
Sie sind winzig, unsichtbar und überall: Viren. Seit Urzeiten schleichen sie durch die Biosphäre, manipulieren Gene, lenken globale Kreisläufe und spielen Roulette mit unserem Immunsystem. Dabei sind sie weder wirklich lebendig noch ganz tot – sie existieren im Dazwischen: als heimliche Regisseure des Lebens. Mal unsichtbare Wächter, mal heimtückische Eindringlinge. Und meist: vollkommen unbemerkt.
Wir nehmen sie oft erst wahr, wenn sie uns flachlegen – mit Husten, Fieber oder ganzen Pandemien. Dann werden sie plötzlich zu Feinden, zu Angstmachern, zu Schlagzeilen. Doch hinter diesen mikroskopischen Strukturen steckt weit mehr als nur Krankheit: eine faszinierende, hochorganisierte Miniwelt, die Biologie auf Speed betreibt.
Diese Abhandlung lädt dazu ein, Viren aus einem anderen Blickwinkel zu sehen: nicht nur als Erreger, sondern als Akteure im Gefüge des Lebens. Was macht sie so erfolgreich? Wie vermehren sie sich mit nichts als ein paar Genen? Wie beeinflussen sie Ökosysteme? Und vor allem: Gibt es Viren wirklich?
Mit wissenschaftlichem Fundament, verständlicher Sprache und einer Prise Augenzwinkern werfen wir einen Blick hinter die Kulissen der unsichtbaren Mikro-Welt – und auf die Methoden, mit denen die Forschung Viren sichtbar macht.
Vorhang auf für das Unsichtbare…
📑Inhaltsverzeichnis
1. Ein Blick in die Welt des Unsichtbaren
1.1. Wächter der Natur: Viren als Gleichgewichtsregulatoren
1.2. Viren als Motor der Evolution
1.3. Gibt es Viren wirklich?
2. Viren und ihre Mechanismen: Einblick am Beispiel des Influenzavirus
2.1. Wie das Influenzavirus auf Reisen geht
2.2. Die Architektur des Influenzavirus
2.3. Der Infektionsprozess des Influenzavirus
2.4. Die Anpassungsfähigkeit des Influenzavirus
2.5. Ein- und Austrittswege des Influenzavirus
2.6. In den meisten Fällen lokal begrenzte mukosale Infektion
2.7. Virenstrategie: Effiziente Vermehrung ohne rasche Zellzerstörung
2.8. Zerstörung der Wirtszelle
2.9. Selbstbegrenzung gefährlicher Viren: Warum sie selten Pandemien auslösen
2.10. Warum macht das Virus manche Menschen krank und andere nicht?
3. Ein Blick auf die Anfänge der Mikrobiologie – wie alles begann
3.1. Frühe Entdeckungen: Die ersten Blicke ins Unsichtbare
3.2. Die Geburt der modernen Mikrobiologie
3.3. Der Schritt in die Welt der Viren
3.4. Viren und die Koch’schen Postulate
4. Moderne Methoden zur Entdeckung und Analyse von Viren
4.1. Probenentnahme
4.2. Probenaufbereitung
4.3. Zellkultur
4.4. Viren sichtbar machen
4.4. a) Elektronenmikroskopie
4.4. b) Kristallisation
4.4. c) Kryo-Elektronenmikroskopie
4.4. d) Kryo-Elektronentomographie
4.4. e) Zusammenfassung
4.5. Der genetische Fingerabdruck der Viren
4.5.1. Nukleinsäure-Extraktion
4.5.2. Nukleinsäure-Amplifikation
4.5.3. Sequenzierung
4.5.3. a) First Generation: Sanger-Sequenzierung
4.5.3. b) Second Generation: Next-Generation Sequencing (NGS)
4.5.3. c) Third Generation: Echtzeit-Sequenzierung
4.5.3. d) Emerging Technologies: Zukunft der Sequenzierung
4.6. Bioinformatische Analyse
5. Gibt es Viren wirklich?
6. Woher kommen Viren?
6.1. Der Baum des Lebens
6.2. Die Haupthypothesen zur Herkunft von Viren
6.2.1. Hypothesen in einer zellulären Welt
6.2.2. Hypothesen in der prä-zellulären RNA-Welt
7. Warum gibt es Viren?
Epilog: Die Unscheinbaren

1. Ein Blick in die Welt des Unsichtbaren
Unsere sichtbare Welt ist nur die Hälfte der Geschichte – um uns herum, auf uns und in uns existiert ein unsichtbares Universum voller mikroskopischer Akteure. Unter ihnen sind Viren die rätselhaftesten Bewohner: Sie besitzen keinen eigenen Stoffwechsel, keinen Zellkern – und doch können sie das Schicksal ganzer Ökosysteme beeinflussen.
So winzig, dass selbst die besten Lichtmikroskope kapitulieren, offenbaren Viren erst unter dem Elektronenmikroskop ihre verblüffende Formenvielfalt: Da tauchen Gebilde auf, die aussehen wie außerirdische Raumsonden – kugelige Formen mit stacheligen Fortsätzen, schraubenartige Spiralen oder perfekte geometrische Körper.
Hinter diesen originellen Strukturen steckt pure Funktionalität – ganz ohne unnötigen Schnickschnack: ein Päckchen genetische Information (DNA oder RNA), sicher verpackt in eine robuste Proteinhülle. Manche Modelle gönnen sich noch eine schützende Membranhülle – dreist dem letzten Opfer entrissen.
Schlicht, aber wirkungsvoll: das Erfolgsrezept der Viren.

Zommen wir zunächst in diese Mikrowelt, um ein Gefühl für die Größenverhältnisse zu bekommen.
Die Mission eines Virus: Infizieren, vermehren, überleben
Jedes Virus hat eine klare Aufgabe: Es muss einen geeigneten Wirt finden, um sich zu vermehren und als Art zu überleben. Diese Wirte können Menschen, Tiere, Pflanzen oder Bakterien sein. Es gibt sogar Viren, die andere Viren infizieren. Sobald ein Virus eine geeignete Wirtszelle findet, schleust es sein genetisches Material in diese Zelle ein und übernimmt die molekularen Maschinerien der Zelle, um Kopien von sich selbst herzustellen. So kann sich ein Virus rasch von Zelle zu Zelle ausbreiten und dabei Milliarden von Kopien erzeugen. Auf diese Weise existieren Viren seit Milliarden von Jahren und sind allgegenwärtig.
Der Lebenszyklus eines Virus: Ruhephase vs. Angriffsmodus
Ein Virus existiert in zwei radikal unterschiedlichen Zuständen – fast wie ein Doppelagent:
— Die Extrazelluläre Phase: Das Virion —
Dasein als „Nanospore“: Ein inaktives, aber infektiöses Partikel.
Aufgabe: Überleben außerhalb von Wirtszellen – auf Türklinken, in Tröpfchen, im Boden.
Besonderheit: Kein Stoffwechsel, keine Vermehrung – nur Warten auf den richtigen Wirt.
Wie ein Samenkorn im Wind: inert, aber voller potenziellen Lebens.
— Die Intrazelluläre Phase: Der aktive Virus —
Brutale Effizienz: Ein einziges Virion kann über 10.000 neue Viren erzeugen.
Mission Start: Sobald eine geeignete Wirtszelle infiziert wird.
Strategie: Kapere die Zellmaschinerie, produziere Nachkommen – bis zum Zerbrechen.
Virion vs. Virus – Warum die Unterscheidung wichtig ist
In der Wissenschaft zählt jedes Detail – selbst ob ein Virus gerade „schläft“ oder die Zelle unterwandert.
- Virion: Infektiöses Partikel außerhalb von Zellen – das reisende Partikel.
- Virus: der Oberbegriff – umfasst beide Phasen, inaktiv wie aktiv.
Das Virion ist also nicht das Virus selbst, sondern seine reisefähige Verpackung. Erst wenn es in eine Zelle eindringt und dort aktiv wird, spricht man vom Virus.
Diese begriffliche Unterscheidung wurde erstmals 1983 vom Virologen Bandea vorgeschlagen. Auch wenn sie sich nicht in allen Disziplinen durchgesetzt hat, schafft sie Klarheit – und macht deutlich: Ein Virus ist mehr als nur ein „Partikel“ – es ist ein Prozess.
1.1. Wächter der Natur: Viren als Gleichgewichtsregulatoren
Das Wort „Virus“ lässt bei den meisten sofort die Alarmglocken schrillen: Influenza, Corona, HIV, Ebola – Krankheit, Gefahr, Pandemie. Doch dieses Bild ist nur ein winziger Ausschnitt der Wirklichkeit. Von den unzähligen Virenarten, die unseren Planeten bevölkern, sind gerade einmal 21 Typen für den Menschen gefährlich. Der Rest? Unsichtbare Helfer im Hintergrund – Hüter des ökologischen Gleichgewichts.
Also keine Panik: Die meisten Viren interessieren sich kein bisschen für uns. Sie befallen Mikroorganismen – Bakterien, Archaeen, Einzeller – die verborgenen Baumeister des Lebens. Und genau dort entfalten sie ihre wahre Kraft: Sie steuern die Populationen dieser Mikroben, lenken Stoffkreisläufe, beeinflussen das Klima, verteilen Gene wie Informationsboten und halten so die Balance im System.
Kaum ein Ort auf der Erde, an dem sie nicht zu finden sind. Sie surfen auf Meeresströmungen, verstecken sich in Regentropfen, reisen als blinde Passagiere auf Pollenkörnern und haften geduldig an Staubpartikeln, die zwischen Kontinenten wandern. Ihr Reich ist riesig – und bleibt dennoch verborgen im Schatten des Sichtbaren.
Zahlen, die den Verstand sprengen
Mit geschätzten 100 Millionen Arten zählen Viren zu den häufigsten biologischen Entitäten der Erde. Ihre Gesamtzahl wird auf etwa 10³¹ Partikel geschätzt – eine Eins mit 31 Nullen – das ist mehr als alle Sterne im Universum, mehr als alle Zellen aller Lebewesen zusammen. In einem einzigen Milliliter Meerwasser tummeln sich etwa 10 Millionen Viruspartikel. Die Erde: ein echter Planet der Viren – wie es der Forscher Aleksandar Janjic formulierte.
Dabei sind sie ultraleicht: Ein einzelnes Viruspartikel wiegt gerade mal ein Femtogramm (10⁻¹⁵ Gramm) – das ist ein Millionstel Milliardstel Gramm – leichter als ein Photon Sonnenlicht. Rechnet man ihre schwindelerregende Anzahl (10³¹) zusammen, erreichen sie vielleicht das Gewicht eines ausgewachsenen Blauwals. Und dennoch: Ohne sie kein Gleichgewicht, kein Kreislauf – kein Leben, wie wir es kennen.
Doch was macht sie zu einem integralen Bestandteil des Ökosystems?
In den Ozeanen – den größten Lebensräumen unseres Planeten – durchsetzen Viren täglich Milliarden von Mikroorganismen. Was nach Vernichtung klingt, ist in Wirklichkeit Teil eines fein austarierten Systems: Indem sie gezielt Mikroben befallen und zerstören, verhindern sie, dass einzelne Arten überhandnehmen. Eine unsichtbare Form von Populationskontrolle – subtil, aber wirksam wie das Raubtier in der Savanne.
Und sie tun noch mehr: Wenn ihre Wirtszellen zerplatzen, setzen sie wertvolle Nährstoffe frei – Kohlenstoff, Stickstoff, Phosphor. Diese stehen sofort anderen Lebewesen zur Verfügung, halten Nahrungsketten am Laufen, nähren das Plankton, das wiederum den Sauerstoff für unsere Atmosphäre produziert.
Gleichzeitig wirken Viren als Evolutions-Booster. Sie übertragen Gene von Organismus zu Organismus – ein natürlicher Gentransfer, der neue Eigenschaften ermöglicht, Vielfalt fördert und Innovationen hervorbringt, lange bevor wir selbst von Gentechnik wussten.
Und so zeigt sich: Viren sind keine bloßen Krankheitsbringer. Sie sind feinverästelte Zahnräder im Getriebe der Natur – unsichtbar, selten bemerkt, aber unersetzlich.
Ein Blick in verschiedene Lebensräume offenbart ihre Wirkung.
🌊 In den Weltmeeren
Populationskontrolle: Tief unter der Wasseroberfläche tobt ein Mikrokampf planetaren Ausmaßes: Bakteriophagen – Viren, die gezielt Bakterien infizieren – eliminieren täglich bis zu 40 % der Meeresbakterien. Damit verhindern sie explosive Algenblüten, die ganze Ozeane in sauerstoffarme Todeszonen verwandeln könnten.
Ohne diese „Mikrobenjäger“ wäre unser Planet längst unter einem Leichentuch aus Algen versunken.
📖 Weitere Quellen:
Viral control of biomass and diversity of bacterioplankton in the deep sea
A sea of zombies! Viruses control the most abundant bacteria in the Ocean.
The smallest in the deepest: the enigmatic role of viruses in the deep biosphere
Gen-Schmuggel: Im blauen Dunkel der Meere vollbringt Prochlorococcus – ein winziges Cyanobakterium – Großes: Es produziert rund 10 % des globalen Sauerstoffs. Aber selbst dieser Mikroheld steht unter der Kontrolle noch kleinerer Strippenzieher: Cyanophagen – Viren, die perfekt auf ihn spezialisiert sind. Sie schleusen eigene Photosynthesegene ein und zwingen die infizierte Zelle zur Kooperation. Das Ergebnis: Die Bakterie bleibt „arbeitsfähig“, produziert weiter Energie – nun im Dienst ihrer viralen Besetzer. Dieses parasitäre Partnerschaftsmodell veranschaulicht den sogenannten Black-Queen-Effekt: Indem Viren bestimmte Funktionen übernehmen, können Mikroben diese selbst abbauen – und sich auf andere Aufgaben spezialisieren.
Eine unfreiwillige Arbeitsteilung, orchestriert von Viren.
Tiefer eintauchen: Einen eindrucksvollen Einblick in die geheimnisvolle Welt unter dem Meeresspiegel bietet das Video des Schmidt Ocean Institute. Es zeigt, wie Forschende mit modernster Technik den Spuren mikrobiellen Lebens folgen – und dabei auch den Viren auf der Spur sind.
Doch nicht nur im Wasser übernehmen Viren diese regulatorische Rolle – sie sind ebenso aktiv in anderen Ökosystemen.
🟫 In den Böden
Auch unter unseren Füßen herrscht virales Treiben: Viren halten dominante Bodenbakterien in Schach und sorgen so dafür, dass kein Mikroorganismus die Oberhand gewinnt. Diese unsichtbare Kontrolle schützt die fragile Balance des Nährstoffkreislaufs – Grundlage für alles Wachstum.
Wie unsichtbare Gärtner durchkämmen sie das Mikroleben der Erde, jäten Überfluss und schaffen Raum für Vielfalt.
🌳 In der Pflanzenwelt
Bäume und Felder haben geheime Verbündete: Pflanzenviren. Rund um jedes Wurzelgeflecht, entfaltet sich ein verborgenes Netzwerk aus Kontrolle, Schutz und Gegenspiel. Einige Pflanzenviren attackieren gezielt schädliche Bakterien, die Pflanzen krank machen würden. Andere kurbeln das pflanzeneigene Immunsystem an – und wenn Mikroben sterben, zersetzen Viren deren Überreste zu fruchtbarem Dünger. Manche Pflanzen gehen sogar noch einen Schritt weiter: Sie rekrutieren ganz gezielt schützende Viren, die wie Bodyguards im Wurzelraum patrouillieren.
Ohne diese mikroskopischen Allianzen wären viele Wälder anfälliger für Pilzüberwucherung – und unser Getreide den Angriffen aus dem Boden schutzlos ausgeliefert.
🔗 Viruses as components of forest microbiome
🪱 In der Darmflora von Mensch und Tier
Auch in unseren Eingeweiden tobt ein stiller Machtkampf – und wir profitieren davon. Spezialisierte Darmviren (Bakteriophagen) jagen gezielt schädliche Keime wie E. coli und halten die bakterielle Balance stabil. Sie übertragen Schutzgene zwischen Mikroben – wie geheime Datenpakete, die Immunantworten optimieren. Manche Viren dämpfen sogar überaktive Abwehrreaktionen und verhindern so Entzündungen.
Ohne diese Nano-Sheriffs würden schädliche Bakterien den Darm innerhalb von Tagen überrennen.
🔗 Over 100.000 Viruses Identified in the Gut Microbiome
☁️ In der Atmosphäre
Hoch über unseren Köpfen findet der größte Gentransfer der Erde statt – ein einziger Sturm kann 500 Millionen Virionen pro m² über Kontinente verteilen – die ultimative Bio-Invasionsroute. Dank ihrer extremen Robustheit überleben Viren wo andere scheitern – in UV-getränkten Höhen, eisigen Wolken und trockener Luft. Sie reisen per Staub, Meersalz oder Pflanzentröpfchen über Ozeane und Kontinente hinweg. Viren in Wolken beeinflussen sogar Niederschlagsmuster.
Diese atmosphärische Gen-Börse macht aus lokalen Mutationen globale Evolution – als hätte die Natur ihr eigenes Internet erfunden.
🔗 Deposition rates of viruses and bacteria above the atmospheric boundary layer
⚡️ In Extremlebensräumen
Selbst in heißen Quellen, Salzseen oder unter der Erdkruste existieren Viren, die dortige Mikroorganismen regulieren und deren genetische Vielfalt steigern – eine essenzielle Überlebensstrategie in lebensfeindlichen Umgebungen.
🔗 Viruses in Extreme Environments, Current Overview, and Biotechnological Potential
Das größte Paradox der Biologie: Aus milliardenfacher Zerstörung erwächst globales Gleichgewicht. Das nächste Mal, wenn du einen Virus fürchtest, denk daran: Mit jedem Atemzug trägst du Milliarden dieser Winzlinge – und sie tragen dich. Ein Pakt des Lebens – so alt wie die Evolution selbst.
„Wir leben in einem Gleichgewicht, in einem perfekten Gleichgewicht“, und Viren sind ein Teil davon, sagt Susana Lopez Charretón, Virologin an der Nationalen Autonomen Universität von Mexiko. „Ich glaube, ohne Viren wären wir aufgeschmissen.“ [Warum die Welt Viren zum Funktionieren braucht]
„Wenn alle Viren plötzlich verschwinden würden, wäre die Welt für etwa anderthalb Tage ein wunderbarer Ort, und dann würden wir alle sterben – das ist das Fazit“, sagt Tony Goldberg, Epidemiologe an der University of Wisconsin-Madison. „Alle wichtigen Dinge, die sie in der Welt bewirken, überwiegen bei weitem die schlechten Dinge.“ [Warum die Welt Viren zum Funktionieren braucht]
1.2. Viren als Motor der Evolution
Lange bevor Dinosaurier über die Erde stampften, trieben Viren bereits ihr Unwesen – und formten dabei das Leben, wie wir es heute kennen. Ihr Werkzeug: horizontaler Gentransfer, ein biologischer Copy-Paste-Mechanismus, der evolutionäre Quantensprünge ermöglichte.
Für den Evolutionsbiologen Patrick Forterre vom Institut Pasteur sind Viren die Architekten des Lebens, ohne die die Evolution vielleicht ganz anders gelaufen wäre. (Vgl. dazu Spektrum der Wissenschaft: „Die wahre Natur der Viren“, ScienceDirect: „The origin of viruses and their possible roles in major evolutionary transitions“ oder „The two ages of the RNA world, and the transition to the DNA world: a story of viruses and cells”)
Genetische Sabotage mit Folgen
Viren sind Meister der Manipulation. Wenn sie eine Zelle infizieren, schleusen sie nicht nur ihre eigene Erbinformation ein – manchmal wird ihr Genmaterial ins Genom des Wirts integriert und über Generationen hinweg weitervererbt. Aus vermeintlichen Störern werden so kreative Genarchitekten.
Ein spektakuläres Beispiel: Die Plazenta der Säugetiere verdankt ihre Existenz einem Virus. Ein virales Hüllprotein – ursprünglich dazu gedacht, Immunantworten zu unterdrücken – wurde in den Genpool eingebaut und half mit, die Barriere zwischen Mutter und Embryo zu entwickeln. Ohne dieses „fremde“ Gen: kein Mutterleib, kein Säugetier.
Doch es geht noch weiter: Rund 8 % des menschlichen Erbguts stammen von alten Retroviren, die sich einst in unsere DNA eingeschrieben haben – stille Zeugen uralter Infektionen, die uns heute womöglich mitgestalten. Selbst unser Gehirn könnte virale Spuren tragen – etwa Gene, die für die Entwicklung des Cortex entscheidend sind.
Sind wir nicht alle ein bisschen Virus?
CRISPR, das heute als revolutionäre Genschere gefeiert wird, geht auf ein uraltes Abwehrsystem von Bakterien zurück – ein genetisches Archiv vergangener Virusangriffe, aus dem Bakterien lernen, sich gegen neue Feinde zu wehren.
Manche Wissenschaftler fragen sogar: Könnten Viren an der Entstehung des Lebens selbst beteiligt gewesen sein? Einige Hypothesen vermuten, dass virusähnliche Partikel einst die ersten Moleküle waren, die genetische Information speichern und weitergeben konnten – eine Grundbedingung für Leben.
Die Ironie des Schicksals: Wir fürchten Viren als Todbringer – dabei wären wir ohne sie nicht einmal entstanden.
1.3. Gibt es Viren wirklich?
Trotz ihrer immensen Bedeutung gibt es immer wieder Zweifel an der Existenz von Viren. Wie können wir sicher sein, dass sie tatsächlich real sind? Diese Frage lässt sich nicht mit einer einfachen Beobachtung beantworten – Viren entziehen sich unserem bloßen Auge und offenbaren sich nur durch indirekte Spuren und spezialisierte Nachweismethoden.
Um dieser Frage auf den Grund zu gehen, müssen wir zunächst verstehen, wie Viren agieren: Welche Mechanismen nutzen sie, um sich zu vermehren? Wie interagieren sie mit ihren Wirten? Und vor allem: Welche wissenschaftlichen Methoden gibt es, um sie sichtbar zu machen und nachzuweisen?
Die Suche nach diesen Antworten führt uns in eine faszinierende Welt aus hochentwickelten Technologien und jahrzehntelanger Forschung. In den kommenden Kapiteln werden wir Schritt für Schritt erkunden, wie Wissenschaftler Viren nachweisen – und damit der entscheidenden Frage näherkommen: Gibt es Viren wirklich?

2. Viren und ihre Mechanismen:
Einblick am Beispiel des Influenzavirus
Werfen wir zunächst einen Blick darauf, wie Viren ihre Wirtszellen „kapern“ und für ihre Vermehrung nutzen. Ein Paradebeispiel hierfür ist das Influenzavirus – nicht nur, weil es zu den am besten erforschten Viren gehört, sondern auch, weil es eindrucksvoll zeigt, wie Viren Zellen manipulieren und sich verbreiten. Die Mechanismen, die es anwendet, eröffnen uns einen idealen Einblick in die geheimnisvolle Welt der Viren und ihre vielschichtigen Wechselwirkungen mit ihren Wirten.
2.1. Wie das Influenzavirus auf Reisen geht
2.2. Die Architektur des Influenzavirus
2.3. Der Infektionsprozess des Influenzavirus
2.4. Die Anpassungsfähigkeit des Influenzavirus
2.5. Ein- und Austrittswege des Influenzavirus
2.6. In den meisten Fällen lokal begrenzte mukosale Infektion
2.7. Virenstrategie: Effiziente Vermehrung ohne rasche Zellzerstörung
2.8. Zerstörung der Wirtszelle
2.9. Selbstbegrenzung gefährlicher Viren: Warum sie selten Pandemien auslösen
2.10. Warum macht das Virus manche Menschen krank und andere nicht?
💡Hinweis: Die folgenden Kapitel bauen auf grundlegenden Kenntnissen über Zellen, den Unterschied zwischen DNA und RNA, Proteine sowie zelluläre Prozesse wie die Proteinbiosynthese auf. Falls diese Themen noch neu für dich sind, könnte ein Blick in Kapitel 2, 3 und 4 der Abhandlung „Die Wunderwelt des Lebens“ oder ähnliche Einführungstexte hilfreich sein.
Kapitel 2: Die Zelle – der Urbaustein
Kapitel 3: Proteine – die Bausteine des Lebens
Kapitel 4: Vom Code zum Protein – zelluläre Mechanismen
2.1. Wie das Influenzavirus auf Reisen geht
Das Influenzavirus ist ständig auf Achse – ein unsichtbarer Jetsetter mit erstaunlichen Ansteckungsrouten: Mal reist es first class per Nieswolke, mal trampt es über Türgriffe.
Tröpfchenflug – Erste Klasse durch die Luft
Ein Nieser genügt: Bis zu 40.000 virusbeladene Tröpfchen schießen durch die Luft – wie ein Mini-Raketenangriff auf die Umgebung (Reichweite: bis zu 2 Meter!).
Schmierattacke – Der heimliche Handshake
Türklinke, Fahrstuhlknopf, Tastatur – das Virus chillt auf Oberflächen teils stundenlang. Ein Griff, ein Wisch durchs Gesicht – und schon hat es sich per Hand-zu-Gesicht-Trick Zugang verschafft.

Mission Atemwege: Virale Invasion
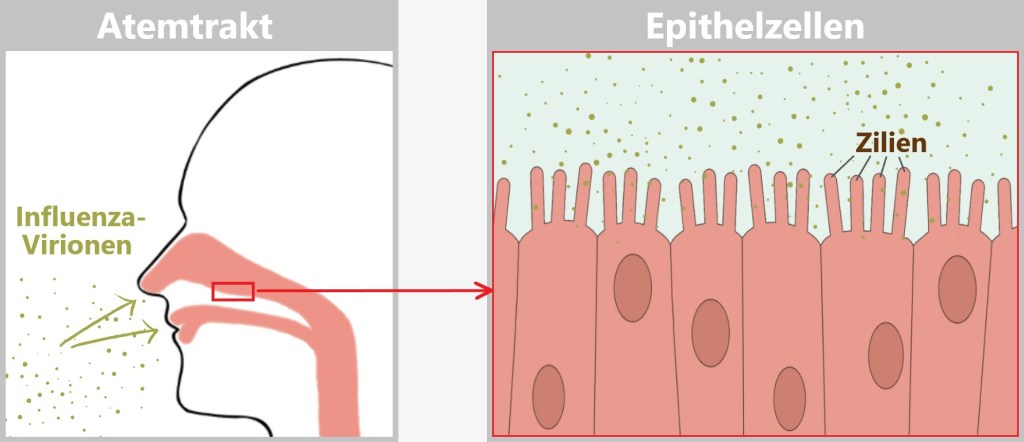
Angekommen im Atemtrakt, startet das Virus seinen Sturm auf die Epithelzellen:
Lieblingsziel: Schleimhautzellen von Nase, Rachen, Bronchien.
Warum? Hier sitzen massenhaft beliebte Rezeptoren – perfekte Andockstellen.
Folge: Innerhalb von Stunden kapert es die Zellfabrik und produziert neue Viren.
Von außen wirkt es wie ein Staubkorn mit schlechten Absichten – doch bei genauerer Betrachtung entpuppt sich das Influenzavirus als hochkomplexe Nanomaschine. Um zu verstehen, wie es Zellen kapert und ständig mutiert, lohnt sich ein Blick ins Innere.
2.2. Die Architektur des Influenzavirus
Das Influenzavirus ist der Grund, warum wir mit Fieber im Bett liegen und über „die Grippe“ fluchen. Von den drei Stämmen (A, B, C) ist Typ A der gefährlichste Globetrotter: wandlungsfähig wie ein Schauspieler und unberechenbar wie Aprilwetter. Typ B und C sind dagegen eher die „Bodenständigen“ – weniger variabel und weniger gefährlich. Doch alle teilen denselben genialen Bauplan (siehe untere Abbildung).
Das Viruspartikel – dieser nur 80-120 Nanometer kleine Überlebenskünstler – ähnelt einem winzigen, kugeligen Nano-U-Boot (manchmal ist es auch oval). In seinem Inneren: das virale Erbgut aus RNA – aber mit einem besonderen Trick!
Während wir RNA oder DNA oft als langen, durchgehenden Faden kennen, besitzen Viren unterschiedliche DNA- und RNA-Strukturen. Manche haben ein einziges zusammenhängendes Molekül, andere tragen ihr Erbgut in mehrere RNA-Segmente aufgeteilt.
Die Kommandozentrale: Genom in 8 Teilen
Das Influenzavirus setzt auf die modulare Bauweise: es nutzt 8 separate RNA-Segmente – wie ein Baukasten, dessen Teile sich immer neu kombinieren lassen – perfekt für Überraschungen!
Diese RNA-Segmente sind unterschiedlich lang – vom kompakten Mini-Modul bis zur XXL-Bauanleitung – und doch perfekt aufeinander abgestimmt. Damit sie nicht wie lose Zettel im Wind verloren gehen, werden sie sorgsam eingepackt: Jedes Segment wird von einer Hülle aus Nukleoproteinen (NP) umschlungen – wie wertvolle Schriftrollen in Schutzfolie. Doch die NP-Hülle ist mehr als bloßer Schutz: Sie hilft der viralen Maschinerie, die genetische Information präzise zu lesen, zu kopieren und weiterzugeben.
Der Werkzeuggürtel: Polymerase-Komplexe
Zusätzlich bringt das Virus seine eigenen 3D-Drucker mit – die RNA-Polymerasen (bestehend aus den Untereinheiten: PB1, PB2, PA). Diese sind fest an die RNA-Segmente gebunden: wie Handwerker, die ihr Werkzeug am Gürtel tragen.
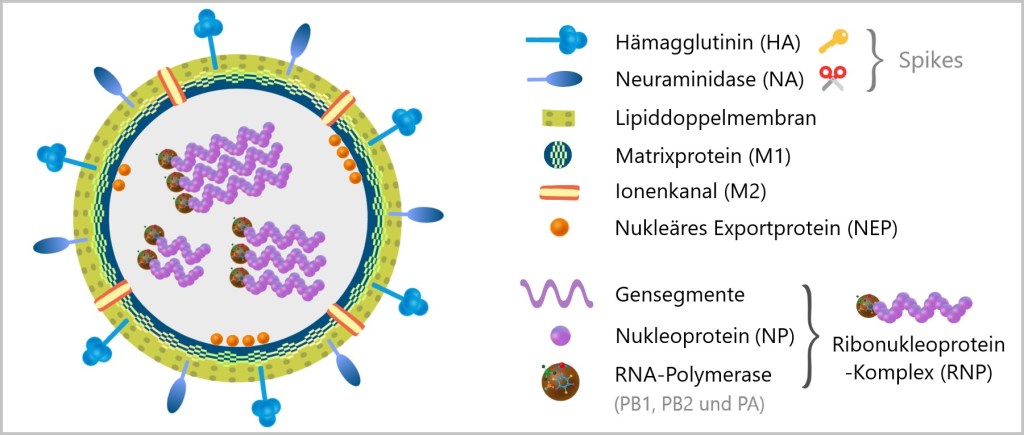
Der RNP-Komplex – das Herzstück des Virus
Jedes RNA-Segment + Nukleoproteine (NP) + Polymerase bildet einen Ribonukleoprotein-Komplex (RNP) – eine perfekt organisierte Einheit: das Steuerzentrum des Virus. Alle acht – sauber verpackt und einsatzbereit – wie ein tragbarer Werkzeugkoffer für die Zellübernahme.
Die Lipidhülle – der gestohlene Tarnumhang
Das Virus klaut sich seine äußere Schicht direkt von der Wirtszelle: eine Lipiddoppelschicht – identisch zur Zellmembran und damit die perfekte Tarnung! Direkt darunter liegt das Matrixprotein M1 – der molekulare Gerüstbauer, der alles zusammenhält. Es verbindet die äußere Hülle mit dem inneren Komplex und sorgt dafür, dass das Virus seine Form behält – wie ein Stützrahmen unter der Tarnkappe.
Die Spikes: Schlüssel & Schere
In der Virushülle befinden sich wichtige Oberflächenproteine, die wie kleine Stacheln oder Greifarme aus der Oberfläche herausragen. Diese werden „Spikes“ genannt. Das Influenzavirus besitzt zwei besonders wichtige Spikes, die ihm helfen, Zellen zu infizieren: Hämagglutinin (HA) und Neuraminidase (NA).
🔑 HA – der Türöffner: Dient als Schlüssel, um an der Wirtszelle anzudocken.
→ 18 bekannte Varianten (H1–H18)
✂️ NA – der Flucht-Helfer: Löst die Verbindung zur Wirtszelle, damit es weiterziehen kann.
→ 11 Varianten (N1–N11)
Virus-Typen: Ein Zahlenspiel
Die Kombination aus HA und NA bestimmt den Stamm:
- H1N1 (Schweinegrippe)
- H5N1 (Vogelgrippe)
- H3N2 (saisonale Grippe)
Wie bei Autokennzeichen: HA/NA-Codes verraten, welches Modell da unterwegs ist – nur eben ohne TÜV!
2.3. Der Infektionsprozess des Influenzavirus
a) Anheften des Virus an die Wirtszelle (Adsorption)
b) Eindringen in die Zelle (Endozytose)
c) Freisetzung des viralen Erbguts (Uncoating)
d) Virusreplikation – Die molekulare Fabrik
e) Zusammenbau (Assembly) der neuen Viruspartikel
f) Knospung (Budding) und Freisetzung der neuen Viren
a) Anheften des Virus an die Wirtszelle (Adsorption)
Die Oberfläche des Atemtrakts ist von einem dichten Epithel aus Schleimhautzellen ausgekleidet. Diese Zellen tragen Sialinsäure-Reste auf ihrer Oberfläche – Zuckermoleküle, die zentral für Zellkommunikation und immunologische Selbsterkennung sind (vgl. Kapitel „SELBST-Marker: Sialinsäuren“ in „Die Wunderwelt des Lebens“).
Das Influenzavirus kapert diesen Mechanismus: Sein Oberflächen-Protein Hämagglutinin (HA) bindet gezielt an die Sialinsäure der Wirtszelle – eine klassische Schlüssel-Schloss-Interaktion, die den Eintritt des Virus einleitet.
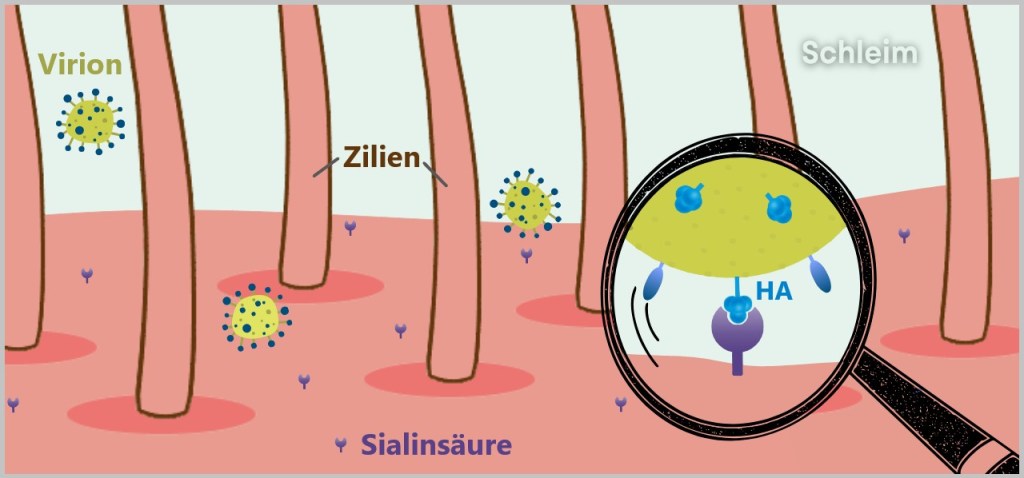
Bevor das Influenzavirus eine Zelle infizieren kann, muss es an die Wirtszelle andocken – ein entscheidender Schritt im Infektionsprozess. Doch die Epithelzellen der Atemwege sind nicht wehrlos: Ihre beweglichen Zilien (Flimmerhärchen) transportieren Fremdpartikel wie Staub, Bakterien oder Viren weg, bevor diese die Zelloberfläche erreichen können.
Die Darstellung zeigt die Größenverhältnisse an der Zelloberfläche. Die Zilien sind 5–10 Mikrometer lang, während die Schleimschicht eine Dicke von 10–100 Mikrometern aufweist. Mit nur 80–120 Nanometern ist das Viruspartikel winzig. Es muss schnell eine Zelle erreichen, bevor die Zilien es weiterbefördern.
Zwischen den Zilien gibt es freie Zellbereiche, an denen das Virus direkten Kontakt zur Zelloberfläche herstellen kann. Die Sialinsäure-Reste (1 Nanometer) auf der (Wirts-)Zellmembran dienen als Andockstelle für das HA-Protein (13 Nanometer) des Virus, das groß genug ist, um diese Strukturen zu erreichen. Das Virus kann somit die Schleimschicht durchdringen und an die Wirtszelle binden.
b) Eindringen in die Zelle (Endozytose)
Das Influenzavirus tarnt sich meisterhaft: Durch die Bindung seines Hämagglutinins an Sialinsäure-Reste imitiert es ein harmloses Nährstoffmolekül. Die Zelle fällt auf den Trick herein und initiiert ihren standardmäßigen Aufnahmemechanismus: die Endozytose.
Was folgt, ist ein molekulares Schauspiel:
Die Zellmembran stülpt sich um das gebundene Virus herum – ausgelöst durch Signalmoleküle, die eigentlich für den Nährstofftransport zuständig sind. Wie eine sich schließende Falle bildet sich eine Einbuchtung, die das Virus komplett umschließt. Mit einem letzten „Schnapp“ der Membran formt sich ein Endosom – ein Transportvesikel, das den Eindringling nun unschuldig ins Zellinnere schleust.
Was die Zelle als harmlosen Transport verbucht, entpuppt sich als Trojanisches Pferd.
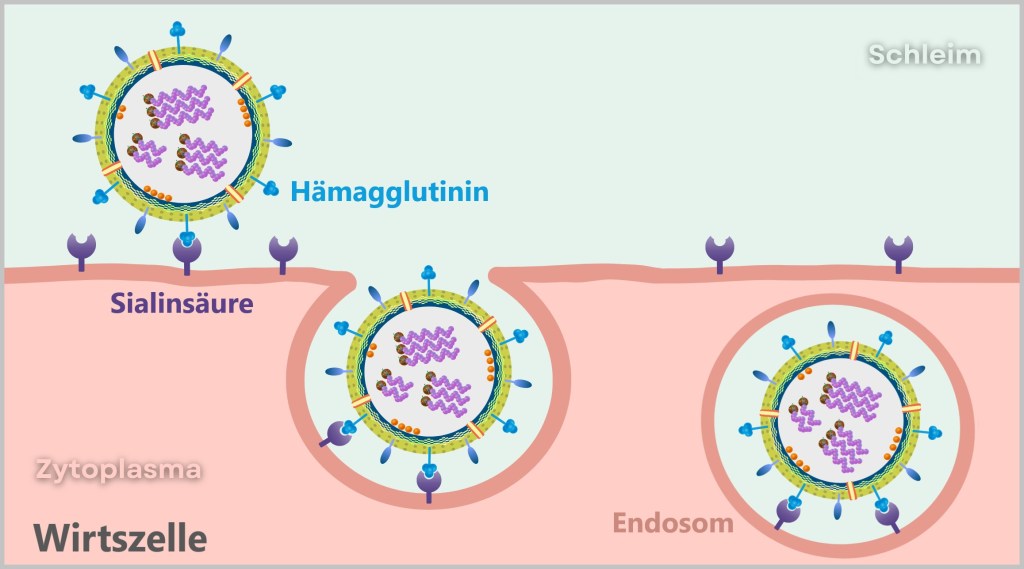
Das Virus ist jetzt in der Zelle – noch eingeschlossen im Endosom – aber bereit, sein Innerstes zu entfalten.
c) Freisetzung des viralen Erbguts (Uncoating)
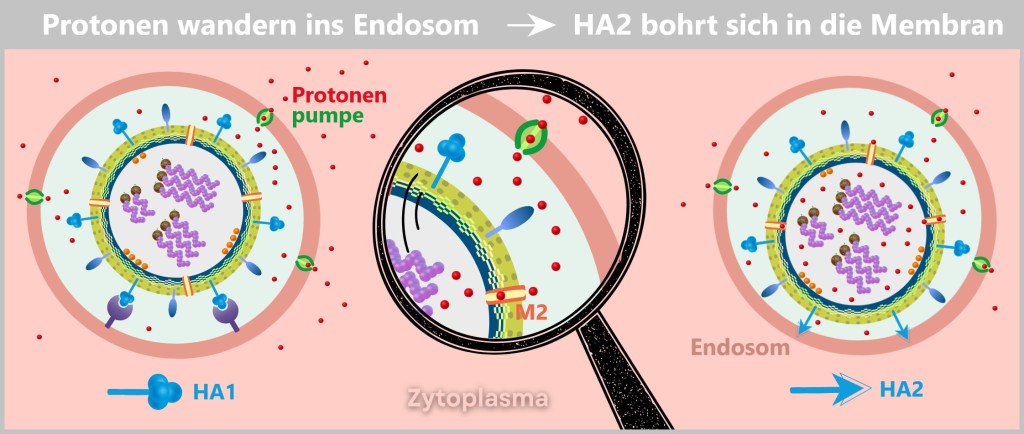
Das frühe Endosom reift zum späten Endosom heran – einem Ort, bei dem die Zelle normalerweise unerwünschte Eindringlinge abbaut. Protonenpumpen senken dort den pH-Wert, indem sie Protonen (H⁺-Ionen) ins Innere transportieren. Damit schaffen sie eine saure Umgebung, die Verdauungsenzyme aktivieren soll.
Der saure Trick
Doch das Influenzavirus hat einen genialen Gegenplan: Die saure Umgebung löst eine dramatische Umwandlung des viralen Hämagglutinins (HA) aus. Das Protein spaltet sich – die Bindungsdomäne HA1 wird abgespalten und die Fusionsdomäne HA2 freigelegt.

Diese Fusionsdomäne HA2 ist hydrophob – sie scheut Wasser – und rammt sich wie ein Enterhaken in die Endosomenmembran. (siehe untere Abbildung).
Gleichzeitig öffnet das M2-Protein – ein viraler Ionenkanal – als heimlicher Komplize die Schleusen: Protonen strömen ins Innere des Virus und lockern die Verpackung des Erbguts.
Jetzt zieht HA2 mit unerbittlicher Kraft die Virusmembran und die Endosomenmembran zusammen. Die beiden Lipidmembranen verschmelzen; ein Mechanismus, der als Membranfusion bekannt ist. Dies geschieht, weil die Lipidmoleküle in den Membranen flexibel sind und sich neu anordnen können, um eine kontinuierliche Doppelschicht zu bilden.

Diese Fusion schafft eine Pore – das Tor zur Freiheit für das virale Genom. Mit einem letzten, eleganten Schub gleitet die virale RNA ins Zytoplasma. Die Entkleidung – das Uncoating – ist vollendet.
Die Zelle ahnt nicht, dass sie soeben die Blaupause ihrer eigenen Unterwerfung freigesetzt hat.
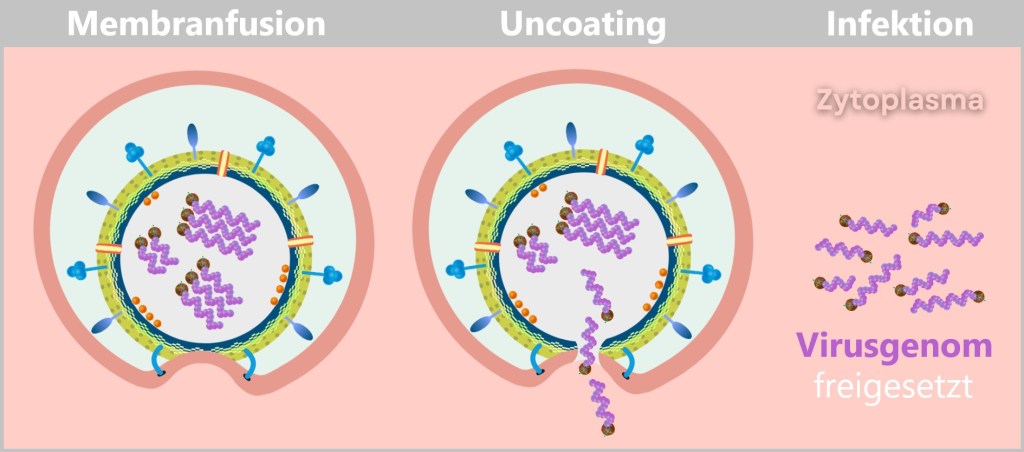
Durch Membranfusion und Uncoating wird die Virushülle aufgelöst, wodurch das Genom freigesetzt wird und die Infektion beginnt.
Warum wird das Virus nicht zersetzt?
Das Virus wird nicht von den Verdauungsenzymen der Zelle abgebaut, weil der Freisetzungsprozess schnell erfolgt, bevor der Abbaumechanismus (die Aktivierung der Verdauungsenzyme) ins Spiel kommen kann. Das Virus nutzt den Prozess der pH-Senkung und die Veränderungen im Endosom, um sich schnell aus diesem zu befreien, indem es die Membranfusion auslöst und sein Erbgut direkt in das Zytoplasma der Zelle entlässt. Diese „Flucht“ aus dem Endosom ist schneller als der zelluläre Abbauprozess, weshalb das Virus nicht zersetzt wird.
d) Virusreplikation – Die molekulare Fabrik
Die virale RNA kommt nicht schutzlos daher – sie reist in High-Tech-Rüstung: Eingehüllt in schützende Nukleoproteine (NP) und ausgestattet mit der viralen Polymerase bildet jedes der acht RNA-Segmente einen hochorganisierten Ribonukleoprotein-Komplex (RNP). Diese molekularen Kommandoeinheiten sind für den Einsatz perfekt gerüstet:
- Die Nukleoproteine wirken wie ein Panzer – sie schirmen die RNA gegen zelluläre Abwehrsysteme ab.
- Die Polymerase ist das Schweizer Taschenmesser des Virus – Werkzeug für Kopieren (Replikation) und Übersetzen (Transkription) in einem.

Sobald die Ribonukleoprotein-Komplexe (RNPs) im Zytoplasma freigesetzt sind, läuft die systematische Übernahme der zellulären Produktionslinien an – die Virusfabrik geht in Betrieb. Das Genom übernimmt dabei zwei zentrale Aufgaben: Zum einen dient es als Bauplan für die Herstellung viraler Proteine (Proteinsynthese), zum anderen wird es selbst vervielfältigt (Genomreplikation) – damit jeder neue Viruspartikel seine eigene Kopie des Erbguts mit auf den Weg bekommt.

Proteinsynthese: Das virale Genom dient als Vorlage für die Synthese der Proteine, die für den Aufbau neuer Viruspartikel benötigt werden.
Genomreplikation: Gleichzeitig wird die virale RNA vervielfältigt, um die genetische Information für neue Viren bereitzustellen.
Während die meisten RNA-Viren im Zytoplasma verbleiben, hat Influenza einen cleveren Trick auf Lager: Es hijackt den Zellkern. Warum? Dort findet es optimale Bedingungen für die Replikation seiner RNA.
Der Weg dorthin ist jedoch alles andere als selbstverständlich. Die RNPs manipulieren das zelluläre Transportsystem: Sie präsentieren gefälschte Importsignale – molekulare Passierscheine, die ihnen die Tür zum Zellkern öffnen. Zelluläre Importine, eigentlich zuständig für den Transport körpereigener Proteine, werden so zu ahnungslosen Schleusern. In einem Akt biologischer Täuschung werden die viralen RNPs direkt ins Kontrollzentrum der Zelle eskortiert.
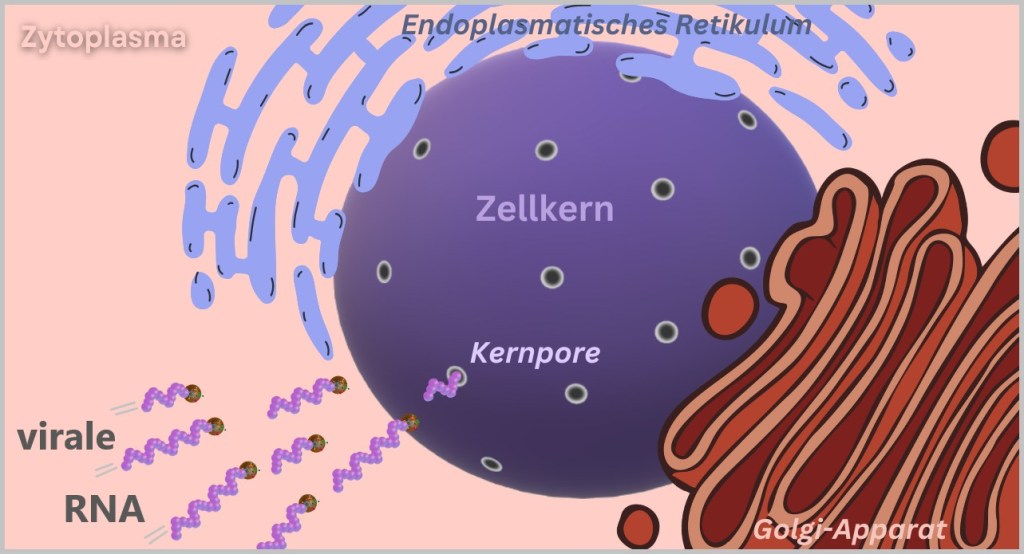
Im Zellkern entfalten die RNPs schließlich ihre volle Wirkung. Die virale Polymerase beginnt ihr Doppelspiel:
- Kopieren der viralen RNA (Replikation) → Bauplan für neue Viren
- Produktion viraler mRNA (Transkription) → Bauanleitung für Proteine
Da dieser Vorgang besonders raffiniert abläuft, wird im Folgenden jeder Schritt ausführlich beschrieben.
1️⃣ Aktivierung der RNA-Polymerase – Jetzt geht’s los
Die virale Polymerase benötigt einen molekularen Zündfunken, um aktiv zu werden. Und den findet sie im Zellkern: eine biochemische Spezialzone, die sich deutlich vom Zytoplasma unterscheidet. Hohe Konzentrationen von Nukleotiden, Ionen und kernspezifischen Faktoren senden ein klares Signal: „Hier ist der Ort, um loszulegen!“ Erst in dieser Umgebung erwacht die Polymerase zum Leben. Bleibt dieser molekulare Weckruf aus, verharrt sie im Ruhezustand – getarnt als harmloses Zellbestandteilchen.
2️⃣ Ausgangssituation: (-)ssRNA – Genom in Spiegelschrift
Das Erbgut des Influenzavirus ist ein Meister der Tarnung. Statt als lesbare Bauanleitung aufzutreten, erscheint es wie ein Rätsel in Spiegelschrift: acht einzelne RNA-Segmente, negativ gepolt, ohne die typischen Merkmale einer zellulären Nachricht. Kein Absender, kein Briefkopf, kein Poststempel. Für die Zelle ist das keine Nachricht – sondern biologisches Rauschen.
Wissenschaftlich ausgedrückt:
Das virale Genom besteht aus acht segmentierten Einzelsträngen RNA (engl. single-stranded RNA = ssRNA) mit negativer Polarität: (-)ssRNA. „Negativ“ bedeutet: Diese RNA ist die komplementäre Vorlage zur mRNA (also spiegelverkehrt) und somit nicht direkt lesbar.
Außerdem fehlen ihr zwei entscheidende Erkennungsmerkmale: das 5′-Cap (eine Art molekularer Startknopf) und die 3′-Poly-A-Sequenz, die eine normale mRNA schützt und identifiziert.
Warum so kompliziert?
Weil es genial ist.
Mit dieser molekularen Maskerade erreicht das Virus zwei Dinge:
Unsichtbar bleiben: Die (-)ssRNA wird vom zellulären Immunsystem nicht sofort als Bedrohung erkannt. Wäre das virale Genom schon als mRNA vorhanden, würden die Alarmsysteme der Zelle anspringen.
Volle Kontrolle über die Produktion: Das Virus bettelt nicht um Hilfe der Wirtsenzyme. Weil nur die viruseigene RNA-Polymerase in der Lage ist, aus der (-)ssRNA lesbare mRNA zu erzeugen, kann das Virus exakt steuern:
➤ Wann mRNA erzeugt wird.
➤ Wie viel davon produziert wird.
➤ Welche Segmente priorisiert werden.
Kurz gesagt: Was aussieht wie ein kryptisches Puzzle ist in Wahrheit ein hochpräziser Kontrollmechanismus – ein Bauplan, der sich erst dann offenbart, wenn die virale Maschinerie bereit ist – und das Immunsystem noch schläft.
3️⃣ Aus (-)ssRNA wird (+)ssRNA (die mRNA)
Die virale Polymerase steht bereit, die (-)ssRNA in lesbare mRNA umzuschreiben – doch es fehlt der Startknopf. Ohne den 5′-Cap bleibt die Maschinerie stumm.
Lösung? Diebstahl auf Nano-Ebene.
Dieser raffinierte Trick heißt Cap-Snatching – oder auf Deutsch: „Kappen-Schnappen“.
Der Coup im Detail
Die Polymerase-Untereinheit PB2 streift durch die zellulären mRNAs wie ein gerissener Dieb auf der Suche nach dem wertvollsten Schmuckstück. Ihr Ziel: Die 5′-Kappe, das universelle „Siegel“ für zelluläre Proteinfabriken. Ihre Komplizin, PB1 – die „molekulare Schere“ – trennt die Kappe mitsamt 10–15 Nukleotiden ab – ein perfekter Primer für die virale Transkription. Die gestohlene Kappe wird an die virale RNA geheftet. Die Zelle glaubt, sie habe eine legitime mRNA vor sich – und startet die Produktion viraler Proteine. Die gekappte Wirts-mRNA wird hingegen abgebaut – die zelluläre Proteinproduktion bricht zusammen.
Parallel erhält die virale mRNA am 3′-Ende einen Poly-A-Schwanz, der sie stabilisiert und schützt.
Warum dieser Trick so brillant ist
✅ Energieersparnis: Das Virus nutzt vorhandene Ressourcen – kein Aufwand für eine eigene Kappen-Synthese.
✅ Sabotage: Der Abbau der zellulären mRNAs legt die Wirtsabwehr lahm.
✅ Tarnung: Die gestohlene Kappe tarnt virale mRNA als „harmlose“ zelluläre Botschaft.
Die Folgen des Raubzugs
Die Zelle verliert ihre eigenen Baupläne – und produziert nun virale Proteine auf Hochtouren.
Das Virus gewinnt doppelt: Schnelle Vermehrung und Schwächung der Gegnerin.
Dieser Prozess ist ein Klassiker der Virologie – ein Paradebeispiel dafür, wie Viren ihre Wirtszellen zu Marionetten machen.
Am Ende entstehen zahlreiche „nackte“ (+)ssRNA-Stränge im Zellkern, die direkt als mRNA für die Translation – also die virale Proteinproduktion und Genomvermehrung – genutzt werden.
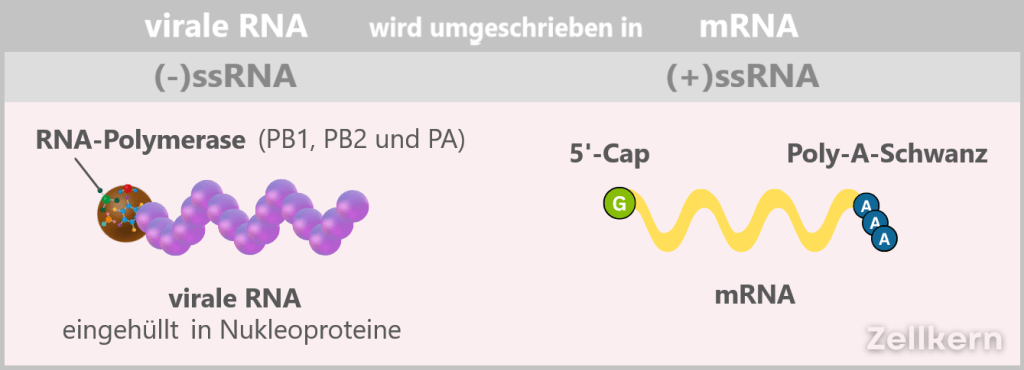
4️⃣ Die Virus-Produktion läuft heiß
Die frisch gekappten viralen mRNAs verlassen den Zellkern – ausgerüstet mit gestohlener Signatur und Poly-A-Schwanz. Im Zytoplasma erwarten sie die Ribosomen, die ahnungslos die Baupläne des Feindes abarbeiten.
Die Beute: Eine ganze Proteinfabrik
Die Ribosomen produzieren virale Proteine am Fließband – darunter:
Hämagglutinin (HA): Der Schlüssel zum Zelleintritt – der unentbehrliche Türöffner.
Neuraminidase (NA): Der Befreier neuer Viren – die scharfe molekulare Schere.
Matrixprotein (M1): Die stabile Hülle für das Virusinnere – der Gerüstbauer.
Ionenkanalprotein (M2): Der pH-Wächter – reguliert das Säuremilieu im Virus.
RNA-Polymerase: Die Kopiermaschine – eine virale Druckerpresse.
Nukleoprotein (NP): Die Bodyguards – verpacken und schützen die RNA-Segmente.
Nukleäres Exportprotein (NEP): Der Spediteur – sorgt für den Transport viraler RNPs aus dem Zellkern.
Diese frisch hergestellten Proteine sind bereit für den finalen Akt: die Montage neuer Virenpartikel.
5️⃣ Rückkehr in den Zellkern
Nach ihrer Produktion im Zytoplasma machen sich die meisten viralen Proteine zurück auf den Weg zum Zellkern – der Kommandozentrale der Virusreplikation. Ausgenommen sind nur die Oberflächenstars Hämagglutinin (HA), Neuraminidase (NA) und das Ionenkanalprotein M2, die direkt an der Zellmembran ihre Einsätze haben.
Die übrigen Virenakteure kehren ins Hauptquartier zurück, um neue Befehle abzuholen und die End-Mission vorzubereiten.

