Die wissenschaftliche Diskussion zu DNA-Rückständen in mRNA-Impfstoffen hat sich seit Veröffentlichung dieses Beitrags deutlich weiterentwickelt.
Ein umfassend erweiterter und methodisch aktualisierter Folgeartikel ordnet die Herstellungsprozesse, Nachweismethoden sowie die bisherigen Studien zu DNA-Rückständen nun ausführlich ein und ersetzt diesen älteren Beitrag inhaltlich weitgehend.
Zur Artikelserie:
DNA-Rückstände in modRNA-basierten Impfstoffen
In den letzten Jahren hat die Entwicklung und Anwendung von mRNA-Impfstoffen einen bedeutenden Fortschritt erlebt. Was einst nur im Labor vorhanden war, ist heute für viele von uns bereits Realität geworden, da wir mehrfach mit mRNA-Impfstoffen geimpft wurden. Diese Technologie wird nun als vielversprechendes Instrument für zukünftige medizinische Maßnahmen betrachtet. Es wird erwartet, dass in naher Zukunft weitere Impfstoffe auf mRNA-Basis auf den Markt kommen, um verschiedene Krankheiten zu bekämpfen. Eine Übersicht dazu findet man hier.
Aktuell sind weltweit sechs mRNA-Impfstoffe zur Covid-19-Impfung zugelassen, davon zwei in der EU. Die Europäische Arzneimittel-Agentur (EMA) hat das Sicherheitsprofil dieser zugelassenen mRNA-Impfstoffe als sehr beruhigend eingestuft. Dennoch haben jüngste Medienberichte, die sich mit der Frage der „Fremd-DNA in Corona-Impfstoffen“ befassen, zu kontroversen Diskussionen geführt und verstärkt Aufmerksamkeit erregt.
Quellen:
- mRNA-Impfstoffe gegen Corona: Wie DNA hineinkommen kann und was das bedeutet
- Stimmungsmache mit angeblichen DNA-Verunreinigungen
- Corona: War der Biontech-Impfstoff verunreinigt?
- „Der Streit lenkt ab von den wahren Problemen beim mRNA-Impfstoff“
- Fremd-DNA im Covid-Impfstoff: „Das Hauptproblem ist, dass Geninformationen in die Zellen gelangen können“
- mRNA-Impfstoffe: Verunreinigung mit DNA?
- KBV warnt vor irreführendem Schreiben: Keine Hinweise auf Verunreinigung von mRNA-Impfstoff
Angesichts der essenziellen Bedeutung der mRNA-Impfstoffe für unsere Gesundheit ist es interessant, den genauen Hintergrund sowie die kritischen Standpunkte näher zu beleuchten.
In den öffentlichen Debatten werden oft wissenschaftliche Argumente aus dem Zusammenhang gerissen und mit allgemeinen Beschreibungen vermischt. Besonders wenn politische und wirtschaftliche Interessen betroffen sind, scheint die Objektivität im öffentlichen Diskurs zunehmend vernachlässigt zu werden. Die Klärung dieses Themas ist nicht nur aus wissenschaftlicher Sicht von Bedeutung, sondern auch im gesellschaftlich-politischen Kontext relevant.
Diese Abhandlung zielt darauf ab, eine Brücke zwischen den hochspezialisierten wissenschaftlichen Erklärungen in der Fachliteratur und den allgemeinverständlichen Beschreibungen in den Medien zu schlagen. Durch die Vermittlung von detailliertem Wissen zum Thema „Fremd-DNA in mRNA-Impfstoffen“ soll dazu beigetragen werden, komplexe Prozesse einem breiteren Publikum verständlich zu machen.
Es wird daher vorgeschlagen, die einzelnen Abschnitte dieses Artikels in aufeinanderfolgender Reihenfolge zu lesen. Auf diese Weise können die Informationen systematisch aufeinander aufbauen und ein umfassenderes Verständnis des Themas ermöglichen.
Eine eingehende Beschäftigung mit dem Thema ermöglicht es interessierten Lesern, ihre Wissensbasis zu erweitern und ein tieferes Verständnis für die zugrunde liegenden Zusammenhänge zu entwickeln. Wissen wird in diesem Zusammenhang zu einer entscheidenden Ressource. Personen, die ein besseres Verständnis der Abläufe in der Impfstoffentwicklung haben, sind eher in der Lage, informierte Entscheidungen zu treffen, im Gegensatz zu Personen ohne dieses Wissen, die dazu neigen, sich auf die Aussagen und Überzeugungen anderer zu verlassen.
1. Entdeckung von Fremd-DNA im Impfstoff
2. Wo liegt die Brisanz der Studienergebnisse?
2.1. Bedeutung der DNA für mRNA-Impfstoffe
2.2. Herstellungsverfahren des BioNTech mRNA-Impfstoffs
3. Die Herstellungsverfahren in a nutchell
3.1. Gewinnung der genetischen Information
3.2. Vervielfältigung der Spike-Protein-DNA
3.3. Transkription zur Herstellung der RNA
3.4. RNA-Prozessierung
3.5. Reinigung der synthetischen mRNA
3.6. Die mRNA wird in Lipid-Nanopartikel verpackt
3.7. Einige Schlussfolgerungen
4. Methoden zur Messung von DNA und RNA
4.1. Fluoreszenz-Assay
4.2. Oxford-Nanopore-Technologie
4.3. UV-Spektroskopie
5. Messergebnisse und deren Auswertung
5.1. Richtlinien zur Begrenzung von Rest-DNA in Impfstoffen
5.2. Die Messmethoden von Pfizer
5.3. DNA-Fragmente in C19 mRNA-Impfstoffen entdeckt
5.4. Reproduktion der Studienergebnisse
5.5. Kurze Zusammenfassung
6. Reaktionen und Wissenstand zur Fremd-DNA auf offizieller Seite
6.1. Die Europäische Arzneimittel-Agentur (EMA)
6.2. Das Paul-Ehrlich-Institut (PEI)
6.3. Kurze Zusammenfassung
7. Sind weitere Untersuchungen notwendig?
7.1. Offizielle Aussagen zu DNA-Resten
7.2. Weitere Studien zum Thema
7.3. SV40 Sequenzen
7.4. SV40-Promotor und das p53-Tumorsuppressor-Gen
7.5. Rest-DNA und die Lipid-Nanopartikel
7.6. Mögliche Integration in die DNA
7.7. Weitere Aufrufe zu Untersuchungen
7.8. Kurze Zusammenfassung
8. Epilog
1. Entdeckung von Fremd-DNA im Impfstoff
Laut einer Studie, die auf der Plattform OSF Preprints am 11. April 2023 veröffentlicht wurde, hat man in Chargen des Pfizer- und Moderna-Impfstoffs, den Covid-19 mRNA-Impfstoffen, DNA-Kontaminationen festgestellt, die die Grenzwerte der Europäischen Arzneimittelagentur (EMA) von 330ng/mg und der FDA von 10ng/Dosis übersteigen. Bei diesen DNA-Verunreinigungen handelt es sich um sogenannte „Fremd-DNA“, also um ein genetisches Material, das nicht natürlicherweise im menschlichen Organismus vorhanden ist. Diese Studie bildet den Auftakt der Untersuchung zu diesem Thema.
Die Untersuchung wurde unter der Leitung von Kevin McKernan und seinem Team durchgeführt. Dr. Kevin McKernan ist ein US-amerikanischer Genetiker und Pflanzenbiologe. Er war zuerst als Leiter der Forschungs- und Entwicklungsabteilung des Humangenomprojekts am Massachusetts Institute of Technology (MIT) tätig. Später gründete er die Firma Medicinal Genomics, bei der er als CSO (Chief Scientific Officer) fungiert. Er hat eine umfassende Erfahrung in der Biotechnologiebranche, insbesondere in Projekten zur DNA-Sequenzierung und Genomik. Seine Fachkenntnisse erstrecken sich auf Next-Generation-Sequenzierungstechnologien, und er war maßgeblich an der Entwicklung von Methoden und Technologien zur Entschlüsselung von DNA beteiligt.
Am 19.10.2023 erschien eine weitere Studie, die erneut die DNA-Kontamination in mRNA-Produkten von Pfizer und Moderna untersuchte. Hauptautor dieser Studie ist Dr. David Speicher, Dr. Kevin McKernan wird hier als Co-Autor genannt.
Gegenüber The Epoch Times sagte Speicher: „In unserer Studie haben wir DNA-Kopien der Gene Spike, ORI (Replikationsursprung) und SV40-Enhancer-Gene gemessen“. „Die Mengen an SV40-Enhancer-Promotor, ORI und Virus-Spike in Pfizer betragen bis zu 186 Milliarden Kopien pro Dosis.“
In den Moderna-Fläschchen wurden auch mehrere Millionen Kopien der ORI- und Spike-DNA gefunden, das SV40-Enhancer-Gen wurde dort nicht nachgewiesen. [Studie]
Dies leitete eine Debatte über die Präsenz von Fremd-DNA in den Corona-Impfstoffen ein.
2. Wo liegt die Brisanz der Studienergebnisse?
2.1. Bedeutung der DNA für mRNA-Impfstoffe
Wie in dem BioNTech-Kurzvideo erläutert, beginnt die Herstellung von mRNA-Impfstoffen mit der Identifizierung der genetischen Information, konkret der DNA, die die Grundlage des Impfstoffs bildet. In diesem Fall handelt es sich um die Spike-Protein-DNA des SARS-CoV-2. Um ausreichend genetisches Material für die Impfstoffproduktion zu gewinnen, wird diese DNA zuerst vervielfältigt. Hierfür stehen verschiedene Methoden zur Verfügung. Die vervielfältigte DNA wird als Schablone genutzt, um die mRNA zu synthetisieren. Die produzierte mRNA trägt die genetische Information für das Spike-Protein.
2.2. Herstellungsverfahren des BioNTech mRNA-Impfstoffs
Im Bewertungsbericht der Europäischen Arzneimittel-Agentur (EMA) für den BioNTech mRNA-Impstoff werden auf Seite 18 zwei verschiedene Herstellungsverfahren erwähnt: Prozess 1, der während der klinischen Studien verwendet wurde, und Prozess 2, der während der Großserienproduktion angewendet wird.
Beim Prozess 1 erfolgt die Vervielfältigung der Spike-Protein-DNA mittels der Polymerase-Kettenreaktion (PCR). Beim Prozess 2 wird mithilfe von Bakterien die Spike-Protein-DNA vervielfältigt. Aufgrund der Dringlichkeit, die Nachfrage nach Impfstoffen schnell zu bedienen, wurde ‚Prozess 2‘ als Standard für die Großserienproduktion festgelegt.
Diesen Sachverhalt brachten erstmals die israelischen Forscher Joshua Guetzkow und Retsef Levi an die Öffentlichkeit. Sie haben Pfizer-Dokumente analysiert, die seit geraumer Zeit Stück für Stück freigeklagt werden (siehe hier: Klage sowie gerichtliche Anordnung). Die Ergebnisse dieser Auswertung wurden im Mai 2022 im British Medical Journal veröffentlicht. Dort kann man folgendes lesen:
„Eine im Oktober 2020 vorgenommene Änderung am Protokoll der zulassungsrelevanten Pfizer/BioNTech BNT162b2 (Comirnaty) klinischen Studie (C4591001) zeigt, dass nahezu alle im Rahmen der Studie verwendeten Impfstoffdosen aus sogenannten ‚klinischen Chargen‘ stammen, die unter dem Begriff ‚Prozess 1‘ hergestellt wurden. Um jedoch die Produktion für eine groß angelegte Verteilung als ‚Notfallversorgung‘ nach der Zulassung zu steigern, wurde eine neue Methode namens ‚Prozess 2‘ entwickelt. Die Unterschiede umfassen Änderungen am DNA-Template, das zur Transkription der RNA verwendet wird, sowie an der Reinigungsphase und dem Herstellungsprozess der Lipid-Nanopartikel. Es wurde festgestellt, dass Chargen von ‚Prozess 2‘ eine deutlich niedrigere mRNA-Integrität aufweisen. Die Protokolländerung gibt an, dass ‚jede Charge von mit Prozess 2‘ hergestelltem BNT162b2 an etwa 250 Teilnehmer im Alter von 16 bis 55 Jahren verabreicht würde, wobei vergleichende Immunogenitäts- und Sicherheitsanalysen mit 250 zufällig ausgewählten Empfängern von ‚Prozess 1‘-Chargen durchgeführt werden. Nach unserem besten Wissen gibt es keinen öffentlich verfügbaren Bericht zu diesem Vergleich zwischen Dosen von ‚Prozess 1‘ und ‚Prozess 2‘.“
Mit anderen Worten, das Zulassungsverfahren basierte auf Präparaten, die aus dem Herstellungsverfahren ‚Prozess 1‘ stammten. Diese mRNA-Impfstoffe wurden an 22.000 Probanden verabreicht, und die Aussagen zur Wirksamkeit und Sicherheit des Impfstoffs beruhen auf den Daten dieser Personen. Für den weltweiten Einsatz der mRNA-Impfstoffe wurde jedoch ‚Prozess 2‘ angewendet, ein vollständig unterschiedliches Herstellungsverfahren. Der damit produzierte Impfstoff wurde nur an 250 Probanden getestet.
In diesem Zusammenhang vermerkt die EMA im Bewertungsberichts für den BioNTech mRNA-Impfstoff auf Seite 32 folgendes:
„Aufgrund der festgestellten Unterschiede zwischen den nach dem Wirkstoffverfahren 1 und 2 hergestellten Chargen in Bezug auf die CQA (Critical Quality Attribute) mRNA-Integrität und der fehlenden Charakterisierungsdaten wurde ein Haupteinwand in Bezug auf die Vergleichbarkeit, Charakterisierung und klinische Eignung des einen vorgeschlagenen Zulassungskriteriums erhoben. Die biologische Charakterisierung des Wirkstoffs war begrenzt, und es wurden zusätzliche Daten und Diskussionen gefordert, um die Funktionalität zu klären.“
Die Untersuchungsergebnisse von McKernan und Speicher beziehen sich auf Impfstoff-Chargen, die nach ‚Prozess 2‘ hergestellt wurden.
Bei der Festlegung des Sicherheitsprofils eines mRNA-Impfstoffs sind bestimmte Merkmale des Impfstoffs von entscheidender Bedeutung, darunter seine Zusammensetzung, Reinheit und Stabilität. Die Reinheit des Impfstoffs ist entscheidend, um sicherzustellen, dass er frei von potenziell schädlichen Verunreinigungen ist. Verunreinigungen könnten unerwünschte immunologische Reaktionen oder andere Nebenwirkungen hervorrufen.
Die Vervielfältigung der Spike-Protein-DNA in Bakterien beinhaltet im ‚Prozess 2‘ eine anschließende Reinigung, um DNA-Fragmente, Bakterienreste und prozessbedingte Verunreinigungen zu entfernen.
Es scheint, dass auch Moderna ein ähnliches Verfahren benutzt. [Studie]
Jedoch legt die Studie von Speicher DJ et al. nahe, dass die Reinigung der Impflösung offenbar nicht immer effektiv ist. In der Studie heißt es: „Diese Daten belegen das Vorhandensein von Milliarden bis Hunderten von Milliarden DNA-Molekülen pro Dosis in diesen Impfstoffen. Mittels Fluorometrie übertreffen alle Impfstoffe die von FDA und WHO festgelegten Richtlinien für Rest-DNA von 10 ng/Dosis um das 188- bis 509-fache.“
Um ein besseres Verständnis dieser Situation und deren potenzielle Auswirkungen zu erlangen, werden in den folgenden Kapiteln die Herstellungsverfahren der mRNA-Impfstoffe sowie die Methoden zur Qualitätssicherung genauer vorgestellt.
3. Die Herstellungsverfahren in a nutchell
Wie bereits beschrieben, unterscheiden sich ‚Prozess 1‘ und ‚Prozess 2‘ vor allem in der Art und Weise, wie die Spike-Protein-DNA vervielfältigt wird, die als Vorlage für die Herstellung der mRNA dient, die den Bauplan für das Spike-Protein des Coronavirus enthält.
‚Prozess 1‘ nutzt PCR-Geräte für die Vervielfältigung der DNA, ein in-vitro-Verfahren, während ‚Prozess 2‘ Bakterien für die Vervielfältigung der DNA verwendet, was ein in-vivo-Verfahren darstellt.
Die folgende Tabelle stellt grob die Gemeinsamkeiten und Unterschiede im Pfizer-Produktionsprozess dar, der in der Realität variieren kann.

(*) Informationen zum Herstellungsverfahren laut EMA „EPAR zum Covid-19-Impfstoff von BioNTech/Pfizer“ (in Abschnitt 2.2., Seite 32)
3.1. Gewinnung der genetischen Information
3.2. Vervielfältigung der Spike-Protein-DNA
3.2.1. Vervielfältigung der Spike-Protein-DNA - im Prozess 1
3.2.2. Vervielfältigung der Spike-Protein-DNA - im Prozess 2
3.3. Transkription zur Herstellung der RNA
3.4. RNA-Prozessierung
3.5. Reinigung der synthetischen mRNA
3.5.1. Reinigung der synthetischen mRNA im Prozess 1
3.5.2. Reinigung der synthetischen mRNA im Prozess 2
3.6. Die mRNA wird in Lipid-Nanopartikel verpackt
3.7. Einige Schlussfolgerungen
3.1. Gewinnung der genetischen Information

Der erste Schritt ist die Identifizierung der genetischen Information (DNA) für das gewünschte Protein, in diesem Fall das Spike-Protein des Coronavirus.

Die identifizierte DNA des Spike-Proteins dient im Herstellungsprozess als Vorlage für die Synthese von mRNA. In diesem Zusammenhang wird die identifizierte DNA-Sequenz oft als „DNA-Matrize“ oder „DNA-Template“ bezeichnet.
Um an die DNA des Spike-Proteins zu gelangen, muss man zunächst das Virus aus einer infizierten Probe, wie zum Beispiel Blut oder Gewebe, isolieren. Anschließend wird das Virus sequenziert, um die exakte Abfolge der Nukleotide (der genetischen ‚Buchstaben‘) in der Virus-DNA zu bestimmen. Die gewonnenen genetischen Informationen werden in öffentliche Datenbanken eingetragen. Dadurch können Forscher und Biotechniker auf bereits veröffentlichte genetische Daten zugreifen, selbst wenn sie das Virus nicht selbst isoliert haben. Mit den Sequenzierungsdaten des Virus sind Biotechniker in der Lage, das Spike-Protein synthetisch herzustellen.
Es ist üblich, dass Forschungsteams oder Unternehmen auf bereits veröffentlichte genetische Daten zurückgreifen, die von anderen Forschern in öffentlichen Datenbanken bereitgestellt wurden. Diese Herangehensweise wird von Francis Aurelio deSouza, CEO von Illumina (2016 bis 2023), in der Diskussion „World Economic Forum Panel on Preparing for the Next Pandemic“ (Minute: 12:20-13:51) im Fall des SARS-CoV-2 kurz beschrieben.
3.2. Vervielfältigung der Spike-Protein-DNA
3.2.1. Vervielfältigung der Spike-Protein-DNA - im Prozess 1
3.2.2. Vervielfältigung der Spike-Protein-DNA - im Prozess 2

3.2.1. Vervielfältigung der Spike-Protein-DNA – im Prozess 1
‚Prozess 1‘ nutzt PCR-Geräte zur Vervielfältigung der DNA, ein in-vitro-Verfahren. Der Begriff ‚in vitro‚ bezieht sich auf Experimente oder Prozesse, die außerhalb eines lebenden Organismus, typischerweise in einem Reagenzglas oder einer Petrischale, durchgeführt werden. Die PCR imitiert die Vervielfältigung von DNA, wie sie auch in lebenden Organismen stattfindet.
Seit Beginn der Corona-Pandemie kennen wir alle die Abkürzung PCR. Was PCR ist und wie PCR funktioniert, wissen jedoch nur die wenigsten. Daher hier ein kurzer Überblick.
Polymerase Kettenreaktion (PCR)
PCR ist die Abkürzung für Polymerase Chain Reaction oder zu deutsch Polymerase-Kettenreaktion. Mit Hilfe der Polymerase-Kettenreaktion können von einem bestimmten DNA-Abschnitt in vitro viele Kopien hergestellt werden.
Um eine Polymerase-Kettenreaktion (PCR) zu starten, benötigt man verschiedene Zutaten und Geräte:
a) Die DNA-Vorlage (engl.: DNA-Template), die vervielfältigt werden soll (siehe Abb.1).
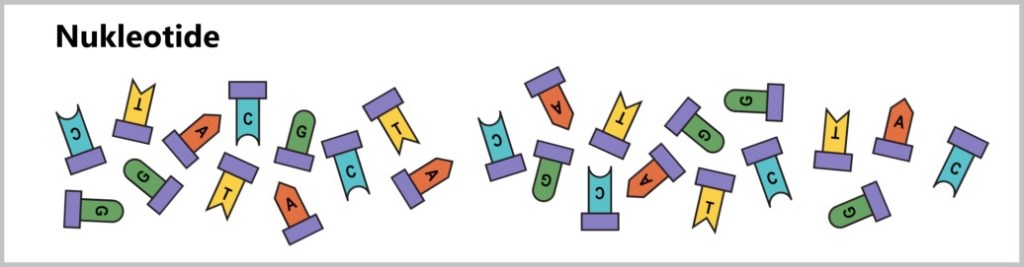
b) Nukleotide: Um eine neue DNA zu synthetisieren, werden viele Nukleotide benötigt, da sie die Grundbausteine der DNA sind. Es gibt insgesamt vier Arten von Nukleotiden, die die genetische Information tragen: Adenin (A), Thymidin (T), Cytosin (C) und Guanin (G).
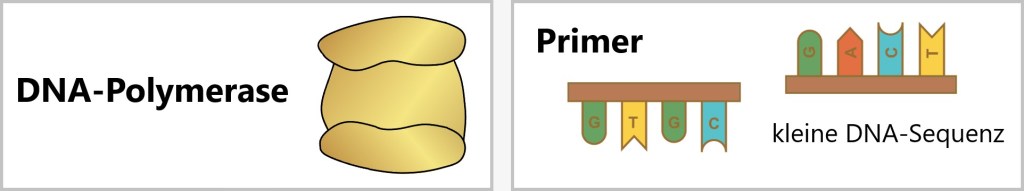
c) DNA-Polymerase: Bei der DNA-Polymerase handelt es sich um ein Enzym, das eine entscheidende Rolle bei der Synthese von DNA spielt. Man kann sie sich wie einen Baumeister vorstellen. Die DNA-Polymerase nimmt die vorhandene DNA als Vorlage und verwendet die Nukleotid-Bausteine, um einen neuen DNA-Strang zu bauen. Sie fügt diese Bausteine in der richtigen Reihenfolge zusammen, um sicherzustellen, dass die Kopie der DNA genauso aussieht wie das Original.
d) Primer: Ein Primer ist eine kurze DNA-Sequenz, die als Startpunkt für die DNA-Synthese dient. Der Primer ist also so etwas wie ein Wegweiser für die DNA-Polymerase. Er sagt der Polymerase, wo sie beginnen soll, die DNA zu kopieren, und in welche Richtung sie gehen soll.
e) Pufferlösung: Die Pufferlösung schafft eine stabile und kontrollierte Umgebung für die PCR, damit die DNA-Polymerase effizient arbeiten kann. Sie optimiert den pH-Wert, stabilisiert die Enzymaktivität und sorgt dafür, dass die notwendigen Ionen (meist Magnesiumionen) vorhanden sind, um eine erfolgreiche DNA-Synthese zu ermöglichen.
Oder vereinfacht: Die Pufferlösung in der PCR ist wie die Brühe in einer Suppe. Denk dir die PCR-Reaktion als das Kochen eines komplexen Gerichts, bei dem alle Zutaten perfekt zusammenspielen müssen. Die Pufferlösung spielt die Rolle der Brühe, die nicht nur den Geschmack beeinflusst, sondern auch sicherstellt, dass alles reibungslos verläuft. Kurz gesagt, die Pufferlösung ist wie die „Brühe“, die die PCR zu einem gelungenen „Gericht“ macht, bei dem die DNA-Polymerase die Hauptzutat ist.
f) Thermocycler: Ein Gerät, das die PCR-Temperaturzyklen automatisch durchführt. Es ermöglicht die präzise Kontrolle der Temperatur für die verschiedenen PCR-Schritte.

Ablauf der Polymerase Kettenreaktion
Alle Zutaten wie die DNA-Vorlage, die Nukleotide, die DNA-Polymerase und die Primer werden in ein Röhrchen mit Pufferlösung gegeben. Das Röhrchen kommt dann in den Thermocycler. Zu den grundlegenden Schritten des Verfahrens gehören die Denaturierung, Primerbindung und DNA-Synthese.
Schritt 1 – Denaturierung (Aufspalten): Die PCR beginnt mit der Erhitzung der DNA-Vorlage im Thermocycler. Dabei wird die DNA auf etwa 94-98°C für etwa 20-30 Sekunden erhitzt. Dadurch trennen sich die beiden Stränge der DNA (Denaturierung), weil die Bindungen zwischen den Nukleotiden aufbrechen. Aus einem DNA Doppelstrang entstehen zwei DNA Einzelstränge, die als Vorlage für ihre Vervielfältigung dienen.

Schritt 2 – Primerbindung: Im zweiten Schritt wird das Reaktionsgemisch auf circa 50-65°C abgekühlt. Jetzt binden sich die Primer an die jeweiligen Einzelstränge der DNA. Die Primer dienen als Startpunkte für die DNA-Synthese. In der PCR werden zwei Primer verwendet, einen für jeden Strang der DNA, um die gewünschte Region zu markieren, die vervielfältigt werden soll. Es gilt das Prinzip der komplementären Basenpaarung (Adenin mit Thymin und Guanin mit Cytosin).
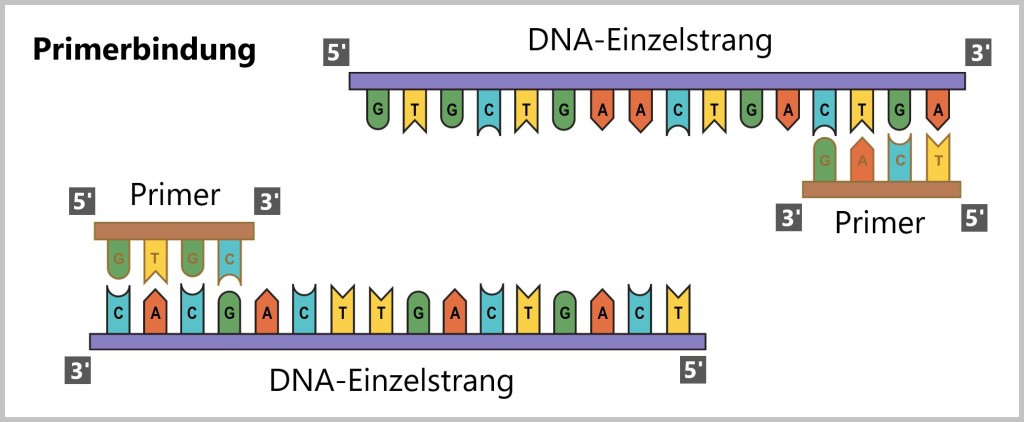
Schritt 3 – DNA-Synthese: Dieser Schritt wird auch als Amplifikation (lat. amplificatio: Verstärkung), Elongation (lat. elongare: verlängern) oder Polymerisation (griech. poly: viele; griech. meros: Teil) bezeichnet. Die Temperatur wird auf die optimale Arbeitstemperatur der DNA-Polymerase (ungefähr 70°C) eingestellt. Die DNA-Polymerase bindet an den Primer und startet die Synthese des neuen DNA-Strangs. Währenddessen liest sie den Einzelstrang der DNA-Vorlage in Richtung 3′ zu 5′ und synthetisiert die neue DNA in Richtung 5′ zu 3′. Dies erfolgt unter Verwendung der Basenpaarungsregeln (A mit T und G mit C), wodurch zwei neue Doppelstränge entstehen.

