstanter Ionenstrom fließt. Die angelegte Spannung zwischen der negativen Elektrode (cis) und der positiven Elektrode (trans) treibt den Ionenfluss an. Der Ionenstrom wird im Well (kleiner Kanal im Chip) gemessen, wie im Strom/Zeit-Diagramm dargestellt. Solange keine DNA durch die Pore wandert, bleibt der Strom konstant.
Und das Beste: Die DNA kann dabei extrem lang sein. ONT hält den Weltrekord für die längste je sequenzierte DNA – über zwei Millionen Basenpaare am Stück. Ganze Chromosomenabschnitte lassen sich so ohne Unterbrechung erfassen – wie ein Roman in einem einzigen Atemzug.
Aus Rauschen wird Sinn – Die Kunst der Signalinterpretation
Die elektrischen Rohsignale, die beim Durchgang der DNA durch die Pore entstehen, ähneln zunächst einem verrauschten Radiokanal. Doch mithilfe spezialisierter Algorithmen – dem sogenannten Basecalling – wird aus dem Stromrauschen eine klare Buchstabenfolge: A, T, G oder C.
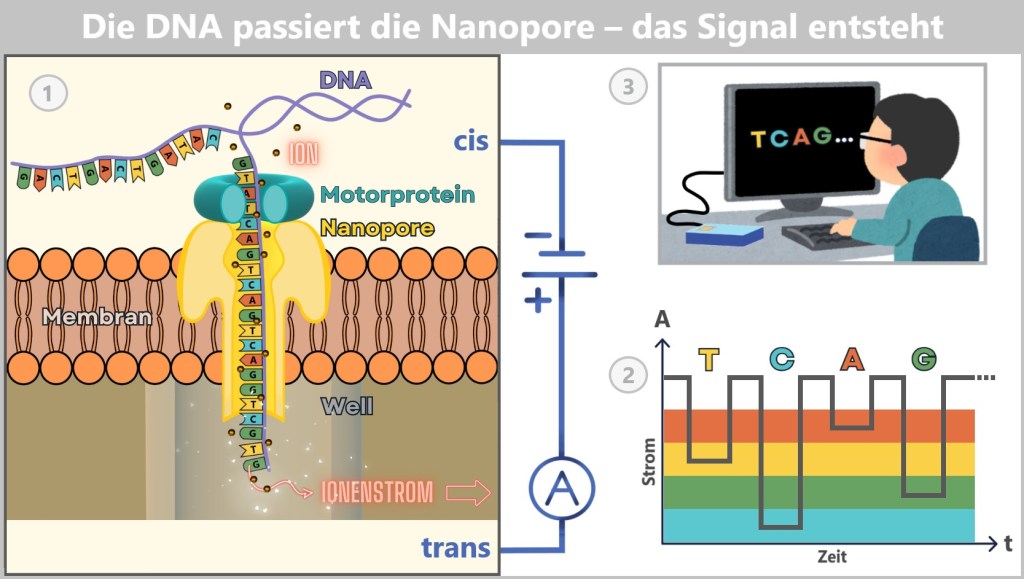
1) Hier sieht man eine schematische Darstellung der Nanopore in einer Membran. Ein Motorprotein zieht die DNA kontrolliert durch die Pore. Die negativ geladene DNA bewegt sich aufgrund der angelegten Spannung von der cis-Seite zur trans-Seite. Dabei beeinflussen die einzelnen Basen (A, T, G, C) den Ionenstrom auf spezifische Weise.
2) Ein Graph stellt die Veränderung des gemessenen Stroms über die Zeit dar. Jede Basen-Kombination erzeugt ein charakteristisches Signal, das durch Algorithmen entschlüsselt wird.
3) Ein Computer analysiert die elektrischen Signale und bestimmt daraus die Reihenfolge der Basen.
Anders als bei PacBio werden bei ONT die Basen nicht einzeln unterschieden, sondern in Gruppen von fünf bis sechs Nukleotiden, die gleichzeitig in der Pore liegen. Jeder dieser „5-Mer“ erzeugt ein charakteristisches Stromsignal. Das erhöht die Lesegeschwindigkeit, macht die Interpretation aber komplexer – ein wenig wie das Erkennen von Wörtern in einem ungewohnten Dialekt. Besonders Homopolymeren (lange Wiederholungen derselben Base, z. B. „AAAAA“) stellen eine Herausforderung dar.
Die KI-gestützte Basecalling-Software verbessert sich kontinuierlich. Die Fehlerquoten sind heute drastisch gesunken, und ONT-Reads erreichen mittlerweile auch eine hohe Genauigkeit – besonders wenn die Datenmenge für eine statistische Mehrfachkontrolle jeder DNA-Base (durch wiederholte, zufällige Erfassung) hoch genug ist.
Eine coole visuelle Erklärung gibt’s im Video How nanopore sequencing works von Oxford Nanopore Technologies.
Wer es etwas ausführlicher mag, findet hier weitere Hintergrundinfos.
Besondere Stärken – Schnell, portabel, vielseitig
Was ONT besonders macht:
Mobilität: Geräte wie der MinION passen in eine Hosentasche. Sie benötigen nur einen Laptop und etwas Strom – ideal für Feldforschung, Raumfahrt oder schnelle Analysen bei Ausbrüchen (z. B. Ebola oder SARS-CoV-2).
Schnelligkeit: Die DNA wird live gelesen. Erste Sequenzdaten liegen oft schon nach wenigen Minuten vor – ein unschätzbarer Vorteil in klinischen Notfällen.
Direkte Epigenetik: Wie bei PacBio erkennt ONT Methylierungen und andere epigenetische Modifikationen direkt, da sie das elektrische Signal subtil beeinflussen – ganz ohne zusätzliche Behandlung oder Reagenzien.
Direkte RNA-Sequenzierung: Auch RNA-Moleküle können ohne vorherige Umwandlung in cDNA gelesen werden – ein echtes Alleinstellungsmerkmal.
Warum direkte RNA-Sequenzierung wichtig ist
Andere Sequenzierverfahren wie PacBio müssen mRNA erst in cDNA umwandeln, um sie lesen zu können. Das ist, als würde man ein Rezept zunächst abschreiben – dabei können kleine Details verloren gehen. Die Oxford-Nanopore-Technologie hingegen liest die mRNA direkt – ohne Umweg über DNA.
So bleiben alle „Randnotizen des Kochbuchs“ erhalten: chemische Modifikationen, Varianten und sogar die exakte Länge der Moleküle – Informationen, die bei einer Kopie oft verschwinden. Dadurch entsteht ein unverfälschtes Bild der Genaktivität – ein unschätzbarer Vorteil, um komplexe Abläufe wie Genregulation oder fehlerhafte RNA-Verarbeitung zu erkennen.
Gerade für die personalisierte Medizin ist das ein Durchbruch: Denn viele Krankheiten entstehen nicht allein durch veränderte DNA, sondern auch durch Abweichungen in der RNA.
Beispiele aus der Praxis:
- Krebsdiagnostik: RNA-Spleißvarianten können helfen, aggressive Tumorformen frühzeitig zu erkennen und gezielt zu behandeln.
- Erbkrankheiten: Bei Erkrankungen wie spinaler Muskelatrophie oder Duchenne-Muskeldystrophie ermöglichen RNA-Profile eine genaue Einschätzung des Verlaufs und personalisierte Therapien, z. B. mit Antisense-Oligonukleotiden.
- Infektionsmedizin: Die direkte Sequenzierung viraler RNA erlaubt eine schnelle Identifikation von Virusvarianten – entscheidend bei Epidemien und für die Wahl der Therapie.
Die direkte RNA-Sequenzierung hilft also, die persönliche Rezeptvariante jedes Menschen zu erkennen – mit all ihren kleinen Abweichungen, Randnotizen und besonderen Zutaten – und dadurch Diagnosen zu präzisieren sowie maßgeschneiderte Behandlungen zu ermöglichen.
Fazit: Der elektrische Sinnesfinger der Genomik
ONT ist der sinnesstarke Grenzgänger der Genomik: Schnell wie ein Rennpferd, portabel wie ein Schweizer Taschenmesser und nah dran am biologischen Geschehen. Wo andere kopieren müssen, fühlt es live – selbst bei RNA oder epigenetischen Spuren.

4.4.3. Genomsequenzierung – Technologien im Vergleich
In der Welt der Genomik gibt es nicht die eine Methode – sondern ein ganzes Ensemble. Jede Sequenzierplattform hat ihren eigenen Stil, ihre Stärken und ihr ideales Einsatzgebiet: vom klassischen Solo-Erzähler bis zum Hochgeschwindigkeitsstreamer.
Auch wenn alle vier Sequenzierer ihre Bühne haben – nicht jeder eignet sich für jede Inszenierung. Wer gezielt ein einzelnes Gen untersuchen will, braucht andere Mittel als jemand, der das gesamte Genom in epischer Breite erfassen möchte. Und wer seltene Varianten aufspüren will, muss genau hinlesen.
Betrachten wir die vier Hauptakteure im Detail, um ihre Stärken, Schwächen und strategischen Rollen zu verstehen.
a) Durchsatz vs. Verwertbarkeit – Masse ist nicht immer gleich Klasse
„Durchsatz“ klingt nach Effizienz – möglichst viele DNA-Buchstaben pro Minute. In der Sequenzierung misst man ihn in Basen pro Lauf: also wie viele Basen ein Gerät in einem einzigen Sequenzierprozess ausliest.
Moderne Plattformen wie Illuminas NovaSeq X Plus erreichen mit kurzen Reads (100–300 bp) Spitzenwerte von bis zu 16 Terabasen – das sind 16 Milliarden Buchstaben in einem einzigen Lauf. Wie ein Hochleistungsdrucker, der über Nacht ganze Genombibliotheken produziert.
PacBio und Oxford Nanopore verfolgen einen anderen Ansatz. Sie erzeugen lange Reads – von 10.000–25.000 bp bei PacBio HiFi-Reads, bis hin zu über 1 Million bp bei ONT. Ihr Durchsatz ist geringer: PacBio (Revio) erreicht bis zu 1.300 Gigabasen, ONT (PromethION) bis zu 7.000 Gigabasen pro Lauf. Klingt nach weniger – und das ist es auch, wenn man nur die Basenzahl betrachtet.
Doch: Mehr Daten bedeuten nicht zwangsläufig mehr Erkenntnis. Entscheidend ist nicht nur, wie viel gelesen wird – sondern wie präzise und wie oft. Zwei zentrale Konzepte helfen, die Aussagekraft von Sequenzdaten einzuordnen:
🔍 Quality Score – das Vertrauen in jeden Buchstaben
Der sogenannte Q-Score (Quality Score) beschreibt, wie wahrscheinlich ein gelesener DNA-Buchstabe korrekt ist. Die Skala ist logarithmisch:
- Q20 = 99 % Genauigkeit → 1 Fehler pro 100 Basen
- Q30 = 99,9 % Genauigkeit → 1 Fehler pro 1.000 Basen
- Q40 = 99,99 % Genauigkeit → 1 Fehler pro 10.000 Basen
In der Forschung ist häufig Q20 ausreichend. In der medizinischen Diagnostik gelten Q30 oder höher als Standard, bei Hochpräzisionsanwendungen (z. B. Tumorgenetik) wird Q40 erwartet.
Bei Rohdaten liegt Illumina typischerweise im Bereich von Q30–Q40, während PacBio (Continuous Long Reads, CLR) und ONT nur Werte zwischen Q10-Q15 aufweisen.
Sanger übertrifft alle – mit Q40–Q50, allerdings nur auf kurzen DNA-Abschnitten. Ideal zur gezielten Bestätigung einzelner Varianten.
Wie also erreichen moderne Sequenzierungsverfahren die hohen Anforderungen der klinischen Diagnostik? Die Antwort: durch Coverage und bioinformatische Analyse.
🔁 Coverage – die Wiederholung macht die Wahrheit
Coverage (Abdeckung) beschreibt, wie oft jeder Bereich des Genoms gelesen wurde.
Beispiel: 30x Coverage bedeutet, dass jeder Buchstabe im Durchschnitt 30-mal gelesen wurde – auf verschiedenen Fragmenten, in unterschiedlichen Konstellationen.
Warum ist das wichtig? Einzelmessungen können Fehler enthalten. Nur durch Wiederholung und statistische Mehrheitsbildung lassen sich echte Varianten von Artefakten unterscheiden.
In der medizinischen Diagnostik ist 30x Coverage der Mindeststandard.
Illumina erreicht bei 30–50x Coverage eine Gesamtgenauigkeit von Q35–Q40+. Es erkennt zuverlässigSingle Nucleotide Variants (SNVs), Insertions/Deletions (Indels) und Copy Number Variants (CNVs).
PacBio HiFi Reads erreichen bei 30x Coverage Q30–Q40 und ONT Q30-35. ONT benötigt 50x Coverage, um eine Präzision von Q44 zu erreichen.
Der große Vorteil von PacBio und ONT: Sie lesen wesentlich längere DNA-Abschnitte – oft 10.000 bis 100.000 Basen, bei ONT sogar über 1 Million. Dadurch erkennen sie komplexe Strukturen, Wiederholungen, epigenetische Signaturen und ermöglichen RNA-Untersuchung und die Phasierung – also die Zuordnung von Varianten zu väterlichen oder mütterlichen Chromosomen. Hier zählt nicht die Masse, sondern das Gesamtbild.
🧬 Je nach Ziel – unterschiedliche Anforderungen an Coverage und Genauigkeit
Die nötige Coverage und Sequenzierqualität hängen vom klinischen Einsatz ab:
Populationsstudien: Q30 bei 30x Coverage reicht meist aus, um statistische Muster zu erkennen.
Krebsdiagnostik: Q35 bei 60–100x zur sicheren Detektion seltener somatischer Mutationen.
Liquid Biopsies, Mosaik-Analysen: Q40 bei 100–300x – zur Unterscheidung von echten Signalvarianten vom Hintergrundrauschen.
Phasierung: Lange Reads (PacBio, ONT) mit 30x Coverage genügen oft, um Varianten zuverlässig zuzuordnen.
📉 Das Dilemma: Mehr Sicherheit – mehr Aufwand
Mehr Coverage erhöht die Sicherheit, bedeutet aber auch mehr Daten, längere Rechenzeiten, größere Infrastruktur – und höhere Kosten.
b) Wirtschaftlichkeit – Kosten pro Base vs. Kosten pro Erkenntnis
Illumina (NovaSeq/NextSeq) bleibt der Preis-Leistungs-Favorit für Whole-Genome-Sequencing:
👉 200–800 € für ein vollständiges Genom bei 30x-Abdeckung – präzise bei Punktmutationen, ideal für Routineanalysen.
PacBio (Revio/Sequel IIe) und Oxford Nanopore (PromethION/MinION) sind teurer:
👉 700–1.200 € für 15–30x-Abdeckung – dafür mit zusätzlichen Informationen wie lange Reads, strukturelle Varianten, Epigenetik und RNA. Das ist kein Luxus, sondern in vielen Fällen diagnostisch entscheidend.
Oxford Nanopore punktet zudem mit niedrigem Einstiegspreis: Ein MinION kostet wenige Tausend Euro, ist mobil und liefert Daten binnen Stunden.
👉 Die einfache Rechnung „mehr Basen = besserer Deal“ greift zu kurz. Entscheidend ist, was aus den Daten gelesen und verstanden werden kann.
🧾 Versteckte Kosten – Das Genom ist günstiger zu lesen als zu verstehen
Ein menschliches Genom für unter 300 €? Technisch ja – aber meist sind nur Reagenzien, Maschinenzeit und Rohdaten abgedeckt.
Was oft fehlt, sind die unsichtbaren Posten:
🔧 Probenaufbereitung
🔍 Qualitätskontrolle & Library-Erstellung
🧠 Bioinformatik & klinische Interpretation
📄 Befundbericht & ärztliche Rücksprache
Diese Bereiche machen häufig 70–80 % der Gesamtkosten aus.