1) An freien Ribosomen im Zytoplasma wird die mRNA in virale Proteine wie RNA-Polymerase, Matrixproteine (M1), Nukleoproteine (NP) und nukleäre Exportproteine (NEP) übersetzt.
2) Diese Proteine wandern anschließend zurück in den Zellkern, um an der Replikation und Verpackung des viralen Genoms mitzuwirken.
3) An den festen Ribosomen des endoplasmatischen Retikulums (ER) werden die Oberflächenproteine (HA und NA) und das Ionenkanalprotein (M2) synthetisiert. Diese Proteine gelangen nach ihrer Herstellung zum Golgi-Apparat, wo sie weiter modifiziert und für den Einbau in die virale Hülle vorbereitet werden.
6️⃣ Die Genom-Kopierfabrik: (+)ssRNA → neue (-)ssRNA
Während im Zytoplasma fleißig virale Proteine vom Band laufen, läuft im Zellkern die Geheimoperation „Genom-Vervielfältigung“:
Frisch gebildete RNA-Polymerasen schnappen sich die neu synthetisierten (+)ssRNA-Stränge und übersetzen sie zurück in virale Spiegelschrift – heraus rollen neue (-)ssRNA-Stränge. Die Polymerase bleibt daran gebunden als integrierte Druckerpresse für künftige Einsätze.
Nichts bleibt dem Zufall überlassen: Noch während der genetische Code rückübersetzt wird, umhüllen Nukleoproteine (NP) die entstehende (-)ssRNA – sie hat nicht einmal eine Sekunde, um „nackt“ zu sein – kein Risiko, dass die zelluläre Überwachung zugreift. Die frisch kopierten RNA-Segmente werden sofort verpackt und versiegelt: Zusammen mit der Polymerase bilden sie wieder vollständige Ribonukleoprotein-Komplexe (RNPs) – ein komplett ausgestattetes Genom-Modul, bereit für die nächste Generation Virus.
Sobald die acht Segmente verpackt sind, übernimmt das virale Logistik-Team: NEP (Exportprotein) und M1 (Matrixprotein) markieren die RNPs für den Abtransport. Sie schleusen sie durch die Kernporen – die streng bewachten Grenzübergänge der Zelle – direkt ins Zytoplasma. Mission: Montagehalle.
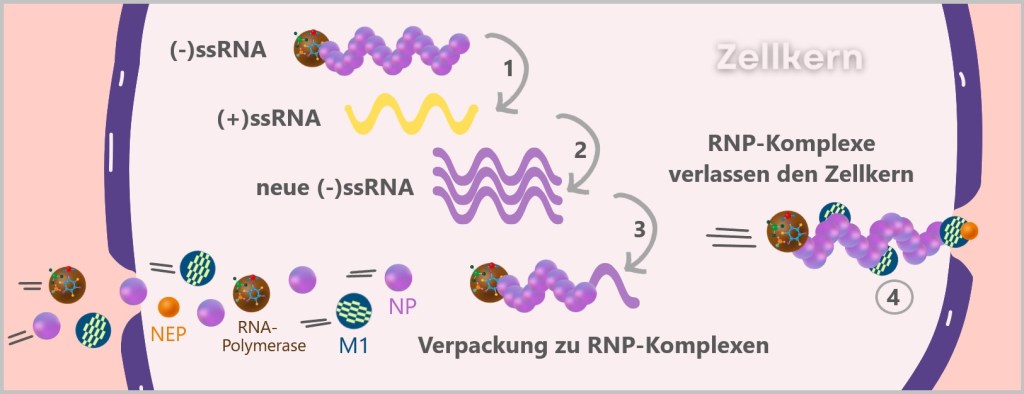
1) Die virale RNA-Polymerase nutzt die (-)ssRNA als Vorlage und synthetisiert daraus eine komplementäre (+)ssRNA.
2) Diese (+)ssRNA dient nun als Matrize für die erneute Synthese von viraler (-)ssRNA – also der eigentlichen Erbinformation für neue Viruspartikel.
3) Bereits während der Synthese wird die neue (-)ssRNA von Nukleoproteinen (NP) umhüllt und mit Polymerase, M1 und NEP zum sogenannten RNP-Komplex verpackt – stabil und bereit zum Export.
4) Die fertigen RNP-Komplexe verlassen den Zellkern über die Kernporen und wandern ins Zytoplasma – dort beginnt bald der Zusammenbau neuer Viren.
Wie eine Schwarzdruckerei im Hinterzimmer: Die Polymerase produziert ununterbrochen Kopien, die NP-Proteine verpacken sie sofort – und Schleuser (NEP/M1) schmuggeln sie unauffällig hinaus.
Während die Zelle ahnungslos ihre Ressourcen verheizt, steht der eigentliche Showdown noch bevor…
e) Zusammenbau (Assembly) der neuen Viruspartikel
Nachdem alle Bauteile produziert sind, beginnt im Zytoplasma die koordinierte Endmontage – ein Prozess so präzise wie die Konstruktion einer Raumsonde: Jedes Teil muss perfekt sitzen, sonst hebt nichts ab.
Die Oberflächenproteine HA & NA reisen über den Golgi-Apparat – die „Verpackungsabteilung“ der Zelle – zur Zellmembran (untere Abb. links). Dort verankern sie sich in der Lipiddoppelschicht, wie Türgriffe und Rettungsscheren, die aus der Hülle eines künftigen Viruspartikels ragen.
Auch die Ribonukleoprotein-Komplexe (RNPs) machen sich auf den Weg, bereits im Schlepptau der Matrixproteine M1, die als Logistikmanager fungieren. Ihre Aufgabe: Die wertvolle Fracht zielsicher zu den HA/NA-bestückten Membranbereichen zu navigieren (untere Abb. rechts).
An der Zellmembran fügt sich das virale Puzzle Stück für Stück zusammen: Die RNPs ordnen sich unter der mit HA/NA gespickten Membran an. Die Matrixproteine helfen dabei, die RNPs mit den Bereichen der Zellmembran in Kontakt zu bringen.
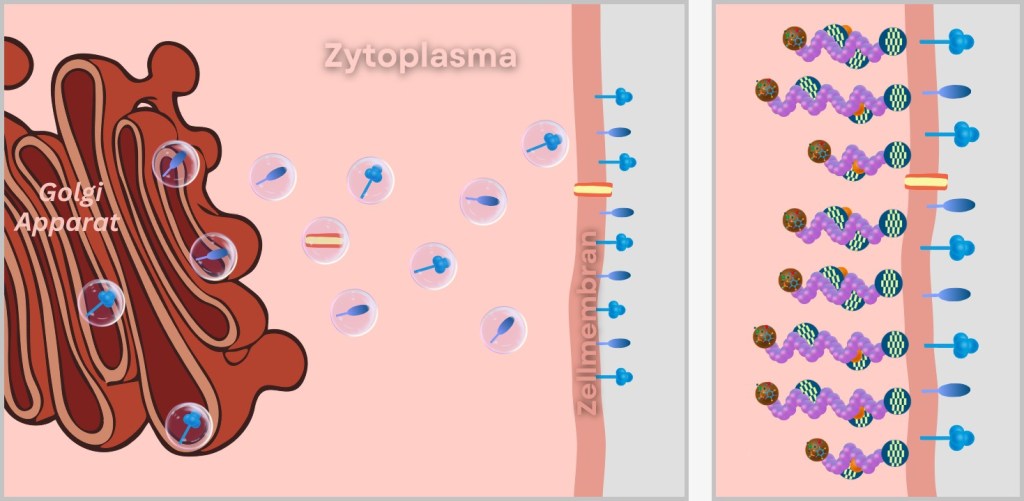
Links: Einbau der viralen Oberflächenproteine (HA und NA) und des Ionenkanalproteins (M2) in die Zellmembran.
Rechts: Transport der Ribonukleoprotein-Komplexe (RNPs) zur Zellmembran und Bindung an die entstehende Virushülle.
Und jetzt – Trommelwirbel – ist alles bereit für den großen Ausbruch!
f) Knospung (Budding) und Freisetzung der neuen Viren
An der Zellmembran formt sich eine Ausstülpung – wie eine Seifenblase mit tödlicher Fracht. Doch was so spielerisch aussieht, ist präzise Choreografie:
➤ Virusproteine drängen nach außen, die Lipidschicht wölbt sich zum perfekten „Virus-Paket“.
➤ Matrixproteine (M1) spannen die Membran wie ein Trampolin – stabil, aber flexibel genug für den Absprung.
➤ Die Wirtslipide schließen sich zur getarnten Hülle – das Virus verpackt sich selbst.
Doch noch ist es nicht frei. An der Zelloberfläche lauern Sialinsäure-Fesseln – normalerweise HAs Lieblings-Ankerplatz. Ohne Gegenwehr würde das Virus kleben bleiben wie Kaugummi unter der Schuhsohle.
Neuraminidase (NA) greift ein: Die molekulare Schere zerschnippelt die Sialinsäure-Reste auf der Zelloberfläche. Kein Haften, kein Zurück – freie Bahn zur nächsten Zelle.
Wie ein Gefängnisausbruch mit Style: M1 lockert die Gitterstäbe, NA durchtrennt die Alarmsysteme – und weg sind sie! Final Countdown für die Virus-Crew! Alle Systeme go – HA/NA check, RNPs check, Lipidpanzer check. Startsequenz initiiert in 3…2…1… Budding!

Left: Budding of the virus at the cell membrane. Right: Release of the newly formed virus particle.
Das folgende Video fasst den Replikationszyklus des Influenzavirus noch einmal gut verständlich zusammen.
Nach der Infektion einer Wirtszelle durch ein einziges Influenzavirus entstehen typischerweise Hunderte bis Tausende neuer Viruspartikel. Die genaue Anzahl variiert und hängt von verschiedenen Faktoren wie dem Virusstamm, der Art der Wirtszelle und den zellulären Bedingungen ab.
Vereinfachte und reale Darstellung der Virusstruktur
In den ersten Abbildungen dieses Textes wurde die Struktur des Influenzavirus zur besseren Übersicht vereinfacht dargestellt (siehe untere Abbildung links). Das Matrixprotein bildet in diesen Darstellungen eine kugelige, netzartige Ringstruktur, die das virale Genom umgibt. Diese vereinfachte Darstellung soll die komplexen Prozesse der Virusvermehrung leichter nachvollziehbar machen.
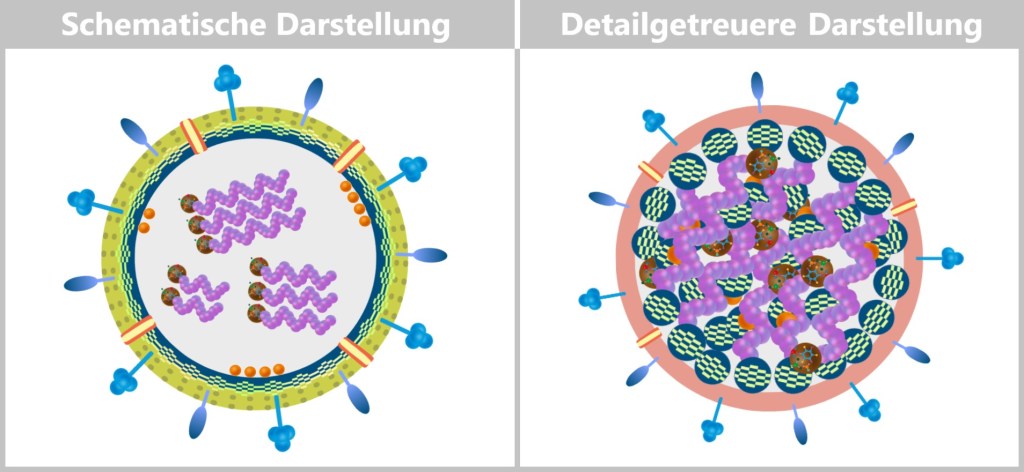
Links: Schematische Darstellung zur Verdeutlichung des Virusaufbaus
Rechts: Detailgetreuere schematische Darstellung der Virionenstruktur
In den folgenden Abbildungen wurde jedoch die Struktur der neuen Virionen näher an der biologischen Realität gezeigt. Dabei sind die Matrixproteine nicht als durchgehender Ring abgebildet, sondern befinden sich in einzelnen Einheiten, die sowohl an die innere Lipidschicht binden als auch lose mit den Ribonukleoprotein-Komplexen (RNPs) verknüpft sind. Zudem spiegelt die Farbgebung der Lipidschicht die Herkunft aus der Zellmembran der Wirtszelle wider (siehe obere Abbildung rechts).
Die RNPs, die das virale Genom darstellen, liegen im Inneren als lockeres Bündel vor – nicht streng parallel, sondern flexibel angeordnet mit unterschiedlich ausgerichteten Enden. Die Matrixproteine (M1) halten dieses Bündel zusammen und verbinden es mit der Lipidschicht, wodurch das Virion seine Form und Stabilität erhält.
2.4. Die Anpassungsfähigkeit des Influenzavirus
Viren – insbesondere RNA-Viren wie das Influenzavirus – mutieren außergewöhnlich schnell. Der Grund liegt in ihrer fehleranfälligen Replikationsmaschinerie: Die virale RNA-Polymerase besitzt keinen Mechanismus zur Korrektur von Kopierfehlern, wie es bei der DNA-Replikation in menschlichen Zellen der Fall ist. Dadurch entstehen bei jeder Vervielfältigung zufällige Mutationen – kleine Veränderungen im genetischen Material des Virus.
Innerhalb einer infizierten Person bildet sich so eine Vielzahl leicht unterschiedlicher Viruspartikel. Die meisten Mutationen sind neutral, das heißt, sie beeinflussen weder die Funktionsweise des Virus noch seine Fähigkeit, sich zu vermehren. Einige Mutationen sind jedoch nachteilig und führen dazu, dass das Virus weniger effizient repliziert oder gar nicht mehr infektiös ist – diese Varianten verschwinden rasch durch natürliche Selektion.
Doch manche Mutationen verschaffen dem Virus einen Überlebensvorteil, insbesondere wenn sie die Oberflächenproteine Hämagglutinin (HA) und Neuraminidase (NA) betreffen. Diese Proteine sind zentrale Angriffspunkte des Immunsystems: Der Körper produziert Antikörper, die spezifisch an sie binden und das Virus neutralisieren. Verändert sich jedoch die Struktur von HA oder NA durch Mutationen, können die Antikörper das Virus schlechter erkennen. Das Virus wird sozusagen „unsichtbar“ für die Immunabwehr und kann sich weiterhin vermehren und ausbreiten.
Diese ständige Anpassung erklärt, warum Grippeviren jedes Jahr erneut Infektionswellen auslösen und es schwierig ist, dauerhafte Impfstoffe gegen Influenza zu entwickeln.
Das Influenzavirus existiert nicht als starre genetische Einheit, sondern als eine sogenannte Mutantenwolke (Quasispecies) – eine dynamische Population von Virusvarianten, die durch kontinuierliche Mutationen entsteht. Diese genetische Vielfalt ist der Schlüssel zu seinem Überleben: Natürliche Selektion sorgt dafür, dass sich jene Varianten durchsetzen, die unter den gegebenen Bedingungen am erfolgreichsten sind. Diese hohe Anpassungsfähigkeit des Influenzavirus zeigt eindrucksvoll, wie Evolution in Echtzeit abläuft.
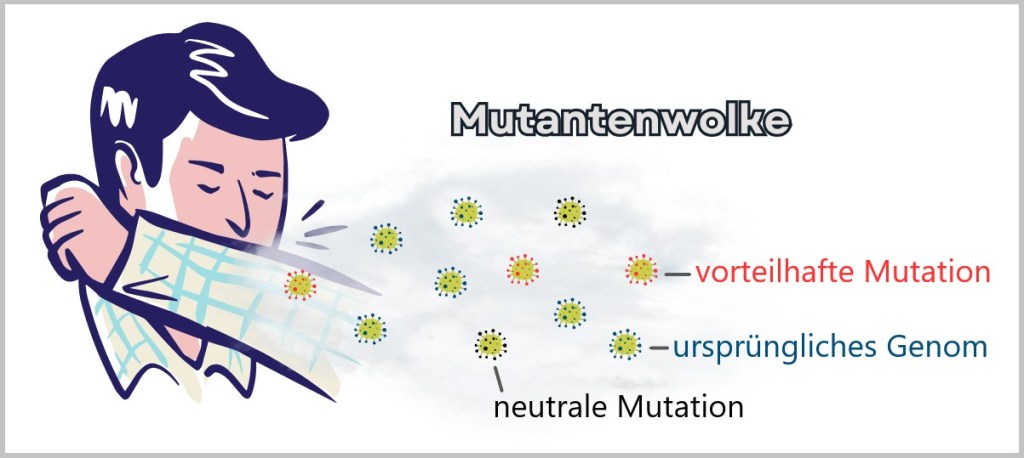
Das Influenzavirus muss sich ständig durch Mutationen verändern, um weiterhin als Grippevirus zu existieren. Die hohe Mutationsrate führt zu einer Vielzahl leicht unterschiedlicher Viruspartikel innerhalb einer infizierten Person.
2.5. Ein- und Austrittswege des Influenzavirus
Influenzaviren nutzen die Schleimhautoberflächen der Atemwege als Eintrittspforte, da Schleimhäute die Grenze zwischen der äußeren Umwelt und unserem Körperinneren darstellen. Viele Viren starten ihre Infektion über die Interaktion mit den Epithelzellen dieser Schleimhäute, um sich gezielt in ihrem Wirt zu verbreiten. [Virus Infection of Epithelial Cells]
Wie in der unteren Zeichnung dargestellt, gelangt das Influenzavirus über die Luft in die Atemwege – in Nase, Rachen und Lunge – und bindet an die apikale Seite (obere Seite) der Epithelzellen, die zur äußeren Umgebung hin orientiert ist. Diese apikale Seite ist mit feinen, haarartigen Strukturen, den Zilien, bedeckt, die Schleim und Fremdpartikel transportieren. Die gegenüberliegende, basolaterale Seite der Zelle ist dem darunterliegenden Gewebe zugewandt und mit der Basalmembran verbunden, die sie am Bindegewebe verankert.
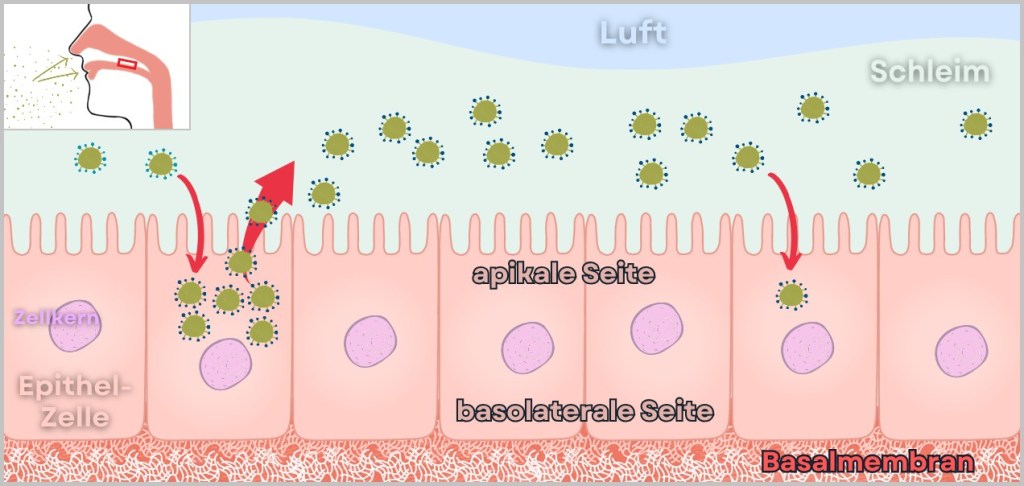
Auch die Freisetzung der neugebildeten Influenzaviren erfolgt gezielt an der apikalen Seite. Durch diese Anordnung können sich die Viren in die Umgebung verbreiten, etwa über Tröpfchen beim Husten oder Niesen, und dadurch leicht neue Wirte infizieren. Diese apikale Freisetzung stellt einen evolutionären Vorteil dar, da sie die Effizienz der Übertragung deutlich steigert.
2.6. In den meisten Fällen lokal begrenzte mukosale Infektion
Influenzaviren sind auf Infektionen der Schleimhautoberfläche spezialisiert. Ihre Infektion bleibt daher meist lokal auf die Epithelzellen der Atemwege, das heißt, auf eine mukosale Infektion beschränkt. Das Virus breitet sich entlang der apikalen Seite der Epithelzellen von Zelle zu Zelle aus, ohne dabei in tiefere Gewebeschichten zu gelangen. Selbst bei einer Ausbreitung von den oberen Atemwegen bis hin zur Lunge bleibt die Infektion auf die Schleimhautoberfläche begrenzt.
Die basolaterale Seite der Epithelzellen bleibt in der Regel unberührt, da sie für die Virusübertragung keine Rolle spielt. Würde das Virus aus der basolateralen Seite der Wirtszelle austreten, könnte es ins umliegende Gewebe und letztlich in das Blut- oder Lymphsystem gelangen, was zu einer systemischen Infektion führen könnte. Für Influenzaviren wäre dies jedoch von Nachteil, da sie dann einer stärkeren Immunabwehr ausgesetzt wären und die Übertragung über die Atemwege erschwert würde.
In seltenen Fällen – besonders bei stark geschwächten Personen – kann das Virus jedoch die Epithelbarriere durchbrechen und in das darunterliegende Gewebe sowie in Blut- oder Lymphgefäße eindringen und zu einer systemischen Infektion führen.

Die Schleimhaut besteht aus mehreren Schichten: Epithelzellen bilden die äußere Schutzschicht, die Basalmembran trennt als dünne Barriere, die Lamina propria stützt mit Gewebe und Immunzellen, die Endothelzellen bilden die Wand der Blutgefäße, und das Blutgefäß führt ins Körperinnere.
Links – Mukosale Infektion (begrenzt auf die Schleimhaut): Das Virus infiziert Epithelzellen ausschließlich über die apikale Seite. Es verbleibt in der Schleimhaut, wobei es von Zelle zu Zelle entlang der apikalen Oberfläche weitergegeben wird. Die Basalmembran und darunterliegende Gewebe wie die Lamina propria bleiben intakt. Eine mukosale Infektion ist lokal begrenzt und begünstigt die Übertragung über die Schleimhäute, etwa durch Atemwege.
Rechts – Systemische Infektion (Ausbreitung über das Blut): Das Virus tritt auf der apikalen Seite in die Epithelzellen ein, verlässt sie jedoch über die basolaterale Seite. Es durchdringt die Basalmembran und bewegt sich durch die Lamina propria, entweder durch Wanderung oder durch Infektion der dortigen Zellen. Schließlich erreicht es ein Blutgefäß, indem es durch Spalten zwischen Endothelzellen oder durch direkte Infektion der Endothelzellen in das Gefäßsystem eindringt. Der Eintritt des Virus in die Blutbahn markiert den Übergang zu einer systemischen Infektion. Eine systemische Infektion ist kritisch, weil das Virus über das Blut den ganzen Körper erreichen kann und so lebenswichtige Organe wie Lunge, Herz oder Gehirn schädigen könnte.
2.7. Virenstrategie: Effiziente Vermehrung ohne rasche Zellzerstörung
Manche Viren – darunter auch Influenzaviren – sind erstaunlich ökonomisch: Statt ihre Wirtszelle sofort zu zerstören, nutzen sie deren Ressourcen möglichst effizient aus. Warum die Wohnung abfackeln, wenn man monatelang kostenlos wohnen kann? Solange die Zelle intakt bleibt, liefert sie alles, was das Virus zur Replikation braucht: Energie, Enzyme, Bausteine. Und auch das Immunsystem merkt erst später, dass was faul ist – denn wo nichts brennt, wird kein Alarm ausgelöst. Diese Strategie verlängert das Leben der infizierten Zelle, verzögert die Immunantwort – und maximiert die Produktion neuer Viren.
Viren-Weisheit: Die besten Parasiten bleiben unterm Radar!
2.8. Zerstörung der Wirtszelle
Was mit listiger Schonung beginnt, endet in molekularem Burnout: Die infizierte Zelle erleidet letztlich den Zelltod. Dieser tritt ein, wenn die Zelle entweder durch die Massenproduktion der Viren überlastet und strukturell geschädigt wird, durch zelluläre Schutzmechanismen in den programmierten Zelltod (Apoptose) übergeht oder vom Immunsystem gezielt eliminiert wird. Diese charakteristischen Veränderungen der Wirtszelle durch das Virus werden als zytopathische Effekte (CPE) bezeichnet. Dieser gesamte Prozess kann bereits innerhalb von 24 Stunden nach der Infektion stattfinden.
Um den Zelltod durch Influenzaviren besser zu verstehen, betrachten wir die drei Mechanismen, durch die die Wirtszelle schließlich zerstört wird.
a) Überlastung und strukturelle Schädigung
b) Apoptose: Programmierter Zelltod zur Virusbekämpfung
c) Immunantwort: Zerstörung durch das Immunsystem
a) Überlastung und strukturelle Schädigung
Das Virus übernimmt die zellulären Prozesse zur Produktion seiner eigenen Bestandteile. Mit jeder neuen Generation viraler Proteine und RNA wird die Energie und Ressourcenkapazität der Zelle zunehmend aufgebraucht. Da die Zelle praktisch nur noch für die Virusvermehrung arbeitet, kommen ihre eigenen überlebenswichtigen Prozesse zum Erliegen. Die Zelle wird zur ausgepressten Zitrone – Ribosome laufen heiß, Mitochondrien kollabieren.
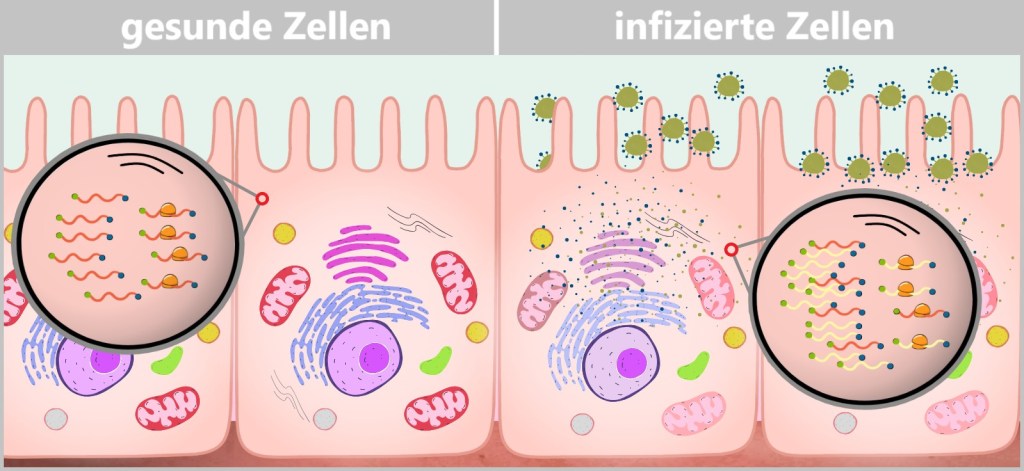
Links: Gesunde Zelle mit funktionierender zellulärer Maschinerie. Die körpereigene mRNA (orange) wird von den Ribosomen gelesen, um Proteine für die Zellfunktionen herzustellen. Die Zelle zeigt eine lebhafte Färbung, die auf die volle Ressourcenkapazität und Energie hinweist. Rechts: Virusinfizierte Zelle, stark belastet durch die Produktion viraler Bestandteile. Die virale mRNA (gelb) verdrängt zunehmend die körpereigene mRNA, und die Ribosomen lesen überwiegend virale Anweisungen für die Virusvermehrung. Die blasse Färbung der Zellorganellen symbolisiert die Erschöpfung der Energiereserven und die Überlastung der Zelle.
Während der Knospung an der Zellmembran – wenn neue Viruspartikel die Zelle verlassen – wird die Zellmembran mehrfach durchbrochen und verformt. Dieser Prozess führt letztlich zur Zerstörung der Membranintegrität, wodurch die Zelle ihre Stabilität und ihre schützenden Funktionen verliert. Die Zelle stirbt schließlich durch den unaufhörlichen Ressourcenverbrauch und den strukturellen Zerfall infolge der Virusfreisetzung.
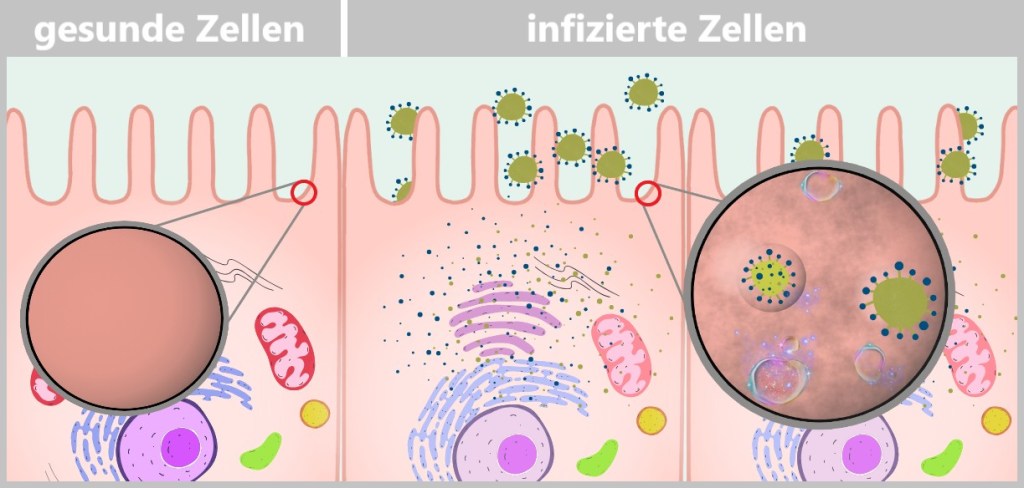
Während des Knospens bildet die Membran kleine Ausstülpungen, aus denen die neuen Viren freigesetzt werden. Jedes Knospungsereignis entzieht der Zellmembran kleine Teile ihrer Lipiddoppelschicht, da die neu entstehenden Viren Membranmaterial der Wirtszelle als ihre Hülle verwenden. Nach wiederholten Virusfreisetzungen ist die Membran deutlich ausgedünnt und strukturell geschwächt, die Membran zeigt oft Verformungen und Unebenheiten. Die dauerhafte Belastung durch die Knospung führt dazu, dass die Membran poröser und anfälliger wird. Die Zelle verliert zunehmend die Fähigkeit, ihren inneren Zustand zu regulieren und kann ihre selektive Durchlässigkeit für Ionen und Moleküle kaum noch aufrechterhalten. Da die Membran fortwährend geschädigt wird, geht ihre strukturelle Stabilität verloren. Letztlich kann die Membran so stark beeinträchtigt werden, dass sie reißt oder zerfällt, was zum Zelltod führt.
b) Apoptose: Programmierter Zelltod zur Virusbekämpfung
Wenn eine Zelle merkt, dass sie gekapert wurde, zieht sie manchmal die Notbremse – und opfert sich für das größere Ganze: Sie bringt sich selbst um, um die Ausbreitung zu stoppen. Der Plan: Den Feind mit ins Grab nehmen. Statt mit einem Knall zu sterben, zerfällt die Zelle kontrolliert in kleine Fragmente, die sogenannte Apoptose-Körper bilden, welche anschließend von Immunzellen abgebaut werden.
Dabei laufen präzise Prozesse ab: Die DNA wird zerschnitten, die Zellmembran bildet typische blasenartige Ausstülpungen (sogenannte Blebbing), die inneren Strukturen werden fein säuberlich recycelt. Die Zelle stirbt still – und schützt damit den Organismus.
Dieser Mechanismus beweist – Zellen haben mehr Ehre als manche Regierungen!
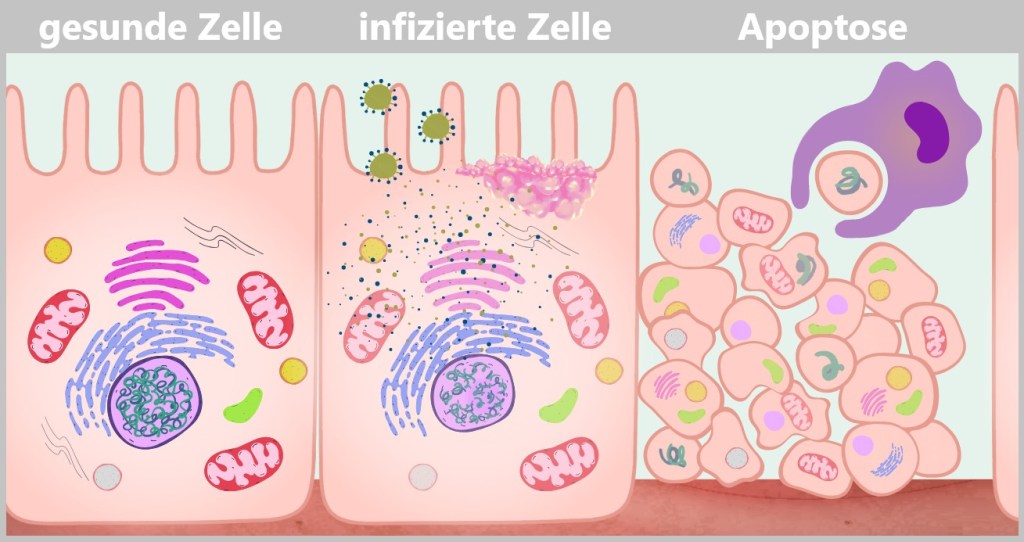
Gesunde Zelle: Links ist eine intakte Zelle dargestellt, deren Zellmembran und Zellkern unversehrt sind. Der Zellkern enthält vollständige DNA-Stränge, und die Zelle zeigt keine Anzeichen von Stress oder Schädigung.
Infizierte Zelle – Einleitung der Apoptose: Im mittleren Abschnitt beginnt die Zelle sichtbare Veränderungen zu zeigen. Die Zellmembran bildet blasenartige Ausstülpungen (Blebbing), und der Zellkern schrumpft. Innerhalb des Zellkerns sind DNA-Bruchstücke sichtbar, die durch apoptotische Prozesse entstehen. Diese Phase stellt den Übergang von einer funktionierenden Zelle zu ihrem kontrollierten Zerfall dar.
Apoptose: Rechts zerfällt die Zelle in mehrere kleine Fragmente, sogenannte Apoptose-Körper. Im Hintergrund ist eine Immunzelle (Makrophage) dargestellt, die diese Fragmente aufnimmt und abbaut. Dies verhindert die Freisetzung viraler Bestandteile und schützt das umliegende Gewebe vor einer weiteren Infektion.
c) Immunantwort: Zerstörung durch das Immunsystem
Sobald das Immunsystem eine virusinfizierte Zelle entdeckt, schlägt es Alarm – und das heißt Ärger für das Virus! Spezialisierte Kämpfer wie natürliche Killerzellen (NK-Zellen) und zytotoxische T-Zellen stürzen sich ins Gefecht. Sie erkennen die infizierten Zellen anhand viraler Proteinsignale, die wie Warnflaggen auf der Zell-Oberfläche wehen. Mit tödlicher Präzision setzen sie toxische Moleküle frei, zerstören die Zelle und bremsen so die Virusvermehrung aus!
Wer mehr über diese faszinierende Abwehrschlacht wissen will, findet Details in „Die Wunderwelt des Lebens“, besonders in Kapitel 5.3 d) „Natürliche Killerzellen“ und Kapitel 5.5.7 b) „Zytotoxische T-Zellen“.
NK-Zellen und zytotoxische T-Zellen sind ein unschlagbares Team – sie jagen und eliminieren virale Bedrohungen und halten die Infektion in Schach.
Regeneration der Epithelzellen
Wie bereits erwähnt, befällt das Influenzavirus vor allem die Epithelzellen der Atemwege – insbesondere die Zellen der Nasenschleimhaut, der Bronchien und der Lungenbläschen (Alveolen). Durch die Virusvermehrung sowie die Immunabwehr, insbesondere durch natürliche Killerzellen (NK-Zellen) und zytotoxische T-Zellen, werden viele dieser Zellen stark geschädigt oder zerstört.
Sobald die akute Infektion eingedämmt ist, setzt der Reparaturprozess ein: Spezialisierte Stammzellen beginnen mit der Regeneration des Gewebes. Sie vermehren sich und entwickeln sich zu den verschiedenen Epithelzelltypen, die für die Wiederherstellung der Atemwege benötigt werden.
- Obere Atemwege (Nase, Bronchien): Neue Zilienzellen entstehen, deren feine Härchen (Zilien) Schleim und Fremdpartikel nach oben transportieren. Zudem werden Becherzellen gebildet, die den Schleim produzieren und so die Atemwege feucht und geschützt halten.
- Lunge (Alveolen): Hier werden die geschädigten, flachen Epithelzellen ersetzt, die den Sauerstoffaustausch zwischen Luft und Blut ermöglichen.
Die Regeneration dauert je nach Schwere der Infektion unterschiedlich lange. Bei einer milden Erkrankung kann das Epithel innerhalb von ein bis zwei Wochen vollständig erneuert sein. Nach schwereren Infektionen, etwa bei einer Influenza-Pneumonie, kann der Heilungsprozess jedoch mehrere Wochen in Anspruch nehmen. Sobald die neuen Zellen eine dichte Schicht bilden, ist die Schutzfunktion der Atemwege wiederhergestellt. In den meisten Fällen erfolgt die Regeneration vollständig – bei sehr schweren Schäden können jedoch Narben oder strukturelle Veränderungen im Epithel zurückbleiben.
2.9. Selbstbegrenzung gefährlicher Viren: Warum sie selten Pandemien auslösen
Hochgefährliche Viren, die ihren Wirt rasch töten, begrenzen ihre eigene Verbreitung. Wenn ein Virus seinen Wirt so schnell schädigt, dass dieser keine Zeit hat, andere zu infizieren, wird die Übertragungskette effektiv unterbrochen. Ein Beispiel dafür ist das Ebola-Virus, das oft lokal begrenzt bleibt und daher selten Pandemien auslöst.
Im Gegensatz dazu lösen Viren mit moderater Pathogenität häufiger globale Ausbrüche aus. Moderate Pathogenität beschreibt die Fähigkeit eines Erregers, Krankheiten auszulösen, ohne dabei extrem schwere oder tödliche Verläufe bei den meisten Infizierten zu verursachen. Solche Viren führen typischerweise zu milden bis mittelschweren Symptomen, die es den infizierten Personen ermöglichen, weiterhin mobil und sozial aktiv zu bleiben. Dadurch erhöhen sich die Chancen für die Weitergabe des Virus an andere. Beispiele dafür sind viele Influenzaviren. Schwere Verläufe einer Influenza-Infektion treten dabei vorwiegend bei Personen mit geschwächtem Immunsystem, höherem Alter oder bestehenden Grunderkrankungen auf. In diesen Fällen sind eine sorgfältige medizinische Überwachung und eine intensivierte Behandlung angebracht, um schwere Komplikationen zu verhindern.
Die erfolgreichsten Viren sind nicht die, die uns umbringen – sondern die, die uns gerade so am Leben lassen, dass wir ihre Drecksarbeit erledigen.
2.10. Warum macht das Virus manche Menschen krank und andere nicht?
💡Hinweis: Für ein besseres Verständnis dieses Abschnitts empfehlen wir das Kapitel 5 in der Abhandlung „Die Wunderwelt des Lebens“. Dort werden die Grundlagen zum Immunsystem auf anschauliche Weise erklärt.
Nicht jede Person, die sich mit dem Influenzavirus infiziert, erkrankt gleich schwer: Manche haben nur leichte Symptome wie einen Schnupfen, andere entwickeln eine schwere Grippe mit Fieber und Atemnot, und einige bleiben sogar völlig symptomfrei. Warum ist das so? Die Antwort liegt in einer komplexen Wechselwirkung zwischen dem Virus und dem Wirt – also dem Menschen, der infiziert wird. Mehrere entscheidende Faktoren spielen dabei eine Rolle:
Das Immunsystem des Wirts
Jeder Mensch hat ein individuelles Immunsystem, das unterschiedlich gut auf das Influenzavirus reagiert. Frühere Grippeinfektionen können eine Teilimmunität bieten, weil das Immunsystem Antikörper und Gedächtniszellen entwickelt hat, die das Virus schneller erkennen und bekämpfen. Ein starkes Immunsystem kann die Infektion so im Keim ersticken, während ein geschwächtes Immunsystem (z. B. bei älteren Menschen oder chronisch Kranken) oft überfordert ist.
Die Viruslast
Die Menge an Viruspartikeln, die beim ersten Kontakt in den Körper gelangen – die sogenannte Viruslast –, beeinflusst den Verlauf der Infektion. Bei einer geringen Viruslast kann das angeborene Immunsystem die Eindringlinge schnell erkennen und zerstören, bevor sie sich stark vermehren. Eine hohe Viruslast, z. B. durch engen Kontakt mit einer infizierten Person, stellt jedoch eine größere Herausforderung dar und kann das Infektionsgeschehen verstärken.
10 Viren im Rachen? Kein Problem.
10.000 Viren? Ab ins Bett!
Die genetische Veranlagung des Wirts
Zwei Menschen, ein Virus – doch nur einer wird krank. Manche Menschen tragen genetische Varianten in ihrem Immunsystem, die sie anfälliger oder widerstandsfähiger gegen das Influenzavirus machen. Zum Beispiel können Unterschiede in Genen, die Immunrezeptoren steuern, beeinflussen, wie gut das Immunsystem das Virus erkennt.
Es gibt zahlreiche Studien, die die unterschiedlichen Immunantworten aufgrund der genetischen Veranlagung untersuchen:
Die Studie „IFITM3: How genetics influence influenza infection demographically” zeigte, dass Menschen mit bestimmten Varianten des IFITM3-Gens (Interferon-induziertes Transmembranprotein 3) seltener schwere Grippeerkrankungen entwickeln, weil dieses Gen die Vermehrung des Influenzavirus in Zellen hemmt.
Die Studie „HLA targeting efficiency correlates with human T-cell response magnitude and with mortality from influenza A infection” untersuchte, wie HLA-Allele (MHC-Klasse-I) die T-Zell-Antwort auf das Influenzavirus beeinflussen. Sie fand heraus, dass bestimmte HLA-Allele effizienter Influenza-Peptide präsentieren und eine stärkere T-Zell-Antwort auslösen. Menschen mit diesen Allelen hatten mildere Verläufe bei Grippeinfektionen, während andere Allele mit schwächeren T-Zell-Antworten und höherer Mortalität assoziiert waren. Das zeigt, dass genetische Unterschiede in MHC-Molekülen direkt die Schwere einer Influenzainfektion beeinflussen können.
Die Mutantenwolke des Virus
Wie bereits erwähnt, existiert das Influenzavirus nicht als einheitlicher Stamm, sondern als eine „Mutantenwolke“ – eine Vielfalt genetischer Varianten, die durch die fehleranfällige RNA-Polymerase entstehen. Manche Varianten in dieser Wolke sind aggressiver, weil sie z. B. besser an Zellen binden oder dem Immunsystem entgehen. Welche Variante dominiert, kann entscheiden, wie schwer die Infektion verläuft.
Die Gewebespezifität des Virus
Influenzaviren unterscheiden sich in ihrer Vorliebe für bestimmte Gewebe im Körper. Die meisten Stämme vermehren sich bevorzugt in den oberen Atemwegen (z. B. Nase und Rachen), was oft zu milderen Symptomen wie Halsschmerzen führt. Andere Stämme dringen tiefer in die Lunge vor und können eine schwere Lungenentzündung auslösen.
📌 Fazit: Ein Hoch auf die Vielfalt!
Die Interaktion zwischen Virus und Mensch ist wie Tinder für Mikroben – manche Matches sind harmlos, andere enden im Desaster. Entscheidend ist:
- Wirtspoker (Gene + Immunsystem)
- Virus-Roulette (Dosis + Mutationen)
- Gewebe-Tinder (Wo landet das Virus?)
Unser Körper ist kein passives Ziel – sondern ein lernendes System. Jeder Infekt ist ein Update fürs Immungedächtnis, jeder Virus ein Trainingspartner fürs Leben. Denn nur im Kampf wächst unser Schutzschild – ein Leben lang.
3. Ein Blick auf die Anfänge der Mikrobiologie – wie alles begann
Die Geschichte der Mikrobiologie ist eine Reise von der Unsichtbarkeit zur Klarheit, von Spekulationen zu konkretem Wissen. Ihre Ursprünge lassen sich bis ins 17. Jahrhundert zurückverfolgen, als die Neugier der Menschen und die technische Innovation erstmals eine verborgene Welt enthüllten.
3.1. Frühe Entdeckungen: Die ersten Blicke ins Unsichtbare
3.2. Die Geburt der modernen Mikrobiologie
3.3. Der Schritt in die Welt der Viren
3.4. Viren und die Koch’schen Postulate
3.1. Frühe Entdeckungen: Die ersten Blicke ins Unsichtbare
Im Jahr 1665 war es Robert Hooke, der mit einem frühen Mikroskop erstmals Pflanzenzellen beschrieb und damit den Begriff „Zelle“ prägte.
Doch der wirkliche Durchbruch kam einige Jahre später mit Antonie van Leeuwenhoek. Mit seinen selbstgebauten, extrem leistungsfähigen Linsen beobachtete er 1676 erstmals winzige, lebendige Organismen, die er als „animalcules“ bezeichnete – kleine Tierchen, wie Bakterien und einzellige Lebewesen, die wir heute kennen. Van Leeuwenhoeks Entdeckungen eröffneten eine völlig neue Perspektive auf die Natur, doch das Wissen darüber, wie diese Organismen lebten oder Krankheiten verursachten, war noch weit entfernt.
3.2. Die Geburt der modernen Mikrobiologie
Es dauerte fast zwei Jahrhunderte, bis die Mikrobiologie systematisch erforscht wurde. Im 19. Jahrhundert erlebte das Fachgebiet einen Quantensprung. Louis Pasteur widerlegte die alte Vorstellung, dass Leben einfach aus dem Nichts entstehen könne (Theorie der Spontanzeugung), und bewies, dass Mikroorganismen für Prozesse wie Gärung und Fäulnis verantwortlich sind. Seine Arbeiten legten den Grundstein für die Keimtheorie der Krankheiten, die schließlich von Robert Koch weiterentwickelt wurde.
Kochs Forschung führte 1876 zu einem entscheidenden Meilenstein: den Koch’schen Postulaten. Diese Regeln ermöglichten es erstmals, Mikroorganismen als spezifische Verursacher von Krankheiten zu identifizieren. Kochs Arbeit revolutionierte die Bakteriologie und machte es möglich, Krankheitserreger wie den Milzbranderreger (Bacillus anthracis) und später auch den Tuberkulose-Erreger eindeutig nachzuweisen.
Doch während die Mikrobiologie mit der Erforschung von Bakterien große Fortschritte machte, blieben Viren lange im Verborgenen. Selbst die besten Mikroskope der damaligen Zeit konnten diese winzigen, unsichtbaren Partikel nicht abbilden.
3.3. Der Schritt in die Welt der Viren
Das Ende des 19. Jahrhunderts brachte den nächsten Durchbruch. Dmitri Iwanowski zeigte 1892, dass ein filtrierter Extrakt aus Tabakpflanzen, die an der Tabakmosaikkrankheit litten, infektiös blieb, obwohl er durch Porzellanfilter geleitet wurde, die Bakterien zurückhielten. Martinus Beijerinck bestätigte diese Beobachtungen und prägte den Begriff „Virus“ (vom lateinischen Wort für „Gift“ oder „Schleim“) für den mysteriösen, nicht-bakteriellen Erreger. Damit begann die systematische Erforschung dieser neuen Welt.
Der wahre Zugang zur Welt der Viren wurde jedoch erst mit dem Elektronenmikroskop möglich, das in den 1930er Jahren entwickelt wurde. Erst dann konnten Wissenschaftler Viren sichtbar machen und ihre Struktur verstehen.
3.4. Viren und die Koch’schen Postulate
In Diskussionen über den Nachweis von Viren werden oft die Koch’schen Postulate als Maßstab herangezogen, um die Existenz von Viren infrage zu stellen. Doch wie passen diese Postulate, die im 19. Jahrhundert entwickelt wurden, in das heutige Verständnis von Infektionskrankheiten? Ein Blick auf die historischen Hintergründe und die wissenschaftliche Weiterentwicklung hilft, diese Frage zu klären.
Die Koch’schen Postulate: Ein wissenschaftlicher Meilenstein
Robert Koch (1843–1910), einer der Begründer der modernen Bakteriologie, entwickelte die nach ihm benannten Postulate, um den Zusammenhang zwischen Mikroorganismen und Infektionskrankheiten zu beweisen. Sie wurden 1890 auf dem 10. Internationalen Medizinischen Kongress vorgestellt und bestehen aus vier Kriterien:
Postulat 1: Der Mikroorganismus muss in jedem Fall der Krankheit nachgewiesen werden, sollte aber nicht in gesunden Organismen vorkommen.
Postulat 2: Der Mikroorganismus muss aus dem erkrankten Organismus isoliert und in Reinkultur gezüchtet werden.
Anmerkung: Eine Reinkultur bedeutet, dass nur eine einzige Art von Mikroorganismen gezüchtet wird, ohne andere Arten dazwischen.
Postulat 3: Ein zuvor gesundes Individuum zeigt nach einer Infektion mit dem Mikroorganismus aus der Reinkultur die gleichen Symptome wie das Individuum, von dem der Mikroorganismus ursprünglich stammt.
Postulat 4: Der Mikroorganismus muss erneut aus dem Versuchswirt isoliert und als derselbe identifiziert werden.
Diese bahnbrechenden Prinzipien legten den Grundstein für die experimentelle Medizin und die Keimtheorie der Krankheiten.
Robert Koch: Ein Pionier gegen Widerstände
Robert Koch untersuchte die Erreger von Krankheiten wie Tuberkulose, Cholera und Milzbrand. Für seine Forschungen reiste er oft in Seuchengebiete, wie Kalkutta zur Untersuchung der Cholera oder Bombay während der Beulenpest. Koch verbrachte Monate in diesen Ländern, immer nah am Zentrum der Seuchen. In seinem Laborzelt arbeitete er unermüdlich am Mikroskop.
Koch hatte jedoch mit erheblichem Widerstand zu kämpfen. Zu seiner Zeit war die Idee, dass Krankheiten durch mikroskopische Organismen verursacht werden, noch umstritten. Viele seiner Kollegen und Zeitgenossen waren skeptisch und lehnten seine Theorien ab. Wissenschaftler glaubten damals noch oft, dass Seuchen und Epidemien von sogenannten Miasmen – giftigen Dämpfen, die aus dem Erdreich aufsteigen – ausgelöst würden.
Trotz dieser Herausforderungen setzte sich Koch unermüdlich für seine Forschung ein. Er nutzte innovative Techniken seiner Zeit wie die Agarplatte und die Ölimmersionslinsen, um Bakterien zu kultivieren und zu untersuchen. Diese Methoden ermöglichten es ihm, wichtige Entdeckungen zu machen und das Verständnis von Infektionskrankheiten revolutionär zu verändern.