Wiederholung der Zyklen
Die so entstandenen DNA-Doppelstränge dienen als Vorlagen für den nächsten Zyklus. Die Schritte 1 bis 3 werden solange wiederholt, bis die erforderliche Menge DNA erreicht ist. Jeder Zyklus verdoppelt die Menge an DNA. Der Begriff „Polymerase-Kettenreaktion“ beschreibt die zyklische Natur dieser Technik. Er verdeutlicht, dass der Prozess in aufeinanderfolgenden Zyklen wiederholt wird, wobei jede Runde zu einer exponentiellen Vermehrung der DNA (1-2-4-8-16-32 usw.) führt.

3.2.2. Vervielfältigung der Spike-Protein-DNA – im Prozess 2
‚Prozess 2‘ nutzt lebende Zellen, genauer Bakterien, zur Vervielfältigung der DNA, ein in-vivo-Verfahren. in vivo (lateinisch für ‚im Lebendigen‘) bezeichnet in der Wissenschaft Prozesse, die im lebendigen Organismus ablaufen.
a) Warum Bakterien?
b) Das Bakterium Escherichia coli (E. coli)
c) Plasmide
d) Plasmide in der Gentechnik
e) Plasmide in der Impfstoffherstellung
f) Einbau der Spike-Protein-DNA in das Plasmid
g) Übertragung der modifizierten Plasmide in Bakterien
h) Vermehrung der Bakterien
i) Ernte der Bakterien
j) Isolierung der modifizierten Plasmide
k) Linearisierung der Spike-Protein-DNA
a) Warum Bakterien?
Der Kern der Evolution liegt in der kontinuierlichen Veränderung des genetischen Materials. Diese Flexibilität ist entscheidend dafür, dass Organismen sich im Laufe der Generationen neuen Umweltbedingungen erfolgreich anpassen können. Es gibt verschiedene Wege, wie Lebewesen ihr Erbgut modifizieren. Mutationen verändern bestehende genetische Elemente, während Gentransfer neue Elemente hinzufügen kann.
Für höhere, mehrzellige Organismen erfolgt die Durchmischung des Erbguts in der Regel durch die Kreuzung artgleicher Individuen. Die Vereinigung von väterlichen und mütterlichen Keimzellen führt zur Entstehung eines neuen Organismus mit einer einzigartigen Genkomposition.
Im Gegensatz dazu haben einzellige Organismen wie Bakterien keine Geschlechterausbildung und vermehren sich durch einfache Teilung. Bei der Teilung bildet die Bakterienzelle zwei identische Tochterzellen aus. Dieser Prozess beginnt mit der Verdopplung des genetischen Materials in der Zelle, gefolgt von der Teilung der Zelle selbst.
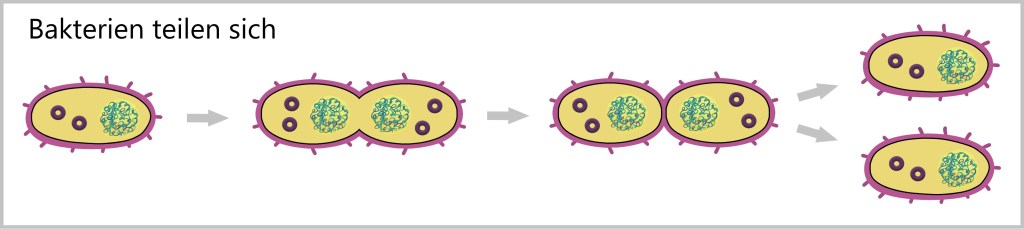
Bei Prokaryoten (wie Bakterien) erfolgt alles, was für das Überleben, die Vermehrung und die Weiterentwicklung notwendig ist, innerhalb der Einzelzelle. Dennoch streben auch sie nach genetischem Austausch und gelten als besonders anpassungsfähig.
Im Gegensatz zu Eukaryoten (wie Menschen) ist die Erbinformation der Bakterien, ihre DNA, nicht in einem Zellkern verpackt. Sie schwimmt frei im Inneren der Zelle, im sogenannten Zytoplasma.

Der Großteil des genetischen Materials ist in einem einzelnen DNA-Molekül, dem Bakterienchromosom, gespeichert. Zusätzlich besitzen Bakterien eigenständige, ringförmige DNA-Moleküle, sogenannte Plasmide. Diese Plasmide spielen eine besondere Rolle beim Gentransfer, da sie häufig wichtige Zusatzinformationen enthalten. [Gentechniken]
Durch Plasmide können Bakterien diese Zusatzinformationen untereinander austauschen. Dieser Gentransfer ermöglicht es Bakterien, rasch neue genetische Informationen zu erlangen, was ihnen Vorteile bei der Anpassung an die Umwelt, der Resistenz gegenüber Antibiotika oder der Fähigkeit zur Nutzung neuer Nahrungsquellen verschaffen kann. Dieser Mechanismus verleiht Bakterien die Fähigkeit, sich effektiv an veränderte Umweltbedingungen anzupassen und erfolgreich zu überleben.
Die Möglichkeit, genetisches Material in Bakterien einzuführen und zu manipulieren, insbesondere durch die Verwendung von Plasmiden, hat einen wichtigen Meilenstein in der Entwicklung der Gentechnik markiert.
Die Geburtsstunde der Gentechnik kann auf das Experiment von Stanley Cohen und Herbert Boyer im Jahr 1973 zurückgeführt werden. In diesem Experiment führten sie ein Plasmid in Bakterien ein und zeigten, dass das eingeschleuste Gen in den Bakterien exprimiert werden konnte; was bedeutet, das Gen wurde in den Bakterien aktiviert. Dies war der erste erfolgreiche Versuch, ein Gen von einer Art auf eine andere zu übertragen. Das verwendete Plasmid trug Antibiotika-Resistenzgene, die es den Wissenschaftlern ermöglichten, die transformierten Bakterien von den nicht transformierten zu unterscheiden. Diese bahnbrechende Arbeit bildete die Grundlage für die moderne Gentechnik. [Das Cohen-Boyer-Experiment]
b) Das Bakterium Escherichia coli (E. coli)
In der Wissenschaft und Forschung wird gern das Bakterium Escherichia coli (E. coli) verwendet und dafür gibt es mehrere Gründe:
- E. coli lässt sich leicht im Labor kultivieren und vermehren.
- Die Vermehrungszeit von E. coli ist relativ kurz, typischerweise im Bereich von 20 bis 30 Minuten.
- Das Genom von E. coli ist gut erforscht und verstanden.
- E. coli lässt sich leicht genetisch manipulieren.
- Die Kultivierung von E. coli ist im Vergleich zu einigen anderen Organismen kostengünstig.

Escherichia coli ist molekularbiologisch und genetisch der am besten untersuchte Organismus und wird deswegen auch das Haustier der Genetiker genannt.
Aus diesen Gründen wird E. coli auch in der Impfstoffentwicklung eingesetzt.
c) Plasmide
Plasmide sind zusätzliche DNA-Moleküle in Bakterien, die unabhängig vom Bakterienchromosom existieren. Sie sind kleiner als das Bakterienchromosom und können in der Zelle variabel in ihrer Anzahl sein. Plasmide liegen als doppelsträngige DNA vor. Sie sind in der Regel ringförmig, was bedeutet, dass die DNA in einem geschlossenen Kreis angeordnet ist. Diese ringförmige Struktur unterscheidet Plasmide von der linearen DNA, die im Zellkern von Eukaryoten (wie Menschen) vorkommt.

Ein Plasmid kann verschiedene genetische Informationen tragen. Es könnte zum Beispiel ein Gen für Antibiotika-Resistenz, ein Gen für die Produktion eines bestimmten Proteins oder andere nützliche Gene enthalten.
Damit Wissenschaftler sehen, welche Gene auf dem Plasmid vorliegen und wie sie angeordnet sind, benutzen sie sogenannte „Plasmidkarten“. Die Plasmidkarte ist eine schematische Darstellung der genetischen Struktur der Plasmid-DNA. Die folgende Abbildung soll grundlegende Begriffe erklären.
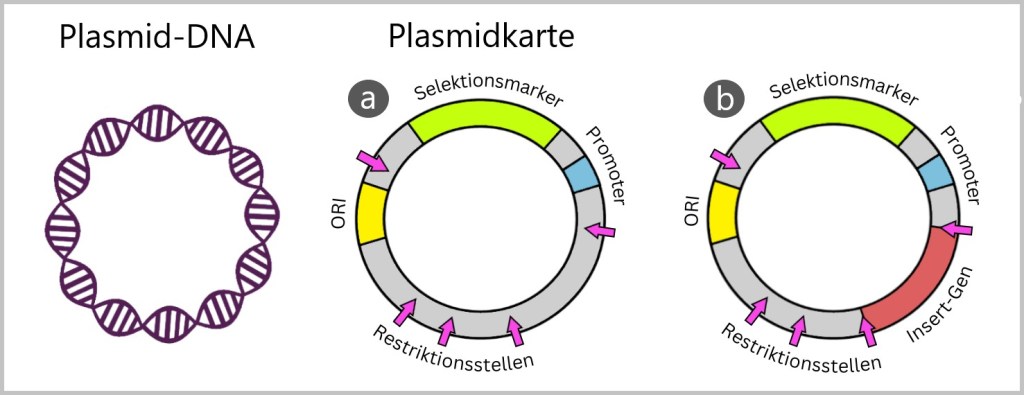
ORI (Origin of Replication): In der Plasmid-DNA gibt es einen besonderen Bereich, den man als „origin of replication“ (Replikationsursprung) oder kurz „ORI“ bezeichnet. Der Replikationsursprung ist wie der Startknopf für die Verdoppelung des Plasmids. Wenn das Bakterium merkt, dass es sich vermehren muss (wenn Umweltbedingungen und interne Signale günstig sind), kann es diesen Startknopf drücken, und das Plasmid macht eine Kopie von sich selbst. Damit das Plasmid sich erfolgreich vermehren kann, muss dieser ORI zum Bakterium passen, ähnlich wie ein Schlüssel zum Schloss. Wenn ORI und Bakterium kompatibel sind, kann sich das Plasmid selbstständig vermehren, auch unabhängig von der Zellteilung.
Selektionsmarker: Das sind besondere Gen-Abschnitte, die den Bakterien einen Überlebensvorteil unter bestimmten Bedingungen geben. Häufig sind dies Antibiotikaresistenzgene, die es den Bakterien erlauben, in einem Medium mit einem bestimmten Antibiotikum zu überleben.
Promoter: Der Promoter ist eine spezielle DNA-Region, die die Aktivität von Genen steuert. Er wirkt wie ein Startsignal, das den Beginn der Genexpression, also die Herstellung von Proteinen aus den Genen, ermöglicht. Der Promoter ist wichtig, weil er den Zeitpunkt und die Menge der Proteinsynthese beeinflusst. Er reguliert, wann und wie stark bestimmte Gene auf dem Plasmid abgelesen werden und steuert somit die Produktion der entsprechenden Proteine.
Restriktionsstellen: Restriktionsstellen sind bestimmte DNA-Sequenzen auf einem Plasmid, die durch Restriktionsenzyme erkannt und geschnitten werden können. Restriktionsenzyme sind Proteine, die dazu neigen, DNA an spezifischen Sequenzen zu schneiden. Bakterien nutzen Restriktionsstellen als Teil ihrer Verteidigungsstrategie. Wenn ein Bakterium auf eine fremde DNA stößt, die nicht zu ihm gehört, kann es Restriktionsenzyme einsetzen, um diese DNA zu schneiden und zu zerstören.
d) Plasmide in der Gentechnik
Obwohl Restriktionsenzyme ursprünglich Teil der bakteriellen Abwehr gegen fremde DNA sind, nutzen Wissenschaftler in der Gentechnik diese Enzyme gezielt für den Gentransfer.
Wenn ein Forscher beispielsweise DNA in ein Plasmid einführen möchte, kann er Restriktionsenzyme verwenden. Die Restriktionsenzyme sind wie winzige molekulare Scheren. Sie erkennen eine bestimmte Abfolge von DNA-Buchstaben (Nukleotiden) in der DNA. Diese Abfolge wird als Restriktionsschnittstelle bezeichnet. Wenn das Restriktionsenzym die spezifische DNA-Sequenz findet, bindet es sich daran und schneidet die DNA an dieser spezifischen Stelle. Es schneidet beide DNA-Stränge, um die DNA an dieser Stelle zu unterbrechen. Durch den Schnitt entstehen zwei getrennte DNA-Fragmente. Diese Fragmente können weiter verwendet werden, indem man eine fremde DNA in diese Lücke einsetzt und die Enden mit einem Enzym namens DNA-Ligase wieder verbindet.
Dieser Mechanismus wird als Restriktionsverdaulichkeit bezeichnet. Er ermöglicht es, fremde DNA in ein Plasmid einzuführen. Es ist eine Schlüsseltechnologie in der Gentechnik, um gezielte genetische Manipulationen durchzuführen und Gene in Bakterien zu übertragen.
Die Abb.13-b zeigt eine Plasmidkarte mit einem eingefügten Gen, auch als Insert-Gen bezeichnet.
Insert-Gen: Das Insert-Gen ist das eigentliche Gen oder die genetische Sequenz, die in das Plasmid eingeführt wird. Dies kann ein Gen sein, das für ein bestimmtes Protein kodiert, oder eine andere genetische Information, die untersucht oder exprimiert werden soll.
Eine kurze, animierte Zusammenfassung darüber, was ein Plasmid ist und wofür es verwendet wird, findet sich hier.
e) Plasmide in der Impfstoffherstellung
Die Fähigkeit des Plasmids, sich selbst zu vervielfältigen, macht es für die Forschung und Biotechnologie sehr interessant. Man kann es nutzen, um eine bestimmte Fremd-DNA, die man vorher in das Plasmid eingebracht hat, in großen Mengen zu produzieren.
f) Einbau der Spike-Protein-DNA in das Plasmid
Die DNA für das Spike-Protein des SARS-CoV-2 wird in das Plasmid eingefügt; manchmal sagt man auch, es wird „einkloniert“. Spezielle Enzyme wirken wie genetische Scheren (Restriktionsenzyme) und Klebstoffe (Ligasen). Sie schneiden das Plasmid an einer bestimmten Stelle auf und fügen das Spike-Protein-Gen ein. Nachdem das Gen an seinem Platz ist, wird das Plasmid wieder geschlossen.
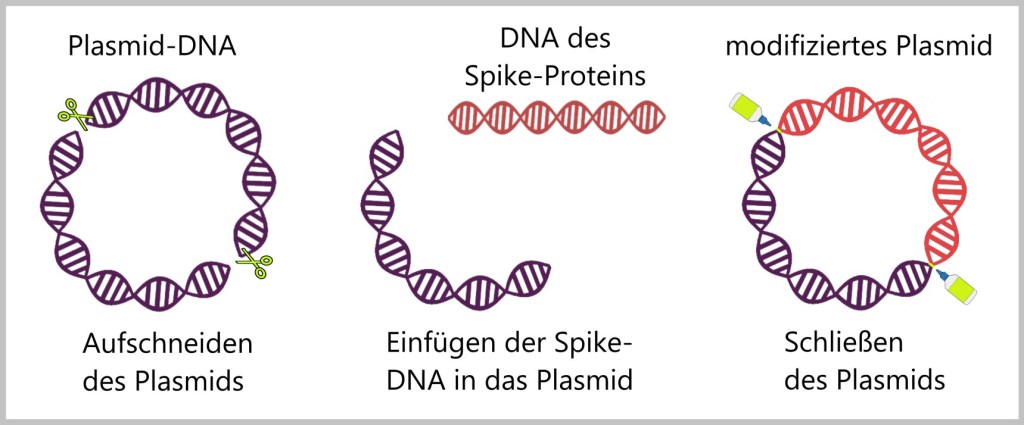
Die Spike-DNA wird jedoch nicht allein in das Plasmid eingebracht, sondern zusammen mit anderen genetischen Elementen.
Wie bereits zu Beginn erwähnt, wurden von Speicher et al. bei der Untersuchung der Impfstoffe von Pfizer und Moderna auch andere Sequenzen gefunden, darunter ORI, SV40-Komponenten und ein Antibiotikaresistenzgen.
Schauen wir uns eine vereinfachte Darstellung der Plasmidkarten von Pfizer und Moderna an. Eine genauere Darstellung findet man hier.
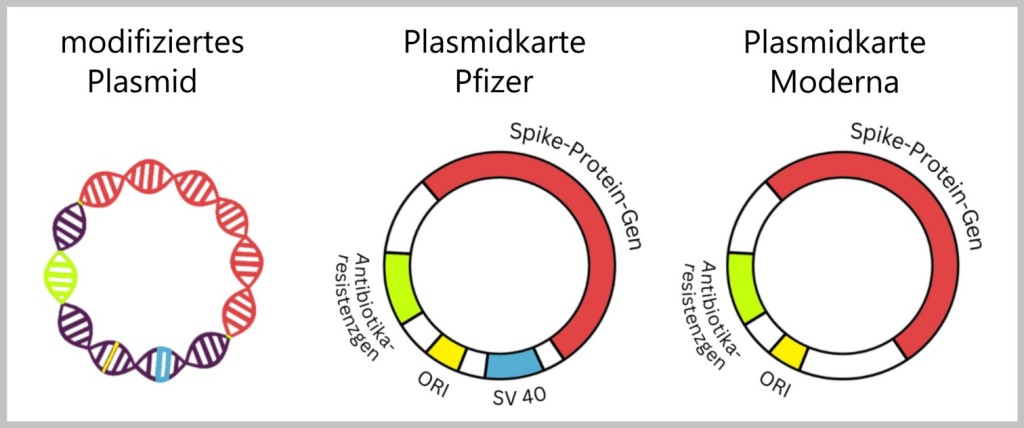
ORI (Origin of Replication): Der ORI dient als Startpunkt für die DNA-Replikation. Er liefert Signale, die dem Replikationsapparat mitteilen, wo er mit der Verdopplung der DNA beginnen soll.
Spike-Protein-Gen: Das Spike-Protein-Gen kodiert für das Spike-Protein, das auf der Oberfläche des SARS-CoV-2-Virus vorhanden ist und eine wichtige Rolle bei der Infektion von Wirtszellen spielt.
SV40-Gen-Komponenten: SV40 steht für Simianes Virus 40, ein Virus, das in Affen gefunden wurde. SV40 kann sowohl Affen als auch Menschen infizieren. Das SV40-Enhancer-Gen und der SV40-Origin of Replication (Ori) werden häufig in der Molekularbiologie als Werkzeuge verwendet, um die Expression von Genen und die Replikation von DNA in Zellen zu fördern. (Die Expression von Genen bezieht sich auf den Prozess, bei dem die genetische Information eines Gens in ein funktionales Protein umgewandelt wird.) SV40-Gen-Komponenten wurde nur im Pfizer-Impfstoff gefunden.
Antibiotikaresistenzgen: Dieses Gen kodiert für eine Eigenschaft, die Bakterien vor Antibiotika schützen. In der Genetik wird es häufig als Selektionsmarker verwendet. Nur diejenigen Bakterien, die das fremde Gen über das Plasmid aufgenommen haben, überleben in einer Umgebung mit dem entsprechenden Antibiotikum. Damit ist es möglich, im Produktionsprozess nur die Bakterien zu bekommen, die das modifizierte Plasmid erfolgreich aufgenommen haben. Moderna verwendet das „Kanamycin-Resistenzgen“ und Pfizer das „Neo/Kan-Resistenzgen“. Neo/Kan-Resistenzgen ist eine Abkürzung für ein Gen, das für die Resistenz gegenüber den Antibiotika Neomycin und Kanamycin verantwortlich ist.
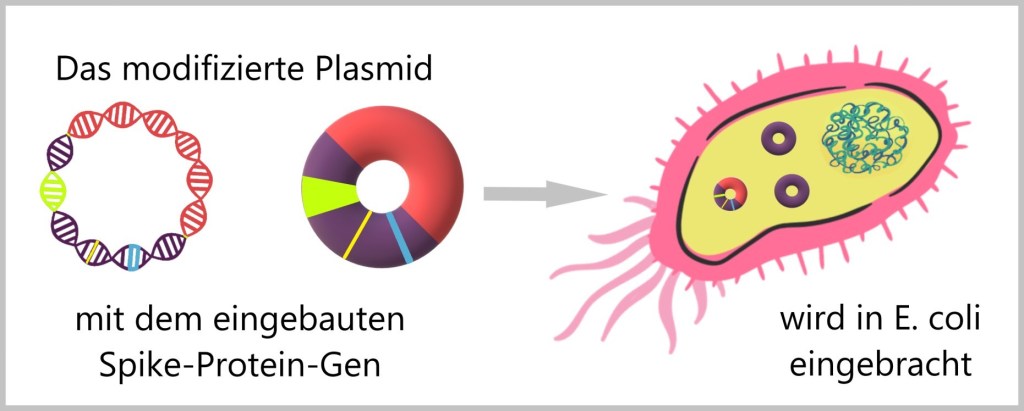
g) Übertragung der modifizierten Plasmide in Bakterien
Die so modifizierten Plasmide, die jetzt das Spike-Protein-Gen tragen, werden dann in die E. coli Bakterien eingeführt. Dieser Prozess wird als Transformation bezeichnet. Die Bakterien nehmen das Plasmid auf und integrieren es in ihre Zellstruktur.
In der Molekularbiologie und Gentechnik werden Plasmide oft als Vektoren bezeichnet, wenn sie dazu verwendet werden, fremde genetische Information zu transportieren und zu vervielfältigen. Vektoren dienen als Transportmittel zur Übertragung von Fremd-DNA in eine Wirtszelle (Bakterium).
h) Vermehrung der Bakterien
Die Bakterien kommen nun in einen Fermenter. Ein Fermenter, auch als Bioreaktor bezeichnet, ist ein Gerät, das in der Biotechnologie verwendet wird, um Produkte wie Antibiotika, Enzyme, Vitamine oder Impfstoffe herzustellen. Mit ihnen ist die großtechnische Herstellung möglich. Ein Fermenter ermöglicht die präzise Kontrolle von Faktoren wie Temperatur, pH-Wert, Belüftung, Rührgeschwindigkeit, Nährstoffzufuhr und anderes.
Das Nährmedium im Fermenter enthält alle notwendigen Nährstoffe für die Bakterien. Unter diesen geeigneten Bedingungen beginnen die Bakterien, sich zu vermehren. Während dieser Vermehrung replizieren sich auch die Plasmide innerhalb der Bakterien.
Damit sich nur die Bakterien vermehren, die das Plasmid mit dem Spike-Protein-Gen aufgenommen haben, wird den Bakterien im Fermenter noch das Antibiotikum zugeführt. Nur die Bakterien überleben, die das modifizierte Plasmid erfolgreich eingefügt haben, da sich das Spike-Protein-Gen im gleichen Plasmid befindet wie die Antibiotikaresistenz (siehe Abb.15 und 16).
E. coli-Bakterien können sich alle 20-30 Minuten einmal teilen. Innerhalb von wenigen Tagen hat sich im Fermenter eine Kolonie von Trillionen Bakterien mit modifizierten Plasmiden entwickelt, die die Erbinformation des Spikeproteins tragen.
i) Ernte der Bakterien
Nachdem die E. coli-Zellen das Plasmid mit der SARS-CoV-2-Spike-Sequenz produziert haben, werden sie geerntet. „Ernten“ bezieht sich auf den Prozess, bei dem Zellen aus dem Kulturmedium entfernt und gesammelt werden. Das Sammeln geschieht normalerweise durch Zentrifugation, wodurch die Zellen am Boden eines Behälters konzentriert werden.
j) Isolierung der modifizierten Plasmide
Die Isolierung der modifizierten Plasmide erfolgt durch Lyse der Bakterien, gefolgt von verschiedenen Reinigungsschritten, um Verunreinigungen zu entfernen. „Lysieren“ bezeichnet den Prozess des gezielten Aufbrechens oder Zerstörens der Zellmembran von E. coli-Zellen, um den zellulären Inhalt freizusetzen.

Das Aufbrechen der Zellmembran während des Lysierungsprozesses führt dazu, dass der gesamte zelluläre Inhalt, bestehend aus DNA, Proteinen und anderen Molekülen, freigesetzt wird. Nach der Lyse befinden sich der zelluläre Inhalt der Bakterien sowie der verbliebene Rest der Zellmembran in der Lösung (Abb.17 und 18).

Daher erfolgt nach der Lyse die Reinigung des extrahierten Materials durch verschiedene Verfahren, um die modifizierten Plasmide zu isolieren. Reinigungsschritte entfernen unerwünschte Verunreinigungen, wie z.B. Proteine und andere zelluläre Bestandteile (siehe dazu Abschnitt 3.5.2. Reinigung der synthetischen mRNA im Prozess 2).
Die Plasmide liegen zu diesem Zeitpunkt in ihrer ringförmigen Struktur vor.
k) Linearisierung der Spike-Protein-DNA
Das isolierte Plasmid wird nun gezielt mithilfe eines Restriktionsenzyms geschnitten. Dieses Restriktionsenzym erkennt die spezifische DNA-Sequenz des SARS-CoV-2-Spike und schneidet es aus dem Plasmid. Durch den Schnitt entsteht eine lineare Form der SARS-CoV-2-Spike-Sequenz, die nun für die Transkription besser zugänglich ist. Lineare DNA wird in der Regel besser von den Enzymen erkannt, die für die Transkription verantwortlich sind.
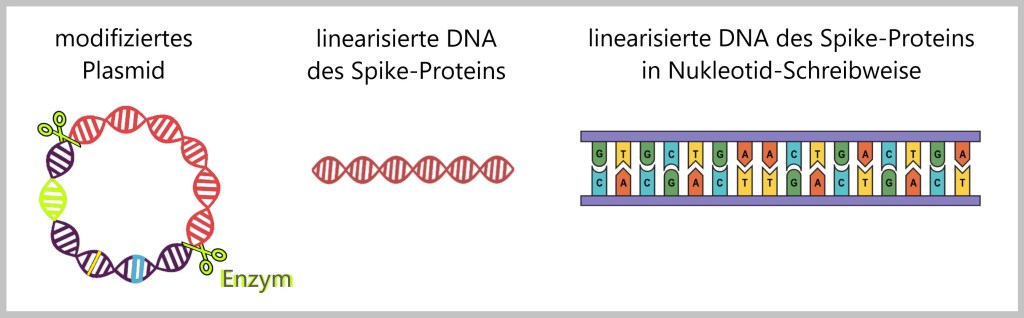
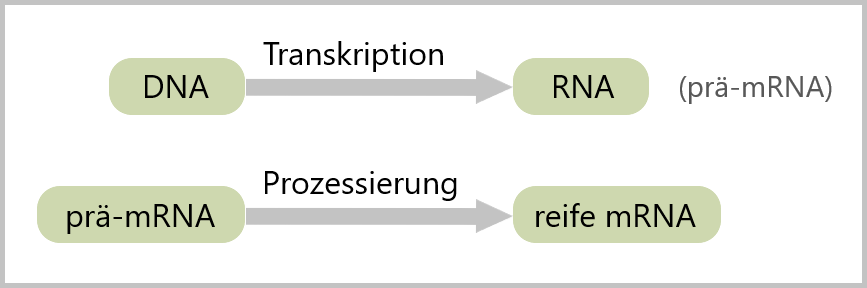
3.3. Transkription zur Herstellung der RNA

Die Transkription ist der Prozess, bei dem aus der DNA die RNA synthetisiert wird oder anders formuliert, Transkription ist das „Umschreiben“ der DNA in RNA. Die in-vitro Transkription (IVT) erfolgt mithilfe von Enzymen, die als RNA-Polymerasen bezeichnet werden (siehe Abb.20).