Denn: Lesen ist heute günstig – Verstehen bleibt anspruchsvoll.
c) Von der Probe zum Befund – Wie schnell spricht das ganze Genom?
Das Genomlesen ist wie eine Aufführung: Der Vorhang hebt sich mit der Probenentnahme – aber wann fällt der letzte Vorhang, der klinische Befund?
Es hängt von Technologie und Infrastruktur ab:
Oxford Nanopore ermöglicht vorläufige Ergebnisse binnen 24–48 Stunden – dank Echtzeit-Analyse während der Sequenzierung.
Illumina liefert mit optimierten Workflows Daten in 3–7 Tagen – der Goldstandard für routinierte Diagnostikabläufe.
PacBio benötigt 4–7 Tage, bietet dafür jedoch besonders hochwertige Langlesungen und epigenetische Informationen.
Sanger liefert Validierungen gezielt binnen Stunden – aber nur für kleine Zielregionen.
Die Plattformwahl ist also nicht nur eine Frage der Datenqualität – sondern auch des klinischen Timings.
Wer in Stunden statt Tagen handeln muss, greift zu Oxford Nanopore. Wer robuste Routinedaten braucht, setzt auf Illumina. Und wer maximale Kontexttiefe wünscht, nimmt sich mit PacBio etwas mehr Zeit – für deutlich mehr Einsicht.
Der Vorhang fällt. Doch hinter den Kulissen balanciert zwischen Hoffnung, Hightech und menschlicher Sorgfalt geht die Suche nach dem perfekten Tempo für jeden Patienten weiter. Denn am Ende zählt nicht die schnellste Maschine – sondern der richtige Takt für die menschliche Geschichte dahinter.
d) Klinische Einsatzfelder – Welche Technologie passt wozu?
Die Genomsequenzierung ist längst mehr als ein Forschungswerkzeug – sie ist zum zentralen Instrument geworden, um Therapien individuell zu planen. Doch welche Methode eignet sich für welche Aufgabe?
Ein Blick auf sechs zentrale Anwendungsfelder zeigt, wie diese Technologien die moderne Medizin prägen.
| Anwendungsfeld | Primäre Methode | Ergänzende Methoden |
|---|---|---|
| Routinediagnostik | Illumina | Sanger |
| Pharmakogenomik | Illumina | Sanger |
| Epigenetik | PacBio, ONT | – |
| Krebsgenomik | Illumina (SNVs), PacBio/ONT (SVs) | Sanger |
| Neonatale Notfalldiagnostik | ONT | Illumina, Sanger |
| Seltene Erkrankungen | PacBio, ONT | Illumina, Sanger |
👉 Erkenntnis: Die optimale Technologie hängt stets von der klinischen Fragestellung ab – Standardanalyse, Suche nach dem Unbekannten oder Notfalldiagnostik? Validierung oder Erstdiagnose?
e) KI als Co-Pilot – Automatisierung ohne Verantwortung abzugeben
Die Integration von Künstlicher Intelligenz (KI) – insbesondere Deep Learning – ist in allen modernen DNA-Sequenzierplattformen (Illumina, PacBio, Oxford Nanopore) unverzichtbar. Ihre Hauptanwendungsfelder sind:
🧬 Basecalling (Umwandlung von Rohdaten in DNA-Sequenzen) & Fehlerkorrektur
🔍 Qualitätskontrolle & Echtzeitanalyse
🧠 De-novo-Assembly, Genomanalyse & Variantenerkennung
📚 Pathogenitätsvorhersage, Literaturrecherche & Klinische Klassifikation
📄 Automatisierte Befundvorlagen
👉 Erkenntnis: KI transformiert die Sequenzierung von einem reinen Datengenerierungs- zu einem intelligenten Interpretationsprozess – und ist damit Kern moderner genomischer Forschung und Diagnostik.
Doch wichtig bleibt: KI denkt (noch) nicht wie ein Arzt. Bei seltenen Erkrankungen, komplexen Verläufen oder neu entdeckten Genveränderungen bleibt die ärztliche Erfahrung zentral. Auch rechtlich ist eine ärztlich validierte Auswertung Pflicht – selbst im Zeitalter digitaler Genomik.
Am Ende ist Genomsequenzierung keine Solovorstellung – sie braucht das ganze Ensemble: Präzise Technik, kluge Bioinformatik und ärztliche Intuition. Denn nur wenn alle mitspielen, wird aus Daten heilsame Erkenntnis.
f) Vergleichstabelle: Sanger, Illumina, PacBio, ONT
| Merkmal | Sanger | Illumina | PacBio | ONT |
|---|---|---|---|---|
| Proben-vorbereitung | DNA-Template, Extraktion, Primer-Design, ddNTPs 🕒 ~6–12 Std. | Fragmentierung, Adapter-Ligation 🕒 ~6–24 Std. | Hohe DNA-Qualität, Fragmentierung, SMRTbell-Adapter 🕒 ~1–2 Tage | Fragmentierung, Adapter mit Helikase (Motorprotein) 🕒 ~6–24 Std. |
| Leselänge | Kurz: 500–1.000 bp | Sehr kurz: 100–300 bp (paired-end möglich) | Lang: 10.000–25.000 bp (HiFi-Reads) | Ultra-lang: 10.000 bp – >1 Mb |
| Durchsatz | Sehr niedrig: Einzelreaktionen (~1–100 kb/Lauf) | Sehr hoch: bis 16.000 Gb (NovaSeq X Plus), >100 Proben gleichzeitig | Mittel: bis 1.300 Gb / SMRT-Cell (Revio) | Flexibel: 100–7.000 Gb (PromethION), bis 20 Gb (MinION) |
| Genauigkeit | Sehr hoch: Q40–Q50 (99,99–99,999 %) | Hoch: Q30–Q40 (99,9–99,99 %), bei 30x: Q35–Q40+ | Hoch: Q30–Q40 (HiFi-Reads), bei 30x | Rohdaten: Q15–Q20+, bei 30x: Q30–Q35, Duplex: Q44 bei 50x |
| Kosten/Genom (EUR) (nur Sequenzierung) | – | ~200–800 (30x) (Großlabore) | ~700–1.200 (15–30x) | ~700–1.200 (15–30x) |
| Zeit bis Ergebnis (WGS, Probe → Befund) | – | 3–7 Tage (inkl. Vorbereitung, Sequenzierung, Analyse) | 4–7 Tage (inkl. Vorbereitung, Sequenzierung, Analyse) | 24–48 Std. (Echtzeit, inkl. Analyse) |
| Varianten-erkennung | SNVs, mitochondriale Varianten (Validierung) | SNVs, Indels, CNVs (indirect), somatische Varianten, Splice-Varianten | Indels, SVs, CNVs, Repeat-Expansionen, Splice-Varianten, somatische SVs | Indels, SVs, CNVs, Repeat-Expansionen, Splice-Varianten, somatische SVs |
| RNA-Sequenzierung | – | mRNA durch Umwandlung in cDNA, Splice-Varianten | mRNA durch Umwandlung in cDNA, Splice-Varianten (lange Reads) | Direkte RNA-Sequenzierung (ohne cDNA), Splice-Varianten, Langzeit-Transkripte |
| Epigenetische Modifikationen | – | Indirekt: nur mit Bisulfit-Sequenzierung (aufwendig) | Direkt: Methylierungserkennung durch Einbaukinetik (HiFi-Reads) & Bioinformatische Tools | Direkt: Methylierung durch Signaländerungen (z. B. 5mC, 6mA) |
| Einsatzort | Labor: Stationäre Geräte | Labor: Stationäre Großgeräte (z. B. NovaSeq) | Labor: Stationäre Großgeräte (Revio) | Flexibel: Labor (PromethION), mobil/Point-of-Care (MinION) |
| Hauptvorteile | Höchste Präzision, ideal zur Validierung | Hoher Durchsatz, kostengünstig, präzise bei SNVs/Indels, routinetauglich | Lange, präzise Reads (HiFi), ideal für SVs, Epigenetik, Phasierung | Ultra-lange Reads, mobil, schnell, direkte RNA- und Epigenetik-Erkennung |
| Limitationen | Niedriger Durchsatz, WGS unpraktisch, teuer | Kurze Reads, eingeschränkt bei SVs/Epigenetik, komplexe Library-Prep | Teurer, mittlerer Durchsatz, längere Vorbereitungszeit | Geringere Rohdatenqualität, abhängig von Bioinformatik |
| Rolle in der personalisierten Medizin | Validierung kritischer SNVs (Pharmakogenomik, Krebs), mitochondriale Analysen | Routinediagnostik, Pharmakogenomik, Krebsgenomik (SNVs/Indels), seltene Erkrankungen | Epigenetik, Krebsgenomik (SVs), seltene Erkrankungen (SVs, Repeat-Expansionen), Phasierung | Neonatale Notfalldiagnostik, Epigenetik, Krebsgenomik (SVs), seltene Erkrankungen, mobile Diagnostik |
g) Key Takeaways: Zwischen Reife und Routine
Die DNA-Sequenzierung befindet sich an einem entscheidenden Wendepunkt: In den vergangenen Jahren hat sie sich von einem rein forschungsbasierten Verfahren zu einem klinisch relevanten Werkzeug der modernen Medizin entwickelt. Ob in der Onkologie, der Diagnostik seltener Erkrankungen oder der Pharmakogenomik – Genomanalysen liefern zunehmend Antworten, wo konventionelle Methoden an ihre Grenzen stoßen.
Und doch: Trotz der beeindruckenden technischen Reife ist die Genomsequenzierung bislang kein flächendeckender Standard in der klinischen Praxis.
Technologie reif – Infrastruktur unvollständig
Moderne Hochdurchsatzverfahren wie Illumina oder die Langlesetechnologien von PacBio und Oxford Nanopore ermöglichen heute hochpräzise Analysen zu vergleichsweise geringen Kosten. Whole-Genome-Sequenzierungen (WGS) sind in spezialisierten Laboren für wenige Hundert Euro pro Fall durchführbar – mit einer Gesamtdauer von unter einer Woche. Mobile Plattformen wie MinION liefern in akuten Situationen – etwa bei schwer erkrankten Neugeborenen – erste genetische Hinweise sogar binnen 48 Stunden.
Trotz dieser Fortschritte bleibt der klinische Einsatz fragmentarisch. Gründe dafür sind vielfältig: Es mangelt an standardisierten bioinformatischen Auswertungsprotokollen, an interdisziplinär geschultem Fachpersonal sowie an einer rechtssicheren und wirtschaftlich tragfähigen Integration in den Versorgungsalltag. Vor allem die Interpretation genetischer Varianten – insbesondere solcher mit unklarer klinischer Bedeutung – erfordert spezialisiertes Know-how, das vielerorts nicht verfügbar ist.
Großbritannien als Blaupause
Während viele Länder noch mit Pilotprogrammen experimentieren oder Einzelfallentscheidungen treffen, institutionalisiert das Vereinigte Königreich die Genomsequenzierung als integralen Bestandteil der öffentlichen Gesundheitsvorsorge. Dort soll im Rahmen eines nationalen Pilotprogramms künftig jedes Neugeborene genomisch untersucht werden – ein Präzedenzfall für die pränatale und frühkindliche Vorsorgemedizin. Ziel ist es, genetisch bedingte Erkrankungen bereits im frühkindlichen Stadium zu erkennen – noch bevor klinische Symptome auftreten. Damit wird DNA-Analytik vom Einzelfall zur bevölkerungsweiten Präventionsstrategie – ein Paradigmenwechsel.
Dieses Modell demonstriert, dass die Hürde für eine breite Implementierung weniger technischer, sondern vor allem systemischer Natur ist: fehlende Datenstandards, ethische und rechtliche Unklarheiten sowie eine zurückhaltende Erstattungspolitik verhindern vielerorts eine flächendeckende Integration. Der britische Vorstoß gilt daher als wegweisendes Beispiel für die institutionalisierte Genommedizin.
Technologischer Ausblick
Mehrere technologische Entwicklungen dürften die Umsetzung in die Breite in naher Zukunft weiter beschleunigen:
🚀 Sinkende Kosten pro Genom machen Sequenzierung auch in kleineren Einrichtungen wirtschaftlich attraktiv.
🚀 Dezentrale, mobile Systeme wie MinION ermöglichen ortsunabhängige Diagnostik, etwa in Notaufnahmen oder ländlichen Regionen.
🚀 Schnellere Time-to-Result – von der Probenentnahme bis zur Befundung – erlaubt den Einsatz auch in zeitkritischen Fällen.
🚀 Die Integration von Genomdaten mit Transkriptomik, Proteomik und Epigenetik (Multi-Omics) verspricht eine kontextualisierte, hochauflösende Diagnostik.
🚀 KI-gestützte Analytik sowie automatisierte Interpretationstools senken die Abhängigkeit von manueller Auswertung und reduzieren Fehlerquellen.
4.5. Genomsequenzierung: Der nächste Technologiesprung
Damit die Entschlüsselung unserer DNA künftig schneller, günstiger und überall möglich wird, arbeitet die Forschung an der nächsten Generation von Sequenzierungstechnologien.
a) FENT: Wie ein Mikrochip die DNA-Analyse revolutioniert
b) SBX: Wenn DNA sich streckt, um verstanden zu werden
c) Das G4X-System: Vom genetischen Rezept zum räumlichen Atlas der Zelle
a) FENT: Wie ein Mikrochip die DNA-Analyse revolutioniert
– und für alle zugänglich macht
Angetrieben von der Vision, genetische Informationen in Echtzeit und patientennah erfassen zu können, entwickelt das Unternehmen iNanoBio ein neues Verfahren, das diese Idee auf ein völlig neues Niveau heben soll.
Es kombiniert die bewährte Methode – das Auslesen von DNA durch winzige Nanoporen – mit einem Bauteil aus der Mikroelektronik: dem MOSFET (Metal-Oxide-Semiconductor-Feldeffekttransistor), Herzstück moderner Computerchips.
Vom Computerchip zum DNA-Sensor
Ein MOSFET ist ein winziger Schalter mit drei Kontakten: Source (Eingang), Drain (Ausgang) und Gate (Steuerung). Liegt am Gate eine Spannung an, öffnet sich ein leitender Kanal zwischen Source und Drain – vergleichbar mit einem Türsteher, der ein Tor freigibt. Bereits minimale Spannungsänderungen beeinflussen den Stromfluss, was MOSFETs extrem empfindlich macht.
Ein MOSFET ist ein winziger Schalter aus der Mikroelektronik mit drei Kontakten: Source (Eingang), Drain (Ausgang) und Gate (Steuerung). Source und Drain sind im Halbleitermaterial verankert, das Gate ist durch eine hauchdünne Oxidschicht isoliert.
Ohne Spannung am Gate bleibt der Kanal zwischen Source und Drain geschlossen – kein Strom fließt. Legt man jedoch Spannung am Gate an, entsteht ein elektrisches Feld, das leitende Teilchen (z. B. Elektronen) anzieht und einen Stromkanal öffnet. Der MOSFET wirkt damit wie ein extrem sensibler Türsteher: Erst wenn er das Signal bekommt, öffnet er das Tor und lässt Strom passieren.
Jetzt kommt der Clou: iNanoBio hat es geschafft, eine winzige Pore direkt in das empfindliche Steuerzentrum des MOSFETs zu integrieren. Wenn ein DNA-Strang hindurchgleitet, verändert jedes einzelne Nukleotid (A, T, C oder G) den Stromfluss zwischen Source und Drain charakteristisch. Das Ergebnis: ein neuartiger Sensor mit dem Namen FENT – kurz für Field-Effect Nanopore Transistor.
Der FENT ist zylindrisch aufgebaut und umschließt die Pore. Dadurch kann er Signale gleichzeitig aus mehreren Richtungen messen, was die Genauigkeit erhöht und Fehler reduziert.