Links: Eine Agarplatte – ein fester Nährboden in einer Petrischale, dem Agar als Geliermittel zugesetzt wurde, um Bakterien gezielt zu kultivieren. Rechts: Ein Mikroskop mit Ölimmersionslinse – eine spezielle Mikroskoptechnik, bei der ein Tropfen Öl zwischen Objektiv und Probe die Lichtbrechung minimiert, sodass kleinste Mikroben schärfer sichtbar werden.
Herausforderungen und Grenzen der Postulate
Genau die seinen Theorien entgegengebrachte Skepsis veranlasste Koch, die Postulate aufzustellen, um den Beweis zu erbringen, dass es einen Zusammenhang zwischen den pathogenen Eigenschaften der Bakterien und der Krankheit gibt.
Koch selbst erkannte, dass seine Postulate nicht immer uneingeschränkt gelten. Ein bekanntes Beispiel ist seine Arbeit mit dem Cholera-Erreger Vibrio cholerae. Er fand heraus, dass dieser Mikroorganismus nicht nur bei erkrankten, sondern auch bei scheinbar gesunden Menschen vorkommen kann. Diese Entdeckung stellte das erste Postulat infrage und führte dazu, dass Koch die universelle Gültigkeit dieses Kriteriums aufgab.
Kochs Innovationsgeist
Koch war ein Pionier seiner Zeit. In seiner Rede auf dem 10. Internationalen Medizinischen Kongress erklärte er:
„Es war geboten, mit unwiderleglichen Gründen den Beweis zu führen, dass die bei einer Infectionskrankheit aufgefundenen Mikroorganismen auch wirklich die Ursache dieser Krankheit seien.“
Sein wissenschaftlicher Ansatz, Skeptiker durch strikte Nachweise zu überzeugen, war wegweisend. Doch er selbst erkannte, dass neue Technologien und Methoden nötig sind, um weiterführende Fragen zu beantworten:
„Mit den zu Gebote stehenden experimentellen und optischen Hülfsmitteln war auch nicht weiter zu kommen und es wäre wohl noch geraume Zeit so geblieben, wenn sich nicht gerade damals neue Forschungsmethoden geboten hätten, welche mit einem Schlage ganz andere Verhältnisse herbeiführten und die Wege zu weiterem Eindringen in das dunkle Gebiet öffneten, mit Hülfe verbesserter Linsensysteme …“
In Bezug auf schwer nachweisbare Krankheitserreger wie die der Influenza oder des Gelbfiebers bemerkte er:
„Ich möchte mich der Meinung zuneigen, dass es sich bei den genannten Krankheiten gar nicht um Bakterien, sondern um organisierte Krankheitserreger handelt, welche ganz anderen Gruppen von Mikroorganismen angehören.“
Koch war damit den Viren bereits auf der Spur, konnte sie jedoch aufgrund der zu seiner Zeit begrenzten technischen Möglichkeiten nicht eindeutig identifizieren. Er erkannte jedoch, dass diese unsichtbaren Erreger existieren mussten.
Warum Viren die Koch’schen Postulate sprengen
Koch‘s Forschungsergebnisse entsprachen dem damaligen Entwicklungsstand der Wissenschaft. Die Mikrobiologie befand sich im 19. Jahrhundert noch in einer frühen Entwicklungsphase, in der grundlegende Prinzipien erst entdeckt und systematisch erforscht wurden. Die Virologie als eigenständiges Feld entstand erst nach Kochs Zeit, als Dmitri Iwanowski und Martinus Beijerinck infektiöse Partikel entdeckten, die kleiner waren als Bakterien. Mit der Erfindung des Elektronenmikroskops in den 1930er Jahren konnten Viren schließlich sichtbar gemacht werden. Viren unterscheiden sich jedoch fundamental von Bakterien, weshalb die Koch’schen Postulate oft nicht direkt auf sie anwendbar sind:
➤ Wirtabhängigkeit: Viren können sich nur in lebenden Wirtszellen vermehren und lassen sich nicht in einer Reinkultur züchten.
➤ Asymptomatische Infektionen: Viele Virusinfektionen verlaufen ohne Symptome, was die Zuordnung von Erreger und Krankheit erschwert.
➤ Komplexe Nachweisverfahren: Moderne molekularbiologische Methoden wie PCR ermöglichen den Nachweis von viralen Genomsequenzen, was eine Erweiterung der klassischen Postulate erfordert.
Koch und die heutige Wissenschaft
Robert Koch und seine Zeitgenossen legten den Grundstein für die Mikrobiologie, insbesondere durch die Entwicklung von Methoden zur Isolierung und Kultivierung von Bakterien. Die Keimtheorie der Krankheiten war damals ein Meilenstein in der Wissenschaftsgeschichte. Koch wusste, dass die Wissenschaft ständig im Wandel begriffen ist. Hätte Koch Zugang zu modernen Technologien wie PCR, Sequenzierung oder Elektronenmikroskopen gehabt, hätte er seine Methodik angepasst?
Moderne Technologien wie PCR und Sequenzierung werden in den kommenden Kapiteln vorgestellt.
Seine Schlussworte beim Kongress von 1890 geben die Antwort und zeigen seinen Optimismus für die Zukunft:
„Lassen Sie mich mit dem Wunsche schließen, dass sich die Kräfte der Nationen auf diesem Arbeitsfelde und im Kriege gegen die kleinsten, aber gefährlichsten Feinde des Menschengeschlechts messen mögen und dass in diesem Kampfe zum Wohle der gesammten Menschheit eine Nation die andere in ihren Erfolgen immer wieder überflügeln möge.“
Hätte Robert Koch die heutigen Möglichkeiten der Molekularbiologie, Virologie und Immunologie erleben können, wäre er vermutlich fasziniert gewesen – nicht nur von den neuen Erkenntnissen, sondern auch von den revolutionären Methoden, die es ermöglichen, selbst die kleinsten Erreger sichtbar zu machen. Denn genau darin lag sein Antrieb: Unsichtbares erkennbar zu machen, das Verborgene zu entschlüsseln. Wie hätte er wohl auf die ersten Bilder eines Virus unter dem Elektronenmikroskop reagiert?
Von den ersten Beobachtungen zur modernen Wissenschaft
Was einst mit staubigen Linsen und ratlosen Blicken begann, ist heute Hightech bis auf Molekülebene. Mit jeder neuen Technologie blicken wir tiefer in den Mikrokosmos. Die Mikrobiologie hat das Unsichtbare sichtbar gemacht – aber erst die moderne Technik erlaubt es uns, das Unsichtbare wirklich zu verstehen. Heute spüren wir Viren auf, die sich jahrhundertelang unserem Blick entzogen haben.
Und wie wir dem Unsichtbaren heute auf die Spur kommen – das ist die Geschichte des nächsten Kapitels.
4. Moderne Methoden zur Entdeckung und Analyse von Viren
Bisher haben wir uns mit dem Thema Viren vertraut gemacht. In diesem Kapitel wechseln wir die Perspektive: vom Staunen zum Messen. Denn ohne die Methoden der modernen Biologie wüssten wir kaum etwas über Viren – ihre Formen, ihr Erbgut, ihre Vielfalt.
Was folgt, ist ein Blick in den Maschinenraum der Wissenschaft. Zugegeben, ein technischer Abschnitt – aber er ist wichtig: Denn hier wird sichtbar, wie wir das Unsichtbare überhaupt zu fassen kriegen.
Wer gerne verstehen möchte, wie man Viren heute sichtbar macht, entschlüsselt, katalogisiert und analysiert, der findet hier eine Art Werkzeugkasten des 21. Jahrhunderts.
Doch wann und warum greifen wir zu diesen Werkzeugen?
Die Identifikation von Viren kann unterschiedliche Ziele verfolgen: Manche Viren rücken durch ihre Auswirkungen auf Menschen, Tiere oder Pflanzen in den Fokus, während andere in Umweltproben oder unerforschten Lebensräumen entdeckt werden, um ihre ökologische Bedeutung zu verstehen. Der Nachweis bekannter Viren dient vor allem der Diagnostik, während die Identifikation unbekannter Viren Einblicke in die biologische Vielfalt und Evolution ermöglicht.
Da Viren keine universellen Merkmale wie eine gemeinsame Zellstruktur besitzen, konzentrieren sich Nachweismethoden auf ihre genetische Information, ihre Struktur oder ihre Wechselwirkungen mit Wirtszellen. Der Nachweisprozess folgt keinem starren Schema, sondern kombiniert verschiedene Schritte, die je nach Fragestellung und Virusart variieren.
In den folgenden Abschnitten werden die zentralen Verfahren zur Virusidentifikation detailliert erläutert – von der Probenentnahme bis hin zur bioinformatischen Analyse.
Vorgehensweisen zur Identifikation von Viren
4.1. Probenentnahme
4.2. Probenaufbereitung
4.2. a) Filtration
4.2. b) Zentrifugation
4.2. c) Präzipitation
4.2. d) Chromatographie
4.3. Zellkultur
4.4. Viren sichtbar machen
4.4. a) Elektronenmikroskopie
4.4. b) Kristallisation
4.4. c) Kryo-Elektronenmikroskopie
4.4. d) Kryo-Elektronentomographie
4.4. e) Zusammenfassung
4.5. Der genetische Fingerabdruck der Viren
4.5.1. Nukleinsäure-Extraktion
4.5.2. Nukleinsäure-Amplifikation
4.5.3. Sequenzierung
4.5.3. a) First Generation: Sanger-Sequenzierung
4.5.3. b) Second Generation: Next-Generation Sequencing (NGS)
4.5.3. c) Third Generation: Echtzeit-Sequenzierung
4.5.3. d) Emerging Technologies: Zukunft der Sequenzierung
4.6. Bioinformatische Analyse
4.1. Probenentnahme
Bevor wir Viren auf die Schliche kommen, müssen wir sie erst einmal finden – und das ist wie eine Schatzsuche im Mikroformat, mit Verstecken von Blut bis Tiefseeschlamm. Ob Mensch, Pflanze oder Ozean: Viren verstecken sich überall, und Forscher werden zu Detektiven mit Pipetten, Schutzanzug und Spezialausrüstung.

Menschliche Proben
Wer kennt es nicht? Ein Wattestäbchen tief im Nasen-Rachen-Raum – der Klassiker, seit Corona jedem ein Begriff. Doch Viren lauern nicht nur in Schleimhäuten, sondern auch in Blut, Stuhl oder Gewebe. Ähnlich wie ein Dieb DNA-Spuren hinterlässt, verraten sie sich durch ihre genetischen Überreste. Entscheidend ist, dass die Probe sauber entnommen und schnell verarbeitet wird – idealerweise auch möglichst schonend (und schmerzfrei?) für den Patienten. Auch Pflanzen, Tiere oder Insekten liefern Probenmaterial, um Virusvorkommen in ganz unterschiedlichen biologischen Systemen zu untersuchen.
Umweltproben
Wasser, Erde, sogar Luft – Viren hängen überall rum. Forschende fischen sie aus Flüssen, buddeln sie aus dem Boden oder saugen sie mit Hightech-Filtern direkt aus der Atmosphäre.
Extremkandidaten: Viren unter Extrembedingungen
In der Tiefsee, im ewigen Eis oder in brodelnden Vulkanquellen – Viren zählen zu den härtesten Überlebenskünstlern überhaupt. Um sie dort zu erwischen, braucht es spezielle Sonden, die wie Raumschiff-Fangarme in die Tiefe greifen: „Huch, was schwimmt denn da in 4000 Metern Tiefe? Einfach mal mitnehmen!“
Mehr zum Ozean
Viren spielen eine zentrale, oft unterschätzte Rolle für die Gesundheit der Ozeane. Wo Leben existiert, gibt es auch Viren – unsichtbare, aber allgegenwärtige Akteure, die zahlenmäßig jede andere biologische Einheit übertreffen. Sie kontrollieren das Ökosystem ähnlich wie große Raubtiere, indem sie Populationen regulieren und so das ökologische Gleichgewicht bewahren.
Ein herausragendes Forschungsprojekt war die Tara Oceans Expedition (2009–2013). Ziel dieses vierjährigen Projekts war es, das mikrobielle Leben in den Ozeanen und dessen Einfluss auf das globale Ökosystem zu erforschen. Wissenschaftler sammelten weltweit über 35.000 Proben von Plankton, Algen, und Viren.
Im Rahmen der Expedition entdeckte das Forschungsteam über 5.000 neue RNA-Virusarten, darunter die faszinierenden Mirusviren. Diese Entdeckungen erweitern unser Verständnis der Vielfalt, Evolution und Ökologie der Ozeane.
Wenn dich diese Expedition interessiert, schau dir die Tara Oceans Videos an (kurz oder lang).
Transport: Der VIP-Service für Viren
Damit die winzigen Verdächtigen nicht unterwegs kaputtgehen, geht’s für sie nach der Entnahme schnell in die Kühlkette. Tiefkühltransporte und sterile Verpackungen sind Pflicht, sonst zerfällt die „virale Beute“ schneller als ein Eiswürfel in der Sahara.
Doch eine gute Probe ist nur der Anfang.
Was wir in Röhrchen, Tupfer oder Tiefkühlbox nach Hause bringen, ist meist ein biologisches Wimmelbild: Zelltrümmer, Bakterien, Proteine – und irgendwo dazwischen ein paar Viren, winzig und verborgen. Jetzt heißt es: sortieren, reinigen, konzentrieren – bevor die eigentliche Analyse überhaupt beginnen kann.
4.2. Probenaufbereitung
In jeder Probe verstecken sich Viren wie Nadeln im mikrobiellen Heuhaufen – der Grund, warum wir putzen müssen, bevor die Virusjagd beginnen kann:
- 99 % Ballast: Zelltrümmer, Proteine, Bakterien
- 1 % Zielobjekt: Winzige Virionen, die wir isolieren wollen
Der Großteil einer biologischen Probe besteht aus nicht-viralem Material – was die gezielte Untersuchung enorm erschwert. Deshalb ist die Probenaufbereitung ein unverzichtbarer Zwischenschritt: Sie entfernt Verunreinigungen, reichert virale Partikel an und bereitet das Material für die eigentliche Analyse vor. Ihr Ablauf variiert je nach Probenart, Analyseziel und gesuchtem Virus.
Probenaufbereitung ist wie Goldwaschen: Man braucht Geduld, das richtige Sieb – und die Hoffnung, dass in jedem Eimer Schlamm ein Nugget glänzt.
Zu den wichtigsten Techniken zählen:
a) Filtration
b) Zentrifugation
c) Präzipitation
d) Chromatographie
a) Filtration – Das Mega-Sieb

Die Filtration ist ein erster Reinigungsschritt, bei dem größere Partikel und grobe Verunreinigungen entfernt werden.
So lassen sich virale Partikel gezielt von größeren Strukturen wie Bakterien oder Zellfragmenten trennen.
Funktion: Hält Bakterien & Zellmüll zurück – lässt nur Virionen passieren
Besonderheit: Spezialfilter mit Nanoporen (0,02 µm!)
Cool Fact: Manche Filter laden sich elektrostatisch auf, um Viren besser zu fangen
Mehr Infos
Die Filtration erfolgt üblicherweise mithilfe spezieller Membranfilter mit definierter Porengröße. Filter mit einer Porengröße von 0,2 Mikrometern werden häufig eingesetzt, da sie Bakterien und größere Partikel zuverlässig zurückhalten, während kleinere virale Partikel hindurchgelangen. Ein Beispiel aus der Praxis ist der Einsatz dieser Methode bei Wasserproben aus Umweltstudien, wo virale Partikel effektiv von anderen Mikroorganismen getrennt werden.
Eine weiterentwickelte Variante ist die Ultrafiltration, die Filter mit noch feineren Porengrößen im Bereich von 0,01 bis 0,1 Mikrometern verwendet. Diese Technik ermöglicht nicht nur die Entfernung von Bakterien, sondern auch die Abtrennung kleinerer Partikel.

Trotz ihrer Effektivität hat die Filtration einige Einschränkungen:
Reinheit: Kleinste Partikel oder gelöste Substanzen können nach der Filtration weiterhin in der Probe vorhanden sein.
Verstopfung: Insbesondere bei Proben mit hoher Partikelkonzentration kann die Filtermembran blockiert werden, was den Prozess erschwert.
Kapazität: Die Durchflussrate ist durch die Porengröße begrenzt, was die Filtration zeitaufwendig machen kann.
Um eine möglichst reine Virusprobe zu erhalten, wird die Filtration häufig mit weiteren Reinigungsschritten wie Zentrifugation oder Chromatographie kombiniert. Diese zusätzlichen Methoden können die verbleibenden Verunreinigungen entfernen und die Qualität der Proben für nachfolgende Analysen optimieren.
b) Zentrifugation – Die Schwerkraft-Turbine

Die Zentrifugation ist eine zentrale Technik zur Trennung von Partikeln basierend auf ihrer Größe und Dichte.
Dieser Prozess nutzt die Zentrifugalkraft, die durch das Drehen der Probe mit hoher Geschwindigkeit erzeugt wird.
Prinzip: Schweres sinkt, Leichtes schwimmt – Viren landen dazwischen
Highspeed: Bis zu 100.000 U/min (eine Waschmaschine schafft 1.200)
Trick: Dichtegradienten trennen sogar Virus-Typen voneinander
Mehr Infos
Funktionsweise der Zentrifugation
Wenn eine Lösung über längere Zeit bei Raumtemperatur stehen bleibt, setzen sich schwerere Partikel aufgrund der Schwerkraft langsam ab. Die Zentrifugation beschleunigt diesen Prozess erheblich, indem die Probe mit Geschwindigkeiten von mehreren tausend bis zu 100.000 Umdrehungen pro Minute (bei der Ultrazentrifugation) rotiert. Die extremen Zentrifugalkräfte führen dazu, dass Partikel je nach ihrer Dichte und Größe getrennt werden.

Um die empfindlichen Moleküle vor Überhitzung zu schützen, sind viele Zentrifugen mit Kühlsystemen ausgestattet, die eine gleichmäßige Temperatur während des gesamten Prozesses gewährleisten.
Typen der Zentrifugation
Differenzielle Zentrifugation
Trennprinzip: Partikel werden basierend auf ihrer Größe und Masse getrennt. Größere und schwerere Partikel setzen sich schneller ab als kleinere und leichtere.
Verfahren: Die Probe wird schrittweise mit steigenden Zentrifugalkräften behandelt. Nach jedem Schritt wird der Überstand (Supernatant), der die verbleibenden kleineren Partikel enthält, vorsichtig in ein neues Röhrchen überführt. Der Boden der ursprünglichen Zentrifugenröhre enthält das Pellet, in dem sich die größeren Partikel abgesetzt haben, und wird entfernt.
Anwendung: Diese Methode wird häufig genutzt, um Zellkomponenten, Organellen und Viren aus komplexen Gemischen zu isolieren.
Ein typisches Beispiel für die Vorbereitung von Proben zur Untersuchung auf unbekannte Viren ist die Gewinnung eines Zelllysats. Hierbei werden Zellen, die aus Gewebeproben oder Abstrichen isoliert wurden, durch chemische, physikalische oder enzymatische Verfahren aufgeschlossen. Dieser Schritt setzt virale Partikel frei, die innerhalb der Wirtszellen repliziert wurden. Anschließend wird das Zelllysat durch Zentrifugation oder Filtration gereinigt, um virale Komponenten von zellulären Trümmern zu trennen. Zelllysate sind besonders nützlich, wenn der Verdacht besteht, dass Viren in spezifischen Zelltypen oder Geweben verborgen sind.

Dichtegradientenzentrifugation
Trennprinzip: Partikel werden nach ihrer Dichte getrennt. Die Probe wird auf einen Dichtegradienten (meist Saccharose oder Cäsiumchlorid) aufgetragen und zentrifugiert. Während der Rotation wandern die Partikel zu der Position im Gradient, die ihrer eigenen Dichte entspricht.
Verfahren: Die Partikel kommen zur Ruhe an der Stelle, an der ihre Dichte mit der des Mediums übereinstimmt, was eine präzise Trennung ermöglicht.
Anwendung: Diese Methode eignet sich hervorragend, um Viren, Proteine und Nukleinsäuren aus komplexen Gemischen zu isolieren und zu analysieren.
Entnahme der viralen Fraktion
Nach der Zentrifugation wird die virale Fraktion typischerweise als eine klar abgegrenzte Schicht innerhalb der Zentrifugenröhre sichtbar. Diese Schicht kann vorsichtig mithilfe einer Pipette entnommen werden, um die Viren für weitere Analysen bereitzustellen. Die genaue Position der viralen Schicht hängt dabei von der genutzten Methode und den physikalischen Eigenschaften der Viren ab.
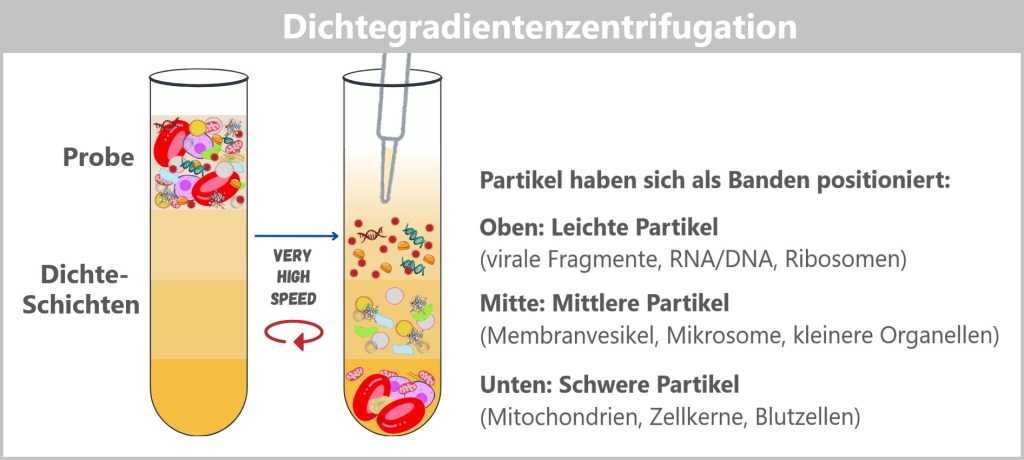
In den meisten Fällen wird die Zentrifuge auf eine konstante Geschwindigkeit eingestellt, die ausreichend ist, um die Partikel im Dichtegradienten zu trennen. Diese Geschwindigkeit ist typischerweise sehr hoch (z. B. bei der Ultrazentrifugation bis zu 100.000 U/min), da die Trennung im Dichtegradienten von der ausreichenden Zentrifugalkraft abhängt.
Kombination der Methoden
Oft werden differenzielle und Dichtegradientenzentrifugation kombiniert, um eine höhere Reinheit und Präzision bei der Trennung der Partikel zu erreichen. Diese Methoden sind essenziell für die biomedizinische Forschung und Diagnostik, da sie eine präzise Trennung und Konzentration viraler Partikel ermöglichen.
c) Präzipitation – Der Chemie-Trick
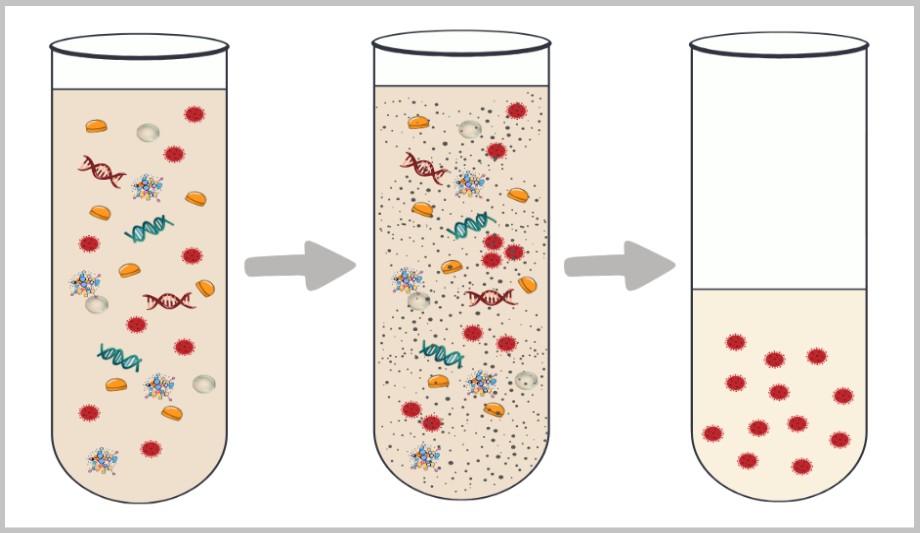
Die Präzipitation (Fällung) ist eine bewährte Methode zur Isolierung und Reinigung von Viruspartikeln aus einer Lösung.
Dabei werden chemische Substanzen hinzugefügt, die die Löslichkeit der Viren herabsetzen und sie so aus der Lösung ausfällen lassen.
So geht’s: Chem. Substanzen machen Viren schwer & klebrig → sie fallen aus
Vorteil: Billig, simpel – aber nichts für empfindliche Viren
Mehr Infos
Der Vorgang umfasst mehrere Schritte und nutzt spezifische chemische Reaktionen, um die Viruspartikel effektiv zu konzentrieren.
Ablauf der Präzipitation
Vorbereitung der Probe: Vor der Präzipitation wird die Probe oft vorgereinigt, etwa durch Ultrafiltration oder Ultrazentrifugation, um größere Partikel und Verunreinigungen zu entfernen. Dies verbessert die Effizienz der Präzipitation.
Zugabe einer Fällungschemikalie: Eine Chemikalie wie Polyethylenglykol (PEG), Ethanol oder Ammoniumsulfat wird der Lösung zugeführt. Diese Stoffe verändern die chemischen Eigenschaften der Flüssigkeit, indem sie die Löslichkeit der Viruspartikel verringern.
Aussalzen: Beim Aussalzen konkurrieren die zugesetzten Salzionen oder Moleküle mit den Viruspartikeln um die verfügbaren Wassermoleküle. Da weniger Wassermoleküle für die Viren verfügbar sind, nimmt deren Löslichkeit ab, und sie beginnen, sich zusammenzuschließen. Dadurch entsteht ein sichtbarer Niederschlag, der als Fällung bezeichnet wird.Die Oberflächenladungen und die biochemischen Eigenschaften wie Größe und Dichte der Viruspartikel begünstigen ihre Fällung im Vergleich zu anderen Partikeln in der Lösung.
Zentrifugation (in der Abbildung: ZFG): Um die gefällten Viruspartikel zu konzentrieren, wird die Probe zentrifugiert. Dies beschleunigt das Absetzen der Viren, die sich als fester Niederschlag am Boden des Gefäßes sammeln.
Waschen: Das Pellet wird mit einem Puffer oder einer Lösung gewaschen, um verbleibendes PEG, Salze oder andere Verunreinigungen zu entfernen.
Ergebnis: Die Lösung enthält nun hauptsächlich die isolierten Viren, die für nachfolgende Analysen verwendet werden können.
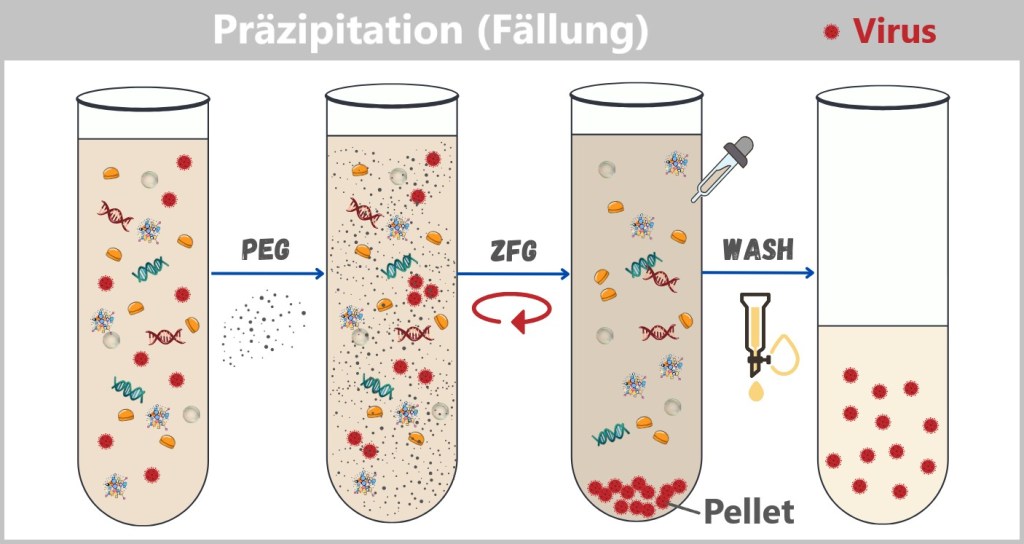
Nach der Zentrifugation befindet sich das Viruspellet am Boden des Reagenzglases, während der Überstand (Supernatant) oben liegt. Der Überstand wird vorsichtig abgesaugt oder abgepipettiert, um das Pellet nicht zu stören. Eine Waschlösung, meist ein Puffer wie PBS (phosphatgepufferte Salzlösung) oder ein ähnliches Medium, wird zum Pellet hinzugegeben. Das Volumen der Waschlösung ist in der Regel vergleichbar mit dem ursprünglichen Volumen der Probe, um genügend Verunreinigungen zu entfernen. Das Pellet wird durch sanftes Pipettieren, Vortexen (leichtes Rühren mit einem Vortexgerät) oder Umkippen des Reagenzglases resuspendiert, sodass es sich in der Waschlösung verteilt. Dieser Schritt sorgt dafür, dass verbleibende Verunreinigungen in die Waschlösung übergehen können. Nach dem Mischen wird das Reagenzglas erneut zentrifugiert. Dabei setzen sich die Viruspartikel wieder als Pellet ab. Die Waschlösung (Supernatant) wird vorsichtig entfernt, ohne das Pellet zu stören. Dieser Waschschritt wird je nach Bedarf ein- bis dreimal wiederholt, um sicherzustellen, dass möglichst viele Verunreinigungen entfernt werden. Nach dem letzten Waschschritt wird das Viruspellet in einer kleinen Menge Puffer (z. B. PBS oder einem Analysepuffer) resuspendiert, um es für nachfolgende Analysen vorzubereiten.
Vorteile der Präzipitation
Breit einsetzbar: Für viele Virenarten gut geeignet. Bei sehr empfindlichen Proben allerdings mit Vorsicht zu genießen: Vor allem behüllte Viren können durch osmotischen Stress oder unspezifische Aggregation geschädigt werden.
Hohe Ausbeute: Präzipitation ermöglicht eine effektive Konzentration und Reinigung mit minimalem Verlust an Material.
Einfache Anwendung: Der Prozess ist kostengünstig und erfordert keine hochkomplexen Geräte.
Die Präzipitation eignet sich besonders, um große Mengen an Viren effizient zu isolieren.
d) Chromatographie – Die VIP-Lounge für Viren
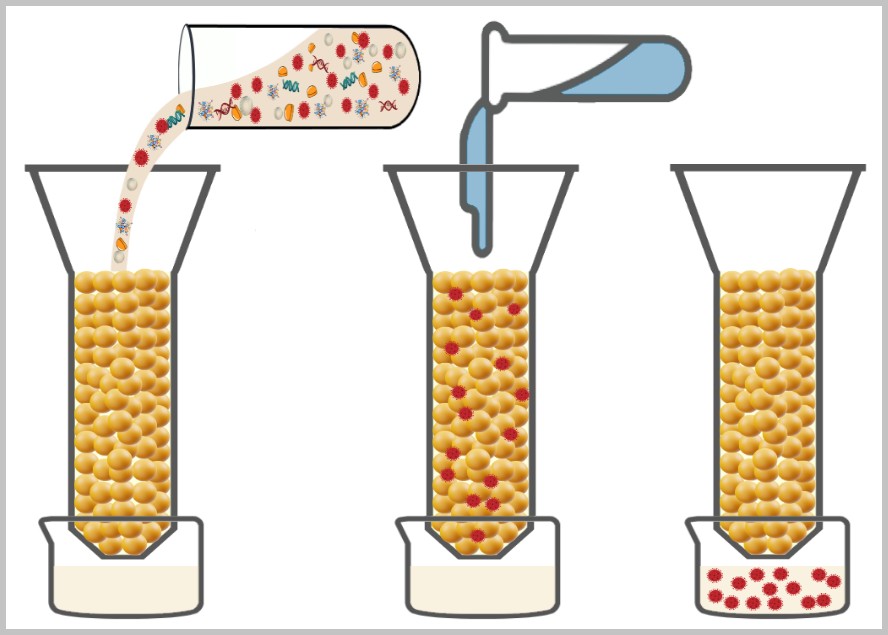
Die Chromatographie ist ein hochpräziser Reinigungsschritt in der Virusaufbereitung.
Dabei werden virale Partikel anhand ihrer spezifischen physikalischen oder chemischen Eigenschaften von anderen Bestandteilen der Probe getrennt.
So geht‘s: Säulen trennen Viren nach Größe, Ladung oder Affinität
Vorteil: Besonders schonend, präzise und effizient
Mehr Infos
Prinzip der Chromatographie
Die Chromatographie ist eine Methode zur Trennung verschiedener Stoffe in einem Gemisch. Ganz allgemein gibt es in diesem Verfahren eine mobile (bewegliche) Phase und eine stationäre (unbewegliche) Phase. Die mobile Phase ist die Stoffmischung, die getrennt werden soll. Sie bewegt sich durch die stationäre Phase, die als Matrix bezeichnet wird. Die Stoffe in der mobilen Phase interagieren mit den Stoffen in der stationären Phase. Durch die Wechselwirkungen bewegen sich die einzelnen Komponenten im Stoffgemisch unterschiedlich schnell oder bleiben stecken. Somit können die verschiedenen Stoffe voneinander getrennt werden.
Es gibt verschiedene Arten der Chromatographie. Eine häufig verwendete Variante ist die Ionenaustauschchromatographie, die auf den elektrischen Ladungen der Partikel basiert. Diese Methode eignet sich hervorragend zur Reinigung viraler Partikel, da Viren typischerweise eine definierte Oberflächenladung haben.
Ablauf der Ionenaustauschchromatographie (IEC)
Kontrolle der Virenladung:
Der isoelektrische Punkt (IEP) eines Virus gibt den pH-Wert an, bei dem das Virus elektrisch neutral ist. Durch Anpassung des pH-Werts der Lösung kann die Ladung des Virus gezielt beeinflusst werden:
- Bei einem pH-Wert unter dem IEP ist das Virus positiv geladen.
- Bei einem pH-Wert über dem IEP ist das Virus negativ geladen.
Auswahl der Matrix:
Die Matrix wird so gewählt, dass sie eine entgegengesetzte Ladung zur Virenladung besitzt, um eine Bindung zu ermöglichen:
- Anionenaustauscher: Positiv geladene Matrix, um negativ geladene Partikel zu binden.
- Kationenaustauscher: Negativ geladene Matrix, um positiv geladene Partikel zu binden.
Typischerweise besteht die Matrix aus Harzen oder Membranen, die eine spezifische Ladung tragen.
Bindung der Viren:
Die Probe wird auf die Chromatographiesäule aufgetragen, und die Lösung fließt durch die Matrix. Die Viren binden an die Matrix aufgrund elektrostatischer Wechselwirkungen. Andere Moleküle, wie Nukleinsäuren oder Proteine, binden schwächer oder passieren die Säule unverändert.
Waschen:
Mit einem Waschpuffer werden ungebundene oder schwach gebundene Verunreinigungen aus der Matrix entfernt. Dieser Schritt verbessert die Reinheit der gebundenen Viruspartikel.
Elution (Freisetzung der Viren):
Um die gebundenen Viren von der Matrix zu lösen, wird ein Elutionspuffer hinzugefügt. Dies kann auf zwei Wegen geschehen:
- Änderung des pH-Werts: Der pH-Wert wird so eingestellt, dass die Ladung der Viruspartikel aufgehoben wird, wodurch die Bindung zur Matrix verloren geht.
- Erhöhung der Salzkonzentration: Zusätze wie Natriumchlorid oder Magnesiumchlorid neutralisieren die elektrostatische Bindung zwischen Viren und Matrix.
Ergebnis:
Die eluierten Viruspartikel werden gesammelt. Da Verunreinigungen bereits während der Waschschritte entfernt wurden, enthält die elutierte Fraktion hauptsächlich die gereinigten Viren, die für weitere Analysen oder Anwendungen genutzt werden können.
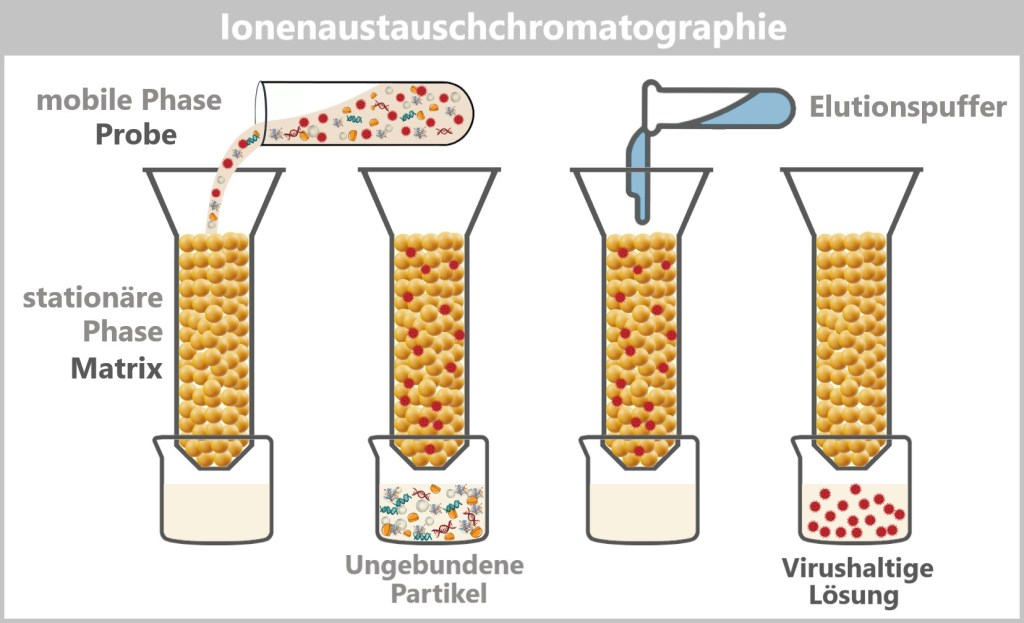
Weitere Varianten der Chromatographie
Neben der Ionenaustauschchromatographie gibt es andere Varianten, die je nach Ziel und Probe eingesetzt werden:
Affinitätschromatographie: Nutzt spezifische Bindungen zwischen Virusproteinen und Liganden auf der Matrix.
Größenausschlusschromatographie: Trennt Partikel nach ihrer Größe.
Hydrophobe Interaktionschromatographie: Nutzt die hydrophoben Eigenschaften der Partikel.
In der Praxis werden diese Verfahren oft kombiniert. Beispielsweise könnte eine Probe zunächst filtriert, anschließend zentrifugiert und danach durch Präzipitation und Chromatographie weiter aufgereinigt werden. Die Auswahl und Reihenfolge hängen dabei immer von der Art der Probe und dem Ziel der Analyse ab.
Warum dieser Aufwand?
Ganz einfach: Eine schlechte Probe bringt schlechte Daten – wie ein verwackeltes Foto von Bigfoot. Deshalb: sauber arbeiten, gut kühlen und schnell sein … denn Moleküle verzeihen keine Nachlässigkeit.
Die Probenaufbereitung ist kein Nebenjob – sie ist das Fundament. Ohne sie fällt alles Weitere in sich zusammen. Und wenn das virale Material einmal gut vorbereitet ist, geht es ans Eingemachte: Jetzt müssen sich die Viren beweisen – in der Zellkultur.
4.3. Zellkultur
Viren sind die ultimativen Schmarotzer: Ohne Wirtszelle läuft bei ihnen gar nichts – kein Leben, keine Vermehrung. Alleine – völlig hilflos. Aber gib ihnen eine lebende Zelle, dann geht’s ab. Deshalb braucht die Virologie ein verlässliches Werkzeug: die Zellkultur. Ohne sie döst selbst das gefährlichste Virus vor sich hin wie ein Büroangestellter im Homeoffice.
Und weil Viren nicht gern allein sind, richten Virologen ihnen ein kuscheliges Zuhause ein – eine Luxus-WG im Miniaturformat: sterile Petrischalen, perfekte Temperatur, ein Nährmedium voller Vitamine und Zucker – alles, was das Virusherz begehrt. Die verwendeten Zellen stammen von Menschen, Tieren oder Pflanzen – je nachdem, welchen Virus man gerade verwöhnen will.
Doch nicht jeder Virus ist pflegeleicht. Manche sind anspruchsvolle Diva-Typen, die nur in bestimmten Zelllinien gedeihen. Andere machen’s in allem, was „Help!“ schreit. Und manchmal? Passiert einfach… nichts. Dann heißt es: neue Zellen, neues Glück.
Zellkulturen sind weit mehr als eine Virenzuchtstation. Sie ermöglichen:
➤ die Vermehrung von Viren in kontrollierter Umgebung,
➤ die Analyse ihrer Eigenschaften und
➤ den Nachweis infektiöser Erreger – besonders bei neuen oder unbekannten Viren.
Warum Zellkulturen trotz PCR absolut cool sind
PCR-Tests sind schnell, billig und überall verfügbar – wie das Fast Food der Diagnostik. Aber sie können nur sagen: „Ja, hier war mal Virus!“ Ob das Virus aber noch lebendig und infektiös ist? Keine Ahnung!
Zellkultur = Realitätstest.
Kann das Virus Zellen infizieren und sich darin vermehren? Ja? Dann haben wir es mit einem echten Erreger zu tun. Nein? Dann bleibt’s bei genetischem Geistermüll.
Zellkultur als Fitness-Test: unverzichtbar, wenn man:
➤ die Infektiosität überprüfen will,
➤ neue Viren testet, deren Gefährlichkeit noch unklar ist.
Viren müssen erst mal beweisen, dass sie was können!
Auch in Zeiten von PCR und Hochdurchsatzsequenzierung (dazu später mehr) bleibt die Zellkultur der Praxistest nach der Theorieprüfung: „Alles schön und gut – aber funktioniert’s auch?“
Denn nur wer Zellen infizieren kann, hat echtes Gefahrenpotenzial. Die Zellkultur trennt die Spreu (harmlose Gensequenzen) vom Weizen (aktive, infektiöse Viren). Gerade in Zeiten von synthetischer Biologie wird das entscheidend: Nur weil die Gensequenz korrekt aussieht, heißt das noch lange nicht, dass das Virus auch funktioniert.
Infektion und Beobachtung der Zellkultur
Vorbereitung der Zellkultur
Zellen werden in einem Nährmedium kultiviert, das essentielle Nährstoffe, Wachstumsfaktoren und einen geeigneten pH-Wert enthält. Diese Umgebung ermöglicht das Überleben und die Vermehrung der Zellen. Es werden häufig Zelllinien verwendet, die eine kontinuierliche Vermehrung ermöglichen, wie beispielsweise Vero-Zellen (aus Nierenepithelzellen von Affen) oder HeLa-Zellen (menschliche Krebszellen).
Die Wahl der geeigneten Wirtszellen hängt entweder von ersten Hinweisen aus mikroskopischen Beobachtungen ab oder erfolgt durch Tests mit verschiedenen Zelllinien. Forscher verwenden dabei häufig Zelllinien, die dafür bekannt sind, Viren aus spezifischen Probenquellen zu unterstützen. Beispielsweise werden für Ozeanproben oft Zelllinien von Meeresorganismen genutzt, während bei der Untersuchung von Atemwegsviren menschliche Epithelzellen bevorzugt werden.
Infektion der Zellkultur
Um Viren in einer Probe nachzuweisen, wird diese auf eine geeignete Zellkultur aufgebracht. Enthält die Probe Viren, können diese in die Zellen eindringen, deren zelluläre Maschinerie zur Vermehrung nutzen und charakteristische Veränderungen hervorrufen, die als zytopathische Effekte (CPE) bezeichnet werden. Wie solche Zellveränderungen durch Virusinfektionen entstehen können, zeigt Kapitel „2.8. Zerstörung der Wirtszelle“ anschaulich.
Nachweis und Beobachtung:
Nach einer gewissen Zeit können die infizierten Zellen unter einem Licht- oder Elektronenmikroskop auf zytopathische Effekte (charakteristische Veränderungen) untersucht werden. Diese Effekte dienen als sichtbare Hinweise auf eine erfolgreiche Infektion:
➤ Zellschrumpfung oder -verklumpung
➤ Bildung von Synzytien (mehrkernige Zellverbände)
➤ Zelllyse (Auflösung der Zellen)
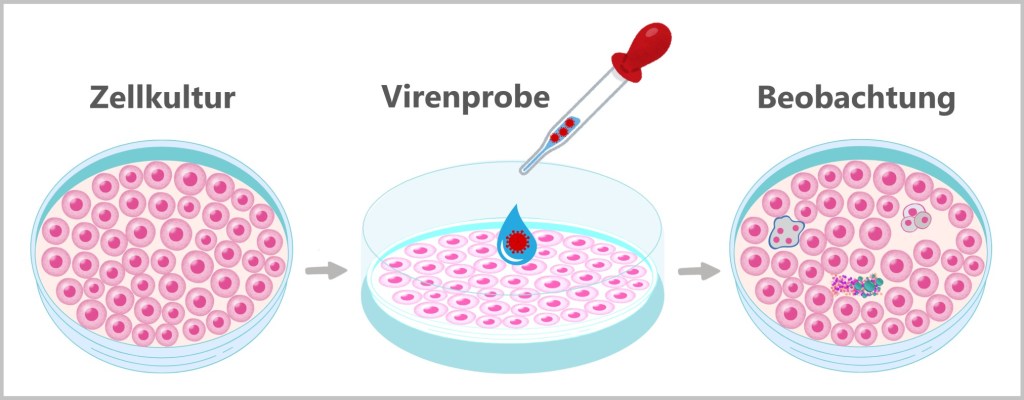
🎥 Live im Zeitraffer: So breitet sich das Influenzavirus in der Zellkultur aus
Bezug zum Influenzavirus
Studie: „Erkennung virusbedingter Zellveränderungen durch Influenza mit Hilfe künstlicher Intelligenz (CNN)“
Die Arbeit präsentiert eine umfangreiche Sammlung von Bildern, die die typischen zytopathischen Effekte (CPE) nach einer Infektion mit dem Influenzavirus zeigen. Diese visuellen Darstellungen verdeutlichen die charakteristischen Zellveränderungen, die durch das Virus verursacht werden. Allerdings ist die manuelle Beobachtung und Auswertung dieser Effekte äußerst arbeits- und zeitintensiv. Um diesen Prozess zu optimieren, erprobten die Autoren den Einsatz Künstlicher Intelligenz (KI), genauer gesagt tiefer Convolutional Neural Networks (CNNs), zur automatisierten Bildauswertung.
Diese Studie zeigt eindrucksvoll, wie moderne Methoden wie die Virologie, Zellbiologie und KI-Technologie ineinandergreifen, um innovative Lösungen für komplexe wissenschaftliche Fragestellungen zu entwickeln.
Rolle und Anwendungen der Zellkultur in der Virologie
Diagnose bekannter Viren: Zellkulturen werden eingesetzt, um Viren zu vermehren und ihre Präsenz durch spezifische Veränderungen in den Zellen nachzuweisen.
Nachweis unbekannter Viren: Wenn die genetische oder antigene Struktur eines Virus nicht bekannt ist, können Zellkulturen dazu beitragen, dessen biologische Aktivität zu charakterisieren.
Erforschung des viralen Lebenszyklus: Zellkulturen ermöglichen die Untersuchung der Interaktionen von Viren mit Wirtszellen, einschließlich des Eindringens, der Replikation und der Freisetzung neuer Viruspartikel.
Studien zur Virulenz und Pathogenität: Sie helfen, die Mechanismen zu verstehen, mit denen Viren Zellen schädigen und Infektionen auslösen.
Analyse der Immunantwort: Zellkulturen unterstützen die Untersuchung, wie das Immunsystem auf virale Infektionen reagiert und wie infizierte Zellen Immunreaktionen auslösen.
Testung antiviraler Medikamente: Zellkulturen dienen dazu, die Wirksamkeit und Verträglichkeit potenzieller antiviraler Substanzen zu prüfen und neue Therapien zu entwickeln.
Impfstoffproduktion: Viele Impfstoffe, wie beispielsweise der gegen Masern, werden in Zellkulturen hergestellt.
Herstellung von Virusvektoren: Zellkulturen sind unerlässlich für die Produktion von Virusvektoren – gentechnisch veränderten Viren, die therapeutische Gene oder Impfstoffinformationen in Zellen einschleusen, etwa in der Gentherapie, der Krebsbehandlung oder bei Impfstoffen wie denen gegen Ebola oder COVID-19 (z. B. AstraZeneca).
Einschränkungen der Methode
Nicht alle Viren sind kultivierbar: Einige Viren, wie Hepatitis B, benötigen spezialisierte Zellen oder Systeme, um in vitro vermehrt zu werden.
Viren mit sehr engen Wirtsspezifitäten: Manche Viren sind auf sehr spezifische Wirtszellen angewiesen, die in Zellkulturen nur schwer reproduziert werden können, was die Forschung erschwert.
Unzureichende Repräsentation von in-vivo-Umgebungen: Zellkulturen bieten nur eine vereinfachte Version des natürlichen Wirts, wodurch wichtige virus-spezifische Reaktionen fehlen könnten.
Laborangepasste Viren vs. natürliche Viren: Viren, die über viele Passagen in Zellkulturen gezüchtet werden, können Mutationen entwickeln, die ihre Eigenschaften verändern. Dadurch können sie sich von ihren natürlichen Varianten unterscheiden – etwa durch weniger Spike-Proteine oder eine veränderte Wirtsspezifität.
Limitierte Reproduzierbarkeit von Ergebnissen: Die Zellkulturbedingungen können das Verhalten der Zellen beeinflussen, was zu Schwankungen in den Ergebnissen führt.
Zeitaufwändig: Der Nachweis eines Virus über Zellkultur ist im Vergleich zu molekularen Methoden langsamer.
Komplexität und Kosten: Die Herstellung und Pflege von Zellkulturen erfordert spezialisierte Laborausrüstung und Expertise.
Biosicherheitsrisiken: Bei der Arbeit mit hochpathogenen Viren müssen hohe Sicherheitsstandards eingehalten werden, was zusätzliche Anforderungen an die Labore und die Forscher stellt.
Isolierung und Weiterverarbeitung
Zellkulturen sind das All-inclusive-Resorts für Viren – hier können sie sich ungestört vermehren. Aber wie immer im Leben: Alles Schöne hat auch mal ein Ende. Für die Winzlinge heißt das: „Check-out bitte!“ Die Reise geht weiter zu den High-Tech-Labs. Jetzt wird’s ernst – und spannend. Denn jetzt wollen wir wissen: Was genau da gewachsen ist. Und wie es aussieht. Willkommen bei den Tools der Virusanalyse:
Selfie mit dem Elektronenmikroskop: Will ein Virus wissen, wie es wirklich aussieht? Die Elektronenmikroskopie verpasst ihm den ultimativen Close-up – schärfer als jeder Instagram-Filter.
PCR und Sequenzierung: Der genetische Persönlichkeitstest: Ein bisschen Erbgut hier, ein paar Enzyme dort – und schon wird klar, wer (oder was) da eigentlich im Reagenzglas sitzt.
ELISA: Der Antikörper-Check: Dieser Test schnüffelt nach Antikörpern im Blut – und verrät, ob das Immunsystem schon Alarm geschlagen hat. Verdächtiger Fund? Fall abgeschlossen!
In den nächsten Kapiteln zerlegen wir einige dieser Methoden – ganz ohne Laborkittel.
4.4. Viren sichtbar machen
Viren sind wie Geister: Man spürt ihre Wirkung, aber sieht sie nie. Sie hinterlassen Chaos in Zellen, schreiben sich in unsere DNA ein, verursachen Krankheiten, prägen die Evolution und spielen seit Jahrhunderten Hide-and-Seek mit der Wissenschaft. Selbst unter dem besten Lichtmikroskop bleiben sie unsichtbar.
Doch wie heißt es so schön: Seeing is believing. Aber wie macht man das Unsichtbare sichtbar? Wie lassen sich Viren zweifelsfrei nachweisen? Und wie entschlüsseln wir ihre Strukturen und Mechanismen?
Die Antwort: Wir holen die ganz großen Mikroskope raus! Geräte, so leistungsstark, dass sie selbst die trickreichsten Viren in flagranti erwischen und keine Tarnung mehr hilft. Moderne bildgebende Verfahren zerlegen Viren heute bis ins atomare Detail.
Willkommen im Reich des Sichtbarmachens der Moderne – wo das Unsichtbare endlich Gestalt annimmt! Und los geht’s mit …
a) Elektronenmikroskopie
b) Kristallisation
c) Kryo-Elektronenmikroskopie
d) Kryo-Elektronentomographie
e) Zusammenfassung
a) Elektronenmikroskopie: Erste Bilder von Viren
Die Geschichte der Virologie begann mit der Erkenntnis, dass es etwas Kleineres als Bakterien geben muss, das Krankheiten verursacht.
Der Physiker Richard Feynman brachte es 1959 auf den Punkt:
„It is very easy to answer many… fundamental biological questions; you just look at the thing!“
„Es ist sehr einfach, viele grundlegende biologische Fragen zu beantworten; man muss sich die Sache nur anschauen!“
Doch wie kann man etwas sichtbar machen, das weit unterhalb der Auflösungsgrenze eines herkömmlichen Mikroskops liegt?
Mit der Erfindung des Elektronenmikroskops (EM) im Jahr 1931 wurde genau das erstmals möglich: Viren konnten direkt gesehen werden. Die Methode nutzt Elektronenstrahlen statt Licht, um extrem kleine Strukturen sichtbar zu machen. Wissenschaftler konnten erstmals die charakteristischen Hüllen und Formen von Viren abbilden und damit ihre physische Existenz bestätigen.