Die Transkription besteht aus drei Hauptphasen:
Initiation (Start): Die RNA-Polymerase setzt sich am Anfang der DNA-Matrize fest. Dabei erkennt sie spezifische DNA-Sequenzen, bekannt als Promotoren, die den Startpunkt der Transkription definieren. Um mit der Transkription zu beginnen, wird die DNA-Matrize zuerst denaturiert. Das bedeutet, dass die Doppelhelix der DNA aufgetrennt wird, um die einzelsträngige DNA für die RNA-Synthese zugänglich zu machen.
Elongation (Verlängerung): Nun bewegt sich die RNA-Polymerase entlang der DNA-Matrize und liest sie ab. Polymerasen bewegen sich von 5′ nach 3′ (mehr dazu hier). Während des Ablesens baut die RNA-Polymerase mit den RNA-Nukleotiden (den Bausteinen der RNA) den RNA-Strang auf. Dies geschieht durch die Bildung komplementärer Basenpaare zwischen den RNA-Nukleotiden und den Basen der DNA-Matrize. Die Basenpaarung erfolgt zwischen Adenin (A) und Uracil (U), sowie zwischen Cytosin (C) und Guanin (G).
Termination (Beendigung): Die Transkription endet an einem spezifischen Stopp-Signal auf der DNA. Die RNA-Polymerase löst sich von der DNA, und die synthetisierte RNA wird freigesetzt.
Aus der doppelsträngigen DNA ist eine einzelsträngige RNA entstanden. DNA und RNA ähneln sich in Zusammensetzung und Struktur, weisen aber entscheidende Unterschiede auf:
Funktion: Die DNA enthält die genetische Information, die für den Aufbau und die Funktion eines Organismus erforderlich ist. Die RNA ist für die Umsetzung der genetischen Informationen in Proteine zuständig.
Struktur: Die DNA ist normalerweise als Doppelhelix angeordnet, wobei zwei Stränge miteinander verflochten sind. Die RNA ist in der Regel als einzelner Strang vorhanden, obwohl sie bestimmte regionale Doppelhelix-Strukturen annehmen kann.
Basen: Beide enthalten Adenin (A), Cytosin (C) und Guanin (G), aber in der DNA wird Thymin (T) verwendet, während in der RNA Uracil (U) anstelle von Thymin vorkommt. Dies bedeutet, dass die Basenpaarung in der DNA Adenin-Thymin (A-T) und Cytosin-Guanin (C-G) ist, während es in der RNA Adenin-Uracil (A-U) und Cytosin-Guanin (C-G) ist.
Stabilität und Lebensdauer: Die DNA ist in der Regel stabiler und hat eine längere Lebensdauer als RNA. RNA-Moleküle können in der Zelle schneller abgebaut werden, und das liegt an der Base Uracil. Uracil ist eine instabile Base, die leicht durch Enzyme abgebaut werden kann.
Warum benutzt der Organismus Uracil in der RNA?
Der schnelle Abbau von RNA hilft der Zelle, sich rasch an Veränderungen anzupassen. Die kurze Lebensdauer von RNA ermöglicht es der Zelle, ihre genetische Information zügig zu verändern, was besonders wichtig ist für Prozesse wie Zellteilung und Anpassung an neue Umweltbedingungen. Uracil ermöglicht es der Zelle auch, Gene schnell ein- oder auszuschalten, was entscheidend ist, um sich an unterschiedliche Bedingungen anzupassen. Zusammengefasst kann man sagen, Uracil in RNA verleiht der Zelle mehr Flexibilität und Kontrolle über ihre genetische Information, was ihre Anpassungsfähigkeit und Überlebensfähigkeit entscheidend beeinflusst.
Uracil ist eine Komponente von Uridin, einer Verbindung, die zusätzlich Zucker enthält. Uridin ist somit ein Molekül, das aus Uracil und Zucker besteht, beide sind Bestandteile der RNA (siehe Abb.21).

Für die Herstellung von mRNA-Impfstoffen nutzt man Pseudouridin anstelle von Uridin, um eine stabile RNA zu gewährleisten, die langsamer abgebaut wird. [transkript]
Sowohl die mRNA-Impfstoffe von Pfizer als auch von Moderna enthalten Pseudouridin mψU anstelle von Uridin. [spektrum]
Während der Transkription wird das synthetische Nukleosid N-Methyl-Pseudouridin (mψU) anstelle des natürlichen Nukleosids Uridin in die künstliche RNA eingebaut (siehe Abb.22).

Bei der Transkription können jedoch neben der gewünschten RNA auch unerwünschte Nebenprodukte wie DNA-RNA-Hybride und doppelsträngige RNA (dsRNA) entstehen.
DNA-RNA-Hybride
Die Bildung von DNA-RNA-Hybriden während der Transkription erfolgt, wenn der entstehende RNA-Strang sich an einen DNA-Einzelstrang bindet. Dieses Nebenprodukt besteht aus einem Teil DNA und einem Teil RNA, was zu einem Hybridmolekül führt.

Nach der in-vitro Transkription (IVT) erfolgt der Abbau dieser DNA-RNA-Hybride nicht spontan wie in lebenden Zellen. Um solche Nebenprodukte zu entfernen, werden spezifische Reinigungsschritte (siehe Abschnitt 3.5.2.) durchgeführt.
Doppelsträngige RNA (dsRNA)
Auch die Bildung von doppelsträngiger RNA (dsRNA) während der in-vitro-Transkription (IVT) von Messenger-RNA (mRNA) ist ein unerwünschtes Nebenprodukt. [NIH – Doppelsträngige RNA…]
Wie bereits oben beschrieben, setzt die Transkription erst ein, sobald die DNA-Matrize denaturiert wird, also in zwei einzelne Stränge aufgetrennt wird. An diesem Punkt bindet die RNA-Polymerase an die entstandene einzelsträngige DNA-Matrize und initiiert die Synthese eines komplementären RNA-Strangs. Unter spezifischen Bedingungen, wie etwa in einer denaturierenden Umgebung, können die frisch synthetisierten RNA-Stränge dazu neigen, miteinander zu interagieren und eine doppelsträngige Struktur zu bilden. Dies kann auf zwei Arten geschehen:
Intermolekulare Basenpaarung: Zwei RNA-Stränge, die von verschiedenen DNA-Matrizen synthetisiert wurden, können miteinander interagieren und Basenpaarungen bilden.
Intramolekulare Basenpaarung: Ein einzelner RNA-Strang kann sich selbst falten und mit sich selbst Basenpaarungen bilden.

Für die Entfernung solcher Nebenprodukte werden spezifische Reinigungsschritte (siehe Abschnitt 3.5.2.) angewendet.
3.4. RNA-Prozessierung

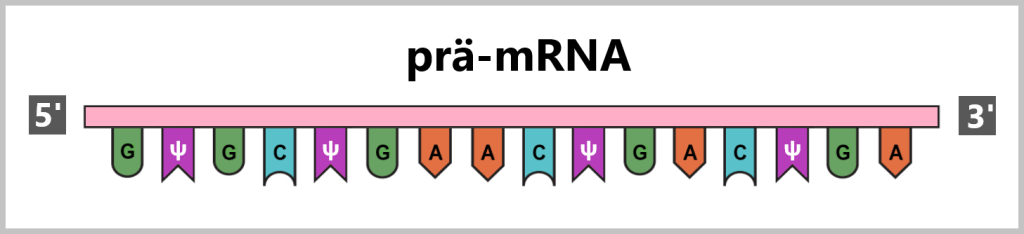
RNA-Prozessierung umfasst eine Reihe von Veränderungen, die nach der Transkription stattfinden, um aus der RNA (prä-mRNA) eine reife, funktionale mRNA zu erzeugen.
Bei der Herstellung der mRNA für Impfstoffe wird versucht, die natürlichen Prozesse der Transkription und post-transkriptionellen Modifikationen nachzuahmen, die normalerweise auch in Zellen bei Eukaryoten (wie Menschen) stattfinden. Die synthetisch hergestellte mRNA wird so gestaltet, dass sie bestimmte Eigenschaften von natürlich vorkommender mRNA imitiert.
Eine funktionelle mRNA, die als Impfstoff dient, muss den Aufbau einer normalen zellulären mRNA aufweisen, einschließlich einer speziellen Schutzkappe am 5′-Ende und einem Stabilisierungsschwanz am 3′-Ende. Diese Modifikationen sind wichtig für die Stabilität der mRNA und ihre effiziente Übersetzung in Proteine.
Daher umfassen die Modifikationen der prä-mRNA das Hinzufügen der Schutzkappe (mRNA-Kapping) und des Stabilisierungsschwanzes (Polyadenylierung).
mRNA-Kapping: Das mRNA-Kapping erfolgt während der Transkription, während die RNA-Polymerase den DNA-Strang abliest und die mRNA synthetisiert. Sobald die mRNA ein kurzes Stück synthetisiert hat, wird die Schutzkappe, bestehend aus 7-Methylguanosin, am 5′-Ende der mRNA hinzugefügt. Diese Modifikation schützt die mRNA vor dem Abbau durch nukleolytische Enzyme.
Polyadenylierung: Die Polyadenylierung erfolgt kurz nach der Transkription, indem eine lange Kette von Adenin-Nukleotiden an das 3′-Ende der mRNA gebunden wird. Dieser Poly-A-Schwanz erhöht die Stabilität der reifen mRNA und schützt sie vor dem Abbau. Der Poly-A-Schwanz kann mehrere hundert Nukleotide lang sein. Wie oft eine mRNA abgelesen wird, ist durch den Poly-A-Schwanz reguliert, da er sich bei der Translation (dem Prozess, bei dem anhand der mRNA-Vorlage das Protein hergestellt wird) kontinuierlich verkürzt. Dies erfolgt so lange, bis der Poly-A-Schwanz vollständig entfernt ist und es zum Abbau der mRNA kommt.
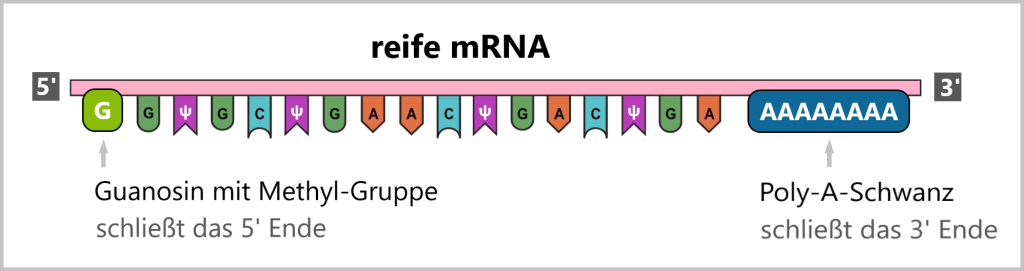
3.5. Reinigung der synthetischen mRNA
3.5.1. Reinigung der synthetischen mRNA im Prozess 1
3.5.2. Reinigung der synthetischen mRNA im Prozess 2

3.5.1. Reinigung der synthetischen mRNA im Prozess 1
Die Reinigung von PCR-Produkten und in-vitro transkribierter RNA ist ein wichtiger Schritt, um Verunreinigungen wie nicht eingebaute Nukleotide, DNA-Matrizen und Enzyme zu entfernen.
Für die Reinigung stehen mehrere Methoden zur Verfügung. Aus den Informationen zum Herstellungsverfahren laut EMA „EPAR zum Covid-19-Impfstoff von BioNTech/Pfizer“ (in Abschnitt 2.2. auf Seite 32) kommt bei ‚Prozess 1‘ die Reinigung mittels Magnetkügelchen zum Einsatz.
Magnetkügelchen-Reinigung
Für die Reinigung von mRNA werden magnetische Kügelchen in der Regel so hergestellt, dass sie selektiv an RNA binden. Das kann durch die Verwendung von speziellen Sonden erreicht werden, die an die Kugeloberfläche gebunden sind. Diese Sonden bestehen aus einer Sequenz von Thyminen (T), die komplementär zu den Adeninen (A) im Poly-A-Schwanz der mRNA sind (siehe Abb.27).

Der Reinigungsprozess läuft dann folgendermaßen ab (siehe Abb.28).
(a) Die Magnetkügelchen, beschichtet mit spezifischen Thymin-Molekülen, werden zur Impfstofflösung hinzugefügt, die die gewünschte mRNA enthält. (b) Die Moleküle auf den Magnetkügelchen haben eine Affinität zu den Zielmolekülen der mRNA, den Adeninen am Poly-A-Schwanz, und binden selektiv an diese. (c) Nachdem die Magnetkügelchen die Zielmoleküle gebunden haben, wird ein Magnetfeld angelegt. Die Magnetkügelchen mit den gebundenen mRNAs werden vom Magnet angezogen und an einen bestimmten Bereich des Gefäßes gezogen. (d) Mehrere Waschschritte werden durchgeführt, um nicht gebundene Verunreinigungen zu entfernen, wobei frische Pufferlösung hinzugefügt wird. Eine Pufferlösung ist eine Flüssigkeit, die dafür sorgt, dass die Bedingungen der Zielmoleküle und den Magnetkügelchen genau richtig sind, damit sie miteinander arbeiten können. Gleichzeitig hilft sie dabei, den pH-Wert der Flüssigkeit stabil zu halten, sodass alles gut funktioniert. (e) Um die gebundene mRNA von den Magnetkügelchen zu trennen, wird eine spezielle Lösung, ein Elutionspuffer, hinzugefügt. Die Bedingungen im Elutionspuffer führen dazu, dass die Magnetkügelchen die gebundenen mRNAs freisetzen. Die freigesetzten mRNAs können nun in einer konzentrierten Form für nachfolgende Verarbeitungsschritte verwendet werden.
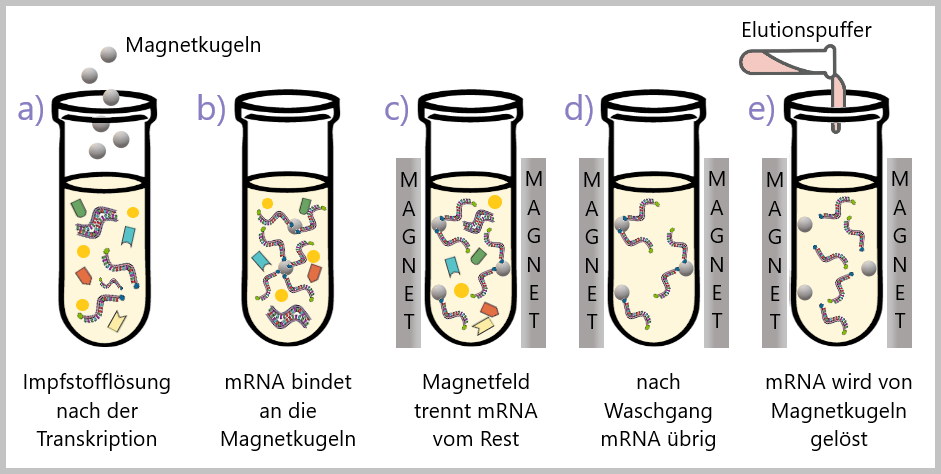
Weitere ausführliche Informationen zur Magnetkügelchen-Reinigung findet man hier und hier.
Nach den offiziellen Angaben wird die Reinigung mittels Magnetkügelchen nur im Rahmen von ‚Prozess 1‘ durchgeführt. Mit der Umstellung auf die Großserienproduktion (Prozess 2) ändert sich auch das Reinigungsverfahren. Diese Änderung könnte mit der Magnetkraft zusammenhängen, die eine entscheidende Rolle für eine präzise Trennung bei großen Volumina spielt. Es wird darauf hingewiesen, dass einfache Magnete als begrenzt angesehen werden, da ihre Magnetkraft mit zunehmender Entfernung schnell abnimmt. [sepmag]
3.5.2. Reinigung der synthetischen mRNA im Prozess 2
Während der Herstellung der mRNA durch Plasmide und der in-vitro-Transkription werden verschiedene Verunreinigungen eingeführt oder erzeugt. [ScienceDirect]
Daher ist es im Anschluss erforderlich, die Impfstofflösung mit der gewünschten mRNA von potenziellen Verunreinigungen und anderen Materialien zu reinigen. Zu den potenziellen Verunreinigungen gehören Überreste des DNA-Templates, doppelsträngige RNA, DNA-RNA-Hybride, Endotoxine, überschüssige RNA-Polymerase und grundlegende Verunreinigungen. [securecell]
Endotoxine: Endotoxine sind toxische Bestandteile von Bakterien, genauer gesagt von gramnegativen Bakterien wie dem Bakterium Escherichia coli (E. coli). Diese Endotoxine sind Bestandteile der äußeren Membran von Bakterien und können starke immunologische Reaktionen im Organismus hervorrufen.
DNA-Templates: Die DNA-Templates (SARS-CoV-2-Spike-Sequenz) werden während der Transkription abgelesen und dienen als Matrize (Vorlage) für die RNA-Synthese. Die Überreste dieser DNA-Templates sind in der Impflösung noch vorhanden und müssen daher sorgfältig entfernt werden, um Reinheit und Sicherheit zu gewährleisten.
Doppelsträngige RNA (dsRNA): Während des Transkriptionsprozesses kann es vorkommen, dass doppelsträngige RNA (dsRNA) als Zwischenprodukt entsteht. [PubMed]
Eine doppelsträngige RNA wird als immunogen betrachtet, weil sie in vielen Organismen, einschließlich Säugetieren, als potenzieller Indikator für eine Virusinfektion erkannt wird. Es gibt verschiedene Arten von Rezeptoren, die doppelsträngige RNA als fremd erkennen und eine Immunantwort auslösen können.
DNA-RNA-Hybride: Während des Transkriptionsprozesses kann es zu Hybridisierung zwischen dem DNA-Template und der neu synthetisierten RNA kommen. Die RNA kann sich teilweise an die DNA binden und so einen DNA-RNA-Hybrid bilden. Wenn solche Hybride in der Impfstofflösung verbleiben, könnten sie möglicherweise unerwünschte immunologische Reaktionen oder andere unvorhergesehene Effekte auslösen.
RNA-Polymerase: Überschüssige RNA-Polymerase, das Enzym, das die Transkription durchführt, kann eine potenzielle Verunreinigung darstellen. Eine Überexpression oder unvollständige Entfernung dieses Enzyms könnte zu unerwünschten Effekten führen. (Enzyme gehören zu den Proteinen.)
Grundlegende Verunreinigungen: Dieser Begriff bezieht sich auf Verunreinigungen, die während der verschiedenen Herstellungsschritte hinzugefügt werden können. Dazu gehören Puffer, Enzyme, Proteine oder andere chemische Substanzen, die während des Herstellungsprozesses verwendet werden.
Es stehen verschiedene Methoden zur Verfügung, um die transkribierte mRNA zu reinigen. Laut den Angaben der Europäischen Arzneimittel-Agentur (EMA) „EPAR zum Covid-19-Impfstoff von BioNTech/Pfizer“ (Abschnitt 2.2, Seite 32) werden im Rahmen des Herstellungsverfahrens von ‚Prozess 2‘ der Proteinase-K-Aufschluss und UFDF-Schritte angewendet. Ein weiterer Reinigungsschritt, der in dem EMA Dokument (auf Seite 17 und 40) aufgeführt wird, ist die DNase I.
Darüber hinaus erwähnen führende Biotechnologieunternehmen wie MERCK und SARTORIUS die Chromatographie als einen weiteren Reinigungsschritt bei der Produktion von mRNA-Impfstoffen.
Die offiziellen Dokumente geben keine detaillierten Einblicke in die genauen Schritte der Reinigung während des gesamten Herstellungsprozesses der aktuellen COVID-19 mRNA-Impfstoffe. Da das Hauptziel dieses Artikels darin besteht, grundlegendes Wissen auf diesem Gebiet allgemein verständlich zu vermitteln, werden im folgenden Abschnitt nur beispielhaft Reinigungsschritte an einem bestimmten Punkt des Prozesses beschrieben. Es ist wichtig zu beachten, dass die genaue Zusammensetzung und die Reihenfolge der Reinigungsschritte unter realen Produktionsbedingungen variieren können und oft viel komplexer sind. [QdB] Weitere vertiefende Informationen zu diesem Thema sind in den Veröffentlichungen von MERCK (Herstellungsstrategien für mRNA-Impfstoffe und Therapeutika) und SARTORIUS (Overview of Sartorius Solutions serving mRNA Processing, Seite 7) zu finden.
Im weiteren Verlauf betrachten wir folgende Reinigungsschritte:
a) DNase I – Behandlung
b) Proteinase K – Behandlung
c) UF/DF-Schritte
d) Chromatographie
a) DNase I-Behandlung
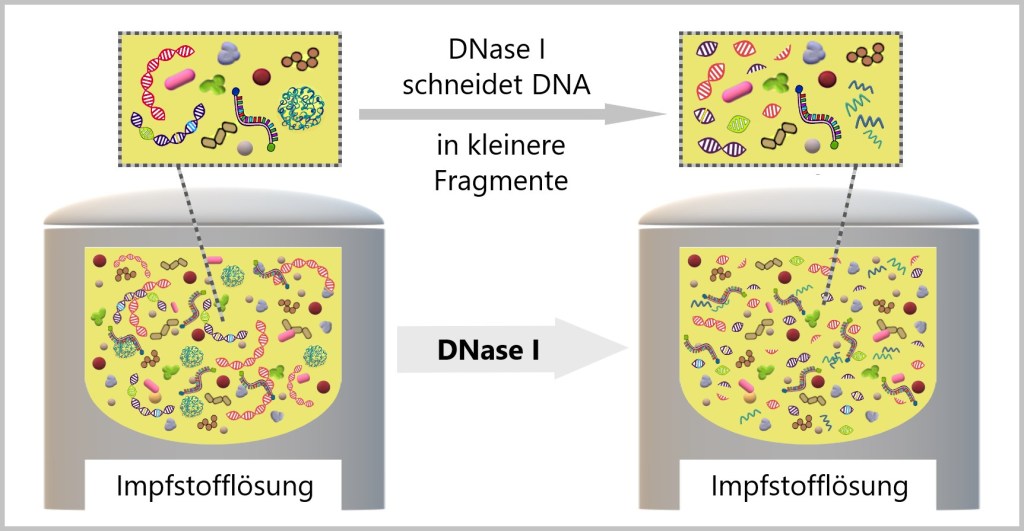
DNasen sind Enzyme, die in der Lage sind, DNA zu zerschneiden. In diesem Kontext wird auch von ‚Spalten‘ und ‚Verdauen‘ der DNA gesprochen. DNasen wirken wie winzige Scheren und schneiden die DNA in kleinere Stücke. Es gibt verschiedene Arten von DNasen, die sich darin unterscheiden, ob sie die DNA an bestimmten oder unspezifischen Stellen schneiden können.
Die DNase I, die oft aus Rinderpankreas gewonnen wird, schneidet die DNA unspezifisch, das heißt, sie kann an verschiedenen Stellen des DNA-Strangs zuschlagen.
In der Impfstoffherstellung wird DNase I verwendet, um die vorhandene DNA in der Lösung in kleinere Fragmente zu schneiden. Diese kleineren DNA-Fragmente können dann leichter durch nachfolgende Reinigungsschritte entfernt werden.
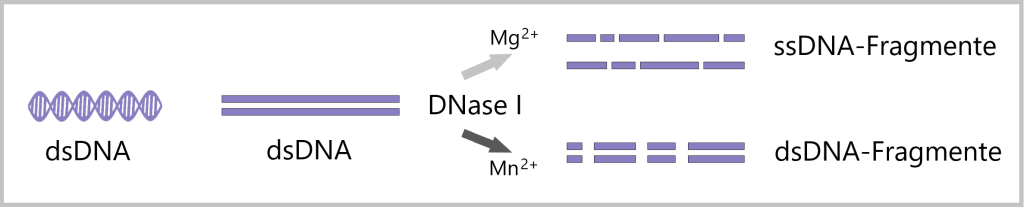
Die Wirkung der DNase I auf DNA kann je nach den Anwesenheiten von Magnesium-Ionen (Mg2+) und Mangan-Ionen (Mn2+) unterschiedlich sein. Die Art der Ionen beeinflusst die spezifischen Schnittmuster der DNase I, was zu verschiedenen Arten von DNA-Fragmenten führen kann, einschließlich sowohl einzelsträngiger (ssDNA) als auch doppelsträngiger (dsDNA) DNA-Fragmente. [bioswisstec, YEASEN]
In Gegenwart von Mg2+ schneidet die DNase I die doppelsträngige DNA unabhängig voneinander an zufällig gewählten Stellen. Dies führt zu einzelsträngigen DNA-Fragmenten (ssDNA-Fragmenten).
In Gegenwart von Mn2+ schneidet die DNase I beide dsDNA-Stränge ungefähr an der gleichen Stelle. Dies kann zur Bildung von doppelsträngigen DNA-Fragmenten (dsDNA-Fragmenten) führen, wenn die geschnittenen Fragmente sich überlappen oder gemeinsame Bereiche haben. Es können auch einzelsträngige DNA-Fragmente (ssDNA-Fragmente) mit überhängenden Enden entstehen.
Der Pfizer-BioNTech-Zulieferer Thermo Fisher Scientific erörtert in einem Online-Artikel die Wirksamkeit von DNase I. Der Text verdeutlicht, dass die Effektivität von DNase I von verschiedenen Faktoren abhängt, wie beispielsweise der Ausgangs-DNA-Kontamination. Es wird erläutert, dass DNase I in der Lage ist, doppelsträngige DNA (dsDNA) effizient zu spalten, während seine Aktivität für einzelsträngige DNA (ssDNA) um das 500-fache geringer ist. Wenn es um die Aktivität auf RNA-DNA-Hybriden geht, beträgt sie weniger als 1–2 % der Aktivität auf dsDNA. Der Beitrag weist darauf hin, dass es höchstwahrscheinlich schwierig ist, bei der Vorbereitung von RNA-Proben jede einzelne DNA-Molekülkette vollständig zu entfernen.
b) Proteinase K – Behandlung
Proteinase K ist ein Enzym, das zur Klasse der Proteinasen gehört. Diese Enzyme haben die Fähigkeit, Proteine zu hydrolysieren, das heißt, sie in kleinere Fragmente wie Peptide oder Aminosäuren zu zerlegen. Die Proteine in der Impfstofflösung können sowohl von bakteriellen Bestandteilen als auch von den während des Herstellungsprozesses verwendeten Hilfsstoffen und Stabilisatoren stammen.
Der Wirkmechanismus von Proteinase K beginnt mit der Bindung an das Protein. Nach dieser Bindung spaltet Proteinase K die Proteine in kleinere Fragmente auf. Anschließend werden die entstandenen Fragmente aus dem Enzym freigesetzt.

Nach der Inaktivierung der Proteinase K erfolgt oft eine Aufreinigung der Impfstofflösung, um die unerwünschten Rückstände zu entfernen. Filtrationsschritte können verwendet werden, um die kleineren Fragmente zu entfernen.
c) Reinigungsschritt: UF/DF
Die Abkürzungen ‚UF‘ und ‚DF‘ stehen für ‚Ultrafiltration‘ und ‚Diafiltration‘. Die Ultrafiltration trennt Moleküle basierend auf ihrer Größe, während die Diafiltration hilft, Verunreinigungen weiter zu entfernen und das gewünschte Produkt zu konzentrieren. Zusammen werden sie als UF/DF-Prozess bezeichnet. Die Kombination von Ultrafiltration/Diafiltration (UF/DF) ist eine gängige Methode zur Aufreinigung von biologischen Produkten wie der mRNA.
Ultrafiltration ist ein Prozess, der eine spezielle Art von Filtration mit einer semipermeablen (halbdurchlässigen) Membran mit definierter Porengröße beinhaltet. Durch diese Membran können Moleküle je nach ihrer Größe getrennt werden. Kleinere Moleküle können die Membran passieren, während größere Moleküle, wie z.B. mRNA, zurückgehalten werden.