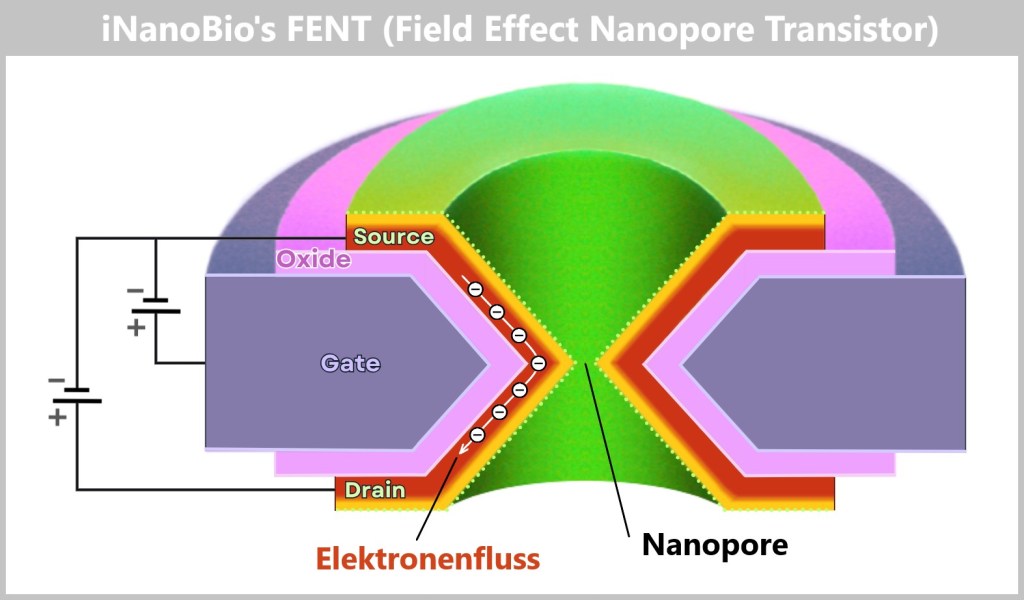
Der FENT ist ein zylindrisch gebauter MOSFET, der eine Nanopore vollständig umschließt. Zwischen Source und Drain wird eine Spannung angelegt, sodass ein kleiner Strom durch den Halbleiter der Porenwand fließt – der sogenannte Source-Drain-Kanalstrom. Die dünne Oxidschicht isoliert das Gate vom Kanal und ermöglicht die präzise Erfassung elektrischer Feldänderungen. Die Nanopore dient als Sensorzone für lokale Feldänderungen, verursacht durch vorbeigleitende DNA-Basen.
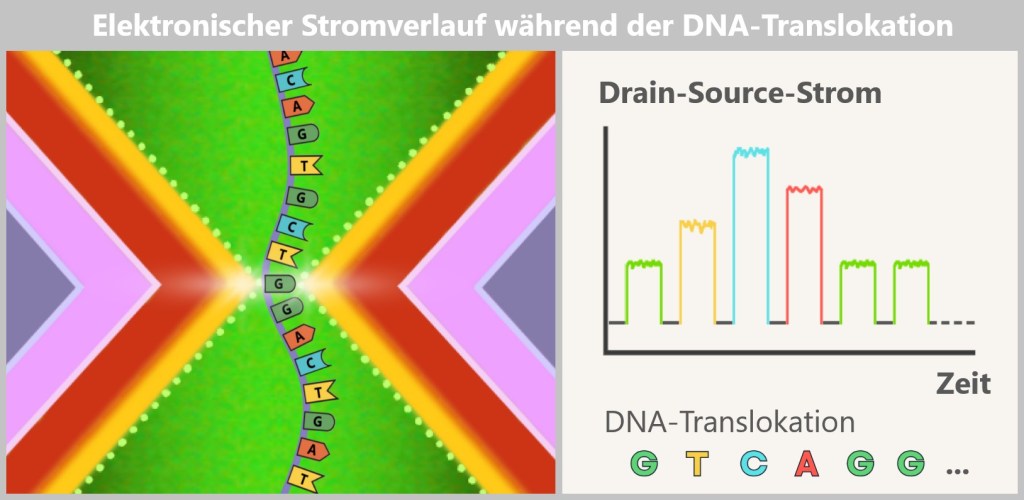
Die Abbildung zeigt das Funktionsprinzip eines FENT während der Passage einer DNA-Einzelstrangs (ssDNA) durch die Nanopore. Vor Beginn der Messung wird die ursprünglich doppelsträngige DNA (dsDNA) durch Hitze oder chemische Behandlung in ihre beiden Einzelstränge getrennt. Nur ein Einzelstrang wird anschließend durch die Nanopore geführt.
Im Ausgangszustand – also ohne DNA in der Pore – fließt zwischen Source und Drain ein konstanter Ruhestrom durch den Halbleiter, der die Porenwand bildet. Dieser Strom wird durch die Spannung zwischen Source und Drain sowie das Gate-Potenzial bestimmt.
Sobald jedoch eine DNA-Base (A, T, C oder G) die Nanopore passiert, beeinflusst sie das elektrische Verhalten des Systems:
Jede Base besitzt aufgrund ihrer spezifischen chemischen Struktur und Atomverteilung ein eigenes, winziges elektrisches Feld. Dieses Feld dringt durch die extrem dünne Porenwand und wirkt auf den Halbleiter wie ein lokales Gate-Signal. Dadurch verändert sich lokal die Ladungsträgerdichte (Elektronenkonzentration) im Transistorkanal, was zu einer messbaren Änderung des Drain-Source-Stroms führt.
Diese Stromänderungen werden als charakteristische Signale für die jeweilige Base aufgezeichnet. So entsteht während der Translokation eine sequenzabhängige Stromspur, die es ermöglicht, die Reihenfolge der DNA-Basen elektronisch zu bestimmen.
Eine allgemeinverständliche Erklärung liefert das Video FENT Nanopore Transistor Explainer Video.
Geschwindigkeit, die Maßstäbe setzt
Laut iNanoBio kann ein FENT bis zu 1 Million DNA-Basen pro Sekunde und Pore auslesen – etwa 100-mal schneller als bisherige Nanopore-Technik. Ein 5 x 5 mm großer Chip mit 100.000 dieser Sensoren könnte ein menschliches Genom in wenigen Minuten sequenzieren. Die Kosten wären deutlich geringer, die Fehlerrate niedriger.
Mögliche Anwendungen
- Krebsfrüherkennung aus Blutproben
- Mobile Diagnostik für Infektionen direkt vor Ort
- Großstudien in der Forschung dank hoher Geschwindigkeit
Langfristig will iNanoBio DNA-Analysegeräte so verbreiten wie heutige Blutdruckmessgeräte – verfügbar in Kliniken, Laboren, Arztpraxen und möglicherweise auch zu Hause. Erste Prototypen existieren, der Weg zur Massenproduktion läuft.
Wenn die Technologie hält, was sie verspricht, könnte die Genomsequenzierung bald so selbstverständlich sein wie eine Routineuntersuchung – und unser Verständnis von Gesundheit grundlegend verändern.
b) SBX: Wenn DNA sich streckt, um verstanden zu werden
Im Wettlauf um die Verbesserung der DNA-Analyse verfolgt der Pharmakonzern Roche mit SBX (Sequencing By eXpansion) einen verblüffend einfachen Ansatz: Er macht DNA künstlich größer, damit ihre „Buchstaben“ A, T, C und G leichter auseinanderzuhalten sind.
Schritt 1 – Vom Original zur vergrößerten Kopie
SBX erstellt aus der Original-DNA eine Surrogat-Kopie, den Xpandomer. Dafür wird das DNA-Fragment an einem winzigen Anker auf einem Substrat fixiert, und mithilfe einer Polymerase wird eine Kopie erstellt – allerdings nicht aus natürlichen Bausteinen, sondern aus Surrogat-Nukleotiden.
Ein Surrogat (von lateinisch surrogatum = „Ersatz“) bezeichnet allgemein einen Ersatz oder Stellvertreter für etwas anderes.
Diese bestehen aus:
- der passenden Base (A, T, C oder G),
- einem für jede Base spezifischen gefalteten Verlängerungsfaden als Signalgeber,
- einer spaltbaren Bindung, die den Faden in Form hält,
- einem Translocation Control Element (TCE), das den Ablauf später präzise steuert.
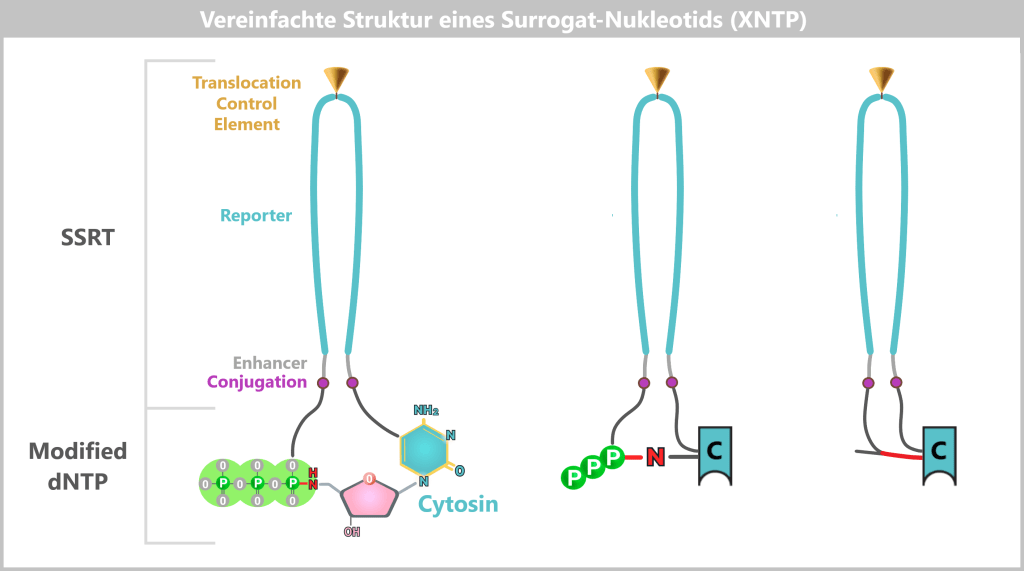
[Sequencing by expansion (SBX) technology]
Das Surrogat-Nukleotid (XNTP) besteht aus zwei funktionellen Teilen: dem modifizierten Nukleotid und dem SSRT (symmetrically synthesized reporter tether).
Das modifizierte dNTP enthält die eigentliche Base (hier beispielhaft Cytosin, C), die über eine säureempfindliche Bindung (rot markiert) mit dem Reporterfaden verbunden ist.
Daran schließt sich das Triphosphat (PPP) an, das die Einbindung des Nukleotids in die wachsende Kette durch die Polymerase ermöglicht.
Der Reporterfaden (SSRT) besteht aus einem gefalteten Verlängerungsfaden mit einem Translocation Control Element (TCE) an der Spitze, das den Durchtritt durch die Nanopore später steuert.
Ein Enhancer-Bereich (grau/lila) erleichtert die Einbindung durch die Polymerase und stabilisiert die Struktur.
Nach der Synthese kann die rote, säureempfindliche Bindung gelöst werden – dadurch „entfaltet“ sich der Reporterfaden und erzeugt beim Sequenzieren das ausgedehnte Xpandomer-Molekül.

Das DNA-Fragment (Template DNA) ist über einen Anker am festen Substrat fixiert. Ein Primer bindet an die DNA-Vorlage, unterstützt vom Leader und Concentrator, die den Startpunkt stabilisieren und die Positionierung erleichtern.
Fixierung – Die DNA-Vorlage ist am Substrat verankert, der Primer-Abschnitt hybridisiert an den passenden Bereich der DNA.
Verlängerung – Eine spezielle Polymerase ergänzt die Basen der Vorlage – aber nicht mit natürlichen Nukleotiden, sondern mit Surrogat-Nukleotiden (XNTPs), die jeweils einen Reporterfaden mitführen.
Xpandomer entsteht – Am Ende liegt ein künstlich erweiterter DNA-Strang vor: eine Surrogat-Kopie (Xpandomer), deren Basen durch charakteristische Reporterfäden ersetzt sind.
Der Xpandomer bleibt zunächst am Substrat fixiert und wird in einem späteren Schritt durch Licht oder chemische Spaltung freigesetzt.
Nach der Synthese wird der Xpandomer von der Original-DNA gelöst, die spaltbaren Bindungen werden getrennt – die Fäden entfalten sich und vergrößern den Abstand zwischen den Basen um das 50-Fache. Das erleichtert die spätere Identifikation erheblich.

Nach der Synthese wird der Xpandomer auf dem Substrat chemisch entfaltet („expandiert“) und anschließend freigesetzt.
Links) Expansion: Eine milde Säurebehandlung löst die spaltbaren Bindungen (rot) zwischen Base und Reporterfaden. Dadurch entfalten sich die Reporterstrukturen – aus der kompakten Doppelstruktur wird ein lang gestreckter Xpandomer, der die Abfolge der Basen repräsentiert.
Rechts) Freisetzung: Ein UV-Lichtimpuls spaltet die Verbindung zwischen dem Substrat und dem Leader-Bereich.
Der nun frei bewegliche Xpandomer bleibt über Leader und Concentrator strukturell markiert – diese Bereiche helfen später bei der Ausrichtung und beim Einzug in die Nanopore.
Aus einer dichten DNA-Vorlage entsteht ein langes, lesbares Molekül, dessen Reporterabschnitte die genetische Information elektrisch sichtbar machen.
Schritt 2 – Die Sequenzierung
Anstelle der DNA selbst wird nun der Xpandomer durch Nanoporen auf einem Sequenzierungschip gezogen – rund acht Millionen davon arbeiten parallel. Der Adapter des Xpandomers führt ihn gezielt in eine Pore.
Wenn ein Verlängerungsfaden die Pore erreicht, bleibt er kurz am TCE hängen. Jede Base erzeugt dabei ein spezifisches elektrisches Signal. Nach 1,5–2 Millisekunden löst ein Spannungspuls das TCE, der nächste Faden folgt. So entsteht eine klare, gut unterscheidbare Signalkurve – präziser als bei Oxford Nanopore, wo die Basen ein eher „verwaschenes“ Signal erzeugen.

[Sequencing By Expansion, Introduction to Sequencing By Expansion]
Der freigesetzte Xpandomer wird durch ein elektrisches Feld zur Nanopore gezogen. Dort kontrolliert das TCE (Transient Capture Element) den Eintritt und hält die Reporter kurzzeitig fest.
Während jeder Reporterabschnitt (die verlängerte „Base“) die Pore passiert, ändert sich der Ionenstrom in charakteristischer Weise: Jede Base erzeugt dabei ein eigenes elektrisches Signal – vergleichbar mit einem individuellen Barcode. Diese Signalfolge wird in Echtzeit aufgezeichnet:
Ein Spannungspuls löst nach jeder Messung den Faden vom TCE, sodass der nächste Abschnitt eingezogen wird.
Folgende Animation veranschaulicht die Grundprinzipien der SBX-Technologie von Roche.
Eine ausführliche Präsentation gibt es hier.
Vorteile und Leistung
- Geschwindigkeit: Millionen Xpandomer werden gleichzeitig und in Echtzeit gelesen.
- Genauigkeit: Signal-Rausch-Verhältnis ähnlich hoch wie bei Illumina.
- Flexibilität: Leselängen bis über 1.000 Basen möglich – schneller und präziser als Nanopore, länger als Illumina.
Erste Tests zeigen: SBX kann bis zu sieben vollständige menschliche Genome pro Stunde mit 30x Abdeckung entschlüsseln. Bei dringenden Fällen dauert der gesamte Ablauf – von der Probenvorbereitung bis zur fertigen Analyse – nur rund 5,5 Stunden.
Die Markteinführung ist für 2026 geplant. Ob sich SBX gegen Illumina und Oxford Nanopore durchsetzen wird, hängt von Preis, Verfügbarkeit und den Anforderungen der Anwender ab.
c) Das G4X-System: Vom genetischen Rezept zum räumlichen Atlas der Zelle
Unser Körper besteht aus Milliarden Zellen, die wie winzige Küchen arbeiten. Die DNA ist ihr Kochbuch – sie enthält alle Rezepte des Lebens. Wird ein Rezept gebraucht, erstellt die Zelle eine RNA-Kopie, die als Bauanleitung für Proteine dient. Diese Proteine übernehmen konkrete Aufgaben: Sie bauen Strukturen, transportieren Signale oder bekämpfen Krankheitserreger.