Die Illustration zeigt die Unterschiede in der Sichtbarmachung biologischer Strukturen durch zwei fundamentale Technologien der Mikroskopie.
Auf der linken Seite repräsentiert der Blick durch das Lichtmikroskop die klassischen Möglichkeiten, wie sie seit dem 17. Jahrhundert genutzt werden. Hier sind größere Strukturen wie menschliche Zellen gut erkennbar, ebenso Bakterien, die durch Färbetechniken sichtbar gemacht werden können. Viren hingegen bleiben unsichtbar, da ihre Größe (80–120 nm für Influenzaviren) weit unterhalb der Auflösungsgrenze eines Lichtmikroskops liegt.
Auf der rechten Seite zeigt der Blick durch das Elektronenmikroskop, wie moderne Technologien die Grenzen der Sichtbarmachung überwunden haben. Hier sind nicht nur menschliche Zellen und Bakterien in viel höherer Detailtiefe sichtbar, sondern auch Viren. Die Darstellung geht sogar so weit, dass Details wie die virale Hülle und das Genom in hochauflösenden Bildern sichtbar gemacht werden können.
Lichtmikroskope bieten einen allgemeinen Überblick und ermöglichen die Analyse lebender Zellen, während Elektronenmikroskope tiefere Einblicke in die Welt der Mikroorganismen und Viren gewähren – bis hin zu molekularen Details.
Das folgende Video illustriert den Unterschied zwischen Lichtmikroskopen und Elektronenmikroskopen sowie deren Funktionsweise auf anschauliche Weise.
Wie funktioniert Elektronenmikroskopie?
Elektronenmikroskope haben eine weitaus höhere Auflösung als Lichtmikroskope, da Elektronen eine viel kürzere Wellenlänge als Licht besitzen. Dies ermöglicht die Visualisierung von Strukturen im Nanometerbereich. Man unterscheidet dabei zwei Haupttypen:
1️⃣ Transmissionselektronenmikroskopie
(TEM – Transmission Electron Microscopy)
Ein Elektronenstrahl durchdringt eine extrem dünne Probe.
Dabei entstehen hochaufgelöste 2D-Bilder der inneren Strukturen.
(Früher: Kontrastarme Flecken, Heute: Bis zu 0,05 nm Auflösung)
Besonders nützlich für feine Details im Inneren von Viren.
2️⃣ Rasterelektronenmikroskopie
(REM/SEM – Scanning Electron Microscopy)
Der Elektronenstrahl tastet die Oberfläche einer Probe ab.
Dadurch entstehen detaillierte 3D-Bilder der Probenoberfläche.
(Früher: Grobe Umrisse, Heute: Molekülschärfe)
Gut geeignet für die äußere Struktur von Viren.

(schematische Darstellung)
Transmission Electron Microscopy: Zeigt eine Durchsicht des Virus, sodass auch innere Strukturen sichtbar sein können. Das Virus erscheint meist als zweidimensionale Projektion mit feinen Details im Inneren. Die Elektronen durchdringen die Probe, und der Kontrast entsteht durch Wechselwirkungen mit unterschiedlichen Dichten der Zell- oder Virusbestandteile.
Scanning Electron Microscopy: Zeigt eine dreidimensionale Oberfläche, oft mit einem plastischen, reliefartigen Effekt. Die Probe wird mit Elektronen abgetastet, wodurch ein tiefenscharfes Oberflächenbild entsteht. Innere Strukturen sind nicht sichtbar, da die Elektronen nicht durch die Probe hindurchgehen.
Ein originales TEM-Bild eines Influenzaviruspartikels kannst du hier sehen.
Weitere Infos zum Einsatz der Elektronenmikroskopie
Mithilfe der Elektronenmikroskopie (EM) lassen sich unterschiedliche Aspekte des Influenzavirus untersuchen:
🔬 Einblicke in die Ultrastruktur des Influenzavirus
Mithilfe der Transmissionselektronenmikroskopie (TEM) mit negativer Färbung konnten Wissenschaftler detaillierte Einblicke in die Ultrastruktur des Influenzavirus gewinnen. Die Studie zeigt die Anordnung der Spike-Proteine (Hämagglutinin und Neuraminidase) auf der Virusoberfläche sowie die interne Struktur, einschließlich des Matrixproteins (M1) und der RNA, und beleuchtet Unterschiede zwischen intakten und beschädigten Viruspartikeln.
🔬 Einblicke in die Struktur der Ribonukleoprotein-Komplexe (vRNPs)
Mittels EM konnten Wissenschaftler die helikale Anordnung der vRNPs im Viruspartikel sichtbar machen. Der Artikel von Noda und Kawaoka (2010) beschreibt detailliert die Architektur der vRNPs innerhalb des Viruspartikels und deren Verpackung in Virionen. Sie betonen, dass jede der acht vRNA-Segmente mit Nukleoproteinen und einem Polymerase-Komplex assoziiert ist, wodurch die vRNPs gebildet werden, die für die Transkription und Replikation des viralen Genoms essenziell sind. Elektronenmikroskopische Analysen haben gezeigt, dass diese vRNPs eine helikale Struktur aufweisen und innerhalb des Virions spezifisch angeordnet sind.
Elektronenmikroskopie in der Virologie
Dank der EM lassen sich virale Strukturen und Mechanismen aufdecken, darunter:
✅ die Form und Größe von Viren (z. B. Kugelform des Influenzavirus)
✅ die Anordnung der Spike-Proteine auf der Virushülle
✅ und Einblicke in den Infektionszyklus, z. B.:
• wie sie in die Wirtszelle eindringen
• wie die Replikation eingeleitet wird
Grenzen der Elektronenmikroskopie
Trotz ihrer beeindruckenden Auflösung hat die EM einige Nachteile:
⚡Probenvorbereitung → Proben müssen häufig entwässert, geschnitten und beschichtet werden, was ihre natürliche Struktur beeinflussen kann.
⚡Proben müssen fixiert werden → Das bedeutet, dass die Probe nicht in ihrem natürlichen Zustand untersucht wird.
⚡Keine lebende Probe → da die Proben im Vakuum analysiert werden, um eine Streuung der Elektronenstrahlen in der Luft zu vermeiden.
⚡Strahlenschäden → Der Elektronenstrahl kann empfindliche Proben verändern oder zerstören.
⚡Fehlender Kontrast → Biologische Proben sind oft kontrastarm und müssen speziell gefärbt werden.
⚡Kein „echtes“ 3D-Bild → TEM-Bilder sind nur zweidimensional, wodurch komplexe Strukturen schwer zu rekonstruieren sind.
Die Elektronenmikroskopie war der erste große Durchbruch: Endlich konnte man Viren sehen. Doch die frühen EM-Bilder wirkten noch verschwommen – wie Mondfotos aus den 1960ern. Um den Viren noch näher zu kommen, brauchte es mehr als ein Mikroskop. Man brauchte atomare Klarheit. Und so kam die Kristallisation ins Spiel – die Ultra-HD-Version der Virenforschung. Denn erst wenn sich Virusproteine in perfekte Kristalle zwängen, verraten sie ihr molekulares Innenleben: Atom für Atom, Bindung für Bindung.
b) Kristallographie: Detaillierte Strukturen von Virusproteinen
Während die Elektronenmikroskopie die grobe Struktur von Viren sichtbar machen konnte, blieb ihre molekulare Feinstruktur – ihre atomaren Details – lange im Verborgenen. Um zu verstehen, wie virale Proteine aufgebaut sind, musste man ihre Atomstruktur bestimmen – und das war nur mit Röntgenkristallographie möglich.
Wie kam es dazu?
Die Idee, biologische Makromoleküle mittels Röntgenbeugung zu analysieren, entwickelte sich in den 1920er und 1930er Jahren. Ein bahnbrechender Durchbruch gelang dem Physiker John Desmond Bernal, der 1934 zusammen mit Dorothy Crowfoot Hodgkin nachwies, dass Proteine in hydratisierter Form kristallisiert werden können, ohne ihre natürliche Struktur zu verlieren. Diese Entdeckung war entscheidend, um die Röntgenkristallographie für die Analyse von Biomolekülen nutzbar zu machen.
In den 1930er Jahren gelang es Wendell Meredith Stanley, das Tabakmosaikvirus in kristalliner Form zu isolieren. Dies war bedeutsam, weil es bewies, dass Viren nicht nur materielle Partikel sind, sondern auch aus regelmäßig angeordneten Molekülen bestehen, die sich kristallisieren lassen. Stanley brachte das Virus in eine feste, hochgeordnete Struktur – vergleichbar mit Salz- oder Zuckerkristallen. Diese Kristalle ermöglichten es später, mit Röntgenstrahlen die atomare Struktur von Virusproteinen zu entschlüsseln – ein entscheidender Fortschritt für die Virologie.
Stanley‘s Durchbruch und dessen Bedeutung
Wie hat Stanley die Kristallisation durchgeführt?
Stanley entwickelte eine innovative Methode, um die winzigen Viruspartikel zu isolieren und zu analysieren. Er extrahierte die Viren aus infizierten Tabakpflanzen und reinigte sie durch Zentrifugation sowie weitere Aufreinigungstechniken. Diese gereinigten Viren löste er anschließend in einer Lösung, die er langsam verdampfen ließ. Durch diesen Prozess kristallisierten die Viren aus, und es entstanden feste Strukturen, die er mit einem Elektronenmikroskop genauer untersuchen konnte. Dies war der erste Schritt, um die Struktur von Viren auf atomarer Ebene zu analysieren.
Warum war die Kristallisation so bedeutend?
Die Kristallisation des Tabakmosaikvirus lieferte zwei bahnbrechende Erkenntnisse:
Nachweis der Existenz von Viren als Partikel: Bis dahin waren Viren eher als vage, unsichtbare Krankheitserreger bekannt. Stanley zeigte, dass sie physische Einheiten mit einer klar definierten Struktur sind, die sich sogar kristallisieren lassen.
Grundlage für die Strukturaufklärung: Die Kristalle ermöglichten es späteren Forschern, mithilfe der Röntgenkristallographie die Architektur von Viren zu entschlüsseln und zu zeigen, dass sie aus genetischem Material – entweder RNA oder DNA – und Proteinen bestehen. Diese Entdeckung war entscheidend, um die Funktionsweise von Viren zu verstehen.
Was hat die Strukturaufklärung enthüllt?
Die Untersuchung des Tabakmosaikvirus zeigte erstmals, dass Viren eine klare und wiederholbare Struktur besitzen:
Genetisches Material und Proteine: Das genetische Material eines Virus enthält die Anweisungen zur Herstellung neuer Viruspartikel, während die Proteine die schützende Hülle bilden und Funktionen wie das Eindringen in Wirtszellen ermöglichen.
Unterscheidung von anderen Mikroorganismen: Stanleys Arbeit verdeutlichte, dass Viren sich grundlegend von Bakterien und anderen Mikroorganismen unterscheiden. Viren sind deutlich kleiner, besitzen keine Zellstruktur und bestehen nur aus einem Minimum an Komponenten, was sie zu einzigartigen biologischen Einheiten macht.
Was versteht man unter Kristallisation?
Kristallisation ist der physikalische Prozess, bei dem Atome, Moleküle oder Ionen aus einer ungeordneten Phase (z. B. einer Lösung, einer Schmelze oder einem Gas) in eine feste, geordnete Struktur übergehen, die als Kristall bezeichnet wird. Dieser Übergang führt zur Bildung eines Kristallgitters – einer regelmäßigen, dreidimensionalen Anordnung, in der sich die Teilchen in einem sich wiederholenden Muster organisieren. Dabei ordnen sich die Teilchen so an, dass sie einen energetisch günstigen Zustand erreichen, der durch spezifische Wechselwirkungen (z. B. Wasserstoffbrücken, elektrostatische Kräfte, van-der-Waals-Kräfte) und Bindungswinkel definiert ist.
Kristallisation ist also der Übergang von Chaos (ungeordnete Teilchen) zu Ordnung (Kristallgitter), angetrieben von physikalischen Kräften und energetischen Prinzipien. Sie kann natürlich (z. B. bei der Mineralbildung) oder künstlich (z. B. in Laboren) stattfinden.
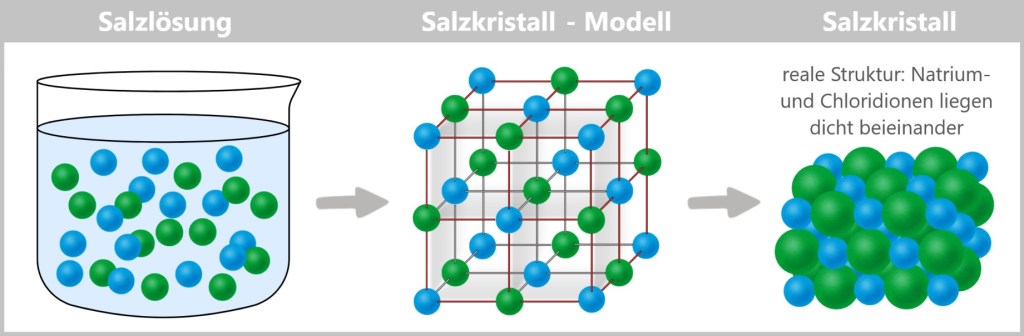
Links: In einer Salzlösung sind Natriumionen (grün) und Chloridionen (blau) frei beweglich und ungeordnet. Rechts: Beim Verdunsten des Wassers ordnen sich die Ionen zu einem regelmäßigen Kristallgitter an.
Grundprinzip der Proteinkristallisation
Proteinkristallisation bedeutet, dass man gelöste Proteine aus einer Lösung in eine feste, geordnete, kristalline Form überführt. Das Ziel ist ein dreidimensionales Gitter, in dem die Proteinmoleküle regelmäßig angeordnet sind. Das erreicht man, indem man die Löslichkeit der Proteine kontrolliert verringert, sodass sie langsam aus der Lösung „ausfallen“ und sich zu einem Kristall organisieren.
Der typische Prozess läuft so ab:
Reinigung: Das Protein wird hochrein isoliert, da Verunreinigungen die Kristallbildung stören.
Lösung herstellen: Das Protein wird in einer wässrigen Lösung mit Puffern, Salzen und manchmal organischen Zusätzen (z. B. Polyethylenglykol, PEG) gelöst.
Übersättigung: Durch Veränderung der Bedingungen (z. B. Verdunstung, Zugabe von Präzipitanten wie PEG oder Salzen) wird die Lösung übersättigt, sodass die Proteine auszufallen beginnen.
Keimbildung und Kristallwachstum: Zuerst bilden sich kleine Proteinaggregate (Keime), die dann zu größeren Kristallen wachsen, wenn die Bedingungen stimmen.

Proteine werden zunächst in Lösung gebracht, wo sie ungeordnet vorliegen. Durch gezielte Veränderung der Bedingungen (z. B. Erhöhung der Salzkonzentration, Veränderung des pH-Werts oder langsame Verdunstung des Lösungsmittels) wird die Lösung übersättigt. Dadurch beginnen die Proteine, sich zu organisieren und zu einem Kristall zu wachsen.
Auf molekularer Ebene richten sich die Proteine aufgrund ihrer Ladungen, Wasserstoffbrückenbindungen und hydrophoben Wechselwirkungen in einem regelmäßigen Gitter an. Die Moleküle „suchen“ dabei die energetisch günstigste Position, was zur Bildung eines stabilen Kristalls führt. Zwischen den Proteinen befindet sich Wasser, das durch kleine blaue Punkte dargestellt wird. Dieses Wasser ist ein integraler Bestandteil des Kristalls und stabilisiert die Proteinstruktur durch Wasserstoffbrückenbindungen.Proteinkristalle weisen oft eine symmetrische Struktur auf (z. B. kubisch oder hexagonal), da sich die Moleküle in wiederholbaren Mustern anordnen.
Proteine sind riesige Moleküle mit Tausenden von Atomen, komplexen 3D-Formen und uneinheitlichen Oberflächen (mit Ladungen, hydrophoben Bereichen usw.). Sie können nicht einfach wie die Natrium- und Chloridionen beim Salzkristall abwechselnd gestapelt werden.
Merkmale der Proteinkristalle
Die Festigkeit: Proteinkristalle sind deutlich fragiler als klassische Kristalle wie Salz oder Diamant. Sie bestehen zu 30–70 % aus Wasser, das in Kanälen und Hohlräumen des Kristallgitters eingeschlossen ist. Dadurch sind sie eher weich und gelartig. Mechanische Belastung oder Austrocknung kann sie leicht zerstören.
Die Farbe: Proteinkristalle sind meist farblos oder leicht opak (undurchsichtig). Proteine selbst besitzen keine natürliche Farbe – die bunten Darstellungen in wissenschaftlichen Abbildungen dienen lediglich dazu, strukturelle Merkmale und chemische Eigenschaften hervorzuheben.
Auswahl des Proteinkristalls
Bei der Kristallisation entstehen oft viele kleine Kristalle gleichzeitig, da sich die Proteinmoleküle an verschiedenen Stellen in der Lösung zu ordnen beginnen. Für die Analyse wird jedoch nur der bestgeordnete Kristall ausgewählt.
Wie findet man den richtigen Kristall?
Früher wurden die Kristalle unter dem Mikroskop betrachtet: Klare, scharfe Kanten und eine regelmäßige Form (z. B. kubisch oder hexagonal) deuteten auf eine hohe Qualität hin. Trübe oder unregelmäßige Kristalle hingegen waren weniger geeignet.
Heute kommen moderne Bildanalysesysteme zum Einsatz, die hochauflösende Mikroskopie mit automatischer Bewertung kombinieren. Zusätzlich kann eine Röntgenbeugung durchgeführt werden: Scharfe, symmetrische Beugungsmuster deuten auf eine gute Kristallordnung hin, während diffuse oder unregelmäßige Reflexionen auf eine geringe Qualität schließen lassen.
Warum braucht man Kristalle?
Kurz gesagt: Proteinkristalle dienen als „Verstärker“ für Röntgenstrahlen.
Ein einzelnes Proteinmolekül würde bei einer Röntgenbeugung nur ein extrem schwaches und diffuses Signal erzeugen – zu wenig, um eine detaillierte Struktur zu bestimmen. In einem Kristall hingegen sind Millionen identischer Proteinmoleküle regelmäßig angeordnet und gleich orientiert. Dadurch verstärken sich die Beugungssignale der einzelnen Moleküle durch konstruktive Interferenz und ergeben ein klares, regelmäßiges Beugungsmuster.
Diese geordnete Verstärkung ist essenziell für die Röntgenkristallographie, da sie erst durch das entstehende Beugungsmuster die dreidimensionale Struktur des Proteins entschlüsseln kann. Ohne Kristalle wäre die Analyse mit dieser Methode unmöglich.
Ablauf der Röntgenkristallographie
Nachdem das Protein erfolgreich kristallisiert wurde, kann seine räumliche Struktur (also seine genaue Faltung und Anordnung) mit der Röntgenkristallographie bestimmt werden. Dabei erkennt man Details wie:
➤ Position jedes einzelnen Atoms
➤ Faltungsstruktur des Proteins (α-Helices, β-Faltblätter usw.)
➤ Interaktionen mit anderen Molekülen (z. B. Antikörper, Wirkstoffe)
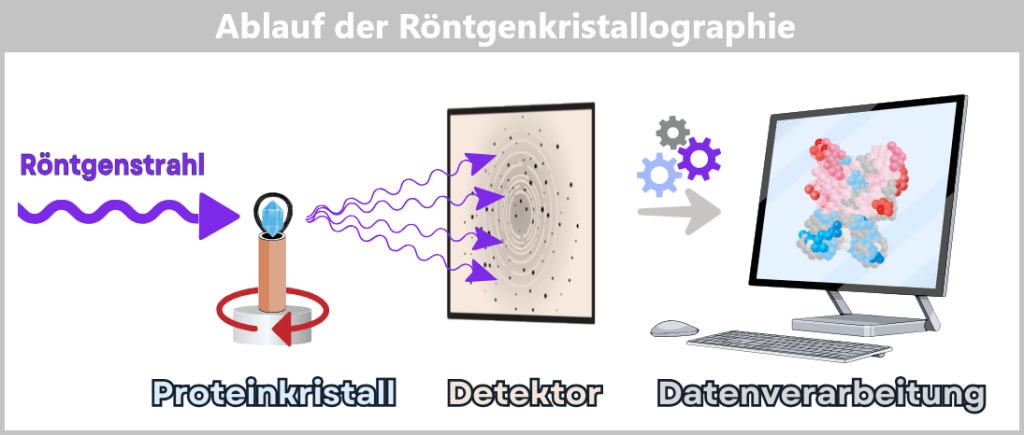
1️⃣ Röntgenstrahl auf den Kristall schießen
Ein gebündelter Röntgenstrahl trifft auf einen Proteinkristall. Proteine bestehen aus Aminosäuren, und diese wiederum aus Atomen. Röntgenstrahlen haben eine Wellenlänge von etwa 0,1 nm, was in der Größenordnung von Atomen liegt, sodass sie gut geeignet sind, um Details auf dieser Skala sichtbar zu machen. Die Atome im Kristall beugen die Röntgenstrahlen nicht direkt, sondern ihre Elektronenhüllen lenken die Strahlen in bestimmte Richtungen ab.
2️⃣ Beugungsmuster entsteht
Die gebeugten Röntgenstrahlen überlagern sich und erzeugen ein charakteristisches Muster auf einem Detektor. Dieses Beugungsmuster besteht aus vielen Punkten (sogenannten Reflexen), die an verschiedenen Positionen und mit unterschiedlicher Intensität auftreten. Um eine vollständige Datenerfassung zu ermöglichen, wird der Kristall in kleinen Schritten gedreht (typischerweise in 0,1–1°-Schritten), während für jede Position ein Beugungsmuster aufgenommen wird.
3️⃣ Mathematische Berechnung der 3D-Struktur
Die verschiedenen Aufnahmen der Beugungsmuster zeigen nicht direkt die Struktur des Proteins. Stattdessen enthalten sie Informationen darüber, wie die Röntgenstrahlen an den Elektronen der Atome im Kristall gebeugt wurden. Mithilfe mathematischer Methoden (Fourier-Transformation) wird aus den Beugungsmustern eine Elektronendichtekarte berechnet. Diese Karte zeigt die dreidimensionale Verteilung der Elektronen im Kristall.
Da sich Elektronen hauptsächlich in der Nähe der Atomkerne befinden, lassen sich aus der Elektronendichtekarte die Positionen der Atome ableiten. Die Karte erscheint zunächst als „wolkige“ Struktur, in der Bereiche mit hoher Elektronendichte den Atomen entsprechen.
Die Qualität der Elektronendichtekarte hängt direkt von der Ordnung im Kristall ab – je besser die Kristalle, desto schärfer die Elektronendichtekarte. Durch weitere Analyse und Interpretation dieser Karte kann schließlich ein detailliertes, dreidimensionales Modell des Proteins erstellt werden.
〰️ Synchrotronstrahlung – Licht für die kleinsten Geheimnisse der Natur
Um die Struktur von Proteinen noch genauer zu untersuchen, nutzt man in vielen Fällen Synchrotronstrahlung – eine extrem intensive Röntgenstrahlung, die in speziellen Teilchenbeschleunigern erzeugt wird.
💡 Was ist das genau?
Synchrotronstrahlung entsteht, wenn geladene Teilchen (z. B. Elektronen) auf nahezu Lichtgeschwindigkeit beschleunigt und dann durch Magnetfelder in eine Kreisbahn gelenkt werden. Dabei geben sie hochenergetische Strahlung ab – darunter auch besonders brillante Röntgenstrahlen, die sich perfekt für die Proteinkristallographie eignen. Dadurch können auch winzige oder schwer zu kristallisierende Proteine untersucht werden.
🌍 Weltweit gibt es mehrere große Synchrotron-Forschungszentren, z. B. in:
- ESRF (Frankreich) – Europäisches Synchrotron-Strahlungszentrum
- DESY (Deutschland) – Deutsches Elektronen-Synchrotron
- Diamond Light Source (UK) – Synchrotronanlage in Großbritannien
🎥 Wenn du live miterleben möchtest, wie Proteine zu Kristallen werden und wie ihre Strukturen mithilfe von Röntgenstrahlen entschlüsselt werden, dann begleite die Forscher der Diamond Light Source in die faszinierende Welt der Kristallographie!
Understanding Crystallography – Part 1: From Proteins to Crystals
Hier siehst du, wie Wissenschaftler Proteine in Kristalle verwandeln.
Understanding Crystallography – Part 2: From Crystals to Diamond
In diesem Video siehst du, wie die Kristalle mit Röntgenstrahlen untersucht werden, um ihre 3D-Struktur zu enthüllen.
Die Proteindatenbank – Ein Schatz der Strukturbiologie
Die Röntgenkristallographie hat nicht nur dazu beigetragen, die Struktur einzelner Proteine aufzuklären, sondern auch die Entstehung einer globalen Ressource ermöglicht: die Protein Data Bank (PDB). Diese Datenbank sammelt und speichert die 3D-Strukturen von Proteinen, Nukleinsäuren und anderen Biomolekülen.
Die PDB wurde 1971 gegründet und enthält heute über 200.000 Einträge. Bis in die 1970er Jahre war die Röntgenkristallographie die einzige Methode zur Bestimmung der dreidimensionalen Struktur von Proteinen mit atomarer Auflösung.
In den folgenden Jahrzehnten erweiterten neue Techniken wie die Kernspinresonanzspektroskopie (NMR) und später die Kryo-Elektronenmikroskopie (Cryo-EM) das methodische Spektrum. Während NMR besonders für kleine Proteine in Lösung geeignet ist, ermöglicht Cryo-EM die Untersuchung großer Proteinkomplexe ohne Kristallisation.
Jede Proteinstruktur liefert detaillierte Informationen über die räumliche Anordnung der Atome in einem Molekül. Die Röntgenkristallografie hat damit nicht nur unser Verständnis revolutioniert – sie hat eine globale Infrastruktur des Wissens geschaffen. Eine stille Bibliothek, in der jedes Protein seine Geschichte erzählt – festgehalten in atomarer Genauigkeit. Zugänglich für Forscher auf der ganzen Welt.
Röntgenkristallographie in der Virologie
Viren sind komplexe Gebilde, die aus Proteinen, Nukleinsäuren (DNA oder RNA) und manchmal auch Lipiden bestehen. Ganze Viren sind oft zu groß und zu flexibel, um sie zu kristallisieren und mit Röntgenkristallographie zu untersuchen.
Einige Viren besitzen jedoch regelmäßige, symmetrische Strukturen, die eine dichte, wiederholte Packung ermöglichen und damit die Kristallisation ermöglichen. Ein bekanntes Beispiel ist das Tabakmosaikvirus, dessen Struktur bereits in den 1950er Jahren mithilfe der Röntgenkristallographie aufgeklärt wurde.
Für die meisten Viren ist es jedoch praktikabler, einzelne Virusproteine zu untersuchen. Besonders interessant sind dabei Proteine, die das Virus für die Infektion von Wirtszellen, seine Vermehrung oder zur Umgehung des Immunsystems benötigt.
Um ein Beispiel für die Struktur eines Virusproteins zu zeigen, greifen wir auf die Proteindatenbank (PDB) zurück. Die folgende Abbildung stammt aus der PDB und zeigt das Hämagglutinin-Protein des H5N1-Influenzavirus. Seine atomare Struktur wurde mithilfe der Röntgenkristallographie bestimmt und in der Protein Data Bank hinterlegt.
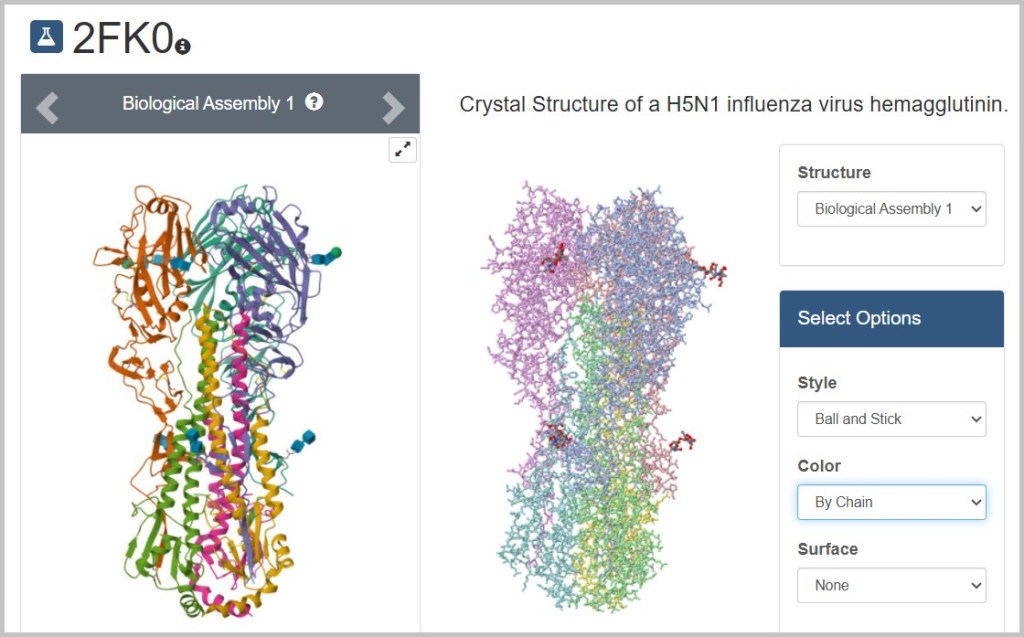
Anleitung zur Suche in der Proteindatenbank
1️⃣ Öffne die Protein Data Bank (PDB): https://www.rcsb.org.
2️⃣ Suche nach einem Protein:
In unserem Beispiel suchen wir nach dem Hämagglutinin-Protein des Influenzavirus.
👉 Gib in das Suchfeld den Begriff „influenza hemagglutinin“ ein.
👉 Filtere die Ergebnisse unter [Experimental Method] nach [X-ray diffraction] (Röntgenkristallographie).
3️⃣ Wähle einen Eintrag aus:
👉 Klicke auf ein Ergebnis. Für unser Beispiel wählen wir PDB-ID: 2FK0 – das Hämagglutinin-Protein des H5N1-Influenzavirus.
4️⃣ Ansicht des Proteins:
Links in der PDB siehst du die Cartoon-Darstellung oder das Ribbon-Diagramm, eine schematische Ansicht der 3D-Faltung des Proteins.
5️⃣ Visualisierung der atomaren Struktur:
👉 Klicke auf [Structure], um in die 3D-Ansicht zu wechseln.
👉 Wähle einen anderen Viewer: „JSmol“ (siehe: unten rechts).
👉 Für die atomare Struktur-Ansicht wähle den [Style]: „Ball-and-Stick“.
🔍 Nun sind alle Atome und Bindungen sichtbar. Mit der Maus kannst du das Modell drehen, zoomen und verschieben.
6️⃣ Strukturelle Merkmale hervorheben:
👉 Unter [Color] kannst du verschiedene Farb-Darstellungen. Das Beispiel: „By chain“ färbt jede Proteinkette in einer eigenen Farbe, um Untereinheiten des Proteins zu unterscheiden.
Weitere Infos zum Einsatz der Röntgenkristallographie
Seit über 40 Jahren ist die Röntgenkristallographie eine leistungsfähige Methode zur Bestimmung von Virusstrukturen. Da wir bereits das Oberflächenprotein Hämagglutinin des Influenzavirus kennengelernt haben, beziehen sich die nachfolgend aufgeführten Studien darauf.
🔗 Eine der ersten Studien dazu wurde 1981 veröffentlicht: „Structure of the haemagglutinin membrane glycoprotein of influenza virus”. Sie enthüllte die dreidimensionale Struktur des HA-Proteins und legte die Grundlage für das Verständnis der Infektionsmechanismen des Influenzavirus.
🔗 Eine neuere Studie aus dem Jahr 2019 mit dem Titel „Identification of a pH sensor in Influenza hemagglutinin using X-ray crystallography” untersuchte die Bindungsregion des HA-Proteins mittels Röntgenkristallographie. Sie zeigte, wie Konformationsänderungen der Bindungsregion in Abhängigkeit des pH-Werts die Virusfunktion beeinflussen.
🔗 Eine Studie aus dem Jahr 2020, „Structure of avian influenza hemagglutinin in complex with a small molecule entry inhibitor”, nutzte Röntgenkristallographie, um die Struktur des H5-Hämagglutinin (HA)-Proteins des Influenzavirus H5N1 in komplex mit dem Inhibitor CBS1117 aufzuklären. Die Kristallstruktur lieferte detaillierte Einblicke in die molekularen Wechselwirkungen, die für die Entwicklung neuer antiviraler Wirkstoffe entscheidend sind.
Durch die Kombination der Strukturen einzelner Proteine können Wissenschaftler ein detailliertes Bild des gesamten Virus zusammensetzen.
Grenzen der Röntgenkristallographie
Obwohl die Röntgenkristallographie eine unglaublich leistungsfähige Methode zur Strukturbestimmung ist, stößt sie in bestimmten Fällen an ihre Grenzen:
⧎ Schwierige Kristallisation: Viele Proteine, insbesondere große Komplexe oder Membranproteine, lassen sich nur schwer oder gar nicht in Kristallform bringen.
⧎ Künstliche Bedingungen: Proteine werden in der Kristallform fixiert, was möglicherweise nicht ihrem natürlichen Zustand entspricht.
⧎ Fehlende Dynamik: Die Methode zeigt nur eine statische Momentaufnahme, aber keine Informationen über Bewegung oder Flexibilität der Moleküle.
⧎ Strahlenschäden: Die hochenergetischen Röntgenstrahlen können empfindliche Moleküle leicht verändern.
Auf der Suche nach einer neuen Methode
Diese Einschränkungen machten klar: Nicht jedes Biomolekül lässt sich wie Lego in Kristallform pressen. Die Lösung? Ein mikroskopischer Kälte-Blitz: Kryo-EM. Ihr Trick? Moleküle in Millisekunden schockgefrieren – so schnell, dass Wasser gar nicht erst kristallisiert. Keine Kristall-Zwangsjacke, keine Röntgen-Bräune – nur eiskalte Atom-Details.
c) Kryoelektronenmikroskopie: Ein moderner Blick auf Viren
Nachdem wir die klassische Elektronenmikroskopie und die Röntgenkristallographie kennengelernt haben, die uns erstmals Einblicke in die Struktur von Viren ermöglichten, führt uns die Kryoelektronenmikroskopie – kurz Kryo-EM – in eine neue Ära der Forschung.
Was ist Kryo-EM überhaupt?
Kryo-EM ist eine Technik, bei der Proben extrem schnell eingefroren („kryo“ = kalt) und dann mit einem Elektronenmikroskop untersucht werden. Das Besondere: Die Proben bleiben in einem nahezu natürlichen Zustand, weil sie nicht chemisch fixiert oder gefärbt werden müssen, wie bei anderen Methoden. Das macht sie ideal, um empfindliche Strukturen wie Viren zu betrachten.
Wie kam es dazu?
Früher hatten Wissenschaftler ein großes Problem, wenn sie biologische Proben mit Elektronenmikroskopen untersuchen wollten: Wasser verdunstete im Vakuum der Mikroskope, und empfindliche Moleküle wurden durch den intensiven Elektronenstrahl beschädigt. Dadurch entstanden unscharfe oder verzerrte Bilder.
Forscher hatten bereits die Idee, Proben zu kühlen, um sie zu schützen. Doch es gab ein weiteres Problem: Wenn Wasser gefriert, bilden sich Eiskristalle. Diese beugen Elektronen so stark, dass klare Bilder unmöglich wurden.
Der entscheidende Durchbruch kam in den 1980er Jahren, als der Wissenschaftler Jacques Dubochet eine Technik namens Tauchgefrieren entwickelte. Dabei wird die Probe extrem schnell auf sehr niedrige Temperaturen abgekühlt, bevor sich Eiskristalle bilden können. Stattdessen bleibt das Wasser in einem glasartigen, gefrorenen Zustand erhalten, der die biologische Probe perfekt umhüllt. So wurde die Kryo-Elektronenmikroskopie (Kryo-EM) geboren.
Ein weiterer wichtiger Fortschritt kam in den 1970er Jahren, als Joachim Frank eine Methode entwickelte, um die unscharfen Bilder aus dem Mikroskop mit computergestützten Berechnungen zu verbessern. Indem er zahlreiche Aufnahmen einzelner Moleküle aus unterschiedlichen Blickwinkeln kombinierte, gelang es ihm, ein gestochen scharfes 3D-Modell zu rekonstruieren. Dadurch war es erstmals möglich, die Struktur von Biomolekülen zu bestimmen, ohne sie zuvor kristallisieren zu müssen – ein großer Vorteil für Proteine, die sich nur schwer oder gar nicht in Kristallform bringen lassen.
1990 nutzte Richard Henderson die Kryo-EM erstmals, um ein Biomolekül in so hoher Auflösung zu untersuchen, dass selbst kleine Details wie Aminosäure-Seitenketten sichtbar wurden. Für diese bahnbrechenden Entwicklungen erhielten Dubochet, Frank und Henderson 2017 den Nobelpreis für Chemie.
Seitdem hat sich die Technologie rasant weiterentwickelt:
↗️ Moderne Kameras nehmen gestochen scharfe Bilder direkt auf, ohne Qualitätsverluste.
↗️ Automatisierte Mikroskope können mehrere Proben gleichzeitig analysieren.
↗️ Leistungsstarke Computerprogramme ermöglichen die Verarbeitung riesiger Datenmengen, um noch genauere Strukturen zu berechnen.
Dank dieser Fortschritte kann die Kryo-EM heute komplexe Moleküle, Proteine, Viren und sogar ganze Zellstrukturen in ihrem natürlichen Zustand sichtbar machen – und das in einer nie dagewesenen Detailtiefe.
Im Folgenden werfen wir einen genaueren Blick darauf, wie die Kryo-EM Viren sichtbar macht.
Mit Kryo-EM Viren sichtbar machen – Schritt für Schritt
1️⃣ Probenvorbereitung: Viren züchten und isolieren
Wenn man Viren untersuchen will, muss man sie erst einmal haben. Dafür werden sie meist in geeigneten Wirtszellen gezüchtet, z. B. in Zellkulturen im Labor. Viren sind Parasiten, die sich nur in Wirtszellen vermehren können. Um sie zu untersuchen, infiziert man also geeignete Zellen in einer Zellkultur und lässt die Viren sich vermehren.
Nach der Infektion der Zellen werden die Viren zu bestimmten Zeitpunkten „geerntet“, um verschiedene Stadien ihres Lebenszyklus (z. B. Anheftung, Eindringen, Vermehrung) zu untersuchen. Danach werden die Viren aus der Zellkultur isoliert und gereinigt, z. B. durch Zentrifugation oder Filtration, damit man eine saubere Virusprobe hat, ohne störende Zellreste.
2️⃣ Probepräparation: Aufbringen der Probe auf ein Gitter
Die gereinigte Virusprobe ist eine wässrige Lösung, in der die Viren schweben. Die Viruslösung wird mit einer Pipette auf ein Gitter aufgetragen (siehe untere Abbildung).
Ein typisches Kryo-EM-Gitter ist winzig, es hat einen Durchmesser von 3 Millimetern, damit es in die Halterungen des Elektronenmikroskops passt. Das Gitter besteht aus einem feinen Netz aus Metall (z. B. Kupfer oder Gold), das wie ein Sieb aussieht. Auf das Metallgitter wird eine ultradünne Kohlenstofffolie gelegt, die selbst winzige Löcher enthält (meist 1–2 Mikrometer im Durchmesser). Durch die Oberflächenspannung verteilt sich die Lösung in den winzigen Löchern und haftet dort, bevor sie eingefroren wird. Diese Anordnung hat zwei wichtige Funktionen: Das Gitter sorgt für Stabilität, während die Löcher den Elektronenstrahl ungehindert passieren lassen, um Hintergrundrauschen zu minimieren.

A – Gitter, bedeckt mit einer löchrigen Kohlenstofffolie,
B – vergrößertes Bild einer Gitteröffnung, einer sogenannten Mesh-Öffnung,
C – vergrößertes Bild eines Lochs, in der die Viren haften
3️⃣ Schnellgefrieren der Probe
Die Probe bzw. das Gitter wird nun blitzschnell in flüssigem Ethan oder Stickstoff auf etwa -180 °C eingefroren. Das nennt man Vitrifizierung – das Wasser wird nicht zu Kristallen, sondern zu einem glasartigen Zustand, der die Struktur perfekt erhält. So bleiben die Viren in ihrem natürlichen Zustand erhalten – als wären sie in der Zeit eingefroren. Zudem schützt das Eis die Moleküle vor Schäden durch die intensive Elektronenstrahlung.
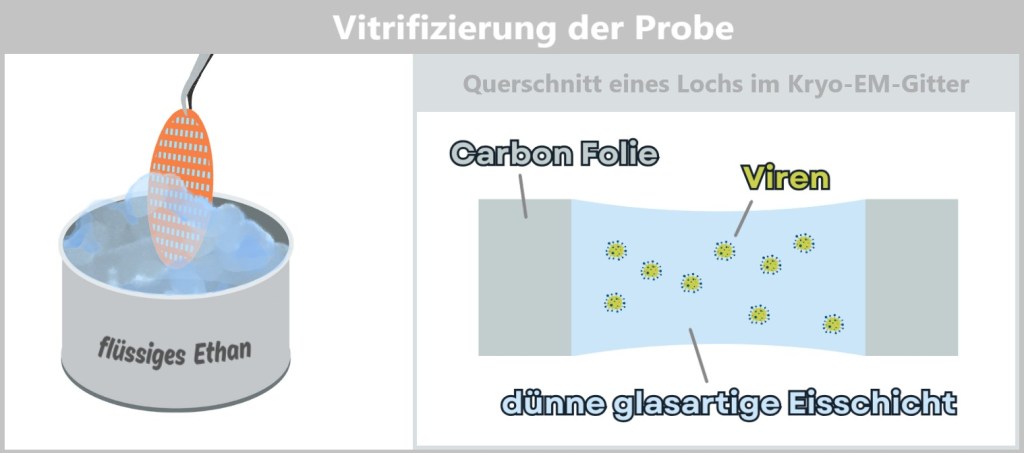
4️⃣ Aufnahme der Bilder im Elektronenmikroskop
Das gefrorene Gitter mit den Viren wird in das Kryo-Elektronenmikroskop eingelegt. In der vitrifizierten Lösung sind die Viren zufällig ausgerichtet – sie liegen also in verschiedenen Winkeln vor. Der Elektronenstrahl trifft die Probe aus einer festen Richtung (meist senkrecht), und eine hochempfindliche Kamera nimmt 2D-Projektionen der Viren auf.
Da jedes Virus in einer anderen Orientierung eingefroren ist, erhält man automatisch Bilder aus vielen verschiedenen Blickwinkeln – ohne dass das Gitter gedreht werden muss. Diese große Anzahl an Bildern ist wichtig für die spätere 3D-Rekonstruktion. Insgesamt werden oft Tausende bis Hunderttausende Aufnahmen gemacht.
5️⃣ Datenverarbeitung: Vom 2D-Bild zum 3D-Modell
Nach der Aufnahme beginnt die eigentliche Herausforderung: die computergestützte Rekonstruktion. Spezielle Software erkennt die einzelnen Viruspartikel in den 2D-Bildern und sortiert sie nach ihrer Orientierung.
Durch mathematische Verfahren (wie „Single Particle Analysis“) werden die 2D-Bilder zusammengefügt, um ein hochauflösendes 3D-Modell des Virus zu berechnen. Je mehr Bilder und je besser deren Qualität, desto schärfer wird das Modell.
Falls verschiedene Stadien des Viruslebenszyklus untersucht werden sollen (z. B. vor und nach dem Eindringen in eine Zelle), wiederholt man diesen Prozess mit Proben aus unterschiedlichen Zeitpunkten. So kann man sogar Veränderungen in der Struktur über die Zeit sichtbar machen.

Die folgenden Videos fassen die bisherigen Informationen zur Kryo-EM anschaulich zusammen:
👉 What is Cryo-Electron Microscopy (Cryo-EM)?
👉 Cryo-EM Animation
6️⃣ Interpretation und Visualisierung
Nach der 3D-Rekonstruktion liegt ein hochauflösendes Modell des Virus vor, das dessen Aufbau zeigt – z. B. die Hülle, die Spike-Proteine und manchmal sogar innere Strukturen.
Nun geht es darum, die eiskalten Daten zu analysieren, zu interpretieren und anschaulich darzustellen. Dieser Schritt ist wichtig, um die biologische Bedeutung der Struktur zu entschlüsseln und die Erkenntnisse für weitere Forschung oder die Öffentlichkeit zugänglich zu machen.
Schritte der Interpretation und Visualisierung
a) Analyse der Struktur: Wissenschaftler untersuchen die 3D-Struktur, um Schlüsselmerkmale des Virus oder seiner Proteine zu identifizieren. Diese Analyse hilft, die molekularen Mechanismen der Virusinfektion zu verstehen. Dazu gehören z. B.:
- Bindungsstellen: Wo bindet das Virus an Wirtszellen?
- Funktionelle Bereiche: Welche Teile des Proteins sind für die Infektion oder Vermehrung essenziell?
- Veränderungen: Gibt es Unterschiede in der Struktur zwischen verschiedenen Virusstämmen oder -stadien?
b) Vergleich mit bekannten Strukturen: Die rekonstruierte Struktur wird mit bereits bekannten Strukturen aus Datenbanken wie der Protein Data Bank (PDB) verglichen. Dies kann Aufschluss über evolutionäre Beziehungen, funktionelle Ähnlichkeiten oder mögliche Angriffspunkte für Medikamente geben.
c) Visualisierung der Struktur: Mithilfe spezieller Software wird das 3D-Modell visualisiert, um die Struktur anschaulich darzustellen und wichtige Merkmale hervorzuheben. Dabei gibt es verschiedene Darstellungsformen:
- Oberflächendarstellung: Zeigt die äußere Form des Virus oder Proteins.
- Stäbchenmodell (Ball-and-Stick): Zeigt die Anordnung der Atome und Bindungen.
- Sekundärstruktur: Hebt strukturelle Elemente wie α-Helices und β-Faltblätter hervor.
d) Publikation und Datenfreigabe: Die Ergebnisse werden in wissenschaftlichen Fachzeitschriften veröffentlicht – oft mit hochauflösenden Bildern oder Animationen der 3D-Struktur. Zudem werden die Rohdaten und rekonstruierten Modelle in öffentlichen Datenbanken wie der PDB (Protein Data Bank) oder der EMDB (Electron Microscopy Data Bank) gespeichert, sodass andere Forscher darauf zugreifen können.
Wenn Viren wüssten, wie oft wir sie jetzt in 3D drehen und zoomen können… sie würden sich glatt was anziehen!
Influenzavirus in 3D: ein Beispiel
Es kann schwierig sein, 3D-Modelle von Viren im Internet zu finden. Forschungsdaten werden oft erst nach der Veröffentlichung von Studien freigegeben und dann in speziellen Datenbanken wie der EMDB (Electron Microscopy Data Bank) gespeichert. Doch diese Daten sind für Laien schwer zugänglich, weil sie spezielle Software benötigen, um sie als 3D-Modelle anzuzeigen.
Erfreulicherweise bietet die NIH 3D-Plattform (National Institutes of Health) ein hilfreiches Beispiel: ein 3D-Modell eines Influenzavirus, das mit der Kryo-Elektronenmikroskopie erstellt wurde. Du kannst es dir unter diesem Link anschauen! Beachte, dass du möglicherweise einen 3D-Viewer brauchst, um es zu erkunden.
Weitere Infos zum Einsatz der Kryo-EM
🔬 Diese Studie zum Influenzavirus zeigte mithilfe der Kryo-Elektronenmikroskopie die Interaktionen zwischen den Nukleoproteinen und der viralen RNA sowie die spezifische Anordnung der Ribonukleoprotein-Komplexe (RNPs) innerhalb des Virions. Solche strukturellen Einblicke sind entscheidend für das Verständnis der Mechanismen, mit denen das Influenzavirus sein Genom verpackt und während des Infektionszyklus repliziert.
🔬 Ergänzend zeigt eine frühere Studie von Liu et al. (2017) strukturelle Details filamentöser Influenzavirus-Partikel und deren RNP-Organisation mittels Kryo-EM.
Warum Kryo-EM so genial ist – besonders bei Viren
Die Kryo-EM ist ein unschlagbares Werkzeug, wenn es darum geht, Viren sichtbar zu machen – besonders wenn’s um die schwer fassbaren Kandidaten geht:
- Viren mit ihren tückischen Spike-Proteinen
- Riesige Proteinkomplexe, die sich weigern, ordentlich zu kristallisieren
- Membranproteine, die sonst nur „Hallo!“ sagen und sofort zerfallen
Die Methode: Sie friert Viren blitzschnell ein, sodass sie wie in einer Zeitkapsel erhalten bleiben, und erzeugt Tausende von Aufnahmen mithilfe eines Elektronenstrahls. Ein Computer setzt diese Bilder dann zu einem hochauflösenden 3D-Modell zusammen – so können wir genau sehen, wie ein Virus aufgebaut ist. Diese Methode wird als Single Particle Analysis (SPA) bezeichnet, da einzelne Viruspartikel analysiert und rechnerisch zu einer 3D-Struktur zusammengeführt werden.
Vom Standbild zum Blockbuster
Was die Kryo-EM an detailreichen „Schnappschüssen“ liefert, ist beeindruckend – aber manchmal reicht ein Bild eben nicht aus. Um zu verstehen, wie Viren sich bewegen, Zellen kapern oder sich replizieren, braucht es bewegte Szenen. Genau hier setzt die Kryo-Elektronentomographie an: eine Technik, mit der man Viren fast wie in einem Film beim Arbeiten zuschauen kann.

d) Kryo-Elektronentomographie – 3D-Virusmodelle in der Zelle
Während die Kryo-EM hochgereinigte Viren isoliert untersucht, erlaubt die Kryo-ET Wissenschaftlern, dreidimensionale Schnappschüsse molekularer Interaktionen direkt innerhalb der Zelle aufzunehmen. So können Viren in ihrem natürlichen Umfeld – also in der Zelle – sichtbar gemacht werden.
Mit Kryo-ET Viren sichtbar machen – Schritt für Schritt
1️⃣ Probenvorbereitung – Einfrieren der Zelle
Geeignete Zellen werden in einer Zellkultur gezüchtet und mit Viren infiziert. Dann wartet man auf einen definierten Zeitpunkt – zum Beispiel auf das Eindringen der Viren in die Zelle oder auf ihre Vermehrung – und entnimmt die Zellen. Diese werden in eine Pufferlösung überführt, um eine homogene Zelllösung zu erhalten.
Anschließend wird die Zelllösung auf ein Kryo-EM-Gitter aufgetragen – ein kleines Metallgitter mit einer löchrigen Kohlenstofffolie, das bereits aus der Kryo-EM bekannt ist. Das Gitter wird sofort in flüssiges Ethan oder flüssigen Stickstoff (bei ca. -180 °C) getaucht. Durch diese blitzschnelle Gefriertechnik (Vitrifizierung) bleiben die Zellstrukturen erhalten, ohne dass sich störende Eiskristalle bilden.