Bei der Diafiltration wird während der Ultrafiltration ständig frischer Puffer (eine spezielle Flüssigkeit oder Impfstofflösung) hinzugefügt. Dies hilft, Verunreinigungen weiter zu verdünnen und beispielsweise die mRNA in einer geeigneten Pufferlösung zu konzentrieren.
Die vorherrschende Technik für die Ultrafiltration/Diafiltration (UF/DF) ist die Tangentialflussfiltration (TFF). [ROCKER]
Tangentialflussfiltration
„Die traditionelle Filtrationsmethode, bekannt als Direktflussfiltration oder Dead-End-Filtration, beinhaltet den vertikalen Fluss der Probe durch die Membran, wodurch kleine Moleküle passieren und abgetrennt werden können. Diese Methode hat jedoch den Nachteil, dass sich Makromoleküle auf der Membranoberfläche ansammeln und eine Filterkuchenschicht bilden können. Mit zunehmender Filtrationszeit wird die Schicht dicker, was zu einer verringerten Filtrationseffizienz und einer kürzeren Lebensdauer der Membran führt.

Im Gegensatz dazu bewegt sich der Probenfluss bei der Tangential-Flow-Filtration (Tangentialflussfiltration) horizontal in einem tangentialen Winkel über die Membranoberfläche. Der Probenfluss wird kontinuierlich gefiltert und spült während der Zirkulation die Membranoberfläche, wodurch die Ansammlung von Makromolekülen verhindert und die durch verringerte Flussraten verursachte Konzentrationspolarisierung verringert wird. Dies gewährleistet eine stabile Durchflussrate und verlängert effektiv die Lebensdauer der Filtermembran. Die Tangentialflussfiltration bietet zusätzliche Vorteile, da sie gleichzeitig Konzentration und Diafiltration ermöglicht.“ [ROCKER]
Hier sind einige Begriffserklärungen aus der Zeichnung:
Retentat: Das Retentat ist das Material, das nach einer Filtration zurückbleibt. In der Impfstoffherstellung repräsentiert das Retentat die gewünschte Substanz, wie die mRNA.
Permeat: Das Permeat ist das Material, das durch den Filter hindurchgeht. In der Impfstoffherstellung repräsentiert das Permeat die durch den Filter hindurchgelassenen Stoffe, die im Impfstoff unerwünscht sind.
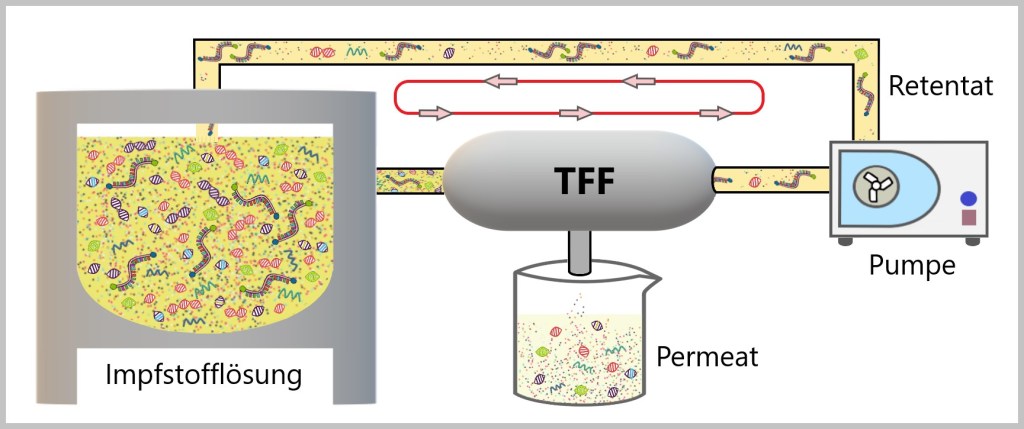
Die Abbildung zeigt den Ablauf einer Ultrafiltration/Diafiltration (UF/DF) mit Tangential Flow Filtration (TFF). Die Impfstofflösung fließt durch die Filteranlage (hier als TFF bezeichnet). Die Ultrafiltration wird eingesetzt, um die mRNA von den Verunreinigungen zu trennen. Dabei werden Membranen mit definierten Porengrößen eingesetzt, die nur Moleküle unterhalb einer bestimmten Größe passieren lassen. Während der Ultrafiltration wird die Diafiltration durchgeführt. Hierbei wird die mRNA mit einem geeigneten Puffer gespült, um die Konzentration unerwünschter Verunreinigungen weiter zu reduzieren und die mRNA in einer geeigneten Pufferlösung zu konzentrieren.
Das Ergebnis dieses Prozesses sind zwei Produkte. Zum einen das Permeat, das die unerwünschten Substanzen enthält, die aus der Impfstofflösung entfernt werden. Und das Retentat, die die gewünschte mRNA enthält, vermengt mit dem zugefügten Puffer.
Um den Kreislauf aufrechtzuerhalten, führt eine Pumpe das Retentat zurück in den Behälter zusammen mit verbliebenen Verunreinigungen. Dies ermöglicht, den Prozess zu wiederholen und die Lösung erneut durch den UF/DF-Zyklus zu führen. Dieser Zyklus wird wiederholt, bis die gewünschte Reinheit und Konzentration der mRNA im Endprodukt erreicht ist.
In einem Online-Beitrag von MERCK zum Thema „Herstellungsstrategien für mRNA-Impfstoffe und Therapeutika“ wird die Tangential-Flow-Filtration (TFF) als eine Methode zur Aufreinigung diskutiert. Dabei wird darauf hingewiesen, dass bei der Anwendung von TFF kleine DNA-Fragmente an die mRNA hybridisieren können, was zusätzliche Verunreinigungen verursacht. Dieses Risiko kann jedoch vermieden werden, wie im Beitrag erklärt wird, indem das DNA-Template durch Abtrennung entfernt wird.
MERCK bezieht sich dabei auf ein Patent aus dem Jahr 2014 von Stéphane Bancel, dem CEO von Moderna, das sich mit „Herstellungsverfahren für die Produktion von RNA-Transkripten“ befasst. Darin heißt es:
[0079] Das linearisierte Plasmid-DNA-Template wird aus der in-vitro-Transkription entfernt, das heißt, die DNA wird von der RNA getrennt. In einer Ausführungsform wird die DNA-Matrize chromatographisch unter Verwendung eines auf Poly-A-Abfang, z.B. Oligo dT, basierenden Affinitätsreinigungsschritts entfernt.
[0080] Es ist typisch, DNase I zu verwenden, um die DNA-Matrize unmittelbar nach der in vitro-Transkription enzymatisch zu verdauen. Bei den erfindungsgemäßen Verfahren wird keine DNase verwendet. Die Entfernung des gesamten Plasmids wird dem enzymatischen Verdau (DNase) vorgezogen, da somit das Risiko der Hybridisierung von degradierten DNA-Fragmenten mit der transkribierten mRNA verringert wird.
[0082] Die Methode zur Herstellung einer RNA-Transkription kann zusätzliche Reinigungsschritte nach der In-vitro-Transkription umfassen, zum Beispiel einen Ionenaustauschchromatographieschritt.
Für eine umfassende Reinigung ist es offensichtlich erforderlich, neben DNase I und TFF auch einen zusätzlichen Chromatographieschritt einzubeziehen.
d) Chromatographie
Die Chromatographie ist eine Methode zur Trennung verschiedener Stoffe in einem Gemisch. Ganz allgemein gibt es in diesem Verfahren eine mobile (bewegliche) Phase und eine stationäre (unbewegliche) Phase. Die mobile Phase ist die Stoffmischung, die getrennt werden soll. Sie bewegt sich durch die stationäre Phase. Die Stoffe in der mobilen Phase interagieren mit den Stoffen in der stationären Phase. Durch die Wechselwirkungen bewegen sich die einzelnen Komponenten im Stoffgemisch unterschiedlich schnell oder bleiben stecken. Somit können die verschiedenen Stoffe voneinander getrennt werden.
In der Abb.35 sind verschiedene Chromatographietechniken wie Umkehrphasen-Ionenpaar, Anionenaustausch- und Affinitätschromatographie dargestellt.

Gemäß den Ausführungen von MERCK wird insbesondere die Poly(dT)-Affinitätschromatographie für die Großserienproduktion hervorgehoben. Daher wird im folgenden Schritt das Prinzip der Affinitätschromatographie näher erläutert.
Poly(dT)-Affinitätschromatographie
In der Affinitätschromatographie ist die Idee, dass die Moleküle, die interessant sind (wie die mRNA), eine besondere Affinität oder Anziehung zu einem bestimmten Material haben. Die Anziehungskräfte zwischen den (mRNA-)Molekülen (mobile Phase) und dem Material (stationäre Phase) wird genutzt, um die gewünschten Moleküle aus einer Mischung zu isolieren.
Die mRNA enthält eine spezielle Sequenz, die als Poly(A) bekannt ist (siehe Abb.36). In der Affinitätschromatographie wird eine Säule mit einem Material, das Poly(dT) enthält, verwendet. Poly(dT) ist eine Kette von Nukleotiden, die alle das Basenpaar „Thymidin“ enthalten. Poly(A) ist hingegen eine Kette von Nukleotiden, die alle das Basenpaar „Adenin“ enthalten. Poly(dT) ist das „Gegenstück“ zu Poly(A) – sie ziehen sich gegenseitig an.

Der Ablauf sieht in etwa so aus: Eine Poly(dT)-Matrix wird in einer Säule platziert. Man kann sich die Säule wie einen zylindrischen Behälter vorstellen, der mit einem speziellen Poly(dT)-Material gefüllt ist.
Die Impfstofflösung, die die zu reinigende mRNA enthält (siehe Abb.37), a) wird durch die Säule geleitet. b) Die mRNA bindet spezifisch an die Poly(dT)-Matrix, c) während andere Bestandteile ungebunden durchlaufen. Dieser Schritt trennt die mRNA von den anderen Komponenten. d) Die gebundene mRNA kann dann durch bestimmte Änderung der Bedingungen oder Zugabe spezifischer Elutionspuffer von der Säule gelöst und freigesetzt werden.
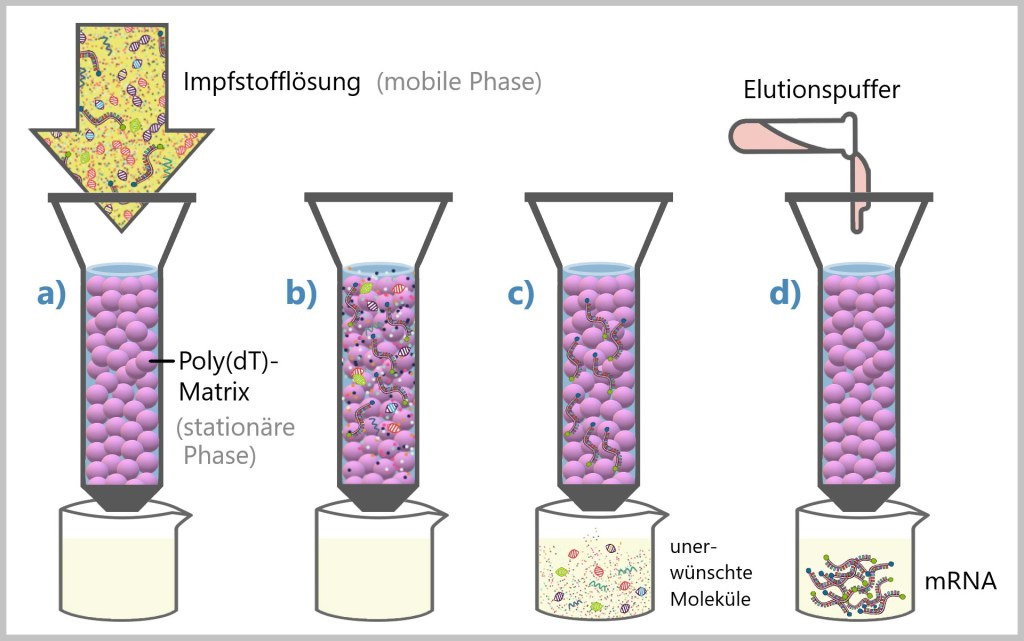
Aufgrund der Einschränkung der Poly(dT)-Affinitätschromatographie, welche keine Unterscheidung zwischen doppelsträngiger RNA (dsRNA) und einfachsträngiger RNA (ssRNA) erlaubt und zudem produktbezogene Verunreinigungen wie DNA-Fragmente, die an die mRNA hybridisiert haben, nicht entfernt, empfiehlt MERCK einen weiteren Chromatographieschritt mittels Anionenaustausch.
Anionenaustausch-Chromatographie
Die Anionenaustausch-Chromatographie (AEX-Chromatographie) ist eine chromatographische Technik zur Trennung von Molekülen basierend auf ihren unterschiedlichen Ladungen. Dabei werden Unterschiede in der Nettooberflächenladung genutzt.
In diesem Verfahren wird als stationäre Phase ein positiv geladenes Anionenaustauschharz verwendet, um Moleküle mit negativen Ladungen selektiv zu binden und zu trennen. Diese stationäre Phase besteht üblicherweise aus kleinen Perlen oder Partikeln. Die mobile Phase, in diesem Fall die Impfstofflösung, enthält das Zielmolekül – die mRNA, doppelsträngige RNA (dsRNA) und andere mögliche Verunreinigungen (siehe Abb.38).
Die Bindung zwischen den negativ geladenen Molekülen und dem Harz hängt von der Stärke der negativen Ladung auf den Molekülen ab. Moleküle mit höherer negativer Ladung binden fester ans Harz, während solche mit niedrigerer negativer Ladung schwächere Wechselwirkungen haben und möglicherweise leichter eluieren (ablösen).
a) Der Prozess erfolgt in einem Säulenformat, bei dem die Impfstofflösung auf die Säule mit dem positiv geladenen Anionenaustauschharz aufgetragen wird. Negativ geladene Moleküle in der Impfstofflösung binden an das Harz, b) während ungebundene Moleküle weggespült werden.
Da sowohl mRNA als auch dsRNA aufgrund ihrer Phosphatgruppen negative Ladungen tragen, erfolgt die Trennung aufgrund ihrer unterschiedlichen Strukturen und Affinitäten zum Anionenaustauschharz. Oft bindet dsRNA stärker an das Harz als mRNA.
Durch Änderung der Pufferbedingungen, wie Erhöhen der Ionenstärke des Elutionspuffers oder Veränderung des pH-Werts, werden die gebundenen RNA-Moleküle schrittweise von der Säule eluiert. Die unterschiedlichen Affinitäten von mRNA und dsRNA zum Anionenaustauschharz führen zu unterschiedlichen Retentionszeiten (Zeit, die ein Molekül für das Passieren der Säule benötigt), wobei c) dsRNA in der Regel stärker ans Harz bindet und d) daher länger gebunden bleibt.

In dem offiziellen EMA-Dokument für BioNTech gibt es keine expliziten Hinweise über den Einsatz der Chromatographie während des Produktionsprozesses. Im Netz findet man jedoch ein „inoffizielles“ Arbeitsdokument von EMA, bei dem auf Seite 29 die Chromatographie als Reinigungsschritt aufgezählt wird.
In einem ScienceDirect-Beitrag (Online-Plattform für wissenschaftliche Fachzeitschriften) wird erwähnt, dass Moderna Covid-19 mRNA-Impfstoffe sowohl durch Anionenaustausch als auch durch Poly dT-Affinitätschromatographie vorbereitet und gereinigt hat.
3.6. Die mRNA wird in Lipid-Nanopartikel verpackt

Die gereinigte mRNA wird in Lipid-Nanopartikel verpackt. Diese Lipidhüllen schützen die mRNA vor dem Abbau im Körper, fördern ihre Aufnahme in Zellen und ermöglichen die Freisetzung der mRNA im Zellinneren. Die formulierten RNA-Impfstoffe sind nun fertig zur Verabreichung.

3.7. Einige Schlussfolgerungen
Durch die anschauliche Darstellung des Produktionsprozesses in diesem Kapitel wurden nicht nur die strukturellen Schritte beim Herstellungsprozess des mRNA-Impfstoffs näher erläutert, sondern auch die technologischen Herausforderungen grob skizziert, denen die Hersteller gegenüberstehen.
Wie aus den Unterlagen von MERCK, SARTORIUS und Thermo Fisher Scientific hervorgeht, befindet sich die Forschung und Entwicklung der einzelnen technologischen Schritte noch in einem fortlaufenden Prozess. Die EMA-Dokumente zeigen, dass der Informationsaustausch zwischen den Herstellern und den Zulassungsbehörden hinsichtlich einiger Herstellungsparameter noch nicht abgeschlossen ist. Der kritische Bericht von Kevin McKernan et al. kann in diesen Kontext eingeordnet werden.
4. Methoden zur Messung von DNA und RNA
Die Testung der RNA-Konzentration und möglicher Rest-DNA (potenzielle Kontaminationen) ist ein entscheidender Schritt im Produktionsprozess von COVID-19 mRNA-Impfstoffen. Diese Maßnahme ist von essenzieller Bedeutung im Rahmen der Produktüberwachung und Qualitätssicherung, um sicherzustellen, dass die hergestellten Impfstoffe den Anforderungen an Reinheit und Wirksamkeit entsprechen.
In diesem Kapitel wird ein Überblick über verschiedene Messverfahren vermittelt, die als Nachweisverfahren von RNA und DNA in den mRNA-Impfstoffen dienen, darunter:
4.1. Fluoreszenz-Assay
4.1.1. quantitative Polymerase-Kettenreaktion - qPCR
4.1.2. Fluorometrie mit DNA- oder RNA-Farbstoffen
4.1.3. Qubit-Fluorometrie
4.2. Oxford-Nanopore-Technologie
4.3. UV-Spektroskopie
Diese Techniken werden eingehender behandelt, um den Lesern ein Verständnis für ihre Funktionsweise, Anwendungen und Einsatzmöglichkeiten zu vermitteln.
4.1. Fluoreszenz-Assay
Ein Fluoreszenz-Assay ist ein biochemischer Test, bei dem Fluoreszenz als Indikator für das Vorhandensein oder die Menge eines spezifischen Zielmoleküls verwendet wird. Dies kann auch die Messung von Nukleinsäuren wie DNA oder RNA umfassen, indem Fluoreszenzsignale durch spezifische Bindung von Fluoreszenzfarbstoffen oder Sonden an die Ziel-Nukleinsäuren erzeugt werden.
Ein Fluoreszenz-Assay wird unter anderem durch Kriterien bestimmt wie
- Spezifität (das Zielmolekül selektiv zu erkennen),
- Empfindlichkeit (auch bei niedriger Konzentration des Zielmoleküls),
- Stabilität und Reproduzierbarkeit,
- Robustheit (widerstandsfähig gegenüber kleinen Variationen),
- Durchführbarkeit,
- Zeit- und Kosteneffizienz.
Fluoreszenz-Assay ist ein übergeordneter Begriff, der verschiedene Anwendungen umfasst, darunter die quantitative PCR (qPCR), die Fluorometrie mit DNA- oder RNA-Farbstoffen, die Qubit-Fluorometrie und viele andere Fluoreszenz-basierte Techniken. Diese Methoden nutzen alle das Prinzip der Fluoreszenz, um bestimmte Moleküle zu detektieren, zu quantifizieren oder zu analysieren.
In der Regel werden die Ergebnisse von Fluoreszenz-Assays wie qPCR, Fluorometrie mit DNA- oder RNA-Farbstoffen und Qubit-Fluorometrie als DNA-Menge pro Volumeneinheit angegeben. Die Einheit ist typischerweise Nanogramm pro Mikroliter (ng/µl), was bedeutet, dass das Ergebnis das Gewicht der DNA pro Volumeneinheit angibt.
4.1.1. quantitative Polymerase-Kettenreaktion – qPCR
Die quantitative PCR (qPCR), auch als quantitative Echtzeit-PCR bekannt, ist eine Erweiterung der herkömmlichen PCR-Technik, die es ermöglicht, die Menge einer Zielsequenz in einer Probe zu bestimmen. Die grundlegenden Schritte der qPCR sind identisch mit denen der PCR, wie im vorherigen Abschnitt (3.2.1.) beschrieben.
In der qPCR wird ein spezieller Farbstoff, der als fluoreszierender Reporter bekannt ist, als eine Art „Lichtsignal“ verwendet, um die Menge der DNA während des PCR-Prozesses zu messen.
Es gibt zwei verschiedene Strategien in der qPCR, die es ermöglichen, die Menge an DNA zu bestimmen: die a) Farbstoff-basierte und die b) Sonden-basierte qPCR.
a) Farbstoff-basierte qPCR
In der Analyse wird der zu untersuchenden Probe eine Mischung aus Nukleotiden, einem Primerpaar (‚forward‘ [vorwärts] und ‚reverse‘ [rückwärts]), einer DNA-Polymerase und einem Fluorophor-Farbstoff zugesetzt. SYBR Green ist ein Farbstoff, der häufig in der quantitativen PCR (qPCR) verwendet wird, um DNA zu quantifizieren.
1) Der erste Schritt ist die Denaturierung, bei der die doppelsträngige DNA durch Erhitzen auf etwa 95 Grad Celsius in zwei Einzelstränge aufgespalten wird. 2) In einem nachfolgenden Schritt binden die Primer bei abgesenkter Temperatur an spezifische Regionen der DNA-Zielsequenz. 3) Unter Mitwirkung der DNA-Polymerase erfolgt dann im dritten Schritt die Synthese eines neuen DNA-Strangs. Der Farbstoff bindet an die neu synthetisierte doppelsträngige DNA und erzeugt Fluoreszenz. Diese Fluoreszenz wird in jedem PCR-Zyklus gemessen. In der Regel werden 35-40 Zyklen durchlaufen. Durch die Amplifikation der DNA-Zielsequenz entstehen mehr Bindungsstellen für den Farbstoff, wodurch die Zunahme der Fluoreszenz direkt mit der Menge der vorhandenen doppelsträngigen DNA korreliert.
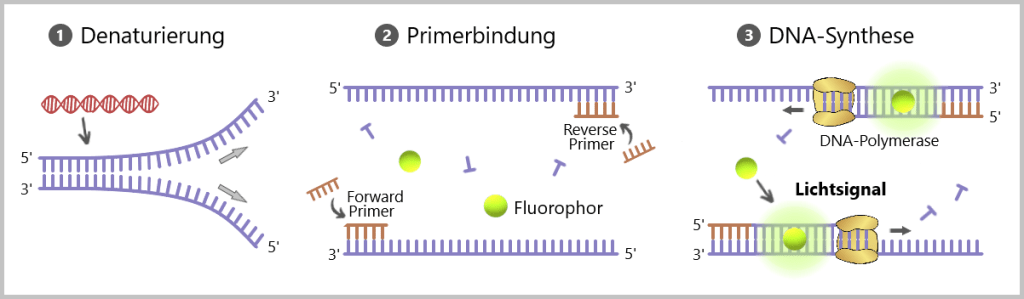
Die Primer sind entscheidend: Die Amplifikation in der Farbstoff-basierten qPCR erfordert die Verwendung von spezifisch designten Primern. Diese Primer binden gezielt an spezifische Sequenzen auf den einzelnen Strängen der Ziel-DNA. Erst durch die Primerbindung startet die DNA-Synthese.
Mit der Farbstoff-basierten Methode kann jeweils nur eine DNA-Zielsequenz erfasst werden. Da lediglich ein spezifisches Primerpaar benötigt wird, macht es diese Methode zu einer schnellen und kostengünstigen Option.
Ein Nachteil besteht jedoch darin, dass sich der Farbstoff nicht ausschließlich an die Zielsequenz bindet, was zu einer ungenauen Messung führen kann. Um sicherzustellen, dass nur die beabsichtigte DNA-Sequenz während der Polymerase-Kettenreaktion (PCR) amplifiziert wurde, wird nach jedem Experiment üblicherweise eine Schmelz-Kurve erstellt. Diese Kurve ist wichtig, weil jede DNA-Sequenz ihren eigenen charakteristischen Schmelzpunkt hat. Ein spezifischer und erwarteter Verlauf der Schmelz-Kurve, deutet darauf hin, dass die Amplifikation erfolgreich und spezifisch war.
b) Sonden-basierte qPCR
In der Analyse wird der zu untersuchenden Probe eine Mischung aus Nukleotiden, einem Primerpaar und einer DNA-Polymerase zugesetzt. Darüber hinaus wird eine Sonde hinzugefügt, die mit einem Fluorophor (fluoreszierender Farbstoff) und einem Quencher (löscht das Fluoreszenzsignal) markiert ist (siehe Abb.41). Solange die Sonde intakt ist und nicht abgebaut wird, wird das Fluoreszenzsignal aufgrund der räumlichen Nähe zum Quencher unterdrückt.
1) Der erste Schritt ist die Denaturierung, bei der die doppelsträngige DNA durch Erhitzen auf etwa 95 Grad Celsius in zwei Einzelstränge aufgespalten wird. 2) In einem nachfolgenden Schritt binden sowohl die Primer wie auch die spezifische Sonde bei abgesenkter Temperatur an spezifische Regionen der DNA-Zielsequenz. 3) Unter Mitwirkung der DNA-Polymerase erfolgt dann im dritten Schritt die Synthese eines neuen DNA-Strangs. Während der DNA-Synthese durch die DNA-Polymerase wird die Sonde abgebaut, was zur Trennung des Fluorophors vom Quencher führt und das Fluoreszenzsignal freisetzt. In der qPCR wird die Menge der fluoreszierenden Signale, die während der DNA-Synthese entstehen, in Echtzeit gemessen.