Proteine wirken jedoch selten allein. Sie arbeiten in fein abgestimmten Netzwerken, deren Aktivität davon abhängt, wo, wann und mit welchen Partnern sie interagieren. Prozesse wie Immunreaktionen oder Tumorwachstum entstehen nicht in einer einzelnen Zelle, sondern aus dem Zusammenspiel vieler spezialisierter Zellen – oft in unterschiedlichen Bereichen eines Gewebes. Biologie ist also nicht nur biochemisch, sondern auch räumlich organisiert. Klassische Sequenzierungsmethoden erfassen jedoch meist nur die genetischen Inhalte, nicht deren räumliche Verteilung.
Genaktivität direkt im Gewebe
Der G4X Spatial Sequencer von Singular Genomics macht sichtbar, wo im Gewebe Gene aktiv sind und Proteine gebildet werden – direkt in konservierten Gewebeproben (FFPE, formalinfixiert und paraffineingebettet).
FFPE-Proben werden häufig in der Pathologie eingesetzt, etwa zur Krebsdiagnostik. Sie wirken wie eine „eingefrorene Momentaufnahme“, nur dass hier chemische Fixierung mit Formalin statt Kälte verwendet wird. Formalin vernetzt Proteine und Nukleinsäuren miteinander und fixiert so die molekularen Strukturen räumlich.
Was dabei erhalten bleibt:
- RNA (oft fragmentiert und chemisch modifiziert) – spiegelt die Genaktivität zum Fixierzeitpunkt wider.
- Proteine – zeigen, welche Strukturen und Signalwege in diesem Moment aktiv waren.
So lässt sich Monate oder Jahre später rekonstruieren, welche Gene und Proteine zu diesem exakten Zeitpunkt im Gewebe präsent und aktiv waren – als hätte man die Zeit angehalten. Mit dem G4X kann diese „eingefrorene“ molekulare Landschaft hochaufgelöst sichtbar gemacht werden – inklusive der exakten Position im Gewebe.
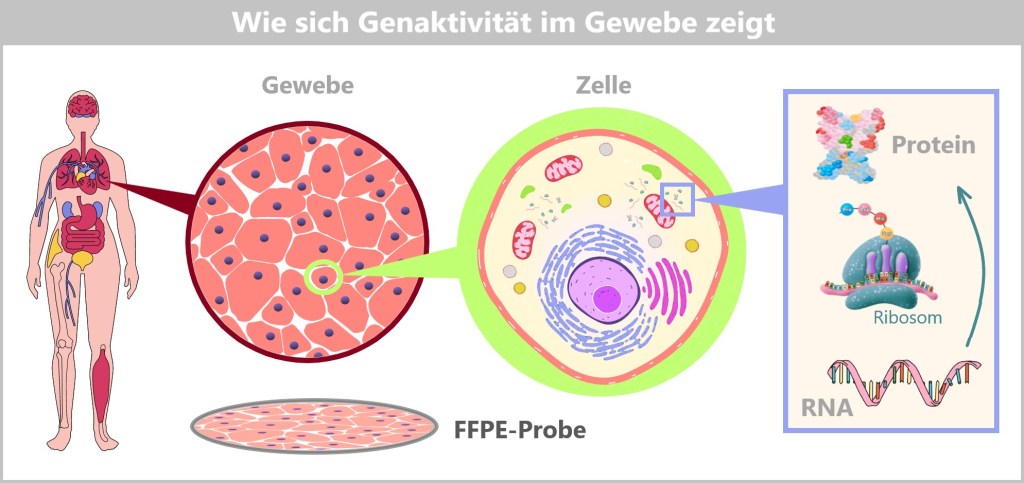
Ein Gewebe besteht aus vielen spezialisierten Zellen. Jede Zelle in einem Gewebe nutzt nur einen Teil ihres genetischen Programms. RNA macht die aktiven Gene sichtbar. RNA-Moleküle werden von Ribosomen abgelesen und in Proteine übersetzt. FFPE-Gewebeschnitte bewahren diese räumliche Anordnung – die Basis, um mit dem G4X-System zu messen, wo welche RNA aktiv ist.
Wie der G4X funktioniert
Man kann sich den G4X wie einen Mikroskop-Scanner mit eingebautem Labor vorstellen, der direkt im Gewebe misst, welche Gene und Proteine aktiv sind – und zwar in ihrem räumlichen Kontext.
Präparation: Ein sehr dünner Schnitt der FFPE-Probe wird auf ein spezielles Trägermaterial gebracht.
RNA-Nachweis: Um die Verteilung bestimmter RNA-Sequenzen im Gewebe zu erkennen, kommen Padlock-Sonden zum Einsatz.
Padlock-Sonden – DNA-Lassos für Zielsequenzen
Stellen Sie sich vor, Sie möchten in einem riesigen Gelände (dem Gewebe) nur ein bestimmtes Objekt (eine RNA-Sequenz) finden. Eine Padlock-Sonde ist ein DNA-Fragment, dessen beide Enden komplementär zu benachbarten Abschnitten der Zielsequenz passen.

Padlock-Sonde: Einzelsträngige DNA (ssDNA) 5’– Arm1 – Linker/Barcode – Arm2 –3′
Arm 1 und Arm 2 sind komplementär zu benachbarten Sequenzen der Ziel-RNA.
Dazwischen liegen Linker und Barcode, die zusätzliche Identifikationsinformationen tragen und nicht binden.
Bindet die Sonde an ihr Ziel, liegen ihre beiden Enden nebeneinander und können durch Ligation zu einem stabilen DNA-Ring geschlossen werden.
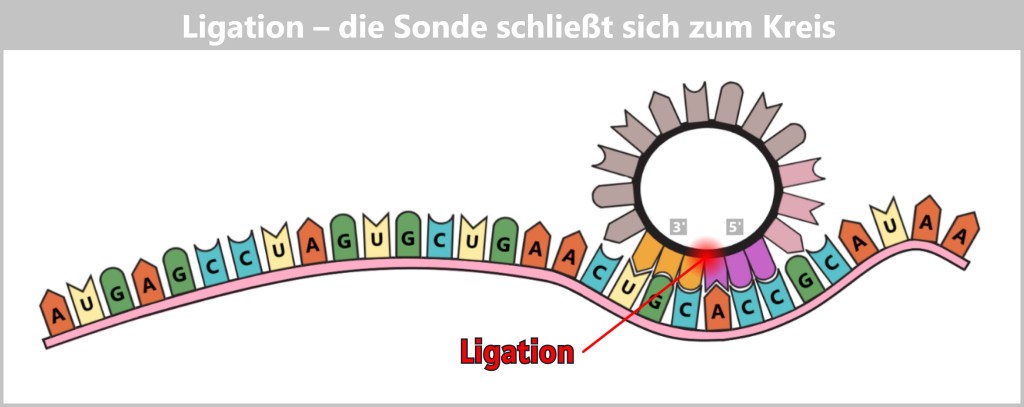
Nachdem beide Arme der Sonde an die Ziel-RNA gebunden haben, verbindet eine Ligase die 5′- und 3′-Enden. Dadurch entsteht ein geschlossener DNA-Ring, der die Ziel-RNA markiert und für die Amplifikation bereit ist.
Liegt zwischen den Bindungsstellen eine variable Sequenz (z. B. eine Mutation), wird diese zuvor mithilfe einer Reverse Transkriptase in DNA umgeschrieben und in die Sonde integriert.
Der entstandene DNA-Kreis dient als Vorlage für die Rolling Circle Amplification (RCA):
- Eine DNA-Polymerase läuft immer wieder im Kreis und erzeugt einen langen DNA-Strang, der die Zielsequenz vielfach wiederholt – wie ein Endlosband oder ein Garnknäuel.
- Diese Signale bleiben exakt an der Stelle im Gewebe, wo die RNA war.
- Mit fluoreszenzmarkierten Bausteinen und Sequenzieren durch Synthese wird die Sequenz direkt im Gewebe entschlüsselt.
Jede Padlock-Sonde trägt einen Barcode, der verrät, welches Gen oder welche Variante sie erkannt hat. Das Ergebnis: eine präzise Karte, die zeigt, welche RNA an welchem Ort vorhanden war und welche Varianten sie trug.
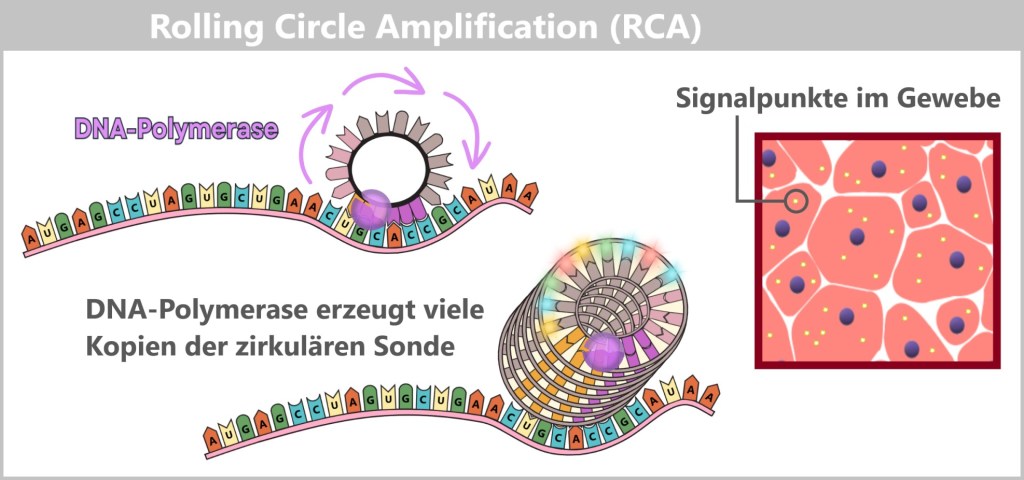
Eine spezielle DNA-Polymerase liest die zirkuläre Padlock-Sonde mehrfach ab und erzeugt einen langen DNA-Strang mit vielen Wiederholungen. Fluoreszente Nukleotide markieren die Amplifikate. Dadurch entsteht ein starkes, fluoreszierendes Signal – sichtbar als leuchtender Punkt im Gewebe, der die Position der gesuchten RNA markiert.
Proteinnachweis im selben Gewebeschnitt
Für den Nachweis von Proteinen nutzt das G4X-System Antikörper, die mit kurzen DNA-Barcodes versehen sind. Nachdem der Antikörper an sein Zielprotein gebunden hat, hybridisiert eine Padlock-Sonde an diesen Barcode. Sie wird geschlossen, amplifiziert und ausgelesen – ganz wie bei der RNA-Analyse. Der Barcode verrät eindeutig, um welches Protein es sich handelt und an welcher Stelle im Gewebe es vorkommt.
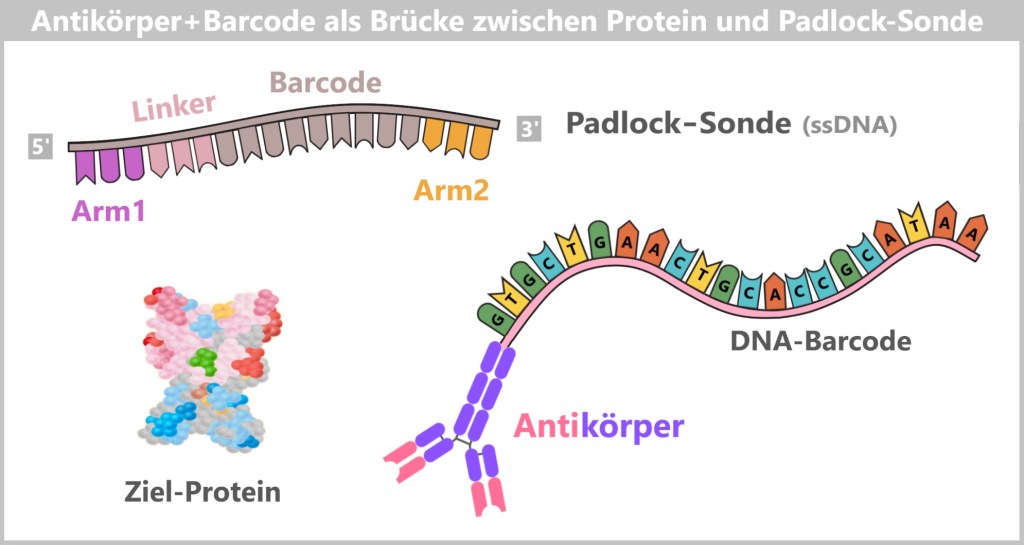
Ein Antikörper, der spezifisch für ein Zielprotein ist, wird mit einem einzigartigen, kurzen einzelsträngigen DNA-Strang (Barcode) gekoppelt.

Der Antikörper bindet in der Gewebeprobe an sein Zielprotein. Eine Padlock-Sonde, deren Enden komplementär zu zwei benachbarten Abschnitten des DNA-Barcodes sind, bindet an diesen und wird durch eine DNA-Ligase zu einem geschlossenen Ring verknüpft. Eine DNA-Polymerase erkennt den Ring und liest ihn fortlaufend ab, wodurch ein lokal verankertes, zirkuläres DNA-Amplifikat entsteht – analog zum RNA-Nachweis. Durch den Einbau fluoreszenzmarkierter Nukleotide wird dieses Amplifikat sichtbar. Die leuchtenden Signalpunkte im Gewebe markieren die Positionen, an denen ein Antikörper mit einem spezifischen DNA-Barcode ein bestimmtes Protein gebunden hat.
Eine ausführliche Präsentation des G4X-Veerfahrens findet man hier und hier.
Warum das wichtig ist
Der G4X kann gezielt Biomarker nachweisen, die Aufschluss über das Vorliegen, den Verlauf oder die Aggressivität einer Erkrankung geben – und zeigt dabei nicht nur, welche aktiv sind, sondern auch wo sie im Gewebe auftreten.
Gerade bei Tumoren macht diese räumliche Auflösung sichtbar, welche Gene „angeschaltet“ sind, in welchen Zellbereichen das geschieht und wie Tumor- und Immunzellen interagieren. So lassen sich Tumorheterogenität besser verstehen, Krankheitsmechanismen aufklären und Therapien gezielter planen.
Die gleichzeitige in-situ Erfassung von RNA und Proteinen liefert ein umfassendes Bild der Gewebeorganisation und eröffnet neue Möglichkeiten für:
- das Verständnis komplexer Krankheitsprozesse
- die präzisere Identifikation von Biomarkern
- die Entwicklung personalisierter Therapien
Bei Erkrankungen wie Krebs, chronischen Entzündungen oder neurodegenerativen Störungen kann diese räumliche Multiomics-Perspektive entscheidend sein – denn oft bestimmt nicht das Ob, sondern das Wo und Wie die beste therapeutische Maßnahme.
Fazit
Die Genomsequenzierung steht vor einem Quantensprung: Technologien wie FENT, SBX und G4X versprechen, das Erbgut schneller, präziser und vielseitiger zu lesen als je zuvor – bis hin zur direkten Analyse im Gewebe. Bald könnten komplette Genome in Minuten entschlüsselt werden. Die wahre Revolution beginnt jedoch danach: wenn wir die Fülle an genetischen Informationen verstehen, in medizinisches Wissen übersetzen und verantwortungsvoll zum Wohl der Menschen einsetzen.
4.6. Vom Code zur Heilung
Bioinformatik als Schlüssel der Medizin von morgen
4.6.1. Vom genetischen Code zur computergestützten Genomanalyse
Noch nie war es so leicht, DNA zu entschlüsseln – doch was nützt der Text, wenn niemand ihn versteht? Ein kleiner Abstrich, ein Tropfen Blut – und schon liegen Millionen genetischer Datenpunkte vor. Die moderne Sequenzierung macht das Erbgut so zugänglich wie nie zuvor: schnell, kostengünstig und nahezu fehlerfrei. Daraus erwächst jedoch eine neue Herausforderung: Wie verwandelt man diese Datenflut in verwertbares medizinisches Wissen?