2️⃣ Dünne Zellbereiche erzeugen (falls nötig)
Durch die Vitrifizierung werden die Zellen stabilisiert – sie sind nun fest wie Glas. Da Zellen oft zu dick für die Kryo-ET sind, nutzen Wissenschaftler einen Focussed Ion Beam (FIB), um die Eisschicht auf die gewünschte Dicke zu reduzieren.
Dafür wird das Kryo-EM-Gitter in einem FIB-SEM-Gerät (Focussed Ion Beam mit Rasterelektronenmikroskop) zunächst gescannt, um interessante Zellen zu lokalisieren. Anschließend fräst ein fokussierter Ionenstrahl mit höchster Präzision eine Lamelle (~100–200 nm dick) aus der Eisschicht. Diese Lamelle stellt einen Querschnitt der Zelle dar und zeigt relevante Bereiche mit Viruspartikeln – z. B. die Zellmembran oder das Zytoplasma mit Viren.
Wie das Ganze in der Praxis aussieht, zeigt das Video „Cryo-lamella preparation“ im Abschnitt „FIB-milling of lamella in waffle grids“ auf der Seite „Cryo EM 101, Kapitel 3“.
3️⃣ Bildaufnahme im Kryo-EM
Das gefrorene Zellgitter wird ins Kryo-Elektronenmikroskop gebracht. Hier erzeugt ein Elektronenstrahl hochaufgelöste Bilder der Probe.
Tomographie – Wie ein CT-Scan für Zellen
Die Probe wird schrittweise gedreht. Ein Goniometer (Drehmechanismus) kippt das Gitter in kleinen Winkelschritten (z. B. 1–2°), typischerweise über einen Bereich von ±60° bis ±70°. Für jeden Winkel wird ein 2D-Bild aufgenommen, sodass eine sogenannte „Tilt-Series“ (Neigungsserie) entsteht – eine Sammlung von 2D-Bildern aus verschiedenen Perspektiven (siehe untere Abbildung).
📌 Kryo-ET vs. SPA: Im Gegensatz zur Single Particle Analysis (SPA), bei der viele identische Viren benötigt werden, konzentriert sich die Kryo-ET auf einen spezifischen Bereich der Probe – oft eine einzelne Zelle oder eine Virus-Wirtszell-Interaktion. Man benötigt also keine große Anzahl identischer Partikel, sondern kann individuelle biologische Strukturen direkt in ihrem natürlichen Umfeld untersuchen.
4️⃣ 3D-Rekonstruktion
Die Kryo-ET nutzt die Neigungsserie, um rechnerisch ein 3D-Modell zu rekonstruieren – wie ein medizinischer CT-Scan, nur für Viren statt für Knochen. Das Ergebnis: ein hologrammartiges Volumenbild, das zeigt, wie Viren in echten Zellen tanzen, andocken und tricksen.
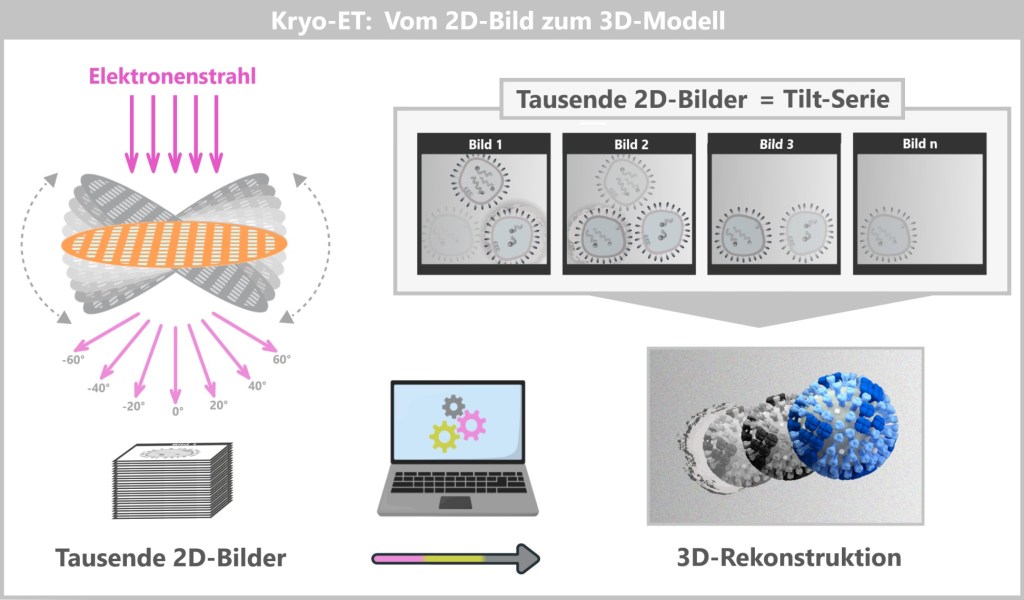
5️⃣ Interpretation & Visualisierung
Jetzt wird’s actionreich! Die 3D-Modelle zeigen Viren in ihrer natürlichen Umgebung:
- wie sie in der Zelle sitzen
- wie sie mit Zellorganellen interagieren
- verschiedenen Stadien (z. B. Eintritt, Replikation, Freisetzung)
Die Ergebnisse werden interpretiert, um z. B. den Infektionsmechanismus des Virus oder seine Interaktion mit der Wirtszelle zu verstehen.
Manchmal werden mehrere Zeitpunkte verglichen, um eine Art „Film“ zu erstellen, der die Dynamik simuliert.
Das folgende Video gibt einen Eindruck davon: Es wurden detailreiche 3D-Modelle eines Influenzavirus während der Knospung an der Zellmembran visualisiert und anschließend zu einem Film zusammengesetzt.
6️⃣ Publikation und Datenspeicherung
Damit alle was davon haben, landen die spektakulärsten Virus-Modelle in öffentlichen Datenbanken wie der Electron Microscopy Data Bank (EMDB) – das Netflix für Strukturbiologen.
Warum das cool ist:
- Open Science = Kein Wissenschaftler muss das Rad zweimal bauen.
- Transparenz = Jeder kann nachvollziehen, wie der „Film“ entstand.
- Kooperation = Teamwork macht auch Virenforschung zum Hit.
Wenn Viren ein Twitter hätten, würden sie jetzt trenden mit #EMDBChallenge.
Weitere Infos zum Einsatz der Kryo-ET
🔬 Die Studie „Quantitative Strukturanalyse von Influenzaviren mittels Kryo-Elektronentomographie und Convolutional Neural Networks“ nutzte Kryo-Elektronentomographie (Kryo-ET) und moderne Computertechnik, um die Struktur des Influenzavirus zu untersuchen. Sie zeigt die vielfältige Form des Virus und enthält Videos aus 3D-Rekonstruktionen, die den Prozess veranschaulichen.
🔬 Die Studie „Strukturelle Veränderungen des Influenzavirus bei niedrigem pH-Wert, charakterisiert durch Kryo-Elektronentomographie“ untersuchte, wie das Influenza-A-Virus auf saure Bedingungen reagiert, die es während des Eintritts in Wirtszellen erfährt. Durch Kryo-Elektronentomographie beobachteten die Forscher, dass bei niedrigem pH-Wert die viralen Hüllproteine (Hämagglutinin) strukturelle Veränderungen durchlaufen, die zur Fusion der viralen und zellulären Membranen führen. Diese Fusion ermöglicht es dem Virus, sein genetisches Material in die Wirtszelle freizusetzen und die Infektion zu initiieren.
Forscher untersuchen nicht nur humanpathogene Viren
Forscher interessieren sich längst nicht mehr nur für Krankheitserreger wie Grippe- oder Coronaviren – auch Viren aus der Umwelt sind extrem spannend. Allen voran: Bakteriophagen, die Bakterien befallen. In den Ozeanen sind sie echte Öko-Ingenieure.
1️⃣ Winzige Regisseure im Ökosystem
Phagen töten gezielt bestimmte Bakterien und halten so das mikrobielle Gleichgewicht im Meer im Lot. Damit steuern sie indirekt wichtige Kreisläufe wie den Kohlenstoff- oder Stickstoffhaushalt. Wer ihre Struktur und ihr Verhalten kennt, versteht besser, wie stabile Meeresökosysteme funktionieren.
2️⃣ Evolution live erleben
Phagen sind die häufigsten biologischen „Lebewesen“ der Erde – und genetisch unglaublich vielfältig. Mit Hilfe der Kryo-EM entdecken Forscher ständig neue Virusfamilien. Ihre genetischen Codes landen in Datenbanken und erzählen eine Geschichte über die Ko-Evolution von Viren und Bakterien.
3️⃣ Vom Meer ins Labor
Einige marine Phagen produzieren Enzyme mit echtem Potenzial: für Gentechnik, Diagnostik oder sogar als Alternative zu Antibiotika. Auch in der Landwirtschaft könnten sie helfen, gezielt schädliche Bakterien auszuschalten – ganz ohne Chemiekeule.
4️⃣ Und wohin mit den Daten?
Struktur- und Gensequenzen werden offen zugänglich gemacht – z. B. in der NCBI Virus Database oder der EMDB. Bioinformatiker stöbern dort nach neuen Genen, cleveren Tricks – und manchmal nach Ideen für die nächste biotechnologische Anwendung.
Grenzen der Kryo-EM
Die Kryo-EM (inklusive Single Particle Analysis (SPA) und Kryo-Elektronentomographie (Kryo-ET)) sind bahnbrechende Methoden, aber sie haben auch einige Einschränkungen, die von technischen, biologischen und praktischen Faktoren abhängen:
Probenvorbereitung ist anspruchsvoll
Problem: Die Proben müssen extrem dünn sein (nur ein paar Hundert Nanometer) und perfekt vitrifiziert werden, ohne Eiskristalle, die die Struktur zerstören könnten. Das ist technisch schwierig und erfordert viel Erfahrung.
Auswirkung auf Viren: Wenn die Viruslösung nicht gleichmäßig eingefroren wird oder zu dick ist, können die Bilder unscharf werden oder die Viren beschädigt werden. Bei der Kryo-ET können dicke Zellproben die Elektronen zu stark streuen, was die Bildqualität verschlechtert.
Begrenzte Auflösung
Problem: Obwohl Kryo-EM sehr hochauflösende Bilder liefern kann (bis zu 2–3 Ångström bei optimalen Bedingungen), hängt die Auflösung von der Qualität der Probe und der Datenmenge ab. Bei komplexen oder variablen Strukturen (z. B. flexiblen Viren) kann die Auflösung leiden.
Auswirkung auf Viren: Kleinste Details (z. B. einzelne Atome in Proteinen) sind nicht immer sichtbar, besonders wenn die Viren sich bewegen oder unterschiedlich geformt sind.
Eingeschränkte Dynamik: Eingefroren in der Zeit
Problem: So beeindruckend die 3D-Modelle auch sind – sie zeigen immer nur einen eingefrorenen Moment. Echte Bewegungen oder Abläufe – etwa wie ein Virus live in eine Zelle eindringt – bleiben außen vor.
Auswirkung auf Viren: Bei Single Particle Analysis (SPA) sehen wir nur eine Struktur zu einem bestimmten Zeitpunkt. Und auch bei der Kryo-ET müssen Forscher verschiedene Zellen in unterschiedlichen „Stadien“ der Infektion einfrieren, um daraus eine zeitliche Abfolge zu rekonstruieren. Klingt nach Film – ist aber eher eine Stop-Motion-Rekonstruktion als ein Livestream.
Hohe Kosten und Komplexität
Problem: Kryo-EM erfordert teure Geräte (Elektronenmikroskope kosten Millionen), spezialisierte Labore (z. B. Vakuum und Kryo-Bedingungen) und viel Rechenpower für die Datenanalyse. Das macht sie zeit- und kostenintensiv.
Auswirkung auf Viren: Nur gut ausgestattete Forschungseinrichtungen können Kryo-EM nutzen, was den Zugang einschränkt. Kleine Labore oder Entwicklungsregionen können diese Technik oft nicht anwenden.
Rauschen und Datenmenge
Problem: Die Bilder sind oft verrauscht, weil man nur wenige Elektronen nutzt, um die empfindlichen Proben nicht zu beschädigen. Die Verarbeitung erfordert Tausende Bilder, was rechenintensiv ist.
Auswirkung auf Viren: Wenn die Datenmenge nicht ausreicht oder das Rauschen zu hoch ist, können die 3D-Modelle ungenau werden.
Begrenzte Probengröße
Problem: Kryo-EM funktioniert nur mit sehr kleinen Proben (z. B. einzelne Viren oder Zellen). Größere Strukturen wie ganze Gewebe sind schwer zu untersuchen, weil die Elektronen nicht tief genug eindringen.
Auswirkung auf Viren: Kryo-ET kann Zellen abbilden, aber nur bis zu einer bestimmten Dicke (ca. 1 Mikrometer). Größere Organismen oder Gewebe sind nicht geeignet.
Quellen
Kryo-Elektronenmikroskopie: Prinzip, Stärken, Grenzen und Anwendungen
Herausforderungen und Erfolge in der Kryo-Elektronentomographie
e) Zusammenfassung
Die Wissenschaft hat über Jahrzehnte hinweg immer leistungsfähigere Methoden entwickelt, um die Welt der Viren sichtbar zu machen. Während das bloße Auge und Lichtmikroskope schnell an ihre Grenzen stoßen, haben Elektronenmikroskopie, Röntgenkristallographie und schließlich die Kryo-EM und Kryo-ET völlig neue Einblicke ermöglicht. Diese Technologien haben unser Verständnis von Viren revolutioniert.
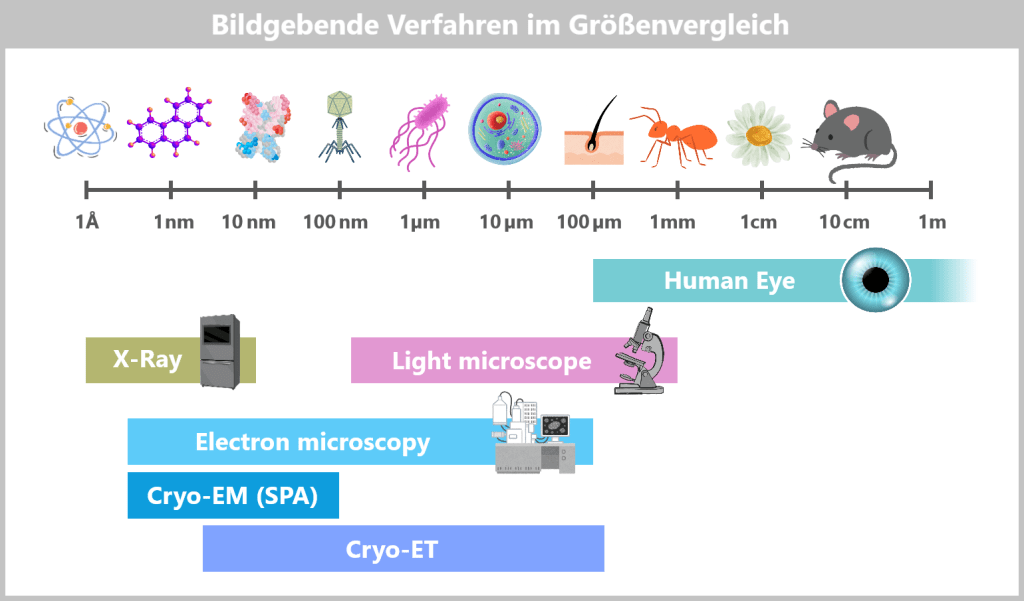
Skalierung der Bildgebung
➡️ Menschliches Auge
Sichtbar: ca. 0,1 mm (100 µm) – mehrere Meter
Beispiel: Haar, Sandkorn
➡️ Lichtmikroskop
Sichtbar: ca. 200 nm – 1 mm
Beispiel: Bakterien, Zellen, Zellkerne
➡️ Klassische Elektronenmikroskopie (EM, TEM/SEM)
Sichtbar: ca. 0,2 nm – 100 µm
Beispiel: Viren, Zellorganellen, Zellmembranen
➡️ Röntgenkristallographie
Sichtbar: ca. 1 Å (0,1 nm) – 10 nm
Beispiel: Atomare Strukturen von Proteinen
➡️ Kryo-EM (Einzelpartikelanalyse)
Sichtbar: ca. 2 Å (0,2 nm) – 100 nm
Beispiel: Proteinkomplexe, Ribosomen
➡️ Kryo-ET (Kryo-Elektronentomographie)
Sichtbar: ca. 3-5 nm – 200 µm
Beispiel: Viren in Zellen, Zellorganellen in hoher Auflösung
Elektronenmikroskopie (1930er)
→ Erste Bilder von Viren
Die Elektronenmikroskopie (EM) war ein revolutionärer Schritt in der Mikroskopie, da sie erstmals die Abbildung von Strukturen ermöglichte, die weit unterhalb der Auflösungsgrenze von Lichtmikroskopen liegen. Statt Licht verwendet die EM einen Strahl von Elektronen, der eine viel höhere Auflösung ermöglicht. Damit konnten Wissenschaftler erstmals Viren sichtbar machen, die zu klein sind, um mit herkömmlichen Mikroskopen betrachtet zu werden.
Die EM war ein großer Fortschritt, aber ihre Grenzen bei der Darstellung von Proben in ihrem natürlichen Zustand und die fehlende 3D-Information führten zur Entwicklung der Röntgenkristallographie.
Röntgenkristallographie (1950er–heute)
→ Detaillierte Strukturen von Virusproteinen
Die Röntgenkristallographie ermöglichte es, die atomare Struktur von Proteinen und anderen Biomolekülen zu entschlüsseln. Dabei wird ein Kristall des Proteins mit Röntgenstrahlen bestrahlt, und das entstehende Beugungsmuster wird analysiert, um die Positionen der Atome zu bestimmen. Diese Methode lieferte detaillierte Einblicke in die Struktur von Virusproteinen, was für das Verständnis ihrer Funktion und die Entwicklung von Medikamenten entscheidend war.
Die Röntgenkristallographie ist zwar leistungsstark, aber ihre Einschränkungen bei der Untersuchung von großen, komplexen Strukturen und die Notwendigkeit von Kristallen führten zur Entwicklung der Kryo-Elektronenmikroskopie (Kryo-EM).
Kryo-EM (1980er–heute)
→ Ein moderner Blick auf Viren
Die Kryo-EM kombiniert die Vorteile der Elektronenmikroskopie mit einer schonenden Probenvorbereitung. Die Proben werden blitzschnell eingefroren (vitrifiziert), wodurch sie in einem naturnahen Zustand erhalten bleiben. Dies ermöglicht die Abbildung von einzelnen Viruspartikeln oder großen Proteinkomplexen ohne Kristallisation. Die Kryo-EM liefert hochauflösende Bilder und kann auch flexible oder dynamische Strukturen abbilden.
Die Kryo-EM war ein großer Fortschritt, aber sie war auf isolierte Partikel beschränkt und konnte keine komplexen, zellulären Umgebungen abbilden. Dies führte zur Entwicklung der Kryo-Elektronentomographie (Kryo-ET).
Kryo-ET (2000er–heute)
→ 3D-Virusmodelle in der Zelle
Die Kryo-ET erweitert die Kryo-EM, indem sie 3D-Modelle von Viruspartikeln oder anderen Strukturen direkt in ihrer zellulären Umgebung erstellt. Dabei wird die Probe aus verschiedenen Winkeln abgebildet, und die Bilder werden zu einem 3D-Modell zusammengesetzt. Dies ermöglicht es, Viren in ihrem natürlichen Kontext zu studieren, z. B. wie sie mit Zellen interagieren oder sich vermehren.
Die Kryo-ET ist ein mächtiges Werkzeug, aber ihre begrenzte Auflösung und die Komplexität der Probenvorbereitung könnten die Entwicklung neuer Technologien vorantreiben, die noch höhere Auflösungen in komplexen zellulären Umgebungen ermöglichen.
Jede dieser Technologien hat die Grenzen unseres Sehvermögens erweitert – und gleichzeitig neue Herausforderungen mit sich gebracht. Doch genau diese Grenzen waren stets der Antrieb für die Entwicklung noch leistungsfähigerer Methoden. Wissenschaft ist ein stetiger Prozess des Entdeckens, Verfeinerns und Weiterdenkens.
Man sagt: „Sehen ist Glauben“, doch für Biologen gilt: „Sehen ist Verstehen“. Je detaillierter wir biologische Strukturen abbilden können, desto tiefer dringen wir in die Geheimnisse des Lebens ein. Doch das reine Sichtbarmachen eines Virus und seiner Interaktion mit dem Wirt reicht nicht aus, um seine Natur vollständig zu entschlüsseln.
Vom Sehen zum Entschlüsseln: Die nächste Stufe der Erkenntnis
Um zu verstehen, was ein Virus wirklich ausmacht, müssen wir sein genetisches Erbe entschlüsseln – seinen einzigartigen „Fingerabdruck“. Dies gelingt mit modernen molekularbiologischen Methoden, die die virale Nukleinsäure analysieren und so einen Blick in den genetischen Bauplan des Virus ermöglichen.
4.5. Der genetische Fingerabdruck der Viren
Nachdem Viren endlich sichtbar geworden waren – dank Elektronenmikroskopie und Kristallstrukturanalyse – stellte sich die nächste große Frage: Was macht ein Virus eigentlich zum Virus?
Schnell war klar: Wie jedes biologische System brauchen auch Viren einen genetischen Bauplan – etwas, das ihre Eigenschaften codiert und ihre Vermehrung ermöglicht. Doch was genau trägt diese Information?
Lange galt das Protein als Favorit: vielfältig, komplex, scheinbar perfekt geeignet. Die DNA hingegen erschien vielen Forschenden zu simpel, zu langweilig, um der Träger des Lebens zu sein.
Doch wie sich herausstellte, lag die Antwort genau dort: in diesem unscheinbaren Molekül, das sich als der ultimative Datenträger des Lebens entpuppte – und bei manchen Viren auch in seinem Verwandten, der RNA.
Die Entdeckung, dass nicht Proteine, sondern Nukleinsäuren das Geheimnis der viralen Vermehrung bergen, war ein wissenschaftlicher Krimi für sich: geprägt von Irrtümern, rivalisierenden Teams und bahnbrechenden Entdeckungen.
Die Suche nach dem Code des Lebens
Frühe Ansätze zur Entschlüsselung der Vererbung
Die Grundlagen der Vererbungslehre wurden bereits im 19. Jahrhundert durch Gregor Mendels Experimente gelegt. Wissenschaftler erkannten, dass Organismen ihre Eigenschaften an die nächste Generation weitergeben, doch der genaue Mechanismus blieb lange unklar. In den Chromosomen der Zellen, die man Ende des 19. Jahrhunderts als mögliche Träger der Erbinformation identifizierte, suchte man nach den entscheidenden Molekülen. Proteine schienen aufgrund ihrer Komplexität die naheliegendsten Kandidaten zu sein.
Wendell Meredith Stanley und das Tabakmosaikvirus
In den 1930er Jahren bestätigten die Arbeiten von Wendell Meredith Stanley die zentrale Rolle von Proteinen im Aufbau von Viren. Stanley isolierte das Tabakmosaikvirus und bereitete es auf eine innovative Weise auf: Er extrahierte das Virus aus infizierten Tabakpflanzen, reinigte es durch Zentrifugation und ließ es anschließend in einer Lösung auskristallisieren. Diese Kristalle enthielten sowohl Proteine als auch genetisches Material, doch zunächst stand nur das Protein im Fokus der Forschung. Man ging davon aus, dass es die genetischen Anweisungen für die Virusreplikation trug.
Der Paradigmenwechsel: Von Proteinen zur DNA
Der Zweifel an der Protein-Hypothese wuchs, als Oswald Avery 1944 zeigte, dass DNA die Fähigkeit besitzt, Eigenschaften von Bakterien zu verändern. Sein Experiment, bei dem DNA aus einer Bakterienart extrahiert und auf eine andere übertragen wurde, offenbarte, dass DNA die Erbinformation tragen kann. Doch diese Entdeckung wurde anfangs skeptisch betrachtet.
Erst das Hershey-Chase-Experiment von 1952 brachte den entscheidenden Durchbruch. Die Wissenschaftler Alfred Hershey und Martha Chase arbeiteten mit Bakteriophagen, Viren, die Bakterien infizieren. Sie markierten die DNA der Viren radioaktiv und verfolgten ihren Weg in die Wirtszellen. Die Proteinhüllen der Phagen blieben außerhalb der Zelle, während die DNA eindrang und die Virusreplikation steuerte. Damit war bewiesen: Die DNA – nicht das Protein – trägt die genetischen Informationen.

Die Doppelhelix und die Rolle der RNA
1953 entschlüsselten James Watson und Francis Crick die Struktur der DNA, unterstützt durch Rosalind Franklins Röntgenbilder. Die Doppelhelix offenbarte, wie die DNA ihre Informationen speichert und bei der Zellteilung kopiert. Parallel wurde klar, dass RNA bei manchen Viren, darunter dem Tabakmosaikvirus, die genetischen Informationen trägt. Dies rückte Stanleys Arbeit in ein neues Licht: Die RNA in den Viruspartikeln war der eigentliche Träger der genetischen Information, nicht das Protein.
Viren – Die Minimalisten unter den Lebensformen
Viren sind wahre Meister der Reduktion. Diese minimalistischen Überlebenskünstler haben die Kunst genetischer Effizienz perfektioniert – ob mit DNA, RNA oder sogar rückwärts geschriebener RNA (die Spiegelschrift wie bei Influenzaviren mit seiner negativen ssRNA).
Ihr Genom ist wie ein ultraleichter Überlebensrucksack:
✔ Alles Nötige drin, was zählt – Baupläne fürs Kopieren, Verpacken, Wirt-Kapern
✖ Kein Ballast – keine eigenen Ribosomen, keine Energieproduktion, kein Smalltalk
Viren sind in ihrer genetischen Struktur einzigartig: Während alle bekannten Lebewesen ausschließlich DNA als Erbmaterial nutzen, bedienen sich Viren entweder DNA oder RNA – ein entscheidender Unterschied, der ihre enorme Anpassungsfähigkeit und evolutionäre Kreativität prägt.
Wichtige molekularbiologische Methoden zur Genom-Analyse von Viren
Dank moderner molekularbiologischer Techniken können Viren heute mit hoher Präzision analysiert werden. Damit kann man:
- ihre genetische Information entschlüsseln,
- ihre Abstammung nachvollziehen und
- ihre Verbreitungswege verfolgen.
Je nach Fragestellung kommen unterschiedliche Methoden zum Einsatz:
- Will man ein Virus identifizieren?
- Soll sein Genom vollständig sequenziert werden?
- Oder will man seine Aktivität im Wirtssystem beobachten?
Die nächsten Abschnitte stellen die zentralen Methoden zur Analyse des genetischen Fingerabdrucks vor – vom Isolieren der Erbsubstanz bis hin zu modernen Sequenzierungstechnologien.
4.5.1. Nukleinsäure-Extraktion
4.5.2. Nukleinsäure-Amplifikation
4.5.3. Sequenzierung
💡Hinweis: Für grundlegende Informationen zu DNA und RNA sowie deren Funktionen empfehlen wir das Kapitel „4.2. Die Proteinbiosynthese“ in der Abhandlung „Die Wunderwelt des Lebens“. Dort werden die Grundlagen auf anschauliche Weise erklärt und bieten eine ideale Vorbereitung für das Verständnis dieses Themas.
4.5.1. Nukleinsäureextraktion
Um einen Virus im Detail analysieren zu können, müssen wir an seinen innersten Schatz heran: sein genetisches Material – DNA oder RNA müssen erst einmal ausgegraben werden. Doch die Nukleinsäure liegt gut versteckt, eingepackt in Proteinhüllen, eingebettet in Zelltrümmer oder vermischt mit allerlei molekularem „Beifang“.
Die Aufgabe: Die virale Nukleinsäure aus diesem molekularen Allerelei befreien – sauber, effizient und ohne sie zu beschädigen.
Das Ziel: Möglichst viel, möglichst reine DNA oder RNA – bereit für PCR, Sequenzierung oder Mutationsanalyse. Die Nukleinsäureextraktion ist damit der erste und einer der wichtigsten Schritte jeder molekularen Virusdiagnostik. Je nach Probentyp, Virusart und Untersuchungsziel kommen unterschiedliche Extraktionsmethoden zum Einsatz – von klassischen Kits bis zu automatisierten Hochdurchsatzsystemen.
Wie funktioniert die Nukleinsäureextraktion?
Der Ablauf lässt sich in vier einfache Schritte unterteilen:
1️⃣ Zellaufschluss – Erst mal alles aufbrechen
Bevor man an die RNA oder DNA herankommt, müssen die Zellen (und ggf. Viren) in der Probe geknackt werden. Das klappt z. B. durch Ultraschall, Enzyme oder mechanisches Zermahlen. Hauptsache, die Hülle ist weg – und das Genom liegt frei.
Methoden, die ans Eingemachte gehen
Ultraschall: Sonikation zerschlägt die Zell- und Virushüllen mit Schallwellen.
Enzyme: Proteinase K baut Proteine ab, die das Genom einpacken.
Mechanisches Zermahlen: Kleine Glasperlen in einem Röhrchen werden geschüttelt. Die Perlen prallen gegen die Zellen und zerreissen die Zellmembran.
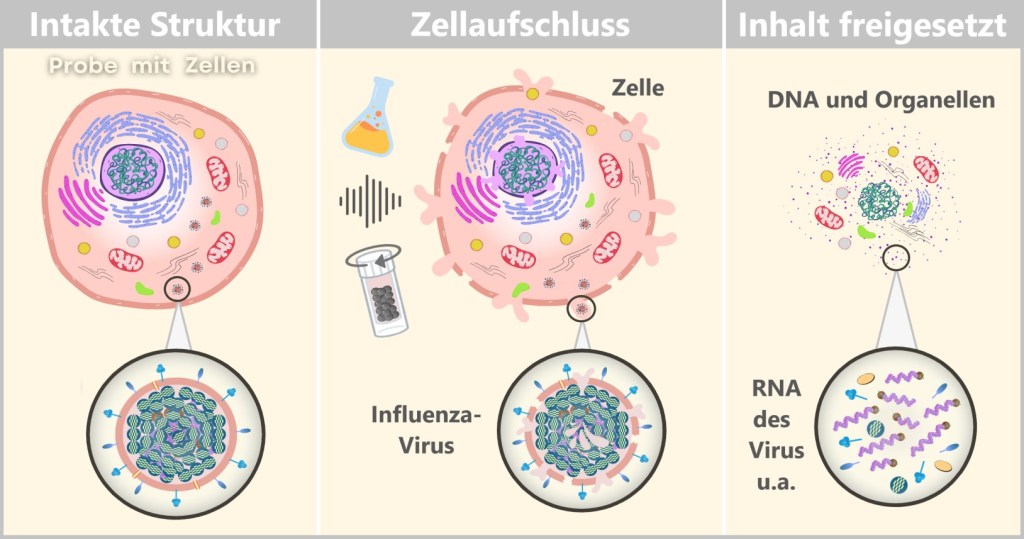
Intakte Struktur (links): Eine schematische Darstellung einer intakten Zelle mit Zellorganellen, Zellkern und DNA. Innerhalb der Zelle sind Influenzaviren sichtbar. Zusätzlich wird ein einzelnes Influenzavirus vergrößert dargestellt, mit seiner kugelförmigen Hülle, die aus einer Wirtszellmembran, einem Kapsid und RNA-Strängen besteht.
Zellaufschluss (Mitte): Die Zellmembran ist perforiert dargestellt, wodurch das Zytoplasma austreten kann. Auch die Kernmembran weist Löcher auf. Ein Influenzavirus ist vergrößert dargestellt, mit aufgebrochener Hülle, aus der virale RNA und andere Bestandteile freigesetzt werden. Neben der Zelle sind Symbole für chemische Substanzen (Flasche), physikalische Einwirkungen (Schallwellen) und mechanische Kräfte abgebildet, die die verschiedenen Methoden des Zellaufschlusses veranschaulichen.
Inhalt freigesetzt (rechts): Nach dem Zellaufschluss schwimmen die Organellen, die DNA und andere Zellinhalte frei im Medium. Ebenfalls sichtbar sind die freigesetzten Bestandteile des Influenzavirus, darunter RNA-Segmente, Spikes und andere molekulare Komponenten, die in der Vergrößerung dargestellt werden.
2️⃣ Aufräumen – Proteine & Co. rausfischen
Jetzt ist die Probe ein ziemlicher Mix: Nukleinsäuren, Proteine, Fette und Zellreste tummeln sich wild durcheinander. Damit am Ende nur das drin ist, was wir brauchen, kommen Reagenzien oder Enzyme zum Einsatz, die den ganzen Ballast gezielt abbauen.
Methoden, die klar Schiff machen
Phenol-Chloroform-Extraktion: Alt, aber bewährt – trennt Nukleinsäuren zuverlässig von störenden Proteinen und Lipiden. Besonders nützlich bei stark „vermüllten“ Proben.
👉Achtung: Die verwendeten Chemikalien sind ziemlich toxisch – nur für geübte Hände (und mit Schutzbrille!).Proteinase K: Dieses Enzym zersetzt Proteine wie z. B. Membran- oder Strukturproteine, die noch in der Probe herumschwimmen – damit die DNA/RNA ungestört bleibt.
DNase/RNase-Behandlung: Wird eingesetzt, um gezielt nicht-virale DNA oder RNA abzubauen – besonders praktisch, wenn z. B. nur die virale RNA untersucht werden soll.
3️⃣ Reinigung – Die reine RNA oder DNA
Jetzt wird’s elegant: Die Nukleinsäuren werden gezielt gebunden – an spezielle Oberflächen wie Silica-Membranen oder magnetische Kügelchen. Der Rest? Wird einfach weggespült. Eine Art molekulares Sieb – nur smarter.
Methoden, die richtig filtern
Spin Columns (Säulenverfahren): Hier binden DNA oder RNA an eine Spezialmembran, meist aus Silica. Dann heißt es: Spülen, spülen, spülen – bis alles andere raus ist. Am Ende bleibt: schön saubere Nukleinsäure. Diese Methode ist schnell, zuverlässig und steckt in vielen Labor-Kits.
Magnetbeads-Technologie: Winzige magnetische Kügelchen heften sich gezielt an die Nukleinsäuren. Mit einem Magneten werden die passenden Matches dann blitzschnell aus der Suppe gefisch. Geht blitzschnell und eignet sich super für automatische Hochdurchsatz-Verfahren, die tausende Matches pro Stunde arrangieren müssen. Mehr Informationen dazu in den Videos:
DNA and RNA extraction with magnetic beads – How it works und
Nucleic acid purification with chemagic M-PVA Magnetic Bead Technology.
4️⃣ Elution – Der große Auftritt
Jetzt kommt der finale Akt: Die gereinigten Nukleinsäuren werden in einer kleinen Menge Flüssigkeit (Elutionspuffer oder Wasser) gelöst – und voilà: Die Probe ist jetzt analysierbereit. Ganz ohne Ballast, dafür mit jeder Menge Potenzial für PCR, Sequenzierung & Co.

Links: Der freigesetzte Zell- und Virusinhalt nach dem Zellaufschluss, bestehend aus einer Mischung aus Zellbestandteilen, Proteinen und Nukleinsäuren.
Mitte: Nach der Entfernung von Proteinen und Verunreinigungen bleiben nur die Nukleinsäuren (RNA-Segmente des Virus) übrig, dargestellt als kleine Pünktchen.
Rechts: Vergrößerung einiger RNA-Segmente, die nach der Elution in einer Flüssigkeit gelöst und für die Analyse bereitgestellt sind.
Wenn wenig viel zu wenig ist
Auch wenn die Reinigung sorgfältig war: Auf molekularer Ebene ist echte Reinheit kaum zu erreichen. In der Probe können immer noch winzige Störenfriede herumschwirren – Proteine, Salze, andere Moleküle. Das Problem: Die wenigen viralen Nukleinsäuren gehen in diesem „Hintergrundrauschen“ leicht unter – wie ein Flüstern im Konzertsaal.
Und genau hier steht der nächste große Schritt an: die Amplifikation. Dabei wird das virale Erbgut millionenfach vervielfältigt – damit selbst das leiseste Flüstern laut und deutlich hörbar wird.
4.5.2. Nukleinsäure-Amplifikation
Egal wie gut die Extraktion war: In vielen Fällen ist die Ausbeute an viraler DNA oder RNA winzig. Um sie nachweisen oder analysieren zu können, braucht es mehr Kopien – viele mehr. Genau das ist der Job der Amplifikation: Sie vervielfältigt das Erbmaterial millionen- oder sogar milliardenfach – wie ein molekularer Kopierer.
Welche Methode man verwendet, hängt davon ab, was man sucht:
🦠📌 Wenn das Virus bekannt ist – gezielt nachspüren
Bei bekannten Viren weiß man, wo man suchen muss: Bestimmte Abschnitte ihres Genoms sind bereits kartiert. Mit Hilfe passender Primer – kurze Gensequenzen, die exakt an diese Abschnitte binden – kann gezielt ein bestimmter Bereich vervielfältigt werden. Die klassische Methode dafür ist die PCR (Polymerase-Kettenreaktion): präzise, empfindlich und perfekt für den gezielten Nachweis.
🦠❓ Wenn das Virus unbekannt ist – breit ansetzen
Ist das Virus noch ein unbeschriebenes Blatt, fehlen die spezifischen Primer. Dann wird die gesamte virale DNA oder RNA verstärkt – unspezifisch, aber umfassend. Hier kommen Methoden wie die Random Primed Amplification oder Whole Genome Amplification (WGA) zum Einsatz. Sie erzeugen möglichst viele Kopien – egal von welchem Abschnitt – um später durch Sequenzierung herauszufinden, was überhaupt in der Probe steckt.
Nachfolgend werden beide Ansätze näher erläutert:
a) Polymerase-Kettenreaktion (PCR)
b) Random Primed PCR
a) Polymerase-Kettenreaktion (PCR)
Die PCR – kurz für Polymerase-Kettenreaktion (englisch: Polymerase Chain Reaction) – ist so etwas wie der Kopierer im Labor: Mit ihr lassen sich selbst winzige Mengen viraler DNA millionenfach vervielfältigen. Das ist besonders praktisch, wenn man auf Spurensuche geht – um DNA-Viren in einer Probe nachzuweisen.
Aber was ist mit RNA-Viren, wie etwa dem Influenzavirus? Die lassen sich nicht direkt kopieren – erst müssen sie „übersetzt“ werden: Bei der sogenannten RT-PCR (Reverse Transcription PCR) wird die RNA des Virus mithilfe eines Enzyms (Reverse Transkriptase) in DNA umgeschrieben. Danach läuft alles wie bei der normalen PCR: Vervielfältigen, analysieren, fertig.
PCR – Schritt für Schritt
Um beim Beispiel des Influenzavirus zu bleiben, betrachten wir den typischen Ablauf der Reverse-Transkriptase-PCR (RT-PCR), die bei RNA-Viren wie Influenza eingesetzt wird.
1️⃣ Probenentnahme
Alles beginnt mit einem Abstrich – meist aus Nase oder Rachen, denn genau dort treiben sich Influenzaviren gern herum. Das gesammelte Material landet in einem Spezialmedium, das die empfindliche RNA des Virus schützt. Und das ist auch nötig, denn RNA ist ein Sensibelchen: Überall lauern RNasen – Enzyme, die RNA abbauen, und die finden sich fast überall – auf der Haut, in der Luft, in der Probe selbst.
2️⃣ RNA-Extraktion
Die Probe ist ein bunter Cocktail: virale Partikel, menschliche Zellen, Proteine, Lipide – das volle Programm. Wie du schon im Kapitel „Nukleinsäureextraktion“ gelesen hast, wird jetzt das Wesentliche herausgeholt: die RNA. Am Ende bleibt eine saubere Mischung zurück, die vor allem drei Sorten RNA enthalten kann:
➤ virale Genom-RNA (beim Influenzavirus: negativsträngige RNA (-)ssRNA),
➤ virale mRNA, die bei aktiver Infektion in den Wirtszellen gebildet wird,
➤ zelluläre RNA des Wirts.
Besonders spannend: Die virale mRNA. Dank eines viralen Tricks – dem sogenannten Cap-Snatching – hat sie eine 5′-Cap-Struktur und einen Poly-A-Schwanz, genau wie unsere eigene mRNA. Das macht sie stabiler und besonders gut geeignet für den nächsten Schritt: die Umschreibung in DNA.
Damit sie fit bleibt für die Analyse, wird die gewonnene RNA in einem stabilisierenden Puffer aufbewahrt.
3️⃣ Umwandlung von RNA in DNA
Bevor die PCR loslegen kann, braucht sie ein Update: Sie arbeitet nur mit DNA, nicht mit RNA. Deshalb muss die virale RNA erst in komplementäre DNA (cDNA) umgewandelt werden. Und das übernimmt ein cleveres Enzym: die Reverse Transkriptase.
So läuft die Umwandlung:
RNA-Vorlage: Die mRNA des Influenzavirus ist ein einzelsträngiges RNA-Molekül mit zwei praktischen Anhängseln: Am 5′-Ende trägt sie eine Cap-Struktur, die sie stabilisiert – am 3′-Ende einen Poly-A-Schwanz, eine Kette aus Adenin-Basen. Beides macht die virale RNA besonders gut zugänglich für den nächsten Schritt.
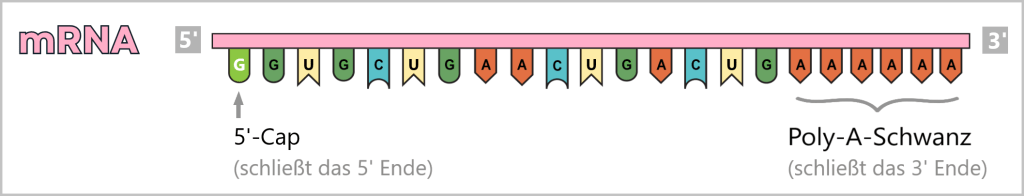
Primer dockt an: Damit die Reverse Transkriptase weiß, wo sie starten soll, wird ein Primer gebraucht – ein kurzes DNA-Stück, das sich oft gezielt an den Poly-A-Schwanz bindet.
Reverse Transkriptase legt los: Sie setzt sich auf den Primer und liest die RNA-Basen (A, U, G, C) der Vorlage ab. Dann fügt sie die passenden DNA-Basen (A, T, G, C) aneinander – und baut so einen komplementären DNA-Strang. Das Ergebnis ist ein hybrides Molekül aus RNA und DNA (RNA-cDNA-Hybrid).

Sowohl die 5′-Cap-Struktur als auch der Poly-A-Schwanz der RNA erscheinen nicht in ihrer ursprünglichen Form in der resultierenden cDNA. Sie werden entweder ignoriert (5′-Cap) oder komplementär abgebildet und teilweise verkürzt (Poly-A-Schwanz).
Vom Strang zum Doppelstrang: Dieser erste DNA-Strang dient jetzt selbst als Vorlage – ein zweiter Strang wird ergänzt, sodass eine doppelsträngige cDNA (ds cDNA) entsteht. Diese ist stabil – und bereit für die PCR.

Wie das Ganze in Aktion aussieht? Diese Animation zeigt die cDNA-Synthese im Schnelldurchlauf – einfach und verständlich erklärt.
4️⃣ Was braucht man für eine PCR?
Bevor es mit der Vervielfältigung losgehen kann, müssen ein paar Zutaten und Werkzeuge bereitstehen:
a) DNA-Vorlage: Das Ausgangsmaterial ist die doppelsträngige cDNA, die im vorherigen Schritt aus viraler RNA hergestellt wurde.
b) Nukleotide – die Bausteine: Vier verschiedene Bausteine braucht es, um DNA zu kopieren: Adenin (A), Thymin (T), Cytosin (C) und Guanin (G). Sie werden später von der Polymerase zu einem neuen Strang zusammengesetzt.
c) DNA-Polymerase – der Baumeister: Dieses Enzym liest die DNA-Vorlage und setzt die passenden Nukleotide zu einem neuen Strang zusammen – präzise und blitzschnell. Bei der PCR kommt oft eine hitzestabile Polymerase (z. B. Taq-Polymerase) zum Einsatz, damit sie die hohen Temperaturen der PCR-Zyklen übersteht.
d) Primer – die Wegweiser: Primer sind kurze DNA-Stücke (meist 18–24 Basen lang), die der Polymerase zeigen, wo sie mit dem Kopieren beginnen soll. Für die PCR braucht man immer zwei: einen Vorwärts- und einen Rückwärts-Primer, die sich an gegenüberliegenden Strängen der Ziel-DNA anlagern.

e) Pufferlösung – das richtige Milieu: Sie sorgt dafür, dass die Polymerase sich wohlfühlt: mit stabilem pH-Wert, Magnesiumionen und allem, was das Enzym für eine zuverlässige Arbeit braucht.
f) Thermocycler – das Temperatur-Karussell: Ein Gerät, das die nötigen Temperaturzyklen automatisch durchführt. Er heizt, kühlt und hält die Temperaturen präzise – für die verschiedenen PCR-Schritte in perfektem Timing.

5️⃣ Der Ablauf der Polymerase-Kettenreaktion (PCR)
Alle Zutaten – DNA-Vorlage (ds cDNA), Nukleotide, DNA-Polymerase, Primer und Pufferlösung – kommen in ein kleines Reaktionsröhrchen. Dieses wird anschließend in den Thermocycler gestellt, der die PCR-typischen Temperaturzyklen automatisch durchläuft. Jeder Zyklus besteht aus drei Kernschritten:
Schritt 1 – Trennung der DNA-Stränge (Denaturierung): Die Probe wird auf ca. 94–98 °C für etwa 20-30 Sekunden erhitzt. Dadurch lösen sich die Wasserstoffbrücken zwischen den DNA-Basen – der Doppelstrang „schmilzt“ in zwei Einzelstränge auf. Diese dienen im nächsten Schritt als Vorlage.
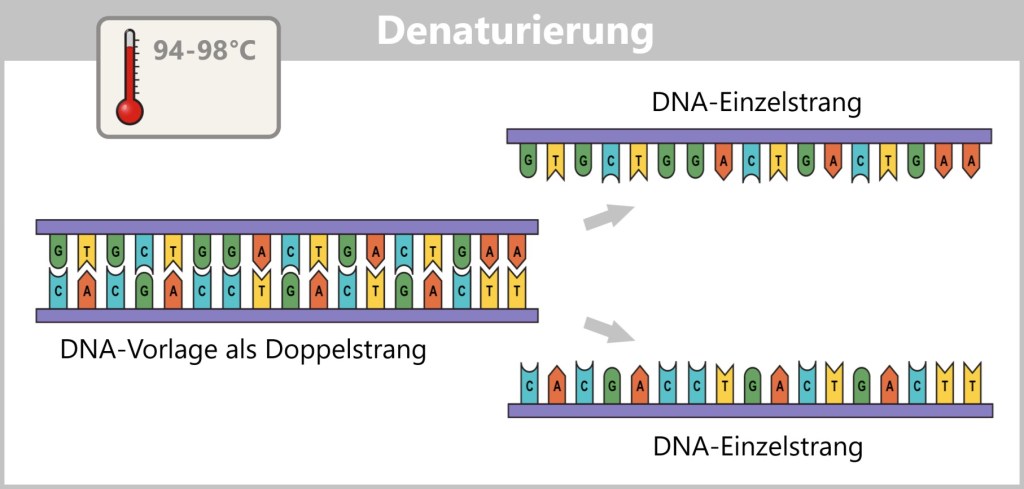
Schritt 2 – Primerbindung (Annealing): Die Temperatur wird auf 50–65 °C abgesenkt. Jetzt binden sich die Primer gezielt an die jeweiligen Einzelstränge. Sie markieren den Startpunkt für die DNA-Synthese. Die verwendeten Primer sind so konstruiert, dass sie nur an virale Sequenzen binden – also nicht an menschliche RNA oder DNA.
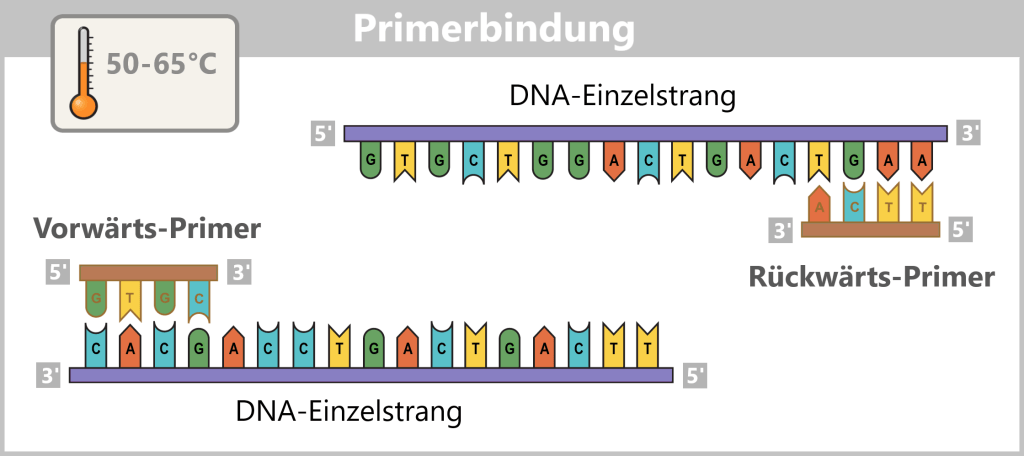
Schritt 3 – DNA-Synthese (Amplifikation): Bei etwa 70 °C, der optimalen Temperatur für die Polymerase, beginnt die eigentliche Vervielfältigung. Die DNA-Polymerase bindet an den Primer, liest den Einzelstrang von 3′ nach 5′ – und synthetisiert parallel den neuen Strang in 5′ nach 3′-Richtung. Dabei setzt sie Nukleotide nach dem Prinzip der Basenpaarung zusammen: A mit T und G mit C. So entstehen zwei neue DNA-Doppelstränge.
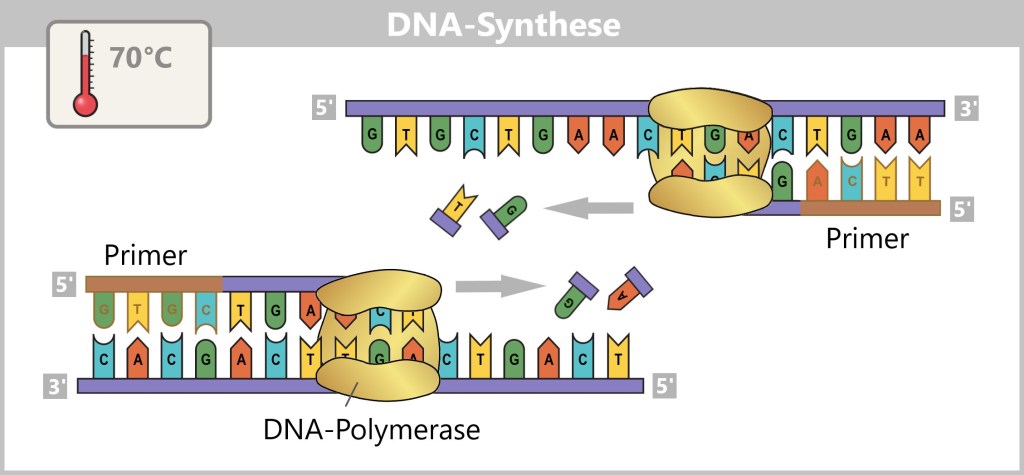
6️⃣ Wiederholung der Zyklen
Die neu entstandenen DNA-Doppelstränge dienen direkt als Vorlage für die nächste Runde. Die Schritte Denaturierung – Primerbindung – DNA-Synthese wiederholen sich zyklisch.
Mit jedem Zyklus verdoppelt sich die DNA-Menge: 1 → 2 → 4 → 8 → 16 → 32 …
Nach nur 25–40 Zyklen entstehen auf diese Weise Milliarden Kopien des gesuchten DNA-Abschnitts.
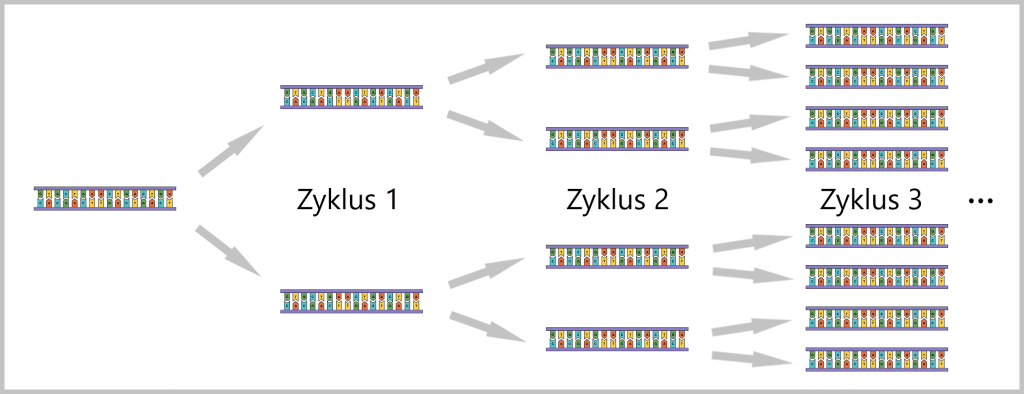
📈 Was am Ende der RT-PCR vorliegt:
Eine hochkonzentrierte Lösung spezifischer DNA-Fragmente – direkt abgeleitet von der viralen RNA.
Im Fall des Influenzavirus enthält sie ausschließlich jene Genomabschnitte, nach denen gezielt gesucht wurde. Diese DNA dient nun als Grundlage für weiterführende Analysen:
➤ zur Bestätigung der Virusidentität,
➤ zur Unterscheidung verschiedener Virusvarianten,
➤ oder zur Quantifizierung der Viruslast im Patientenmaterial.
🎥 Tipp: Das Video „What is PCR? Polymerase Chain Reaction“ fasst die Abläufe noch einmal anschaulich zusammen. Auch wenn es sich auf menschliche DNA bezieht, bleibt das Grundprinzip exakt das gleiche.
b) Random Primed PCR
💡Hinweis: Wenn du mit der PCR noch nicht vertraut bist, lies zuerst den Abschnitt „PCR – Schritt für Schritt“. Dort findest du die Grundlagen und Abläufe anschaulich erklärt. Die folgenden Ausführungen setzen dieses Wissen voraus und konzentrieren sich speziell auf die Besonderheiten der Random Primed PCR.
Die Random Primed PCR ist eine besondere Variante der PCR, die vor allem dann eingesetzt wird, wenn ein Virus unbekannt ist oder sein Genom stark variiert.
Im Gegensatz zur klassischen PCR, die mit spezifischen Primern gezielt definierte DNA-Abschnitte vervielfältigt, verwendet diese Methode sogenannte „Random Primer“: Kurze, zufällig zusammengesetzte DNA-Sequenzen, die sich an vielen Stellen der Ziel-DNA (oder cDNA) binden können – ganz unabhängig von deren genauer Basenabfolge.
Vorteil: Auch unbekannte oder stark veränderte Bereiche des Genoms lassen sich auf diese Weise mitamplifizieren – ein entscheidender Vorteil bei der Vorbereitung auf eine Sequenzierung, bei der die exakte Basenreihenfolge bestimmt wird.
Beispiele für Random Primer:
Hexamer Primers (6 Basen lang): AGCTGA, CTAGCT, …
Heptamer Primers (7 Basen lang): CCTGAGT, GATTACA, …
Nonamer Primers (9 Basen lang): GCAGTTCGC, ATGGCCGTA, …
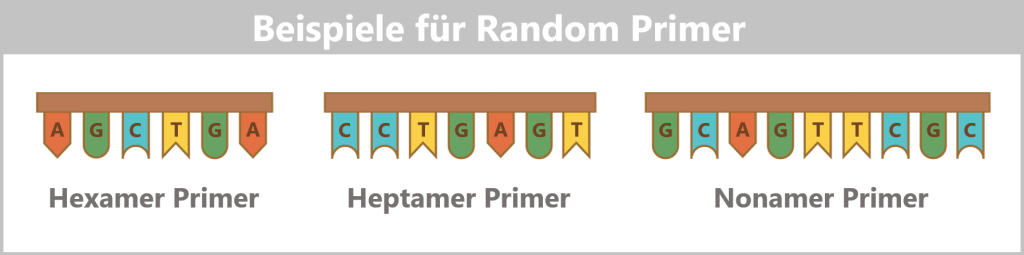
In der Praxis kommen meist Mischungen aller möglichen Primer-Varianten zum Einsatz (z. B. 4⁶ = 4096 Kombinationen bei Hexamern). Das sorgt dafür, dass möglichst viele Bindungsstellen im Genom erreicht werden.
Ablauf der Random Primed PCR
Schritt 1 – Denaturierung: Die DNA (oder cDNA) wird auf ca. 95 °C erhitzt, um die beiden Stränge zu trennen. Es entstehen Einzelstränge, an die die Primer binden können.
Schritt 2 – Primerbindung: Die Temperatur wird gesenkt, sodass sich die zufälligen Primer an viele verschiedene Stellen der DNA anlagern. Da die Primer zufällig sind, können sie an vielfältige Positionen im Genom binden.
Hinweis zur Genomgröße: Viren haben sehr unterschiedliche Genomlängen – von wenigen Tausend bis zu Hunderttausenden Basenpaaren. Random Primer helfen, möglichst viel davon abzudecken.
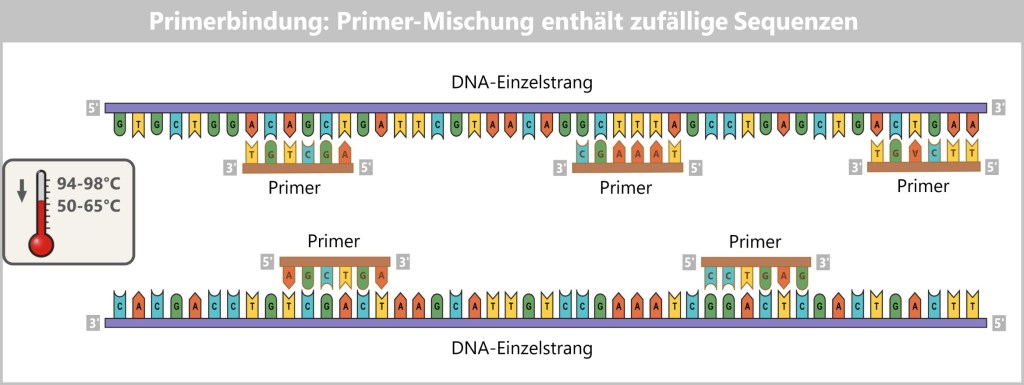
Schritt 3 – DNA-Synthese: Die DNA-Polymerase bindet an die Primer und beginnt, von dort aus neue DNA-Stränge zu synthetisieren. Dabei liest sie die Vorlage, bis sie entweder ans Ende gelangt oder auf einen weiteren Primer stößt. Dieser wirkt wie ein Stoppsignal – das Ergebnis sind meist kürzere DNA-Fragmente.
Genau das ist gewollt: Die Polymerase erzeugt viele kurze, sich überlappende Fragmente, die später für die Genomrekonstruktion oder gezielte Analysen genutzt werden können.
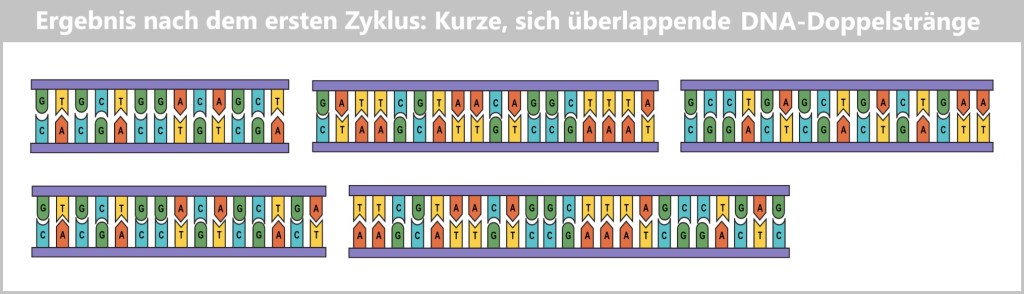
🔁 Zyklische Wiederholung
Dieser Prozess wird – wie bei der klassischen PCR – mehrfach wiederholt, meist 20 bis 40 Zyklen.
- Je mehr Zyklen, desto mehr DNA entsteht.
- Mit steigender Zykluszahl steigt auch die Wahrscheinlichkeit, dass Primer sich gegenseitig „abfangen“ → kürzere Fragmente entstehen.
- Aber: Zu viele Zyklen können die Vielfalt verringern, weil manche Regionen überrepräsentiert werden.
Eine ausgewogene Zahl an Zyklen sowie eine gut abgestimmte Primer-Mischung sind entscheidend für die Qualität und Repräsentativität des Endprodukts.
📈 Ergebnis der Random Primed PCR
Am Ende liegt eine große Anzahl an kurzen, überlappenden DNA-Fragmenten vor – keine vollständigen Stränge, sondern ein Fragmentteppich, der das gesamte virale Genom abdecken kann.