Die Zunahme dieser Lichtsignale erfolgt in direktem Verhältnis zur Menge der DNA, die während jeder Runde der PCR vervielfältigt wird. Mit anderen Worten, die gemessene Fluoreszenz ist proportional zur Menge der DNA in der Probe.
Sowohl Primer als auch Sonden sind entscheidend: Die Primer binden gezielt an spezifische Sequenzen auf den einzelnen Strängen der Ziel-DNA. Erst durch die Primerbindung startet die DNA-Synthese. Die Sonden dienen als zusätzliches Werkzeug zur Erkennung und Quantifizierung der Ziel-DNA. Die Sonden in der qPCR sind so konzipiert, dass sie spezifisch an eine bestimmte DNA-Sequenz binden können. Diese Sequenz wird so ausgewählt, dass sie eindeutig und charakteristisch für das gesuchte DNA-Fragment ist. Die Sonden binden nur an die Ziel-DNA, wenn die Sequenz in der Probe vorhanden ist. Dies ermöglicht es, gezielt nach einer bestimmten DNA-Sequenz zu suchen und deren Menge zu quantifizieren. Daher ist die Messung in der Sonden-basierten qPCR tendenziell genauer und spezifischer im Vergleich zur Farbstoff-basierten qPCR.
Detaillierte Informationen zur Funktionsweise der beiden qPCR-Methoden sind hier zu finden.
Die qPCR kann auch zur Quantifizierung von RNA verwendet werden.
Hierzu ist ein zusätzlicher Schritt erforderlich, das Umschreiben der RNA in komplementäre DNA (cDNA). Dieser Schritt wird als Reverse Transkription (RT) bezeichnet, und daher nennt man die Anwendung von qPCR auf RNA RT-qPCR oder Reverse-Transkriptase-qPCR. Diese cDNA dient dann als Vorlage für die PCR-Amplifikation. Die qPCR wird dann wie oben beschrieben durchgeführt, um die Menge an spezifischer RNA in einer Probe zu bestimmen.
c) Beschränkungen der qPCR
In der Polymerase-Kettenreaktion (PCR) hängt die effiziente Amplifikation von DNA von ihrer spezifischen Länge ab. Es ist bekannt, dass bei der quantitativen PCR (qPCR) Amplikone (amplifizierte DNA-Fragmente) von mehr als 100 bp (Basenpaaren) bevorzugt werden. Ein Basenpaar bezeichnet die Bindung zwischen zwei Nukleotiden (Adenin-Thymin und Cytosin-Guanin) innerhalb der DNA.
Grund dafür ist, dass längere Amplikone eine stabilere Amplifikation ermöglichen und die spezifische Detektion des Zielgens verbessern können. Amplikone mit weniger als 100 bp (Basenpaaren) können weniger spezifisch sein und dazu neigen, unspezifische Produkte zu generieren, was zu Fehlinterpretationen der qPCR-Ergebnisse führen kann.
4.1.2. Fluorometrie mit DNA- oder RNA-Farbstoffen
Die fluorometrische Messung von Nukleinsäuren basiert auf der Verwendung von spezifischen Fluoreszenzfarbstoffen, die selektiv an DNA oder RNA binden. Diese Farbstoffe geben nur dann ein Fluoreszenzsignal ab, wenn sie an das Ziel gebunden sind. Die Konzentrationen der Nukleinsäuren werden anhand der Fluoreszenzsignale der Proben gemessen.
Grundsätzliche Schritte der Fluorometrie zur DNA- und RNA-Quantifizierung:
Die DNA oder RNA wird aus der Probe extrahiert und anschließend gereinigt. Verunreinigungen wie Proteine, Lipide oder andere chemische Verbindungen können die Bindung der Fluoreszenz-Farbstoffe an die DNA oder RNA stören oder zusätzliche Signale in der Fluorometrie erzeugen, die nicht von den Nukleinsäuren stammen.
Ein Fluoreszenzfarbstoff wird gezielt ausgewählt, um eine spezifische Bindung an DNA oder RNA zu ermöglichen. Diese Bindung markiert die Nukleinsäuren in der Probe und erleichtert die spätere Quantifizierung ihrer Menge. Die Auswahl des Farbstoffs erfolgt aufgrund seiner Affinität zu bestimmten Strukturen oder Eigenschaften der Nukleinsäuren. Beispiele:
RiboGreen für RNA: RiboGreen ist darauf ausgelegt, spezifisch an Ribonukleinsäuren (RNA) zu binden. Dieser Farbstoff macht keine Unterscheidung zwischen verschiedenen Arten von RNA, sondern eher zwischen RNA und DNA im Allgemeinen.
PicoGreen für DNA: PicoGreen zeigt eine hohe Sensitivität gegenüber DNA. Dieser Farbstoff macht keine Unterscheidung zwischen verschiedenen Arten von DNA, sondern eher zwischen DNA und RNA im Allgemeinen.
1) Nachdem die spezifischen Farbstoffe zur Probe hinzugefügt wurden, erfolgt die Interaktion mit den Nukleinsäuren (DNA oder RNA). 2) Der Farbstoff bindet an die Ziel-Nukleinsäuren in einer spezifischen Weise. Diese Bindung führt zur Freisetzung von Fluoreszenz, wodurch die markierten Nukleinsäuren lichtemittierend werden. 3) Die Probe wird in den Fluorometer geladen, ein Gerät, das für die Fluoreszenzmessung ausgelegt ist. Das Fluorometer regt die Farbstoffe in der Probe mit Licht einer bestimmten Wellenlänge (λex) an. Der Fluoreszenzfarbstoff absorbiert dieses Licht und emittiert Licht mit einer längeren Wellenlänge (λem). Die emittierte Fluoreszenz wird gemessen und die Intensität des Signals wird registriert.

Um genaue Ergebnisse zu gewährleisten, werden Kontrollen durchgeführt, um sicherzustellen, dass die gemessene Fluoreszenz ausschließlich von der Ziel-Nukleinsäure stammt. Zur Quantifizierung wird oft im Vorfeld eine Standardkurve erstellt. Die Konzentration der DNA oder RNA in der Probe wird dann anhand dieser Standardkurve berechnet.
4.1.3. Qubit-Fluorometrie
Die Qubit-Fluorometrie ist eine spezielle Form der Fluorometrie, die in der quantitativen Analyse von Nukleinsäuren, insbesondere DNA und RNA, verwendet wird. Sie basiert auf der Verwendung von Fluoreszenzfarbstoffen, die spezifisch mit Nukleinsäuren interagieren und ein Fluoreszenzsignal abgeben, wenn sie gebunden sind.
Im Gegensatz zu herkömmlichen Fluorometrie-Techniken, die oft auf der Messung von Fluoreszenzintensität beruhen, nutzt die Qubit-Fluorometrie die Messung von Fluoreszenzsignalen, der an Nukleinsäuren gebundenen Farbstoffe. Dies ermöglicht eine hochsensitive und präzise Bestimmung der Konzentration von DNA oder RNA in einer Probe, selbst in sehr niedrigen Konzentrationen.
DNA-Messung
Zur Probenvorbereitung wird ein nukleinsäurebindender Fluoreszenzfarbstoff verwendet, der spezifisch an DNA bindet und ein Fluoreszenzsignal emittiert.
Das Qubit-Fluorometer wird kalibriert, um sicherzustellen, dass es korrekt auf die Fluoreszenzsignale der DNA-Farbstoff-Lösung reagiert und genaue Messungen durchführt. Eine Reihe von Standardproben mit bekannten Konzentrationen an DNA wird vorbereitet. Diese Proben werden verwendet, um eine Standardkurve zu erstellen, die die Beziehung zwischen der Fluoreszenzintensität und der DNA-Konzentration darstellt.
1) Die Probe wird mit der DNA-Farbstoff-Lösung gemischt und in das Qubit-Fluorometer geladen. 2) Der Farbstoff bindet an die DNA und gibt ein Fluoreszenzsignal ab. 3) Das Fluorometer misst die Fluoreszenzsignale der Proben und vergleicht sie mit der Standardkurve, um die Konzentration der DNA in den Proben zu bestimmen. Die gemessenen DNA-Konzentrationen werden anhand der Standardkurve berechnet und als Ergebnisse der Qubit-Fluorometrie erhalten.

Die RNA-Messung erfolgt analog mit einem spezifischen RNA-Farbstoff.
Qubit-Fluorometrie – eine genauere Methode zur Quantifizierung von DNA oder RNA
Die Qubit-Fluorometrie wird oft als eine genauere Methode zur Quantifizierung von DNA oder RNA im Vergleich zu anderen Methoden wie der UV-Spektroskopie oder der Gelelektrophorese betrachtet. Der Hauptgrund für die Genauigkeit der Qubit-Fluorometrie liegt in ihrer Fähigkeit, spezifisch und selektiv die Fluoreszenz der Nukleinsäuren zu messen, ohne dass andere Substanzen in der Probe störend wirken. Dies ermöglicht eine präzise Quantifizierung der Nukleinsäuren, auch bei geringen Konzentrationen und in komplexen Proben.
Obwohl die Qubit-Fluorometrie eine präzise Methode zur Messung der Gesamtmenge an DNA oder RNA in einer Probe ist, kann sie nicht zwischen verschiedenen Sequenzen unterscheiden. Sie misst lediglich die gesamte DNA-Konzentration oder RNA-Konzentration. Wenn spezifische Informationen über die DNA- oder RNA-Sequenz benötigt werden, müssen andere Methoden wie qPCR verwendet werden.
4.2. Oxford-Nanopore-Technologie
Die Nanoporen-Sequenzierung ermöglicht eine Echtzeitanalyse langer DNA- oder RNA-Fragmente.
Nanoporen sind winzige Öffnungen, die nur wenige Nanometer groß sind. Sie sind in eine Membran eingebettet, die aus einem leitfähigen Material besteht. Diese spezielle Membran ist in einem Chip platziert.

Die DNA- oder RNA-Probe wird zunächst in kleinere Stücke fragmentiert. Diese Fragmentierung erleichtert den Transport der Moleküle durch die Nanoporen. Die fragmentierte DNA oder RNA wird dann in eine Flüssigkeit gegeben, die als Elektrolytlösung bezeichnet wird. Diese Flüssigkeit enthält gelöste Salze und ermöglicht es den geladenen Molekülen, sich frei zu bewegen. Die Elektrolytlösung wird in Kontakt mit den Nanoporen gebracht, um den Sequenzierungsprozess zu starten.
Eine konstante elektrische Spannung wird angelegt, um einen Ionenstrom durch die Nanoporen zu erzeugen. Dies bewirkt, dass die geladenen DNA- oder RNA-Moleküle zu den Nanoporen wandern und durch sie hindurch passieren (siehe Abb. 45).
Die Helicase, ein Enzym, bindet sich an die Doppelstränge der DNA, trennt und wickelt sie in einzelsträngige Formen auf, um sie für den Transport durch die Nanopore vorzubereiten. Für die RNA, die bereits einzelsträngig ist, ist die Aktion der Helicase nicht erforderlich.

Das Motorprotein, das Teil der Helicase ist, schiebt dann die einzelsträngige DNA oder RNA durch die Nanopore. Während die DNA oder RNA durch die Nanopore wandert, verändert sie den elektrischen Strom, der durch die Pore fließt. Diese elektrische Signatur hängt von der Sequenz der DNA ab, die gerade durch die Pore passiert. Jede Base in der DNA (Adenin, Cytosin, Thymin oder Guanin) hat eine charakteristische Wirkung. Diese Änderung des Stroms wird dann als Änderung der elektrischen Spannung wahrgenommen, die an der Nanopore gemessen wird. Eine Software berechnet aus der Spannungsveränderung die Sequenz der DNA.
Das Ergebnis der Oxford-Nanopore-Technologie ist die Sequenz der untersuchten DNA- oder RNA-Probe. Diese Sequenz kann Informationen über die genetische Zusammensetzung, Länge oder andere wichtige genetische Merkmale liefern.
Zur besseren Veranschaulichung der Nanoporen-Sequenzierung wird dieses Video empfohlen.
4.3. UV-Spektroskopie
Die UV-Spektroskopie ist eine gängige Technik zur Analyse von Nukleinsäuren, die verwendet wird, um die Konzentration von DNA oder RNA in einer Probe zu bestimmen. Das Prinzip basiert auf der Absorption elektromagnetischer Strahlung durch Nukleinsäuren, insbesondere im UV-Bereich, was zu charakteristischen Absorptionsmustern führt. Im folgenden Abschnitt wird die Anwendung dieser Technik auf RNA erläutert.
In der UV-Spektroskopie kommen spezielle Geräte zum Einsatz, bekannt als UV/VIS-Spektrophotometer. Diese umfassen eine Strahlungsquelle, einen Monochromator, der aus einem Prisma und einer Blende besteht, sowie einen Detektor. Die zu untersuchende RNA wird auf einen transparenten Probenträger gegeben, der UV-Licht durchlässt. Während der Messung wird die Probe zwischen dem Monochromator und dem Detektor platziert. Der Monochromator sorgt dafür, dass nur Strahlung einer bestimmten Wellenlänge auf die Probe trifft. RNA-Moleküle absorbieren UV-Licht bei bestimmten Wellenlängen, insbesondere im Bereich von etwa 240 bis 300 nm (Nanometer), aufgrund der Anwesenheit von den Basen wie Adenin, Guanin, Cytosin und Uracil.
Die RNA-Lösung wird bestrahlt und absorbiert einen Teil des Lichts, wodurch die Strahlung abgeschwächt wird. Je mehr RNA enthalten ist, desto höher ist die Absorption.
Die Absorption des UV-Lichts durch die RNA-Moleküle führt zu einem charakteristischen Absorptionsmuster. Durch die Messung der Absorption bei verschiedenen Wellenlängen kann die UV-Spektroskopie Informationen über die Konzentration der RNA in der Probe sowie über deren Reinheit und Struktur liefern.
Detaillierte Informationen zu diesem Thema gibt es hier.
Obwohl die UV-Spektroskopie eine äußerst nützliche Technik zur Analyse von Nukleinsäuren ist, hat sie auch einige Nachteile, wie zum Beispiel eine Empfindlichkeit gegenüber Verunreinigungen. Um genaue Messergebnisse zu erhalten, erfordert die UV-Spektroskopie eine hohe Reinheit der Proben.
Wenn eine Probe sowohl RNA als auch DNA enthält, kann dies die Messergebnisse der UV-Spektroskopie beeinflussen. Da sowohl RNA als auch DNA UV-Licht absorbieren, kann die Anwesenheit von DNA in der Probe zu einer Überschätzung der gemessenen RNA-Konzentration führen. Dies liegt daran, dass das UV-Signal nicht nur von der RNA, sondern auch von der DNA stammt, was zu einem erhöhten Absorptionswert führt.
5. Messergebnisse und deren Auswertung
Die detaillierte Darstellung der grundlegenden Produktionsschritte bei der Herstellung und Qualitätssicherung von mRNA-Impfstoffen in den Kapiteln 3 und 4 legt den Grundstein für ein tieferes Verständnis der Analyseergebnisse der unabhängigen Labore und den daraus resultierenden Diskussionen.
5.1. Richtlinien zur Begrenzung von Rest-DNA in Impfstoffen
5.2. Die Messmethoden von Pfizer
5.3. DNA-Fragmente in C19 mRNA-Impfstoffen entdeckt
5.3.1. Messmethoden und Messergebnisse der Studie
5.3.2. Ein unerwarteter Fund: SV-40-Promotor-Enhancer
5.3.3. Größenprofil der Rest-DNA
5.4. Reproduktion der Studienergebnisse
5.4.1. Dr. Phillip Buckhaults
5.4.2. Dr. Jürgen O. Kirchner und Prof. Dr. Brigitte König
5.5. Kurze Zusammenfassung
5.1. Richtlinien zur Begrenzung von Rest-DNA in Impfstoffen
Derzeit empfiehlt die US-amerikanische FDA den Herstellern von Virusimpfstoffen, die Menge an Rest-DNA im Endprodukt bei parenteralen Impfungen auf unter 10 ng/Dosis und die Größe der DNA auf unter die Größe eines funktionellen Gens oder ~200 Basenpaare zu begrenzen.[1] Dies steht auch im Einklang mit den Empfehlungen der Weltgesundheitsorganisation (WHO). [2,3] Frühere DNA-Restwerte wurden 1985 von der FDA auf 10 pg/Dosis festgelegt (10 pg = 0,01 ng). Eine Studiengruppe der WHO kam 1986 zu dem Schluss, dass das Risiko bis zu 100 pg/Dosis vernachlässigbar ist, und 1996 erhöhte die WHO die Werte weiter auf bis zu 10 ng pro Dosis. [2] [Studie]
[1] https://www.fda.gov/media/113760/download
[2] https://pubmed.ncbi.nlm.nih.gov/17321940/
[3] https://cdn.who.int/media/docs/default-source/biologicals/documents/trs_978_annex_3.pdf
5.2. Die Messmethoden von Pfizer
Zur Gewährleistung der Einhaltung der von der Weltgesundheitsorganisation (WHO) definierten Grenzwerte während des Produktionsprozesses verwendet Pfizer das quantitative Polymerase-Kettenreaktions (qPCR)-Messverfahren zur Messung des DNA-Restgehalts (siehe Abb.47).
Die Abbildung zeigt auch, dass der Hersteller die „UV Spectroscopy“ und Fluoreszenz-Assay-Messverfahren zur Bestimmung der RNA-Menge einsetzt. Der Begriff „Fluoreszenz-Assay“ ist ein Überbegriff, der verschiedene Messmethoden umfasst, die auf der Fluoreszenz basieren. Es könnte sich um Fluorometrie mit einem bestimmten Fluoreszenzfarbstoff handeln, da weder qPCR noch Qubit-Fluorometrie im Protokoll explizit genannt werden.
Der Hersteller verwendet selektiv unterschiedliche Messmethoden zur Bestimmung des RNA-Inhalts und DNA-Inhalts (siehe auch hier, hier und hier).

Es ist wichtig zu beachten, dass die gemessenen Werte für den RNA- und DNA-Inhalt von der verwendeten Messmethode abhängen. Ein höherer gemessener RNA-Gehalt und eine niedrigere Rest-DNA-Menge deuten üblicherweise auf eine höhere Produktqualität hin. Es sei darauf hingewiesen, dass es keine regulatorischen Vorgaben gibt, welche Messverfahren für die Qualitätssicherung verwendet werden sollen.
5.3. DNA-Fragmente in C19 mRNA-Impfstoffen entdeckt
In der Studie mit dem Titel „DNA fragments detected in COVID-19 vaccines in Canada“ wurde die RNA-Sequenzierung von Covid-19 mRNA-Impfstoffen durchgeführt. Dr. David Speicher ist der Hauptautor dieser Studie, während Dr. Kevin McKernan als Co-Autor aufgeführt ist. Die Untersuchung ergab DNA-Fragmente, die von den im Herstellungsprozess verwendeten Plasmiden stammen. Alle hier bereitgestellten Informationen stammen aus der von Speicher et al. durchgeführten Studie.
Untersucht wurden 27 COVID-19-Impfstofffläschchen, die aus Kanada stammen und 12 verschiedenen Chargen von Moderna und Pfizer angehören. Hier eine detaillierte Auflistung:
Anzahl der Fläschchen pro Impfstofftyp:
- Pfizer-BioNTech BNT162b2: 8 Fläschchen (ungeöffnet, abgelaufen)
- Moderna Spikevax mRNA-1273: 16 Fläschchen (ungeöffnet, abgelaufen)
- Moderna XBB.1.5: 3 Fläschchen (benutzt, nicht abgelaufen)
Details zu den untersuchten Chargen:
- 5 Chargen monovalenter Moderna-Impfstoffe für Kinder/Erwachsene
- 1 Charge bivalenter BA.4/5-Impfstoff für Erwachsene
- 1 Charge bivalenter Wuhan-BA.1-Impfstoff für Kinder und Erwachsene
- 1 Charge monovalenter Impfstoff von Moderna XBB.1.5
- 3 Chargen monovalenter Impfstoffe von Pfizer für Erwachsene
- 1 Charge bivalenter Wuhan-BA.4/5-Impfstoffe von Pfizer für Erwachsene
5.3.1. Messmethoden und Messergebnisse der Studie
Die Studie nutzte etablierte und standardisierte Methoden zur Detektion von DNA und RNA, darunter quantitative Polymerase-Kettenreaktion (qPCR) und Qubit-Fluorometrie-Techniken, wobei zuvor veröffentlichte Primer- und Sondensequenzen verwendet wurden. Mittels Oxford Nanopore-Sequenzierung wurde die Größenverteilung der DNA-Fragmente bestimmt. Die verwendeten Methoden wurden im Kapitel 4 bereits vorgestellt.
Die Messergebnisse sind in den folgenden Tabellen dargestellt, wobei jeweils die Spannen der gemessenen Werte angegeben sind. Die Werte in ng/Dosis (Nanogramm pro Dosis) repräsentieren die gemessenen Mengen an DNA pro Impfstoffdosis.


In der qPCR werden spezifische Primer verwendet, um nur die DNA-Sequenzen zu amplifizieren, die für den Plasmidursprung der Replikation (ori) und die Spike-Sequenzen spezifisch sind. Daher können die Werte für jede Sequenz separat gemessen und quantifiziert werden.
Bei der Qubit-Fluorometrie hingegen wird die Gesamtmenge der DNA in der Probe gemessen, unabhängig von der spezifischen Sequenz. Daher wird bei der Qubit-Fluorometrie keine Unterscheidung zwischen den einzelnen Sequenzen gemacht, sondern die Gesamtmenge der DNA in der Probe quantifiziert.
Schlussfolgerung
Die vorliegenden Daten zeigen, dass in den getesteten mRNA-COVID-19-Impfstoffen Milliarden bis Hunderte von Milliarden DNA-Molekülen pro Dosis vorhanden sind. Unter Verwendung der Qubit-Fluorometrie überschreiten alle geprüften Impfstoffe die von der WHO festgelegten Richtlinien für Rest-DNA von 10 ng/Dosis um das 188- bis 509-fache. Interessanterweise lag der DNA-Restgehalt, der durch die Methode qPCR gemessen wurde, in allen getesteten Produkten jedoch unter diesen Richtlinien. Diese Ergebnisse zeigen, dass es sehr wichtig ist, bei der Interpretation von Richtlinien für die DNA-Menge klar und einheitlich vorzugehen. [Studie]
An dieser Stelle ist es wichtig festzustellen, dass die gemessenen Daten stark von dem verwendeten Messverfahren abhängen. Damit weist die Studie auf eine derzeit existierende Schwachstelle in den regulatorischen Vorgaben hin, die es ermöglicht, verschiedene Messverfahren gezielt auszuwählen, um gewünschte Messergebnisse zu erhalten (siehe Abb.47).
Im Patent US10077439B2 von Moderna, verfasst unter anderem von Stéphane Bancel, CEO von Moderna, wird explizit darauf hingewiesen, dass die quantitative Polymerase-Kettenreaktion (qPCR) die Gesamtmenge an vorhandener DNA unterschätzt.
[00103] … Quantitative PCR wird häufig zur Messung der Rest-DNA eingesetzt, erkennt jedoch nur die DNA-Moleküle, die beide qPCR-Primer enthalten, und misst daher nicht alle anderen kleineren DNA-Moleküle, die teilweise verdaut sind. …
Siehe dazu auch Abschnitt 4.1.1.c) Beschränkungen der qPCR. Für mehr Informationen wird der Artikel „Fluorometrie im Detail“ von McKernan empfohlen.
„Ich denke, das Wichtigste für die Regulierungsbehörden ist, dass es nicht eine einzelne Technologie gibt, die eine Antwort gibt“, sagte McKernan in einem Interview mit REBEL News Canada.
Das bedeutet, mehrere Messmethoden zur Bestimmung von DNA- und RNA-Werten würde die Zuverlässigkeit der Ergebnisse erhöhen und potenzielle Abweichungen zwischen den Methoden identifizieren. Die Verwendung von mehreren Methoden ermöglicht eine interne Validierung der Ergebnisse und kann helfen, mögliche Fehlerquellen zu erkennen. Beispielsweise könnte die qPCR-Methode verwendet werden, um DNA- und RNA-Werte zu messen, und die Ergebnisse könnten dann mit den Werten verglichen werden, die durch das Qubit-Fluorometrie-Messverfahren erhalten werden. Durch den Vergleich der Ergebnisse aus verschiedenen Methoden können mögliche Unterschiede oder Inkonsistenzen identifiziert werden.
5.3.2. Ein unerwarteter Fund: SV-40-Promotor-Enhancer
Speicher et al. fanden SV40-Promotor-Enhancer-ori-Sequenzen und SV40-PolyA-Sequenzen in den Pfizer-Impfstoffen. Siehe dazu auch Tabelle 2 der Studie.
SV40 steht für Simianes Virus 40, ein Virus, das 1960 in Nierenzellen von Affen entdeckt wurde. Es kann sowohl Affen als auch Menschen infizieren. SV40 ist für seine Verwendung in der biomedizinischen Forschung und als Kontaminant in einigen früheren Chargen von Polio-Impfstoffen bekannt.
„Es ist nicht das vollständige Simian-Virus 40 (SV40), das vor vielen Jahren in den Polio-Impfstoffen beobachtet wurde.“ [McKernan]
SV40 enthält bestimmte genetische Abschnitte, die eine wichtige Rolle dabei spielen, wie Gene aktiviert werden. Zu diesen Abschnitten gehören der SV40-Enhancer und der SV40-Promotor.
Der SV40-Promotor ist wie eine Startsequenz für ein Gen. Wenn ein Gen exprimiert wird (aktiv ist), bedeutet das, dass die genetische Information dieses Gens in ein Protein umgesetzt wird. Der SV40-Promotor ist eine besonders starke Startsequenz, die in der Lage ist, die Expression eines Gens effizient zu initiieren. Stell dir vor, die Genexpression ist wie das Starten eines Motors in einem Auto. Der SV40-Promotor ist wie der Zündschlüssel, der den Motor einschaltet. Wenn der Zündschlüssel gedreht wird, wird der Motor aktiviert und das Auto beginnt zu fahren.
Der SV40-Enhancer befindet sich in der Nähe des Promotors und kann die Aktivität des Promotors erhöhen. Der SV40-Enhancer kann dann als Turbo-Boost betrachtet werden, der dem Motor zusätzliche Leistung verleiht. Wenn der Turbo-Boost aktiviert wird, beschleunigt das Auto schneller und erreicht eine höhere Geschwindigkeit. Ähnlich verstärkt der SV40-Enhancer die Aktivität des SV40-Promotors und erhöht die Expression des Gens, ähnlich wie der Turbo-Boost die Leistung des Motors steigert.
McKernan: „Der Pfizer-Vektor, den sie verwenden, hat einen SV40-Promotor. Es handelt sich also um etwas, das die Antibiotikaresistenz fördert, die sich im Rückgrat dieser Vektoren befindet. … Es besteht jedoch immer noch die Sorge, dass, wenn diese DNA in diesen Impfstoffen in hohen Konzentrationen vorhanden ist, dieser Promotor in das Genom integrieren könnte und wenn es sich vor einem Onkogen integriert, könnte dies die Expression eines Onkogens fördern.“
SV40-Komponenten und die Zulassungsbehörden
In der unteren Abbildung wird links die Plasmidkarte von Pfizer gezeigt, die mithilfe der Messergebnisse von McKernan rekonstruiert wurde. Auf der rechten Seite ist die Plasmidkarte zu sehen, die Pfizer den Zulassungsunterlagen für die Europäische Arzneimittelagentur (EMA) beigefügt hat. Deutlich ist zu erkennen, dass die SV40-Komponenten in der rechten Plasmidkarte fehlen.

Da die Richtlinien der Weltgesundheitsorganisation (WHO) und der US-amerikanischen Arzneimittelbehörde FDA vorschreiben, dass alle offenen Leserahmen und Promotoren, wie z.B. die SV40-Komponenten, offengelegt werden müssen, hat das Team von McKernan die Behörden mit seinen Messergebnissen konfrontiert. Als Ergebnis bestätigten daraufhin die EMA, die FDA und Health Canada, dass tatsächlich SV40-Komponenten im Plasmid verwendet werden. [Powerpoint-Aufzeichnung Massachusetts-Folien, Folie 17]
In einer E-Mail an The Epoch Times (Oktober 2023) schreibt Health Canada: „Obwohl die vollständige DNA-Sequenz des Pfizer-Plasmids zum Zeitpunkt der Erstanmeldung bereitgestellt wurde, hat der Sponsor die SV40-Sequenz nicht spezifisch identifiziert.“
Kurz darauf teilte auch die Europäische Arzneimittel-Agentur (EMA) The Epoch Times in einer E-Mail mit: „Während im ursprünglichen Antrag auf Marktzulassung für Comirnaty die vollständige DNA-Sequenz des Plasmid-Ausgangsmaterials angegeben wurde, hat der Antragsteller die SV40-Sequenz nicht ausdrücklich hervorgehoben“. … BioNTech habe die Einbeziehung des Verstärkers in seinen Impfstoff nicht hervorgehoben, da „er als nicht funktioneller Teil des Plasmids angesehen wurde. Sie haben diese Informationen seitdem als Antwort auf die von der EMA gestellten Fragen geklärt.“
Wir erinnern uns, die EMA empfahl Ende 2020 die bedingte Zulassung für den COVID-19-Impfstoff Comirnaty von BioNTech und Pfizer. Erst drei Jahre später haben die Behörden nun die SV40-Sequenz im Comirnaty- Marktzulassungsantrag gefunden.
Ein Sprecher der EMA erklärte daraufhin, dass die Agentur „bestätigen kann, dass sie keine zuverlässigen Beweise für DNA-Rückstände gesehen haben, die die zugelassenen/sicheren Werte überschreiten“ für die COVID-19 Impfstoffe von Pfizer/BioNTech oder Moderna. Der EMA „sind auch keine wissenschaftlichen Beweise bekannt, die zeigen, dass die sehr geringen Mengen an Rest-DNA, die in Impfstoffchargen vorhanden sein können, in die DNA von geimpften Personen integriert werden könnten.“
Welche potentiellen Gefahren mit dem DNA-Restgehalt verbunden sind, wird im Kapitel 7 „Sind weitere Untersuchungen notwendig?“ diskutiert.
5.3.3. Größenprofil der Rest-DNA
Um die unterschiedlichen Größen oder Längen der Rest-DNA-Moleküle zu erfassen, wurde von Speicher DJ et al. der Impfstoff BNT162b2 mit Oxford Nanopore (ONT) sequenziert (Abb.49).