Die Antwort kommt aus einer Disziplin, die lange im Hintergrund agierte und heute unverzichtbar geworden ist: Bioinformatik – die Brücke zwischen A, T, G, C und der Diagnose, zwischen rohen Sequenzdaten und einer konkreten Therapieentscheidung. Um zu verstehen, warum sie so zentral ist, lohnt ein kurzer Blick zurück.
Vom Erbgesetz zum genetischen Code
Bereits 1860 entdeckte Gregor Mendel, dass es „vererbbare Einheiten“ geben muss – Gene, lange bevor DNA bekannt war. Erst Mitte des 20. Jahrhunderts zeigte Oswald Avery, dass diese Informationsträger tatsächlich DNA sind. Kurz darauf enthüllten Watson und Crick die Doppelhelix-Struktur mit ihrer komplementären Basenpaarung (A–T, G–C). 1958 formulierte Francis Crick das „Zentrale Dogma“: Information fließt von DNA über RNA zu Proteinen – den funktionellen Bausteinen des Lebens.
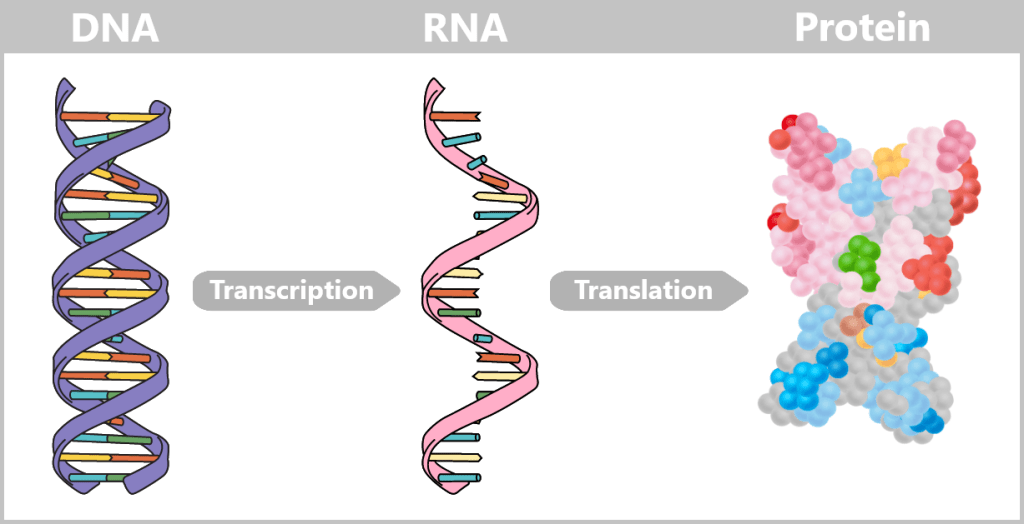
Die genetische Information wird durch den Prozess der Transkription von der DNA in RNA umgewandelt. Die RNA wird dann in das endgültige Protein übersetzt, das eine Vielzahl von Funktionen hat. [An overview of artificial intelligence in the field of genomics]
Ein Gen ist also eine Anleitung, wie ein Protein hergestellt wird. Doch wie liest man diese Anleitung – und wie interpretiert man sie richtig? Die Antwort kam zunächst über den Umweg der Reverse Genetik.
In den 1960er Jahren war DNA noch schwer direkt zugänglich – Proteine dagegen waren funktional sichtbar (etwa als Enzyme, Transportmoleküle oder Strukturbausteine) und konnten mit den damaligen Methoden isoliert und analysiert werden. Forscher wussten bereits seit Anfang des 20. Jahrhunderts, dass Proteine aus Aminosäureketten bestehen. Ab den 1960er Jahren wurde klar, dass jede Aminosäure einem sogenannten Codon auf der RNA entspricht – also einer Dreierkombination aus RNA-Basen (A, U, G, C), die wiederum auf eine DNA-Sequenz zurückführbar ist.
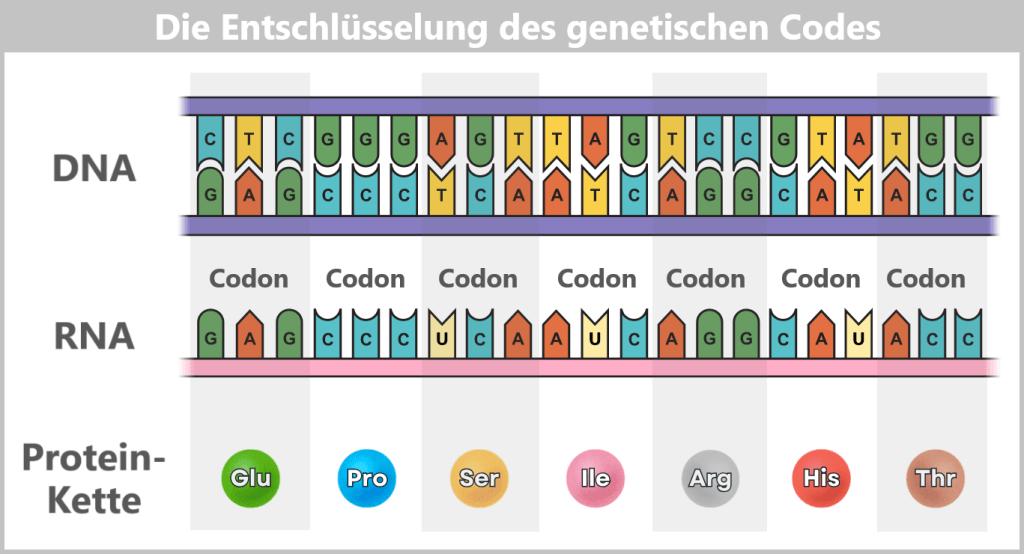
Forscher starteten bei einem bekannten Protein und arbeiteten sich zur RNA- und DNA-Sequenz zurück. Der genetische Code wurde zum ersten Mal wie ein Rezeptbuch lesbar:
Protein = fertiges Gericht
Aminosäure = Zutat
Codon = Wort für diese Zutat im handgeschriebenen Rezept (RNA)
DNA = Seite im Kochbuch, von der das Rezept abgeschrieben wurde
Erste Muster und die Geburt der Genomannotation
Noch vor der eigentlichen DNA-Sequenzierung entdeckte man typische Strukturelemente in Genen:
Promotoren – Startsignal, an dem das „Lesen“ eines Gens beginnt.
Start- und Stopcodons – erstes und letztes Signal im Rezept.
Transkriptionsfaktoren – Proteine, die Gene ein- oder ausschalten.
Codierende Sequenzen – Zutatenliste (Codons) und deren Reihenfolge für das Protein.
Nicht-codierende DNA – Abschnitte ohne direkte Protein-Codierung, deren Funktion zunächst unklar war.
Diese Erkenntnisse legten den Grundstein der heutigen Genomannotation – der computergestützten Erkennung biologischer Signale in DNA-Sequenzen.
Der Sanger-Moment: DNA wird lesbar (ab 1977)
Mit Frederick Sangers Methode konnten DNA-Sequenzen erstmals direkt bestimmt werden – zunächst nur für kurze Abschnitte. Die Analyse konzentrierte sich auf die Identifikation von Genen und einfache Vergleiche. Erste Programme wie FASTA (1985) und BLAST (1990) ermöglichten den Abgleich neuer Sequenzen mit Datenbanken wie GenBank (1982).
Auch das Verständnis der Genstruktur entwickelte sich weiter: Man entdeckte, dass nur bestimmte Teile eines Gens (Exons) für die Proteinsynthese genutzt werden, während andere Abschnitte (Introns) aus der transkribierten RNA herausgeschnitten werden. Zudem identifizierte man DNA-Abschnitte, die die Genexpression verstärken (Enhancer) oder abschwächen (Silencer) – oft weit entfernt vom eigentlichen Gen.
Der Prozess der Genomannotation begann sich zu entwickeln, war jedoch noch rudimentär: DNA wurde hauptsächlich als lineare Abfolge von Nukleotiden betrachtet, mit Fokus auf proteinkodierende Gene. Regulatorische Elemente und nicht-codierende DNA wurden erst ansatzweise erkannt und verstanden. Die Bioinformatik war geboren – zunächst als Werkzeug für Forschungslabore.
Das Humangenomprojekt: Daten im Gigantenmaßstab (1990–2004)
Das Ziel, alle 3,2 Milliarden Basen des menschlichen Genoms zu entschlüsseln, brachte völlig neue Probleme: Millionen kurzer Sequenzfragmente mussten zu langen DNA-Strängen zusammengesetzt werden. Tools wie Phrap für die Assemblierung und später Ensembl für die Annotation wurden entwickelt. Die Datenmengen explodierten – und mit ihnen die Notwendigkeit leistungsfähiger Rechnerinfrastruktur.
Doch während das Humangenomprojekt noch lief, zeichnete sich bereits eine neue Technologie ab, die Geschwindigkeit und Datenmenge in völlig neue Dimensionen katapultieren sollte.
NGS-Revolution und klinischer Durchbruch (2005–2015)
Die Next-Generation Sequencing (NGS)-Technologien machten Genome günstiger und schneller zugänglich – allerdings als gewaltige Datenlawinen aus Milliarden kurzer Fragmente. Neue Werkzeuge wie Bowtie, BWA oder STAR ordneten diese blitzschnell zu, GATK half, Mutationen zu erkennen, und Tools wie SnpEff oder VEP interpretierten deren Bedeutung.
Gleichzeitig entstanden Sequenzierstrategien für unterschiedliche klinische Fragen:
- Whole Genome Sequencing (WGS) für vollständige Analysen,
- Whole Exome Sequencing (WES) für die 1–2 % proteinkodierender DNA,
- Targeted Sequencing für definierte Genpanels.
Damit wurde die Bioinformatik erstmal zu einem Diagnosetool – etwa bei erblichen Krebsformen, seltenen Erkrankungen oder personalisierter Pharmakotherapie.
Künstliche Intelligenz und moderne Bioinformatik (2015-heute)
Die Verbindung von Künstlicher Intelligenz (KI) und Bioinformatik stellt einen der dynamischsten und vielversprechendsten Forschungsbereiche unserer Zeit dar. Gemeinsam entschlüsseln sie die Komplexität des Lebens und beschleunigen biomedizinische Entdeckungen in nie dagewesenem Tempo.
Künstliche Intelligenz: Mehr als nur Automatisierung
KI umfasst Techniken, bei denen Maschinen menschenähnliche Intelligenzleistungen erbringen – insbesondere Lernen, Problemlösen und Mustererkennung. Im biomedizinischen Kontext sind vor allem folgende Teilbereiche relevant:
Maschinelles Lernen (ML): Algorithmen lernen aus Daten (z.B. Genomsequenzen, Proteinstrukturen, Patientendaten), um Vorhersagen zu treffen oder Muster zu erkennen, ohne explizit programmiert zu sein. Beispiel: Erkennung von Tumorzellen in Gewebeproben.
Deep Learning (DL): Eine Untergruppe des ML, die künstliche neuronale Netze mit vielen Schichten („deep“) nutzt, um hochkomplexe Muster in riesigen Datenmengen zu erfassen. Beispiel: Vorhersage der 3D-Struktur von Proteinen allein aus ihrer Aminosäuresequenz (AlphaFold).
Natürliche Sprachverarbeitung (NLP): Ermöglicht Computern, wissenschaftliche Literatur, Krankenakten oder klinische Studienberichte zu verstehen und daraus Wissen zu extrahieren.
4.6.2. Moderne Bioinformatik – Der digitale Werkzeugkasten der Biologie
Bioinformatik ist die Wissenschaft der Speicherung, Analyse und Interpretation biologischer Daten mit computergestützten Methoden. Ihre moderne Ausprägung ist durch drei Faktoren geprägt:
Explosion biologischer Daten: Next-Generation Sequencing (NGS) generiert Terabytes an Genom-, Transkriptom- und Epigenomdaten pro Experiment.

– wie die Kosten der Genomsequenzierung fielen und die Datenmengen explodierten
Vor gut zwanzig Jahren war das Entschlüsseln eines menschlichen Genoms noch ein gigantisches Unterfangen: Das Human Genome Project (2003) kostete rund 3 Milliarden US-Dollar und lieferte nur etwa 1 Gigabyte an Daten. Mit immer schnelleren Sequenziermaschinen und neuen Technologien wie Nanoporen-Sequenzierung hat sich das Bild völlig verändert. 2012 lag der Preis pro Genom bereits bei rund 6000 Dollar, 2018 bei 1000 Dollar – und heute (2023) kann man ein Genom für etwa 100 Dollar analysieren.
Parallel dazu stieg das erzeugte Datenvolumen rasant: von wenigen Gigabyte über Terabyte und Petabyte bis hin zu den hunderten Petabytes, die in weltweiten Projekten mit Millionen von Genomen entstehen. Schätzungen zufolge könnten bis 2025 rund 40 Exabyte an menschlichen Genomdaten anfallen – das entspricht etwa dem achtfachen Speicherbedarf aller jemals gesprochenen Worte der Menschheitsgeschichte. [How AI Is Transforming Genomics]
Komplexität biologischer Systeme: Das Verständnis von Krankheiten erfordert die Integration von Daten über Gene, Proteine, Stoffwechselwege und Zellinteraktionen.
Bedarf an prädiktiver Medizin: Ziel ist die Vorhersage von Krankheitsrisiken, Therapieansprechen und individueller Behandlung (Präzisionsmedizin).
Bereits bei vielen Schlüsselanwendungen kann man mittlerweile von einer Verschmelzung zwischen Bioinformatik und Künstlicher Intelligenz (KI) sprechen. Hier sind einige zentrale Anwendungsgebiete:
a) Genomanalyse und Sequenzierung
KI-Algorithmen werden genutzt, um DNA-Sequenzen schneller und präziser zu analysieren. Modelle wie DeepVariant, entwickelt von Google, nutzen tiefe neuronale Netze, um genetische Varianten mit hoher Genauigkeit zu identifizieren. Dies ist entscheidend für die Erkennung von Mutationen, die mit Krankheiten wie Krebs oder seltenen genetischen Störungen assoziiert sind.
b) Proteinfaltung und Strukturvorhersage
Ein Meilenstein der KI in der Bioinformatik ist AlphaFold von DeepMind, dass die Struktur von Proteinen mit beispielloser Präzision vorhersagt. Dies hat weitreichende Implikationen für die Arzneimittelentwicklung, da die 3D-Struktur eines Proteins seine Funktion bestimmt.
c) Multi-Omics Datenanalyse
Das ganzheitliche Verständnis biologischer Systeme erfordert die Integration von Multi-Omics-Daten (Genomik, Proteomik, Transkriptomik, Epigenetik, Metabolomik), um umfassendes Bild ihrer Funktionsweise zu gewinnen. KI hilft, die komplexen Wechselwirkungen zwischen diesen Ebenen zu entschlüsseln.
Biologische Systeme sind ein komplexes Zusammenspiel verschiedener Informationsebenen: Von der DNA (Genomik) über epigenetische Steuerung, RNA (Transkriptomik/Epitranskriptomik) und Proteine (Proteomik) bis hin zu Stoffwechselprodukten (Metabolomik). Erst ihr dynamisches Wechselspiel definiert Gesundheit und Krankheit.