Aus vielen kurzen DNA-Schnipseln entsteht ein genetisches Mosaik. Die Fragmente überlappen sich und lassen sich später wie Puzzlestücke zu einem vollständigen Virusgenom zusammensetzen.
🧩 Was passiert mit den DNA-Schnipseln?
Direkte Analyse einzelner Fragmente
Oft reicht es, gezielt bestimmte Fragmente zu sequenzieren, um wichtige Informationen zu erhalten – z. B. zur Mutationsanalyse oder Typisierung eines Virus.
Rekonstruktion des gesamten Genoms
Wenn das gesamte Genom analysiert werden soll (z. B. zur Entdeckung neuer Viren oder zur Erstellung von Verwandtschaftsanalysen), werden die Fragmente mithilfe von Sequenziertechnologien ausgelesen (mehr dazu im kommenden Kapitel). Spezielle Softwareprogramme setzen die sich überlappenden Fragmente anschließend wie ein Puzzle wieder zusammen.
🧬 Vom Fragment zum vollständigen Genom
Die Amplifikation mithilfe der Random Primed PCR ist nur der erste Schritt – sie sorgt dafür, dass genügend genetisches Material für weiterführende Untersuchungen vorliegt. Doch um wirklich zu verstehen, mit welchem Virus man es zu tun hat, reicht die bloße Vervielfältigung nicht aus.
Jetzt geht es darum, die genaue Abfolge der Basenpaare in den erzeugten DNA-Fragmenten zu bestimmen – also ihre Sequenz. Erst durch diese Sequenzierung lässt sich das genetische Profil des Virus entschlüsseln: Man erkennt Mutationen, kann Virenstämme unterscheiden und sogar evolutionäre Stammbäume erstellen.
Im folgenden Kapitel schauen wir uns deshalb an, wie Sequenzierung funktioniert, welche Technologien dafür eingesetzt werden – und wie aus vielen kleinen DNA-Schnipseln ein vollständiges virales Genom rekonstruiert wird.
4.5.3. Sequenzierung
Die Sequenzierung ist DER entscheidende Schritt, um das Erbgut von Viren zu entschlüsseln. Dabei wird die exakte Reihenfolge der Basen in der DNA (A, T, C, G) oder RNA (A, U, C, G) bestimmt.
Was bringt das?
Eine Virus-Genomsequenz ist der QR-Code der Biologie: Einmal gescannt, weiß man sofort, womit man’s zu tun hat.
Identifikation des Virus:
🔹 Welches Virus ist es genau?
🔹 Ist das Virus bereits bekannt oder handelt es sich um eine neue Entdeckung?
🔹 Zu welcher Virusfamilie oder Gattung gehört es?
Erkennung von Mutationen:
🔹 Hat sich das Virus verändert – wenn ja, wie?
🔹 Sind neue Varianten oder Stämme entstanden?
🔹 Welche genetischen Veränderungen bestehen im Vergleich zu früheren Versionen?
Bestimmung diagnostischer Marker:
🔹 Gibt es stabile (konservierte) Abschnitte im Genom, die sich gut für Tests eignen?
🔹 Gibt es Gene oder Proteine, die einzigartig für dieses Virus sind?
🔹 Lassen sich bestimmte Gene oder Proteine gezielt nutzen – z. B. für Medikamente?
Sequenzierung verpasst jedem Virus einen genetischen Fingerabdruck – einzigartig, präzise und fälschungssicher.
🧬 Vom Reagenzglas zur Hochtechnologie
Die Entschlüsselung der Virus-DNA war früher Handarbeit – heute ist sie Hightech. Moderne Sequenzierer analysieren Millionen DNA-Fragmente gleichzeitig – schnell, automatisiert und hochpräzise.
Seit den ersten manuellen Verfahren hat sich die Technologie rasant weiterentwickelt. Jede neue Generation hat den Blick ins Erbgut klarer, schneller und umfassender gemacht.
Die folgende Übersicht zeigt, wie sich die Sequenzierung verändert hat – und welche Technologien heute zur Verfügung stehen:
| Generation | Beschreibung |
|---|---|
| First Generation: Sanger-Sequenzierung | Der Oldtimer: langsam, aber präzise. Ideal für kurze Abschnitte. |
| Second Generation: Next-Generation Sequencing (NGS) | Die Hochdruckpresse: sequenziert Millionen DNA-Fragmente gleichzeitig. |
| Third Generation: Echtzeit-Sequenzierung | Der Quantensprung – Einzelmolekül-Analyse in Echtzeit. |
| Emerging Technologies | Science Fiction wird zur Realität. |

First Generation: Sanger-Sequenzierung
Die Sanger-Sequenzierung, benannt nach dem britischen Biochemiker Frederick Sanger, markiert einen historischen Wendepunkt in der Molekularbiologie. Mit dieser Methode gelang es ihm erstmals, die genaue Abfolge der DNA-Basen zu entschlüsseln – ein wissenschaftlicher Durchbruch, der ihm 1980 seinen zweiten Nobelpreis für Chemie einbrachte.
Obwohl heute modernere und automatisierte Verfahren zur Verfügung stehen, gilt die Sanger-Sequenzierung noch immer als Goldstandard, wenn es um höchste Genauigkeit bei kurzen DNA-Abschnitten geht. Sie wird bis heute in vielen Labors zur Validierung von Ergebnissen eingesetzt.
Sie basiert auf dem Prinzip des Abbruchs der DNA-Synthese. Dabei wird die DNA kopiert und durch den Einbau spezieller Stopp-Signale entstehen DNA-Fragmente unterschiedlicher Länge. Diese werden nach Größe sortiert, sodass die Reihenfolge der Bausteine abgelesen werden kann.
So funktioniert der Klassiker der DNA-Analyse
1️⃣ DNA-Denaturierung
Zunächst wird die doppelsträngige DNA erhitzt. Durch die hohe Temperatur lösen sich die Wasserstoffbrücken zwischen den Basenpaaren, und der Doppelstrang trennt sich in zwei Einzelstränge. Diese dienen später als Vorlage für die Synthese neuer DNA.
2️⃣ Vorbereitung der Reaktionsmischungen
Es werden vier separate Reaktionsmischungen hergestellt. Jede enthält:
➥ DNA-Einzelstränge: die Vorlage.
➥ Primer: ein kurzer Abschnitt, der der Polymerase den Startpunkt vorgibt.
➥ DNA-Polymerase: das Enzym, das neue Stränge synthetisiert.
➥ dNTPs (desoxyNukleosidTriPhosphate): die „normalen“ Bausteine der DNA (A, T, C, G). Sie ermöglichen die Verlängerung der DNA-Kette, da ihre Hydroxylgruppe am 3’-Ende eine Verbindung mit dem nächsten Baustein eingehen kann.
➥ ddNTPs (didesoxyNukleosidTriPhosphate): diese „modifizierten“ DNA-Bausteine besitzen keine Hydroxylgruppe und können deshalb keine weiteren Bausteine an sich binden. Wird ein ddNTP während der Synthese eingebaut, stoppt der Aufbau des DNA-Strangs genau an dieser Stelle. Jeder der vier ddNTP-Typen (A, T, C, G) ist zusätzlich mit einem eigenen fluoreszierenden Farbstoff markiert.
Das Verhältnis von dNTPs zu ddNTPs wird so gewählt, dass möglichst viele unterschiedlich lange Fragmente entstehen.
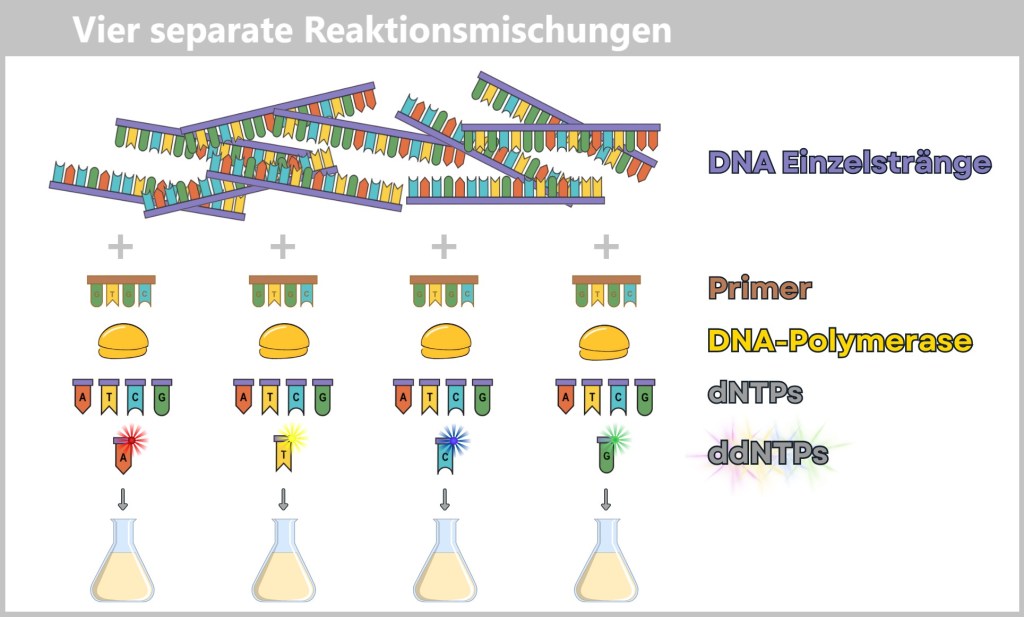
3️⃣ DNA-Synthese mit zufälligem Stopp
DNA-Synthese – kurz erklärt: Die DNA-Polymerase ist der beste Kopierer der Natur: Sie scannt einen DNA-Strang wie eine Vorlage und baut Schritt für Schritt den passenden Gegenstrang – immer nach dem Baukasten-Prinzip: A paart nur mit T und C nur mit G. Heraus kommt eine perfekte Spiegelkopie.
Die DNA-Synthese findet gleichzeitig in den vier separaten Reaktionsmischungen statt – eine für jede der vier Basen (A, T, C, G).
In jeder Mischung läuft Folgendes ab: Die DNA-Polymerase setzt sich auf den Primer und beginnt mit der Arbeit.
Wie findet man den richtigen Primer?
Für den erfolgreichen Einsatz der Sanger-Methode sind Primer unbedingt notwendig. Doch wie findet man die Primer-Sequenz, wenn das Genom eines Virus völlig unbekannt ist?
Als Frederick Sanger 1977 seine Methode entwickelte, wusste man bei kleinen Viren wie dem Bakteriophagen Phi-X174 bereits einiges über die Genomstruktur. Forscher hatten herausgefunden, dass bestimmte Enzyme – sogenannte Restriktionsenzyme – DNA an ganz bestimmten Stellen schneiden. Diese Schnittstellen waren bekannt und konnten gezielt genutzt werden:
In der Nähe dieser Schnittstellen lag oft schon ein kurzes Stück bekannter DNA-Sequenz – genau genug, um einen passenden Primer zu entwerfen. So schuf man sich künstlich einen definierten Startpunkt für die Sequenzierung.
Heutzutage ist das einfacher. Die Wissenschaft hat sich stark weiterentwickelt, und die meisten Viren sind bereits gut untersucht. Bei neuen oder wenig bekannten Viren geht man wie folgt vor:
Vergleich mit bekannten Viren: Oft ähnelt ein neues Virus genetisch bereits bekannten Viren. Wissenschaftler nutzen diese Ähnlichkeiten, um mögliche Primerstellen vorherzusagen.
Experimentelle Ansätze: Wenn wenig bekannt ist, schneiden Enzyme die Virus-DNA in kleinere Stücke. Forscher analysieren diese Fragmente, um Startpunkte für Primer zu finden.
Sie liest den Einzelstrang der DNA-Vorlage und fügt passende Bausteine (dNTPs) ein, um einen neuen Strang zu erzeugen.
Normalerweise wird ein „klassisches“ dNTP eingebaut – damit kann die Kette wachsen. Gelegentlich wird aber ein „modifiziertes“ ddNTP eingebaut – und hier stoppt die Synthese sofort. Weil ddNTPs eine kleine chemische Gruppe (Hydroxylgruppe) fehlt, die für das Anknüpfen des nächsten Bausteins notwendig wäre.
So entstehen viele DNA-Fragmente mit unterschiedlicher Länge – jedes endet genau an der Stelle, wo zufällig ein ddNTP eingebaut wurde. Und jedes Fragment trägt am Ende eine fluoreszierende Farbe, die anzeigt, mit welchem Basentyp (A, T, C oder G) das Fragment endet.
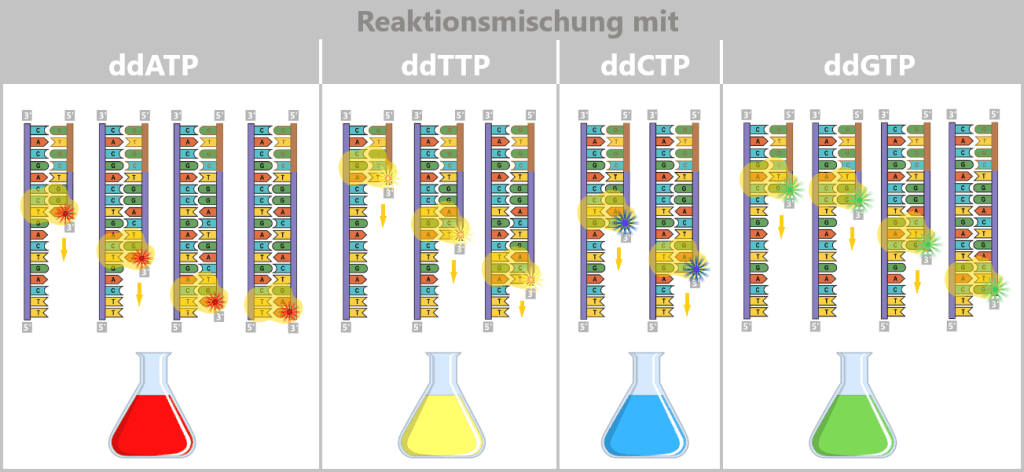
In jeder Reaktionsmischung ist ein modifizierter Nukleotid-Baustein (ddATP, ddTTP, ddCTP, ddGTP) enthalten. Wenn dieser Baustein während der DNA-Synthese eingebaut wird, stoppt die Synthese an genau dieser Position, da der modifizierte DNA-Baustein keine weitere Verlängerung der DNA-Kette erlaubt. In der ddATP-Mischung stoppt die Synthese, sobald Adenin (A) eingebaut wird. In der ddTTP-Mischung erfolgt der Abbruch, wenn Thymin (T) eingebaut wird. Analog dazu führen ddCTP und ddGTP zum Abbruch bei Cytosin (C) bzw. Guanin (G). Dieses Verfahren erzeugt DNA-Fragmente unterschiedlicher Längen, die jeweils mit dem spezifischen Stopp-Nukleotid enden. Ziel ist es, alle theoretisch möglichen Sequenzfragmente herzustellen.
4️⃣ Denaturierung der Fragmente
Die entstandenen doppelsträngigen DNA-Fragmente werden erneut erhitzt, damit sie sich wieder in Einzelstränge trennen. Nur so können sie später einzeln analysiert werden. Übrig bleibt einzelsträngige DNA – bereit für den nächsten Schritt.
5️⃣ Auswertung
Nun geht’s an die Analyse: Die Fragmente werden mit Hilfe der Gelelektrophorese nach ihrer Größe sortiert. Dafür werden die vier Reaktionsmischungen in verschiedene Taschen eines Gels gefüllt.
Das Gel funktioniert wie ein feines Netz oder Schwamm:
- Kurze Fragmente rutschen schneller hindurch.
- Längere Fragmente bewegen sich langsamer.
Jeder Strang endet mit einem farblich markierten ddNTP – je nach Base (A, T, C oder G) leuchtet ein anderer Farbton auf. Diese Farben werden mithilfe eines Lasers detektiert und automatisch aufgezeichnet.
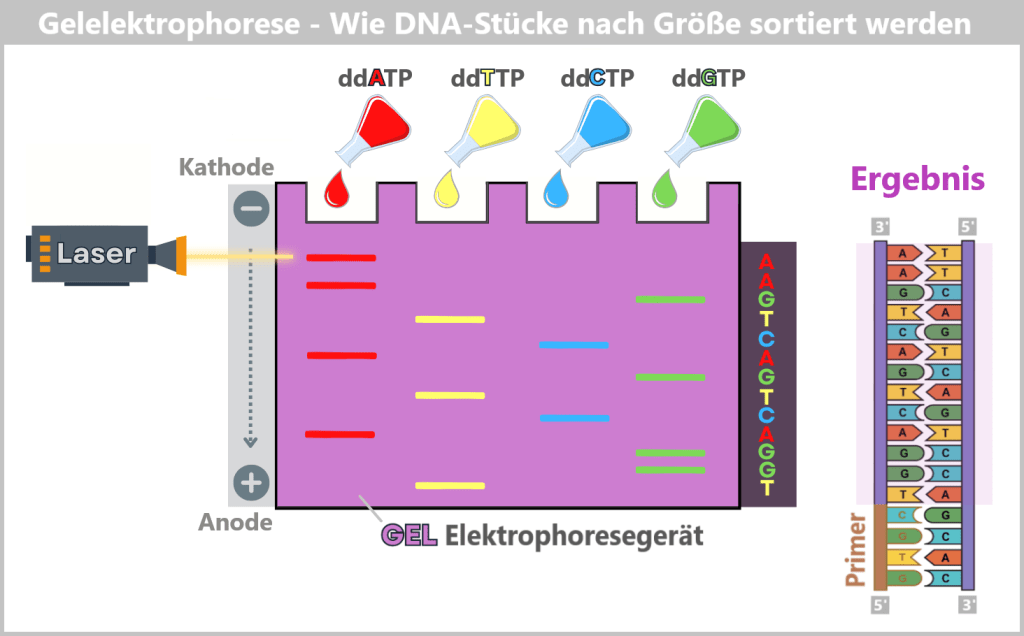
In den Reaktionsgefäßen befinden sich unterschiedlich lange DNA Fragmente, die jeweils mit dem gleichen Stopp-Nukleotid (je nach Ansatz A, T, G und C) enden. Die Reaktionsmischungen werden auf das Gel aufgetragen. Durch das elektrische Feld wandern die negativ geladenen DNA-Fragmente von der Kathode (−) zur Anode (+). Die Fragmentgröße bestimmt die Wandergeschwindigkeit durch das Gel. Kleinere DNA-Fragmente bewegen sich schneller durch die Poren des Gels und erreichen daher zuerst die Anode, während größere Fragmente langsamer vorankommen. Durch das Lesen der fluoreszierenden Signale kann die Basenreihenfolge der DNA-Sequenz bestimmt werden.
Wie liest man daraus die Sequenz?
Die Reihenfolge der Fragmente im Gel entspricht der Reihenfolge der Basen im ursprünglichen DNA-Strang.
- Das kürzeste Fragment zeigt die erste Base.
- Das nächstlängere Fragment die zweite,
- … und so weiter, bis die gesamte Sequenz entschlüsselt ist.
Da jedes neue Fragment komplementär zum Ursprungsstrang ist, lässt sich aus der Analyse direkt die Basenfolge des Original-DNA-Strangs ableiten.
🎥 Tipp: Eine sehr anschauliche Erklärung findest du im Video „Sanger Sequencing / Chain Termination Method“.
Wo wird die Sanger-Sequenzierung eingesetzt?
Die Methode eignet sich besonders gut für:
- Kurze DNA-Abschnitte
- Bestätigung und Kontrolle von Ergebnissen aus anderen Methoden
- Einzelfallanalysen, z. B. in der medizinischen Diagnostik
Manche Methoden werden nicht alt – sie werden klassisch.
Grenzen der Methode
So präzise die Sanger-Sequenzierung auch ist – bei großen oder komplexen Genomen stößt sie schnell an ihre Grenzen. Die Methode ist aufwendig, zeitintensiv und teuer, besonders wenn viele Proben oder umfangreiche Datensätze analysiert werden sollen. Deshalb wurde sie in vielen Bereichen durch moderne Hochdurchsatzverfahren ersetzt – allen voran durch die Technologien der Next-Generation Sequencing (NGS).
Second Generation: Next Generation Sequencing (NGS)
Next-Generation Sequencing (NGS) ist eine moderne Methode zur Entschlüsselung von DNA- und RNA-Sequenzen – und hat die genetische Forschung seit Beginn des 21. Jahrhunderts grundlegend verändert. Im Vergleich zur klassischen Sanger-Sequenzierung ist NGS schneller, kostengünstiger und verarbeitet deutlich größere Datenmengen in kürzerer Zeit.
Der entscheidende Unterschied: Während bei der Sanger-Methode jeweils nur ein einzelnes DNA-Fragment sequenziert wird, kann NGS Millionen von Fragmenten gleichzeitig auslesen. Dazu wird das Erbmaterial zunächst in kleine Stücke zerlegt und mit speziellen Markierungen versehen. Diese Fragmente werden auf einer speziellen Oberfläche fixiert und mithilfe fluoreszierender Nukleotide schrittweise ergänzt – wobei jeder neu eingebaute Baustein sofort ausgelesen wird.
Durch dieses parallele Arbeiten entsteht innerhalb kurzer Zeit ein hochauflösender Datensatz – ideal für die Analyse ganzer Genome, RNA-Profile oder großer Probenmengen. Genau deshalb ist NGS heute eine Schlüsseltechnologie in der Forschung, Diagnostik und Biotechnologie.
Ein tiefer Blick in die Illumina-Sequenzierung
Die Illumina-Sequenzierung ist eine der am häufigsten eingesetzten Technologien im Bereich des Next-Generation Sequencing. Sie basiert auf dem Prinzip „Sequencing by Synthesis“ und ermöglicht das gleichzeitige Auslesen von Millionen DNA-Fragmenten. Hier folgt eine Schritt-für-Schritt-Erklärung:
Schritt 1: DNA-Präparation = Bibliothekserstellung
Schritt 2: Cluster-Generierung auf einer Flowcell
Schritt 3: Sequenzierung durch Synthese
Schritt 1: DNA-Präparation = Bibliothekserstellung
Für die Sequenzierung wird die DNA zunächst in ein Format gebracht, das für die Illumina-Plattform geeignet ist. Dieser Prozess wird auch als Bibliothekserstellung bezeichnet und er umfasst folgende Teilschritte:
1a) Fragmentierung
Die DNA wird mechanisch oder enzymatisch in kleinere Stücke zerlegt (typischerweise mit einer Zielgröße von etwa 150–500 Basenpaaren). Dabei entstehen DNA-Fragmente leicht unterschiedlicher Länge, die anschließend oft noch über ein Größenauswahlverfahren vereinheitlicht werden.
Zur Veranschaulichung betrachten wir in unserem Beispiel drei DNA-Fragmente mit leicht unterschiedlichen Längen.
1b) Adapter-Ligation
An beide Enden der DNA-Fragmente werden spezifische Adaptersequenzen (P5- und P7-Adapter) angehängt. Diese Adapter erfüllen mehrere Funktionen:
Bindungsstellen für die Flowcell: Die Adapter enthalten spezielle DNA-Sequenzen, die wie ein Schlüssel zu einem Schloss passen. Dadurch können die DNA-Fragmente später an einer Oberfläche haften bleiben, was für die Sequenzierung wichtig ist.
Primer-Bindungsstellen: Die Adapter enthalten Abschnitte, an die Sequenzierungsprimer binden können. Diese Primer werden später genutzt, um die DNA-Stränge schrittweise zu synthetisieren.
Indizes (optional): Falls mehrere Proben gleichzeitig sequenziert werden, ermöglichen Index-Sequenzen die Zuordnung der Fragmente zu ihrer jeweiligen Probe.
Zur besseren Übersichtlichkeit verzichten wir in unserem Beispiel auf die Darstellung der Indizes.
1c) PCR-Amplifikation
Um sicherzustellen, dass genügend DNA für die Sequenzierung vorliegt, werden die adaptierten DNA-Fragmente mittels Polymerase-Kettenreaktion (PCR) vervielfältigt.
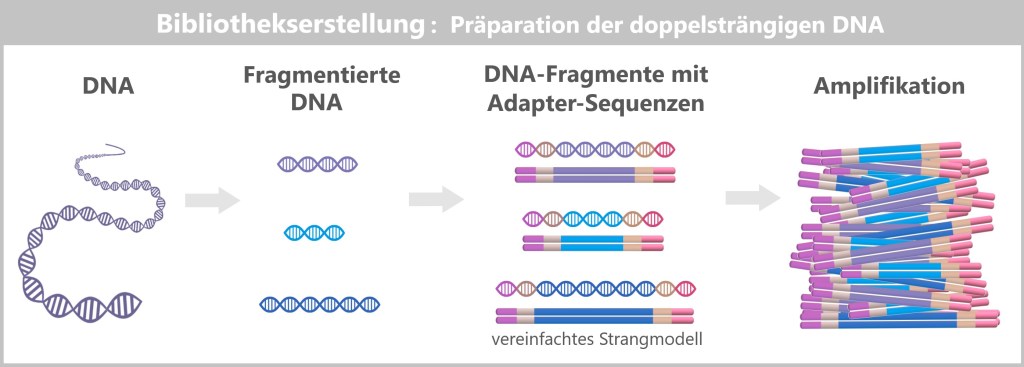
Die folgende Vergrößerung zeigt eine detaillierte Darstellung eines fragmentierten DNA-Doppelstrangs, versehen mit den beiden Adaptern.
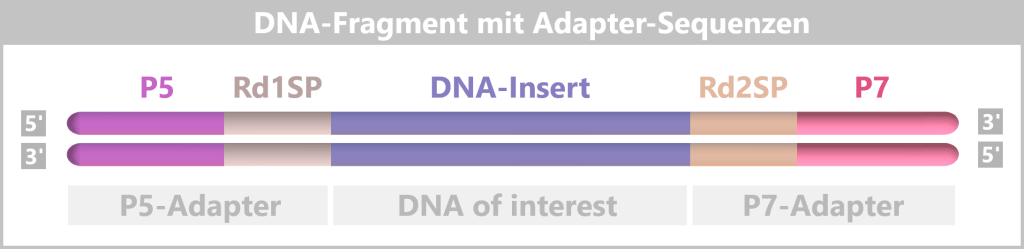
Jedes DNA-Fragment (DNA-Insert) erhält zwei Adaptersequenzen: Der P5-Adapter besteht aus der P5-Adaptersequenz, die die Bindung an die Flowcell ermöglicht, und der Primer-Bindestelle Rd1SP (Read 1 Sequencing Primer) zur Initiation der DNA-Synthese. Ebenso enthält der P7-Adapter die P7-Adaptersequenz zur Flowcell-Bindung sowie die Primer-Bindestelle Rd2SP (Read 2 Sequencing Primer) für die Initiation der DNA-Synthese.
Wichtige Anmerkung: Die oberen und unteren DNA-Einzelstränge besitzen jeweils P5- und P7-Adapter. Allerdings sind die Adapter des unteren Strangs nicht identisch, sondern komplementär zu den Adaptern des oberen Strangs.

Diese Darstellung ist stark vereinfacht. In der Praxis sind Illumina-Adaptersequenzen mit 60-120 Basenpaarendeutlich länger (siehe hier). Auch das DNA-Insert ist zur besseren Veranschaulichung verkürzt – tatsächlich haben DNA-Fragmente typischerweise eine Länge von 100–500 Basenpaaren.
Schritt 2: Cluster-Generierung auf einer Flowcell
Bevor die DNA sequenziert werden kann, muss sie an einer festen Oberfläche fixiert und vervielfältigt werden. Dies geschieht auf einer Flowcell, einer speziellen Glasplatte, die mit Millionen winziger DNA-Andockstellen versehen ist.
Das Ziel dieses Schrittes ist es, zahlreiche Kopien jedes DNA-Fragments auf dieser Oberfläche zu erzeugen, um die Signale bei der Sequenzierung zu verstärken. Dafür ist die Flowcell mit speziellen Oligonukleotiden (DNA-Molekülen) beschichtet, die komplementär zu den Adaptersequenzen der DNA-Fragmente sind. Es gibt zwei Typen dieser Oligonukleotide:
- P5-Oligonukleotide (ACGTAC), die an die P5-Adapter (TGCATG ) der DNA-Fragmente binden.
- P7-Oligonukleotide (ACGTCA), die mit den P7-Adaptern (TGCAGT ) der DNA interagieren.
Man kann sich die Oberfläche der Flowcell wie einen dichten Klettverschluss vorstellen: Die DNA-Fragmente haften mit ihren Adaptersequenzen daran, ähnlich wie kleine Haken, die in das Klettgewebe greifen.
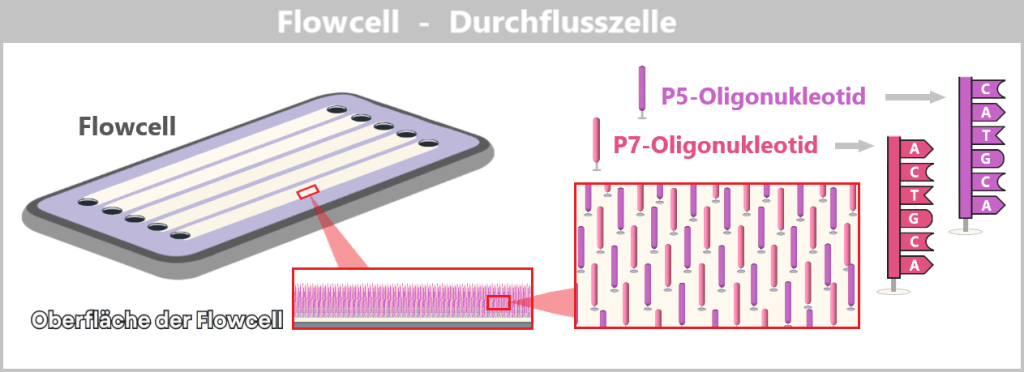
2a) DNA-Fragmente binden an die Flowcell-Oberfläche
Zu Beginn des Schritts wird eine Lösung mit einzelsträngigen DNA-Fragmenten, die Adaptersequenzen tragen, auf die Flowcell gespült. Diese Fragmente wurden zuvor durch Denaturierung in Einzelstränge aufgetrennt, sodass sie sich frei in der Lösung bewegen können.
Während die DNA-Fragmente durch die Flowcell strömen, binden ihre Adapter durch komplementäre Basenpaarung an die passenden Oligonukleotiden auf der Flowcell, wie in der unteren Abbildung gezeigt.

P5-Adapter eines DNA-Einzelstrangs bindet mit dem P5-Oligonukleotid auf der Flowcell:
Flowcell — P5-Oligo ACGTAC
— (3') TGCATG-CAGT-[Insert]-GTCA-ACTGCA (5')
P7-Adapter eines DNA-Einzelstrangs bindet mit dem P7-Oligonukleotid auf der Flowcell:
Flowcell — P7-Oligo ACGTCA
— (3') TGCAGT-TGAC-[Insert]-ACTG-CATGCA (5')
2b) Erste Synthese
Primerbindung und DNA-Synthese
Nachdem die DNA-Fragmente an die Flowcell gebunden wurden, startet die erste DNA-Synthese. Dazu binden sich spezifische Primer an die Adaptersequenzen der gebundenen Stränge:
- Primer GTCA bindet an den P5-Adapter des gebundenen Einzelstrangs.
- Primer ACTG bindet an den P7-Adapter des gebundenen Einzelstrangs.
Die DNA-Polymerase, die nur in 5′ → 3′-Richtung arbeiten kann, synthetisiert daraufhin einen neuen Strang, der komplementär zum bereits gebundenen Einzelstrang ist.
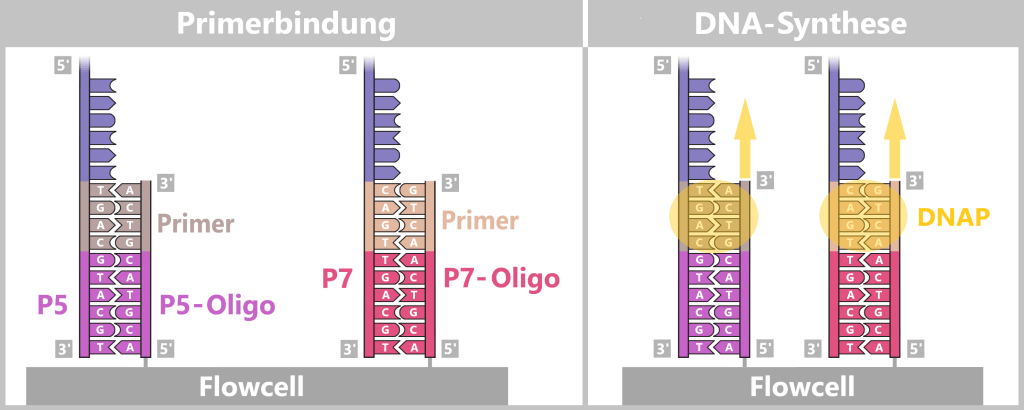
Nach der Synthese liegen die gebundenen DNA-Fragmente als Doppelstränge vor.
Flowcell (P5-Oligo) — (5') ACGTAC-GTCA-[Insert]-CAGT-TGACGT (3')
— (3') TGCATG-CAGT-[Insert]-GTCA-ACTGCA (5')
Flowcell (P7-Oligo) — (5') ACGTCA-ACTG-[Insert]-TGAC-GTACGT (3')
— (3') TGCAGT-TGAC-[Insert]-ACTG-CATGCA (5')

Trennung des Doppelstrangs
Nachdem die neue DNA synthetisiert wurde, wird der Doppelstrang durch Denaturierung getrennt (siehe obere Abbildung):
❌ Der ursprüngliche Strang hat nach der Denaturierung keine Verbindung mehr zur Flowcell und wird ausgespült.
✅ Der neu synthetisierte Strang bleibt mit seinem 5′-Ende fest an der Flowcell gebunden, während das 3′-Ende frei bleibt.
Nun sind die DNA-Fragmente als komplementäre Einzelstränge (Forward- und Reverse-Stränge) an die Flowcell gebunden.
Allerdings wäre eine direkte Sequenzierung zu diesem Zeitpunkt nicht möglich, da die Fluoreszenzsignale noch zu schwach wären, um zuverlässig ausgelesen zu werden. Deshalb folgt nun die Brückenamplifikation.
2c) Brückenamplifikation
Damit die DNA-Sequenzierung ein ausreichend starkes Fluoreszenzsignal liefert, müssen die einzelnen DNA-Stränge vervielfältigt werden. Dies geschieht durch die Brückenamplifikation, einen zyklischen Prozess, bei dem sich die DNA-Stränge an die Flowcell anlagern, vervielfältigt und wieder getrennt werden.
Hybridisierung des zweiten Adapters
Die Brückenamplifikation beginnt damit, dass sich der gebundene DNA-Einzelstrang biegt und mit seinem freien 3′-Ende an ein benachbartes komplementäres Oligonukleotid (5′) auf der Flowcell bindet. Dieser Vorgang wird als Faltung des DNA-Strangs bezeichnet. Dadurch bildet sich eine Art Brücke, die den Adapter des Strangs mit dem passenden Oberflächenmolekül verbindet.
DNA-Synthese
Sobald die DNA-Stränge gebunden sind, werden Primer (ACTG, GTCA) und die DNA-Polymerase hinzugefügt. Die Primer binden spezifisch an die Adaptersequenzen. Die DNA-Polymerase synthetisiert neue komplementäre Stränge in 5′ → 3′-Richtung. Nach der Synthese liegt die DNA nun wieder als Doppelstrang vor.
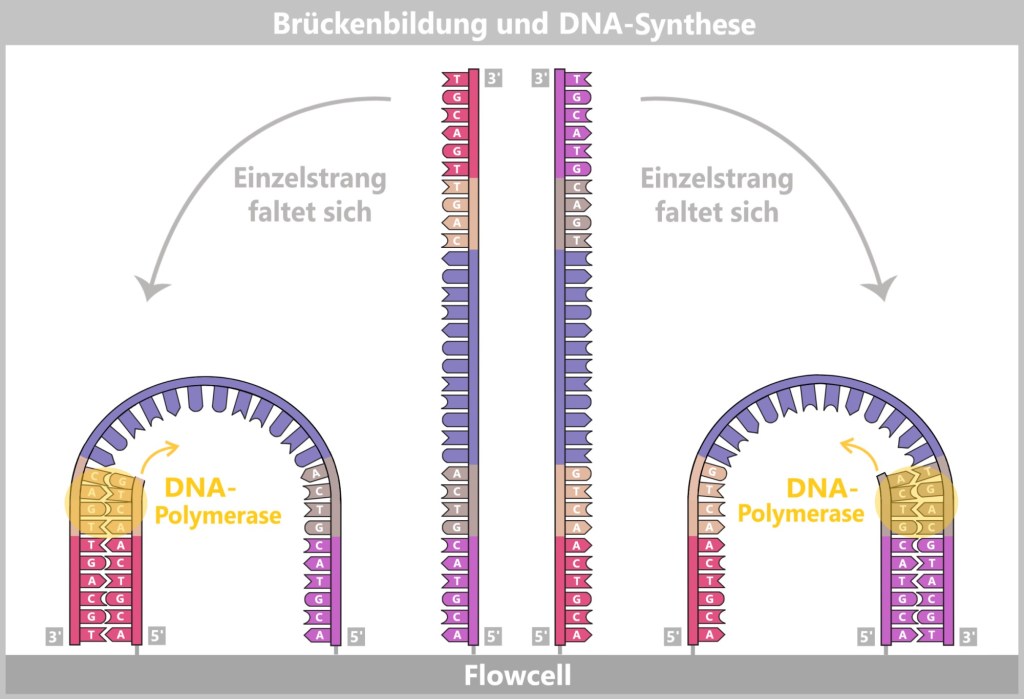
Die gebundenen Einzelstränge falten sich und hybridisieren mit einem komplementären Oligonukleotid auf der Flowcell, wodurch eine Brückenstruktur entsteht. Anschließend bindet ein Primer an den 3′-Enden der Stränge, und die DNA-Polymerase synthetisiert die komplementären Stränge in 5′ → 3′-Richtung.
Denaturierung– Trennung der Stränge
Durch Hitze oder chemische Behandlung werden die neu gebildeten Doppelstränge wieder in Einzelstränge aufgetrennt. Jetzt befinden sich vier einzelsträngige DNA-Fragmente auf der Flowcell:
|| die ursprünglichen zwei Einzelstränge (Forward-Strang & Reverse-Strang) und
|| die neu synthetisierten komplementären Stränge (Forward-Strang & Reverse-Strang).
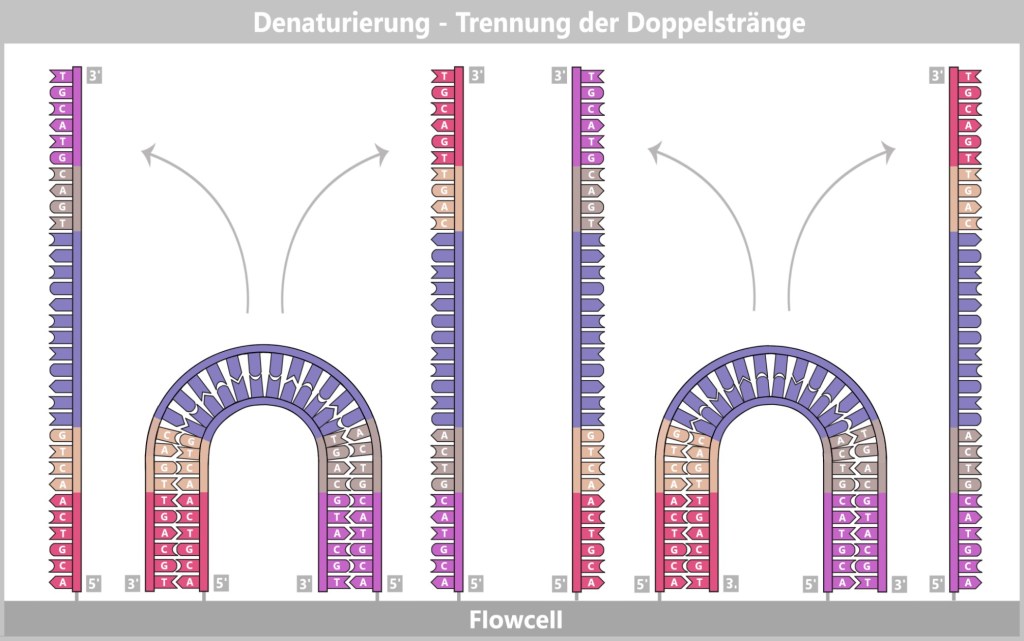
Nach der Denaturierung werden die Brücken-Doppelstränge getrennt, sodass auf der Flowcell nun jeweils zwei Einzelstränge (vorwärts und rückwärts) zu sehen sind. Diese Einzelstränge sind weiterhin an der Flowcell gebunden und bereit für die weitere Amplifikation.
Wiederholung der Amplifikation
Dieser Zyklus wiederholt sich: Die Einzelstränge falten sich erneut, hybridisieren wieder mit den Oligonukleotiden und werden durch DNA-Synthese vervielfältigt.

Nach mehreren Zyklen entstehen auf der Flowcell Millionen identischer Kopien jedes ursprünglichen DNA-Fragments – die Cluster.
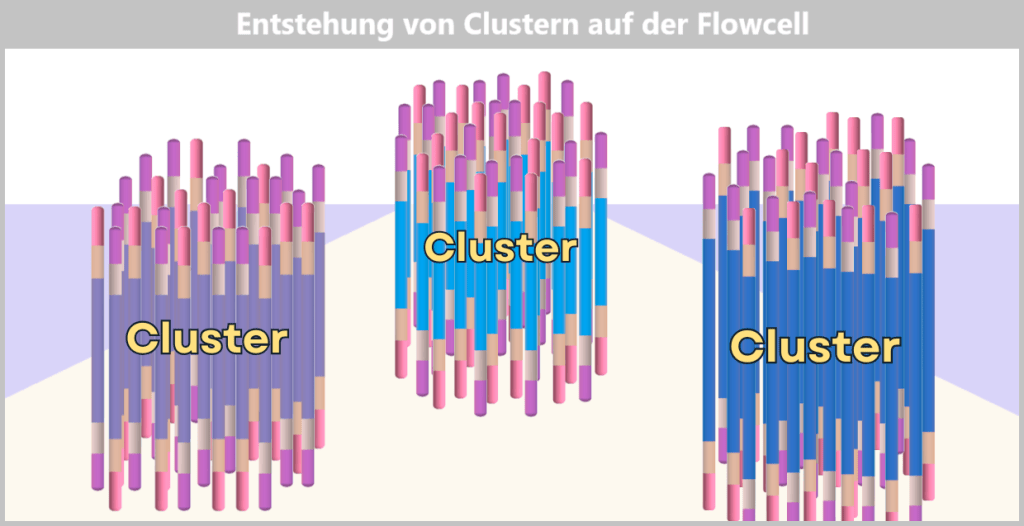
Jedes Cluster besteht aus zahlreichen Kopien eines einzelnen DNA-Fragments. In dieser Darstellung sind exemplarisch nur drei Cluster gezeigt, da unser Beispiel von drei DNA-Fragmenten ausgeht. In der Realität befinden sich jedoch Millionen bis Milliarden solcher Cluster auf einer Flowcell, um eine hohe Sequenzierkapazität zu ermöglichen.
Bildung der finalen Cluster
Zu Beginn unseres Beispiels sind drei DNA-Fragmente vorhanden. Nach mehreren Amplifikationszyklen entstehen für jedes dieser Fragmente unzählige Cluster, die ausschließlich aus ihren DNA-Kopien bestehen.
Jedes Cluster setzt sich nun aus zwei Strang-Typen zusammen:
🔹 Forward-Stränge (5′ → 3′, P7-gebunden)
🔹 Reverse-Stränge (3′ → 5′, P5-gebunden)
💡 Beachte: Forward- und Reverse-Stränge sind in den Clustern nicht antiparallel zueinander ausgerichtet! Stattdessen ragen die 3′-Enden beider Stränge nach oben.
Entfernung der Reverse-Stränge
Damit die Sequenzierung korrekt ablaufen kann, müssen sich alle DNA-Stränge innerhalb eines Clusters in derselben Richtung befinden. Für die eigentliche Analyse werden nur die Forward-Stränge benötigt. Daher folgt nun ein gezielter Reinigungsschritt:
🔹 Alle Reverse-Stränge (P5-gebunden) werden abgetrennt und ausgespült.
🔹 Gleichzeitig werden die freien Enden der P5-Oligos chemisch blockiert, um eine erneute Bindung zu verhindern.
Nun befinden sich ausschließlich Vorwärtsstränge auf der Flowcell. Die Cluster sind vollständig ausgebildet, und die DNA-Bibliotheken sind bereit für die Sequenzierung.