Die Sequenzierung ergab eine Gesamtlänge des Plasmids von 7810 bp (Basenpaaren). Wie in der Abbildung (links) zu sehen, ist ein Großteil der Masse der gemessenen DNA-Moleküle klein. Die Analyse ergab eine mittlere DNA-Fragmentlänge von 214 bp.
„Eines von fünfzig Molekülen war mehr als 1.000 Basen lang.“ [McKernan, Covid-19 Vaccine Expert Panel Briefing to the Massachusetts Legislature and Public Health Officials, 1:29:30]
Sie fanden auch ein Fragment, das eine Länge von 3,2 kb (Kilobasenpaaren) oder 3200 bp (Basenpaaren) aufwies und die gesamte Rückseite des Plasmids abdeckte (siehe Abb.49 rechts, dargestellt als blaue Vektorregion). In einem Interview erläuterte McKernan, dass dieses 3.200 Basenpaare umfassende Fragment das Gen für die Resistenz gegen die Antibiotika Kanamycin und Neomycin enthielt, einschließlich des entsprechenden SV40-Promotors. Weiter sagte er: „Andere Fragmente ähnlicher Länge umfassten die Sequenz für das Spike-Protein.“
Gemäß den Richtlinien der Weltgesundheitsorganisation (WHO) wird empfohlen, die Größe der Rest-DNA auf unter die Größe eines funktionellen Gens oder etwa ~200 Basenpaare zu begrenzen.
5.4. Reproduktion der Studienergebnisse
5.4.1. Dr. Phillip Buckhaults
Die Ergebnisse von McKernan und Speicher wurden von Dr. Phillip Buckhaults, Professor an der University of South Carolina, bestätigt. Dr. Phillip Buckhaults ist ein erfahrener Molekularbiologe und Krebsgenetiker der über umfangreiche Erfahrung mit Next-Gen-Sequenzierungsanwendungen für globale Gene Expressionsanalyse und Erkennung von Genmutationen verfügt. Er präsentierte seine Ergebnisse zur DNA-Kontamination im Zusammenhang mit den mRNA-COVID-19-Impfstoffen bei einer Anhörung im Senat von South Carolina im September 2023. Der Originalbeitrag wurde von YouTube entfernt, ist aber hier und hier noch zu finden.
Dr. Buckhaults und seine Kollegen identifizierten Fremd-DNA in Chargen von Pfizer-BioNTech (Comirnaty) und Moderna (Spikevax). [@P_J_Buckhaults]
In der Anhörung beschreibt Dr. Buckhaults seine Untersuchungsergebnisse:
„Dies ist also ein Bild des Sequenzierungsergebnisses, des Sequenzierungslaufs, den ich im Labor von ein paar Chargen des Pfizer-Impfstoffs durchgeführt habe. Und all diese kleinen Linien hier (Abb.50) sind die winzigen DNA-Stücke, die im Impfstoff enthalten sind. Sie gehören dort nicht hin. Sie sind nicht Teil des Verkaufsgesprächs oder der Marketingkampagne. Und es sind eine Menge davon.“

„Dieses kleine Diagramm hier (Abb.51 links) zeigt die Größenverteilung (für den Pfizer-Impfstoff). Die DNA-Stücke, die im Impfstoff enthalten sind, sind kurze, kleine Stücke von 100 bis 120 Basenpaaren. Es gibt einige, die etwa 500 Basenpaare lang sind, einige wenige, die sogar 5.000 Basenpaare lang sind, aber die meisten sind um die 100 Basenpaare lang. Warum ist das wichtig? Weil die Wahrscheinlichkeit, dass sich ein DNA-Stück in das menschliche Genom integriert, nicht von seiner Größe abhängt. Das Risiko für Ihr Genom hängt nur davon ab, wie viele Teilchen es gibt. … All diese kleinen DNA-Stücke, die in dem Impfstoff enthalten sind … ergeben viele, viele tausend Möglichkeiten, eine Zelle einer geimpften Person zu verändern. Die (DNA-)Stücke sind sehr klein, weil sie während des (Impfstoff-Herstellungs-)Prozesses zerhackt wurden, um sie verschwinden zu lassen, aber sie haben die Gefahr der Genomveränderung erhöht.“

„Wir haben also all diese DNA-Stücke genommen und sie dazu benutzt, um sie zusammenzukleben, was die Ausgangs-DNA gewesen sein muss. Das ist sozusagen wieder das, was wir im Labor die ganze Zeit machen, und all diese kleinen roten und grünen Linien hier (Abb.50), das sind alles unabhängige kleine DNA-Stücke. In diesem Sequenzierungslauf müssen es 100.000 DNA-Stücke gewesen sein, und Sie können sie alle wieder zusammensetzen und sehen, woher sie stammen, nämlich von diesem Kreis hier (Abb.51 rechts).“
„Es handelt sich um ein Plasmid, das Sie online bei Agilent kaufen können. Und es ist klar, dass Pfizer dieses Plasmid genommen hat und und dann Spike hinein geklont hat und es für einen Prozess namens in-vitro-Transkription/Translation verwendet hat, bei dem man eine RNA-Polymerase mit diesem Plasmid füttert und sie eine ganze Reihe von mRNA-Kopien für einen herstellt. Dann nimmt man diese mRNA und mischt sie mit dem Lipid-Nanopartikel-Transkriptionsreagenz, und schon hat man seinen mRNA-Impfstoff, aber die DNA hat man vorher nicht herausbekommen. Sie haben das so gemacht. Diese kleinen Stücke haben sie etwas zerkleinert, so dass alle diese kleinen Plasmastücke mit der RNA verpackt wurden. Es ist völlig klar, was passiert ist, wenn man sich nur die forensische Analyse der DNA-Sequenzierung anschaut.“
„Wir können die Menge dieses Materials im Labor ziemlich einfach messen. Wir sind gut darin, so etwas zu tun … Wir können genau quantifizieren, wie viel von diesem Zeug in einem Impfstoff oder einem anderen Gewebe ist, und ich schätze, dass in jeder Dosis etwa zwei Milliarden Kopien von dem einen Teil, nach dem wir suchen, enthalten sind. … Aber wenn man 2 Milliarden Kopien von diesem Teil sieht, gibt es etwa 200 Milliarden von allem anderen. Das bedeutet also, dass in jeder Dosis des Impfstoffs wahrscheinlich etwa 200 Milliarden Stücke dieser Plasma-DNA enthalten sind, die in diese Lipid-Nanopartikel eingekapselt sind, damit sie in die Zelle gelangen können. Das ist eine schlechte Idee.“
5.4.2. Dr. Jürgen O. Kirchner und Prof. Dr. Brigitte König
Der deutsche Biologe Dr. Jürgen O. Kirchner warnte schon 2022 vor der Problematik der DNA-Kontamination des BioNTech-Impfstoffs in dem Buch „Die mRNA-Maschine“, dass er unter dem Pseudonym David O. Fischer veröffentlichte.
Kirchner ist promovierter Biologe und bringt umfassende Berufserfahrung in der Pharmabranche mit. Er war für verschiedene Pharmaunternehmen tätig und hat sich im Rahmen seiner Aufgaben intensiv mit Studien befasst, die die Wirksamkeit von Medikamenten belegen sollten. In seiner Tätigkeit kümmerte er sich auch um das Marketing für Arzneien und deren Zulassung.
Dr. Kirchner hat Chargen des BioNTech-Impfstoffs, die nach Deutschland geliefert wurden, zur Untersuchung an das Magdeburger Labor von Prof. Dr. Brigitte König geschickt.
Frau Prof. Dr. König hat Biologie, Chemie und Medizin studiert, sich im Fach „Medizinische Mikrobiologie und Infektionsimmunologie“ habilitiert und gehört als externe Professorin dem Lehrstuhl der Medizinischen Fakultät an der Otto-von-Guericke-Universität in Magdeburg an.
Aus einem Interview Dr. Kirchners mit The Epoch Times stammen die folgenden Passagen, die Einblicke in seine Erkenntnisse gewähren.
„Die DNA-Verunreinigungen in den mRNA-Impfstoffen sind ein sehr schwerwiegendes Problem, was zunächst gar nicht so erkennbar war, obwohl in den Berichten der Europäischen Arzneimittel Agentur EMA drin steht, schon frühzeitig im Februar 2021, dass diese Impfstoffe mit DNA verunreinigt sind. Das Ausmaß war nicht bekannt. Das hat ein amerikanischer Wissenschaftler mit seinem Team, Kevin McKernan, herausgefunden und im April dieses Jahres (2023) veröffentlicht.“
„Daraufhin habe ich ein Labor gefunden und gebeten, deutsche Chargen dieses Impfstoffes zu untersuchen wie es bei uns aussieht. Und die Ergebnisse waren eine Katastrophe – hochgradige Verunreinigungen mit DNA.“
„Dieser Impfstoff – Sie hatten ja gefragt, woher kommt das – der ist deshalb mit DNA belastet, weil er mit der Hilfe von Bakterien hergestellt wird. Diese Bakterien liefern die DNA-Matrize, über die die mRNA dann gebildet wird, vermehrt wird. Bei den klinischen Studien hat man das ohne Bakterien gemacht, das heißt die Impfstoffe der klinischen Studien waren bei weitem nicht so hoch mit DNA belastet wie die Verkaufsware. Das bedeutet gleichfalls, dass die Gefahr, die von der DNA in den Impfstoffen ausgeht, überhaupt nicht klinisch überprüft worden ist, weil der Impfstoff in den klinischen Prüfungen hatte das nicht. Somit ist diese ganze Thematik ein riesiges schwarzes Loch, von dem keiner weiß, was sich dahinter verbirgt. Und das muss ganz schnell geändert werden.“
„Geprüft wurden Chargen des Impfstoffs BioNTech. Aktuell verwenden wir die Daten von fünf Chargen, von den wir verschlossene Chargen haben bekommen können. Der Grenzwert der Verunreinigungen ist von der WHO selbst auf 10 ng (Nanogramm) pro Dosis festgelegt worden. Die niedrigste Konzentration, die gefunden wurde in einem Impfstoff, war das 83fache und das höchste das 355fache. … Wir reden hier nicht, da ist der Grenzwert etwas überschritten, sondern hier ist es so, wir haben 3500 statt 10. Das muss man sich einfach mal vor Augen führen.“
Die Ergebnisse ihrer Analysen sind in der nachfolgenden Tabelle zusammengefasst.

Die Resultate wurden am 12. Dezember 2023 in einer MDR-Reportage der breiten Öffentlichkeit präsentiert. Der MDR-Beitrag wurde jedoch kurz darauf aus der MDR-Mediathek entfernt. Unter diesem Link ist der Beitrag weiterhin einsehbar.
Auf Anfrage von The Epoch-Times, warum der MDR-Beitrag gelöscht wurde, erhielt Epoch-Times von einer MDR-Sprecherin die folgende Antwort: „Der MDR hat am 12.12.2023 um 20.15 Uhr in der Sendung ‚Umschau‘ einen Beitrag zu Corona-Impfstoffen ausgestrahlt. Aufgrund sorgfältiger interner Prüfung steht fest: Dabei wurden unsere publizistischen Sorgfaltskriterien nicht eingehalten. Der Beitrag wurde am 17.12.2023 depubliziert.“ Welche „publizistischen Sorgfaltskriterien“ genau beim Beitrag nicht eingehalten worden sein sollen, teilte der MDR jedoch nicht mit.
5.5. Kurze Zusammenfassung
- Die gemessenen Werte für DNA und RNA sind von der spezifischen Messmethode abhängig, die zur Analyse verwendet wird.
- Es existiert kein einheitlicher Regulierungsstandard seitens der Behörden, der vorschreibt, welche Messverfahren von den Herstellern zur Prüfung von RNA und DNA einer Substanz verwendet werden müssen.
- Es wurden große Mengen an kleinen DNA-Fragmenten gefunden, die über dem von der WHO empfohlenen Grenzwert 10 ng/Dosis liegen.
- Es wurden auch vereinzelt sehr lange DNA-Moleküle gefunden, die über den von der WHO empfohlenen Länge von 200 Basenpaaren liegen.
- Es wurden funktionale Bestandteile des SV40-Virus gefunden, der SV40-Promotor und der SV40-Enhancer.
- Die Rest-DNA, die möglicherweise bei der Verpackung der mRNA in Lipid-Nanopartikel vorhanden ist, könnte ebenfalls in die Lipid-Nanopartikel verpackt werden. Diese Lipid-Nanopartikel tragen dazu bei, die Stabilität und den Schutz der eingekapselten Nukleinsäuren zu erhöhen, indem sie sie vor dem Abbau durch Enzyme wie Nukleasen schützen, die normalerweise in biologischen Systemen vorhanden sind.
6. Wissenstand und Reaktionen zur Fremd-DNA von offizieller Seite
6.1. Die Europäische Arzneimittel-Agentur (EMA)
6.1.1. Was ist die EMA und welche Aufgabe hat sie?
6.1.2. Was wusste die EMA über die DNA-Rückstände?
6.2. Das Paul-Ehrlich-Institut (PEI)
6.2.1. Was ist das PEI und welche Aufgabe hat es?
6.2.2. Fragen an das Paul-Ehrlich-Institut (PEI)
6.3. Kurze Zusammenfassung
6.1. Die Europäische Arzneimittel-Agentur (EMA)
6.1.1. Was ist die EMA und welche Aufgabe hat sie?
Die EMA (European Medicines Agency, Europäische Arzneimittel-Agentur) ist eine Agentur der Europäischen Union mit Sitz in Amsterdam, Niederlande. Die EMA wurde gegründet, um einen einheitlichen Zulassungsprozess für Arzneimittel in der gesamten Europäischen Union zu ermöglichen und somit einen schnelleren Zugang zu neuen Medikamenten für die Patienten zu gewährleisten. Hier sind einige der Hauptaufgaben der EMA:
Zulassung von Arzneimitteln: Die EMA spielt eine zentrale Rolle bei der Bewertung von Anträgen für die Zulassung neuer Arzneimittel in der Europäischen Union. Sie prüft die Qualität, Sicherheit und Wirksamkeit der Arzneimittel, bevor sie auf dem europäischen Markt verfügbar gemacht werden.
Überwachung der Sicherheit von Arzneimitteln: Die EMA überwacht kontinuierlich die Sicherheit von bereits zugelassenen Arzneimitteln. Dies umfasst die Erfassung von Daten zu Nebenwirkungen und anderen sicherheitsrelevanten Informationen. Bei Bedarf können Maßnahmen ergriffen werden, um die Sicherheit der Patienten zu gewährleisten, wie beispielsweise die Änderung von Packungsbeilagen oder der Widerruf von Zulassungen.
Qualitätskontrolle: Die EMA überprüft die Qualität von Arzneimitteln und stellt sicher, dass sie den erforderlichen Standards entsprechen. Dies bezieht sich auf die Herstellung, Prüfung und Verpackung von Arzneimitteln.
6.1.2. Was wusste die EMA über die DNA-Rückstände?
Im Folgenden eine Überprüfung einiger EMA-Dokumente sowie entsprechende Erläuterungen. Die geschwärzten Bereiche in den Dokumenten wurden von der EMA selbst vorgenommen.
Ein „Assessment Report“ ist ein Bewertungsbericht, der von der EMA erstellt wird, um die Bewertung eines Arzneimittels durch den Ausschuss für Humanarzneimittel (CHMP) zu dokumentieren. Dieser Bericht enthält eine umfassende Analyse aller verfügbaren Daten. Darüber hinaus kann er auch Empfehlungen und Anforderungen für die weitere Überwachung des Arzneimittels nach der Zulassung enthalten.
EMA-Dokument vom Februar 2021

„Die Robustheit des DNase-Verdauungsschritts gilt nicht als umfassend nachgewiesen, obwohl es eine routinemäßige Kontrolle von Rest-DNA-Verunreinigungen auf Wirkstoffebene gibt. Es wurde bestätigt, dass Studien zur Verbesserung der Robustheit dieses Schrittes durchgeführt werden, über die berichtet werden sollte (REC7).“
Im Assessment Report der EMA vom Februar 2021 wird festgestellt, dass die Robustheit des DNase-Verdauungsschritts nicht umfassend nachgewiesen wurde, obwohl es eine routinemäßige Kontrolle von residualen DNA-Verunreinigungen auf Ebene des Wirkstoffs gibt. Es wurde bestätigt, dass Studien zur Verbesserung der Robustheit dieses Schrittes im Gange sind und dass diese Studien gemäß der Verpflichtung (REC7) des Inhaber der Marktzulassung (BioNTech) berichtet werden sollen.
EMA-Dokument vom Mai 2021

„REC7: Der Zulassungsinhaber sollte die Ergebnisse der Studien vorlegen, die zur Verbesserung der Robustheit des DNase-Verdauungsschritts im Wirkstoffherstellungsprozess durchgeführt wurden.“
In diesem Auszug des EMA-Dokuments bezieht sich „MAH“ auf den „Marketing-Authorisation Holder“, also den Inhaber der Marktzulassung (BioNTech) für das Arzneimittel Cormirnaty.
Die Passage besagt, dass der MAH eine Verpflichtung zur Erfüllung nach der Zulassung des Arzneimittels übernommen hat, nämlich die Bereitstellung der Ergebnisse von Studien, die durchgeführt wurden, um die Robustheit des DNase-Verdauungsschritts im Herstellungsprozess des Wirkstoffs zu verbessern.
Um das zu verdeutlichen:
- Wie im Kapitel 3.5.2. beschrieben, ist DNase ein Enzym, das DNA abbaut.
- Der Herstellungsprozess des Wirkstoffs bezieht sich auf den Prozess, durch den der eigentliche Wirkstoff des Arzneimittels hergestellt wird.
- Der DNase-Verdauungsschritt ist ein spezifischer Schritt im Herstellungsprozess, bei dem DNase verwendet wird, um die DNA zu verdauen und zu entfernen. (siehe dazu 3.5.2. Reinigung der synthetischen mRNA im Prozess 2)
- Die Robustheit dieses Schrittes bezieht sich darauf, wie stabil und zuverlässig dieser Schritt im Herstellungsprozess ist, und wie gut er in verschiedenen Situationen funktioniert.
Die EMA fordert den MAH auf, die Ergebnisse von Studien bereitzustellen, die durchgeführt wurden, um sicherzustellen, dass dieser spezifische Schritt im Herstellungsprozess des Wirkstoffs zuverlässig und stabil ist. Dies dient dazu, die Qualität und Sicherheit des Arzneimittels zu gewährleisten, insbesondere in Bezug auf potenzielle Verunreinigungen durch DNA-Rückstände.

„Es wird kein detaillierter Bericht über die Studien vorgelegt, die zur Verbesserung der Robustheit des DNase-Aufschlusses durchgeführt wurden.“
„Es wird erwartet, dass eine detaillierte Zusammenfassung der Ergebnisse der Studien, die zur Verbesserung der Robustheit des DNase-Verdauss durchgeführt wurden, bis Ende des zweiten Quartals 2021 in Modul 3.2.S.2.5 des Dossiers aufgenommen wird.“
„PAM nicht erfüllt“
In diesem Abschnitt des EMA-Dokuments wird darauf hingewiesen, dass der MAH keine ausführlichen Berichte über die durchgeführten Studien zur Verbesserung der Robustheit des DNase-Verdauungsschritts vorgelegt hat.
„PAM“ steht für „Post-Authorisation Measure“, was bedeutet, dass eine Maßnahme nach der Zulassung eines Arzneimittels ergriffen werden muss. In diesem Fall bedeutet „PAM not fulfilled“ also, dass die Maßnahme, die der MAH (BioNTech) bezüglich der Berichterstattung über die Studien zur Verbesserung der DNase-Verdauung durchführen musste, nicht erfüllt wurde.
Die Passage besagt, dass erwartet wird, dass der MAH eine detaillierte Zusammenfassung der Ergebnisse aus den durchgeführten Studien zur Verbesserung der Robustheit des DNase-Verdauungsschritts bis zum Ende des zweiten Quartals 2021 im Modul 3.2.S.2.5 des Dossiers einreicht.
EMA-Dokument vom August 2021
Die Diskussion um die „Verbesserung der Robustheit des DNase-Verdauungsschritts“ wird fortgeführt.


Die Empfehlung Nr. 7 (REC7), die Ergebnisse der zur Verbesserung der Robustheit des DNase-Verdauungsschritts durchgeführten Studien vorzulegen, wurde nur teilweise erfüllt. Zur Erfüllung der Empfehlung Nr. 7 (REC7) sind weitere Maßnahmen erforderlich, darunter die Vorlage einer detaillierten Zusammenfassung der Studienergebnisse und die Aufnahme dieser Daten in Modul 3.2.5.2.5 des Dossiers bis zum Ende des zweiten Quartals 2021.
Im Anschluss an den während der ACMF PPQ-Kampagne beobachteten Anstieg der Rest-DNA wurden Versuche in kleinem Maßstab eingeleitet, um die Robustheit des DNase I-Verdauungsschritts zu verbessern. Es wurden Studien durchgeführt, um die Auswirkungen von Reaktionskomponenten, Prozess- und Betriebsparametern auf die Menge der Rest-DNA-Matrize besser zu verstehen. Die Studien im kleinen Maßstab sind nicht schlüssig und es werden keine Anpassungen des DNase-Schrittes empfohlen, daher werden die Daten aus diesen Studien nicht bereitgestellt.
Der Text besagt, dass die Empfehlung Nr. 7 (REC7) bezüglich der Vorlage von Studienergebnissen zur Verbesserung der Robustheit des DNase-Verdauungsschritts nur teilweise erfüllt wurde. Es wird festgestellt, dass weitere Maßnahmen erforderlich sind. Die Antwort von BioNTech auf diese Empfehlung beinhaltet Versuche in kleinem Maßstab, um die Robustheit des DNase I-Verdauungsschritts zu verbessern. Diese Versuche sollen die Auswirkungen verschiedener Faktoren auf die Menge der Rest-DNA besser verstehen helfen. Es wird jedoch festgestellt, dass die durchgeführten Studien nicht schlüssig sind und daher keine Anpassungen am DNase-Schritt empfohlen werden. Als Folge werden die Daten aus diesen Studien nicht bereitgestellt.

Im EMA-Bericht auf Seite 3 wurde der „GMP -Inspection check“ mit „No“ bewertet. Diese Bewertung zeigt an, dass bei einer Inspektion der Good Manufacturing Practice (GMP) Mängel oder Unzulänglichkeiten in Bezug auf die Einhaltung bestimmter Anforderungen oder Standards festgestellt wurden. Das Produkt Cormirnaty war zu diesem Zeitpunkt NICHT GMP konform.
EMA-Dokument vom März 2022

„REC 7: „Der MAH sollte die Ergebnisse der Studien vorlegen, die zur Verbesserung der Robustheit des DNase-Verdauungsschritts durchgeführt wurden“ ist teilweise erfüllt.“

„Weitere Maßnahmen sind erforderlich, um die Empfehlung 7 zu erfüllen, einschließlich der Vorlage einer detaillierten Zusammenfassung der Studienergebnisse“
„VAR IB-55: Es werden keine Ergebnisse vorgelegt, da der Antragsteller die Studie im kleinen Maßstab für nicht schlüssig hält und keine Anpassung des DNase-Verdauungsschritts empfohlen wird. Dies wird nicht akzeptiert, und es sollten Daten vorgelegt werden, die belegen, dass keine Änderung erforderlich ist. Darüber hinaus sollte die Korrelation zwischen der DNase-I-Aktivität und dem Gehalt an Rest-DNA-Template, wie er mit den firmeneigenen Methoden gemessen wird, ausreichend bewertet werden.“
Dieser Abschnitt aus dem EMA-Dokument bezieht sich auf die Überprüfung der Erfüllung bestimmter Verpflichtungen (Recommendation, REC) des Marketing-Authorisation Holders (MAH) im Zusammenhang mit der Herstellung des Arzneimittels „tozinameran“.
„Tozinameran“ ist der Wirkstoffname für den COVID-19-Impfstoff von Pfizer-BioNTech. Der Impfstoff wird unter dem Markennamen „Comirnaty“ vermarktet. „Tozinameran“ ist der internationale, nicht-proprietäre Name (INN) für den Wirkstoff, der von der Weltgesundheitsorganisation (WHO) festgelegt wurde. Der Begriff wird verwendet, um den spezifischen Wirkstoff des Impfstoffs unabhängig von Markennamen zu identifizieren.
Teilweise erfüllt: Es wird festgestellt, dass die Verpflichtung, die Ergebnisse der durchgeführten Studien zur Verbesserung der Robustheit des DNase-Verdauungsschritts vorzulegen, teilweise erfüllt wurde. Es wird jedoch darauf hingewiesen, dass weitere Maßnahmen erforderlich sind, um die Empfehlung 7 (REC 7) vollständig zu erfüllen.
Besonderheiten zu den einzelnen Variablen (VAR):
VAR IB-55: Es werden keine Ergebnisse bereitgestellt, da der Antragsteller die Studie als nicht schlüssig betrachtet und keine Anpassung des DNase-Verdauungsschritts empfohlen wird. Dies wird jedoch nicht akzeptiert, und es werden Daten gefordert, um zu belegen, dass keine Änderung erforderlich ist. Darüber hinaus sollte die Korrelation zwischen der DNase I-Aktivität und der Rest-DNA, wie sie mit den firmeneigenen Methoden gemessen werden, ausreichend bewertet werden.
VAR IB-106-G: Um die Empfehlung 7 (REC7) abzuschließen, sollten Daten von den beiden zusätzlichen Standorten Mainz/Rentschler und Marburg bereitgestellt werden. Abschnitt 3.2.S.2.5 des Dossiers sollte mit Daten zu dem Rest-DNA-Gehalt von allen drei Standorten aktualisiert werden. Darüber hinaus sollte Abschnitt 3.2.S.2.5 um Informationen zur verbesserten Prozesskontrolle aktualisiert werden. Wenn möglich, sollten die geringfügigen Anpassungen in den entsprechenden Akzeptanzkriterien reflektiert werden.
Insgesamt zeigt das behördliche EMA-Dokument, dass im März 2022 – über ein Jahr nach dem Masseneinsatz des BioNTech-Impfstoffs – dieser Punkt nicht sauber geklärt ist.
Trotz der Feststellung „Partly Fulfilled“ (teilweise erfüllt) befindet die EMA die vom MAH (BioNTech) vorgeschlagenen Änderungen für „is/are approvable“ genehmigungsfähig:

Um den Disput zwischen EMA und Pfizer zur Robustheit der DNase I besser zu verstehen, liefert der Artikel von ThermoFisher SCIENTIFIC weitere Hinweise. Dort wird darauf hingewiesen, dass es schwierig sein kann, alle DNA-Kontaminationen vollständig zu entfernen.
6.2. Das Paul-Ehrlich-Institut (PEI)
6.2.1. Was ist das PEI und welche Aufgabe hat es?
Das Paul-Ehrlich-Institut (PEI) ist eine Bundesoberbehörde in Deutschland, die dem Bundesministerium für Gesundheit unterstellt ist. Es wurde nach dem deutschen Medizinforscher Paul Ehrlich benannt und hat seinen Sitz in Langen, Hessen. Das PEI hat eine zentrale Rolle im Bereich der Arzneimittelsicherheit und Impfstoffzulassung in Deutschland. Hier sind einige der Hauptaufgaben des PEI:
Zulassung: Das PEI ist für die Zulassung von Arzneimitteln und Impfstoffen verantwortlich. Es prüft die Qualität, Wirksamkeit und Sicherheit von neuen Medikamenten und Impfstoffen, bevor sie auf dem Markt zugelassen werden.
Überwachung: Das PEI überwacht kontinuierlich die Sicherheit und Qualität von Arzneimitteln und Impfstoffen, die bereits auf dem Markt sind. Es stellt sicher, dass die Produkte gemäß den geltenden Standards hergestellt werden und den Anforderungen an Sicherheit, Reinheit und Wirksamkeit entsprechen. Es sammelt und bewertet Berichte über Nebenwirkungen und unerwünschte Ereignisse, um potenzielle Risiken frühzeitig zu erkennen und geeignete Maßnahmen zum Schutz der öffentlichen Gesundheit zu ergreifen.
Forschung und Entwicklung: Das PEI betreibt auch Forschung auf dem Gebiet der Arzneimittelsicherheit und Impfstoffentwicklung. Es arbeitet eng mit anderen nationalen und internationalen Behörden sowie mit akademischen Einrichtungen und der Industrie zusammen, um neue Erkenntnisse zu gewinnen und innovative Ansätze zur Verbesserung der Gesundheit zu fördern.
6.2.2. Fragen an das Paul-Ehrlich-Institut (PEI)
27.09.2023 – Brief von ÄFI an das PEI
ÄFI (Ärztinnen und Ärzte für individuelle Impfentscheidung e.V)
Aufgrund der Studie von Kevin McKernan und angesichts der Verantwortlichkeit des PEI für die Überwachung und Qualitätssicherung von Impfstoffen in Deutschland formulieren „Ärztinnen und Ärzte für individuelle Impfentscheidung e.V“ (ÄFI) in einem Brief an das PEI am 27.09.2023 wichtige Fragen zum Thema „DNA-Verunreinigung in den mRNA-Impfstoffen von Pfizer/BioNTech und Moderna“.
Frage 1: Sind die vom PEI durchgeführten oder veranlassten Sicherheitsprüfungen von Impfstoffen darauf ausgelegt, derartige Fremdstoffe (DNA-haltige Fragmente/Plasmide) nachzuweisen, zu klassifizieren und zu quantifizieren?
Frage 2: Falls Ja: Zu welchen Ergebnissen ist das PEI gekommen? Hat das PEI Kenntnisse darüber erlangt, dass es zu signifikanten DNA-Verunreinigungen der genannten mRNA-Impfstoffen gekommen ist?
Frage 3: Falls Nein: Hat das PEI nach Bekanntwerden der oben zitierten Ergebnisse im Frühjahr des Jahres 2023 Vorkehrungen im Sicherheitsprüfprotokoll getroffen, eine Verunreinigung für die in Deutschland eingesetzten Chargen beider Impfstoffhersteller auszuschließen?
19.10.2023 – Antwort des PEI an ÄFI
In der Antwort des PEI ist folgendes zu lesen:
„Das Paul-Ehrlich-Institut ist ein amtliches Kontrolllabor im europäischen OMCL-Netzwerk und hat die meisten Chargen des von der EU-Kommission zugelassenen COVID-19-Impfstoffprodukts Comirnaty (in allen Indikationen und Konzentrationen) entsprechend OMCL-Leitfaden und Zulassungsvorgaben geprüft und ggf. die staatliche Chargenfreigabe für Deutschland erteilt.“
„Bei den Parametern wie dem Rest-DNA-Gehalt im Impfstoff, die nur vom Hersteller experimentell geprüft werden, überprüft das OMCL die Testergebnisse des Herstellers daraufhin, ob die in der Zulassung festgelegten Grenzwerte in jeder Charge eingehalten wurden.“
Die von den OMCL durchzuführenden Chargenfreigabetests für die COVID-19-Impfstoffe sind hier in der Tab. 2 zusammengefasst.
„Die vom Paul-Ehrlich-Institut geprüften und freigegebenen Chargen wiesen keine Beanstandungen auf. Daher waren keine Maßnahmen wie weitere Sicherheitsvorkehrungen erforderlich. Chargen, die nicht alle Kriterien erfüllen, werden nicht für den deutschen Markt zugelassen.“
Zusammengefasst bestätigt die Antwort des PEI: Es gibt einen Rest-DNA-Gehalt und die experimentelle Überprüfung der Impfstoff-Chargen erfolgt ausschließlich beim Hersteller.
Die Diskussion gewinnt an Fahrt, was zu einer Vielzahl weiterer Anfragen aus medizinischen Fachkreisen zum Thema führt. Als Reaktion darauf veröffentlicht das Paul-Ehrlich-Institut (PEI) am 22.12.2023 einen Brief mit Informationen für medizinische Fachkreise.
22.12.2023 – Brief des PEI mit Informationen für medizinische Fachkreise
Aus diesem Brief werden die wichtigsten Aussagen des PEI analysiert. Um die Übersichtlichkeit zu verbessern, werden die Aussagen in separate Abschnitte unterteilt.
Abschnitt I
„Vielen der in der Öffentlichkeit kursierenden Daten und Untersuchungen zu mutmaßlichen Kontaminationen der COVID-19-mRNA-Impfstoffe liegen methodische Mängel zu Grunde. Auch stellt sich das Problem der möglicherweise unsachgemäßen Lagerung der untersuchten Impfstoffdosen.“
Das PEI unterstellt damit den Wissenschaftlern, die es gewagt haben, den Impfstoff auf Rest-DNA-Gehalt zu überprüfen: Inkompetenz. Zu diesen Wissenschaftlern gehören u.a.:
Dr. Kevin McKernan leitete das Team für Forschung und Entwicklung beim Human Genome Project. Er gründete Medicinal Genomics und war Mitbegründer der Agencourt Bioscience Corporation. Er ist ein renommierter Genomiker und entwickelte bahnbrechende Sequenzierungstechnologien.
Dr. David J. Speicher ist Assistenzprofessor für Biologie und Gesundheitswissenschaften an der Redeemer University in Hamilton, Ontario. Er hat in Virologie promoviert und verfügt über umfassende Erfahrung in der Erkennung von Infektionskrankheiten mithilfe molekularbiologischer Verfahren. Seine Forschung konzentriert sich auf den Nachweis von Infektionskrankheiten mit Hilfe von Speichel-Biomarkern, und er war Leiter einer Covid-19-Testanlage.
Dr. Phillip Buckhaults, Professor an der University of South Carolina, ist ein erfahrener Molekularbiologe und Krebsgenetiker mit umfangreicher Erfahrung in der Anwendung von Next-Gen-Sequenzierungstechnologien für die globale Genexpressionsanalyse und Erkennung von Genmutationen. Er ist zudem ein Impfbefürworter.
Dr. Jessica Rose ist eine kanadische Forscherin mit einem Bachelor-Abschluss in angewandter Mathematik und einem Master-Abschluss in Immunologie von der Memorial University of Newfoundland. Sie hat außerdem einen Doktortitel in Computerbiologie von der Bar-Ilan-Universität und zwei Post-Doc-Abschlüsse: einen in Molekularbiologie von der Hebräischen Universität Jerusalem und einen in Biochemie vom Technion Institute of Technology.
Prof. Dr. König hat Biologie, Chemie und Medizin studiert, sich im Fach „Medizinische Mikrobiologie und Infektionsimmunologie“ habilitiert und gehört als externe Professorin dem Lehrstuhl der Medizinischen Fakultät an der Otto-von-Guericke-Universität in Magdeburg an.
… und so weiter. Die Liste ist lang.
Abschnitt II
„Experimentelle Bestimmungen z. B. von Rest-DNA durch Dritte in auf dem Markt verfügbaren Impfstoffdosen müssen folgende Kriterien erfüllen, um wissenschaftlich haltbare Ergebnisse zu erbringen:“
„(i) Sie dürfen nicht an Proben erfolgen, die aus abgelaufenen (Haltbarkeitsdatum überschritten) Impfstoff-Fläschchen oder aus angebrochenen oder nicht sachgemäß gelagerten Impfstoff-Fläschchen stammen.“
a) Zugang zu Impfstoffdosen
Zu beachten ist, dass Impfstoffe in der Regel von staatlichen Gesundheitsbehörden an medizinische Einrichtungen, Hausärzte, Fachärzte, Apotheken und Impfzentren verteilt werden. Dies bedeutet, dass diese Institutionen eine Art Monopolstellung beim Zugang und der Verteilung dieser Produkte haben.
In diesem Zusammenhang ist der unten gelistete Aufruf des PEI an „Ärztinnen und Ärzte sowie Apothekerinnen und Apotheker“ vom 05.12.2023 bemerkenswert.

Gleichzeitig werden Millionen der wertvollen Impfstoffdosen entsorgt. Nur fünf Tage zuvor, am 01.12.2023, titelte ZEIT ONLINE „Jetzt rächen sich die Hamsterkäufe“. In dem Artikel heißt es:
„Allein in diesem Jahr sind 95 Millionen Coronaimpfdosen in den Zentrallagern verfallen und mussten vernichtet werden… Damit ist rund ein Drittel der 458 Millionen bisher an Deutschland ausgelieferten Dosen verfallen… Und die Zahl der verfallenen Impfdosen wird aller Voraussicht nach weiter steigen, denn in den Zentrallagern des Bundes liegen aktuell weitere 56 Millionen Impfdosen, wie das Gesundheitsministerium mitteilt.“
Das wirft Fragen auf. Warum wird bei einem offensichtlichen Überangebot an Impfstoffdosen die Bereitstellung einzelner Exemplare zur Überprüfung verhindert? Welche Bedenken hat das Paul-Ehrlich-Institut (PEI) diesbezüglich? Diese zusätzlichen Untersuchungen könnten doch potenziell die Ergebnisse der unabhängigen Studien und Labore widerlegen. Warum testet das PEI nicht selbst? Inwiefern trägt diese Reaktion des PEI zur Vertrauensbildung bei?
b) Klaus Cichutek über „Regulatorische Anforderungen an die Impfstoffqualität“
In einem Paper mit dem Titel „Beschleunigte Entwicklung von COVID-19-Impfstoffen: Technologieplattformen, Vorteile und damit verbundene Risiken“ verfasst und befürwortet Klaus Cichutek, der von 2009 bis Ende 2023 Präsident des Paul-Ehrlich-Instituts war, zusammen mit anderen folgende Aussagen:
Regulatorische Anforderungen an die Impfstoffqualität
Wie bei gängigen Impfstoffen ist die Etablierung eines vollständig qualitätsgesicherten Herstellungsprozesses von grundlegender Bedeutung für die Entwicklung von COVID-19-Impfstoffen. Dies erfordert detaillierte prozessspezifische Entwicklungen und Spezifikationen sowie die Implementierung geeigneter Kontrollmaßnahmen einschließlich In-Prozess-Kontrollen. Die gesamte Produktion von Impfstoffen muss den Anforderungen der „Good Manufacturing Practice“ (GMP) entsprechen … Für COVID-19-Impfstoffe wurde ein gewisses Maß an Flexibilität in Bezug auf das Herstellungsspektrum dieser Chargen zur Qualifizierung der Prozessleistung oder durch die Berücksichtigung der jeweiligen Daten für ähnliche Produkte derselben Plattformtechnologie angewandt. …
Zu diesen Tests gehören auch Kontrollen und die Minimierung möglicher Kontaminationen. …
Zugelassene Impfstoffe unterliegen zudem einer Chargenprüfung durch das Paul-Ehrlich-Institut oder ein anderes amtliches Kontrolllabor des europäischen OMCL-Netzwerks. Damit wird sichergestellt, dass für die Qualitätssicherung besonders wichtige Freigabetests, wie z.B. die Potenz, vor der offiziellen Chargenfreigabe für den Markt einer zusätzlichen experimentellen Prüfung durch ein qualifiziertes öffentliches Prüflabor unterzogen werden müssen.
Anforderungen an klinische Prüfungen
Auch nach der Zulassung werden weitere Studien zur Wirksamkeit („Wirksamkeit im Alltag“) und zur Sicherheit der Impfstoffe durch die Bundesbehörden durchgeführt, um sicherzustellen, dass die Impfstoffe nach der Zulassung in ihrem vorgesehenen Anwendungsbereich in breiten Bevölkerungsschichten ihr positives Nutzen-Risiko-Profil behalten.
Für die erneute Überprüfung der Nutzen-Risiko-Abwägung des untersuchten Arzneimittels gehören beispielsweise:
1. individuelle Berichte über erwartete schwerwiegende unerwünschte Nebenwirkungen mit unerwartetem Ausgang,
2. eine klinisch relevante Zunahme der Häufigkeit zu erwartender schwerwiegender unerwünschter Nebenwirkungen,
3. Ereignisse im Zusammenhang mit der Durchführung der Studie oder der Entwicklung des untersuchten Arzneimittels, die die Sicherheit der betroffenen Personen beeinträchtigen könnten.
Inwieweit stehen diese Aussagen im Widerspruch zu der oben zitierten Aufforderung im PEI-Brief vom 05.12.2023?
c) Kann sich der Rest-DNA-Gehalt in einer abgelaufenen und verschlossenen mRNA-Impfstoffflasche erhöhen?
Die Halbwertszeit der DNA beträgt 521 Jahre, was bedeutet, dass sich die Hälfte der DNA-Moleküle in einem Probenmaterial nach dieser Zeit zersetzt. Auch wenn die DNA zerfällt und in kleinere Fragmente zerbricht, bleibt die Gesamtmenge der DNA in einer Probe unverändert. Die Qubit-Fluorometrie und qPCR messen die DNA-Konzentration pro Volumeneinheit, jedoch bleibt die Gesamtmenge der DNA in der Probe gleich, selbst wenn sie in kleinere Fragmente zerfällt. Daher erhöht sich der Rest-DNA-Gehalt in einer mRNA-Impfstoffflasche auch nach einer kurzen Zeitspanne von 2 Jahren nicht, selbst unter nicht optimalen Lagerbedingungen.
Abschnitt III
„(ii) Die angewandte Methodik zur Bestimmung der Rest-DNA-Menge muss nachgewiesenermaßen geeignet und nachvollziehbar sein – insbesondere sollten Testinterferenzen durch das Vorhandensein von Lipid-Nanopartikeln in den Impfstoff-Fläschchen ausgeschlossen worden sein (was bei Testung am finalen Impfstoff-Fläschchen nicht garantiert werden kann).“
In der Studie von Speicher et al. ist folgendes zu lesen: „Der Impfstoff in diesen drei Fläschchen wurde seriell um das 10-fache verdünnt, um die LNP-Hemmung (Hemmung von Lipidnanopartikeln) in der qPCR zu beurteilen. Wir beobachteten die erwartete ~3,3 Cq-Antwort nach der Verdünnung von 1:10 (1:10, 1:100, 1:1000), was darauf hindeutet, dass es eine LNP-Hemmung gibt, die die Quantifizierung der DNA bei diesen Verdünnungen beeinflussen könnte. Daher wurden die Daten aus den 1:10-Verdünnungen für die weitere Analyse verwendet. Diese Verdünnung sowie die Tatsache, dass einige der Dosen so konzipiert waren, dass sie vor der Anwendung verdünnt werden konnten, wurde in unseren Berechnungen berücksichtigt.“
Dies deutet darauf hin, dass Maßnahmen ergriffen wurden, um potenzielle Testinterferenzen durch das Vorhandensein von Lipid-Nanopartikeln in den Impfstofffläschchen zu berücksichtigen.
Abschnitt IV
(iii) Die angewandte Methode muss validiert worden sein, um verlässliche und überprüfbare Ergebnisse zu liefern.
„In den häufig zitierten Preprint-Veröffentlichungen von McKernan et al. (April 2023) und Speicher et al. (Oktober 2023) fehlen ausreichende Angaben, ob die genannten Bedingungen eingehalten wurden, sowie Angaben zur Nachvollziehbarkeit der gewählten Methodik. Eine Methoden-Validierung ist unerlässlich, um sicherzustellen, dass mit der Durchführung der eingesetzten Methode zu jedem Zeitpunkt und unabhängig von der ausführenden Person verlässliche und reproduzierbare Ergebnisse erzielt werden und dass die Methode für ihren Einsatzzweck geeignet ist. Die o. g. Bedingungen für den Erhalt wissenschaftlich haltbarer Messergebnisse bei der Rest-DNA-Bestimmung werden von den Herstellern eingehalten.“
Zu diesem Thema äußert sich Dr. Kevin McKernan in einem Interview mit Prof. Dr. Paul Cullen und Prof. Dr. Henrieke Stahl:
„Unsere Methoden sind im öffentlichen Raum und wurden von unabhängigen Laboratorien reproduziert. Das PEI jedoch führt gar keine eigenen Prüfungen auf DNA am Impfstoff durch, sondern überlässt diese Prüfung den Herstellern. Die Hersteller wiederum haben, soweit wir wissen, keine detaillierte Einsicht in ihre Prüfverfahren gewährt; nur ein durch eine Cyberattacke offengelegtes EMA-Dokument gibt einen gewissen Einblick in die Prüfmethoden. Würden die Hersteller oder auch das PEI ihre eigenen Methoden und selbst erhobenen Daten transparent veröffentlichen, wären wir gern bereit, Vergleiche anzustellen und möglicherweise voneinander zu lernen.“
„Bis zu diesem Zeitpunkt sind unsere Methoden nicht nur die einzigen öffentlich zugänglichen Messverfahren, sondern auch die am besten validierten. Denn sie wurden gleich mit verschiedenen „Stress-Tests“ traktiert, wie etwa einer Gradienten-PCR, mit Feststellungen der unteren Nachweisgrenzen, einer Analyse der PCR-Primer-Konzentrationen und einer PCR-Inhibitionsanalyse. Darüber hinaus wurden die vermehrten DNA-Fragmente auch von dritter Seite mittels eines Goldstandards, der sogenannten Sanger-Methode, sequenziert. Außerdem wurde eine Standard-Sequenzierung mit der Illumina- und Oxford Nanopore-Methode in mehreren unabhängigen Labors durchgeführt.“
„Die EMA hat, wie dem oben genannten Dokument „Rolling Review“ zu entnehmen ist, zur DNA-Messung qPCR, und zur RNA-Messung Fluorometrie und Spektrometrie im ultravioletten Lichtspektrum akzeptiert. Dieses Vorgehen steht im Widerspruch zum Moderna-Patent US 10.077.439, welches vom Firmenchef Stéphane Bancel eingereicht wurde. Hier wird gezeigt, dass qPCR die Menge an DNA unterschätzt, weil sie Fragmente mit einer Länge von weniger als etwa 100 Basenpaare nicht erfassen kann.“
6.3. Kurze Zusammenfassung
- Die Europäische Arzneimittel-Agentur (EMA) ist sich der Problematik der „herstellungsbedingten Rest-DNA-Verunreinigungen“ seit Beginn bewusst. Bereits im Februar 2021 forderte sie gezielt zusätzliche Daten vom Hersteller an, um die Robustheit (d.h. Zuverlässigkeit) des DNase-Verdauungsschritts (siehe REC7) zu klären, da dieser verwendet wird, um die DNA zu entfernen.
- Im März 2022 stellte die EMA fest (VAR IB-55), dass der Antragsteller keine Ergebnisse vorlegte, da er die Schlüssigkeit der Studie im kleinen Maßstab in Frage stellte und keine Anpassung des DNase-Verdauungsschritts empfahl. Diese Haltung wurde von der EMA nicht akzeptiert, und der Antragsteller wurde aufgefordert, Daten vorzulegen, die belegen, dass keine Änderung erforderlich ist. Darüber hinaus wurde betont, dass die Korrelation zwischen DNase-Verdauungsschritt und dem Gehalt an Rest-DNA, ausreichend bewertet werden sollte. Trotz dieser Bedenken wurde seitens der EMA die formelle Genehmigung erteilt.
- Das Paul-Ehrlich-Institut (PEI) bestätigt die Existenz von Rest-DNA im Impfstoff sowie die Tatsache, dass die experimentelle Überprüfung der Impfstoffchargen ausschließlich beim Hersteller erfolgt.
- Das PEI stellt die von unabhängigen Laboren berichteten Messergebnisse bezüglich der Rest-DNA im Impfstoff in Frage. Die Behörde zweifelt die verwendeten Messmethoden und die Qualität der verwendeten Impfstoffproben an.
- Das Paul-Ehrlich-Institut (PEI) hat in einem „Brief an Ärztinnen und Ärzte sowie Apothekerinnen und Apotheker“ darum gebeten, keine Impfstoffdosen für weitere Untersuchungen an unabhängige Labore zu schicken. Dieser Appell erfolgt zu einem Zeitpunkt, an dem ein Überschuss an Impfstoffdosen vorhanden ist.
- Obwohl die experimentellen Daten von McKernan et al. und Speicher et al. auf etablierten Verfahren beruhen, die den gängigen wissenschaftlichen Standards entsprechen und auch von Pfizer/BioNTech und Moderna verwendet werden, unternimmt das PEI keine Bemühungen, den Disput durch weitere unabhängige experimentelle Untersuchungen mit Beteiligung aller Parteien zu klären.
7. Sind weitere Untersuchungen notwendig?
Die Ausführungen im Kapitel 6 erwecken den Eindruck, dass sowohl die Hersteller der mRNA-Impfstoffe als auch die verantwortlichen Regulierungsbehörden an einer weiteren fachlichen Auseinandersetzung mit den Befunden aus Kapitel 5 nicht interessiert sind.
Die Hauptargumente der Regulierungsbehörden wurden von McKernan zusammengefasst:

Es gilt zu untersuchen, ob die Schlussfolgerungen der Behörden, wonach die DNA zu kurz, die DNA-Menge zu gering oder die DNA nicht funktionsfähig sei, eindeutig und unbestritten sind. Es ist wichtig, diese Schlussfolgerungen kritisch zu hinterfragen und die Argumente näher zu beleuchten bzw. zu untersuchen, um festzustellen, ob sie tatsächlich zutreffen oder ob weitere Untersuchungen erforderlich sind.
7.1. Offizielle Aussagen zu DNA-Resten
7.2. Weitere Studien zum Thema
7.3. SV40 Sequenzen
7.4. SV40-Promotor und das p53-Tumorsuppressor-Gen
7.5. Rest-DNA und die Lipid-Nanopartikel
7.6. Mögliche Integration in die DNA
7.7. Aufrufe zu weiteren Untersuchungen
7.8. Kurze Zusammenfassung
7.1. Offizielle Aussagen zu DNA-Resten
An dieser Stelle sind zwei Patente von Moderna hervorzuheben.
Patent US 10077439 B2 „Beseitigung von DNA-Fragmenten bei der Herstellung von MRNA“
In diesem Patent von Moderna (2014), verfasst unter anderem von Stéphane Bancel (CEO von Moderna), wird explizit darauf hingewiesen, dass verbleibende DNA in Arzneimittelprodukten die Aktivierung der angeborenen Immunantwort auslösen und potenziell onkogene Wirkungen bei bestimmten Patientenpopulationen verursachen könnte.
[0003] Die im mRNA-Herstellungsprozess verwendete DNA-Matrize muss entfernt werden, um die Wirksamkeit von Therapeutika und die Sicherheit zu gewährleisten, da restliche DNA in Arzneimittelprodukten die Aktivierung der angeborenen Reaktion induzieren kann und bei Patientenpopulationen möglicherweise onkogen wirkt. Regulatorische Richtlinien erfordern möglicherweise auch die Quantifizierung, Kontrolle und Entfernung der DNA-Matrize in RNA-Produkten. Derzeit verfügbare oder gemeldete Methoden beheben diesen Mangel nicht.
Patent US 10898574 B2 „Lieferung und Formulierung von manipulierten Nukleinsäuren“
In einem weiteren Patent von Moderna (2021) wird eindeutig formuliert:
„Es gibt zahlreiche Probleme mit früheren Methoden zur Abgabe pharmazeutischer Zusammensetzungen, um eine wirksame Proteinexpression sowohl für therapeutische als auch für biotechnologische Anwendungen zu erreichen. Beispielsweise kann sich eingeführte DNA mit einer gewissen Häufigkeit in die genomische DNA der Wirtszelle integrieren, was zu Veränderungen und/oder Schäden an der genomischen DNA der Wirtszelle führt. Alternativ kann die in eine Zelle eingeführte heterologe Desoxyribonukleinsäure (DNA) an Tochterzellen (unabhängig davon, ob sich die heterologe DNA in das Chromosom integriert hat oder nicht) oder an Nachkommen vererbt werden.“
In dem „FDA-Leitfaden zu prophylaktischen DNA-Impfstoffen: Analyse und Empfehlungen“ von 2009 ist zu lesen:
„DNA gilt als Kontamination „herkömmlicher“ (nicht DNA-)Impfstoffe. Normalerweise minimieren oder entfernen Hersteller während des Produktionsprozesses fremde DNA, um die Exposition des Menschen gegenüber potenziell schädlichem Material zu minimieren. Es überrascht nicht, dass Bedenken geäußert wurden, dass sich Plasmid-DNA in das Wirtsgenom integrieren und die Wahrscheinlichkeit einer malignen Transformation, einer genomischen Instabilität oder einer Dysregulation des Zellwachstums erhöhen könnte, als die ersten DNA-Impfstoffe für den klinischen Einsatz vorgeschlagen wurden.“
7.2. Weitere Studien zum Thema
7.2.1. Issues associated with residual cell-substrate DNA in viral vaccines
In dieser Studie mit dem Titel „Probleme im Zusammenhang mit restlicher Zellsubstrat-DNA in viralen Impfstoffen“ wird darauf hingewiesen, dass das Vorhandensein einiger restlicher zellulärer DNA, die aus dem Produktionszellsubstrat in viralen Impfstoffen stammt, unvermeidlich ist. Ob diese DNA ein Sicherheitsrisiko darstellt, ist unbekannt. DNA hat zwei biologische Aktivitäten, die berücksichtigt werden müssen. Erstens kann DNA onkogen sein; zweitens kann DNA infektiös sein.
7.2.2. High spontaneous integration rates of end-modified linear DNAs upon mammalian cell transfection
In dieser Studie mit dem Titel „Hohe spontane Integrationsraten von endmodifizierten linearen DNAs bei der Transfektion von Säugetierzellen“ wurde die Häufigkeit der Integration von Genmaterial ins Erbgut untersucht, nachdem man Zellen mit verschiedenen Formen von DNA behandelte. Es stellte sich heraus, dass nach der Behandlung mit verschiedenen Formen von linearer DNA etwa 10 bis 20% der Zellen stabil transfiziert wurden. Mit anderen Worten, ein beträchtlicher Prozentsatz der ursprünglich behandelten Zellen nahm das genetische Material auf. Die Integration von linearen DNAs erfolgte trotz veränderter DNA-Enden mit ähnlicher Häufigkeit. Diese Ergebnisse deuten darauf hin, dass die Zellen Mechanismen haben, um nicht-natürliche DNA-Enden zu eliminieren und die DNA anschließend durch Integration zu retten.
Die Studie legt nahe, dass nicht-integrierende Gentherapiemethoden möglicherweise weiterentwickelt werden müssen, bevor sie breiter eingesetzt werden können.
7.2.3. Double-stranded DNA induces a prothrombotic phenotype in the vascular endothelium
Diese Studie mit dem Titel „Doppelsträngige DNA induziert einen prothrombotischen Phänotyp im Gefäßendothel“ untersucht die Auswirkungen von doppelsträngiger DNA (dsDNA) auf die Blutgefäß-Innenwand, auch bekannt als vaskuläres Endothel. Die Forscher haben menschliche Endothelzellen im Labor mit synthetischer dsDNA behandelt. Dabei stellten sie fest, dass dies zu einer verstärkten Produktion von Molekülen führte, die das Blutgerinnungssystem aktivieren. Dies wiederum führte in vitro zu einer beschleunigten Blutgerinnung. Auch eine Injektion von dsDNA in Mäuse (in vivo) beschleunigte die Bildung von Blutgerinnseln in den Blutgefäßen, was zu einer verkürzten Zeit bis zur vollständigen Gefäßokklusion mit Stillstand des Blutflusses führte. Diese Studie zeigt, dass doppelsträngige DNA (dsDNA), sowohl viraler Herkunft als auch endogener (die natürlicherweise im Körper vorhanden ist) Herkunft, direkte prothrombotische Effekte im vaskulären Endothel auslöst.
7.3. SV40 Sequenzen
Die SV40-Komponenten spielen in der Gentherapie eine bedeutende Rolle. Der Promotor und der Enhancer sind zwei Schlüsselelemente der SV40-DNA, die in der Gentherapie eingesetzt werden, um die Expression von Fremdgenen in Zielzellen zu verstärken.
Der Promotor ist eine spezifische DNA-Sequenz, die die Transkription eines benachbarten Gens initiiert. In SV40 umfasst der Promotor sowohl den Replikationsursprung als auch die Bereiche für die frühen und späten Promotoren. Diese Struktur ermöglicht es dem Promotor, die Expression von Fremdgenen effizient zu regulieren.
Der Enhancer ist eine regulatorische DNA-Sequenz, die die Aktivität eines Promotors verstärken kann, unabhängig von der Orientierung oder Entfernung relativ zum Promotor. Der SV40-Enhancer ist bekannt für seine starke Enhancer-Aktivität und wird oft in Gentherapie-Vektoren verwendet, um die Expression von eingeführten Genen zu erhöhen.
Ein Schlüsselaspekt der SV40-Komponenten in der Gentherapie ist ihre Fähigkeit, den Import in den Zellkern zu unterstützen. Der SV40-Enhancer kann spezifische Signalwege nutzen, um die Kernporen zu passieren und effizient in den Zellkern zu gelangen. In diesem Zusammenhang sind die folgenden Studien erwähnenswert:
- SV40-basierte Gentherapie-Vektoren: Aus einem Gegner einen Freund machen
- Wirkung einer DNA-Kern-Targeting-Sequenz auf den Gentransfer und die Expression von Plasmiden im intakten Gefäßsystem
- Kern-Targeting von Plasmiden und Protein-DNA-Komplexen
Bereits 1997 wurde in einer Studie festgestellt, dass spezifische SV40-Sequenzen wichtig für den Transport von Molekülen in den Zellkern sind. In einer späteren Studie von 1999 wurde entdeckt, dass der SV40-Enhancer das Plasmid erfolgreich in die Zellkerne verschiedener Zelltypen befördert. Diese Forschungsergebnisse zeigen, wie bestimmte SV40-Sequenzen den Transport in den Zellkern steuern können.