Beispielsweise zeigt das neue KI-Tool AlphaGenome, wie selbst kleinste Veränderungen in der DNA die Genaktivität sowie die Produktion von RNA und Proteinen beeinflussen können. Es sagt präzise voraus, welche Folgen der Austausch eines einzelnen DNA-Bausteins für Gene und ihre Produkte hat. Während frühere Systeme meist nur die rund 2 % des Genoms analysierten, die Proteine codieren, erfasst AlphaGenome erstmals das gesamte Genom. Grundlage ist eine Multi-Omics-Analyse, die DNA-Veränderungen direkt mit ihren Auswirkungen auf RNA und Proteine verknüpft. Damit entsteht ein umfassendes Bild der Genexpression – ein entscheidender Schritt hin zu präziserer Genforschung und personalisierter Medizin.
d) Metagenomik und Mikrobiomanalyse
KI hilft, die komplexen Wechselwirkungen im menschlichen Mikrobiom zu verstehen, indem sie Muster in den Daten von Mikroorganismen erkennt. Dies ist wichtig für die Erforschung von Krankheiten wie Diabetes oder Darmerkrankungen.
e) Arzneimittelentwicklung
KI beschleunigt nahezu jede Phase des langwierigen und teuren Drug-Discovery-Prozesses: Zielidentifikation, virtuelles Screening von Millionen von Molekülen, Vorhersage von Wirkstoffeffekten und Nebenwirkungen, Optimierung von Molekülstrukturen. Unternehmen wie BenevolentAI oder Recursion nutzen KI-Plattformen, um vielversprechende Wirkstoffkandidaten in Rekordzeit zu identifizieren, die bereits in klinischen Studien getestet werden.
f) Analyse von biomedizinischen Bildern
KI wird in der Bildanalyse eingesetzt, etwa bei der Auswertung von MRT- oder CT-Scans, um Tumore oder andere Anomalien zu erkennen. Convolutional Neural Networks (CNNs), die die Funktionsweise des menschlichen visuellen Kortex nachahmen, haben sich hier als besonders effektiv erwiesen.
g) KI-Genomik in der Gesundheitsforschung
Die Genexpression im menschlichen Körper folgt keinem festen Muster – und Krankheiten wie Krebs machen sie noch schwerer durchschaubar. Genau hier setzt die räumliche Biologie an: Mit modernster Mikroskopie und genetischer Sequenzierung zeigt sie, wie Gene in einzelnen Zellen und sogar in ihren Organellen aktiv sind.
Doch nicht nur die sichtbaren Genabschnitte sind entscheidend. In den lange als „Junk-DNA“ abgetanen Regionen vermuten Forscher heute zentrale Schalter der Genregulation. Dort entscheidet sich, welche Proteine produziert werden – und welche nicht. KI und Deep-Learning-Modelle können in diesen bislang verborgenen Bereichen Muster erkennen, die mit Krankheiten verknüpft sind. So erhofft man sich neue Biomarker und Therapieansätze. Ein Beispiel ist die Plattform des Biotech-Unternehmens Deep Genomics:
Die Kombination aus räumlicher Biologie, intelligenter Datenanalyse und KI ermöglicht damit einen bislang unerreichten Blick in die molekulare Logik des Lebens – bis auf die Ebene einzelner Zellen.
Neue Methoden der Genbearbeitung könnten das Verhalten einzelner Gene gezielt steuern – etwa um krebsfördernde Gene zu deaktivieren oder die Regeneration von Knorpelzellen bei Arthrose zu fördern. Noch ist dieses Feld stark experimentell, doch KI verspricht, die Verfahren präziser und sicherer zu machen.
Mit KI-gestützten CRISPR-Techniken eröffnen sich neue Möglichkeiten, Gene nicht nur gezielter zu identifizieren und zu verändern, sondern auch deren Zusammenspiel in komplexen biologischen Netzwerken besser zu verstehen. Erste Ansätze deuten darauf hin, dass sich dadurch Nebenwirkungen künftig verringern und die Wirksamkeit neuer Therapien steigern ließe. Auch moderne Verfahren zum systematischen Testen und Optimieren von Genvarianten könnten – sofern sie sich bewähren – wichtige Schritte in Richtung maßgeschneiderter Behandlungen darstellen.
Wer sich dafür interessiert, mehr über CRISPR-Techniken zu erfahren, findet hier weiterführende Informationen.
Die Kombination von Fortschritten in KI und Genomik gilt daher als vielversprechend und könnte langfristig die Entwicklung neuer Zell- und Gentherapien beschleunigen. Da diese Behandlungen jedoch auf lebenden Zellen beruhen, die stark variieren und spezielle Bedingungen erfordern, befindet sich vieles noch in der Erprobung. KI könnte hier helfen, Prozesse stabiler, reproduzierbarer und klinisch besser anwendbar zu machen – doch ob und in welchem Umfang sich diese Hoffnungen erfüllen, bleibt abzuwarten.
h) Onkologie
KI-Modelle analysieren genetische Profile, um personalisierte Behandlungspläne zu erstellen. Beispielsweise können Algorithmen Vorhersagen treffen, wie Patienten auf bestimmte Medikamente reagieren, basierend auf ihrem genetischen Make-up. Dies ist besonders in der Onkologie relevant, wo KI-basierte Systeme wie IBM Watson Oncology Behandlungsempfehlungen geben.
Fazit
Bioinformatik ist heute das Gehirn der modernen Medizin. Gemeinsam mit Künstlicher Intelligenz verwandelt sie Datenfluten aus dem Erbgut in handfeste Erkenntnisse – von der Diagnose seltener Krankheiten bis zur Entwicklung neuer Medikamente.
Ob bei der Analyse von Genomen, der Vorhersage von Proteinstrukturen oder der Planung personalisierter Therapien: KI-gestützte Bioinformatik macht sichtbar, was bislang verborgen blieb. Sie eröffnet den Weg zu einer Medizin, die nicht nur Krankheiten behandelt, sondern sie voraussieht – maßgeschneidert für jeden einzelnen Patienten.

5. Eine Erfolgsgeschichte
Wie sehr diese Entwicklungen schon heute Realität sind, zeigt der Fall von KJ Muldoon.
Als der kleine Junge wenige Stunden nach seiner Geburt die Diagnose erhielt, schien sein Schicksal besiegelt. Er leidet an einem extrem seltenen Stoffwechseldefekt, der durch eine einzelne DNA-Punktmutation (SNV) verursacht wird und dazu führt, dass sein Körper giftiges Ammoniak nicht abbauen kann. Ohne funktionierendes Enzym sammeln sich toxische Stoffwechselprodukte rasch an – lebensbedrohlich schon im Säuglingsalter. Bisher galt: Nur eine riskante Lebertransplantation konnte das Überleben sichern.
Doch in KJs Fall wagten Ärzte und Forscher einen neuen Weg. Innerhalb von nur sechs Monaten entwarfen sie eine Therapie, die ausschließlich für ihn bestimmt war: eine maßgeschneiderte Base-Editing-Behandlung.
Das Prinzip dahinter ist ebenso bestechend wie elegant: Ein RNA-Suchsystem tastet das Erbgut ab, findet gezielt die Stelle mit KJs Punktmutation und bindet dort. Anstatt den DNA-Strang komplett zu durchtrennen – wie es beim klassischen CRISPR-Cas9 der Fall wäre – wird mithilfe eines Enzyms eine einzelne Base chemisch verändert: der Austausch eines DNA-Buchstabens, der die krankheitsverursachende Mutation korrigiert. Anschaulich gesagt: Es ist, als würde man in einem Buch einen einzigen Tippfehler berichtigen, ohne den ganzen Absatz aufzuschneiden oder neu zu schreiben – äußerst präzise und schonend für das Erbgut. Die Behandlung erfolgte direkt im Körper („in vivo“), über mehrere aufeinanderfolgende Behandlungsrunden, die gezielt die Leber erreichten.
Die ersten Ergebnisse sind ermutigend. KJ kann inzwischen mehr Proteine verdauen, Infekte verlaufen milder, er nimmt an Gewicht zu. Für seine Eltern bedeutet das: ein Stück Alltag ohne ständige Lebensgefahr – und vor allem eine neue Hoffnung auf die Zukunft ihres Kindes.
Hinter diesem medizinischen Durchbruch steckt eine Erfolgsgeschichte gleich mehrerer Disziplinen. Ohne moderne Genomsequenzierung wäre die exakte Mutation nicht sichtbar geworden. Ohne Bioinformatik ließe sich das passende Base-Editing-Werkzeug nicht in so kurzer Zeit und mit dieser Präzision entwerfen. Und ohne das Konzept der personalisierten Medizin wäre es undenkbar gewesen, ein Heilmittel ausschließlich für einen einzigen Patienten zu entwickeln.
Noch ist die Therapie experimentell, und niemand weiß, ob die Wirkung dauerhaft anhält oder ob langfristige Spätfolgen auftreten. Doch der Fall von KJ zeigt, wohin die Reise geht: zu einer Medizin, die nicht mehr auf Standardschemata setzt, sondern auf individuelle Lösungen – gestützt auf Daten, Molekularbiologie und maßgeschneiderte Therapien. Eine Medizin, die Krankheiten nicht nur behandelt, sondern sie im Idealfall gezielt an ihrer genetischen Wurzel packt.
Weitere Hintergründe zu dieser Geschichte gibt es in diesem Video.
6. Ein Blick in die Zukunft: Das Synthetic Human Genome Project
Die Biologie steht vor einem nächsten Quantensprung. Nach dem Entziffern und Editieren von Genomen kommt nun die Vision, sie von Grund auf neu zu schreiben. Genau das ist das Ziel des Synthetic Human Genome Project (SynHG) – ein ambitioniertes Vorhaben, das die Grenzen von Biologie und Medizin neu ausloten könnte.
Mit einer Anschubfinanzierung von £10 Millionen durch den Wellcome Trust und unter der Leitung von Professor Jason Chin am Generative Biology Institute in Oxford arbeiten führende Universitäten wie Cambridge, Manchester und das Imperial College London an diesem Projekt. Die Mission: Werkzeuge und Technologien entwickeln, um synthetische menschliche Genome herzustellen – ein Schritt, der unser Verständnis des Lebens selbst verändern könnte.
Ein Genom neu komponieren
Im Unterschied zur Genom-Editierung, die bestehende DNA nur anpasst, geht SynHG deutlich weiter: Es will das Genom Buchstabe für Buchstabe neu zusammensetzen. Während CRISPR einzelne Tippfehler im Text des Lebens korrigiert, geht es hier um die komplette Neuschreibung eines Kapitels.
Der erste Meilenstein: die Synthese eines vollständigen menschlichen Chromosoms als Machbarkeitsnachweis. Es ist der Anfang eines Weges, der Jahrzehnte dauern könnte – und doch an frühere Pionierleistungen anknüpft, wie das Humangenomprojekt (2003) oder das Synthetic Yeast Project, bei dem 2023 erstmals synthetische Hefe-Chromosomen entstanden.
Moderne Methoden – generative KI, maschinelles Lernen und robotergestützte Assemblierung – sollen die gewaltige Aufgabe meistern.
Mögliche Anwendungen – zwischen Laborvision und Klinikalltag
Die Chancen sind groß. Auf Ebene somatischer Zellen – also solcher, deren Veränderungen nicht an Nachkommen weitergegeben werden – könnten sich neue Wege für die regenerative Medizin eröffnen:
- maßgeschneiderte Ersatzgewebe und Organe aus dem Labor,
- virusresistente Zellen gegen HIV oder SARS-CoV-2,
- Gentherapien gegen Krebs oder Erbkrankheiten wie Mukoviszidose.
Solche Anwendungen sind keine ferne Science-Fiction – sie könnten in den nächsten 10 bis 20 Jahren Realität werden, da Technologien wie CRISPR und Stammzellkulturen bereits etabliert sind.
Ganz anders sieht es bei der Keimbahn-Editierung aus – also Eingriffen in Ei- oder Samenzellen. Hier ließen sich theoretisch schwere Erbkrankheiten wie Huntington dauerhaft verhindern. Vorstellungen von „Designer-Babys“ mit gewünschten Eigenschaften wie Aussehen oder Intelligenz bleiben jedoch wissenschaftlich und ethisch hoch problematisch, da solche Merkmale von Hunderten Genen abhängen.
Das Projekt selbst beschränkt sich auf Experimente in Reagenzgläsern und Zellkulturen. Es zielt nicht darauf ab, künstliches Leben zu erschaffen. Dennoch eröffnet es Forschenden eine beispiellose Möglichkeit, in grundlegende menschliche Lebenssysteme einzugreifen – mit weitreichenden Konsequenzen, die kritisch hinterfragt werden müssen.
Risiken und offene Fragen
Die technischen Hürden sind gewaltig. Bei somatischen Zellen drohen Mosaizismus (nicht alle Zellen übernehmen das neue Genom) oder Tumorbildung durch unbeabsichtigte Genveränderungen. In der Keimbahn sind die Risiken noch größer: Entwicklungsstörungen, epigenetische Effekte oder unvorhersehbare Folgen über Generationen hinweg.
Forscher diskutieren deshalb Sicherheitsmechanismen wie genetische „Kill-Switches“, mit denen synthetische Zellen im Ernstfall abgeschaltet werden könnten.
Ethik: Zwischen Heilung und Hybris
Somatische Therapien erinnern an Organtransplantationen: umstritten, aber gesellschaftlich akzeptiert, wenn sie heilen. Keimbahn-Eingriffe dagegen berühren Tabuzonen – aus Angst vor sozialer Ungleichheit oder gar eugenischen Szenarien. Viele Länder, darunter Deutschland, verbieten sie durch Gesetze wie das Embryonenschutzgesetz. Internationale Organisationen wie WHO und UNESCO fordern strikte Regulierung.
„Um Vertrauen zu schaffen, müssen wir die Gesellschaft aktiv einbeziehen“, betont die Soziologin Professorin Joy Zhang von der University of Kent, die die ethischen Dimensionen des Projekts untersucht. Mit ihrer Initiative Care-full Synthesis versucht sie, einen offenen Dialog über Chancen, aber auch über die Grenzen dieser Forschung anzustoßen.
Der renommierte Genetiker Professor Bill Earnshaw von der Universität Edinburgh, der Verfahren zur Herstellung künstlicher menschlicher Chromosomen entwickelt hat, formuliert es drastischer: „Der Geist ist aus der Flasche. Selbst wenn wir jetzt Regeln und Beschränkungen festlegen – jede Organisation mit den nötigen Maschinen könnte im Prinzip synthetisieren, was sie will. Aufhalten ließe sich das kaum.“
In diesem Licht wirkt die Forderung zahlreicher Fachorganisationen – darunter die International Society for Cell and Gene Therapy (ISCT) – nach einem zehnjährigen Moratorium für den Einsatz von CRISPR und verwandten Verfahren in der menschlichen vererbbaren Keimbahn noch dringlicher. Ein solcher Aufschub erscheint nicht nur als Vorsichtsmaßnahme, sondern als notwendige politische Reaktion, um gesellschaftliche Debatten, ethische Leitplanken und internationale Kontrollmechanismen überhaupt erst zu ermöglichen.
Der Ethik-Experte Arthur Caplan von der New York University beschreibt das Dilemma aus wissenschaftlicher Sicht:
„… Wenn Sie mich also fragen, werden wir eine Genmanipulation von Kindern sehen, die auf ihre Verbesserung abzielt? Ich sage ja, ohne Zweifel. Wann? Ich bin mir nicht sicher, was die Antwort darauf ist.“
„… Es wird kommen. Es gibt Eigenschaften, die die Menschen in Zukunft eifrig versuchen werden, ihren Kindern beizubringen. Sie werden versuchen, genetische Krankheiten zu entschlüsseln, sie loszuwerden. Sie werden versuchen, Kapazitäten und Fähigkeiten aufzubauen, von denen sie zustimmen, dass sie wirklich wunderbar sind. Werden wir diese Interventionen aus ethischen Gründen an den Nagel hängen? Größtenteils nein, wäre meine Vorhersage, aber nicht innerhalb der nächsten 10 Jahre. Die Werkzeuge sind noch zu grob.“
Ausblick
SynHG öffnet ein Fenster in eine Zukunft, in der wir das Genom nicht nur lesen und editieren, sondern schreiben können. Die somatischen Anwendungen – personalisierte Therapien, regeneratives Gewebe, maßgeschneiderte Zellen – könnten schon bald die Medizin prägen.
Eingriffe in die Keimbahn dagegen bleiben vorerst ein ferner Horizont – technisch, rechtlich und ethisch. Das Projekt stellt nicht nur eine wissenschaftliche, sondern auch eine gesellschaftliche Gratwanderung dar.