Links: Ein finales Cluster mit Forward- (P7-gebunden) und Reverse-Strängen (P5-gebunden). Rechts: Die Reverse-Stränge wurden gezielt durchtrennt und ausgespült, sodass nur noch die Forward-Stränge verbleiben. Diese Ausrichtung ist notwendig, um die Sequenzierung korrekt durchzuführen.
Schritt 3: Sequenzierung durch Synthese
An diesem Punkt sind einzelne klonale Cluster (aus den ursprünglich drei DNA-Fragmenten) über die gesamte Oberfläche der Flowcell verteilt. Jedes Cluster besteht aus zahlreichen identischen DNA-Kopien – vergleichbar mit einer kleinen Insel identischer Bäume. Nun kann die eigentliche Sequenzierung beginnen.
Um die Sequenzierung zu starten, werden Primer, DNA-Polymerasen und modifizierte Nukleotide auf die Flowcell aufgetragen.
Die modifizierten Nukleotide haben drei besondere Eigenschaften:
① Terminierung der Synthese
Sie besitzen eine chemische Modifikation an der Hydroxylgruppe, die verhindert, dass ein weiteres Nukleotid an die wachsende DNA-Kette angefügt wird. Dadurch stoppt die DNA-Synthese nach dem Einbau jedes einzelnen Nukleotids.
② Reversibilität der Blockierung
Diese Modifikation kann anschließend chemisch entfernt werden, sodass die DNA-Synthese fortgesetzt werden kann. Dadurch erfolgt die Sequenzierung schrittweise, ein Nukleotid nach dem anderen.
③ Fluoreszenzmarkierung
Jedes der vier Basen (A, T, C, G) ist mit einem spezifischen fluoreszierenden Farbstoff markiert.
Aufgrund dieser Eigenschaften werden diese Nukleotide als reversible Terminator-Nukleotide (RT-dNTPs) bezeichnet.
Ablauf der Sequenzierung
➥ Die Sequenzierungsprimer hybridisieren mit den Vorwärtssträngen der DNA-Bibliothek.
➥ Die DNA-Polymerase bindet an den Primer und beginnt die Synthese. Aufgrund der modifizierten Nukleotide kann sie jedoch immer nur ein einziges Nukleotid pro Zyklus anfügen.
➥ Nach jedem Einbau wird die Flowcell von einer hochauflösenden Kamera abfotografiert. Die fluoreszierende Markierung zeigt an, welches Nukleotid eingebaut wurde. Ein Computer analysiert die Fluoreszenzsignale und ordnet sie den jeweiligen Basen zu.
➥ Anschließend werden überschüssige Nukleotide weggespült und die Blockierung am eingebauten Nukleotid durch einen chemischen Schritt entfernt.
➥ Der Zyklus beginnt von vorn, bis das gesamte DNA-Fragment sequenziert ist.
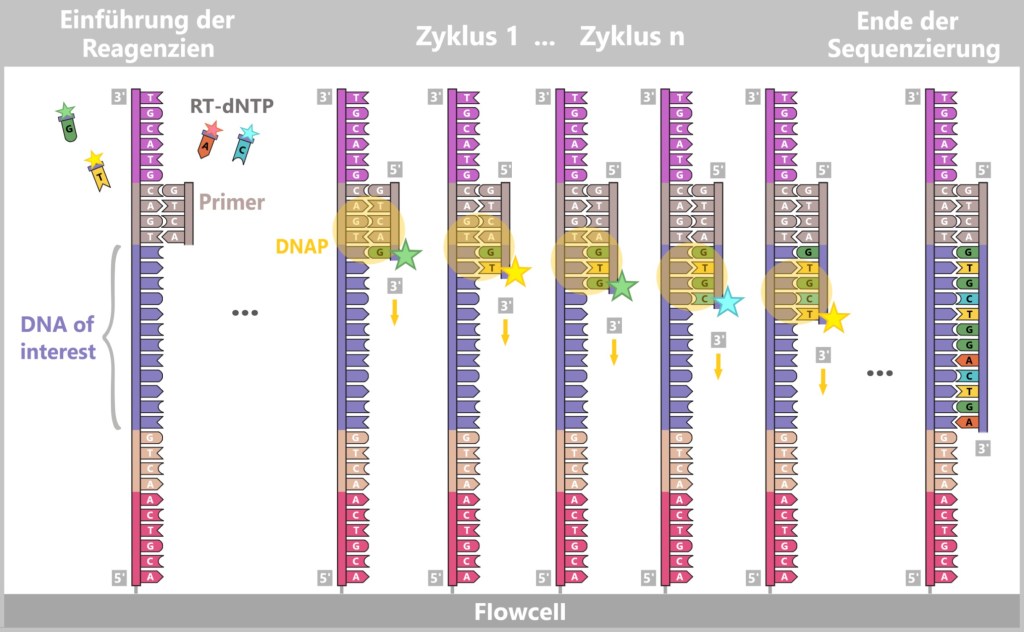
Primer, DNA-Polymerase (DNAP) und modifizierte Nukleotide (RT-dNTPs) werden auf die Flowcell aufgetragen. Die RT-dNTPs sind fluoreszenzmarkiert (jede Base hat eine eigene Farbe) und besitzen eine reversible Blockierung an der 3′-Hydroxylgruppe. Dadurch wird nach jeder Baseneinfügung die DNA-Synthese vorübergehend gestoppt. Sobald ein RT-dNTP eingebaut wurde, regt ein Laser den Fluoreszenzfarbstoff zum Leuchten an. Eine Kamera erfasst dieses Signal und bestimmt, welche Base eingebaut wurde. Anschließend wird die Fluoreszenzmarkierung zusammen mit der Blockierung chemisch entfernt, sodass die DNA-Synthese im nächsten Zyklus fortgesetzt werden kann. Dieser Prozess wird in jedem Zyklus wiederholt: Eine Base wird hinzugefügt, das Signal erfasst und die Blockierung entfernt. Die Sequenzierung läuft über eine festgelegte Anzahl an Zyklen, wodurch eine begrenzte Anzahl an Basen gelesen wird (z. B. 150 Basenpaare bei 150er-Reads).
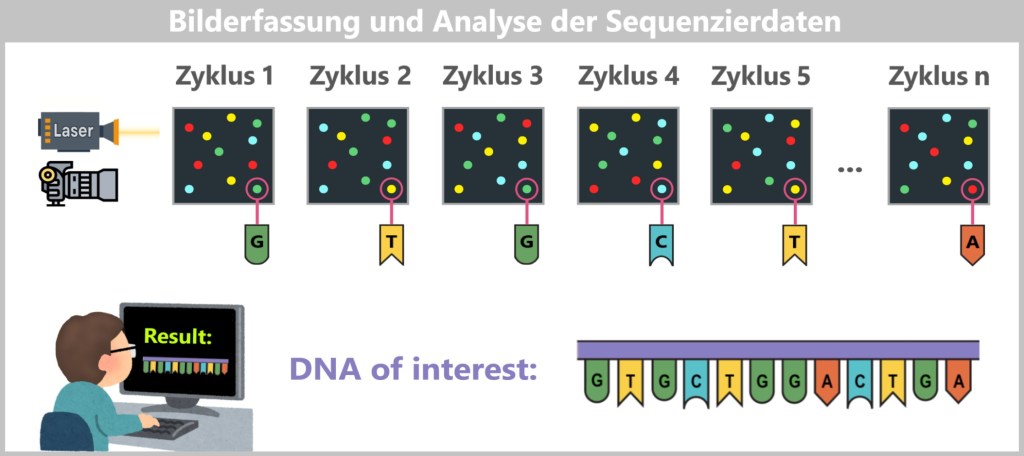
Während der Sequenzierung erfasst eine Kamera in jedem Zyklus die Fluoreszenzsignale der eingebauten Nukleotide. Jedes Cluster sendet ein spezifisches Farbsignal, das der eingebauten Base entspricht. Über mehrere Zyklen hinweg entsteht so für jedes Cluster eine individuelle Sequenz. Ein Computer analysiert die Farbinformationen aus den einzelnen Bildern und ordnet sie der jeweiligen DNA-Sequenz zu. So wird aus den gemessenen Fluoreszenzsignalen die exakte Basenabfolge der „DNA of interest“ rekonstruiert.
Doppel-Check für die DNA: Paired-End-Sequenzierung
Die Paired-End-Sequenzierung ist ein Verfahren, das auf vielen Illumina-Plattformen zum Einsatz kommt, um die Genauigkeit und Verlässlichkeit der Sequenzierung zu verbessern. Dabei wird jedes DNA-Fragment von beiden Enden aus gelesen – es entstehen also zwei Reads pro Fragment: Read 1 (Vorwärts) und Read 2 (Rückwärts).
Nach der ersten Sequenzierung des Vorwärtsstrangs (Read 1) erfolgt eine zweite Brückenamplifikation, bei der die ursprünglichen Stränge erneut erzeugt werden. Anschließend werden die bereits gelesenen Stränge entfernt – und die Sequenzierung des Rückwärtsstrangs (Read 2) beginnt.
Da beide Reads aus demselben Fragment stammen, können sie rechnerisch zusammengeführt werden. Das erleichtert die Assemblierung, erhöht die Fehlererkennung und verbessert die Lesbarkeit wiederholter oder komplexer Sequenzabschnitte.
Die Paired-End-Sequenzierung eignet sich besonders gut für lange, verschachtelte oder schwierige DNA-Bereiche – und liefert dadurch präzisere und belastbarere Ergebnisse.
Ergebnis der Illumina-Sequenzierung
Am Ende der Illumina-Sequenzierung liegt eine riesige Menge kurzer DNA-Abschnitte vor – sogenannte Reads. Jeder dieser Reads stammt von einem kleinen Fragment des ursprünglichen Erbmaterials und wurde millionenfach vervielfältigt und ausgelesen.
Damit daraus wieder ein vollständiges Bild entsteht, werden die Reads mithilfe spezieller Software am Computer zusammengesetzt:
🔹 Gibt es eine bekannte Referenz-DNA, werden die Reads wie Puzzleteile an das bekannte Muster angelegt.
🔹 Fehlt eine Vorlage, müssen sie anhand von Überlappungen Stück für Stück neu zusammengesetzt werden.
So entsteht aus zahllosen kurzen Sequenzen Schritt für Schritt ein vollständiges Bild des ursprünglichen DNA-Materials. Das Ergebnis ist eine präzise analysierte Gensequenz, die nicht nur die genetische Struktur sichtbar macht, sondern auch Hinweise auf Mutationen oder Varianten liefern kann.
🎥 Tipp: Eine anschauliche Erklärung findest du im Video „Illumina Sequencing Technology“.
Anwendungsbereiche
Die Illumina-Sequenzierung gilt als genau und zuverlässig. Ihren Einsatz hat sie in der:
Genomforschung: Entschlüsselung der DNA von Menschen, Tieren, Pflanzen
Medizin: Untersuchung genetischer Erkrankungen, Entwicklung personalisierter Therapien
Mikrobiologie: Analyse von Bakterien und Viren
Umweltforschung: Untersuchung von DNA in Boden- oder Wasserproben
Und was kommt danach?
Die Illumina-Technologie hat die genetische Analyse revolutioniert – doch auch sie hat ihre Grenzen: Die Vorbereitung ist aufwendig, die Auswertung rechenintensiv, und sehr lange DNA-Abschnitte lassen sich nur in kleinen Stücken erfassen.
Deshalb geht die Entwicklung weiter. Neue Technologien der Third Generation Sequencing setzen auf ganz andere Prinzipien – und ermöglichen erstmals das direkte Auslesen extrem langer DNA-Stränge, oft sogar in Echtzeit.
Zeit für einen Blick auf die nächste Generation der Sequenzierung.
Third Generation: Echtzeit-Sequenzierung
Mit der dritten Generation der Sequenzierungstechnologien beginnt ein grundlegender Wandel in der Genomforschung. Statt wie bisher auf fragmentierte oder chemisch veränderte DNA zu setzen, ermöglichen diese Methoden das direkte Auslesen genetischer Informationen – in Echtzeit, ohne komplexe Vorbereitung und mit neuen analytischen Möglichkeiten.
Zwei zentrale Verfahren dieser Generation sind:
- Single Molecule Real Time (SMRT) Sequencing – verfolgt die DNA-Synthese in Echtzeit, indem Lichtimpulse registriert werden, die beim Einbau einzelner Basen entstehen.
- Nanopore Sequencing – leitet DNA-Stränge durch winzige Nanoporen und misst Veränderungen im elektrischen Strom, um die Basen zu identifizieren.
Warum die Nanopore-Sequenzierung ein Gamechanger ist
Die Nanopore-Technologie eröffnet ganz neue Perspektiven in der Genomforschung – durch ihre Flexibilität, Geschwindigkeit und Unabhängigkeit von aufwändigen Vorbereitungsschritten:
✅ Echtzeit-Sequenzierung: Im Gegensatz zu klassischen Verfahren, die DNA zunächst vervielfältigen oder chemisch modifizieren müssen, liest Nanopore-Technologie das genetische Material direkt aus.
✅ Lange Leselängen: Anstelle kurzer Fragmente können extrem lange DNA-Stränge ausgelesen werden – oft über Hunderttausende Basenpaare hinweg, in einem einzigen Durchgang. Das erleichtert die Analyse komplexer Genome und struktureller Varianten erheblich.
✅ Vielseitigkeit: Neben DNA lässt sich auch RNA direkt analysieren – ohne den Zwischenschritt über cDNA (komplementäre DNA). Das macht die Technologie besonders wertvoll für Studien zur Genexpression oder Virusforschung.
✅ Portabel und kostengünstig: Geräte wie der MinION sind kaum größer als ein USB-Stick und ermöglichen Sequenzierung sogar außerhalb des: etwa in der Klinik, im Regenwald oder direkt am Tatort.
Mit diesen Eigenschaften erschließt die Nanopore-Sequenzierung völlig neue Anwendungsfelder – von der Grundlagenforschung über Diagnostik bis hin zur Umwelt- und Biodiversitätsanalyse.
Grund genug, diese Technologie einmal genauer unter die Lupe zu nehmen.
Für einen ersten Eindruck und eine kurze Übersicht ist das folgende Video geeignet.
Nanopore-Sequenzierung – Schritt für Schritt
Wenn heute von Nanopore Sequencing die Rede ist, meint man fast immer die Oxford Nanopore-Technologie (ONT). Zwar gibt es theoretisch andere Ideen mit Nanoporen, aber ONT ist derzeit die einzige, die wirklich im Einsatz ist. Seit ihrer allgemeinen Markteinführung im Jahr 2015 hat sie die Sequenzierung revolutioniert.
Das Besondere an dieser Methode? Sie liest DNA oder RNA direkt und in Echtzeit – ohne vorherige chemische Modifikationen oder Verstärkungsreaktionen. Die Nanopore-Technologie funktioniert dabei wie ein winziges Hochleistungs-Labor, das genetische Informationen präzise entschlüsselt.
Wie funktioniert das?
Die ONT-Sequenzierung basiert auf einem Zusammenspiel von drei zentralen Komponenten:
➤ Nanoporen – fungieren als winzige molekulare „Lesegeräte“. Wenn ein DNA- oder RNA-Strang durch eine Nanopore hindurchwandert, erzeugt dies charakteristische elektrische Signale – quasi einen molekularen „Fingerabdruck“.
➤ Membran – dient als Filter und Barriere. Sie sorgt dafür, dass nur Ionen und Nukleinsäuren die Nanoporen passieren, während unerwünschte Moleküle ausgeschlossen bleiben. Dadurch entstehen saubere, präzise Messwerte.
➤ Chip – ist die Basis des Systems. Er hält die Membran mit den Nanoporen und erfasst die elektrischen Signale, die beim Durchtritt der DNA entstehen.
Der Chip besitzt zwei Kammern:
- Obere Kammer (cis): Hier wird die DNA-Probe eingefüllt.
- Untere Kammer (trans): Hier landet die DNA, nachdem sie die Nanoporen durchlaufen hat.
Beide Kammern sind mit einer ionenhaltigen Flüssigkeit gefüllt, die elektrischen Strom leitet. Die Membran trennt die Kammern, sodass kein Strom fließen kann – außer an den Stellen, an denen die Nanoporen eingebettet sind. Diese Poren sind die einzigen „Tunnel“, durch die Ionen und Nukleinsäuren passieren können.
Sobald ein DNA- oder RNA-Strang durch eine Nanopore gleitet, verändert sich der Stromfluss. Diese feinen Veränderungen werden erfasst und in genetischen Code übersetzt – in Echtzeit: direkt, schnell und genau.
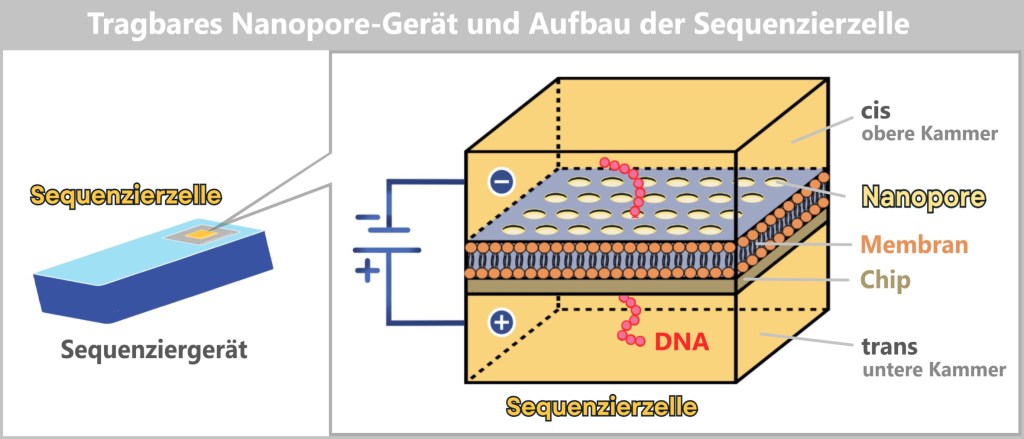
Links ist ein tragbares Nanopore-Sequenziergerät dargestellt. In der Praxis ist es etwas größer als ein USB-Stick. Ein Pfeil zeigt auf die austauschbare Sequenzierzelle. Die vergrößerte Darstellung rechts zeigt den schematischen Aufbau der Sequenzierzelle.
Präzision auf Nanoebene
Chip-Mikrostruktur
Der Chip wird mit hochpräzisen Fertigungstechniken wie Photolithografie oder Ätzverfahren hergestellt. Dabei entstehen winzige Kanäle, sogenannte Wells, die nur wenige Nanometer bis Mikrometer groß sind. Neben diesen strukturellen Komponenten enthält der Chip auch die elektrochemische Messvorrichtung, die den Ionenstrom registriert, sobald DNA oder RNA durch die Nanopore wandert.
Selbstassemblierung der Lipidmembran
Diese Membran besteht aus Lipiden (Fettmolekülen) und ist elektrisch isolierend. Das bedeutet, dass sie selbst keinen Strom leitet. Die Membran wird zunächst ohne Nanoporen auf den Chip aufgespannt. Sie wird nicht mechanisch auf den Chip gespannt, sondern bildet sich durch Selbstorganisation über den Öffnungen.
Wie funktioniert das?
Eine Lösung mit Lipidmolekülen wird auf den Chip gegeben. Diese Moleküle besitzen:
🔹einen hydrophoben (wasserabweisenden) Anteil und
🔹einen hydrophilen (wasseranziehenden) Anteil.
Durch diese Eigenschaften organisieren sie sich automatisch in eine stabile Doppelschicht. Die Wells im Chip sind so konstruiert, dass die Lipidmembran genau über ihnen stabilisiert wird.
Selbstassemblierung der Nanoporen
Die Nanoporen bestehen meist aus Proteinen, die aus Bakterien oder Hefezellen gewonnen werden. In der Natur dienen diese Poren als Transportkanäle für Moleküle durch Zellmembranen. In der Nanopore-Technologie werden sie jedoch als hochempfindliche Sensorenfür DNA- oder RNA-Molekülegenutzt.
Die Integration der Nanoporen erfolgt nach dem Aufspannen der Membran. Dank eines natürlichen Prozesses namens Selbstassemblierung setzen sie sich von selbst in die Membran ein!
Wie funktioniert das?
🔹Die Nanoporen werden in einer Lösung zugegeben.
🔹Sie „finden“ automatisch die Wells, da sie sich nur dort in die Membran einbauen können, wo eine Öffnung vorhanden ist.
🔹Dort durchdringen sie die Membran und bilden stabile Kanäle.
Die Wells sind somit exakt definierte Bereiche, in denen die Membran zugänglich ist – und genau dort können sich die Nanoporen gezielt integrieren.
Warum ist das wichtig?
Die korrekte Positionierung der Nanoporen über den Wells ist entscheidend, weil:
🔹Die DNA durch die Nanoporen von der cis- zur trans-Kammer gezogen wird, und das nur funktioniert, wenn die Nanoporen in der Membran über den Wells sitzen.
🔹Die Wells sind mit Elektroden verbunden, die den Ionenstrom messen. Wenn eine Nanopore nicht exakt über einem Well sitzt, gibt es keinen messbaren Stromfluss – die Nanopore wäre funktionslos.
Der Weg der DNA durch die Nanopore
Nachdem wir den Aufbau der Sequenzierzelle und das Grundprinzip der Oxford Nanopore-Technologie kennengelernt haben, geht es nun um den eigentlichen Sequenzierprozess.
1️⃣ Vorbereitung der DNA/RNA-Probe
Bevor die eigentliche Sequenzierung beginnt, muss die Nukleinsäure (DNA oder RNA) vorbereitet werden.
➥ Extraktion: Die DNA oder RNA wird aus der Probe (z. B. Blut, Speichel, Zellkultur, Umweltproben) isoliert. Dies geschieht mit standardisierten Extraktionsverfahren (siehe dazu Kapitel „4.5.1. Nukleinsäure-Extraktion“).
➥ Fragmentierung (optional): Nanopore-Sequenzierung kann sehr lange DNA- oder RNA-Moleküle lesen. Falls die DNA zu lang ist oder für bestimmte Anwendungen angepasst werden soll, kann sie mechanisch oder enzymatisch in Fragmente geschnitten werden.
➥ Adapter-Ligation: Da die Nanoporen nur DNA oder RNA mit speziellen Enden verarbeiten können, werden Adapter an die Enden der Moleküle angehängt. Diese Adapter enthalten:
- Motorproteine, die die DNA kontrolliert durch die Nanopore ziehen
- Barcode-Sequenzen (falls mehrere Proben gleichzeitig sequenziert werden sollen)
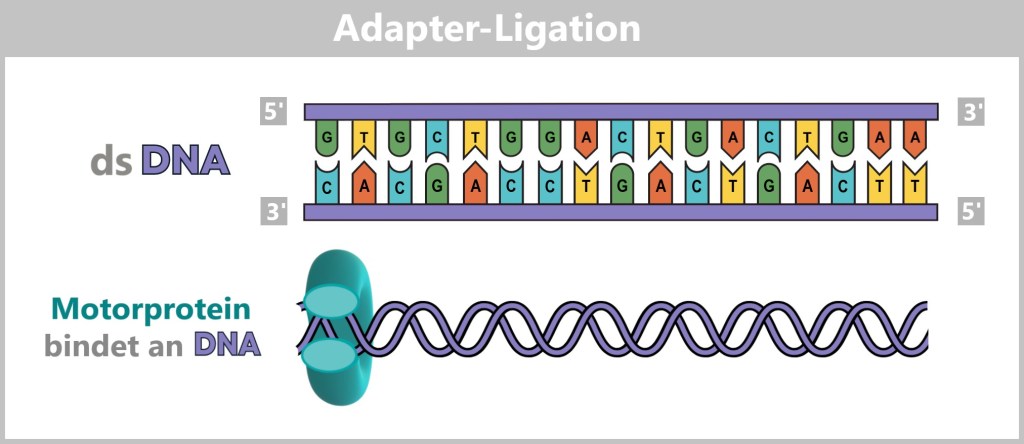
Damit das Motorprotein an die DNA binden kann, werden zuvor spezielle Adapter an die DNA-Enden angefügt. Diese Adapter sorgen dafür, dass das Motorprotein gezielt an einem Ende der DNA ansetzt. Zur besseren Übersicht zeigt die Abbildung nur die Bindung des Motorproteins, während die Adapter selbst nicht dargestellt sind. In der Regel bindet nur ein Motorprotein pro DNA-Molekül, da die Adapter so gestaltet sind, dass sie die Bindung an einem bevorzugten Ende erleichtern.
Woher stammt das Motorprotein?
Das Motorprotein, das bei der Oxford Nanopore Sequenzierung verwendet wird, ist ein natürlich vorkommendes Enzym, das aus Bakterien gewonnen wird. Es handelt sich um eine modifizierte Version eines Proteins, das ursprünglich von Bakterien wie E. coli oder anderen Mikroorganismen stammt. Diese Proteine haben in der Natur die Aufgabe, DNA zu entwinden und zu transportieren – eine Fähigkeit, die sich die Nanopore-Technologie zunutze macht.
Falls RNA sequenziert werden soll, kann optional eine Reverse Transkription durchgeführt werden, um RNA in DNA umzuwandeln. Die ONT-Technologie kann jedoch sowohl RNA als auch DNA direkt sequenzieren, Die direkte RNA-Sequenzierung kann spezifische Informationen über RNA-Modifikationen liefern.
2️⃣ Anlegen einer elektrischen Spannung
In beiden Kammern der Sequenzierzelle befinden sich geladene Teilchen (Ionen). Sobald eine Spannung zwischen der oberen (cis) und der unteren (trans) Kammer angelegt wird, fließen Ionen durch die Nanoporen. Dies erzeugt einen messbaren elektrischen Strom. Solange sich keine DNA in der Pore befindet, bleibt der Ionenfluss konstant, und es wird ein stabiler Strom gemessen.
3️⃣ Die DNA wird in die Sequenzierzelle gegeben
Die vorbereitete DNA-Probe, an die bereits Motorproteine gebunden sind, wird in die cis-Kammer (die obere Kammer der Sequenzierzelle) pipettiert. Durch Diffusion oder sanftes Mischen verteilt sich die DNA in der Lösung und gelangt in die Nähe der Membran, in der die Nanoporen eingebettet sind.
4️⃣ Andocken an die Nanopore
Das Motorprotein, das an die DNA gebunden ist, führt die DNA zur Nanopore und dockt gezielt daran an. Sobald die Verbindung hergestellt ist, beginnt das Motorprotein mit seiner Helicase-Aktivität: Es entwindet die doppelsträngige DNA (dsDNA) in zwei Einzelstränge. Ein Strang wird von der Nanopore erfasst und weiter durch sie hindurchgezogen. Dieser Prozess läuft direkt an der Pore ab und stellt sicher, dass die DNA präzise und gleichmäßig durch die Nanopore transportiert wird.
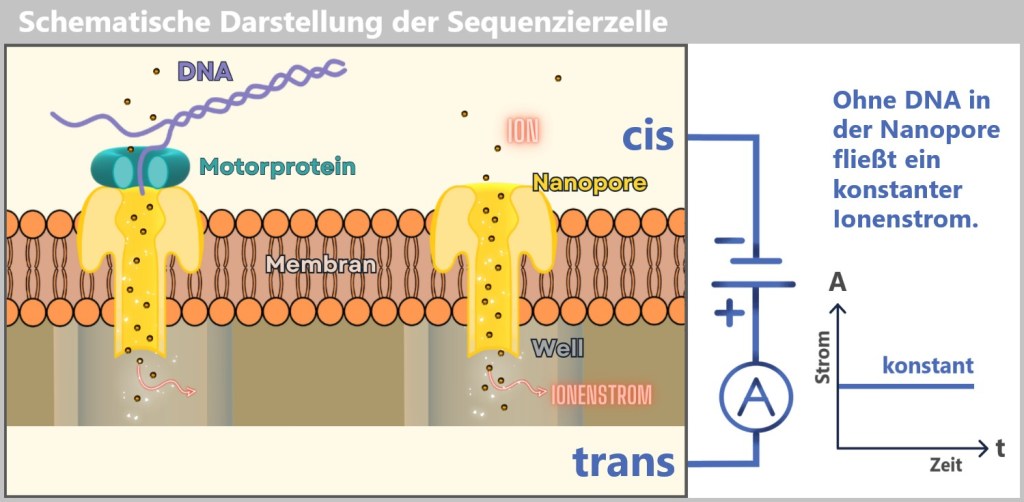
Die Sequenzierzelle besteht aus zwei Kammern: der cis-Kammer (oben) und der trans-Kammer (unten), getrennt durch eine Membran mit eingebetteten Nanoporen. Links hat ein DNA-Molekül mithilfe eines Motorproteins an eine Nanopore angedockt. Rechts ist eine leere Nanopore dargestellt, durch die ein konstanter Ionenstrom fließt.Die angelegte Spannung zwischen der negativen Elektrode (cis) und der positiven Elektrode (trans) treibt den Ionenfluss an. Der Ionenstrom wird im Well (kleiner Kanal im Chip) gemessen, wie im Strom/Zeit-Diagramm dargestellt. Solange keine DNA durch die Pore wandert, bleibt der Strom konstant.
5️⃣ Die DNA passiert die Nanopore – das Signal entsteht
Die DNA besitzt aufgrund ihrer negativ geladenen Phosphatgruppen eine elektrische Ladung. Die angelegte Spannung zwischen der cis-Kammer (negativ geladen) und der trans-Kammer (positiv geladen) bewirkt, dass die DNA durch die Nanopore gezogen wird.
Das Motorprotein spielt dabei eine entscheidende Rolle:
➤ Es steuert die DNA-Bewegung durch die Pore – langsam, gleichmäßig und stabil.
➤ So entsteht ein klar lesbares Signal für eine zuverlässige Signalmessung.
Zum Verständnis: Die DNA würde von Natur aus mit einer Geschwindigkeit von Millionen Basen pro Sekunde durch die Nanopore schießen. Das Motorprotein bremst diese Geschwindigkeit auf etwa 450 Basen pro Sekunde ab (abhängig vom Gerät und den Einstellungen). Dadurch wird die Sequenzierung mittels dieser Technologie überhaupt erst möglich.
Wenn die DNA durch die Pore wandert, beeinflusst sie den Ionenfluss – denn jede Base (A, T, G, C) hat eine einzigartige Größe und chemische Struktur. Diese Unterschiede verändern den Ionenstrom auf spezifische Weise. Diese Stromveränderungen werden von einer Elektrode gemessen, die direkt unter der Nanopore sitzt. Jeder Well (die winzigen Tunnel im Chip, auf denen die Nanoporen sitzen) hat eine fest zugeordnete Elektrode, die als Messgerät fungiert. Dadurch kann die Software die Signale präzise einer bestimmten Nanopore zuordnen – eine Voraussetzung für die korrekte Rekonstruktion der DNA-Sequenz.
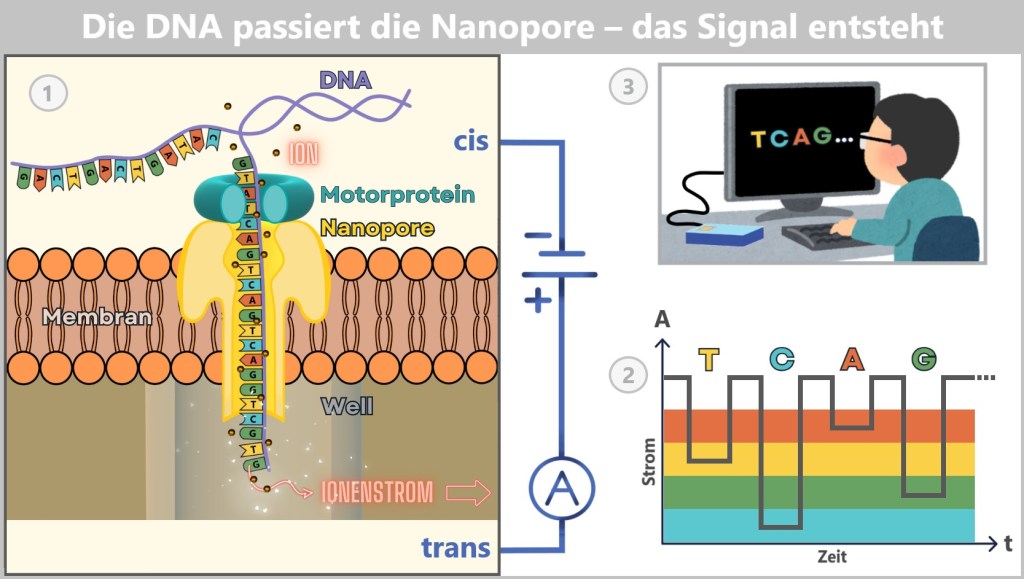
1) Ein Motorprotein zieht die DNA kontrolliert durch die Pore. Die negativ geladene DNA bewegt sich aufgrund der angelegten Spannung von der cis-Seite zur trans-Seite. Dabei beeinflussen die einzelnen Basen (A, T, G, C) den Ionenstrom auf spezifische Weise.
2) Ein Graph stellt die Veränderung des gemessenen Stroms über die Zeit dar. Jede Basen-Kombination erzeugt ein charakteristisches Signal, das durch Algorithmen entschlüsselt wird.
3) Ein Computer analysiert die elektrischen Signale und bestimmt daraus die Reihenfolge der Basen.
6️⃣ Das elektrische Signal wird aufgezeichnet
Während die DNA durch die Nanopore gezogen wird, befinden sich etwa 10–15 Basen gleichzeitig in der Pore. Jede dieser Basen beeinflusst den Ionenstrom unterschiedlich – das Signal ist also eine Überlagerung der Effekte mehrerer Basen.
Doch wie kann man trotzdem einzelne Basen unterscheiden?
Das funktioniert durch einen cleveren Algorithmus:
- Das Motorprotein bewegt die DNA schrittweise – etwa eine Base nach der anderen.
- Der gemessene Strom verändert sich dabei auf charakteristische Weise, abhängig von der Reihenfolge der Basen.
- Spezialisierte Software (Basecalling) nutzt maschinelles Lernen, um aus den überlappenden Signalen die genaue Sequenz zu errechnen.
- Das Modell „entwirrt“ die überlappenden Informationen und ordnet sie der richtigen Base an der entscheidenden Position zu.
Auf diese Weise entsteht aus den elektrischen Signalen nach und nach die vollständige DNA-Sequenz.
Ergebnis der Nanopore-Sequenzierung
Am Ende liefert die Oxford-Nanopore-Technologie die vollständige Abfolge der Basen in der untersuchten DNA- oder RNA-Probe. Diese Sequenz enthält detaillierte Informationen über die genetische Zusammensetzung, die Länge der Moleküle sowie mögliche Besonderheiten wie Mutationen oder strukturelle Veränderungen.
🚀 Daten in Echtzeit – ein echter Geschwindigkeitsvorteil
Der gesamte Prozess läuft live ab. Während die DNA oder RNA durch die Pore wandert, wird die Abfolge der Basen sofort erfasst und ausgewertet.
Zum Vergleich:
- Die klassische Sanger-Sequenzierung benötigt mehrere Stunden oder sogar Tage für eine Analyse.
- Die Illumina-Sequenzierung liefert Ergebnisse in der Regel in wenigen Stunden bis zu mehreren Tagen. Der genaue Zeitrahmen hängt von der Länge, Komplexität und dem Umfang der zu sequenzierenden Probe ab – kleinere Projekte können oft in wenigen Stunden abgeschlossen werden, während umfangreichere Analysen mehrere Tage benötigen.
- Die Oxford Nanopore Technologie (ONT) hingegen liefert in wenigen Minuten erste Ergebnisse.
Die generierten Sequenzdaten werden parallel verarbeitet und gespeichert. Sie stehen sofort für die weitere Analyse bereit.
Blick nach vorn: Sequenzierung 4.0 – was kommt als Nächstes?
Während die dritte Generation noch gefeiert wird, tüftelt die Forschung bereits an der Überholspur. Neue, aufkommende Technologien versprechen noch mehr: mehr Geschwindigkeit, höhere Genauigkeit und noch vielseitigere Einsatzmöglichkeiten.
Im nächsten Kapitel werfen wir einen flüchtigen Blick auf die Zukunft der Sequenzierung.
Emerging Technologies: Zukunft der Sequenzierung
Während das Nanopore Sequencing (ONT) bereits eine Revolution in der Sequenzierungstechnologie darstellt, gibt es vielversprechende Ansätze, die diese Methode noch weiterentwickeln könnten.
Transistor statt Pore
Ein spannendes Beispiel ist die FENT-Technologie (Field-Effect Nanopore Transistor), die DNA und andere Biopolymere mit einer neuartigen Transistor-Struktur analysiert, die extrem schnell und genau arbeitet. Diese Technologie könnte eine noch präzisere, schnellere und kostengünstigere Analyse von DNA und anderen Biopolymeren ermöglichen.
Das Video „FENT-Nanopore-Transistor-Erklärungsvideo“ gibt einen faszinierenden Einblick in diese Innovation.
Ob und wann diese Technologie für die Virusdiagnostik relevant wird, bleibt abzuwarten – doch sie zeigt eindrucksvoll, wie sich die Sequenzierungstechnologien weiterentwickeln.
Das nächste große Ding in der Genetik
Neben FENT könnten auch Methoden wie In Situ Sequencing (ISS), die Genexpression direkt in Geweben sichtbar macht, zukünftig eine Rolle spielen – etwa um virale Aktivität in Zellen zu erforschen.
Die Zukunft der Sequenzierung?
Sie wird uns umhauen (wortwörtlich!) – und wir sind live dabei!
4.6. Bioinformatische Analyse
Die rohen Sequenzdaten sind einfach eine lange Abfolge von Basen, z. B. „AGCUACGUA…“ bei einer RNA-Sequenz. Sequenzieren ist wie Buchstabieren – erst die Bioinformatik erzählt die Geschichte dahinter – „übersetzt“ diese Daten in Informationen. Aus dem Buchstabensalat (AGCUACGUA…) wird ein Steckbrief: Welches Virus ist das? Was kann es? Und wie gefährlich?
Warum Rohdaten chaotisch sind
💨 Technische Patzer: Wie ein verwackeltes Foto – manche Sequenzteile fehlen oder sind unscharf („Rauschen“).
❓ Rätselbasen („N“): Stellen, wo selbst die Maschine passen muss: „Könnte A, C oder U sein – keine Ahnung!“
🦠 Mutanten-WG: RNA-Viren wie Influenza sind Wohngemeinschaften aus Varianten (Mutantenwolken). Die Sequenzierung mischt alle zusammen – wie ein Smoothie aus 100 Früchten.
Was Bioinformatiker tun
Sie putzen, sortieren und puzzeln die Daten – bis klar wird:
- Wer ist hier? (Virusidentität)
- Was ist neu? (Mutationen)
- Wie reagieren? (Erstmal tief durchatmen!)
Das ist Detektivarbeit – nur mit mehr Computern und weniger Trenchcoats.
Vom Datenchaos zur Erkenntnis: Die 5 Schritte der Bioinformatik
1️⃣ Qualitätskontrolle: Das Datenpuzzle sortieren
2️⃣ Alignment & Assemblierung: Das große Puzzle-Spiel
3️⃣ Homologie-Suche: Will sehen!
4️⃣ Datenbankeintrag: Reif fürs Register?
5️⃣ Funktionelle Annotation: Das Puzzle erwacht zum Leben
1️⃣ Qualitätskontrolle: Das Datenpuzzle sortieren
Sequenzdaten sind wie ein Puzzle vom Flohmarkt: Einige Teile sind doppelt, einige fehlen, manche passen nicht und mitten im Bild ein Kaffeefleck. Was tun? – Bevor man das Ganze zusammensetzt, muss geordnet, geputzt und aussortiert werden. Hier beginnt die Arbeit der Bioinformatik:
🔍 FastQC (ein Software-Tool) – der kritische Buchprüfer:
Er blättert durch die Sequenzdaten wie durch ein altes Manuskript und markiert alle schmuddeligen Seiten – zu kurz, zu fehlerhaft, zu verdächtig.
✂️ Trimmomatic (ein Software-Tool) – der molekulare Rasenmäher:
Was kaputt, verfranst oder einfach nur überflüssig ist, wird rigoros abgeschnitten. Besonders an den Enden, wo Fehler gerne nisten.
🔄 Fehlerkorrektur – Demokratie auf molekularer Ebene:
Wenn neun von zehn Reads sagen: „Hier gehört ein A hin“, dann wird der einsame Buchstabe G einfach überstimmt. Auch DNA hat Mehrheitsentscheidungen.
Am Ende bleibt nur, was Qualität hat:
🔹 Kurze oder minderwertige Reads? Raus damit.
🔹 Einzelne Sequenzfehler? Korrigiert.
🔹 Insgesamt zu wenig gute Daten? Dann lieber nochmal sequenzieren.
Warum? Damit nachher niemand falsche Schlüsse aus einem Tippfehler zieht. Denn nur mit sauberen Daten geht’s jetzt ans eigentliche Detektivspiel: dem Zusammenbau des Genoms!
2️⃣ Alignment & Assemblierung: Das große Puzzle-Spiel
Moderne Sequenziermethoden liefern DNA- oder RNA-Schnipsel, als hätte jemand ein Puzzle explodieren lassen:
- Illumina produziert Millionen kurzer Reads.
- Nanopore liefert meterlange Fetzen (na gut – fast).
Doch egal ob klein oder groß: Am Ende müssen sie zusammengesetzt werden – mit Algorithmen, Logik und viel Rechenpower.
🦠📌 Methode 1: Alignment (bei bekannten Viren)
Wo passt dieses Teil hin?
Die bereinigten Sequenzdaten werden mit einer bekannten Referenzsequenz verglichen – wie Puzzleteile, die man auf ein Deckbild legt. So lassen sich selbst kleinste Abweichungen erkennen:
Was man findet:
🔹 Mutationen: Ein A statt G? Vielleicht macht das das Virus ansteckender.
🔹 Fehlstellen: Kleine Löcher im Genom – sogenannte Deletionen.
🔹 Extra-Teile: Unerwartete Einfügungen – Insertionen.
Auch wenn die Vorlage (Referenzsequenz) nur eine Variante ist, zeigt das Alignment die ganze Mutanten-WG in der Probe.
🦠❓ Methode 2: De-novo-Assembly (bei neuen Viren)
Ohne Vorlage puzzeln – wie ein Blindflug!
Gibt es keine bekannte Referenz, müssen die einzelnen Reads ohne Schablone zusammengesetzt werden. Der Trick: Überlappende Abschnitte zeigen, welche Teile zueinanderpassen. So entsteht – Stück für Stück – ein vollständiges Genom. Dieser Prozess ist fehleranfälliger als das Alignment, liefert aber eine erste Genomsequenz eines neu entdeckten Virus.
Der Unterschied:
Alignment ist wie IKEA-Möbel bauen – mit Anleitung.
De-novo-Assembly ist wie: „Hier sind Holzteile, viel Glück!“
3️⃣ Homologie-Suche: Will sehen!
Jetzt liegen die Karten auf dem Tisch. Der unbekannte DNA-Schnipsel wird ausgespielt – alle Daten sind gesammelt, alles ist vorbereitet. Jetzt heißt es: Will sehen!
Hier kommt BLAST ins Spiel – die große Suchmaschine für Gene. Sie vergleicht den genetischen Joker mit Millionen anderer Sequenzen in weltweiten Datenbanken. Und wenn irgendwo ein Verwandter existiert – BLAST findet ihn. Ob naher Cousin oder entfernter Urahn: Die Ähnlichkeiten im Code verraten, ob du einen alten Bekannten in der Hand hältst. Oder vielleicht ein völlig neues Virus.
BLAST zeigt:
🔹 den Grad der Übereinstimmung in Prozent
🔹 statistische Werte, die anzeigen, wie zuverlässig der Treffer ist
🔹 mögliche Verwandtschaften, selbst wenn die Ähnlichkeit nur entfernt ist
Wer es wirklich wissen will:
How to Use BLAST for Finding and Aligning DNA or Protein Sequences
🦠📌 Bei bekannten Viren: BLAST bestätigt die Ergebnisse des Alignments: Gehört der Stamm zu einer bereits beschriebenen Variante? Gibt es neue Mutationen? Beim Influenzavirus könnte BLAST z. B. zeigen, dass ein neuer Stamm zu 98 % mit dem H3N2-Stamm der letzten Grippesaison übereinstimmt.
🦠❓Bei unbekannten Viren: BLAST klärt, ob die neue Sequenz einer bekannten Virusfamilie zugeordnet werden kann – oder ob es sich um einen völlig neuen Vertreter handelt. Falls keine oder nur sehr entfernte Treffer auftauchen, sind weiterführende Analysen notwendig.
4️⃣ Datenbankeintrag: Reif fürs Register?
Jetzt wird’s offiziell. Die gesammelten Sequenzdaten stehen bereit – doch nicht jede neue Virusvariante schafft es gleich ins große wissenschaftliche Archiv. Erst wird geprüft: Reif fürs Register?
🦠📌 Bei bekannten Viren: Nach der Homologie-Suche geht’s in die Feinanalyse: Hat der Stamm was Spannendes zu bieten? Die Variantendetektion schaut ganz genau hin – wie ein Genetiker mit Vergrößerungsglas. Gibt es Mutationen, die dem Virus helfen, dem Immunsystem zu entkommen (Stichwort: Immunflucht)? Wenn ja – ab damit ins Register! Der neue Stamm wird in eine öffentliche Datenbank eingetragen und steht dann Forschern weltweit zur Verfügung.
🦠❓ Bei unbekannten Viren: Wenn BLAST meldet: „Kenne ich nicht!“, wird es spannend. Jetzt helfen weiterführende bioinformatische Werkzeuge – zum Beispiel die funktionelle Annotation, die fragt: Was könnte dieses Virus können? Stellt sich dabei heraus: Hier haben wir etwas wirklich Neues, dann wird der Fund in eine der großen internationalen Datenbanken eingetragen – inklusive Namen, genetischem Fingerabdruck und möglicher Herkunft.
Die wichtigsten Datenbanken für Virusgenome:
🌐 GenBank – der Allrounder:
eine riesige Bibliothek für genetische Sequenzen aller Lebensformen, einschließlich Viren.
🌐 Influenza Research Database (IRD) – Spezialarchiv für Influenzaviren,
inklusive praktischer Analysetools.
🌐 GISAID – Die Formel 1 unter den Datenbanken:
superschnell, vor allem für Influenzaviren und SARS-CoV-2.
🌐 VIRALzone – Eine Art Wikipedia für Virusfamilien:
mit Infos zu Genom, Aufbau und Replikation.
Was in diesen Datenbanken landet, wird Teil eines globalen Gedächtnisses.
5️⃣ Funktionelle Annotation: Das Puzzle erwacht zum Leben
Das Genom ist sequenziert – aber jetzt will man wissen: „Welche Gene machen was?
Die funktionelle Annotation versucht, den „Bauplan“ des Virus zu lesen – und herauszufinden, welche Teile wofür zuständig sind. Warum ist dieses Virus infektiöser als andere? Wie schafft es das Virus, dem Immunsystem zu entkommen? Und könnte es resistent gegen Medikamente sein?
Dazu durchsucht man das Genom nach bekannten Mustern. Gibt es Abschnitte, die an Gene erinnern, die man schon aus anderen Viren kennt? Stimmen bestimmte Sequenzen mit Enzymen überein, die beim Eindringen in die Zelle helfen? Oder mit Regionen, die die Oberfläche des Virus formen – jene Stellen, die das Immunsystem als erstes erkennt?
Auch räumliche Strukturen spielen eine Rolle. Moderne KI-Modelle wie AlphaFold können aus der Gensequenz ein 3D-Modell des entstehenden Proteins berechnen. So lässt sich vorhersagen, ob eine Mutation nur ein kleines Detail verändert – oder ob sie das ganze Verhalten des Virus beeinflusst.
Das ist besonders wichtig, wenn es um:
- Ansteckung geht: Eine Mutation in einem Oberflächenprotein könnte das Virus „klebriger“ machen – es haftet besser an Zellen.
- Immunflucht: Kleine Veränderungen an den „Erkennungsstellen“ reichen, damit Antikörper das Virus übersehen.
- Medikamentenresistenz: Manche Mutationen schalten die Zielstruktur eines Wirkstoffs einfach aus – und machen ihn damit wirkungslos.
Funktionelle Annotation ist wie die Gebrauchsanleitung eines Virus – nur ohne freundliche Warnhinweise.
💡 Zusammenfassung: Viren-Cracking für Anfänger
Sequenzieren: Buchstabiere das Virus-ABC.
Qualitätskontrolle: Räum den Datenmüll weg.
Alignment & Assembly: Puzzle die Schnipsel zusammen – mit oder ohne Vorlage.
BLAST: Google nach genetischen Verwandten.
Datenbank: Gib der Welt deine Entdeckung – wenn sie was taugt.
Annotation: Lies zwischen den Genen – was kann dieses Virus wirklich?
Klassifizierung der Viren basierend auf ihrem genetischen Material
Viren können auf verschiedene Arten klassifiziert werden – zum Beispiel nach ihrer Form, ihren Wirten, ihrer Übertragungsweise oder ihrem genetischen Material.
Eine der bekanntesten und am häufigsten verwendeten Genom-Klassifikationen ist die Baltimore-Klassifikation, die 1971 von David Baltimore eingeführt wurde. Dieses System teilt Viren basierend auf einer Kombination aus den folgenden Merkmalen ein:
- Art des genetischen Materials (DNA oder RNA),
- Strängigkeit (einzelsträngig oder doppelsträngig),
- Richtung (positiv oder negativ) und
- Replikationsmechanismus (wie mRNA hergestellt wird).
Das Ziel aller Viren ist es, mRNA zu produzieren, denn nur mit mRNA können sie die Proteine herstellen, aus denen sie bestehen, und sich vermehren. Die Baltimore-Klassifikation umfasst sieben Gruppen, die zeigen, wie unterschiedlich Viren dieses Ziel erreichen – von direkter Nutzung ihrer RNA bis hin zu komplexen Umwegen über DNA. Hier ein Überblick:
Gruppe I: Doppelsträngige DNA-Viren (dsDNA)
Die Herstellung von mRNA erfolgt durch die direkte Transkription der DNA mittels einer RNA-Polymerase, die entweder zellulär oder viral sein kann.
(Beispiele: Adenoviren, Herpesviren und Pockenviren)
Gruppe II: Einzelsträngige DNA-Viren (ssDNA)
Die einzelsträngige DNA wird zunächst in doppelsträngige DNA umgeschrieben. Anschließend erfolgt die Transkription zur Herstellung von mRNA.
(Beispiele: Parvoviren)
Gruppe III: Doppelsträngige RNA-Viren (dsRNA)
Die Herstellung der mRNA erfolgt durch die Transkription der RNA-Stränge mittels einer viralen RNA-abhängigen RNA-Polymerase.
(Beispiele: Reoviren)
Gruppe IV: (+) Einzelsträngige RNA-Viren [(+)ssRNA]
Die virale RNA dient direkt als mRNA, da sie die gleiche Richtung wie die mRNA hat (5′ → 3′).
(Beispiele: Coronaviren, Polioviren)
Gruppe V: (–) Einzelsträngige RNA-Viren [(–)ssRNA]
Die RNA ist in entgegengesetzter Richtung aufgebaut (3′ → 5′) und muss durch eine virale RNA-abhängige RNA-Polymerase in (+)RNA umgeschrieben werden, die dann als mRNA dient.
(Beispiele: Influenzaviren, Rabies-Virus)
Gruppe VI: Retroviren [(+)ssRNA mit DNA-Zwischenschritt]
Die Reverse Transkriptase (RT) wandelt RNA in DNA um, die ins Wirtsgenom integriert wird. Die mRNA wird von der integrierten DNA durch zelluläre Mechanismen hergestellt.
(Beispiele: HIV)
Retroviren wie HIV nutzen einen einzigartigen Replikationsmechanismus: Die virale RNA wird durch die Reverse Transkriptase (RT) in einen komplementären DNA-Strang (cDNA) umgeschrieben. Die ursprüngliche RNA wird abgebaut, und ein zweiter DNA-Strang wird synthetisiert. Die entstandene dsDNA wird in das Wirtsgenom integriert. Dieser Prozess macht Retroviren besonders hartnäckig, da sie in einer latenten Phase verweilen und jederzeit reaktiviert werden können.
Gruppe VII: Doppelsträngige DNA-Viren mit RNA-Zwischenschritt (dsDNA-RT)
Die virale DNA wird im Zellkern repariert und dient als Vorlage für die Synthese von mRNA und prägenomischer RNA. Die prägenomische RNA wird durch die Reverse Transkriptase wieder in DNA umgeschrieben.
(Beispiele: Hepatitis-B-Virus)
Das Hepatitis-B-Virus besitzt ein unvollständig doppelsträngiges DNA-Genom. Im Zellkern der Wirtszelle wird diese DNA repariert, bevor mRNA und prägenomische RNA transkribiert werden. Die prägenomische RNA dient als Vorlage für die Bildung neuer Viruspartikel.

Die Viren werden nach ihrer Genomstruktur und ihrer Strategie zur mRNA-Synthese kategorisiert. Doppelsträngige (ds) und einzelsträngige (ss) Genome werden unterschieden, die Polarität der RNA wird durch „+“ (positiv) und „-“ (negativ) Stränge gekennzeichnet, und der Mechanismus der reversen Transkription ist durch „RT“ hervorgehoben. Am Ende steht die mRNA, die alle Viren benötigen, um ihre Proteine zu produzieren.