1997: „Am wichtigsten ist, dass der Kernimport sequenzspezifisch war: Eine Region der SV40-DNA, die den Replikationsursprung und die frühen und späten Promotoren enthielt, unterstützte den Import, während bakterielle Sequenzen allein und andere von SV40 abgeleitete Sequenzen dies nicht taten.“
1999: „Im Gegensatz dazu wird ein ähnliches Plasmid, das den SV40-Enhancer trägt, in die Zellkerne aller getesteten Zelltypen transportiert.“
7.4. SV40-Promotor und das p53-Tumorsuppressor-Gen
Kevin McKernan erwähnte in einer Präsentation [Massachusetts-Folien, Folie 23] eine Studie zur Interaktion zwischen dem SV40-Promotor und p53.
p53 ist ein Schlüsselprotein im Tumorsuppressor, oft als „Wächter des Genoms“ bezeichnet. Es reguliert den Zellzyklus, die DNA-Reparatur, die Apoptose (programmierter Zelltod) und die Seneszenz (Zellalterung). Mutationen im p53-Gen sind häufig in vielen Krebsarten zu finden und führen zu Funktionsverlust und gestörter Regulation von Zellwachstum und -teilung.
Die Studie ergab, dass p53 mit einem spezifischen Bereich des SV40-Virusgenoms, bekannt als „Frühpromotor“, interagiert. Die Aussage in der Studie lautet: „Das p53-Protein bindet an den SV40-Frühpromotor.“
Rein hypothetisch könnte dies verschiedene Auswirkungen haben:
p53-Funktionsverlust: Wenn der SV40-Frühpromotor an p53 bindet und seine normale Funktion beeinträchtigt, könnte dies zu einem Funktionsverlust von p53 führen. p53 ist ein wichtiger Tumorsuppressor, der die Zelle normalerweise vor unkontrolliertem Wachstum schützt. Ein Funktionsverlust von p53 könnte daher zu einem erhöhten Risiko von Krebs oder anderen unerwünschten zellulären Veränderungen führen.
Störung der p53-vermittelten Signalkaskade: p53 reguliert die Expression vieler Gene, die an der Zellzyklusregulation, Apoptose (programmierter Zelltod) und DNA-Reparatur beteiligt sind. Wenn der SV40-Frühpromotor an p53 bindet und seine normale Transkriptionsaktivität beeinträchtigt, könnte dies zu einer Störung dieser wichtigen zellulären Prozesse führen, was potenziell zu genetischen Instabilitäten oder anderen zellulären Problemen führen könnte.
Interferenz mit anderen p53-Interaktionen: p53 interagiert mit vielen anderen Proteinen und regulatorischen Elementen im Zellkern, um seine Funktionen auszuüben. Wenn SV40-DNA-Sequenzen in den Zellkern gelangen und dort mit p53 interagieren, könnten sie andere wichtige p53-Interaktionen stören, was zu weitreichenden Auswirkungen auf die zelluläre Physiologie führen könnte.
Dies sind hypothetische Szenarien und daher wären weitere experimentelle Untersuchungen erforderlich, um die tatsächlichen Auswirkungen einer SV40-p53-Interaktion zu verstehen.
Darauf verweist auch McKernan in seiner Präsentation [Massachusetts-Folien, Folie 23]. Er sagte, dass die SV40-p53-Interaktion auf Genotoxizität untersucht werden müsste und das es bisher keine Studien zur Genotoxizität gibt. Es müsse analysiert und bewertet werden, ob es Integrationsrisiken oder onkogene Risiken gibt.
7.5. Rest-DNA und die Lipid-Nanopartikel
Speicher et al. stellten in der Studie fest, dass die Plasmid-DNA wahrscheinlich in Lipid-Nanopartikel verpackt ist. Die Aufgabe der Lipid-Nanopartikel ist es, ihren Inhalt vor dem Abbau durch Enzyme im Körper zu schützen. Die Lipid-Nanopartikel böten einen Schutz für die eingeschlossene DNA und würden es ihr ermöglichen, die Zellmembran zu durchqueren und in die Zellen einzudringen. Sobald die DNA in der Zelle angekommen ist, könnte der SV40-Enhancer, wenn er Teil der DNA-Sequenz ist, dazu beitragen, die DNA-Sequenz in den Zellkern zu transportieren. Diese würde das Risiko der Intergration der Plasmid-DNA in das Genom erhöhen.
Dr. Phillip Buckhaults beschreibt dies mit folgender Analogie:
20 griechische Soldaten, die vor den Mauern von Troja herumlaufen, sind keine große Sache.
20 griechische Soldaten, die in ein großes Holzpferd verpackt sind, sind eine andere Sache.
Die Schlussfolgerung von Speicher et al. in ihrer Studie lautet wie folgt: „Die FDA- und WHO-Richtlinien berücksichtigten nicht die Verpackung von DNA in Lipid-Nanopartikeln, was wahrscheinlich zu einer längeren DNA-Persistenz sowie einer erhöhten Transfektionseffizienz (genetisches Material erfolgreich in eine Zielzelle zu transportieren) führte. Darüber hinaus wurde in den Leitlinien eine kumulative Dosierung mit LNP-basierter modRNA nicht berücksichtigt.“
„Unsere Ergebnisse erweitern bestehende Bedenken hinsichtlich der Sicherheit von Impfstoffen und stellen die Relevanz von Leitlinien in Frage, die vor der Einführung einer effizienten Transfektion mit LNPs (Lipid-Nanopartikeln) konzipiert wurden. Angesichts mehrerer offensichtlicher Einschränkungen drängen wir darauf, dass unsere Arbeit unter forensischen Bedingungen repliziert wird und dass die Richtlinien überarbeitet werden, um eine hocheffiziente DNA-Transfektion und kumulative Dosierung zu berücksichtigen.“
7.6. Mögliche Integration in die DNA
7.6.1. Eine weitere Studie
Die folgende Studie untersuchte zunächst das „Vorhandensein von viralem Spike-Protein und vakzinalem Spike-Protein im Blutserum von Patienten mit Long-COVID-Syndrom“. Von den 81 analysierten Long-COVID-Patienten wurden bei 2 Patienten Fragmente des Impfstoff-Spike-Proteins gefunden, zwei Monate nach der Impfung. Die Studienverfasser stellten sich die Frage, ob eine dauerhafte Produktion von Spike-Proteinen auf DNA-Veränderungen beruhen könnte. Um diese Frage zu klären, führten sie eine ergänzende Studie durch.
In der ergänzenden Studie wurden die Blutproben von Patienten mit dem Long-COVID-Syndrom auf das „Vorhandensein von Spike-Proteinsequenzen des BNT162b2-Impfstoffs in der DNA“ überprüft. Dafür setzten sie spezifische DNA-Tests ein, um den genetischen Code des COVID-Impfstoffs in den Zellgenomen der Studienteilnehmer nachzuweisen. Die Ergebnisse deuteten auf das Vorhandensein von Spike-Protein-Sequenzen hin, die denen des Impfstoffs ähnelten.
In der Diskussion wurde darauf aufmerksam gemacht, dass die Ergebnisse auf eine mögliche Integration des Impfstoff-Spike-Proteins in die DNA der Patienten hinweisen könnten. Es wurden auch Verbindungen zu anderen Studien hergestellt, die ähnliche Ergebnisse zeigten. Die Studie weist jedoch auch auf ihre eigenen Grenzen hin, wie z.B. die Notwendigkeit weiterer Untersuchungen, um die Integration des Impfstoffs zu bestätigen und mögliche Fehlerquellen auszuschließen.
Insgesamt empfehlen die Autoren weitere Studien mit größeren Stichproben und Kontrollgruppen, um die Spezifität und Prävalenz der Impfstoffintegration bei Long-COVID-Patienten zu bewerten.
7.6.2. Kleinere DNA-Fragmente
Um die Patientensicherheit im Hinblick auf die DNA-Kontamination von Wirtszellen zu gewährleisten, hat die Food and Drug Administration (FDA) Branchenleitlinien für die Herstellung von Zell- und Gentherapien veröffentlicht:
„Da einige Zellsubstrate auch tumorigene genetische Sequenzen oder retrovirale Sequenzen beherbergen, die eine Infektion übertragen können, empfehlen wir Ihnen, Maßnahmen zu ergreifen, um die biologische Aktivität jeglicher Rest-DNA in Verbindung mit Ihrer Viruspräparation zu minimieren. Dies kann erreicht werden, indem die Größe der DNA unter die Größe eines funktionalen Gens reduziert wird und die Menge der Rest-DNA verringert wird. Wir empfehlen, die Menge an Rest-DNA bei kontinuierlichen nicht-tumorigenen Zellen auf weniger als 10 ng/Dosis und die DNA-Größe auf weniger als ca. 200 Basenpaare zu begrenzen.
Wenn Sie Zellen verwenden, die von Tumoren abstammen oder tumorigene Phänotypen oder andere Merkmale aufweisen, die Anlass zu besonderen Bedenken geben können, kann die Begrenzung bestimmter Rest-DNA-Mengen erforderlich sein, um die Produktsicherheit zu gewährleisten. …“
Auch wenn die FDA Richtlinien die Begrenzung von Rest-DNA in Impfstoffen vorschreibt, bedeutet dies nicht zwangsläufig, dass DNA-Fragmente kleiner als 200 bp (Basenpaare) keine Auswirkungen haben oder keine Rolle spielen können.
Selbst kleine DNA-Fragmente können theoretisch in das Genom eingebaut werden, wenn sie in die Zellen gelangen und durch die zellulären Mechanismen erkannt werden. Ob diese Fragmente eine funktionelle Rolle im Genom spielen können, hängt von verschiedenen Faktoren ab, einschließlich ihrer Sequenz, ihrer Position im Genom und der Verfügbarkeit von Reparaturmechanismen. Daher könnten selbst sehr kleine DNA-Fragmente, einschließlich solcher unter 200 Basenpaare, in das Genom integriert werden und biologische Effekte haben.
Kevin McKernan schreibt in einem Substack:
Einige Richtlinien legen den Fokus ausschließlich auf DNA-Fragmente, die größer als 200 Basenpaare sind. Die Annahme, dass Fragmente unter 200 Basenpaaren keine Relevanz haben, beruht jedoch auf einer fehlerhaften Logik. Diese Annahme geht davon aus, dass DNA in diesem Größenbereich nicht in der Lage ist, einen Open Reading Frame (ORF), also einen spezifischen Abschnitt in der DNA oder RNA, der Informationen für die Übersetzung in ein Protein enthält, zu kodieren. Sollte dies dennoch möglich sein, wird angenommen, dass das resultierende Peptid so kurz (66 Aminosäuren) wäre, dass es nicht von Bedeutung ist, oder dass die kurze DNA schnell abgebaut wird.
„Es gibt einige Gründe, warum dieses Argument fehlerhaft ist.
1) Die Vorschriften sahen nicht vor, dass sich diese DNA in Nuklease-resistenten Lipid-Nanopartikeln befindet, daher bedeutet eine kurze Länge keinen schnellen Abbau.
2) Auch kleine DNA-Fragmente können funktionsfähig sein. Der 72 Basenpaar lange SV40 Enhancer ist ein solches Element, von dem bekannt ist, dass es Transkriptionsfaktoren rekrutiert und die DNA in den Zellkern hinein bewegt. Die Arbeit von David Dean deckt dies ab.
3) Selbst kleine Fragmente, wenn sie integriert werden, können ein menschliches Gen aus dem Rahmen werfen und Probleme verursachen.“
Kevin McKernan hob bei einem Vortrag hervor, dass die Menge der DNA in Nanogramm nicht allein ausschlaggebend ist, sondern auch die Molarität, also die Anzahl der DNA-Moleküle in einer bestimmten Lösungsmenge (Dosis). „Ein 200-Basen-Fragment ist wie eine Schrotkugel, die in Ihr Genom eindringt und eine viel höhere Neigung hat, sich zu integrieren und Ihr Genom zu schädigen.“
7.6.3. Größere DNA-Fragmente
Das Vorhandensein von größeren DNA-Fragementen ist ein weiteres Thema, dass untersucht werden sollte. In einem Interview erläutert McKernan diese Sachlage:
„Sutton und sein Team haben gezeigt, dass Hybride, also Paare aus DNA und RNA, das Enzym DNase blockieren können, das für den Abbau von DNA verantwortlich ist. Solche Hybride könnten auch in mRNA-Impfstoffen vorhanden sein. Das N1-Methyl-Pseudouridin in der modifizierten RNA des Impfstoffs könnte dazu führen, dass diese mit der DNA interagiert und sich daran bindet. Die Sequenzierung der Plasmid-DNA war in der Region des Spike-Gens weniger effektiv, was darauf hindeutet, dass DNA:RNA-Hybride die Sequenzierungsreaktion stören können. Es ist wahrscheinlich, dass solche Hybride enzymatische Reaktionen blockieren, da die DNase normalerweise auf doppelsträngige DNA (also DNA:DNA-Hybride) abzielt. In meinem Substack habe ich dieses Problem ausführlich beschrieben.“
7.6.4. Dr. Phillip Buckhaults
In seiner Anhörung im Senat von South Carolina September 2023 sagte Buckaults:
„Wir machen das ständig im Labor. Wir nehmen DNA-Stücke und vermischen sie mit einem Lipidkomplex wie in dem Pfizer-Impfstoff. Wir gießen ihn auf Zellen, und ein großer Teil davon gelangt in die Zellen und ein großer Teil in die DNA dieser Zellen, und er wird zu einem festen Bestandteil der Zellen. Es ist nicht nur eine vorübergehende Sache, es ist in dieser Zelle und in all ihren Nachkommen von nun an und für immer. Deshalb bin ich sozusagen beunruhigt über diese DNA im Impfstoff, denn sie unterscheidet sich von der RNA, weil sie permanent sein kann. Das ist eine echte Gefahr für die Genomveränderung von langlebigen Zellen wie Stammzellen, und sie könnte theoretisch, das ist alles eine theoretische Sorge – aber auf der Grundlage der Molekularbiologie, eine anhaltende Autoimmunreaktion auf dieses Gewebe hervorrufen. Es ist auch ein sehr reales theoretisches Risiko für zukünftige Krebserkrankungen bei manchen Menschen, je nachdem, wo im Genom dieses fremde Stück DNA landet, kann es einen Tumorsuppressor unterbrechen oder ein Endogen aktivieren.“ …
„Die DNA ist ein langlebiger Informationsspeicher. Mit dem, womit Sie geboren wurden, werden Sie sterben und es an Ihre Kinder weitergeben. Die DNA hält Hunderttausende von Jahren. Und sie kann Generationen überdauern, wenn du sie an deine Kinder weitergibst. Veränderungen der DNA bleiben also bestehen. Die RNA ist von Natur aus vorübergehend, sie ist nicht von Dauer, und diese Eigenschaft der RNA war Teil des Verkaufsarguments für den Impfstoff.“ ….
„Das Pseudo-Uridin sollte dafür sorgen, dass die RNA etwas länger hält, aber es handelt sich immer noch um ein vorübergehendes Phänomen, das Stunden bis Tage dauert. Und wenn Proteine einmal hergestellt sind, halten sie auch nicht ewig, sondern nur Stunden bis Tage. Aber etwas, das seinen Weg in die DNA findet, hat das Potenzial, sehr lange zu halten, vielleicht ein Leben lang.“
„Meiner Meinung nach sollte jemand DNA-Proben von Stammzellen geimpfter Menschen sequenzieren und herausfinden, ob dieses theoretische Risiko eingetreten ist oder nicht.“
7.6.5. Dr. Jürgen O. Kirchner
Auf die Frage von The Epoch Times „Welches Risiko besteht aus Ihrer Sicht, wenn man fremde DNA gespritzt bekommt?“, antwortet Kirchner:
„Das ist eine ganz wichtige Frage. Zunächst mal, was macht die DNA? Wenn die DNA eingeschleust wird in die Zellen, dann kann sie in den Zellkern wandern und sich integrieren in die menschliche DNA und dort entweder Gene ausschalten, Gene einschalten – unter anderem auch Krebs-Gene einschalten. Und wenn das DNA-Stücke sind, die zu groß dafür sind, in den Zellkern zu kommen, es gibt auch sogenannte Bakterien-Plasmide, die ebenfalls in den Impfstoffen gefunden wurden, dann kann dieses Plasmid außerhalb des Zellkerns schon Aktivität zeigen, das heißt es wird abgelesen. Und das fatale ist, dass das Plasmid, was in den Impfstoffen gefunden wurde, das Spike-Gen beinhaltet und ein Gen für eine Antibiotikaresistenz. … Das muss wissenschaftlich untersucht werden.“
7.6.6. Vorläufige Hinweise auf die Integration von DNA
Auf dem International Crisis Summit-5 (ICS-5) vom 23.02.2024 in Washington DC hat Kevin McKernan neueste Untersuchungen zu einer möglichen DNA-Integration vorgestellt. Der ganze Beitrag ist unter diesem Link zu sehen.
Ab Minute 9:50 werden Ergebnisse der Untersuchungen mit Impfstoff behandelten OVCAR3- und MCF7-Zellen präsentiert.
OVCAR3-Zellen: Diese Zelllinie stammt von einem Ovarialkarzinom ab, das ein bösartiger Tumor der Eierstöcke ist. MCF7-Zellen: Diese Zelllinie stammt von einem Brustkrebs (Mammatumor) ab. Beide Zelllinien sind etablierte Modelle für die Untersuchung von Krebsbiologie, das Testen von Medikamenten und die Entwicklung von Therapien.
Kevin McKernan berichtete, dass die DNA in die so behandelten Zellkulturen nachgewiesen wurde. „Wir können die DNA dort sehen. Es ist eine sehr hohe Kopienzahl… Es handelt sich um eine etwa 3000-fache Abdeckung des gesamten Impfstoffgenoms.“
Mit einfachen Worten bedeutet das, dass die genetische Information des Impfstoffs in die Zellen gelangt ist. Die Forscher konnten die DNA im Inneren der Zellen sichtbar machen und feststellen, dass sie in sehr hoher Anzahl vorhanden war.
Die Angabe „etwa 3000-fache Abdeckung des gesamten Impfstoffgenoms“ bezieht sich darauf, wie oft die DNA-Sequenzen des Impfstoffs in den Zellen gefunden wurden. Eine „3000-fache Abdeckung“ bedeutet, dass die DNA-Sequenzen im Durchschnitt etwa 3000 Mal in den untersuchten Zellen vorhanden waren. Dies deutet darauf hin, dass eine große Menge der genetischen Information des Impfstoffs in die Zellen eingedrungen ist.
Kevin McKernan berichtete weiter: „Wir haben auch zwei Genomintegrationen gefunden. Diese sind noch nicht repliziert worden, wie alle unsere qPCR-Daten, es ist also noch sehr früh, aber wir haben Fusionen von DNA zwischen der Spike-Sequenz und Chromosom 12 und zwischen der Spike-Sequenz und Chromosom 9. Dies muss mit Long-Read-Sequenzierern bestätigt werden, mit denen wir das gesamte Integrationsereignis abdecken können… Aber wir sollten so etwas nicht sehen… Wir müssen also noch ein wenig mehr Validierung betreiben, aber es ist immer noch ein wenig besorgniserregend.“
Nähere Informationen dazu hier.
7.7. Weitere Aufrufe zu Untersuchungen
Auch der WDR berichtete am 18.01.2024 über die Thematik der DNA-Verunreinigungen in den mRNA-Impfstoffen:
Hier nochmal eine Zusammenfassung der Aussagen.
Dr. Thomas Voshaar, Lungenfacharzt am Klinikum Moers, beantwortete dem WDR folgende Fragen:
„Teilen Sie die Meinung, dass es Zeit wird, für eine Neubewertung dieser mRNA-Impfstoffe?“
„Ja, absolut. Es gibt ja zwei Dinge, die jetzt aufgefallen sind. Das eine ist, DNA darf herstellungsbedingt in ganz geringer Menge da drin sein. Das ist eine WHO Spezifikation. Warum das vernünftig ist und warum man das zulässt, dass muss man auch mal hinterfragen. Weil man sagt zwar, da geht kein relevantes Risiko von aus und deshalb bin ich im Moment auch beruhigt. Aber wir wissen das nicht. Es gibt dazu überhaupt gar keine Untersuchungen, wie wenig ungefährlich ist und ab wann es vielleicht gefährlich ist.“
„Aber warum nicht? Warum gibt es diese Untersuchungen nicht?“
„Ja, das ist wirklich eine Frage. Man hat natürlich diesen neuen Impfstoff mit dieser neuen Technologie unter relativ hohem Druck, zeitlichem Druck und das wird natürlich auch immer wieder positiv betont, entwickelt und deshalb ist vielleicht manches auf der Strecke geblieben. Aber nochmal, es war ein Konsens, auch bei der WHO, dass man bedingt durch den Herstellungsprozess es akzeptiert, das bestimmte DNA-Mengen da drin sein dürfen. Aber es bleibt dabei, es ist eine Verunreinigung. Lieber würde man das nicht da drin haben. Und jetzt kommt noch hinzu, dass einige Labore wohl festgestellt haben, dass in bestimmten Chargen auch mehr drin ist und größere DNA-Stücke als eigentlich zulässig wäre.“
„Das heißt, was fordern Sie konkret?“
„Man muss auf jeden Fall das Paul-Ehrlich-Institut (PEI) nochmal in die Pflicht nehmen. Das Paul-Ehrlich-Institut beschränkt sich überwiegend darauf, die Protokolle der Überprüfung des Herstellers anzugucken. Und man kann das nicht dem Hersteller überlassen, sondern man muss andere Labore beauftragen und muss das nochmal komplett hinterfragen.“
„Wir haben dazu (zum Thema DNA-Verunreinigungen) die Firma BioNTech um Stellungnahme gebeten, aber KEINE Antworten erhalten.“
7.8. Kurze Zusammenfassung
- Studien zeigen, dass das Vorhandensein von Plasmid-DNA, die aus dem Produktionsprozess von mRNA-Impfstoffen stammt, unvermeidbar ist.
- Das Vorhandensein von Plasmid-DNA im Impfstoff ist nicht unbedenklich. Ob diese DNA-Reste ein Sicherheitsrisiko darstellen, muss gezielt untersucht werden.
- Laboruntersuchungen belegen die Möglichkeit der Integration linearisierter DNA-Sequenzen in Säugetierzellen sowie die potenziell kritischen Auswirkungen von dsDNA auf die Funktionalität menschlicher Endothelzellen.
- Studien weisen darauf hin, dass Rest-DNA möglicherweise in den Lipid-Nanopartikeln eingeschlossen ist. Dies könnte es ihr ermöglichen, die Zellmembran zu durchqueren und in die Zellen einzudringen.
- Das Vorhandensein von SV40-Komponenten wie Promoter und Enhancer birgt die potenzielle Gefahr, DNA-Fragmente in den Zellkern zu transportieren und könnte auch die ordnungsgemäße Funktionalität des p53-Tumorsuppressor-Gens beeinträchtigen.
- Weltweit weisen immer mehr Wissenschaftler auf diese möglichen Gefahren hin und betonen die Notwendigkeit zusätzlicher sorgfältiger Untersuchungen.
8. Epilog
Die mRNA-Technologie ist zweifellos ein beeindruckendes Zeugnis menschlicher Intelligenz. Gleichzeitig verdeutlicht sie die extreme Komplexität nicht nur der einzelnen technologischen Produktionsschritte, sondern auch der Wechselwirkungen dieser Präparate mit dem menschlichen Körper. Wie bei jeder hochkomplexen Technologieplattform weist sie Vor- und Nachteile auf.
Die Debatte über DNA-Verunreinigungen im mRNA-Impfstoff, die in dieser Abhandlung zusammengefasst wurde, unterstreicht die dringende Notwendigkeit weiterer Untersuchungen im Hinblick auf die Zuverlässigkeit des technologischen Produktionsprozesses, der Standardisierung der Messverfahren und Grenzwerte für die Qualitätssicherung sowie zur Bewertung der Langzeitwirkungen solcher Therapeutika.
Bei Produkten, die für den zukünftigen weltweiten Einsatz konzipiert sind, sollten Sorgfalt und Gründlichkeit im Vordergrund stehen.
Und es bleiben Fragen:
Warum schweigen die Hersteller von mRNA-Impfstoffen zu diesem Thema?
Warum werden die Behörden nicht aktiv, um die Ergebnisse und Bedenken unabhängiger Labore und Wissenschaftler durch eigene, herstellerunabhängige experimentelle Untersuchungen zu widerlegen bzw. um mehr Klarheit in der Diskussion zu erlangen?
Erinnern wir uns an die eindringlichen Worte der ehemaligen Bundeskanzlerin Frau Merkel zu Beginn der Corona-Pandemie:
„Wir sind eine Gemeinschaft, in der jedes Leben und jeder Mensch zählt.“
Danksagung
Ich möchte meinen aufrichtigen Dank an die zahlreichen Wissenschaftler, Forscher und engagierten Personen aussprechen, die durch ihre Studien, Artikel und Vorträge kontinuierlich dazu beitragen, verschiedene Aspekte dieses komplexen Themas zu erforschen und der Öffentlichkeit näherzubringen. Ihre Hingabe und ihre Bemühungen sind von unschätzbarem Wert und tragen maßgeblich zum Fortschritt unseres Verständnisses bei.
Quellen (Stand vom 02.03.2024)