Vor dem Hintergrund des möglichen Missbrauchs der Technologie stellt sich die Frage, weshalb Wellcome überhaupt die Finanzierung entschied. Dr. Tom Collins, der diesen Schritt verantwortete, erklärte gegenüber BBC News:
„Wir haben uns gefragt, was die Kosten der Untätigkeit wären. Diese Technologie wird eines Tages entwickelt werden. Indem wir sie jetzt einsetzen, versuchen wir zumindest, so verantwortungsvoll wie möglich vorzugehen und uns den ethischen und moralischen Fragen so offen wie möglich zu stellen.“
Während man in Europa noch darum ringt, das Thema in der öffentlichen Debatte behutsam auszubalancieren, geht man auf der anderen Seite des Atlantiks deutlich unbefangener mit dem Begriff „neue Lebensformen“ um. Eric Nguyen, Biowissenschaftler an der renommierten Stanford University, skizziert in seinem TED-Talk „How AI Could Generate New Life-Forms“ eine weit visionärere Perspektive.
Auf eindrückliche Weise zeigt er, wie künstliche Intelligenz (KI) die Biologie transformieren könnte – nicht nur durch das „Lesen“ und „Schreiben“ von DNA, sondern auch durch die gezielte Erschaffung neuartiger Organismen. KI-Modelle wie maschinelles Lernen und generative Algorithmen sind bereits heute in der Lage, komplexe Muster in biologischen Daten zu erkennen: in Genomen, Proteinstrukturen oder Stoffwechselwegen. Nguyen geht jedoch weiter und argumentiert, dass KI künftig auch völlig neue DNA-Sequenzen entwerfen könnte – der Ausgangspunkt bislang unbekannter Lebensformen.
Diese Vision ist zweifellos zukunftsweisend, wirft aber grundlegende Fragen auf: Welche Prioritäten sollen gesetzt werden? Wie lässt sich der Einsatz solcher Technologien in einer Welt mit massiver Ressourcenungleichheit verantworten? Wer finanziert die Forschung, wer kontrolliert die Ergebnisse – und nach welchen Kriterien wird entschieden, welche Lebensformen „nützlich“ oder „sicher“ sind?
Nguyens Vortrag verdeutlicht: KI-gestützte Biotechnologie könnte nicht nur Lösungen für globale Herausforderungen ermöglichen, sondern zwingt uns zugleich, über die ethischen, sozialen und philosophischen Dimensionen der „Erzeugung von Leben“ nachzudenken.
Die Botschaft: KI-gestützte Biotechnologie verspricht enorme Fortschritte, wirft jedoch zugleich tiefgreifende ethische und gesellschaftliche Fragen auf – und stellt damit die Grundlagen unseres Menschseins zur Diskussion.
Klaus Schwab – Gründer und ehemaliger Vorsitzender des Weltwirtschaftsforums
„Wir müssen, sowohl individuell als auch kollektiv, die moralischen und ethischen Fragen angehen, die durch Spitzenforschung in den Bereichen künstliche Intelligenz und Biotechnologie aufgeworfen werden, die eine erhebliche Lebensverlängerung, Designerbabys und das Extrahieren von Erinnerungen ermöglichen wird.“
[BrainyQuote]
7. Personalisierte Medizin und Smart Governance
Die biomolekulare Revolution der Regierungsführung
7.1. Macht, Governance und Smart Governance
7.2. Gouvernementalität
7.3. Personalisierte Medizin als Katalysator biomolekularer Gouvernementalität
7.4. Der weltweite Trend: Globale biomolekulare Gouvernementalität
7.5. Schlussfolgerung: Globale Ambivalenz der biomolekularen Macht
Die Entschlüsselung des menschlichen Genoms markierte nicht nur einen medizinischen Wendepunkt, sondern auch den Beginn einer neuen Epoche politischer Steuerung. Was als wissenschaftliches Großprojekt in Laboren und Rechenzentren begann, ist längst in den Steuerungsapparaten moderner Staaten angekommen: die präzise Vermessung, Kategorisierung und Nutzung unserer biologischen Grundlagen.
Die Vision der personalisierten Medizin verspricht individuelle Therapien, präzise Vorhersagen über Krankheitsrisiken und eine effizientere Nutzung knapper Ressourcen. Doch diese Fortschrittserzählung greift zu kurz, wenn wir sie nur medizinisch, ethisch oder datenschutzrechtlich betrachten. Tatsächlich sind wir Zeugen einer grundlegenden Transformation von Macht und Gouvernementalität – einer biomolekularen Revolution der Regierungsführung.
Bevor wir tiefer einsteigen, gilt es einige zentrale Begriffe zu klären.
7.1. Macht, Governance und Smart Governance
Governance bezeichnet die Kunst, gesellschaftliche Prozesse zu steuern, Entscheidungen zu treffen und Ordnung zu sichern. Klassisch geschieht dies im Zusammenspiel von Staat, Märkten und Zivilgesellschaft.
Smart Governance transformiert dieses Verständnis: Sie operiert mit digitalen Technologien, Datenanalysen und algorithmischen Prozessen. Steuerung wird dadurch kontinuierlich, prozessual und in Echtzeit möglich. Macht zeigt sich hier nicht mehr nur als politische Autorität, sondern als Geflecht aus Informationsflüssen, Rückkopplungen und Prognosen.
Macht selbst ist kein Besitz, sondern eine Beziehung: die Fähigkeit, Verhalten zu beeinflussen, Handlungen zu lenken oder Entscheidungen zu rahmen. Sie wirkt sowohl sichtbar – über Institutionen und Gesetze – als auch subtil, indem sie Erwartungen, Normen und Wissensbestände formt.
7.2. Gouvernementalität
Michel Foucault, ein bedeutender Denker in Bezug auf die Analyse von Machtstrukturen, prägte den Begriff der Gouvernementalität, um die Gesamtheit der Wissensformen, Praktiken und Techniken zu beschreiben, durch die moderne Gesellschaften regiert werden. Regierung bedeutet hier nicht nur die Ausübung von Zwang, sondern auch die Gestaltung von Handlungsräumen, die Menschen dazu bringen, sich selbst im Sinne gesellschaftlicher Zielsetzungen zu steuern.
Zentrale Dimensionen der Gouvernementalität sind:
Verknüpfung von Wissen und Macht
Wissen ist nie neutral. Es entsteht durch die Sammlung und Deutung von Daten, ist in Machtprozesse eingebettet und strukturiert soziale Ordnungen. Mit der Digitalisierung wird diese Verbindung zentral: Big Data und Künstliche Intelligenz liefern Prognosen, Mustererkennungen und Entscheidungsgrundlagen, die tief in gesellschaftliche Prozesse eingreifen. Smart Governance setzt genau hier an: Daten werden zum Fundament politischer Legitimation und Machtausübung.
Biopolitik
Biopolitik bezeichnet die Regulierung des Lebens selbst – Gesundheit, Reproduktion, Arbeit, Bildung, Migration oder Sicherheit. Ziel ist die Ordnung ganzer Bevölkerungen durch Gesetze, Standards und politische Programme. Heute verbindet sich Biopolitik mit digitaler Steuerung: Smart Health, Smart Cities oder Smart Economy verdeutlichen, wie Wissen durch Daten in konkrete Regierungspraktiken umgesetzt wird.
Dispositive der Macht
Biopolitik wirkt über Dispositive: Netzwerke aus Institutionen, Gesetzen, Diskursen und Praktiken. Krankenhäuser, Schulen, Medien oder digitale Plattformen sind Teil dieser Strukturen. Sie normieren Verhalten nicht durch offenen Zwang, sondern durch subtile Rahmungen dessen, was als „normal“ oder „wünschenswert“ gilt. So werden Entscheidungen indirekt gelenkt und Verhalten in den Alltag eingebettet.
Disziplinarmacht
Neben der Biopolitik, die ganze Bevölkerungen reguliert, richtet sich Disziplinarmacht auf das Individuum. Sie wirkt über Überwachung, Belohnung, Strafe und Training. Ziel ist nicht nur Gehorsam, sondern die produktive Formung von Körpern und Verhaltensweisen. Externe Vorgaben verwandeln sich durch Sanktionen und Anreize in innere Motivationen, die Menschen zur Selbstdisziplin bewegen.
Technologien der Selbstführung
Technologien der Selbstführung bezeichnen Praktiken, durch die Individuen gesellschaftliche Normen verinnerlichen und ihr Verhalten eigenständig anpassen. Digitale Anwendungen – etwa Fitness-Apps, Lernplattformen oder Finanz-Tools – übersetzen gesellschaftliche Vorgaben in persönliche Ziele. So entsteht eine Machtform, die nicht nur von außen wirkt, sondern von innen weitergeführt wird: Menschen folgen Regeln, weil sie sie als sinnvoll und nützlich für sich selbst empfinden.
Wer sich näher für die Kunst des Regierens interessiert, findet hier weiterführende Informationen.
7.3. Personalisierte Medizin als Katalysator biomolekularer Gouvernementalität
Die personalisierte Medizin, gestützt auf moderne Genomik und Bioinformatik, erscheint auf den ersten Blick als rein medizinisch-technischer Fortschritt. Doch betrachtet man sie durch die Linse der Gouvernementalität, zeigt sie sich als mächtiger Katalysator für eine tiefgreifende Transformation von Machtstrukturen und Regierungspraktiken. Durch die präzise Analyse individueller genetischer Profile und deren Verknüpfung mit umfassenden Datenbanken wird nicht nur die Gesundheitsversorgung individualisiert, sondern auch die Art und Weise, wie Gesellschaften gesteuert und Individuen reguliert werden.
Wissen & Macht: Vom Genom zum regierbaren Körper
Die Entschlüsselung des Genoms war die initiale Wissensexplosion, doch erst die personalisierte Medizin macht dieses Wissen regierbar. Bioinformatik und moderne Genomik generieren nicht nur Daten, sondern schaffen prädiktives Wissen, das die Grundlage für Interventionen bildet. Dieses Wissen ist keineswegs neutral: Es kategorisiert Individuen anhand genetischer Risikoprofile (z. B. „hohes Risiko für Brustkrebs“, „prädestiniert für Herz-Kreislauf-Erkrankungen“). Diese Kategorisierung wird zum Fundament neuer Machtstrukturen. Staaten wie auch private Akteure nutzen diese Daten, um Krankheitsrisiken vorherzusagen, Gesundheitsbudgets zu optimieren und präventive Maßnahmen zu steuern. Die soziale Ordnung wird dadurch neu strukturiert – vom Zugang zu Versicherungen und Arbeitsmärkten bis hin zur Priorisierung präventiver Ressourcen.
Smart Governance manifestiert sich hier in algorithmischen Entscheidungsprozessen, die scheinbar objektiv, tatsächlich jedoch normativ sind. Wer Zugang zu welchen Therapien erhält oder welche Bevölkerungsgruppen als „risikobehaftet“ gelten, wird zunehmend durch datenbasierte Modelle bestimmt, die Machtverhältnisse unsichtbar reproduzieren („Wir handeln auf Basis der besten verfügbaren Evidenz“).
Die CMS Health Tech Ecosystem Initiative, die offiziell am 30. Juli 2025 während der Veranstaltung „Make Health Tech Great Again“ im Weißen Haus angekündigt wurde, verkörpert den Trend zur Herausbildung neuer Machtstrukturen. CMS (Centers for Medicare & Medicaid Services) ist eine US-Bundesbehörde im Department of Health and Human Services (HHS), die Gesundheitsprogramme wie Medicare und Medicaid verwaltet und den nationalen Gesundheitsmarkt reguliert. Ziel der Initiative ist es, eine intelligentere, sicherere und stärker personalisierte Gesundheitsversorgung zu schaffen, die durch Partnerschaften mit innovativen Unternehmen des privaten Sektors vorangetrieben wird. Während der Veranstaltung sicherte sich die US-Regierung Zusagen von großen Gesundheits- und IT-Unternehmen – darunter Amazon, Anthropic, Apple, Google und OpenAI –, um den Grundstein für ein digitales Gesundheitsökosystem der nächsten Generation zu legen.
Laut US-Präsident Donald Trump erhalten die US-Gesundheitssysteme dadurch ein High-Tech-Upgrade und treten ein in „eine Ära der Bequemlichkeit, Rentabilität, Schnelligkeit und – offen gesagt – einer besseren Gesundheit für die Menschen“. (Man beachte die Reihenfolge…)
Biopolitik 2.0: Die Optimierung der Bevölkerung auf molekularer Ebene
Die klassische Biopolitik regulierte Bevölkerungen durch Statistiken über Geburtenraten oder Sterblichkeit. Die personalisierte Medizin hebt dies auf eine neue, molekulare Stufe. Genomik fragmentiert die Bevölkerung in immer kleinere, biomolekular definierte Untergruppen. Die Bevölkerung wird nicht mehr nur nach Alter oder Geschlecht, sondern nach Genotyp, Risikoallelen und Biomarkern kategorisiert („die BRCA1-Träger“, „die Menschen mit LCT-Gen für Laktoseintoleranz“). Dies ermöglicht eine hyperpräzise und individualisierte Regulierung – Biopolitik wird granular.
Der „gesunde“, „normale“ Körper ist nicht länger allein derjenige ohne Symptome, sondern der, der ein „normales“, „risikoarmes“ Genom aufweist. Abweichung beginnt nicht mit Krankheit, sondern mit genetischer Prädisposition. Damit entsteht eine neue Klasse von „präsymptomatisch Kranken“ oder „Risikosubjekten“, die biopolitisch gemanagt werden: durch häufigere Screenings, Präventionsprogramme, Impfkampagnen, prophylaktische Operationen, Lebensstilempfehlungen oder reproduktive Entscheidungen, die auf genetischen Prognosen basieren.
Während Foucault den Nationalstaat als Hauptakteur der Biopolitik sah, verlagert sich die Macht heute zunehmend auf private Akteure: Big-Tech- und Biotech-Unternehmen, die Daten, Algorithmen und Technologien kontrollieren. Sie definieren de facto, was genetische „Normalität“ und „Risiko“ bedeutet.
Man spricht mittlerweile von einer Ökonomisierung des biologischen Lebens. Genetische Daten werden zu einer wertvollen Handelsware – einer Bio-Ressource. Große Technologie- und Pharmaunternehmen akkumulieren Biodaten, um daraus Gewinne zu generieren: in der Medikamentenentwicklung, bei Therapieempfehlungen, in der personalisierten Werbung. Der menschliche Körper und seine biomolekularen Daten werden so Teil kapitalistischer Verwertungslogiken. Das Individuum selbst wird zum „Humankapital“, das verpflichtet scheint, sein genetisches Potenzial zu optimieren.
In einem NBCNews-Beitrag vom 26. August 2025 erklärte der US-Gesundheitsminister RFK Jr., die 60 größten Technologieunternehmen würden den Amerikanern künftig den Zugriff auf ihre persönlichen Gesundheitsdaten gestatten – Daten, die jahrelang ohne ihre Zustimmung ökonomisch genutzt worden seien: „Sie werden ab nächstem Jahr alle Ihre Gesundheitsdaten auf Ihrem Mobiltelefon einsehen können.“
Was hier als politischer Erfolg statuiert wird, bestätigt nur den Status Quo.
Dispositive der Macht: Das Netzwerk der biomolekularen Überwachung
Das dispositive Netzwerk, das diese neue Form der Biopolitik trägt, ist vielschichtig: Pharmakonzerne entwickeln zielgenaue Therapien; Biobanken und genetische Datenbanken liefern das Rohmaterial; Algorithmen analysieren und interpretieren es; Ärzte vermitteln Ergebnisse und Empfehlungen; digitale Gesundheits-Apps überwachen die compliance. Jedes Glied in dieser Kette fungiert als Machtdispositiv: Es definiert, was als „gesund“ oder „krank“ gilt, und vermittelt subtil, was „verantwortungsvolle Gesundheitsvorsorge“ im Zeitalter der Genetik bedeutet. Genetische Tests werden etwa als „verantwortungsbewusstes“ Handeln vermarktet – und bringen Individuen so dazu, sich unbemerkt an biopolitischen Zielen wie Prävention oder Kosteneffizienz zu beteiligen.