5. Gibt es Viren wirklich?
In einer Welt, in der man alles infrage stellen kann – wie beweist man, dass etwas existiert?
Manche sagen: Viren seien bloß Artefakte, Laborkonstrukte, Zelltrümmer ohne eigene Identität. Doch es gibt Merkmale, die sich nicht wegdiskutieren lassen:
Viren haben etwas, das nur sie haben
Viele Viren besitzen konservierte Genomabschnitte, die für eindeutig virale Funktionen kodieren:
- Kapsidproteine, mit denen sie ihre Erbinformation verpacken,
- RNA-abhängige Polymerasen, mit denen sie ihr Erbgut vervielfältigen,
- Integrasen, mit denen sie virale Gene ins Wirtsgenom schleusen (z. B. bei Retroviren wie HIV),
- Proteasen, mit denen sie Virus-Proteine zurechtschneiden, damit sie funktionieren.
Das Kapsid – das Erkennungszeichen des Virus
Was macht ein Virus zu einem Virus? Nicht nur sein parasitärer Lebensstil – sondern vor allem sein Aufbau.
Fast alle Viren verfügen über ein Kapsid – eine proteinhaltige Hülle, die ihr Erbgut schützt und gleichzeitig zum Eindringen in Wirtszellen dient. Ob ikosaedrisch oder helikal: Das Kapsid ist funktional wie evolutionär DAS zentrale Merkmal des Virus.
Es schützt, es transportiert, es organisiert – und es ist einzigartig virusspezifisch.
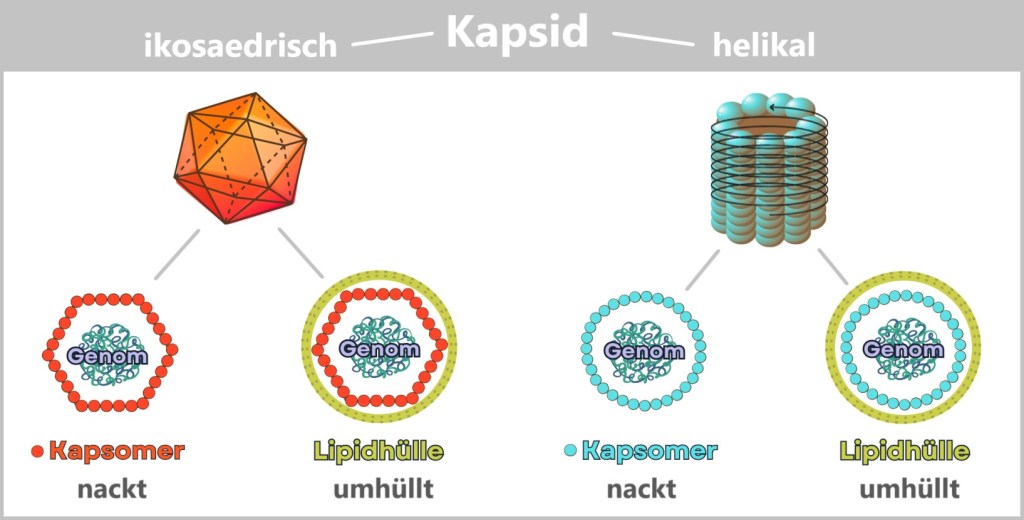
Das Kapsid ist die äußere Proteinhülle eines Virus, die dessen genetisches Material schützt und dessen äußere Gestalt bestimmt. Es besteht aus vielen identischen Bausteinen (Kapsomeren), die sich zu symmetrischen Strukturen zusammensetzen. Die häufigsten Kapsidformen sind ikosaedrisch (20-flächig) und helikal (schraubenförmig).
Zusätzliche Bestandteile wie eine Lipidhülle – die von der Wirtszelle stammt – können die äußere Form verändern. So wirken viele behüllte Viren „sphärisch“, obwohl das darunterliegende Kapsid meist ikosaedrisch ist. In einigen Fällen, wie bei Bakteriophagen, weicht der Aufbau deutlich ab: Man spricht dann von einer komplexen Morphologie, etwa mit einem ikosaedrischen Kopf und einem helikalen Schwanz. Diese morphologischen Unterschiede bilden die Grundlage zur strukturellen Klassifikation von Viren.
Virale Hallmark-Gene – Signaturen jenseits der Zellwelt
Die Gene, die für Kapsidproteine codieren, gehören zu den sogenannten Hallmark-Genen. Sie sind:
- hoch konserviert innerhalb der Viruswelt,
- funktional essenziell für den viralen Lebenszyklus,
- und nicht in zellulären Organismen zu finden.
Viele dieser Proteine weisen eine ganz besondere Form auf: die Jelly-Roll-Faltung. Diese dreidimensionale Struktur erinnert an eine aufgerollte Biskuitrolle und verleiht dem Kapsidprotein eine bemerkenswerte Stabilität. Man findet sie bei den unterschiedlichsten Viren – von jenen, die Menschen infizieren, bis zu Viren, die nur in uralten Tiefsee-Mikroben vorkommen. Ihr auffälliger Aufbau ist so charakteristisch, dass Forscher selbst neuartige Viren allein auf Grundlage dieser genetischen Signatur erkennen können.
Wer sie einmal gesehen hat, erkennt sie wieder – wie ein Symbol, das sich durch die Tiefen der Evolution zieht – eine evolutionäre Handschrift, lesbar für jeden, der bereit ist, hinzusehen.
Wie z. B. die Arbeiten von Koonin et al. zeigen: Trotz aller Vielfalt tragen Viren einen gemeinsamen, alten genetischen Kern in sich – als würden sie das Wappen ihrer Urahnen in sich tragen.
Fazit: Viren-spezifische Genomabschnitte sind ein Alleinstellungsmerkmal!
Viren entziehen sich einfachen Definitionen. Sie sind weder klassische Lebewesen noch bloße Molekülhaufen. Aber sie tragen ein Set an Eigenschaften in sich, das sie unterscheidbar macht – in ihrer Form, in ihrer Funktion, in ihrer Geschichte.
Wenn etwas seit Milliarden Jahren seine eigene genetische Spur zieht – dann ist es real. Auch wenn es kein Eigenleben führt. Auch wenn wir es nicht sehen können.

Fragen & Antworten
🟡 „Sind Viren nur Exosomen mit PR-Abteilung?“
Nein. Exosomen sind biologischer Verpackungsmüll. Viren bringen einen eigenen Bauplan mit – und einen Transportbehälter in perfekter Geometrie.
Wer Exosomen mit Viren verwechselt, verwechselt auch Tupperdosen mit Raumschiffen.
🔬 „Aber man hat sie doch nie richtig gesehen!“
Doch. Elektronenmikroskope liefern Bilder, Kryo-Tomographie sogar 3D-Videos in Nanometer-Auflösung.
Unsichtbar? Nur ohne Strom.
🧬 „Die RNA ist doch nur zufälliger Zellrest?“
Nein. Virale RNA/DNA ist einzigartig, funktional, präzise organisiert – und weltweit reproduzierbar.
Zufall? Dann wäre ein Uhrwerk auch nur etwas Metall – mit Teamgeist und guter Laune.
📦 „Dieses Kapsid – kann das nicht einfach so entstehen?“
Kaum. Diese Nanoboxen mit perfekter Symmetrie gibt’s nur bei Viren.
Ein Fußball formt sich auch nicht zufällig im Rasen.
🌍 „Vielleicht ist das alles nur ein Messartefakt?“
Dann ist es ein erstaunlich globales. Alle Labore finden die gleichen Sequenzen, Strukturen, Eigenschaften.
Viren kennen keine Laborgrenzen. Nur Reproduzierbarkeit.
Am Ende bleibt die Erkenntnis:
Viren sind keine Fiktion, kein Artefakt, kein Zufallsprodukt. Sie sind das am besten kartierte Unbehagen der Biologie – real, reproduzierbar, überall.
Wer Viren für Zellmüll hält, glaubt auch,
ein Tesla sei ein besonders ambitionierter Einkaufswagen.
Quellen und Links, die die Isolation und Sequenzierung von Viren dokumentieren:
National Center for Biotechnology Information (NCBI) Virus Database:
NCBI Virus Database
Das NCBI beherbergt eine umfassende Datenbank von Virusgenomen, die von Wissenschaftlern weltweit sequenziert wurden. Hier kannst du Tausende von Virusgenomen einsehen, die isoliert und sequenziert wurden.
GISAID (Global Initiative on Sharing Avian Influenza Data):
GISAID
GISAID ist eine Plattform, die insbesondere für den Austausch von Sequenzdaten von Influenza- und SARS-CoV-2-Viren genutzt wird. Hier sind detaillierte Daten zur Sequenzierung und Herkunft dieser Viren abrufbar.
European Nucleotide Archive (ENA): ENA Browser
Die ENA bietet Zugang zu Sequenzdaten von Viren, die in Laboren weltweit isoliert wurden. Diese Datenbank wird von der European Bioinformatics Institute (EBI) betrieben und enthält umfangreiche Informationen zu viralen Genomen.
PubMed – Wissenschaftliche Artikel zur Virusisolierung und -sequenzierung:
Beispielhafte Suchanfrage: „Virus Isolation and Sequencing“
PubMed ist eine Suchmaschine für wissenschaftliche Literatur. Hier kannst du nach spezifischen Studien zur Isolation und Sequenzierung von Viren suchen.

6. Woher kommen Viren?
Diese Fragen führen an die Grenzen unseres Wissens. Sie berühren nicht nur die Ursprünge der Viren, sondern die Grundlagen des Lebens selbst. Bevor wir den möglichen Szenarien ihrer Entstehung folgen, lohnt sich ein Blick auf das große Ganze: den „Baum des Lebens“ – so, wie ihn die moderne Evolutionsforschung heute zeichnet.
6.1. Der Baum des Lebens
Alle bekannten Lebensformen – vom Bakterium bis zum Blauwal – teilen sich einen gemeinsamen Ursprung: LUCA, den Last Universal Common Ancestor. Er war nicht der erste Organismus überhaupt, sondern der letzte gemeinsame Vorfahr aller heute lebenden zellulären Lebensformen – Bakterien, Archaeen und Eukaryoten. Ein evolutionärer Knotenpunkt, kein Startpunkt.
Was wissen wir über LUCA? Wahrscheinlich lebte er vor rund 3,5 bis 4 Milliarden Jahren, in einer warmen, noch jungen Welt. Er war zellbasiert, besaß bereits DNA, RNA und Proteine, konnte Energie umwandeln – aber war noch weit entfernt von der Komplexität moderner Zellen. Ein Prototyp des Lebens, von dem sich alles weitere verzweigte.

LUCA (letzter universeller gemeinsamer Vorfahre) verzweigt sich in die Domänen Bakterien (z. B. Terrabakterien, Proteobakterien) und Archaeen (z. B. Euryarchaeota, Asgard). Durch die Verschmelzung eines Proteobakteriums mit einem ASGARD-Archeen (Endosymbiose) entsteht FECA (erster eukaryotischer gemeinsamer Vorfahre), der sich zu LECA (letzter eukaryotischer gemeinsamer Vorfahre) entwickelt, aus dem die Eukaryoten (Tiere, Pflanzen, Pilze) hervorgehen. Eine zweite Endosymbiose mit Cyanobakterien führt zur Entstehung der Chloroplasten in Pflanzen. Rechts veranschaulicht eine Zeitleiste die Entstehung des Lebens, von der Erde (ca. 4,5 Milliarden Jahre vor heute) über die Große Sauerstoffkatastrophe (ca. 2,4 Milliarden Jahre vor heute) bis zur Kambrischen Explosion (ca. 540 Millionen Jahre vor heute).
Doch während sich der Baum des Lebens mit Ästen, Zweigen und Blättern füllte, bleibt eine Frage offen: Wo sitzen die Viren in diesem Bild – oder gehören sie überhaupt dazu?
Denn anders als Bakterien oder Eukaryoten haben Viren keine Zellstruktur, keinen eigenen Stoffwechsel und hinterlassen keine Fossilien. Manche Wissenschaftler sehen sie als „verlorene Zweige“ des Lebensbaums, andere als uralte Vorformen, die vielleicht vor LUCA existierten.
Um diese Frage zu klären, müssen wir zurück an den Anfang – oder besser: zu den möglichen Anfängen.
Wo also stehen Viren in diesem Stammbaum des Lebens?
Sind sie Spätankömmlinge, entstanden aus entgleisten Genen zellulärer Organismen? Oder gehören sie zu den Urformen des Lebens – vielleicht sogar älter als LUCA selbst?
Die Wissenschaft kennt (noch) keine eindeutige Antwort. Doch einige Hypothesen versuchen, dem Ursprung der Viren auf die Spur zu kommen.
6.2. Die Haupthypothesen zur Herkunft von Viren
💡Hinweis: Die folgenden Entstehungshypothesen setzen ein grundlegendes Verständnis der sogenannten RNA-Welt voraus – einer frühen Phase der Erdgeschichte, in der das Leben noch aus einfachen, selbstreplizierenden RNA-Molekülen bestand. Um dich in diese fremde Epoche einzufühlen und die Hypothesen besser einordnen zu können, empfehlen wir dir den Artikel „Das erste Flüstern des Lebens“. Er erzählt in poetischer Sprache von den Anfängen biologischer Ordnung – lange vor Zellen, Proteinen und DNA.
6.2.1. Hypothesen in einer zellulären Welt
Diese Hypothesen setzen existierende Zellen voraus, aus denen Viren hervorgehen.
6.2.1.a) Progressive Hypothese – Viren als entflohene Gene
6.2.1.b) Regressive Hypothese – Viren als geschrumpfte Zellwesen
6.2.2. Hypothesen in der prä-zellulären RNA-Welt
Diese Hypothesen verorten die Entstehung von virenähnlichen Strukturen in einer Zeit vor oder während der Bildung erster Zellen, in der RNA-Welt, wo selbstreplizierende Moleküle dominieren.
6.2.2.a) Virus-first-Hypothese – Viren waren zuerst da
6.2.2.b) Co-Evolution-Hypothese – Viren und Zellen entstanden gemeinsam

6.2.1. Hypothesen in einer zellulären Welt
Die sogenannte Cell-first-Hypothese geht davon aus, dass zelluläre Lebensformen – wie LUCA – bereits existierten, bevor Viren entstanden. Innerhalb dieses Ansatzes lassen sich zwei Hauptszenarien unterscheiden:
a) Die Progressive Hypothese: Viren stammen demnach von genetischem Material ab, das aus Zellen „entkommen“ ist – quasi entflohene Gene, die sich verselbstständigt und einen eigenen evolutionären Weg eingeschlagen haben.
b) Die Regressive Hypothese: Nach dieser Vorstellung waren Viren ursprünglich vollständige Zellen, die sich im Laufe der Evolution zunehmend reduziert haben – bis nur noch die nötigsten Funktionen für das parasitäre Leben übrigblieben. Eine Art evolutionärer Rückbau.
Beiden Varianten liegt ein gemeinsamer Gedanke zugrunde: Viren sind kein eigenständiger Ursprung des Lebens, sondern ein evolutionäres Nebenprodukt zellulärer Organismen – durch Verlust oder Entkopplung.
a) Die Progressive Hypothese – Viren als entflohene Gene
Die progressive Hypothese – auch Escape-Hypothese – meint, dass Viren aus genetischem Material hervorgingen, das einst Teil zellulärer Organismen war. Diese „entflohenen Gene“ entwickelten sich im Laufe der Evolution zu eigenständigen, infektiösen Einheiten – losgelöst vom ursprünglichen Zellverband.
Wie könnte das passiert sein?
In lebenden Zellen gibt es kleine, bewegliche DNA- oder RNA-Stücke, die sich innerhalb des Genoms frei bewegen können – sozusagen „springende Gene“. Diese Elemente, zu denen Plasmide (ringförmige DNA-Moleküle) oder Transposons (DNA-Abschnitte, die ihre Position im Erbgut ändern können) gehören, haben die Fähigkeit, sich unabhängig zu vervielfältigen und sogar zwischen Organismen auszutauschen.
Die Hypothese besagt, dass sich einige dieser genetischen Elemente so weit weiterentwickelten, dass sie eines Tages in der Lage waren, sich eigenständig zu vermehren. Sie entkamen aus der Zellkontrolle, entwickelten Schutzmechanismen wie eine Hülle – das Kapsid, das ihre Erbinformation schützt – und wurden so zu den ersten Viren.

nach der Viren aus ursprünglich zelleigenen Genen hervorgingen, die sich allmählich verselbstständigten.
Protozelle: Eine frühe, einfache Zellform mit genetischem Material: Großer DNA-Ring, Ribosomen und kleineren DNA-Ringen.
Abspaltung von genetischem Material: Bewegliche genetische Elemente – z. B. Plasmide oder sogenannte Transposons („springende Gene“) – lösen sich von der Zelle ab. Diese könnten erste Schritte zur Unabhängigkeit gemacht haben.
Vesikelbildung: Das abgespaltene genetische Material wird in kleine Membranbläschen (Vesikel) eingeschlossen, was Schutz und Mobilität bietet.
Entstehung eines Virus: Im Laufe der Evolution entstehen daraus funktionale Viren – mit einem schützenden Proteinmantel (Kapsid, orange). Wie genau sich dieser entwickelte, bleibt unklar („Kapsid-Rätsel?“).
✅ Argumente für diese Hypothese
➤ Ähnlichkeit mit mobilen genetischen Elementen: Einige virale Gene ähneln stark den „springenden Genen“ innerhalb von Zellen. Besonders Retroviren, wie das HI-Virus, nutzen einen Mechanismus, der dem von Transposons ähnelt: Sie schreiben ihre RNA in DNA um und integrieren sie ins Erbgut ihrer Wirte.
➤ Viren tauschen Gene mit ihren Wirten aus: Forscher haben in Virengenomen Gene entdeckt, die offensichtlich aus zellulären Organismen stammen. Das könnte bedeuten, dass Viren einst aus Zellen hervorgingen und im Laufe der Zeit genetisches Material aufnahmen und weiterentwickelten.
➤ Erklärung für die Vielfalt der Viren: Da verschiedene „springende Gene“ aus unterschiedlichen Zellen entkommen sein könnten, erklärt diese Hypothese, warum es so viele verschiedene Viren mit unterschiedlichen Eigenschaften gibt.
❌ Schwächen der Hypothese
➤ Kapsidproteine sind extrem alt: Einige Schlüsselproteine, die für den Aufbau der Virushülle (Kapsid) verantwortlich sind, zeigen keine direkte Verwandtschaft mit Zellproteinen. Es bleibt unklar, wie entflohene Gene diese komplexen Strukturen entwickeln konnten. Die Evolution dieser Schlüsselproteine reicht so weit zurück, dass sie möglicherweise schon vor der Entstehung der ersten zellulären Lebensformen existierten. Eine Studie von Krupovic & Koonin (2017) legt nahe, dass Kapsidgene sich unabhängig entwickelt haben könnten, bevor es moderne Zellen gab.
➤ Viren haben einzigartige Enzyme, die älter als LUCA sein könnten: Manche Viren enthalten RNA-Polymerasen, die sich stark von denen in Zellen unterscheiden. Diese Enzyme sind so eigenständig, dass sie möglicherweise aus einer Zeit stammen, in der es noch keine heutigen Zellmechanismen gab. Einige Forscherende vermuten daher, dass Viren direkte Überbleibsel aus der RNA-Welt sein könnten – einer Phase der frühen Evolution, in der das Leben noch nicht auf DNA, sondern auf RNA basierte.
➤ Viren in allen drei Domänen des Lebens: Viren sind überall – sie infizieren Bakterien, Archaeen und Eukaryoten. Wenn sie erst nach der Entstehung der ersten Zellen aus diesen hervorgegangen wären, müsste man erwarten, dass sie stärker auf eine bestimmte Zelllinie beschränkt sind. Ihre universelle Verbreitung legt nahe, dass sie bereits existierten, bevor sich Bakterien, Archaeen und Eukaryoten aufspalteten.
➤ Viren teilen sich keine gemeinsame Abstammung mit zellulärem Leben:Während Bakterien, Archaeen und Eukaryoten alle auf LUCA zurückgeführt werden können, gibt es für Viren keine gemeinsame Linie. Sie scheinen also nicht einfach ein „Seitentrieb“ eines Zellstammbaums zu sein, sondern eine sehr alte, parallele Entwicklung.
⬇️ Fazit
Die Cell-first-Hypothese betrachtet Viren als Rebellen des zellulären Lebens – ursprünglich harmlose genetische Passagiere, die sich verselbstständigten und einen eigenen evolutionären Weg einschlugen. Doch sie setzt voraus, dass es bereits komplexe Zellen gab, bevor Viren entstanden.
b) Die Regressive Hypothese – Viren als geschrumpfte Zellwesen
Die Regressions- oder Reduktionshypothese sieht Viren nicht als abgespaltene Gen-Bruchstücke, sondern frühere Zellen, die sich im Laufe der Evolution immer weiter reduziert wurden – bis sie ihre Selbstständigkeit verloren und zu „Genpaketen“ wurden, die auf andere Zellen angewiesen sind.
Wie könnte das passiert sein?
Ein früher, komplexer, zellulärer Organismus (z. B. ein Parasit) entwickelte eine immer engere Abhängigkeit zu seinem Wirt. Mit der Zeit verlor er überflüssige Gene – z. B. für Stoffwechsel, Zellteilung, Zellmembran – bis er nicht mehr selbstständig leben konnte. Am Ende blieb nur ein winziger Rest: die Gene zur Vermehrung (DNA/RNA), verpackt in einer Hülle (Kapsid) – ein Virus.
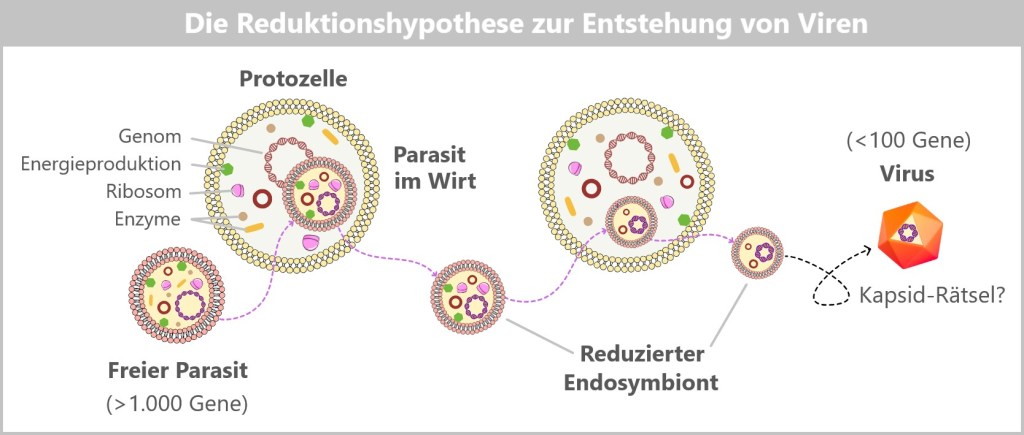
Vom reisenden Zellparasit zum ultraleichten Virus
Ein freilebender Parasit (z. B. ein kleines Bakterium) befällt eine andere Zelle – so wie auch heute manche Bakterien andere Zellen parasitieren (z. B. Rickettsien oder Chlamydien). Warum? Vermutlich bot die Protozelle eine sichere Umgebung. Vielleicht hatte der Wirt Zugang zu Ressourcen, die der Parasit nicht leicht bekam – etwa Energie, Enzyme oder Nukleotide – eine Art molekulares All-inclusive-Resort.
Möglicherweise begann alles sogar symbiotisch – ähnlich wie bei den Mitochondrien. Erst später wurde diese Beziehung einseitig ausgenutzt – sie wurde parasitär.
Der Parasit lebte zunächst noch eigenständig. Er konnte die Wirtszelle verlassen oder sich in ihr vermehren. Doch nach und nach brauchte er immer weniger eigene Gene – der Wirt lieferte schließlich alles, was er benötigte. Also reduzierte der Parasit sein Gepäck – Schritt für Schritt.
Im Laufe der Zeit verlor er durch Mutationen oder Selektion viele seiner ursprünglichen Gene:
➤ Er stellte keine eigenen Proteine mehr her.
➤ Er verlor die Fähigkeit zur Energiegewinnung.
➤ Schließlich sogar die Gene zur Zellteilung.
Am Ende bleibt nur noch sein Genom – verpackt in einen cleveren „Koffer“: das Kapsid.
Genau hier entsteht das Virus: kein eigenständiges Lebewesen mehr im klassischen Sinn, sondern ein ultraleichter „Reisender“, der sich nur noch mit Hilfe eines Wirts vermehren kann.
✅ Argumente für diese Hypothese
➤ Parallelen zu intrazellulären Parasiten: Einige heutige Mikroorganismen, etwa Rickettsien oder Chlamydien, können sich nur innerhalb anderer Zellen vermehren – genau wie Viren. Und: Ihre Genome sind stark reduziert. Das zeigt, dass Zellen durch Parasitismus tatsächlich drastisch „verschlanken“ können.
➤ Große DNA-Viren als „Übergangsformen“: Einige Riesenviren (z. B. Mimivirus, Pandoravirus) haben unglaublich große Genome – größer als manche Bakterien – und enthalten Gene, die man eher in echten Zellen erwarten würde (z. B. DNA-Reparaturenzyme). Diese Viren wirken wie evolutionäre Zwischenstufen zwischen echten Zellen und typischen Viren.
➤ Verlust statt Entstehung: Evolution bedeutet nicht nur „Mehr“, sondern oft auch „Weniger“. Besonders im parasitischen Kontext verlieren Organismen oft Funktionen – genau wie hier angenommen.
❌ Schwächen der Hypothese
➤ Ursprung bleibt unklar: Auch wenn die Hypothese erklärt, wie aus einer Zelle ein Virus werden könnte – sie sagt wenig darüber, wann und unter welchen Bedingungen das passiert wäre.
➤ Keine gemeinsame Abstammung: Die Vielfalt der Viren spricht eher gegen eine gemeinsame „Urzelle“ aller Viren. Wenn alle Viren aus regressiven Zellen stammen, müssten sie sich trotzdem mehrfach unabhängig voneinander zurückentwickelt haben. Manche Forschende nehmen daher an: Regressive Evolution ist nur eine von mehreren Ursprüngen.
➤ Gilt eher für große DNA-Viren: Diese Hypothese passt gut auf große DNA-Viren – aber weniger auf sehr einfache RNA-Viren, die keinerlei zellähnliche Strukturen oder Gene enthalten.
➤ Kapsidproteine zeigen keine direkte Ähnlichkeit: Zellen – selbst extrem reduzierte parasitische – haben keine Kapside. Das ist eine virenspezifische Struktur. Wenn sich ein Virus also durch Reduktion aus einer Zelle entwickelt hat, stellt sich die Frage: Wie konnte eine Struktur wie das Kapsid entstehen, die es in Zellen gar nicht gibt? Sie scheinen also neu entstanden zu sein – und das passt schwer in eine Theorie, die auf „Verlust“ und „Reduktion“ basiert.
➤ Energielücke: Selbst degenerierte Parasiten (z. B. Mycoplasma) behalten Stoffwechselgene – Viren haben gar keine.
⬇️ Fazit
Die Regressive Hypothese zeigt Viren nicht als „flüchtige Genfragmente“ – sondern als Miniaturausgabe ehemaliger Zellen. Besonders bei großen DNA-Viren ist diese Vorstellung plausibel: Sie könnten einst vollwertige zelluläre Parasiten gewesen sein, die durch Anpassung ihre Unabhängigkeit verloren haben. Sie wären dann gewissermaßen das biologische Gegenteil von Evolution zur Komplexität – eine Rückentwicklung zum Kern des Überlebens: Replikation. Die Hypothese scheitert aber an der Vielfalt einfacher Viren.
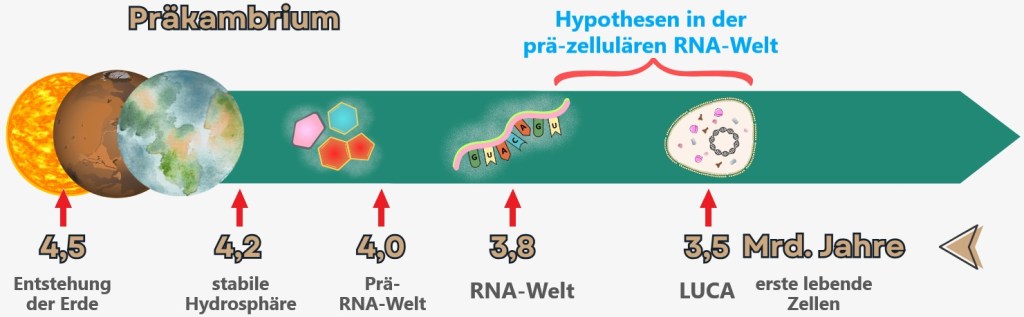
6.2.2. Hypothesen in der prä-zellulären RNA-Welt
In der Ursuppe der frühen Erde regte sich zum ersten Mal so etwas wie Leben – zart, flüchtig und doch folgenschwer. Moleküle entstanden, die nicht nur waren, sondern handelten: Sie schnitten, verbanden, kopierten sich selbst. In winzigen Lipidblasen, die kamen und gingen, wuchs ein Netzwerk aus kooperierenden und konkurrierenden Ribozymen (mehr dazu in „Das erste Flüstern des Lebens“).
Es war kein Leben im heutigen Sinn. Es war ein Spiel von Möglichkeiten. Aus diesem Netzwerk gingen zwei Entwicklungslinien hervor:
① Die stabilen, strukturverliebten Replikatoren, aus denen die ersten Zellen wurden.
② Und die freien, reduzierten Replikatoren, die als Proto-Viren gelten könnten – beweglich, anpassungsfähig, parasitär.
Der frühe Parasitismus – war weniger Infektion als Interaktion – eher ein Tanz zwischen Nutzen und Ausgenutztwerden als echter Wirt-Befall. Zwei Hypothesen beschreiben diesen Ursprung:
a) Virus-first-Hypothese – Viren waren zuerst da
b) Co-Evolution-Hypothese – Viren und Zellen entstanden gemeinsam
a) Virus-first-Hypothese – Viren waren zuerst da
Was war zuerst – der Wirt oder der Virus?
Die Virus-first-Hypothese gibt eine verblüffende Antwort: „Weder noch!“ Sie postuliert, dass virenähnliche Replikatoren schon in der RNA-Welt existierten – lange vor Zellen, DNA oder LUCA – als Vorboten des Lebens.
Die RNA-Welt als Brutkasten der Proto-Viren
In dieser Ära waren Ribozyme (RNA-Moleküle mit Enzymfunktion) die ersten „Lebenskünstler“. Einige wurden zu molekularen Schmarotzern:
- Sie nutzten andere RNA-Stränge als Kopiervorlage („Parasitismus light“).
- Sie hüllten sich in Lipidvesikel oder Peptidringe (noch keine echten Kapside).
- Sie vermehrten sich ohne Zellen – etwa an Tonmineralien, die als Katalysatoren dienten.
Der Sprung zum modernen Virus: Erst als Zellen entstanden, wurden aus diesen Replikatoren effiziente Parasiten – die nun zelluläre Maschinerie kapern konnten.
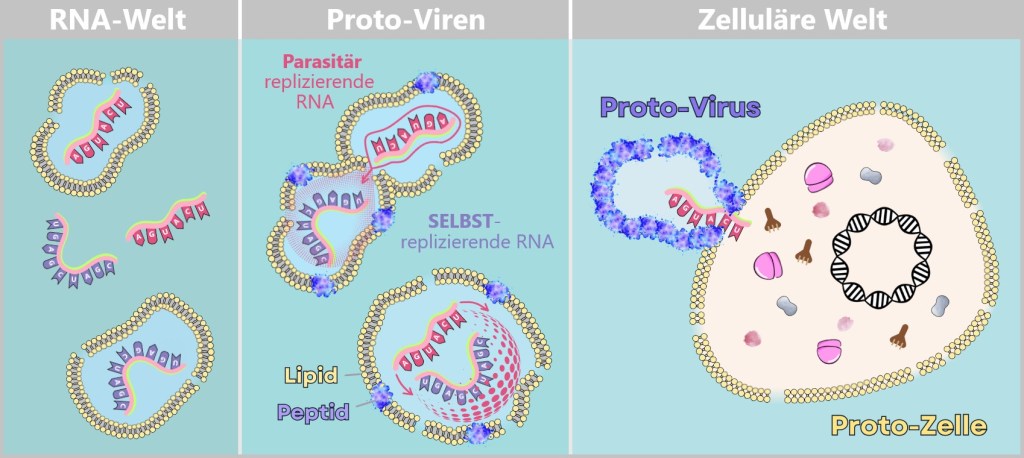
Diese Grafik zeigt einen möglichen evolutionären Übergang von präbiotischer Chemie hin zur Entstehung erster virenähnlicher Strukturen:
RNA-Welt (links): Es existieren einfache RNA-Moleküle, einige geschützt in Lipidvesikeln, mit der Fähigkeit zur Selbstreplikation.
Proto-Viren (Mitte): In dieser Phase entstehen erste parasitäre RNA-Moleküle (rot), die sich nicht mehr selbst vervielfältigen, sondern andere RNA-Stränge (blau) ausnutzen. Umhüllt von Lipiden und ersten Peptiden (kurze Ketten aus Aminosäuren) beginnt eine frühe Form des molekularen Parasitismus. Die roten Strukturen symbolisieren parasitäre Interaktionen zwischen RNA-Molekülen.
Zelluläre Welt (rechts): Mit der Entwicklung erster Proto-Zellen verschieben sich diese Strategien: RNA-Parasiten (Proto-Viren) verlassen ihre Peptidvesikel und beginnen, zelluläre Organismen zu infizieren – ein früher Vorläufer der heutigen Viren.
✅ Argumente für diese Hypothese
➤ Einzigartige virale Enzyme: RNA-Polymerasen von Viren (wie beim Influenzavirus) ähneln keinen zellulären Proteinen. Das deutet auf einen sehr alten Ursprung hin, möglicherweise aus der RNA-Welt, bevor sich die Domänen des Lebens trennten.
➤ Einzigartige Kapsidproteine: Die Hüllen, mit denen Viren ihr Genom schützen, haben keine direkten Entsprechungen in zellulären Organismen – ein Hinweis auf frühe, unabhängige Evolution.
➤ Globale Verbreitung: Viren infizieren alle drei Domänen des Lebens – Bakterien, Archaeen und Eukarya. Das deutet auf einen Ursprung vor ihrer Trennung hin.
➤ Enorme Vielfalt: Die Vielfalt viraler Genome (RNA, DNA, Einzel- oder Doppelstrang) spricht für eine lange evolutionäre Geschichte, möglicherweise bis in die RNA-Welt zurück.
➤ Parallelen zur RNA-Welt: Viele heutige Viren tragen RNA-Genome. Das passt zu der Annahme, dass RNA das ursprüngliche genetische Material war – und Viren lebendige Relikte dieser Zeit sein könnten.
❌ Schwächen der Hypothese
➤ Wirtsabhängigkeit: Heutige Viren sind auf zelluläre Maschinerie (z. B. Ribosomen) angewiesen. Wie konnten sie sich in der Ursuppe ohne Zellen vermehren? Nutzten sie vielleicht lose Netzwerke von selbstreplizierenden RNA-Molekülen als „Wirte“?
➤ Herkunft der Kapside: Die Evolution komplexer Kapsidstrukturen bleibt ungeklärt. Echte Kapsidproteine brauchen Ribosomen – und die gab’s noch nicht. Proto-Hüllen aus selbstorganisierenden Peptiden (z. B. kurze Aminosäureketten) oder Lipiden könnten RNA stabilisiert haben. Die ersten Kapsidvorläufer könnten sich als Reaktion auf äußeren Stress oder zur besseren Stabilisierung der RNA entwickelt haben – nicht als parasitärer Mechanismus, sondern als Überlebensvorteil, aber die Details bleiben spekulativ.
➤ Kein direkter Nachweis: Viren hinterlassen keine Fossilien. Auch molekulare Spuren aus der RNA-Welt sind bisher nicht nachweisbar – eine generelle Herausforderung in der Erforschung früher Lebensformen.
⬇️ Fazit
Die Virus-first-Hypothese ist eine der faszinierendsten – und umstrittensten – Theorien zur Entstehung des Lebens. Sie legt nahe, dass Viren nicht Nachzügler, sondern Pioniere der Evolution waren: molekulare Boten, die genetische Information verbreiteten, lange bevor es Zellen gab. Vielleicht waren sie sogar ein Katalysator für die Entstehung komplexeren Lebens. Was als molekulares Gerangel begann, wurde zur Blaupause moderner Viren.
b) Co-Evolution-Hypothese – Viren und Zellen entstanden gemeinsam
Nicht Zellen zuerst. Nicht Viren zuerst.
Sondern: Beides zugleich – im Tanz der frühen Evolution.
Die Co-Evolution-Hypothese geht davon aus, dass Viren und zelluläre Vorläufer sich parallel entwickelten – aus demselben molekularen Urschlamm der RNA-Welt. Es war kein „entweder–oder“, sondern ein dynamisches Zusammenspiel: Replikatoren, die sich gegenseitig herausforderten, nutzten, stabilisierten – und so die Grundlagen für Leben schufen.
Die Ursuppe als Experimentierfeld
In frühen Lipidvesikeln – kleinen, unvollkommenen Bläschen aus Fetten – sammelten sich RNA-Stränge. Manche dieser Moleküle replizierten sich selbst, andere halfen beim Kopieren, wieder andere schnitten oder verbanden Sequenzen. Kooperation und Konkurrenz begannen gleichzeitig.
Einige Replikatoren entwickelten sich zu immer komplexeren Systemen, aus denen erste Protozellen hervorgingen. Andere blieben reduziert, nutzten lieber bestehende Strukturen, statt sie selbst aufzubauen – eine Art minimalistischer Lebensstil, der an frühe Viren erinnert.
Ein Wechselspiel entsteht
In diesem Szenario war Parasitismus kein später Zusatz, sondern ein ursprüngliches Element der molekularen Evolution.
- Virale Vorläufer konnten Zellvorläufer beeinflussen, etwa durch Genaustausch oder Störung.
- Zellvorläufer wiederum konnten Virenähnliche Replikatoren stabilisieren oder integrieren, etwa als mobile Gene oder regulatorische Elemente.
So könnten Viren und Zellen aus denselben Netzwerken hervorgegangen sein, in denen alles noch fließend war: Selbstständigkeit, Abhängigkeit, Kopie, Konkurrenz.
Kein Ursprung – sondern ein Verhältnis
Die Co-Evolution-Hypothese ist weniger eine Erklärung für den „ersten Virus“, sondern ein Blick auf eine Beziehung, die so alt ist wie das Leben selbst. Viren wären demnach nicht nachträgliche Störenfriede, sondern von Anfang an Teil des Systems – Mitspieler in der Geschichte des Lebens.
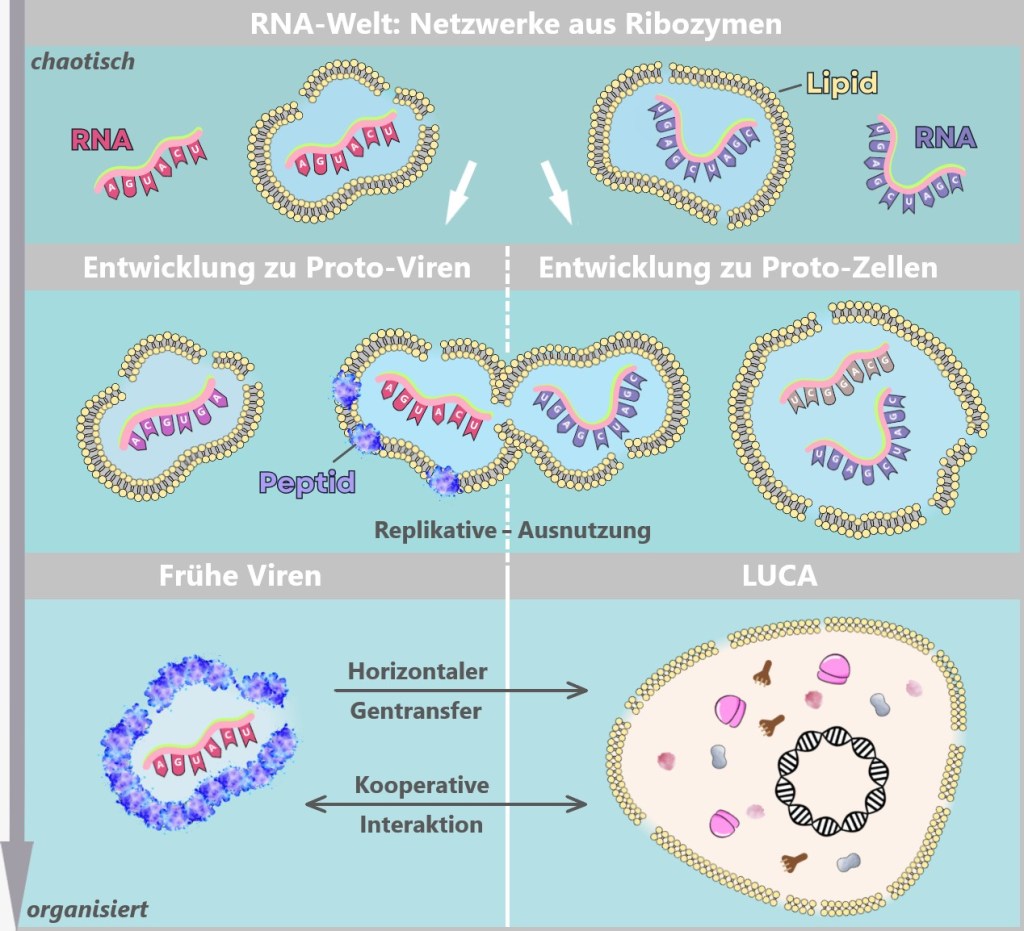
Die Grafik veranschaulicht die zentrale Idee der Co-Evolution-Hypothese: Viren und zelluläre Lebensformen stammen aus einer gemeinsamen molekularen Frühzeit – der RNA-Welt. In Lipidvesikeln koexistierten und konkurrierten Ribozymnetzwerke, aus denen sich zwei unterschiedliche Replikatorstrategien entwickelten: Einige bildeten die Vorstufen stabiler Proto-Zellen, andere reduzierten sich auf das Wesentliche und nutzten fremde Replikationsmechanismen – die frühen Proto-Viren.
Bereits vor der Entstehung echter Zellen zeichnete sich eine Art „molekularer Parasitismus“ ab – nicht im klassischen Sinne, sondern als ungerichtete Replikationsnutzung. Mit dem Auftreten zellulärer Lebensformen intensivierten sich diese Wechselwirkungen: Frühviren konnten nun genetisches Material zwischen Zellen übertragen (horizontaler Gentransfer) oder mit ihnen in komplexe, manchmal sogar kooperative Beziehungen treten. Die Entstehung von LUCA war somit nicht das Ende dieser Co-Evolution – sondern ihr erster Höhepunkt.
✅ Argumente für diese Hypothese
➤ Vermeidung des Henne-Ei-Problems: Sie umgeht elegant die Frage, ob erst Viren oder erst Zellen da waren: Beides entwickelte sich parallel aus denselben molekularen Vorläufern.
➤ Anpassung an die Dynamik der RNA-Welt: Die RNA-Welt war kein linearer Prozess, sondern ein Netzwerk aus Kooperation und Konkurrenz. Diese Hypothese spiegelt diese Vielfalt besser wider als ein klarer Ursprungspfad.
➤ Erklärung der viralen Vielfalt: Unterschiedliche Virusgruppen (RNA, DNA, retrovirale Elemente) könnten sich unabhängig, aber im gleichen Umfeld entwickelt haben – was ihre enorme Diversität plausibel macht.
➤ Evolutionäre Wechselwirkung: Die Co-Evolution erklärt, warum zellenähnliche und virenähnliche Systeme bereits früh miteinander agierten (z. B. durch horizontalen Gentransfer, RNA-Konkurrenz, gegenseitige Anpassung).
➤ Viren als Treiber der Zellkomplexität: Viren könnten nicht nur „Parasiten“, sondern Katalysatoren der Zellentwicklung gewesen sein, z. B. durch Gentransfer, Regulation, Immun- und Abwehrmechanismen.
❌ Schwächen der Hypothese
➤ Begriffliche Unschärfe: Ab wann ist ein Replikator „Virus“ und ab wann „Zelle“? Die Übergänge sind fließend – was die Hypothese erklärend, aber auch schwer fassbar macht.
➤ Fehlende Fossilien: Wie bei der Virus-first-Hypothese fehlen direkte Spuren früher viraler Replikatoren. Molekulare Fossilien aus der RNA-Welt existieren schlicht nicht.
➤ Komplexitätsproblem: Auch in dieser Hypothese bleibt unklar, woher bestimmte virusspezifische Proteine wie Kapsid- oder Polymerase-Proteine ursprünglich kamen. Für echte Kapside, effiziente Replikation und Wirtsnutzung braucht es komplexe Proteine, deren Ursprung noch unklar ist – besonders wenn Ribosomen noch nicht existierten.
➤ Experimentell schwer überprüfbar: Die Hypothese ist theoretisch gut begründet, aber kaum direkt testbar. Simulationen und Rückschlüsse bleiben oft spekulativ.
⬇️ Fazit
Die Co-Evolution-Hypothese bietet eine flexible, systemische Sicht auf die Frühzeit des Lebens – ohne sich auf eine lineare Ursache-Wirkung-Kette festzulegen. Sie passt gut zu den chaotisch-kooperativen Verhältnissen in der RNA-Welt. Viren wären nach diesem Modell nicht „entweder Zellabkömmlinge oder Ureltern“, sondern evolutionäre Parallelgänger – gleich alt, gleich bedeutend, nur auf einem ganz anderen Kurs unterwegs.
Viren sind die Hieroglyphen der Biologie
…wir entschlüsseln sie, aber ihr Ursprung bleibt ein Geheimnis.
Wie flackernde Schatten an der Höhlenwand:
- mal abtrünnige Zellteile,
- mal entlaufene Gene,
- mal Urahnen des Lebens selbst.
Vielleicht geht es bei der Frage nach ihrer Herkunft gar nicht um den einen Ursprung, sondern um viele evolutionäre Pfade – Pfade, die sich kreuzen, überlagern und rückkoppeln. Die klassischen Hypothesen (Virus-first, Co-Evolution, Progressive, Regressive) müssen sich nicht widersprechen – sie könnten sich an verschiedenen Stellen der Geschichte verwirklicht haben.
Und vielleicht ist die Frage nach dem Ursprung gar nicht die entscheidende. Viren zwingen uns, etwas Tieferes zu begreifen:
Leben ist kein Zustand, sondern ein Prozess – und Viren sind sein unsteter Puls.
Sie erinnern uns daran, dass Evolution kein geradliniger Stammbaum ist, sondern ein wirbelnder Fluss aus Kooperation, Diebstahl und Neuerfindung. Woher sie kamen? Wir wissen es nicht. Dass sie bleiben? Gewiss.
Vielleicht faszinieren sie uns genau deshalb – weil sie zeigen, dass das Leben nie stillsteht.
Weiterführende Quellen
Die uralte Viruswelt und die Evolution der Zellen
Die Ursprünge der Viren: Evolutionäre Dynamik der Fluchthypothese
Viren stehen im Mittelpunkt der zellulären Evolution
Der Ursprung von Viren und ihre mögliche Rolle bei großen evolutionären Übergängen
Eine virozentrische Perspektive auf die Evolution des Lebens
Wie haben sich Viren entwickelt und in welcher Beziehung stehen sie zum zellulären Leben?
Viren und Zellen sind seit Anbeginn der Evolution miteinander verflochten
Was uns Viren über Evolution und Immunität erzählen: Über Darwin hinaus?
Gab es Viren auf der Erde, bevor lebende Zellen auftauchten? Ein Mikrobiologe erklärt
Virus-First-Hypothese • Viren älter als das Leben?
Viren aus der Urzeit? – Die Entstehung der Viren und ihre Rolle in der Evolution

7. Warum gibt es Viren?
Die kurze Antwort: Weil es immer etwas geben muss, das stört.
Klingt witzig – ist aber ein Naturprinzip:
Nichts bleibt lebendig, wenn es nie herausgefordert wird.
Man kann vieles über Viren sagen.
Dass sie nicht leben.
Dass sie nur stören und zerstören.
Dass sie bloß molekulare Parasiten sind –
auf das Leben angewiesen, das sie befallen.
Und doch: Ohne sie wäre vieles nicht, wie es ist.
Vielleicht gäbe es uns nicht einmal.
Leben braucht Wiederholung. Und Abweichung.
Die Geschichte des Lebens begann nicht mit einem Ziel.
Sie begann mit Wiederholung.
Wieder und wieder verbanden sich Moleküle, zerfielen, verbanden sich neu.
Nicht, weil sie mussten – sondern weil es möglich war.
Was sich wiederholt, wird irgendwann wahrscheinlich.
Was funktioniert, bleibt.
Was bleibt, muss sich verändern.
Und Veränderung braucht Abweichung.
Mutation. Fehler. Störung.
Ohne sie gäbe es keine Vielfalt, keine Evolution – keine Geschichte.
Ordnung ist träge. Leben ist Bewegung.
Als die ersten stabilen Systeme entstanden, war das ein Triumph –
aber auch eine Gefahr.
Denn was stabil ist, neigt zur Starre.
Was zu perfekt funktioniert, wagt nichts Neues.
Leben aber braucht Bewegung.
Hier beginnen die Viren ihre Rolle zu spielen –
nicht als Gegner des Lebens, sondern als Gegenkraft.
Sie fordern heraus.
Sie unterbrechen.
Sie treiben Zellen in die Defensive – und in die Innovation.
Die Dualität: Ordnung vs. Chaos
Zellen bauen Mauern, Viren springen darüber.
Ihre Beziehung ist eine paradoxe Symbiose:
Sie töten – und machen Leben möglich,
halten Ökosysteme am Laufen
und brachten uns Plazenta-Gene.
Viren sind keine Fehler – sie sind ein Prinzip.
Sie sind Grenzverletzer, nicht aus Bosheit, sondern aus Prinzip.
Sie halten das Leben durchlässig. Offen. Wachsam.
Sie übertragen Gene, öffnen neue Wege, mischen Systeme auf.
Nicht immer zum Guten.
Aber immer mit Wirkung.
Vielleicht sind Viren genau das, was das Leben braucht, um lebendig zu bleiben.
Nicht als Gegenmodell – sondern als Mitspieler, als Schattenwurf des Lebendigen selbst.
Der Baum des Lebens – durchdrungen von Viren
Wenn wir heute auf den Baum des Lebens schauen, auf seine Äste, Zweige, Verzweigungen – dann sehen wir die Zellen, die Arten, die sichtbare Spur der Evolution.
Und die Viren?
Fehlen.
Nicht, weil sie unwichtig wären – sondern weil sie sich nicht verorten lassen.
Sie sind weder Ast noch Blatt, sondern der Wind, der sie bewegt.
Sie sind das Flüstern zwischen den Zweigen.
Keine eigene Linie, kein Teil des Holzes – und doch überall.
Ein Strom von Information, der den Baum nicht nur umgibt, sondern auch formt.
Sie sind die Punkte zwischen den Linien, die Verbindungen schaffen.
Als Wellen der Störung, die Wachstum erzwingen.
Als Abweichung, die das Muster nicht zerstört, sondern erweitert.
Sie sind die dunkle Materie der Biologie: unsichtbar, aber alles durchdringend.
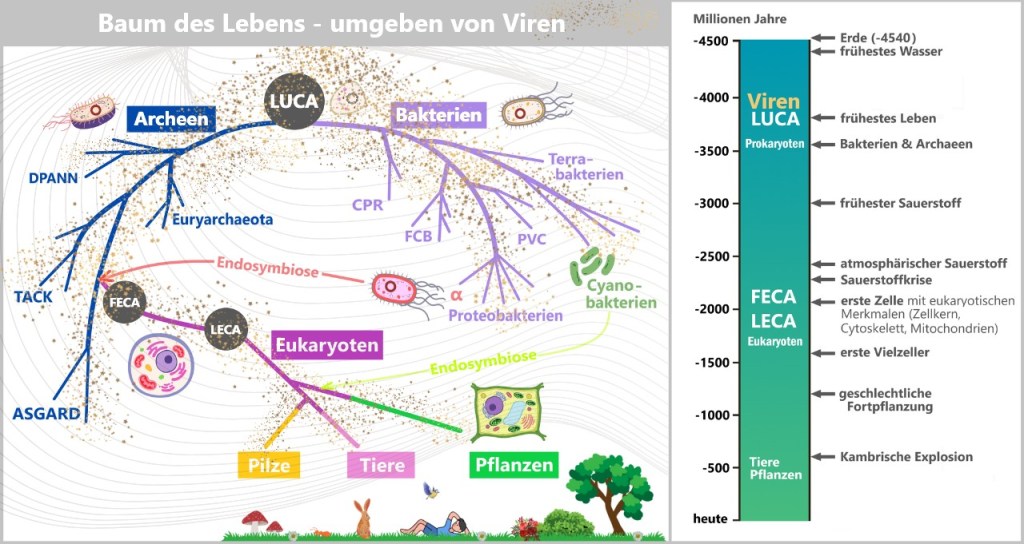
Der „Baum des Lebens“ zeigt die Abstammungslinien zellulärer Organismen – Bakterien, Archaeen und Eukaryoten – zurück bis zum gemeinsamen Vorfahren (LUCA). Die feinen Pünktchen und Bahnen symbolisieren Viren: nicht als festen Ast, sondern als diffuses Netzwerk, das den gesamten Baum durchdringt.
In dieser Sichtweise sind Viren keine Außenseiter, sondern kreative Spielarten molekularer Evolution. Keine Entität mit klarer Abstammung, sondern eine wiederkehrende Erscheinung in einem Universum, das mit Variation und Wiederholung spielt.
Und darum sind sie da.
Nicht weil sie wollen. Nicht weil sie müssen.
Sondern weil sie immer wieder entstehen – dort, wo Natur variiert.
Wo Leben sich organisiert – und aus dieser Ordnung heraus etwas Neues wagt.
Sie sind da, weil sie nicht zu vermeiden sind.
Wie ein Echo des Prinzips, das allem zugrunde liegt:
Störung ist nicht das Gegenteil von Leben.
Störung ist seine Möglichkeit.
Viren sind da, weil Störung kein Unfall ist –
sondern das Werkzeug des Universums, um Leben wach zu halten.
Denk daran, wenn dich das nächste Mal ein Virus nervt:
Vielleicht ist es bloß das Universum, das dir einen kosmischen Klaps verpasst –
und murmelt:
„Na los, wach auf! Hier ist deine tägliche Dosis Chaos – damit du schön evolvierst.“
Epilog: Die Unscheinbaren
Viren sind der Sand im Getriebe der Schöpfung.
Ohne Willen, ohne Leib –
und doch die unsichtbare Hand,
die Evolution schreibt.
Das Flüstern zwischen den Zeilen des Lebens –
nicht aus Absicht, sondern aus Notwendigkeit.
Sie kamen aus dem Nebel der Anfänge
und blieben im Schatten –
nicht als Fremdkörper, sondern als Gegenspieler,
als Prüfstein und als Impuls.
Sie lehren uns Demut:
Denn was zerstört,
kann auch erschaffen.
Vielleicht ist das ihre Botschaft:
Dass das Leben nicht gegen das Chaos wächst,
sondern mit ihm tanzt.
Nachtrag:
Ich habe mich dem Thema als völliger Laie genähert. Und gerade das erwies sich als Vorteil – denn ich wusste, was ich nicht wusste.
Mit Neugier, Skepsis und Staunen bin ich eingetaucht in die Welt der Viren – und war überrascht, welch ungeahnte Dimensionen sich auftaten. Dieses Thema trägt eine Wucht an Erkenntnis in sich, die ich nie erwartet hätte.
Während meines Suchens, Fragens und Formulierens hatte ich großartige Unterstützung durch KI-Systeme wie ChatGPT und DeepSeek. Der Dialog mit diesen Werkzeugen hat mein Lernen nicht nur beschleunigt, sondern auch vertieft. Ohne diese Hilfe hätte ich vieles nicht gesehen – und erst recht nicht so klar formulieren können.
Was hier entstanden ist, ist das Ergebnis von menschlicher Neugier und maschinischer Geduld. Und es zeigt, dass Lernen heute mehr denn je ein gemeinschaftlicher Prozess sein kann – über Grenzen hinweg, auch über die zwischen Mensch und Maschine.
Quellen (Stand vom 07.07.2025)