Diese Entwicklung spiegelt sich in nationalen Gesundheitsstrategien, die genetische Screenings fördern, um Krankheitslasten zu minimieren. Ziel ist die Maximierung der „Gesundheitsdividende“ bei gleichzeitiger Kostenreduktion – eine biomolekulare Rationalisierungsstrategie.
Initiativen wie das 100.000 Genome Project oder das Newborn Genomes Programme von Genomics England gehen in die gleiche Richtung. Im Vergleich dazu ist der Europäische Raum für Gesundheitsdaten (EHDS) – eine EU-weite Initiative zur Vernetzung und sicheren Nutzung von Gesundheitsdaten – deutlich tiefgreifender. Der EHDS ist die Daten- und Regulierungsinfrastruktur, die personalisierte Medizin in großem Maßstab überhaupt erst ermöglicht.
Für die meisten Bürger klingt das nach Aufbruch: mehr Mitsprache, bessere Therapien, präzisere Prävention. Doch die verheißungsvolle Vision wirft einen Schatten. In der Praxis stehen sich ungleiche Gegner gegenüber: auf der einen Seite der einzelne Patient, der sein genetisches Profil, seine Laborwerte und seine Krankengeschichte beisteuert; auf der anderen Seite globale Pharmakonzerne, Datenplattformen und Technologieanbieter mit Milliardenbudgets, die diese Daten veredeln, patentieren, vermarkten. Cui bono? – wem nützt das Ganze? Die Antwort fällt ernüchternd aus: Der schnelle Profit landet bei den Großen, die Versprechen bleiben beim Kleinen.
Zwar betont der EHDS Bürgerrechte – mehr Kontrolle, mehr Transparenz, mehr Selbstbestimmung. Doch wer je erlebt hat, wie unübersichtlich Einwilligungsprozesse bei Gesundheits-Apps, Versicherungen oder elektronischen Patientenakten sind, weiß: Zwischen formaler Option und tatsächlicher Handlungsfähigkeit klafft eine Lücke. Während Goliath über Lobbykanäle, Fachjuristen und Datenzentren verfügt, bleiben David Informationsbroschüren und Vertrauen.
Der EHDS ist damit beides: Regierungstechnologie und Machtprojekt. Er entscheidet, ob Daten als öffentliches Gut oder als Rohstoffmarkt behandelt werden. Profitieren wird Goliath in jedem Fall. Die offene Frage ist, ob David mehr bleibt als die Rolle des Feigenblatts. Wirklich emanzipatorisch wird der Gesundheitsdatenraum erst, wenn Nutzen und Risiken fair verteilt werden – durch verbindliche Gemeinwohlauflagen, transparente Rechenschaft und reale Rückflüsse für die Patienten. Andernfalls bleibt das große Versprechen von „Empowerment“ ein Narrativ, das bestehende Asymmetrien lediglich kaschiert.
Disziplinarmacht: Die internalisierte biomolekulare Überwachung
Während die Biopolitik die Bevölkerung im Blick hat, richtet die personalisierte Medizin ihre disziplinierende Wirkung auf das Individuum. Das genetische Risikoprofil fungiert wie ein ständiger, internalisierter Überwacher. Wer weiß, dass er ein erhöhtes Risiko für Darmkrebs trägt, reguliert sein Verhalten: Ernährungsgewohnheiten, Vorsorgetermine und Sportroutinen werden der genetischen Prognose angepasst.
Die elektronische Patientenakte (ePA) verschärft diese Dynamik. Indem sie genetische und medizinische Daten zentral speichert und jederzeit abrufbar macht, verwandelt sie medizinische Empfehlungen in eine allgegenwärtige, digitale Instanz. Persönliche Hinweise, Erinnerungen an Medikamenteneinnahmen, personalisierte Berichte oder Schnittstellen zu Gesundheits-Apps verstärken die Selbstüberwachung: Die externe Anweisung („Du musst dich screenen lassen“) wird zur inneren Stimme („Ich muss auf mich achten, weil mein Genom es verlangt“). Die ePA strukturiert und institutionell legitimiert diese Selbstdisziplinierung, sodass sie unsichtbar und scheinbar freiwillig wirkt – gesteuert durch digitale Infrastruktur, soziale Normen und medizinische Autorität zugleich.
Die Folge ist eine subtile, aber umfassende Form der Macht: Sie manifestiert sich nicht in Sanktionen, sondern in der permanenten, digitalen Sorge um die eigene – genetisch vorgestellte – Zukunft. Die ePA macht das Individuum zum disziplinierten Akteur seines eigenen Risikoprofils, während die Überwachung formal verschwindet, aber technisch und institutionell allgegenwärtig bleibt.
Technologien der Selbstführung: Der genetisierte Selbstoptimierer
Hier zeigt sich die personalisierte Medizin in ihrer reinsten Form als Technologie der Selbstführung. DNA-Tests für zuhause, Fitness-Tracker und Gesundheits-Apps sind die Werkzeuge, mit denen Individuen gesellschaftliche Normen von Gesundheit und Leistungsfähigkeit verinnerlichen und aktiv an sich selbst arbeiten. Sie übersetzen das abstrakte Konzept der genetischen Prädisposition in konkrete, tägliche Handlungen: eine Extra-Portion Gemüse, 10.000 Schritte, Meditation zur Stressreduktion. Der Einzelne führt sich selbst im Sinne einer biomolekularen Logik – nicht weil der Staat es befiehlt, sondern weil er sein genetisches Schicksal optimieren will. Die Machtverhältnisse werden so unsichtbar und erscheinen als Ausdruck individueller Souveränität und Selbstfürsorge.
7.4. Der weltweite Trend: Globale biomolekulare Gouvernementalität
Die personalisierte Medizin ist längst kein nationales Phänomen mehr, sondern Teil einer globalen Transformation von Macht und Governance. Weltweit entstehen Initiativen, die Genomdaten, Gesundheitsinformationen und digitale Technologien bündeln, um Bevölkerung und Individuen in immer präzisere Risikoprofile zu übersetzen. Die Logik von Foucaults Gouvernementalität – die Verbindung von Wissen, Macht und Selbststeuerung – manifestiert sich hier auf transnationaler Ebene.
In den Vereinigten Staaten sammeln Programme wie das All of Us Research Program der NIH oder das Million Veteran Program genetische und medizinische Daten von Millionen Bürgern. Die Erhebung diverser Gesundheitsdaten dient nicht nur der wissenschaftlichen Forschung, sondern bildet die Grundlage für algorithmische Risikoprofile, präventive Interventionen und ressourcenoptimierte Gesundheitsstrategien.
Kanada verfolgt mit der Canadian Precision Health Initiative und dem Pan-Canadian Genomics Network ein vergleichbares Ziel: die systematische Sammlung und Vernetzung biomolekularer Daten, kombiniert mit einer Infrastruktur, die sichere Verwaltung, internationale Kooperation und wirtschaftliche Nutzung ermöglicht.
Hier zeigt sich Biopolitik 2.0: Bevölkerungen werden auf molekularer Ebene fragmentiert, gesteuert und optimiert.
Europa bildet ein orchestriertes Netzwerk transnationaler Biopolitik. Der Europäische Gesundheitsdatenraum (EHDS), unterstützt durch Initiativen wie ICPerMed und ERA PerMed, bündelt genetische, klinische und Lebensstildaten über Ländergrenzen hinweg.
Nationale Biobanken wie die UK Biobank oder der französische Health Data Hub fungieren als Dispositive, die sowohl Wissen als auch Macht generieren: Sie bestimmen, welche Daten genutzt werden, wie Populationen klassifiziert werden und welche gesundheitspolitischen Interventionen legitim erscheinen.
Durch Plattformen wie SHIFT-HUB oder die Helmholtz-Initiative iMed wird die internationale Vernetzung von Wissenschaft, Wirtschaft und Gesellschaft institutionalisiert – eine transnationale Form von Smart Governance, die Foucaults Konzept der Technologien der Selbstführung in den globalen Maßstab überträgt.
Auch in Asien und im Pazifikraum zeigt sich die globale Logik: Das GenomeAsia 100k Project, Singapurs National Precision Medicine Strategy und Chinas National Health Data Platform verbinden biomolekulare Daten mit KI-gestützten Analysen, um präventive, individualisierte und populationsbasierte Steuerung zu ermöglichen.
Australien und Israel setzen auf digitale Infrastruktur, die die Selbstdisziplinierung der Bürger verstärkt – elektronische Patientenakten, genetische Datenbanken und personalisierte Apps fungieren als subtile, aber allgegenwärtige Machtdispositive.
Selbst globale Forschungsprojekte wie der Human Cell Atlas illustrieren die transnationale Dimension: Indem die Vielfalt menschlicher Zellen kartiert wird, entsteht ein universelles Wissens- und Machtnetzwerk, das Grundlagen für personalisierte Therapien weltweit bereitstellt. Die Grenzen zwischen staatlicher Steuerung, privatwirtschaftlicher Macht und wissenschaftlicher Expertise verschwimmen: Foucaults klassische Trennung zwischen Nationalstaat und Disziplinarmacht wird durch globale Netzwerke und internationale Kooperationen ersetzt.
7.5. Schlussfolgerung: Globale Ambivalenz der biomolekularen Macht
Die weltweite Expansion personalisierter Medizin zeigt eindrücklich, dass die biomolekulare Revolution nicht mehr auf einzelne Staaten beschränkt ist. Sie manifestiert sich als transnationales Netzwerk aus Daten, Algorithmen und Governance, das sowohl Bevölkerungen als auch Individuen auf molekularer Ebene steuert. Globale Initiativen – von den USA über Europa bis Asien, Australien und Israel – verbinden Biopolitik, Disziplinarmacht und Technologien der Selbstführung auf internationaler Ebene.
Die Ambivalenz bleibt dabei zentral: Auf der einen Seite eröffnen diese Entwicklungen Chancen für präzisere Therapien, individualisierte Prävention und potenziell mehr Gesundheitsgerechtigkeit. Auf der anderen Seite entstehen Machtasymmetrien, bei denen Staaten, internationale Organisationen und private Akteure den Zugang zu Daten, Ressourcen und Technologien kontrollieren. Individuen werden zunehmend zu disziplinierten Subjekten ihres eigenen genetischen Profils, während globale Dispositive unbemerkt normative und ökonomische Ziele durchsetzen.
Damit verbindet sich die lokale Erfahrung personalisierter Medizin nahtlos mit einer globalen Gouvernanzlogik: Smart Governance und biomolekulare Biopolitik sind längst internationalisiert, und die Herausforderung besteht darin, Nutzen und Risiken fair zu verteilen. Die weltweite Dimension dieser Transformation verdeutlicht, dass die biomolekulare Revolution nicht nur medizinisch, sondern auch politisch und sozial unumkehrbar ist – ein globales System der Steuerung, das zugleich Chancen für Gesundheit und Risiken für Machtkonzentration birgt.

8. Epilog
Der Wunsch, Gesundheit und Lebenszeit zu optimieren, ist ein Urinstinkt der Menschheit. Schon in antiken Hochkulturen galt Gesundheit nicht als Zufall, sondern als erstrebenswerter Zustand, erreichbar durch Lebensführung und Philosophie. Die griechische „Diätetik“ – eine ganzheitliche Praxis aus Ernährung, Bewegung und Seelenhygiene – und das Leitbild „mens sana in corpore sano“ zeugen von einem 2500 Jahre alten Optimierungswillen. Dieser Drang, gespeist aus dem Überlebensinstinkt, der Angst vor Kontrollverlust und dem Streben nach Lebensqualität, ist nicht nur uralt, sondern ewig.
In der Moderne verkörpert die personalisierte Medizin den neuesten Ausdruck dieses Traums. Sie verspricht, den Menschen nicht als Durchschnitt, sondern als Individuum zu behandeln, basierend auf genetischem Code, Biomarkern und Lebensstil-Daten. Doch ist sie die Erfüllung dieses Traums oder eine moderne Fata Morgana? In ihrer utopischen Vision suggeriert sie die vollständige Beherrschbarkeit der Biologie, eine Überwindung der „biologischen Lotterie“. Die Realität ist nüchterner: Sie ermöglicht präzisere Diagnosen und Therapien, verhindert Behandlungsfehlschläge – aber sie schafft Krankheit nicht ab.
Denn die Idee einer krankheitsfreien Existenz bleibt ein Trugbild. Evolutionäre Biologie und Biophysik setzen Grenzen: Pathogene Keime entwickeln sich weiter, Krebs ist eine statistische Folge der Zellteilung, und Alterung ein unvermeidbarer degenerativer Prozess. Das realistische Ziel ist daher nicht die Elimination von Krankheit, sondern ihre Transformation: tödliche Erkrankungen in chronische Zustände umzuwandeln und die „healthspan“ – die Jahre in guter Gesundheit – zu maximieren. Dieses Ziel ist radikal, aber erreichbar, wie Fortschritte in der Behandlung von HIV oder Diabetes zeigen.
Doch wie weit ist der Mensch bereit, für dieses Ziel zu gehen? Die Geschichte zeigt, dass im Angesicht von Leid und Tod ethische Grenzen oft verschwimmen. Von alltäglicher Disziplin über finanzielle Opfer bis hin zu experimentellen Therapien reicht das Spektrum der Bereitschaft. Hier stellt sich eine unbequeme Frage: Welche Kompromisse ist die Gesellschaft bereit einzugehen?
Die personalisierte Medizin erfordert Daten – genetische, medizinische, persönliche. Diese Datensammlung birgt das Risiko, Privatsphäre und Autonomie zu untergraben. Die COVID-19-Pandemie war ein Stresstest, der zeigte, dass große Teile der Bevölkerung bereitwillig Freiheitsrechte für Sicherheit handelten. Modelle wie das chinesische Sozialkreditsystem illustrieren ein mögliches Zukunftsmodell, in dem Transparenz und Kontrolle im Namen von Gesundheit und Sicherheit belohnt werden. Doch solche Szenarien sind nicht zwangsläufig: Sie sind ein Extrem, kein unvermeidbares Schicksal.
Die personalisierte Medizin bietet zwei Gesichter. Ihr pragmatisches Versprechen ist eine effizientere, präzisere und menschlichere Medizin, die Leben rettet und Leid lindert – etwa durch maßgeschneiderte Krebstherapien oder frühzeitige Diagnosen. Ihr utopisches, aber riskantes Versprechen ist die Illusion einer totalen Kontrolle über Leben und Tod. Sie ist damit der Höhepunkt einer jahrtausendealten Reise. Ob diese Reise in einer emanzipierten Selbstoptimierung oder einer dystopischen „Biokratie“ endet, hängt nicht von der Technologie ab, sondern von der ethischen und politischen Rahmensetzung, die die Gesellschaft ihr gibt.
Der uralte Traum von Gesundheit erscheint heute greifbarer denn je, doch er fordert uns heraus, kluge Antworten zu finden. Die personalisierte Medizin kann ein Werkzeug der Emanzipation sein, wenn wir sie mit Bedacht nutzen. Die entscheidende Frage der Moderne lautet daher nicht nur, wie viel Freiheit wir für ein längeres Leben opfern, sondern wie wir Gesundheit und Freiheit in Einklang bringen. Nur so wird das ewige Streben nach Gesundheit zu einem Fortschritt für alle.
Die Welt wird sich nicht auf einen Schlag verändern; das ist noch nie geschehen. Kurzfristig wird das Leben weitgehend unverändert weitergehen, und die Menschen werden 2025 ihre Zeit größtenteils genauso verbringen wie 2024. Wir werden uns weiterhin verlieben, Familien gründen, uns online streiten, in der Natur wandern usw.
Doch die Zukunft wird unaufhaltsam auf uns zukommen, und die langfristigen Veränderungen für unsere Gesellschaft und Wirtschaft werden gewaltig sein.
Sam Altman (CEO von OpenAI)
Quellen (Stand vom 22.11.2025)
